Note: Descriptions are shown in the official language in which they were submitted.
CA 02577543 2007-01-31
DEMANDES OU BREVETS VOLUMINEUX
LA PRESENTE PARTIE DE CETTE DEMANDE OU CE BREVETS
COMPREND PLUS DIN TOME.
=
CECI EST LE TOME DE A_
NOTE: Pour les tomes additionels, veillez contacter le Bureau Canadien des
Brevets.
JUMBO APPLICATIONS / PATENTS
THIS SECTION OF THE APPLICATION / PATENT CONTAINS MORE
THAN ONE VOLUME.
THIS IS VOLUME OF
NOTE: For additional volumes please contact the Canadian Patent Office.
. .
CA 02577543 2007-01-31
WO 9951642
PCT/US99/06858
ANTIBODY VARIANTS AND FRAGMENTS THEREOF
BACKGROUND OF THE INVENTION
Field of the Invention
The present invention concerns variants of polypeptides comprising an Fc
region. More
particularly, the present invention concerns Fc region-containing polypeptides
that have altered effector
function as a consequence of one or more amino acid substitutions in the Fc
region of the nonvariant
polypeptide. The invention also relates to novel immune complexes and an assay
for determining
binding of an analyte, such as an Fc region-containing polypeptide, to a
receptor.
Description of Related Art
Antibodies are proteins, which exhibit binding specificity to a specific
antigen. Native antibodies
are usually heterotetramericglycoproteins of about 150,000 dattons, composed
of two identical light (L)
chains and two identical heavy (H) chains. Each light chain is linked to a
heavy chain by one covalent
disulfide bond, while the number of disulfide linkages varies between the
heavy chains of different
immunoglobulin isotypes. Each heavy and fight chain also has regularly spaced
intrachain disulfide
bridges. Each heavy chain has at one end a variable domain (VH) followed by a
number of constant
domains. Each light chain has a variable domain at one end (VL) and a constant
domain at its other end;
the constant domain of the light chain is aligned with the first constant
domain of the heavy chain, and
the light chain variable domain is aligned with the variable domain of the
heavy chain. Particular amino
acid residues are believed to form an interface between the light and heavy
chain variable domains.
The term "variable" refers to the fact that certain portions of the variable
domains differ
extensively in sequence among antibodies and are responsible for the binding
specificity of each
particular antibody for its particular antigen. However, the variability is
not evenly distributed through the
variable domains of antibodies. It is concentrated in three segments called
complementarity determining
regions (CDRs) both in the light chain and the heavy chain variable domains.
The more highly
conserved portions of the variable domains are called the framework regions
(FR). The variable
domains of native heavy and light chains each comprise four FR regions,
largely adopting a 0-sheet
configuration, connected by three CDRs, which form loops connecting, and in
some cases forming part
of, the p-sheet structure. The CDRs in each chain are held together in close
proximity by the FR regions
and, with the CDRs from the other chain, contribute to the formation of the
antigen binding site of
antibodies (see Kabat et al. Sequences of Proteins of Immunological Interest,
5th Ed. Public Health
Service, National Institutes of Health, Bethesda, MD. (1991)).
The constant domains are not involved directly in binding an antibody to an
antigen, but codlibit
various effector functions. Depending on the amino acid sequence of the
constant region of their heavy
chains, antibodies or immunoglobulins can be assigned to different classes.
There are five major
classes of immunoglobutins:IgA, IgD, IgE, IgG and IgM, and several of these
may be further divided into
subclasses (isotypes), e.g. IgG1, IgG2, IgG3, and 1gG4; IgAl and IgA2. The
heavy chain constant
regions that correspond to the different classes of immunoglobulins are called
a-8, c, y, and p,
CA 02577543 2007-01-31
WO 99/51642
PCT/US99/(105.55
respectively. Of the various human immunoglobulin classes. only human IgG1,
IgG2, IgG3 and IgM are
known to activate complement
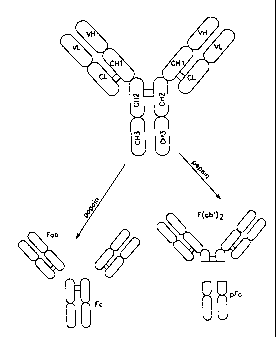
A schematic representation of the native IgG1 structure is shown in Fig. 1,
where the various
portions of the native antibody molecule are indicated. Papain digestion of
antibodies produces two
identical antigen binding fragments, called Feb fragments, each with a single
antigen binding site, and
a residual "Fc" fragment. whose name reflects its ability to crystallize
readily. The crystal structure of
the human lgG Fc region has been determined (Deisenhofer, Biochemistry 20:2361-
2370 (1981)). In
human IgG molecules the Fc region is generated by papain cleavage N-terminal
to Cys 226. The Fc
region is central to the effector functions of antibodies.
The effector functions mediated by the antibody Fc region can be divided into
two categories:
(1) effector functions that operate after the bin-ding of antibody to an
antigen (these functions involve the
participation of the complement cascade or Fc receptor (FcR)-bearing cells);
and (2) effector functions
that operate independently of antigen binding (these functions confer
persistence in the circulation and
the ability to be transferred across cellular barriers by transcytosis). Ward
and Ghetie, Therapeutic
Immunology 2:77-94 (1995).
While binding of an antibody to the requisite antigen has a neutralizing
effect that might prevent
the binding of a foreign antigen to its endogenous target (e.g. receptor or
ligand), binding alone may not
remove the foreign antigen. To be efficient in removing and/or destructing
foreign antigens, an antibody
should be endowed with both high affinity binding to its antigen, and
efficient effector functions.
Cla binding
Clq and two serene proteases, C1r and Cis, form the complex Cl, the first
component of the
complement dependent cytotoxicity (CDC) pathway. Clq is a hexavalent molecule
with a molecular
weight of approximately 460,000 and a structure likened to a bouquet of tulips
in which six collagenous
"stalks" are connected to six globular head regions. Burton and Woof, Advances
in immunol. 51:1-84
(1992). To activate the complement cascade, it is necessary for C1q to bind to
at least two molecules
of IgG1, IgG2, or IgG3 (the consensus is that IgG4 does not activate
complement), but only one
molecule of 1gM, attached to the antigenic target Ward and Ghetie, Therapeutic
Immunology 2:77-94
(1995) at page 80.
Based upon the results of chemical modifications and crystallographic studies,
Burton et al.
(Nature, 288:338-344 (1980)) proposed that the binding site for the complement
subcomponent Clq on
IgG involves the last two (C-terminal) 13-strands of the CH2 domain. Burton
later suggested (Motec.
immunot, 22(3):161-206 (1985)) that the region comprising amino acid residues
318 to 337 might be
involved in complement fixation.
Duncan and Winter (Nature 332:738-40(1988)), using site directed mutagenesis,
reported that
G1u318, Lys320 and Lys322 form the binding site to Clq. The data of Duncan and
Winter were
generated by testing the binding of a mouse IgG2b isotype to guinea pig C1q.
The role of G1u318,
Lys320 and Lys322 residues in the binding of Clq was confirmed by the ability
of a short synthetic
peptide containing these residues to inhibit complement mediated lysis.
Similar results are disclosed
-2-
CA 02577543 2007-01-31
WO 99/51642
rk. 161.'77/woo,'
in U.S. Patent No. 5,648,260 issued on July 15, 1997, and U.S. Patent No.
5,624,821 issued on April
29, 1997.
The residue Pro331 has been implicated in C1q binding by analysis of the
ability of human IgG
subdasses to carry out complement mediated cell iysis. Mutation of Ser331 to
Pro331 in IgG4 conferred
the ability to activate complement. (Tao etal., J. Exp. Med., 178:661-
667(1993); Brekke etal., Eur. J.
lmmunot , 24:2542-47 (1994)).
From the comparison of the data of the Winter group, and the Tao at al. and
Brekke at al.
papers, Ward and Ghetie concluded in their review article that there are at
least two different regions
involved in the binding of Cl q: one on the a-strand of the CH2 domain bearing
the Glu318, Lys320 and
Lys322 residues, and the other on a turn located in close proximity to the
same 0-strand, and containing
a key amino acid residue at position 331.
Other reports suggested that human IgG1 residues Lys235, and G1y237, located
in the lower
hinge region, play a critical role in complement fixation and activation. Xu
et at,J. Immunot 150:152A
(Abstract) (1993). W094/29351 published December 22, 1994 reports that amino
acid residues
necessary for C1q and FcR binding of human IgG1 are located in the N-terminal
region of the CH2
domain, le. residues 231 to 238.
It has further been proposed that the ability of IgG to bind Ciq and activate
the complement
cascade also depends on the presence, absence or modification of the
carbohydrate moiety posVoned
between the two CH2 domains (which is normally anchored at Asn297). Ward and
Ghetie. Therapeutic
Immunology 2:77-94(1995) at page 81.
Fc receotor bindina
Effector functions can also be mediated by the interaction of the Fc region of
an antibody with
Fc receptors (FcRs), which are specialized cell surface receptors on
hematopoietic cells. Fc receptors
belong in the immunoglobulin superfamily, and have been shown to mediate both
the removal of
antibody-coated pathogens by phagocytosis of immune complexes, and the lysing
of erythrocytes and
various other cellular targets (e.g. tumor cells) coated with the
corresponding antibody, via antibody
dependent cell mediated cytotoxicity (ADCC). Van de Winkel and Anderson, J.
Leuk Bid. 49:511-24
(1991).
FcRs are defined by their ipecificity for immunoglobulin isotypes; Fc
receptors for IgG
antibodies are referred to as FcyR, for IgE as FceR, for IgA as FcaR and so
on. Three subclasses of
gamma receptors have been identified: FcyRl (CD64), FcyRII (C032) and FcyRIII
(CD16). Because
each FcyR subclass is encoded by two or three genes, and alternative RNA
spicing leads to multiple
transcripts, a broad diversity in FcyR isoforms exists. The three genes
encoding the FcyRI subclass
(FcyRIA, FcyRIB and FcyRIC) are clustered in region 1q21.1 of the long arm of
chromosome 1; the
genes encoding FcyRII isoforms (FcyRIIA, FcyRIIB and FcyRIIC) and the two
genes encoding FcyRIII
(FcyRIIIA and FcyRIIIB) are all clustered in region 1q22. FcRs are reviewed In
Ravetch and Kinet, Annu.
Rev. Immunol 9:457-92 (1991); Capel etal., Immunomethods 4:25-34(1994); and de
Haas at at, J. Lab.
Clin. Med. 126:330-41 (1995).
-3-
CA 02577543 2007-01-31
WO 99/51642
1-1.- it 11J07,,, vvu.ns
While FcyRI binds monomeric IgG with a high affinity, FcyRII and FcyRIII are
low-affinity
receptors, interacting with complexed or aggregated IgG. The classical method
for detecting these low-
affinity receptors is by "rosetting" using antibody-coated erythrocytes (EA)
sensitized with IgGs. Bredius
at al. evaluated rosette formation between IgG-sensitized red blood cells and
polymorphonuclear
leukocytes (PMN) which express FcyRIla and FcyRIllb at their cell-surface.
Rosette was defined as
three of more EA bound per PMN (Bredius etal. Immunology 83:624-630(1994)).
See, also, Tax etal.
J. lmmunot 133(3):1185-1189 (1984); Nagarajan etal. J. Biol. Chem.
270(43):25762-25770(1995); and
Warmerdam et a/. J. Immunot 147(4):1338-1343(1991) concerning rosette assays.
However, binding
of these EA "immune complexes" to FcR is not easily quantified. Accordingly,
more defined complexes
with detectable affinity for these FcRs have been developed. For example, IgG
dimers have been
formed using anti-light chain monoclonal antibodies (Nagarajan et al., supra
and Warmerdam at al.,
supra) or chemical cross-linking agents (Hogarth etal. lmmunomethods 4:17-24
(1994); and Tamm at
at J. Biol. Chem. 271(7):3659-3666(1996)). Heat-aggregated immune complexes
have also been
evaluated for binding to cells expressing FcRs (Tax at al., supra and Tam at
al., supra).
The binding site for the FcrRs on human IgG was found to reside in the lower
hinge region,
primarily involving residues at amino acid positions 233-238, all of which
were found to be necessary
for full FcyR binding activity. Canfield and Morrison, J. Exp. Med. 173:1483-
91 (1991); Chappel etal.,
Proc. Natl. Acad. Sci. LISA, 88:9036-40(1991); Lund et at, J. Immunot,
147:2657-62(1991); Lund at
Molec. Immunot, 29:53-59 (1992); Jefferis et al., Molex. lmmunot, 27:1237-40
(1990): and Sarmay
et at, Molec. Immunot, 29:633-639 (1992).
Pro331 in IgG3 was changed to Ser, and the affinity of this mutant to target
cells analyzed. The
affinity was found to be six-fold lower than that of unmutated Ig33,
Indicating the involvement of Pro331
in FcyRI binding. Morrison etal., Immunologist, 2:119-124(1994); and Canfield
and Morrison, J. Exp.
Med. 173:1483-91 (1991).
In addition, G1u318 was identified as being involved in binding to FcyRII.
Ward and Ghetie,
Therapeutic Immunology 2:77-94(1995).
SUMMARY OF THE INVENTION
The present invention provides a variant of a polypeptide comprising a human
IgG Fc region,
which variant comprises an amino acid substitution at amino acid position 270
or 329, or at two or more
of amino acid positions 270, 322, 329, and 331 of the human IgG Fc region,
where the numbering of the
residues in the IgG Fc region is that of the Eli index as in Kabat.
The invention further relates to a variant of a polypeptide comprising a human
IgG Fc region,
which variant binds FcyRI, FcyRII, FcyRIII and FcRn but does not activate
complement and comprises
an amino acid substitution at amino acid position 322 or amino acid position
329, or both amino acid
positions of the human IgG Fc region, where the numbering of the residues in
the IgG Fc region is that
of the EU index as in Kabat.
The invention also pertains to a variant of a parent polypeptide comprising a
human IgG Fc
region, which variant has a better binding affinity for human C1q than the
parent polypeptide and
-4-
CA 02577543 2007-01-31
W099/51642 "IN.
===.o
comprises an amino acid substitution in the IgG Fc region. For example, the
binding affinity of the
variant for human C1q may be about two-fold or more improved compared to the
binding affinity of the
parent polypeptide for human Cl q. Preferably the parent polypeptide binds C1q
and mediates CDC (for
example, the parent polypeptide may comprise a human IgG1, IgG2 or IgG3 Fc
region). The variant with
improved C1q binding preferably comprises an amino acid substitution at one or
more of amino acid
positions 326, 327, 333 and 334 of the human IgG Fc region, where the
numbering of the residues in
the IgG Fc region is that of the EU index as in Kabat
In another aspect, the invention provides a variant of a polypeptide
comprising a human IgG Fc
region, which variant comprises an amino acid substitution at amino acid
position 326, 327, 333 or 334
of the human IgG Fc region, where the numbering of the residues in the IgG Fc
region is that of the EU
Index as in Kabat
In yet a further aspect, the invention provides a method for modifying a
polypeptide comprising
a human IgG Fc region comprising substituting an amino acid residue at amino
acid position 270 or 329,
or at two or more of amino acid positions 270, 322, 329, and 331 of the human
IgG Fc region, where the
numbering of the residues in the IgG Fc region is that of the EU index as in
Kabat.
The invention further provides a method for modifying a polypeptide comprising
a human IgG
Fc region comprising substituting an amino acid residue at amino acid position
326, 327, 333 or 334 of
the human IgG Fc region, where the numbering of the residues in the IgG F..-
region is that of the EU
Index as in Kabat. The method optionally further comprises a step wherein a
variant with improved
binding affinity for human C1q is identified.
The invention also provides a composition comprising the polypeptide variant
and a
physiologically acceptable carrier or diluent This composition for potential
therapeutic use is sterile and
may be lyophilized.
Diagnostic and therapeutic uses for the polypeptide variant are contemplated.
In one diagnostic
application, the invention provides a method for determining the presence of a
protein of interest
comprising exposing a sample suspected of containing the protein to the
polypeptide variant and
determining binding of the polypeptide variant to the sample. In one
therapeutic application, the invention
provides a method of treating a mammal suffering from a disorder comprising
administering to the
mammal a therapeutically effective amount of a variant of a polypeptide
comprising a human IgG Fc
region, which variant binds FcyRI, FcyRII, FcyRIII and FcRn but does not
activate complement and
comprises an amino acid substitution at amino acid position 270, 322, 329 or
331 of the human IgG Fc
region, where the numbering of the residues in the IgG Fc region is that of
the EU index as in Kabat
The invention further provides: isolated nucleic acid encoding the polypeptide
variant a vector
comprising the nucleic acid, optionally, operably linked to control sequences
recognized by a host cell
transformed with the vector: a host cell comprising the vector, a process for
producing the polypeptide
variant comprising culturing this host cell so that the nucleic acid is
expressed and, optionally, recovering
the polypeptide variant from the host cell culture (e.g. from the host cell
culture medium).
The invention also pertains to an immune complex comprising: (a) an Fc region-
containing
polypeptide; (b) a first target molecule which comprises at least two binding
sites for the Fc region-
-5-
CA 02577543 2007-01-31
WO 99/51642
ra.. f 0,J07,1vvwftw
containing polypeptide; and (c) a second target molecule comprises at least
two binding sites for the first
target molecule. The immune complex may be used in an FcR-binding assay,
particularly where the FcR
has a low affinity for the Fc region-containing polypeptide. Other uses for
the immune complex are
disclosed herein.
Moreover, the invention provides a method for determining binding of an
analyte, such as an Fc
region-containing polypeptide, to a receptor (e.g. a low affinity FcR)
comprising the following steps
performed sequentially: (a) forming a molecular complex between the analyte
and a first target molecule,
wherein the first target molecule comprises at least two binding sites for the
analyte; and (b) determining
binding of the molecular complex of step (a) to the receptor (e.g. to a
binding domain of the receptor
coated on an assay plate). Optionally, the molecular complex of step (a)
further comprises a second
target molecule which comprises at least two. binding sites for the first
target molecule.
The invention also relates to an assay kit, such as a kit useful for
determining binding of an
analyte to a receptor comprising: (a) a first target molecule which comprises
at least two binding sites
for the analyte; and (b) a second target molecule which comprises at least two
binding sites for the first
target molecule.
