Note: Descriptions are shown in the official language in which they were submitted.
CA 02573931 2011-09-06
WO 2006/007722 PCT/CA2005/001142
METHOD AND DEVICE TO OPTIMIZE ANALYTE AND ANTIBODY
SUBSTRATE BINDING BY LEAST ENERGY ADSORPTION
Field of the Invention
The invention relates to assay devices and methods for detecting the presence
of
antibody in a test sample and algorithmically creating internal
standard/calibration
curves, for detecting the presence of an analyte in the test sample and to
measure the
quantity of same.
Background of the Invention
Methods of analysis for immunodiagnostic assays incorporate techniques to
measure
errors resulting from faulty assay technique when assays are completed. For
example,
the measure of assay sensitivity characterizes sensitivity by classical
statistical
analysis based on repeated measurement of low concentration samples to confirm
that
the sample result is not statistically different from zero. As the standard
error
incurred is inversely proportional to the square root of the number of actual
measurements, this method does not measure the inherent assay sensitivity.
Known in the art analytical sensitivity, is the minimal detectable, or change
in
concentration, wherein the zero standard is measured several times and the
limit of
sensitivity becomes a concentration equating to 2 - 3 SD (Standard Deviation)
from
M (the MEAN). However, the precision, known in the art as the closeness of
individual measures of an analyte in multiple aliquots of a single and
homogeneous
volume of matrix, may be incorrect by an order of magnitude. The concomitant
fitting of any derived calibration curve does not create a true values dose
response
curve. This results in considerable error in the actual sensitivity of an
assay.
To measure the accuracy, known in the art as the closeness of mean test
results
obtained by the method for the true concentration of the analyte, accuracy is
used to
define how close the average measured value is to the true value. The
difference in
measurement is known as the bias or degree of accuracy. Bias may vary over the
range of the assay. It is known in the art that methods for measuring this
true value
need to be developed.
CA 02573931 2007-01-12
WO 2006/007722 PCT/CA2005/001142
The repeatability or precision of an assay, defined as closeness of individual
measures
of an analyte when the procedure is applied repeatedly to multiple aliquots of
a single
homogeneous volume of matrix or the estimated error in an analytical assay, is
known
in the art as the percentage coefficient of variation (%CV). Automated assay
analysis
machines can be affected by variations in sample concentration, temperature,
heat and
edge effects, incomplete suspension of particles and solid phase
precipitation.
Precision effects also result from fraction separation and counting errors. In
optical
systems error is due to effects of turbidity, presence of fluorophores,
deterioration of
lamps and detectors and the deterioration, over time, of reagents. These
factors
generally lead to significant decreases in signal to noise ratio. Mechanical
manipulation errors can result from poor pipetting and instrument stand-by
periods.
Therefore, the assessment for precision of any analytical method requires the
measurement of variability at known and relevant concentrations by using
defined or
standard control solutions to create baseline calibration standards. Accurate
determination of such calibrators is based on measurement of known
concentrations
in dilution series at predetermined intervals, which are then interpolated.
Commercially available, as well as in-house prepared reference solutions or
reference
standards are available, but are often calibrated with standard, external or
pooled
matrices, which may vary considerably from actual patient test samples. Part
of the
solution to overcome these errors is to plot the precision against a wide
range of
concentrations to obtain a precision profile, or quantitative calibration of
the assay.
Cross reactivity, assay specificity, bias causing interference, alterations in
antigen,
antibody, binding sites, low dose (competitive assay) and high dose (sandwich
assay)
hook effects, heterophilic antibody interference, endogenous interfering auto-
antibodies, complement, rheumatoid factor, interference in solid phase
antibody
binding, endogenous signal generating substances, enzyme inhibitors, catalysts
and
co-factors have also been shown to express confounding activity in assays,
including
cross reactivity, matrix effects and carry over of sample in automated
immunoassay
instruments and samplers.
For clinical applications, the quality control samples may not reflect actual
concentrations in the patient, may not reflect the spectrum of present
analytes and
-2-
CA 02573931 2011-09-06
WO 2006/007722 PCT/CA2005/001142
interfere with the sample matrix to no longer reflect the content of the
patient
samples. The quality control samples may measure performance at discrepant
intervals of concentration which may not reflect clinical decision points.
The applicants have developed microarray assays that provide rapid detection
of the
presence of analytes in a sample. These are described in US Patent Application
No.
10/856,785 filed on May 28, 2004 entitled "Method and Device for Rapid
Detection
and Quantitation of Macro and Micro Matrices"
The assays permit rapid quantitative and qualitative measurements of
analyte concentration in a sample. The analyte is labeled with a first
antibody that is
conjugated with a detectable marker. A typical assay device defines a chamber
between the loading portion and the reading portion such that a liquid portion
of the
sample moves from the loading portion to the reading portion by capillary
action.
Site-specific immobilized arrays of test dots are printed on the reading
portion. The
test spots include a second antibody that is bound to the surface of the assay
device
and that is adapted to bind and label analyte. The assay device also has site-
specific
immobilized arrays of calibration dots containing predetermined amounts of
bound
analyte for reaction with excess amounts of the first antibody labeled with
the
detectable marker. Once the conjugated analyte is bound to the test dots, the
measure
of the analyte in the test dots can be determined by comparison of test dot
label
intensity with the label intensity of the calibration dots and reading
concentration of
analyte from appropriate calibration curves.
