Note: Descriptions are shown in the official language in which they were submitted.
CA 02566213 2006-11-08
WO 2005/113544 PCT/US2005/016381
-1-
SUBSTANTIALLY PURE 2-{[2-(2-METHYLAMINO-PYRIMIDIN-4-YL)-1H-
INDOLE-5-CARBONYL]-AMINO}-3-(PHENYLPYRIDIN-2-YL-AMINO)-PROPIONIC ACID
AS AN IxB KINASE INHIBITOR
FIELD OF THE INVENTION
This invention is directed to an indole derivative, its preparation, a
pharmaceutical composition
comprising the compound, its use, and intermediates thereof.
BACKGROUND OF THE INVENTION
NF-KB is a heterodimeric transcription factor that regulates the expression of
multiple inflanunatory
genes. The expression of more than 70 known proteins is transcriptionally
regulated by the binding of
NF-xB to specific sequence elements in the promoter region of these genes
(Baeuerle and Baichwal,
Advances in Immunology 65:111-137, 1997). NF-KB has been implicated in many
pathophysiologic
processes including angiogenesis (Koch et al., Nature 376:517-519, 1995),
atherosclerosis (Brand et
al., J Clin Inv. 97:1715-1722, 1996), endotoxic shock and sepsis (Bohrer et
al., J. Clin. Inv. 100:972-
985, 1997), inflammatory bowel disease (Panes et al., Am J Physiol. 269:H1955-
H1964, 1995),
ischemia/reperfusion injury (Zwacka et al., Nature Medicine 4:698-704, 1998),
and allergic lung
inflammation (Gosset et al., Int Arch Allergy Immunol. 106:69-77, 1995). Thus
the inhibition of NF-
KB by targeting regulatory proteins in the NF-KB activation pathway represents
an attractive strategy
for generating anti-inflammatory therapeutics due to NF-KB's central role in
inflammatory conditions.
The IKB kinases (IKKs) are key regulatory signaling molecules that coordinate
the activation of NF-
KB. Two IKKs, IKK-1 (IKK-a) and IKK-2 (IKK-0), are structurally unique kinases
containing an N-
terminal kinase domain with a dual serine activation loop, a leucine zipper
domain, and a C-terminal
helix-loop-helix domain and serine cluster. - IKK enzymes show relatively low
sequence homologies
with other kinases, and early profiles with known kinase inhibitors have not
identified compounds with
striking potency. Kinetic analysis shows that IKK-2 binds to and
phosphorylates IKBa, and IKBs with
high and relatively equal affinities (Heilker et al. 1999). Recombinant IKK-2
phosphorylates IKBa
CA 02566213 2006-11-08
WO 2005/113544 PCT/US2005/016381
-2-
peptide 26-42 with near equal affinity to full length IKBa, however the native
IKK enzyme complex
phosphorylates full length IKBa 25,000 fold more efficiently, suggesting
important regulatory
sequences in the C-terminal region of IKBa, or additional regulatory proteins
in the IKK enzyme
complex that accelerate the rate of catalysis (Burke et al., Journal of
Biological Chemistry 274:36146-
36152, 1999). Phosphorylation of IKBa occurs via a random sequential kinetic
mechanism, meaning
either ATP or IKBa may bind first to IKK-2, that both must be bound before
phosphorylation of IKBa
can take place (Peet and Li, Journal of Biological Chemistry 274:32655-32661,
1999). IKK-2 binds
ATP with uniquely high affinity (Ki=130 nM) compared to other serine-threonine
kinases such as p38
and JNK perhaps indicating a unique ATP binding pocket that reflects the
relatively poor activity to
many broad specificity kinase inhibitors when tested against IKK-2. To date,
no crystal structure of
IKK-2 has been reported. However homology modeling has identified 3 structural
domains including
an N-terminal kinase domain with an activation loop, a leucine zipper domain
that likely mediates the
formation of IKK-1 and IKK-2 homo/heterodimers, and a C-terminal helix-loop-
helix with serine rich
tail. Activation of IKK-2 is dependent upon phosphorylation of serine 177 and
181 in the activation or
T loop. Alanine mutations abolish activity, while glutamate mutations result
in a constitutively active
enzyme (Mercurio et al. Science 278:860-866, 1997; Delhase et al., Science
284:30 313, 1999).
IKK-1 and IKK-2 occur both as heterodimers and IKK-2 homodimers, and are
associated with a 700-
900 kDa cytoplasmic enzyme complex called the "IKK Signalsome" (Mercurio et
al., Science
278:860-866, 1997). Another component, IKKAP-1 or NEMO/IKKy has no apparent
catalytic
function but will associate directly with IKK-2 and is necessary for full
activation of NF-xB (Mercurio
et al., Mol Cell Biol 19:1526-1538, 1999). Many immune and inflammatory
mediators including
TNFct, lipopolysaccharide (LPS), IL-10, CD3/CD28 (antigen presentation),
CD40L, FasL, viral
infection, and oxidative stress have been shown to lead to NF-xB activation.
Although the receptor
complexes that transduce these diverse stimuli appear very different in their
protein components, it is
understood that each of these stimulation events leads to activation of the
IKKs and NF-KB.
The IKK complex appears to be the central integrator of diverse inflammatory
signals leading to the
phosphorylation of IKB. IKKs are activated at dual serine residues by upstream
kinases including NF-
xB inducing kinase, NIK (Malinin et al., Nature 385:540-544, 1997), and MEKK-1
(Yujiri et al.,
Science 282:1911-1914, 1998). The differential activities of NIK and MEKK-1
remain unclear
although initial data indicates these kinases may preferentially activate IKK-
1 and IKK-2, respectively.
Activated IKK phosphorylates a cytoplasmic inhibitor protein, IKB that binds
NF-xB, thereby masking
a nuclear localization signal present in Rel proteins (Cramer et al.,
Structure 7: R1-R6, 1999). IKK
phosphorylation of IKB on serines 32 and 36 forms a structural motif
recognized by the E3 ligase,
CA 02566213 2006-11-08
WO 2005/113544 PCT/US2005/016381
-3-
(3TRcP (Yaron et al., Nature 396:590-594, 1998). Docking of (3TRcP results in
the formation of a
ligase complex which polyubiquitinates IKB thus targeting it for degradation
by the 26S proteosome.
Free NF-KB is then identified by nuclear transport proteins, which translocate
it to the nucleus where it
can associate with sequence specific regulatory elements on gene promoters.
Although both kinases can phosphorylate IKB in vitro, early studies using
genetic mutants indicated
that IKK-2, but not IKK-1, was essential for activation of NF-KB by pro-
inflammatory stimuli such as
II.-1P and TNFa. Furthermore, only catalytically inactive mutants of IKK-2
blocked the expression of
NF-KB regulated genes such as monocyte chemotactic protein (MCP-1) and
intercellular adhesion
molecule (ICAM-1) (Mercurio et al, Science 278:860-866, 1997). Studies of
knockout animals for
IKK-1 and IKK-2 substantiate these initial findings (Hu et al., Science
284:316-320, 1999; Li et al.,
Genes & Development 13:1322-1328, 1999; Li et al., Science 284:321-324, 1999;
Takeda et al.,
Science 84:313-316, 1999; Tanaka et al., Immunity 10:421-429, 1999). IKK-1"1"
animals were born
alive but died within hours. Pups showed abnormalities of the skin due to
defective proliferation and
differentiation, but showed no gross deficiency in cytokine induced activation
of NF-KB. In contrast,
IKK-2-'- embryos died at day 14-16 of pregnancy from liver degeneration and
apoptosis that bore a
striking resemblance to that observed in Rel A knock-out animals (Beg et al.,
Nature 376:167-170,
1995). Furthermore, embryonic fibroblasts from IKK-2-1" animals exhibited
markedly reduced NF-KB
activation following cytokine stimulation, while IKK-1"" did not.
