Note: Descriptions are shown in the official language in which they were submitted.
CA 02570820 2006-12-04
WO 2006/001795 PCT/US2004/018620
BASIC SALTS AND MONOHYDRATES OF CERTAIN
ALPHA, BETA-PROPIONIC ACID DERIVATIVE
BACKGROUND OF THE INVENTION
Formulation of a convenient pharmaceutical dosage form can be very
complicated. While there are many factors that contribute to the design
criteria, perhaps
the most important is the active pharmaceutical ingredient ("API") that will
be
delivered. The API's solubility, route of administration, dosage size, taste,
absorption
target or cite of application, metabolic properties and the like often must
all be
considered. And if the API has stability and/or handling issues, the
complexity rises
accordingly. Therefore, where possible, it is highly desirable to develop APIs
that can
be conveniently handled and processed. Chemical stability, solid-state
stability and
shelf life of the active ingredients are also important considerations. To the
extent that
the API is stable, non-reactive with common excipients under normal processing
and
storage conditions and the like, dosage form development can be greatly
facilitated.
The API and compositions containing it should be capable of being
effectively stored over appreciable periods of time, without exhibiting a
significant
change in the physico-chemical characteristics of active compounds.
Crystalline
materials, for example, can in certain cases be less difficult to handle and
to formulate
when compared to amorphous forms. But stable crystalline forms that are
suitable for
formulation and provide sufficient solubility and bioavailabilty are neither
necessarily
available nor predictable, especially when complex molecules are involved.
Moreover,
some crystalline materials are not sufficiently stable to ensure that they
will not convert
to another form, crystalline or not, during manufacturing or storage.
Salt formation may improve certain properties such as stability, water
solubility and bioavailability. Salts may also influence hygroscopicity and
crystallinity.
Of course, there is a vast array of possible salts (See WO 03/048 1 1 6 at 21,
line 18-22,
line 4) not all salts can be made are equally easy to make or equally
advantageous. And
not all salts permit the same crystalline forms. The nature of the salts can
also influence
can the properties of the API and the ability to make a stable dosage form
using modem,
high speed equipment in an efficient and cost effective manner. And, perhaps
most
importantly, there must be no unnecessary impediment to the safety and
efficacy of the
API.
SUMMARY OF THE INVENTION
The present invention includes the basic salts of ((S)-2-methoxy-3-[4-{3-
(4-methanesulfonyloxyphenyl) propylamino}phenyl]propionic acid) (also referred
to
CA 02570820 2006-12-04
WO 2006/001795 PCT/US2004/018620
-2-
herein as the basic salts of "(S)MP") and, in particular, the amino acid salts
thereof. A
particularly preferred amino acid salt in accordance with the present
invention is the
arginine salt of (S)MP (the "Arginine salt" or "(S)MP-Arg".). The invention
also
encompasses these salts in pure or substantially pure forms.
The present invention also includes the monohydrate form of (S)MP as
well as the monohydrate of the basic salts of (S)MP, preferably the
monohydrate of the
amino acid salts of (S)MP, and most preferably (S)MP arginine monohydrate
(also
referenced as "Arginine salt monohydrate" or "(S)MP-Arg-1H20"). The invention
also
encompasses these monohydrates in a pure or substantially pure forms.
Another aspect of the present invention is crystalline forms of the basic
salts of (S)MP, whether in monohydrated form or not.
A particularly preferred embodiment of the present invention is a
crystalline form of (S)MP monohydrate or (S)MP-Arg-1H20.
Another aspect of the present invention provides one or more of the
foregoing salts and/or monohydrates of (S)MP as the API in a novel
pharmaceutical
product which is stable and can be efficiently manufactured using traditional
high speed
equipment. These pharmaceutical products include one of the foregoing as the
API salts
and/or monohydrates of (S)MP and a pharmaceutically acceptable carrier,
diluent,
excipient or solvent.
Another particularly preferred aspect of the present invention is a
pharmaceutical composition that contains, as the API, one or more of the
forgoing salts
and/or monohydrates of (S)MP and, in particular an Arginine salt and/or
Arginine salt
monohydrate, in crystalline form, wherein the API is present in an amount of
at least
about 0.01 % by weight of the pharmaceutical composition.
Another aspect of the present invention provides a process for the
preparation of one or more of the foregoing salts and/or monohydrates of (S)MP
and, in
particular an (S)MP monohydrate Arginine salt and/or Arginine salt
monohydrate, in a
crystalline form.
DESCRIPTION OF THE DRAWINGS
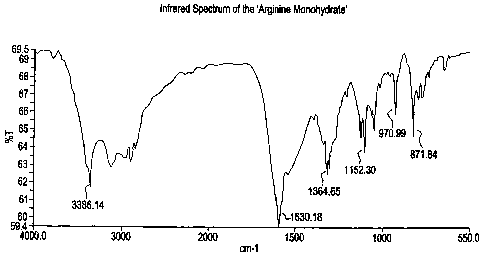
Figure 1 is an infrared spectrum of one batch of Arginine monohydrate of
((S)-2-methoxy-3-[4-{3-(4-methanesulfonyloxyphenyl)
propylamino}phenyl]propionic
acid).
Figure 2 is an X-ray powder diffractogram of one batch of Arginine
monohydrate of ((S)-2-methoxy-3-[4-{3-(4-methanesulfonyloxyphenyl)
propylamino}phenyl]propionic acid).
CA 02570820 2006-12-04
WO 2006/001795 PCT/US2004/018620
-3-
Figure 3 is a DSC thermogram of one batch of Arginine monohydrate of
((S)-2-methoxy-3-[4-{3-(4-methanesulfonyloxyphenyl)
propylamino}phenyl]propionic
acid).
Figure 4 is an infrared spectrum of one batch of Arginine salt of ((S)-2-
methoxy-3-[4-{3-(4-methanesulfonyloxyphenyl) propylamino}phenyl]propionic
acid).
Figure 5 is an X-ray powder diffractogram of one batch of Arginine salt
of ((S)-2-methoxy-3-[4-{3-(4-methanesulfonyloxyphenyl)
propylamino}phenyl]propionic acid).
Figure 6 is a DSC thermogram of one batch of Arginine salt of ((S)-2-
methoxy-3-[4-{3-(4-methanesulfonyloxyphenyl) propylamino}phenyl]propionic
acid).
