Note: Descriptions are shown in the official language in which they were submitted.
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
1
BENZOXAZOCINES AND THEIR THERAPEUTIC USE AS MONOAMINE REUPTAKE INHIBITORS
Field of the Invention
This invention relates to novel benzoxazocine compounds which inhibit
monoamine reuptake. In particular, compounds of the present invention exhibit
activity
as analgesic agents and also as anti-emetics but also may find utility in a
range of other
therapeutic indications.
Background of the Invention
Nefopam, i.e. 5-methyl-1-phenyl-3,4,5,6-tetrahydro-1 H-2,5-benzoxazocine
hydrochloride, is a centrally acting non-narcotic analgesic.
W003/092689 discloses that the single enantiomers of nefopam are useful for
the treatment of pain and emesis.
W02004/056788 discloses derivatives of nefopam. Its publication date is later
than the priority dates now claimed.
Summary of the Invention
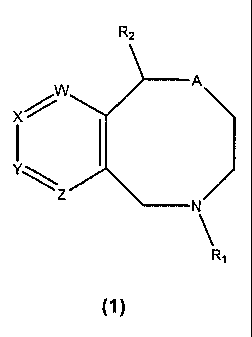
Novel compounds according to this invention are of general formula (1):
Ra
W
X
Y\
\'Z
R~
(1)
wherein R~ is H, C~-C6 alkyl optionally substituted with F or C3-C6 cycloalkyl
or C~-C4
alkenyl;
A is O,' CHZ or S(O)S where n is 0-2;
one of W, X, Y, and Z is N, CH or CR3 and the others are CH;
R2 is C~-Cs heteroaryl, C5-Coo cycloalkyl or cycloalkenyl optionally
containing one
or more heteroatoms selected from O, N and S(O)~ where n is 0-2, and
optionally
substituted with R3; or a phenyl group optionally substituted in one or more
positions with
one or more substituents independently selected from halogen, CN, CF3, C~-Cs
alkyl and
ORS, or the phenyl group is fused to a five or six membered ring which may be
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
2
carbocyclic, heterocyclic (containing 1-2 heteroatoms selected from O, N and
S),
aromatic or heteroaromatic (containing 1-2 heteroatoms selected from O and N);
R3 is selected from halogen; CF3; CN; ORS; S02N(RS)2; CORS; C02R5; CON(R5)2;
NR~COR4; NR~S02R4; NR~C02R4; NR~CON(RS)2; OCR-C6 alkyl substituted with R3; C~-
C6
alkyl optionally substituted with unsubstituted R3; C3-C6 cycloalkyl
optionally substituted
with unsubstituted R3; CZ-C6 alkenyl optionally substituted with unsubstituted
Ra; C2-C6
alkynyl optionally substituted with unsubstituted R3; aryl optionally
substituted with
unsubstituted R3; and five or six membered aromatic heterocycles containing 1-
4
heteroatoms selected from N and O;
R4 is C~-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, C3-C6 cycloalkyl, aryl and
heteroaryl; and
R5 is H, C~-Cs alkyl, C2-C6 alkenyl, CZ-Cs alkynyl, C3-C6 cycloalkyl, aryl or
heteroaryl and is the same as or different to another R5;
or a pharmaceutically acceptable salt thereof.
Compounds disclosed in WO2004/056788 may be excluded, e.g. those of the
general formula:
0
x~
Y\
N
R~
wherein
R~ is H, C,-Cs alkyl, optionally substituted with F or C3-Cs cycloalkyl or C2-
C6
alkenyl;
either R2 and R3 are the same or different and are H, a halogen, CN, CF3, C~-
Cs
alkyl or ORS, or RZ and R3 form a five or six membered ring which may be
carbocyclic,
heterocyclic (containing 1-2 heteroatoms taken from O, N or S), aromatic or
heteroaromatic (containing 1-2 heteroatoms taken from O and N) ;
one of W, X, Y and Z is N, or CR4 and the others are each CH;
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
3
R4 is a halogen atom, CF3, CN, ORS, SOzN(Rs)z, CORs, COZRs, CON(Rs)z,
NR~CORS, NR~S02R5, NR~COZRS, NR~CON(Rs)z, OCR-Cs alkyl optionally substituted
with
R4, C~-Cs alkyl optionally substituted with R4, C3-Cs cycloalkyl optionally
substituted with
R4, Cz-Cs alkenyl optionally substituted with R4, CZ Cs alkynyl optionally
substituted with
R4, aryl optionally substituted with R4, or a five or six membered aromatic
heterocycle
containing 1-4 heteroatoms selected from N and O, linked either through carbon
or
nitrogen;
R5 is C~-Cs alkyl, Cz-Cs alkenyl, Cz-Cs alkynyl, C3-Cs cycloalkyl, aryl or
heteroaryl;
each Rs (which may be the same or different) is H, C~-Cs alkyl, Cz-Cs alkenyl,
Cz
Cs alkynyl, C3-Cs cycloalkyl, aryl or heteroaryl; and
R~ is aryl or heteroaryl;
or a pharmaceutically acceptable salt thereof.
Compounds of the invention have utility as therapeutic agents. Compositions
containing them and their therapeutic uses are further aspects of the
invention. Further,
compounds of formula (1 ) wherein R3 is a halogen atom such as Br are useful
as
intermediates.
Description of Preferred Embodiments
It will be appreciated that compounds according to the invention contain an
asymmetrically substituted carbon atom. The presence of this asymmetric centre
in a
compound of formula (1 ) can give rise to stereoisomers, and in each case the
invention
is to be understood to extend to all such stereoisomers, including enantiomers
and
diastereomers, and mixtures including racemic and non-racemic mixtures
thereof.
As used in this specification, the term "C~-Cs alkyl" refers to a straight or
branched
chain alkyl moiety having from one to six carbon atoms, including, for
example, methyl,
ethyl, propyl, isopropyl, butyl, tent butyl, pentyl, hexyl and the like.
_ The term "C5-Cs heteroaryl" refers to an aryl system of five or six atoms,
of which
at least one atom is selected from O, N and S. This term includes, for
example, pyridine,
tetrahydropyran etc.
The term "Cz-Cs alkenyl" refers to a straight or branched chain alkyl moiety
having two to six carbon atoms and having in addition one double bond, of
either E or ~
stereochemistry where applicable. This term includes, for example, vinyl, 1-
propenyl, l-
and 2-butenyl, 2-methyl-2-propenyl etc.
The term "Cz-Cs alkynyl" refers to a straight or branched chain alkyl moiety
having
two to six carbon atoms and having in addition one triple bond. This term
includes, for
example, ethynyl, 1-propargyl, 1- and 2-butynyl etc.
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
4
The term "Ca-Cs cycloalkyl" refers to a saturated alicyclic moiety having from
three to six carbon atoms and includes, for example, cyclopropyl, cyclobutyl,
cyclopentyl,
cyclohexyl and the like.
The term "Cs-C~o cycloalkenyl" refers to an unsaturated alicyclic moiety
having
from five to ten carbon atoms and at least one double bond and includes, for
example,
phenyl and the like.
The term "aryl" means an optionally substituted phenyl or naphthyl group.
The term "carbocyclic" refers to a saturated alicyclic moiety having five or
six
carbon atoms and includes, for example, benzofused cyclopentyl and cyclohexyl
and the
like.
The term "heterocyclic" refers to a saturated heterocyclic moiety having from
five
or six atoms but containing one or more heteroatom selected from N, O and S,
and
includes, for example, benzofused pyrrolidinyl, tetrahydrofuranyl,
piperidinyl, dioxalane
and the like.
The term "heteroaromatic" refers to aromatic ring systems of five or six atoms
of
which at least one atom is selected from O, N and S, and includes, for
example, pyrrolyl,
pyridinyl, diazolyl, diazinyl, triazolyl, triazinyl, tetrazolyl, furanyl,
oxazolyl, isoxazolyl or
oxadiazolyl as well as benzofused furanyl, thiophenyl, pyridyl, indolyl,
pyridazinyl,
piperazinyl, pyrimidinyl and the like, e.g. benzofuranyl, quinolinyl,
isoquinolinyl or
quinazolinyl. Such rings can be linked either through carbon or nitrogen.
The term "halogen" means fluorine, chlorine, bromine or iodine.
Compounds of the general formula (1 ) may be prepared by any suitable method
known in the art andlor by the processes described below. It will be
appreciated that
where a particular stereoisomer of formula (1) is required, the synthetic
processes
described herein may be used with the appropriate homochiral starting material
and/or
isomers maybe resolved from mixtures using conventional separation techniques
(eg.
H PLC).
In the description and formulae below, variables such as R~, R2, R3, R4, R5,
A, W,
X, Y and Z are as defined above, except where otherwise indicated. It will be
appreciated that functional groups, such as amino, hydroxyl .or carboxyl
groups, present
in the various compounds described below, and which it is desired to retain,
may need to
be in protected form before any reaction is initiated. In such instances,
removal of the
protecting group may be the final step in a particular reaction. Suitable
protecting groups
for such functionality will be apparent to those skilled in the art. For
specific details, see
"Protective Groups in Organic Synthesis", Wiley Interscience, T W Greene, PGM
Wuts.
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
A process for preparing compounds of general formula (1), where W, X, Y or Z
is
N or C-Br and A is O or S(O)S, comprises cyclisation with acid (for instance
with p-
toluenesulphonic acid) of a diol (2) which can in turn be obtained by
reduction of ketone
(3) with a suitable reducing agent.
5
RZ R
AH AH
X
Y
R~ O R~
~2) ~3)
Reduction of a keto amide of general formula (3) can be carried out with
reagents
well known to those familiar in the art of synthetic organic chemistry. An
example of a
highly reactive reducing agent is lithium aluminium hydride, although reagents
based on
borane (e.g. borane.tetrahydrofuran complex) or modified sodium borohydride
reduction
(e.g. with a nickel or cobalt salt enhancer) may be equally effective.
Equally, reduction of the ketone in (3), for example with sodium borohydride,
followed by acid cyclisation, for example with p-toluenesulphonic acid, then
ultimate
reduction of the amide group, for example with borane, also leads to compounds
of
general formula (1).
Ketones (3) can be prepared by condensation of a carboxylic acid (4) or an
active
derivative thereof, with an amine (5). Active derivatives of acids of formula
(4) include,
for example, acid anhydrides or acid halides, such as acid chlorides.
R~
N
x R ~ AH
Y\ . H
\z
~4) ~5)
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
6
The coupling reaction may be performed using standard conditions for amidation
reactions of this type. Thus, the reaction may be achieved in a solvent, for
example an
inert organic solvent such as an ether, e.g. a cyclic ether such as
tetrahydrofuran, an
amide, e.g. a substituted amide such as dimethylformamide, or a halogenated
hydrocarbon such as dichloromethane at low temperature, e.g. -30°C to
ambient
temperature, such as -20°C to 0°C, optionally in the presence of
as base, e.g. an organic
base such as an amine, e.g. triethylamine or a cyclic amine such as N
methylmorpholine. Where an acid (4) is used directly, the reaction may
additionally be
performed in the presence of a condensing agent, for example a diimide such as
N,N'-
dicyclohexylcarbodiimide, advantageously in the presence of a triazole such as
1-
hydroxybenzotriazole. Alternatively, the acid may be reacted with a
chloroformate, for
example ethyl chloroformate, prior to reaction with the amine (5).
Acids (4) may be prepared by Friedel-Crafts acylation of RZ with an anhydride
(7).
This reaction is carried out in an inert solvent (such as dichloromethane) in
the presence
of a Lewis acid catalyst (such as aluminium trichloride).
O
.W
X ~ \
II 0
Y~ ~
z
O
(7)
It is well recognised by those skilled in the art that such reactions may
provide
mixtures of products and in turn that these mixtures can often be separated by
flash
column chromatography. For example, where Y = C-Br, W = X = Z = CH and R2 is
phenyl, Friedel-Crafts acylation under aluminium trichloride catalysis
provides two
isomeric bromides (4a) and (4b). These can be readily separated by column
chromatography and independently progressed to compounds of general formula
(1),
wherein X or Y is C-Br, by the route described above.
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
7
Br
)H H
(4a) (4b)
Compounds of general formula (1) where one of W, X, Y and Z is C-hal such as
C-Br represent flexible intermediates that may be used for the preparation of
other
compounds of general formula (1 ). For instance, such compounds can be
smoothly
converted into the corresponding nitrite (R3 = CN) either by reaction with
cuprous
cyanide in a dipolar aprotic solvent such as N methylpyrrolidinone (NMP) or
under
palladium-catalysed conditions.
The nitrite can be readily converted, by hydrolysis, into the primary amides
(R3= CONHz), esters and the corresponding carboxylic acids (COZRS) or into the
corresponding tetrazoles, by treatment with a suitable azide donor such as
sodium azide
or trimethylsilylazide.
In addition, compounds of general formula (1) where one of W, X, Y and Z is C-
hal such as C-Br can be lithiated with n-, sec or tent-butyllithium in an
inert organic
solvent such as an ether, e.g. a cyclic ether such as tetrahydrofuran, at very
low
temperature, e.g. -78 °C. Treatment with either a carbon (e.g. carbon
dioxide, N,N-
dimethylformamide or paraformaldehyde), sulphur (e.g. SOzCIz, followed by
amidation,
such as with ammonia) or nitrogen (diphenylphosphoryl azide, followed by
reduction,
such as with REDAL) provides access, by subsequent derivatisation, to
derivatives
where R3 is COZRs, CON(R5), CHzORS, SOZN(R4)z, NR~COR4, NR~C02Rs or
NR~CON(R5)z.
In addition, compounds of general formula (1) where one or W, X, Y and Z is C-
hat such as C-Br can undergo palladium-catalysed coupling reactions with
carbon-based
coupling partners. Thus, such compounds can be coupled to alkenes of the
general type
CHz=CHR3 under Heck conditions, to alkynes of the general type CH=CHR3 under
Sonogoshira conditions, or to metalloheterocycles, e.g. where the metal is
tin, under
Stille coupling conditions. This gives access to compounds where R3 is
optionally
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
8
substituted CZ-C6 alkenyl or C2-C6 alkynyl, or a five-membered aromatic
heterocycle
containing 1-4 heteroatoms selected from N (such as in pyrrole, diazoles,
triazoles or
tetrazoles) and O (such as in furan, oxazoles or oxadiazoles). Such coupling
reactions
ensure that chains and rings are linked through carbon.
In addition to the examples described above, additional compounds of formula
(1)
may be prepared by interconversion of other compounds of formula (1). Thus,
for
example, a compound wherein R3 is alkyl may be prepared by hydrogenation
(using
palladium on carbon in a suitable solvent, such as an alcohol, e.g. ethanol)
of a
corresponding compound wherein R3 is alkenyl.
Any mixtures of final products or intermediates obtained can be separated on
the
basis of the physico-chemical differences of the constituents, in known
manner, into the
pure final products or intermediates, for example by chromatography,
distillation,
fractional crystallization, or by formation of a salt if appropriate or
possible under the
circumstances.
The compounds according to the invention exhibit in vitro inhibiting
activities with
respect to monoamine (i.e. noradrenaline, serotonin and dopamine) reuptake.
The
activity and selectivity of the compounds may be determined by use of an
appropriate
monoamine reuptake assay.
This invention also relates to a method of treatment for patients (including
man
andlor mammalian animals raised in the dairy, meat or fur industries or as
pets) suffering
from disorders or diseases which can be attributed to monoamine reuptake as
previously
described, and more specifically, a method of treatment involving the
administration of
the monoamine reuptake inhibitor of formula (1 ) as the active constituents.
Accordingly, the compounds of formula (1 ) can be used in the treatment of
pain
and emesis. Pain and related conditions that can be treated include syndromes
characterised by chronic pain and fatigue, fibromyalgia, chronic fatigue
syndrome,
complex regional pain syndrome, irritable bowel syndrome, myofacial pain and
atypical
chest pain. The compounds may also find utility in a range of other
therapeutic
indications such as depression, post-traumatic stress disorders, attention-
deficit
disorders, obsessive-compulsive disorders, pre-menstrual syndrome, substance
abuse
and sexual dysfunction; a method of management (by which is meant treatment or
prophylaxis) of disease or conditions mediated by monoamine reuptake in
mammals, in
particular in humans, which method comprises administering to the mammal an
effective,
amount of a compound of formula (1 ) above, or a pharmaceutically acceptable
salt
thereof; and a compound of formula (1 ) for use in human or veterinary
medicine,
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
9
particularly in the management (by which is meant treatment or prophylaxis) of
diseases
or conditions mediated by monoamine reuptake; and the use of a compound of
formula
(1 ) in the preparation of an agent for the management (by which is meant
treatment or
prophylaxis) of diseases or conditions mediated by monoamine reuptake.
The disease or conditions referred to above include pain, emesis, depression,
post-traumatic stress disorders, attention-deficit disorders, obsessive-
compulsive
disorders, pre-menstrual syndrome, substance abuse and sexual dysfunction.
Compounds . of formula (1 ) may be administered orally, topically, buccally,
ocularly, rectally, vaginally, parenterally, intra-nasally, sublingually or by
inhalation spray,
e.g. in dosage unit formulations containing non-toxic pharmaceutically
acceptable
carriers, adjuvants and vehicles. The term parenteral as used herein includes
subcutaneous injections, intravenous, intramuscular, intrasternal injection or
infusion
techniques. In addition to the treatment of warm-blooded animals such as mice,
rats,
horses, cattle, sheep, dogs, cats etc, the compounds of the invention are
effective in the
treatment of humans.
The pharmaceutical composition containing the active ingredient may be in a
form suitable for oral use, for example, as tablets, troches, lozenges,
aqueous or oily
suspensions, dispersible powders or granules, emulsions, hard or soft
capsules, or
syrups or elixirs. The composition may be in immediate or controlled release
form.