Brief Description of the Drawings
Figure 1 is a schematic representation of a native IgG. Disulfide bonds are
represented by
heavy lines between CHI and CL domains and the two CH2 dsmains. V is variable
domain; C is
constant domain; L stands for light chain and H stands for heavy chain.
Figure 2 shows C1q binding of wild type (wt) C288 antibody; C268 antibody with
a human IgG2
constant region (IgG2); and mutants K322A, K320A and E318A.
Figure 3 depicts C1 q binding of mutants P331A, P329A and K322A.
Figures 4A (SEQ ID NO:1) and 4B (SEQ ID NO:2) depict the amino acid sequences
of E27 anti-
IgE antibody light chain (Fig. 4A) and heavy chain (Fig. 4B).
Figure 5 is a schematic diagram of the "immune complex" prepared for use in
the FcR assay
described in Example 1. The hexamer comprising three anti-IgE antibody
molecules (the "Fc region-
containing polypeptide") and three IgE molecules (the "first target molecule")
is shown. IgE has two
"binding sites" for the anti-IgE antibody (E27) in the Fc region thereof. Each
IgE molecule in the complex
is further able to bind two VEGF molecules ("the second target polypeptide").
VEGF has two "binding
sites" for igE.
Figure 6 shows C1q binding results obtained for mutants D270K and D270V
compared to wild
type C2B8.
Figure 7 depicts complement dependent cytotoxicity (CDC) of mutants D270K and
D270V,
compared to wild type C2B8.
Figure 8 shows Cl q binding ELISA results for 293 cell-produced wild type C2B8
antibody (293-
Wt-C2B8), CHO-produced wild type C2B8 antibody (CHO-Wt-C2B8) and various
mutant antibodies.
Figure 9 shows Clq binding ELISA results obtained for wild type (wt) C288 and
various mutant
antibodies as determined in Example 3.
-6-
CA 02577543 2009-04-01
WO 99/51642
PCI7U599/06858
Figure 10 depicts the three-dimensional structure of a human IgG Fc,
highlighting residues:
Asp270, Lys326, Pro329, Pro331, Lys322 and 01u333.
Figure 11 shows Clq binding ELISA results obtained for wild type C2B8 and
various mutant
antibodies as determined in Example 3.
Figure 12 shows CI q binding ELISA results obtained for wild type C2B8 and
double mutants,
K326M-E333S and K326A-E333A.
Figure 13 shows CDC of wild type C2B8 and double mutants, K326M-E333S and
K326A-E333A
Figure 14 depicts C1q binding ELISA results obtained for C2B8 with a human
IgG4 heavy chain
constant region (IgG4), wild type C2B8 (Wt-C2B8), C2B8 with a human IgG2 heavy
chain constant
region (1g02), and mutant antibodies as described in Example 3.
Detailed Description of the Preferred Embodiments
I. Definitions
Throughoutthe present specification and claims, the numbering of the residues
in an IgG heavy
chain is that of the EU index as in Kabat etal., Sequences of Proteins of
Immunological Interest, 5th Ed.
Public Health Service, National Institutes of Health, Bethesda, MD (1991)
. The "EU index as In Kabat" refers to the residue numbering of the human IgG1
EU
antibody.
The term "Fc region" is used to define a C-terrninal region of an IgG heavy
chain as shown in
Figure 1. Although the boundaries of the Fc region of an 1gG heavy chain might
vary slightly, the human
IgG heavy chain Fc region Is usually defined to stretch from amino acid
residue at position Cys226 to
the carboxyl-terminus. The term "Fc region-containing polypeptide" refers to a
polypeptide, such as an
antibody or immunoadhesin (see definitions below), which comprises an Fc
region.
The Fc region of an IgG comprises two constant domains, CH2 and CH3, as shown
in Figure
1. The "CH2" domain of a human IgG Fc region (also referred to as "CY2"
domain) usually extends from
amino acid 231 to amino acid 340. The CH2 domain is unique in that it is not
closely paired with another
domain. Rather, two N-linked branched carbohydrate chains are interposed
between the two CH2
domains of an intact native IgG molecule. It has been speculated that the
carbohydrate may provide
a substitute for the domain-domain pairing and help stabilize the CH2 domain.
Burton, Molec.
immuno1.22:161-206 (1985).
"Hinge region" is generally defined as stretching from G1u216 to Pro230 of
human IgG1 (Burton,
Molec. immunoL22:161-206 (1985)) Hinge regions of other IgG isotypes may be
aligned with the IgG1
sequence by placing the first and last cysteine residues forming inter-heavy
chain S-S bonds in the se
positions.
"C1 q" is a polypeptide that Includes a binding site for the Fc region of an
immunoglobulin. C1q
= 35 together with two serine proteases, C1 r and Cis, forms the complex
Cl, the first component of the
complement dependent cytotoxicity (CDC) pathway. Human Clq can be purchased
commercially from,
e.g. Quidel, San Diego, CA.
The term "Fc receptor or "FcR" is used to describe a receptor that binds to
the Fc region of an
antibody. The preferred FcR Is one, which binds an IgG antibody (a gamma
receptor) and includes
-7-
CA 02577543 2007-01-31
WO 99/51642
re.. e ruayyluvoo
receptors of the FcyRI, FcyRII, and FcyRIII subclasses, including allelic
variants and alternatively spliced
forms of these receptors. FcRs are reviewed in Ravetch and Kinet, Annu. Rev.
Immunol 9:457-92
(1991); Capel et at, Immunomethods 4:25-34(1994); and de Haas etal., J. Lab.
Clin. Med. 126:330-41
(1995). Other FcRs, including those to be identified in the future, are
encompassed by the term "FcR"
herein. The term also includes the neonatal receptor, FcRn, which is
responsible for the transfer of
maternal IgGs to the fetus (Guyer etal., J. Immunot 117:587(1976) and Kim
etal., J. Immunot 24:249
(1994)).
The term "binding domains refers to the region of a polypeptide that binds to
another molecule.
In the case of an FcR, the binding domain can comprise a portion of a
polypeptide chain thereof (e.g.
the a chain thereof) which is responsible for binding an Fc region. One useful
binding domain is the
extracellular domain of an FcR a chain.
The term "antibody" is used in the broadest sense and specifically covers
monoclonal antibodies
(including full length monoclonal antibodies), polyclonal antibodies,
multispecific antibodies (e.g.,
bispecific antibodies), and antibody fragments so long as they exhibit the
desired biological activity.
"Antibody fragments", as defined for the purpose of the present invention,
comprise a portion
of an intact antibody, generally including the antigen binding or variable
region of the intact antibody, so
long as they retain at least the CH2 region of an IgG immunoglobulin heavy
chain constant domain,
comprising amino acid residues 322, 329 E...d 331, and have the ability, alone
or in combination with
another antibody fragment, to specifically bind a selected antigen. Examples
of antibody fragments
include linear antibodies; single-chain antibody molecules; and multispecific
antibodies formed from
antibody fragments. The antibody fragments preferably retain at least part of
the hinge and optionally
the CH1 region of an IgG heavy chain. More preferably, the antibody fragments
retain the entire
= constant region of an IgG heavy chain, and include an IgG light chain.
The term "monoclonal antibody" as used herein refers to an antibody obtained
from a population
of substantially homogeneous antibodies, i.e., the individual antibodies
comprising the population are
identical except for possible naturally occurring mutations that may be
present in minor amounts.
Monoclonal antibodies are highly specific, being directed against a single
antigenic site. Furthermore,
in contrast to conventional (polyclonal) antibody preparations that typically
include different antibodies
directed against different determinants (epitopes), each monoclonal antibody
is directed against a single
determinant on the antigen. The modifier "monoclonal" indicates the character
of the antibody as being
obtained from a substantially homogeneous population of antibodies, and is not
to be construed as
requiring production of the antibody by any particular method. For example,
the monoclonal antibodies
to be used in accordance with the present invention may be made by the
hybridoma method first
described by Kohler etal., Nature 256:495(1975), or may be made by recombinant
DNA methods (see,
e.g., U.S. Patent No. 4,816,567). The "monoclonal antibodies" may also be
isolated from phage
antibody libraries using the techniques described in Clackson at al., Nature
352:624-628 (1991) and
Marks etal., J. Mot Biol. 222:581-597(1991), for example.
The monoclonal antibodies herein specifically include "chimeric" antibodies
(immunoglobulins)
in which a portion of the heavy and/or light chain is identical with or
homologous to corresponding
-8-
CA 02577543 2007-01-31
WO 99/51642
rt.,-171159Y/U05S5
sequences in antibodies derived from a particular species or belonging to a
particular antibody class or
subclass, while the remainder of the chain(s) is identical with or homologous
to corresponding
sequences in antibodies derived from another species or belonging to another
antibody class or
subclass, as well as fragments of such antibodies, so long as they exhibit the
desired biological activity
(U.S. Patent No. 4,816,567; and Morrison et al., Proc. Natl. Acad. Sci. USA
81:6851-6855(1984)).
"Humanized" forms of non-human (e.g., murine) antibodies are chimeric
antibodies that contain
minimal sequence derived from non-human immunoglobulin. For the most part,
humanized antibodies
are human immunogbbulins (recipient antibody) in which residues from a
hypervariable region of the
recipient are replaced by residues from a hypervariable region of a non-human
species (donor antibody)
such as mouse, rat, rabbit or nonhuman primate having the desired specificity,
affinity, and capacity.
In some instances, Fv framework region (FR) residues of the human
immunoglobulin are replaced by
corresponding non-human residues. Furthermore, humanized antibodies may
comprise residues that
are not found in the recipientantibody or in the donor antibody. These
modifications are made to further
refine antibody performance. In general, the humanized antibody will comprise
substantially all of at
least one, and typically two, variable domains, in which all or substantially
all of the hypervariable loops
correspond to those of a non-human immunoglobulin and all or substantially all
of the FR regions are
those of a human immunoglobulin sequence. The humanized antibody optionally
also will comprise at
least a portion of an imrnunogbbulin constant region (Fc), typically that of a
human immunoglobulin. For
further details, see Jones et at., Nature 321:522-525 (1986); Riechmann et
al.. Nature 332:323-329
(1988); and Presta, Cuir. Op. Struct. Bid. 2:593-596(1992).
The term uhypervariable region" when used herein refers to the amino acid
residues of an
antibody which are responsible for antigen-binding. The hypervariable region
comprises amino acid
residues from a "completrentaritydetermining region" or "CDR" (i.e. residues
24-34 (L1), 50-56 (U) and
89-97 (L3) in the light chain variable domain and 31-35 (H1), 50-65 (H2) and
95-102 (H3) in the heavy
chain variable domain; Kabat et al., Sequences of Proteins of Immunological
Interest, 5th Ed. Public
Health Service, National Institutes of Health, Bethesda, MD. (1991)) and/or
those residues from a
"hypervariable loop" (i.e. residues 26-32 (L1), 50-52 (L2) and 91-96 (L3) in
the light chain variable
domain and 26-32 (H1), 53-55 (H2) and 96-101 (H3) in the heavy chain variable
domain; Chothia and
Lesk J. WI. Biol. 196:901-917 (1987)). "Framework" or "FR" residues are those
variable domain
residues other than the hypervariable region residues as herein defined.
As used herein, the term "immunoadhesin" designates antibody-like molecules
which combine
the "binding domain" of a heterologous "adhesin" protein (e.g. a receptor,
ligand or enzyme) with an
immunoglobulin constant domain. Structurally, the immunoadhesins comprise a
fusion of the adhesin
amino acid sequence with the desired binding specificity which is other than
the antigen recognition and
binding site (antigen combining site) of an antibody (i.e. is "heterologous")
and an immunoglobulin
constant domain sequence.
The term "ligand binding domain" as used herein refers to any native cell-
surface receptor or
any region or derivative thereof retaining at least a qualitative ligand
binding ability of a corresponding
native receptor. In a specific embodiment, the receptor is from a cell-surface
polypeptide having an
-9-
CA 02577543 2007-01-31
WO 99/51642
PCT/U599/06858
extracellular domain that is homologous to a member of the immunoglobulin
supergenefamily. Other
receptors, which are not members of the immunoglobulin supergenefamily but are
nonetheless
specifically covered by this definition, are receptors for cytokines, and in
particular receptors with
tyrosine kinase activity (receptor tyrosine kinases), members of the
hematopoietin and nerve growth
factor receptor superfamilies, and cell adhesion molecules, e.g. (E-, L- and P-
) selectins.
The term "receptor binding domain" is used to designate any native ligand for
a receptor,
including cell adhesion molecules, or any region or derivative of such native
ligand retaining at least a
qualitative receptor binding ability of a corresponding native ligand. This
definition, among others,
specifically includes binding sequences from ligands for the above-mentioned
receptors.
An "antibody-imrnunoadhesin chimera" comprises a molecule that combines at
least one binding
domain of an antibody (as herein defined) with at least one immunoadhesin (as
defined in this
application). Exemplary antibody-immunoadhesin chimeras are the bispecific CD4-
IgG chimeras
described in Berg etal., PNAS (USA) 88:4723-4727 (1991) and Chamow etal., J.
Immunol. 153:4268
(1994).
An "isolated" polypeptide is one that has been identified and separated and/or
recovered from
a component of its natural environment. Contaminant components of its natural
environment are
materials that would interfere with diagnostic or therapeutic uses for the
polypeptide, and may include
enzymes, hormones, and other proteinaceous or nonproteinaceous solutes. In
preferred embodiments,
the polypeptide will be purified (1) to greater than 95% by weight of
polypeptide as determined by the
Lowry method, and most preferably more than 99% by weight, (2) to a degree
sufficient to obtain at least
15 residues of N-terminal or internal amino acid sequence by use of a spinning
cup sequenator, or (3)
to homogeneity by SDS-PAGE under reducing or nonreducing conditions using
Coomassie blue or,
preferably, silver stain. isolated polypeptide includes the polypeptide in
situ within recombinant cells
since at least one component of the polypeptide's natural environment will not
be present. Ordinarily,
however, isolated polypeptide will be prepared by at least one purification
step.
"Treatment" refers to both therapeutic treatment and prophylactic or
preventative measures.
Those in need of treatment include those already with the disorder as well as
those in which the disorder
is to be prevented.
A "disorder is any condition that would benefit from treatment with the
polypeptide variant. This
includes chronic and acute disorders or diseases including those pathological
conditions which
predispose the mammal to the disorder in question.
The word "laber when used herein refers to a detectable compound or
composition which is
conjugated directly or indirectly to the polypeptide. The label may be itself
be detectable (e.g.,
radioisotope labels or fluorescent labels) or, in the case of an enzymatic
label, may catalyze chemical
alteration of a substrate compound or composition which is detectable.
An "isolated" nucleic acid molecule is a nucleic acid molecule that is
identified and separated
from at least one contaminant nucleic acid molecule with which it is
ordinarily associated in the natural
source of the polypeptide nucleic acid. An isolated nucleic acid molecule is
other than in the form or
setting in which it is found in nature. Isolated nucleic acid molecules
therefore are distinguished from
-10-
CA 02577543 2007-01-31
WO 99/51642
PCT/US99/06855
the nucleic acid molecule as it exists in natural cells. However, an isolated
nucleic acid molecule
includes a nucleic acid molecule contained in cells that ordinarily express
the polypeptide where, for
example, the nucleic acid molecule is in a chromosomal location different from
that of natural cells.
The expression "control sequences" refers to DNA sequences necessary for the
expression of
an operably linked coding sequence in a particular host organism. The control
sequences that are
suitable for prokaryotes, for example, include a promoter, optionally an
operator sequence, and a
ribosome binding site. Eukaryotic cells are known to utilize promoters,
polyadenylation signals, and
enhancers.
Nucleic acid is "operably Nnked" when it is placed into a functional
relationship with another
nucleic acid sequence. For example, DNA for a presequence or secretory leader
is operably linked to
DNA for a polypeptide if it is expressed ai a preprotein that participates in
the secretion of the
polypeptide; a promoter or enhancer is operably linked to a coding sequence if
it affects the transcription
of the sequence; or a ribosome binding site is operably linked to a coding
sequence if it is positioned so
as to facilitate translation. Generally, "operably linked" means that the DNA
sequences being linked are
contiguous, and, in the case of a secretory leader, contiguous and in reading
phase. However,
enhancers do not have to be contiguous. Linking is accomplished by ligation at
convenient restriction
sites. If such sites do not exist, the synthetic oligonucleotide adaptors or
linkers are used in accordance
with conventinna! practice.
As used herein, the expressions "cell," "cell line," and "cell culture" are
used interchangeably
and all such designations include progeny. Thus, the words "transformants" and
"transformed cells"
include the primary subject cell and cultures derived therefrom without regard
for the number of
transfers. It is also understood that all progeny may not be precisely
identical in DNA content, due to
deliberate or inadvertent mutations. Mutant progeny that have the same
function or biological activity
as screened for in the originally transformed cell are included. Where
distinct designations are intended,
it will be clear from the context.
The term "molecular complex" when used herein refers to the relatively stable
structure which
forms when two or more heterologous molecules (e.g. polypeptides) bind
(preferably noncovalently) to
one another. The preferred molecular complex herein is an immune complex.
"Immune complex" refers to the relatively stable structure which forms when at
least one target
molecule and at least one heterologous Fc region-containing polypeptide bind
to one another forming
a larger molecular weight complex. Examples of immune complexes are antigen-
antibody aggregates
and target molecule-immunoadhesin aggregates. The term "immune complex" as
used herein, unless
indicated otherwise, refers to an ex vivo complex (i.e. other than the form or
setting in which it may be
found in nature). However, the immune complex may be administered to a mammal,
e.g. to evaluate
clearance of the immune complex in the mammal.
