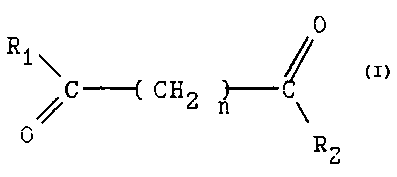Note: Descriptions are shown in the official language in which they were submitted.
CA 02574103 2007-O1-30
wu 95/s1977 . Y(:17UJY'/VO"4
NOVEL POTENT INDUCERS OF TERMINAL
DIFFERENTIATION AND METHODS OF USE THEREOF
This application is a continuation~in-part of U.S. Serial No.
07/771,760, filed October 4, 1991, now issued as U.S. Patent No.
5,369,108. The invention described herein was made in
the course of work under Grant Number CA-'S7227-O1 from
the National institutes of Health. The United States
Government has certain rights in this invention.
Background of the Invention
Throughout this application various publications are
referenced by arabic numerals within parentheses. Full
citations for these publications may be found at the end
of the .specification ,immediately preceding the claims.
The disc_~.osures of these publications in their entireties
are hereby incorporated by reference into this
application in order to more fully describe the state of
the art to which this invention pertains.
Cancer is a disorder in which a population of cells has
become, in varying degrees, unresponsive to the control
mechanisms which noimally govern proliferation and
differentiation. For many years thE~~ have been two
principal strategies for chemotherapeutic treatment of
.cancer: .a) blocking hornnone-dependent tumor cell
proliferation by interference with the production or
peripheral action of sex hormones; and b) killing cancer
cells directly by exposing them to cytotoxic substances,
which injure both neoplastic and normal cell populations.
Relatively recently, cancer therapy is also being
attempted by the induction of terminal differentiation of
the neoplastic cells (1). In cell culture models
differentiation has been reported by exposure of cells to
CA 02574103 2007-O1-30
WO 95131977 ~ ~ PCT/US95/06554
-2-
a variety of stimuli, including: cyclic AMP and retinoic
acid (2,3), aclarubicin and other anthracyclines (4).
There is abundant evidence that neoplast?~ transformation
does not necessarily destroy the potential of cancer
cells to differentiate (1,5,F). There are many examples
of tumor cells which do not respond to the normal
regulators of proliferation and appear to be blocked in
the expression of their differentiation program, and yet
can be induced to differentiate and cease replicating.
A variety of agents, including some relatively simple
polar compounds (5,7-9), derivatives of vitamin D and
retinoic acid (10-12), steroid hormones (13), growth
factors (6,14), proteases (15,16), tutor promoters
(17,18), and inhibitors of DNA or RNA synthesis (4,19-
24), can induce various transformed cell lines and
primary human tumor explants to expres ~ more
differentiated characteristics.
Early studies by the present inventors identified a
series of polar compounds that were effective inducers of
differentiation in a number of transformed cell lines
(8,9). Of these, the most effective inducer, was the
hybrid polar/apolar compound N,N~-hexamethylene
bisacetamide (HI~A) (9). The use of this polar/apolar
compound to induce murine erythroleukemia cells (MELC) to
undergo erythroid differentiation with suppression of
oncogenicity has proved a useful model to study inducer-
mediated differentiation of transformed cells (5,7-9).
~A-induced MELC terminal erythroid differentiation is
a multistep process. Upon addition of HI~A to MELC
(745A-DS19) in culture, there is a latent period of ZO to
1~ hours before commitment to terminal differentiation is
detected. Commitment is defined as the capacity of cells
to express terminal differentiation despite removal of
inducer (25). Upon continued exposure to HI~A there is
progressive recruitment of cells to differentiate. The
CA 02574103 2007-O1-30
WO 95131977 ~- PCT/OS95/06554
-3-
present inventors have reported that MELC cell lines made
resistant to relatively low levels of vincristine become
markedly more sensitive to the inducing action of HN~A
and can be induced to differentiate with little or no
latent period (26).
HI~A is capable of inducing phenotypic changes consistent
with differentiation in a broad variety of cells lines
(5). The characteristics of the drug induced effect have
been most extensively studied in the murine
erythroleukemia cell system (MELC) (5,25,27,28). MELC
induction of differentiation is both time . and
- concentration dependent. The minimum concentration
required to demonstrate an effect ~n_ grit o in most
strains is 2 to 3 mM; the minimum duration of continuous
exposure generally required to induce differentiation in~
a substantial portion (~20%) of the population without
- continuing drug exposure is about 36 hours.,
The primary target of action of HI~A is not known. There
is evidence that protein kinase C is involved in the
pathway of induces-mediated differentiation (29). The in
vitro studies provided a basis for evaluating the
potential of HI~A as a cytodifferentiation agent in the
treatment of human cancers (30). Several phase i
clinical trials with HI~A have been completed (31-36).
Clinical trials have shown that this compound can induce
therapeutic response in patients with cancer (f5,36).
However, these phasa I clinical trials also have
demonstrated that the potential efficaey of HI~A is
limited, in Bart, by dose-related toxicity which prevents
achieving optimal blood levels and by the need for
intravenous administration of large quantities of the
agent, over prolonged periods. -
Recently, the present inventors have reported a number of
compounds related to HI~3A with polar groups separated by
CA 02574103 2007-O1-30
WO 95/31977 PCT/US95/06554
-4-
apolar linkages that, on~a molar basis, are as active
(37) or 100 times more active than HI~A (38) . As a
class, however, it has been found that the symmetrical
dimers such as HI~A and related compounds are not the
best cytodifferentiating agents.
It has unexpectedly been found that the best compounds
comprise two polar end groups separated by a flexible
chain of methylene groups, wherein one or both of the
polar end groups is a large hydrophobic group.
Preferably, the polar end groups are different and only
one is a large hydrophobic group. These compounds are
unexpectedly a thousand times more active than HI~A and
ten times more active than HI~A related co3npounds.
This new class of compounds of the present invention may
be useful for selectively inducing terminal
differentiation of neoplastic cells and therefore aid in
treatment of tumors in patients.
CA 02574103 2007-O1-30
~WO 95131977 PCT/US95/06554
-5-
Summary of the Invention
The present invention provides the compound having the
structure:
O
R1 ~
C ---( C H 2 n3~-- \
herein each of RI and RZ are independently the same as or
different from each other; when R1 and R2 are the same,
each is a substituted or unsubstituted arylamino,
cycloalkylamino, pyridineamino, piperidiri~, 9-purine-6-
amine, or thiazoleamino group; when R1 and R2 are
different, R1 - R3-N-R4, wherein each of R3 and R4 are
independently the same as or different from each other
and are a hydrogen atom, a hydroxyl group, a substituted
or unsubstituted, branched or unbranched alkyl, alkenyl,
cycloalkyl, aryl, alkyloxy, aryloxy, arylalkyloxy, or
pyridine group, or R3 and R4 bond together . to form a
piperidine group and R2 is a hydroxylamino, hydroxyl,
amino, alkylamino, dialkylamino or alkyloxy group; and
n is an integer from about 4 to about 8.
The present invention also provides the compound above
having the structure:
~R4
G
R3 N\
C-'t GH
2 n \
R2
wherein each of R3 and R4 are independently the same as or
different from each other and are a hydrogen atom, a
hydroxyl group, a substituted ~or unsubstituted, branched
CA 02574103 2007-O1-30
'WO 95/31977 PCTIUS95106554
-6-
or unbranched alkyl, alkenyl, cycloalkyl, aryl, alkyloxy,
aryloxy, arylalkyloxy, or pyridine group, or R3 and R4
bond together to form a piperidine group; RZ is a
hydroxylamino, hydroxyl, amino, alkylamino, dialkylamino
or alkyloxy group; and n is an integer from about 4 to
about 8.
The present invention also provides the compound above
having the structure:
R\
C ----( G H ~
2 n \
~ R w
wherein R is a substituted or unsubstituted arylamino,
cycloalkylamino, pyridineamino, piperidino, 9-purine-6-
amine, or thiazoleamino group; and n is an integer from
about 4 ~o about e.
The present invention also provides the compound having
the structure:
0
G-'-( Cg ~--C N C CH
Y
R
wherein each of X and Y are independently the same as or
different from each other and are a hydroxyl, amino or
hydroxylamino group, a substituted or unsubstituted
alkyloxy, alkylamino, dialkylamino, arylamino,
alkylarylamino, alkyloxyamino, aryloxyamino,
alkyloxyalkylainino, or aryloxyalkylamino group; R is a
hydrogen atom, a hydroxyl group, a substituted or
unsubstituted alkyl, aryl, alkyloxy, or aryloxy group;
and each of m and n are independently the same as or
CA 02574103 2007-O1-30
WO 95/31977 .. ' PGT/US95/06554
different from each other and are each an integer from
about 0 to about 8.
The present invention further provides the compound
having the structure:
;,
II II '
C ( CH2 -~ 0- ~ (0g2 ~ i -C- ( CHI )o C
% ~Y
X
8i R2
wherein each of X and Y are independently the same as or
.. different from each other and are a hydroxyl,. amino or
hydroxylamino group, a substituted or unsubstituted
alkyloxy, alkylamino, dialkylamino, ~ arylamino,
alkylarylamino, alkyloxyamino, aryloxyamino;
alkyloxyalkylamino, or aryloxyalkylamino group; each of
R, and R2 are independently the same as or different from
each other and are a hydrogen atom, a hydroxyl group, a
substituted or unsubstituted alkyl, aryl, alkyloxy, or
aryloxy group; and each of m, n, and o are independently
the same as or different from each other and are each an
integer from about 0 to about 8.
The present invention still further provides the compound
having the structure:
~~ II II ii
X,c ccaz ~, i c ~c- i -tca2;~c~Y
R1 R2
wherein each of X and Y are independently the same as or
different from each other and are a hydroxyl, amino or
hydroxylamino .group; a substituted or unsubstituted
alkyloxy, alkylamino, dialkylamino, arylamino,
alkylarylamino, alkyloxyamino, aryloxyamino,
CA 02574103 2007-O1-30
WO 95131977 PCT/I1S95I06554
_g_
alkyloxyalkylamino, or aryloxyalkylamino group; each of
R, and Rz are independently the same as or different from
each other and are a hydrogen atom, a hydroxyl group, a
substituted or unsubstituted alkyl, aryl, alkyloxy, or
aryloxy group; and each of m and n are independently the
same as or different from each other and are each an
integer from about 0 to about 8.
The present invention also provides the compound having
the structure:
°w ~~ II II
c- c c$2 ~-c-~rx-c ~c-NH-c- t cHZ ~-c
~Y
wherein each of X and Y are independently the same as or
different from each other and are a hydroxyl, amino or
hydroxylamino group, a substituted or unsubstituted
alkyloxy, alkylamino, dialkylamino, arylamino,
alkylarylamino, alkyloxyamino, aryloxyamino,
alkyloxyalkylamino, or aryloxyalkylamino group; and each
of m and n are independently the same as or different
from each other and are each an integer~from about 0 to
about 8.
The present invention also provides the compound having
the structure:
0
//
O~C.-- (CH C-N1 ~ ~ Nz C (CH C
x~ 2~ ~~ (~ Z~ ~Y
0 0
wherein each of X and Y are independently the same as or
different from each other and are a hydroxyl, amino or
hydroxylamino group, a substituted or unsubstituted
alkyloxy, alkylamino, dialkylamir~o, arylamino,
alkylarylamino, alkyloxyamino, aryloxyamino,
CA 02574103 2007-O1-30
WO 95131977 PGT/U593/06554 .
_g_
alkyloxyalkylamino, or aryloxyalkylamino group; each of
R, and R: are independently the same as or different from
each ot=r and are a hydrogen atom, a hydroxyl group, a
substituted or unsubstituted alkyl, aryl, alkyloxy, or
aryloxy group; and each of m and n are independently the
same as or different from each other and are each an
integer from about 0 to about 8.
The present. invention further provides the compound
having the structure:
CH3 . ~g~
I I /j
c c ~cx" ~
g/ I ~ I Y
H , h
wherein each of X and Y are independently the same as or
dif f erent from each other and are a- hydroxyl , amino or
hydroxylamino group, a substituted or unsubstituted
alkyloxy, alkylamino, dialkylamino, arylamino,
2.0 alkylarylamino, alkyloxyamino, aryloxyamino,
alkyloxyalkylamino, or aryloxyalkylamirio group; and n is
an integer from about 0 to about 8.
The present invention still further provides the compound
having the structure.:
\\ ( s /%
/C ~CHZ ~ C (CH2 k1
I Y
R2
wherein each of X and Y are independently the same as or
different from each other and are a hydroxyl, amino or
hydroxylamino group, a substituted or unsubstituted
alkyloxy, alkylamino, dialkylamino, arylamino,
alkylarylamino, alkyloxyamino; aryloxyamino,
alkyloxyalkylamino, or aryloxyalkylamino group; each of
CA 02574103 2007-O1-30
WO 95131977 PCT/US95/06554
-10-
R, and R~ are independently the same as c~r different from
each other and are a hydrogen atom, a hydroxyl group, a
substituted or unsubstituted alkyl, aryl, alkyloxy,
aryloxy, carbonylhydroxylamino, or fluoro group; and
each of m and n are independently the same as or
different from each other and are each an integer from
about 0 to about 8.
The present invention also provides the compound having
the structure:
//
C C
J
R1 R2
wherein each of R~ and R2 are independently the same as or
different from each other and are a hydroxyl, alkylbxy,
amino, hydroxylamino, alkylamino, dialkylamino,
arylamino, alkylarylamino, alkyloxyamino, aryloxyamino,
alkyloxyalkylamino, or aryloxyalkylamino group.
The present invention also provides the compound having
the structure:
R 1 ~~
~C GH CH CH . CH C~
. R2
wherein each of R, and RZ are independently the same as or
different from each other and are a hydroxyl, alkyloxy,
amino, hydroxylamino, alkylamino, dialkylamino,
arylamino, alkylarylamino, alkyloxyamino, aryloxyamino,
alkyloxyalkylamino, or aryloxyalkylamino group.
The present invention further provides the compound
CA 02574103 2007-O1-30
WO 95!31977 -~ ~ PCT/LIS95I06554 ,
-11-
having the structure:
0
R1~
C ~ C~: CH C \
. ~ . R2
wherein each of R,~and R2 are independently the same as or
different from each other and are a hydroxyl, alkyloxy,
amino, hydroxylamino, alkylamino, dialkylamino,
arylamino, alkylarylamino, alkyloxyamino, aryloxyamino,
alkyloxyalkylamino, or aryloxyalkylamino group.