Accordingly, cell and animal experiments indicate that IKK-2 is a central
regulator of the pro-
inflammatory role of NF-KB, wherein the IKK-2 is activated in response to
immune and inflammatory
stimuli and signaling pathways. Many of those immune and inflammatory
mediators, including II.-1(3,
LPS, TNFa, CD3/CD28 (antigen presentation), CD40L, FasL, viral infection, and
oxidative stress,
play an important role in respiratory diseases. Furthermore, the ubiquitous
expression of NF-KB, along
with its response to multiple stimuli means that almost all cell types present
in the lung are potential
target for anti-NF-xB/IKK-2 therapy. This includes alveolar epithelium, mast
cells, fibroblasts,
vascular endothelium, and infiltrating leukocytes, including neutrophils,
macrophages, lympophocytes,
eosinophils and basophils. By inhibiting the expression of genes such as
cyclooxygenase-2 and 12-
lipoxygenase (synthesis of inflammatory mediators), TAP-1 peptide transporter
(antigen processing),
MHC class I H-2K and class II invariant chains (antigen presentation), E-
selectin and vascular cell
adhesion molecule (leukocyte recruitment), interleukins-1, 2, 6, 8
(cytokines), RANTES, eotaxin, GM-
CSF (chemokines), and superoxide dismutase and NADPH quinone oxidoreductase
(reactive oxygen
species), inhibitors of IKK-2 are believed to display broad anti-inflammatory
activity.
CA 02566213 2006-11-08
WO 2005/113544 PCT/US2005/016381
-4-
Patent application WO 94/12478, the content of which is incorporated herein by
reference, describes,
inter alia, indole derivatives that inhibit blood platelet aggregation. Patent
applications WO 01/00610
and WO 01/30774, the content of each of which is incorporated herein by
reference, describe indole
derivatives and benzimidazole derivatives, which are able to modulate NF-xB.
As described above,
NF-xB is a heterodimeric transcription factor that is able to activate a large
number of genes that
encode, inter alia, proinflammatory cytokines such as IL-l, IL-2, TNFa or IL-
6. NF-xB is present in
the cytosol of cells, where it is complexed with its naturally occurring
inhibitor IxB. Stimulation of
the cells, for example by cytokines, leads to the IxB being phosphorylated and
subsequently broken
down proteolytically. This proteolytic breakdown leads to the activation of NF-
xB, which then
migrates into the nucleus of the cell, where it activates a large number of
proinflammatory genes.
In diseases such as rheumatoid arthritis, osteoarthritis, asthma, chronic
obstructive pulmonary disorder
(COPD), rhinitis, multiple sclerosis, cardiac infarction, Alzheimer's
diseases, diabetes Type II,
inflammatory bowel disease or atherosclerosis, NF-xB is activated beyond its
normal extent. The
inhibition of NF-xB is also described as being useful for treating cancer on
its own or in addition to
cytostatic therapy. Inhibition of the NF-xB-activating signal chain at various
points or by interfering
directly with the transcription of the gene by glucocorticoids, salicylates or
gold salts, has been shown
as being useful for treating rheumatism.
The first step in the abovementioned signal cascade is the breakdown of IxB.
This phosphorylation is
regulated by the specific IxB kinase. Thus far, inhibitors of IicB kinase are
known to frequently suffer
from the disadvantage of being non-specific for inhibiting only one class of
kinases. For example,
most inhibitors of IxB kinase inhibit several different kinases at the same
time because the structures
of the catalytic domains of these kinases are similar. Consequently, the
inhibitors act, in an
undesirable manner on many enzymes, including those that possess the vital
function.
Chronic obstructive pulmonary disease (COPD) is a debilitating inflammatory
disease of the lungs
characterized by the progressive development of airflow limitation that is not
fully reversible (Pauwels
et al., 2001). The airflow limitation is associated with an abnormal
inflammatory response of the lungs
to noxious particles or gases, primarily caused by cigarette smoking. Although
COPD affects the lungs,
it also produces significant systemic consequences. The term COPD encompasses
chronic obstructive
bronchitis, with obstruction of small airways, and emphysema, with enlargement
of air spaces and
destruction of lung parenchyma, loss of lung elasticity, and closure of small
airways. Chronic
bronchitis, by contrast, is defined by the presence of a productive cough (due
to hypersecretion of
mucus) of more than three months' duration for more than two successive years.
There is some
CA 02566213 2006-11-08
WO 2005/113544 PCT/US2005/016381
-5-
epidemiologic evidence that mucus hypersecretion is accompanied by airflow
obstruction, perhaps as a
result of obstruction of peripheral airways. Most patients with COPD have all
three pathologic
conditions (chronic obstructive bronchitis, emphysema, and mucus plugging),
but the relative extent of
emphysema and obstructive bronchitis within individual patients can vary,
Vestbo et al., 1996; Barnes,
2004a, Barnes, 2004b; Hogg, 2004.
In industrialized countries, cigarette smoking accounts for most cases of
COPD, but in developing
countries other environmental pollutants (particularly with sulfur dioxide and
particulates) and
certain occupational chemicals (such as cadmium), are important causes.
Passive smoking is also a
risk factor.
COPD patients are predisposed to exacerbations, that is, an acute worsening of
their respiratory
symptoms. An exacerbation of COPD is an event in the natural course of the
disease characterized by a
change in the patient's baseline dyspnea, cough and/or sputum beyond day-to-
day variability sufficient
to warrant a change in management (Rodriguez-Roisin, 2000; Burge and Wedzicha,
2003).
Tracheobronchial infections are believed to be a common cause of exacerbation
in COPD, although
controversy exists regarding the nature of the infectious agent as well as its
exact role (Wedzicha,
2002; White et al., 2003). In addition, exacerbations of COPD are clearly
associated with the levels
of respirable particles and environmental air pollutants, and these have been
linked to hospital
admission rates (Rennard and Farmer, 2004).
The frequency of exacerbations is linked to disease severity in COPD.
Exacerbations, may adversely
affect the natural history of these disorders, perhaps by contributing to
increased rates of lung
function decline, systemic effects, and premature mortality. Unfortunately, to
date, there is no widely
accepted definition of what constitutes an exacerbation of COPD (Rodriguez-
Roisin, 2000). The
intensity and duration of increased symptoms required to qualify as an
"exacerbation" are difficult to
define. Indeed, several definitions co-exist, and many clinical trials employ
substantially different
criteria or describe poorly the definition(s) used to diagnose exacerbation.
The most widely quoted
clinical criteria used in the characterization of acute exacerbation of COPD
are those described by
Anthonisen et al., (1987). In that study exacerbations were divided into three
groups: type 1
exacerbations were characterized by increased breathlessness, increased sputum
volume, and new or
increased sputum purulence; type 2 included any two of these symptoms; and
type 3 consisted of any
one of the symptoms together with at least one additional feature, including
sore throat or nasal
discharge within the last five days; unexplained fever; increased wheeze;
increased cough; or a 20%
increase in respiratory or heart rate compared with baseline. These criteria
have been used as a
CA 02566213 2006-11-08
WO 2005/113544 PCT/US2005/016381
-6-
benchmark ever since, and all proposed etiologies of exacerbation need to
establish their relationship
to these key features.