Compositions intended for oral use may be prepared according to any method
known to the art for the manufacture of pharmaceutical compositions and such
compositions' may contain one or more agents selected from the group
consisting of
sweetening agents, flavouring agents, colouring agents and preserving agents
in order to
provide pharmaceutically elegant and palatable preparations. Tablets contain
the active
ingredient in admixture with non-toxic pharmaceutically acceptable excipients
which are
suitable for the manufacture of tablets. These excipients may be for example,
inert
diluents, such as calcium carbonate, sodium carbonate, lactose, calcium
phosphate or
sodium phosphate; granulating and disintegrating agents, for example corn
starch, or
alginic acid; binding agents, for example starch, gelatin or acacia, and
lubricating agents,
for example magnesium stearate, stearic acid or talc. The tablets may be
uncoated or
they may be coated by known techniques to delay disintegration and absorption
in the
gastrointestinal tract and thereby provide a sustained action over a longer
period. For
example, a time delay material such as glyceryl monostearate or glyeryl
distearate may
be employed. They may also be coated by the techniques described in US4256108,
US4166452 and US4265874 to form osmotic therapeutic tablets for control
release.
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
Formulations for oral use may also be presented as hard gelatin capsules where
in the active ingredient is mixed with an inert solid diluent, for example
calcium
carbonate, calcium phosphate or kaolin, or as soft gelatin capsules wherein
the active
ingredient is mixed with water or an oil medium, for example peanut oil,
liquid paraffin or
5 olive oil. Aqueous suspensions contain the active materials in admixture
with excipients
suitable for the manufacture of aqueous suspensions. Such excipients are
suspending
agents, for example sodium carboxymethylcellulose, methylcellulose,
hydroxypropylmethylcellulose, sodium alginate, polyvinylpyrrolidone, gum
tragacanth
and gum acacia; dispersing or wetting agents may be a naturally occurring
phosphatide,
10 for example lecithin, or condensation products of an alkylene oxide with
fatty acids, for
example pofyoxyethyfene stearate, or condensation products of ethylene oxide
with long
chain aliphatic alcohols, for example heptadecaethyleneoxycetanol, or
condensation
products of ethylene oxide with partial esters dervied from fatty acids and a
hexitol such
a polyoxyethylene with partial esters derived from fatty acids and hexitol
anhydrides, for
example polyoxyethylene sorbitan monooleate. The aqueous suspensions may also
contain one or more preservatives, for example ethyl or n-propyl p-
hydroxybenzoate, one
or more colouring agents, one or more flavouring agents, and one or more
sweetening
agents, such as sucrose or saccharin.
Oily suspensions may be formulated by suspending the active ingredient in a
vegetable oil, for example arachis oil, olive oil, sesame oil or coconut oil,
or in a mineral
oil such as liquid paraffin. The oily suspensions may contain a thickening
agent, for
example beeswax, hard paraffin or cetyl alcohol. Sweetening agents such as
those set
forth above, and flavouring agents may be added to provide a palatable oral
preparation.
These compositions may be preserved by the addition of an anti-oxidant such as
ascorbic acid.
Dispersible powders and granules suitable for preparation of an aqueous
suspension by the addition of water provide the active ingredient in admixture
with a
dispersing or wetting agent, suspending agent and one or more preservatives.
Suitable
dispersing or wetting agents and suspending agents are exemplified, for
example
sweetening, flavouring and colouring agents, may also be present.
The pharmaceutical compositions of the invention may also be in the form of
oil-
in-water emulsions. The oily phase may be a vegetable oil, for example olive
oil or
arachis oil, or a mineral oil, for example liquid paraffin or mixtures of
these. Suitable
emulsifying agents may be naturally-occuring gums, for example gum acacia or,
gum
tragacanth, naturally occurring phosphatides, for example soya bean, lecithin,
and esters
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
11
or partial esters derived from fatty acids and hexitol anhydrides, for example
sorbitan
monooleate and condensation products of the said partial esters with ethylene
oxide, for
example polyoxyethylene sorbitan monooleate. The emulsions may also contain
sweetening and flavouring agents.
Syrups and elixirs may be formulated with sweetening agents, for example
gycerol, propylene glycol, sorbitol or sucrose. Such formulations may also
contain a
demulcent, a preservative and flavouring and colouring agents. The
pharmaceutical
compositions may be in the form of a sterile injectable aqueous or oleagenous
suspension. This suspension may be formulated according to the known art using
those
suitable dispersing or wetting agents and suspending agents which have been
mentioned above. The sterile injectable preparation may also be in a sterile
injectable
solution or suspension in a non-toxic parenterally-acceptable diluent or
solvent, for
example as a solution in 1,3-butanediol. Among the acceptable vehicles and
solvents
that may be employed are water, Ringer's solution and isotonic sodium chloride
solution.
In addition, sterile, fixed oils are conventionally employed as a solvent or
suspending
medium. For this purpose any bland fixed oil may be employed including
synthetic
mono- or diglycerides. In addition, fatty acids such as oleic acid find use in
the
preparation of injectables.
The compounds of formula (1) may also be administered in the form of
suppositories for rectal administration of the drug. These compositions can be
prepared
by mixing the drug with a suitable non-irritating excipient which is solid at
ordinary
temperatures but liquid at the rectal temperature and will therefore melt in
the rectum to
release the drug. Such materials are cocoa butter and polyethylene glycols.
For topical use, creams, ointments, jellies, solutions or suspensions, etc
containing the compounds of Formula (1) are employed. For purposes of this
specification, topical application includes mouth washes and gargles.
Dosage levels of the order of from about 0.05 mg to about 140 mg per kilogram
of body weight per day are useful in the treatment of the above-indicated
conditions
(about 2.5 mg to about 7 g per patient per day). For example, emesis may be
effectively
treated by the administration of from about 0.01 to 50 mg of the compound per
kilogram
of body weight per day (about 0.5 mg to about 3.5 g per patient per day).
The amount of active ingredient that may be combined with the carrier
materials
to produce a single dosage form will vary depending upon the host treated and
the
particular mode of administration. For example, a formulation intended for the
oral
administration of humans may vary from about 5 to about 95 percent of the
total
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
12
composition. Dosage unit forms will generally contain between from about 1 mg
to about
500 mg of an active ingredient.
It will be understood, however, that the specific dose level for any
particular
patient will depend upon a variety of factors including the activity -of the
specific
compound employed, the age, body weight, general health, sex, diet time of
administration, route of administration, rate of excretion, drug combination
and the
severity of the particular disease undergoing therapy.
The following Examples illustrate the invention. They include compounds which
are in the following Table.
Example- R1 R2 W,X,Y,Z
1 benzyl phenyl W=X=Y=Z=CH
13 methyl (3-tosyloxy)phenylW=X=Z=CH
Y=C-Br
20 methyl (2-methyl-3- W=X=Z=CH
methoxy) henyl Y=C-Br
21 methyl (2-methyl-3- W=X=Z=CH
methoxy)phenyl Y=C-CN
22 methyl (2-methyl-3- W=X=Z=CH
methoxy)phenyl Y=C-cyclopropyl
44 methyl 2-methylphenyl W=Y=Z=CH
X=C-Br
45 methyl 2-methylphenyl W=Y=Z=CH
X=C-CN
46 methyl 2-methylphenyl W=Y=Z=CH
X=C-c clopropyl
47 methyl 2-methylphenyl W=X=Z=CH
Y=C-Br
48 methyl 2-methylphenyl W=X=Z=CH
Y=C-C N
49 methyl 2-methylphenyl W=X=Z=CH
Y=C-cyclopropyl
50 methyl 3-pyridyl W=Y=Z=CH
X=C-Br
51 methyl 3-pyridyl W=X=Z=CH
Y=C-Br
52 methyl 3-pyridyl W=X=Z=CH
Y=C-CN
All 'H NMR recorded on a Bruker AC250. All chemical shifts (8) have been
rounded to 2 decimal places, and coupling constants (,~ are measured in Hz and
to the
nearest 0.1 decimal place.
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
13
Example 1
5-Benzyl-1-phenyl-1,3,4,6-tetrahydro-5H-benz[fj-2,5-oxazocine
To a stirring solution of desmethylnefopam (96 mg, 0.41 mmol) in DMF (3 ml)
were added sodium hydride (15 mg, 0.49 mmol) and then benzyl bromide (0.7 ml,
0.61
mmol). The resulting blue solution was heated at 65°C for 2 hours. The
reaction was
quenched by the addition of water and the organics were extracted into
dichloromethane,
dried (MgS04), filtered and concentrated in vacuo to provide the crude
material.
Purification by silica gel column chromatography (1-~2%MeOH / DCM) furnished
the
target product (45 mg, 34%) as a brown oil.
8H (CDCI3; 250MHz) 2.74-2.80 (1 H, m, one of CH ), 2.96-3.00 (1 H, m, one of
CH ), 3.88-
4.07 (4H, m, four of Cue), 4.27-4.36 (1 H, m, one of CHz), 4.92 (1 H, d,
J12.7, one of
ArCH NRz), 5.82 (1 H, s, CHOR), 7.05-7.07 (1 H, m, aromatic ~, 7.26-7.42 (11
H,
aromatic H~, 7.56-7.60 (2H, m, aromatic
Example 2
5-Allyl-1-phenyl-1,3,4,6-tetrahydro-5H-benz[f]-2,5-oxazocine
Sodium hydride (24 mg, 1.01 mmol) was added to a solution of
desmethylnefopam (200 mg, 0.84 mmol) at RT. The resulting mixture was stirred
for 2
hours before allyl bromide (0.12 ml, 1.33 mmol) was added dropwise. The blue
solution
was heated at 65°C for a further two hours, then quenched with water.
The aqueous
mixture was extracted with dichloromethane and the combined extracts dried
(MgS04),
filtered and concentrated in vacuo to provide the target product (66 mg, 28%).
8H (CDCI3; 250MHz) 2.90-3.13 (2H, m, two of CH ), 3.64 (2H, d, J6.1, two of CH
), 4.01
(1 H, bd, J13.6, one of CHz), 4.26 (1 H, d, J12.1, one of ArCHzNR2), 4.38-4.49
(1 H, m, one
of Cue), 5.01 (1 H, d, J12.1, one of ArC,H~NR2), 5.42-5.49 (2H, m, two
terminal alkenic ~,
5.77 (1 H, s, CHOR), 6.27-6.37 (1 H, m, alkenic ~, 7.11-7.39 (8H, aromatic Ha,
7.51 (1 H,
bs, aromatic ~.
Example 3
5-Cyclopropyl-1-phenyl-1,3,4, 6-tetrahydro-5H-benz[f]-2,5-oxazocine
Sodium cyanoborohydride (106 mg, 1.69 mmol) was added to a stirring
suspension of desmethylnefopam (100 mg, 0.42 mmol), acetic acid (0.24 ml,
4.22 mmol), (1-ethoxycyclopropyloxy)trimethylsilane (0.51 mL, 2.53 mmol) and
4A
molecular sieves in methanol (1.5 ml). The resulting brown solution was heated
at reflux
temperature overnight. The reaction mixture was quenched by the addition of
sodium
bicarbonate solution and extracted into dichloromethane. The combined organic
phases
were dried (MgS04), filtered and concentrated in vacuo. The crude material was
purified
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
14
by silica gel column chromatography (DCM~2%MeOH/DCM) to provide the target
product (84 mg, 68%).
8H (CDCI3; 250MHz) 0.56 (4H, bs, cyclopropyl CHzCHz), 1.90-1.94 (1 H, m,
cyclpropyl
Cue, 2.88 (1 H, ddd, J14.3, 6.1, 2.7, one of C~CH ), 3.00 (1 H, ddd, J14.3,
7.3, 2.1, one
of CHzCH ), 3.87 (1H, ddd, J12.5, 6.1, 2.1, one of CH CHz), 3.96 (1H, d,
J12.8, one of
ArCHzNR2), 4.15 (1 H, ddd, J12.5, 7.3, 2.7, one of CH CH ), 4.86 (1 H, d, one
of
ArCHzNRz), 5.84 (1 H, s, CHOR), 6.99-7.00 (1 H, m, aromatic ~, 7.18-7.30 (8H,
aromatic
Example 4
5-Propyl-1-phenyl-1,3,4,6-tetrahydro-5H-benz[f]-2,5-oxazocine
5-Allyl-1-phenyl-1,3,4,6-tetrahydro-5H-bent[fj-2,5-oxazocine (66 mg, 0.24
mmol)
was dissolved in THF (4 ml) and 10% PdIC (2 micro-spatulas) added. The
reaction
vessel was shaken under a hydrogen atmosphere for 3 hours. After this time,
the
mixture was filtered and the filtrate concentrated in vacuo. Purifcation of
the crude
mixture by silica gel column chromatography (DCM-~5%MeOH/DCM) furnished the
target product as a pale yellow oil (42 mg, 62%).
SH (CDCI3; 250MHz)1.03 (3H, t, J7.3, CH3), 1.99-2.10 (2H, m, two of propyl
CH~CH2),
2.96-3.22 (4H, m, two of propyl CH CHz and two of ring CHzCHz), 4.02-4.10 (1
H, m,
ArCHzNR2), 4.37-4.51 (2H, m, two of C,H~CHZ) 5.03-5.31 (1H, bs, one of ArCH
NRZ),
5.77 (1 H, CHOR), 7.15-7.18 (3H, m, aromatic ~, 7.27-7.40 (5H, m, aromatic ~,
7.63
(1 H, bs, aromatic H).
Example 5
5-Methylcyclopropyl-1-phenyl-1,3,4,6-tetrahydro-5H-bent[f]-2,5-oxazocine
Desmethylnefopam (110 mg, 0.43 mmol) was dissolved in anhydrous DMF (2 ml)
under a nitrogen atmosphere and sodium hydride (as a 60% suspension in mineral
oil,
21 mg, 0.52 mmol) was added. The resulting mixture was heated to 80°C.
(Bromomethyl)cyclopropane (0.06 ml, 0.65 mmol) was added and the reaction
mixture
kept between 80 and 90°C. Once the starting material was shown to have
been
consumed by TLC, the reaction was quenched by the addition of water and brine.
The
aqueous mixture was extracted three times with ethyl acetate, and the combined
organics were washed with brine, before being dried (MgS04), filtered and
concentrated
in vacuo to give a red/pink oil. Purification by silica gel column
chromatography
(DCM-X1.5%MeOH/DCM) furnished the target product as a red oil (35 mg, 26%).
8H(CDCI3; 250MHz) 0.08-0.20 (2H, m, two of cyclopropyl CH C,H~), 0.57-0.63
(2H, m, two
of cyclopropyl CH CHz), 0.99-1.04 (1 H, m, cyclopropyl Cue, 2.41 (1 H, dd,
J12.5, 7.0, one
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
of CHz), 2.70 (1 H, dd, J12.5, 5.8, one of CH,z), 2.83-3.00 (2H, m , two of
C_H2C,H~), 3.90
(1 H, ddd, J12.5, 5.8, 2.4, one of C~CH ), 3.95 (1 H, d, J12.5, one of ArCH
NR2), 4.22
(1 H, ddd, J12.5, 7.6, 3.2, one of CH CH ), 4.77 (1 H, d, J12.5, one of
ArC,H~NR2), 5.83
(1 H, s, CHOR), 7.01 (1 H, d, J7.0, aromatic ~, 7.16-7.33 (8H, m, aromatic ~.
5 Example 6
5-Isopropyl-1-phenyl-1,3,4,6-tetrahydro-5H-bent[f~-2,5-oxazocine
Desmethylnefopam (116 mg, 0.45 mmol) was dissolved in acetone (3m1) over 4A
molecular sieves (spatula tip). A drop of acetic acid was added and the
resulting mixture
was stirred at RT for one hour. After this time, sodium cyanoborohydrde (114
mg,
10 1.82 mmol) was added and the mixture was stirred overnight at RT, after
which time,
TLC showed that starting material had been consumed. The reaction mixture was
filtered and the collected solids washed with ethyl acetate. The filtrate was
concentrated
in vacuo to remove the acetone, providing a pale yellow residue. This was re-
dissolved
in dichloromethane and washed with a sat. aq. solution of sodium bicarbonate.
The
15 organic layer was separated, dried (MgSO4), filtered and concentrated in
vacuo to
provide the crude product. Purification by silica gel column chromatography
(DCM-~1 %MeOH/DCM) furnished the desired compound (34 mg, 30%).
8H (CDCI3; 250MHz) 1.13 (3H, d, J6.7, one CH3 of N(CH3)2), 1.25 (3H, d, J6.4,
one CH3
of N(CH3)2), 1.73 (1 H, dd, J4.6, 1.5, Cf~, 2.77-3.11 (2H, m, two of C,H~CH ),
3.71 (1 H, d,
J12.8; one of ArCH NR2), 3.87 (1 H, ddd, J12.2, 8.9, 1.8, one of CH CHz), 4.11
(1 H, ddd,
J12.2; 5.0, 2.3, one of CH CHz), 4.34 (1 H, d, J12.8, one of ArCHzNR2), 5.90
(1 H, s,
CHOR), 7.02 (1 H, d, J7.3, aromatic ~, 7.16-7.30 (8H, aromatic ~.
Example T
8-(4-Pyridyl)-5-methyl-1-(3-methoxy)phenyl-1,3,4,6-tetrahydro-5H-benz[fj-2,5-
oxazocine
8-Bromo-5-methyl-1-(3-methoxy)phenyl-1,3,4,6-tetrahydro-5H-Benz[fi]-2, 5-
oxazocine, i.e. compound 21b in W02004/056788 (117 mg, 0.32 mmol), pyridine-4-
boronic acid (52 mg, 0.42 mmol), Pd(PPh3)4 (10 mol%) and ICOH (2M solution in
water,
0.48 ml) were suspended in DME (2 ml). The mixture was degassed and then
purged
with nitrogen before heating at reflux temperature under a nitrogen atmosphere
for 17
hours. The mixture was allowed to cool to RT and diluted with ethyl acetate
(20 ml) and
washed with water (20 ml). The aqueous phase was extracted into ethyl acetate
(2 x 20
ml) and the combined organic extracts were dried (MgS04), filtered and
concentrated in
vacuo. Purification by silica gel column chromatography (EtOAc~10°lo
MeOHIEtOAc)
furnished the target compound as a pale yellow oil (25 mg, 22%).
