Note: Descriptions are shown in the official language in which they were submitted.
79806-13D
CA 02574055 2007-O1-30
1
COMPOSITION FOR PREPARATION OF AN ORGANOZINC COMPOUND
This is a divisional application of Canadian
Patent Application No. 2,273,942, filed December 15, 1997.
This invention relates to a process for the
preparation of an organozinc reagent, more specifically to
the preparation of organozinc reagents comprising an
aromatic moiety, to compositions comprising organozinc
reagents, and to processes for the use of such reagents and
compositions.
The subject matter of this divisional application
is directed to novel compsitions of matter which may result
in the formation of compounds of formula (II), as discussed
herein.
The subject matter of the parent application has
been restricted to processes for preparation of an
organozinc compound comprising an aromatic moiety and a
process for the introduction of an organic moiety into a
substrate compound by reaction of the substrate compound
with an organozinc compound. However, it should be
understood that the expression "the invention" and the like,
when used herein, encompass the subject matter of both the
parent and this divisional application.
Organozinc reagents may be made by reacting a zinc
halide with an organic compound of another metal. The co-
product of such a reaction is a halide of that other metal.
To purify the organozinc compound, it has been proposed in
the case of aromatic organozinc compounds, and when the
other metal is magnesium, to precipitate its halide by
adding 1,4-dioxan, (e.g. Nutzel in Houben-Weyls Methoden der
CA 02574055 2007-O1-30
79806-13D
2
organischen Chemie 1973, XIII/2a, 197-198, 592-599 and Soai
et al. J. Chem. Soc. Perkin Trans. 1991. 1613-1615, based on
Schlenk et al. Ber. 1929, LXII, 920-924). However, the
organozinc compounds so produced have been found to be
unsuitable for use in asymmetric synthesis.
We have now devised a simpler process for
purifying aromatic organozinc compounds. Our process is
applicable to a wider range of 'other' metals and to
purifying aromatic organozinc reagents in which the organic
groups are such that distillation would not be convenient.
The organozinc reagents so produced are believed to be in a
state different in point of purity from that resulting from
the dioxan process, and in at least some aspects, are
believed to be more suitable for enantioselective synthesis.
According to a first aspect of the present
invention, there is provided a process for the preparation
of an organozinc compound comprising an aromatic moiety by
reaction between a zinc chloride, bromide or iodide and an
organometallic compound of another metal comprising an
aromatic moiety, thereby producing~a reaction product
comprising an organozinc compound and a halide salt of the
other metal, the reaction product being contacted with a
liquid in which the organozinc compound is soluble and the
halide salt of the other metal is of low solubility, and
separating the halide salt of the other metal from the
liquid, characterised in that the liquid is a hydrocarbon.
According to one aspect of the invention of the
parent application, there is provided a process for the
preparation of an organozinc compound comprising an aromatic
moiety by reaction between a zinc chloride, bromide or
iodide and an organometallic compound of another metal
comprising an aromatic moiety, thereby producing a reaction
CA 02574055 2007-O1-30
79806-13D
2a
product comprising an organozinc compound and a halide salt
of the other metal, the reaction product being contacted
with a liquid in which the organozinc commpound is soluble
and the halide salt of the other metal is of low solubility,
and separating the halide salt of the other metal from the
liquid, wherein the liquid is a hydrocarbon provided that
the organometallic compound of another metal comprising an
aromatic moiety is not 2,4,6-(trifluoromethyl)phenyl
lithium.
According to another aspect of the invention of
the parent application, there is provided a process for the
introduction of an organic moiety into a substrate compound
wherein the substrate compound is reacted with an organozinc
compound prepared by reaction between a zinc chloride,
bromide or iodide and an organometallic compound of another
metal comprising an aromatic moiety, thereby producing a
reaction product comprising the organozinc compound and a
halide salt of the other metal, the reaction product being
contacted with a liquid in which the organozinc compound is
soluble and the halide salt of the other metal is of low
solubility, and separating the halide salt of the other
metal from the liquid, wherein the liquid is a hydrocarbon.
According to one aspect of the invention of the
present divisional application, there is provided a
composition of matter obtained by mixing a Lewis acid, a
chelator selected from aminoalcohol, aminothiol, diamine or
diol, in which the chelating groups are separated by 2 or 3
carbon atoms and an organozinc compound of formula ZnRllRiz
wherein R11 and R12 are each independently organic groups,
provided that at least one of R11 and R12 comprises an
aromatic moiety.
79806-13D
CA 02574055 2007-O1-30
2b
The organozinc compound can be recovered from the
liquid, or can be employed as a reagent as a solution in the
liquid.
The hydrocarbon in which the organozinc compound
is soluble and the co-product halide is of low solubility
can be linear, branched or cyclic. Aliphatic and
particularly aromatic hydrocarbons are most commonly
employed. Examples of suitable aliphatic hydrocarbons
include petroleum ethers; linear or branched alkanes,
particularly those having from 5 to 22 carbon atoms, and
preferably from 6 to 14 carbon atoms; kerosenes; and cyclic
hydrocarbons, particularly those comprising a 5 to 8
membered alicyclic ring. Many suitable hydrocarbons have a
boiling point at atmospheric pressure in the range of
from 60 to 130°C. Examples of suitable aromatic
hydrocarbons include those comprising from 6 to 10 carbon
atoms, and include benzene and alkyl-substituted benzenes.
Preferred hydrocarbons include hexane and cyclohexane, and
particularly preferred hydrocarbons are toluene, xylene and
mesitylene. The hydrocarbon need not itself be a liquid at
ambient temperature, provided it forms a liquid system at
processing temperatures in presence of other materials
present, and so for example, butane may be employed under
suitable pressure conditions. The liquid may be a mixture
of one or more hydrocarbons. The liquid may also comprise
one or more other compounds, providing that the solubility
of the undesired metal halide in the liquid is not increased
to an unacceptable level thereby.
If desired, the contact between the reaction
product and the hydrocarbon may be preceded by a
conventional dioxan addition and removal of the resulting
metal halide precipitate as a first crude purification step,
with the resultant dioxan-zinc solution being contacted with
CA 02574055 2007-O1-30
79806-13D
2c
the hydrocarbon as a polishing step to precipitate futher
metal halide.
At least one of the organic groups in the
organozinc compound prepared by the process of the present
invention comprises an aromatic moiety. Examples of
suitable aromatic moieties include particularly phenyl and
naphthyl groups. The aromatic moiety may be a
heteroaromatic group, especially a furyl, pyridyl, quinolyl
or thienyl group; a metalloaromatic group such as
ferrocenyl; or an araliphatic group, particularly a benzyl
group. The aromatic groups are optionally substituted with
optionally substituted phenyl being a preferred group. The
second of the organic groups in the organozinc compound can
be a second aromatic group, or may be a non-aromatic group.
Where a non-aromatic group is present, the group can be an
aliphatic group especially a C1-2o, particularly a C1-12,
aliphatic, and preferably alkyl, group and most preferably a
methyl, trihalomethyl, such as trifluoromethyl, ethyl,
pentahaloethyl, such as pentafluoroethyl, n- or iso-propyl,
or n-, iso- or tert-butyl group; a cycloaliphatic,
especially a C3_8 cycloaliphatic group, preferably a
cyclopropyl, cyclobutyl, cyclopentyl or cyclohexyl group; a
carbon-linked heterocyclic group, or a hydrogenated
derivative of an aromatic group. The non-aromatic groups
may be saturated or unsaturated. The organic groups in the
organozinc compound may carry one or more substituents.