The inhibition of NF-xB is also described as being useful for treating
hypoproliferative diseases, e.g.,
solid tumor and leukemias, on its own or in addition to cytostatic therapy.
Inhibition of the NF-xB-
activating signal chain at various points or by interfering directly with the
transcription of the gene by
glucocorticoids, salicylates or gold salts, has been shown as being useful for
treating rheumatism.
Patent application WO 01/30774 discloses indole derivatives and US Application
Serial No.
10/642,970, discloses indole and benzimidazole derivatives which are able to
modulate NF-xB and
which exhibit a strong inhibitory effect on IxB kinase. Particularly, US
Application Serial No.
10/642,970 discloses indole and benzimidazole derivatives of formula (I),
their preparation,
qR R1
4
JN
O
X R 11
R2 N \ N
N M-~
H ,N-H
R3
pharmaceutical compositions containing these compounds, and methods for the
prophylaxis and
therapy of a disease associated with an increased activity of IxB kinase
comprising administering such
compounds. Furthermore, US Application Serial No. 10/642,970 discloses the
following compounds
of formulae (B), (C), and (D):
CA 02566213 2006-11-08
WO 2005/113544 PCT/US2005/016381
-7-
\ I ~ I
O
O~N
~NH H N N-/
MeO H NH
H3C
(B) (compound 43),
O)NO
~H I N
OMe ~ H N~
NH
H3C
(C) (compound 32); and
\ I J~ I
N N
O
ON -
OH H N N{
H NH
H3C
(D) (compound 48).
However, US Application Serial No. 10/642,970 does not specifically disclose
the compound of
Formula (I) wherein M is N; R1 is hydrogen, R2 is carboxyl (-COOH), R3 is
methyl, R4 is pyridin-2-
yl, R11 is hydrogen, and X is CH.
In view of the aforesaid, there is a need for an inhibitor of IKB kinase that
operates through the
selective inhibition of IKK, particularly an IKK-2 inhibitor. Also desired
would be to have such an
inhibitor that exhibits a localized activity as opposed to a systemic
activity. Such an inhibitor should
have a utility in treating a patient suffering from or subject to IKK-2
mediated pathological (diseases)
conditions, e.g., asthma, or chronic obstructive pulmonary disorder (COPD),
that could be ameliorated
by the targeted administering of then inhibitor.
CA 02566213 2006-11-08
WO 2005/113544 PCT/US2005/016381
-8-
SUMMARY OF THE INVENTION
The present invention is directed to a compound having activity as an
inhibitor, preferably a selective
inhibitor, of IKB (IKK), particularly IKK-2, and to a composition and methods
related thereto.
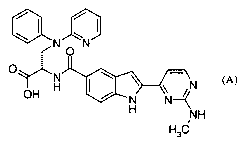
In particular, the present invention is directed to the substantially pure
compound of formula (A):
tN 0 N
a n
O
H N ~N
OH H N-~
NH
H3C
(A)
or a pharmaceutically acceptable salt, or solvate of said compound.
Furthermore, the present invention is directed to a pharmaceutical composition
comprising a
pharmaceutically effective amount of the compound of formula (A), and a
pharmaceutically acceptable
carrier.
Furthermore, the present invention is directed to the use of a compound of
formula (A) as an inhibitor
of IKB kinase.
BRIEF DESCRIPTION OF THE DRAWINGS
FIGURE 1 shows a picture of mouse imaging studies in the NF-KB-Luciferase
Reporter Mouse Model
wherein animals are treated with the Vehicle/PBS solution (negative control
animals) and wherein
animals have IL- 1 0-induced NF-KB activation without any compound present
[Vehicle/[L-1(3] or with
increasing doses (0.3 mpk, 1 mpk, 3 mpk, and 10 mpk) of Compound (A) or
Compound (B).
FIGURE 2 shows a graph of mouse imaging studies in the NF-KB-Luciferase
Reporter Mouse Model
showing bioluminescence levels for animals treated with the Vehicle/PBS
solution (negative control
animals) and wherein animals have IL-1(3-induced NF-KB activation without any
compound present
[Vehicle/IL-1(3] or with increasing doses (0.3 mpk, 1 mpk, 3 mpk, and 10 mpk)
of Compound (A) or
Compound (B).
CA 02566213 2006-11-08
WO 2005/113544 PCT/US2005/016381
-9-
FIGURE 3, on the left, shows a graph of Compound (A) levels after i.t.
instillation of 0.3 mg/kg of
Compound (A) in lung and plasma tissue; and on the right, shows a graph of
Compound (A) and
Compound (B) levels after i.t. instillation of 0.3 mg/kg of Compound (B) in
lung and plasma tissue.
FIGURE 4, on the left, shows a graph of the lung exposure of Compound (A)
after administering
increasing doses (0.01 mpk, 0.03 mpk, 0.10 mpk, and 0.30 mpk) of Compound (A);
and on the right,
shows a graph of the plasma exposure of Compound (A) after administering
increasing doses (0.01
mpk, 0.03 mpk, 0.10 mpk, and 0.30 mpk) of Compound (A).
FIGURE 5, on the left, shows a graph of the lung exposure of Compounds (A) and
(B) after
administering increasing doses (0.01 mpk, 0.03 mpk, 0.10 mpk, and 0.30 mpk) of
Compound (B); and
on the right, shows a graph of the plasma exposure of Compounds (A) and (B)
after,administering
increasing doses (0.01 mpk, 0.03 mpk, 0.10 mpk, and 0.30 mpk) of Compound (B).
DETAILED DESCRIPTION
List of Abbreviations
As used above, and throughout the description of the invention, the following
abbreviations, unless
otherwise indicated, shall be understood to have the following meanings:
BoczO di-tert-butyl dicarbonate
DIEA N,N-diisopropylethylamine
DMAP 4-dimethylaminopyridine
DMF dimethylformamide
DMSO dimethylsulfoxide
ESI-MS electrospray ionization mass spectrometry
FAB-MS fast-atom bombardment mass spectrometry
HATU O-(7-azabenzotriazol-1-yl)-N,N,N',N'-tetramethyluronium
hexaflurophosphate
HPLC high performance liquid chromatography
PBS phosphate-buffered saline
i.n. intranasally
PO oral
i.p. intraperitoneal
i.t. intra-tracheally
Microcystin-LR liver toxin produced by certain cyanobacteria of the genera
Anabaena
and Oscillatoria
mbr millibar
CA 02566213 2006-11-08
WO 2005/113544 PCT/US2005/016381
-10-
mpk mg/kg
HEPES 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
DTT dithiothreitol
ATP adenosine triphosphate
streptavidin-HRP Streptavidin-Horseradish Peroxidase Conjugate
TMB tetramethylbenzidine
pfu plaque-forming units
MDI metered-dose inhaler
DPI dry powder inhaler
Definitions
As used above, and throughout the description of the invention, the following
terms, unless otherwise
indicated, shall be understood to have the following meanings.
"Compound of the invention", and equivalent expressions, means the compound of
formula (A), as
hereinbefore described, which expression includes the pharmaceutically
acceptable salt and the
solvate, e.g., hydrate. Similarly, reference to intermediates, whether or not
they themselves are
claimed, is meant to embrace the salts, and solvates, where the context so
permits. For the sake of
clarity, particular instances when the context so permits are sometimes
indicated in the text, but these
instances are purely illustrative and they are not intended to exclude other
instances when the context
so permits.
