Note: Descriptions are shown in the official language in which they were submitted.
DEMANDE OU BREVET VOLUMINEUX
LA PRESENTE PARTIE DE CETTE DEMANDE OU CE BREVET COMPREND
PLUS D'UN TOME.
CECI EST LE TOME 1 DE 2
CONTENANT LES PAGES 1 A 21
NOTE : Pour les tomes additionels, veuillez contacter le Bureau canadien des
brevets
JUMBO APPLICATIONS/PATENTS
THIS SECTION OF THE APPLICATION/PATENT CONTAINS MORE THAN ONE
VOLUME
THIS IS VOLUME 1 OF 2
CONTAINING PAGES 1 TO 21
NOTE: For additional volumes, please contact the Canadian Patent Office
NOM DU FICHIER / FILE NAME:
NOTE POUR LE TOME / VOLUME NOTE:
CA 02577180 2007-02-14
WO 2006/055292 PCT/US2005/040107
SINGLE PROTEIN PRODUCTION IN LIVING CELLS FACILITATED BY A
MESSENGER RNA INTERFERASE
CROSS-REFERENCE TO RELATED APPLICATIONS
[0001] This application claims priority to U.S. Provisional Application No.
60/624,976,
entitled "Single Protein in Living Cells Facilitated by an mRNA Interferase"
by Inouye et al.,
filed on November 4, 2004. The entire disclosure of this application is
incorporated herein
by reference.
FIELD OF THE INVENTION
[0002] The present invention relates to a system for producing a single-
protein in living
cells facilitated by an mRNA interferase that is a single-stranded RNA- and
sequence-specific
endoribonuclease.
BACKGROUND OF THE INVENTION
[0003] Most bacteria contain suicidal genes whose expression leads to growth
arrest and
eventual death upon exposure to cellular stress (reviewed by Elenberg-Kulka
and Gerdes,
Ann. Rev. Microbiol. 53: 43-70 (1999); Engelberg-Kullca et al., Trends
Microbiol. 12: 66-71
(2004)). These toxin genes are usually co-expressed with their cognate
antitoxin genes in the
same operon (referred to as an addiction module or antitoxin-toxin system). E.
coli has five
addiction modules (Christensen et al., J. Mol. Biol. 332: 809-19 (2003)) among
which the
MazE/MazF module has been most extensively investigated. The x-ray structure
of the
MazE/MazF complex (Kamada et al., Mol. Cell 11: 875-84 (2003)) is known and
the
enzymatic activity of MazF has been recently characterized (Zhang et al, J.
Biol. Chem. 278:
32300-306 (2003)).
[0004] MazF is a sequence-specific endoribonuclease that specifically cleaves
single-
stranded RNAs (ssRNAs) at ACA sequences. An endonuclease is one of a large
group of
enzymes that cleave nucleic acids at positions within a nucleic acid chain.
Endoribonucleases
or ribonucleases are specific for RNA. MazF is referred to as an mRNA
interferase since its
primary target is messenger RNA (mRNA) in vivo. Transfer RNAs (tRNAs) and
ribosomal
RNAs (rRNAs) appear to be protected from cleavage because of either their
secondary
structure or association with ribosomal proteins, respectively. Therefore,
MazF expression
CA 02577180 2007-02-14
WO 2006/055292 PCT/US2005/040107
causes nearly complete degradation of mRNA, leading to severe reduction of
protein
synthesis and ultimately, to cell death (Zhang et al., Mol. Cell 12: 913-23
(2003)). MazF is
found in selected bacteria, and recently the E. coli protein PemK (encoded by
plasmid R100)
was also shown to be a sequence-specific endoribonuclease (Zhang et al., J.
Biol. Chem.
279: 20678-20684 (2004)). PemK cleaves RNA with high specificity at a specific
nucleic
acid sequence, i.e., UAX, wherein X is C, A or U. See PCT/US2004/018571, which
is
incorporated herein by reference. These sequence-specific endoribonucleases
are conserved,
underscoring their essential roles in physiology and evolution. We refer to
this family of
sequence-specific endoribonuclease toxins as "mRNA interferases" (Zhang et
al., J. Biol.
Chem. 279: 20678-20684 (2004)).
[0005] In the present study, we have exploited the unique cleavage properties
of MazF to
design a single-protein production (SPP) system in living E. coli cells. Upon
expression of a
gene engineered to express an ACA-less mRNA without altering its amino acid
sequence,
high levels of individual target protein synthesis were sustained for at least
for 96 hours while
background cellular protein synthesis was virtually absent. Therefore, the
toxic effect of
MazF is directed at mRNA with minimal side effects on cellular physiology. In
fact, despite
their state of growth arrest, these cells retain essential metabolic and
biosynthetic activities
for energy metabolism (ATP production), amino acid and nucleotide biosynthesis
and
transcription and translation. In addition to demonstrating the efficacy of
the SPP system for
human and yeast proteins, the technology was also effective for overexpression
of an integral
inner membrane protein whose natural levels of expression are relatively low.
The SPP
system yields unprecedented signal to noise ratios that both preclude any
protein purification
steps for experiments that require recovery of proteins in isolation, and,
more importantly,
enable structural and functional studies of proteins in intact, living cells.
BRIEF DESCRIPTION OF THE FIGURES
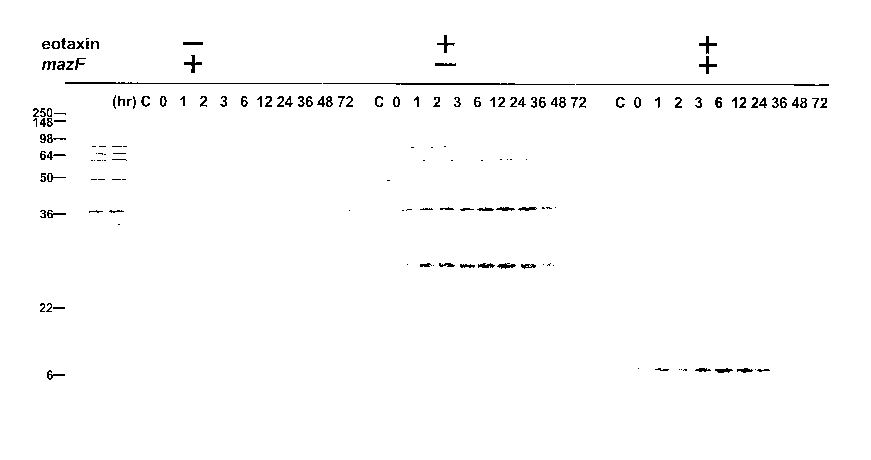
[0006] Figure 1. Expression of Human Eotaxin with Use of pColdI(SP-1) and
pCo1dI(SP-2) with and without MazF Coexpression
[0007] Figure 2. Effect of ACA Sequences on Eotaxin Expression
[0008] Figure 3. Effect of Removal of All ACA Sequences in the MazF ORF on
Eotaxin Expression
[0009] Figure 4. Expression of Yeast Proteins in the SPP System
Page 2 of 30
CA 02577180 2007-02-14
WO 2006/055292 PCT/US2005/040107
[0010] Figure 5. Expression of LspA, an Inner Membrane Protein in the SPP
System
Using pColdIV(SP-2).