DETAILED DESCRIPTION
A compound (also referred to herein as a molecule or chemical) that may
be used as a starting material for the preparation of salts and hydrates of
the present
invention, can be prepared according to known procedures, such as those
disclosed in,
inter alia, International Publication No. WO 03/048 1 1 6, which is
incorporated herein by
reference both in its entirety and for the purpose stated. See in parkicular
Example 14.
As used herein, the salts and monohydrates of the present invention refer
to a molecule, per se, unless expressly stated otherwise or as is appropriate
under the
circumstances. When reference is made to a crystal or crystalline material, a
sufficient
number of molecules necessary to make a single crystal is contemplated. In the
context
of a pharmaceutical dosage form, pharmaceutical product or pharmaceutical
composition, at least 0.10% by weight of the product is a basic salt of (S)MP
or a
monohydrate thereof as disclosed herein.
The present invention provides basic salts of (S)MP, including those in
crystalline form. Examples of basic salts include salts of alkali and earth
alkali metals,
and amino acid salts (both naturally occurring and synthetic amino acids).
Preferably,
the salt is an arginine salt of compound (I) ("Arginine salt"), which was
found to be
particularly suitable.
The present invention encompasses monohydrates of (S)MP and its basic
salts as novel materials. Monohydrates ideally include a ratio of one molecule
of (S)MP
or its basic salt with one molecule of bound water of crystallization.
Preferred are
monohydrates of basic salts and more preferably Arginine salts of (S)MP. Most
preferred are (S)MP monohydrate and Arginine salt monohydrate. Even more
preferred
is the Arginine salt monohydrate having the characteristics of at least one of
Figures 1-3.
CA 02570820 2006-12-04
WO 2006/001795 PCT/US2004/018620
-4-
The present invention also provides a process for preparing basic salts
and monohydrates of (S)MP and in particular amino acid salt monohydrates
thereof. In
particular, Arginine monohydrate of (S)MP or a salt thereof is preferably
dispersed or
dissolved in a suitable solvent, and is reacted with a suitable source of
arginine ion in
the presence of water, which comprises:
(i) refluxing (S)MP, DM water and Arginine';
(ii) fizrther refluxing with additional quantities of a suitable reaction
solvent, preferably alcoholic solvent, more preferably, isopropanol;
(iii) cooling and filtering the reaction mixture to obtain the Arginine
salt hydrate.
The concentration of (S)MP is preferably in the range of about 60 to 70%
weight/weight, more preferably in the range of about 65 to 69%. The
concentration of
arginine is preferably in the range of about 21 to 39% weight/weight, more
preferably in
the range of about 26 to 33%. The concentration of water is in the range of
about 1.0 to
9.0%.
Formation of a monohydrate requires the presence of water at some
stage; water may be added as a co-solvent in the above process, e.g., about 5
to 100%
water. However, it is also possible to provide sufficient water for
monohydrate
formation by canying out the reaction with exposure to atmospheric moisture,
or by use
of non-anhydrous solvents.
A suitable reaction solvent for the preparation of arginine salt
monohydrate is a ketone, such as acetone, ethyl methyl ketone, ether such as
tetrahydrofuran, dioxane, isopropyl ether, diethyl ether, an alcohol such as
methanol,
ethanol, propan-2-ol, isopropanol or mixtures thereof and the like. The
temperature used
in the reaction is maintained in the range of about 30 to 120 C, preferably
about 30 to
70 C. The duration of the reaction is maintained in the range of about 3 to
12 h,
preferably, for approximately 5 hours.
In one aspect of this invention, the Arginine salt monohydrate has been
reproducibly isolated, containing water ranging from about 2.6-3.4% by
weight/weight,
preferably from about 2.9-3.4% by weight/weight, preferably from about 3.0% by
weight/weight. This is consistent with a monohydrate formation (1:1 ratio of
Arginine
salt of compound (I) and water) (1 molar equivalent of HZ0 = 3.00 % by
weight/weight)
has been isolated.
The crystalline form may be characterized by X-ray powder diffraction
pattern (XRPD). X-ray powder diffraction pattern analysis was performed on
Rigaku
CA 02570820 2006-12-04
WO 2006/001795 PCT/US2004/018620
-5-
D/Maz-2200 model diffractometer equipped with horizontal goniometer in 0/20
geometry.
The Copper Ka(k= 1.5418 A) radiation was used. The tube voltage and amperage
were set
at 50 KV and 34 mA respectively. The divergence and scattering slits are set
at 1/2
degree, receiving slit at 015 mm and scattering slit at 1/2 degree. Diffracted
radiation is
detected by scintillation counter detector, 0 to 29 continuous scan at 3
degrees/minutes
from 3 to 45 degrees be used. A stand'ard was analyzed to check the
instrumental
alignment the data was collected and analyzed using.
Differential scanning calorimetry was performed using a Shimadzu DSC-
50 calorimeter. The sample was placed into aluminum pan, the weight was
accurately
recorded, and the pan was covered with lid and left unclamped. Each sample was
equilibrated and heated (10 C/minute) under nitrogen atmosphere.
The crystalline form may also characterized by Fourier transform infra
red spectra and recorded in solid state as KBr dispersion. Perkin-Ehner 1600
Fourier
transform infra red spectrophotometer was used for characterization.
In one suitable embodiment, there is provided an Arginine salt
monohydrate of (S)MP (1 molar equivalent of H20 = 3.00 % by weight/weight),
characterized by one or more of the following:
(i) Infra red spectrum containing peaks at about 1630, 1364, 1152,
971, 871 cni ';
(ii) X-Ray powder diffraction (XRPD) pattern containing peaks at
about 8.44, 17.48, 17.60, 18.84, 21.02, 21.18, 22.02 degrees 20; and/or
(iii) Endotherms at about 92.60, 132.53, 231.80 and 272.22 C in
Differential Scanning Calorimeter (DSC).
In one preferred aspect, Arginine monohydrate has infrared spectrum
containing peaks at 1630, 1364, 1152, 971, 871 cm"1. Preferably, the infrared
spectruin
is substantially in accordance with Figure 1.
In one preferred aspect, an Arginine monohydrate provides X-Ray
powder diffraction (XRPD) pattern substantially as shown in Figure 2, and
preferably
has peaks at 8.44, 17.48, 17.60, 18.84, 21.02, 21.18, 22.02 degrees 20--j: 0.2
20.
In one preferred aspect, an Arginine monohydrate provides a Differential
Scanning Calorimeter (DSC) spectrum substantially as shown in accordance with
Figure
3, and preferably has endotherms at about 92.60, 132.53, 231.80 and 272.22 C.