"Treating" or "treatment" means prevention, partial alleviation, or cure of
the disease. The compound
and compositions of this invention are useful in treating conditions that are
characterized by the
activation of NF-KB and/or enhanced levels of cytokines and mediators that are
regulated by NF-KB
including, but not limited to TNFa and IL-1(3. Inhibition or suppression of NF-
KB and/or NF-KB-
regulated genes such as TNFa may occur locally, for example, within certain
tissues of the subject, or
more extensively throughout the subject being treated for such a disease.
Inhibition or suppression of
NF-KB and/or NF-KB-regulated genes such as TNFa may occur by one or more
mechanisms, e.g., by
inhibiting or suppressing any step of the pathway(s) such as inhibition of
IKK. The term "NF-KB-
associated condition" refers to diseases that are characterized by activation
of NF-KB in the cytoplasm
(e.g., upon phosphorylation of IKB). The term "TNFa-associated condition" is a
condition
characterized by enhanced levels of TNFa. In the instant specification, the
term NF-KB-associated
condition will include a TNFa-associated condition but is not limited thereto
as NF-KB is involved in
the activity and upregulation of other pro-inflammatory proteins and genes.
The term "inflammatory
or immune diseases or disorders" is used herein to encompass both NF-KB-
associated conditions and
CA 02566213 2006-11-08
WO 2005/113544 PCT/US2005/016381
-11-
TNFa-associated conditions, e.g., any condition, disease, or disorder that is
associated with release of
NF-xB and/or enhanced levels of TNFa, including conditions as described
herein.
"Patient" includes both human and other mammals.
"Pharmaceutically effective amount" is meant to describe an amount of a
compound, composition,
medicament or other active ingredient effective in producing the desired
therapeutic effect.
"Substantially pure" is meant to refer to the compound where it is
substantially free of biological or
chemical constituents, e.g., isolated from a biological or chemical
composition where biological or
chemical components are co-isolated therewith, and wherein the analytical
purity for the compound is
preferably at least 70%. More preferred is where the analytical purity is at
least 90%; even further
preferred is where the analytical purity is at least 95%; also "substantially
pure" is meant to refer to the
compound where it is substantially free of prodrugs, e.g., Compound (B).
The invention also relates to a process for preparing the compound of the
formula (A), as shown
in the following Scheme.
CA 02566213 2006-11-08
WO 2005/113544 PCT/US2005/016381
-12-
OH OH gpCxp O
O II
BOCZO ' I O O N. DMAP ~O~N~/O"
-- /X' ~ ~ III'
n O
H-CI HZN O (~) O H O ()
1 ?
N N_
N N O N \/ O N \/
chiral separation ~
O O
\
(iv) O N
Oi0 O 0 0 O
3 3S
0
\ Ho \ ~ - I
NH \ _
/
N_ H N rN O /
I I / ' /N N /
deprotection N ~ ~
O
( ) HZN (vi) H I \ ~ ~ /N
v f~o
O / O
NH
9 10 /
I \
_
N/~
NaOH/Methanol /N O /
(vii) OY~
I / N N ~N
H
OH
~
NH
A /
Starting compounds for the chemical reactions are known or they can be readily
prepared using
methods known from the literature. US Application Serial No. 10/642,970
describes the preparation of
the indole carboxylic acid intermediate (compound 8) used in the coupling step
(vi) above. The
compounds of the formulae (B), (C) and (D) are prepared as described in US
Application Serial No.
10/642,970, which is incorporated herein by reference.
Coupling methods of peptide chemistry that are well known to one skilled in
the art (see, e.g., Houben-
Weyl, Methoden der Organischen Chemie [Methods of Organic Chemistry], volumes
15/1 and 15/2,
Georg Thieme Verlag, Stuttgart, 1974, the content of which is incorporated
herein by reference), are
advantageously used for condensing the compounds. Compounds such as
carbonyldiimidazole,
carbodiimides such as dicyclohexylcarbodiimide or diisopropylcarbodiimide
(DIC), 0- ((cyano
(ethoxycarbonyl)methylene)-amino)-N, N, N', N'-tetramethyluronium
tetrafluoroborate (TOTU) or
polyphosphoric acid (PPA) are suitable for use as condensing agents or
coupling reagents.
CA 02566213 2006-11-08
WO 2005/113544 PCT/US2005/016381
-13-
The condensations can be carried out under standard conditions. In the
condensation, it is necessary
for the non-reacting amino groups that are present to be protected with
reversible protecting groups.
The same applies to carboxyl groups that are not involved in the reaction,
with these groups preferably
being present, during the condensation, as (C1-C6)-alkyl esters, benzyl esters
or tert-butyl esters. An
amino group protection is not necessary if the amino groups are still present
in the form of precursors
such as nitro groups or cyano groups and are only formed by hydrogenation
after the condensation.
After the condensation, the protecting groups that are present are eliminated
in a suitable manner. For
example, NOZ groups (guanidino protection in amino acids), benzyloxycarbonyl
groups and benzyl
groups in benzyl esters can be eliminated by hydrogenation. The protecting
groups of the tert-butyl
type are eliminated under acidic conditions while the 9-fluorenylmethyloxy-
carbonyl radical is
removed using secondary amines.
The invention also relates to a pharmaceutical composition comprising a
pharmaceutically effective
amount of the compound of the formula (A) and a pharmaceutically acceptable
carrier.
Embodiments
Because of the pharmacological properties of the compound according to the
invention, it is suitable
for the treatment of all those patients suffering from or subject to
conditions that can be ameliorated by
the targeted administration of an inhibitor of IKB kinase to a site where the
treatment is better effected
by localized versus systemic activity, e.g., asthma, or chronic obstructive
pulmonary disorder (COPD).
In practice, the compound of the present invention may be administered in
pharmaceutically
acceptable dosage form to humans and other animals by topical or systemic
administration, including
oral, inhalational, rectal, nasal, buccal, sublingual, vaginal, colonic,
parenteral (including
subcutaneous, intramuscular, intravenous, intradermal, intrathecal and
epidural), intracistemal and
intraperitoneal. It will be appreciated that the preferred route may vary with
for example the condition
of the recipient..
Intranasal, intratracheal, or inhalational administration, as well as
aerosolization, are particular
methods of administering the compound according to the invention.
Combination therapies may improve efficacy and decrease the risk of side
effects compared with
increasing the dose of a single agent. IKK inhibitors can be combined with
bronchodilators including
but not limited to short-acting B2-agonists; long-acting B2-agonists such as
salmeterol and formoterol;
anticholinergic agents such as ipratropium bromide and tiotropium bromide. IKK
inhibitors can also
be combined with methylxanthines such as theophylline.
CA 02566213 2006-11-08
WO 2005/113544 PCT/US2005/016381
-14-
Inhibitors of IKK2 can be combined with several anti-inflammatory therapies
including but not limited
to immunomodulators directed at various stages of the inflammatory cascade and
directed to
ameliorating inflainmatory processes. Such therapies include, but are not
limited to:
(A) Inhibitors of cellular recruitment and toxic inflammatory mediators
including but not limited to
phosphodiesterase-4 inhibitors; inhibitors of p38 mitogen-activated protein
kinase; biopharmaceuticals
such as anti-tumor necrosis factor-alpha, anti-interleukin-8, and anti-
monocyte chemoattractant
protein-1; inhibitors of adhesion molecules and chemotactic factors; and
molecules that interfere with
cell survival and clearance/apoptosis;
(B) Inhibitors of proteolytic enzymes including but not limited to inhibitors
of neutrophil-derived
serine proteases such as neutrophil elastase; and inhibitors of matrix
metalloproteinases (MMPs) such
as MMP-2, MMP-9 and MMP-12;
(C) Antioxidants including but not limited to N-acetylcysteine and inhibitors
or scavengers of reactive
oxygen species; and toxic peptides such as defensins that can directly cause
cell injury;
(D) Inhibitors of mucus production including but not limited to inhibitors of
mucous genes; and also
mucus clearing agents such as expectorants, mucolytics, and mucokinetics; and
(E) Antibiotic therapy such as with a ketolide, for example Ketek .