The term "target molecule" refers to a molecule, usually a polypeptide, which
is capable of being
bound by a heterologous molecule and has one or more binding sites for the
heterologous molecule. The
term `binding site" refers to a region of a molecule to which another molecule
can bind. The "first target
molecule" herein comprises at least two distinct binding sites (for example,
two to five separate binding
-11-
CA 02577543 2007-01-31
WO 99/51642
rt. 1 lIJOY7/1100.10
sites) for an analyte (e.g. an Fc region-containing polypeptide) such that at
least two analyte molecules
can bind to the first target molecule. In the preferred embodiment of the
invention, the two or more
binding sites are identical (e.g. having the same amino acid sequence, where
the target molecule is a
polypeptide). In Example 1 below, the first target molecule was IgE and had
two separate binding sites
in the Fc region thereof to which the Fc region-containing polypeptide (an
anti-WE antibody, E27) could
bind. Other first target molecules include dimers of substantially identical
monomors (e.g. neurotrophins,
IL8 and VEGF) or are polypeptides comprising two or more substantially
identical polypeptide chains
(e.g. antibodies or immunoadhesins). The "second target molecule" comprises at
least two distinct
binding sites (for example, two to five separate binding sites) for the first
target molecule such that at
least two first target molecules can bind to the second target molecule.
Preferably, the two or more
binding sites are identical (e.g. having the sari* amino acid sequence, where
the target molecule is a
polypeptide). In Example 2, the second target molecule was VEGF, which has a
pair of distinct binding
sites to which the variable domain of the IgE antibody could bind. Other
second target molecules are
contemplated, e.g. other dimers of substantially identical monomers (e.g.
neurotrophins or IL8) or
polypeptides comprising two or more substantially identical domains (e.g.
antibodies or
immunoadhesins).
An "analyte" is a substance that is to be analyzed. The preferred analyte is
an Fe region-
cortaining polypeptide that is to be analyzed for its ability to bind to an Fc
receptor.
A "receptor is a polypeptide capable of binding at least one ligand. The
preferred receptor is
a cell-surface receptor having an extracellular gand-binding domain and,
optionally, other domains (e.g.
transmembrane domain, intracellular domain and/or membrane anchor). The
receptor to be evaluated
in the assay described herein may be an intact receptor or a fragment or
derivative thereof (e.g. a fusion
protein comprising the binding domain of the receptor fused to one or more
heterologous polypeptides).
Moreover, the receptor to be evaluated for its binding properties may be
present in a ceii or isolated and
optionally coated on an assay plate or some other solid phase.
The phrase low affinity receptor denotes a receptor that has a weak binding
affinity for a gand
of interest, e.g. having a binding constant of about 50nM or worse affinity.
Exemplary low affinity
receptors include FcyRII and FcyRIII as well as adhesion molecules, such as
integrins.
A "parent polypeptide" is a polypeptide comprising an amino acid sequence
which lacks one or
more of the Fc region substitutions disclosed herein and/or which differs in
effector function compared
to a polypeptide variant as herein disclosed. The parent polypeptide may
comprise a native sequence
Fc region or an Fc region with existing amino acid sequence modifications
(such as additions, deletions
and/or substitutions).
Modes for Carrying Out the invention
The invention herein relates to a method for making a polypeptide variant The
"parent',
"starting" or "nonvariant" polypeptide is prepared using techniques available
in the art for generating
polypeptides comprising an Fc region. In the preferred embodiment of the
invention, the polypeptide is
an antibody and exemplary methods for generating antibodies are described in
more detail in the
following sections. The polypeptide may, however, be any other polypeptide
comprising an Fc region,
-12-
CA 02577543 2007-01-31
WO 99/51642
rt.. 1 PUDYY/UutiDo
e.g. an immunoadhesin. Methods for making immunoadhesins are elaborated in
more detail
hereinbelow.
The starting polypeptide of particular interest herein is usually one that
binds to C1q and
displays complement dependent cytotoxicity (CDC). The amino acid substitutions
described herein will
generally serve to alter the ability of the starting polypeptide to bind to
Clq and/or modify its complement
dependent cytotodcity function, e.g. to reduce and preferably abolish these
effector functions. However,
polypeptides comprising substitutions at one or more of the described
positions with improved effector
functions are contemplated herein. For example, the starting polypeptide may
be unable to bind C1q
and/or mediate CDC and may be modified according to the teachings herein such
that it acquires these
effector functions. Moreover, polypePtides with pre-existing C1q binding
activity, optionally further
having the ability to mediate CDC may be 'modified such that one or both of
these activities are
enhanced.
To generate the polypeptide variant one or more amino acid alterations (e.g.
substitutions) are
introduced in the Fc region of the starting polypeptide. The amino acid
positions to be modified are
generally selected from heavy chain positions 270, 322, 326, 327, 329, 331,
333, and 334, where the
numbering of the residues in an IgG heavy chain is that of the EU index as in
Kabat et al., Sequences
of Proteins of Immunological Interest, 5th Ed. Public Health Service, National
Institutes of Health,
Bethesda, MD (1991). The Fc region is preferably from a human IgG and mcst
preferably a human IgG1
or human IgG3. The human IgG1 Fc region may be a human A or non-A allotype.
Proline is conserved at position 329 in human IgG's. This residue is
preferably replaced with
alanine, however substitution with any other amino acid is contemplated, e.g.,
serine, threonine,
asparagine, glycine or valine.
Proline is conserved at position 331 in human IgG1, IgG2 and 1903, but not
IgG4 (which has
a serine residue at position 331). Residue 331 is preferably replaced by
alanine or another amino acid,
e.g. serine (for IgG regions other than IgG4), glycine or valine.
Lysine 322 is conserved in human IgGs, and this residue is preferably replaced
by an alanine
residue, but substitution with any other amino acid residue is contemplated,
e.g. serine, threonine,
glycine or valine.
0270 is conserved in human IgGs, and this residue may be replaced by another
amino acid
residue, e.g. alanine, serine, threonine, glycine, valine, or lysine.
K326 is also conserved in human IgGs. This residue may be substituted with
another residue
Including, but not limited to, valine, glutamic acid, alanine, glycine,
aspartic acid, methionine or
tryptophan, with tryptophan being preferred.
Likewise, E333 is also conserved in human IgGs. E333 is preferably replaced by
an amino acid
residue with a smaller side chain volume, such as valine, glycine, alanine or
serine, with serine being
preferred.
K334 is conserved in human IgGs and may be substituted with another residue
such as alanine
or other residue.
-13-
CA 02577543 2007-01-31
WO 99/51642
PCT/US99/1/6553
In human IgG1 and IgG3, residue 327 is an alanine. In order to generate a
variant with
improved C1q binding, this alanine may be substituted with another residue
such as glycine. In IgG2
and IgG4, residue 327 is a glycine and this may be replaced by alanine (or
another residue) to diminish
C1q binding.
Preferably, substitutions at two, three or all of positions 326, 327, 333 or
334 are combined,
optionally with other Fc region substitutions, to generate a polypeptide with
improved human C1q binding
and preferably improved CDC activity in vitro or in vivo.
In one embodiment, only one of the eight above-identified positions is altered
in order to
generate the polypeptide variant Preferably only residue 270,329 or 322 is
altered if this is the case.
Alternatively, two or more of the above-identified positions are modified. If
substitutions are to be
combined, generally substitutions which enhance human C1q binding (e.g. at
residue positions 326, 327,
333 and 334) or those which diminish human C1q binding (e.g., at residue
positions 270, 322, 329 and
331) are combined. In the latter embodiment, all four positions (i.e., 270,
322, 329 and 331) may be
substituted. A variant may be generated in which the native amino acid residue
at position 329 of the
human heavy chain constant region is substituted with another amino acid,
optionally in combination with
a substitution of the amino acid residue at position 331 and/or substitution
of the amino acid residue at
position 322. Otherwise, the native amino acid residue at position 331 and the
native amino acid residue
at position 322 of the human IgG Fc region may both be substitute with another
amino acid residue.
=
One may also combine an amino acid substitution at position 270 with further
substitution(s) at
position(s) 322, 329 and/or 331.
DNA encoding amino acid sequence variant of the starting polypeptide is
prepared by a variety
of methods known in the art. These methods include, but are not limited to,
preparation by site-directed
(or oligonucleotide-mediated)rnutagenesis, PCR mutagenesis, and cassette
mutagenesis of an earlier
prepared DNA encoding the polypeptide.
Site-directed mutagenesis is a preferred method for preparing substitution
variants. This
technique is well known in the art (see, e.g.,Carter etal. Nucleic Acids Res.
13:4431-4443 (1985) and
Kunkel etal., Proc. Natl. Acad.Sci.USA 82:488(1987)). Briefly, in carrying out
site-directed mutagenesis
of DNA, the starting DNA is altered by first hybridizing an oligonucleotide
encoding the desired mutation
to a single strand of such starting DNA. After hybridization, a DNA polymerase
is used to synthesize
an entire second strand, using the hybridized oligonucleotide as a primer, and
using the single strand
of the starting DNA as a template. Thus, the oligonucleotide encoding the
desired mutation is
Incorporated in the resulting double-stranded DNA.
PCR mutagenesis is also suitable for making amino acid sequence variants of
the starting
polypeptide. See Higuchi, in PCR Protocols, pp.177-183 (Academic Press, 1990);
and Vallette etal.,
Nuc. Acids Res. 17:723-733(1969). Briefly, when small amounts of template DNA
are used as starting
material in a PCR, primers that differ slightly in sequence from the
corresponding region in a template
DNA can be used to generate relatively large quantities of a specific DNA
fragment that differs from the
template sequence only at the positions where the primers differ from the
template.
-14-
CA 02577543 2007-01-31
WO 99/51642
PCT/US99/06555
Another method for preparing variants, cassette mutagenesis, is based on the
technique
described by Wells etal., Gene 34:315-323 (1985). The starting material is the
plasmid (or other vector)
comprising the starting polypeptide DNA to be mutated. The codon(s) in the
starting DNA to be mutated
are identified. There must be a unique restriction endonuclease site on each
side of the identified
mutation site(s). If no such restriction sites exist, they may be generated
using the above-described
oligonucleotide-mediated mutagenesis method to introduce them at appropriate
locations in the starting
polypeptide DNA. The plasmid DNA is cut at these sites to linearize it A
double-stranded
oligonucleotide encoding the sequence of the DNA between the restriction sites
but containing the
desired mutation(s) is synthesized using standard procedures, wherein the two
strands of the
oligonucleotide are synthesized separately and then hybridized together using
standard techniques.
This double-stranded oligonucleotide is referred to as the cassette. This
cassette is designed to have
5' and 3' ends that are compatible with the ends of the linearized plasmid,
such that it can be directly
ligated to the plasmid. This plasmid now contains the mutated DNA sequence.
Alternatively, or additionally, the desired amino acid sequence encoding a
polypeptide valiant
can be determined, and a nucleic acid sequence encoding such amino acid
sequence variant can be
generated synthetically.
The polypeptide variant(s) so prepared may be subjected to further
modifications, oftentimes
depending on the intended use of the polypeptide. Such modifications may
involve further alteration of
the amino acid sequence, fusion to heterologous polypeptide(s) and/or covalent
modifications.
For example, it may be useful to combine the above amino acid substitutions
with one or more
further amino acid substitutions that reduce or ablate FcR binding. For
example, the native amino acid
residues at any one or more of heavy chain positions 233-238,318 or 331 (where
the numbering of the
residues in an IgG heavy chain is that of the EU index as in Kabat et al.,
supra) may be replaced with
non-native residue(s), e.g. alanine.
With respect to further amino acid sequence alterations, any cysteine residue
not involved in
maintaining the proper conformation of the polypeptide variant also may be
substituted, generally with
serine, to improve the oxidative stability of the molecule and prevent
aberrant cross linking.
Another type of amino acid substitution serves to alter the glycosylation
pattern of the
polypeptide. This may be achieved by deleting one or more carbohydrate
moieties found in the
polypeptide, and/or adding one or more glycosylation sites that are not
present in the polypeptide.
Glycosylation of polypeptides is typically either N-linked or 0-linked. N-
linked refers to the attachment
of the carbohydrate moiety to the side chain of an asparagine residue. The
tripeptide sequences
asparagine-X-serine and asparagine-X-threonine, where X is any amino acid
except proline, are the
recognition sequences for enzymatic attachment of the carbohydrate moiety to
the asparagine side
chain. Thus, the presence of either of these tripeptide sequences in a
polypeptide creates a potential
glycosylation site. 0-linked glycosylation refers to the attachment of one of
the sugars N-
aceylgalactosamine, galactose, or xylose to a hydroxyamino acid, most commonly
serine or threonine,
although 5-hydroxyproline or 5-hydroxylysine may also be used. Addition of
glycosylation sites to the
polypeptide is conveniently accomplished by altering the amino acid sequence
such that it contains one
-15-
CA 02577543 2007-01-31
WO 99/51642
rt.. I ILIJYYMNS,15
or more of the above-described tripeptide sequences (for N-finked
glycosylation sites). The alteration
may also be made by the addition of, or substitution by, one or more serine or
threonine residues to the
sequence of the original polypeptide (for 0-linked glycosylation sites). An
exemplary glycosylation
variant has an amino acid substitution of residue Asn 297 of the heavy chain.
The polypeptide variant may be subjected to one or more assays to evaluate any
change in
biological activity compared to the starting polypeptide. For example, the
ability of the variant to bind
Clq and mediate complement dependent cytotoxicity (CDC) may be assessed.
To determine Clq binding, a Clq binding ELISA may be performed. Briefly, assay
plates may
be coated overnight at 4 C with polypeptide variant or starting polypeptide
(control) in coating buffer.
The plates may then be washed and blocked. Following washing, an aliquot of
human Clq may be
added to each well and incubated for 2 hrs at room temperature. Following a
further wash, 100 I of a
sheep anti-complementC1q peroxidase conjugated antibody may be added to each
well and incubated
for 1 hour at room temperature. The plate may again be washed with wash buffer
and 100 p1 of
substrate buffer containing OPD (0-phenylenediamine dihydrochloride (Sigma))
may be added to each
well. The oxidation reaction, observed by the appearance of a yellow color,
may be allowed to proceed
for 30 minutes and stopped by the addition of 100 I of 4.5 N H2SO4. The
absorbance may then read
at (492-405) nm.
An exemplary polypeptide variant is one that displays a "significant reduction
in Clq binding'
in this assay. This means that about 100 g/mlof the polypeptide variant
displays about 50 fold or more
reduction in Clq binding compared to 100 g/m1 of a control antibody having a
nonmutated IgG1 Fc
region. In the most preferred embodiment, the polypeptide variant "does not
bind Clq", i.e. 100 g/m1
of the polypeptide variant displays about 100 fold or more reduction in C1q
binding compared to
100 g/m1 of the control antibody.
Another exemplary variant is one which "has a better binding affinity for
human Clq than the
parent poiypeptide". Such a molecule may display, for example, about two-fold
or more, and preferably
about five-fold or more, improvement in human C1q binding compared to the
parent polypeptide (e.g.
at the IC5c, values for these two molecules). For example, human Clq binding
may be about two-fold
to about 500-fold, and preferably from about two-fold or from about five-fold
to about 1000-fold improved
compared to the parent polypeptide.
To assess complement activation, a complement dependent cytotoxicity (CDC)
assay may be
performed, e.g. as described in Gazzano-Santoro etal., J. Immunot Methods
202:163(1996). Briefly,
various concentrations of the polypeptide variant and human complement may be
diluted with buffer.
Cells which express the antigen to which the polypeptide variant binds may be
diluted to a density of
-1 x 106 cells /ml. Mixtures of polypeptide variant, diluted human complement
and cells expressing the
antigen may be added to a flat bottom tissue culture 96 well plate and allowed
to incubate for 2 hrs at
37 C and 5% CO2 to facilitate complement mediated cell lysis. 50 I of Warner
blue (Accumed
International) may then be added to each well and incubated overnight at 37 C.
The absorbance is
measured using a 96-well fluorometer with excitation at 530 nm and emission at
590 nm. The results
-16-
CA 02577543 2007-01-31
WO 99/51642
ri.. i /1U07,/%1U0O0
may be expressed in relative fluorescence units (RFU). The sample
concentrations may be computed
from a standard curve and the percent activity as compared to nonvariant
polypeptide is reported for the
polypeptide variant of interest
Yet another exemplary variant "does not activate complement". For example, 0.6
pg/ml of the
polypeptide variant displays about 0-10% CDC activity in this assay compared
to a 0.6 pg/mlof a control
antibody having a nonmutated IgG1 Fc region. Preferably the variant does not
appear to have any CDC
activity in the above CDC assay.
The invention also pertains to a polypeptide variant with enhanced CDC
compared to a parent
polypeptide, e.g., displaying about two-fold to about 100-fold improvement in
CDC activity in vitro or in
vivo (e.g. at the ICK, values for each molecule being compared).
Preferably the polypeptide variant essentially retains the ability to bind
antigen compared to the
nonvariant polypeptide, i.e. the binding capability is no worse than about 20
fold, e.g. no worse than
about 5 fold of that of the nonvariant polypeptide. The binding capability of
the polypeptide variant may
= be determined using techniques such as fluorescence activated cell
sorting (FACS) analysis or
radioimmunoprecipitation (RIA), for example.
The ability of the polypeptide variant to bind an FcR may also be evaluated.
Where the FcR is
a high affinity Fc receptor, such as FcyRI or FcRn, binding can be measured by
titrating monomeric
polypeptide variant and measuring bound polypeptide variant using an antibody
which specifically binds
to the polypeptide variant in a standard ELISA format (see Example 2 below).
Another FcR binding
assay for low affinity FcRs is elaborated in more detail in the following
section.
Preferably the variant retains the ability to bind one or more FcRs, e.g. the
ability of the
polypeptide variant to bind FcrRI, FcyRII, FcyRIII and/or FcRn is no more than
about 20 fold reduced,
preferably no more than about 10 fold reduced, and most preferably no more
than about two fold
reduced compared to the starting polypeptide as determined in the FcyR1 or
FcRn assays of Example
2 or the FcyRII or FcyRIII assays described in the following section.
A. Receptor Binding Assay and Immune Complex
A receptor binding assay has been developed herein which is particularly
useful for determining
binding of an analyte of interest to a receptorwhere the affinity of the
analyte for the receptor Is relatively
weak, e.g. in the micromolar range as is the case for Fc-rRIla, FcyRlib,
FerRlila and FcyR111b. The
method involves the formation of a molecular complex that has an improved
avidity for the receptor of
Interest compared to the noncomplexed analyte. The preferred molecular complex
is an immune
complex comprising: (a) an Fc region-containing polypeptide (such as an
antibody or an
immunoadhesin); (b) a first target molecule which comprises at least two
binding sites for the Fc region-
containing polypeptide; and (c) a second target molecule which comprises at
least two binding sites for
the first target molecule.