It is to be understood that the X-ray diffraction (XRD) patterns, IR
spectra and endotherms reported herein, while reported as absolute numbers in
tables
CA 02570820 2006-12-04
WO 2006/001795 PCT/US2004/018620
-6-
and absolute positions in the figures, are intended to include the normal
amount of
positional variation due to experimental error, operator error, differences in
equipment,
technique, packing, contamination and the like. It is understood by one
skilled in the art
that there may be substantial variation in the measured values. For example,
it is
believed the measurement of XRD peaks may have variability of.f0.2 20.
However,
based on these techniques, particularly when two or more of them are used in
conjunction, and the overall spectrum and/ or patterns reported in the
figures, one of
ordinary skill in this art will be able to identify whether or not a compound
is a basic salt
or basic salt monohydrate of (S)MP in accordance with the invention.
It should be kept in mind that slight variations in the observed 2 theta
angles values are to be expected based on the specific diffractometer
employed, the
analyst and the sample preparation technique. More variation is expected for
the relative
peak intensities, which is largely affected by the particle size of the
sample. Thus,
identification of the exact crystalline form of a compound should be based
primarily on
observed 2 theta angles with lesser importance attributed to relative peak
intensities. The
peaks reported herein are listed in order of their peak intensities. Thus, the
first listed
peak has stronger intensity than the second listed peak in the pattern. For
example, the 2
theta diffraction angles and corresponding d-spacing values account for
positions of
various peaks in the X-ray powder diffraction pattern. D-spacing values are
calculated
with observed 2 theta angles and wavelength using the Bragg equation well
known to
those of skill in the art.
In a further preferred aspect of the invention, an Arginine salt
monohydrate has a melting point in the range of 125 to 140 C, such as 127 to
135 C,
for example 132.5 C.
In a further preferred aspect of the invention, the invention provides
Arginine salt monohydrate, characterized in that it provides at least one of
an:
(i) Infrared spectrum substantially in accordance with Figure 1;
(ii) X-Ray powder diffraction (XRPD) pattern substantially in
accordance with Table 1 or Figure 2; and/or
(iii) Endotherm at about 132.5 C in Differential Scanning Calorimeter
(DSC) spectrum substantially in accordance with Figure 3.
The arginine salt of (S)MP can be produced by reacting (S)MP or some
other salt thereof, preferably dispersed or dissolved in a suitable solvent,
is reacted with
a suitable source of arginine ion in the presence of a solvent in the 1:1
ratio of (S)MP
and arginine, which comprises:
CA 02570820 2006-12-04
WO 2006/001795 PCT/US2004/018620
-7-
(i) refluxing compound (I), DM water and arginine
(ii) further refluxing with suitable reaction solvent,
(iii) azeotropically separating the product obtained by using suitable
solvent cooling and filtering the resultant product to obtain arginine salt of
(S)MP.
The concentration of (S)MP is preferably in the range of about 60 to 80%
weight/weight, more preferably in the range of about 65 to 75%. The
concentration of
arginine is preferably in the range of about 20 to 40% weight/weight, more
preferably in
the range of about 25 to 35%.
A suitable reaction solvent for the preparation of anhydrous form is a
ketone, such as acetone, ethyl methyl ketone, ether such as tetrahydrofuran,
dioxane,
diisopropyl ether, diethylether, an alcohol, such as methanol, ethanol, propan-
2-ol. The
solvent used during the azeotropic removal of water is selected from a
hydrocarbon such
as xylene, toluene, an ester such as ethyl acetate, a nitrile such as
acetonitrile, or a
halogenated hydrocarbon such as dichloromethane, dichloroethane and the like.
The
temperature used in the reaction is maintained in the range of about 30 to 120
C,
preferably about 30 to 70 C. The duration of the reaction is maintained in the
range of
about 3 to 12 h.
Accordingly, the present invention provides arginine salt of (S)MP in
nonhydrated form ((S)MP-Arg) as a novel compound, which is characterized by at
least
one of an:
(i) Infra red spectrum containing peaks at 1638, 1364, 1173, 1152,
971, 871 cni ';
(ii) X-Ray powder diffraction (XRPD) pattern containing peaks at
16.40, 16.82, 17.38, 19.28, 19.70, 20.22, 20.56 degrees 20; and/or
(iii) Endotherm at about 192.9 C and 228.7 C.
In one preferred aspect, the (S)MP-Arg provides an infrared spectrum
substantially in accordance with Figure 4.
In one preferred aspect, (S)MP-Argis characterized by an infrared
spectrum containing peaks at about 1638, 1364, 1173, 1152, 971, 871 cm t.
In one preferred aspect, the (S)MP-Arg provides X-Ray powder
diffraction (XRPD) substantially in accordance with Table 2 or Figure 5.
In one preferred aspect, the (S)MP-Arg characterized by an X-ray
powder diffraction(XR.PD) pattem containing peaks at about 16.40, 16.82,
17.38, 19.28,
19.70, 20.22,20.56 degrees 20.t0.2 20.
CA 02570820 2006-12-04
WO 2006/001795 PCTIUS2004/018620
-8-
In one preferred aspect, the (S)MP-Arg provides a Differential Scanning
Calorimeter (DSC) spectrum substantially in accordance with Figure 6.
In one preferred aspect, the (S)MP-Arg is characterized by a Differential
Scanning Calorimeter (DSC) spectram containing endotherm at about 192.9 and
228.7 C.
In a further preferred aspect the (S)MP-Arg has a melting point in the
range of about 185 to 200 C, such as 187 to 195 C, for example, 192.9 C.
In a further preferred aspect, the invention provides a (S)MP-Arg
characterized in that it provides at least one of an:
(i) Infrared spectrum substantially in accordance with Figure 4;
(ii) X-Ray powder diffraction (XRPD) pattern substantially in
accordance with Figure 5; and/or
(iii) Endotherm at about 192.9 C in Differential Scanning Calorimeter
(DSC) spectrum substantially in accordance with Figure 6.
The invention also provides a process for the conversion of the Arginine
salt monohydrate to (S)MP-Arg, wherein the Arginine salt monohydrate is
suspended in
a suitable solvent and refluxed to remove water azeotropically, which
comprises:
(i) refluxing Arginine salt monohydrate with a suitable solvent to
azeotropically remove water content,
(ii) cooling and filtering the resultant product to obtain Arginine salt
of compound (I).