Immuno-assays in general, depend implicitly on the direct detection and
measurement
of the signal generated by the number of antigen to antibody adsorption sites.
Non-
competitive assays identify these adsorption sites by using a secondary
labeled
antibody, whereas competitive assays measure unoccupied adsorption sites. As
immuno-assays are a function of antibody concentration, volume and affinity
constants, only if these values are held constant will it be possible to
obtain
comparatively accurate measurements. The actual quantity of analyte in the
sample
still needs to be measured. The analyte concentration is measured by
comparison to
the pre-calibrated concentration site-specific immobilized arrays of internal
reference
standards.
-3-
CA 02573931 2007-01-12
WO 2006/007722 PCT/CA2005/001142
If pre-calibrated, external reference standard concentrations are used to
create external
calibration curves, they provide a consistent source of error in the
conversion of
interpolated detection signal into analyte concentration assumed to be present
in the
test sample. Further error in antibody to analyte measurement may be induced
by the
use of a solid support or substrate on which either antigen or antibody is
adsorbed.
Although enhancing binding charge on the substrate further compounds external
reference errors, ambient analyte assays are equally distorted.
Although adsorption to solid support surfaces increases by addition of
hydrophobic
binding forces, enhancing surface modification can cause changes in protein
analyte
structure, as well as antibody structure, to significantly alter complex
formation.
Structural change leading to alteration in complex formation has a direct
effect on the
quanta of signal e.g. photon counting errors, measured by a detector to
determine a
label to concentration ratio.
There is therefore a need for a method when testing fluids for analyte,
produced under
certain conditions, to optimize the diagnostic value of site specific
immobilized
arrays. There is a need for such a device having secure, repeatable,
quantifiable,
reproducible, modulated and reliable attachment of site-specific immobilized
arrays to
a solid support without aberration to improve the sensitivity of such immuno-
assays to
have a dynamic detection range of femtomol to nanomol per ml analyte
concentrations. Modulated substrate surface modification enhances reliable
dynamic
internal calibration as requested by regulatory agencies, such as the FDA
(U.S. Food
and Drug Administration).
Summary of the Invention
The invention relates to a method for test site-specific immobilized arrays,
having a
captive antibody for binding to an analyte and calibration site-specific
immobilized
arrays, containing known concentrations of analyte to be printed onto a common
surface of an assay device. The assay device is preferably surface modified to
enable
modulated and site specific quantifiable attachment of requisite molecular
species and
their aggregates to a surface. A proportion of hydrophilic, hydrophobic and
covalent
linking sites to binding sites on the surface of the assay substrate in a
device are
modulated in order to optimize the sensitivity of the assay on an assay
specific basis.
-4-
CA 02573931 2007-01-12
WO 2006/007722 PCT/CA2005/001142
In the case of multiplexing the modulation of the surface energy is done on an
assay
pair i.e. antigen/antibody basis.
The immuno-diagnostic device has a solid support, the surface of which has
been
modified to allow modulation and therefore optimization of countervailing
hydrophilic and hydrophobic forces. This modification permits the optimal
adsorption
of analyte protein and antibody. Printing of site-specific immobilized arrays
with
diagnostic dots from 5 micrometers to 500 micrometers in diameter, with a
preferred
range of 50 micrometers to 125 micrometers in diameter, is also a function of
constant
relative humidity. Surprisingly, internal calibration of a test by printing
analyte of
known concentrations in site-specific immobilized arrays allows determination
of the
proportional measurement by use of surplus antibody-detectable marker
complexes.
The method pertains to this combination of parameters to establish the optimal
configuration for obtaining more accurate diagnostic test results.
According to one aspect of the present invention there is provided a method of
making an assay device for conducting an assay to detect a concentration of an
analyte in a sample fluid, said assay device having a surface, the surface
having a site
specific immobilized calibration dot including a pre-determined quantity of
the
analyte printed thereon and a test dot including a capture antibody for
binding said
analyte, said method including the following steps:
= modifying said surface to provide hydrophobic binding sites, hydrophilic
linking sites and covalent linking sites;
= printing said test spots and said calibration dots on said surface;
= applying the sample test fluid to the assay device;
= testing a sensitivity of the assay; and
= modulating the ratio of said hydrophobic binding sites to said hydrophilic
linking to said covalent sites in order to optimize the sensitivity of the
assay.
-5-
CA 02573931 2007-01-12
WO 2006/007722 PCT/CA2005/001142
Brief Description of the Drawings
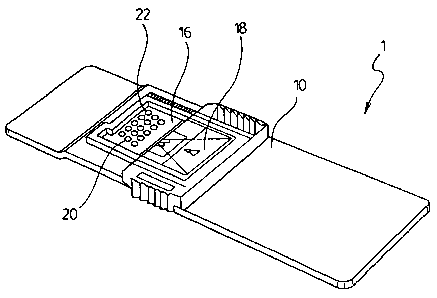
Figure 1 is a perspective view of an assay device of the present invention for
carrying
out fixed array tests;
Figure 2 is a photograph of a top surface of an assay device of the present
invention
showing a spot morphology of ten different print buffers on epoxy substrate;
Figure 3 is a photograph of a top surface of an assay device of the present
invention
showing a mean fluorescence intensity of spotted BSA.RB conjugate dilutions.