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
16
8H(CDC13; 250MHz) 2.50 (3H, s, NC_H3), 2.60 (1 H, ddd, J14.2, 5.8, 2.6, one of
CHzCHz),
2.86 (1 H, ddd, J14.2, 8.3, 2.1, one of CHzCHz), 3.75 (1 H, d, J12.8, one of
ArCHzNR2),
3.78 (3H, s, OCH3), 3.88 (1 H, ddd, J12.5, 5.8, 2.1, one of CH CH ), 4.22 (1
H, ddd, J12.5,
8.3, 2.6, one of CH CHz), 4.87 (1 H, d, J12.8, one of ArCH NR2), 5.81 (1 H, s,
CHOR),
6.79-6.91 (4H, m, aromatic ~, 7.14 (1 H, d, J7.6, aromatic ~, 7.22-7.28 (1 H,
m, aromatic
~, 7.44-7.62 (4H, m, aromatic ~, 8.65 (1 H, d, J5.8, aromatic ~.
5-Pyrimidineboronic acid
"BuLi (3.02 ml, 2M solution in hexane, 7.55 mmol) was added dropwise to a
stirring solution of 5-bromopyrimidine (1 g, 6.29 mmol) and triisopropylborate
(1.46 ml,
7.55 mmol) in anhydrous toluene (16 ml) and anhydrous THF (4 ml) at -
70°C under a
nitrogen atmosphere. The reaction mixture was stirred at -70°C for 30
mins and then
removed from the cold bath. When the internal temp. reached -20°C, the
reaction was
quenched by the dropwise addition of 2M HCI (10 ml). The mixture was allowed
to warm
to RT and then separated. The aqueous phase was taken to pH 5.5 with 2M KOH
and
extracted into THF (3 x 25 ml). The combined organic extracts were dried
(MgSO4),
filtered and concentrated in vaeuo to provide a colourless solid. The solid
was slurried in
acetonitrile (2 ml), collected by filtration and dried on the sinter to give
the target boronic
acid as a brilliant white solid (340 mg, 44%).
8H(MeOD; 250MHz) 8.98 (2H, s), 9.14 (1 H, s).
Example 8
8-(5-Pyrimidine)-5-methyl-1-(3-methoxy)phenyl-1,3,4,6-tetrahydro-5H-bent[fj-
2,5-
oxazocine
8-Bromo-5-methyl-1-(3-methoxy)phenyl-1,3,4,6-tetrahydro-5H-bent[fj-2,5-
oxazocine, i.e. compound 21 b in WO2004/056788 (118 mg, 0.3 mmol), CsF (148
mg,
0.98 mmol), 5-pyrimidine boronic acid (52 mg, 0.42 mmol) and Pd(PPh3)4 (10
mol%)
were suspended in DME (2 ml) and water (100 wL). The mixture was degassed and
purged with nitrogen then heated to reflux temperature for 17 hours. The
reaction
mixture was allowed to cool to RT before diluting with ethyl acetate (20 ml)
and washing
with water (20 ml). The aqueous phase was extracted with ethyl acetate (2 x 20
ml) and
the combined organic extracts were dried (MgS04), filtered and evaporated to
dryness to
provide crude material. Purification by silica gel column chromatography
(EtOAc~10%
MeOH/EtOAc) furnished the target product as a pale yellow/brown solid (36 mg,
30%).
8H (CDCI3; 250MHz) 2.53 (3H, s, NCH3), 2.68 (1 H, ddd, J14.2, 5.8, 2.6, one of
CH CH ),
2.88 (1 H, ddd, J14.2, 8.2, 2.2, one of CH CH ), 3.79 (1 H, d, J12.8, one of
ArCHzNR2),
3.79 (3H, s, OCH3), 3.89 (1 H, ddd, J12.8, 5.8, 2.2, one of CH Cue), 4.24 (1
H, ddd, J12.8,
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
17
8.2, 2.6, one of CH~CH ), 4.89 (1 H, d, J12.8, one of ArCH NR2), 5.82 (1 H, s,
CHOR),
6.79-6.91 (3H, m, aromatic H~, 7.17-7.29 (2H, m, aromatic ~, 7.40-7.43 (2H, m,
aromatic ~, 8.95 (s, 2H, pyrimidine ~, 9.20 (1 H, s, pyrimidine ~.
Example 9
5-Methyl-1-(3-methoxy)phenyl-1,3,4,6-tetrahydro-5H-benz[fi]-2,5-oxazocine
8-Bromo-5-methyl-1-(3-methoxy)phenyl-1,3,4,6-tetrahydro-5H-benz[f]-2, 5-
oxazocine, i.e. compound 21 b in W02004/056788 (206 mg, 0.57mmol), was
suspended in toluene (3 ml) and degassed under vacuum, then purged with
nitrogen.
Palladium tetrakis (33 mg) was added to the mixture, which was then subjected
to the
degassing/nitrogen purge sequence again. A degassed solution of sodium
carbonate
(2M, 12 ml) and boronic ester (117 mg, 0.57 mmol) were added and the whole
mixture
was then degassed, purged with nitrogen and heated to 80°C overnight.
The reaction
mixture was allowed to cool, diluted with water (10 ml) and the resulting
mixture
extracted into ethyl acetate (3 x 50 ml). The combined organic phases were
dried
(MgS04), filtered and concentrated in vacuo to provide the crude product.
Purification by
silica gel column chromatography [EtOAc] was carried out twice to give a
mixture of
products.
Mass spec analysis suggested the debrominated species had been formed. m/z
(ES+) 284 (MH+). bH (CDCI3; 250MHz) 2.51 (3H, s, N CH3), 2.68 (1 H, ddd,
J14.2, 5.2, 2.9,
one of CH CH ), 2.88 (1 H, ddd, J14.2, 9.0, 2.6, one of CHzCH ), 3.78 (3H, s,
OCH3),
3.81 (1 H, d, J12.8, one of ArCHzNRz), 3.88 (1 H, ddd, J12.8, 5.2, 2.6, one of
CH C,H~),
4.23 (1 H, ddd, J12.8, 9.0, 2.9, one of CHzCHz), 4.86 (1 H, . d, J12.8, one of
ArCH NRZ),
5.76 (1 H, s, CHOR), 6.78-6.86 (3H, m, aromatic ~, 7.03-7.05 (1 H, m, aromatic
~, 7.20-
7.27 (4H, m, aromatic ~.
Example 10
5-Methyl-1-(3-methoxy)phenyl-1,3,4,6-tetrahydro-5H-benz[fj-2,5-oxazocine-8-
carboxamide
8-Cyano-5-methyl-1-(3-methoxy)phenyl-1,3,4,6-tetrahydro-5H-benz[f]-2,5
oxazocine, i.e. compound 21 b in W02004/056788 (73 mg, 0.24 mmol), was heated
to
reflux temperature in tBuOH (3 mL) with KOH (20 mg, 0.36 mmol) for 90 mins. At
the end
of this time, the reaction mixture was allowed to cool to RT. The mixture was
then
washed with brine (20 ml) and extracted into dichloromethane (3 x 20 ml). The
combined organic extracts were dried (MgS04) and concentrated in vacuo to
provide the
crude amide as a yellow solid. The solid was slurried in ethyl acetate,
collected by
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
18
suction filtration, washed with heptane and dried on the sinter to provide
amide (24 mg,
31 %).
b (MeOD; 250MHz) 2.44 (3H, s, NCH3), 2.51-2.60 (1 H, m (poorly resolved ddd),
one of
CH CH ), 2.72-2.81 (1 H, m (poorly resolved ddd), one of CH CHz), 3.75 (4H, s,
OCH3
and one of ArCH ), 3.84-3.90 (1 H, m (poorly resolved ddd), one of CH CHz),
4.17-4.27
(1 H, m (poorly resolved ddd), one of CH CH ), 5.01 (1 H, d, J12.5, one of
ArCHz), 5.83
(1 H, s, CHOR), 6.80-6.83 (3H, m, aromatic ~, 7.14-7.21 (2H, m, aromatic ~,
7.70-7.76
(2H, m, aromatic ~.
MS (ESf) 325 ([M-H]'); (ESI+) 349 ([M+Na]+)
2-Benzoyl-4-fluorobenzoic acid and 2-Benzoyl-5-fluorobenzoic acid
3-Fluorophthalic anhydride (5 g, 33 mmol) was suspended in benzene (17.7 ml)
at RT under nitrogen. Aluminium chloride (8.8 g, 66 mmol) was added in
portions,
turning the mixture orange in colour. The reaction was heated at reflux
temperature for 4
hours before cooling to RT. The yellow slurry was added carefully to 10%HCI
(100 ml),
producing a white precipitate. The aqueous mixture was extracted into ethyl
acetate (3 x
100 mL) and the combined organic extracts were dried (MgS04), filtered and
concentrated in vacuo to provide a mixture of the keto-acids 2-benzoyl-4-
fluorobenzoic
acid and 2-benzoyl-5-fluorobenzoic acid as colourless crystals (8 g,
quantitative yield).
8H (MeOD; 250MHz) 7.17-7.25 (3H, m, Ark, 7.31-7.42 (2H, m, ArH), 7.48 (2H, d,
J8.0,
Ark, 7.69 (1 H; d, J8.0, Ark.
2-Benzoyl-6-fluoro-N-(2-hydroxyethyl)-N-methylbenzamide and 2-Benzoyl-3-fluoro-
N-(2-hydroxyethyl)-N-methylbenzamide
A crude mixture of 2-benzoyl-6-fluorobenzoic acid and 2-benzoyl-3-
fluorobenzoic
acid (7 g, 28,mmol) was suspended in dichloromethane (70 ml) at RT. A few
drops of
DMF, followed by oxalyl chloride (3 ml, 34 mmol) were added dropwise and the
resulting
mixture was stirred until gas evolution had ceased, and the suspended solid
had
dissolved. The mixture was concentrated in vacuo and co-evaporated with
dichloromethane (2 x 40 mL) to provide the crude acid chloride. (2-
methylamino)-
ethanol (2.24 ml, 28 mmol) and triethylamine (3.9 ml, 28 mmol) were dissolved
in
dichloromethane (50 ml) and cooled in an ice bath. The acid chloride was
dissolved in
dichloromethane (50 ml) and added dropwise to the cooled solution of amine.
The
mixture was allowed to warm to RT and stirred for 4.5 hours, after which point
the
reaction was quenched by the addition of sat. aq ammonium chloride solution
(200 ml).
The organics were extracted into dichloromethane (3 x 100 ml), and the
combined
extracts were dried (MgS04), filtered and concentrated in vacuo. Purification
was
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
19
achieved by gravity silica gel column chromatography (50% EtOAc/heptane to
100%
EtOAc) with pre-absorption of the crude amide onto silica.
2-Benzoyl-6-fluoro-N (2-hydroxyethyl)-N methylbenzamide (490 mg) was
obtained as a 3:4 mixture of two rotamers:
sH (CDCI3; 250MHz) 3.04 (3H, s, CH3, major rotamer), 3.10 (3H, s, CH3, minor
rotamer),
3.33-3.40 (2H, two of CHzCH , major rotamer), 3.52-3.64 (2H, two of CH CH ,
minor
rotamer), 3.78-3.96 (4H, two of CH CH,z for major and minor rotamer), 7.22-
7.27 (4H, m,
ArH both rotamers), 7.28-7.42 (6H, m, ArH both rotamers), 7.55-7.61 (2H, ArH
both
rotamers), 7.75-7.83 (4H, m, ArH both rotamers).
2-Benzoyl-3-fluoro-N (2-hydroxyethyl)-N methylbenzamide (2.74 g) was obtained
as two rotamers:
sH (CDCI3; 250MHz) 3.03 (3H, s, CH3, one rotamer), 3.05 (3H, s, CH3, one_
rotamer),
3.55-3.59 (4H, m, two of CHzCHz for each rotamer), 3.75-3.79 (4H, m, two of CH
CH for
each rotamer), 7.16-7.27 (4H, m, ArH both rotamers), 7.44-7.63 (4H, m, ArH
both
rotamers), 7.84 (2H, d, J7.9, ArH both rotamers).
2-~[3-Fluoro-2-(hydroxyphenyl-methyl)-benzyl]-methylamino}-ethanol
2-Benzoyl-3-fluoro-N-(2-hydroxyethyl)-N-methylbenzamide (2.58 g, 8.57 mmol)
was dissolved in anhydrous THF (25m1) at RT under a nitrogen atmosphere.
Borane-
dimethylsulfide complex (2.0M in THF, 18.8 ml, 37.7 mmol) was added dropwise
and the
resulting mixture was stirred at RT overnight. The reaction was carefully
quenched by
the addition of HCI (10%, 36m1). The mixture was then heated to reflux
temperature for
approx. 1 hour before cooling to ambient temperature. The THF was removed in
vacuo
and the remaining solution was partitioned between MTBE (40 ml) and water (60
ml).
The aqueous phase was separated, basified with 2M NaOH(aq) and extracted into
ethyl
acetate (4 x 50 ml). The combined organic extracts were dried (MgS04),
filtered and
concentrated in vacuo to give 2-~[3-fluoro-2-(hydroxyphenyl-methyl)-benzyl]-
methylamino]~-ethanol as a viscous oil which later crystallised.
8H (CDCI3; 250MHz) 2.17 (3H, s, CH3), 2.35-2.42 (1 H, m, one of CH CHz), 2.52-
2.62 (1 H,
m, one of CH CHz), 2.88 (1 H, d, J12.5, one of ArCHzNR2), 3.42 (1 H, d, J12.5,
one of
ArCHzNR2), 3.62-3.65 (2H, m, two of CH CH ), 6.38 (1 H, s, CHOH), 6.96 (1 H,
d, J7.3,
Ark, 7.10-7.37 (7H, m, Ark.
Example 11
10-Fluoro-5-methyl-1-phenyl-1,3,4, 6-tetrahydro-5H-benz[f]-2,5-oxazocine
Crude 2-~[3-fluoro-2-(hydroxyphenylmethyl)benzyl]methylamino}ethanol (234 mg,
0.81 mmol) was heated to reflux temperature with pTSA (231 mg, 1.21 mmol) in
toluene
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
(4 ml). The reaction was kept open to allow the toluene/water to evaporate.
After
approx. 1.5 hours, a gummy residue remained in the reaction flask. It was
allowed to
cool to RT at which point, sat. aq sodium bicarbonate solution and ethyl
acetate were
added to solubilise the residue. The aqueous phase was repeatedly extracted
with ethyl
5 acetate, and the combined extracts were dried (MgS04), filtered and
evaporated to
dryness to provide the crude cyclised product. Purification by gravity silica
gel column
chromatography [EtOAc] furnished the target compound, (90 g, 25%).
8H (CDCI3; 250MHz) 2.47 (3H, s, CH3), 2.59 (1 H, ddd, J13.9, 5.6, 3.3, one of
C,H~CH ),
2.78 (1 H, ddd, J13.9, 8.5, 2.6, one of CHzCH ), 3.58 (1 H, d, J12.4, one of
ArC,H~NR2),
10 3.83 (1 H, ddd, J12.3, 5.6, 2.6, one of CHzCHz), 4.30 (1 H, ddd, J12.3,
8.5, 3.3, one of
CH CHz), 5.09 (1 H, d, J12.4, one of ArCH NRZ), 5.94 (1 H, d, J1.9, CHOR),
6.89-6.96
(1 H, m, Ark, 7.04 (1 H, d, J7.6, Ark, 7.19-7.32 (6H, m, Arty
2-([2-Fluoro-6-(hydroxylphenylmethyl)benzyl]methylamino~ethanol
2-Benzoyl-6-fluoro-N-(2-hydroxyethyl)-N-methylbenzamide (440 mg, 1.46 mmol)
15 was dissolved in dry THF (4 ml) under nitrogen. Borane-dimethylsulfide
(2.0M solution in
THF, 3.2 ml, 6.43 mmol) was added dropwise and the resulting mixture was
stirred
overnight at RT. The reaction was quenched by the addition of 2M HCI (6m1) and
heated
at reflux temperature for 1 hour. The acidic mixture was then cooled and
partitioned
between MTBE (30 ml) and water (30 ml). The aqueous phase was separated and
20 extracted with ethyl acetate (4 x 30 ml). The combined extracts were dried
(MgS04),
filtered and concentrated in vacuo to give the crude amine. Purification by
gravity silica
gel column chromatography [EtOAc] furnished 2-([2-fluoro-6-
(hydroxylphenylmethyl)benzyl]methylamino}ethanol (19 mg, 5%).
sH (250MHz; CDCI3) 2.72 (3H, s, NCH3), 2.54-2.73 (2H, m, two of CH CH ), 3.36
(1 H, dd,
J12.8, 2.1, one of ArCH NR2), 3.63-3.72 (3H, two of CHZCH~ and one of ArCH
NRZ),
5.91 (1 H, s, CHOH), 6.97-7.07 (2H, m, aromatic ~, 7.22-7.40 (6H, m, aromatic
~.
Example 12
7-Fluoro-5-methyl-1-phenyl-1,3,4,6-tetrahydro-5H-bent[f]-2,5-oxazocine
Crude 2-~[2-fluoro-6-(hydroxyphenyl-methyl)benzyl]methylamino~ethanol (20 mg;
0.069 mmol) was dissolved in toluene and pTSA (20 mg, 0.1 mmol) added. The
mixture
was heated to 120°C in an open vessel to allow evaporation of the
solvent. After 30
mins, a gummy residue remained in the reaction vessel. The vessel was allowed
to cool
and the residue dissolved in ethyl acetate (5 ml) and sat. aq. sodium
bicarbonate
solution (10 ml). The aqueous phase was washed with ethyl acetate and the
combined
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
21
organic extracts dried (MgSO4), filtered and concentrated in vacuo to provide
the product
as a crude light brown oil (10 mg, 53%).
8H (CDCI3, 250MHz) 2.50 (3H, s, NCH3), 2.68 (1 H, ddd, J13.9, 6.3, 2.8, one of
CH~C,H~),
2.84 (1H, ddd, J13.9, 7.8, 2.1, one of CH Cue, 3.90 (1H, ddd, J 12.5, 6.3,
2.1, one of
CH CH ), 4.05 (1 H, d, J13.4, one of ArCH ), 4.21 (1 H, ddd, J12.5, 7.8, 2.8,
one of
CH CH ), 4.71 (1 H, dd, J13.4, 3.4, one of ArCH ), 5.81 (1 H, s, CHOR), 6.82
(1 H, d, J7.6,
aromatic ~, 7.37 (1 H, t, J8.5, aromatic ~, 7.10-7.23 (2H, m, aromatic ~, 7.25-
7.35 (3H,
m, aromatic ~, 7.37-7.45 (1 H, m, aromatic ~.
Purity measured at 50%.