The solvent used during the azeotropic removal of water is selected from
a hydrocarbon such as xylene, toluene, an ester such as ethyl acetate, a
nitrile such as
acetonitrile, or a halogenated hydrocarbon such as dichloromethane,
dichloroethane and
the like. The temperature used in the reaction is maintained in the range of
about 30 to
80 C, preferably about 30 to 70 C. The duration of the reaction is maintained
in the
range of about 3 to 12 h.
The invention also provides a process for the conversion of the (S)MP-
Arg to Arginine monohydrate, wherein the (S)MP-Argis suspended in a suitable
solvent
in the presence of water, and optionally thereafter as required:
(i) refluxing (S)MP-Arg and DM water,
(ii) further refluxing with suitable reaction solvent,
(iii) cooling and filtering the reaction mixture to obtain the Arginine
monohydrate.
CA 02570820 2006-12-04
WO 2006/001795 PCT/US2004/018620
-9-
A suitable reaction solvent for the preparation of Arginine monohydrate
is a ketone, such as acetone, ethyl methyl ketone an ether such as
tetrahydrofuran,
dioxane, isopropyl ether, diethyl ether, an alcohol such as methanol, ethanol,
propan-2-
ol, isopropanol or mixtures thereof and the like. The temperature used in the
reaction is
maintained in the range of about 30 to 80 C, preferably about 30 to 70 C. The
duration
of the reaction is maintained in the range of about 3 to 12 h.
Recovery of the desired compound may comprise crystallization from a
solvent, conveniently the reaction solvent, usually assisted by cooling. For
example,
from an alkanol such as propan-2-ol or a hydrocarbon such as toluene. An
improved
yield of the badic salts may be obtained by evaporation of some or all of the
solvent or
by crystallization at elevated temperature, for example about 75 C, followed
by slow
cooling. Co-solvents can be added to reduce the solubility of the product in
the solvent
system to provide a good yield, e.g. diethyl ether, diisopropyl ether and
heptane. Careful
control of precipitation temperature and seeding may be used to improve the
reproducibility of the product form.
Crystallization may also be initiated by, in one non-limiting example,
seeding.
When used herein the term "Tonset" is generally determined by
Differential Scanning Calorimetry and has a meaning generally understood in
the art, as
for example expressed in "Pharmaceutical Thermal Analysis, Techniques and
Applications", Ford and Timmins, 1989 as "The temperature corresponding to the
intersection of the pre-transition baseline with the extrapolated leading edge
of the
transition".
"Substantially pure" means the material that contains about 95-99.8%, at
least about 95%, preferably about 98%, more preferably about 99.8% of the
compounds
of the present invention. "Pure" means greater than 99.8% of the compound is
the
desired compound. This is, of course, not counting excipients, diluents,
carriers or
solvents and other pharmaceutically active molecules in the case of a
pharmaceutical
composition.
As mentioned above the compounds of the invention have useful
therapeutic properties of the type: The present invention accordingly provides
(S)MP in
monohydrated form, as a basic salt or as a basic salt monohydrate for use as
an active
therapeutic substance disclosed for other members of the claim described in WO
03/048 1 1 6. These molecules form may be administered per se or, preferably,
as a
CA 02570820 2006-12-04
WO 2006/001795 PCT/US2004/018620
-10-
phannaceutical composition also comprising a pharmaceutically acceptable
carrier,
diluent or excipient.
Accordingly, the present invention also provides pharmaceutical
compositions comprising the basic salts of (S)MP, (S)MP monohydrate and
monohydrated forms of basic salts of (S)MP, and more preferably (S)MP-Arg and
(S)MP-1H20 mixed with, solubilized or dispersed in a pharmaceutically
acceptable
carrier, diluent, solvent and /or the excipient. Preferably, the basic salt of
(S)MP, (S)MP
monohydrate or monohydrated form of a basic salt of (S)MP (collectively the
"API") is
in a crystalline form. When present in such pharmaceutical compositions of
pharmaceutical products, the API should be present in an amount of at least
about
0.10% by weight of the pharmaceutical product. When the API is in crystalline
form, at
least one crystal should be present, although preferably at least about 0.10%
by weight
should be present.
These APIs and in particular, Arginine salt monohydrate or Arginine salt
are normally administered in unit dosage form.
The API may be administered by any suitable route but usually by the
oral or parenteral routes. For such use, the compound will normally be
employed in the
form of a pharmaceutical composition in association with a pharmaceutical
carrier,
diluent and/or excipient, although the exact form of the composition will
naturally
depend on the mode of administration.
The pharmaceutical compositions are prepared by admixture and are
suitably adapted for oral, parenteral or topical administration, and as such
may be in the
form of tablets, capsules, oral liquid preparations, powders, granules,
lozenges, pastilles,
reconstitutable powders, injectable and infusible solutions or suspensions,
suppositories
and transdermal devices. Orally administrable compositions are preferred.
Tablets and capsules for oral administration are usually presented in a
unit dose, and contain conventional excipients sttch as binding agents,
fillers, diluents,
tableting agents, lubricants, disintegrants, colorants, flavorings, and
wetting agents. The
tablets may be coated according to methods known in the art.
Suitable fillers for use include cellulose, mannitol, lactose and other
similar agents. Suitable disintegrants include starch, polyvinylpyrrolidone
and starch
derivatives such as sodium starch glycolate. Suitable lubricants include, for
example,
magnesium stearate. Suitable pharmaceutically acceptable wetting agents
include
sodium lauryl toluenesulfonate.
CA 02570820 2006-12-04
WO 2006/001795 PCTIUS2004/018620
-11-
Solid oral compositions may be prepared by conventional methods of
blending, filling, tableting or the like. Repeated blending operations may be
used to
distribute the active agent throughout those compositions employing large
quantities of
fillers. Such operations are, of course, conventional in the art.
Oral liquid preparations may be in the form of, for example, aqueous or
oily suspensions, solutions, emulsions, syrups, or elixirs, or may be
presented as a dry
product for reconstitution with water or other suitable vehicle before use.
Such liquid
preparations may contain conventional additives such as suspending agents, for
example
sorbitol, syrup, methyl cellulose, gelatin, hydroxyethylcellulose,
carboxymethyl
cellulose, aluminum stearate gel or hydrogenated edible fats, emulsifying
agents, for
example lecithin, sorbitan monooleate, or acacia; non-aqueous vehicles (which
may
include edible oils), for example, almond oil, fractionated coconut oil, oily
esters such as
esters of glycerine, propylene glycol, or ethyl alcohol; preservatives, for
example methyl
or propyl p-hydroxybenzoate or sorbic acid, and if desired conventional
flavoring or
coloring agents.