Figure 4A is a plot of a mean fluorescence intensity of spotted BSA.RB
conjugate
dilutions with Erie Superchip Epoxy;
Figure 4B is a plot of a mean fluorescence intensity of spotted BSA.RB
conjugate
dilutions with Erie Superchip Epoxy with Epoxy Batch 40309;
Figure 4C is a plot of a mean fluorescence intensity of spotted BSA.RB
conjugate
dilutions with Erie Superchip Epoxy with Epoxy Batch 40406;
Figure 4D is a plot of a mean fluorescence intensity of spotted BSA.RB
conjugate
dilutions with Erie Superchip Epoxy with Epoxy Batch 40309;
Figure 5 is a plot of the ratios of mean fluorescence intensities of spotted
BSA.RB
conjugate dilutions post washing (W) over the mean fluorescence intensities
intervening blanks post washing (WC) for ERIE and three batches of Epoxysilane
of
the present invention;
Figure 6 is a photograph of a top surface of an alternate embodiment of the
assay
device of the present invention;
Figure 7 is a graph depicting dose/response curves to realize an internal
dynamic
calibration curve;
Figure 8 is a graph showing a correlation of charge density on binding to
modified
solid support surface;
-6-
CA 02573931 2007-01-12
WO 2006/007722 PCT/CA2005/001142
Figure 9 is a photograph of an assay device showing adequate surface
modification of
a support surface of the device;
Figure 10 is a photograph showing a site specific immobilization of array dots
comparing clumped, variegated, annular and evenly spaced distribution of
analyte
within a dot;
Figure 11 is a photograph showing mouse embryo site specific immobilized
hybridized oligonucleotides;
Figure 12 is a photograph showing mouse spleen site specific immobilized
hybridized
oligonucleotides; and
Figure 13 is a photograph showing a patient. IgG and IgM serum responses to
ten
pathogen microarray plus IgG and IgM calibration standards.
Detailed Description of the Invention
The method of the present invention is preferably carried out in association
with an
assay device having site-specific immobilized arrays of calibration dots and
site-
specific immobilized arrays of test dots printed thereon. A preferred assay
device is
shown in Figure 1. The assay device has a substantially planar surface 16. The
substantially planar surface 16 has printed thereon at least one and
preferably at least
three of test capture 20 and calibration site-specific immobilized arrays 22
printed in
the reading area. More preferably a multiplex of site-specific immobilized
arrays of
test spots for detecting the presence of analytes are printed on the reading
area 16. The
site-specific immobilized array test spots 20 preferably include bound
antibodies that
specifically bind to a protein analyte. The bound antibodies are preferably
spaced
apart as to make each bound antibody available for binding to the test antigen
free of
stearic hindrance from adjacent antigen complexes. Preferably, a non-reactive
protein
separates the bound antibodies in each of the site-specific immobilized array
test dots.
The reading area 16 has site-specific immobilized arrays of calibration dots
22 printed
thereon. The calibration dots contain a pre-determined quantity of said
analyte for
reacting with unreacted reagent from the vessel that is bound to a detectable
marker.
The site-specific immobilized arrays of calibration spots allow the intensity
of the
-7-
CA 02573931 2007-01-12
WO 2006/007722 PCT/CA2005/001142
label to be correlated to the amount of the antigen present. The intensity of
label in the
site-specific immobilized arrays of test spots is then used to derive the
quantity of
antigen present in a tested sample volume.
The assay immobilization substrate may be selected from materials which permit
surface modification, including silicon, glass and polymeric materials, but in
a
preferred embodiment is made of plastic support substrate such as polystyrenes
and
polypropylenes. The polymer surface can be readily modified to adsorb antigen
and
antibody. Protein adsorption requires hydrophobic binding sites for optimal
attachment to the surface of the assay device, whereas antibody binds well to
hydrophilic charge linkages, including covalent bonds. These site-specific
immobilized arrays of test spots are proportionally adsorbed to the reading
area
surface as a function of surface modification resulting from increase of unit
area
effective charge density. Increase or changes in the balance of forces
associated with
Van der Waal's interactions, hydrophilic and hydrophobic forces and modulation
in
covalent bond density allows selective enhancement of hydrophilic to
hydrophobic
properties of the reading area surface and site-specific immobilization of
test spot
arrays.
The enhanced spread of spot fluid volume, resulting as a modification of
contact
angle, also is a function of relative humidity and temperature. As the
relative humidity
increases, drying time of the spots increases which allows larger spot
formation on the
surface for a similar dispensed volume of test fluid. The net effect results
in much
thinner spots, thereby approaching the ideal single molecular layer adsorbate
which
forms a more sensitive molecular layer for optimal analyte capture by the
capture
antibody. Surprisingly, the even dispersion of analyte throughout the site
specific
immobilized test spot arrays optimizes test dot morphology in three
dimensions.
The analyte to be captured and measured is identified by a label conjugated to
an
analyte specific marker antibody. Fluorescent dyes are known in the art as
detectable
markers for providing accurate labeling. Surprisingly, the analyte-antibody-
dye
complex provides a measure of analyte concentration when equated to a baseline
derived from the site-specific immobilized arrays used as standard internal
analyte
-8-
CA 02573931 2007-01-12
WO 2006/007722 PCT/CA2005/001142
calibrators. In contrast, the use of external calibrators is a major source of
accepted
error commonly found in immuno-assays.
The excess dye conjugated anti-analyte antibody, which is not bound to the
respective
analyte, is normally considered to be redundant and is washed away as is known
in
the art. Surprisingly, this method of the present invention uses the anti-
analyte
antibody, to create multiple internal, in device calibration lines directly
from the site-
specific immobilized calibration arrays.