The drug combinations of the present invention can be provided to a cell or
cells, or to a human
patient, either in separate pharmaceutically acceptable formulations
administered simultaneously or
sequentially, formulations containing more than one therapeutic agent, or by
an assortment of single
agent and multiple agent formulations. However administered, these drug
combinations form a
pharmaceutically effective amount of components.
The treatment regimen/dosing schedule can be rationally modified over the
course of therapy so that
the lowest amounts of each of the pharmaceutically effective amount of
compounds used in
combination which together exhibit satisfactory pharmaceutical effectiveness
are administered, and so
that administration of such pharmaceutically effective amount of compounds in
combination is
continued only so long as is necessary to successfully treat the patient.
A pharmaceutical composition according to the invention is preferably produced
and administered in
dosage units, with each unit containing, as the active constituent, a
particular dose of the compound.
Pharmaceutically acceptable salts of the compound of formula (A) are within
the scope of this
invention. The term "salt(s)" means acid or base addition salts formed with
acids and bases. In
addition, the term "salt(s)" include zwitterion salts (inner salts), i.e., as
the compound of formula (A)
CA 02566213 2006-11-08
WO 2005/113544 PCT/US2005/016381
-15-
contains both a basic moiety, such as an amine or a pyridine or imidazole
ring, and an acidic moiety,
such as a carboxylic acid. Pharmaceutically acceptable (i.e., non-toxic,
physiologically acceptable)
salts are preferred, such as, for example, acceptable metal and amine salts in
which the cation does not
contribute significantly to the toxicity or biological activity of the salt.
However, other salts may be
useful, e.g., in isolation or purification steps that may be employed during
preparation, and thus, are
contemplated within the scope of the invention. Salts of the compounds of the
formula (A) may be
formed, for example, by reacting a compound of the formula (A) with an amount
of acid or base, such
as an equivalent amount, in a medium such as one in which the salt
precipitates or in an aqueous
medium followed by lyophilization.
Acid addition salts are formed with'the compound of the invention bearing
basic moiety(ies) such as
an imino nitrogen, amino or mono or disubstituted group is present. Particular
acid addition salts are
the pharmaceutically acceptable acid addition salts, i.e., salts whose anion
is non-toxic to a patient at a
pharmaceutical dose of the salt, and so that the beneficial effects inherent
in the free form of the
compound are not vitiated by side effects ascribable to the anion. The salts
chosen are chosen
optimally to be compatible with the customary pharmaceutical vehicles and
adapted for the form of
applicable administration. Acid addition salts of the compound of this
invention can be prepared by
reaction of the free form of the molecule bearing the basic moiety with the
appropriate acid, by the
application or adaptation of known methods. For example, the acid addition
salts of the compound of
this invention can be prepared either by dissolving the free form of the
molecule bearing the base
moiety in water or aqueous alcohol solution or other suitable solvents
containing the appropriate acid
and isolating the salt by evaporating the solution, or by reacting the free
form of the molecule bearing
the base moiety and acid in an organic solvent, in which case the salt
separates directly or can be
obtained by concentration of the solution. Some suitable acids for use in the
preparation of such salts
are hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid,
various organic carboxylic and
sulfonic acids, such as acetic acid, citric acid, propionic acid, succinic
acid, benzoic acid, tartaric acid,
fumaric acid, mandelic acid, ascorbic acid, malic acid, methanesulfonic acid,
toluenesulfonic acid,
mandelic acid, ascorbic acid, malic acid, fatty acids, adipate alginate,
ascorbate, aspartate,
benzenesulfonate, benzoate, cyclopentanepropionate, digluconate,
dodecylsulfate, bisulfate, butyrate,
lactate, laurate, lauryl sulfate, maleate, hydroiodide, 2-hydroxy-
ethanesulfonate, glycerophosphate,
picrate, pivalate, palmoate, pectinate, persulfate, 3-phenylpropionate,
thiocyanate, 2-
naphthalenesulfonate, undecanoate, nicotinate, hemisulfate, heptanoate,
hexanoate, camphorate,
camphorsulfonate, and others.
The acid addition salts of the compound of this invention can also be used to
regenerate the parent
compound of the invention from the salts by the application or adaptation of
known methods. For
CA 02566213 2006-11-08
WO 2005/113544 PCT/US2005/016381
-16-
example, the parent compound of the invention can be regenerated from their
acid addition salts by
treatment with an alkali, e.g., aqueous sodium bicarbonate solution or aqueous
ammonia solution.
Base addition salts are formed with the compound of the invention bearing the
carboxy moiety.
Particular base addition salts are the pharmaceutically acceptable base
addition salts, i.e., salts whose
cation is non-toxic to a patient at a pharmaceutical dose of the salt, so that
the beneficial effects
inherent in the free form of the compound are not vitiated by side effects
ascribable to the anion. The
salts chosen are chosen optimally to be compatible with the customary
pharmaceutical vehicles and
adapted for the form of applicable administration. Base addition salts of the
compound of this
invention can be prepared by reaction of the free form of the molecule bearing
the acid moiety with the
appropriate base, by the application or adaptation of known methods. For
example, the base addition
salts of the compound of this invention can be prepared either by dissolving
the free form of the
molecule bearing the acid moiety in water or aqueous alcohol solution or other
suitable solvents
containing the appropriate base and isolating the salt by evaporating the
solution, or by reacting the
free form of the molecule bearing the acid moiety and base in an organic
solvent, in which case the salt
separates directly or can be obtained by concentration of the solution. Some
suitable bases for use in
the preparation of such salts are those derived from alkali and alkaline earth
metal salts or amines such
as: sodium hydride, sodium hydroxide, potassium hydroxide, calcium hydroxide,
aluminum hydroxide,
lithium hydroxide, magnesium hydroxide, zinc hydroxide, ammonia,
ethylenediamine, N-methyl-
glucamine, lysine, arginine, ornithine, choline. N,N'-dibenzylethylenediamine,
chloroprocaine,
diethanolamine, procaine, N-benzylphenethylamine, diethylamine, piperazine,
tris(hydroxymethyl)-
aminomethane, tetramethylammonium hydroxide, and the like.
The base addition salts of the compound of this invention can also be used to
regenerate the parent
compound of the invention from the salts by the application or adaptation of
known methods. For
example, the parent compound of the invention can be regenerated from their
base addition salts by
treatment with an acid, e.g., hydrochloric acid.
In practice, the compound of the present invention is administered in a
suitable formulation to patients
such that its activity is particularly localized. It will be appreciated that
the preferred route can be
varied depending on the site of the condition for which administration is
directed.
Pharmaceutically acceptable dosage forms refers to dosage forms of the
compound of the invention,
and includes, for example, powders, suspensions, sprays, inhalants, tablets,
emulsions, and solutions,
particularly suitable for inhalation. Techniques and formulations generally
may be found in
Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, PA, latest
edition.