There may be present organic groups of more than one type,
as the result for example of using a mixture of starting
materials or mixing single-type reagents without or with
disproportionation.
Organometallic compounds which can be reacted with
zinc halides in the process according to the present
invention include organic compounds of alkaline metals and
CA 02574055 2007-O1-30
79806-13D
2d
alkaline earth metals, particularly organomagnesium
compounds and organolithium compounds, organoaluminium,
organoborane, organotin, organocuprate, organocerium
organocadmium and organomercury compounds. The nature of
the organic groups) in the organometallic compound will be
selected so as to introduce the desired organic groups into
the organozinc compound. Accordingly, an organometallic
compound comprising at least one aromatic moiety must be
employed. Correspondingly, where an organozinc compound
comprising a non-aromatic group is to be produced, an
, CA 02574055 2007-O1-30
79806-13D
3
organometallic compound comprising such a group can also be employed. Example
of
suitable organomagnesium compounds inctude organomagnesium halides,
particularty
optionally substituted alkyl and aryl magnesium halide compounds, and
particularly
optionally substituted C,_s alkyl or optionally substituted phenyl magnesium
halides.
Examples of suitable organolithium compounds include optionally substituted
alkyl and
aryl lithium compounds, and particularly optionally substituted C,_6 alkyl or
optionally
substituted phenyl lithium compounds. Further suitable organolithium compounds
are
organolithium halides, such as optionally substituted alkyl and aryl lithium
halide
compounds, especially optionally substituted C,_6 alkyl or optionally
substituted phenyl
I o lithium halide compounds, and particularly organolithium chlorides and
bromides.
Preferred organolithium compounds include methyllithium,
trifluoromethyltithium,
ethylfithium, n- and iso-propyllithium, and n-, iso- and tert-butyllithium,
phenyllithium and
the chlorides and bromides of methyllithium, trifluoromethyllithium,
ethyllithium, n- and iso-
propyllithium, and n-, iso- and tert-butyllithium and phenyltithium.
When the said 'other' metal is magnesium, the organometallic compound is
conveniently a Grignard reagent, that is, introduced as an ethereal solution
of a
compound of stoichiometry R-Mg-X, where R is the required organic group and X
is
chlorine, bromine or iodine. The solvent is commonly an ether, such as a
di(C,~ alkyl)
ether, for example diethyl ether, dibutyl ether, diisoamyl ether and glyme.
Asymmetric
2 o dialkyl ethers can also be employed, such as t-butylmethylether. Other
ethers that may
be employed include diglyme and tetrahydrofuran, THF being usually preferred
because
of its capacity to dissolve compounds having a greater range of groups R. The
ether
quantity may if desired be less than sufficient to dissolve the whole of the
Grignard
reactant.
In certain embodiments, a mixture of different organometallic compounds can be
employed to produce a mixed organozinc compound. Approximately equal amounts
of
such different organometallic compounds would often be employed in such
embodiments.
The mole ratio of organometallic compound to zinc chloride, bromide or iodide
is
often selected to be in the range of from 1 : 1 to 3 :1, and preferably from 2
: 1 to 2.5 : 1.
3o When a mixture of different organometallic compounds is employed, the total
amount of
such compounds is selected to be within this range.
The zinc chloride, bromide or iodide employed is preferably zinc bromide.
The reaction between the zinc chloride, bromide or iodide and the
organometallic
compound conveniently takes place in the presence of a solvent. Examples of
suitable
solvents include ethers, commonly di(C,_e alkyl) ethers, including diethyl
ether, dibutyl
ether, diisoamyl ether and glyme. Asymmetric dialkyl ethers can also be
employed, such
as t-butylmethylether. Other ethers that may be employed include diglyme and
tetrahydrofuran. In certain embodiments of the present invention, especially
when an
organolithium compound is employed as organometallic compound, the sotverit
CA 02574055 2007-O1-30
79806-13D
4
comprises a hydrocarbon, thereby achieving the reaction and contact of the
reaction
product with a hydrocarbon in a single step. Further contacts with hydrocarbon
may
additionally be employed.
Advantageously, the organometailic compound is introduced into the reaction
mixture in the form of a solution in the solvent in which it has been
prepared. This solvent
will depend on the exact nature of the organometallic compound, but is
conveniently an
ether, particularly one or more of the ethers listed above a suitable reaction
solvents.
Organolithium compounds particularly can be introduced as a solution in a
mixture of an
ether and a hydrocarbon solvent. When such a mixed solvent is employed, the
1 o hydrocarbon conveniently comprises at least 50% by weight of the solvent.
The reaction temperature is typically in the range of from -30 to 150, and
especially 10 to 120°C. Conveniently, a temperature of from ambient up
tp the reflux
temperature of any solvents) present may be employed. Reaction times are
typically in
the range 0.5 to 100 hours, especially 1 to 48 hours and preferably from 2 to
15 hours.
Times and temperatures should be sufficient to disproportionate any organozinc
halide
which may be formed during the reaction. Atmospheric pressure is suitable,
unless
components of unusually low or high volatility are being used.
Before contacting the reaction product with the liquid, any solvent already
present,
such as ether solvent in which the compound of the said other metal is
introduced may be
2 o removed, for example by distillation, possibly at subatmospheric pressure.
However,
advantageously, such solvent is not removed prior to contact with the liquid.
Preferably,
the liquid employed has a higher boiling point than any other solvent present.
Accordingly, after addition of an aliquot of the liquid, the more volatile
other solvent can
be removed by distillation or by azeotroping. A further aliquot of the liquid
can be added if
2 5 desired. This procedure may be repeated one or more times. Removal of the
other
solvent followed by addition of a further charge of liquid is most preferably
employed
when the other metal halide is a magnesium halide. The resulting solution of
organozinc
reagent may be passed to reaction with a substrate compound as hereinafter
defined, or
stored as such, or concentrated, possibly with crystallisation if its
structure so permits.
3 o If desired, the reaction can be carried out on a one-pot basis from
magnesium or
other reactive metal, zinc halide and organic halide, possibly aided by
ultrasound (Luche
et al. J Org Chem 1983 48 (21 ) 3837-3839).
In the ensuing description, definitions of carbon chain length by reference to
the
numerical extremities of a range are to be understood as including all the
members of the
3 5 range.
The organozinc reagent product preferably contains less than 20, especially
less
than 5, especially less than 1, % w/w of other metal, calculated as % of total
metal. Such
a content of metal halide appears to correspond to a substantial absence of
material
effective to cause competing achirai catalysis to form a racemic product. Zinc
reagent
CA 02574055 2007-O1-30
79806-13D
having the so-defined content of metal halide is provided by the process of
the first aspect
of the invention as a solution or as a solid, possibly crystalline. If the
reagent is in
solution, the solvent is preferably one or more of the liquids defined above.
The organozinc reagent may if desired be further purified by contacting with
active
5 carbon or a cheiating ligand such as a 2,2'-bipyridyl that forms an
insoluble complex with
metal halide and/or possibly by vacuum distillation. The reagent, whether or
not so
further purified, may be used as an intermediate for producing a
semiconductor, for
example by vapour deposition or epitaxy.