The present invention further provides the compound
having the structure: ~
0
_, ~ C~ R2
Rs _ C \
wherein each of R, a'nd RZ are independently the same as or
different from each other and are a hydroxyl, alkyloxy,
amino, hydroxylamino, alkylamino, dialkylamino;
arylamino, alkylarylami~na, alkyloxyamino, aryloxyamino,
alkyloxyalkylamino, or aryloxyalkylamino group.
The present invention further provides the compound
having the structure:
0
3=CH-C-R2
0
~~
3s . Rs-C
CA 02574103 2007-O1-30
WO 95!31977 PCT/US95/06554
-12-
wherein each of R, and R2 are independently the same as or
different from each other and are a hydroxyl, alkyloxy,
amino, hydroxylamino, alkylamino, dialkylamino,
arylamino, alkylarylamino, alkyloxyamino, aryloxyamino,
alkyloxyalkylamino, or aryloxyalkylamino group.
The present invention further provides the compound
having the structure:
0
CH=CH-C-RZ
0
II
R1 C-CH=CH
wherein each of R1 and Rz are independently the same as or
different from each other and are a hydroxyl, alkyloxy,
amino, hydroxylamino, alkylamino, dialkylamino,
arylamino, alkylarylamino, alkyloxyamino, aryloxyamino,
alkyloxyalkylamino, or aryloxyalkylamino group.
The present invention alSO provides the pharmaceutically
acceptable salts of any of the compounds defined above.
The present invention further provides a compound having
the structure:
0 0
R-C-N- (CHZ) n -C-NH-OH
I
H
wherein R is a substituted or unsubstituted phenyl,
piperidine or thiazole. group and n is an integer from
about 4 to about 8 or a pharmaceutically acceptable salt
thereof .
CA 02574103 2007-O1-30
WO 95131977 . ~ PCT/L1S95/06554
-13-
The present invention also provides a compound having the
structure:
O O
R-C-N- (CHy) n -C-NH-OH
H
wherein R is a substituted or unsubstitued 2-pyridine, 3-
pyridine, or 4-pyridine and n is an integer from about 4
to about 8 or a pharmaceutically acceptable salt thereof.
The present invention further provides a compound having
the structure:
0 0
R_~_C_N_ (CHZ)n -C-1~I~.pH
H
3
wherein R is a substituted or unsubstituted phenyl,
pyridine, piperidine or thiazole group and n is an
. integer from about 4 to about 8 or a pharmaceutically
acceptable salt thereof.
In addition, the present invention provides a method of
selectively inducing terminal differentiation of
neoplastic cells and thereby inhibiting proliferation of
such cells which comprises contacting the cells under
suitable conditions with an effective .amount~of. any of
the compounds above, effective to sE~ectively induce
terminal differentiation.
The present invention also provides a method of treating
a patient having a tumor characterized by proliferation
of neoplastic cells which comprises administering to the
patient an effective amount of any of the compounds
above, effective to selectively induce terminal
differentiation of such neoplastic cells and thereby
inhibit their proliferation.
CA 02574103 2007-O1-30
WO 95/31977 PCTlUS95/06554
-14-
The present invention also provides a pharmaceutical
composition comprising a therapeutically acceptable
amount of any of . the compounds above, or oharn~aceutically
acceptable salts thereof, and a pharmaceutically
acceptable carrier.
Lastly, the present invention provides the
pharmaceutical composition defined above, alone or in
combination with, an antitumor agent, in sustained rel-ease
form.
CA 02574103 2007-O1-30
WO 95/31977 PGT/US95/06554 .
-15-
Detailed Description of the Invention
The present invention provides the compound having the
structure:
. //C (CH2.~C~
R
IO wherein each of R, and RZ are independently the same as or
different from each other; when R, and RZ are the same,
each is a substituted or unsubstituted arylamino,
cycloalkyl-amino, pyridineamino, piperidino, 9-purine-6-
amine, or thiazoleamino group; when R, and R, are
different, R, - R~-N-R4, wherein each of R3 and R4 are
independently the same as or different from each other
and are a hydrogen.atom, a hydroxyl group, a substituted
. or unsubstituted, branched or unbranched alkyl, alkenyl,
cycloalkyl, aryl, alkyloxy, aryloxy, arylalkyloxy, or
pyridino group, or R3 and R4 bond together to form a
piperidine group .and RZ is a hydroxylamino, hydroxyl,
amino, alkylamino, dialkylamino or alkyloxy group; and
n is an integer from about 4 to about 8.
The present. invention also provides the compound above
having the structure:
g4
R3 N~ 0
~l
~0 ~CHZ~C~
0 R2
wherein each of R3 and R4 are independently the same as or
different from each other and are a hydrogen atom,-a
_ hydroxyl group, a substituted or unsubstituted, branched
or unbranched alkyl, alkenyl, cycloalkyl, aryl, alkyloxy;
aryloxy, arylalkyloxy, or pyridine group, or R3 and R4
CA 02574103 2007-O1-30
WO 95/31977 PCT/US95/06554
-16-
bond together to form a piperidine group; R= is a
hydroxylamino, hydroxyl, amino, alkylamino, dialkylamino
or alkyloxy group; and n is an integer from about 4 to
about 8.
In the preferred embodiment of the compound above, R~ is
a hydroxylamino, hydroxyl, amino, methylamino,
dimethylamino, or methyoxy group and n is 6. Most
preferably, R4 is a hydrogen atom and R3 is a substituted
or unsubstituted phenyl group.
The phenyl group may be substituted with a methyl, cyano,
vitro, trifluoromethyl, amino, aminocarbonyl,
methylcyano, chloro, fluoro, bromo, iodo,~i2,3-difluoro,
2,4-difluoro, 2,5-difluoro, 3,4-difluoro, 3,5-difluoro,
2,6-difluoro, 1,2,3-trifluoro, 2,3,6-trifluoro, 2,4,6-
trifluoro, 3,4,5-trifluoro, 2,3,5,6-tetrafluoro,
2,3,4,5,6-pentafluoro, azido, hexyl, t-butyl, phenyl,
carboxyl, hydroxyl, methyoxy, benzyloxy, phenylaminooxy,
phenylmethoxy, phenylamino-carbonyl, methyoxycarbonyl,
methylaminocarbonyl, dimethylamino,
dimethylaminocarbonyl, or hydroxylamino-carbonyl group.
In other preferred embodiments of the compound above, R4
is a hydrogen atom and R3 is a cyclohexyl group; R, is a
hydrogen atom and R3 is a methyoxy group; R3 and R4 each
bond together to form a piperidine .group; R4 is a
hydrogen atom and R3 is a hydroxyl group; R4 is a
hydrogen atom and R3 is a benzyloxy group; R4 is a
hydrogen atom and R3 is a b-pyridine group; R4 is a
hydrogen atom and R3 is a i3-pyridine group; R4 is a
hydrogen atom and R3 is a a-pyridine group; R3 and R4 are
both methyl groups; or R, is a methyl group and R3 is a
phenyl group.
CA 02574103 2007-O1-30
WO 95131977 ~ PCT/US95/06554
-17-
The present invention also provides the compound having
the structure:
0
\
0 B
wherein R is a substituted or unsubstituted arylamino,
cycloalkylamino, pyridineamino, piperidino, 9-purine-6-
amine, or thiazoleamino group; and n is an integer from
about 4 to about 8.
3
In the preferred embodiment of the compound above, R is
a substituted-or unsubstituted phenylamino group. The
phenylamino group may be substituted with a cyano,
. methylcyano, nitro, carboxyl, aminocarbonyl,
methylaminocarbonyl, dimethylaminocarbonyl,
trifluoromethyl, hydroxylamin.ocarbonyl, N-
hydroxylaminocarbonyl, methoxycarbonyl, ctrloro, fluoro,
methyl, methoxy, 2,3-difluoro, 2,3-difluoro, 2,4-
difluoro, 2,5-difluoro, 2,6~-difluoro, 3,~-difluoro, 2,6-
difluoro, 2,3,6-trifluoro, 1,2,3-trifluoro, 3,4,5-
trifluoro, 2,3,4,5-tetrafluoro, or 2,3,4;5,6-pentafluoro
group.
.In another embodiment of the compound.above, R is a
cyclohexylamino group.
The present invention also provides the compound having
the structure:
0 O 0 0
11 II II
.35 . C L CH ~--C N C.--( CH
X~ Z m ~ (
Y
g
CA 02574103 2007-O1-30
WO 95/31977 ' ~ PCT/L1S95/06554
-18-
wherein each of X and Y are independently the same as or
different from each other and are a hydroxyl, amino or
hydroxylamino group, a substituted or unsubstituted
alkyloxy, alkylamino, dialkylamino, arylamino,
alkylarylamino, alkyloxyamino, aryloxyam.ino,
alkyloxyalkylamino, or aryloxyalkylamino group; R is a
hydrogen atom, a .hydroxyl group, a substituted or
unsubsti.tuted alkyl, aryl, alkyloxy, or aryloxy group;
and each of m and n are independently the same as or
different from each other and are each an integer from
about 0 to about 8.
In the preferred embodiment of the compound above, each
of X, Y, and R is a hydroxyl group and each~of m and n is
5.
The present invention also provides the compound having
the structure:
o\ II ~~ ____ I~
~c tcHZ m c- l ~c$2 ~ l -c t cg2 o c
R1. g2
wherein each.of X and Y are independently the same as or
different from each other and are a hydroxyl, amino or
hydroxylamino group, a substituted or unsubstituted
alkyloxy, alkylamino, dialkylamino, arylamino,
aikylarylamino, alkyloxyamino, aryloxyamino,
alkyloxyalkylamino, or aryloxyalkylamino group; each of
R~ and RZ are independently the same as or different from
each other and are a hydrogen atom, a.hydroxyl group, a
substituted or unsubstituted alkyl, aryl, alkyloxy, or
aryloxy group; and each oi: m, n, and o are independently
the same as or different from each other and are each an
integer from about 0 to about 8.
CA 02574103 2007-O1-30
WO 95/31977 ~ PCT/LTS95/06554
-19-
10
In the preferred embodiment of the compound above, each
of X an~i Y is a hydroxyl group and each of R, and RZ is a
methyl group. Most preferably, each of n and o is 6, and
m is 2.
The present invention also provides the compound having
the structure
oy II II to
(CH2 ) N-C y-C N L CH
.. i m .. I ~ I 2~ \
g ~ Y
Hi R2
3
wherein each of X and Y are independently the same as or
different from each other and are a hydroxyl, amino or
hydroxylamino group, a substituted or unsubstituted
alkyloxy, alkylamino, dialkylamino, arylamino,
alkylarylamino, alkyloxyamino, aryloxyamino,
alkyloxyalkylamino, or aryloxyalkylamino group; each of
R, and Rz are independently the same as or different from
each other and are a hydrogen atom, a hydroxyl group, a
substituted or unsubstituted alkyl, aryl, alkyloxy, or
aryloxy group; and each of m and n are independently the
same as or different from each other and are each an
integer from about O to about 8.
The present invention also provides the compound having
the structure:
II II (I fl ~I
~c- ccH2 ~m c-xH-c c-xa- c-tcaz~-c\
x
wherein each of X and Y are independently the same as or
CA 02574103 2007-O1-30
WO 95/31977 PCT/US95/06554
-20-
different from each other and are a hydroxyl, amino or
hydroxylamino group, a substituted or unsubstituted
alkyloxy, alkylamino, dialkylamino, arylamino,
alkylarylamino, alkyloxyamino, aryloxyamino,
alkyloxyalkylamino, or aryloxyalkylamino group; and each
of m and n are independently the same as or different
from each other and are each an integer from about 0 to
about 8.
In the preferred embodiment of the compound above, each
of X and~Y is a hydroxyl group and each of m and n is 5.
The present invention also provides the compound having
the structure:
I1 IZ. II
C (CHZ ) C-N N-C (C$2 ) C
~~ n II II
0 0
wherein each of X and Y are independently the same as or
different from each other and are a hydroxyl, amino or
hydroxylamino group, a substituted or unsubstituted
_ 2S alkyloxy, alkylamino, dialkylamino, arylamino,
alkylarylamino, alkyloxyami.no, aryloxyamino,
alkyloxyalkylamino, or aryloxyalkylannino group; each of
R1 and R., are independently the same as or different f rom
each other and are a hydrogen atom, a hydroxyl group, a
substituted or unsubstituted alkyl, aryl, .alkyloxy, or
aryloxy group; and each of m and n are independently the
same as or different from each other and are each an
integer from about 0 to about 8.
CA 02574103 2007-O1-30
WO 95/31977 PCT/US95/06554 .
-21-
The present invention also provides the compound having
the structure:
0 _i H3 ~ g3
C C tCH2 )n
X/ ~
H H
wherein each of X and Y are independently the same as or
different from each other and are a hydroxyl, amino or
~hydroxylamino group, a substituted or unsubstituted
alkyloxy, alkylamino, dialkylamino, arylamino,
alkylarylamino, alkyl.oxyamino, aryloxyamin8,
alkyloxyalkylamino, or aryloxyalkylamino group; and n is
an integer from about 0 to about 8.
In the preferred embodiment of the compound above,, each
of X and Y is a dimethylamino group and n is 4 or 5.
The present invention also provides the compound having
the structure:
0 ' R1 ~ 0
. ~ I i
. ./0 ~Cg2'~ 0 .(CH2 ~C\
I y
2
wherein each of X and Y are independent3y the same as or
different from each other and are a hydroxyl, amino or
hydroxylamino group, a substituted or unsubstituted
alkyloxy, alkylamino, dialkylamino, arylamino,
' alkylarylamino, alkyloxyamino, aryloxyamino,
alkyloxyalkylamino, or aryloxyalkylamino group; each of
R, and RZ are independently the same as or different from
each other and are a hydrogen atom, a hydroxyl group, a
substituted or unsubstituted alkyl, aryl, alkyloxy,
CA 02574103 2007-O1-30
i
WO 95131977 PCTIUS95/06554
-22-
aryloxy, carbonylhydroxylamino, or fluoro group; and
each of m and n are independently the same as or
different from each other and are each an integer from
about 0 to about 8.