In Example 1 below, the Fc region-containing polypeptide is an anti-IgE
antibody, such as the
E27 antibody (Figs. 4A-413). E27, when mixed with human IgE at an 1:1 molar
ratio, forms a stable
hexamer consisting of three E27 molecules and three IgE molecules. In Example
1 below, the "first
-17-
CA 02577543 2007-01-31
WO 99/51642 Ilar
target molecule" is a chimeric form of IgE in which the Fab portion of an anti-
VEGF antibody is fused to
the human IgE Fc portion and the "second target molecule" is the antigen to
which the Fab binds (i.e.
VEGF). Each molecule of IgE binds two molecules of VEGF. VEGF also binds two
molecules of IgE
per molecule of VEGF. When recombinant human VEGF was added at a 2:1 molar
ratio to IgE:E27
hexamers, the hexamers were linked into larger molecular weight complexes via
the IgE:VEGF
interaction (Fig. 5). The Fc region of the anti-IgE antibody of the resultant
immune complex binds to FcR
with higher avidity than either uncomplexed anti-IgE or anti-IgE:IgE hexamers.
Other forms of molecular complexes for use in the receptor assay are
contemplated. Examples
comprising only an Fc region-containing polypeptide:first target molecule
combination include an
immunoadhesin:ligand combination such as VEGF receptor (KDR)-
immunoadhesin:VEGF and a full-
length bispecific antibody (bsAb):first target molecule. A further example of
an Fc region-containing
polypeptide:first target molecule:second target molecule combination include a
nonblocking
antibody:soluble receptor ligand combination such as anti-Irk antibody:soluble
Tric receptorneurotrophin
(Urfer of al. J. Biol. Chem. 273(10):5829-5840 (1998)).
Aside from use in a receptor binding assay, the immune complexes described
above have
further uses including evaluation of Fc region-containing polypeptide function
and immune complex
clearance in vivo. Hence, the immune complex may be administered to a mammal
(e.g. in a pre-clinical
animal study) and evaluated for its half-life etc.
To determine receptor binding, a polypeptide comprising at least the binding
domain of the
receptor of interest (e.g. the extracellular domain of an a subunit of an FcR)
may be coated on solid
phase, such as an assay plate. The binding domain of the receptor alone or a
receptor-fusion protein
may be coated on the plate using standard procedures. Examples of receptor-
fusion proteins include
receptor-glutathione S-transferase (GST) fusion protein, receptor-chitin
binding domain fusion protein,
receptor-hexaHis tag fusion protein (coated on glutathione, chitin, and nickel
coated plates, respectively).
Alternatively, a capture molecule may be coated on the assay plate and used to
bind the receptor-fusion
protein via the non-receptor portion of the fusion protein. Examples include
anti-hexaHis F(ab,2 coated
on the assay plate used to capture receptor-hexaHis tail fusion or anti-GST
antibody. coated on the
assay plate used to capture a receptor-GST fusion. In other embodiments,
binding to cells expressing
at least the binding domain of the receptor may be evaluated. The cells may be
naturally occurring
hematopoietic cells that express the FcR of interest or may be transformed
with nucleic acid encoding
the FcR or a binding domain thereof such that the binding domain is expressed
at the surface of the cell
to be tested. =
The immune complex described hereinabove is added to the receptor-coated
plates and
incubated for a sufficient period of time such that the analyte binds to the
receptor. Plates may then be
washed to remove unbound complexes, and binding of the analyte may be detected
according to known
methods. For example, binding may be detected using a reagent (e.g. an
antibody or fragment thereof)
which binds specifically to the analyte, and which is optionally conjugated
with a detectable label
(detectable labels and methods for conjugating them to polypeptides are
described below in the section
entitled "Non-Therapeutic Uses for the Polypeptide Variant").
-18-
CA 02577543 2007-01-31
W099/51642
As a matter of convenience, the reagents can be provided in an assay kit,
i.e., a packaged
combination of reagents, for combination with the analyte in assaying the
ability of the analyte to bind
to a receptor of interest. The components of the kit will generally be
provided in predetermined ratios.
The kit may provide the first target molecule and/or the second target
molecule, optionally complexed
together. The kit may further include assay plates coated with the receptor or
a binding domain thereof
(e.g. the extracellular domain of the a subunit of an FcR). Usually, other
reagents, such as an antibody
that binds specifically to the analyte to be assayed, labeled directly or
indirectly with an enzymatic label,
will also be provided in the kit. Where the detectable label is an enzyme, the
kit will include substrates
and cofactors required by the enzyme (e.g. a substrate precursor which
provides the detectable
chromophore or fiuorophore). In addition, other additives may be included such
as stabilizers, buffers
(e.g. assay and/or wash lysis buffer) and the like. The relative amounts of
the various reagents may be
varied widely to provide for concentrations in solution of the reagents that
substantially optimize the
sensitivity of the assay. Particularly, the reagents may be provided as dry
powders, usually lyophilized,
including excipients that on dissolution will provide a reagent solution
having the appropriate
concentration. The kit also suitably includes instructions for carrying out
the assay.
B. Antibody Preparation =
In the preferred embodiment of the invention, the Fc region-containing
polypeptide which is
modified according to the teachings herein is an antibody. Techniques for
producing antibodies follow:
(Q Antigen selection and preparation
Where the polypeptide is an antibody, it is directed against an antigen of
interest. Preferably,
the antigen is a biologically important polypeptide and administration of the
antibody to a mammal
suffering from a disease or disorder can result in a therapeutic benefit in
that mammal. However,
antibodies directed against nonpolypeptide antigens (such as tumor-associated
glycolipid antigens; see
US Patent 5,091,178) are also contemplated.
Where the antigen is a polypeptide, it may be a transmembrane molecule (e.g.
receptor) or
ligand such as a growth factor. Exemplary antigens include molecules such as
renin; a growth hormone,
including human growth hormone and bovine growth hormone; growth hormone
releasing factor;
parathyroid hormone; thyroid stimulating hormone; lipoproteins; alpha-1-
antitrypsin; insulin A-chain;
insulin B-chain; proinsulin; follicle stimulating hormone; calcitonin;
luteinizing hormone; glucagon; clotting
factors such as factor VIIIC, factor IX, tissue factor, and von Wdlebrands
factor; anti-clotting factors such
as Protein C; atrial natriuretic factor; lung surfactant a plasminogen
activator, such as urokinase or
human urine or tissue-type plasminogen activator(t-PA); bombesin; thrombin;
hemopoielic growth factor;
tumor necrosis factor-alpha and -beta; enkephalinase; RANTES (regulated on
activation normally T-ced
expressed and secreted); human macrophage inflammatory protein (MIP-1-alpha);
a serum albumin
such as human serum albumin; Muellerian-inhibiting substance; retaxin A-chain;
relaxin B-chain;
prorelaxin; mouse gonadotropin-associated peptide; a microbial protein, such
as beta-lactamase;
DNase; igE; a cytotoxic T-Iymphocyte associated antigen (CTLA), such as CTLA-
4; inhibin; activin;
vascular endothelial growth factor (VEGF); receptors for hormones or growth
factors; protein A or D;
rheumatoid factors; a neurotrophic factor such as bone-derived neurotrophic
factor (BDNF),
-19-
CA 02577543 2007-01-31
WO 99/51642
neurotrophin-3, -4, -5, or -6 (NT-3, NT-4, NT-5, or NT-6), or a nerve growth
factor such as NGF-6;
platelet-derived growth factor (PDGF); fibroblast growth factor such as aFGF
and bFGF; epidermal
growth factor (EGF); transforming growth factor (TGF) such as TGF-alpha and
TGF-beta, including TGF-
01, TGF-62, TGF-63, TGF-p4, or 1GF-65; insulin-likegrowth factor-I and -H (IGF-
I and IGF-II); des(1-3)-
IGF-I (brain IGF-I), insulin-like growth factor binding proteins; CD proteins
such as CD3, CD4, CD8,
CD19 and CD20; erythropoietin; osteoinductive factors; immunotoxins; a bone
morphogenetic protein
(BMP); an interferon such as interferon-alpha, -beta, and -gamma; colony
stimulating factors (CSFs),
e.g., M-CSF, GM-CSF, and G-CSF; interieukins (ILs), e.g., IL-1 to IL-10;
superoxide dismutase; T-cell
receptors; surface membrane proteins; decay accelerating factor; viral antigen
such as, for example, a
portion of the AIDS envelope; transport proteins; homing receptors;
addressins; regulatory proteins;
integrins such as CD11a, CD11b, CD11c, CD18, an !CAM. VLA-4 and VCAM; a tumor
associated
antigen such as HER2, HER3 or HER4 receptor, and fragments of any of the above-
listed polypeptides.
Preferred molecular targets for antibodies encompassed by the present
invention include CD
proteins such as CD3, CD4, CD8, CD19, CD20 and CD34; members of the Erb13
receptor family such
as the EGF receptor, FIER2, HER3 or HER4 receptor; cell adhesion molecules
such as LFA-1, Mad ,
p150.95, VLA-4, ICAM-1, VCAM and av/P3 integrin including either a or p
subunits thereof (e.g. anti-
CD11a, anti-CD18 or anti-CD11 b antibodies); growth factors such as VEGF; IgE;
blood group antigens;
filc2/flt3 receptor; obesity (013) receptor; mpi receptor, CTLA-4; protein C
etc. .
Soluble antigens or fragments thereof, optionally conjugated to other
molecules, can be used
as immunogens for generating antibodies. For transmembrane molecules, such as
receptors, fragments
of these (e.g. the extraceikilar domain of a receptor) can be used as the
immunogen. Alternatively, cells
expressing the transmembrane molecule can be used as the immunogen. Such cells
can be derived
from a natural source (e.g. cancer cell lines) or may be cells which have been
transformed by
recombinant techniques to express the transmembrane molecule. Other antigens
and forms thereof
useful for preparing antibodies will be apparent to those in the art.
(k) Polydonal antibodies
Polyclonal antibodies are preferably raised in animals by multiple
subcutaneous (Sc) or
intraperitoneal (ip) Injections of the relevant antigen and an adjuvant. It
may be useful to conjugate the
relevant antigen to a protein that is immunogenic in the species to be
immunized, e.g., keyhole limpet
hemocyanin, serum albumin, bovine thyroglobulin, or soybean trypsin inhibitor
using a bifunctional or
derivatizing agent, for example, maleimidobenzoyl sulfosuccinimide ester
(conjugation through cysteine
residues), N-hydroxysuccinirnide (through lysine residues), glutaraldehyde,
succinic anhydride, SOCl2,
or R1N=C=NR, where R and 121 are different alkyl groups.
Animals are immunized against the antigen, immunogenic conjugates, or
derivatives by
combining, e.g., 100 ug or 5 g of the protein or conjugate (for rabbits or
mice, respectively) with 3
volumes of Freund's complete adjuvant and injecting the solution intradermally
at multiple sites. One
month later the animals are boosted with 1/5 to 1/10 the original amount of
peptide or conjugate in
Freund's complete adjuvant by subcutaneous injection at multiple sites. Seven
to 14 days later the
-20-
CA 02577543 2007-01-31
W099131642
animals are bled and the serum is assayed for antibody titer. Animals are
boosted until the titer
plateaus. Preferably, the animal is boosted with the conjugate of the same
antigen, but conjugated to
a different protein and/or through a different cross-linking reagent.
Conjugates Also can be made in
recombinant cell culture as protein fusions. Also, aggregating agents such as
alum are suitably used
to enhance the immune response.
(110 Monoclonal antibodies
Monoclonal antibodies may be made using the hybridoma method first described
by Kohler et
al., Nature, 256:495(1975), or may be made by recombinant DNA methods (U.S.
Patent No. 4,816,567).
In the hybridoma method, a mouse or other appropriate host animal, such as a
hamster or
macaque monkey, is immunized as hereinabove described to elicit lymphocytes
that produce or are
capable of producing antibodies that will specifically bind to the protein
used for immunization.
Alternatively, lymphocytes may be immunized in vitro. Lymphocytes then are
fused with myeloma cells
using a suitable fusing agent, such as polyethylene glycol, to form a
hybridoma cell (Goding, Monoclonal
Antibodies: Principles and Practice, pp.59-103 (Academic Press, 1986)).
The hybridoma cells thus prepared are seeded and grown in a suitable culture
medium that
preferably contains one or more substances that inhibit the growth or survival
of the unfused, parental
myeloma cells. For example, if the parental myeloma cells lack the enzyme
hypoxanthine guanine
phosphoribosyl transferase (HGPRT or HPRT), the culture medium for the
hybridornas typically will
include hypoxanthine, aminopterin, and thymidine (HAT medium), which
substances prevent the growth
of HGPRT-deficient cells.
Preferred myeloma cells are those that fuse efficiently, support stable high-
level production of
antibody by the selected antibody-producingcells, and are sensitiveto a medium
such as HAT medium.
Among these, preferred myeioma cell lines are murine myeloma lines, such as
those derived from
MOPC-21 and MPC-11 mouse tumors available from the Salk Institute Cell
Distribution Center, San
Diego, California USA, and SP-2 or X63-Ag8-653 cells available from the
American Type Culture
Collection, Rockville, Maryland USA. Human myeloma and mouse-human
heteromyeloma cell lines also
have been described for the production of human monoclonal antibodies (Kozbor,
J. lmmunol., 133:3001
(1984); Brodeur et at., Monoclonal Antibody Production Techniques and
Applications, pp. 51-63 (Marcel
Dekker, Inc., New York, 1987)).
Culture medium in which hybridoma cells are growing is assayed for production
of monoclonal
antibodies directed against the antigen. Preferably, the binding specificity
of monoclonal antibodies
produced by hybridoma cells is determined by immunoprecipitationor by an in
vitro binding assay, such
as radioimmunoassay (R1A) or enzyme-linked immunoabsorbent assay (EL1SA).
After hybridoma cells are identified that produce antibodies of the desired
specificity, affinity,
and/or activity, the clones may be subcloned by limiting dilution procedures
and grown by standard
methods (Goding, Monoclonal Antibodies: Principles and Practice, pp.59-103
(Academic Press, 1986)). .
Suitable culture media for this purpose Include, for example, D-MEM or RPMI-
1640 medium. In
addition, the hybridoma cells may be grown in vivo as ascites tumors in an
animal.
-21-
CA 02577543 2007-01-31
W099/51642 - - - - - -
The monoclonal antibodies secreted by the subclones are suitably separated
from the culture
medium, ascites fluid, or serum by conventional irnmunoglobulin purification
procedures such as, for
example, protein A-Sepharose, hydroxylapatite chromatography, gel
electrophoresis, dialysis, or affinity
chromatography.
DNA encoding the monoclonal antibodies is readily isolated and sequenced using
conventional
procedures (e.g., by using oligonucleotide probes that are capable of binding
specifically to genes
encoding the heavy and light chains of the monoclonal antibodies). The
hybridoma cells serve as a
preferred source of such DNA. Once isolated, the DNA may be placed into
expression vectors, which
are then transfected into host cells such as E. coil cells, simian COS cells,
Chinese hamster ovary
(CHO) cells, or myeloma cells that do not otherwise produce immunoglobulin
protein, to obtain the
synthesis of monoclonal antibodies in the recombinant host cells. Recombinant
production of antibodies
will be described in more detail below.
In a further embodiment, antibodies or antibody fragments can be isolated from
antibody phage
libraries generated using the techniques described in McCafferty of al.,
Nature, 348:552-554 (1990).
Clackson etal., Nature, 352:624.628(1991) and Marks et al., J. MoL Biol.,
222:581-597(1991) describe
the isolation of murine and human antibodies, respectively, using phage
libraries. Subsequent
publications describe the production of high affinity (nM range) human
antibodies by chain shuffling
(Marks at al., Bio/rechnology, 10:779-783 (1992)), as well as combinatorial
infection and in vivo
recombination as a strategy for constructing very large phage libraries
(Waterhouse at a/.. Nuc. Acids.
Res., 21:2265-2266 (1993)). Thus, these techniques are viable alternatives to
traditional monoclonal
antibody hybridoma techniques for isolation of monoclonal antibodies.
The DNA also may be modified, for example, by substituting the coding sequence
for human
heavy- and light-chain constant domains in place of the homologous murine
sequences (U.S. Patent No.
4,816,567; Morrison, et aL, Proc. Net! Acad. Sci. USA, 81:6851 (1984)), or by
covalently joining to the
immunoglobulin coding sequence all or part of the coding sequence for a non-
immunoglobulin
polypeptide.
Typically such non-immunoglobulin polypeptides are substituted for the
constant domains of an
antibody, or they are substituted for the variable domains of one antigen-
combining site of an antibody
to create a chimeric bivalent antibody comprising one antigen-combining site
having specificity for an
antigen and another antigen-combining site having specificity for a different
antigen.
(iv) Humanized and human antibodies
A humanized antibody has one or more amino acid residues introduced into it
from a source
which is non-human. These non-human amino acid residues are often referred to
as "import" residues,
which are typically taken from an "import" variable domain. Humanization can
be essentially performed
following the method of Wnter and co-workers (Jones of at, Nature, 321:522-525
(1986); Riechmann
at aL, Nature, 332:323-327 (1988); Verhoeyen at al, Science, 239:1534-1536
(1988)), by substituting
rodent CDRs or CDR sequences for the corresponding sequences of a human
antibody. Accordingly,
such "humanized" antibodies are chimeric antibodies (U.S. Patent No.
4,816,567) wherein substantially
less than an intact human variable domain has been substituted by the
corresponding sequence from
-22-
CA 02577543 2007-01-31
W099/51642 a
.11===========÷=-......--
a non-human species. In practice, humanized antibodies are typically human
antibodies in which some
CDR residues and possibly some FR residues are substituted by residues from
analogous sites in rodent
antibodies.
The choice of human variable domains, both light and heavy, to be used in
making the
humanized antibodies is very important to reduce antigenicity. According to
the so-called sbest-fit"
method, the sequence of the variable domain of a rodent antibody is screened
against the entire library
of known human variable-domain sequences. The human sequence which is closest
to that of the
rodent is then accepted as the human framework (FR) for the humanized antibody
(Sims et al., J.
lmmunot, 151:2296 (1993); Chothia et at, J. Mot Biol., 196:901 (1987)). Mother
method uses a
particular framework derived from the consensus sequence of all human
antibodies of a particular
subgroup of light or heavy chains. The same framework may be used for several
different humanized
antibodies (Carteret al., Proc. Natl. Acad. Sc!. USA, 89:4285(1992); Presta et
al., J. Immnot, 151:2623
(1993)).