It is believed that the organozinc reagent prepared in the process of the
present
1 o invention can form complexes with chelating compounds catalytic for its
reaction with an
aldehyde. The chelate compound is derived for example from a chelator selected
from
aminoalcohol, diamine or diol, in which the chelating groups are separated by
2 or 3
carbon atoms, as in a 1,2- or 1,3-hydroxy- or mercapto-amine. The chelate
compound
may comprise a Lewis acid, for example lithium, boron, titanium or zinc, and
preferably
zinc. If an optically active product is to be made, the chelating compound
should itself be
scalemic.
The chelate compound can for example have the formula I or analogue thereof
with zinc replaced by another Lewis acid former:
R'
R' Y- N-R4
(I)
Zn - RS
2 0 ' Rz Q /
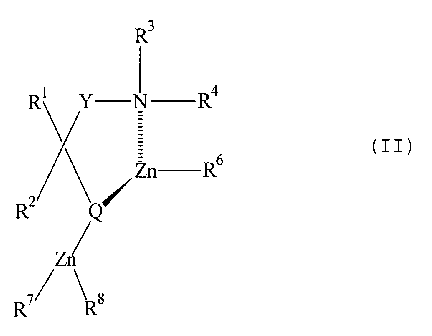
It is believed that the composition formed between the organozinc compound
produced by the process of the first aspect of the present invention and the
chelate
compound can for example have the formula II or its analogue having another
intra-
annular Lewis acid former in place of zinc:
R'
R' Y- N-R,
Zn - Rs
Rz Q /
Zn
R~ Ra
CA 02574055 2007-O1-30
79806-13D
6
In these formulae:
Y is methylene or ethylene optionally carrying one or more substituents inert
to
organozinc compounds;
R' and RZ are groups inert to organozinc compounds. Typically they are
selected
from hydrogen and the organic groups which may be present in the organozinc
compound produced in the first aspect of the present invention, particularly
hydrocarbon
groups, such as C,_,2 alkyl and preferably phenyl, heterocyclyl, ether,
thioether and
secondary amino, and may be substituted. They may be joined externally to form
a ring
or rings;
R3 and R4 are independently selected from hydrogen and the organic groups
which
may be present in the organozinc compound produced in the first aspect of the
present
invention, particularly hydrocarbon, such as C,.,2 alkyl, preferably C,.~
alkyl, and may
together form a ring; or a group of formula -CORx, -C02Rx, or -S02RX wherein
Rx is a C,.°
alkyl group. R3 and R' (except hydrogen) may carry one or more substituents
inert to
organozinc compounds; either or both of R' or R4 may cant' a substituent -Z-
NR9R'°
where Z is ethylene or trimethylene optionally carrying one or more
substituents inert to
organozinc and R9 and R'° each independently are as defined for R3 but
need not be the
same as R3;
RS can be any of the organic groups which may be present in the organozinc
compound produced in the first aspect of the present invention. Note that R5
occurs only
in I and is present normally as the product of reacting an organozinc~compound
with an
amino alcohol. Preferably RS is aryl or heterocyclyi;
R6 can be any of the organic groups which may be present in the organozinc
compound produced in the first aspect of the present invention. Note that R6
occurs only
2 5 in II. It may be the same as RS in I but may with advantage be lower
aliphatic (C,_s), since
the zinc alkyls leading to such a value for R6 are readily available in a pure
state and need
not be made by the process of the invention;
R' and Ra are as defined for the organic groups of the organozinc compound
produced in the first aspect of the present invention; and
3 o Q represents O, S or a group of formula -NR''R=, wherein Ry and RZ are
each
independently as defined for R3 and R4.
it is believed that some association of molecules of I can take place, but
that
reaction with ZnR'Re reverses such association.
When a group of formula -Z-NR9R'° is present, the nitrogen of -Z-
NR9R'° may
35 coordinated to the annular zinc atom.
The composition of matter which may result in the formation of compounds of
Formula (II) is believed to be new. Accordingly, a second aspect of the
present invention
provides a composition of matter obtained by mixing a Lewis acid, a chelator
selected
from aminoalcohols, aminothiols, diamines or diols, in which the chefating
groups are
CA 02574055 2007-O1-30
79806-13D
7
separated by 2 or 3 carbon atoms and an o~ganozinc compound of formula ZnR"R'2
wherein R" and R'2 are each independently organic groups as defined for the
organic
groups of the organozinc compound produced in the first aspect of the present
invention.
The Lewis acid may be an organolithium compound, an organoboron compound
or an organotitanium compound, but is preferably an organozinc compound of
formula
ZnR'3R'4, wherein R'3 and R'4 are each independently organic groups, and can
be any of
the organic groups which may be present in the organozinc compound produced in
the
first aspect of the present invention, but provided that the compound ZnR'3R"
is different
from the compound of formula ZnR"R'2. Preferably R" and R'" are each C,~ alkyl
groups.
If a composition according to the second aspect of the present invention is to
be
Wised to prepare an optically active product, the chelator compound
°from which the
composition is prepared should itself be scatemic.
In the composition of matter according to the present invention, in the
organozinc
compound of formula ZnR"R'2, R" and R'2 are preferably phenyl, tolyl, xylyl or
mesityl
groups.
The mole ratio of chelator to Lewis acid ira the composition at point of
mixing is
often from 0.1 : 1 to 1 : 0.1, commonly from : 0.5 : 1 to 1 : 0.5 and
preferably from 0.8 : 1
to 1 : 0.8. The mole ratio of chelator and/or Lewis acid to organozinc
compound of
2o formula ZnR"R'Z is often from 1 : 1 to 1 : 100, commonly from 1 : 5 to 1 :
50, and
preferably from 1 : 10 to 1 : 25.
Examples of chelating compounds suitable for use in the compositions of the
second aspect of the present invention are those compounds numbered 50-65b in
the
chapter Organozinc, organocadmium and organomercury reagents, pages 211-229,
especially 223-224, of Comprehensive Organic Synthesis, Volume 1, first
edition 1991,
Pergamon Press. These chelating compounds and also the article by Evaris, in
Science
1988, 240, 420-426, are incorporated herein by reference and are authority for
structures
I and II. The chelator is preferably one of cyclohexanediamine
bis(triflamide),
dimethytaminoisobomeot, a,a-diphenyt-2-pyrollidinemethanol, and especially
diethyl or
3 o dibutylnorephedrine. Should other structures be proposed, the compositions
are of
course still within the scope of the invention.
The compositions according to the second aspect of the present invention
preferably additionally comprise a hydrocarbon, most preferably the liquid
employed in the
process according to the first aspect of the present invention.
Organozinc compounds can be employed as reagents in a number of reactions,
particularly in reactions generating asymmetry. However, many organozinc
compounds
conventionally employed suffer because of the relatively high concentrations
of metal
halides, which function as contaminant Lewis acids. It is believed that the
presence of
such a contaminant Lewis acid is undesirable owing to one or more of the
mechanisms:
CA 02574055 2007-O1-30
79806-13D
8
(a) it competes with the organozinc compound for complexation with the
catalyst;
(b) it reacts with organozinc compounds to form organozinc halides which are
less
selective reagents affording substantially lower enantiomeric excess;
(c) it coordinates to the substrate compound and activates it for addition of
free
organozinc compound affording substantially lower enantiomeric excess; and
(d) it catalyses the non-enantioselective reaction at a rate sufficient to
substantially
lower the purity of the product.
According to a third aspect of the present invention, there is provided a
process
for the introduction of an organic moiety into a substrate compound wherein
the substrate
1o compound is reacted with an organozinc compound prepared according to the
process of
the first aspect of the present invention.