CA 02566213 2006-11-08
WO 2005/113544 PCT/US2005/016381
-17-
If desired, and for more effective distribution, the compound can be
microencapsulated in, or attached
to, a slow release or targeted delivery systems such as biocompatible,
biodegradable polymer matrices
(e.g., poly(d,l-lactide co-glycolide)), liposomes, and microspheres and
subcutaneoasly or
intramuscularly injected by a technique called subcutaneous or intramuscular
depot to provide
continuous slow release of the compound(s) for a period of 2 weeks or longer.
The compound may also be sterilized, for example, by filtration through a
bacteria retaining filter, or
by incorporating sterilizing agents in the form of sterile solid compositions
which can be dissolved in
sterile water, or some other sterile medium immediately before use.
Formulations suitable for nasal or tracheal administration means formulations
that are in a form
suitable to be administered nasally or by inhalation to a patient. The
formulation may contain a carrier,
in a powder form, having a particle size for example in the range 1 to 500
microns (including particle
sizes in a range between 20 and 500 microns in increments of 5 microns such as
30 microns, 35
microns, etc.). Suitable formulations wherein the carrier is a liquid, for
administration as for example a
nasal spray or as nasal drops, include aqueous or oily solutions of the active
ingredient. Formulations
suitable for aerosol administration may be prepared according to conventional
methods and may be
delivered with other therapeutic agents. MDI and DPI are feasible means for
effecting inhalation
therapy by administering a dosage form of the compound of the present
invention.
Actual dosage levels of active ingredient(s) in the compositions of the
invention may be varied so as to
obtain an amount of active ingredient(s) that is (are) effective to obtain a
desired therapeutic response
for a particular composition and method of administration for a patient. A
selected dosage level for any
particular patient therefore depends upon a variety of factors including the
desired therapeutic effect,
on the route of adniinistration, on the desired duration of treatment, the
etiology and severity of the
disease, the patient's condition, weight, sex, diet and age, the type and
potency of each active
ingredient, rates of absorption, metabolism and/or excretion and other
factors.
Total daily dose of the compounds of this invention administered to a patient
in single or divided doses
to about 1000 mg, more particularly from about 50 mg to 300 mg, and, further
particularly from about
10 mg to 100 mg. However, higher or lower daily doses can also be appropriate.
The daily dose can be
administered either by means of a once-only administration in the form of a
single dosage unit, or of
several smaller dosage units, or by means of the multiple administration of
subdivided doses at
predetermined intervals. The percentage of active ingredient in a composition
may be varied, though it
should constitute a proportion such that a suitable dosage shall be obtained.
Obviously, several unit
dosage forms can be administered at about the same time. A dosage may be
administered as frequently
CA 02566213 2006-11-08
WO 2005/113544 PCT/US2005/016381
-18-
as necessary in order to obtain the desired therapeutic effect. Some patients
may respond rapidly to a
higher or lower dose and may find much weaker maintenance doses adequate. For
other patients, it
may be necessary to have long-term treatments at the rate of 1 to 4 doses per
day, in accordance with
the physiological requirements of each particular patient. It goes without
saying that, for other patients,
it will be necessary to prescribe not more than one or two doses per day.
The formulations can be prepared in unit dosage form by any of the methods
well known in the art of
pharmacy. Such methods include the step of bringing into association the
active ingredient with the
carrier that constitutes one or more accessory ingredients. In general the
formulations are prepared by
uniformly and intimately bringing into association the active ingredient with
liquid carriers or finely
divided solid carriers or both, and then, if necessary, shaping the product.
Experimental
Mass-spectroscopic methods (FAB-MS, ESI-MS) are used for analyzing.
Temperatures are given in
degrees Celsius; RT denotes room temperature (from 22 C to 26 C).
Abbreviations used are either
explained or correspond to customary conventions to one skilled in the art.
The invention is exemplified through the following Examples.
EXAMPLES
PREPARATION 1
Synthesis of Methyl (3-(N-phenyl-N-2-pyridyl)amino)-2-(di-tert-
butoxycarbonylamino)propionate
(Scheme 1, Compound
Scheme 1
/O O O ~ BoczO ~ ~ \ O~
Nx0 DMAP O Ny
O H ~O O
1 ~
2
\ \
I~ N N I I / I /
H N O N enantiomer separation N
CsC03, CH3CN O'k N O1" ~O'N
O O
3 3R
Methyl 2-(di-tert-butoxycarbonylamino)acrylate (Scheme 1, Compound 2)
CA 02566213 2006-11-08
WO 2005/113544 PCT/US2005/016381
-19-
50 g (0.228 mol) of (tert-butoxycarbonyl)serine (j) were dissolved in 300 mL
of acetonitrile. 107 g
(0.493 mol) of di-tert-butyl dicarbonate and 2.64 g (22 mmol) of 4-
(dimethylamino)pyridine (DMAP)
were added. The mixture was stirred at room temperature overnight, after which
the solvent was
removed under reduced pressure and the residue was taken up in 500 mL of ethyl
acetate. The organic
phase was washed with 500 mL of 1 N HC1, dried using magnesium sulfate and the
organic solvents
were removed under reduced pressure. 23 g of acrylate 2 were obtained by
crystallizing the residue
from 200 mI. of heptane at -30 C and then filtering with suction. The mother
liquor was concentrated
and the residue was dissolved in 140 mL of acetonitrile. 31 g (0.142 mol) of
di-tert-butyl dicarbonate
and 1.26 g (10 nunol) of DMAP were added. After the mixture had been heated at
50 C for 8 h, the
solvent was removed in vacuo and the residue was taken up in 500 mL of ethyl
acetate. The organic
phase was washed with 400 n1i. of 1 N HC1 and dried over magnesium sulfate.
After the solvent had
been removed in vacuo, a further 31.5 g of the acrylate 2 were obtained by
crystallizing from heptane.
Yield: 54.5 g(0.181 mol) 79%. Empirical formula C14H23NO6; M.W. = 301.34; MS
((2M*)+Na+)
625.7. 'H NMR (DMSO-d6) 1.40 (s, 18 H), 3.74 (s, 3 H), 5.85 (s, 1 H), 6.28 (s,
1 H).
Methyl (3-(N-phenyl-N-2-pyridyl)amino)-2-(di-tert-butoxycarbonylamino)-
propionate (Scheme 1,
Compound 3)
4.96 g (16.5 mmol) of acrylate 2 were mixed with 5.6 g (33 mmol) of 2-
anilinopyridine and 32.16 g
(98.7 mmol) of cesium carbonate. 50 mL of acetonitrile were added and the
mixture was stirred at
45 C for 2 days. The solid was filtered off with suction through kieselguhr
and washed 3 times with
100 mL portions of acetonitrile. The combined organic phases were evaporated
and the residue was
chromatographed on silica gel using 1:1 heptane/diethyl ether. 5.66 g (73%) of
the ester 3 were
obtained. Empirical formula C25H33N306; M.W. = 471.56; MS (M+H) 472.2.
Separation of the enantiomers (Scheme 1, Compound 3(S) and Compound 3R
Racemic amino ester 3 was prepared from the corresponding acrylic ester 2 and
then resolved into
enantiomers 3(S) and 3R) by means of preparative BPLC using chiral stationary
phases such as
Chiralpak AD (Daicel) 100 x 380, RT, flow rate 300 mL/min. The purity of the
enantiomers were
determined by analytical HPLC such as Chiralpak-AD-H (Daicel) 4.6 x 250, 30 C,
flow rate I mUmin,
room temperature.