Along with the known capture antibody site-specific immobilized array test
dots to
capture the analyte, a series of site-specific immobilized array calibration
dots is
to printed directly adjacent to the site-specific immobilized arrays of test
dots, as shown
in Figure 1. This second set of site-specific immobilized calibration dots 22
consists
of decreasing per dot concentration of known concentration of analyte. Thus
the
assay, when carried out on the device, has now been reduced to a single step
assay,
with no mandatory need for intermediary washing steps to remove excess anti-
analyte
antibody, as the excess of conjugated label antibody background concentration
is
effectively reduced by binding to the predominant site-specific immobilized
array
spots containing known concentrations of calibration antigen.
Surprisingly, the process of the present invention provides epoxysilane
substrates for
immobilized arrays with modulated, covalent, high capacity binding of amino-
terminal oligonucleotide libraries, cDNA libraries, proteins, peptides,
glycoproteins,
lipopolysaccharides, as well as small molecules such as Biotinyl-3, 6-
dioxaoctanediamine (Pierce, EZ-Link Biotin-PEO-Amine). No organic solvents are
used in the process. A fume hood is not required, neither are special disposal
precautions. Optimal results are obtained when using printing buffers 3X to 6X
SSC
pH 7, 0.15M Sodium Phosphate Buffer 8.5, 0.1M Sodium Phosphate Buffer pH 7.2,
and 0.1 M Sodium Bicarbonate pH 8.6. Spot size can be adjusted by adding a
small
amount of detergent (0.01% to 0.05% SDS).
When the test fluid containing the analyte-antibody-dye complex as well as
surplus
anti-analyte-antibody-dye complex, is flowed over the two types of site-
specific
immobilized arrays of test spots printed in the assay device, the surplus
complex binds
with the known analyte spots to create the reference concentrations for
calibration
-9-
CA 02573931 2007-01-12
WO 2006/007722 PCT/CA2005/001142
when the dye concentration is read in a reader. The analyte-antibody-dye
complex
binds with the capture antibody site-specific immobilized arrays of test spots
and
provides a reading for the concentration of analyte in the test solution. This
method of
the present invention for Internal Dynamic Calibration (IDCTM), integrates a
comparative internal calibration standard curve with the comparative test
curve as
shown in Figure 8. The actual concentration of test fluid analyte is
accurately
determined because the test specific internal calibration standard is
simultaneously
provided.
The method when used in conjunction with the device provides an empirical
determination of the correct enhancement of charges induced on the assay
device for
sufficiently binding assay components without causing loss of detection signal
as a
result of high density binding events, i.e. number of bonds per unit surface
area of the
assay device.
In order to attain maximum sensitivity in the assay device of the present
invention, the
assay device has known concentrations of antigen in the calibration dots. The
increasing antigen concentrations are read during the assay as a result of
binding with
surplus antigen-antibody plus fluorescent label calibration complex. The
antigen,
whether a requisite protein and or nucleic acid or derivative, needs to be
firmly
attached to the immobilizing substrate surface of the assay device to prevent
becoming soluble and being washed away during the course of the assay process.
Attachment of analyte site-specific immobilized arrays of test spots is
optimized with
a balance of hydrophobic, hydrophilic and covalent linkage, modulated by
analyte
suspension buffer containing molecular spacer for even dispersion of analyte
throughout the printed test spot. Even dispersion ensures even illumination
level per
test spot and minimizes stearic hindrance.
Surprisingly, the degree of surface modification maximal for protein
adsorption, may
act in reverse for antibody adsorption as shown in Figure 9. Optimal density
of
surface charges can attach antibody so firmly to the charge-modified surface
to such
an extent that although antibody is present, no antigen binding events are
found. As a
direct consequence, no free antigen-label complex can be captured.
-10-
CA 02573931 2007-01-12
WO 2006/007722 PCT/CA2005/001142
Direct binding of antibodies to a bare surface which has not been substrate
modified
according to the present invention, may result in conformational antibody
changes
that reduce their affinity for the analyte. Analytes also passively adsorb
onto
energized surfaces. The proportion of analyte bound can range from 5% to 95%,
so
careful optimization of a surface modification and coating process is
important.
Binding of protein to plastic for example, occurs because water molecules have
a
much stronger affinity for each other than for hydrophobic regions. The
exclusion of
hydrophobic sites from the solution causes parts of proteins to adsorb to
substrates.
The solid support surface of the assay device is modified to have a balance of
,
hydrophilic enhancement for antibody binding and test fluid flow
characteristics
versus maintenance of hydrophobic and covalent binding for protein adsorption.
Printing, on the modified surface, of the calibration and test dot analytes
suspended in
modulation buffer containing molecular spacer, is carried out under constant
humidity
control to ensure that the spots tend to minimal thickness leading to the
formation of
molecular layer thickness site-specific immobilized arrays of test spots. Key
attributes
of the modular surface resulting from the present invention, includes
maintenance of
assay precision, sensitivity and repeatability.
As shown in Figure 6, an internal calibration method of the assay consists of
multiple
repeats to support high confidence limits for accurate diagnosis. An effective
balance
of the applied parameters for attachment of the dots to the modified surface
is
confirmed by performing immuno-diagnosis of test sample fluid. When optimal
conditions for a specific assay have been confirmed, mass production commences
with quality control tests incorporated at required intervals.