Preferably, the org~nozinc compound is employed as a composition according to
....
the second aspect of the present invention.
The substrate compound contains at least one of:
(a) a hetero-atom multiply linked to carbon;
(b) a multiple carbon-carbon bond conjugated with a hetero-atom multiply
finked to
carbon;
(c) an activated halogen; and
(d) an ethylene oxide
2 o and at least one organic group as hereinbefore defined.
Category (a) includes especially aldehydes and aldimines. Category (b)
includes
especially a,(3-unsaturated compounds containing such groups or also possibly
other
electron-withdrawing groups such as CF3, sulphone or nitro. Category (c)
includes allyl
halides, benzyi halides, aryl halides and acyl halides.
2 5 The reaction involving addition of an organozinc compound to a substrate
from
category (a) is believed to proceeds by way of an intermediate of formula II
above in
which intra-annular zinc is present if more than one mole of organozinc
compound is used
per mole of substrate. Since the reaction of II regenerates I the excess of
organozinc can
be for example 5-25% in molar terms.
3o A preferred process according to the third aspect of the present invention
comprises reacting a prochiral aldehyde or aldimine with either:
a) an organozinc compound prepared by the process of the first aspect of the
present
invention in the presence of a scalemic chelating compound catalytic for the
reaction;
preferably an aminoalcohol, diamine, aminothiol or a diol, in which the
chelating groups
35 are separated by 2 or 3 carbon atoms; or
b) a composition according to the second aspect of the present invention.
Aldehydes and aldimines that can be employed in the process according to the
third aspect of the present invention have the formulae:
CA 02574055 2007-O1-30
79806-13D
9
Ra----CHO or Re-- --CH===N----Rb
wherein Ra is an aromatic or aliphatic group, particularly an optionally
substituted phenyl,
pyridyl or thienyl group, and Rb is H or an optionally substituted alkyl,
preferably C,_6 alkyl,
aryl or benzyl group. It will be recognised that to generate a chiral product,
the organic
group introduced by the organozinc compound should be different from that
represented
by Re.
Substituents which may substitute the organometallic compounds, organozinc
compounds, groups R'''°, groups RX, Ry, RZ, Ra and Rb and Y, especially
Ra and Rb, are
1 o generally selected to be inert under the reaction conditions under the
prevailing reaction
conditions. Commonly, the substituents are selected from halogen, alkyl,
particularly C,_s
.- alkyl, aryl, particularly phenyl, -OR, -OCOR, -COR, -C02R, -CN, -SH, -SR,--
S02R, -S03R,
-SONR2, -SCSR, -PR3, -POR, -POZR, -NR2 and -SiR3, wherein R represents H or an
alkyl,
preferably a C,.6 alkyl, or aryl, preferably a phenyl, group. When R
represents an alkyl or
aryl group, further substituents may be present. The substituents may also be
selected
from metal carbonyl groups, such as iron carbonyl.
In a particular process the substrate is a substituted benzaldehyde,
especially
halo, particularly chloro, fluoro or bromo ( each 2, ,3- or 4-), alkoxy,
especially C,.4 alkoxy,
( preferably 4-) or trifluoromethyl, or heterocyciic aldehyde such as pyridine-
3-aldehyde.
The reaction preferably occurs in the presence of a solvent. Preferably, the
solvent has no basic or acidic functional groups present, and is most
preferably a
hydrocarbon as defined herein for use as the liquid. The mole ratio of
organozinc
compound to aldehyde or aldimine is often selected to be in the range of from
0.8 : 1 to 3
1, and is preferably from 1 : 1 to 1.5 : 1, based on the number of aldehyde or
aidimine
2 5 functionalities to which an organic moiety is desired to be introduced.
Commonly, there is
a single such functionality.
The reaction may take place with catalysis by transmetallation, in presence of
a
compound of a transition metal such at titanium, copper, nickel or palladium.
Thus the
effective reactant may be a compound of such metal, formed as a distinct
component of
3 o the reaction mixture or transiently.
The organozinc composition may be introduced as such or formed in a distinct
process step or formed transiently in a reaction mixture containing organozinc
compound,
catalytic chelator compound and substrate compound. If the addition reaction
is required,
the process preferably comprises the separate steps:
35 (i) reacting an organozinc compound with the chelating compound;
(ii) reacting the product of (i) with an organozinc compound produced by the
process
according to the first aspect of the present invention, an organic group,
commonly
an aromatic group of which is to be added to the substrate; and
(iii) reacting the product of (ii) with the substrate.
79806-13D
CA 02574055 2007-O1-30
In this process, step (l) should be controlled to avoid the first organozinc
compound from occupying the reactive sites in the chelator which may be
occupied by the
second organozinc compound if the first organozinc compound does not provide
the
group to be added to the substrate compound. Such control may be for example
by
5 reference to mixing procedures or by allowing sufficient time for
redistribution of the first
organozinc compound into the ring position.
The processes of the various aspects of the invention are carried out using
anhydrous reactants and in anhydrous oxygen-free conditions. When reactant
metals
other than organozinc compounds are present, carbon dioxide should also be
excluded.
1 o In the process of the third aspect, after the reaction with the substrate,
the
reaction mixture is worked up by procedures established in this branch of
technology,
usually by reacting it with acid, such as hydrochloric or sulphuric acid and
separating the
required product as a water-insoluble layer. The temperature is kept low,
usually below
20°C, to avoid or limit possible racemisation, although if racemisation
is not a problem,
higher temperatures may be employed.
The invention is further illustrated by the following examples.
(a) Preparation of D(+) Diethylnorephedrine (D(+)DENE) (1-phenyl-1-hydroxy-2-
methyl-2-diethylaminoethane) Materials:
Material Amt(g) Strength Moles Mol Eq
IS,2R norephedrine 30.8 98% 0.2 1
ethanol 130mi
potassium carbonate 55.7 99% 0.4 2
iodoethane 78 99% 0.5 2.5
Equipment:
An oven dried 500m1 multinecked RBQF with thermometer, pressure equalising
addition funnel with nitrogen bubbler and PTFE paddle stirrer.
2 5 Procedure:
The dry glassware was purged with nitrogen. The potassium carbonate,
norephedrine and absolute ethanol were charged together and stirred while
heating to
reflux at 70°C to give a thin pale yellow slurry. The iodoethane was
mixed with ethanol,
charged to the addition funnel and then added dropwise to the reaction mixture
over 3
3o hours. Samples were taken and analysed by GC at approximately hourly
intervals.
From the rate of reaction it was decided to continue refluxing overnight.
After 19 hours
the reaction mixture was cooled to ambient temperature and the inorganic
solids filtered
CA 02574055 2007-O1-30
' 79806-13D
11
using a sintered glass filter. The solids were slurry washed with two 25m1
portions of
ethanol. The filtrates were combined and stripped of solvent on the rotary
evaporator at
40°C and 20 mmHg to give a creamy white oily solid, weight 48.68. This
solid was
dissolved in 150mf methylene chloride and washed with 75mi water. The pH of
the
aqueous phase was increased from 10 to 11 using dilute sodium hydroxide and
the
phases stirred together then allowed to separate. The well separated organic
phase
was run-off and the aqueous phase re-washed tvvice with 25m1 methylene
chloride. The
combined organics were dried over solid anhydrous magnesium sulphate, filtered
over
paper and concentrated to dryness on a rotary evaporator to give 50.3g of an
off-white
oil, part of which solidified overnight. The insolubles were separated using a
sintered
glass filter and washed with two 25m1 portions of hexane. The washings and
filtrates
were combined and the solvent was stripped on--the rotary evaporator to give
36.3g of a
pale yellow oil. By GC this gave a strength based on area % of 96.7%, that is
85% yield
based on norephedrine. The optical purity was determined by chiral HPLC to be
'96.7%.