For parenteral administration, fluid unit dose forms are prepared
containing a compound of the present invention and a sterile vehicle. The
compound,
depending on the vehicle and the concentration, can be either suspended or
dissolved.
Parenteral solutions are normally prepared by dissolving the active compound
in a
vehicle and filter sterilizing before filling into a suitable vial or ampoule
and sealing.
Advantageously, adjuvants such as a local anesthetic, preservatives and
buffering agents
are also dissolved in the vehicle. To enhance the stability, the composition
can be frozen
after filling into the vial and the water removed under vacuum.
Parenteral suspensions are prepared in substantially the same manner
except that the active compound is suspended in the vehicle instead of being
dissolved
and sterilized by exposure to ethylene oxide before suspending in the sterile
vehicle.
Advantageously, a surfactant or wetting agent is included in the composition
to facilitate
uniform distribution of the active compound.
As is common practice, the compositions will usually be accompanied by
written or printed directions for use in the medical treatment concerned. The
unit dose
compositions of the invention comprise the API in an amount providing up to
about 12
mg, including about 0.1-12 mg.
The invention is illustrated by examples which are not intended to limit
the invention in any way.
CA 02570820 2006-12-04
WO 2006/001795 PCT/US2004/018620
-12-
EXAMPLE 1
Preparation of [(S) 3-Methoxy -3-[4-{3-(4-methanesulfonyoxyphenyl)
propylamino}phenyl]propionic acid] ((S)MP).
/ C02H
H OMe
lik
O;S~ I H
Me 0
Step (i)
To a suspension of LAH (22.1 g, 2.5 eq, 583 mmol) in dry THF (1.0 L),
was added dropwise a THF (50 mL) solution of methyl 3-(4-
hydroxyphenyl)propionate
(21 g, 1.0 eq, 116 mmol) at RT. The reaction mixture was refluxed for 4-5 h.
It was
worked up by quenching with excess ethyl acetate followed by addition of water
(23
mL), 15% aq. NaOH (23 mL) and water (70 mL) under controlled stirring and
maintaining RT. To the workup mixture conc. HCI was added to adjust the pH at
7Ø It
was then filtered through celite and washed with ethyl acetate. Combined
filtrate was
dried (Na2SO4) and condensed. Obtained residue was chromatographed (ethyl
acetate/hexanes) to obtain 3-(4-hydroxyphenyl)propanol (17 g, 100%) as white
solid.
Mp: 52-54 C.
1H NMR (CDC13, 200 MHz S: 1.78-1.86 (m, 2H); 2.63 (t, J = 7.9 Hz,
2H); 3.67 (t, J = 6.3 Hz, 2H); 6.74 (d, J = 8.8 Hz, 2H); 7.05 (d, J = 8.8 Hz,
2H).
IR (neat) cm"1: 3485, 3029, 2940, 1505.
Mass m/z (CI): 152 [M + 1].
Step (ii)
To a DCM (550 mL) solution of 3-(4-hydroxyphenyl)propanol (17 g, 1.0
eq, 111.8 mmol), obtained in the step (i) and triethylamine ( 93.3 mL, 6.0 eq,
670.8
mmol) was added methanesulfonyl chloride ( 26 mL, 3.0 eq, 335.4 mmol) dropwise
at
0 C. The reaction mixture was stin~ed at RT for 16 h, after that it was worked
up by
diluting with excess DCM and washing the organic layer with dil. HCI, water
and brine.
The organic layer was dried (Na2SO4) and concentrated. Desired product from
the crude
mass was purified by recrystallization from diisopropylether. The remaining
mother
liquor was condensed and was chromatographed (ethyl acetate/hexanes) to obtain
further amount desired compound (total yield 20.8 g, 61%) as white solid.
Mp: 60-62 C.
'H NMR (CDC13, 200 MHz: S 2.00-2.18 (m, 2H); 2.77 (t, J = 7.8 Hz,
2H); 3.00 (s, 3H); 3.13 (s, 3H); 4.23 (t, J= 6.3 Hz, 211); 7.22 (aromatics,
4H).
CA 02570820 2006-12-04
WO 2006/001795 PCTIUS2004/018620
-13-
IR (neat) cm 1: 3029, 2935, 1504.
Mass m/z (Cl): 309 [M + 1].
Step (iii)
Preparation of (S)-2-Hydroxy-3-(4-nitrophenyl)propionic acid
~ CO2H
~ ~ H'OH
02N
To a solution of (S)-(4-nitrophenyl) glycine (10g, 47.6 mmol) in a
mixture of water (50 mL), H2SO4 (1M, 60 mL) and acetone (150 mL) at-5 C, was
added under stirring, a solution of sodium nitrite (9.85g, 142.8 mmol) in
water (40 mL)
drop wise over a period of 30 min. The reaction mixture was stirred at -5 to 0
C for
another 1.5 h, followed by stirring at room temperature for 16 h. Acetone was
removed
and then the reaction mixture was diluted with 500 mL ethyl acetate. Organic
layer was
washed with brine, dried over anhydrous Na2SO4, and concentrated. The crude
mass
was purified by crystallization from isopropyl acetate (9.0 g, 96 %).
Mp: 134-136 C.
[a]D: -25 (c 1.0, MeOH)
1H NMR (CDC13) S:
3.04 (dd, J=14, 7.8 Hz, 1H), 3.24 (dd, J=14, 4, Hz, 1H), 4.39 (dd, J 7.3, 4.1
Hz, 1H),7.42(d,J=8.7Hz,2H), 8.16(d,J=8.7Hz,2H).
IR (neat) cm 1: 3485, 3180, 2927, 1715, 1515, 1343.
Mass m/z (CI): 212 (M+1).
Step (iv)
Preparation of (S)-ethyl-2-hydroxy-3-(4-nitrophenyl) propionate
, CO2Et
O N ~ I W OH
2
(S)-2-Hydroxy-3-(4-nitrophenyl) propionic acid (9.0 g, 42.6 mmol),
obtained from step (iii) above, was dissolved in dry EtOH (300 mL). To this
solution
was added conc. H2SO4 (326 L, 5.9 mmol), and refluxed for 5 to 6 h. The
reaction
mixture was neutralized with aqueous sodium bicarbonate. Ethanol was condensed
on
rotavapor, and the residue was dissolved in ethyl acetate. Organic layer was
washed
with aqueous sodium bicarbonate, water, brine, and then dried over anhydrous
Na2SO4,
and concentrated. Desired product was obtained from the crude mass by
crystallizing
from diisopropylether (8.0 g, 78.5 %).