It is further important that antibodies be humanized with retention of high
affinity for the antigen
and other favorable biological properties. To achieve this goal, according to
a preferred method,
humanized antibodies are prepared by a process of analysis of the parental
sequences and various
conceptual humanized products using three-dimensional models of the parental
and humanized
sequences. Three-dimensional immunoglobulin nvyiels are commonly available and
are familiar to
those skilled in the art. Computer programs are available which illustrate and
display probable three-
dimensional conformational structures of selected candidate immunoglobulin
sequences. Inspection of
these displays permits analysis of the likely role of the residues in the
functioning of the candidate
immunoglobulin sequence, i.e., the analysis of residues that influence the
ability of the candidate
immunoglobulin to bind its antigen. In this way, FR residues can be selected
and combined from the
recipient and import sequences so that the desired antibody characteristic,
such as increased affinity
for the target antigen(s), is achieved. In general, the CDR residues are
directly and most substantialiy
involved in influencing antigen binding.
Alternatively, it is now possible to produce transgenic animals (e.g., mice)
that are capable,
upon immunization, of producing a full repertoire of human antibodies in the
absence of endogenous
immunoglobulin production. For example, it has been described that the
homozygous deletion of the
antibody heavy-chain joining region (JH) gene in chimeric and germ-line mutant
mice results in complete
inhibition of endogenous antibody production. Transfer of the human germ-line
immunoglobulin gene
array In such germ-fine mutant mice will result in the production of human
antibodies upon antigen
challenge. See, e.g., Jakobovits et at, Proc. Natl. Acad. Sci. USA, 90:2551
(1993); Jakobovits etal.,
Nature, 362:255-258 (1993); Bruggermann et at., Year in immuno., 7:33 (1993);
and Duchosal at a/.
Nature 355:258 (1992). Human antibodies can also be derived from phage-display
libraries
(Hoogenboom at al., J. Mot Biot, 227:381 (1991); Marks et al., J. Mot Biol.,
222:581-597 (1991);
Vaughan et at. Nature Biotech 14:309 (1996)).
-23-
CA 02577543 2007-01-31
W099/51642 .
(v) Multispecific antibodies
Multispecific antibodies have binding specificities for at least two different
antigens. While such
molecules normally will only bind two antigens (i.e. bispecific antibodies,
BsAbs), antibodies with
additional specificities such as trispecific antibodies are encompassed by
this expression when used
herein. Examples of BsAbs include those with one arm directed against a tumor
cell antigen and the
other arm directed against a cytotoxic trigger molecule such as anti-
FcyRI/anti-CD15, anti-
p185HER2/FcyRIII (CD16), anti-CD3/anti-malignantB-cell (1D10), anti-CD3/anti-
p185', anti-CD3/anti-
p97, anti-CD3/anti-renalcell carcinoma, anti-CD3/anti-OVCAR-3,anti-CD3/L-D1
(anti-colon carcinoma),
anti-CD3/anti-melanocyte stimulating hormone analog, anti-EGF receptor/anti-
CD3, anti-CD3/anti-
CAMA1, anti-CD3/anti-CD19, anti-CD3/MoV113, anti-neural cell ahesion molecule
(NCAM)/anti-CD3, anti-
folate binding protein (FBP)/anti-CD3, anti-pan carcinoma associated antigen
(AMOC-31)/anti-CD3;
BsAbs with one arm which binds specifically to a tumor antigen and one arm
which binds to a toxin such
as anti-saporintanti-Id-1, anti-CD22/anti-saporin, anti-CD7/anti-saporin, anti-
CD38/anti-saporin, anti-
CEA/anti-ricin A chain, anti-interferon-a (IFN-a)Ianti-hybridoma idiotype,
anti-CEA/anti-vinca alkaloid;
BsAbs for converting enzyme activated prodrugs such as anti-CD30/anti-alkaline
phosphatase (which
catalyzes conversion of mitomycin phosphate prodrug to mitomycin alcohol);
BsAbs which can be used
as fibrinolytic agents such as anti-fibrin/anti-tissueplasminogen activator
(tPA), anti-fibrin/anti-uroldnase-
type plasminogen activator (u PA); BsAbs for targeting immune complexes to
cell surface receptors such
as anti-low density lipoprotein (LOW/anti-Fe receptor (e.g. FcyRI, FcyRII or
FcyRIII); BsAbs for use in
therapy of infectious diseases such as anti-CD3/anti-herpes simplex virus
(HSV), anti-T-cell
receptor:CD3 complex/anti-influenza, anti-FcyR/anti-HIV; BsAbs for tumor
detection in vitro or in vivo
such as anti-CENanti-E0TUBE, anti-CEA/anti-DPTA, anti-p185HER2/anti-hapten;
BsAbs as vaccine
adjuvants; and BsAbs as diagnostic tools such as anti-rabbit IgG/anti-
ferritin, anti-horse radish
peroxidase (HRP)/anti-hormone,anti-somatostatin/anti-substanceP, anti-HRP/anti-
F1TC,anti-CEA/anti-
6-galactosidase. Examples of trispecific antibodies include anti-CD3lanti-
CD4/anti-CD37,anti-CD3/anti-
CD5/anti-0037 and anti-CD3/anti-CD8/anti-CD37. Bispecific antibodies can be
prepared as full length
antibodies or antibody fragments (e.g. F(ab1)2bispecific antibodies).
Methods for making bispecific antibodies are known in the art. Traditional
production of full
length bispecific antibodies is based on the coexpression of two
immunoglobulin heavy chain-light chain
pairs, where the two chains have different specificities (Milstein et at.,
Nature, 305:537-539 (1983)).
Because of the random assortment of immunoglobulin heavy and light chains,
these hybridomas
(quadromas) produce a potential mixture of 10 different antibody molecules, of
which only one has the
correct bispecific structure. Purification of the correct molecule, which is
usually done by affinity
chromatography steps, is rather cumbersome, and the product yields are low.
Similar procedures are
disclosed in WO 93/08829, and in Traunecker etal., EMBO J., 10:3655-3659
(1991).
According to a different approach, antibody variable domains with the desired
binding
specificities (antibody-antigen combining sites) are fused to immunoglobulin
constant domain
sequences. The fusion preferably is with an immunoglobulin heavy chain
constant domain, comprising
-24-
CA 02577543 2007-01-31
W099/51642
at least part of the hinge, CH2, and CH3 regions. it is preferred to have the
first heavy-chain constant
region (CHI) containing the site necessary for light chain binding, present in
at least one of the fusions.
DNAs encoding the immunoglobulin heavy chain fusions and, if desired, the
immunoglobulin light chain,
are inserted into separate expression vectors, and are co-transfected into a
suitable host organism. This
provides for great flexibility in adjusting the mutual proportions of the
three polypeptide fragments in
embodiments when unequal ratios of the three polypeptide chains used in the
construction provide the
optimum yields. It is, however, possible to insert the coding sequences for
two or all three polypeptide
chains in one expression vector when the expression of at least two
polypeptide chains in equal ratios
results in high yields or when the ratios are of no particular significance.
In a preferred embodiment of this approach, the bispecific antibodies are
composed of a hybrid
immunoglobulin heavy chain with a first binding specificity in one arm, and a
hybrid immunoglobulin
heavy chain-light chain pair (providing a second binding specificity) in the
other arm. It was found that
this asymmetric structure facilitates the separation of the desired bispecific
compound from unwanted
immunoglobulin chain combinations, as the presence of an immunoglobulin light
chain in only one half
of the bispecific molecule provides for a facile way of separation. This
approach is disclosed in WO
94/04690. For further details of generating bispecific antibodies see, for
example, Suresh et Methods
In Enzymology, 121:210(1986).
According to another approach described in W096/27011, the interface between a
pair of
antibody molecules can be engineered to maximize the percentage of
hebsrodimers which are recovered
from recombinant cell culture. The preferred interface comprises at least a
pert of the CH3 domain of an
antibody constant domain. In this method, one or more small amino acid side
chains from the interface
of the first antibody molecule are replaced with larger side chains (e.g.
tyrosine or byptophan).
Compensatory "cavities" of Identical or similar size to the large side
chain(s) are created on the interface
of the second antibody molecule by replacing large amino acid side chains with
smaller ones (e.g.
alanine or threonine). This provides a mechanism for increasing the yield of
the heterodimer over other
unwanted end-products such as homodimers.
Bispecific antibodies include cross-linked or "heteroconjugate" antibodies.
For example, one
of the antibodies in the heteroconjugate can be coupled to avidin, the other
to biotin. Such antibodies
have, for example, been proposed to target immune system cells to unwanted
cells (US Patent No.
4,676,980), and for treatment of HIV infection (WO 91/00360, WO 92/200373, and
EP 03089).
Heteroconjugate antibodies may be made using any convenient cross-linking
methods. Suitable cross-
linking agents are well known In the art, and are disclosed in US Patent No.
4,676,980, along with a
number of cross-linking techniques.
Antibodies with more than two valencies are contemplated. For example,
trispecific antibodies
can be prepared. Tuft etal. J. Immune!. 147: 60 (1991).
While the polypeptide of interest herein is preferably an antibody, other Fc
region-containing
polypeptides which can be modified according to the methods described herein
are contemplated. An
example of such a molecule is an immunoadhesin.
-25-
CA 02577543 2007-01-31
WO 99/51642 rt.stu
077/Wu...nip
C. Immunoadhesin Preparation
=
The simplest and most straightforward immunoadhesin design combines the
binding domain(s)
of the adhesin (e.g. the extracellular domain (ECD) of a receptor) with the Fc
region of an
immunoglobulin heavy chain. Ordinarily, when preparing the immunoadhesins of
the present invention,
nucleic acid encoding the binding domain of the adhesin will be fused C-
terminally to nucleic acid
encoding the N-terminus of an immunoglobulin constant domain sequence, however
N-terminal fusions
are also possible.
Typically, in such fusions the encoded chimeric polypeptide will retain at
least functionally active
hinge, C142 and CH3 domains of the constant region of an immunoglobulin heavy
chain. Fusions are also
made to the C-terminus of the Fc portion of a constant domain, or immediately
N-terminal to the CHI of
the heavy chain or the corresponding region of the light chain. The precise
site at which the fusion is
made is not critical; particular sites are well known and may be selected in
order to optimize the
biological activity, secretion, or binding characteristics of the
immunoadhesin.
In a preferred embodiment, the adhesin sequence is fused to the N-terminus of
the Fc region
of immunoglobulin G, (IgG1). It is possible to fuse the entire heavy chain
constant region to the adhesin
sequence. However, more preferably, a sequence beginning in the hinge region
just upstream of the
papain cleavage site which defines IgG Fc chemically (i.e. residue 216, taking
the first residue of heavy
chain constant region ici be 114), or analogous sites of other immunoglobulins
is used in the fusion. In =
a particularly preferred embodiment, the adhesin amino acid sequence is fused
to (a) the hinge region
and CH2 and 6H3 or (b) the CHI, hinge, CH2 and CH3 domains, of an IgG heavy
chain.
For bispecffic immunoadhesins, the immunoadhesins are assembled as multimers,
and
particularly as heterodimers or heterotetramers. Generally, these assembled
immunoglobulinswill have
known unit structures. A basic four chain structural unit is the form in which
IgG, IgD, and lgE exist. A
four chain unit is repeated in the higher molecular weight immunoglobulins;
Ig114 generally exists as a
pentamer of four basic units held together by disulfide bonds. lgA globulin,
and occasionally IgG globulin,
may also exist in multimeric form in serum. In the case of multimer, each of
the four units may be the
same or different.
Various exemplary assembled immunoadhesins within the scope herein are
schematically
diagrammed below:
(a) ACL-ACL;
(b) ACH-(ACH, ACL-ACH, ACL-VHCH, or VLCL-ACH);
(c) ACL-ACH-(ACL-ACH, ACL-VHCH, VLCL-ACH, or VLCL-VHCH)
(d) ACL-VHCH-(ACH, or ACL-VHCH, or VLCL-ACH);
(e) VLCL-ACH-(ACL-VHCH, or VLCL-ACH); and
(f) (A-Y),-(VLCL-VHCH)2,
wherein each A represents identical or different adhesin amino acid sequences;
VL is an immunoglobulin light chain variable domain;
-26-
CA 02577543 2007-01-31
W099/51642 _
VH is an immunoglobulin heavy chain variable domain;
CL is an immunoglobulin light chain constant domain;
CH is an immunoglobulin heavy chain constant domain;
n is an integer greater than 1;
Y designates the residue of a covalent cross-linking agent
In the interests of brevity, the foregoing structures only show key features;
they do not indicate
joining (J) or other domains of the immunoglobulins, nor are disulfide bonds
shown. However, where
such domains are required for binding activity, they shall be constructed to
be present in the ordinary
locations which they occupy in the immunoglobulin molecules.
Alternatively, the adhesin sequences can be inserted between immunoglobulin
heavy chain and
light chain sequences, such that an immunoglobulin comprising a chimeric heavy
chain is obtained. In
this embodiment, the adhesin sequences are fused to the 3' end of an
immunoglobulin heavy chain in
each arm of an Immunoglobulin, either between the hinge and the CH2 domain, or
between the CH2 and
CH3 domains. Similar constructs have been reported by Hoogenboom, etal.. Mol.
Immunot 28:1027-
1037 (1991).
Although the presence of an immunoglobulin light chain is not required in the
immunoadhesins
of the presFat invention, an immunoglobulin light chain might be present
either covalently assr.liated to
an adhesin-immunoglobulin heavy chain fusion polypeptide, or directly fused to
the adhesin. In the
former case, DNA encoding an immunoglobulin light chain is typically
coexpressed with the DNA
encoding the adhesin-immunoglobulin heavy chain fusion protein. Upon
secretion, the hybrid heavy
chain and the light chain will be covalently associated to provide an
immunoglobulin-like structure
comprising two disulfide-linked immunoglobulin heavy chain-light chain pairs.
Methods suitable for the '
preparation of such structures are, for example, disclosed in U.S. Patent No.
4,816,567, issued 28 March
1989.
Immunoadhesins are most conveniently constructed by fusing the cDNA sequence
encoding
the adhesin portion in-frame to an immunoglobulin cDNA sequence. However,
fusion to genomic
immunoglobulin fragments can also be used (see, e.g. Aruffo at al., Cell
61:1303-1313 (1990); and
Stamenkovic at al.. Cell 66:1133-1144 (1991)). The latter type of fusion
requires the presence of Ig
regulatory sequences for expression. cDNAs encoding IgG heavy-chain constant
regions can be
isolated based on published sequences from cDNA libraries derived from spleen
or peripheral blood
lymphocytes, by hybridization or by polymerase chain reaction (PCR)
techniques. The cDNAs encoding
the madhesin" and the immunoglobulin parts of the immunoadhesin are inserted
in tandem into a plasmid
vector that directs efficient expression in the chosen host cells.
D. Vectors, Host Cells and Recombinant Methods
The invention also provides isolated nucleic acid encoding a polypeptide
variant as disclosed
herein, vectors and host cells comprising the nucleic acid, and recombinant
techniques for the production
of the polypeptide variant
-27-
CA 02577543 2007-01-31
WO 99/51642
II I %Ma. = ..=======ar.....=
For recombinant production of the polypeptide variant, the nucleic acid
encoding it is isolated
and inserted into a replicable vector for further cloning (amplification of
the DNA) or for expression. DNA
encoding the polypeptide variant is readily isolated and sequenced using
conventional procedures (e.g.,
by using oligonucleotide probes that are capable of binding specifically to
genes encoding the
polypeptide variant). Many vectors are available. The vector components
generally include, but are not
limited to, one or more of the following: a signal sequence, an origin of
replication, one or more marker
genes, an enhancer element, a promoter, and a transcription termination
sequence.
Signal sequence component
The polypeptide variant of this invention may be produced recombinandy not
only directly, but
also as a fusion polypeptide with a heterologous polypeptide, which is
preferably a signal sequence or
other polypeptide having a specific cleavage site at the N-terminus of the
mature protein or polypeptide.
The heterologous signal sequence selected preferably is one that is recognized
and processed (i.e.,
cleaved by a signal peptidase) by the host cell. For prokaryotic host cells
that do not recognize and
process the native polypeptide variant signal sequence, the signal sequence is
substituted by a
prokaryotic signal sequence selected, for example, from the group of the
alkaline phosphatase,
penicillinase, Ipp, or heat-stable enterotoxin II leaders. For yeast secretion
the native signal sequence
may be substituted by, e.g., the yeast invertase leader, a factor leader
(including Saccharomyces and
Kluywoomyces a-factor leaders), or acid phosphatase leader, the C. Whims
glucoamy:ase leader, or
the signal described in WO 90/13646. In mammalian cell expression, mammalian
signal sequences as
well as viral secretory leaders, for example, the herpes simplex gD signal,
are available.
The DNA for such precursor region is ligated in reading frame to DNA encoding
the polypeptide
variant.
(ii) Origin of replication component
Both expression and cloning vectors contain a nucleic acid sequence that
enables the vector
to replicate in one or more selected host cells. Generally, in cloning vectors
this sequence is one that
enables the vector to replicate independently of the host chromosomal DNA, and
includes origins of
replication or autonomously replicating sequences. Such sequences are well
known for a variety of
bacteria, yeast, and viruses. The origin of replication from the plasmid
pEIR322 is suitable for most
= Gram-negative bacteria, the 2 plasmid origin is suitable for yeast, and
various viral origins (SV40,
polyoma, adenovirus, VSV or BPV) are useful for cloning vectors in mammalian
cells. Generally, the
origin of replication component is not needed for mammalian expression vectors
(the SV40 origin may
typically be used only because it contains the early promoter).
(iii) Selection gene component
Expression and cloning vectors may contain a selection gene, also termed a
selectable marker.
Typical selection genes encode proteins that (a) confer resistance to
antibiotics or other toxins, e.g.,
ampicillin, neomycin, methotrexate, or tetracycline, (b) complement
auxotrophic deficiencies, or (c)
supply critical nutrients not available from complex media, e.g., the gene
encoding D-alanine racemase
for Bacilli.