(b) Preparation of D(+) di-n-butyl norephedrine D(+)DBNE
The same procedure was followed using n-butyl bromide.
EXAMPLE 1 : Preparation of Diphenylzinc via Phenyl-magnesium Bromide
Materials
Material Actual 1 CO% Gram Molar
Wt(g) Wt(g) Moles Ratio
Anhydrous zinc bromide 11.37 11.26 0.05 1.0
Phenylmagnesium bromide
(3.0M solution in diethylether)33.3m1 18.13 0.1 2.0
Diethyiether (dried over 75m1
Na)
Toluene 100m1
Tetrahydrofuran (dried) 50m1
D(+)DENE 0.64 0.62 0.003 0.06
Nicotinaidehyde (pyridine-3-5.4 5.35 0.05 1.0
aldehyde)
Hydrochloric acid (6M 50m1
HCI)
Diethylether 1 OOmI
2 o Apparatus:
CA 02574055 2007-O1-30
79806-13D
12
An oven dried, multinecked 250m1 round bottom Quickfit (RTM) flask (RBQF)
fitted
with a PTFE paddle stirrer, thermometer, reflux condenser with nitrogen
bubbler and a
port with a Suba-Seal (RTM) septum cap was used.
To filter the inorganic solids the condenser was replaced by an in-line No 3
s sintered glass filter connected with flexible PTFE tubing under inert
atmosphere. The
filtrates were collected in a second dry 250m1 RBQF fitted with PTFE paddle
stirrer,
pressure equalising addition funnel, thermometer and inlet port from the in-
line filter, all
under an inert nitrogen atmosphere.
Method
l0 (a) Preparation of diphenylzinc
The zinc bromide was dried overnight at 120°C and about 14 mmHg,
then
dissolved in 75m1 dry diethylether (stored over Na) in a nitrogen atmosphere
in the flask
cooled to 4°C using an ice bath. The inert atmosphere, was re-
established and the
phenylmagnesium bromide introduced from a 'Sure-Seal' (RTM) bottle via the
septum
15 using a dry 20m1 syringe. The reaction mixture was held below 10°C
during this addition, .
then allowed to warm to room temperature (19°C) and stirred for 30 min.
The mixture at
the end of the addition was a grey/brown solution with a granular white
precipitate; The
reffux condenser was re-set for distillation and the mixture heated to
76°C; 74m1 ether
were distilled off at 39°C atmospheric pressure. 100m1 of dry toluene
were then added by
20 syringe, upon which the grey crystalline slurry lightened in colour.
Distillation was
continued until the head temperature reached 113°C; then the mixture
was cooled to
21 °C and allowed to stand overnight. A further 50m1 of toluene were
added, then the
mixture warmed to 105°C and filtered using the apparatus described
above. The filter
cake was blown dry with nitrogen. The filtrate was a pale yellow solution of
substantially
25 pure diphenyizinc (88 mol % Zn, 2 mol % Mg). The flask was sealed and
stored in a
refrigerator.
(b) Preparation of phenyl-3-pyridyl carbinol
(i) invention process
To the pale yellow filtrates the D(+)DENE was added against a counter-current
of
30 nitrogen. The temperature rose from 16 to 31°C and was then cooled
back to 17°C. To
the resulting solution of complexed reagent Ii 40m1 of dry THF were added in
one portion
by syringe; the temperature rose to 30°C. The nicotinaldehyde in 10g
THF was charged.
to the addition funnel and added dropwise to the mixture at 21 °C over
6 hours. During
this period samples were taken and, after quenching in acid, neutralising and
extracting
35 with methylene chloride, analysed by chiral capillary GC (see below). After
2I3 of the
aldehyde had been added the GC showed accumulation of aldehyde and the
addition was
thus stopped.
Work-up was by addition of the HCI to the chilled reaction mixture, keeping
the
temperature below 20°C. The organic layer was separated and washed with
25m1 water.
CA 02574055 2007-O1-30
.' 79806-13D
13
The aqueous layer was washed with 25m1 diethyJether and 25m1 methylene
chloride. The
aqueous phases were combined and neutralised to pH 6I7 with sodium hydroxide
solution
in the presence of 50m1 methylene chloride. The organic phase was separated
and the
aqueous layer washed with a further 25m1 of methylene chloride. This was
combined with
the previous extract, then distilled on a rotary evaporator to give 8.7g of a
pale yellow
viscous oil which solidified on cooling.
The product was analysed by GC and shown to be 88.5% strength based on area
%. The optical purity was determined by both chiral shift HNMR (73% ee) and
chiral
capillary electrophoresis (73.4% ee).
(ii) Comparative runs
Run (i) was repeated twice with the difference that the diphenylzinc was a
commercially obtained- sample purified by sublimation and that the
diphenylzinc, --..
D(+)DENE and aldehyde were reacted together in a single stage, and (for one of
these
runs) magnesium chloride 6 mol% on the aidehyde was present. The results were:
1 5 % Yield % e.e.
No MgCl2 86 54
MpCh 47 18
Example 2
Example 1 (b) was repeated but using 0.06 mol of diethylzinc, 0.06 mol of
2 o D(+)DENE and 1 mol of diphenyi- zinc. The diethylzinc and DENE were
reacted together
before adding the diphenylzinc. The products were
phenylpyridylcarbinol 36% yield, 46% e.e.
pyridylethyl carbinol 14% yield.
' 79806-13D
CA 02574055 2007-O1-30
14
Comparison 3 : use of Schtenk procedure
Materials:
Material Actual 100% Gram Molar
Wt(g) Wt(g) Moles Ratio
Zinc chloride
(1.0M solution in diethylether)20m1 2.73 0.02 1.0
Phenylmagnesium bromide
(3.0M solution in diethylether)13.3m1 7.25 0.04 2.0
Tetrahydrofuran (THF)
(distilled
.and dried over molecular60m1
sieves)
Tetrahydrofuran (dried)
50m1
1,4-dioxane 7.05 7.05 0.08 4.0
D(+)DBNE 0.324 0.316 0.0012 0.06
Nicotinaldehyde 2.16 2.14 0.02 1.0
Hydrochloric acid (6M) 20m1
~
Apparatus:
' As in Example 1.
Method
(a) Preparation of diphenylzinc
The zinc chloride solution was charged to the flask by syringe and diluted
with
60m1 THF atl under a nitrogen atmosphere. The stirrer was started and
1o phenylmagnesium bromide introduced using a dry 20m1 syringe dropwise over 5
min initially forming a precipitate which dissolved by the end of the
addition. The
reaction mixture was held at 20-25°C during addition and stirred for a
total of 2
hours. The dioxan was then added by syringe causing a fine, white solid to
precipitate. The slung was stirred a further 45 min at 25°C, then
filtered via the in-
line sintered glass filter. The filter cake was washed with 1 Oml THF and
blown dry
with nitrogen. The filtrate was a pale yellow solution in THF of diphenyizinc
(57
mol % Zn, 10.2 mol % Mg). The flask was sealed and stored in a refrigerator.