In order to produce consistent, modulating epoxysilane coatings for substrate
surfaces,
the substrate is cleaned in detergent solution, washed for several hours, then
transferred into bleach. The epoxysilane coating is applied as fresh 2-
Morpholinoethanesulfonic acid, monohydrate (MES) solution with added 3-
Glycidyloxypropyltrimethoxysilane. Excess water is removed from the
substrates, and
the substrates transferred into Epoxysilane/MES solution. The substrates are
washed
in preferably 3 but alternatively 1 or 2 changes of DI water and dried.
Immediately
after epoxysilane coating the substrate surface is hydrophilic. Baking
dehydrates the
- 11 -
CA 02573931 2007-01-12
WO 2006/007722 PCT/CA2005/001142
surface without degrading the reactive epoxy groups on the surface yet makes
the
surface less hydrophilic. The substrates are best stored at room temperature.
This covalent epoxysilane process of the present invention has proven to be
the
optimal method to produce surface energy modulation as the covalent reaction
thermodynamically favours energy minimization and the epoxysilne surface can
be
modulated using baking time and temperature, to have controlled surface
energies
(contact angles) from very hydrophilic to moderately hydrophobic.
If the surface tension of the coating is greater than the surface energy of
the substrate,
the coating will not spread out and form a film. As the surface energy of the
substrate
is increased, the coating will spread out and form a film but, when dry, will
have poor
adhesion. Further increases in the surface energy of the substrate will result
in easier
wet-film formation and better dry-film adhesion.
Examples
Example 1. Effect of printing buffer on spot morphology and binding capacity.
With respect to the Epoxy surface, figure 3, 0.150 M Phosphate buffer again
produced
the best results. 10% Glycerol gave the lowest signals. For this surface also
50%
DMSO showed false signals in the blank spots. Overall signal intensities and
binding
capacities are quite evidently less than those for the corresponding Hydrogel-
Epoxy
surface at all BPA concentrations, and in all buffers.
Figure 2 shows a dot morphology of 10 different print buffers on epoxy
substrate.
Eight dilutions (5x) of BPA were printed in triplicate from the maximum of
1000
gg/ml at left to 0.0 at right. Substrate was blocked with blocking buffer for
2 hours.
Bound BPA was reported by treatment with 4 g/ml Streptavidin-Cy3 in the same
blocking buffer for 1 hour. Phosphate pH 8.5 10% DMSO/Water Blank 50%
DMSO/Water 10% Glycerol/Water Blank MES pH 4.7 Borate pH 9.3 Carbonate pH
9.6 6 x SSC pH 7.2 3 x SSC pH 7.2 Water
-12-
CA 02573931 2007-01-12
WO 2006/007722 PCT/CA2005/001142
Example 2. Printing Optimization and Quality Control for Covalent Binding
Microarray Substrates
A number of methods are generally accepted for quality control of pin-printed
microarrays. In a printed library of oligo or cDNA arrays, spot morphology and
missing spots can be visualized with DNA intercalating dyes such as SYBR 555
(Invitrogen/Molecular Probes P32930), and by hybridisation with AlexaFluor dye-
labeled random 9-mers (Invitrogen/Molecular probes P32934, and P32937). Trace
DNA with a fluorescent label that is not excited by either of the standard
green or red
laser wavelengths may be added to every oligo or cDNA of the library. This is
ideal
and provides quantitation of the DNA in each spot concurrently with the
hybridisation
signals, read in a confocal microscope with a third laser and interference
filter for the
labeled tracer.
Prior to taking the effort and expense of printing a library on batches of
substrates it is
preferred to check the batches themselves for binding capacity, spot
morphology, and
batch-to-batch uniformity.
Whether printing solutions of oligonucleotides, cDNA, proteins or small
molecules on
microarray substrates there is an optimal concentration for the solution. Each
surface
has a maximum binding capacity. Printing in excess of this wastes precious
material,
and may cause higher background levels plus increased carryover or cross-
contamination of the library. Printing less than the optimum amount produces
lower
signals and potentially lower signal to noise ratios.
BSA.Rhodamine B conjugate (BSA.RB). Rhodamine B Isothiocyanate (RBITC,
Sigma-Aldrich #283984) is a fluorophor that absorbs the green laser wavelength
used
for Cy3, and emits over a broader spectrum in both Cy3 and Cy5 channels of
standard
scanning confocal microscopes. Six mg of RBITC was dissolved in 600 microL
Ethyl
Lactate. Three 1 ml aliquots of Bovine Serum Albumin (BSA, Sigma-Aldrich
#A7517), 20 mg/ml, were prepared in Carbonate/Bicarbonate buffer pH 9.6. Each
aliquot was mixed with 100, 200 or 300 microL of the RBITC solution and
allowed to
react overnight. The reaction was quenched by adding 10 mg of Lysine to each
of the
aliquots and letting it react for 2 hours. A minicolumn was packed with
Sephadex
G50 and washed with 3X SSC. Each aliquot was gel filtered on the G50 column,
and
- 13 -
CA 02573931 2007-01-12
WO 2006/007722 PCT/CA2005/001142
eluted in 3X SSC, the intended microarray printing buffer. The first colored
peak (the
BSA.RB conjugate) was collected and the later peaks discarded. The conjugation
ratios resulting from the increasing ratios of RBITC were estimated from the
comparative absorbance spectra of pure BSA, pure RBITC and the conjugates to
be
approximately 2:1, 5.5:1 and 12.8:1 dye molecules per BSA molecule.