SUMMARY OF THE INVENTION
[0011] The present invention describes a single-protein production (SPP)
system in living
E. coli cells that exploits the unique properties of an mRNA interferase, for
example, MazF, a
bacterial toxin that is a single stranded RNA- and ACA-specific
endoribonuclease, which
efficiently and selectively degrades all cellular mRNAs in vivo, resulting in
a precipitous
drop in total protein synthesis. In one embodiment of the present invention, a
system for
expressing a single target protein in a transformable living cell while
reducing non-target
cellular protein synthesis includes: (a) an isolated transformable living cell
comprising
cellular mRNA having at least one first mRNA interferase recognition sequence;
(b) a first
expression vector comprising an isolated nucleic acid sequence encoding an
mRNA
interferase polypeptide, wherein the isolated nucleic acid sequence encoding
the mRNA
interferase polypeptide is mutated by replacing at least one second mRNA
interferase
recognition sequence with an alternate triplet codon sequence to produce a
mutated nucleic
acid sequence encoding a mutated mRNA interferase polypeptide; and (c)
optionally, a
second expression vector comprising an isolated nucleic acid sequence encoding
a target
protein, wherein the isolated nucleic acid sequence encoding the target
protein is mutated by
replacing at least one third mRNA interferase recognition sequence with an
alternate triplet
codon sequence to produce a mutated nucleic acid sequence encoding a mutated
target
protein; wherein the isolated cell is transformed with the first expression
vector and the
second expression vector; and wherein the isolated cell is maintained under
conditions
permitting expression of the mutant target protein in the cell.
[0012] In another embodiment, the present invention provides a method of
increasing
expression of a target protein in an isolated living cell including the steps:
(a) mutating an
isolated nucleic acid sequence encoding an mRNA interferase polypeptide to
replace at least
one first mRNA interferase recognition sequence with an alternate triplet
codon sequence to
produce a mutated nucleic acid sequence encoding a mutated mRNA interferase
polypeptide,
(b) mutating an isolated nucleic acid sequence encoding the target protein to
replace at least
one second mRNA interferase recognition sequence with an alternate triplet
codon sequence
to produce a mutated nucleic acid sequence encoding a mutated target protein;
(c) providing a
Page 3 of 30
CA 02577180 2007-02-14
WO 2006/055292 PCT/US2005/040107
first expression vector comprising the mutated nucleic acid sequence of step
(a) and a second
expression vector comprising the mutated nucleic acid sequence of step (b);
(d) providing an
isolated living transformable cell having cellular messenger RNA sequences
comprising at
least one of a third mRNA interferase recognition sequence, (e) introducing
the first
expression vector and the second expression vector into the isolated living
transformable cell;
(f) expressing the mutated mRNA interferase polypeptide, and (g) maintaining
the isolated
cell under conditions permitting expression of the mutant target protein in
the cell.
DETAILED DESCRIPTION OF THE INVENTION
[0013] The following definitions set forth the parameters of the present
invention.
[0014] The abbreviation "ACA" refers to the sequence Adenine-Cytosine-Adenine.
[0015] As used herein, the terms "encode", "encoding" or "encoded", with
respect to a
specified nucleic acid, refers to information stored in a nucleic acid for
translation into a
specified protein. A nucleic acid encoding a protein may comprise non-
translated sequences
(e.g., introns) within translated regions of the nucleic acid, or may lack
such intervening non-
translated sequences (e.g., as in cDNA). The information by which a protein is
encoded is
specified by the use of codons. Typically, the amino acid sequence is encoded
by the nucleic
acid using the "universal" genetic code.
[0016] The term "codon" as used herein refers to triplets of nucleotides that
together
specify an amino acid residue in a polypeptide chain. Most organisms use 20 or
21 amino
acids to make their polypeptides, which are proteins or protein precursors.
Because there are
four possible nucleotides, adenine (A), guanine (G), cytosine (C) and thymine
(T) in DNA,
there are 64 possible triplets to recognize only 20 amino acids plus the
termination signal.
Due to this redundancy, most amino acids are coded by more than one triplet.
The codons
that specify a single amino acid are not used with equal frequency. Different
organisms often
show particular "preferences" for one of the several codons that encode the
same given amino
acids. If the coding region contains a high level or a cluster of rare codons,
removal of the
rare codons by resynthesis of the gene or by mutagenesis can increase
expression. See J.
Sambrook and D.W. Russell, Molecular Cloning: A Laboratory Manual, Third
Edition, Cold
Spring Harbor Press, Cold Spring Harbor, N.Y. (2001), at 15.12; which is
incorporated herein
by reference. "Codon selection" therefore may be made to optimize expression
in a selected
Page 4 of 30
CA 02577180 2007-02-14
WO 2006/055292 PCT/US2005/040107
host. The most preferred codons are those which are frequently found in highly
expressed
genes. For "codon preferences" in E. coli, see Konigsberg, et al., Proc.
Nat'l. Acad. Sci.
U.S.A. 80:687-91 (1983), which is incorporated herein by reference.
[0017] One of skill will recognize that individual substitutions, deletions or
additions to a
nucleic acid, peptide, polypeptide, or protein sequence which alters, adds or
deletes a single
amino acid or a small percentage of amino acids in the encoded sequence is a
"conservatively
modified variant" where the alteration results in the substitution of an amino
acid with a
chemically similar amino acid. The term "conservatively modified variants"
applies to both
amino acid and nucleic acid sequences. With respect to particular nucleic acid
sequences,
conservatively modified variants refers to those nucleic acids which encode
identical or
conservatively modified variants of the amino acid sequences. Because of the
degeneracy of
the genetic code, a large number of functionally identical nucleic acids
encode any given
protein. For instance, the codons UUA, UUG, CUU, CUC, CUA, and CUG all encode
the
amino acid leucine. Thus, at every position where a leucine is specified by a
codon, the codon
can be altered to any of the corresponding codons described without altering
the encoded
polypeptide. Such nucleic acid variations are "silent variations" and
represent one species of
conservatively modified variation. Every nucleic acid sequence herein which
encodes a
polypeptide also, by reference to the genetic code, describes every possible
silent variation of
the nucleic acid. One of ordinary skill will recognize that each codon in a
nucleic acid (except
AUG, which is ordinarily the only codon for methionine; and UGG , which is
ordinarily the
only codon for tryptophan) can be modified to yield a functionally identical
molecule.