In the preferred embodiment of the compound above, each
of X and Y is a hydroxylamino group, R1 is a methyl group,
R2 is a hydrogen atom, and each of m and n is 2. In
another preferred embodiment, each of X and Y is a
hydroxylamino group, R, is a carbonylhydroxylamino group,
R, is a hydrogen atom, and each of m and n is 5. In a
further preferred embodiment, each of X and Y is a
hydroxylamino group, each of R, and Rz is a fluoro group,
and each of m and n is 2. ~
The present invention also provides the compound having!
the Structure:
// _
C C
~2 . R2
wherein each of R, and R2 are independently the same as or
different from each other and are a hydroxyl, alkyloxy,
amino, hydroxylamino, alkylamino,. dialkylamino,
arylarnim,~ alkylarylamino, alkyloxyamino, arylo3cyamino,
alkyloxyalkylamino, or aryloxyalkylamino group.
Preferably, R, is a phenylamino group and RZ is a
hydroxylamino group.
CA 02574103 2007-O1-30
WO 95!31977 PGT/US95106554
-23-
The present invention also provides the compound having
the structure:
.R 1 \. . %0 .
C CH CH CH CH C
//
0 R2
wherein each of R, and R, are independently the same as or
different from each other and are a hydroxyl, alkyloxy,
amino, hydroxylamino, alkylamino, dialkylamino,
- arylamino, alkylarylamino, alkyloxyamino, aryloxyamino,
alkyloxyalkylamino, or aryloxyalkylamino group.
Preferably, R1 is phenylamino group and RZ is
hydroxylamino group:
The present invention also provides the compound having
the structure:
R1\ ' /0
. /~ CH CH C~
0 R2
wherein each of R, and Rz are independently the same as or
different from each other and are a hydroxyl, alkyloxy,
amino, hydroxylamino, alkylamino, dialkylamino,
arylamino, alkylarylamino, alkyloxyamino, aryloxyamino;
alkyloxyalkylamino, or aryloxyalkylamino group.
In the preferred embodiment, either R, or RZ is ~ a
hydroxylamino group.
The present invention also provides the compound having
CA 02574103 2007-O1-30
WO 95131977 PGT/US95106554
-24-
the structure:
0
~2
R1-C . ,
wherein each of R, and R2 are independently the same as or
different from each other and are a hydroxyl, alkyloxy,
amino, hydroxylamino, alkylamino, dialkylamino,
arylamino, alkylarylamino, alkyloxyamino, aryloxyamino,
alkyloxyalkylamino, or aryloxyalkylamino g''roup.
Tn a preferred embodiment; the compound above has the
structure:
0
i1
p C-NH-OH
..
HO-NH-C
The present invention also provides a compound having the
structure:
~i= C H- C - RZ
0
R1- C
CA 02574103 2007-O1-30
W0 95131977 . PGT/US95106554
-25-
wherein each of R, and R= are independently the same as or
different from each other and are a hydroxyl, alkyloxy,
amino, hydroxylamino, alkylamino, dialkylamino,
arylamino, alkylarylamino, alkyloxyamino, aryloxyamino;
alkyloxyalkylamino~, or aryloxyalkylamino group.
In a preferred embodiment, the compound above has the
structure:
~~
~ CH=CH-C-NH-OH
HO-NH-C
The present invention also provides a compound having the
structure:
~~
CH= CH" C"- R2
Ii _
~1 C- CH= C H
wherein each of R, and RZ are independently the same as or
di f f event ~f rom each other and are a hydroxyl , a~kyloxy ,
amino, hydroxylamino, alkylamino, dialkylamino,
arylamino, alkylarylamino, alkyloxyamino, aryloxyamino,
alkyloxyalkylamino, or aryloxyalkylamino group.
In the preferred embodiment , the compound def fined above
has the structure:
CA 02574103 2007-O1-30
WO 95/31977 PC'T/US95/06554
-26-
~G
CH=CH-~~-NH-GH
0 ;
II
HG-NH-C-CH='Cii ~/ ~)
-_%
The present invention also provides the pharmaceutically
acceptable salts of any of the compounds defined above.
The present invention further provides a compound having
the structure:
0 O
Il II
R-C-N- (CH2) o -C-NFi-OH
H
wherein R is a substituted or unsubstituted phenyl,
piperidine or ehiazole group and n is an integer from
about 4 to about 8 or a phazmaceutically acceptable salt
thereof.
In .a pref erred embodiment of the .compound def fined above
R is a substituted phenyl group. In a more preferred
embodiment the phenyl group is substituted with a methyl,
cyano, nitro, thio, trifluoromethyl, amino,
aminocarbonyl, methylcyano, chloro, fluoro, bromo, iodo,
2,3-difluoro, 2,4-difluoro, 2,5-difluoro, 3,4-difluoro,
3,5-difluoro, 2,6-difluoro, 1,2,3-trifluoro, 2,3;6-
trifluoro, 2,4,6-trifluoro, 3,4,5-trifluoro, 2,3,5,6-
tetrafluoro, 2,3,4,5,6-pentafluoro, azido, hexyl, t-
butyl., phenyl, carboxyl, hydroxyl, methyoxy, phenyloxy,
benzyloxy, phenylaminooxy, phenylaminocarbonyl,
methyoxycarbonyl, methylaminocarbonyl, dimethylamino,
dimethylamino-carbonyl, or hydroxylaminocarbonyl group.
CA 02574103 2007-O1-30
WO 95/31977 PGT/US95/06554
-27-
The present invention also provides a compound having the
structure:
O 0
R-C-N- (CHZ) p -C-NH-OH
H
wherein R is a substituted or unsubstitued 2-pyridine, 3-
pyridine, or 4-pyridine and n is an integer from about 4
to about 8 or a pharmaceutically acceptable salt thereof .
The present invention further provides a compound having
the structure:
0 0
~~ ~~
R=NH-C-N-(CHZ)Q -C-NH-OH
H
wherein R is a substituted or unsubstituted phenyl,
pyridine, piperidine or thi.azole group and n is an
integer from about 4 to about 8 or a pharmaceutically
acceptable salt thereof.
In a preferred embodiment of the compound defined above,
R is a substituted phenyl group. In a more preferred
embodiment, the phenyl group is substituted with a
methyl, cyano, vitro, thio, trifluoromethyl, amino,
aminocarbonyl, methylcyano, chloro, fluoro, bromo,.iodo,
2,3-difluoro, 2,4-difluoro, 2,5-difluoro, 3,4-difluoro,
3,5-difluoro, 2,6-difluoro, 1,2;3-trifluoro, 2,3,6-
trifluoro, 2,4,6-trifluoro, 3,4,5-trifluoro, 2,3,5,6-
tetrafluoro; 2,3,4,5,6-pentafluoro, azido, hexyl, t-
butyl, phenyl, carboxyl, hydroxyl, methyoxy, phenyloxy,
benzyloxy, phenylam'inooxy, phenylaminocarbonyl_,
methyoxycarbonyl, methylarninocarbonyl, dimethylamino,
dimethylamino-carbonyl, or hydroxylaminocarbonyl group.
In a further preferred embodiment the compound defined
CA 02574103 2007-O1-30
WO 95/319?? PCTlUS95/06554
-28-
above has the structure:
N-H 0
i-NH- (CH,) 5 -C-NH-OH
O
or a pharmaceutically acceptable salt thereof.
In a further preferred embodiment the compound defined
above has the structure:
_ ~ ~ -N-H 0
Cl '- \4 -NH- (CHZ)s _~_~_Og
y
or a pharmaceutically acceptable salt thereof.
The present invention further provides a method of
selectively inducing terminal differentiation of
neoplastic cells and thereby inhibiting proliferation of
such cells which comprises contacting~the cells under
suitable conditions with an effective amount of any of
the compounds above, effective to selectively induce
terminal differentiation.
The contacting must be performed continuously for a
prolonged period of time, i.e. for at least 48 hours,
preferably~for about 4-5 days or longer.
The method may be practiced i~ vivo or ~ vi ro. If the
method is practiced 'fin vitro, contacting may be effected
by incubating the cells with the compound. The
concentration of the compound in contact with the cells
should be from about T EcM to about 25 mM, preferably from
4 ~.cM to about 5 mM. The concentration, depends upon the
individual compound and the state of the neoplastic
cells.
CA 02574103 2007-O1-30
a
WO 95!31977 FCT/US95l06554
-29-
The method may also comprise initially treating the veils
with an antitumor agent so as to~render them resistant to
an antitumor agent and subsequently contacting the
resulting resistant cells under suitable conditions with
an effective amount of any of the compounds above,
effective to selectively induce terminal differentiation
of such cells. .
The antitumor agent may be one of numerous chemotherapy
agents such as.an alkylating agent, an antimetaboiite, a
hormonal agent, an antibiotic, colchicine, a vinca
alkaloid, L-asparaginase, procarbazine, hydroxyurea,
mitotane, nitrosoureas or an imidazole carboxamide.
Suitable agents are 'those agents which promote
depolarization of tubulin. preferably the antitumor
agent is colchicine or a vinca alkaloid; especially
preferred are vinblastine and vincristine. In
embodiments where the antitumor agent is vincristine, the
cells preferably are treated so that they are resistant
to vincristine at a concentration of about 5 mg/ml. The
treating of the cells to render them resistant to an
antitumor agent may be effected by Contacting the cells
with the agent for a period of at least 3-5 days. The
contacting of the resulting cells with any of the
compounds above is performed as described previously.
The present invention also provides a method of treating
a patient having a tumor characterized by proliferation
of neoplastic cells which comprises administering to the
patient an effective amount of any of the compounds
above, or pharmaceutically acceptable salts thereof,
effective to selectively induce terminal differentiation
of such neoplastic cells and thereby inhibit their
proliferation.
The method of the present invention is intended for the
treatment of human patients with tumors. However, it is
CA 02574103 2007-O1-30
WO 95/31977 ~ ~ PCT/US95/06554
-30-
also likely that the method would be effective in the
treatment of tumors in other mammals. '.'he term t~.:mor is
intended to include any cancer :.aused by the
proliferation of neoplastic cells, such as lung cancer,
acute lymphoid myeloma, bladder melanoma, renal
carcinoma, breast carcinoma, or colorectal carcinoma.
The administration of the compound to the patient may be
effected orally or parenterally. To date, administration
intravenously has proven to be effective. The
administration of the compound must be performed
continuously for a prolonged period of time, such as for
at least 3 days and preferably more than 5 days. In the
most preferred embodiments, the administration is
effected continuously for at least 10~ days and is
repeated at intervals wherein at each interval the
administration is continuously effected for a,t .least 10
days. For example, the administration may be effected at
intervals as short as 5-10 days, up to about 25-35 days
and continuously for at least 10 days during each such
interval. The optimal interval period will vary
depending on the type of patient and tumor. For example,
in the incidence of acute leukemia, the so called
myelodysplastic syndrome, continuous infusion would seem
to be indicated so long as the patient tolerated the drug
without toxicity and there was a positive response.
The amount of the compound administered to the patient is
less than an amount which would cause toxicity in the
patient. In the certain embodiments, the amount of the
compound which is administered to the patient is less
than the amount Which causes a concentration of the
compound in the patient's plasma to egual or exceed the
toxic level of the compound. Preferably, the
concentration of the compound in the patient's plasma is
maintained at about 1.0 mM. It has been found with HI~A
that administration of the compound in an amount from
about 5 gm/m2/day to about 30 gm/mi/day, particularly
CA 02574103 2007-O1-30
WO 95/31977 ~ PCT/US95106554
-3i-
about 20 gm/m=/day, is effective without producing
toxicity in the patient. The optimal amount of the
compound which should be administered to the patient i:.
the..practice of the present invention will depend on the
particular compound used and the type of cancer being
treated.
This invention, in addition to the above listed
compounds, is intended to encompass the use of homologs
and analogs of such compounds. In this context, homologs
are molecules having substantial structural similarities
to the above-described compounds and analogs are
molecules having substantial biological similarities
regardless of structural similarities.
The method may also comprise initially administering to
the patient an amount of an antitumor agent to render the
cells resistant to an antitumor agent and subsequently
administering to the patient an effective amount of any
of the compounds above, or pharmaceutically acceptable
salts thereof,~effective to selectively induce terminal
differentiation of such neoplastic cells and thereby
inhibit their proliferation.
The antitumor agent may be one of numerous chemotherapy
agents such as an alkylating agent, an antimetabolite, a
.. hormonal agent, an antibiotic, colchicine, a vinca
alkaloid, L-asparaginase, procarbazine, hydroxyurea,
"- mitotane, ni:trosoureas or an imidazole carboxamide.
Suitable agents are those agents which promote
depolarization of tubulin. Preferably the antitumor
agent is colchicine or a vinca alkaloid; especially
preferred are vinblastine and vincristine. In
embodiments where the antitumor agent is vincristine, an
35~~ amount is administered to render the cells are resistant
to vincristine at a concentration of about 'S mg/ml. The
administration of the agent is performed essentially as
CA 02574103 2007-O1-30
WO 95131977 PCT/US95/06554
-32-
described above for the administration of any of the
compounds. Preferably, the administration of the agent
is for a period of at least 3-5 days. The admiristraticn
of any of the compounds above is performed as descr ibed
previously.
The present invention also provides a pharmaceutical
composition comprising a therapeutically acceptable
amount of~any of the compounds above, or pharmaceutically
3.0 acceptable salts thereof, and a pharmaceutically
acceptable carrier, such as sterile pyrogen-free water.
preferably, the therapeutically acceptable amount is an
amount effective to selectively induce terminal
differentiation of suitable neoplastic ells and less
than an amount which causes toxicity in a patient.
The present invention provides the pharmaceutical
composition above in combination with an antitumor agent.
The antitumor agent may be any of the agents previously
described.
Lastly, the present invention provides the pharmaceutical
composition above, alone or in combination with an
antitumor agent, in sustained release form. By
"sustained release form" applicants mean incorporation of
the pharmaceutical compositions in a pharmaceutically
acceptable formulation which provides for the sustained
release of a therapeutically effective amount of the
compounds of this invention over a period of time
necessary to derive the intended therapeutic effect.
Sustained release formulations of pharmaceutical
compositions allow for less frequent administration of
the compound and provide for administration of the
pharmaceutical composition at or near the target area in
a subject's system. Sustained release formulations and
methods of incorporating pharmaceutical compositions
therein are well known to those of ordinary skill in the
CA 02574103 2007-O1-30
WO 95131977 PCT/US95/06554
-33-
art. Examples include, but are not limited to, suc:~
formulations as incorporation into ion exchange resins
cU.S. Patent No. 5,296,228 to Chang et al.), xanthan gums
(U. S. Patent No. 5,292,534 to Valentine at al.;,
mic=ospheres (U.S. Patent No. 5,288,502 to McGinity et
al.)~ hydrogels (U.S~. Patent No. 5;266,325 to Kuzma et
al.) and solid forms such as wax-like or fat-like
hydrophobic substances containing water insoluble
polymers'(U.S. patent No. 5,270,055 to Moest). Methods
of administering compounds for sustained-release are also
known in the art and include, but are not limited to,
surgical implantation of microencapsulated pharmaceutical
compounds near the intended target site iU.S. Patent No.
- 5,290,271 to Jernberg) and incorporation of compound into
transdertnal patches (U.S. Patent No. 5,298,256 to
Flockhart et sal. and U.S. Patent No. 5,290,561 to
Farhadieh et al.). The text of the above cited patents
and the references disclosed therein are hereby
encorporated by reference in their entirety into this
disclosure.
The invention is. illustrated in the Experimental Details
section which follows. This section is set Earth to aid
in an understandirig of the invention but is not intended
0, and should not be construed to, limit in any way the
' invention as aet. forth in the claims which follow
thereafter.
CA 02574103 2007-O1-30
WO 95131977 ~ PCTlUS95106554
-34-
Experimental Details
Cells and Materials
MELC 745A-DS19 cells and the variants of MELC derived
from this cell line, namely, the vincristine-resistant
MELC V3.17 and VCR.C(2)15 cell lines (26i, and the
dimethylsulfoxide-resistant cell line, DR10 (39), were
maintained in alpha minimal essential medium containing
10% fetal calf serum (16). Cell cultures for all
experiments were initiated with cells in logarithmic
growth phase (day 2 cultured cells) at a density of 105
cells/ml. Inducer compounds were added in the final
concentrations indicated below, dissolved in culture
medium without fetal calf serum unless otherwise
indicated. Cell density and benzidine reactively were
determined as described (16).
Commitment to terminal differentiation, characterized by
limited cell division (colony size <32 cells) and
accumulation of hemoglobin (benzidine reactive colonies)
was assayed by a colony cloning assay using 2%
methylcellulose as described (25) (see Table I for
results). . -
HL-60 human leukemia cells, derived from peripheral blood
leukocytes of a patient with acute promyelocytic leukemia
(40). Induced differentiation of HL-50~cells assayed by
deterniining the proportion of cells that developed the
capacity to reduce nitroblue tetrazolium (NBT) (41) (see
Table 2 for results).
CA 02574103 2007-O1-30
WO 95/31977 ' PCT/US95/06554
-35-
Chemistrv
The compounds having the structure:
C.
6 //
R~ vfi-. , ~~
- NHGI
Preparation of PhCH.ONHOCtCH,)bC00CH3:
A solution of suberic acid monomethyl ester (1.9 g; 0.01
mol), oxaloyl chloride (1.75 mL; 2.54 g; 0.02 mol) and
0.1 mL DMF in benzene (200 mL) was stirred overnight at
room temperature. The solvent was evaporated and oily
residue was dissolved in chloroform (--20 mL) and mixed
together with ,chloroform so3ution (100 mL) of, 0-
benzylhydroxylamine (2.46 g; 0.02 mol) and pyridine (1.6
mL; 1.68 g; 0.02 mol). The reaction mixture was stirred
at room temperature overnight. The chloroform solution
was washed with water (50 mL), 10% hydrochloric acid, and
again with water (2 x 50 mL). The organic layer was
dried over anhydrous magnesium sulfate and evaporated.
The solid residue was slurried in hexanes (--100 mL) and
filtered. The yield of PhCH20NIi0C (CH2) bC00CH3 was 2 : 61 g
(89%).
o\
\C (CH2 ?-C
OHN/ 6 \4CH3
The above suberic acid monobenzyloxyamide monomethyl
ester (l g; 3.4 mol) was dissolved in dry methanol (50
CA 02574103 2007-O1-30
WO 95/31977 ' PGTlUS95/06554
-36-
mL) and S% Pd-C ;50 mg) was added. Tfe black suspensio:
was shaken under hydrogen pressure (--50 psi) overnight at
room temperature. The catalyst was separated by
=i!tratior_, and filtrate .was evaporated. The soli~3
residue was slurried ir_ hexanes ~:~20 mL) and filtered.
The yield of the monomethyl ester monohydroxamic acid of
suberic acid was 900 mg (95%).
~ H NNl~t ( DMSO - d6 , 2 0 0 Ngiz ) , b ( ppm ) 10 . 31 ( s , NHOH , 1 H ) ;
8.89 (s, broad, NHOH, 1H); 3.57 (s, CH3, 3H); 2.27 (t,
J=7.4Hz, CH:COOCH3, 2H); 1.91 (t, J=7.4Hz, CH=CONHOH, 2H);
1.49 (m, 4H) , 1.24 (m, 4H) .
a
1~ ~~ //
/~' (CH2 6 C\
HOHN OH
Suberic acid monobenzyloxyamide monomethyl ester (1g; 3.4
mmol) and potassium hydroxide (210 mg; 3.75.mmo1) were
dissolved in 10 mL of methanol-water (4:1) mixture. The
reaction mixture was refluxed two hours and solvent was
evaporated. The solid residue was dissolved in 5 mL
water and acidified with cone, hydrochloric acid to pH--5.
White precipitate was filtered, dried and crystallized
from ethyl acetate-hexanes. The yield of suberic acid
monobenzyloxyamide was 820 mg (86%). The product was
dissolved in methanol (50 mL) and 5% Pd-C (50 mg) was
added. The reaction mixture was shaken under hydrogen
pressure (50 psi) overnight. The catalyst was separated
by filtration and filtrate was evaporated. The solid
residue was slurried in hexanes and filtered. The yield
of suberic acid monohydroxamic acid was 520 mg (81%). 'H
NMR (DMSO-d6, 200 NlEiz) , b (ppm) 11.96 (s, broad, COOH, 1H) ;
10.31 (s, NHOH, 1H); 8.63 (s, broad, NHOH, 1H); 2.17 (s,
J=7.4Hz, CHZCOOH, 2H) ; 1.91 (s, CHZCONHOH, 2H) ; 1.46 (m,
CA 02574103 2007-O1-30
WO 95131977 PG'T/US95/06554
-37-
4H); 1.22 (m, 4H).
Compounds having the structure:
J
ri_, a t~ .~3~ ~-.\
I J ~ ?~i HO H
G
General Procedure
A' pyridine (500 mL) solution of 0-benzylhydroxylamine
(2.46 g; 0.02 mol), the corresponding amine (0.02 mol)
and suberoyl chloride was stirred at room temperature
overnight. The solvent was evaporated and the-semisolid
residue was dissolved in 1000 mL chloroform-methanol
(4:1); the resulting solution was washed with water (2 x
100 mL) , 10% hydrochloric acid (3 x 100 mL) , and again
2.0 with water (2 x 100 mL) . Organic layer was dried over
anhydrous magnesium sulfate and evaporated. The solid
residue was dissolved in methanol (100 ~mL) and 5% Pd-C
was added. The black suspension was shaken under
hydrogen pressure (~50 ps~.) overnight. The catalyst was
separated by filtration, and the filtrate was evaporated.
The target products were isolated by vcolumn
chromatography on silica gel with ethyl acetate-
tetrahydrofuran.
3: 0 '
0 ~ . //
/~ ~Cg2 ?.'C
HOHN 6 \ NHOCH3
Yield 1.1 g (26%) , 1H NN~t (DMSO-D6, 200 Ngiz) , b tppm)
CA 02574103 2007-O1-30
WO 95/31977 " PCT/US95/06554
-38-
10.93 (s, NHOCH3, 1H); 16.32 (s, NHOH, 1H); 8.66 (s, NHOH,
1H); 3.55 (s, CH;, 3H); 1.91 !t, J=7.6Hz, CH,CO-,4H); 1.45
(m, 4H); 1.20 (m, 4H).
~ \_
s. ~ ~..11 J : C i~~
Yield 1.2 g (21%) . 'H NMR (DMSO-db, 200 MHz) , b (ppm)
.10.31 (s, NHOH, 1H); 8.60 (s, broad, NHOH, 1H); 7.57 (d,
J=7.6Hz, NH-C6H", 1H), 3.40 (m, CH-NH, 1H); 1.99 (t,
J=7Hz , CHZCONHC6H1~ ; 2H) ; 1. 91 ( t , J=7 . 6Hz , G~H.CONHOH, 2H) ;
1.63 (m, 4H); 1.44 (m, 6H); 1.20 (m, 8H).
0
0 ~ //
/C (CHZ ?--C
HOHN 6 \
N (CH3 )
Yield 870 mg ( 20% ) . 1H NNQt (DMSO-D~, 200 N~Iz ) , b (ppm)
10.31 (s, NHOH, 1H); 8.67 (s, broad, NHOH, 1H); 2.85 (d,
J=30Hz, N(CH3)2, 6H) ; 2.24 (t, J=7.4Hz, CHzCON(CH3) , 2H) ;
1.91 (t, J=7.4Hz, CHzC00NHOH, 2H); 1.50 (m, 4H); 1.20 (m,
4H) .
3 0 ~ ~ //
C (CH2 ~--C
6
~ N NHO H
Yield 1.4 g (27%); 'H NMR iDMSO-db, 200 MHz); b(ppm) 10.31
(s, NHOH, 1H) ;. 8.67 (s, NHOH, 1H) ; 3.40 (2t, CHIN, 4H) ;
2.20 (t, J=7.4 Hz, CHzCON(CH2)s, 2H) ; 1.91 (t, J=7.4Hz,
CA 02574103 2007-O1-30
WO 95/31977 _ ~ PCTlIJS95/06554
-39-
CH,CONHOH, 2H); 1.10-1.60 ;m, broad, 14 H).
Compound having structure:
0 ~ ~f
' u,~ )..-~
.. 1.
~ L 5
HOgN N~ocx2c~~J
to
The chloroform (500 mL) solution of 0-benzylhydroxylamine
(1.23 g; 0.01 mol), 0-(trimethylsilyl>hydroxylamine~(1.1
g; 0.01 mol), pyridine t1.6 mL; 1.7 g; 0.02 mol) and
suberoyl chloride (1.8 mL; 2.11 g; 0.01 mQl) was stirred
at room temperature overnight. The reaction suspension
was diluted with methanol (100 mL), washed with 10%
hydrochloric acid ( 3 x 100 mL) : The organic layer was
dried over anhydrous magnesium sulfate and evaporated.
The solid residue was subjected to chromatography on
silica gel in ethyl acetate-tetrahydrofuran (4:1). The
yield was 500 mg (17%) . 'H NI~2 (DMSO-db, 200 l~iz) , 8 (ppm)
11.09 (s, NFiOCHZC6Hs, 1H) ; 10.31 (s, NHOH, 1H) ; 8.67 (s,
broad, NHDH, 1H) ; 7.36 (s, CbHs, 5H) , 4.76 (s, CHZC6H5, 2H) ;
1.92 (t, Jg7.4Hz, CHzCO-, 4H); 1.45 (m, 4H); 1.20 (m, 4H).
2~
compound having the structure:
3 0 /C (CH2 ). CN
S
HO HN
Into a cooled solution 'of potassium hydroxide (2.24 g;
35 0.04 mol) and O-benzylhydroxylamine hydrochloride in 30
mL of tetrahydrofuran-water (1:1) mixture, 6-
bromohexanoyl chloride (3.1 mL; 4.27 g; 0.02 mol) was
CA 02574103 2007-O1-30
WO 95/31977 PCT/US95I06554
-40-
added. The reaction mixture was stirred at room
temperature for one hour. The solvent was evaporated and
solid residue was partitioned between chloroform (200 mL)
and water (100 mL>. Chloroform layer was washed with 10%
hydrochloric acid (3 x 50 mL) and water (2 x 50 mLi. The
organic layer was dried over anhydrous magnesium sulfate
and evaporated. The product was purified by
crystallization from ethyl acetate-hexanes. The yield of
N-benzyloxy-6-bromohexanoyl amide was 4.7 g (?8%). A
dimethylsulfoxide (250 mL) solution of N-benzyloxy-6-
bromohexanoyl amide (4.5 g; 15 mmol) and sodium cyanide
(7.35 g; 0.15 mol) was heated at 130°C overnight. The
solvent was evaporated and solid residue was partitioned
between chloroform (300 mL) and water (~00 mL). The
chloroform layer was washed with water (5 x 100 mL),
dried over anhydrous magnesium sulfate, and evaporated.
The oily residue was purified by column chromatography on
silica gel. in ethyl acetate-tetrahydrofuran (4:1) as an
eluent. The yield of N-benzyloxy-6-cyanohexanoylamide
was 1.62 g (43%). The product was dissolved in methanol
(50 mL) and 5% Pd-C (100 mg) Was added. The black
suspension was shaken under hydrogen pressure (-50 psi)
overnight. The catalyst was isolated by filtration and
filtrate was evaporated. ~ The solid-residue was slurried
in hexanes (-20 mL) and filtered. The yield of N-
hydroxy-6-cyanohexanoylamide was 900 mg (overall yield
30%) : 'H NI~t (DMSO-db, 200 I~iz) , a (ppm) 10.32 (s, NHOH,
1H); 8.65 (s, NHOH, 1H); 2.45 (t,J=7Hz, CHZCN, 2H) 1.93
(t, J=?Hz, CHZCONHOH, 2H);'1.49 (m, 4H); 1.33 (m, 2H).
Compounds ha~ring the structure:
. ~
R C---~(CH2 j--C
n \
OH
CA 02574103 2007-O1-30
WO 95131977 ,. . ~ PCT/US95I06554
-41-
General Procedure
A diacid dichloride ( 0 . O1 mol ) was added into a cooled
(0°C) solution of potassium hydroxide (1.12 g; 0.02 mol)
and corresponding amine (0.01 mol) ,in 30 mL of
tetrahydrofuran-water (i:1) mixture. The reaction
mixture was stirred at room temperature about one hour.
Solvent Was evaporated and the solid residue was
partitioned between chloroform (300 mL) and water (300
mL). In some eases a small amount of methanol is
necessary to dissolve all solid. The organic layer was
washed with 10% potassium hydroxide (3 x 30 mL). The
basic water extract was acidified with 10% hydrochloric
acid: The precipitate was collected by filtration, dried
and purified by crystallization from ethyl acetate or by
column chromatography on silica gel iw ethyl acetate-
tetrahydrofuran (4:1). The yields are from 20-3?%.
2 0 II , /%
NH C (CH2 ) C ~.