The 5.5:1 conjugate was selected for printing on the following basis. BSA has
about
the same molecular weight as a 200-mer strand of cDNA to occupy about the same
surface area as a 70-mer oligo probe hybridized to a 200-mer target. In
addition, the
recommended nucleotide/dye ratio for hybridizing labeled transcripts is about
40:1, or
5 fluorophors per 200-mer. Thus, the BSA.RB conjugate molecule simulates the
binding surface area per molecule of a probe oligo and the fluorescence
labeling of
the target expected in a microarray. The 70-mer oligos of the Qiagen/Operon
libraries
are amino-terminated for covalent reaction with active surfaces such as Epoxy,
Aldehyde and NHS ester. The superficial lysine residue amino groups of BSA
that
have not reacted with RBITC are available to react with the surface via the
same
chemistry.
The recommended printing concentration for the Qiagen/Operon 70-mer oligo
libraries is 15 microMolar (15 pM/microL). A 2X serial dilution of the BSA.RB
conjugate in 3X SSC was prepared in a 384 well plate such that blanks (just
the 3X
SSC buffer alone) alternated between conjugate dilutions. This was designed to
test
the washing of the array printer pins and determine the severity of well-to-
well
carryover or cross-contamination. The starting concentration was therefore
adjusted to
be slightly higher than recommended, 18 pM/microL.
Epoxysilane covalent coated slides of the present invention and samples of
Erie
SuperChip Epoxy provided by the manufacturer were printed from the 384 well
serial
dilution plate with Point Technologies tungsten split pins by a Perkin Elmer
SpotArray. The wash solution was 10% Ethanol in RO water, the SpotArray being
set
up for 3 cycles of 2 seconds of jet wash followed by 2 seconds of vacuum
drying
between wells. Dots were spaced 340 microns apart 12 spots per row, 6
replicates of
each BSA.RB dilution followed by 6 replicates of the corresponding blank on
the
same line, as shown in Figure 3.
-14-
CA 02573931 2007-01-12
WO 2006/007722 PCT/CA2005/001142
Figure 3 shows commercial epoxysilane covalent binding microarray substrate
(left)
and three in-house made batches printed with serially diluted BSA.Rhodamine B
conjugate imaged before (top) and after (bottom) washing for 24 hours with I %
ethanolamine +I% BSA pH 8.5 blocking buffer. The top and bottom images of each
set were scanned at the same laser and gain settings but are not the same for
all sets.
The BSA.RB dilution arrays were scanned on a Perkin Elmer ScanArray Express
confocal laser microscope. Typically settings were laser 60%, photomultiplier
55%
but were not exactly the same for all slides. These pre-wash of files were
saved for
subsequent analysis. The slides were washed for 24 hours in blocking buffer
(1%
ethanolamine + I% BSA adjusted to pH 8.5 with HC1), then scanned again at the
same settings to obtain the post wash image files. Before and after washing
images
were analyzed with ScanAlyze (EisenLabs, Stanford). Grids and circles were
adjusted
optimally for each array such that the circles exactly enclosed the
fluorescent spots.
Analysis of Protein Retention and Carryover:
ver:
Depending on the batch, spots ranged from 160 microns to 250 microns in
diameter as
seen in Figure 3. Washing of the pins was not complete as one can clearly see
six
fainter spots to the right of the six spots of each dilution of BSA.RB
conjugate. This
can cause cross-contamination of the libraries, limiting the number of
printings that
can be performed before the library must be discarded. It underlines the
importance of
making aliquots of the libraries to extend their useful life and reduce costs.
Figures 4 A,B,C and D show the plotted fluorescence intensities of the dots
versus the
printed (pre-wash) concentration of the BSA.RB conjugate (upper purple curve
in
each chart) and their corresponding neighboring blanks (lower purple curve in
each
chart). The figures also show the post wash conjugate retention curve (upper
dark)
and blank (lower faint). At the concentration recommended by Qiagen/Operon for
the
printing of their 70-mer libraries (15 pM/ L) the carryover is quite high.
Analysis
shows that printing at this concentration yields lower retained protein than
printing at
about 5 pM/ L (1/3 the concentration). Thus library material is wasted.
Further, the
carryover/cross-contamination is higher at the higher concentrations as more
protein
or oligonucleotide is carried from well to well on the pins even after
thorough
washing.
-15-
CA 02573931 2007-01-12
WO 2006/007722 PCT/CA2005/001142
Substrate % Retention %contamination
100*(P/W) I00*(WC/W)
etween 2.5 - 10 pM/ ^ between 2.5 - 10 M/
ESCE 47-58 .7-5.8
40309 33 - 37 6.1-7.4
40406 16-47 .5-7.7
10721 35-48 .5-2.7
Table 2: Summary of numerical analysis from the charts in figure 2, from which
the
optimal printing concentration range can be seen to be between 2.5 and 10 pM/
L.
Over this range maximum retained protein (signal) is maintained while
carryover is
reduced.
Figure 5 shows plots of the ratios of mean fluorescence intensities of spotted
BSA.RB conjugate dilutions post washing (W) over the mean fluorescence
intensities
intervening blanks post washing (WC) for ERIE and 3 batches of Epoxysilane of
the
present invention, as analyzed by ScanAlyze. Averages are of 6 replicates
before and
after washing as shown the images of figure 1. (Key: P - printed BSA.RB
dilution, PC
- printed blank carryover, W- BSA.RB dilution post wash, WC - blank carryover
post
wash.)