Accordingly, each silent variation of a nucleic acid which encodes a
polypeptide of the
present invention is within the scope of the present invention.
[0018] The term "eotaxin" as used herein refers to a chemotactic factor
consisting of 74
amino acid residues that belongs to the C-C (or beta) chemokine family and has
been
implicated in animal and human eosinophilic inflammatory states.
[0019] The present invention includes active portions, fragments, derivatives,
mutants,
and functional variants of mRNA interferase polypeptides to the extent such
active portions,
fragments, derivatives, and fiulctional variants retain any of the biological
properties of the
mRNA interferase. An "active portion" of an mRNA interferase polypeptide means
a peptide
that is shorter than the full length polypeptide, but which retains measurable
biological
activity. A "fragment" of an mRNA interferase means a stretch of amino acid
residues of at
Page 5 of 30
CA 02577180 2007-02-14
WO 2006/055292 PCT/US2005/040107
least five to seven contiguous amino acids, often at least about seven to nine
contiguous
amino acids, typically at least about nine to thirteen contiguous amino acids
and, most
preferably, at least about twenty to thirty or more contiguous amino acids. A
"derivative" of
an mRNA interferase or a fragment thereof means a polypeptide modified by
varying the
amino acid sequence of the protein, e.g.., by manipulating the nucleic acid
encoding the
protein or by altering the protein itself. Such derivatives of the natural
amino acid sequence
may involve insertion, addition, deletion, or substitution of one or more
amino acids, and may
or may not alter the essential activity of the original mRNA interferase.
[0020] The term "gene" refers to an ordered sequence of nucleotides located in
a
particular position on a segment of DNA that encodes a specific functional
product (i.e, a
protein or RNA molecule). It can include regions preceding and following the
coding DNA
as well as introns between the exons.
[0021] The term "induce" or inducible" refers to a gene or gene product whose
transcription or synthesis is increased by exposure of the cells to an inducer
or to a condition,
e.g., heat.
[0022] The terms "inducer" or "inducing agent" refer to a low molecular weight
compound or a physical agent that associates with a repressor protein to
produce a complex
that no longer can bind to the operator.
[0023] The term "induction" refers to the act or process of causing some
specific effect,
for example, the transcription of a specific gene or operon, or the production
of a protein by
an organism after it is exposed to a specific stimulus.
[0024] The terms "introduced", "transfection", "transformation",
"transduction" in the
context of inserting a nucleic acid into a cell, include reference to the
incorporation of a
nucleic acid into a prokaryotic cell or eukaryotic cell where the nucleic acid
may be
incorporated into the genome of the cell (e.g., chromosome, plasmid, plastid
or mitochondrial
DNA), converted into an autonomous replicon, or transiently expressed (e.g.,
transfected
mRNA).
[0025] The term "isolated" refers to material, such as a nucleic acid or a
protein, which is
substantially free from components that normally accompany or interact with it
as found in its
naturally occurring environment. The isolated material optionally comprises
material not
found with the material in its natural environment; or, if the material is in
its natural
environment, the material has been synthetically (non-naturally) altered by
deliberate human
Page 6 of 30
CA 02577180 2007-02-14
WO 2006/055292 PCT/US2005/040107
intervention. For example, an "isolated nucleic acid" may comprise a DNA
molecule
inserted into a vector, such as a plasmid or virus vector, or integrated into
the genomic DNA
of a prokaryotic or eukaryotic cell or host organism. When applied to RNA, the
term
"isolated nucleic acid" refers primarily to an RNA molecule encoded by an
isolated DNA
molecule as defined above. Alternatively, the term may refer to an RNA
molecule that has
been sufficiently separated from other nucleic acids with which it is
generally associated in
its natural state (i.e., in cells or tissues). An isolated nucleic acid
(either DNA or RNA) may
further represent a molecule produced directly by biological or synthetic
means and separated
from other components present during its production.
[0026] The abbreviation "IPTG" refers to isopropyl-beta-D-
thiogalactopyranoside, which
is a synthetic inducer of beta-galactosidase, an enzyme that promotes lactose
utilization, by
binding and inhibiting the lac repressor. For example, IPTG is used in
combination with the
synthetic chromogenic substrate Xgal to differentiate recombinant from non-
recombinant
bacterial colonies in cloning strategies using plasmid vectors containing the
lacZ gene.
[0027] The term "MazF" as used herein refers to the general class of
endoribonucleases,
to the particular enzyme bearing the particular name, and active fragments and
derivatives
thereof having structural and sequence homology thereto consistent with the
role of MazF
polypeptides in the present invention.
[0028] The abbreviation "1spA" refers to the gene responsible for signal
peptidase II
activity in E. coli.
[0029] The abbreviation "LspA" refers to the gene responsible for Lipoprotein
Signal
Peptidase activity in E. coli.
[0030] The family of enzymes encompassed by the present invention is referred
to as
"mRNA interferases". It is intended that the invention extend to molecules
having structural
and functional similarity consistent with the role of this family of enzymes
in the present
invention.
[0031] As used herein, the term "nucleic acid" or "nucleic acid molecule"
includes any
DNA or RNA molecule, either single or double stranded, and, if single
stranded, the molecule
of its complementary sequence in either linear or circular form. In discussing
nucleic acid
molecules, a sequence or structure of a particular nucleic acid molecule may
be described
herein according to the normal convention of providing the sequence in the 5'
to 3' direction.
Unless otherwise limited, the term encompasses known analogues.
Page 7 of 30
CA 02577180 2007-02-14
WO 2006/055292 PCT/US2005/040107
[0032] The term "oligonucleotide" refers to a nucleic acid molecule comprised
of two or
more ribo- or deoxyribonucleotides, preferably more than three, joined by
phosphodiester
bonds.
[0033] The term "operator" refers to the region of DNA that is upstream (5')
from a
gene(s) and to which one or more regulatory proteins (repressor or activator)
bind to control
the expression of the gene(s)
[0034] As used herein, the term "operon" refers to a functionally integrated
genetic unit
for the control of gene expression. It consists of one or more genes that
encode one or more
polypeptide(s) and the adjacent site (promoter and operator) that controls
their expression by
regulating the transcription of the structural genes. The term "expression
operon" refers to a
nucleic acid segment that may possess transcriptional and translational
control sequences,
such as promoters, enhancers, translational start signals, polyadenylation
signals, tenninators,
and the like, and which facilitate the expression of a polypeptide coding
sequence in a host
cell or organism.
[0035] The phrase "operably linked" includes reference to a functional linkage
between a
promoter and a second sequence, wherein the promoter sequence initiates and
mediates
transcription of the DNA sequence corresponding to the second sequence.