-28-
CA 02577543 2007-01-31
WO 99/51642
One example of a selection scheme utilizes a drug to arrest growth of a host
cell. Those cells
that are successfully transformed with a heterologous gene produce a protein
conferring drug resistance
and thus survive the selection regimen. Examples of such dominant selection
use the drugs neomycin,
mycophenolic acid and hygromycin.
Another example of suitable selectable markers for mammalian cells are those
that enable the
identification of cells competent to take up the polypeptide variant nucleic
acid, such as DHFR, thymidine
kinase, metallothionein-I and -II, preferably primate metallothionein genes,
adenosine deaminase,
omithine decarboxylase, etc.
For example, cells transformed with the DHFR selection gene are first
identified by culturing all
of the transformants in a culture medium that contains methotrexate (Mbc), a
competitive antagonist of
DHFR. An appropriate host cell when wild-type DHFR is employed is the Chinese
hamster ovary (CHO)
cell line deficient in DHFR activity.
Alternatively, host cells (particularly wild-type hosts that contain
endogenous DHFR)
transformed or co-transformed with DNA sequences encoding polypeptide variant,
wild-type DHFR
protein, and another selectable marker such as aminoglycoside 3'-
phosphotransferase (APH) can be
selected by cell growth in medium containing a selection agent for the
selectable marker such as an
aminoglycosidic antibiotic, e.g., kanamycin, neomycin, or G418. See U.S.
Patent No. 4,965,199.
A suitable selection gene for use in yeast is the trpl gene 7-esent in the
yeast plasrnid YRp7
(Stinchcomb at al., Nature, 282:39 (1979)). The bp1 gene provides a selection
marker for a mutant
strain of yeast lacking the ability to grow in tryptophan, for example, ATCC
No. 44076 or PEP4-1. Jones, =
Genetics, 85:12(1977). The presence of the bp1 lesion in the yeast host cell
genome then provides an
effective environment for detecting transformation by growth in the absence of
tryptophan. Similarly,
Leu2-deficient yeast strains (ATCC 20,622 or 38,626) are complemented by known
plasmids bearing
the Leta gene.
In addition, vectors derived from the 1.6 ttm circular plasmid p1<1)1 can be
used for
transformation of Kluyveromyces yeasts. Alternatively, an expression system
for large-scale production
of recombinant calf chymosin was reported for K lactis. Van den Berg,
Bioffechnology, 8:135(1990).
Stable multi-copy expression vectors for secretion of mature recombinant human
serum albumin by
industrial strains of Kluyveromyces have also been disclosed. Fleer et al.,
Bioffechnology, 9:968-975
(1991).
(iv) Promoter component
Expression and cloning vectors usually contain a promoter that is recognized
by the host
organism and is operably linked to the polypeptide variant nucleic acid.
Promoters suitable for use with
prokaryotic hosts include the pholt promoter, B-lactamase and lactose promoter
systems, alkaline
phosphatase, a tryptophan (trp) promoter system, and hybrid promoters such as
the tac promoter.
However, other known bacterial promoters are suitable. Promoters for use in
bacterial systems also will
contain a Shine-Dalgamo (S.D.) sequence operably linked to the DNA encoding
the polypeptide variant.
Promoter sequences are known for eukaiyotes. Virtually all eukaryotic genes
have an AT-rich
region located approximately 25 to 30 bases upstream from the site where
transcription is initiated.
-29-
CA 02577543 2007-01-31
WO 99/51642
PCT/US99/U6355
Another sequence found 70 to 80 bases upstream from the start of transcription
of many genes is a
CNCAAT region where N may be any nucleotide. At the 3' end of most eukaryotic
genes is an AATAAA
sequence that may be the signal for addition of the poly A tail to the 3' end
of the coding sequence. All
of these sequences are suitably inserted into eukaryotic expression vectors.
Examples of suitable promoting sequences for use with yeast hosts include the
promoters for
3-phosphoglycerate kinase or other glycolytic enzymes, such as enolase,
glyceraidehyde-3-phosphate
dehydrogenase, hexokinase, pyruvate decarboxylase, phosphofructokinase,
glucose-6-phosphate
isomerase, 3-phosphoglycerate mutase, pyruvate kinase, triosephosphate
isomerase, phosphoglucose
isomerase, and glucokinase.
Other yeast promoters, which are inducible promoters having the additional
advantage of
transcription controlled by growth conditions, are the promoter regions for
alcohol dehydrogenase 2,
isocytochrome C, acid phosphatase, degradative enzymes associated with
nitrogen metabolism,
metallothionein, glyaeraldehyde-3-phosphate dehydrogenase, and enzymes
responsible for maltose and
galactose utilization. Suitable vectors and promoters for use in yeast
expression are further described
in EP 73,657. Yeast enhancers also are advantageously used with yeast
promoters.
Polypeptide variant transcription from vectors in mammalian host ceNs is
controlled, for example,
by promoters obtained from the genonies of viruses such as polyoma virus,
fowlpox virus, adenovirus
(such as Adenovirus 2), bovine papilloma virus, avian szrcoma virus,
cytomegaiovirus, a retrovirus,
hepatitis-B virus and most preferably Simian Virus 40 (SV4U), from
heterologous mammalian promoters,
e.g., the actin promoter or an immunoglobulin promoter, from heat-shock
promoters, provided such
promoters are compatible with the host cell systems.
The early and late promoters of the SV40 virus are conveniently obtained as an
8V40 restriction
fragment that also contains the SV40 viral origin of replication. The
immediate early promoter of the
human cytomegalovirus is conveniently obtained as a Hindi II E restriction
fragment. A system for
expressing DNA in mammalian hosts using the bovine papilloma virus as a vector
is disclosed in U.S.
Patent No. 4,419,446. A modification of this system is described in U.S.
Patent No. 4,601,978. See also
Reyes et at., Nature 297:598-601 (1982) on expression of human 6-interferon
cDNA in mouse cells
under the control of a thymidine kinase promoter from herpes simplex virus.
Alternatively, the rous
sarcoma virus long terminal repeat can be used as the promoter.
(v) Enhancer element component
Transcription of a DNA encoding the polypeptide variant of this invention by
higher eukaryotes
is often increased by inserting at enhancer sequence into the vector. Many
enhancer sequences are
now known from mammalian genes (globin, eiastase, albumin, a-fetoprotein, and
insulin). Typically,
however, one will use an enhancer from a eukaryotic cell virus. Examples
include the SV40 enhancer
on the late side of the replication origin (bp 100-270), the cytomegalovirus
early promoter enhancer, the
polyoma enhancer on the late side of the replication origin, and adenovirus
enhancers. See also Yaniv,
Nature 297:17-18(1982) on enhancing elements for activation of eukaryotic
promoters. The enhancer
may be spliced into the vector at a position 5' or 3' to the polypeptide
variant-encoding sequence, but
is preferably located at a site 5' from the promoter.
-30-
CA 02577543 2007-01-31
WO 99/51642
rt.-1/UbYYM0438
(w) Transcdption termination component
Expression vectors used in eukaryotic host cells (yeast, fungi, insect, plant,
animal, human, or
nucleated cells from other multicellular organisms) will also contain
sequences necessary for the
termination of transcription and for stabilizing the mR NA. Such sequences are
commonly available from
the 5' and, occasionally 3', untranslated regions of eukaryotic or viral DNAs
or cDNAs. These regions
contain nucleotide segments transcribed as polyadenylated fragments in the
untransiated portion of the
mRNA encoding the polypeptide variant. One useful transcription termination
component is the bovine
growth hormone polyadenylation region. See VV094/11026 and the expression
vector disclosed therein.
(vii) Selection and transformation of host cells
Suitable host cells for cloning or expressing the DNA in the vectors herein
are the prokaryote,
yeast, or higher eukaryote cells described 'above. Suitable prokaryotes for
this purpose include
eubacteria, such as Gram-negative or Gram-positive organisms, for example,
Enterobacteriaceae such
as Escherichia, e.g., E coli, Enterobacter, Erwinia, Klebsiella, Proteus,
Salmonella, e.g., Salmonella
typhimurium, Serratia, e.g., Serratia marcescans, and Shigella, as well as
Bacilli such as B. subtilis and
B. licheniformis (e.g., B. lichenifonnis 41P disclosed in DD 266,710 published
12 April 1989),
Pseudomonas such as P. aeruginosa, and Streptomyces. One preferred E. coil
cloning host is E. coli
294 (ATCC 31,446), although other strains such as E. coil B, E. coli X1776
(ATCC 31,537), and E. coil
W3110 (ATCC 27,325) are suitable. These examples are illustrative rather than
limiting.
In addition to prokaryotes, eukaryotic microbes such as filamentous fungi or
yeast are suitable
cloning or expression hosts for polypeptide variant-encoding vectors.
Saccharomyces cerevisiae, or
common baker's yeast is the most commonly used among lower eukaryotic host
microorganisms.
However, a number of other genera, species, and strains are commonly available
and useful herein,
such as Schizosaccharomyces pombe; Kluyveromyces hosts such as, e.g., K.
lactis, K. fragilis (ATCC
12,424), K. bulgaricus (ATCC 16,045), K. wickeramil (ATCC 24,178), K. waltil
(ATCC 56,500), K.
drosophilarum (ATCC 36,906), K. thermotolerans, and K. marxianus; yarrowia (EP
402,226); Pichia
pastoris (EP 183,070); Candida; Trichoderma reesia (EP 244,234); Neurospora
crassa;
Schwanniomyces such as Schwanniomyces occidentalis; and filamentous fungi such
as, e.g.,
Neurospora, Penicillium, Tolypocladium, and AspergHlus hosts such as A.
nidulans and A. niger.
Suitable host cells for the expression of glycosylated polypeptide variant are
derived from
multicellular organisms. Examples of invertebrate cells include plant and
insebt cells. Numerous
baculoviral strains and variants and corresponding permissive insect host
cells from hosts such as
Spodoptera fwgiperda (caterpillar), Aedes aegypti (mosquito), Aedes albopictus
(mosquito), Drosophila
melanogaster (fruitfly), and Bombyx mon have been identified. A variety of
viral strains for transfection
are publicly available, e.g., the L-1 variant of Autographs califomica NPV and
the Bm-5 strain of Bombyx
mori NPV, and such viruses may be used as the virus herein according to the
present invention,
particularly for transfection of Spodoptera fnigiperda cells.
Plant cell cultures of cotton, corn, potato, soybean, petunia, tomato, and
tobacco can also be
utilized as hosts.
-31-
CA 02577543 2007-01-31
WO 99/51642 PCT/US99/06858
However, interest has been greatest in vertebrate cells, and propagation of
vertebrate cells in
culture (tissue culture) has become a routine procedure. Examples of useful
mammalian host cell lines
are monkey kidney CV1 line transformed by SV40 (COS-7, ATCC CRL 1651); human
embryonic kidney
line (293 or 293 cells subcloned for growth in suspension culture, Graham at
al., J. Gen Vim!. 36:59
(1977)); baby hamster kidney cells (BHK, ATCC CCL 10); Chinese hamster ovary
cells/-DHFR (CHO,
Urlaub at al., Proc. Natl. Acad. Sci. USA 77:4216 (1980)); mouse sertoli cells
(TM4, Mather, Biol.
Reprod. 23:243-251 (1980)); monkey kidney cells (CV1 ATCC CCL 70); African
green monkey kidney
cells (VERO-76, ATCC CRL-1587); human cervical carcinoma cells (HELA, ATCC CCL
2); canine
kidney cells (MDCK, ATCC CCL 34); buffalo rat liver cells (BRL 3A, ATCC CRL
1442); human lung cells
(W138, ATCC CCL 75); human liver cells (Hep G2, HB 8065); mouse mammary tumor
(MMT 060562,
ATCC CCL51); TRI cells (Mather at al., Annals N.Y. Acad. Sci. 383:44-68
(1982)); MRC 5 cells; F54
cells; and a human hepatoma line (Hep G2).
Host cells are transformed with the above-described expression or cloning
vectors for
polypeptide variant production and cultured in conventional nutrient media
modified as appropriate for
inducing promoters, selecting transformants, or amplifying the genes encoding
the desired sequences.
(viii) Culturing the host cells
The host cells used to produce the polypeptide variant of this invention may
be cultured in a
variety of media. Commercially available media such as Ham's F10 (Sigma),
Minimal Essential Medium
((MEM), (Sigma), RPMI-1640 (Sigma), and Dulbecco's Modified Eagle's Medium
((DMEM), Sigma) are =
suitable for culturing the host cells. In addition, any of the media described
in Ham at al., Meth. Enz.
58:44 (1979), Barnes et al., Anal. Biochem.102:255 (1980), U.S. Pat. Nos.
4,767,704; 4,657,866;
4,927,762; 4,560,655; or 5,122,469; WO 90/03430; WO 87/00195; or U.S. Patent
Re. 30,985 may be
used as culture media for the host cells. Any of these media may be
supplemented as necessary with
hormones and/or other growth factors (such as insulin, transferrin, or
epidermal growth factor), salts
(such as sodium chloride, calcium, magnesium, and phosphate), buffers (such as
HEPES), nucleotides
(such as adenosine and thymidine), antibiotics (such as GENTAMYCINTm drug),
trace elements (defined
as inorganic compounds usually present at final concentrations in the
micromolar range), and glucose
or an equivalent energy source. Any other necessary supplements may also be
included at appropriate
concentrations that would be known to those skilled in the art. The culture
conditions, such as
temperature, pH, and the like, are those previously used with the host cell
selected for expression, and
will be apparent to the ordinarily skilled artisan.
(ix) Polypeptide variant purification
When using recombinant techniques, the polypeptide variant can be produced
intracellularly,
in the periplasmic space, or directly secreted into the medium. If the
polypeptide variant is produced
intracellularly, as a first step, the particulate debris, either host cells or
lysed fragments, is removed, for
example, by centrifugation or ultrafiltration. Carter at al., Bioffechnology
10:163-167 (1992) describe
a procedure for isolating antibodies which are secreted to the periplasmic
space of E. coll. Briefly, cell
paste is thawed in the presence of sodium acetate (pH 3.5), EDTA, and
phenylmethylsulfonylfluoride
-32-
CA 02577543 2007-01-31
WO 99/51642
IMAIUSYWU0535
=
(PMSF) over about 30 min. Cell debris can be removed by centrifugation. Where
the polypeptide variant
is secreted into the medium, supernatants from such expression systems are
generally first concentrated
using a commercially available protein concentration fitter, for example, an
Amicon or Millipore PeIli=
ultrafiltration unit. A protease inhibitor such as PMSF may be included in any
of the foregoing steps to
inhibit proteolysis and antibiotics may be included to prevent the growth of
adventitious contaminants.
The polypeptide variant composition prepared from the cells can be purified
using, for example,
hydroxylapatite chromatography, gel electrophoresis, dialysis, and affinity
chromatography, with affinity
chromatography being the preferred purification technique. The suitability of
protein A as an affinity
ligand depends on the species and isotype of any immunoglobulin Fc region that
is present in the
. polypeptide variant. Protein A can be used to purify polypeptide variants
that are based on human yl,
y2, or 74 heavy chains (Lindmarlc etal., J. Immunot Meth. 62:1-13(1983)).
Protein G is recommended
for all mouse isotypes and for human y3 (Guss etal., EMBO J.
5:15671575(1986)). The matrix to which
the affinity ligand is attached is most often agarose, but other matrices are
available. Mechanically
stable matrices such as controlled pore glass or poly(styrenedivinyl)benzene
allow for faster flow rates
and shorter processing times than can be achieved with agarose. Where the
polypeptide variant
comprises a CH3 domain, the Bakerbond ABXTM resin (J. T. Baker, Phillipsburg,
NJ) is useful for
purification. Other techniques for protein purification such as fractionation
on an ion-exchange column,
ethanol precipitation, Reverse Phacio HPLC, chromatography on silica,
chromatography on heparin
= = SEPHAROSETh chromatography on an anion or cation exchange resin (such
as a polyaspartic acid
column), chromatofocusing, SDS-PAGE, and ammonium sulfate precipitation are
also available
depending on the polypeptide variant to be recovered.
Following any preliminary purification step(s), the mixture comprising the
polypeptide variant of
interest and contaminants may be subjected to low pH hydrophobic interaction
chromatography using
an elution buffer at a pH between about 2.5-4.5, preferably performed at low
salt concentrations
(e.g.,from about 0-0.25M salt).
E. Pharmaceutical Formulations
Therapeutic formulations of the polypeptide variant are prepared for storage
by mixing the
polypeptide variant having the desired degree of purity with optional
physiologically acceptable carriers,
excipients or stabilizers (Remington's Pharmaceutical Sciences 16th edition,
Osol, A Ed. (1980)), in the
form of lyophilized formulations or aqueous solutions. Acceptable carriers,
excipients, or stabilizers are
nontoxic to recipients at the dosages and concentrations employed, and include
buffers such as
phosphate, citrate, and other organic acids; antioxidants including ascorbic
acid and methionine;
preservatives (such as octadecyldimethylbenzyl ammonium chloride;
hexamethonium chloride;
benzalkonium chloride, benzethonium chloride; phenol, butyl or benzyl alcohol;
alkyl parabens such as
methyl or propyl paraben; catechol; resorcinol; cyciohexanol; 3-pentanol; and
m-cresol); low molecular
weight (less than about 10 residues) polypeptide; proteins, such as serum
albumin, gelatin, or
immunoglobulins; hydrophilic polymers such as polyvinylpyrrolidone; amino
acids such as glycine,
glutamine, asparagine, histidine, arginine, or lysine; monosaccharides,
disaccharides, and other
-33-
CA 02577543 2007-01-31
WO 99/51642
PalUS99/068511
carbohydrates including glucose. mannose, Of dextrins; cheiating agents such
as EDTA; sugars such
as sucrose, mannitol, trehalose or sorbitol; salt-forming counter-ions such as
sodium; metal complexes
(e.g., Zn-protein complexes); and/or non-ionic 'surfactants such as TWEENT,A,
PLURONICSTM or
polyethylene glycol (PEG).
The formulation herein may also contain more than one active compound as
necessary for the
particular indication being treated, preferably those with complementary
activities that do not adversely
affect each other. Such molecules are suitably present in combination in
amounts that are effective for
the purpose intended.