TBS-protected 3-bromophenol
To a solution of 3-bromophenol (100 g, 0.6 mol) in dichloromethane (600 ml)
was
added imidazole (100 g, 1.46 mol) at 0°C. After 10 min, TBDMS-CI (96 g,
0.64 mol) was
added, and the mixture was stirred at RT overnight. The mixture was then
diluted with
MTBE (1 L) and filtered. The filtrate was washed with sat. ammonium chloride
(4 x 200
ml), sat. bicarb. (200 ml) and brine (200 ml), before being dried (MgS04),
filtered and
concentrated in vacuo to give the product as a yellow oil (156.6 g) that was
used without
further purification.
8H (CDC13; 250MHz) 7.10-6.77 (4H, m, aromatics), 0.99 (9H, s, C(CH3)3), 0.21
(6H, s, 2 x
CH3).
4-Bromo-2-(3 tbutyldimethylsilyloxy-benzoyl)-benzoic acid and 5-Brom~-2-(3-
tbutyldimethylsilyloxy-benzoyl)-benzoic acid
A solution of TBS-protected 3-bromophenol (156.6 g, 0.54 mol) in anhydrous
THF (160 ml) was added dropwise to a stirring suspension of magnesium turnings
(13 g,
0.54 mol) in dry THF (240 ml). The Grignard formation initiated following the
addition of
1 ml of solution. The resulting mixture was left stirring until the reflux had
stopped, and
then this was added in a single portion to a stirred solution of 4-
bromophthalic anhydride
(122.4 g, 0.54 mol) in anhydrous THF (240 ml). The reaction mixture was heated
at
reflux temperature overnight. After cooling to RT, the reaction was quenched
by the
addition of sat. aq. solution of ammonium chloride (250 ml) and the organics
were
extracted into ethyl acetate (3 x 250 ml). The combined organic extracts were
dried
(MgS04), filtered and concentrated in vacuo to provide the crude acid as a
brown oil
(226.6 g). The crude material was carried through to the next step without
further
purification.
8H (CDCI3; 250MHz) 8.07-6.75 (7H, m, aromatics), 0.99-0.94 (9H, m, C(CH3)3),
0.20-0.13
(6H, m, 2 x CH3).
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
22
5-Bromo-N-(2-hydroxyethyl)-N-methyl-2-(3 tbutyldimethylsilyloxy-benzoyl)-
benzamide
To a solution of a mixture of 4-bromo-2-(3 tbutyldimethylsilyloxy-benzoyl)-
benzoic
acid and 5-bromo-2-(3 tbutyldimethylsilyloxy-benzoyl)-benzoic acid (226.6 g,
0.52 mol) in
anhydrous d'ichloromethane (900 ml), under an atmosphere of nitrogen, was
added
anhydrous N,N-dimethylformamide (1 ml) followed by the slow addition of oxalyl
chloride
(50.8 ml, 0.57 mol), and the reaction stirred at RT overnight. The
dichloromethane was
then removed in vacuo. Dichloromethane (2 x 500 ml) was added to the crude oil
and
evaporated in vacuo. The crude acid chloride was dissolved in dichloromethane
(900. ml), cooled to 0°C (ice bath) and triethylamine (81 ml, 0.57 mol)
added dropwise. N-
methylethanolamine (42.8 g, 0.57 mol) was then added dropwise and the mixture
left to
warm to RT overnight under an atmosphere of nitrogen. The mixture was then
quenched with brine (800 ml) and the layers separated. The aqueous layer was
then
separated and washed with dichloromethane (800 ml), and the combined organics
were
dried (MgS04), filtered and concentrated in vacuo to yield the mixture of
regioisomers
(255.5 g) as a dark brown oil.
100 g of the above brown oil was purified by silica gel column chromatography
(3:1 EtOAc: Heptane). This afforded 14 g of 5-bromo-N (2-hydroxyethyl)-N-
methyl-2-(3-
tbutyldimethylsilyloxy-benzoyl)-benzamide.
8H (CDCI3; 250MHz) 7.59-7.25 (7H, m, aromatics), 3.91-3.57 (4H, m, CHz-CH ),
3.11
and 2.99 (3H, 2 x s, NCH3), 0.98 (9H, s, C(CH3)3), 0.21 (6H, s, 2 x CH3).
5-Bromo-N (2-hydroxyethyl)-N methyl-2-(3-tosyloxy-benzoyl)-benzamide
TBAF (10.1 ml, 1 M solution in THF, 10.14 mmol) was added to a stirred
solution
of 5-bromo-N-(2-hydroxyethyl)-N methyl-2-(3 tbutyldimethylsilyloxy-benzoyl)-
benzamide
(5g, 10.14 mmol) in THF (60 ml) at RT.. A solution of tosyl chloride (1.9 g,
10.14 mmol)
in THF (60 ml) was then added dropwise and the mixture heated to reflux
temperature
for 3 h. A further 10mmol of triethylamine followed by 7 mmol of TsCI were
added over a
further hour to drive the reaction to completion. After cooling to RT the
mixture was
diluted with ethyl acetate (100 ml), washed with water (2 x 100 ml), dried
(MgS04),
filtered and concentrated in vacuo to give 7 g of an orange oil. Purification
by silica gel
column chromatography [2:1 EtOAc:Heptane] afforded the title compound (3.14 g,
58%)
as a pale yellow oil.
8H (CDCI3; 250MHz) 7.74-7.20 (11 H, m, aromatics), 3.86-3.53 (4H, m, CH2CH2),
3.07
and 2.97 (3H, 2 x s, NCH3), 2.47 (3H, s, Ar-CH3).
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
23
2-(~5-Bromo-2-[hydroxyl-(3-tosyloxy-phenyl)-methyl-benzyl~-methylamino)-
ethanol
Borane dimethylsulfide complex (2.0M in THF, 12.94 ml, 25.88 mmol) was added
dropwise to a solution of 5-bromo-N (2-hydroxyethyl)-N methyl-2-(3-tosyloxy-
benzoyl)-
benzamide (3.14 g, 5.88 mmol) in anhydrous THF (20 ml) at 0°C. The
reaction mixture
was left to stir at RT overnight. The crude mixture was then quenched
carefully by
addition of an aqueous 6M HCI solution (20 ml) and then heated to reflex
temperature for
2h. After cooling to RT, the THF was removed in vacuo. Water (10 ml) was added
and
the mixture was then basified to pH 10 by addition of an aqueous solution of
sodium
hydroxide solution (50 %), and then extracted with ethyl acetate (2 x 20 mL).
The
combined organic layers were then dried (MgS04), filtered and concentrated in
vacuo to
give a white oil/solid (3.17 g). Purification by silica gel column
chromatography
[5:1,EtOAc:Heptane] afforded the title compound (0.74g, 24%) as a colourless
oil.
8H (CDCI3; 250MHz) 7.67-6.83 (11 H, m, aromatics), 5.72 (1 H, s, CHOH), 3.73-
3.67 (2H,
m, CH2), 3.39-3.27 (2H, m, CH2), 2.63-2.53 (2H, m, CHz), 2.44 (3H, s, Ar-CH3),
2.22 (3H,
s, NCH3).
Example 13
8-Bromo-5-methyl-1-(3-tosyloxy)phenyl-1,3,4,6-tetrahydro-5H-bent[f]-2,5-
oxazocine
2-({5-Bromo-2-[hydroxyl-(3-tosyloxyphenyl)methyl]benzyl)methylamino)ethanol
(0.74 g, 1.43 mmol) was heated to reflex temperature under Dean-Stark
conditions with
pTSA (0.41 g, 2.14 mmol) in toluene (20 ml) for 2 h. The reaction mixture was
allowed to
cool and then diluted with ethyl acetate (10 ml). The mixture was washed with
sat. aq.
sodium bicarbonate solution (2 x 10 ml). The combined organic extracts were
dried
(MgSO4), filtered and concentrated in vacuo to provide 0.65 g of product that
was used
without further purification.
8H (CDCI3; 250MHz) 7.67-6.72 (11 H, m, aromatics), 5.67 (1 H, s, CH-O), 4.55
(1 H, d, J
13, ArCHAHsN), 4.11 (1 H, ddd, J 13, 8 and 2.5, CHp,HBCH2), 3.83 (1 H, ddd, J
13, 8 and
2.5, CH,~ HsCH2), 3.60 (1 H, d, J 13, ArCH~HBN), 2.79-2.59 (2H, m, CH CH2),
2.48 and
2.46 (6H, 2 x s, 2 x CH3).
Example 14
8-Bromo-5-methyl-1-(3-hydroxy)phenyl-1,3,4,6-tetrahydro-5H-benz[fi]-2,5-
oxazocine
Potassium hydroxide (0.30 g, 5.33 mmol) was added to a stirred solution of 8-
bromo-5-methyl-1-(3-tosyloxy)phenyl-1,3,4,6-tetrahydro-5H-bent[f]-2,5-
oxazocine (0.65
g, 1.3 mmol) in MeOH (5 ml) and DMF (5 ml) and the mixture heated at 60
°C for 3h.
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
24
After cooling to RT, the crude mixture was poured into water (10 ml) and the
mixture
extracted with dichloromethane (2 x 10 ml). The aqueous layer was then
acidified to pH
9 and extracted with dichloromethane (2 x 10 ml). The combined organics were
dried
(MgS04), filtered and concentrated in vacuo to give 0.42 g of a yellow oil.
Purification by
silica gel column chromatography (5% MeOHIDCM) afforded the title compound
(0.20 g,
44%) as a white solid.
8H (CDCI3; 250MHz) 7.34-6.67 (7H, m, aromatics), 5.60 (1 H, s, CH-O), 5.17 (1
H, d, J 13,
ArCH~,HeN), 4.20 (1 H, ddd, J 13, 8 and 2.5, CH o,HBCH2), 3.80 (1 H, ddd, J
13, 8 and 2.5,
CH,~HBCHZ), 3.53 (1 H, d, J 13, ArCH, HBN), 2.75-2.56 (2H, m, CH CHZ), 2.36
(3H, s,
NCH3).
Example 15
8-Cyano-5-methyl-1-(3-hydroxy)phenyl-1,3,4,6-tetrahydro-5H-benz[f]-2,5-
oxazocine
8-Bromo-5-methyl-1-(3-hydroxy)phenyl-1,3,4,6-tetrahydro-5H-benz[f]-2,5-
oxazocine (100 mg, 0.29 mmol), Zn (1 mg, 0.02 mmol), Zn(OAc)2 (7 mg, 0.034
mmol),
Zn(CN)2 (25 mg, 0.22 mmol), Pd2(dba)3 (37 mg, 0.04 mmol) and 1,1'-
bis(diphenylphosphino)ferrocene (18 mg, 0.034 mmol) were heated to
140°C in DMF (3
ml) and H20 (30 ~,I) for 4h. The black reaction mixture was allowed to cool to
RT and
diluted with water (20 ml). The resulting brown precipitate was collected by
filtration and
washed with water then brine. The filtrate was extracted with ethyl acetate (2
x 60 ml).
The combined organic extracts were dried (MgS04), filtered and concentrated in
vacuo
to provide the crude product as a brown oil. Purification by gravity silica
gel column
chromatography [dichloromethane] furnished the desired product (25 mg, 29%) as
a
yellow oil.
8H(CDCI3; 250MHz) 7.45-6.70 (7H, m, aromatics), 5.65 (1 H, s, CHO), 5.03 (1 H,
d, J 13,
ArCH~,HB-N), 4.26-4.18 (1 H, m, CHAHBCHZ), 3.81-3.75 (1 H, m, CH~HBCHZ), 3.61
(1 H, d,
J 13, ArCH~HB-N), 2.70-2.53 (2H, m, CHzCH2), 2.41 (3H, s, NCH3).
Example 16
8-Cyclopropyl-5-methyl-1-(3-hydroxy)phenyl-1,3,4,6-tetrahydro-5H-bent[fj-2,5-
oxazocine
8-Bromo-5-methyl-1-(3-hydroxy)phenyl-1,3,4,6-tetrahydro-5H-benz[f]-2,5-
oxazocine (100 mg, 0.29 mmol), cyclopropylboronic acid (33 mg,0.36 mmol),
K3P04 (217
mg, 1.01 mmol), Pd(OAc)2 (4 mg, 0.015 mmol) and P(cyclohexane)3 (9 mg, 0.029
mmol)
were heated to 100°C in toluene (2.5 mL) and H20 (125 w1) for 4 hours.
After cooling to
RT, water (2.5 ml) was added and the mixture extracted with ethyl acetate (2 x
5 ml),
washed with brine, dried (MgS04), filtered and concentrated in vacuo.
Purification by
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
gravity silica gel column chromatography [dichloromethane] furnished the
desired
product (40 mg, 44%) as a colourless oil.
8H(CDCI3; 250MHz) 7.27-6.69 (7H, m, aromatics), 5.61 (1 H, s, CHO), 5.04 (1 H,
d, J 13,
ArCHAHB-N) 4.23-4.15 (1 H, m, CH ,HBCHZ), 3.83-3.78 (1 H, m, CH~HBCHZ), 3.66
(1 H, d, J
5 13, ArCH,~He-N), 2.78-2.58 (2H, m, CH CH2), 2.41 (3H, s, NCH3), 1.84-1.78 (1
H, m,
CHCHZ), 0.95-0.90 (2H, m, CH CHZ), 0.64-0.62 (2H, m, CHZCHz).
Example 17
8-Bromo-5-methyl-1-(3-allyloxy)phenyl-1,3,4,6-tetrahydro-5H-benz[fJ-2, 5-
oxazocine
8-Bromo-5-methyl-1-(3-hydroxy)phenyl-1,3,4,6-tetrahydro-5H-Benz[f]-2,5
10 oxazocine (50 mg, 0.14 mmol), allyl alcohol (33 mg, 0.57 mmol), Pd(OAc)Z (1
mg, 0.0035
mmol), triphenylphosphine (2 mg, 0.0075 mmol), Ti(O'Pr)4 (10 mg, 0.035 mmol),
molecular sieves (4 A, 28 mg) were heated at 50°C in benzene (1 ml)
overnight. After
cooling to RT, the black reaction mixture poured into water (5 ml), extracted
with MTBE
(2 x 5 ml). The combined organic extracts were dried (MgS04), filtered and
concentrated
15 in vacuo to provide the crude product as a brown oil. Purification by
gravity silica gel
column chromatography [dichloromethane] furnished the desired product (16 mg,
30%)
as a pale yellow oil.
sH(CDCI3; 250MHz) 7.38-7.19 (3H, m, aromatics), 6.90-6.80 (4H, m, aromatics),
6.09
5.96 (1 H, m, CH=CH2), 5.71 (1 H, s, CHO), 5.42-5.25 (2H, m, CH=CHz), 4.76 (1
H, d, J
20 13, ArCH ,HB-N), 4.51 (2H, d, J 5, OCHZ), 4.21-4.12 (1 H, m, C,H~HBCH2),
3.88-3.80 (1 H,
m, CH .,~HBCHZ), 3.63 (1 H, d, J 13, ArCH~He-N), 2.87-2.77 (1 H, m, CH2CH
,NB), 2.69-2.60
(1H, m, CH2CHpHe) 2.47 (3H, s, NCH3).
Example 18
8-Cyano-5-methyl-1-(3-allyloxy)phenyl-1,3,4,6-tetrahydro-5H-bent[f]-2,5-
oxazocine
25 8-Cyano-5-methyl-1-(3-hydroxy)phenyl-1,3,4,6-tetrahydro-5H-benz[fJ-2,5-
oxazocine (50 mg, 0.17 mmol), allyl alcohol (49 mg, 0.68 mmol), Pd(OAc)2 (1
mg, 0.0035
mmol), triphenylphosphine (2 mg, 0.0075 mmol), Ti(O'Pr)4 (20 mg, 0.07 mmol),
molecular sieves (4 A, 40 mg) were heated at 50°C in benzene (2 ml)
overnight. After
cooling to RT, the black reaction mixture poured into water (5 ml) and
extracted with
MTBE (2 x 5 ml). The combined organic extracts were dried (MgS04), filtered
and
concentrated in vacuo to provide the crude product as a brown oil.
Purification by gravity
silica gel column chromatography [dichloromethane] furnished the desired
product (4
mg, 7%) as a pale yellow oil.
8H(CDCI3; 250MHz) 7.65-6.81 (7H, m, aromatics), 6.08-5.97 (1 H, m, CH=CH2),
5.74 (1 H,
s, CHO), 5.42-5.25 (2H, m, CH=CHz), 4.98 (1 H, d, J 13, ArCH~Hg-N), 4.51 (2H,
d, J 5,
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
26
OCH~), 4.31-4.23 (1 H, m, CH~HBCHZ), 3.90-3.82 (1 H, m, CH,~HBCH2), 3.74 (1 H,
d, J 13,
ArCH,~HB-N), 2.81-2.70 (2H, m, CHZCH ), 2.51 (3H, s, NCH3).
Example 19
8-Cyclopropyl-5-methyl-1-(3-allyloxy)phenyl-1,3,4,6-tetrahydro-5H-benz[fj-2,5-
oxazocine
8-Cyclopropyl-5-methyl-1-(3-hydroxy)phenyl-1, 3,4,6-tetrahydro-5H-benz[f]-2,5-
oxazocine (90 mg, 0.29 mmol), allyl alcohol (0.1 ml, 1.16 mmol), Pd(OAc)2 (1
mg, 0.0035
mmol), triphenylphosphine (3 mg, 0.011 mmol), Ti(O'Pr)4 (0.03 mL, 0.07 mmol),
molecular sieves (4 A, 64 mg) were heated at 50°C in benzene (3 ml)
overnight. After
cooling to RT, the black reaction mixture was poured into water (5 ml),
extracted with
MTBE (2 x 5 ml). The combined organic extracts were dried (MgS04), filtered
and
concentrated in vacuo to provide the crude product as a brown oil.
Purification by gravity
silica gel column chromatography [dichloromethane] furnished the desired
product (3
mg, 3%) as a pale yellow oil.