(b) Preparation of phenyl-3-pyridyl carbinol
To a sample of the filtrates at 5°C a premixed solution of
D(+)DBNE,
2 0 nicotinaldehyde and 1 Oml THF was added dropwise over 10 min. By the end
of
CA 02574055 2007-O1-30
' 79806-13D
the addition a light, white sturry had forded. The reaction mixture was warmed
to
ambient temperature, stirred 18 hours, then cooled to 7°C and quenched
with
dilute hydrochloric acid. The organic layer was separated and washed with 25m1
water. The aqueous layer was washed with 25m1 diethylether and 25m1
5 methylene chloride. The aqueous phases were combined and neutralised to pH
6/7 with sodium hydroxide solution in the presence of 50m1 methylene chloride.
The organic phase was separated and the aqueous layer washed with a further
25m1 methylene chloride. This was combined with the previous extract and
distilled on a rotary evaporator to give 4.11g of a pale yellow solid.
1 o The product was analysed by GC and shown to be 74.1 % strength based on
area
%. This gives a yield of 84.5% based on the aldehyde. The optical purity was
w determined by both chiral shift HNMR and chiral HPLC and found to be 0% ee,
i.e.
racemic.
Example 4 : Preparation of diphenylzinc via phenyllithium
15 Materials:
MWt Qnty 100% Moles
~(g)
Anhydrous zinc chloride136.6 2.78g 2.73 0.02
Phenyilithium (1.8M 84.05 22.2m1 3.36 0.04
solution in
30:70 cyclohexane:diethyl-
ether)
Diethylether (dried 75m1
over Na)
Tetrahydrofuran (dried 50m1
over
Na)
D(+)DENE 207 0.2568 0.248 0.0012
Nicotinaldehyde 107 2.168 2.14 0.02
Hydrochloric acid ~ 50m1
Methylene chloride 75m1
Apparatus
As in Example 1.
2 o Method:
(a) Preparation of diphenylzinc
CA 02574055 2007-O1-30
79806-23D
16
The zinc chloride was dried overnight at_120°C and _14 mmHg and
dissolved in
75m1 of dry diethylether at reflux under nitrogen. The solution was cooled to
3°C using
an ice bath placed under the flask. The phenyllithium was charged by syringe
via a
septum into the addition funnel, and then added dropwise over 15 min to the
zinc
chloride solution. Initially a white precipitate was formed which became
darker and
heavier and after 15 min further stirring was difficult. The mixture was
heated under
reflux (39°C) using an oil bath for 2'/Z hours, dining which the solid
turned dark grey.
The slurry was filtered hot using the in-line filter. The filter-cake was
blown dry using
nitrogen. The filtrates were somewhat cloudy due to an insoluble brown oil;
this was
1 o allowed to settle and removed by pipette. The ether solution was diluted
with THF,
cooled to >5°C using an ice bath and stored in a refrigerator.
(b) Preparation of phenyl-3-pyridyl carbinol. .
To a sample of the solution was added the solid D(+)DENE, followed by the
solid
aldehyde. The resulting cloudy yellow solution was warmed to 20°C,
during which a
white precipitate formed, then stirred overnight. 17 hours later the reaction
mixture was
worked-up by adding 50m1 methylene chloride and then slowly 50m1 hydrochloric
acid
maintaining the temperature below 15°C. The organic layer was separated
and washed
with 25m1 water. The washing water combined with the aqueous layer was
neutralised
to pH 6!7 using sodium hydroxide solution and extracted twice with methylene
chloride,
50m1 then 25m1. The organic extracts were 1 combined and distilled on a rotary
evaporator to give 4.31 g of a viscous, dark straw-coloured oil.
The oil was analysed by GC and shown to be 86% strength, based on area %.
This equates to 3.7g @ 100% and gives a 100% yield based on the aldehyde.
The product was characterised by HNMR: CHOH,1H,5.83s; ArH 7.24, 5H;
ArH:Ha7.37,J=5.26,7.21; ArH:Hb7.80,J=8.0; ArH:Hc8.55,J=1.37,3.89;
ArH:Hd8.77,J=1.8.
The optical rotation is a2° _ - 13.6 c=2.0 CH2CIZ. The optical purity
was measured by chiral
shift 1 HNMR and chiral GC (60% ee)and chiral capilliary electrophoresis as
60.6% ee.
Example 5
3o Preparation of diphenylzinc by reaction of phenyllithium with zinc bromide
Example 4(a) was repeated using zinc bromide in place of zinc chloride and in
process conditions varied as follows:
(l) reaction mixture allowed to stand overnight at 0-5°C, then refluxed
at 41 °C for 2
hours;
(ii) reaction mixture refluxed 1 hour at 43°C, then diluted with
toluene (25% wlw); the
ether was distilled off and reflux resumed at 82°C, to a total reflux
time of 3 hours;
(iii) the zinc bromide was added as a solution (10.8%
w/w) in toluene; refluxing was at 90°C and was carried on for 16 hours.
CA 02574055 2007-O1-30
79806-13D
17
The resulting solutions of diphenylzinc were reacted with substituted
benzaldehydes and the products analysed for yield and enantiomeric excess.
Results
are shown in the Table.
Table
Source of Benzaldehyde Yield % Enantiomeric
diphenylzinc substituent w/w excess,
on the
aldehyde
i o - CF3 80.6 0
ii o - CF3 83.3 43.5
ii m - CF3 94.4 55.8
iii p - CF3 100.0 65.7
By comparison with Example 4 it appears that the solubility of lithium bromide
in
the ether-cyclohexane mixture is too great to give diphenylzinc pure enough to
afford
enantiomeric excess. However, when as in (ii) and (iii), the diphenylzinc is
brought into
ether-free solution, the solubility of lithium bromide is low enough to give
diphenylzinc
capable of substantial enantiomeric excess.
Example 6
Reaction of diphenylzinc, itself prepared from phenyimagnesiumbromide and zinc
bromide, with substituted benzaldehydes
Anhydrous zinc bromide (6.82g) was charged to a dry, stirred 250m1 flask
filled with
nitrogen. Dried diethylether (75m1) was added and the slurry heated to reflux
until a
homogenous solution. A 3molar ethereal solution of phenylmagnesium bromide
(ZOmI)
was slowly added. Solvent was distilled from the reaction mixture at ambient
pressure,
whilst maintaining a constant volume by addition of toluene until a distillate
temperature of
at feast 108'C was reached. The hot slurry was screened under a nitrogen
atmosphere
2 o using an in-fine sintered glass filter. The filtrates were received into a
dry, stirred 250m1
flask and cooled to ambient temperature. L(-) diethylnorephedrine was added
and the
substituted benzaldehyde charged from a dropping funnel. The reaction mixture
was
stirred for a total of 12 hours then cooled on an ice bath and quenched by
addition of
dilute hydrochloric acid. The organic layer was separated, washed with an
equal volume
2 5 of water and concentrated to yield a crude product. Unless indicated
otherwise, weights of
products are on crude material. For characterisation purposes materials were
further
purified by recrystallisation. Optical purities were measured by chiral GC and
chiral shift
HNMR.