= The active ingredients may also be entrapped in microcapsule prepared,
for example, by
coacervation techniques or by interfacial polymerization, for example,
hydroxymethylcelluloseor gelatin- .
microcapsule and poly-(methylmethacylate) microcapsule, respectively, in
colloidal drug delivery
systems (for example; liposomes, albumin microspheres, microemulsions, nano-
particles and
nanocapsules) or in macroemulsions. Such techniques are disclosed in
Remington's Pharmaceutical
Sciences 16th edition, Osol, A. Ed. (1980).
The formulations to be used for in vivo administration must be sterile. This
is readily
accomplished by filtration through sterile filtration membranes.
Sustained-release preparations may be prepared. Suitable examples of sustained-
release
= preparations include setrOermeable matrices of solid hydrophobic polymers
containing the polypeptide
variant, which matrices are in the form of shaped articles, e.g., films, or
microcapsule. Examples of
sustained-release matrices include polyesters, hydrogels (for example, poly(2-
hydroxyethyl-
methacrylate), or poly(vinylalcohol)), polylactides (U.S. Pat. No. 3,773,919),
copolymers of L-glutamic
acid and y ethyl-L-glutamate, non-degradableethylene-vinyl acetate, degradable
lactic acid-glycolic acid
copolymers such as the LUPRON DEPOTTm (injectable microspheres composed of
lactic acid-glycolic
acid copolymer and leuprolide acetate), and poly-D-0-3-hydroxybutyric acid.
While polymers such as
ethylene-vinyl acetate and lactic acid-glycolic acid enable release of
molecules for over 100 days, certain
hydrogels release proteins for shorter time periods. When encapsulated
antibodies remain in the body
for a long time, they may denature or aggregate as a result of exposure to
moisture at 37 C, resulting
In a loss of biological activity and possible changes in immunogenicity.
Rational strategies can be
devised for stabilization depending on the mechanism involved. For example, if
the aggregation
mechanism is discovered to be intermolecular S-S bond formation through thio-
disulfide interchange,
stabilization may be achieved by modifying sulfhydryl residues, lyophilizing
from acidic solutions,
controlling moisture content, using appropriate additives, and developing
specific polymer matrix
=
compositions.
F. Non-Therapeutic Uses for the Polypeptide Variant
The polypeptide variant of the invention may be used as an affinity
purification agent in this
process, the polypeptide variant is immobilized on a solid phase such a
Sephadex resin or filter paper,
using methods well known in the art. The immobilized polypeptide variant is
contacted with a sample
containing the antigen to be purified, and thereafter the support is washed
with a suitable solvent that
-34-
CA 02577543 2007-01-31
WO 99/51642
11µ.. I /V WY/110WD
will remove substantially all the material in the sample except the antigen to
be purified, which is bound
to the immobilized polypeptide variant. Finally, the support is washed with
another suitable solvent, such
as glycine buffer, pH 5.0, that will release the antigen from the polypeptide
variant
The polypeptide variant may also be useful in diagnostic assays, e.g., for
detecting expression
of an antigen of interest in specific cells, tissues, or serum.
For diagnostic applications, the polypeptide variant typically will be labeled
with a detectable
moiety. Numerous labels are available which can be generally grouped into the
following categories:
(a) Radioisotopes, such as 35S, 14c, 125., 3H, and 1311. The polypeptide
variant can be
labeled with the radioisotope using the techniques described In Current
Protocols in Immunology,
Volumes 1 and 2. Co!igen etal., Ed. Wiley-lnterscience, New York, New York,
Pubs. (1991) for example
and radioactivity can be measured using scintillation counting.
(b) Fluorescent labels such as rare earth chelates (europium chelates) or
fluorescein and
its derivatives, rhodamine and its derivatives, dansyl, Ussamine,
phycoerythrin and Texas Red are
available. The fluorescent labels can be conjugated to the polypeptide variant
using the techniques
disclosed in Current Protocols in Immunology, supra, for example. Fluorescence
can be quantified using
a fluorimeter.
(c) Various enzyme-substrate labels are available and U.S. Patent No.
4,275,149 provides
a review of some of these. The enzyme generally catalyzes a chemical
alteration of the chron .,genic
substrate that can be measured using various techniques. For example, the
enzyme may catalyze a
color change in a substrate, which can be measured spectrophotometrically.
Alternatively, the enzyme
may alter the fluorescence or chemiluminescenceof the substrate. Techniques
for quantifying a change
in fluorescence are described above. The chemiluminescent substrate becomes
electronically excited
by a chemical reaction and may then emit light which can be measured (using a
chemiluminometer, fnr
example) or donates energy to a fluorescent acceptor. Examples of enzymatic
labels include luciferases
(e.g., firefly luciferase and bacterial luciferase; U.S. Patent No.
4,737,456), luciferin, 2,3-
dihydrophthalazinediones, malate dehydrogenase, urease, peroxidase such as
horseradish peroxidase
(HRPO), alkaline phosphatase, 8-galactosidase, glucoamylase, lysozyme,
saccharide oxidases (e.g.,
glucose oxidase, galactose oxidase, and glucose-6-phosphate dehydrogenase),
heterocyclic oxidases
(such as uricase and xanthine oxidase), lactoperoxidase, microperoxidase, and
the like. Techniques for
conjugating enzymes to antibodies are described in O'Sullivan et W., Methods
for the Preparation of
Enzyme-Antibody Conjugates for use in Enzyme Immunoassay, in Methods in Enzym.
(ed J. Langone
& H. Van Vunakis), Academic press, New York, 73:147-166(1981).
Examples of enzyme-substrate combinations include, for example:
(i) Horseradish peroxidase (HRPO) with hydrogen peroxidase as a substrate,
wherein the
hydrogen peroxidase oxidizes a dye precursor (e.g.,orthophenyiene diamine
(OPD) or 3,31,5,5'-
tetramethyl benzidine hydrochloride (TM B)):
(ii) alkaline phosphatase (AP) with para-Nitrophenyl phosphate as
chromogenic substrate;
and
-35-
CA 02577543 2007-01-31
WO 99/51642
/tMYY/1105341
(iii)
p-D-galactosidase (13-D-Gal) with a chrornogenic substrate (e.g., p-
nitropheny143-0-
galacbosidase) or fiuorogenic substrate 4-methylumbeiliferyl-fi-D-
galactosidase.
Numerous other enzyme-substrate combinations are available to those skilled in
the art. For a
general review of these, see U.S. Patent Nos. 4,275,149 and 4,318,980.
Sometimes, the label is indirectly conjugated with the polypeptide variant.
The skilled artisan
will be aware of various techniques for achieving this. For example, the
polypeptide variant can be
conjugated with biotin and any of the three broad categories of labels
mentioned above can be
conjugated with avidin, or vice verse. Biotin binds selectively to avidin and
thus, the label can be
conjugated with the polypeptide variant in this indirect manner.
Alternatively, to achieve indirect
conjugation of the label with the polypeptide variant, the polypeptide variant
is conjugated with a small
hapten (e.g., digoxin) and one of the different types of labels mentioned
above is conjugated with an anti-
hapten polypeptide variant (e.g., anti-digoxin antibody). Thus, indirect
conjugation of the label with the
polypeptide variant can be achieved.
In another embodiment of the invention, the polypeptide variant need not be
labeled, and the
presence thereof can be detected using a labeled antibody which binds to the
polypeptide variant.
The polypeptide variant of the present invention may be employed in any known
assay method,
such as competitive binding assays, direct and indirect sandwich assays, and
immunoprecipitation
assay,.. Zola, Monoclonal Antibodies: A Manual .of Techniques, pp.147-158 (CRC
Pre, Inc. 1987).
The polypeptide variant may also be used for in vivo diagnostic assays.
Generally, the
polypeptide variant is labeled with a radionuclide (such as "11n, 99Tc, 14C,
1311, 1291, 3H, 32P or 35S) so that
the antigen or cells expressing it can be localized using immunoscintiography.
G. in Vivo Uses for the Polypeptide Variant
It is contemplated that the polypeptide variant of the present invention may
be used to treat a
mammal e.g. a patient suffering from a disease or disorder who could benefit
from administration of the
polypeptide variant. The conditions which can be treated with the polypeptide
variant are many and
include cancer (e.g. where the polypeptide variant binds the HER2 receptor or
CD20); allergic conditions
such as asthma (with an anti-IgE antibody); and LFA-mediated disorders (e.g.
where the polypeptide
variant is an anti-LFA-1 or anti-ICAM-1 antibody) etc. Where the polypeptide
variant does not bind
complement, but retains FcR binding capability, exemplary diseases or
disorders to be treated include:
cancer (e.g. where ADCC function is desirable, but complement activation would
lead to amplified side
effects, such as vasculitis in the blood vessels at the tumor site); disorders
treated with an agonist
antibody; disorders wherein the polypeptide variant binds a soluble antigen
and wherein stoichiometry
leads to immune complexes which activate the complement cascade and result in
unwanted side effects;
conditions employing an antagonist antibody which downmodulates receptor
function without damaging
tissue or organ function; intravenous immunoglobulin treatment for, e.g.,
immunodeficient individuals with
autoimmune disorders.
The polypeptide variant is administered by any suitable means, including
parenteral,
subcutaneous, intraperitoneal, intrapulmonary, and intranasal, and, if desired
for local
-36-
CA 02577543 2009-04-01
WO 99/51642 PCT/US99/06858
=¨
immunosuppressive treatment, intralesional administration. Parenteral
infusions include intramuscular,
intravenous, intraarterial, intraperitoneal, or subcutaneous administration.
In addition, the polypeptide
variant is suitably administered by pulse infusion, particularly with
declining doses of the polypeptide
variant Preferably the dosing is given by injections, most preferably
intravenous or subcutaneous
injections, depending in part on whether the administration is brief or
chronic.
For the prevention or treatment of disease, the appropriate dosage of
polypeptide variant will
depend on the type of disease to be treated, the severity and course of the
disease, whether the
. potypeptide variant is administered for preventive or therapeutic
purposes, previous therapy, the patients
clinical history and response to the polypeptide variant, and the discretion
of the attending physician.
The polypeptide variant is suitably administered to the patient at one time or
over a series of treatments.
Depending on the type and severity of the disease, about 1 pg/kg to 15 mg/kg
(e.g., 0.1-
20mg/kg) of polypeptide variant is an initial candidate dosage for
administration to the patient, whether,
for example, by one or more separate administrations, or by continuous
infusion. A typical daily dosage
might range from about 1 pg/kg to 100 mg/kg or more, depending on the factors
mentioned above. For
repeated administrations over several days or longer, depending on the
condition, the treatment is
sustained until a desired suppression of disease symptoms occurs. However,
other dosage regimens
may be useful. The progress of this therapy is easily monitored by
conventional techniques and assays.
The polypeptide variant composition will be formulated, dosed, and admi
istered In a fashion
consistent with good medical practice. Factors for consideration in this
context include the particular
disorder being treated, the particular mammal being treated, the clinical
condition of the individual
patient, the cause of the disorder, the site of delivery of the agent, the
method of administration, the
scheduling of administration, and other factors known to medical
practitioners. The "therapeutically
effective amount' of the polypeptide variant to be administered will be
governed by such considerations,
and the minimum amount necessary to prevent, ameliorate, or treat a
disease or disorder. The
polypeptide variant need not be, but is optionally formulated with one or more
agents currently used to
prevent or treat the disorder in question. The effective amount of such other
agents depends on the
amount of polypeptide variant present in the formulation, the type of disorder
or treatment, and other
factors discussed above. These are generally used in the same dosages and with
administration routes
as used hereinbefore or about from 1 to 99% of the heretofore employed
dosages.
The invention will be more fully understood by reference to the following
examples. They should
not, however, be construed as limiting the scope of this invention.
EXAMPLE 1
Low Affinity Receptor Binding Assay
This assay determines binding of an IgG Fc region to recombinant FeyRita,
FcyRIlb and
FcyfilIla a subunits expressed as His6-glutathione S transferase (GST)-tagged
fusion proteins. Since
the affinity of the Fc region of IgG1 for the FcyRI Is in the nanomolar range,
the binding of IgG1 Fc
mutants can be measured by titrating monomeric IgG and measuring bound IgG
with a polyclonal anti-
-37-
CA 02577543 2007-01-31
WO 99/51642
PCT/99/6858
IgG in a standard ELISA format (Example 2 below). The affinity of the other
members of the FcyR family,
i.e. FcyRlia, FcyRIlb and FcyRIlla for IgG is however in the micromolar range
and binding of monomeric
IgG1 for these receptors can not be reliably measured in an ELISA format
The following assay utilizes Fc mutants of recombinant anti-IgE E27 (Figures
4A and 4B) which,
when mixed with human IgE at a 1:1 molar ratio, forms a stable hexamer
consisting of three anti-IgE
molecules and three IgE molecules. A recombinant chimeric form of IgE
(chimeric IgE) was engineered
and consists of a human IgE Fc region and the Fab of an anti-VEGF antibody
(Presta et al. Cancer
Research 57:4593-4599 (1997)) which binds two VEGF molecules per mole of anti-
VEGF. When
recombinant human VEGF is added at a 2:1 molar ratio to chimeric IgE:E27
hexamers, the hexamers
are linked into larger molecular weight complexes via the chimeric IgE
Fab:VEGF interaction. The E27
component of this complex binds to the FcyRIla, FcyRIlb and FcyRIlla a
subunits with higher avidity to
permit detection in an ELISA format.
MATERIALS AND METHODS
Receptor Coat Fcy receptor a subunits were expressed as GST fusions of His6
tagged
extracellular domains (ECDs) in 293 cells resulting in an ECD-6His-GST fusion
protein (Graham et al.
J. Gen. Vint 36:59-72(1977) and Gorman etal. DNA Prot. Eng. Tech. 2:3-
10(1990)) and purified by
NI-NTA column chromatography (Oiagen, Australia) and buffer exchanged into
phosphate buffered
= = saline (PBS). Concentrations were determined by absorption at 280nm
u:='ng extinction coefficients
derived by amino acid composition analysis. Receptors were coated onto Nunc
F96 maxisorb plates
(cat no. 439454) at 10Ong per well by adding 100 jtl of receptor-GST fusion at
1 pig/m1 in PBS and
incubated for 48 hours at 4 C. Prior to assay, plates are washed 3x with 250
plof wash buffer (PBS pH
7.4 containing 0.5% TWEEN 20m) and blocked with 250 MI of assay buffer (50mM
Iris buffered saline,
0.05% TWEEN 20TN, 0.5% RIA grade bovine albumin (Sigma A7888), and 2mM EDTA pH
7.4).
Immune Complex Formation: Equal molar amounts (1:1) of E27 and recombinant
chimeric IgE
which binds two moles recombinant human VEGF per mole of chimeric IgE are
added to a 12 x 75mm
polypropylene tube in PBS and mixed by rotation for 30 minutes at 25 C. E27
(anti-IgE) /chimeric IgE
(19E) hexamers are formed during this incubation. Recombinant human VEGF (165
form, MW 44,000)
is added at a 2:1 molar ratio to the IgE concentration and mixed by rotation
an additional 30 minutes at
25 C. VEGF- chimeric IgE binding links E27:chimeric IgE hexamers into larger
molecular weight
complexes which bind FcyR a subunit ECD coated plates via the Fc region of the
E27 antibody.
E27:chimerIc IgE:VEGF (1:1:2 molar ratio) complexes are added to FcyR a
subunit coated
plates at E27 concentrations of 5 jig and 1 pa total IgG in quadruplicate in
assay buffer and Incubated
for 120 minutes at 25 C on an orbital shaker.
Complex Detection: Plates are washed 5x with wash buffer to remove unbound
complexes
and IgG binding is detected by adding 100 I horse radish peroxidase (HRP)
conjugated goat anti-
human IgG (y) heavy chain specific (Boehringer Mannheim 1814249) at 1:10,000
in assay buffer and
incubated for 90min at 25 C on an orbital shaker. Plates are washed 5x with
wash buffer to remove
unbound HRP goat anti-human 1gG and bound anti-IgG is detected by adding 100
1 of substrate
-38-
CA 02577543 2007-01-31
WO 99/51642
PCT/U599/116355
solution (0.4mg/m1 o-phenylenedaimine dihydrochloride, Sigma P6912, 6 mM H202
in PBS) and
incubating for 8 min at 25 C. Enzymatic reaction is stopped by the addition of
100 pl 4.5N H2SO4 and
colorimetric product is measured at 490 nm on a 96 well plate densitometer
(Molecular Devices). Binding
of E27 mutant complexes is expressed as a percent of the wild type E27
containing complex.
EXAMPLE 2
Identification of Unique C1q Binding Sites in a Human IgG Antibody
In the present study, mutations were identified in the CH2 domain of a human
IgG1 antibody,
"C2B8" (Reff at al., Blood 83:435 (1994)), that ablated binding of the
antibody to C1q but did not alter
the conformation of the antibody nor affect binding to each of the FcyRs. By
alanine scanning
mutagenesis, five mutants in human IgG1 were identified, D270K, 0270V, K322A
P329A, and P331, that
were non-lytic and had decreased binding to C1q. The data suggested that the
core C1q binding sites
In human IgG1 is different from that of murine IgG2b. In addition, K322A,
P329A and P331A were found
to bind normally to the CO20 antigen, and to four Fc receptors, FcyRI, FcyRII,
FcyRIII and FcRn.
MATERIALS AND METHODS
Construction of C288 Mutants: The chimeric light and heavy chains of anti-CD20
antibody
C2B8 (Reff at al., Blood 83:435 (1994)) subcloned separately into previously
described PRK vectors
(Gorman at al., DNA Protein Eng. Tech. 2:3(1990)) were used. By site directed
mutagenesis (Kunkel
etal., Proc. Natl. Acad.ScL(ISA 82:488(1985)), alanine scan variants of the Fc
region in the heavy chain
were constructed. The heavy and light chain plasmids were co-transfected into
an adenovirus
transformed human embryonic kidney cell line as previously described (Werther
at al., J. lmmunol.
157:4986(1996)). The media was changed to serum-free 24 hours after
transfection and the secreted
antibody was harvested after five days. The antibodies were purified using
Protein A-SEPHAROSECL-
4BTM (Pharmacia), buffer exchanged and concentrated to 0.5 ml with PBS using a
Centricon-30
(Amicon), and stored at 4 C. The concentration of the antibody was determined
using total Ig-binding
ELISA.