8H(CDCI3; 250MHz) 7.24-6.76 (7H, m, aromatics), 6.04-5.97 (1 H, m, CH=CHZ),
5.41 (1 H,
s, CHO), 5.41-5.24 (2H, m, CH=CHI), 4.70 (1 H, d, J 13, ArC,H~HB-N), 4.49 (2H,
d, J 5,
OCHz), 4.19-4.11 (1 H, m, CHpHBCH2), 3.88-3.82 (1 H, m, CH,o HBCH~), 3.71 (1
H, d, J 13,
ArCH~HB-N), 2.92-2.82 (1 H, m, CH2CH,o,HB), 2.69-2.66 (1 H, m, CH~CH,.~HB),
2.49 (3H, s,
NCH3), 1.89-1.83 (1 H, m, CHCHZ), 0.98-0.90 (2H, m, CHC,H~), 0.71-0.65 (2H, m,
CHCH ).
The following Examples were prepared in a similar manner.
The substituted 'bromophenyl' starting materials, used for the Grignard
formation,
were either sourced commercially or generated synthetically via known organic
chemistry, e.g. as in the following example where a nitro group was converted
to a
bromo via the intermediate amine.
2-Methoxy-6-aminotoluene
2-methyl-3-nitroanisole (20 g, 0.12 mol) was suspended in methanol (200 ml) at
RT under nitrogen. Pd/C (5%, 2 g) was added and the system was shaken under an
atmosphere of hydrogen overnight, until TLC indicated one product. The
reaction
mixture was filtered through celite, and the celite washed with methanol (3 x
100 ml) and
the filtrate concentrated in vacuo. This afforded 18 g of a red oil that was
used without
further purification. ,
SH (CDCI3; 250MHz) 7.01 (1 H, t, J 8, aromatics), 6.38 (2H, d, J 8,
aromatics), 3.83 (3H, s,
OCH3), 2.08 (3H, s, CH3).
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
27
2-Methoxy-4-bromotoluene
Crude 2-methoxy-6-amino-toluene (max. 120 mmol) was suspended in HBr
(48%, 48 ml) and water (120 ml) and cooled to 0°C in an ice bath.
Sodium nitrite (9.2 g)
in water (24 ml) was added dropwise to the cold mixture, which turned yellow
then
brown. After.10 min, excess nitrite was destroyed by addition of urea (0.08 g)
and the
mixture was rapidly filtered into cold (0°C) acetone (480 ml) to give a
bright yellow
solution. CuBr (99.999%, 18.89, 131 mmol) was then added portionwise and the
resulting mixture stirred at 0°C for 3h. Gas evolution was observed.
The mixture was
allowed to warm to ambient temperature, and then concentrated in vacuo.
Dichloromethane was then added and the mixture washed with sat. aq. sodium
bicarbonate solution. The organic layer was separated, dried over magnesium
sulphate
and concentrated in vacuo to give the product (23.1 g, 96%) as a red oil.
8H (CDCI3; 250MHz) 7.18-6.77 (3H, m, aromatics), 3.83 (3H, s, OCH3), 2.32 (3H,
s, CH3).
5-Bromo-2-(3-methoxy-2-methylbenzoyl)-benzoic acid and 4-Bromo-2-(3-methoxy
2-methylbenzoyl)-benzoic acid
2-methoxy-4-bromo-toluene (23.1 g, 115 mmol) in dry THF (35 ml) was added
dropwise
to a stirred suspension of magnesium turnings (2.77 g, 115 mmol) and a crystal
of iodine
in dry THF (40 ml) under a nitrogen atmosphere. Upon addition, the reaction
mixture
reached reflux. After cooling to RT, this Grignard solution was added dropwise
to a
stirred solution of 4-bromophthalic anhydride (26.1 g, 115 mmol) in dry THF
(45 ml) and
the resulting mixture heated to reflux temperature overnight. After cooling,
the reaction
was quenched with sat. aq. ammonium chloride solution, and extracted with
ethyl
acetate. The organics were dried (MgS04), filtered and concentrated in vacuo
to give
the crude mixture of keto-acids (34 g) as a brown solid. This material was
carried
straight on to the subsequent step.
5-Bromo-N (2-hydroxyethyl)-N methyl-2-(3-methoxy-2-methyl-benzoyl)-benzamide
A crude mixture of 5-bromo-2-(3-methoxy-2-methylbenzoyl)-benzoic acid and 4-
bromo-2-(3-methoxy-2-methylbenzoyl)-benzoic acid (34 g, 98.5 mmol) was
dissolved in
dichloromethane (200 ml) at RT. DMF (1 ml) followed by oxalyl chloride (9.67
ml, 13.98
g, 108.35 mmol) were added dropwise and the reaction stirred at RT overnight.
The
reaction mixture was then concentrated in vacuo and co-evaporated with
dichloromethane (2 x 200 ml) to provide the crude acid chloride. This was
dissolved in
dichloromethane (200 ml), cooled to 0°C, and triethylamine (15.35 ml,
11.22 g, 108.35
mmol) was added dropwise, followed by N-methylethanolamine and the mixture
left to
warm to RT overnight. The reaction was then quenched with brine and extracted
with
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
28
dichloromethane. The combined organic extracts were dried (MgS04), filtered
and
concentrated in vacuo to give the crude amide as a mixture of regioisomers.
The 5-
bromo-N-(2-hydroxyethyl)-N methyl-2-(3-methoxy-2-methyl-benzoyl)-benzamide (10
g,
25 °l°) was separated by silica gel column chromatography [4:1
EtOAciheptane].
8h, -(CDCI3; 250MHz) 7.55-6.91 (6H, m, aromatics), 3.98-3.69 (7H, m, OCH3 and
CH CH_z), 3.15 and 2.99 (3H, 2 x s, NCH3), 2.20 and 2.17 (3H, 2 x s, CHI).
2-(~5-Bromo-2-[hydroxyl-(3-methoxy-2-methylphenyl)methyl]benzyl}methylamino)-
ethanol
Borane-dimethyl sulphide complex (2.0 M, 55 ml, 0.11 mol) was added dropwise
to a stirring solution of 5-bromo-N (2-hydroxyethyl)-N-methyl-2-(3-methoxy-2-
methyl-
benzoyl)-benzamide (10 g, 0.025 mol) in anhydrous THF (80 ml) at RT under
nitrogen,
and allowed to stir overnight. The reaction was quenched by the careful
addition of 6M
HCI (80 ml), and the resulting mixture was heated to reflux temperature for 2
hours.
After cooling to RT, the THF was removed in vacuo and the mixture partitioned
between
water (40 ml) and MTBE (80 ml). The aqueous phase was separated, basified with
aq.
2M NaOH, and extracted into ethyl acetate (3 x 90 ml). The combined organic
extracts
were dried (MgS04), filtered and concentrated in vacuo to provide the desired
amine as
a viscous oil, which slowly crystallised (6.2 g, 63 %). The material was used
in the next
step without further purification.
sH (CDCI3; 250MHz) 7.41-7.25 (4H, m, aromatics), Ca.89 (1 H, d, J 8,
aromatics), 6.61 (1 H,
d, J 8, aromatics), 6.10 (1 _H, s, CHOH), 4.33 (1 H, d, J 13, CH ,HBN), 3.84
(3H, s, OCH3),
3.84-3.62 (2H, m, C,H~), 3.26 (1 H, d, J 13, CH,~HBN), 2.77-2.73 (2H, m, CHz),
2.33 (3H, s,
NCH3), 1.83 (3H, s, CH3).
Example 20
8-Bromo-5-methyl-1-(2-methyl-3-methoxy)phenyl-1,3,4,6-tetrahydro-5H-Benz[fj-
2,5-
oxazocine
2-({5-bromo-2-[hydroxyl-(3-methoxy-2-methylphenyl)-methyl]-benzyl}-
methylamino)-ethanol (5 g, 12.6 mmol) was heated to reflux temperature in
toluene (250
ml) with pTSA (3.65 g, 18.95 mmol) under Dean-Stark conditions for 4 hours.
The
reaction mixture was allowed to cool to RT, diluted with ethyl acetate (200
ml) and
washed with a sat. aq. solution of sodium bicarbonate (300 ml). The aqueous
layer was
extracted with ethyl acetate (3 x 200 ml) and the combined organic extracts
were dried
over magnesium sulfate, filtered and concentrated in vacuo. Purification by
silica gel
column chromatography [DCM] gave the target product as a pale straw coloured
oil (2.8
g, 59 °!o).
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
29
8H (CDC13; 250MHz) 7.42-6.70 (6H, m, aromatics), 5.97 (1 H, s, CHO), 4.69 (1
H, d, J 13,
CH ,HBN), 4.14-4.05 (1H, m, CH,4HB), 3.89-3.73 (5H, m, CH,~HB, OCH3, and
CH,oHBN),
2.92-2.82 (1 H, m, CH,4HB), 2.69-2.59 (1 H, m, CH .,FHB), 2.45 (3H, s, CH3N),
2.15 (3H, s,
CH3).
Example 21
8-Cyano-5-methyl-1-(2-methyl-3-methoxy)phenyl-1,3,4,6-tetrahydro-5H-bent[f]-
2,5-
oxazocine
8-Bromo-5-methyl-1-(2-methyl-3-methoxy)phenyl-1,3,4,6-tetrahydro-5H-Benz[f]-
2,5-oxazocine (250 mg, 0.66 mmol), Zn (4 mg, 0.04 mmol), Zn(OAc)~ (14 mg, 0.07
mmol), Zn(CN)2 (50 mg, 0.44 mmol), 1,1'-bis(diphenylphosphino)ferrocene (36
mg, 0.07
mmol) and Pd2(dba)3 (74 mg, 0.08 mmol) were heated to 140 °C in
degassed DMF (3
ml) and H20 (301) overnight. The mixture was allowed to cool to RT and water
(10 ml)
was added, producing a brown precipitate. The precipitate was collected by
filtration and
washed with water and brine. The filtrate was then extracted with ethyl
acetate (3 x 20
mL). The combined organic extracts were dried (MgSO4), filtered and
concentrated in
vacuo to provide 100 mg of the crude product. Purification by gravity silica
gel column
chromatography [DCM] furnished the target product as a yellow oil (70 mg, 33
%).
8H (CDCI3; 250MHz) 7.60 (1 H, d, J 1.5, aromatics), 7.48 (1 H, dd, J 1.5 and
8, aromatics),
7.12-7.01 (2H, m, aromatics), 6.82 (1 H, d, J 8, aromatics), 6.46 (1 H, d, J
8, aromatics),
5.97 (1 H, s, CHO), 5.02 (1 H, d, J 13, CH~HBN), 4.30-4.24 (1 H, m, CH
,HBCHZ), 3:96-3.80
(2H, m, CHAHBN and CH~HBCHZ), 3.82 (3H, s, OCH3), 2.96-2.85 (1 H, m,
CHZCHAHB),
2.76-2.65 (1H, m, CH2CH,oHB), 2.53 (3H, s, NCH3), 2.20 (3H, s, CH3).
Example 22
8-Cyclopropyl-5-methyl-1-(2-methyl-3-methoxy)phenyl-1,3,4,6-tetrahydro-5H-
benz[fj-2,5-oxazocine
8-Bromo-5-methyl-1-(2-methyl-3-methoxy)phenyl-1,3,4,6-tetrahydro-5H-Benz[f]-
2,5-oxazocine (295 mg, 0.71 mmol), cyclopropylboronic acid (89 mg, 0.96 mmol),
P(cyclohexane)3 (24 mg, 0.08 mmol), K3P04 (586 mg, 2.73 mmol) and Pd(OAc)Z
(10.4
mg, 0.04 mmol) were heated at 100 °C in toluene (7 ml) and water (336
w1) overnight.
The reaction mixture was allowed to cool and then diluted with ethyl acetate
(30 ml).
The organic mixture was washed with water (30 ml) and the aqueous phase
extracted
into ethyl acetate (3 x 20 ml). The combined organic extracts were dried
(MgS04),
filtered and concentrated in vacuo to provide 0.29 g of the crude product.
Purification by
silica gel gravity column chromatography [DCM] furnished the target product
(100 mg,
38%) as a yellow oiUsolid. '
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
8H (CDC13; 250MHz) 7.11 (1 H, t, J 8, aromatics), '6.97 (1 H, d, J 1.5,
aromatics), 6.88-6.71
(4H, m, aromatics), 5.98 (1 H, s, CH_O), 4.63 (1 H, d, J 13, CHJ,. HBN), 4.18-
4.06 (1 H, m,
CH ,HBCHZ), 3.89-3.82 (5H, m, CH, HBN, CH H~CH2 and OCH3), 2.98-2.88 (1 H, m,
CH2CHaHB), 2.72-2.64 (1H, m, CHZCH,~HB), 2.50 (3H, s, NCH3), 2.16 (3H, s,
CHa), 1.91
5 1.84 (1 H, m, CHCH2), 0.99-0.91 (2H, m, CH CH2), 0.73-0.66 (2H, m, CHzCH ).
Example 23
8-Bromo-5-methyl-1-(3-methoxy-4-methyl)phenyl-1,3,4,6-tetrahydro-5H-benz[f]-
2,5-
oxazocine
2-({5-bromo-2-[hydroxyl-(3-methoxy-4-methylphenyl)-methyl]-benzyl}
10 methylamino)-ethanol (879 mg, 2.23 mmol) was heated to reflux temperature
in toluene
(9m1) with pTSA (635 mg, 3.34 mmol) under Dean-Stark conditions for 3 hours.
The
reaction mixture was allowed to cool to RT, diluted with ethyl acetate (50 ml)
and washed
with a saturated solution of sodium bicarbonate (60 ml). The aqueous layer was
extracted into ethyl acetate (3 x 50 ml) and the combined organic extracts
were dried
15 (MgS04), filtered and concentrated in vacuo. Purification by gravity silica
gel column
chromatography [EtOAc] gave the target product as a pale straw coloured oil
(23 g,
28%). A second impure batch of product was obtained from the column.
bH (CDCI3; 250MHz) 2.18 (3H, s, CH3), 2.48 (3H, s, NCH3), 2.66 (1 H, ddd,
J14.2, 5.6,
2.7, one of C,H~CHZ), 2.84 (1 H, ddd, J14.2, 8.2, 2.1, one of CH2C,H~), 3.67
(1 H, d, J12.8,
20 one of ArCH~NR~), 3.80 (3H, s, OCH3), 3.86 (1H, ddd, J12.7, 5.6, 2.1, one
of CHzCH ),
4.18 (1 H, ddd, J12.8, 8.2, 2.7, one of CH~CH ), 4.80 (1 H, d, J12.8, one of
ArC,H~NR~),
5.71 (1 H, s, CHOR), 6.68 (1 H, dd, J7.6, 1.2, one of ArH), 6.76 (1 H, bs, one
of Ark, 6.89
(1 H, d, J 8.2, Ark, 7.06 (1 H, d, J7.6, one of Ark, 7.32 (1 H, dd, J8.2, 1.8,
one of Ark,
7.39 (1 H, d, J1.8, one of Ark.
25 Example 24
8-Cyano-5-methyl-1-(3-methoxy-4-methyl)phenyl-1,3,4,6-tetrahydro-5H-bent[fij-
2,5-
oxazocine
8-Bromo-5-methyl-1-(3-methoxy-4-methyl)phenyl-1,3,4,6-tetrahydro-5H-benz[f]-
2,5-oxazocine (154 mg, 0.41 mmol), Zn (2 mg, 0.03 mmol), Zn(OAc)2 (10 mg, 0:05
30 mmol), Zn(CN)2 (29 mg, 0.25 mmol), 1,1'-bis(diphenylphosphino)ferrocene (30
mg, 0.05
mmol) and Pd2(dba)3 (55 mg, 0.06 mmol) were heated to 140°C in DMF (3
ml) and H20
(30 ~I) overnight. The mixture was allowed to cool to RT and water (30 ml) was
added,
producing a brown precipitate. The precipitate was collected by filtration and
washed
with water and ethyl acetate. The filtrate was then washed with brine (2 x 40
ml) and the
combined aqueous phases were extracted with ethyl acetate (3 x 20 ml). The
combined
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
31
organic extracts were dried (MgS04), filtered and concentrated in vacuo to
provide the
crude product. Purification by silica gel gravity column chromatography
[EtOAc]
furnished the target product as a brown oil (75 mg, 57%).
sH (CDCI3; 250MHz) 2.17 (3H, s, CH3), 2.46 (3H, s, NCH3), 2.65 (1 H, ddd,
J14.3, 5.1, 3.0,
one of CH CHz), 2.78 (1 H, ddd, J14.3, 8.6, 2.5, one of CH CH ), 3.71 (1 H, d,
J13.0, one
of ArCHzNR2), 3.79 (3H, s, OCH3), 3.84 (1H, ddd, J12.8, 5.1, 2.5, one of
CHzCHz), 4.26
(1 H, ddd, J12.8, 8.6, 3.0, one of CH CH ), 4.99 (1 H, d, J13.0, one of
ArCHzNR2), 5.73
(1 H, CHOR), 6.63 (1 H, d, J1.5, Ark, 6.72 (1 H, s, Ark, 7.06 (1 H, d, J7.6,
ArH), 7.14 (1 H,
d, J8.2, Ark, 7.47 (1 H, dd, J8.2, 1.8, Ark, 7.53 (1 H, d, J1.5, Ark.
Example 25
5-Methyl-1-(3-methoxy-4-methyl)phenyl-1,3,4,6-tetrahydro-5H-bent[f]-2,5-
oxazocine-8-carboxamide
8-Cyano-5-methyl-1-(3-methoxy-4-methyl)phenyl-1,3,4,6-tetrahydro-5H-benz[fj-
2,5-oxazocine (1.1 g, 3.56 mmol) was heated to reflux temperature in tBuOH (24
ml) with
potassium hydroxide (0.29 mg, 5.34 mmol) for 2 h. At the end of this time, the
reaction
mixture was allowed to cool to RT. The mixture was then washed with brine (50
ml) and
extracted into dichloromethane (3 x 50 ml). The combined organic extracts were
dried
(MgS04), filtered and concentrated in vacuo to provide the crude amide as a
light brown
solid. Purification by silica gel column chromatography [1 % MeOH in DCM to
12%
MeOH in DCM] afforded the title compound (385 mg, 32%).
s (CDCI3; 250MHz) 2.16 (3H, s, CH3), 2.47 (3H, s, NCH3), 2.52-2.62 (1 H, m
(poorly
resolved ddd), one of C,H~CH ), 2.75-2.88 (1 H, m (poorly resolved ddd), one
of C,H~CH ),
3.77 (4H, s, OCH3 and one of ArCHz), 3.81-3.92 (1 H, m (poorly resolved ddd),
one of
CHzCHz), 4.15-4.28 (1 H, m (poorly resolved ddd), one of CHZC,H~), 4.89 (1 H,
d, J12.8,
one of ArCH ), 5.77 (1 H, s, CHOR), 6.76-6.75 (3H, m, aromatic ~, 7.03-7.12
(2H, m,
aromatic ~, 7.59-7.68 (2H, m, aromatic ~.