Mp: 74-76 C.
CA 02570820 2006-12-04
WO 2006/001795 PCT/US2004/018620
-14-
[a]D: -13 (c 1.0, MeOH)
'H NMR (CDCl3) 1.30 (t, J = 7 Hz, 3H), 3.06 (dd, J= 14, 7, Hz, 1H),
3.25 (dd, J=14, 4.3, Hz, 1 H), 4.25 (q, J= 7 Hz, 2H), 4.25 (dd, J = 7, 4.3 Hz,
1 H), 7.42
(d,J=8.7Hz,2H),8.16(d,J=8.7Hz,2H).
IR (neat) cm"1: 3432, 2924, 1736, 1518, 1347.
Mass m/z (CI): 240 (M+1).
Step (v)
Preparation of (S)-ethyl-2-methoxy-3-(4-nitrophenyl)propionate
/ COZEt
ON ~ I H~OEt
2
To a mixture of (S)-ethyl-2-hydroxy-3-(4-nitrophenyl)propionate (12.5 g,
52.3 mmol), obtained in step (iv) above, and powdered Ag20 (36.3 g, 157 mmol)
in dry
acetonitrile (260 mL) was added methyl iodide (13 mL, 209.2 mmol) at room
temperature. Activated molecular sieves (4 A) (12.5 g) were added and then the
reaction
mixture was stirred at room temperature for 16 h. The reaction mixture was
filtered
through celite, and concentrated. The crude mass was chromatographed using
ethyl
acetate and hexanes to obtain the desired product as viscous liquid (10.0 g,
75%).
[a]D: -30.1 (c 1.0, MeOH)
'H NMR (CDC13) S:
1.24(t,J=7.1Hz,3H);3.09(d,J=5.4Hz,111);3.12(d,J=2.7Hz,
111); 3.35 (s, 3H); 3.96 (dd, J= 7.5, 5.1 Hz, 111); 4.19 (q, J= 7.1 Hz, 2H);
7.39 (d, J=
8.6Hz,2H);8.13(d,J=8.6Hz,2H).
IR (neat) cm": 2995, 1747, 1604, 1521, 1343.
Mass m/z (CI): 254 (M+1).
Step (vi)
Preparation of (S)-ethyl 2-methoxy-3-(4-aminophenyl)propionate
/ CO2Et
H2N ~ I OMe
(S)-Ethyl 2-methoxy-3-(4-nitrophenyl)propionate (8.0, 31.6 mmol),
obtained in step (v) above, was dissolved in dry methanol (200 mL). To this
solution
was added 10% Pd/C (2.5 g), and hydrogenated using hydrogen gas (20 psi) for 3-
4 h.
The reaction mixture was filtered through celite, and concentrated to a syrupy
mass.
After column chromatography using ethyl acetate / hexanes the desired product
was
isolated as thick liquid (7.0 g, quantitative).
CA 02570820 2006-12-04
WO 2006/001795 PCTIUS2004/018620
-15-
[a]D: -14.1 (c 1.0, MeOH).
Chiral HPLC: >98 % ee.
'H N1VIIZ (CDC13) S:
1.23 (t, J = 7.2Hz, 3H), 2.91 (d, J = 6.1Hz, 2H), 3.30 (bs, 2H, NH2), 3.34
(s, 311), 3.88 (t, J = 6.2Hz, 1H), 4.17 (q, J= 7.2Hz, 2H), 6.62 (d, J= 8.3Hz,
2H), 7.01 (d,
J = 8.1Hz, 2H).
IR (neat) cm"': 3372, 2985, 2932, 1739, 1627, 1519.
Mass m/z (CI): 223 (M), 234 (M+1), 192 (M - OMe).
Step (vii)
Preparation of (S) Ethy12-methoxy-3-[4-{3-(4-methanesulfonyloxy phenyl)
propylamino}phenyl] propionate
COzEt
OMe
;S,
Me O
A mixture of 4-(3-methanesulfonyloxypropyl)phenylmethanesulfonate
(5.5 g, 1.0 eq, 17.9 mmol), obtained in step (ii), (S) ethy12-methoxy-3-(4-
aminophenyl)propionate (4.0 g, 1.0 eq, 17.9 mmol), obtained in step (vi),
tetrabutylammonium bromide (2.8 g, 0.5 eq, 9.0 mmol) and anhydrous K2C03 (7.4
g,
3.0 eq, 53.7 mmol) in dry toluene (90 mL) was heated with stirring at 90 C for
7-9 h.
The reaction mixture was diluted with ethyl acetate (300 mL) and washed with
water
and brine. Organic layer was dried (Na2SO4), condensed, and the residue was
chromatographed using ethyl acetate and hexanes to obtain the title compound
as
viscous liquid (3.4 g, 44%).
[a]25D: -6.5 (c 1.0, MeOH).
'H 1V1VIR (CDC13, 200 MHz): S 1.26 (t, J= 7.0 Hz, 3H); 1.98 (quintet, J
7.2 Hz, 2H); 2.75 (t, J= 7.6 Hz, 2H); 2.93 (d, J= 5.9 Hz, 2H); 3.02-3.22 (m,
5H); 3.37
(s, 3H); 3.91 (t, J= 6.4 Hz, 1H); 4.20 (q, J= 7.0 Hz, 2H); 6.65 (d, J= 8.0 Hz,
2H); 7.08
(d, J = 8.3 Hz, 2H); 7.15-7.3 (aromatics, 4H).
IR (neat) cm1: 3405, 2934, 2934, 1739, 1617, 1522, 1367.
Mass m/z (CI): 435 [M], 436 [M + 1].
CA 02570820 2006-12-04
WO 2006/001795 PCT/US2004/018620
-16-
Step(viii)
Preparation of (S) 3-Methoxy -3-[4-{3-(4-methanesulfonyoxy phenyl)propylamino}
phenyl]propionic acid]
(S) Ethyl 2-methoxy-3-[4- {3-(4-methanesulfonyloxyphenyl)
propylamino}phenyl] propionate (3.4 g, 1.0 eq, 7.8 mmol), obtained in step
(vii) above,
was hydrolyzed by treating with LiOH.H20 (492 mg, 1.5 eq, 11.7 mmol) in MeOH-
THF-water solvent mixture at RT for 3-4h. The reaction mixture was condensed,
diluted
with water and acidified (pH at 3) with aq. HCI. Desired acid was precipitated
out from
aqueous layer, which was then filtered out. If the precipitated acid was not
pure enough
by TLC, it was chromatographed using MeOH and CHC13 as eluents to obtain the
pure
acid as white solid (2.5 g, 79%).