CA 02574055 2007-O1-30
79806-13D
18
R catalyst product ee (%) az HNMR & Mass Spectra
weight weight
(g) (g)
o-CF 0.76 3.8 20 +12.67 CHOH,1 H,6.2Ds;ArH7.1-
3
c=2.05 7.60, 9H
252, 233, 214, 195,
183,
166, 153, 105, 77,
51
m- 0.76 3.6 84 +25.05 CHOH,1H,5.78s;ArH7.1-
CF3 c=2.83 7.65, 9H
252, 233, 214, 195,
183,
166, 153, 105, 77,
51
p- 0.91 . .. 3.5 24 + 9.38 CHOH,1 H,5.78s;ArH7.26
CF3 .
- -
c=1.7 5H
ArH.AB;7.45,4H,J=8.46,9.
83Hz
252, 233, 214, 195,
183,166, 153, 105,
77, 51
o-C1 - -CHOH,1H,6.10s;ArH7.1-
7.90,9H
m-CI 0.76 3.3 23 + 6.45 CHOH,1 H, 5.59s;ArH7.05-
c=2.D 7.28,9H
218.5, 183, 166, 153;
105, 77, 51
p- 0.37 1.3 14 + 3.49 CHOH,1 H, 5.76s;ArH7.25-
CI - -
c=2.0 7.37,9H
218.5, 183, 166, 153,
139, 113, 105, 77,
51
o- 0.32 1.7 +I-
OCH3
m- 0.64 4.1 41 - 4.00 c=2.0CHOH,1 H,5.79s;ArH6.78-
OCH3
7.45,9H;
OCH ,3.80s,3H
p- 0.37 0.9 +/- CHOH,1 H,5.74s;ArH6.8-
- -
OCH3 ~ 7.4,9H;
OCH ,3.76s,3H
214, 213, 197,181,
167,
153, 135, 105, 77,
51
CA 02574055 2007-O1-30
79806-13D
19
Example 7 ,
Reaction of diphenytzinc, itself prepared from phenyllithium and zinc bromide,
with
substituted benzatdehydes
Anhydrous zinc bromide (5.73g) was charged to a dry, stirred 250m1 flask
filled with
nitrogen. Dried diethylether (52m1) was added and the slurry heated to reflux
until a
homogenous solution. The solution was cooled to. ambient temperature and a 1.8
molar
cyclohexane/ether (7:3) solution of phenyilithium (28m1) (Aldrich) was slowly
added from a
dropping funnel over 2 hours. The reaction mixture was heated to reflux for 1
hour then
toluene (30m1) added. Solvent was distilled at ambient pressure, from the
reaction until
1 o the distillate temperature reached at feast 85'C. The slurry was cooled to
ambient
temperature, allowed to settle and the liquid carefully decanted using.a
syringe and
needle. This soiution._was charged to a dry, stirred 250m1 flask, and cooled
to 5'C. L(-)
diethylnorephedrine (0.124g) and tetrahydrofuran 1(2m1) was added and the
substituted
benzaldehyde (0.00715moies) in tetrahydrofuran (8ml) charged from a dropping
funnel
over 2 hours. The reaction mixture was stirred for a total of 12 hours then
cooled on an
ice bath and quenched by addition of dilute hydrochloric acid. The organic
layer was
separated, washed with an equal volume of water and concentrated to yield a
crude
product. Unless indicated otherwise, weights of products are on crude
material. For
characterisation purposes materials were further purified by
recrystallisation. Optical
2o purities were measured by chiral GC and chiral shift HNMR.
R product ee (%) a2o HNMR & Mass Spectra
weight (g)
o-CF3 1.5 44 (+) CHOH,1H,6.20s;ArH7.1-7.60,
9H
252, 233, 214, 195, 183,
166, 153,
105, 77, 51
m-CF3 1.7 56 +7.57 CHOH,1H,5.78s;ArH7.1-7.65,
9H
c=2.4 252, 233, 214, 195, 183,
166, 153,
105, 77, 51
p-CF3 1.8 66 + 33.5 CHOH,1 H,5.78s;ArH7.26
5H
c=1.5 ArH.AB,7.45,4H,J=8.46,9.83Hz
252, 233, 214, 195, 183,166,
153,
105, 77, 51
Example 8
Reaction of diphenylzinc, itself prepared from phenyllithium and zinc bromide,
with
substituted benzaldehydes
Anhydrous zinc bromide (10.45g} was charged to a dry, stirred 250m1 flask
filled with
nitrogen. Dried toluene (97m1) was added and the slurry heated to 80'C until a
homogenous solution. The solution was cooled to 0-5'C and a 1.8 molar
CA 02574055 2007-O1-30
79806-13D
cyciohexane/ether (7:3) solution of phenyllithiurrl (50.5m1) (Aldrich) was
slowly added from
a dropping funnel over 2 hours. The reaction mixture was heated to reflux for
12 hour
then cooled to ambient temperature, allowed to settle and the liquid carefully
decanted
using a syringe and needle into a storage jar and kept at ambient temperature
as a stock
5 solution. Diphenylzinc solution (30m1) was charged to a dry, stirred 250m1
flask, and
cooled to 5'C. L(-) diethylnorephedrine (0.124g) and tetrahydrofuran (2m1) was
added and
the substituted benzaldehyde (0.00715moles) in tetrahydrofuran (8m1) charged
from a
dropping funnel over 2 hours. The reaction mixture was stirred for a total of
12 hours then
cooled on an ice bath and quenched by addition of dilute hydrochloric acid.
The organic
10 layer was separated, washed with an equal volume of wafer and concentrated
to yield a
crude product. Unless indicated otherwise, weights of products are on crude
material. For
characterisation purposes materials were further purified by
recrystailisation. Optical
purifies were measured by chiral GC and chiral shift HNMR.. '
CA 02574055 2007-O1-30
79806-13D
21
R product ee (%) aZO HNMR ~ Mass Spectra
weight (gj
o-CI 2.4 54 - 1.39 CHOH,1 H,6.10s;ArH7.1-7.90,9H
c=2.15 218.5, 183, 165, 152, 139,112,105,
77, 51
m-Cl 1.8 72 + 6.59 CHOH,1 H,5.59s;ArH7.05-7.28,9H
c=2.3 218.5, 183, 165, 152,139,
111, 105,
77, 51
p-CI 1.8 28 + 9.48 CHOH,1 H,5.76s;ArH7.25-7.37,9H
c=2.74 218.5, 183, 166,165, 152,
139, 111,
.~.,.._ 105, 77, 51 . ., ..
o-Br 2.0 28 - 5.16 263, 183, 165, 152, 105,
77, 51
c=2.3
m-Br 3.6 40 + 10.85 CHOH,1H,5.80s;ArH7.15-8.09,9H
c=2.2 263, 183, 166, 153, 105,
77, 51
p-Br 2.0 24 + 6.63 CHOH,1 H,5.76s;ArH7.2-7.65,9H
c=2.11 263, 183, 166, 153, 105,
77, 51
o-OCH3 1.9 +/- -1.66 214, 198, 183, 165, 151,
135, 105,
c=2.4 77, 51
'
m-OCH3 1.7 68 - 7.49 CHOH,1 H,5.79s;ArH6.78-7.45,9H
c=2.1 OCH ,3.80s,3H
214, 197, 183, 181, 165,
152, 135,
. 105, 77, 51
p-OCH3 1.9 1 0.00 CHOH,1 H,5.74s;ArH6.8-7.4,9H
c=2.8 OCH ,3.76s,3H
.