Example 26
8-Cyclopropyl-5-methyl-1-(3-methoxy-4-methyl)phenyl-1,3,4,6-tetrahydro-5H-
benz[f]-2,5-oxazocine
8-Bromo-5-methyl-1-(3-methoxy-4-methyl)phenyl-1,3,4,6-tetrahydro-5H-Benz[f]-
2,5-oxazocine (154 mg, 0.41 mmol), cyclopropylboronic acid (46 mg, 0.53 mmol),
P(cyclohexane)3 (11 mg, 0.041 mmol), I~P04 (304 mg, 1.43 mmol) and Pd(OAc)2 (5
mg,
0.02 mmol) were heated at 100°C in toluene (3 ml) and water (150 p,1)
for 3 hours 15
mins. The reaction mixture was allowed to cool and then diluted with ethyl
acetate
(30m1). The organic mixture was washed with water (30 ml) and the aqueous
phase
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
32
extracted into ethyl acetate (3 x 20 ml). The combined organic extracts were
dried
(MgS04), filtered and concentrated in vacuo to provide the crude product.
Purification by
silica gel gravity column chromatography [EtOAc] furnished the target product
and
recovered starting material.
8H (CDCI3; 250MHz) 0.64-0.71 (2H, m, two of cyclopropyl CHzCH ), 0.89-0.97
(2H, m,
two of cyclopropyl CHzCHz), 1.80-1.89 (1 H, m, cyclopropyl Cue, 2.16 (3H, s,
CH3), 2.48
(3H, s, NCH3), 2.63 (1 H, ddd, J14.2, 5.6, 2.6, one of CH CHz), 2.85 (1 H,
ddd, J14.2, 8.4,
2.3, one of CHzCHz), 3.70 (1 H, d, J12.7, one of ArCHzNRz), 3.78 (3H, s,
OCH3), 3.84
(1 H, ddd, J12.5, 5.6, 2.3, one of C~CHz), 4.17 (1 H, ddd, J12.5, 8.4, 2.6,
one of
CH~CHZ), 4.70 (1 H, d, J12.7, one of ArCH2NR~), 5.73 (1 H, s, CHOR), 6.69 (1
H, dd, J7.6,
1.2, one of ArH~, 6.79 (1 H, s, Ark, 6.86 (2H, d, J1.2, Ark, 6.94 (1 H, s,
ArH), 7.03 (1 H,
d, J7.6, Ark.
Example 27
9-Bromo-5-methyl-1-(3,4-methylenedioxy)phenyl-1,3,4,6-tetrahydro-5H-bent[fJ-
2,5-
oxazocine
2-{[2-(Benzo-[1,3]-dioxol-yl-hydroxymethyl)-4-bromobenzyl]-methylamino}-
ethanol (148 mg, 0.38 mmol) was heated to reflux temperature with pTSA (107
mg, 0.56
mmol) in toluene (3 ml) under Dean-stark conditions for 3 hours. The mixture
was then
cooled, diluted with ethyl acetate and washed with sat. aq. sodium bicarbonate
solution.
The organic phase was dried (MgS04), filtered and concentrated in vacuo to
provide the
crude product. Purification by silica gel gravity column chromatography
[3%MeOHIEtOAc to 10%MeOH/EtOAc] furnished product (89 mg, 62%).
8H(CDCI3; 250MHz) 2.43 (3H, s, CH3), 2.60 (1 H, ddd, J14.2, 5.3, 2.9, one of
CHZC,H~),
2.78 (1 H, ddd, J14.2, 8.7, 2.4, one of CH CH ), 3.61 (1 H, d, J12.7, one of
ArCHzN), 3.81
(1 H, ddd, J12.5, 5.3, 2.4, one of CH CH ), 4.20 (1 H, ddd, J12.5, 8.7, 2.9,
one of
CHzCHz), 4.84 (1 H, d, J12.7, one of ArCH N), 5.61 (1 H, s, CHOR), 5.93 (2H,
s,
ROCHzOR), 6.71-6.75 (3H, m, ArH), 7.07-7.14 (2H, m, Ark, 7.36 (1 H, dd, J8.0,
4.0,
Ark.
Example 28
9-Cyano-5-methyl-1-(3,4-methylenedioxy)phenyl-1,3,4,6-tetrahydro-5H-bent[fj-
2,5-
oxazocine
A solution of 9-bromo-5-methyl-1-(3,4-methylenedioxy)phenyl-1,3,4,6-tetrahydro-
5H-benz[f]-2,5-oxazocine (89 mg, 0.23 mmol), Zn (1 mg, 0.012 mmol), Zn(CN)2
(16 mg,
0.138 mmol), Zn(OAc)2 (2 mg, 0.011 mmol), 1,1'-bis(diphenylphosphino)ferrocene
(18
mg, 0.05 mmol) and Pd2(dba)3 (11 mg, 0.06 mmol) in DMF (2 ml) and H20 (20 ~I)
was
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
33
degassed under vacuum for 15 minutes and then heated at 140°C
overnight. The
mixture was then allowed to cool and water added. The organics were extracted
into
ethyl acetate and then washed with brine to remove the DMF. The combined
organic
washings were dried (MgS04), filtered and concentrated in vacuo to provide the
crude
product. Purification by silica gel column chromatography [EtOAc] furnished
product (21
mg, 27%).
8H (CDCI3; 250MHz) 2.45 (3H, s, NCH3), 2.63 (1 H, ddd, J14.3, 5.2, 2.8, one of
CH CH ),
2.76 (1 H, ddd, J14.3, 8.4, 2.4, one of CH CHz), 3.70 (1 H, d, J12.8, one of
ArC,H~NR2),
3.81 (1 H, ddd, J12.6, 5.2, 2.4, one of CH~CH ), 4.20 (1 H, ddd, J12.6, 8.4,
2.8, one of
C,H~CHz), 4.90 (1 H, d, J12.8, one of ArCH NRZ), 5.69 (1 H, s, CHOR), 5.94
(2H, s,
ROCHzOR), 6.69-6.78 (3H, m, Ark, 7.27-7.33 (2H, m, Ark, 7.51 (1 H, dd,- ,17.9,
1.5,
ArH~.
Example 29
9-Cyclopropyl-5-methyl-1-(3,4-methylenedioxy)phenyl-1,3,4,6-tetrahydro-5H-
benz[f]-2,5-oxazocine
A mixture of 9-bromo-5-methyl-1-(3,4-methylenedioxy)phenyl-1,3,4,6-tetrahydro-
5H-benz[f]-2,5-oxazocine (280 mg, 0.75 mmol), cyclopropylboronic acid (84 mg,
0.97
mmol), K3P04 (555 mg, 2.61 mmol), Pd(OAc)2 (8.4 mg, 0.04 mmol) and
P(cyclohexane)3
(21 mg, 0.075 mmol) in toluene (5 ml) and HZO (250 ~,I) was heated at
100°C for 3 hours,
during which time it turned black. After cooling, water (12 ml) was added and
the
organics were extracted into ethyl acetate. The combined organic extracts were
dried
(MgS04), filtered and concentrated in vacuo to provide the crude product.
Purification
(repeated) by silica gel column chromatography [EtOAc] furnished product (56
mg,
22%).
8H (CDCL3; 250MHz) 0.58-0.63 (2H, m, two of cyclopropyl CH C,H~), 0.86-0.94
(2H, m,
two of cyclopropyl CH CH ), 1.75-1.84 (1 H, m, cyclopropyl CH), 2.44 (3H, s,
CH3), 2.55-
2.63 (1 H, m, one of CHzCH ), 2.75-2.84 (1 H, m, one of CH C,H~), 3.62 (1 H,
d, J12.5, one
of ArC~NRz), 3.81 (1 H, ddd, J12.5, 5.6, 2.4, one of CHzCH ), 4.14-4.22 (1 H,
m, one of
CHzCH ), 4.79 (1 H, d, J12.5, one of ArCH NRZ), 6.63 (1 H, s, CHOR), 5.91 (2H,
s,
ROC~OR), 6.74 and 6.75 (4H, 2 x s, Ark, 6.87 (1 H, dd, J7.6, 1.9, Ark, 7.10 (1
H, d,
J7.9).
Example 30
8-Bromo-5-methyl-1-(3,4-methylenedioxy)phenyl-1,3,4,6-tetrahydro-5H-benz[f]-
2,5-
oxazocine
2-{[2-(Benzo-[1,3]-dioxol-5-yl-hydroxymethyl)-5-bromobenzyl]-methylamino~-
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
34
ethanol (715 mg, 1.82 mmol) and pTSA (519 mg, 2.73 mmol) were heated to reflux
temperature in toluene (8 ml) under Dean-Stark conditions for 2 hours. Once
cooled to
RT, the reaction mixture was diluted with ethyl acetate and washed with sat.
aq. sodium
bicarbonate solution. The organic phases were dried (MgS04), filtered and
concentrated
in vacuo to provide the crude product. Purification by silica gel column
chromatography
[100% EtOAc to 5%MeOH/EtOAc] yielded 214 mg, (31 %).
8H (CDCI3; 250MHz) 2.45 (3H, s, CH3), 2.61 (1 H, ddd, J14.1, 5.8, 2.8, one of
C,H~CH2),
2.79 (1 H, ddd, J14.1, 8.2, 2.2, one of CH CH ), 3.58 (1 H, d, J12.8, one of
ArCHzNRz),
3.81 (1 H, ddd, J12.5, 5.8, 2.2, one of CHzCH ), 4.78 (1 H, ddd, J12.5, 8.2,
2.8, one of
CHzCHz), 4.80 (1 H, d, J12.8, one of ArCHzNRz), 5.64 (1 H, s, CHOR), 5.91 (2H,
s,
ROCHzOR), 6.70-6.77 (3H, m, Ark, 6.87 (1 H, d, J8.3, Ark, 7.29-7.37 (2H, m,
Ark.
Example 31
8-Cyano-5-methyl-1-(3,4-methylenedioxy)phenyl-1,3,4,6-tetrahydro-5H-bent[f]-
2,5-
oxazocine
8-Bromo-5-methyl-1-(3,4-methylenedioxy)phenyl-1,3,4,6-tetrahydro-5H-bent[f]-
2,5-oxazocine (99 mg, 0.26 mmol), Zn (1 mg, 0.015 mmol), Zn(OAc)2 (6 mg,
0'.033
mmol), Zn(CN)2 (18 mg, 0.156 mmol), 1,1'-bis(diphenylphosphino)ferrocene (18
mg,
0.054 mmol) and Pd2(dba)3 (33 mg, 0.036 mmol) were degassed and then heated at
140°C overnight. The reaction mixture was cooled and water added. The
oraanics were
extracted three times with ethyl acetate,and the combined organic extracts
were washed
with brine, dried (MgS04), filtered and concentrated in vacuo to provide the
crude target
as a black oil. Purification by silica gel column chromatography [EtOAc]
furnished
product 21 mg, (25%).
&H (CDCI3; 250MHz) 2.46 (3H, s, CH3), 2.63 (1 H, ddd, J14.3, 5.4, 3.1, one of
CHzCH ),
2.75 (1 H, ddd, J14.3, 8.3, 2.6, one of CH CHz), 3.64 (1 H, d, J12.8, one of
ArCHzNRz),
3.81 (1 H, ddd, J12.7, 5.4, 2.6, one of CH C,H~), 4.24 (1 H, ddd, J12.7, 8.3,
3.1, one of
CH CHz), 4.97 (1 H, d, J12.8, one of ArCHzNR2), 5.68 (1 H, CHOR), 5.93 (2H, s,
ROCH OR), 6.69-6.78 (3H, m, Ark, 7.12 (1 H, d, J7.9, Ark, 7.45- 7.52 (2H, m,
Ark.
Example 32
8-Cyclopropyl-5-methyl-1-(3,4-methylenedioxy)phenyl-1,3,4,6-tetrahydro-5H-
benz[f]-2,5-oxazocine
8-Bromo-5-methyl-1-(3,4-methylenedioxy)phenyl-1, 3,4,6-tetrahydro-5H-benz[f]-
2,5-oxazocine (90 mg, 0.24 mmol), cyclopropylboronic acid (26.8 mg, 0.312
mmol),
143P04 (178 mg, 0.84 mmol), Pd(OAc)2 (3 mg, 0.012 mmol) and P(cyclohexane)3 (7
mg,
0.024 mmol) were heated to 100°C in toluene (2 ml) and water (100 ~,I)
for 3 hours. The
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
reaction mixture was cooled, diluted with water and extracted with ethyl
acetate. The
combined organic extracts were dried (MgSO~), filtered and concentrated in
vacuo to
provide the crude product. Purification was by silica gel column
chromatography [100
%EtOAc to 5%MeOH/EtOAc] furnished 8-cyclopropyl-(3,4-methylenedioxy)-nefopam
49
5 mg,(61%).
8H (CDCI3; 250MHz) 0.67-0.72 (2H, m, two of cyclopropyl CH_zCH2), 0.91-0.99
(2H, m,
two of cyclopropyl C_HzCH ), 2.48 (3H, s, CH3), 2.61 (1 H, ddd, J14.2, 5.7,
2.6, one of
CH2CH~), 2.83 (1 H, ddd, J14.2, 8.4, 2.5, one of CH2CH ), 3.64 (1 H, d, J12.8,
one of
ArCH2NR2), 3.83 (1 H, ddd, J12.5, 5.7, 2.5, one of CH_zCHz), 4.42 (1 H, ddd,
J12.5, 8.4,
10 2.6, one of CHzCHz), 4.74 (1 H, d, J12.8, one of ArCH2,NR2), 5.67 (1 H, s,
CHOH), 5.91
(2H, s, ROCH,aOR), 6.75 (3H, s, ArH), 6.88-6.9 (3H, m, ArH~
Example 33
8-Bromo-5-methyl-1-(3-ethoxy)phenyl-1,3,4,6-tetrahydro-5H-benz[f]-2,5-
oxazocine
2-(~5-Bromo-2-[hydroxyl-(3-ethoxy-phenyl)-methyl]-benzyl~-methylamino)-ethanol
15 (1.06 g, 2.69 mmol) was heated to reflux temperature under Dean-Stark
conditions with
pTSA (0.78 g, 4.03 mural) in toluene (40 ml) for 2.5 h. The reaction mixture
was allowed
to cool and then diluted with ethyl acetate (20 ml). The mixture was washed
with sat. aq
sodium bicarbonate solution (2 x 20 ml) and the combined organic extracts were
dried
(MgS04), filtered and concentrated in vacuo to provide 0.94 g of crude
product.
20 Purification by silica gel gravity column chromatography [EtOAc] gave the
product (0.60
g, 59%) as a colourless oil.
~H (CDC13; 250MHz) 7.37-6.87 (7H, m, aromatics), 5.71 (1 H, s, CH-O), 4.77 (1
H, d, J 13,
ArCH~HeN), 4.14 (1 H, ddd, J 13, 8 and 2.5, C"H~HBCH2), 4.00 (2H, q, J 7,
C,H~CH3), 3.84
(1 H, ddd, J 13, 8 and 2.5, CHAHBCHZ), 3.62 (1 H, d, J 13, ArCH,qHBN), 2.85-
2.79 (1 H, m,
25 C,H~HBCHZ), 2.68-2.60 (1H, m, CH4HBCH2), 2.46 (3H, s, NCH3), 1.39 (3H, t, J
7,
CH2GHs).
Example 34
8-Cyano-5-methyl-1-(3-ethoxy)phenyl-1,3,4, 6-tetrahydro-5H-bent[f]-2,5-
oxazocine
8-Bramo-5-methyl-1-(3-ethoxy)phenyl-1,3,4,6-tetrahydro-5H-benz[f]-2, 5
30 oxazocine (250 mg, 0.65 mmol), Zn (3 mg, 0.05 mmol), Zn(OAc)2 (15 mg, 0.077
mmol),
Zn(CN)2 (46 mg, 0.4 mmol), Pd2(dba)3 (93 mg, 0.1 mmol) and 1,1'-
bis(diphenyfphosphino)ferrocene (43 mg, 0.077 mmol) were heated to
140°C in DMF (5 '
ml) and Hz0 (100 ~I) for 4h. The black reaction mixture was allowed to cool to
RT and
diluted with water. The resulting brown precipitate was collected by
filtration and washed
35 with water then brine. The filtrate was extracted with ethyl acetate (2 x
60m1). The
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
36
combined organic extracts were dried (MgS04), filtered and concentrated in
vacuo to
provide the crude product as a brown oil. Purification by silica gel gravity
column
chromatography [DCM] gave clean product (56 mg, 27%).
8H(CDCI3; 250MHz) 7.50-6.78 (7H, m, aromatics), 5.72 (1 H, s, CHO), 4.96 (1 H,
d, J 13,
ArCH ,HB-N), 4.39-4.19 (1 H, m, CHAHBCHZ), 3.98 (2H, q, J 7, CHzCH3), 3.85-
3.79 (1 H,
m, CH~HBCHZ), 3.67 (1 H, d, J 13, ArCH,~HB-N), 2.82-2.58 (2H, m, CHzCH2), 2.43
(3H, s,
NCH3), 1.38 (3H, t, J 7, CHZCH3).
Example 35
8-Cyclopropyl-5-methyl-1-(3-ethoxy)phenyl-1,3,4,6-tetrahydro-5H-benz[f]-2,5-
oxazocine
8-Bromo-5-methyl-1-(3-ethoxy)phenyl-1,3,4,6-tetrahydro-5H-benz[fj-2, 5-
oxazocine (100 mg, 0.26 mmol), cyclopropylboronic acid (29 mg,0.33 mmol),
IC3PO4 (193
mg, 0.91 mmol), Pd(OAc)2 (3 mg, 0.013 mmol) and P(cyclohexane)3 (8 mg, 0.026
mmol)
were heated to 100°C in toluene (2 ml) and HBO (100 w1) for 4 hours.