Mp: 90-92 C.
[a]d: -16 (c 1.0, MeOH).
'H NMR (CDC13, 200 MHz): S 1.25 (s, 1H, N-H); 1.94 (quintet, 7.2 Hz,
2H); 2.72 (t, J= 7.8 Hz, 2H); 2.82-3.02 (m, 2H); 3.02-3.18 (m, 5H); 3.38 (s,
3H); 3.97
(t, J = 4.8 Hz, 1H); 4.90 (bs, CO2H)); 6.58 (d, J = 8.1 Hz, 2H); 7.05 (d, J =
8.3 Hz, 2H);
7.15-7.24 (aromatics, 4H).
13C NMR (CDC13, 50 MHz) 8: 29.53, 32.34, 37.15 (2C), 46.48, 57.58,
82.07, 116.31, 121.90, 129.75, 130.39, 140.57, 142.48, 147.33, 175.87.
IR (neat) cm 1: 3046, 2932, 1732, 1615, 1520, 1365.
Mass m/z (Cl): 408 [M + 1], 407 [M].
See also Example 14 of WO 03/048 1 1 6, which is hereby incorporated by
reference as part of Example 1.
EXAMPLE 2
L-Arginine salt of (s)-2-methoxy-3-[4-{3-(4-Methanesulfonyloxyphenyl)
propylamino} phenyl]propionicacid in monohydrated form
(Arginine salt monohydrate)
To a mixture of (S)-2-Methoxy-3-[4-{3-(4-methanesulfonyloxyphenyl)
propylamino} phenyl] propionicacid (10 g) and DM water (30 ml), L-Arginine
(0.426 g)
were refluxed added to the reaction mixture at 25-30 C in about 5 minutes
under stiiring
and maintained the reaction mixture at 50-70 C for 4-5 hours. Isopropanol (120
mL)
was added to the reaction mixture at same temperature and heated to reflux
temperature
of the soivent, and maintained for 1-2 hours to get clear solution. Then
cooled to 25-
C in about 5-6 hours and maintained for 2-4 hours under stirring at 25-30 C.
The
CA 02570820 2006-12-04
WO 2006/001795 PCT/US2004/018620
-17-
precipitated product was filtered, dried at 60 C for 8-10 hours to afford pure
L-Arginine
salt of (S)-2-methoxy-3-[4-{3-(4-Methanesulfonyloxyphenyl)propylamino} phenyl]
propionic acid in monohydrated form, as off white crystalline solid. (l Og,
90%)
Melting point: 125-13 5 C
Purity: 98.15% by HPLC.
Mass: 408 (M+ 1).
The infrared spectrum of a KBr pellet of the product was obtained using
a FT- IR spectrometer at 4 cm"1 resolution (Figure 1). Bands were observed at
3386,
1630, 1364, 1152, 971,871.
The X-Ray Powder Diffractogram pattern of the product (Figure 2) was
recorded using the following acquisition conditions: Tube anode: Cu, Generator
tension:
50 kV, Generator current: 34 mA, Start angle: 3.0 29, End angle: 45 20, Step
size:
0.02 20, speed: 3 deg./min. Characteristic XRPD angles and relative
intensities are
recorded in Table 1.
TABLE 1
Angle Relative intensity
2-Theta
7.20( 11
8.44 43
12.68 20
14.22 9
14.80 8
15.96 32
17.02 11
17.48 49
17.60 68
18.02 11
18.84 40
19.22 9
20.00 6
21.02 100
21.18 56
21.66 38
22.02 78
22.38 23
22.98 23
23.14 17
23.80 18
24.40 28
26.36 11
26.92 14
28.68 6
29.12 17
CA 02570820 2006-12-04
WO 2006/001795 PCT/US2004/018620
-18-
Angle Relative intensity
2-Theta
29.68 7
29.96 15
33.56 9
35.74 7
36.20 7
36.56 8
Tonset of 'Arginine hydrate': The Tonset was determined by Differential
Scanning
Calorimetry. Melting endotherm was observed at 92.6 C, 132.53 C, 231.80 C,
272.22 C
EXAIVIPLE 3
L-Arginine salt of (S)-2-methoxy-3-[4-{3-(4-methanesulfonyloxyphenyl)
propylamino}phenyl}propionicacid (Arginine salt)
(S)-2-Methoxy-3-[4- {3-(4-methanesulfonyloxyphenyl)propylamino}
phenyl} propionicacid, (10 grams) and DM water (30 mL) were refluxed. L-
Arginine
(0.426 grams) was added to the reaction mixture at 25-30 C in about 5 minutes
and
maintained at the same temperature for 4-5 hours. Isopropanol (120 mL) was
added to
the reaction mixture and continued stirring further for 2 to 4 hours. The
precipitated
product was filtered, dried at 60 C for 8-10 hours and further refluxed with
toluene to
remove water azeotropically. Reaction mass cooled to 25-30 C and filtered and
washed
with toluene, dried at 60-65 C for 8-10 hours to afford the pure form of L-
Arginine salt
of (s)-2-methoxy-3-[4-{3-(4-methanesulfonyloxyphenyl) propylamino}phenyl}
propionicacid, as off white crystalline solid. (10 grams, 90%)
Melting point 190-194 C
Purity: 98.15% by HPLC.
Mass: 408 (M + 1).
The infrared spectram of a KBr pellet of the product was obtained using
a FT-IR spectrometer at 4 cm"1 resolution (Figure 4). The X-Ray Powder
Diffractogram pattern of the product (Figure 5) was recorded using the
following
acquisition conditions: Tube anode: Cu, Generator tension: 50 kV, Generator
current: 34
mA, Start angle: 3.0 20, End angle: 45 20, Step size: 0.02 20, speed: 3
deg./min.
Characteristic XRPD angles and relative intensities are recorded in Table 2.
CA 02570820 2006-12-04
WO 2006/001795 PCT/US2004/018620
-19-
TABLE 2
Angle Relative intensity
2-Theta
4.06 8
8.10 9
11.72 9
12.10 16
12.62 18
13.66 14
14.06 12
15.16 8
15.28 7
16.40 56
16.80 100
17.38 68
18.02 10
18.64 28
19.28 56
19.70 70
20.22 96
20.56 42
20.92 37
21.30 32
22.10 13
22.56 12
22.86 18
23.50 21
23.66 30
24.10 38
25.28 23
25.82 6
26.02 8
26.42 6
27.70 8-
28.34 9
29.34 6
29.88 7
30.34 9
30.74 6
30.96 10
31.32 7
31.68 6
32.96 7
33.14 10
36.16 6
Tonset of'Arginine hydrate': The Tonset was determined by Differential
Scanning
Calorimetry. Melting endotherm was observed at 192.93 C, 228.69 C, 272.22 C.