Generally,
operably linked means that the nucleic acid sequences being linked are
contiguous and, where
necessary to join two protein coding regions, contiguous and in the same
reading frame.
[0036] The abbreviation "ORF" stands for "open reading frame, a portion of a
gene's
sequence that contains a sequence of bases, uninterrupted by internal stop
sequences, and
which has the potential to encode a peptide or protein. Open reading frames
start with a start
codon, and end with a termination codon. A termination or stop codon
determines the end of
a polypeptide.
[0037] The terms "polypeptide", "peptide" and "protein" are used
interchangeably herein
to refer to a polymer of amino acid residues. The terms apply to amino acid
polymers in
which one or more amino acid residue is an artificial chemical analogue of a
corresponding
naturally occurring amino acid, as well as to naturally occurring amino acid
polymers.
[0038] The abbreviation "PCR" refers to polymerase chain reaction, which is a
technique
for amplifying the quantity of DNA, thus making the DNA easier to isolate,
clone and
sequence. See, e.g., U.S. Pat. No. 5,656,493, 5,33,675, 5,234,824, and
5,187,083, each of
which is incorporated herein by reference.
Page 8 of 30
CA 02577180 2007-02-14
WO 2006/055292 PCT/US2005/040107
[0039] As used herein the term "promoter" includes reference to a region of
DNA
upstream (5') from the start of transcription and involved in recognition and
binding of RNA
polymerase and other proteins to initiate transcription. The term "inducible
promoter" refers
to the activation of a promoter in response to either the presence of a
particular compound,
i.e., the inducer or inducing agent, or to a defined external condition, e.g.,
elevated
temperature.
[0040] The phrase "site-directed mutagenesis" refers to an in vitro technique
wlzereby
base changes i.e., mutations, are introduced into a piece of DNA at a specific
site, using
recombinant DNA methods.
[0041] The term "untranslated region" or UTR, as used herein refers to a
portion of DNA
whose bases are not involved in protein synthesis.
[0042] The terms "variants", "inutants" and "derivatives" of particular
sequences of
nucleic acids refer to nucleic acid sequences that are closely related to a
particular sequence
but which may possess, either naturally or by design, changes in sequence or
structure. By
"closely related", it is meant that at least about 60%, but often, more than
85%, of the
nucleotides of the sequence match over the defined length of the nucleic acid
sequence.
Changes or differences in nucleotide sequence between closely related nucleic
acid sequences
may represent nucleotide changes in the sequence that arise during the course
of normal
replication or duplication in nature of the particular nucleic acid sequence.
Other changes
may be specifically designed and introduced into the sequence for specific
purposes. Such
specific changes may be made in vitro using a variety of mutagenesis
techniques. Such
sequence variants generated specifically may be referred to as "mutants" or
"derivatives" of
the original sequence.
[0043] A skilled artisan likewise can produce protein variants having single
or multiple
amino acid substitutions, deletions, additions or replacements. These variants
may include
inter alia: (a) variants in which one or more amino acid residues are
substituted with
conservative or non-conservative amino acids; (b) variants in which one or
more amino acids
are added; (c) variants in which at least one amino acid includes a
substituent group; (d)
variants in which amino acid residues from one species are substituted for the
corresponding
residue in another species, either at conserved or non-conserved positions;
and (d) variants in
which a target protein is fused with another peptide or polypeptide such as a
fusion partner, a
protein tag or other chemical moiety, that may confer useful properties to the
target protein,
Page 9 of 30
CA 02577180 2007-02-14
WO 2006/055292 PCT/US2005/040107
such as, for example, an epitope for an antibody. The techniques for obtaining
such variants,
including genetic (suppressions, deletions, mutations, etc.), chemical, and
enzymatic
techniques are known to the skilled artisan.
[0044] As used herein, the terms "vector" and "expression vector" refer to a
replicon, i.e.,
any agent that acts as a carrier or transporter, such as a phage, plasmid,
cosmid, bacmid,
phage or virus, to which another genetic sequence or element (either DNA or
RNA) may be
attached so as to bring about the replication of the attached sequence or
element and so that
sequence or element can be conveyed into a host cell. The E. coli SPP system
described
herein utilizes pColdI vectors, which induce protein production at low
temperatures.
[0045] It must be noted that as used herein and in the appended claims, the
singular forms
"a", "and", and "the" include plural referents unless the context clearly
dictates otherwise.
All technical and scientific terms used herein have the same meaning.
[0046] Unless defined otherwise, all technical and scientific terms used
herein have the
same meaning as commonly understood by one of ordinary skill in the art to
which this
invention belongs. Although any methods and materials similar or equivalent to
those
described herein can also be used in the practice or testing of the present
invention, the
preferred methods and materials are now described. All publications mentioned
herein are
incorporated herein by reference to disclose and describe the methods and/or
materials in
connection with which the publications are cited.
EXAMPLES
[0047] The following examples are put forth so as to provide those of ordinary
skill in the
art with a complete disclosure and description of how to make and use the
present invention,
and are not intended to limit the scope of what the inventors regard as their
invention nor are
they intended to represent that the experiments below are all or the only
experiments
performed. Efforts have been made to ensure accuracy with respect to numbers
used (e.g.
amounts, temperature, etc.) but some experimental errors and deviations should
be accounted
for. Unless indicated otherwise, parts are parts by weight, molecular weight
is weight
average molecular weight, temperature is in degrees Centigrade, and pressure
is at or near
atmospheric.
[0048] Strains and Plasmids
Page 10 of 30
CA 02577180 2007-02-14
WO 2006/055292 PCT/US2005/040107
[0049] E. coli BL21(DE3) cells were used in the experiments described below.
The mazF
gene was cloned into the Ndel-Xhol sites of pACYCDuet (Novagen) to create
plasmid
pACYCmazF. pACYCmazF(-9ACA) was constructed by site-directed mutagenesis using
pACYCmazF as template. The eotaxin gene was synthesized on the basis of the
optimal E.
coli codon usage (See Figure 2A) and cloned into the Ndel-Hindlll sites of
pColdI(SP-1) to
create plasmid pColdl(SP-1)eotaxin. pCo1dI(SP-1)eotaxin was constructed as
described in the
text by site-directed mutagenesis using pCo1dl(eotaxin) as template.