After cooling to RT,
water (2 ml) was added and the mixture extracted with ethyl acetate, washed
with brine,
dried (MgS04), filtered and concentrated in vacuo. Purification by silica gel
gravity
column chromatography [DCM] furnished clean product (20 mg, 23%) as a
colourless oil.
8H(CDC13; 250MHz) 7.26-6.76 (7H, m, aromatics), 5.71 (1 H, s, CHO), 4.75 (1 H,
d, J 12.5,
ArCH,o,HB-N) 4.22-4.12 (1 H, m, CH ,HBCHZ), 3.99 (2H, q, J 7, CHZCH3), 3.88-
3.79 (1 H, m,
CH~HBCH2), 3.81 (1 H, d, J 12.5, ArCH .,FHB-N), 2.90-2.61 (2H, m, CH CHZ),
2.50 (3H, s,
NCH3), 1.89-1.82 (1 H, m, CHCH2), 1.39 (3H, t, J 7, CHZCH3), 0.96-0.92 (2H, m,
CHzCHz), 0.70-0.67 (2H, m, CHZCHz).
Example 36
9-Bromo-5-methyl-1-(3-ethoxy)phenyl-1,3,4,6-tetrahydro-5H-bent[fi]-2,5-
oxazocine
2-({4-Bromo-2-[hydroxyl-(3-ethoxy-phenyl)-methyl]-benzyl}-methylamino)-ethanol
(1.20 g, 3.04 mmol) was heated to reflux temperature under Dean-Stark
conditions with
pTSA (0.88 g, 4.57 mmol) in toluene (50 ml) for 4 h. The reaction mixture was
allowed to
cool and then diluted with ethyl acetate (25 ml). The mixture was washed with
sat. aq
sodium bicarbonate solution (2 x 25 ml), and the combined organic extracts
were dried
(MgS04), filtered and concentrated in vacuo to provide 1.4 g of crude product.
Purification by silica gel gravity column chromatography [EtOAc] gave product
(0.59 g,
51 %) as a colourless oil.
8H (CDCI3; 250MHz) 7.37-6.76 (7H, m, aromatics), 5.67 (1 H, s, CH-O), 4.79 (1
H, d, J 13,
ArC,H~HeN), 4.16 (1 H, ddd, J 13, 8 and 2.5, CH ,HBCH2), 4.01 (2H, q, J 7,
CH~CH3), 3.81
(1 H, ddd, J 13, 8 and 2.5, CH,~HBCHZ), 3.63 (1 H, d, J 13, ArCH,~HeN), 2.84-
2.78 (1 H, m,
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
37
CH o,HBCH2), 2.66-2.63 (1 H, m, CH,~HBCHZ), 2.44 (3H, s, NCH3), 1.40 (3H, t, J
7,
CHZCHs). .
Example 37
9-Cyano-5-methyl-1-(3-ethoxy)phenyl-1,3,4,6-tetrahydro-5H-benz[f]-2,5-
oxazocine
9-Bromo-5-methyl-1-(3-ethoxy)phenyl-1,3,4,6-tetrahydro-5H-Benz[f]-2,5-
oxazocine (150 mg, 0.39 mmol),. Zn (2 mg, 0.03 mmol), Zn(OAc)2 (9 mg, 0.046
mmol),
Zn(CN)2 (28 mg, 0.24 mmol), Pd2(dba)3 (56 mg, 0.06 mmol) and 1,1'-
bis(diphenylphosphino)ferrocene (25 mg, 0.046 mmol) were heated to
140°C in DMF (3
ml) and HZO (30 ~,I) overnight. The black reaction mixture was allowed to cool
to RT and
diluted with water. The resulting brown precipitate was collected by
filtration and washed
with water then brine. The filtrate was extracted with ethyl acetate (2 x 80
ml), and the
combined organic extracts dried (MgS04), filtered and concentrated in vacuo to
provide
the crude product. Purification by silica gel gravity column chromatography
(DCM] gave
clean product (12 mg, 10%).
8H(CDCI3; 250MHz) 7.53-6.80 (7H, m, aromatics), 5.75 (1 H, s, CHO), 4.87 (1 H,
d, J 13,
ArCH o,HBN), 4.29-4.14 (1 H, m, CH 4HBCH2), 4.01 (2H, q, J 7, CHZCH3), 3.88-
3.78 (1 H, m,
CH,o HBCH2), 3.73 (1 H, d, J 13, ArCH,~HBN), 2.82-2.71 (1 H, m, CH~HB-CH2),
2.68-2.59
(1 H, m, CH,~ HeCH2), 2.46 (3H, s, N-CH3), 1.39 (3H, t, J 7, CHZCH3).
Example 38
9-Cyclopropyl-5-methyl-1-(3-ethoxy)phenyl-1,3,4,6-tetrahydro-5H-benz[fi]-2,5-
oxazocine
9-Bromo-5-methyl-1-(3-ethoxy)phenyl-1,3,4,6-tetrahydro-5H-bent[f]-2,5-
oxazocine (140 mg, 0.36 mmol), cyclopropylboronic acid (41 mg,0.45 mmol),
I~P04 (269
mg, 1.26 mmol), Pd(OAc)Z (5 mg, 0.02 mmol) and P(cyclohexane)3 (11 mg, 0.04
mmol)
were heated to 100°C in toluene (3 ml) and H20 (150 ~I) for 4 hours.
After cooling to RT,
water (2 ml) was added and the mixture extracted with ethyl acetate, washed
with brine,
dried (MgS04), filtered and concentrated in vacuo. Purification by silica gel
gravity
column chromatography [DCM) gave product (61 mg, 50%) as a colourless oil.
8H(CDCI3; 250MHz) 7.25-6.76 (7H, m, aromatics), 5.69 (1 H, s, CHO), 4.79 (1 H,
d, J 13,
ArC,H~HBN), 4.23-4.16 (1 H, m, CHJ,. HBCHZ), 4.01 (2H, q, J 7, CHzCH3), 3.89-
3.81 (1 H, m,
CH~HBCH2), 3.70 (1 H, d, J 13, ArCH,o HBN), 2.88-2.78 (1 H, m, CHI. HB-CH2),
2.68-2.60
(1 H, m, CH, HBCH2), 2.48 (3H, s, N-CH3), 1.83-1.72 (1 H, m, CHCH2), 1.39 (3H,
t, J 7,
CHzCH3), 0.92-0.88 (2H, m, CHzCH2), 0.62-0.58 (2H, m, CHZCHz).
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
38
Example 39
8-Bromo-5-methyl-1-(3-trifluoro methoxy)phenyl-1,3,4,6-tetrahydro-5H-bent[fj-
2,5-
oxazocine
2-(~5-bromo-2-[hydroxyl-(3-trifluoromethoxy-phenyl)-methyl]-benzy1}
methylamino)-ethanol (635 mg, 1.47 mmol) was heated to reflux under Dean-Stark
conditions with pTSA (420 mg, 2.21 mmol) in toluene (6 ml) for 2 hours. The
reaction
mixture was allowed to cool to RT and then diluted with ethyl acetate (30 ml).
The
mixture was washed with sat. aq. sodium bicarbonate solution (50 ml) and was
extracted
into ethyl acetate. The combined organic extracts were dried (MgS04), filtered
and
concentrated in vacuo to provide the crude cyclised product. Purification by
silica gel
gravity column chromatography [EtOAc] furnished the target product (116 mg,
19%).
8H (CDCI3; 250MHz) 2.47 (3H, s, CH3), 2.67 (1 H, ddd, J14.3, 6.1, 2.5, one of
CH~C~),
2.84 (1 H, ddd, J14.3, 7.8, 2.2, one of CE~CH ), 3.67 (1 H, d, J12.8, one of
ArCHzNR2),
3.86 (1 H, ddd, J12.8, 6.1, 2.2, one of CHzCH ), 4.13 (1 H, ddd, J12.8, 7.8,
2.5, one of
CH CHz), 4.61 (1 H, d, J12.8, one of ArCf~NR2), 5.79 (1 H, s, CHOR), 6.85 (1
H, d, J8.2,
Arf~, 7.11-7.19 (3H, m, Arf~, 7.31-7.36 (3H, m, ArHa.
Example 40
8-Cyano-5-methyl-1-(3-trifluoromethoxy)phenyl-1,3,4,6-tetrahydro-5H-benz[fJ-
2,5-
oxazocine
8-Bromo-5-methyl-1-(3-trifluoromethoxy)phenyl-1,3,4,6-tetrahydro-5H-benz[f]-
2,5-
oxazocine (116 mg, 0.28 mmol), Zn (1 mg, 0.02mmol), Zn(OAc)2 (7 mg, 0.038
mmol),
Zn(CN)Z (20 mg, 0.17 mmol), Pd2(dba)3 (40 mg, 0.04 mmol) and 1,1'-bis-
(diphenylphosphine)ferrocene (20 mg, 0.036 mmol) were heated to 140°C
in DMF (3 ml)
and H2O (30 ~,I) overnight. The black reaction mixture was allowed to cool and
diluted
with water. The resulting brown precipitate v~ras collected by filtration and
washed with
ethyl acetate. The filtrate was washed with brine (2 x 60 ml) and the combined
aqueous
phases were then washed with ethyl acetate (2 x 80 ml). The combined organic
extracts
were dried (MgS04) and concentrated in vacuo to provide the crude product.
Purification
by silica gel gravity column chromatography [1:1 ethyl acetate/heptane)
furnished the
desired product (9 mg, 9%).
SH(CDCI3; 250MHz) 2.50 (3H,s, CH3), 2.61-2.87 (2H, m, two of CH CH ), 3.76 (1
H, d,
J13.1, one of ArCHzNR2), 3.87 (1 H, ddd, J12.7, 5.4, 2.5, one of C~CHZ), 4.25
(1 H, ddd,
J12.7, 8,4, 3.1, one of CHzCHz), 4.86 (1H, d, J13.1, one of ArCHzNRz), 5.82
(1H, s,
CHOR), 7.11-7.16 (4H, m, ArE~, 7.33-7.39 (1 H, m, Ark, 7.52 (1 H, dd, J7.9,
1.8, Arf~,
7.57 (1 H, bs, Ark.
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
39
Example 41
8-Cyclopropyl-5-methyl-1-(3-trifluoromethoxy)phenyl-1,3,4,6-tetrahydro-5H-
benz[f]-
2,5-oxazocine
8-Bromo-5-methyl-1-(3-trifluoromethoxy)phenyl-1,3,4,6-tetrahydro-5 H-benz[f]-
2,5-
oxazocine (160 mg, 0.39 mmol), cyclopropylboronic acid (43 mg,0.5 mmol), K3PO4
(288
mg, 1.36 mmol), Pd(OAc)2 (4 mg, 0.019 mmol) and P(cyclohexane)3 (11 mg, 0.039
mmol) were heated to 100°C in toluene (2 ml) and H20 (100 ~,I) for 4
hours. The
reaction mixture, now black in colour, was allowed to cool to RT. Water (50
ml) was
added and the mixture extracted into ethyl acetate (3 x 80 ml). The combined
organic
extracts were dried (MgS04), filtered and concentrated in vacuo to provide the
crude
cyclopropyl analogue. Purification by silca gel column chromatography [1:1
EtOAc/heptane, then 100% EtOAc] furnished 8-cyclopropyl-(m-trifluoromethoxy)-
nefopam.
sH (CDCI3; 250MHz) 0.67-0.74 (2H, m, two of cyclopropyl C,H~C~), 0.89-1.00
(2H, m,
two of cyclopropyl CH CHz), 1.82-1.93 (1 H, m, cyclopropyl Cue, 2.51 (3H, s,
CH3), 2.68
(1 H, ddd, J14.2, 5.7, 2.5, one of CHzCHz), 2.89 (1 H, ddd, J14.2, 8.2, 2.0,
one of
CH,zCHz), 3.74 (1 H, d, J12.8, one of ArCH NRZ), 3.87 (1 H, ddd, J12.7, 5.7,
2.0, one of
CH CH ), 4.13 (1 H, ddd, J12.7, 8.2, 2.5, one of CH CH ), 4.58 (1 H, d, J12.5,
one of
ArC,H~NR2), 5.81 (1 H, s, CHOR), 6.84-6.97 (3H, m, Ark, 7.09-7.20 (3H, m, Ark,
7.29
7.37 (1 H, m, Ark.
Example 42
9-Bromo-5-methyl-1-(3-trifluoromethoxy)phenyl-1,3,4,6-tetrahydro-5H-bent[f]-
2,5-
oxazocine
Crude 2-(~4-bromo-2-[hydroxyl-(3-trifluoromethoxy-phenyl)-methyl]-benzy1}-
methylamino)-ethanol (1.159 g, 2.69 mmol) was heated to reflux with pTSA (768
mg,
4.04 mmol) in xylene (12m1) with Dean-Stark for 4 hours, 45 mins. The reaction
mixture
was allowed to cool and then washed with a saturated solution of sodium
bicarbonate
(100 ml). The organics were extracted into ethyl acetate (2 x 100 ml) and the
combined
organic extracts dried (MgS04), filtered and concentrated in vacuo to provide
the crude
product as a 2:1 mixture with starting material. Purification by silica gel
colurrin
chromatography provided the target product (189 mg, 17%).
8H (CDCI3; 250MHz) 2.44 (3H, s, CH3), 2.64 (1 H, ddd, J15.0, 5.0, 2.5, one of
CH CH ),
2.81 (1 H, ddd, J15.0, 7.5, 2.5, one of CHzCH ), 3.67 (1 H, d, J12.5, one of
ArC,H~NRz),
3.84 (1 H, ddd, J12.5, 5.0, 2.5, one of CHzCH ), 4.14 (1 H, ddd, J12.5, 5.0,
2.5, one of
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
CHzCHz), 4.65 (1 H, d, J12.5, one of ArCHaNR2), 5.75 (1 H, s, CHOR), 7.09-7.19
(5H, m,
Ark, 7.34 (1 H, d, J8.0, one of Ark, 7.40 (1 H, dd, J8.0, 1.9, one of Ark.
Example 43 ,
9-Cyclopropyl-5-methyl-1-(3-trifluoromethoxy)phenyl-1,3,4,6-tetrahydro-5H-
Benz[fj-
5 2,5-oxazocine
9-Bromo-5-methyl-1-(3-trifluoromethoxy)phenyl-1,3,4,6-tetrahydro-5H-benz[f]-
2,5-
oxazocine (95 mg, 0.28 mmol), I~P04 (205 mg, 0.97 mmol), cyclopropylboronic
acid (31
mg, 0.36 mmol), P(cyclohexane)3 (8 mg, 0.028 mmol) and Pd(OAc)2 (3 mg, 0.014
mmol)
were heated at 100°C in toluene (2m1) and water (100 ~,I). The reaction
mixture turned
10 black in colour after about 20 mins. At the end of 3 hours, the mixture was
allowed to
cool to RT, and water (30 ml) was added. The organics were extracted into
ethyl acetate
(2 x 30 ml) and the combined extracts dried (MgS04), filtered and concentrated
in vacuo.
Purification by silica gel gravity column chromatography [EtOAc] furnished
product.
8H (CDCI3; 250MHz) 0.58-0.63 (2H, m, two of cyclopropyl CH CH ), 0.90-0.94
(2H, m,
15 two of CH CH ), 1.73-1.79 (1 H, m, cyclopropyl Cue, 2.45 (3H, s, CH3), 2.68
(1 H, ddd,
J15.0, 5.0, 2.5, one of CH2C~,, 2.87 (1 H, ddd, J15.0, 7.5, 2.5, one of
CHZC,H~), 3.75
(1 H, d, J12.5, one of ArCH NR2), 3.88 (1 H, ddd, J 12.5, 5.0, 2.5, one of
CHzCHz), 4.19
(1 H, ddd, J 12.5, 7.5, 2.5, one of CH CHI), 4.67 (1 H, J12.5, one of ArCH
NRZ), 5.75 (1 H,
s, CHOR), 6.72 (1 H, d, J2.5, one of Ark, 6.93 (1 H, dd, J7.5, 2.5, Ark, 7.10-
7.19 (4H, m,
20 Ark, 7.29-7.38 (1 H, Ark
Example 44
9-Bromo-5-methyl-1-(2-methyl)phenyl-1,3,4,6-tetrahydro-5H-Benz[f]-2,5-
oxazocine
2-[(4-bromo-2-(hydroxyl-o-tolylmethyl)-benzyl)-methylamino]-ethanol (790 mg,
2.18 mmol) was dissolved in toluene (8 ml) at RT. pTSA (623 mg, 3.27 mmol) was
added
25 and the resulting mixture was heated to reflux temperature under Dean-Stark
conditions
for 1.5 hours. Once cooled to RT, the mixture was diluted with ethyl acetate
and washed
with sat. aq. solution of sodium bicarbonate. The organic extracts were dried
(MgS04),
filtered and concentrated in vacuo to provide the crude material as an oil.
Purification by
silica gel gravity column chromatography [20%EtOAc /heptane to 30%EtOAc]
furnished
30 the product (357 mg, 55% corrected yield)
8H (CDCI3; 250MHz) 2.31 (3H, s, ArCH3), 2.44 (3H, s, NCH3), 2.63 (1 H, ddd,
J14.4, 5.3,
2.4, one of CH CHz), 2.86 (1H, ddd, J14.4, 8.9, 2.1, one of CHzCH ), 3.76 (1H,
d, J,13.0,
one of ArCH NR2), 3.85 (1 H, ddd, J12.7, 5.3, 2.1, one of CHzCH ), 4.12 (1 H,
ddd, J12.7,
8.9, 2.4, one of CH CHz), 4.68 (1 H, d, J13.0, one of ArCH NRZ), 5.95 (1 H, s,
CHOR),
35 7.02-7.22 (6H, m, Ark, 7.38 (1 H, dd, J8.2, 2.1, Ark.