PREPARATION 2
Synthesis of 2-(2-Methylaminopyrimidin-4-yl)-1H-indole-5-carboxylic acid
(Scheme 2, Compound i3
Scheme 2
CA 02566213 2006-11-08
WO 2005/113544 PCT/US2005/016381
-20-
methylgaunidine I I
hydrochloride, O O
0 0 ~ NaOEt/EtOH,
120 C ~ 6.5h, reflux
'~O)y + "o'J~'i/ - "'o N 4 0 5 0 6 NN
H 7
4-Hydrazino- 0
benzoic acid,
50% H2SO411
130 C I \ JJ
HO ~HN
8 /JJH
1-Dimethylamino-4,4-dimethoxypent-l-en-3-one (Scheme 2, Compound 6)
100 g (0.76 mol) of 3,3-dimethoxy-2-butanone (4) were stirred together with
90.2 g (0.76 mol) of N,N-
dimethylformamide dimethyl acetal (~ at 120 C for 48 h. The methanol formed in
the reaction was
removed continuously from the reaction solution by means of distillation.
Crystallization occurred
spontaneously when the solution was cooled, with the crystallization being
brought to completion Uy
addition of a small amount of heptane. This resulted in 128.24 g of crude
product 6 (yield 90%), which
was reacted without any further purification. Empirical formula C9H17NO3; M.W.
= 187.24; MS
(M+H) 188.2. 'H NMR (DMSO-d6) 1.22 (s, 3 H), 2.80 (s, 3 H), 3.10 (s, 9 H),
5.39 (d, J= 15 Hz, 1 H),
7.59 (d, J= 15 Hz, 1 H).
[4-(1,1-Dimethoxyethyl)pyrimidin-2-yl]methylamine (Scheme 2, Compound 7)
1.22 g (53 nunol) of sodium was dissolved in 100 mL of absolute ethanol. 5.8 g
(53 mmol) of
methylguanidine hydrochloride and 10 g (53 mmol) of 1-dimethylamino-4,4-
dimethoxypent-l-en-3-
one (6) were added to this solution, while stirring, and the whole was heated
at reflux for 4 h. To
terminate the reaction, the ethanol was evaporated. The resulting product 7
was used for the
subsequent reaction without any further purification. Yield: 11.5 g (58 mmol,
quantitative); Empirical
formula C9H15N302; M.W. = 197.24; MS (M+H) 198.2. 'H NMR (DMSO-d6) 1.45 (s, 3
H), 2.78 (s, 3
H), 3.10 (s, 6 H), 6.75 (d, J 3 Hz, 1 H), 7.0-7.1 (s(b), 1 H), 8.30 (d, J = 3
Hz, 1 H).
2-(2-Methylaminopyrimidin-4-yl)-1H-indole-5-carboxylic acid (Scheme 2,
Compound 8)
5 g (25 mmol) of [4-(1,1-dimethoxyethyl)pyrimidin-2-yl]methylamine (7) and
3.85 g of
4-hydrazinobenzoic acid were added, at room temperature and while stirring, to
150 mL of 50%
sulfuric acid, and the mixture was heated at 130 C for 4 h. The methanol that
was formed in the
reaction was removed continuously from the reaction solution by means of
distillation. After it had
been cooled down to 10 C, the reaction mixture was poured onto 200 mL of ice
and adjusted to a pH
of about 5.5 with concentrated sodium hydroxide solution. The precipitate of
sodium sulfate and
product mixture formed was filtered off and the filter residue was extracted
several times with
methanol. The combined methanol extracts were concentrated and the product 8
was purified by
CA 02566213 2006-11-08
WO 2005/113544 PCT/US2005/016381
-21-
means of flash chromatography (CH2C12/methano19:1). Yield: 0.76 g(11%);
Empirical formula
C14H12N402; M.W. = 268.28; MS (M+H) 269.1. 'H NMR (DMSO-d6) 2.95 (s, 3 H),
6.90-7.10 (s(b), 1
H), 7.18 (d, J= 3 Hz, 1 H), 7.4 (s, 1 H), 7.58 (d, J = 4.5 Hz, 1 H), 7.80 (d,
J = 4.5 Hz, 1 H), 8.30 (s, 1
H), 8.38 (d, J'= 3 Hz, 1 H), 11.85 (s, 1 H), 12.40-12.60 (s(b), 1 H).
EXAMPLE 1
Synthesis of 2-{ [2-(2-Methylamino-pyrimidin-4-yl)-1H-indole-5-carbonyl]-
amino}-3-(phenyl-pyridin-
2-yl-amino)-propionic acid (A)
Scheme 3
I N N I/ I N N I/
>~O N O HCI H N~O~ HCI 8 HATU, DIEA
~O O 2 O
aus 9
\ I \ I an"~N
O N O 1~0 aq NaOH, MeOH
"~ N N
101 H N N4N OH H N N~
H JJH A H ~H
10 /N
2-{ [2-(2-Methylamino-pyrimidin-4-yl)-1H-indole-5-carbonyl]-amino}-3-(phenyl-
pyridin-2-yl-
amino)-propionic acid, methyl ester (Scheme 3, Compound 10)
2.9 g of the S enantiomer of 3(3 (S)) were dissolved in 30 mL of dioxane and
the solution was cooled
down to 0 C. 30 mL of 4 N HCI in dioxane were added, after which the mixture
was allowed to come
to room temperature and was then stirred for 12 h. The solvent was removed in
vacuo. The residue was
taken up in 30 mL of DMF (solution A). 2.47 g (9.2 mmol) of the acid 8 were
dissolved in 50 mL of
DMF and cooled down to 0 C. 4.21 g of HATU and 6.4 mL of DIEA were added.
After the mixture
had been stirred at 0 C for 45 minutes, it was allowed to come to RT and
solution A was added. The
mixture was stirred at RT for 12 h. The solvent was removed in vacuo and the
residue was partitioned
between 300 mL of a saturated solution of NaHCO3 and 300 mL of ethyl acetate.
The aqueous phase
was extracted 3 times with 100 mL portions of ethyl acetate and the combined
organic phases were
washed with 400 mL of a saturated solution of NaCI. The organic phase was
dried with magnesium
sulfate. The solvents were removed under reduced pressure and the residue was
chromatographed on
silica gel using 1:3 heptane/ethyl acetate. 1.78 g (55%) of the ester 10 was
obtained. Empirical
formula C29H27N703; M.W. = 521.58; MS (M+H) 522.2.
CA 02566213 2006-11-08
WO 2005/113544 PCT/US2005/016381
-22-
2-{ [2-(2-Methylamino-pyrimidin-4-yl)-1H-indole-5-carbonyl]-amino}-3-(phenyl-
pyridin-2-yl-
amino)-propionic acid (Scheme 3, Compound A)
2.0 g (3.8 mmol) of the methyl ester 10 were dissolved in 200 mL of methanol.
1 mL of 2 N aqueous
NaOH was added and the mixture was stirred at room temperature for 12 h. After
the solvents had
been evaporated, the residue was dissolved in water and the pH was adjusted to
-5 using a saturated
solution of NaH2PO4. The resulting precipitate was filtered off and washed
with water. After drying
under reduced pressure of about 1 mbar at 40 C, 1.95 g (quantitative yield) of
the acid A was isolated.