6 . OH
~H l~l~Ht (DN1S0-ds, 200 MHz) ; b (ppm) 11.9? _(s, COOH, 1H) ;
9.84 (s, NH, 1H); 7.57 (d, J=7.4Hz, ortho aromatic
protons, 2H); 7.26 (t, J=8.4Hz, meta aromatic protons,
2H); 6.99 (t; J=7.4Hz, para aromatic proton, iH), 2.27
ft, J=7Hz, CHiCONHPh, 2H); 2.18 (t, J=7.2Hz, 2H); 1.52 (m,
4H): 1.28 (m, 4H).
NC
11 /j
NH C (CH2 ) C~
6 OH
'H NMR (DMSO-db, 200 MHz), b(ppm) 11.95 (s, COON, 1H);
10.20 (s, NH, 1H); 8.10 (s, aromatic proton, 1H); 7.75
CA 02574103 2007-O1-30
WO 95/31977 ~ PCT/US95/06554
-42-
(m, aromatic proton, 1H); 7.45 ;m, aromatic proton, 2H~;
2.28 (t,J=7.4Hz, CH,CONHAr, 2Hi ; 2.21 l~,J=7.2Hz,~ CH,COOH,
2H) ; 1.45 (m, 4H) ; 1.20 (m, 4H) .
C
...
IAA . ~;~_ _ )' y~C-
'H N1~IR (DMSO-db,. 200 NN~iz) , 8 (ppm) 11.95 (s, COOH, 1H) ;
10.29 (s, NH, 1H) ; 7.75 ~s, aromatic protons, 4H) ; 2.33
(t, J=7.2Hz, CHZCONHAr, 2H); 2.18 (t, J=7.4Hz, CH,COOH,
2H) ; 1.53 (m, 4H) ; 1.27 (m, 4H) .
~J 3
II /j
O.~N ~ NH C f,CH2 ) C v
6 OOH
'H NN,Bt (DNtSO-db, 200N~iZ) , 11.98 (s, broad, COOH, 1H) ;
10.48 (s, NH, 1H); 8.21 (d, J=9.2Hz, aromatic protons,
2H); 7.82 (d, J=9.2Hz, aromatic proton, 2H)'; 2.36 (t,
J=7.4Hz, CHzCONHAr, 2H) ; 2.18 (t, J=7.2Hz, CH~COOH, 2H) ;
1. 55 (m, 4H) ; 1.29 (m, 4H) .~
3 0 ~~ //
N NH C (CH2 ) C
6 OH
'H NNHt (DMSO-db, 200 Ngiz) , b (ppm) 12.00 (s, broad COOH,
1H); 1Ø24 (s, NH, 1H); 8.38 (d, J=5.8Hz, aromatic
protons, 2H); 7.55 (d, J=5.8Hz, aromatic proton, 2H);
2.33 (t, J=7.2Hz, CH2CONHAr, 2H); 2.18 (t, J=7.2Hz,
CHZCOOH); 1.52 (m, 4H); 1.27 (m, 4H).
1l ~°
NH
6 ~OH
CA 02574103 2007-O1-30
WO 95/31977 ' PCT'/US95I06554
-43-
'H NMR (DMSO-db, 200MHz), b(ppm) 11.95 (s, COON, 1H); 7.58
i.d, J=8Hz); 3.50 (m, CH, 1H); 2.17 (t, J=7.2Hz, CH.COOH,
2H); 2.00 (t, J=7Hz, CH,CONH-, 2H); 1.60 (m, 4H); 1.46 m,
6H); 1.20 (m, 8H).In the same way the following compounds
were prepared and characterized:
H
I
n r
~J
_ N II
- \C ( CHZ ) C
// n \ o
~ ,
~
wherein n = 4 , 5 , 6 , 7 , and 8 ; R is hydrogen; 2 - , 3 - ,
and 4-cyano; 2-, 3-, and 4-vitro; 2-, 3-, and' 4-
methylcyano; 2-, 3-, and 4-trifluoromethyl; 2-, 3-, and
4-fluoro;
H
I _ .0
II
~C (CHZ n C\
. 0 OH
wherein n = 4, 5, 6, 7, and 8;
H
I 0
N\ II
/C (CH ) C
0 2 n \0H
wherein n = 4, 5, 6; 7, and 8;
/ o
N N\ II
~C (CH2 ~ C\
0 OH
CA 02574103 2007-O1-30
WO 95131977 ~ PCT/US95106554
-44-
wherein n = 4, 5, 6, 7, and 8;
g
N //_
.~ ~ r r'
//J mg~ ,n
~ ~o H
wherein n = 4, 5, 6, 7, and 8;
3
//
(CHZ n C\
0 OH
wherein n = 4, 5, 6, 7, and 8; _
R /H
. ~ . 0
//
/~ (CHZ )--C\
OH
0
wherein R is 2-, 3-, and 4-carboxy; 2-, 3-, and 4-
aminocarbonyl; 2-, 3-, and 4-methylaminocarbonyl; 2-,
3-, and 4-dimethylaminocarbonyl; 2-, 3-, and 4-chloro;
2-, 3-, and 4-bromo; 2-, 3-, and 4-iodo; 2-, 3, and 4-
methyl; 2-, 3-, and 4 methoxy; 2-, 3-, and 4-hydroxy;
2-, 3-, and 4-amino; and 2-, 3-, and 4-dimethylamino.
CA 02574103 2007-O1-30
WO 95/31977 ~ PCT/US95/06554
-45-
Compounds havinS the a.eneral structure:
C
II II //
s \ .=H-, "c ~t C
i~ _ :. I . ~c
.. .
:..
wherein n = 4, 5, ~, and 7.
General Procedure A
A pyridine (500 mL) suspension of 0-benzylhydroxylamine
hydrochloride (3~.2 g; 0.02 mol) and they corresponding
diacid dichloride (0.04 mol) was stirred at room
temperature for three days. Water,(10 mL) was added and
stirring was continued overnight. The solvent was
evaporated and solid residue was purified by column
chromatography on silica gel in tetrahydrofuran-methanol.
The diacid product was dissolved in methanol (100 mL) and
5% Pd-C (100 mg) was added. The reaction suspension was
shaken overnight under hydrogen pressure (DSO psi). The
catalyst was separated by filtration, solid residue was
washed with hot methanol (5 x 50 ml). The combined
methanolic filtrates were evaporated. Tne solid residue
was slurried in acetone and filtered. The yield was iti-
20%.
General~ocedure B
A pyridine (S00 ml) solution of 0-benzylhydroxylamine
(2.46 g; 0.02 mol) and the corresponding dicarboxylic
acid monobenzyl ester monoacid chloride (0.04 mol) was
stirred at room temperature overnight. The solvent was
evaporated. The semisolid residue was dissolved in
chloroform (300 mL) and extracted with 5% hydrochloric
acid (2 x 50 mL) , 10%potassium hydroxide (3 x 100 mL) ,
CA 02574103 2007-O1-30
WO 95/31977 PGT/L1S95106554
-46-
. and water t2 x 100 mL). The organic layer was dried over
anhydrous magnesium sulfate and evaporated. The sold
residue was purified by column chromatography on silica
gel in ethyl acetate. The tribenzyl produce was
S dissolved in methanol 1100 mL) and 5% Pd-C ;100 mg; was
added. The reaction suspension was shaken under hydrogen
pressure (-50 psi) at room temperature overnight. The
solid was separated by filtration and washed with hot
methanol (5 x 50 mL). The combined methanol filtrates
were evaporated to solid residue. The solid residue was
slurried in cooled acetone and filtered. The yield of
target product was 30-60%.
0 0 ~ 0
C (CH C N C C CH2 ;-.-C .
)
/ 2 ~ 5 \
5
H0 O i~
OH
~H NMR (DMSO-d,b, 200MHz) b (ppm)11.53 (s, COOH, .1H) ;
, 2.~1
(t, J=7.2Hz, CH2CON(OH)COCHZ, 4H); 2:18 (t, J=7.OHz,
CHzC00H, 4H); 1.52 (m, 8h); 1.22 (m, H). MS (FAH,
glycerin) 346(M +
1)
Compounds having the structure:
//
~C ~ CCH2 m C' l (CH2 )n ; -C ' (CH2~-C
HO
CH3 CH3 .
A pyridine (500 mL) solution of the monomethyl ester
monoacid chloride of dicarboxylic acid (0.02 mol) and
N, N ~ - dimethyl -1, cv- dieniinoalkane ( 0 . 01 mol ) was stirred at
room temperature overnight. Solvent was evaporated and
oily residue was dissolved in chloroform (300 mL).
Chloroform solution was washed with water (3 x 50 mL),
CA 02574103 2007-O1-30
WO 95/31977 PCT/US95I06554
-47-
10% potassium hydroxide i3 x 50 mL), 10o hydrochloric
acid (3 x 50 mLi, and again with water !3 x 50 mL). The
organic layer was dried and evaporated. The oily residue
was dissolved in potassium hydroxide (1.2 g; 0.021 mol)
in 80% methanol (100 mL). The reaction mixture was
refluxed two hours. The solvent was~evaporated and solid
residue was dissolved in water (50 mL) and extracted with
chloroform (3 x 50 mL). Water solution was acidified to
p8--5 and concentrated (to volume of about 10 mL) . The
water solution or suspension was cooled down and
precipitate was separated by filtration. The solid
product was purified by crystallization from -ethyl
acetate. The yield was 40-60%.
0 II II I/
~
C (CH ) C- N (CHZ ) N-C (CH2)--
2
/ ( 2 I 6 \
6
. CH3 CH3 ~H
H~
~H NN~t (CDC13, 200 Mtiz)b (ppm) 8.15 (s, broad, COOH.,2H)
, ;
3.52 + 3.45 (2s, CHIN,4H) ; 3 .O1 + 2.93 (2s, CH3N, 6H)
;
2.30 (4t, CHZCO, 8 H) .60 (m, 8H) ; 1.32 (m, 8H)
; .
1
1H NNat (DMSO-db, 200 Nffiz) C3s,
, b (ppm) 3.44 + 3.336 + 3
.36
CH2N, 4H); 2.94 + 2.90 + 2.79 (3s, CH3N, 6H); 2.27 2.23
+
+ 2.1.2 (3t, CHZCO; 8H) 1.46 (m, 8H) : 1.23 (m, 8H)
; .
Compounds having the structure:
0
II II II II
3 0 /C- (CHZ m C-11H-C C-NH-C-(CHZ n C\
xo ~ off
A pyridine (500 mL) solution of 6-aminocapric acid (2.6
g; 0 . 02 mol ) and terephthaloyl chloride (2 g; 0 . O1 mol )
was stirred at room temperature overnight (~12 hours),
and at 90°C for 23 hours. The solvent was evaporated,
and the solid residue was crystallized from water (1O mL)
four times. The yield was 800 mg (19%) . 'H NNB2 (DMSO-db,
200 N~i) , 6 (ppm) 12.8 (s, broad, COOH, 2H) ; 8.54 + 7.72
CA 02574103 2007-O1-30
WO 95/31977 PCT/L1S95/06554
-48-
(2t, NH, 2Hi ; 3 .24 + 2.98 (2m, NHCH,, 4H) ; 2.20 - 2 . 03
(2m, CH,CO, 4H); 1.50 (m, 8H); 1.32 (m, 4H).
Compound having the structure:
0
/-
N~
v \ ~i Hc~:
~. o
into a mixture of aniline (2.75 g; 0.03 mol),
hydroxylamine hydrochloride (2.08 g; 0.03 mol), and
potassium hydroxide (5.508; 0.09 mol) in 50%
tetrahydrofuran (100 mL) was slowly bedded at room
temperature a tetrahydrofurane (20 mL) solution of
terephthaloyl chloride (6 g,; 0:03 moI). The .reaction
suspension Was stirred at room temperature for thirty
minutes. The solvent was evaporated. The solid residue
was slurried in hot methanol (1000 mL) and dried over
anhydrous magnesium sulfate: The methanol solution was
separated by filtration and filtrate was evapora$xd. The
solid residue was slurried in 20 mL cooled methanol and
filtered. The white crystals were washed with ether (5
x 50 mL) and dried. The' yield was 4.6 g. (39%) . 'H NMR
(DMSO-db, 200 Ngiz) , b (ppm) 11.35 (s, broad, NHOH, 1H) ;
10.35 (s, NHPh, 1H); 9.19 (s, NHOH, 1H); 8.03 (d, J=BHz,
terephthalic protons, 2H); 7.89 (d, J=BHz, terephthalic
protons, 2H); 7,82 (d, J=7.4Hz, ortho anilide protons,
2H); 7.34 (t, J=7.4Hz, meta anilide protons, 2H); 7.10
(t, J=7.4Hz, para anilide proton, 1H).
Compound having the structure:
i~
3 5 ~ ~ ~-c$Z-c-c~~ce ~ ~ cg=ca-c~
xaoH
CA 02574103 2007-O1-30
WO 95/319'77 PCf/US95I06554
-49-
A solution of 1,4-pherylenediacrylic acid (2.18 g; x.01
mol) in thionyl chloride (50 mL; 81.558; 0:68 mol) was
refluxed overnight. The excess of thionyl chloride was
evaporated. The solid was dissolved in tetrahydrofuran
(20 mL), and added to a cooled (0°C) solution of
potassium hydroxide (1.12 g; 0.02 mol) and aniline in 50%
tetrahydrofuran. The reaction mixture was stirred at
room temperature for thirty minutes. The solvent was
evaporated. The solid residue was slurried in water and
filtered. White crystals were dissolved in a small
amount of methanol and purified on a silica gel column in
tetrahydrofuran. The yield was 315 mg (10%) . 'H NN~2
(DMSO-d6, 200 MHz); b(ppm) 10.80 (s, NHOH, 1H); 10.23 (s,
NFiPh, 1H); 9.09 (s, NHOH, 1H); 7.69 (d, ~T=7.6Hz, ortho
anilide protons, 2H); 7.64 (s, phenylene protons, 4H),
7.5,5 (d, J=15.8Hz, PhNHOCCH=CH-, 1H); 7..40 (d, J=15.8Hz,
HONFiOCCH=CH-, 1H); 7.33 (t, J='7.8Hz, mesa anilide
protons, 2H); 7.06 (t, J=7.2Hz, para ani.lide protons,
1H); 6.89 (d, J=15.8Hz, PhNHOCCH=CH-, 1H) 6.51 (d,
J=15.8Hz, HOHNOCCH=CH-, 1H).
Compounds having the stricture:
0 ~ CH
R/C ~ 2 ~ C\
R
wherein n = 4, 5, 6, 7, and 8.