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
41
Example 45
9-Cyano-5-methyl-1-(2-methyl)phenyl-1,3,4,6-tetrahydro-5H-benz[f]-2,5-
oxazocine
9-Bromo-5-methyl-1-(2-methyl)phenyi-1, 3,4,6-tetrahydro-5H-bent[f]-2, 5-
oxazocine (156 mg, 0.45 mmol), Zn dust (2 mg, 0.03 mmol), Zn(CN)2 (32 mg, 0.27
mmol), Zn(OAc)2 (10 mg, 0.054 mmol), 1,1'-bis(diphenylphosphino)ferrocene (30
mg,
0.054 mmol) and Pd2(dba)3 (55 mg, 0.06 mmol) were heated to 140°C in
DMF (3 ml) and
water (30 w1) under a nitrogen atmosphere over-night. After cooling, water was
added
and the resulting blacklbrown precipitate was collected by suction filtration
and washed
well with ethyl acetate. The filtrate was washed with brine and extracted into
ethyl
acetate. The combined organic extracts were dried (MgS04), filtered and
concentrated
in vacuo to provide the crude material as a very dark brown oil. Purification
by silica gel
gravity column chromatography [ EtOAc] furnished the product (79 mg, 60%).
sH (CDCI3; 250MHz) 2.28 (3H, s, ArCH3), 2.45 (3H, s, ArCH3), 2.65 (1 H, ddd,
J14.4, 5.5,
2.4, one of CHzCHa), 2.85 (3H, ddd, J14.4, 8.9, 2.1, one of C,H~CH ), 3.82-
3.90 (2H, m,
one of CH CHz and one of ArC,H~NRz), 4.07-4.17 (1 H, m, one of CH~CH ), 4.74
(1 H, d,
J12.8, one of ArCH~NR2), 6.02 (1 H, s, CHOR), 7.05 (1 H, d, J6.7, Ark, 7.14-
7.27 (4H, m,
Ark, 7.34 (1 H, d, J7.9, Ark, 7.54 (1 H, dd, J7.9, 1.7, Ark.
Example 46
9-Cyclopropyl-5-methyl-1-(2-methyl)phenyl-1,3,4,6-tetrahydro-5H-bent[f]-2,5-
oxazocine
9-Bromo-5-methyl-1-(2-methyl)phenyl-1,3,4,6-tetrahydro-5H-bent[f]-2,5-
oxazocine (188 mg, 0.55 mmol) was dissolved in toluene (4 ml) and water (200
~,I) at RT.
Cyclopropylboronic acid (61 mg, 0.71 mmol), I~P04 (409 mg, 1.93 mmol),
Pd(OAc)2 (6
mg, 0.03 mmol) and P(cyclohexane)3 (15.4 mg, 0.055 mmol) were added. The
mixture
was heated to 100°C for a total of 3 hours, during which time the
reaction mixture
became black. After cooling to RT, the mixture was diluted with water and
extracted into
ethyl acetate. The organic phases were dried (MgS04), filtered and
concentrated in
vacuo to provide the crude product. Purification by silica gel gravity column
chromatography [EtOAc] furnished the product (129 g, 81 %).
8H (CDCI3; 250MHz) 0.54-0.60 (2H, m, two of cyclopropyl CHzCH ), 0.86-0.89
(2H, m,
two of cyclopropyl CH CHz), 1.69-1.80 (1 H, m, cyclpropyl Cue, 2.34 (3H, s,
ArCH3), 2.45
(3H, s, ArCH3), 2.62 (1 H, ddd, J14.3, 5.5, 2.4,'one of CHzCHz), 2.87 (1 H,
ddd, J14.3, 8.5,
2.1, one of C~CHz), 3.75 (1 H, d, J13.0, one of ArCH NRZ), 3.85 (1 H, ddd,
J12.7, 5.5,
2.1, one of CHzCHz), 4.11 (1 H, ddd, J12.7, 8.5, 2.4, one of CHzCHz), 4.64 (1
H, d, J13.0,
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
42
one of ArCHzNR2), 5.96 (1 H, s, CHOR), 6.62 (1 H, d, J1.8, Ark, 6.89 (1 H, dd,
J7.9, 1.8,
ArHa, 7.05-7.20 (5H, Ark.
Example 47
8-Bromo-5-methyl-1-(2-methyl)phenyl-1,3,4,6-tetrahydro-5H-benz[f]-2,5-
oxazocine
2[(5-bromo-2-(hydroxyl-o-tolylmethyl)-benzyl)methylamino]-ethanol (777 mg,
2.16
mmol) was dissolved in toluene (8 ml) at RT. pTSA (616 mg, 3.24 mmol) was
added, and
the resulting mixture heated at reflux temperature under Dean-Stark conditions
for 4
hours. The reaction was allowed to cool to RT and then diluted with ethyl
acetate, and
washed with sat. aq. sodium bicarbonate solution. The organic washings were
dried
(MgS04), filtered and concentrated in vacuo to provide the impure product.
Purification
by silca gel gravity column chromatography [1:1 EtOAc /heptane to 100% EtOAc]
gave
the desired product (504 mg, 64%).
8H (CDCI3; 250MHz) 2.29 (3H, s, ArCH3), 2.46 (3H, s, NCH3), 2.66 (1 H, ddd,
J14.4, 5.6,
2.4, one of CHzCH ), 2.89 (1H, ddd, J14.4, 8.5, 2.1, one of C,H~CH2), 3.77
(1H, d, J13.1,
one of ArCH NR2), 3.86 (1 H, ddd, J12.7, 5.6, 2.1, one of CHzCH ); 4.09 (1 H,
ddd, J12.7,
8.5, 2.4, one of C,H~CHZ), 4.64 (1 H, d, J13.1, one of ArC,H~NR~), 5.97 (1 H,
s, CHOR),
6.74 (1 H, d, J8.2, Ark, 7.07-7.20 (4H, m, Ark, 7.29 (1 H, dd, J8.6, 2.1,
ArHJ, 7.41 (1 H,
d, J2.1, Ark.
Example 48
8-Cyano-5-methyl-1-(2-methyl)phenyl-1,3,4,6-tetrahydro-5H-benz[f]-2,5-
oxazocine
8-Broriio-5-methyl-1-(2-methyl)phenyl-1,3,4,6-tetrahydro-5H-benz[f]-2,5-
oxazocine (207 mg, 0.60 mmol), Zn (2 mg,0.03 mmol), Zn(OAc)~ (13 mg, 0.071
mmol),
Zn(CN)Z (42 mg, 0.36 mmol), 1,1'-bis(diphenylphosphino)ferrocene (40 mg, 0.072
mmol)
and Pd2(dba)3 (75 mg, 0.082 mmol) were heated to 140°C in DMF (5 ml)
and HZO (50 w1)
overnight. The black reaction mixture was cooled and water added, producing a
brown
precipitate. The precipitate was washed with ethyl acetate and the filtrate
washed with
brine to remove the DMF. The organic phase was dried (MgSO4), filtered and
concentrated in vacuo to provide the crude product. Purification by silica gel
gravity
column chromatography [EtOAc] furnished the desired product (39 g, 22%).
8H (CDCI3; 250MHz) 2.33 (3H, s, ArCH3), 2.46 (3H, s, NCH3), 2.64 (1H, ddd,
J14.9, 5.1,
2.6, one of CHzCH ), 2.77 (1 H, ddd, J14.9, 8.9, 2.4, one of CH~CH ), 3.78 (1
H,'d, J13.1,
one of ArCHzNR2), 3.86 (1 H, ddd, J12.7, 5.1, 2.4, one of C,H~CHZ), 4.18 (1 H,
ddd, J12.7,
8.9, 2.6, one of C,H~CH ), 4.89 (1 H, d, J13.1, one of ArCHzNRz), 5.99 (1 H,
s, CHOR),
6.91-6.98 (2H, m, Ark, 7.12-7.22 (3H, Ark, 7.46 (1 H, dd, J7.9, 1.8, ArHa,
7.55 (1 H, d,
J1.5, ArH~. '
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
43
Example 49
8-Cyclopropyl-5-methyl-1-(2-methyl)phenyl-1,3,4,6-tetrahydro-5H-Benz[fj-2,5-
oxazocine
8-Bromo-5-methyl-1-(2-methyl)phenyl-1, 3,4,6-tetrahydro-5H-benz[f]-2, 5-
oxazocine (205 mg, 0.60 mmol) was dissolved in toluene (4 ml) and H20 (200
p,1) at RT.
Cyclopropylboronic acid (67 mg, 0.77 mmol), I~P04 (446 mg, 2.1 mmol), Pd
(OAc)2 (7
mg, 0.03 mmol) and P(cyclohexane)3 (17 mg, 0.06 mmol) were added and the
resulting
mixture heated at 100°C for 3 hours. The mixture was then allowed to
cool to RT before
partitioning the mixture between water and ethyl acetate. The organic phases
were dried
(MgS04), filtered and concentrated in vacuo to yield the crude product.
Purification by
silica gel gravity column chromatography [EtOAc] provided the desired target
(117 g,
64%).
8H(CDCI3; 250MHz) 0.67-0.74 (2H, m, two of CH C,H~), 0.92-1.0 (2H, m, two of
CH CH ),
1.83-1.93 (1 H, m, cyclopropyl Cue, 2.28 (3H, s, ArCH3), 2.48 (3H, s, NCH3),
2.66 (1 H,
ddd, J14.4, 5.6, 2.3, one of CHzCHz), 2.95 (1 H, ddd, J14.4, 8.5, 2.1, one of
CH CHI),
3.81 (1 H, d, J12.8, one of ArCH ), 3.87 (1 H, ddd, J12.8, 5.6, 2.1, one of
CHzCH ), 4.08
(1 H, ddd, J12.8, 8.5, 2.3, one of CH~CH ), 4.56 (1 H, d, J12.8, one of ArCH
NRZ), 5.98
(1 H, s, CHOR), 6.73 (1 H, d, J7.9, Ark, 6.85 (1 H, dd, J7.9, 1.8, Ark, 6.97
(1 H, d, J1.8,
Ark, 7.14-7.18 (4H, m, ArH).
Example 50
9-Bromo-5-methyl-1-(3-pyridyl)-1,3,4,6-tetrahydro-5H-benz[f]-2,5-oxazoci ne
2-{[4-Bromo-2-(hydroxypyridin-3-yl-methyl)-benzyl]-methylamino}-ethanol (100
mg, 0.29 mmol) was dissolved in xylene (2 mL) at RT. pTSA (434 mg, 2.28 mmol)
was
added, and the resulting mixture was heated at 120°G in an open vessel
until all solvent
and water had evaporated. The reaction vessel was removed from the heat and
allowed
to cool. Ethyl acetate (20 ml) and a sat. aq. solution of sodium bicarbonate
(20 ml) were
added to partition the gummy mixture. The organic phase was separated, and the
aqueous phase was extracted into ethyl acetate (2 x 20 ml). Combined extracts
were
dried (MgS04), filtered and concentrated in vacuo to give the cyclised product
as a pale
brown oil. Purification by silica gel column chromatography with a stepped
gradient,
[100%EtOAc to 3%/5%/10% MeOH/EtOAc)], furnished product (23 mg, 24%).
sH(CDCI3, 250MHz) 2.46 (3H, s, NCH3), 2.66 (1 H, ddd, J14.4, 5.5, 2.8, one of
C,H~CH ),
2.83 (1 H, ~ ddd, J14.4, 8.2, 2.1, one of CHzCHz), 3.67 (1 H, d, J13.1, one of
ArCHzNRZ),
3.86 (1 H, ddd, J12.7, 5.5, 2.1, one of C~CH ), 4.19 (1 H, ddd, J12.7, 8.2,
2.8, one of
CH CHz), 4.73 (1 H, d, J13.1, one of ArCHzNR2), 5.77 (1 H, s, CHOR), 7.11-7.14
(2H, m,
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
44
aromatic ~, 7.24-7.29 (1 H, m, aromatic H~, 7.39 (1 H, dd, J7.9, 1.8, aromatic
~, 7.56
(1 H, d, J7.9, aromatic ~, 8.53-8.57 (2H, m, aromatic ~.
Example 51
8-Bromo-5-methyl-1-(3-pyridyl)-1,3,4,6-tetrahydro-5H-benz[fJ-2,5-oxazocine
2-{[5-Bromo-2-(hydroxypyridin-3-yl-methyl)-benzyl]-methylamino}-ethanol (84
mg,
0.24 mmol) was dissolved in xylene (2 ml). pTSA (364 mg, 1.9 mmol) was added
and the
mixture was heated to reflux temperature in an open vessel to allow
evaporation of the
solvent and water. When the mixture had concentrated down to a dark gum, the
vessel
was removed from the heat and allowed to cool. The residue was partitioned
between
ethyl acetate (20 ml) and sat. aq. sodium bicarbonate solution (20 ml). The
aqueous
phase was extracted further with ethyl acetate (2 x 20 ml), and the combined
organic
extracts dried (magnesium sulphate), filtered and evaporated to dryness to
give crude
product. Purification was by silica gel column chromatography [Ethyl acetate]
to provide
the target compound as an oil/solid.
8H (CDC13; 250MHz) 2.46 (3H, s, CH3), 2.58 (1H, ddd, J14.3, 6.1, 2.6, one of
CH~C~,
2.83 (1H, ddd, J14.3, 7.9, 2.1, one of CH~C~, 3.63 (1H, d, J13.1, one of
ArCH~NR~),
3.86 (1H, ddd, J12.6, 6.1, 2.1, one of CH~C~, 4.16 (1H, ddd, J12.6, 7.9, 2.6,
one of
CH~CH~), 4.68 (1 H, d, J13.1, one of ArCH~NR2), 5.80 (1 H, s, CHOR), 6.84 (1
H, d, J8.3,
aromatic ~, 7.21-7.27 (2H, m, aromatic ~, 7.32-7.40 (2H, m, aromatic ~, 7.55
(1 H, dd,
J7.8, 1.6, aromatic ~, 8.52 (1 H, dd, J4.6, 1.6, aromatic H~, 8.54 (1 H,d ,
J8.6, aromatic
Example 52
8-Cyano-5-methyl-1-(3-pyridyl)-1,3,4,6-tetrahydro-5H-benz[f]-2,5-oxazocine
8-Bromo-5-methyl-1-(3-pyridyl)-1,3,4,6-tetrahydro-5H-Benz[t7-2,5-oxazocine (50
mg, 0.15 mmol), Zn (1 mg, 0.01 mmol), Zn(OAc)2 (4 mg, 0.02 mmol), Zn(CN)2 (13
mg,
0.11 mmol), Pd2 (dba)3 (18 mg 0.02 mmol), and 1,1'-
bis(diphenylphosphine)ferrocene (9
mg 0.02 mmol) were heated to 140 °C in DMF (2 ml) and H20 (20 w1)
overnight. The
black reaction mixture was allowed to cool and diluted with water (20 ml). The
resulting
brown precipitate was collected by filtration and washed with water then
brine. The
filtrate was extracted with ethyl acetate (2 x 50 ml). The combined organic
extracts were
dried (magnesium sulphate), filtered and concentrated in vacuo to provide the
crude
product as a brown oil. Purification was by silica gel column chromatography
[dichloromethane] to furnish the desired product (10 mg, 24%) as a yellow oil.
8H (CDCI3; 250MHz) 2.47 (3H, s, Cue, 2.58-2.84 (2H, m, CH~CHZ), 3.68 (1H, d,
J13.1,
one of ArCH~NRz), 3.82-3.90 (1 H, m, one of CHZC~, 4.19-4.29 (1 H, m, one of
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
CH2C~, 4.88 (1 H, d, J13.1, one of ArCH~NR2), 5.84 (1 H, s, CHOR), 7.11 (1 H,
d, J8.3,
aromatic ,H~, 7.22-7.30 (2H, m, aromatic H), 7.49-7.55 (3H, m, aromatic H~,
8.52 (1 H, dd,
J4.6, 1.6 aromatic H~, 8.55 (1 H, d, J8.6, aromatic H~.
Biological Assay
5 The assay was carried out according to the method described by Perovic et al
(1995), Arzneim-Forsch. Drug Res., 45: 1145.
Assay for Inhibition of Noradrenaline Reuptake Activity
Synaptosomes (100 pg) are incubated for 20 min at 37°C with 0.1
pCi
[3H]norepinephrine in the absence (control) or presence of the test compound
or the
10 reference compound in a buffer containing 118 mM NaCI, 5 mM KCI, 2.5 mM
MgS04, 1.2
mM NaH2P04, 25 mM NaHCOs, 11 mM glucose, 10 NM EGTA and 50 pM ascorbic acid
(pH 7.4). Basal control activity is determined by incubating the same mixture
for 20 min
at 0°C in the presence of 10 NM protriptyline to block the uptake.
Following incubation,
the samples are filtered rapidly under vacuum through glass fiber filters
(GF/B, Packard)
15 and rinsed twice with ice-cold incubation buffer using a 96-sample cell
harvester
(Unifilter, Packard) to eliminate free [3H]norepinephrine. The filters are
dried and the
retained radioactivity is measured in a scintillation counter (Topcount,
Packard) using a
scintillation cocktail (Microscint 0, Packard).
The results are expressed as a percent inhibition of the control uptake of
20 [3H]norepinephrine. The standard inhibitory reference compound is
protriptyline, which is
tested in each experiment at several concentrations to obtain an inhibition
curve from
which its ICso value is calculated.
Assay for Inhibition of Serotonin Reuptake Activity
Synaptosomes (100 pg) are incubated for 15 min at 37°C with 0.1
NCi
25 [3H]serotonin in the absence (control) or presence of the test compound or
the reference
compound in a buffer containing 118 mM NaCI, 5 mM KCI, 2.5 mM MgS04, 1.2 mM
NaH2PO4, 25 mM NaHC03, 11 mM glucose, 10 NM EGTA and,50 pM ascorbic acid (pH
7.4). Basal control activity is determined by incubating the same mixture for
15 min at
4°C in the presence of 10 pM imipramine to block the uptake. Following
incubation, the
30 samples are filtered rapidly under vacuum through glass fiber filters
(GF/B, Packard) and
rinsed twice with ice-cold incubation buffer using a 96-sample cell harvester
(Unifilter,
Packard) to eliminate free [3H]serotonin. The filters are dried and the
retained
radioactivity is measured in a scintillation counter (Topcount, Packard) using
a
scintillation cocktail (Microscint 0, Packard).
CA 02558741 2006-09-06
WO 2005/103019 PCT/GB2005/001519
46
The results are expressed as a percent inhibition of the control uptake of
[3H]serotonin. The standard inhibitory reference compound is imipramine, which
is
tested in each experiment at several concentrations to obtain an inhibition
curve from
which its ICso value is calculated.