Mutagenesis was carried
out using Pfu DNA polymerase (Stratagene) according to the instructions for
the
QuickChange Site-Directed Mutagenesis Kit (Stratagene). pCo1dl(SP-2)eotaxin
was also
constructed by site-directed mutagenesis using pCo1dl(SP-1)eotaxin as
template. pCo1dl(SP-
1)eotaxin(+ACA) was constructed by site-directed mutagenesis using pColdl(SP-
1)eotaxin as
template. The wild-type HsplO gene was amplified by PCR with Yeast chromosome
as
template and cloned into the Ndel-BamHI sites of pCo1dI(SP-2) to create
plasmid pCo1dI(SP-
2)Hsp10. The ACA-less HsplO gene was amplified by two-step PCR with Yeast
chromosome as template and cloned into the Ndel-Ban1HI sites of pCo1dl(SP-2)
to create
plasmid pColdI(SP-2)Hsp10(-ACA). The wild-type and ACA-less Rpbl2 gene was
amplified
by PCR with wild type Rpb 12 plasmid as template and 5' and 3'
oligonucleotides containing
the altered sequence cloned into the Ndel-BamHI sites of pColdI(SP-2) to
create plasmid
pCo1dI(SP-2)Rpb12 and pCo1dI(SP-2)Rpb12(-ACA), respectively. The ACA-less LspA
gene
was amplified by two-step PCR and cloned into the NdeI-BamHI sites of
pColdIV(SP-2) to
create plasmid pCo1dIV(SP-2)1spA(-ACA).
[0050] Assays of Protein Synthesis in Vivo
[0051] E. coli BL21(DE3) carrying plasmids was grown in M9-glucose medium.
When
the OD600 of the culture reached 0.5, the culture was shifted to 15 C for 45
min and 1 mM of
IPTG was added to the culture. At the indicated time intervals, 1 ml of
culture was added to
a test tube containing 10 mCi [35S]-methionine. After incubation for 15 min
(pulse), 0.2 ml
of 40 mg/ml methionine was added and incubated for another 5 min (chase). The
labeled
cells were washed with M9-glucose medium and suspended in 100 l of SDS-PAGE
loading
buffer. 10 l of each sample was analyzed by SDS-PAGE followed by
autoradiography.
[0052] Preparation of the Membrane Fraction
[0053] The cells harvested from 1 ml of culture by centrifugation (10,000 x g
for 5min)
were suspended in the 10 mM Tris-HCI (pH 7.5) and disrupted by sonication. The
total
Page 11 of 30
CA 02577180 2007-02-14
WO 2006/055292 PCT/US2005/040107
membrane fraction was obtained by centrifugation (100,000 x g, for 60 min)
after the
removal of unbroken cells.
[0054] Examnle 1. Effects of MazF Induction of Cellular Protein Synthesis
[0055] E. coli BL21(DE3) carrying pACYCmazF was transformed either with
pCo1dI(SP-1)eotaxin (A and left panel in B) or pColdI(SP-2)eotaxin (right
panel in B and C).
Cells were grown in M9 medium at 37 C. At OD6o of 0.5, the cultures were
shifted to 15 C
and after incubation at 15 C for 45 min to make cells acclimate low
temperature, IPTG (1
mM) was added to induce both eotaxin and MazF expression (0 time). Cells were
pulse-
labeled with 35S-methionine for 15 min at the time points indicated on top of
each gel and
total cellular proteins were analyzed by SDS-polyacrylaminde gel
electrophoresis (PAGE)
followed by autoradiography.
[0056] The mazF gene was cloned into pACYC, a low copy number plasmid
containing
an IPTG inducible phage T7 promoter, yielding pACYCmazF. Cloning techniques
generally
may be found in J. Sambrook and D.W. Russell, Molecular Cloning: A Laboratory
Manual,
Third Edition, Cold Spring Harbor Press, Cold Spring Harbor, N.Y. (2001),
which is
incorporated herein by reference. E. colt BL21 (DE3) transformed with
pACYCmazF was
sensitive to IPTG, a lac inducer, as no colonies were formed on agar plates
containing IPTG
(not shown).
[0057] Figure 1 shows the expression of Human Eotaxin with Use of pColdI(SP-1)
and
pColdI(SP-2) with and without MazF coexpression by SDS-PAGE. Figure 1B shows
the
results for cells transformed with pCo1dI(SP-1)eotaxin (left panel); and
transformed with
pColdI(SP-2)eotaxin (right panel). Figure 1C shows the results for cells
transformed with
pACYCmazF and pCo1dI(SP-2)eotaxin were incubated in LB (left panel) or M9
medium(right panel). Cells were treated in the same manner as in Figure lA and
Figure 1B,
and, at the time points indicated, total cellular proteins were analyzed by
SDS-PAGE
followed by Coomassie Blue staining. Note that the same volumes of the
cultures were taken
for the analysis. Positions of molecular weight markers are shown at the left
hand side of the
gels and the position of eotaxin is indicated by an arrow. As MazF effectively
cleaves
mRNAs at ACA sequences, cellular protein synthesis was dramatically inhibited
at 37 C
upon MazF induction (Zhang et al., Mol. Cell 12: 913-23 (2003)) or at 15 C as
shown in
Figure 1A. In this cold-shock experiment, cells carrying pACYCmazF were first
incubated
Page 12 of 30
CA 02577180 2007-02-14
WO 2006/055292 PCT/US2005/040107
for 45 min at 15 C to induce cold-shock proteins required for cold-shock
acclimation (see
Thieringer et al., Bioassays 20(1): 49-57 (1998)). Then IPTG was added to the
culture to
induce MazF (0 time in Figure lA, left panel). Cells were pulse-labeled with
[35S]
methionine for 15 min at the time points indicated on top of the gel. Panel A
left panel shows
the results for cells transformed only with pACYCeotaxin; panel A middle panel
shows the
results for cells transformed only with pCold(SP-1)eotaxin; and Panel A right
panel shows
the results for cells transformed with both plasmids.
[0058] At 0 time, a very similar protein pattern was observed as that of the
cells in the
absence of IPTG (control, indicated as C), while cellular protein synthesis
was dramatically
inhibited at 1 hr after the addition of IPTG. After 6 hr, the synthesis of
almost all cellular
proteins was ahnost completely blocked.
[0059] Example 2. Expression of an ACA-less mRNA in MazF-induced Cells
[0060] We speculated that if an mRNA that is engineered to contain no ACA
sequences
is expressed in MazF-induced cells, the mRNA might be stably maintained in the
cells so that
the protein encoded by the mRNA may be produced without producing any other
cellular
proteins. To test this possibility, we synthesized the gene for human eotaxin,
eliminating all
ACA sequences in the gene without altering the amino acid sequence. Fig. 2A
shows the
amino acid sequence of human eotaxin and the nucleotide sequences of its gene.
The
nucleotide sequence was designed using preferred E. coli codons and those
triplets underlined
were changed to ACA in the experiment below. The ACA sequence is unique among
64
possible triplet sequences, as it can be altered to other MazF-uncleavable
sequences without
changing the amino acid sequence of a protein regardless of the position of an
ACA sequence
in a reading frame.