Clq Binding ELISA: Costar 96 well plates were coated overnight at 4 C with the
indicated
concentrations of C2B8 in coating buffer (0.05 M sodium carbonate buffer), pH
9. The plates were then
washed 3x with PBS/ 0.05% TWEEN 20Tm, pH 7.4 and blocked with 200 1 of ELISA
diluent without
thimerosal (0.1M NaPO4 / 0.1M NaCi / 0.1% gelatin / 0.05% TWEEN 207m/ 0.05%
ProClin300) for ihr
at room temperature. The plate was washed 3x with wash buffer, an aliquot of
100 I of 2 pg/mIC1q
(0:Wide!, San Diego, CA) was added to each well and incubated for 2 hrs at
room temperature. The plate
was then washed 6x with wash buffer. 100 pl of a 1:1000 dilution of sheep anti-
complement C1q
peroxidase conjugated antibody (Biodesign) was added to each well and
incubated for 1 hour at room
temperature. The plate was again washed 6x with wash buffer and 100 id of
substrate buffer
(PBS/0.012% H202) containing OPD (0-phenylenediaminedihydrochloride (Sigma))
was added to each
well. The oxidation reaction, observed by the appearance of a yellow color,
was allowed to proceed for
30 minutes and stopped by the addition of 100 pl of 4.5 N H2SO4. The
absorbance was then read at
(492-405) nm using a microplate reader (SPECTRA MAX 250TM, Molecular Devices
Corp.). The
-39-
CA 02577543 2007-01-31
WO 99/51642
PCT/U599/06858
appropriate controls were run in parallel (i.e. the ELISA was performed
without C1q for each
concentration of C2B8 used and also the EUSA was performed without C2B8). For
each mutant, C1q
binding was measured by plotting the absorbance (492-405) rim versus
concentration of C2B8 in g/m1
using a 4-parameter curve fitting program (KALEIDAGRAPHTM) and comparing EC 30
values.
Complement Dependent Cytotoxicity (CDC) Assay. This assay was performed
essentially
as previously described (Gazzano-Santoro et al., J. lmmunol. Methods 202:163
(1997)). Various
concentrations of C2B8 (0.08-20 g/m1) were diluted with RHB buffer (RPMI
1640/20mM HEPES (pH
7.2)/2mM Glutamine/0.1% BSA/100 g/mIGentamicin). Human complement (Quidel)
was diluted 1:3
in RHB buffer and VVII.2-S cells (available from the ATCC, Manassas, VA) which
express the CD20
antigen were diluted to a density of 1 x 106 cells /ml with RHB buffer.
Mixtures of 150 I containing equal
volumes of C2B8, diluted human complement and WI12-S cells were added to a
flat bottom tissue
culture 96 well plate and allowed to Incubate for 2 hrs at 37 C and 5% CO2 to
facilitate complement
mediated cell lysis. 50 I of alamar blue (Accumed International) was then
added to each well and
incubated overnight at 37 C. The absorbance was measured using a 96-well
fluorometer with excitation
at 530 nm and emission at 590 nm. As described by Gazzano-Santoro etal., the
results are expressed
in relative fluorescence units (RFU). The sample concentrationswere computed
from a C2B8 standard
curve and the percent activity as compared to wild type C2B8 is reported for
each mutant.
CD20 Binding Potency of the C288 Mutants: The binding of C2B8 and mutants to
the CD20
antigen were assessed by a method previously described (Reff et al., (1994),
supra; reviewed in
Gazzano-Santoro etal., (1996), supra). WIL2-S cells were grown for 3-4 days to
a cell density of 1 x
105 cells/mt. The cells were washed and spun twice in FACS buffer (PBS/0.1 A
BSA/0.02% NaN3) and
resuspended to a cell density of 5 x 106 cells/mt. 200 I of cells (5 x 106
cells/ml) and 20 I of diluted
C2B8 samples were added to a 5 ml tube and incubated at room temperature for
30 minutes with
agitation. The mixture was then washed with 2 ml of cold FACS buffer, spun
down and resuspended
in 200 I of cold FACS buffer. To the suspension, 10 I of goat anti-human IgG-
FITC (American Qualex
Labs.) was added and the mixture was incubated in the dark at room temperature
for 30 minutes with
agitation. After incubation, the mixture was washed with 2 ml of FACS buffer,
spun down and
resuspended in 1 ml of cold fixative buffer (1% formaldehyde in PBS). The
samples were analyzed by
flow cytometry and the results expressed as relative fluorescence units (RFU)
were plotted against
antibody concentrations using a 4-parameter curve fitting program
(KALEIDAGRAPHTm). The EC30
values are reported as a percentage of that of the C2B8 reference material.
FcyR Binding EUSAs: FcyRI a subunit-GST fusion was coated onto Nunc F96
maxisorb plates
(cat no. 439454) by adding 100 pl of receptor-GST fusion at 1 g/m1 in PBS and
incubated for 48 hours
at 4 C. Prior to assay, plates are washed 3x with 250 plot wash buffer (PBS pH
7.4 containing 0.5%
TWEEN 20Th) and blocked with 250 I of assay buffer (50mM Iris buffered
saline, 0.05% TWEEN 20w,
0.5% RIA grade bovine albumin (Sigma A7888), and 2mM EDTA pH 7.4). Samples
diluted to 10 g/m1
in 1 ml of assay buffer are added to FcyRI a subunit coated plates and
incubated for 120 minutes at 25 C
-40-
CA 02577543 2007-01-31
WO 99/51642
PCT/US99/06858
on an orbital shaker. Plates are washed 5x with wash buffer to remove unbound
complexes and IgG
binding is detected by adding 100 pd horse radish percoddase (HRP) conjugated
goat anti-human IgG
ft) heavy chain specific (Boehringer Mannheim 1814249) at 1:10,000 in assay
buffer and incubated for
90min at 25 C on an orbital shaker. Plates are washed 5x with wash buffer to
remove unbound HRP
goat anti-human IgG and bound anti-NG is detected by adding 100 pl of
substrate solution (0.4mg/m1
o-phenylenedaimine dihydrochloride, Sigma P6912, 6 mM H202 in PBS) and
incubating for 8 min at
25 C. Enzymatic reaction is stopped by the addition 01 100 mi 4.5N H2SO4 and
colorimetric product is
measured at 490 nm on a 96 well plate densitometer (Molecular Devices).
Binding of variant is
expressed as a percent of the wild type molecule.
FcyRil and III binding ELISAs were performed as described in Example 1 above.
For measuring FcRn binding activity of IgG variants, EL1SA plates were coated
with 2 pg/m1
streptavidin (Zymed, South San Francisco) in 50 mM carbonate buffer, pH 9.6,
at 4 C overnight and
blocked with PBS-0.5% BSA, pH 7.2 at room temperature for one hour.
Biotinylated FcRn (prepared
using biotin-X-NHS from Research Organics, Cleveland, OH and used at 1-2
pig/m1) in PBS-0.5% BSA,
0.05% polysorbate 20, pH 7.2, was added to the plate and incubated for one
hour. Two fold serial
dilutions of IgG standard (1.6-100 ng/m1) or variants in PBS-0.5% BSA, 0.05%
polysorbate 20, pH 6.0,
were added to the plate and incubated for two hours. Bound IgG was detected
using peroxidase Istieled
goat F(ab1)2 anti-human IgG F(ab1)2 in the above pH 6.0 buffer (Jackson
ImmunoResearch, West Grove,
PA) followed by 3,3',5,54etramethylbenzidine (Kirgaard & Perry Laboratories)
as the substrate. Plates
were washed between steps with PBS-0.05% polysorbate 20 at either pH 7.2 or
6Ø Absorbance was
read at 450 nm on a Vmax plate reader (Molecular Devices, Menlo Park, CA).
Titration curves were fit
with a four-parameter nonlinear regression curve-fitting program
(KaleidaGraph, Synergy software,
Reading, PA). Concentrations of IgG variants corresponoing to the mid-point
absorbance of the titration
curve of the standard were calculated and then divided by the concentration of
the standard
corresponding to the mid-point absorbance of the standard titration curve.
RESULTS AND DISCUSSION
By alanine scanning mutagenesis, several single point mutations were
constructed in the CH2
domain of C2B8 beginning with E318A, K320A and K322A. All the mutants
constructed bound normally
to the CD20 antigen (Table 1).
Table 1
wt E318A K320A K322A P329A P331A
FcRn
CD20
FcyRI +
FcyRII
Fcy12111 + +
*C1q +++ ++ +++
-41-
CA 02577543 2007-01-31
WO 99/31642
PC171.1599/110555
CDC
(+) indicates binding and (-) signifies binding abolished
1/1fith respect to Clq binding, each + sign is equivalent to approximately 33%
binding.
Where binding of human complement to an antibody with a human Fc was analyzed,
the ability
of E318A and K320A to activate complement was essentially identical to that of
wild type C2B8 (Table
1). When compared to wild type C2B8, there appears to be little difference in
the binding of E318A and
K320A to Clq. There is only a 10% decrease in the binding of K320A and about a
30% decrease in the
binding of E318A to Clq (Fig. 2). The results indicate that the effect of the
E318A and the K320A
substitution on complement activation and Clq binding is minimal. Also, the
human IgG1 of C2B8 was
substituted for human IgG2 and used as a negative control in the C1q binding
studies. The IgG2 mutant
appears to have a much lower affinityfor C1q=than the E318A and K320A mutants
(Fig. 2). Thus, the
results demonstrate that E318 and K320 do not constitute the core C1q binding
sites for human IgG1.
Conversely, the K322A substitution had a significant effect on both complement
activity and Clq
binding. The K322A mutant did not have CDC activity when tested in the above
CDC assay and was
more than a 100 fold lower than wild type C2B8 in binding to Clq (Fig. 2). In
the human system, K322
is the only residue of the proposed core Clq binding sites that appeared to
have a significant effect on
complement activation and C1q binding.
Since the Duncan and Winter study was performed using mouse IgG2b and the
above results
reveal that K320 and E318 in human IgG1 ;Ire not involved in C1q binding, and
without being bound to
any one theory, the above data suggest that the C1q binding region in murine
IgGs is different from that
of the human. To investigate this further and also to identify additional
mutants that do not bind to Clq
and hence do not activate complement, several more point mutations in the
vicinity of K322 were
constructed as assessed from the three dimensional structure of the C258 Fc.
Mutants constructed,
K274A, N276A, Y278A, S324A, P329A, P331A. K334A, and T335A, were assessed for
their ability to
bind C1q and also to activate complement. Many of these substitutions had
little or no effect on C1q
binding or complement activation. In the above assays, the P329A and the P331A
mutants did not
activate complement and had decreased binding to Clq. The. P331A mutant did
not activate
complement and was 60 fold lower in binding to Clq (Fig. 3) when compared to
wild type C2B8 (Fig. 2).
The concentration range of the antibody variants used in Fig. 3 is expanded to
1001.1g/m1 in order to
observe saturation of C1q binding to the P331A variant. The mutation P329A
results in an antibody that
does not activate complement and is more than a 100 fold lower in binding to
C1q (Fig. 3) when
compared to wild type C2B8 (Fig. 2).
Mutants that did not bind to C1q and hence did not activate complement were
examined for their
ability to bind to the Fc receptors: FcyRI, FcyRIla, FcyRilb, FcyRilla and
FcRn. This particularstudy was
performed using a humanized anti-IgE antibody, an IgG1 antibody with these
mutations (see Example
1 above). The results revealed the mutants, K322A and P329A, bind to all the
Fc receptors to the same
extent as the wild type protein (Table 2). However, there was a slight
decrease in the binding of P331A
to FcyRIlb.
-42-
CA 02577543 2007-01-31
WO 99/51642
PCT/U599/06858
In conclusion, two amino acid substitutions in the COOH terminal region of the
CH2 domain of
human IgG1, K322A and P329A were identified that result in more than 100 fold
decrease in C1q binding
and do not activate the CDC pathway. These two mutants, K322A and P329A, bind
to all Fc receptors
with the same affinity as the wild type antibody. Based on the results,
summarized in Table 2, and
without being bound to any one theory, it is proposed that the C1q binding
epicenter of human IgG1 is
centered around K322, P329 and P331 and is different from the rnurine IgG2b
epicenter which
constitutes E318, K320 and K322.
Table 2
wt E318A K320A K322A P329A P331A
C D20 100 89 102 86 112 103
TcyRI 100 93 102 r 90 104 74
=FcyRI la 100 113 94 109 111
es
=FcyRI lb 100 106 83 101 96
58
sFcyRIII 100 104 72 90 85 73
CDC 100 108 108 None none
none
'For binding to the Fcyfis the mutants were made in the E27 background (anti-
IgE).
The results are presented as a percentage of the wild type.
A further residue involved in binding human C1q was identified using the
methods described in
the present example. The residue D270 was replaced with lysine and vaiine to
generate mutants D270K
and D270V, respectively. These mutants both showed decreased binding to human
C1q (Fig. 6) and
were non-lytic (Fig. 7). The two mutants bound the CD20 antigen normally and
recruited ADCC.
EXAMPLE 3
Mutants with improved Clq Binding
The following study shows that substitution of residues at positions K326,
A327, E333 and K334
resulted in mutants with at least about a 30% increase in binding to C1q when
compared to the wild type
antibody. This indicated K326, A327, E333 and K334 are potential sites for
improving the efficacy of
antibodies by way of the CDC pathway. The aim of this study was to improve CDC
activity of an
antibody by increasing binding to Gig. By site directed mutagenesis at K326
and E333, several mutants
with increased binding to C1q were constructed. The residues in order of
increased binding at K326 are
K<V<E<A<G<D<M<W, and the residues in order of increased binding at E333 are
E<Q<DW<G<A<S.
Four mutants, K326M, K326D, K326E and E333S were constructed with at least a
two-fold increase in
binding to C1q when compared to wild type. Mutant K326W displayed about a five-
fold increase in
binding to C1q.
Mutants of the wild type C2B8 antibody were prepared as described above in
Example 2. A
further control antibody, wild type C2B8 produced in Chinese hamster ovary
(CHO) cells essentially as
described in US Patent 5,736,137, was included in a C1q binding EUSA to
confirm that wt C2B8
produced in the 293 kidney cell line had the same C1q binding activity as the
CHO-produced antibody
-43-
CA 02577543 2007-01-31
WO 99/51642
PCT/US99/06858
(see CHO-wt-C2B8 in Fig. 8). The C1q binding ELISA, CDC assay, and CO20
binding potency assay
in this example were performed as described in Example 2 above.
As shown in Fig. 8, alanine substitution at K326 and E333 in C2B8 resulted in
mutants with
about a 30% increase in binding to C1q.
Several other single point mutants at K326 and E333 were constructed and
assessed for their
ability to bind Clq and activate complement All the mutants constructed bound
normally to the CD20
antigen.
Wth respect to K326, the other single point mutants constructed were K326A,
K326D, K326E,
K326G, K326V, K326M and K326W. As shown in Fig. 9, these mutants all bound to
C1q with a better
affinity than the wild type antibody. K326W, K326M, K326D and K326E showed at
least a two-fold
increase in binding to C1q (Table 3). Among the K326 mutants, 1(326W had the
best affinity for C1q.
Table 3
'Mutant EC.) value
Wild type 1.53
K326V 1.30
K326A 1.03
K326E 1.08
K32 G 0.95
1(326D 0.76
K326M 0.67
K326W 0.47
=
E333S 0.81
E333A. 0.98
E333G 1.14
E333V 1.18
E333D 1.22
E3330 1.52
K334A 1.07
Substitutions with hydrophobic as well as charged residues resulted in mutants
with increased
binding to Cl q. Even substitution with glycine which is known to impart
flexibility to a chain and is well
conserved in nature, resulted in a mutant with higher affinity for C1q when
compared to the wild type.
It would appear that any amino acid substitution at this site would result in
a mutant with higher affinity
for C1q. As assessed from the three-dimensionalstructure, 1(326 and E333 are
in the vicinity of the core
C1q binding sites (Fig. 10).
In addition to alanine, E333 was also substituted with other amino acid
residues. These
mutants, E333S, E333G, E333V, E333D, and E333Q, all had increased binding to
C1q when compared
to the wild type (Fig. 11). As shown in Table 3, the order of binding affinity
for C1q was as follows:
-44-
CA 02577543 2007-01-31
WO 99/51642
PCIYUS99/06858
E333S>E333A>E333G>E333V>E333D>E333Q. Substitutionswith amino acid residues
with small side
chain volumes, Le. serine, alanine and glycine, resulted in mutants with
higher affinity for C1q in
comparison to the other mutants, E333V, E333D and E3330, with larger side
chain volumes. The mutant
E333S had the highest affinity for Clq, showing a two-fold increase in binding
when compared to the
wild type. Without being bound to any one theory, this indicates the effect on
C1q binding at 333 may
also be due in part to the polarity of the residue.
Double mutants were also generated. As shown in Figs. 12 and 13, double
mutants K326M-
E333S and K326A-E333Awere at least three-fold better at binding human C1q than
wild type C2B8 (Fig.
12) and at least two-fold better at mediating CDC compared to wild type C2B8
(Fig. 13). Addidvity
indicates these are independently acting mutants.
An additional double mutant K326VV-E333S was generated which was six-fold
better at binding
human C1q and three-fold better at mediating CDC compared to wild type C2B8.
This double mutant
was deficient in ADCC activity in a cell-based assay.
As shown in Fig. 14, a further mutant with improved C1q binding (50% increase)
was made by
changing A327 in a human IgG1 constant region to glycine. Conversely, in a
human IgG2 constant
region, changing G327 to alanine reduced C1q binding of the IgG2 antibody.
=
-45-
CA 02577543 2007-01-31
DEMANDES OU BREVETS VOLUMINEUX
LA PRESENTE PARTIE DE CETTE DEMANDE OU CE BREVETS
= COMPREND PLUS D'UN TOME.
CECI EST LE TOME I DE
NOTE: Pour les tomes additionels, veillez contacter le Bureau Canadien des
Brevets.
JUMBO APPLICATIONS / PATENTS
THIS SECTION OF THE APPLICATION / PATENT CONTAINS MORE
THAN ONE VOLUME.
THIS IS VOLUME I OF
NOTE: For additional volumes please contact the Canadian Patent Office.
- -