A chloroform solution of triethylamine (1.4 mL; 1.0 g;
0.01 mol), the corresponding amine (0.01 mol) and diacid
dichloride (0.005 mol) was stirred at room temperature
for five hours. If ~ the reaction mixture was clear, it
35. was washed with water (5 x 100 mL) . The organic layer
was dried over anhydrous magnesium sulfate and evaporated
to a solid residue. If in the course of reaction a
CA 02574103 2007-O1-30
WO 95/31977 PCT/US95/06554
-SO-
precipitate was formed, the precipitate was separated by
filtration. White crystals from Tilt=ation or solid
residue from evaporation were crystallized from ethyl
acetate, tetrahydrofuran, methanol, or their mixture.
The yields were from 60-90%.
f aC ,,\ ,'/ ~ KH_~ ~! CHZ , C~.''-
.Yv o , l
. 0 Nli----,.y. //,-- a 3
~~
'H Nit (DMSO-db, 200 MHz) , b (ppm) 10.23 (s, NH, 2H) ; 7.82
(d, J=9Hz, aromatic protons, 4H), 7.60 (d, J=9Hz,
aromatic protons, 4H) , 2.31 (t, J=7.4Hz, C,HZCO, 4H) ; 2.61
(m, 4H); 1.32 (m, 4H).
(CHZ 6 C\
'H NNBt (DMSO-db, 200 MHz) , b (ppm) 10.48 (s, ° NH, 2H) ; 8.18
(d, J=9.2Hz, aromatic protons, 4H); 7.81 (d, J=9.2Hz,
aromatic protons, 4H0; 2.37 (t, J=7.2Hz,~ CH2C0-, 4H)~; 1.60
(m, 4H); 1.33 (m, 4H).
o
NCCHZ ~ ~ NHS //
// (CH2 6 C\
O ~ ~ ~ CH2CN
'H Nl~t (DMSO-db, 200 MHz) , 69 .91 (s, NH, -2H) , 7.58 (d,
J=8.6Hz, aromatic protons, 4H); 7.26 (d, J=8.6 Hz,
aromatic protons, 4H); 3.94 (s, CH2CN, 4H); 2.29 (t,
J=7.4Hz, CH2C0-, 4H): 1.60 (m, 4H); 1.31 (m, 4H).
0
H3c~roc ~ ~ xH~ //
~/C ( CHZ 6 C \
C IiH ~ ~ COFHCB3
CA 02574103 2007-O1-30
wo 95131977 PCT/US95106554
-51-
'H NMR (DMSO-db, 200 MHz), b(ppm) 10.08 (s, CONHAr, iH;;
7.79 (d, J=8.6Hz, aromatic protons, 4H); 7.63 id, J=BHz,
aromatic protons, 4H) , 7.22 (s, H3CHNC0-, 2H) ; 3.32 ;s,
CH3, 6H); 2.31 (t, J=7Hz, CH,C-), '6H); 1.59 (m, 4H); 1.31
(m, 4H) .
V
H~~ 3~IC~C --~ ~ , j--:ZH ~
1~ w :; ~C (CH., i ~C ,
;; y 6 '.NH --~% -~OH::O~
'~> i
~H NMR (DMSO-db, 200 MHz) , b (ppm) 10.90 (s, broad, NHOH,
2H>; 10.05 (s, NFiAr, 2H); 8.90 (s, broad, NHOH, 2H); T.68
(d, J=9Hz, aromatic protons, 4H); '7.62 (d, J=9Hz,
aromatic protons, 4H) ; 2.31 (t, J=7.2Hz, CHiCO-, 4H) ; 1.59
(m, 4H); 1.30 (m, 4H).
~ 0
~NIi\ . //
C (CHZ ) C . N .
N // 6 \
0 NH ~ \
'H NMR (DMSO-db, 200 MHz) , a (ppm) 10.06 (s, broad, NH,
2H); 8.71 (d, J=2:6Hz, aromatic protons, 2H); 7.31 (d +
d, aromatic protons;. 2H); 2.32 (t, J=7.4Hz, CH2C0-, 4H),-
1. 59 (m, 4H)~ ; 1. 33 (m, 4H) .
NH \ ~l
S ~/~ (CHZ 6 .C\ S
p NH
N
'H NNgt (DMSO-db, 200 MHz) , b (pgm) 12.00 (s, broad, NH;
2H),; 7.43 (d, J=3.6Hz, aromatic protons, 2H); 7.16 (d,
' J=3.6Hz, aromatic protons; 2H) ; 2.41 (t, J=7.2Hz, CHZCONH-
, 4H) 1.58 (m, 4H) ; 1.28 (m; 4H) .
CA 02574103 2007-O1-30
WO 95131977 PCT/US95/06554
-52-
In the similar manner, the following compounds wer--__
prepared and characterized:
;;
NH
', ' C (,~:,a-.
i.. .. :.
:J
1~ N
wherein n = 4, 5, 6, 7, and 8;
all compounds are symmetrical wherein R is 2-, 3-, and 4
cyano; 2-, 3-, and 4-methylcyano; 2-, 3-, and 4-vitro,
2-, 3-, and 4-carboxy; 2-, 3-, and 4-aminocarbonyl; 2-,
3- and 4-methylaminocarbonyl; 2-, 3-, and 4
dimethylaminocarbonyl; and 2-, 3-, and 4-trifluoromethyl;
R p
NHS //
(CHZ )6 C ~
0 NH ~ ~ R .
wherein R is 4-hydroxylaminocarbonyl; 4-methoxycarbonyl;
2-, 3-, and 4-chloro; 2-, 3-, and 4-fluoro; 2-, 3-, and
4-methyl; 2-, 3-, and 4-methoxy; 2,3-difluoro; 2,4-
difluoro; 2,5-difluoro; 2,6-difluoro; 1,2,3,-
trifluoro, 3,4,5-triflubro; 2,3,5,6-tetrafluoro;
2,3,4,5,6-pentafluoro.
p
p
NH ~ //
CH Z )6 C ~
0 . . NH \
N
0
NH \ . . //
(CH2 )~ C~
p HN
CA 02574103 2007-O1-30
WO 95/31977 ~ . PCT/US95106554
-53-
,. C
.a \ //
C:-i - ) f
// _ ~ ~ _ \
~, ~_r
:~ -~
:-i"~1
to
N~N\
;' ;CH., ) C~
// ~ a N \\
C
15 N
N ~
N . NHS
compounds having the structure:
p\ . ~ //
~C (eH2 ~ C~
HO RN Ft
wherein n = 4, 5, 6, 7, and 8.
general ~,rocedure A
A diacid dichloride (0.01 mol) was added to a stirred
solution of potassium hydroxide (1.68 g; 0.03. mol),
hydroxylamine hydrochloride (0.7 g; 0.01 mol), and the
corresponding aniline (0.01 molj in 50% tetrahydrofuran
(1.00 mL). The resulting reaction n~iixture was stirred at
CA 02574103 2007-O1-30
-WO 95/31977 ~ PCT/LTS95/06554
-54-
room temperature thirty minutes, and solvent was
evaporated to solid residue. The solid r-esidue was
slurried in methanol (--100 mL) and dried over anhydrous
magnesium sulfate. The methanol solution was separated
by filtration and evaporated to a solid residue.. The
product was purified by column chromatography on silica
gel in ethyl acetate-tetrahydrofuran (in most cases 3:1;.
The yields were 15-30%.
General ,procedure B
A solution of corresponding monomethyl ester of
dicarboxylic acid (0.01 mol), oxaloyl chloride (0.03
mol), and a few drops DMF.in benzene (500 mL) was stirred
at room temperature overnight. The solvent .was
evaporated and the oily residue was dissolved in dry
benzene (3 x 50 mL) and evaporated again. The
tetrahydrofuran .(SO mL) solution of monoester monoacid
chloride of the corresponding di.carboxylic acid was
slowly added to a cooled solution of the corresponding
amine (0.01 mol) and pyridine (1.6 mL; 1..6 g.; 0.02 mol)
in tetrahydrofuran f200 mL). The reaction mixture was
stirred at room temperature for an hour. The solvent was
evaporated, the reside was dissolved in chlorofo~n (300
mL), and the chloroform solution was washed with 10%
hydrochloric acid (3 x 50 mL), 10% potassium hydroxide (3
x 50 mL), and water (3 x 50 mL). The organic layer was
dried over anhydrous magnesium sulfate and evaporated,
yielding the pure monoester monoamide of dicarboxylic
acid. The product was dissolved in 80% methanol with
potassium hydroxide (0.56 g; 0.01 mol). The reaction
mixture was refluxed two hours and evaporated to solid
residue. The residue was dissolved in water (-20 mL) and
acidified to ~pH 5 with 10% hydrochloric acid. The
monoacid monoamide of the dicarboxy7:ic acid was isolated
by filtration of precipitate or extraction water solution
with chloroform. The isolated monoacid monoamide of the
CA 02574103 2007-O1-30
WO 95/31997 , _ PCT/US95106554
-55-
dicarboxylic acid was mixed together with an equivalent
amount of 0-benzylhydroxylamine and 1,3-dicyclohexyl-
carbodiimide in pyridine '--100 mL per 0.01 mol of 0-
benzylhydroxylamine) and was stirred at room temperature
S overnight. The solvent was evaporated and the solid
residue was partitioned between chloroform (500 mL) 'and.
10% hydrochloric acid (300 mL). The crganic layer was
washed with water (3 x 100 mL) and dried over anhydrous
magnesium sulfate. The solvent was evaporated to solid
residue. The solid residue was dissolved in large
amounts of tetrahydrofuran and filtered through a short
column of silica gel. The crude product was dissolved in
methanol (100 mL) .and 5% Pd-C was added. The reaction
suspension was shaken under hydrogen pressure (-50 psi)
overnight. The catalyst was separated by filtration and
filtrate was evaporated to solid residue. The solid
residue~was slurried in hexanes and filtered. Mostly
pure product was isolated in this way. If necessary
further purification was achieved by column
chroma ography on silica gel with ethyl acetate
tetrahydrofuran: The yields were from 35% ao 65%.
GenFral grocedurg,C
A pyridine (500 mL solution of 0-benxylhydroxylamine
(1.23; 0.01 mol), the corresponding amine (0.01 mol), and
the dichloride of the dicarboxylic acid (0.01 mol) was
stirred at room temperature overnight. The solvent was
evaporated and the white solid residue contains, judged
by 'H NN~t, two symmetrical amides and a target
unsynanetrical one. The solid residue was slurried in
methanol and dried over anhydrous magnesium sulfate. The
filtrate was evaporated and the solid residue was
dissolved in methanol 0100 mL). Into the methanol
' solution 5% Pd-C (100 mg) was added and the black
suspension was shaken under hydrogen prAssure (~50 psi)
overnight$x The catalyst was separated by filtration and
CA 02574103 2007-O1-30
WO 95/31977 PCT/US95/06554
-56-
the filtrate was evaporated. The product was isolated by
column chromatography on silica with ethyl acetate-
tetrahydrofuran. The yields were from 20% to 35%.
General ,procedure D
A chloroform solution of triethylamine (3 mL; 2.18 g;
0.0215 mol), the corresponding amine (0.01 mol;,
0-trimetYiylsilyl)hydroxylamine (1.05 g, 0.01 mol), and
the corresponding diacid chloride of the dicarboxylic
acid (0.01 mol) was stirred at room temperature
overnight. The solvent was evaporated, the residue was
dissolved in methanol (--10 mL), and into the methanol
solution 10% ammonium chloride (~10 mL) was added. The
resulting suspension was stirred at 50°C for two hours.
The solvent was evaporated. The solid residue was
slurried in methanol (300 mL) and dried over anhydrous
magnesium sulfate. The methanol solution was separated
by filtration and evaporated to a solid residue. The
product was isolated by silica gel column chromatography
with ethyl acetate-tetrahydrofuran. The yields were 20-
33%.
0
~I . /%
, NH G (GH2 )6
NHOH
C H N
Elemental analysis: Calc. 63.62 7.63 10.60
Found 63.58 7.59 10.48
'H NN~2 (DMSO-ds, 200 Ngiz) , b (ppm) 10.31 (s, NHOH, 1H) ;
9.83 fs, NHPh, 1H); 8.64 (s, NHDH, 1H); 7.57- (d, J=8.2Hz,
ortho aromatic protons, 2H); 7.26 (t, J=8.4Hz, meta
aromatic protons, 2H), 6.99 (t, J=?.4Hz, para aromatic
protons, 1H) ; 2.27 (t, 'J=7.4Hz, CHZCONHPh, 2H) ; 1.93 (t,
CA 02574103 2007-O1-30
WO 95/31977 PCT/L1S95/06554
-57-
J=7.2Hz, CH=CONHOH, 2H); 1.52 (m, 4H); 1.26 (m, 4H). MS
(Fab, Glycerin) 172, 204, 232, 249, 265, (100%, M + 1).
n
I ~/.
. Nu _ .~ti_.
.~ E \
_'; HC
CN
~H NMR (DMSO-db, 200 MHz), b(ppm) 10.31 (s, NHOH, 1H);
10.08 (s, NHPh, 1H); 8.64 (s, NHOH, 1H); 7.78 (d,
J=7.6Hz, aromatic protons, 1H); 7.66 (t, J=7.4Hz,
aromatic protons, 1H); 7.48 (d, J=7.8Hz, aromatic
3
protons, 1H); 7.29 (t, J=7.4Hz, aromatic protons, 1H);
2.34 (t, J=7Hz, CHZCONHiAr, 2H) ; 1.93 (t, J=7.4Hz,
CH~CONHOH, 2H); 1.58 (m, 4H); 1.27 (m, 4H).
NC
I I
~~NHTC (CHZ ~6 C
NHOH
1H NMR (DMSO-d6, 200 MHz) , b (ppm) 10.31 (s, NHOH, 1H) ;
10.21 (s, NHPh, 1H) ; 8:65 (s, NFiOH, ~ 1H) ; 8.09 (s,
aromatic proton, 1H); 7.77 (m, aromatic proton, 1H); 7.49
(m, aromatic proton, 1H); 2.31 (t, J=7.2Hz, CHZCONHAr,
2Ii) ; 1.93 (t, J=7.2Hz, CHZCONI30H, 2~I) ; 1.51 (m, 4H) .
02N
(~ /%
NH c (cx2) c
6 \
.. v NHOIi
'H NN~t (DMSO-ds, 200 N~iz) , b (ppm) 10.35 (s, NFiAr, 1H) ;
10.31 (s, NHOH, 1H); 8.63 (s, NHOH + aromatic proton 2H);
7.88 (d, J=BHz, aromatic protons, 2H); 7.57 (t, J=BHz,
CA 02574103 2007-O1-30
WO 95I319T! ~ PGT/US95/06554
-5a-
10
aromatic proton, 1H) ; 2.33 (t, J=?.6Hz, CH,CONHAr, 2H; ;
1.93 (t, J=?.4Hz, CH,CONHOH, 2H) , 1.52 (m, 4H) ; 1.2? (m,
4H) .
.. , ;, -
n ,.
' ~ NH - _ NH - _ t=H..' C
- c
uHVH
'H NMR (DMSO-da, 200 MHz), b(ppm) 10.33 (s, NHOH, 1H);
10.15 (s, NHAr, 1H); 10.09 (s, NHPh, 1H); 8.66 (s, NHOH,
1H); ?.91 (d, J=8.6Hz, aromatic protons, 2H); ?.76 (d,
J=?.8Hz, ortho aniline protons, 2H); 7.71 (d, J=8.6Hz,
aromatic protons, 2H); ?.33 (t, J=?.6Hz, meta anilide
protons, 2H); ?.0? (t, J=?.4Hz, para anilide protons);
2.33 (t, J=7.5Hz, CHZNHAr, 2H); 1.93 (t, J=?.2Hz, CHZCNHH,
2H) ; 1.51 (m, 4H) ; 1.28 (m, 4H) .
0
/j .
NH 0 (CH2 )6 C\
NHOH
F
'H N~1R (DMSO-d~, . 200 Liz) , b (ppm) 10.32 (s. NHOH, 1H) ;
10.21 (s, NHAr, 1H); 8.65 (s, NHOH, 1H); ?.31 (d of d,
J=lOHz(2.2Hz), aromatic proton , 2H); 6.84 (t of t,
J=9 .4Hz (2.4Hz) , aromatic protons, 1H) ; 2.29 (t, CH2CONHAr,
2H); 1.93 ~(t, J=7.2Hz, CHzCONHOH, 2H); 1.51 (m, 4H); 1.26
(m, 4H) .
In the same manner the following compounds were prepared
and characterized:
R
NH . 0
l/
C (.CH2 ) C
// n
0 NHOH
CA 02574103 2007-O1-30
WO 95131977 PGT/US95/06554
wherein n = 4 , 5 , 6 , 7 , and 8 ; and R is 2 - , 3 - , and 4 -
cyano; 2-, 3-, and 4-methylcyano; 2-, 3-, and 4-vitro;
2-, 3-, and 4-carboxy; 2-, 3-, and 4-aminocarbanyl; 2-,
3-, and 4-methylaminocarbonyl; 2-,~ 3-, and 4-
dimethylaminocarbonyl; and 2-~, 3-, and 4-
trifluoromethyl; '
r
. NHS //_.
'/C -r--- ( C H 2 ) C
s ~
' 0 NHOH
~5 wherein R is 4-hydroxylaminooarbonyl; 4-methoxycarbonyl;
4-tetrazoyl; 2-, 3-, and 4-chloro; 2-, 3-, and 4-
fluoro; 2-, 3-, and 4-methyl; 2-, 3-, and 4-methoxy;
2,3-difluoro; 2,4-difluoro; 2,5-ditluoro; 2,6-
difluoro; 1,2,3-trifluoro; 3,4,5-trifluoro; 2,4,5-
trifluoro; 2,4,6-trifluoro; 2,3,6-trifluoro; 2,3,5,6-
tetrafluoro; 2,3,4,5,6-pentafluoro; 2-,_ 3-, and 4-
phenyl; 2-, 3-, and 4-benzyloxy; 4-hexyl; and 4-t-
butyl;
~ 0
//
C (CH 2 ~--- C
'~ N NHO H
NH\ /~
~ ~ ~~ (CH2 ) C
6
~ NHOH
CA 02574103 2007-O1-30
WO 95/31977 ~ PGT/US95I06554
'0J-
Compounds having ~he structure:
F
..,\., ., //_
~., ~i~. ;
// ' r. ,. ~ _ ,
C
wherein n = 4, 5, 6, 7, and 8; and R is hydrogen or
methyl.
A diacid dichloride (0.01 mol) was added into a stirred
solution of potassium hydroxide (1.68 g; 0.03 mol),
aniline or N-methylaniline (0.01 mol), and3dimethylamine
hydrochloride. (0.805 g; 0.01 mol) in 50% tetrahydrofuran
(100 mL). The reaction mixture was stirred. thirty
minutes at room temperature. The solvent was partitioned
between chloroform (400 mL) and water (300 mL). The
organic layer was washed with 10% hydrochloric acid (3 x
100 mL), 10% potassium hydroxide (3 x 100 mL), and water
(2 x100 mL). The organic layer was dried over anhydrous
magnesium sulfate and evaporated. The solid residue was
slurried in hexanes and filtered. The yield were 25-34%.
g
/.
N \ //
//C (GHZ ~ G\
0 N (CH3 )2
~H NN~t (DMSO-db, 200 Ngiz) , 6 (ppm) 9 . 82 (s, NHPh, 1H) ; 7.58
(d, J=7.6Hz, ortho aromatic protons, 2H); 7.26 (t,
J=7.4Hz, meta aromatic protons, 2H); 6.99 (t, J=7.4Hz,
para aromatic proton, 1H); 2.85 (d, J=28Hz, N(CH3)Z, 6H);
2.28 (t, J=7.2Hz, CH2C0, 2H); 2.24 (t, J=7.4Hz, CHZCO,
2H); 1.51 (m, 4H); 1.29 (m, 4H).
CA 02574103 2007-O1-30
WO 95131977 PCT/US95/06554
-61-
y
J N \ ' //
fC:i2
// r:
,-
'H NMR (DMSO-db, 200 MHz) , b (ppm) 7. 30 (m, CoHs, 5H) ; 3 . i3
(s, H3CNPh, 3H) ; 2.83 (d, J=26Hz, N(CH3),, 6H) ; 2.17 (t,
J=7.6Hz, CH.,CON(CH3)2, 2H); 1.98 (t, J=7.4Hz, CH,CON(CH3)Ph,
2H); 1.41 (m, 4H); 1.11 (m, 4H).
Compounds havincr the structure
25 wherein R" R2 are NHOH.
A solution of 18.4g (175 mmol) of HZN-OSiMe3 in 100 ml
abs . CH2Clz was slowly added to a stirred solution of the
corresponding diacid chloride of the dicarboxylic acid
( 10g, 43 . '~ mmol ) in 250 ml abs . CHZC12 which was kept at -
78°C under Argon. After the addition was complete, the
mixture was allowed to warm to' room temperature with
stirring. A white precipitate formed during this
process. After 2h at room temperature, the mixture was
heated to reflex for 30 min. to complete the substitution
reaction. It was then agaiw cooled at -78°C, whereupon
10 ml of abs. MeOH were added with stirring. The cooling
was then removed and the mixture was .allowed to come to
room temperature; during which period much more white
CA 02574103 2007-O1-30
WO 95/31977 ~ PGTILJS95I06554
-62-
precipitate appeared. After an additional 10 ml of MeCH
had been added, the reaction was again heated to refiux
for 30 min. The precipitate was filtered off and stirred
with 100 ml of 0.2 N HC1 for 2h. . The product was ther_
filtered, washed with water and dried in a vacuum X0.2
torn room temperature) over CaCl,. As the nmr spectrum
(in db-DMSO) still indicated, the presence of water in the
product after this process, the product was stirred with
40 ml of ~ dry acetone, filtered again and dried in the
same fashion. The water peak in the nmr spectrum then
decreased to the normal size expected for commercial db
.DMSO. Yield: 8.8g (91%).
~H-NMR (db-DMSO, 200 MHz) b (ppm) 11.25 (b~. s, 1H) and
10.75 (br. s, 1H) (N-$); 9.1 (br. s, 2H, O-H); 7.9 (s,
1H, CZ-_H) ; ? . 7 (~m, 2H, C4-_H, C6-_H) ; 7 _ 5 (m, 2H, Cs-H, Ar-
CH=CH_-CONHOH); 6.5 (d, J=16 Hz, 1H, Ar-CH= )
MS (C1) . M+1 223, 179, 161. Found: C, 54.96; calc.:
C, 54.05%.
In a similar manner the known dicarboxylic acids
corresponding to compounds having the following
structures, wherein R, and Ri are OH, were converted to
their acid chlorides and then to the bis-hydroxamic acids
and were also characterized by NMR and mass spectroscopy:
0
0 C R2
' II
Rl - C
and
CA 02574103 2007-O1-30
WO 95131977 PCT/US95/06554
_6~_
~_- _ _'-' C.- F:.:
~~
1 =~ ~1 T T
~y N
JJ1~~,..
CA 02574103 2007-O1-30
pCT/(TS95106554
WO 951319??
-64-
Compounds having the structure:
0 0
R-C-N- (CH,)" -C-NH-OH
H
7-Henzoylamidoheptanoylhydroxamic acid, R _- phenyl, n=6.
In a 25 mL Mask, a solution of 0.571 g of o-
aminoheptanoic acid with 0.3145 g NaOH in 12 mL water was
chilled to 0°C, and than 0.5 mL of benzoyl chloride in 8
mL dry THF was added dropwise over 30 minutes. After 3.5
hrs stirring the THF was evaporated and the solution was
acidified to pH 1. The resulting precipitate of 7-
benzolylaminoheptanoic acid was collected and washed with
ether. It was characterized by NMR and mass spectroscopy
24 (M+1=250). Then 0.20 g of this amide acid was treated
f or 3 hours with 0.1750 g of carbonyl diimidazole in 10
mL dry THF. To this stirring solution was added 0.1114
g of hydroxylamine hydrochloride, and ~he..solution was
stirred overnight at zoom temperature. Then 3 mI of 0.1
N HC1 was added, the THF was evaporated, and the residue
was taken up in 5 mL ethyl acetate 'and 3 mL brine. The
produce amide hydroxamic acid was preset as an ivory
colored solid in the organic layer; it was collected by
filtration in 60% yield. It was characterized by NMR and
mass spectrum fM+1=265) and had m.p. =105°C.
In a similar fashion analogs were~prepared with n=5 or 6,
and with R=p-cyanophenyl, m-cyanophenyl, and thiophenyl,
by the use of the appropriate carboxylic acid chloride
and 7-aminoheptanoic acid or 6-aminohexanoic acid in the
first step.
CA 02574103 2007-O1-30
WO 95131977 ~ PGT/US95106554
-65-
Compounds having the structure:
0 0
R-C-N- (CH;)" -C-NH-OH
H
Suberoyl-(4-pyridyl)-amide hydroxamic acid, R - 4-
pyridyl, n=6.
To an ice-cold solution of 6 mL suberoyl chloride in 20
mL THF was added 1.37 mL methanol and 4.7 mL
triethylamine in 40 mL THF dropwise with stirring. After
19 hours a solution of 3.2032 g 4-aminopyridine and 4.'7
mL triethylamine in 250 ~mL THF was added ~dropwise with
stirring and ice cooling. After 24 hours a small amount
of white solid was removed by filtration, the THF was
evaporated, and the crude product was chromatographed to
.20 afford 2.8879 g of the methyl ester of this amide ester
was added to a solution of 0.9866 g hydroxylamine
hydrochloride in 17 mL methanol with 0.8887 g NaOH, and
the filtered solution was allowed to stand at room
temperature for two days. The precipitated salt to the
hydroxamic acid was washed. with a little ethanol and
stirred in 0.1242 g acetic 'acid in 10 mL water. After 48
hours 0.2291 g of the hydroxamic acid had crystallized,
and it was collected and recrystalli2ed from methanol to
afford the pure product, m:p. 202-203°C. It was
characterized by NNgt and mass spectrum (M+1=266).
In a similar fashion the 2-pyridyl and 3-pyridyl analogs
were prepared, using the appropriate amines.
CA 02574103 2007-O1-30
WO 95/31977 ~ PCT/US95/06554
-66-
Compounds having the formula:
c o
s i1 II
R-NH-~-N-(CH_in -C-NH-OH
H
m-Chloropheaylureido-6-hexanohydroxamic acid, R - m-
chlorophenyl, n=5.
To 3.0 g of 6-aminocaproic acid in 150 mL THF was added
3.5 mL triethylamine, then 3 mL m-chlorophenyl
isocyanate. After overnight standing the solution was
filtered and concentrated by evaporation. Then
partitioning between water and ether, followed by
acidification of the aqueous layer to pH 3.0, afforded a
precipitate of the ureidocarboxylic acid in 35% yield,
characterized by NMR and mass spectrum (M+1=285). This
was then converted to the hydroxamic acid product by
treating 0.0418 g of the acid with 0.321 g carbonyl
diimidazole in 25 mL THF. After 2 hours at room
temperature, the solution was treated with 0.1948 g
hydroxylamine hydrochloride- and stirred for 20 hours.
Then 15 mL 0.1 N HC1 and 2'S mL ethyl acetate were added
and the THF was evaporated. The product appeared as
crystals is the organic layer, and wag collected in 38~c
yield. It had m.p. 162-163°C, and was characterized by
NMR and elemental analysis: C, 51.62; H, 5.82; N, 13.47.
Calc'd C, 52.0; H, 6.05; N, 14.00.
In a similar fashion the unsubstituted phenyl analog was
prepared from phenyl isocyanate.
CA 02574103 2007-O1-30
WO 95131977 . PCT/US95/06554
-67-
TABLE 1
Benzidine
Mol. Optimal Reactive
CPD Structure Weight Conc.luM) Cells l%)
H
O
C /
C
- ( CHI ) o-
/
O
OH
iVH
1 n = 4 (known 236 80 70
compound)
2 ' n = 5 250 20 . 84
3 n = 6 264 2.5 70
4 n = 7~ 278 20 8
n = 8 292 20 15
H
~ ~ C
~
6 274 31 44
- (CHI) 6-
cN ~/ ~
OH
.NC ~
/~
C- 274 31 52
7
(CHI)b-
\
~,
OH
H
/%
8 ~ - (CHI) 6- 294 12 . 5 32
~ .
O OH
CA 02574103 2007-O1-30
WO 95/31977 PCT/US95I06554
-68-
TABLE 1 (continued)
Benzidine
Mol. Optimal Reactive
CPD Structure Weictht Conc.luM) Cells l%)
H
\
- -
9 /C (CHi)6 \ 225 50 20
OH
H
CHZO
355 250 26
(CHI) 6
0/
~
OH
(H3C) zN' /O
/
11 // - ( CHI ) 6-C 216 6 0 5 3
\
O
NHOH
HOC
C~
12 189 250 35
- (CHZ) 6-
/ ~
O
NHOH
H3C /O
13 203 60 17
-(CHi)6-C~
/
NHOH
O
~
14 NC ( CHZ ) s-C 15 6 12 5 3 0
'NHOH
H3COH
/ - (CHz) 6-~ 218 20 43
O NHOH
CA 02574103 2007-O1-30
WO 95131977 ~ PCT/US95106554
-69-
TABLE 1 (continued)
Benzidine
Mol. Optimal Reactive
CPD Structure Weight Conc.fuM) Cells f%)
/O
16 ~~ - (CHI) 6-C\ 270 8 35
O NHOH
17 C-(CHZ)b-C\/ . 256 62 30
O/ \NHOH
( cH, ) 3c° ~ ~~
18 /~-(CHI)6- \ 260 31 38
O NHOH
CHI /
19~ ~ ~ - (CHI) 6-C~ 278 5 24
0 1VHOH -
CH -C /
-( I)6 ~
O NHOH
20 R = 4-methyl 273 20 52
21 R = 4-cyano 289 7 70
22 R = 3-cyano 289 5 55
23 R = 2-cyano ' 289 16 65
24 R = 3-nitro 309 5 30
CA 02574103 2007-O1-30
WO 95/31977 PCT/US95/06554
-70-
TABLE 1 (continued)
Benzidine
Mol. Optimal Reactive
Cep Structure Wei t Conc.luM) Cells (%)
25 R = 4-nitro 309 0.8 30
26 R = 3-trifluoromethyl 332 30 30
27 R = 4-trifluoromethyl 332 5 47
28 R =~2-amino 279 20 54
29 R = 4-cyanomethyl 303 1 30
30 R = 3-chloro 298.5 2 33
31 R = 4-azido (Nj) 304 2 47
32 R 2-fluoro 282 4 65
=
33 R 3-fluoro 282 1 25
=
34 R 4-fluoro 282 4 43
=
35 R 4-benzyloxy 370 4 20
=
36 R 4-methyoxycarbonyl 322 4 28
=
37 R 4-methylaminocarbonyl 321 30 16
=
38 R 2-bromo 343 8 45
=
39 R 2-chloro 298.5 4 34
_
40 R 4-bromo 343 1.6 47
=
CA 02574103 2007-O1-30
WO 95/31977 ~ PCTNS95/06554
-71-
TAB- (continued)
Benzidine
Mol. Optimal Reactive
CPD Structure Weight Conc.(uM) Cells (%)
g
--
41 R = 2,3-difluoro . 300 8 24
42 R = 2,4,5-trifluoro 318 8 36
43 R = 2,3,6-trifluoro 318 31 53
44 R = 2,4,6-trifluoro 318 16 47
3
45 R = 2,4-difluoro 300 6 60
' 46 R = 2,3,4,5,6-pentafluoro 354 31 53
47 R = 3,4-difluoro 300 4 ~ 61-
48 R = 3,4,5-trifluoro 318 8 55
49 R = 2,5-difluoro . 300 4 70
50 R = 3,.5-difluoro 30Q 2 ~3
51 R = 2-methoxy 294 8 36
52 R = 3-methoxy 294 6 38
53 R = 4-methoxy 294 6 37
CH3
. .
54 ~- (CHi) 6-C 290 20 40
p I'lHOH
CA 02574103 2007-O1-30
WO 95/31977 PCT/L1S95/06554
-?2-
TABLE 1 (continued)
Benzidine
Mol. Optimal Reactive
CPD Structure Weight Conc.luM) Cells (%1
H
N O
55 ~ C- ~ 256 30 53
v
O NHOH
H
R \ v //°
~~ C CHZ ) s-
O R
,_
56. R - 4-trifluoromethyl 460 50 20
5? R = 4(N)-hydroxylamino- 442 8 10
carbonyl
58 R = 4-cyanomethyl 402 50 25
59 R = 2,4-difluoro 396 500 ' 54
60 R = 2,6-difluoro 396 100 21
61 R = 3,5-difluoro 396 125 31
62 R = 2,3,6-trifluoro 432 250 28
63 R = 2,4,6-trifluoro 432 125 35
64 R = 2,3,4,5,6-pentafluoro 504 125 13
65 R = 4-vitro , 414 25 14
CA 02574103 2007-O1-30
WO 95/31977 PCT1US95/06554
-73-
TABLE 1 (continued)
Benzidine
Mol. Optimal Reactive
CPD Structure Weight Conc.fuM) Cells l%)
\~ CH3 CH3
66 ~ -CH- (CHz) s-CH-C 270 1250 80
( H3C ) zN ~ ( CHI ) z
CH3 CH3 O
6 7 ~ -CH- ( CHz ) 4-CH- // 2 5 6 2 5 0 0 9 0
('H3C ) zN ~ N t CHa ) z
3
68 \\-(CH ~ -CH-(CH ) -C/ 204 125 ~ 56
z~ 2 2 z
HO ~ ~ NHOH
CONHOH
6 9 ~- ( CHz ) 5-CH- ( CHz ) s-C 3 3 3 6 0 ~ 4 0
HOHN ~ NHOH
70 ~C- (CHz) ~~ HF.(CHz) z-C ~ 226 160 19
HOHN \ F \ NHOH
CA 02574103 2007-O1-30
WO 95/31977 ~ PCT/US95/06554
-74-
TABLE 1 (continued)
Benzidine
Mol. Optimal Reactive
CPD Structure Weight Conc.(uM1 Cells (%)
N
/ H ~ . // ..
~~ - ( CHZ ) o-~
O NH~
s
71 n = 4 310 100 8
72 n = 5 324 250 ' 10
73 n = 6 338 50 7
74 n = 7 352 100 10~
75 n = 8 366 100 10
O ~ -NHOH
HONH-C-
76 196 0
O
II
H=CH-C-NHOH
HONH-
77 222 4 73
CH=CH-C-NHOH
HONH-~-CH=CH
78 248 20 45
i
i -NH- ( CHI ) s-~ NH-OH
O
79 283.3 3 45
CA 02574103 2007-O1-30
WO 95131977 ' P~/pS95/06554
TABLE 1 (continued)
Benzidine
Mol. Optimal Reactive
CPD Structure Weight Conc.(uM) Cells l$)
N-H ~~
C \ i-NH- ( CHZ ) 5-C-NH-OH
O
80 284.74 3 32
3
CA 02574103 2007-O1-30
WO 95131977
-- PCT/US95I06554
_7
TABLE 2
Induction of Differentiation of HL-60
Mol. Optimal NBT
CPD Weight Conc.tuM? Positive
2 250 7 22
3 264 1 21
6 ~ 274 20 30
274 20 21
22 ~ 289 1.? ~ 28
21 289 2 6
26 332 6 27
25 309 3 18
3.6 322 1 32
31 . 304. 2.5 ?
29 ~ 303 1 ~ 15
43 ' 318 2 20
?? 222 4 20
78 248 . 20 12
CA 02574103 2007-O1-30
WO 95131977 PCT/US95/06554
TAHLE 3
Induction of Diff.~rentiaticn of MELC
Mol. Optimal NHT
CPD Wei,3ht ;.onc . f~~~ positive ~ % )
3 264 3 65
77 222 4 61
CA 02574103 2007-O1-30
WO 95/31977 pGT/US95/06554
-78-
Ref erences:
1. Sporn, M.H., Roberts, A.H., and Driscoll, ,:.5.
(1985; in Cancer: Principles and Praccic~ c=
S Oncolocrv, eds. Hellman, S., Rosenberg, S.A., and
I3eVita, V.T. , Jr. , Ed. 2, (J.H. Lippi::cott,
Philadelphia), P. 49.
2. Breitman, T.R., Selonick, S.E., and Collins, S.J.
(1980) Proc. Natl. Acad. Sci. USA 77: 2936-2940.
3. Olsson, I.L. and Hreitman, T.R. (1982) Cancer Res.
42: 3924-3927.
4. Schwartz, E.L. and Sartorelli, A.C. (1982) Cancer
Res. 42: 2651-2655. ,
5. Marks, P.A., Sheffery, M., and Rifkind, R.A. (1987)
Canr~ Res. 47: 659.
6. Sachs, L. (1978) Nature (Load.) 274: 535.
7. Friend, C., Scher; W.,. Holland, J.W., and Sato, T.
(1971) Proc. Natl. Acad S i (USA) 6~8: 378-382.
8. Tanaka, M., Levy, J., Terada, M., Hreslow, R.,
Rifkind, R.A., and Marks, P.A. (1975) Proc. Natl.
Acad. Sci. (USA) T2: 1003-1006.
9, Reuben, R.C., Wife, R.L., Breslow, R., Rifkind,
R.A., and Marks, P.A. (1976) Proc. Natl. Acad. Sci.
(USA) 73: 862-866.
10. Abe; E., Miyaura, C., Sakagami, H., Takeda, M.,
Konno, K. , Yamazaki:, T. , Yoshika, S. , and Suda, T.
(1981) Proc. Natl. Acad Sci (USA) 78: 4990-4994.
CA 02574103 2007-O1-30
WO 95131977 . PCT/US95/06554
_79_
11. Schwartz, E.L., Snoddy, J.R., Kreutter, D.,
Rasmussen, H., and Sartorelli, A.C. !1983) Proc. Am.
Assoc. Cancer Res. 24: 18.
12. Tanenaga, K., ~Hozumi, M., and Sakagami, Y. ;i98C
Cancer Res. 40: 914-919.
13. Lotem, J. and Sachs, L. (1975) Int. J. Cancer 15:
731-740.
14. Metcalf, D. (1985) Science, 229: 16-22.
15. Scher, W., Scher, H.M., and Waxman, S. (1983) Exp-
Hematol. i1: 490-498:
16. Scher, W., Scher, B.M., and Waxman, S. (1982)
Biochem. & Hiophys. Res. Comm. 109: 348-354.
17. Huberman, E. and Callaham, M.F. (1979) _Proc. Natl.
Acad. Sci. (USA) 76: 1293-1297.
18. Lottem, J. and Sachs, L. (1979) Proc. Natl. Acad.
Sci. (USA) 76: 5158-5162.
19. Terada, M., Epner, E., Nudel, U., Salmon, J.,
Fibach, E., Rifkind, R.A., and Marks, P.A. (1978)
Proc. Natl. Acad. Bci. (USA) 75: 2795-2799.
20. Morin, M.J. and Sartorelli, A.C. (1984) Cancer Res.
44: 2807-2812
21. Schwartz, E.L., Brown, H.J., Ni:erenberg, M., Marsh,
J.C., and Sartorelli, A.C. (1983) dancer Res. 43:
2725-2730.
22. Sugano, H., Furusawa, M., Kawaguchi, T.; and Ikawa,
Y. (1973) ~ibl. Hematol. 39: 943-954.
CA 02574103 2007-O1-30
WO 95/31977 . PGT/US95/06554
-eo-
23. Ebert, P.S., Wars, I., and Buell, D.N. (1976; Cancer
Res. 36: 1809-1813.
24. Hayashi, M., Okabe, J., and Hozumi, M. (1979; Gann
70: 235-238.
25. Fibach, E., Reuben, R.C., Rifkind, R.A., and Marks,
P.A.~(1977) Cancer Res. 37: 440-444.
26. Melloni, E., Pontremoli, S., Damiani, G., Viotti,
P., Weich, N., Rifkind, R.A., and Marks, P.A. (1988)
Proc. Natl. Acad. Sci. (USA) 85: 3835-3839.
2?. Reuben, R., Khanna, P.L., Gazitt, Y., Breslow, R.,
Rifkind, R.A., and Marks, P.A. (1978) J. Biol. Chem.
253: 4214-4218.
28. Marks, P.A. and Rifkind, R.A. (1988) Lnternational
Journal of Cell Cloning 6: 230-240.
29. Melloni, E., Pontremoli, S., Michetti, M., Saceo,
0., Cakiroglu, A.G., Jackson, J.F.,. Rifkind, R.A.,
and Marks, P.A. (1987) Proc Natl Ac~,~ad Sciences
(USA) 84: 5282-5286.
30. Marks, P.A. and Rifkind, R.A. (1984) Cancer 54:
2766-2769.
31. Egorin, M.J., Sigman, L.M. VanEcho, D.A., Forrest,
A., Whitacre, M.Y., and Aisner, J. (1987) Cancer
Res. 47: 617-623.
32. Rowinsky, E.W., Ettinger, D.S., Grochow, L.H.,
Brundrett, R.B., Cates, A.E., and Donehower, R.C.
(1986) J. Clin. Oncol. 4: 1835-1844.
CA 02574103 2007-O1-30
WO 95131977 PCT/OS95106554
-81-
33. Rowinsky, E.L. Et~irlc.,j~?~', D.S., McGuire, W.P., N~ve,
D.A., Grochow, L.B., and Donehower, R.C. ~i787'
Cancer Res. 47: 5788-5795.
34. Gallery, P.S., Egorin, M.J., GeAlhaar, L.A., a:~d
~Nayer, M.S.B..(1986) Cancer Res~. 46: 4900-4903.
35. Young, C.W. Fanucchi, M.P., Walsh, T.B., Blatzer,
L., ~Yaldaie, S., Stevens, Y.W., Gordon, C., Tong,
W., Rifkind, R.A., and Marks, P.A. (1988) Cancer
Res. 48: 7304-7309. .
36. Andreeff, M., Young, C., Clarkson, H., Fetten, J.,
'Rifkind, R.A., and Marks, P.A. (1988) Blood 72:
186a.
37. Marks, P.A., Breslow, R., Rifkind, R.A., Ng'o, L.,
and Singh, R. (1989) Proc. Natl. Acad. Sci. (USA)
86: 6358-6362.
38. Hreslow, R., Jursic, B., Yan, Z.F., Friedman, E.,
Leng, L., Ngo, L., Rifkind, R.A., and Marks, P.A.
(1991) Proc; Natl: Acad. Sci. (USA), 88: 5542-5546.
39. Ohta, Y., Tanaka, M., Terada, M:, Miller, O.J'.,
Bank, A., MarkS,~P.A., and Rifkind, R.A. (1976)
Proc. Natl. Acad. Sci: (USA) 73: 1232-1236.-
40. Collies, S.J., Gallo, R.C., and Gallagher, R.E.
(I978) Nature (London) 2?0; 405-409.
41. Syr~der, S.W., Egorin, M.J., Geelhaar, L.A.,
Haarburger, A.W., and Gallery, P.S. (1988) n r
Res. 48; 3613-3616.