[0061] The eotaxin gene shown in Figure 2A was fused with a 17-residue
sequence
consisting of a sequence from a translation enhancing element from the cspA
gene for the
major cold-shock protein, CspA (Qing et al, Nat. Biotechnol. 22: 877-882
(2004)), 6 His
residues, factor Xa cleavage site and the His-Met sequence derived from the
Ndel site for
gene insertion. The entire coding region for the fusion protein was inserted
into pColdI(SP-
1) and pColdI(SP-2) vectors, cold-shock vectors allowing a high protein
expression upon
cold shock (Qing et al, Nat. Biotechnol. 22: 877-882 (2004)). In pCold(SP-1)
two ACA
sequences, one between the Shine-Dalgarno sequence and the initiation codon
and the other
in the translation enhancing element were converted to AUA. In pColdl(SP-2) in
addition to
Page 13 of 30
CA 02577180 2007-02-14
WO 2006/055292 PCT/US2005/040107
the two ACA sequences in pColdI(SP-1) three other ACA sequences in the 5'-
untranslated
region (5'-UTR) also were altered to MazF-uncleavable sequences by base
substitutions (to
GCA, AUA and GCA from the 5' ACA to the 3' ACA, respectively). The resulting
constructs, pColdI(SP-1) eotaxin and pColdl(SP-2)eotaxin, respectively, were
transformed
into E. coli BL21 (DE3) cells.
[0062] After the cells transformed with pColdI(SP-1)eotaxin were cold-shocked
at 15 C
and acclimated to the low temperature for 1 hr, IPTG was added to induce
eotaxin
production. Cells then were pulse-labeled with [35S]methionine for 15 min (0
time; Figure
1A, middle panel). Eotaxin was produced almost at a constant level from 0 time
during 72 hr
incubation together with other cellular proteins. The production of eotaxin at
the 12 hr time
point was approximately 11% of total cellular protein synthesis as judged from
[35S]methionine incorporation.
[0063] When both eotaxin and mazF genes were coexpressed using E. coli BL21
(DE3)
harboring both pACYCmazF and pColdI(SP-1)eotaxin, background cellular protein
synthesis
was dramatically reduced after 3 hr induction, while eotaxin production
continued for 72 hr at
an almost constant level (Figure 1A, right panel). Interestingly the level of
eotaxin production
in this experiment was higher (Figure 1A, right panel; 11% of total protein
production at 12
hr) than that in the absence of MazF induction (Figure 1A, middle panel; 47%
at 12 hr). This
approximately 5 fold enrichment is likely due to the fact that more ribosomes
became
available for eotaxin mRNA translation as cellular mRNAs were degraded by
MazF. Notably,
no specific protein bands were observed after the 12 hr time point.
[0064] When the identical experiment was carried out with the cells harboring
both
pACYCmazF and pColdI(SP-2)eotaxin, eotaxin was almost exclusively
produced(Figure 1B,
right panel). Notably, eotaxin production was substantially higher than that
with pColdl(SP-
1)eotaxin (Figure 1B, left panel). This higher production of eotaxin is likely
due to the
stabilization of the eotaxin mRNA by further removal of ACA sequences in the
5'-UTR in
pColdI(SP-1). Approximately 90% of [35S]methionine was incorporated into
eotaxin at 12 hr
after MazF induction and notably no distinct cellular protein bands were
discernible (Figure
1B, right panel) indicating that the signal-to-noise ratio of eotaxin was
dramatically improved
by the present SPP system. It is interesting to note that the high level of
eotaxin production
did not diminish even 96 hr after induction. Furthermore, background cellular
protein
Page 14 of 30
CA 02577180 2007-02-14
WO 2006/055292 PCT/US2005/040107
synthesis diminished sooner (at 3 hr) than that with pColdl(SP-1)eotaxin (at 6
hr) (compare
the left panel with the right panel in Figure 1B).
[0065] With both vectors (Figure 1A and B), cell growth was completely blocked
upon
MazF induction as judged by OD600 and also by [35S]methionine incorporation
into cellular
proteins. These results indicate that growth-arrested cells by MazF induction
are not
physiologically dead and instead are fully capable of synthesizing proteins if
their mRNAs
have no ACA sequences. This in turn indicates that the cellular integrity of
the E. coli BL21
(DE3) cells is kept intact for a long period of time so that not only energy
metabolism but
also biosynthetic functions for amino acids and nucleotides are fully active
in the growth-
arrested cells. Furthermore, transcriptional and translational machineries are
also well
maintained including RNA polymerase, ribosomes, tRNA, and all the other
factors required
for protein synthesis.
[0066] The production of eotaxin with pColdl(SP-2) eotaxin appears as a major
band by
Coomassie Blue staining after SDS polyacrylamide gel electrophoresis (Figure 1
C). At the 0
hr time point, the eotaxin band was barely discemable while at 12 hr it became
the major
band and its density increased even more after 24 hr. However, longer
incubation did not
significantly enhance the level of its production, suggesting that there is a
threshold level of
eotaxin production in MazF-induced cells. Since the [35S]methionine
incorporation was
constantly maintained for 96 hr (Figure. 1B), its seems that eotaxin
production and
degradation in the SPP system may equilibrate after 24 hr. It is important to
note that the
density of the bands for cellular proteins remained constant as expected from
complete
growth inhibition upon MazF induction. We examined if eotaxin production is
affected by
rich media such as LB medium and found that the use of LB medium did not
enhance eotaxin
production any more than the level obtained with defined M9 medium if
pColdI(SP-2) was
used.
[0067] Example 3. The Negative Effect of ACA Sequences on Protein Production
[0068] In order to confirm that the exclusive eotaxin production in MazF-
induced cells
observed in Figure 1 is due to the ACA-less mRNA for eotaxin, the five native
ACA
sequences were added to the eotaxin gene without altering its amino acid
sequence as shown
in Figure 2A. The eotaxin genes were expressed with use of pColdI(SP-2) and
cells were
treated and labeled with [35S] -methionine in the same manner as described in
Figure 1. The
Page 15 of 30
CA 02577180 2007-02-14
WO 2006/055292 PCT/US2005/040107
left panel shows the results for the ACA-less eotaxin gene (same as the left
panel of Figure 1
B) and the right panel shows the results for the eotaxin gene with 5 ACA
sequences.
[0069] When this gene was expressed with use of pCo1dl(SP-1) together with
pACYCmazF under the same condition as described for Figure 1, only a low level
of eotaxin
production was observed for the first 2 hours after which point the production
was further
reduced to a background level (Figure 2B, right panel) in comparison with the
expression
with the ACA-less mRNA (Figure 2B, left panel).
[0070] Curiously, the mazF gene encodes an mRNA that has an unusually high ACA
content (9 ACA sequences for a 111 residue protein)--in a dramatic contrast to
MazE (82
amino acid residues with only 2 ACA sequences)--suggesting that mazF
expression is
negatively regulated in cells. Therefore, we constructed the mazF gene with no
ACA
[pACYCmazF(-9ACA)] and tested whether the removal of these ACA sequences from
the
mazF coding region may cause more effective reduction of background cellular
protein
production.