Empirical formula C28H25N703; M.W. = 507.56; MS (M+H) 508.3. 'H NMR (DMSO-d6)
2.95 (s, 3 H),
4.22-4.50 (m, 2 H), 4.65-4.72 (m, 1 H), 6.29 - 6.36 (d, 1 H), 6.70 - 6.79 (m,
1 H), 6.90 - 7.10 (sb, 1
H), 7.13-7.19 (m, 1 H), 7.22-7.38 (m, 5 H), 7.40-7.48 (m, 3 H), 7.50-7.55 (m,
1 H), 7.57-7.60 (m, 1 H),
7.96 (bs, 1 H), 8.34 - 8.40(m, 2 H), 8.80 - 8.90 (d, 1 H), 11.80 (s, 1 H).
IN VITRO TEST PROCEDURE
IKK-Enzyme ELISA
The assay buffer has the following composition (50 mM HEPES, 10 mM MgC12, 10
mM Li-
Glycerophosphate, 2 M Microcystin-LR, 0,01% NP-40, 5 mM DTT).
The IKK enzyme preparation was diluted 1:50 (in-house-made preparation) plus
test compound in
DMSO (final concentration in well: 2 %).
The assay procedures were as follows:
Incubation of enzyme and compound for 30 min;
Addition of 1mM ATP or 50 M ATP;
pSer36-IkB Peptide (Substrate): 40 M;
Incubation for 45 min and Addition of anti- pSer32-pSer36-IkB Peptid -
antibody;
Incubation for 45 min and transfer to protein-G-coated plate;
Incubation for 90 min followed by 3x washing;
Addition of streptavidin-HRP, then incubation 45 min followed by 6x washing;
Addition of TMB and Incubation for 15 min; and
Stop solution and read using photometer.
CA 02566213 2006-11-08
WO 2005/113544 PCT/US2005/016381
-23-
The results from the in vitro profiling are shown in Table I below.
Table I
Compound IC50 (nM) IC50 (nM)
1mM ATP 50 M ATP
0.4
B 56.4 ]
_ C 378 ~ 16.8 ~
D 3.8
In the IKK-enzyme ELISA described above, at a 1 mM ATP, the compound of
formula (A) exhibits a
705, 4,725 and 47.5 times greater IKB kinase IC50 than Compounds (B), (C) and
(D), respectively. This
data demonstrates an unexpectedly significantly superior activity for Compound
(A) relative to
Compounds (B), (C) and (D).
IN VIVO TEST PROCEDURES COMPARISON BETWEEN COMPOUNDS A AND B
NF-KB-induced gene expression contributes significantly to the pathogenesis of
inflammatory diseases
such as asthma and arthritis. IKB kinase (IKK) is the converging point for the
activation of NF-xB by a
broad spectrum of inflammatory agonists.
IKK is a multisubunit complex that contains two catalytic subunits, IKK-1
(also known as IKK-a) and
IKK-2 (also known as IKK-(3), and the regulatory subunit IKK-y. Gene knock out
studies have clearly
demonstrated that IKK-2 or IKK-0 subunits of the IKK complex are required for
NF-KB activation by
all known pro-inflammatory stimuli including lipopolysaccharide (LPS), and IL-
10. Accordingly, IKK-
P-deficient cells are defective in activation of IKK and NF-KB in response to
either tumor necrosis
factor alpha (TNFa) or interleukin-1(3 (IL-10).
In house bio-imaging data have also shown that IL-10-induced NF-xB activity in
the lung is inhibited
by the administration of a dominant negative form of IKK(3 (Adv-IKK-2 DN).
Thus a selective
inhibitor of IKK-(3 would not only be of great interest as a potential anti-
inflanunatory agent but also as
a valuable tool to understand the mechanisms regulating NF-KB activation by
these inflammatory
agonists.
CA 02566213 2006-11-08
WO 2005/113544 PCT/US2005/016381
-24-
A. NF-icB-Luciferase Reporter Mouse Model
Methods employed for mouse imaging studies with Compound (A) and Compound (B).
General Description
Compounds were used as nanomilled suspensions in 0.2% Tween-80 in PBS and were
dosed through
the intranasal route.
Intranasal drug and inflammatory stimulus administration: Mice are
anesthetized in 4% isoflurane gas
in oxygen. A volume of 25 gl is applied to each nostril and the mice are
allowed to breathe in the
suspension.
Balb/c female mice at 6-8 weeks of age were used for studies in which AdV-NFKB-
luciferase reporter
was instilled in the lung. To image mice, they are anesthetized with 4%
isoflurane in oxygen. Luciferin
is delivered i.p. at a dose of 150 mg/kg. 10 minutes after luciferin
injection, the animals are imaged in
an IVIS200 system (Xenogen) with a one-minute bioluminescent exposure.
Alternatively, 10-15
minutes post luciferin adnunistration, mice are rapidly euthanized and
internal tissues are dissected and
imaged ex vivo.
B. Dose Response Determination
1-2 x 108 pfu adenovirus-NFKB-luciferase is delivered intranasally 3-5 days
prior to stimulation (i.n.)
with an inflammatory stimulus (IL-1(3 or LPS). Animals are dosed i.n. with 0.3-
10 mg/kg of compound
min- 1 h prior to challenge with 0.5 gg LPS or 50 ng IL-1(3. Animals are then
imaged once or
several times from lh to 24 h after the inflammatory stimulus is administered.
25 The results from the in vivo procedures are as follows.
The effects of Compound (A) and Compound (B) on IL-1(3-induced NF-xB
activation are shown in
Figures 1 and 2. While both compound inhibited in a dose-dependent manner NF-
xB activity,
Compound (A) showed superior efficacy with an estimated ED50 around lmg/kg.
30 C. Pharmacokinetic Procedures
Male Hartley guinea pigs (450-550 g) previously sensitized with ovalbumin were
used for the
determination of compound levels in lung and plasma. Nanomilled suspensions of
Compound (A) and
Compound (B) were dosed via intra-tracheal instillation at 0.01, 0.03, 0.1 and
0.3mg/kg. One hour
after dosing, animals were euthanized (Euthasol), and 1 mL blood samples were
obtained by cardiac
puncture and collected into heparin-coated syringes. Plasma was separated from
the cellular
component of the blood by centrifugation, and stored at -80 C until assayed.
Lungs were dissected out,
CA 02566213 2006-11-08
WO 2005/113544 PCT/US2005/016381
-25-
blotted dry, weighted and stored in 20-25 mL glass vials individually at -80 C
until assayed for
compound levels.
Key differences between Compound (A) and Compound (B) pharmacokinetic profiles
are illustrated in
Figure 3 using the finding from the highest dose group (0.3 mg/kg).
Figure 3 shows that lung exposure to either Compound (A) or Compound (B) after
i.t. instillation of
Compound (B) is low relative to Compound (A) after i.t. administration,
suggesting that Compound
(B) is rapidly absorbed from the lung.
Figures 3 to 5 also show that Compound (B) would be a weaker candidate for
inhalation because 1) It
is highly systemically distributed upon exposure, when administered
intratracheally; 2) Compound (B)
is a prodrug of Compound (A) having a different exposure profile; and 3)
Compound (B) produces
reduced exposure to Compound (A) in the lung than is obtained by dosing
directly with Compound
(A).
Compound (A) is a stronger inhalation candidate than Compound (B) because 1)
Compound (A) has
low systemic exposure after i.t. and PO administration; and 2) Compound (A)
should have longer lung
residency time.
In addition, support is found that Compound (B) is highly systemically
available when administered
orally.
The lung to plasma ratio for Compound (A) ranges from 143 to 284 (depending on
the dose) whereas
the lung to plasma ratio for Compound (B) ranges from 13 to 44 (depending on
the dose). These ratios
were obtained by dividing the lung compound levels to that of the
corresponding plasma levels at the
same dose.