CA 02570820 2006-12-04
WO 2006/001795 PCT/US2004/018620
-20-
EXAMPLE 4
L-Arginine salt of (S)-2-methoxy-3-[4-{3-(4-methanesulfonyloxyphenyl)
propylamino}phenyl]propionic acid (conversion from Arginine salt
monohydrate to Arginine salt)
L-Arginine salt of (S)-2-Methoxy-3-[4-{3-(4-methanesulfonyloxyphenyl)
propylamino)phenyl] propionic acid in monohydrated form (Arginine monohydrate)
(10
grams) and toluene (100 mL) was refluxed for 3-4 hours to remove water
azeotropically.
Reaction mass cooled to 25-30 C and filtered and washed with toluene, dried at
60-
65 C for 8-10 hours to afford the pure form of L-Arginine salt of (S)-2-
methoxy-3-[4-
{3-(4-methanesulfonyloxyphenyl)propylamino} phenyl]propionic acid, as off
white
crystalline solid. (10 grams, 90%)
Melting point.: 190-194 C
Purity: 98.15% by HPLC.
Mass: 408 (M' + 1).
X-ray diffraction (I/Io): 16.920 (100), 20.340 (90), 20.200(83),
17.480(72)19.800(69),19.680(60),19.360(55),21.080(55),20.700(40)24.160(38).
EXAMPLE 5
L-Arginine salt of (S)-2-methoxy-3-[4-{3-(4-methanesulfonyloxyphenyl)
propylamino}phenyl] propionic acid in monohydrated form
(conversion from Arginine salt to Arginine salt monohydrate)
L-Arginine salt of (S)-2-Methoxy-3-[4-{3-(4-methanesulfonyloxyphenyl)
propylamino}phenyl] propionic acid (10 grams) and water (40 mL) were heated at
50-
60 C for 4-5 hours. Isopropanol (120 mL) was added to the reaction mixture at
same
temperature and heated to reflux temperature and maintained for 1-2 hours to
get clear
solution. Then cooled to 25-35 C in about 5-6 hours and maintained for 2-4
hours under
stirring at 25-30 C. The precipitated product was filtered, dried at 60 C for
8-10 hours
to afford pure L-Arginine salt of (S)-2-methoxy-3-[4-{3-(4-
methanesulfonyloxyphenyl)propylamino}phenyl}propionic acid in monohydrate
form,
as off white crystalline solid. (10 grams, 90%)
Melting point:125-135 C
Purity: 98.15% by HPLC.
Mass: 408 (M++ 1).
X-ray diffraction (I/Io): 22.02 (100), 21.02 (98), 8.440 and 17.600
(64),21.2 (60),17.480(46), 21.640(40).
CA 02570820 2006-12-04
WO 2006/001795 PCT/US2004/018620
-21-
EXAMPLE 6
a) Determination of hPPARa activity
Ligand binding domain of hPPARa was fused to DNA binding domain
of Yeast transcription factor Ga14 in eucaryotic expression vector. Using
superfect
(Qiagen, Germany) as transfecting reagent HEK-293 cells are transfected with
this
plasmid and a reporter plasmid harboring the luciferase gene driven by a GAL4
specific
promoter. Compound can be added at different concentrations after 42 hrs of
transfection and incubated overnight. Luciferase activity as a function of
compound
binding/activation capacity of PPARa will be measured using Packard Luclite
kit
(Packard, USA) in Top Count (Ivan Sadowski, Brendan Bell, Peter Broag and
Melvyn
Hollis. Gene. 1992. 118: 137-141; Superfect Transfection Reagent Handbook.
February
1997. Qiagen, Germany).
b) Determination of hPPARr activity
Ligand binding domain of hPPARyl is fused to DNA binding domain of
Yeast transcription factor GAL4 in eucaryotic expression vector. Using
lipofectamine
(Gibco BRL, USA) as transfecting reagent HEK-293 cells are transfected with
this
plasmid and a reporter plasmid harboring the luciferase gene driven by a GAL4
specific
promoter. Compound can be added at 1 M concentration after 48 hrs of
transfection
and incubated overnight. Luciferase activity as a function of drug
binding/activation
capacity of PPARy1 will be measured using Packard Luclite kit (Packard, USA)
in
Packard Top Count (Ivan Sadowski, Brendan Bell, Peter Broag and Melvyn Hollis.
Gene. 1992. 118: 137 -141; Guide to Eukaryotic Transfections with Cationic
Lipid
Reagents. Life Technologies, GIBCO BRL, USA).
c) Determination of HMG CoA reductase inhibition activity
Liver microsome bound reductase is prepared from 2% cholestyramine
fed rats at mid-dark cycle. Spectrophotometric assays are carried out in 100
mM
KH2PO4, 4 mM DTT, 0.2 mM NADPH, 0.3 mM HMG CoA and 125 g of liver
microsomal enzyme. Total reaction mixture volume was kept as 1 ml. Reaction
was
started by addition of HMG CoA. Reaction mixture is incubated at 37 C for 30
min and
decrease in absorbance at 340 nm was recorded. Reaction mixture without
substrate was
used as blank (Goldstein, J. L and Brown, M. S. Progress in understanding the
LDL
receptor and HMG CoA reductase, two membrane proteins that regulate the plasma
cholesterol. J. Lipid Res. 1984, 25: 1450 - 1461). The test compounds will
inhibit the
HMG CoA reductase enzyme.
CA 02570820 2006-12-04
WO 2006/001795 PCT/iTS2004/018620
-22-
Unless stated to the contrary, words and phrases such as "including,"
"containing," "comprising," "having", "for example", "i.e.", "in particular"
and the like,
means "including without limitation" and shall not be construed to limit any
general
statement that it follows to the specific or similar items or matters
immediately
following it. Except where the context indicates to the contrary, all
exemplary values
are intended to be used for purposes of illustration. Most of the foregoing
alternative
embodiments are not mutually exclusive, but may be implemented in various
combinations. As these and other variations and combinations of the features
discussed
above can be utilized without departing from the invention as defined by the
claims, the
foregoing description of the embodiments should be taken by way of
illustration rather
than by way of limitation of the invention as defmed by the appended claims.