[0071] Fig. 3 shows the effect of removal of all ACA sequences in the mazF ORF
on
eotaxin expression. Panel A shows the amino acid sequence of MazF and the
nucleotide
sequence of its ORF. The triplet sequences underlined (a total of nine) were
originally ACA
in the wild-type mazF gene, which were changed to MazF-uncleavable sequences.
Panel B
shows the expression of eotaxin with pColdl(SP-2)eotaxin using the wild-type
mazF gene
(left panel) and ACA-less mazF gene (right panel). The experiments were
carried as
described in Figure 1.
[0072] As shown in Figure 3A, none of the base substitutions alter the amino
acid
sequence of MazF. Although cells harboring pYCACmazF(-9ACA) grew a little
slower than
cells harboring pYCACmazF in M9 medium, the background protein synthesis was
further
reduced without significant effects on the eotaxin production (Figure 3B).
These results
clearly demonstrate that ACA sequences in mRNAs play the crucial role in
protein
production in MazF-induced cells.
[0073] Example 4. Application of the SPP System to Yeast Proteins
[0074] We applied the SPP system to two yeast proteins: HsplO, a heat-shock
factor and
Rpbl2, an RNA polymerase subunit. The ORFs for HsplO and Rpbl2 contain 3 and 1
ACAs, respectively, which were converted to MazF-uncleavable sequences without
altering
their amino acid sequences (Figure 4A). They, together with the wild-type
sequences, then
Page 16 of 30
CA 02577180 2007-02-14
WO 2006/055292 PCT/US2005/040107
were inserted into pColdI(SP-2). The resulting plasmids were termed pCo1dI(SP-
2)Hsp10 for
the wild-type HsplO, pColdl(SP-2)Hsp10(-lACA) for the mutant Hsp10, pCo1dl(SP-
2)Rpb12
for the wild-type Rpbl2 and pColdI(SP-2)Rpbl2(-3ACA), respectively. These
plasmids
were individually transformed into E. coli BL21(DE3) harboring pACYCmazF.
Protein
expression patterns then were examined for 48 hours at 15 C.
[0075] The expression of Yeast Proteins in the SPP System is shown in Figure
4. Using
pCo1dI(SP-2), yeast Hsp10 and Rpb12 were expressed in the SPP system in the
presence and
the absence of ACA sequences in their genes. Experiments were carried out as
described
supra for Figure 1. Figure 4A shows the expression of HsplO using the wild-
type and ACA-
less HsplO genes. The hsp 10 ORF consisting of 106 codons contains 3 ACA
sequences;
GCA-CAA for A25-Q26, ACA for T29 and CCA-CAG for P76-Q77, which were converted
to GCC-CAA, ACC and CCC-CAG, respectively (altered bases are in bold). These
base
substitutions do not alter the amino acid sequence of Hsp 10. Figure 4B shows
the expression
of Rpbl2 using the wild-type and ACA-less genes. The rpbl2 ORF consisting of
70 codons
contains one ACA for T10, which was converted to ACC for threonine.
[0076] Figure 4A shows that Hsp10 can be expressed with its native 3 ACA
sequences
(WT) at a reasonably high level. However when all the ACA sequences were
removed,
Hsp10 synthesis significantly enhanced a few fold. Noticeably, the background
was also
significantly reduced with the ACA-less Hsp10, likely because more ribosomes
were
dedicated for the production of HsplO. Figure 4B shows that although Rpbl2
contains only
one ACA, it causes a devastating effect on its production in the SPP system,
as little 355-
methionine incorporation was observed in the WT panel while reasonable
incorporation was
seen in the ACA-less Rpbl2. These results suggest that mRNA sensitivity to
MazF may be
governed, not only by the number of ACA sequences in an mRNA, but also by
effective
susceptibility of an ACA sequence to MazF. It is likely that the ACA sequence
susceptibility
is determined by its location in a single-stranded region of an inRNA as well
as the effective
translation of an mRNA by ribosomes, as ribosomes are assumed to protect the
inRNA from
its cleavage by MazF.
[0077] Example 5. Application of the SPP System to an Integral Membrane
Protein
[0078] We attempted to apply the SPP system to a minor integral membrane
protein. We
chose the gene 1spA for signal peptidase II in E. coli, which is specifically
required for
cleavage of the signal peptides of lipoproteins (Tokuda and Matsuyarna,
Biochem. Biophys.
Page 17 of 30
CA 02577180 2007-02-14
WO 2006/055292 PCT/US2005/040107
Acta 1693: 5-13 (2004)). E. coli contains a total of 96 lipoproteins, which
are known to
assemble either in the inner membrane or in the outer membrane depending upon
the nature
of the second amino acid residue (acidic or neutral) of the mature
lipoproteins (Yamaguchi
and Inouye, Cell 53: 423-432 (1988); Tokuda and Matsuyama, Biochem. Biophys.
Acta
1693: 5-13 (2004)). The signal peptides of all the other secreted proteins are
cleaved by
signal peptidase I (leader peptidase) , which is estimated to exist only at a
level of 500
molecules per cell in E. coli (Wolfe et al., J. Biol. Chem. 257: 7898-7902
(1982)).
[0079] Lipoprotein Signal Peptidase (LspA) also is considered to be a very low
abundant
protein in the inner membrane. It consists of 164 amino acid residues and
contains four
presumed transmembrane domains, indicating that LspA is an integral inner
membrane
protein. Three ACA sequences in the IspA ORF were altered to non-MazF-
cleavable
sequences without changing its amino acid sequence and the ACA-less LspA was
expressed
using pColdl(SP-2) in the SPP system using mazF(-9ACA).
[0080] The expression of LspA, an inner membrane protein in the SPP system
using
pColdL(SP-2) are shown in Fig. 5. LspA, signal peptidase II or lipoprotein
signal peptidase
was expressed in the SPP system as described in Figure 1. Panel A shows total
cellular
proteins; and Panel B shows the membrane fraction: The position of LspA is
shown by an
arrow.
[0081] As shown in Figure 5A, the expression of LspA in the SPP system
apparently is
toxic to the cells, as 35S-methionine incorporation lasted only 1 hour after
IPTG induction.
Nevertheless, as shown in Figure 5B, a reasonable 35S-methionine incorporation
into LspA
appears to be achieved as the LspA band densities at 0 and 1 hr time points
were the highest
comparing them with other cellular protein bands (compare with the C lane in
Figure 5A).
The background cellular protein synthesis observed at 0 and 1 hr was easily
removed by
ultracentrifugation, and 35S-methionine incorporation was highly enriched in
the membrane
fraction.