Direct printing of fluorophor-labeled proteins or 70-mer oligos is a rapid
method of
checking the binding capacity or percent retention of least energy adsorption
substrate
binding for pin-printed microarrays. Different batches of slides may be
compared for
binding and spot morphology before being accepted for printing of precious
libraries.
In addition it is a valuable method of assessing both the quality of pin
washing, and
setting the optimal printing concentration for libraries. Determining optimal
printing
concentration or maximum signal for minimum concentration and carryover
conserve
libraries and extends their life by minimizing cross-contamination. Based on
these
studies our 600 pM Operon/Qiagen human 70-mer library was split into 4
aliquots of
150 pM each. Three were preserved for printing the whole 22K (version 2.1.2)
library, and one was used for picking focused subsets of 200 to 800 oligos for
-16-
CA 02573931 2007-01-12
WO 2006/007722 PCT/CA2005/001142
protease, breast cancer-related, ring finger protein, clock (circadian rhythm-
related),
and HOX genes. These subsets were further split into 75 pM duplicates.
Three batches of epoxysilane of the present invention and a commercial
Epoxysilane
slide (Erie SuperChip Epoxy) are presented as examples of this method. Spot
morphologies can be significantly out of tolerance (e.g. batch 40406) even
though
binding capacity may be acceptable.
Maximum signal and protein retention falls in the printing concentration range
2.5 to
pM/microL for all slides in this experiment. This approximate optimal range
has
also been found in other experiment with oligonucleotides from 9-mers to
70mers
10 (data not shown). Printing at higher concentrations in fact reduces signals
and
retention presumably by stearic hindrance. Epoxy substrates of the present
invention
provided the optimum signal to background ratio, an up to 2 times improvement
over
the commercial substrate in the optimal printing range, and this is evident in
the
images of figure 3.
Example 3. Multiple site specific immobilized calibration and test arrays.
Multiple arrays of spots have been adsorbed onto the modified solid support
surface
substrate.
As shown in Figure 6, an embodiment of the assay device shows ten printed
columns
of calibration dots 22 of the same analyte concentration. The final two
columns of test
dots 20, on the right, are capture antibody dots with adsorbed test samples of
concentration to be determined. Each row represents a single assay with
internal
dynamic calibration (IDB). The test row is repeated ten times. The entire test
matrix is
10 dots x 10 dots for IDB and 2 x 10 dots for test samples. The measured
fluorescence
intensity determines the relative concentrations of analyte.
Example 4. Dynamic Internal Calibration
As shown in Figure 7, curve A represents the typical variations in dose
response when
test samples are measured using an internal, generated concentration curve.
Curve B
is the derived external calibration line, erroneously interpolated as
determined by a
measured low dose external calibration reading and a high dose external
calibration
-17-
CA 02573931 2007-01-12
WO 2006/007722 PCT/CA2005/001142
reference reading. The respective values in between the low and high points
are then
integrated by drawing a line i.e. curve B. The Internal Dynamic Calibration
produces
a line plot based on several plots resulting in a curve that represents the
actual
calibrated response of the test sample as in Figure 8.
Example 5. Device tested to compare the effect of high density surface charge
with
optimal surface modification with a balanced charge.
To compare dose response of optimized device solid support (upper "Optimal"
curve)
against highly charged solid support modified surface (lower "Charged" curve).
The
dynamic spot CK-MB analyte protein concentration ranged from 26 picogram/ml to
26,200 picogram/ml as shown in Figure 8. The highly charged surface area
effectively prevented analyte binding at concentrations less than 2620
picogram/ml.
Example 6: Method Applied to Production Test Device
Figure 9 illustrates a typical device surface, modified to show comparative
print array
matrices and to confirm adequate surface modification of solid support for
both
analyte and antibody adsorption. The method compares relative dot sizes with
increment in dispensed dot volume.
Example 7. Humidity Dot Size
Surface modified solid supports were tested for hydrophilic flow effects at 24
C at
18% humidity and 47% humidity. Dots were measured for comparative increase in
diameter. Surface area increased by 51 % at higher humidity for the same
volume of
spot fluid, approximating a 50% reduction in spot thickness.
Example 8. Site specific immobilization of array dots containing evenly spaced
distribution of analyte within a dot.
Figure 10 character A, shows the effect of using bicarbonate buffer at pH 9.0
to form
a central aggregate of concentrated analyte which causes an emission peak and
erroneous data results. Character B shows the effect of additional guanidine
hydrochloride which causes dispersed analyte aggregates. Character C shows the
use
of borate and guanidine hydrochloride at pH 5Ø The formation of an annulus
of
-18-
CA 02573931 2007-01-12
WO 2006/007722 PCT/CA2005/001142
analyte at the perimeter of the spot, leaving the centre a minimal
concentration is
shown. Surprisingly, the molecular spacer component of the present invention,
Figure
at character D forms the best spot morphology and signal. The analyte is
evenly
dispersed throughout the spot using borate (50 mM H3BO3, pH 5.0), but adding
5 Magnesium Chloride (50 mM MgCl2) to actively space the analyte throughout
the
spot.
The modulated epoxysilane substrate of the present invention, when used in
conjunction with analyte and substrate incompatible buffers, immobilizes array
spots
containing defective, definitive patterns of analyte and analyte aggregates as
shown in
10 Figure 10. Molecular aggregation and dispersion of analyte in array spots
is critical
when obtaining comparative measurement of analyte concentration. The CV% (per-
cent co-efficient of variance) acceptable in clinical diagnoses is only
obtained when
the array spots contain an even analyte dispersion, as shown in Figure 10 at
character
D. The best dot morphology and signal on nearly every surface was most often
obtained with 50 mM H3BO3, pH 5.0, in most cases with 50 mM MgC12 added.
Example 9. Immobilization of nucleic acid on the epoxysilane substrate of the
present
invention.
The arrays are a focused set of 72 oligonucleotides from the Operon mouse
genome
library, site specific localized on the epoxysilane substrate of the present
invention.
The substrate was blocked in 2mg/m12-aminoethanol + 1% BSA for 24 hours, then
washed with de-ionized water and spun dry.
RNA extracted from whole mouse embryo at 9 days, as shown in Figure 11, and
from
adult mouse spleen tissue, as shown in Figure 12,,using Promega's SV total RNA
extraction system, was quality checked using an Agilent Bioanalyzer. The RNA
was
amplified by NuGen's Ovation method to yield about a 5000 times amplified
aminoallyl-c-DNA. This was conjugated with Cy5-NHS ester to yield the
fluorophor-
labeled c-DNA. C-DNA yield was quantitated and the label incorporation ratio
confirmed using a Nanodrop 1000 fiber-optic spectrophotometer. The Cy5-c-DNA
(0.5 micrograms total c-DNA in 80 microliters) was hybridized on the site
specific
immobilized arrays 20 hours in 5 X SSC + 0.1% SDS. The arrays were washed
successively in 3 X SSC twice, 1 X SSC twice and 0.1 X SSC once, then spun
dry.
-19-
CA 02573931 2007-01-12
WO 2006/007722 PCT/CA2005/001142
They site specific immobilized spot arrays were read using a PE ScanArray
confocal
scanning microscope at relatively low settings of laser <65% and gain <60%.
Obtaining good results at these low settings indicates the c-DNA to be well
labeled
and that the epoxysilane substrate of the present invention retains optimal
probe
oligonucleotides for the target to hybridize.
Example 10. The Method of the present invention applied to an APT Device of
the
present invention; Microwell devices to optimize analyte and antibody
measurement
by least energy adsorption.
A proteomic microarray suitable for serodiagnosis of antipathogen antibody
titer
(APT Device) was prepared as follows. Serial dilutions of human IgG and IgM
were
printed as calibration standards, and bacterial and viral antigens were
printed in
triplicate as shown in the table below.
IgM 900 ug/ml H.pylori T. gondii IgG 900 ug/ml
IgM 300 ug/ml IgG 300 ug/ml
IgM 100 ug/ml Herpes 1 Rubella IgG 100 ug/ml
IgM 33.3 ug/ml IgG 33.3 ug/ml
IgM 11.1 ug/ml Herpes 2 Rubeola IgG 11.1 ug/ml
IgM 3.7 ug/ml IgG 3.7 ug/ml
IgM 1.2 ug/ml CMV C. trachomatis IgG 1.2 ug/ml
IgM 0.4 ug/ml IgG 0.4 ug/ml
IgM 0.14 ug/ml EBV C. jejuni IgG 0.14 ug/ml
IgM 0 ug/ml IgG 0 ug/ml
Table 10: Printed calibration standards and bacterial and viral antigens
The surface was blocked with 1% BSA in Tris-Buffered saline pH 8 (blocking
buffer)
for 18 hours. Serum samples from patients known to have been infected with
Helicobacter pylori (H. pylori), and tested for their antibody titer by
reference
methods, were diluted 1 in 10 by volume in blocking buffer. These diluted
samples
were then incubated, 200 uL of each sample per well, on the microarrays for 15
minutes. The samples were decanted and the wells washed by two cycles of
filling the
wells with Phosphate-buffered saline pH 7.4, shaking for 20 seconds, and
decanting.
A third wash of 20 seconds was done with blocking buffer. The wells were then
filled
with 200 uL of a combination of goat-anti-humanIgG.Cy3 conjugate 3 ug/ml and
goat-anti-human IgM.Dy647 conjugate in blocking buffer and incubated for 15
-20-
CA 02573931 2011-09-06
WO 2006/007722 PCT/CA20051001142
minutes. The conjugates were decanted and the wells washed by two cycles of
filling
the wells with Phosphate-buffered saline pH 7.4, shaking for 20 seconds, and
decanting. The plastic sides of the wells were removed with a wedge leaving
the flat
slide glass bottom, which was washed for 20 seconds in deionized water and
dried by
'spinning it in the centrifuge at 500 RPM for 1 minute.
A Perkin Elmer ScanArray laser scanning confocal microscope was used to image
the
arrays at the following settings: Cy3 (green) channel (IgG) laser 70%
photomultiplier
70%, Cy5 (red) channel (IgM) laser 75% photomultiplier 75%. An array image
representative of the experiment is shown in Figure 13. A multiplex proteomic,
diagnostic assay, conducted in approximately 35 minutes, is thus provided that
gives
quantitative titers of IgG and IgM in patient sera against ten human
pathogens.
Those skilled in the art will recognize, or be able to ascertain using no more
than
routine experimentation, many equivalents to the embodiments of the invention
described specifically above.
-21-