214, 197, 181, 165, 151,
135, 105,
77, 51
Example 9
Reaction of diarylzinc, itself prepared from arylmagnesiumbromide and zinc
bromide, with benzaldehyde
Magnesium powder (50mesh)(1.23g), anhydrous zinc bromide (5.75g) and . ..,
tetrahydrofuran (50m1) were charged to a dry stirred 250m1 flask filled with
nitrogen. The
mixture was heated to reffux at 65'C and a solution of substituted arylbromide
(0.05
moles) in tetrahydrofuran (10m1) was added dropwise. The mixture was
maintained at
reflux for a further 2 hours. Toluene (100m1) was added to the mixture and
solvent
distilled at ambient pressure, whilst maintaining a constant volume by
addition of toluene
until a distillate temperature of at least 108'C was~reached. The hot slurry
was screened
CA 02574055 2007-O1-30
79806-13D
22
under a nitrogen atmosphere using an in-line sintered glass filter. The
filtrates were
received info a dry, stirred 250m1 flask and cooled to 1'C. L(-)
diethylnorephedrine (0.33g)
in tetrahydrofuran (15m1) was added and benzaldehyde (2.7g) in tetrahydrofuran
(5m1)
charged from a dropping funnel. The reaction mixture was warmed to ambient
temperature, stirred for a total of 12 hours then cooled on an ice bath and
quenched by
addition of dilute hydrochloric acid. The organic layer was separated, washed
with an
equal volume of water and concentrated to yield a crude product. Unless
indicated
othervvise, weights of products are on crude material. For characterisation
purposes
materials were further purified by recrystallisation. Optical purities were
measured by
chiral GC and chirai shift HNMR.
R product ee (%) HNMR & Mass Spectra
weight (g)
o-CH3 3.8
m-CH3 5.1
p-CH3 1.8
o-OCH3 1.7 +I- 214, 198, 183, 165, 151, 135, 105, 77,
51
m-OCH3 4.1 40 CHOH,1H,5.79s;ArH6.78-7.45,9H
OCH ,3.80s,3H
214, 197, 183, 181, 165, 152, 135, 105, 77,
51
p-OCH3 3.3 CHOH,1H,5.74s;ArH6.8-7.4,9H
OCH ,3.76s,3H
214, 197, 181, 165, 151, 135, 105, 77, 51
rn-CF3 6.2 8 CHOH,'1 H, 5.78s;ArH7.1-7.65, 9H
252, 233, 214, 195, 183, 166, 153, 105, 77,
51
Example 10
Preparation of dibutyizinc prepared from butylmagnesiumchforide and zinc
bromide.
Anhydrous zinc bromide (8.6g) was charged to a dry, stirred, 250m1 flask
filled with
nitrogen. Dried diethylether (100m1) was added and the slurry heated to reflux
until a
homogenous solution..A 2 molar ethereal solution of butylmagnesium chloride
(37.5m1)
was slowly added. The reaction mixture was refluxed for a further 3 hours.
Solvent was
distilled from the reaction mixture at ambient pressure, whilst maintaining a
constant
volume by addition of toluene until a distillate temperature of at least
108°C was reached.
The hot slurry was screened under a nitrogen atmosphere using an in-line
sintered glass
filter.
CA 02574055 2007-O1-30
79806-13D
,
23
Example 11 .
Reaction of diphenylzinc, itself prepared from phenylmagnesiumbromide and zinc
bromide, with acetaldehyde
Anhydrous zinc bromide (11.37g) was charged to a dry, stirred 250m1 flask
filled with
nitrogen. Dried diethylether (75m1) was added and the slurry heated to reflex
until a
homogenous solution. A 3moiar ethereal solution of phenylmagnesium bromide
(33.3m1)
was slowly added. Solvent was distilled from the reaction mixture at ambient
pressure,
whilst maintaining a constant volume by addition of toluene until a distillate
temperature of
110'C was reached. The hot slurry was screened under a nitrogen atmosphere
using an
1 o in-line sintered glass filter. The filtrates were received into a dry,
stirred 250m1 flask and
cooled to -5'C. D(+) diethylnorephedrine (0.64g) was added and the
acetaldehyde (2.2g)
in toluene (5ml) charged from a dropping funnel. The reaction mixture was
warmed to
ambient temperature, stirred for a total of 12 hours, then cooled on an ice
bath and
quenched by addition of dilute hydrochloric acid. The organic layer was
separated,
washed with an equal volume of water and concentrated to yield a crude product
(4.62g).
This was further purified by chromatography to give 3.868 (R)-phenylethanol of
17%ee,
measured by chiral GC. The product was identified by gas chromatographic
comparison
with authenticated material.
2 o Example 12
Reaction of o-di(1',1',1'-trifiuorotoiyl)zinc, itself prepared from o-
(1',1',1'-
trifluorotolyl)lithium and zinc bromide, with 3-nicotinaldehyde
benzotrifluoride (14.8g) and n-hexane (50m1) were charged to.a dry, stirred,
250m1 flask
filled with nitrogen and cooled to 4'C. A 1.6 molar hexane solution of n-
butyllithium
(68.75m1)(Aldrich) was charged by syringe and needle. This solution was heated
to reflex
for 18 hours, then cooled to ambient temperature. A solution of anhydrous zinc
chloride
(6.95g) in diethylether (100m1) was added and the slurry heated to 43'C.
Solvent was
distilled at ambient pressure, from the reaction until the distillate
temperature reached at
least 108'C. The hot slurry was screened under a nitrogen atmosphere using an
in-line
3o sintered glass filter. The filtrates were received into a dry, stirred
250m1 flask and cooled
to ambient temperature. D(+) diethylnorephedrine (0.64g) and 3-nicotinaldehyde
were
charged from a dropping funnel. The reaction mixture was stirred for a total
of 72 hours
then cooled on an ice bath and quenched by addition of dilute hydrochloric
acid. The
organic layer was separated, washed with an equal volume of water and
concentrated to
yield a crude product. The crude product was shown by GC/mass spec to contain
6% of
the desired product of 49% ee.
CA 02574055 2007-O1-30
79806-13D
c
24
Example 13
Reaction of diphenylzinc, itself prepared from phenylmagnesiumbromide and zinc
bromide, with p-1',1',1'-triffuoromethylbenzaldehyde using S-(-)-a,a-diphenyl-
2-
pyrollidinemethanol catalyst
Anhydrous zinc bromide (6.82g) was charged to a dry, stirred 250m1 flask
filled with
nitrogen. Dried diethyiether (75m1) was added and the slurry heated to reflux
until a
homogenous solution. A 3molar ethereal solution of phenylmagnesium bromide
(20m1)
was slowly added. Solvent was distilled from the reaction mixture at ambient
pressure,
whilst maintaining a constant volume by addition of toluene until a distillate
temperature of
t o at least 108'C was reached. The hot slurry was screened under a nitrogen
atmosphere
using an in-line sintered glass filter. The filtrates were received into a
dry, stirred 250m1
flask and cooled to 2'C. S-(-)-a,a-Biphenyl-2-pyrolfidinemethanol (0.912g)
(Aldrich) was
added and p- 1',1',1'-trifluoromethylbenzaldehyde~ (2.61 g) charged Born a
dropping funnel.
The reaction was warmed to ambient temperature, stirred for a total of 16
hours then
z 5 cooled on an ice bath and quenched by addition of dilute hydrochloric
acid. The organic
layer was separated, washed with an equal volume of water and concentrated to
yield a
crude product. Weight of crude product (3.94g). Chiral GC showed the product
to be 24%
ee. HNMR and mass spec. confirmed the identity of the product.