[0082] Discussion
[0083] The present work demonstrates that complete inhibition of cellular
protein
synthesis by an mRNA interferase does not cause deteriorating effects on the
cellular
physiology. As a result of fragmentation of almost all cellular mRNAs by MazF
at ACA
sequences, cellular protein synthesis is completely blocked, which in turn
leads to complete
cell growth arrest. However, to our surprise, growth arrested cells by MazF
induction were
Page 18 of 30
CA 02577180 2007-02-14
WO 2006/055292 PCT/US2005/040107
found to be fully capable of synthesizing proteins at a high level for a long
period of time (at
least 96 hr at 15 C) if their mRNAs are engineered to have no ACA sequences.
In this
fashion we have achieved for the first time to establish the single-protein
production (SPP) in
vivo.
[0084] Our results demonstrate that MazF-induced cells are not dead. MazF
induction
does not hamper cellular integrity maintaining energy metabolism producing
enough ATP
required various cellular functions including RNA and protein synthesis. In
addition
biosynthesis of amino acids and nucleotides are also maintained intact. It is
quite surprising
to find that in the complete absence of new cellular protein synthesis, all
the protein factors
required for these cellular functions (for example protein factors required
for protein
synthesis) and cellular metabolisms are stably maintained at least 96 hours at
15 C. It
remains to be determined how long these cellular functions could be retained
without
affecting the SPP capability. Although at a glance they appear to be in a
dormant state, they
are fully capable of RNA and protein synthesis and distinctly different from
the dormancy
caused by the stationary phase due to nutritional deprivation. We propose to
term the
physiological state created by MazF induction "quasi-dormant" state. It
remains to be
determined if the quasi-dormant cells are dead or undead. Bacterial viability
is often
determined by the colony forming ability of cells after various treatments.
The viability of E.
coli cells after MazF induction has been examined in this fashion and shown to
be resumed
during limited time incubation after MazF induction if MazE is induced
(Pedersen et al., Mol.
Microbiol. 45: 501-10 (2002); Amitai et al., J. Bacteriol. 186: 8295-8300
(2004)).
Therefore, the effect of MazF is reversible to a certain extent, however it
has been argued that
there is 'a point of no return', from which point all cells are destined to
die (Amitai et al., J.
Bacteriol. 186: 8295-8300 (2004)). Importantly, the MazE gene used by both
group contains
two ACA sequence in its ORF. The present results clearly indicate that in
order for any
genes to be expressed in MazF-induced cells, ACA sequences in these genes have
to be
converted to MazF-uncleavable sequences. Therefore it is highly possible that
the quasi-
dormant cells expressing MazF cannot express MazE unless all the ACA sequences
are
eliminated from its ORF.
[0085] The ability to produce only a single protein of interest in living
cells or undead
cells provides a novel approach for studying the various aspects of proteins
in living cells
previously unattainable. Since by using the SPP system a protein of interest
can be
Page 19 of 30
CA 02577180 2007-02-14
WO 2006/055292 PCT/US2005/040107
exclusively labeled with isotopes (15N and 13C) in living cells, it may be
even possible to
examine NMR structures of proteins in living cells. Recently we have shown
that NMR
structural determination of a protein can be achieved using cell lysates
without protein
purification by expressing a protein of interest by high expression cold-shock
vectors, pCold
(Qing et al., Nat. Biotechnol. 22: 877-882 (2004)). We now demonstrate that
the use of
MazF together with pCold vectors dramatically reduces the signal-to-noise
ratio as the
background cellular protein synthesis can be almost completely blocked by MazF
induction.
In these experiments we showed that the removal of ACA sequences from pCo1dI
vector
itself is also very important by which 5 fold improvement of eotaxin
production was
observed. When combined with MazF, the rate of eotaxin synthesis was at the
leve190% of
the total cellular protein synthesis as judged by 35S-methionine
incorporation. The remaining
10% consisted of a general background without incorporation into any specific
protein bands.
This in turn enables one to perform the structural study of very low abundant
proteins, whose
production is limited because of their toxicity when expressed in a large
quantity. We indeed
demonstrated in the present paper that LspA, a very low abundant inner
membrane protein,
can be exclusively expressed in the membrane fraction. Some proteins may be
folded only in
living cells, whose structural study may be achieved only by the use of the
SPP system.
[0086] Another unique advantage of the SPP system is that a protein of
interest can be
produced or labeled with isotopes in a highly concentrated culture as cell
growth is
completely blocked upon MazF induction. It is possible that the SPP system can
be applied
for the production of not only proteins but also other non-protein compounds.
Furthermore
the SPP system may not be limited only to bacteria, and MazF and other mRNA
interferases
may be applied for eukaryotic cells to create the SPP systems in yeast and
mammalian cells.
[0087] Where a range of values is provided herein, it is understood that each
intervening
value, to the tenth of the unit of the lower limit unless the context clearly
dictates otherwise,
between the upper and lower limit of that range and any other stated or
intervening value in
that stated range is encompassed within the invention. The upper and lower
limits of these
smaller ranges which may independently be included in the smaller ranges is
also
encompassed within the invention, subject to any specifically excluded limit
in the stated
range. Where the stated range includes one or both of the limits, ranges
excluding either both
of those included limits are also included in the invention.
Page 20 of 30
CA 02577180 2007-02-14
WO 2006/055292 PCT/US2005/040107
[0088] The publications discussed herein are provided solely for their
disclosure prior to
the filing date of the present application. Nothing herein is to be construed
as an admission
that the present invention is not entitled to antedate such publication by
virtue of prior
invention. Further, the dates of publication provided may be different from
the actual
publication dates which may need to be independently confirmed.
[0089] While the present invention has been described with reference to the
specific
embodiments thereof, it should be understood by those skilled in the art that
various changes
may be made and equivalents may be substituted without departing from the true
spirit and
scope of the Invention. In addition, many modifications may be made to adapt a
particular
situation, material, composition of matter, process, process step or steps, to
the objective,
spirit and scope of the present invention. All such modifications are intended
to be within the
scope of the claims appended hereto.
Page 21 of 30
DEMANDE OU BREVET VOLUMINEUX
LA PRESENTE PARTIE DE CETTE DEMANDE OU CE BREVET COMPREND
PLUS D'UN TOME.
CECI EST LE TOME 1 DE 2
CONTENANT LES PAGES 1 A 21
NOTE : Pour les tomes additionels, veuillez contacter le Bureau canadien des
brevets
JUMBO APPLICATIONS/PATENTS
THIS SECTION OF THE APPLICATION/PATENT CONTAINS MORE THAN ONE
VOLUME
THIS IS VOLUME 1 OF 2
CONTAINING PAGES 1 TO 21
NOTE: For additional volumes, please contact the Canadian Patent Office
NOM DU FICHIER / FILE NAME:
NOTE POUR LE TOME / VOLUME NOTE: