Note: Descriptions are shown in the official language in which they were submitted.
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
MULTI-LAYER TABLETS AND BIOADHESIVE DOSAGE FORMS
RELATED APPLICATIONS
This application claims the benefit of U.S. Provisional Application Nos.
60/604,990, filed August 27, 2004, 60/604,991, filed August 27, 2004,
60/605,198,
filed August 27, 2004, 60/605,199, filed August 27, 2004, 60/605,200, filed
August
27, 2004, 60/605,201, filed August 27, 2004, 60/607,905, filed September 8,
2004,
60/635,812, filed December 13, 2004, 60/650,191, filed February 4, 2005,
60/650,375, filed February 4, 2005 and 60/676,383, filed April 29, 2005. This
application is a continuation-in-part of U.S. Application No. 11/009,327,
filed
December 9, 2004 and a continuation-in-part of International Application No.
PCT/US2005/007525, filed March 3, 2005 in English and designating the U.S. The
entire teachings of the above-referenced applications are incorporated herein
by
reference.
BACKGROUND OF THE INVENTION
Controlled release systems for drug delivery are often designed to administer
drugs in specific areas of the body. In the case of drug delivery to or via
the
gastrointestinal tract, it is critical that the drug not be delivered
substantially beyond
the desired site of action or absorption, respectively, before it has had a
chance to
exert a topical effect or to pass into the bloodstreain. A drug delivery
system that
adheres to the lining of the appropriate viscus, will deliver its contents to
the targeted
tissue as a function of proximity and duration of contact.
An orally ingested product can adhere to either the epithelial surface or the
inucus lining of the gastrointestinal tract. For the delivery of bioactive
substances, it
can be advantageous to have a polymeric drug delivery device adhere to the
epithelium or to the mucous layer. Bioadhesion in the gastrointestinal tract
proceeds
in two stages: (1) viscoelastic deformation at the point of contact of the
synthetic
material into the mucus substrate, and (2) formation of bonds between the
adhesive
synthetic material and the mucus or the epithelial cells. In general, adhesion
of
polymers to tissues may be achieved by (i) physical or mechanical bonds, (ii)
primary
or covalent chemical bonds, and/or (iii) secondary cliemical bonds (i.e.,
ionic).
1
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
Physical or mechanical bonds can result from deposition and inclusion of the
adhesive
material in the crevices of the mucus or the folds of the mucosa. Secondary
chemical
bonds, contributing to bioadhesive properties, consist of dispersive
interactions (i.e.,
Van der Waals interactions) and stronger specific interactions, which include
hydrogen bonds. The hydrophilic functional groups primarily responsible for
forming
hydrogen bonds are the hydroxyl and the carboxylic acid groups.
Duchene et al., in Drug Dev. Ind. Pharm., 14:283-318 (1988), review the
pharmaceutical and medical aspects of bioadhesive systems for drug delivery.
Polycarbophils and acrylic acid polymers were noted as having the best
adhesive
properties. Others have explored the use of bioadhesive polymers, however, the
extent
of bioadhesion achieved in these studies has been limited. In addition, these
studies do
not demonstrate how to prepare larger bioadhesive drug delivery devices, such
as
tablets. WO 93/21906 discloses methods for fabricating bioadhesive
microspheres and
for measuring bioadhesive forces between microspheres and selected segments of
the
gastrointestinal tract in vitro. Lehr et al. screened microparticles formed of
copolylners of acrylic acid using an in vitro system and determined that the
copolymer "Polycarbophil" has increased adhesion.
Although bioadhesive-coated microparticles are known, larger oral
fomlulations such as tablets with the ability to adequately adhere to the
gastrointestinal tract mucosa are not known. The larger oral formulations
differ from
microparticles in that dosage forms such as tablets, capsules and drug-eluting
devices
cannot enter into an invagination in the mucosa, whereas microparticles are
generally
small enough to fit into an invagination. As a result, larger oral
formulations contact a
smaller surface area of the gastrointestinal tract (particularly as a function
of the ratio
of contact surface area to volume of the formulation), which is expected to
weaken
the interaction between the larger formulation and the gastrointestinal tract.
Separately or in addition to the need to control the location at which a drug
is
released, there is also a need to control the duration over which a drug is
released
from a pharmaceutical formulation. In particular, certain drugs, especially
neuroactive
drugs, have side effects and lower efficacy if blood serum concentrations vary
considerably. Standard immediate release formulations typically cause such
fluctuations in blood serum concentrations, because they dump large quantities
of
drug at one time into the patient's gastrointestinal tract.
2
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
Thus, there is a need for methods for controlling or increasing the absorption
of pharmaceutical agents from drug delivery systems such as tablets through
mucosal
membranes. There also is a need for methods for delaying transit of
pharmaceutical
formulations through gastrointestinal passages.
SUMMARY OF THE INVENTION
In one aspect, the present invention provides pharmaceutical dosage forms for
oral delivery of a drug, comprising a drug to be delivered gastrointestinally,
and a
bioadhesive polymeric coating applied to at least a fraction of one surface of
the
dosage form. The coating provides the dosage form with a fracture strength of
at least
100 N/m2 as measured on rat intestine, and the dosage form has a
gastrointestinal
retention time of at least 4 hours in a fed beagle dog model during which the
drug is
released from the dosage form.
In a first embodiment, the present invention is a tablet for oral delivery of
a
drug, comprising a core including a drug to be delivered gastrointestinally,
and a
bioadhesive polymeric coating applied to at least one surface of the tablet.
The
coating provides the tablet with a fracture strength of at least 100 N/m2 as
measured
on rat intestine, and the tablet has a gastrointestinal retention time of at
least 4 hours
in a fed beagle dog model during which the drug is released from the tablet.
In certain
embodiments, the bioadhesive polymer coating further includes metal compounds,
low molecular weight oligomers or a combination thereof that enhance the
mucosal
adhesion of the synthetic polymer coating. In a preferred embodiment, the
bioadhesive polymeric coating does not substantially swell upon hydration.
In one embodiment, the present invention is a tablet for oral delivery of a
drug,
comprising a core including a drug to be delivered gastrointestinally, and a
bioadhesive polymeric coating applied to at least one surface of the tablet.
The
coating provides the tablet with a fracture strength of at least 100 N/mZ as
measured
on rat intestine, and the tablet has a gastrointestinal retention time of at
least 3 hours
in a fasted beagle dog model (see Example 1) during which the drug is released
from
the tablet. In certain embodiments, the bioadhesive polymer coating further
includes
metal compounds, low molecular weight oligomers or a combination thereof that
enhance the mucosal adhesion of the synthetic polymer coating. In a preferred
3
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
embodiment, the bioadhesive polymeric coating does not substantially swell
upon
hydration.
In another embodiment, the invention is a drug-eluting device for oral
delivery
of a drug, which includes a reservoir having a drug-containing core contained
therein,
one or more orifices or exit ports through which drug from the core can elute
from the
device, and a bioadhesive polymeric coating, applied to at least one surface
of the
device. The coating provides the device with a fracture strength of at least
100 N/m2
as measured on rat intestine, and the device has a gastrointestinal retention
time of at
least 3 hours in a fasted beagle dog model during which the drug is released
from the
device. In a preferred embodiment, the bioadhesive polymeric coating does not
substantially swell upon hydration.
In yet another embodiment, the invention is a drug-eluting device for oral
delivery of a drug, which includes a reservoir having a drug-containing core
contained
therein, one or more orifices or exit ports through which drug from the core
can elute
from the device, and a bioadhesive polyineric coating, applied to at least one
surface
of the device. The coating provides the device with a fracture strength of at
least 100
N/m2 as measured on rat intestine, and the device has a gastrointestinal
retention time
of at least 4 hours in a fed beagle dog model during which the drug is
released from
the device. In a preferred embodiment, the bioadhesive polymeric coating does
not
substantially swell upon hydration.
The present invention provides methods for improving the bioadhesive
properties of drug delivery systems such as tablets, capsules and drug-eluting
devices.
The invention also provides methods for improving the adhesion of drug
delivery
systems to mucosal membranes including membranes of the gastrointestinal
tract. The
polymeric drug delivery systenis of the invention have an improved ability to
bind to
mucosal meinbranes, and thus can be used to deliver a wide range of drugs or
diagnostic agents in a wide variety of therapeutic applications, and/or
improve uptake
of the active agent across the intestinal mucosa. In certain embodiments, the
drug
delivery system comprises particles ranging in size from 0.1-10 m.
Bioadhesive dosage forms of the invention generally have the advantages,
inter alia, of allowing for decreasing dosage levels and/or dosing frequencies
of
drugs. Typically, the bioadhesive and/or mucoadhesive systems for the local
and
4
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
sustained delivery of therapeutic agents allow for more efficient targeting of
drugs to
the required sites on the luminal surface of the gastrointestinal tract. With
the
reduction of dosage level and/or dosing frequencies, several potential
problems
relating to antirnicrobial agents may be reduced or avoided altogether, such
as
gastrointestinal irritation in some patients. Reduction of dosage level and/or
dosing
frequencies can also reduce or avoid disturbances of the normal enteric flora,
which
are caused by certain drugs, that may lead to drug-resistant bacterial
enteritis or
bacterial super-infection. The potential reduction in side effects and the
overall ease
of administration should greatly increase patient compliance, is expected to
further
improve the therapeutic outcome
In another embodiment, the present invention is an orally administrable, multi-
layer, pharmaceutical tablet having an inner and one or more outer layers,
each
comprising a drug (e.g., a drug including a valproic moiety such as sodium
valproate,
divalproex sodium, valproic acid, etc.) admixed with one or more excipients.
At least
one of the excipients is hydrophobic, although such excipient is not required
in each
layer. Additional outer layers (i.e., layers other than the inner and outer
layers
specified above) are optionally free of the drug.
BRIEF DESCRIPTION OF THE DRAWINGS
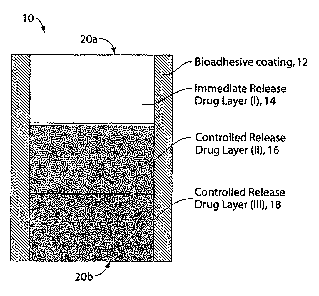
FIG. 1 is an illustration of a trilayer tablet with a bioadhesive coating.
FIGS. 2A-2D show that the trilayer tablets described in Example 1 were
retained in the stomach of beagle dogs at A) 2.5 hours (fasted animal and
tablet with
SpheromerTM III, a poly(butadiene-co-maleic acid) functionalized with DOPA,
outer
layers), B) 3.5 hours (fasted animal and tablet with SpheromerTM III outer
layers), C)
5.25 hours (fed animal and tablet with SpheromerTM I, poly(fumaric-co-sebacic
anhydride 20:80), outer layers) and D) 6 hours (fed animal and tablet with
SpheromerTM I outer layers).
FIG. 3 shows the pharmacokinetics of the 5-layer tablet in fed beagle dogs, as
described in Example 2.
FIGS. 4A and 4B show the release profile of sodium valproate from the tablets
prepared in Example 3.
5
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
FIG. 5 shows the weight gain experienced by sodiuni valproate trilayer tablet
formulations of Example 3(n=6) and a single-layer, matrix tablet formulation
(n=6)
consisting of only the core layer of the trilayer tablets when incubated at 45
C and
60% relative humidity for up to 56 hours.
FIG. 6 shows the release profile of levodopa from the tablet prepared in
Example 4.
FIG. 7 shows the effect of repeat dosing of the trilayer tablet of Example 5
(400 mg acyclovir, administered once per 12 hrs, 2 administrations) compared
to
Zovirax (200 mg, administered once per 6 hrs, 4 administrations) in 6 dogs.
FIG. 8 shows the release profile of itraconazole from the tablet prepared in
Example 6.
FIG. 9 shows the plasma levels of itraconazole following administration of
tablets of Example 6 and Sporanox to beagle dogs in the fed state, as
measured using
LC/MS/MS.
FIG. 10 is a bar graph showing the fracture strength of bonds (mN/cm2)
forlned with the bioadhesive materials, SpheromerTM II and SpheromerTM III, as
compared to Carbopo1934P and Gantrez AN polymers and control (uncoated
substrate).
FIG. 11 is a bar graph of the tensile work (nJ) required to rupture the bonds
formed with the bioadhesive materials, SpheromerTM II and SpheromerTM III, as
compared to Carbopol 934P and Gantrez AN polymers and control (uncoated
substrate).
DETAILED DESCRIPTION OF THE INVENTION
Bioadlzesive Pharnaaceutical Dosage Fonms
In one aspect, the present invention is directed to pharmaceutical dosage
forms
(e.g., tablets and drug-eluting devices) having increased gastrointestinal
retention
time. For purposes of this invention, gastrointestinal residence time is the
time
required for a pharmaceutical dosage form (e.g., tablet or drug-eluting
device) to
transit through the stomach to the pyloric sphincter. For example, a
pharmaceutical
dosage form (e.g., tablet or drug-eluting device) of the invention has a
gastrointestinal
6
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
residence time of at least 3 hours, at least 4 hours, at least 6 hours, at
least 8 hours, or
at least 12 hours. This time can be measured in eitlier a fed or a fasted
state, typically
a fed state. The pharmaceutical dosage forms (e.g, tablets and drug-eluting
devices) of
the invention may also have an increased retention time in the small and/or
large
intestine, or in the area of the gastrointestinal tract that absorbs the drug
contained in
the pharmaceutical dosage form (e.g., tablet or drug-eluting device). For
example,
pharmaceutical dosage forms (e.g., tablets or drug-eluting devices) of the
invention
can be retained in the small intestine (or one or two portions thereof,
selected from the
duodenum, the jejunum and the ileum) for at least 6 hours, at least 8 hours or
at least
12 hours, such as from 16 to 18 hours. For pharmaceutical dosage forms (e.g.,
tablets
and drug-eluting devices) having an enteric coating or an equivalent, the
increased
gastrointestinal residence time may not be applicable, as the bioadhesive may
not be
exposed until the dosage form enters the small intestine or lower. These
pharmaceutical dosage forms (e.g., tablets and drug-eluting devices), as a
whole,
include a bioadhesive polymeric coating that is applied to at least one
surface of the
dosage form.
"Bioadhesion" is defined as the ability of a material to adhere to a
biological
tissue for an extended period of time. Bioadhesion is one solution to the
problem of
inadequate residence time resulting from stomach emptying and intestinal
peristalsis,
and from displacement by ciliary movement. For sufficient bioadhesion to
occur, an
intimate contact must exist between the bioadhesive and the receptor tissue,
the
bioadliesive must penetrate into the crevice of the tissue surface and/or
mucus, and
mechanical, electrostatic, or chemical bonds must form. Bioadhesive properties
of
polymers are affected by both the nature of the polymer and by the nature of
the
surrounding media.
One example of a bioadhesive delivery system of the invention is for the local
delivery of antimicrobial and acid lowering agents to eradicate
Helicobacterpylori.
Eradication of H. pylori not only cures both the gastric and duodenal ulcers
but also
has the potential to prevent a substantial proportion of gastric
adenocarcinoma and
lymphomas. In one embodiment, one or more therapeutic agents including acid
suppressants (ranitidine bismuth citrate, lansoprazole), mucosal defense
enhancing
agent (bismuth salts) and/or mucolytic agents (megaldrate) are incorporated in
the
bioadhesive delivery system and then administered to patients with or at risk
of H.
7
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
pylori infection or ulcers. Preferably, the bioadhesive formulation includes a
multi-
layer core enveloped by a bioadhesive coating.
Polymers
Suitable bioadhesive polymeric coatings are disclosed in U.S. Patent
Nos.6,197,346, 6,217,908 and 6,365,187, the contents of which are incorporated
herein by reference, and include soluble and insoluble, biodegradable and
nonbiodegradable polymers. These can be hydrogels or thermoplastics,
homopolymers, copolymers or blends, and/or natural or synthetic polymers. The
preferred polymers are synthetic polymers, with controlled synthesis and
degradation
characteristics. Particularly preferred polymers are anhydride copolymers of
fumaric
acid and sebacic acid (P(FA:SA)), which have exceptionally good bioadhesive
properties when administered to the gastrointestinal tract. Examples of
P(FA:SA)
copolymers include those having a 1:99 to 99:1 ratio of fumaric acid to
sebacic acid,
such as 5:95 to 75:25, for example, 10:90 to 60:40 or at least 15:85 to 25:75.
Specific
examples of such copolymers have a 20:80 or a 50:50 ratio of fumaric acid to
sebacic
acid.
Polymers used in bioadhesive pharmaceutical dosage forms (e.g., tablets and
drug-eluting devices) of the invention produce a bioadhesive interaction
(fracture
strength) of at least 100 N/m2 (10 mN/cmZ) when applied to the mucosal surface
of rat
intestine. The fracture strength of the pharmaceutical dosage forms (e.g.,
tablets and
drug-eluting devices) is advantageously at least 250 N/m2, at least 500 N/m2
or at
least 1000 N/m2. For example, the fracture strength of a polymer-containing
phannaceutical dosage form (e.g., tablet or drug-eluting device) can be from
100 to
500 N/m2. The forces described herein refer to measurements made upon rat
intestinal
mucosa, unless otherwise stated. The same adhesive measurements made on other
species of animal may differ from those obtained using rats. This difference
is
attributed to both compositional and geometrical variations in the mucous
layers of
different animal species as well as cellular variations in the mucosal
epithelium.
However, the data shows that the same general trends prevail across animals
studied
(i.e., P(FA:SA) produces stronger adhesions than polylactic acid (PLA) in
rats, sheep,
pigs, etc.). For example, the fracture strength of pharmaceutical dosage forms
(e.g.,
tablets and drug-eluting devices) of the invention on rat intestine is
generally at least
8
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
125 N/m2, such as at least 150 N/m2, at least 250 N/m2, at least 500 N/m2 or
at least
1000 N/m2.
The fracture strength of a pharmaceutical dosage form (e.g., tablet or drug-
eluting device) can be measured according to the methods disclosed by Duchene
et al.
Briefly, the tablet is attached on one side to a tensile tester and is
contacted with a
testing surface (e.g., a mucosal membrane, such as rat or pig intestine) on
the opposite
surface. The tensile tester measures the force required to displace the
pharmaceutical
dosage form (e.g., tablet or drug-eluting device) from the testing surface.
Common
tensile testers include a Texture Analyzer and the Instron tensile tester.
In the preferred method for mucoadhesive testing, tablets are pressed using
flat-faced tooling, 0.3750" (9.525 mm) in diameter. Tablet weight will depend
on
composition; in most cases, the tablets have a final weight of 200 mg. These
tablets
are then glued to a plastic 10 mm diameter probe using a common, fast-drying
cyanoacrylate adhesive. Once the tablets are firmly adhered to the probe, the
probe is
attached to the Texture Analyzer. The Texture Analyzer is fitted with a 1 kg
load cell
for maximum sensitivity. The following settings are used:
Pre-Test Speed 0.4 mm / sec Stop Plot At Final Position
Test Speed 0.1 mm / sec Tare Mode Auto
Post-Test Speed 0.1 mm / sec Delay Acquisition Off
Applied Force 20.0 g Advanced Options On
Return Distance 0 inm Proportional Gain 0
Contact Time 420 s Integral Gain 0
Trigger Type Auto Differential Gain 0
Trigger Force 0.5 g Max. Tracking Speed 0 mm / see
The Test and Post-Test Speeds are advantageously as low as the instrument
permits, in order to allow capture of a maximum number of data points. The Pre-
Test
speed is used only until the probe encounters the Trigger Force; i.e., prior
to
contacting the tissue.
The Proportional, Integral, and Differential Gain are set to 0. These
settings,
when optimized, maintain the systeni at the Applied Force for the duration of
the
Contact Time. With soft tissue as a substrate, however, the probe and tablet
are
constantly driven into the deformable surface. This results in visible damage
to the
tissue. Thus, the probe and tablet are allowed to relax gradually from the
Applied
9
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
Force by setting these parameters to 0. The tracking speed, which is a measure
of how
rapidly the feedback is adjusted, is also set to 0.
The tissue on which the tablets are tested is secured in the Mucoadhesive Rig;
the rig is then completely immersed in a 600 mL Pyrex beaker containing 375 mL
of
PBS. The tissue is maintained at approximately 37 C for the duration of the
test; no
stirring is used as the machine can detect the oscillations from the stir bar.
Smart et al., J. Pharm. Pharmacol., 36:295-299 (1984), report another method
to test adhesion to mucosa using a polymer coated glass plate contacting a
dish of
mucosa. A variety of polymeric materials were tested, including sodium
alginate,
sodium carboxymethyl-cellulose, gelatin, pectin and polyvinylpyrrolidone.
Gurney et al., Biomaterials, 5:336-340 (1984) report that adhesion may be
affected by physical or mechanical bonds; secondary chemical bonds; and/or
primary,
ionic or covalent bonds. Park et al., "Alternative Approaches to Oral
Controlled Drug
Delivery: Bioadhesives and In-Situ Systems," in J. M. Anderson and S. W. Kim,
Eds.,
"Recent Advances in Drug Delivery," Plenum Press, New York, 1984, pp. 163-183,
report a study of the use of fluorescent probes in cells to determine
adhesiveness of
polymers to mucin/epithelial surface, which indicates that anionic polymers
with high
charge density appear to be preferred as adhesive polyniers. Mikos et al., in
J. Colloid
Interface Sci., 143:366-373 (1991) and Lehr et al., J. Controlled Rel. Soc.,
13:51-62
(1990) report a study of the bioadhesive properties of polyanhydrides and
polyacrylic
acid, respectively, in drug delivery.
In the past, two classes of polymers have shown useful bioadhesive properties,
hydrophilic polymers and hydrogels. In the large class of hydrophilic
polymers, those
containing carboxylic groups (e.g., poly[acrylic acid]) exhibit the best
bioadhesive
properties. It is thus expected that polymers with the highest concentrations
of
carboxylic groups are preferred materials for bioadhesion on soft tissues. In
other
studies, the most promising polymers were sodium alginate,
carboxyniethylcellulose,
hydroxymethylcellulose and methylcellulose. Some of these materials are water-
soluble, while others are hydrogels.
Rapidly bioerodible polymers such as poly[lactide-co-glycolide],
polyanhydrides, and polyorthoesters, whose carboxylic groups are exposed on
the
external surface as their smooth surface erodes, are particularly suitable for
bioadhesive drug delivery systems. In addition, polymers containing labile
bonds,
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
such as polyanhydrides and polyesters, are well known for their hydrolytic
reactivity.
Their hydrolytic degradation rates can generally be altered by simple changes
in the
polymer backbone.
Representative natural polymers suitable for the present invention include
proteins (e.g., hydrophilic proteins), such as zein, modified zein, casein,
gelatin,
gluten, serum albumin, or collagen, and polysaccharides such as cellulose,
dextrans,
polyhyaluronic acid, polymers of acrylic and methacrylic esters and alginic
acid.
These are generally less suitable for use in bioadhesive coatings due to
higher levels
of variability in the characteristics of the final products, as well as in
degradation
following administration. Syntlletically modified natural polymers include
alkyl
celluloses, hydroxyalkyl celluloses, cellulose ethers, cellulose esters, and
nitrocelluloses.
Representative synthetic polymers for use in bioadhesive coatings include
polyphosphazines, poly(vinyl alcohols), polyamides, polycarbonates,
polyalkylenes,
polyacrylamides, polyalkylene glycols, polyalkylene oxides, polyalkylene
terephthalates, polyvinyl ethers, polyvinyl esters, polyvinyl halides,
polyvinylpyrrolidone, polyglycolides, polysiloxanes, polyurethanes and
copolymers
thereof. Other polymers suitable for use in the invention include, but are not
limited
to, methyl cellulose, ethyl cellulose, hydroxypropyl cellulose, hydroxypropyl
methyl
cellulose, hydroxybutyl methyl cellulose, cellulose acetate, cellulose
propionate,
cellulose acetate butyrate, cellulose acetate phthalate, carboxymethyl
cellulose,
cellulose triacetate, cellulose sulfate sodium salt, poly(methyl
methacrylate),
poly(ethyl methacrylate), poly(butyl methacrylate), poly(isobutyl
methacrylate),
poly(hexyl metliacrylate), poly(isodecyl methacrylate), poly(lauryl
methacrylate),
poly(phenyl methacrylate), poly(methyl acrylate), poly(isopropyl acrylate),
poly(isobutyl acrylate), poly(octadecyl acrylate) polyethylene, polypropylene,
poly(ethylene glycol), poly(ethylene oxide), poly (ethylene terephthalate),
poly(vinyl
acetate), polyvinyl chloride, polystyrene, polyvinyl pyrrolidone, and
polyvinylphenol.
Representative bioerodible polymers for use in bioadhesive coatings include
polylactides, polyglycolides and copolymers thereof, poly(ethylene
terephthalate),
poly(butyric acid), poly(valeric acid), poly(lactide-co-caprolactone),
poly[lactide-co-
glycolide], polyanhydrides (e.g., poly(adipic anhydride)), polyorthoesters,
blends and
copolymers thereof.
11
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
Polyanhydrides are particularly suitable for use in bioadhesive delivery
systems because, as hydrolysis proceeds, causing surface erosion, more and
more
carboxylic groups are exposed to the external surface. However, polylactides
erode
more slowly by bulk erosion, which is advantageous in applications where it is
desirable to retain the bioadhesive coating for longer durations. In designing
bioadhesive polyineric systems based on polylactides, polymers that have high
concentrations of carboxylic acid are preferred. The high concentrations of
carboxylic
acids can be attained by using low molecular weight polymers (MW of 2000 or
less),
because low molecular weight polymers contain a high concentration of
carboxylic
acids at the end groups.
The polymers listed above can be obtained from sources such as Sigma
Chemical Co., St. Louis, Mo., Polysciences, Warrenton, Pa., Aldrich,
Milwaukee,
Wis., Fluka, Ronkonkoma, N.Y., and BioRad, Richmond, Calif., or can
alternatively
be synthesized from monomers obtained from these suppliers using standard
techniques.
When the bioadhesive polymeric coating is a synthetic polymer coating, the
synthetic polymer is typically selected from polyamides, polycarbonates,
polyalkylenes, polyalkylene glycols, polyalkylene oxides, polyalkylene
terephthalates,
polyvinyl alcohols, polyvinyl ethers, polyvinyl esters, polyvinyl halides,
polyvinylpyrrolidone, polyglycolides, polysiloxanes, polyurethanes,
polystyrene,
polymers of acrylic and methacrylic esters, polylactides, poly(butyric acid),
poly(valeric acid), poly(lactide-co-glycolide), polyanhydrides,
polyorthoesters,
poly(fumaric acid), poly(maleic acid), and blends and copolymers of thereof.
In an
exemplary embodiment, the synthetic polymer is poly(fumaric-co-sebacic)
anhydride.
Another group of polymers suitable for use as bioadhesive polymeric coatings
are polymers having a hydrophobic backbone with at least one hydrophobic group
pendant from the backbone. Suitable hydrophobic groups are groups that are
generally
non-polar. Examples of such hydrophobic groups include alkyl, alkenyl and
alkynyl
groups. Preferably, the hydrophobic groups are selected to not interfere and
instead to
enhance the bioadhesiveness of the polymers.
A further group of polymers suitable for use as bioadhesive polymeric
coatings are polymers having a hydrophobic backbone with at least one
hydrophilic
12
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
group pendant from the backbone. Suitable hydrophilic groups include groups
that are
capable of hydrogen bonding or electrostatically bonding to another functional
group.
Example of such hydrophilic groups include negatively charged groups such as
carboxylic acids, sulfonic acids and phosponic acids, positively charged
groups such
as (protonated) amines and neutral, polar groups such as amides and imines.
Preferably, the hydrophilic groups are selected to not interfere and instead
to enhance
the bioadl7esiveness of the polymers. The hydrophilic groups can be either
directly
attached to a hydrophobic polymer backbone or attached through a spacer group.
Typically, a spacer group is an alkylene group, particularly a C1-C8 alkyl
group such
as a C2-C6 alkyl group. Preferred compounds containing one or more hydrophilic
groups include amino acids (e.g., phenyalanine, tyrosine and derivatives
thereof) and
amine-containing carbohydrates (sugars) such as glucosamine.
Polymers can be modified by increasing the nuinber of carboxylic groups
accessible during biodegradation, or on the polymer surface. The polymers can
also
be modified by binding amino groups to the polymer. The polymers can be
modified
using any of a number of different coupling chemistries available in the art
to
covalently attach ligand molecules with bioadhesive properties to the surface-
exposed
molecules of the polymeric microspheres.
Lectins can be covalently attached to polymers to render them target specific
to the mucin and mucosal cell layer. Useful lectin ligands include lectins
isolated
from: Abrus precatroius, Agaricus bisporus, Anguilla anguilla, Arachis
hypogaea,
Pandeiraea sinaplicifolia, Bauhinia purpurea, Caragan arobrescens, Cicer
arietinum,
Codium fr=agile, Datura stramonium, Dolichos biflonus, Erythrina
corallodendron,
Enytlzrina cristagalli, Euonymus europaeus, Glycine max, Helix aspersa, Helix
pomatia, Lathyrus odoratus, Lens culinaris, Limuluspolyphetnus, Lysopersicon
esculentum, Maclura ponzifera, Momordica charantia, Mycoplasma gallisepticurn,
Naja inocambique, as well as the lectins Concanavalin A, Succinyl-Concanavalin
A,
Triticum vulgaris, Ulex europaeus I, II and III, Sambucus nigra, Maackia
amurensis,
Limaxfluvus, Homarus anaenicanus, Cancer antennarius, and Lotus
tetragonolobus.
The attachment of any positively charged ligand, such as polyethyleneimine or
polylysine, to a polymer may improve bioadhesion due to the electrostatic
attraction
of the cationic groups coating the beads to the net negative charge of the
mucus. The
mucopolysaccharides and mucoproteins of the mucin layer, especially the sialic
acid
13
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
residues, are responsible for the negative charge coating. Any ligand with a
high
binding affinity for mucin could also be covalently linked to most polymers
with the
appropriate chemistry, such as with carbodiimidazole (CDI), and be expected to
influence the binding to the gut. For example, polyclonal antibodies raised
against
components of mucin or else intact mucin, when covalently coupled to a
polymer,
would provide for increased bioadhesion. Similarly, antibodies directed
against
specific cell surface receptors exposed on the lumenal surface of the
intestinal tract
would increase the residence time when coupled to polymers using the
appropriate
chemistry. The ligand affinity need not be based only on electrostatic charge,
but
other useful physical parameters such as solubility in mucin or specific
affinity to
carbohydrate groups.
The covalent attachment of any of the natural coiuponents of mucin in either
pure or partially purified form to the polymers generally increases the
solubility of the
polymer in the mucin layer. The list of useful ligands include but are not
limited to the
following: sialic acid, neuraminic acid, n-acetyl-neuraminic acid, n-
glycolylneuraminic acid, 4-acetyl-n-acetylneuraminic acid, diacetyl-n-
acetylneuraminic acid, glucuronic acid, iduronic acid, galactose, glucose,
mannose,
fucose, any of the partially purified fractions prepared by chemical treatment
of
naturally occurring mucin, e.g., mucoproteins, mucopolysaccharides and
mucopolysaccharide-protein complexes, and antibodies immunoreactive against
proteins or sugar structure on the mucosal surface.
The attachment of polyamino acids containing extra pendant carboxylic acid
side groups, such as polyaspartic acid and polyglutamic acid, may also
increase
bioadhesiveness. The polyamino chains would increase bioadhesion by means of
chain entanglement in mucin strands as well as by increased carboxylic charge.
Polymer-Metal Complexes
As disclosed in U.S. Patent Nos. 5,985,312, 6,123,965 and 6,368,586, the
contents of which are incorporated herein by reference, polymers, such as
those
named above, having a metal compound incorporated therein have a further
improved
ability to adhere to tissue surfaces, such as mucosal membranes, and are
suitable for
use in the invention. The metal compound incorporated into the polymer can be,
for
14
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
example, a water-insoluble metal oxide. The incorporation of metal compounds
into a
wide range of different polymers, even those that are not normally
bioadhesive, often
improves their ability to adhere to tissue surfaces such as mucosal membranes.
Metal coinpounds that can be incorporated into polymers to improve their
bioadhesive properties preferably are water-insoluble metal compounds, such as
water-insoluble metal oxides and metal hydroxides, which are capable of
becoming
incorporated into and associated with a polymer to improve the bioadhesiveness
of the
polymer. As defined herein, a water-insoluble metal compound is defined as a
metal
compound with little or no solubility in water, for example, less than about
0.0 to 0.9
mg/ml.
The water-insoluble metal compounds can be derived from a wide variety of
metals, including, but not limited to, calcium, iron, copper, zinc, cadmiuin,
zirconiuin
and titanium. The water-insoluble metal compound preferably is a metal oxide
or
hydroxide. Water-insoluble metal compounds of multivalent metals are
preferred.
Representative metal oxides suitable for use in the compositions described
herein
include cobalt (II) oxide (CoO), cobalt (III) oxide (Co203), selenium oxide
(Se02),
chromium (IV) oxide (Cr02), manganese oxide (Mn02), titanium oxide (Ti02),
lanthanum oxide (La203), zirconium oxide (ZrO2), silicon oxide (Si02),
scandium
oxide (Sc203), beryllium oxide (BeO), tantalum oxide (Ta2O5), cerium oxide
(CeO2),
neodymium oxide (Nd203), vanadiuin oxide (V205), molybdenum oxide (Mo203),
tungsten oxide (WO), tungsten trioxide (W03), samarium oxide (Smz03), europium
oxide (Euz03), gadolinium oxide (Gd203), terbium oxide (Tb407), dysprosium
oxide
(Dya03), holmium oxide (Ho203), erbium oxide (Er203), thulium oxide (Tm203),
ytterbium oxide ('YbZ03), lutetiuin oxide (Lu2O3), aluminum oxide (A1203),
indium
oxide (InO3), germanium oxide (Ge02), antimony oxide (Sba03), tellurium oxide
(Te02), nickel oxide (NiO), and zinc oxide (ZnO). Other oxides include barium
oxide
(BaO), calcium oxide (CaO), nickel (III) oxide (Ni203), magnesium oxide (MgO),
iron (II) oxide (FeO), iron (III) oxide (Fe203), copper (II) oxide (CuO),
cadmium
oxide (CdO), and zirconium oxide (ZrOa).
Preferred properties defining the metal compound include: (a) substantial
insolubility in aqueous environments, such as acidic or basic aqueous
environments
(such as those present in the gastric lumen); and (b) ionizable surface charge
at the pH
of the aqueous environment.
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
The water-insoluble metal compounds can be incorporated into the polymer by
one of the following mechanisms: (a) physical mixtures which result in
entrapment of
the metal compound; (b) ionic interaction between metal compound and polymer;
(c)
surface modification of the polymers which would result in exposed metal
compound
on the surface; and (d) coating techniques such as fluidized bed, pan coating,
or any
similar methods known to those skilled in the art, which produce a metal
compound
enriched layer on the surface. In one embodiment, nanoparticles or
microparticles of
the water-insoluble metal compound are incorporated into the polymer.
In one embodiment, the metal conlpound is provided as a fine particulate
dispersion of a water-insoluble metal oxide e.g., incorporated throughout the
polymer
or disposed on the surface of the polymer which is to be adhered to a tissue
surface.
The metal compound also can be incorporated in an inner layer of the polymer
and
exposed only after degradation or else dissolution of a "protective" outer
layer. For
example, a tablet core containing a polymer and metal may be covered with an
enteric
coating designed to dissolve when exposed to gastric fluid. The metal compound-
enriched core then is exposed and becomes available for binding to GI mucosa.
Fine metal oxide particles can be produced for example by micronizing a
metal oxide by mortar and pestle treatment to produce particles ranging in
size, for
example, from 10.0 to 300 nm. The metal oxide particles can be incorporated
into the
polymer, for example, by dissolving or dispersing the particles into a
solution or
dispersion of the polymer.
Advantageously, metal compounds which are incorporated into polymers to
improve their bioadhesive properties can be metal compounds which are already
approved by the FDA as either food or pharmaceutical additives, such as zinc
oxide.
Suitable polymers that can be used and into which the metal compounds can
be incorporated include soluble and water-insoluble, and biodegradable and
nonbiodegradable polymers, including hydrogels, thermoplastics, and
homopolymers,
copolymers and blends of natural and synthetic polymers, provided that they
have the
requisite fracture strength when mixed with a metal compound. In additional to
those
listed above, representative polymers which can be used in conjunction with a
metal
compound include hydrophilic polymers, such as those containing carboxylic
groups,
including polyacrylic acid. Bioerodible polymers including polyanhydrides,
16
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
poly(hydroxy acids) and polyesters, as well as blends and copolymers thereof
also can
be used. Representative bioerodible poly(hydroxy acids) and copolymers thereof
which can be used include poly(lactic acid), poly(glycolic acid), poly(hydroxy-
butyric
acid), poly(hydroxyvaleric acid), poly(caprolactone), poly(lactide-co-
caprolactone),
and poly(lactide-co-glycolide). Polymers containing labile bonds, such as
polyanhydrides and polyorthoesters, can be used optionally in a modified form
with
reduced hydrolytic reactivity. Positively charged hydrogels, such as chitosan,
and
thermoplastic polymers, such as polystyreile also can be used.
Representative natural polymers which also can be used include proteins, such
as zein, modified zein, casein, gelatin, gluten, serum albumin, or collagen,
and
polysaccharides such as dextrans, polyhyaluronic acid and alginic acid.
Representative synthetic polymers include polyphosphazenes, polyamides,
polycarbonates, polyacrylamides, polysiloxanes, polyurethanes and copolymers
thereof. Celluloses also can be used. As defined herein the term "celluloses"
includes
naturally occurring and synthetic celluloses, such as alkyl celluloses,
cellulose ethers,
cellulose esters, hydroxyalkyl celluloses and nitrocelluloses. Exemplary
celluloses
include ethyl cellulose, methyl cellulose, carboxymethyl cellulose,
hydroxymethyl
cellulose, hydroxypropyl cellulose, hydroxypropyl methyl cellulose,
hydroxybutyl
methyl cellulose, cellulose acetate, cellulose propionate, cellulose acetate
butyrate,
cellulose acetate phthalate, cellulose triacetate and cellulose sulfate sodium
salt.
Polymers of acrylic and methacrylic acids or esters and copolymers thereof
can be used. Representative polymers which can be used include poly(methyl
methacrylate), poly(ethyl methacrylate), poly(butyl methacrylate),
poly(isobutyl
methacrylate), poly(hexyl methacrylate), poly(isodecyl methacrylate),
poly(lauryl
methacrylate), poly(phenyl methacrylate), poly(methyl acrylate),
poly(isopropyl
acrylate), poly(isobutyl acrylate), and poly(octadecyl acrylate).
Other polymers which can be used include polyalkylenes such as polyethylene
and polypropylene; polyarylalkylenes such as polystyrene; poly(alkylene
glycols),
such as poly(ethylene glycol); poly(alkylene oxides), such as poly(ethylene
oxide);
and poly(alkylene terephthalates), such as poly(ethylene terephthalate).
Additionally,
polyvinyl polymers can be used, which as defined herein includes polyvinyl
alcohols,
polyvinyl ethers, polyvinyl esters and polyvinyl halides. Exemplary polyvinyl
polymers include poly(vinyl acetate), polyvinyl phenol and
polyvinylpyrrolidone.
17
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
Water soluble polymers can also be used. Representative examples of suitable
water soluble polymers include polyvinyl alcohol, polyvinylpyrrolidone, methyl
cellulose, hydroxypropyl cellulose, hydroxypropylmethyl cellulose and
polyethylene
glycol, copolymers of acrylic and methacrylic acid esters, and mixtures
thereof. Water
insoluble polymers also can be used. Representative examples of suitable water
insoluble polyniers include ethylcellulose, cellulose acetate, cellulose
propionate
(lower, medium or higher molecular weight), cellulose acetate propionate,
cellulose
acetate butyrate, cellulose acetate phthalate, cellulose triacetate,
poly(methyl
metliacrylate), poly(ethyl methacrylate), poly(butyl methacrylate),
poly(isobutyl
methacrylate), poly(hexyl methacrylate), poly(isodecyl methacrylate),
poly(lauryl
methacrylate), poly(phenyl methacrylate), poly(methyl acrylate),
poly(isopropyl
acrylate), poly(isobutyl acrylate), poly(octadecyl acrylate), poly(ethylene),
poly(ethylene) low density, poly(ethylene) high density, poly(propylene),
poly(ethylene oxide), poly(ethylene terephthalate), poly(vinyl isobutyl
ether),
poly(vinyl acetate), poly(vinyl chloride), polyurethanes, and mixtures
thereof. In one
embodiment, a water insoluble polymer and a water soluble polymer are used
together, such as in a mixture. Such mixtures are useful in controlled drug
release
formulations, wherein the release rate can be controlled by varying the ratio
of water
soluble polymer to water insoluble polymer.
Polymers varying in viscosity as a function of temperature or shear or other
physical forces also may be used. Poly(oxyalkylene) polymers and copolymers
such
as poly(ethylene oxide)-poly(propylene oxide) (PEO-PPO) or poly(ethylene
oxide)-
poly(butylene oxide) (PEO-PBO) copolymers, and copolymers and blends of these
polymers with polymers such as poly(alpha-hydroxy acids), including but not
limited
to lactic, glycolic and hydroxybutyric acids, polycaprolactones, and
polyvalerolactones, can be syntliesized or coinmercially obtained. For
example,
polyoxyalkylene copolymers are described in U.S. Patent Nos. 3,829,506,
3,535,307,
3,036,118, 2,979,578, 2,677,700 and 2,675,619. Polyoxyalkylene copolymers are
sold, for example, by BASF under the tradename PLURONICSTM. These materials
are applied as viscous solutions at room temperature or lower which solidify
at the
higher body temperature. Other materials with this behavior are lcnown in the
art, and
can be utilized as described herein. These include KLUCELTM (hydroxypropyl
cellulose), and purified konjac glucomannan gum.
18
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
Other suitable polymers are polymeric lacquer substances based on acrylates
and/or methacrylates, commonly called EUDRAGITTm polymers (sold by Rohm
America, Inc.). Specific EUDRAGITTM polymers can be selected having various
permeability and water solubility, which properties can be pH dependent or pH
independent. For example, EUDR.AGITTM RL and EUDRAGITTM RS are acrylic
resins comprising copolymers of acrylic and methacrylic acid esters with a low
content of quaternary ammonium groups, which are present as salts and give
rise to
the permeability of the lacquer films, whereas EUDRAGITTM RL is freely
permeable
and EUDRAGITTM RS is slightly permeable, independent of pH. In contrast, the
permeability of EUDRAGITTM L is pH-dependent. EUDRAGITTM L is an anionic
polymer synthesized from methacrylic acid and methacrylic acid methyl ester.
It is
insoluble in acids and pure water, but becomes increasingly soluble in a
neutral to
weakly alkaline solution by forming salts with alkalis. Above pH 5.0, the
polymer
becomes increasingly permeable.
Polymer solutions that are liquid at an elevated teinperature but solid or
gelled
at body temperature can also be utilized. A variety of thermoreversible
polymers are
known, including natural gel-forming materials such as agarose, agar,
furcellaran,
beta-carrageenan, beta- 1,3 -glucans such as curdlan, gelatin, or
polyoxyalkylene-
containing compounds, as described above. Specific examples include
thermosetting
biodegradable polymers for in vivo use described in U.S. Patent No. 4,938,763,
the
contents of which are incorporated herein by reference.
Polymer Blends with Monomers and/or Oligomers
Polymers with enhanced bioadhesive properties are provided by incorporating
anhydride monomers or oligomers into one of the polymers listed above by
dissolving, dispersing, or blending, as taught by U.S. Patent Nos. 5,955,096
and
6,156,348, the contents of which are incorporated herein by reference. The
polymers
may be used to form drug delivery systems which have improved ability to
adhere to
tissue surfaces, such as mucosal membranes. The anhydride oligomers are
generally
formed from organic diacid monomers, preferably the diacids normally found in
the
Krebs glycolysis cycle. Anhydride oligomers that enhance the bioadhesive
properties
of a polymer have a molecular weight of about 5000 or less, typically between
about
19
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
100 and 5000 daltons, or include 20 or fewer diacid units linked by anhydride
linkages and terminating in an anhydride linkage with a carboxylic acid
monomer.
The oligomers can be blended or incorporated into a wide range of hydrophilic
and hydrophobic polymers including proteins, polysaccharides and synthetic
biocompatible polymers, including those described above. In one embodiment,
anhydride oligomers may be combined with metal oxide particles, such as those
described above, to improve bioadhesion even more than with the organic
additives
alone. Organic dyes, because of their electronic charge and hydrophobicity or
hydrophilicity, can either increase or decrease the bioadhesive properties of
polymers
when incorporated into the polymers.
As used herein, the term "anhydride oligomer" refers to a diacid or polydiacid
linked by anhydride bonds, and having carboxy end groups linked to a monoacid
such
as acetic acid by anhydride bonds. The anhydride oligomers have a molecular
weight
less than about 5000, typically between about 100 and 5000 daltons, or are
defined as
including between one to about 20 diacid units linked by anhydride bonds. In
one
embodiment, the diacids are those normally found in the Krebs glycolysis
cycle.
The oligomers can, for example, be formed in a reflux reaction of the diacid
with excess acetic anhydride. The excess acetic anhydride is evaporated under
vacuum, and the resulting oligomer, which is a mixture of species which
include from
about one to twenty diacid units linked by anhydride bonds, is purified by
recrystallizing, for example, from toluene or other organic solvents. The
oligomer is
collected by filtration, and washed, for example, in ethers. The reaction
produces
anhydride oligomers of mono and poly acids with terminal carboxylic acid
groups
linked to each other by anhydride linkages.
An anhydride oligomer is hydrolytically labile. As analyzed by gel permeation
chromatography, the molecular weight may be, for example, on the order of 200-
400
for fumaric acid oligomer (FAPP) and 2000-4000 for sebacic acid oligomer
(SAPP).
The anhydride bonds can be detected by Fourier transform infrared spectroscopy
by
the characteristic double peak at 1750 cm 1 and 1820 cm 1, with a
corresponding
disappearance of the carboxylic acid pealc normally at 1700 crn 1.
In one embodiment, the oligomers can be made from diacids described, for
example, in U.S. Patent Nos. 4,757,128, 4,997,904 and 5,175,235, the
disclosures of
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
which are incorporated herein by reference. For example, monomers such as
sebacic
acid, bis(p-carboxy-phenoxy)propane, isophthalic acid, fumaric acid, maleic
acid,
adipic acid or dodecanedioic acid can be used.
Organic dyes, because of their electronic charge and hydrophilicity or
hydrophobicity, can alter the bioadhesive properties of a variety of polymers
when
incorporated into the polymer matrix or bound to the surface of the polymer. A
partial
listing of dyes that affect bioadhesive properties include, but are not
limited to: acid
fuchsin, alcian blue, alizarin red s, auramine o, azure a and b, Bismarck
brown y,
brilliant cresyl blue ald, brilliant green, carmine, cibacron blue 3GA, congo
red, cresyl
violet acetate, crystal violet, eosin b, eosin y, erythrosin b, fast green
fcf, giemsa,
hematoylin, indigo cannine, Janus green b, Jenner's stain, malachite green
oxalate,
methyl blue, methylene blue, methyl green, methyl violet 2b, neutral red, Nile
blue a,
orange II, orange G, orcein, paraosaniline chloride, phloxine b, pyronin b and
y,
reactive blue 4 and 72, reactive brown 10, reactive green 5 and 19, reactive
red 120,
reactive yellow 2,3, 13 and 86, rose bengal, safranin o, Sudan III and IV,
Sudan black
B and toluidine blue.
Polymers Functionalized with Hydroxy-Substituted Aromatic Groups
Polymers having an aromatic group which contains one or more hydroxyl
groups grafted onto them or coupled to individual monomers are also suitable
for use
in the bioadhesive coatings of the invention, as described in U.S. Provisional
Application No. 60/528,042, filed December 9, 2003, U.S. Application No.
11/009,327, filed December 9, 2004, and WO 2005/056708, the contents of which
are
incorporated herein by reference. Such polymers can be biodegradable or non-
biodegradable polymers. The polymer can be hydrophobic. Preferably, the
aromatic
group is catechol or a derivative thereof and the polymer contains reactive
functional
groups, so that a hydroxyl-substituted aromatic group can be readily attached.
Typically, the polymer is a polyanhydride and the aromatic compound is the
catechol
derivative DOPA. These materials display bioadhesive properties superior to
conventional bioadhesives used in therapeutic and diagnostic applications.
The molecular weight of the suitable polymers and percent substitution of the
polymer with the aromatic group may vary greatly. The degree of substitution
varies
based on the desired adhesive strength, it may be as low as 10%, 25% or 50%,
or up
21
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
to 100% substitution. Generally, about 10 to about 40%, such as about 20% to
about
30% of the monomers in the polymeric backbone are substituted with at least
one
aromatic group. The resulting polymer typically has a molecular weight ranging
from
about 1 to 2,000 kDa.
The polymer that forms that backbone of the bioadhesive material can be a
biodegradable polymer. Examples of preferred biodegradable polymers include
synthetic polyiners such as poly hydroxy acids, such as polymers of lactic
acid and
glycolic acid, polyanhydrides, poly(ortho)esters, polyesters, polyurethanes,
poly(butyric acid), poly(valeric acid), poly(caprolactone),
poly(hydroxybutyrate),
poly(lactide-co-glycolide) and poly(lactide-cocaprolactone), and natural
polymers
such as alginate and other polysaccharides, collagen and chemical derivatives
thereof
(substitutions, additions of chemical groups, for example, alkyl, alkylene,
hydroxylations, oxidations, and other modifications routinely made by those
skilled in
the art), albumin and other hydrophilic proteins, zein and other prolamines
and
hydrophobic proteins, copolymers and mixtures thereof. In general, these
materials
degrade either by enzymatic hydrolysis or exposure to water in vivo and by
surface or
bulk erosion. The foregoing materials may be used alone, as physical mixtures
(blends), or as co-polymers.
Suitable polymers can formed by first coupling the aromatic compound to the
monomer and then polymerizing. In this example, the monomers may be
polymerized
to form a polymer backbone, including biodegradable and non-biodegradable
polymers. Suitable polymer backbones include, but are not limited to,
polyanhydrides,
polyainides, polycarbonates, polyallcylenes, polyalkylene oxides such as
polyethylene
glycol, polyalkylene terephthalates such as poly(ethylene terephthalate),
polyvinyl
alcohols, polyvinyl ethers, polyvinyl esters, polyethylene, polypropylene,
poly(vinyl
acetate), poly(vinyl chloride), polystyrene, polyvinyl halides,
polyvinylpyrrolidone,
polyhydroxy acids, polysiloxanes, polyurethanes and copolymers thereof, alkyl
cellulose, hydroxyalkyl celluloses, cellulose ethers, cellulose esters,
nitrocellulloses,
polymers of acrylic and methacrylic esters, methyl cellulose, ethyl cellulose,
hydroxypropyl cellulose, hydroxy-propyl methyl cellulose, hydroxybutyl methyl
cellulose, cellulose acetate, cellulose propionate, cellulose acetate
butyrate, cellulose
acetate phthalate, carboxyethyl cellulose, cellulose triacetate, cellulose
sulfate sodium
salt, and polyacrylates such as poly(methyl methacrylate),
poly(ethylmethacrylate),
poly(butylmethacrylate), poly(isobutylmethacrylate), poly(hexy.lmethacrylate),
22
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
poly(isodecylmethacrylate), poly(lauryl methacrylate), poly(phenyl
methacrylate),
poly(inethyl acrylate), poly(isopropyl acrylate), poly(isobutyl acrylate),
poly(octadecyl acrylate).
A suitable polymer backbone can be a known bioadhesive polymer that is
hydrophilic or hydrophobic. Hydrophilic polymers include CARBOPOLTM,
polycarbophil, cellulose esters, and dextran.
Non-biodegradable polymers, especially hydrophobic polymers, are also
suitable as polymer backbones. Examples of preferred non-biodegradable
polymers
include ethylene vinyl acetate, poly(methacrylic acid), copolymers of maleic
anhydride with other unsaturated polymerizable monomers, e.g., poly(butadiene
maleic anhydride), polyamides, copolymers and mixtures thereof and dextran,
cellulose and derivatives thereo~
Hydrophobic polymer backbones include polyanhydrides, poly(ortho)esters,
and polyesters such as polycaprolactone. Preferably, the polymer is
sufficiently
hydrophobic that it is not readily water soluble. For example, the polymer may
be
soluble up to less than about 1% w/w in water, preferably about 0.1% w/w in
water at
room temperature or body temperature. In the most preferred embodiment, the
polymer is a polyanhydride, such as a poly(butadiene maleic anhydride) or
another
copolymer of maleic anhydride. Polyanhydrides may be formed from dicarboxylic
acids, as described in U.S. Patent No. 4,757,128 to Domb et al., incorporated
herein
by reference. Suitable diacids include aliphatic dicarboxylic acids, aromatic
dicarboxylic acids, aromatic-aliphatic dicarboxylic acid, combinations of
aromatic,
aliphatic and aromatic-aliphatic dicarboxylic acids, aromatic and aliphatic
heterocyclic dicarboxylic acids, and aromatic and aliphatic heterocyclic
dicarboxylic
acids in combination with aliphatic dicarboxylic acids, aromatic-aliphatic
dicarboxylic acids, and aromatic dicarboxylic acids of more than one phenyl
group.
Suitable monomers include sebacic acid (SA), fumaric acid (FA), bis(p-
carboxyphenoxy)propane (CPP), isophthalic acid (IPh), and dodecanedioic acid
(DD).
A wide range of molecular weights are suitable for the polymer that forms the
baclcbone of the bioadhesive material. The molecular weight may be as low as
about
200 Da (for ojigomers) up to about 2,000 kDa. Preferably the polymer has a
molecular weight of at least 1,000 Da, more preferably at least 2,000 Da, most
preferably the polymer has a moecular weight of up to 20 kDa or up to 200 kDa.
The
23
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
molecular weight of the polymer may be up to 2,000 kDa (e.g., 20 kDa to 1,000
kDa
or 2,000 kDa).
The range of substitution on the polymer can vary greatly and depends on the
polymer used and the desired bioadhesive strength. For example, a butadiene
maleic
anhydride copolymer that is 100% substituted with DOPA will have the same
number
of DOPA molecules per chain length as a 67% substituted ethylene maleic
anhydride
copolymer. Typically, the polymer has a percentage substitution ranging from
10% to
100%, more typically ranging from 20% to 30%.
The polymers and copolymers that form the backbone of the bioadhesive
material typically include reactive functional groups that interact with the
functional
groups on the aromatic compound.
It is important that the polymer or monomer that forms the polymeric
backbone contains accessible functional groups that easily react or interact
with
molecules contained in the aromatic compounds, such as aniines and thiols. In
a
preferred embodiment, the polymer contains amino reactive moieties, such as
aldehydes, ketones, carboxylic acid derivatives, cyclic anhydrides, alkyl
halides, aryl
azides, isocyanates, isothiocyanates, succinimidyl esters or a combination
thereof.
Preferably, the aromatic coinpound containing one or more hydroxyl groups is
catechol or a derivative thereof. Optionally the aromatic compound is a
polyhydroxy
aromatic compound, such as a trihydroxy aromatic compound (e.g.,
phloroglucinol) or
a multihydroxy aromatic compound (e.g., tannin). The catechol derivative may
contain a reactive group, such as an amino, thiol, or halide group. A
preferred
catechol derivative is 3,4-dihydroxyphenylalanine (DOPA), which contains a
primary
amine. Tyrosine, the immediate precursor of DOPA, which differs only by the
absence of one hydroxyl group in the aromatic ring, can also be used. Tyrosine
is
capable of conversion (e.g., by hydroxylation) to the DOPA form. A
particularly
preferred aromatic compound is an amine-containing aromatic compound, such as
an
amine-containing catechol derivative (e.g., dopamine).
Two general methods are used to form the polymer product. In one example, a
compound containing an aromatic group which contains one or more hydroxyl
groups
is grafted onto a polymer. In this example, the polymeric backbone is a
biodegradable
polymer. In a second example, the aromatic compound is coupled to individual
monomers and then polymerized.
24
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
Any chemistry which allows for the conjugation of a polymer or monomer to
an aromatic compound containing one or more hydroxyl groups can be used, for
example, if the aromatic compound contains an amino group and the monomer or
polymer contains an amino reactive group, this modification to the polymer or
monomer is performed through a nucleophilic addition or a nucleophilic
substitution
reaction, such as a Michael-type addition reaction, between the amino group in
the
aromatic compound and the polymer or monomer. Additionally, other procedures
can
be used in the coupling reaction. For example, carbodiimide and mixed
anhydride
based procedures form stable amide bonds between carboxylic acids or
phosphates
and amino groups, bifunctional aldehydes react with primary amino groups,
bifunctional active esters react with primary amino groups, and divinylsulfone
facilitates reactions with amino, thiol, or hydroxy groups.
The aromatic compounds are grafted onto the polymer using standard
techniques to form the bioadhesive material. In one example, L-DOPA is grafted
to
maleic anhydride copolymers by reacting the free amine in L-DOPA with the
maleic
anhydride bond in the copolymer.
A variety of different polymers can be used as the backbone of the
bioadhesive material, as described above. Additional representative polymers
include
1:1 random copolymers of maleic anhydride with ethylene, vinyl acetate,
styrene, or
butadiene. In addition, a number of other compounds containing aromatic rings
with
hydroxy suhstituents, such as tyrosine or derivatives of catechol, can be used
in this
reaction.
In another embodiment, the polymers are prepared by conjugate addition of a
compound containing an aromatic group that is attached to an amine to one or
more
monomers containing an amino reactive group. In a preferred method, the
monomer is
an acrylate or the polymer is acrylate. For example, the monomer can be a
diacrylate
such as 1,4-butanediol diacrylate, 1,3-propanediol diacrylate, 1,2-ethanediol
diacrylate, 1,6-hexanediol diacrylate, 2,5-hexanediol diacrylate or 1,3-
propanediol
diacrylate. In an example of the coupling reaction, the monomer and the
compound
containing an aromatic group are each dissolved in an organic solvent (e.g.,
THF,
CHZC12, methanol, ethanol, CHC13, hexanes, toluene, benzene, CC14, glyme,
diethyl
ether, etc.) to form two solutions. The resulting solutions are combined, and
the
reaction mixture is heated to yield the desired polymer. The molecular weight
of the
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
synthesized polymer can be controlled by the reaction conditions (e.g.,
temperature,
starting materials, concentration, solvent, etc) used in the synthesis.
For example, a monomer, such as 1,4-phenylene diacrylate or 1,4-butanediol
diacrylate having a concentration of 1.6 M, and DOPA or another primary amine
containing aromatic molecule are each dissolved in an aprotic solvent such as
DMF or
DMSO to form two solutions. The solutions are mixed to obtain a 1:1 molar
ratio
between the diacrylate and the amine group and heated to 56 C to form a
bioadhesive
material.
Coatings
Preferred bioadhesive coatings do not appreciably swell upon hydration, such
that they do not substantially inhibit or block movement (e.g., of ingested
food)
through the gastrointestinal tract, as compared to the polymers disclosed by
Duchene
et al. Generally, polyiners that do not appreciably swell upon hydration
include one or
more hydrophobic regions, such as a polymethylene region (e.g., (CH2)n, where
n is 4
or greater). The swelling of a polymer can be assessed by measuring the change
in
volume when the polymer is exposed to an aqueous solution. Polyiners that do
not
appreciably swell upon hydration expand in volume by 50% or less when fully
hydrated. Preferably, such polymers expand in volume by less than 25%, less
than
20%, less than 15%, less than 10% or less than 5%. Even more preferably, the
bioadhesive coatings are mucophilic.
In one embodiment, a polymer that does not appreciably swell upon hydration
(e.g., a hydrophobic polymer) is mixed or blended with a polymer that does
swell or a
hydrophilic substance (e.g., CarbopolTM, poly(acrylic acid), small organic
acids such
as citric acid, maleic acid, fumaric acid, hydrophilic drugs, ionic and non-
ionic
detergents, sugars, salts such as NaCl, disintegrants), provided that the
amount of
swelling or hydration in the polymer does not substantially interfere with
bioadhesiveness. Generally, the amount of swellable polymer or hydrophilic
substance is selected to sufficiently hydrate the non-swellable polymer to
enhance its
bioadhesiveness. The weight ratio of swellable to non-swellable polymer or
hydrophilic substance to non-swellable polymer can be varied in order to
obtain a
coating that combines a desired amount of swelling (e.g., for faster adhesion)
with
26
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
longer-lasting adhesion, such as from 5:1 to 1:5 or 2:1 to 1:2. For example,
the
swellable polymer and/or hydrophilic substance can comprise about 1% to about
30%
by weight of a bioadhesive coating.
In one embodiment, the bioadhesive polymeric coating consists of two layers,
an inner bioadhesive layer that does not substantially swell upon hydration
and an
outer bioadhesive layer that is readily hydratable and optionally bioerodable,
such as
one comprised of CarbopolTM.
The bioadhesive polymers discussed above can be mixed with one or more
plasticizers or tl7ermoplastic polymers. Such agents typically increase the
strength
and/or reduce the brittleness of polymeric coatings. Examples of plasticizers
include
dibutyl sebacate, polyethylene glycol, triethyl citrate, dibutyl adipate,
dibutyl
fumarate, diethyl phthalate, ethylene oxide-propylene oxide block copolymers
such as
PluronicTM F68 and di(sec-butyl) fumarate. Example of thermoplastic polymers
include polyesters, poly(caprolactone), polylactide, poly(lactide-co-
glycolide), methyl
methacrylate (e.g., ELTDRAGITTM), cellulose and derivatives thereof such as
ethyl
cellulose, cellulose acetate and hydroxypropyl methyl cellulose (HPMC) and
large
molecular weight polyanhydrides. The plasticizers and/or thermoplastic
polymers are
mixed witli a bioadhesive polymer to achieve the desired properties.
Typically, the
proportion of plasticizers and thermoplastic polymers, when present, is from
0.5% to
40% by weight.
In one embodiment, the bioadhesive polymer coating, in a dry packaged form
of a tablet, is a hardened shell.
A pharmaceutical dosage form (e.g., tablet or a drug-eluting device) can have
one or more coatings in addition to the bioadhesive polymeric coating, e.g.,
covering
the surface of the bioadhesive coating. These coatings and their thickness
can, for
example, be used to control where in the gastrointestinal tract the
bioadhesive coating
becomes exposed. In one example, the additional coating prevents the
bioadhesive
coating from contacting the mouth or esophagus. In another example, the
additional
coating remains intact until reaching the small intestine (e.g., an enteric
coating).
Examples of coatings include methylmethacrylates, zein, cellulose acetate,
cellulose phthalate, HMPC, sugars, enteric polymers, gelatin and shellac.
Premature
27
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
exposure of a bioadhesive layer or dissolution of a tablet in the mouth can be
prevented with a layer or coating of hydrophilic polymers such as HPMC or
gelatin.
Coatings used in tablets of the invention typically include a pore former to
render the coating permeable to the drug.
Pharmaceutical dosage forms (e.g., tablets and drug-eluting devices) of the
invention can be coated by a wide variety of methods. Suitable methods include
compression coating, coating in a fluidized bed or a pan and hot melt
(extrusion)
coating. Such methods are well known to those skilled in the art.
Mislti-LayeY Tablets
The invention also includes multi-layer tablets comprising a first, a second
and
a third layer, where each layer includes one or more drugs and one or more
excipients,
where the first layer forms the core of the table, the second layer is
adjacent to one
side of the first layer and the third layer is adjacent to the opposite side
of the first
layer. At least one layer of the tablet includes a hydrophobic excipient and
at least one
drug in the tablet is hygroscopic, deliquescent or botli. Preferably, at least
one
hygroscopic and/or deliquescent drug and at least one hydrophobic excipient
are
present (e.g., blended together) in at least one layer of a tablet.
Exemplary hydrophobic excipients include celluloses, particularly cellulose
acetate and ethyl cellulose, stearic acid, magnesiuin stearate, glycerol
monostearate,
fatty acids and salts thereof, monoglycerides, diglycerides, triglycerides,
oil, colloidal
silicon dioxide and talc.
Such tablets optionally include one excipient present in an amount sufficient
to be at least partially rate-controlling with respect to release of the drug
from the
tablet. Typically, tablets that include a rate-controlling excipient (e.g., a
rate-
controlling polymer) contain about 30% to about 60% by weight of the rate-
controlling excipient. Alternatively, the amount of rate-controlling excipient
is
selected relative to the amount of drug in the tablet. In such cases, the
weight of the
rate-controlling excipient is about two times to about five times, such as
about two
times to about three times greater than the weight of the drug.
Typically, the inner and outer layers contain different proportions of each
component (including the drug(s)), thereby establishing a gradient-type
composition.
28
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
In an exemplary embodiment, the first (inner) layer contains the greatest
weight
percentage of the drug(s). Accordingly, the second and third layers and any
additional
layers present contain lesser amounts of drug. In multi-layer tablets having
more than
three layers (e.g., those having a fourth and optionally a fifth layer), the
additional
layers can, for example, contain no drug or contain successively lesser
amounts of
drug. In general, layers the same distance away from the first or inner layer
will
contain approximately equal amount of drug, such that the tablet is
essentially
symmetrical about the inner layer. For tablets containing two or more drugs,
the drugs
can both be present in one or more layers or the different drugs are present
in separate
layers (i.e., the drugs are not mixed together in one layer).
For drugs requiring absorption in buccal and sublingual regions of the GIT,
bioadhesive tablets and particularly bioadhesive multiparticulates and
nanoparticles
are desirable. Drugs absorbed in these sites avoid first-pass metabolism by
liver and
degradation by GIT enzymes and harsh pH conditions typically present in the
stomach
and small intestine. Drugs absorbed in the buccal and sublingual compartments
benefit from rapid onset of absorption, typically within minutes of dosing.
Particularly
suitable are bioadhesive particulates in fast-dissolving dosage forms, e.g.,
OraSolv
(Cima Labs) that disintegrate within 30 sec after dosing and release the
bioadhesive
particules. Target release profiles include immediate release (IR) and
combinations of
zero-order controlled release (CR) kinetics and first-order CR kinetics.
Preferably,
pharmaceutical formulations targeting the buccal and sublingual regions are
constructed such that the formulation disintegrates before passing into the
esophagus.
For drugs requiring absorption in the stomach and upper small intestine and/or
topical delivery to these sites, particularly drugs with narrow absorption
windows,
bioadhesive, gastroretentive drug delivery systems are the option of choice.
Bioadhesive tablets and multiparticulates are formulated to reside for
durations
greater than 3 hrs and optimally greater than 4, 5 or even 6 hrs in the fed
state. Drug
release profiles from these systems are tailored to match the gastric
residence times,
so that greater than 85% of the encapsulated drug is released during the
gastric
residence time. Target release profiles include zero-order CR kinetics, first-
order CR
kinetics and combinations of IR and CR kinetics.
For drugs requiring absorption or topical delivery only in the small
intestine,
enteric-coated, bioadhesive drug delivery systems are preferred. Such systems
are
particularly well suited for topical delivery of therapeutics to Crohn's
disease patients.
29
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
Enteric-coated, bioadhesive tablets and multiparticulates are formulated to
reside in
the stomach for durations less than 3 hrs in the fed state and less than 1 hr
in the
fasted state, during which time less than 10% of the encapsulated drug is
released, due
to the enteric coating. Following gastric emptying, the enteric coating
dissipates,
revealing the underlying bioadhesive coating. Dissipation of the enteric
coating is
typically controlled by pH and/or time duration. Typical enteric polymers
utilizing pH
as a control are Eudragit polymers manufactured by Rohm America: Eudragit L100-
55 dissolves at pH values greater than 5.5, typically found in duodenum;
Eudragit
L100 dissolves at pH values exceeding 6.0, typically found in j ejunuin;
Eudragit S 100
dissolves at pH values exceeding 7.0, typically found in ileum and the
ileocecal
junction.
Time may be used to control unmasking of the bioadhesive coating. Coatings
that dissolve after 3 hrs when the dosage form is administered in the fed
state and
after 1-2 hrs when the dosage form is administered in the fasted state are
suitable for
bioadhesive delivery systems to small intestine. Erosion of soluble polymer
layers is
one means to achieve a time-triggered, enteric dissolution. Polymers such as
HPMC,
HPC, PVP, PVA or combinations of the above may be used as time-delayed,
enteric
coatings and timing of the dissolution of the coating can be increased by
applying
thicker coating weights.
Alternately, non-permeable coatings of insoluble polymers, e.g., cellulose
acetate, ethylcellulose, can be used as enteric coatings for delayed/modified
release
(DR/MR) by inclusion of soluble pore formers in the coating, e.g., PEG, PVA,
sugars,
salts, detergents, triethyl citrate, triacetin, etc., at levels ranging from
0.5 to 50% w/w
of the coating and most preferably from 5 to 25% w/w of the coating.
Also suitable are rupturable coating systems, e.g., Pulsincap, that use
osmotic
forces of swelling from hydrophilic polymers to rupture enteric membranes to
reveal
underlying bioadh.esive coatings.
Target release profiles for the small intestine include: no more than 10% drug
release during the first 3 hrs post-dosing followed by either IR kinetics,
zero-order CR
kinetics, first-order CR kinetics and combinations of IR and CR kinetics.
For drugs requiring absorption or topical delivery only in the lower small
intestine and colon enteric-coated, bioadhesive drug delivery systems are
preferred.
Such systems are particularly well suited for topical delivery of therapeutics
to
patients with Inflammatory Bowel Disease (IBD) including Crohn's disease and
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
Ulcerative Colitis. Enteric-coated, bioadhesive tablets and inultiparticulates
are
formulated to reside in the stomach for durations less than 3 hrs in the fed
state and
less than 1 hr in the fasted state, during which time less than 10% of the
encapsulated
drug is released, due to the enteric coating. Following gastric emptying, the
enteric
coating dissipates, revealing the underlying bioadhesive coating. Suitable
means of
controlling dissipation include pH, time duration and enzymatic action of
colonic
bacteria. Typical of enteric polymers for delivery to the lower
gastrointestinal tract
utilizing pH as a control are Eudragit polymers manufactured by Rohm America:
Eudragit S 100 and FS dissolves at pH values exceeding 7.0, typically found in
ileum
and the ileocecal junction.
Time may be used to control unmasking the bioadhesive coating. Coatings that
dissolve after 4-5 hrs when the dosage form is administered in the fasted
state and
after 5-8 hrs when the dosage form is administered in the fed state are
suitable for
bioadhesive delivery systems to the lower small intestine and colon. Erosion
of
soluble polymer layers is one means to achieve a time-triggered, enteric
dissolution.
Polymers such as HPMC, HPC, PVP, PVA or combinations of the above may be used
as time-delayed, enteric coatings and timing of the dissolution of the coating
can be
increased by applying thicker coating weights.
Alternately, non-permeable coatings of insoluble polymers, e.g., cellulose
acetate, ethylcellulose, can be used as enteric coatings for delayed/modified
release
(DR/MR) by inclusion of soluble pore formers in the coating, e.g., PEG, PVA,
sugars,
salts, detergents, triethyl citrate, triacetin, etc., at levels ranging from
0.5 to 50% w/w
of the coating and most preferably from 5 to 25% w/w of the coating.
Also, coatings of polymers that are susceptible to enzyinatic cleavage by
colonic bacteria are another means of ensuring release to distal ileum and
ascending
colon. Materials such as calcium pectinate can be applied as coatings to
tablets and
multiparticulates and disintegrate in the lower gastrointestinal tract, due to
bacterial
action. Calcium pectinate capsules for encapsulation of bioadhesive
multiparticulates
are also available.
Target release profiles for the lower gastrointestinal tract include: no more
than 10% drug release during the first 4-5 hrs (fasted state) and 5-8 hrs (fed
state) hrs
post-dosing followed by either IR kinetics, zero-order CR kinetics, first-
order CR
kinetics and combinations of IR and CR kinetics.
31
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
In certain aspects, multi-layer tablets of the invention exhibit an
approximately
zero-order release of drug in in vitro testing and/or in vivo administration.
For
formulations where delivery to the stomach is desired, zero-order release
advantageously occurs over about 6-12 hours, particularly 8-10 hours. For
formulations where delivery to the stomach and small intestine are desired,
zero-order
release advantageously occurs over about 8-16 hours, particularly 10-14 hours.
For
formulations where delivery to the small intestine and colon are desired, zero-
order
release advantageously occurs over about 16-30 hours, particularly 22-26
hours.
Multi-layer or gradient tablets can be assembled in several different ways. In
one embodiment, the tablet comprises at least one solid inner layer and two
solid
outer layers, each comprising one or more drugs and one or more pharmaceutical
polymers and/or pharmaceutical excipients. In order to produce a gradient
effect, the
amount of drug and/or excipient differs among the inner and outer layers. For
example, the one or more inner layers can comprise at least 34% of the total
amount
of the drug in the tablet and one or more polymer(s) and/or excipients(s), and
each of
the two outer layers can comprise not more than 33% of the total amount of
drug in
the tablet and one or more polymer(s) and/or excipients(s). Such tablets can
also be
used to commence release of different drugs at different times, by inclusion
of
different drugs in separate layers.
In another embodiment, the multi-layer tablet consists of a solid inner layer
and two solid outer layers, each comprising a drug and one or more
pharmaceutical
polymers or pharmaceutical excipients, wherein at least one polymer or
excipient is
hydrophobic. Tablets of this embodiment preferably provide approximately zero-
order or linear release kinetics. In still another embodiment, the multi-layer
tablet is
enteric coated.
One or more layers of the tablet can contain permeation enhancers to provide
permeability enhancement of drugs through mucosal lining of the
gastrointestinal tract
(GIT). An absorption enhancer facilitates the uptake of a drug across the
gastrointestinal epithelium. Absorption enhancers include compounds that
improve
the ability of a drug to be solubilized in the aqueous environment in which it
is
originally released and/or in the lipophilic environment of the mucous layer
lining of
the intestinal walls. Absorption enhancers further include compounds that
increase
disorder of the hydrophobic region of the membrane exterior of intestinal
cells,
promote leaching of membrane proteins that results in increased transcellular
32
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
transport, or widen the pore radius between cells for increased paracellular
transport.
Examples of absorption enhancers include sodium caprate, ethylenediamine
tetra(acetic acid) (EDTA), citric acid, lauroylcarnitine, palmitoylcarnitine,
tartaric
acid and other agents known to increase GI permeability. Other suitable
absorption
enhancers include sodium salicylate, sodium 5-methoxysalicylate, indomethacin,
diclofenac, polyoxyethylene ethers, sodium laurylsulfate, quaternary ammonium
compounds, sodium deoxycholate, sodium cholate, octanoic acid, decanoic acid,
glyceryl-l-monooctanoate, glyceryl-l-monodecanoate, DL-phenylalanine
ethylacetoacetate enamine, chlorpromazine, D-myristoyl-L-propyl-L-prolyl-
glycinate,
concanavaline A, DL-a-glycerophosphate, and 3-amino-l-hydroxypropylidene-1,1-
diphosphanate.
Alternatively, or in addition, the tablet is coated to provide additional
control
over diffusion of the drug or exposure of the tablet to the gastrointestinal
tract (e.g.,
with an enteric coating). The diffusion-limiting coating can be a
pharmaceutically-
accepted polymeric coating material, such as methylmethacrylates (EudragitsTM,
Rolun and Hass; KollicoatTM, BASF), zein, cellulose acetate, cellulose
phthalate and
hydroxypropylmethylcellullose. The coatings can be applied using a variety of
techniques including fluidized-bed coating, pan-coating and dip-coating.
Multi-layer tablets of the invention can include a bioadhesive coating, as
described above.
Separately or in combination with the bioadhesive coating, a bioadhesive
(such as those described above) can be included in one or more layers of the
tablet.
Multi-layer tablets of the invention are readily prepared. In one example, the
drug(s) is/are mixed with a compressible sugar and granulated with a binder
solution
of compressible sugar in purified water. Subsequent to drying, the granules
are mixed
with different amounts of colloidal silicon dioxide (CabosilTM) and magnesium
stearate. The granules are mixed in different proportions with stearic acid or
monosterate (30, 50, 70%, for example) and then fed into a multilayer
tableting
machine (such as a Korsch or Fette tableting machine) to yield a trilayer
tablet.
Additional layers, often with varying amount of drug granules (e.g., greater
drug
concentration in the center layer and decreasing in each subsequent outer
layer), can
readily be added. In certain embodiments, the outermost layers do not include
a drug.
FIG. 1 illustrates a trilayer capsule shape tablet (10) including a first drug
layer (14), second drug layer (16) and third drug layer (18). The capsule
shape tablet
33
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
(10) is partially enveloped in a bioadhesive polymeric plug (12) such that
drug layer-
ends (20a and 20b) remain exposed for drug release. A drug concentration
gradient
between the three drug layers allows a predetermined hybrid release profile to
be
achieved.
General Characteristics
Exci ip ents
The cores of pharmaceutical dosage forms (e.g., tablets and drug-eluting
devices) of the invention contain one or more excipients, carriers or
diluents. These
excipients, carriers or diluents can be selected, for example, to control the
disintegration rate of a pharmaceutical dosage form (e.g., tablet or drug-
eluting
device). In particular, for bioadhesive tablets, it is advantageous for the
time taken to
release the contents of a tablet to be less than the gastrointestinal
retention time or less
than the retention time in the small and/or large intestines. In one
embodiment, the
release time of a tablet is at least 25% of the gastrointestinal, small
intestine and/or
large intestine retention time, at least 50% of the gastrointestinal, small
intestine
and/or large intestine retention time or at least 75% of the gastrointestinal,
small
intestine and/or large intestine retention time.
It will be understood by those skilled in the art that any vehicle or carrier
conventionally employed and which is inert with respect to the active agent,
and
preferably does not interfere witli bioadhesiveness in embodiments where that
characteristic is desired, may be utilized for preparing and administering the
pharmaceutical compositions of the present invention. Illustrative of such
vehicles and
carriers are those described, for example, in Remington's Pharmaceutical
Sciences, 18th
ed. (1990), the disclosure of which is incorporated herein by reference.
The formulations of the present invention for use in a subject comprise the
drug,
optionally together with one or more acceptable carriers or diluents therefor
and
optionally other therapeutic ingredients. The carriers or diluents must be
"acceptable"
in the sense of being compatible with the other ingredients of the formulation
and not
deleterious to the recipient thereof. The formulations can conveniently be
presented in
unit dosage form and can be prepared by any of the methods well known in the
art of
pharmacy. All methods include the step of bringing into association the drug
with the
34
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
carrier or diluent which constitutes one or more accessory ingredients. In
general, the
formulations are prepared by uniformly and intimately bringing into
association the
agent with the carriers and then, if necessary, dividing the product into unit
dosages
thereof.
Examples of carriers and diluents include phannaceutically accepted hydrogels
such as alginate, chitosan, methylmethacrylates, cellulose and derivatives
thereof
(microcrystalline cellulose, hydroxypropyl cellulose, hydroxypropyl methyl
cellulose,
carboxymethylcellulose, ethylcellulose), agarose and PovidoneTM, kaolin,
magnesium
stearate, starch, lactose, sucrose, density-controlling agents such as barium
sulfate and
oils, dissolution enhancers such as aspartic acid, citric acid, glutamic acid,
tartartic acid,
sodium bicarbonate, sodium carbonate, sodium phosphate, glycine, tricine and
TRIS.
For multi-layer tablets in particular, the tablet typically includes at least
one
polymer or excipient. The polymer may be degradable or non-degradable.
Suitable
degradable polymers include polyesters, such as poly(lactic acid) (p[LA]),
poly(lactide-co-glycolide) (p[LGA]), poly(caprolactone) (p[CL]);
polyanhydrides
such as poly(fumaric-co-sebacic anhydride) (p[FASA]) in molar ratios of 20:80
to
90:10, poly(carboxyphenoxypropane-co-sebacic anhydride) (p[CPPSA]),
poly(adipic
anhydride) (p[AA]); polyorthoesters; polyamides; and polyimides. Other
suitable
polymers include hydrogel-based polymers such as agarose, alginate, and
chitosan.
Suitable non-degradable polymers include polystyrene, polyvinylphenol, and
polymethylmethacrylates (EudragitsTM).
The excipients, carriers or diluents can also be selected to control the time
until
a pharmaceutical dosage form (e.g., tablet or drug-eluting device) detaches
from a
mucosal membrane. In particular, the addition of one or more disintegrating
agents will
reduce the time until a pharmaceutical dosage form (e.g., tablet or drug-
eluting device)
detaches. Alternatively or in combination with the disintegrating agents, an
agent that
interferes with the mucosa-tablet/device adhesion can be used to control the
time until
detachment occurs.
Suitable excipients include stabilizers, plasticizers, wetting agents,
antitack
agents, tack agents, foam agents, antifoam agents, binders, fillers,
extenders,
flavorants, dispersants, surfactants, solubilizers, solubilization inhibitors,
glidants,
lubricants, antiadherents, adherents, coatings, protective agents, sorbents,
suspending
agents, crystallization inhibitors, recrystallization inhibitors,
disintegrants, acidulants,
diluents, allcalizing agents, antioxidants, preservatives, colorants,
electrolytes,
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
solvents, antisolvents, accelerating agents, andlor retarding agents. Examples
include
alginate, chitosan, methylmethacrylates (EudragitsTM), celluloses (especially
microcrystalline cellulose, hydroxypropylmethylcellulose, ethylcellulose etc),
agarose, PovidoneTM, lactose, microcrystalline cellulose, kaolin starch,
magnesium
stearate, stearic acid, glycerol monostearate, sucrose, compressible sugar,
lactose and
barium sulfate.
Drugs and Active Agents
A wide variety of drugs can be included in pharmaceutical dosage forms (e.g.,
tablets and drug-eluting devices) of the invention. Such pharmaceutical dosage
forms
typically contain at least 1 mg of a drug. These pharmaceutical dosage forms
can also
contain at least 2 mg, at least 5 mg, at least 10 mg, at least 25 mg, at least
50 mg, at
least 100 mg, at least 500 mg or at least 1000 mg of a drug (e.g., 2 mg to
1000 mg).
Drugs suitable for use herein can be small organic molecules (e.g., non-
polymeric molecules having a molecular weight of 2000 Da or less, such as 1000
Da
or less), peptides or polypeptides and nucleic acids.
Drugs may be classified using the Biopharmaceutical Classification System
(BCS), which separates pharmaceuticals for oral administration into four
classes
depending on their solubility aiid their absorbability through the intestinal
cell layer.
According to the BCS, drug substances are classified as follows:
Class I - High Permeability, High Solubility
Class II - High Permeability, Low Solubility
Class III - Low Permeability, High Solubility
Class N- Low Permeability, Low Solubility.
Drugs from these four classes can be used in the invention.
The interest in this classification system stems largely from its application
in
early drug development and then in the management of product change through
its
life-cycle. In the early stages of drug development, knowledge of the class of
a
particular drug is an important factor influencing the decision to continue or
stop its
development.
Class I drugs of the BCS system are highly soluble and highly permeable in
the gastrointestinal (GI) tract. Sometimes BCS Class I drugs may be micronized
to
sizes less than 2 microns to increase the rate of dissolution.
36
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
Class II drugs are drugs that are particularly insoluble, or slow to dissolve,
but
that readily are absorbed from solution by the lining of the stomach and/or
the
intestine. Therefore, prolonged exposure to the lining of the GI tract is
required to
achieve absorption.
Many of the known Class II drugs are hydrophobic, and have historically been
difficult to administer. Moreover, because of their hydrophobicity, there
tends to be a
significant variation in absorption depending on whether the patient is fed or
fasted at
the time of taking the drug. This in turn can affect the peak level of serum
concentration, making calculation of dosage and dosing regimens more coinplex.
Class III drugs include biologic agents that have good water solubility and
poor GI permeability, such as proteins, peptides, polysaccharides, nucleic
acids,
nucleic acid oligomers and viruses.
Class IV drugs are lipophilic drugs with poor GI permeability. Both Class III
and IV drugs are often problematic or unsuitable for sustained release or
controlled
release. Class III and Class IV drugs are characterized by poor biomembrane
permeability and are commonly delivered parenterally. Traditional approaches
to
parenteral delivery of poorly soluble drugs include using large volumes of
aqueous
diluents, solubilizing agents, detergents, non-aqueous solvents, or non-
physiological
pH solutions. These formulations, however, can increase the systemic toxicity
of the
drug composition or damage body tissues at the site of administration.
In one example, the drug is selected from hormones, eiizymes, antigens,
digestive aids, ulcer treatments (e.g., bismuth subsalicylate optionally in
combination
with antibiotics effective against H. pylori), antihypertensives, enzyme
inhibitors,
antiparasitics (e.g., antimalarials such as atovaquone), spermicides, anti-
hemorrhoidal
treatments, and radiopaque compounds. In another example, the drug is an
antifungal
agent (e.g., itraconazole, fluoconazole, terconazole, ketoconazole,
saperconazole,
griseofulvin, griseoverdin). In a further example, the drug is an
antineoplastic agent.
In yet another example, the drug is an antiviral agent (e.g., acyclovir).
Other classes of
drug suitable for inclusion in pharmaceutical dosage forms (e.g., tablets and
drug-
eluting devices) of the invention include steroids (e.g, danazol),
immunosuppressants
(e.g., cyclosporine), CNS active agents, cardiovascular agents, anti-
depressant agents,
anti-psychotic agents, anti-epileptic agents (e.g., carbamazepine), agents for
treating a
37
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
movement disorder (e.g., valproic acid and salts thereof) and anti-migraine
agents
(e.g., triptans such as sumatriptan).
The preferred materials to be incorporated into the bioadhesive pharmaceutical
dosage forms (e.g., tablets or drug-eluting devices) are drugs and imaging
agents.
Drugs advantageously incorporate include antibiotics, antivirals (especially
protease
inhibitors alone or in combination with nucleosides for treatment of HIV or
Hepatitis
B or C), anti-parasites (helminths, protozoans), anti-cancer (referred to
herein as
"chemotherapeutics", including cytotoxic drugs such as cisplatin and
carboplatin,
BCNU, 5FIJ, methotrexate, adriamycin, camptothecin, and taxol), antibodies and
bioactive fragments thereof (including humanized, single chain, and chimeric
antibodies), antigen and vaccine formulations, peptide drugs, anti-
inflammatories, and
oligonucleotide drugs (including antisense, aptamers, ribozymes, external
guide
sequences for ribonuclease P, and triplex forming agents).
Examples of other useful drugs for use in bioadhesive pharmaceutical dosage
forms (e.g., tablets and drug-eluting devices) include ulcer treatments such
as
CarafateTM from Marion Pharmaceuticals, neurotransmitters such as L-DOPA,
antihypertensives or saluretics such as Metolazone from Searle
Pharmaceuticals,
carbonic anhydrase inhibitors such as Acetazolamide from Lederle
Pharmaceuticals,
insulin like drugs such as glyburide, a blood glucose lowering drug of the
sulfonylurea class, synthetic hormones such as Android F from Brown
Pharmaceuticals and Testred (methyltestosterone) from ICN Pharmaceuticals, and
antiparasitics such as mebendzole (VermoxTM, Jannsen Pharmaceutical).
Antigens can be microencapsulated in one or more types of bioadhesive
polymer, and subsequently compressed into a tablet or filled into a capsule or
the
reservoir of a drug-eluting device, to provide a vaccine. The vaccines can be
produced
to have different retention times in the gastrointestinal tract. The different
retention
times, among other factors, can stimulate production of more than one type
(IgG,
IgM, IgA, IgE, etc.) of antibody.
In a preferred method for imaging, a radio-opaque material such as barium is
coated with polymer. Radioactive materials or magnetic materials could be used
in
place of or in addition to the radio-opaque materials. Examples of other
materials
include gases or gas-emitting conlpounds that are radioopaque.
38
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
Bioadhesive pharmaceutical dosage forms (e.g., tablets and drug-eluting
devices) of the invention are especially useful for treatment of inflammatory
bowel
diseases such as ulcerative colitis and Crohn's disease. In ulcerative
colitis,
inflammation is restricted to the colon, whereas in Crohn's disease,
inflammatory
lesions may be found throughout the gastrointestinal tract, from the mouth to
the
rectum. Sulfasalazine is one of the drugs that is used for treatment of the
above
diseases. Sulfasalazine is cleaved by bacteria within the colon to
sulfapyridine, an
antibiotic, and to 5-amino salicylic acid, an anti-inflammatory agent. The 5-
amino
salicylic acid is the active drug and it is needed locally. Direct
administration of the
degradation product (5-amino salicylic acid) may be more beneficial. A
bioadhesive
drug delivery system could improve the therapy by retaining the drug for a
prolonged
time in the intestinal tract. For Crohn's disease, retention of 5-
aminosalicylic acid in
the upper intestine is of great importance, since bacteria cleave the
sulfasalazine in the
colon, the only way to treat inflammations in the upper intestine is by local
administration of 5-aminosalicylic acid.
Drugs particularly useful in the treatment of H. pyloNi include antibiotics
such
as ainoxicillin, tetracycline, metronidazole and clarithromycin; H2 blockers
such as
cimetidine, ranitidine, famotidine, and nizatidine; proton pump inhibitors
such as
omeprazole, lansoprazole, rabeprazole, esomeprazole, and pantoprozole; and
stomach-lining protectors such as bismuth subsalicylate.
Multi-layer tablets of the invention are broadly useful for drug delivery, as
they are compatible with a large number of different drugs. Suitable drugs
include
sodium valproate, valproic acid, divalproex sodium, antibiotics, non-steroidal
anti-
inflammatory drugs ("NSAIDS"), such as methyl salicylate, antiulcerative
agents
such as bismuth subsalicylate alone or in combination with antibiotics
effective
against organisms such as H. pylori, digestive supplements and cofactors, and
vitamins.
In a preferred embodiment, the drug contains a valproic moiety. Sodium
valproate is used for the treatment of generalized, partial or other epilepsy.
Valproic
acid is used for the treatment of generalized and partial seizures. Valproic
acid and
sodium valproate typically have a 1:1 dosing relationship. Side effects of
treatment
include occasional sedation (especially if given as part of polytherapy),
ataxia and
tremor and liver dysfunction; increased appetite with associated weight gain
is the
most common side effect. Nausea has been reported but is alleviated by taking
the
39
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
dose after food. The normal monotherapy dosage in adults is 600 mg daily in
divided
doses increased by 200 mg every 3 days until control is achieved up to a
maximum of
2.5 g daily in divided doses. The usual dose range is 1-2 g daily in divided
doses. The
normal dose in children over 20 kg is 400 mg daily in divided doses,
irrespective of
weight, increased until control is achieved up to a maximum of 35 mg/kg/day in
divided doses. The usual dose range 20-30 mg/kg /day in divided doses. The
normal
dose in children up to 20 kg is 20 mg/kg daily in divided doses. Preferably,
multi-
layer tablets of the invention (optionally coated with a bioadhesive) reduce
or
eliminate the need to administer inultiple daily doses of valproate drugs.
Generally,
doses of valproate drugs are increased only if plasma concentrations are
monitored.
Because drugs such as phenytoin, carbamazepine and phenobarbitone increase the
metabolism of sodium valproate, the dose required will be higher by 5-10
mg/kg/day.
Once these agents have been withdrawn, the dose of sodium valproate can be
reduced
slightly as long as seizure control is maintained. Sustained release sodium
valproate
formulations are interchangeable with other dosage forms only when seizure
control
has been achieved, as long as the same total daily dose is given.
A class of drugs that is suitable for use in pharmaceutical dosage forms
(e.g.,
tablets and drug-eluting devices), particularly the multi-layer tablets of the
invention
that include a hydrophobic excipient, are hygroscopic and/or deliquescent
drugs. The
term "hygroscopic" as used herein refers to substances that absorb significant
amounts
of atmospheric moisture when exposed to conditions of normal ambient relative
humidity (RH), for example 10-50% RH. The term "deliquescent" refers to
substances
that tend to undergo gradual dissolution and/or liquefaction due to attracti
and/or
absorption of moisture from air when exposed to these conditions. Those
skilled i the
art will appreciate that over the usual range of ambient temperatures used in
dra
formulation, hygroscopicity and the state of deliquescence are largely
temperature-
independent, and that there are varying degrees of hygroscopicity and
deliquescence.
Non-limiting examples of hygroscopic and/or deliquescent drugs suitable for
use in the present invention include acetylcholine chloride, acetylcamitine,
actinobolin, aluminum methionate, aminopentamide, aminopyrine hydrochloride,
ammonium bromide, ammonium valerate, amobarbital sodium, anthiolimine,
antimony sodium tartrate, antimony sodium thioglycollate, aprobarbital,
arginine,
aspirin, atropine N-oxide, avoparcin, azithromycin monohydrate, betahistine
mesylate, betaine, bethanechol chloride, bismuth subnitrate, bupropion,
butamirate,
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
buthalital sodium, butoctamide, cacodylic acid, calcium chloride, calcium
glycerophosphate, calcium iodide, carbachol, camitine, caspofungin,
ceruletide,
chlorophyllin sodium-copper salt, choline alfoscerate, choline salicylate,
choline
theophyllinate, cilastatin, citicoline, cobalt dichloride, cromolyn disodium,
cupric
sulfate pentahydrate, cyanocobalamin, cyclobutyrol, cysteine hydrochloride,
deaminooxytocin (L-isomer, anhydrous), deanol hemisuccinate, demecarium
bromide,
dexamethazone phosphate disodium salt, DL-dexpanthenol, dibucaine
hydrochloride,
dichlorophenarsine hydrochloride, diclofenac sodium, diethylcarbamazine
citrate,
dimethyl sulfoxidem, drotebanol, echinomycin, ephedrine (anhydrous),
ergotamine,
ethanolamine, fencamine hydrochloride, ferric chloride, ferrous iodide, ficin,
gadobenate dimeglumine, gentainicin C complex sulfate, guanidine, heparin,
hexadimethrine bromide, hexamethonium tartrate, hexobarbital sodium,
histamine,
hydrastine hydrochloride, hyoscyamine hydrobromide, S-[2-[(1-
iminoethyl)amino]ethyl]-2-methyl-L-cysteine, imipramine N-oxide, isometheptene
hydrochloride, isosorbide, levothyroxine sodium, lichenifonnins, lobeline
sulfate,
magnesium chloride hexahydrate, magnesium trisilicate, menadione,
mercaptomerin
sodium, mersalyl, metaraminol, methacholine chloride, methantheline bromide,
methantheline chloride, methitural sodium, L-methyldopa sesquihydrate,
inethylmethioninesulfonium chloride, mildiomycin, minocycline hydrochloride,
mitoxantrone dihydrochloride, morpholine, muscarine chloride, nafronyl acid
oxalate,
narceine, nicotine, nicotinyl alcohol, nolatrexed dihydrochloride, omeprazole,
oryzacidin, oxalic acid, oxophenarsine hydrochloride, panthenol, pantothenic
acid
(sodium salt), papain, penicillamine hydrochloride, penicillin G (potassium
salt),
pentamethonium bromide, pentamidine isethionate, pepsin, perazine
dihydrochloride,
phenobarbital, sodium 5,5-diphenyl hydantoinate, phethenylate sodium,
phosphocreatine (calcium salt tetrahydrate), physostigmine sulfate,
pilocarpine
hydrochloride, pipemidic acid, podophyllotoxin-beta-D-glucoside, potassium
carbonate, potassium iodide, pralidoxime mesylate, prednisolone sodium
phosphate,
procainamide hydrochloride, procaine butyrate, L-proline, promazine
hydrochloride,
propamidine isethionate, prostacyclin sodium, pyridostigmine bromide,
pyronaridine,
quinacillin disodium, quinoline, radioactive sodium iodide, reserpilic acid
dimethylaminoethyl ester dihydrochloride, secobarbital sodium, silver
fluoride,
sodium acetate, sodium bromide, sodium propionate, sodium dibunate, sodium
dichromate(VI), sodium nitrite, sodium pentosan polysulfate, sodium valproate,
41
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
soluble sulfamerazine, stibocaptate, streptomycin, succinylcholine bromide,
succinylcholine iodide, sulfaquinoxaline, sulisatin disodium, suramin sodium,
tamoxifen citrate, taurocholic acid, terazosin hydrochloride, thiobutabarbital
sodium,
thiopental sodium, ticarcillin disodium, 2,2,2-trichloroethanol, trientine,
triethanolamine, triftazin, tolazoline hydrochloride, vinbarbital sodium,
viomycin,
vitamin B12, zinc iodide, and combinations thereof, and pharinaceutically
acceptable
hygroscopic and/or deliquescent salts and variants tllereof.
More than one type of drug can be present in a pharmaceutical dosage form
(e.g., tablet or a drug-eluting device) of the invention. The drugs can be
evenly
distributed throughout a medicament or can be heterogeneously distributed in a
medicament, such that one drug is fully or partially released before a second
drug.
Pharmaceutical Dosage Forms
Pharinaceutical dosage forms (e.g., tablets, capsules, drug-eluting devices)
of
the invention typically weigh at least 5 mg. Tablets, capsules and drug-
eluting devices
can also weigh at least 10 mg, at least 15 mg, at least 25 mg, at least 50 mg,
at least
100 mg, at least 500 mg or at least 1000 mg. Typically, such objects weigh 10
mg to
500 mg.
The pharmaceutical dosage forms (e.g., capsules or tablets) typically contain
between 10 and 70% of therapeutic, diagnostic or prophylactic agent (referred
to
generally as "drug") by weight of a dosage form, or between 30 and 90% by
weight of
the core of a dosage form, where each coating makes up between 1-10%,
preferably
5-6%, by weight of the dosage form, up to a total of about 30% by weight. The
coating can include drug, in ratios of, for example, from 5 and 50% by weight
of the
coating, preferably between 20 and 40% by weight of the coating, while still
retaining
rate control.
Pharmaceutical dosage forms (e.g., tablets, capsules, drug-eluting devices) of
the invention typically measure at least 2 mm in one direction. For example,
pharmaceutical dosage forms can measure at least 5 mm, at least 10 mm, at
least 15
mm or at least 20 mm in one direction. Typically, the diameter of the
pharmaceutical
dosage forms is 2 to 40 mm, preferably 10 to 30 mm such as 20 to 26 mm. Mini-
tablets have a diameter of 2 mm to about 5 mm. Such pharmaceutical dosage
forms
42
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
can measure at least 2 mm, at least 5 mm, at least 10 mm, at least 15 mm or
least 20
mm in a second direction and, optionally, a third direction. For example,
pharmaceutical dosage forms of the invention ranges in size from about 2 to
about 50
mm in length, from about 2 mm to about 15 mm in depth, and from about 2 mm to
about 15 mm in width. Preferably, the pharmaceutical dosage form is of a size
that
facilitates swallowing by a subject.
The volume of a typical pharmaceutical dosage form of the invention is at
least 0.008 mL, at least 0.01 mL, at least 0.05 mL, at least 0.1 mL, at least
0.125 mL,
at least 0.2 mL, at least 0.3 mL, at least 0.4 mL or at least 0.5 mL, such as
from 0.008
mL to 0.5 mL.
Suitable types of tablets are discussed in U.S. Provisional Applications Nos.
60/605,199, filed on August 27, 2004, 60/605,198, filed on August 27, 2004,
and
60/635,812, filed on December 13, 2004, the contents of which are incorporated
by
reference.
In one example, the tablet is a trilayer tablet having an inner core that
includes
one or more drugs in an appropriate matrix of excipients (e.g., HPMC, MCC,
lactose)
and is surrounded on two sides by a bioadhesive polymeric coating, which
optionally
is mixed with the one or more drugs. Preferred bioadhesive polymeric coatings
are a
DOPA-BMA (poly(butadiene-co-maleic acid)) polymer and a mixture of
poly(fumaric-co-sebacic) anhydride and EudragitTM RS PO.
In another example, the tablet is a longitudinally compressed tablet
containing
precompressed inserts of the drug and excipients and optionally a permeation
enhancer. Drug is only released at the edge of this tablet, which can result
in zero-
order kinetics.
In yet another example, the tablet is comprised of a multiplicity of
bioadhesive-coated microspheres that have been compressed into a tablet core
and
subsequently coated with a bioadhesive coating and one or more additional
coatings
(e.g., enteric coatings).
In a further example, the tablet includes a cavity through all or part of the
tablet. A cavity extending through a tablet creates a channel open at both
ends. Such
tablets can be coated, for example, with a compression coating (e.g., enteric,
43
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
bioadhesive, combinations, etc.) on all or selected surfaces. One example is a
tablet
having a channel through the tablet where the channel is uncoated.
In another example, the subject dosage formulations comprise an inner core,
which comprises one or more drugs, excipients, and/or aborption enhancers that
have
been compressed to a form a solid, such as a tablet. For example, powdered
drug
formulations of the invention can be compressed to form a solid. In other
embodiments, a drug can be used that in its pure form, under ambient
conditions, is a
liquid. In some embodiments, the liquid drug that is incorporated into a
compressed
inner core of the invention is present as a free base or free acid. In
embodiments
where the drug is a liquid drug (e.g., nicotine, valproic acid), the drug is
preferably
incorporated into a dosage form of the invention after it has been absorbed
onto an
absorbent material, such as kaolin clay or Cabosil (colloidal silicon
dioxide).
In other embodiments, a solubilized form of an insoluble drug is incorporated
into a dosage form of the invention. Solubilized forms of insoluble drugs may
be
aqueous-based or oil-based. For example, a water-insoluble drug may be
dissolved in
an organic solvent and then absorbed onto an absorbent material, such as a
synthetic
aluminosilicate or silicate, which can absorb certain organic solvents while
still
retaining the properties of a solid.
Capsules of the invention can be constructed in a multitude of ways. For
examples, capsules can be filled with liquid, paste, powder, granules and/or
beads.
Granules and beads are optionally coated with a bioadhesive and/or other
coating
described herein. A capsule coated with a bioadhesive can either have the
bioadhesive
on the surface, or the bioadhesive can be coated with one or more layers that
delay
exposure of the bioadhesive to ambient conditions (e.g., to prevent the
capsule from
adhering to an upper portion or upper portion of the gastrointestinal tract).
Capsules
can include one or more excipients disclosed herein.
Capsules or tablets can be incorporated into standard pharmaceutical dosage
forms such as gelatin capsules and tablets. Gelatin capsules, available in
sizes 000, 00,
0, 1, 2, 3, 4, and 5, from manufacturers such as Capsugel , may be filled with
capsules or tablets and administered orally. Similarly, capsules or tablets
may be dry
blended or wet-granulated with diluents such as microcrystalline cellulose,
lactose,
colloidal silicon dioxide (e.g., CabosilTM) and binders such as
44
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
hydroxypropylmethylcellulose, hydroxypropylcellulose, carboxymethylcellulose
and
directly compressed to form tablets.
Various drug-eluting devices are described in U.S. Patent Nos. 4,290,426,
5,256,440, 5,378,475, 5,773,019 and 6,797,283, the contents of which are
incorporated herein by reference.
In one example, the drug-eluting device includes an inner reservoir comprising
the effective agent; a first coating layer, which is essentially impermeable
to the
passage of the effective agent; and a second coating layer, which is permeable
to the
passage of the effective agent. The first coating layer covers at least a
portion of the
inner reservoir; however, at least a small portion of the inner reservoir is
not coated
with the first coating layer (e.g., there are one or more pores in the first
coating layer).
The second coating layer essentially coinpletely covers the first coating
layer and the
uncoated portion of the inner reservoir. Typically, the first coating layer is
a non-
bioerodable or a slowly bioerodable polynier (e.g., a polymer having a
polymethylene
backbone). For the present invention, the second coating can be either a
bioadhesive
polymeric coating or a coating between the first coating and the bioadhesive
polymeric coating.
In another example, the drug-eluting device includes a multilayer core, often
a
bilayer, formed of polymer matrices that swell upon contact with the fluids of
the
stomach. At least one layer of the multilayer core includes a drug. A portion
of the
polymer matrices are surrounded by a band of insoluble material that prevents
the
covered portion of the polymer matrices from swelling and provides a segment
of the
dosage form that is of sufficient rigidity to withstand the contractions of
the stomach.
As a result, release of the drug is regulated by escape of the drug through
one or more
pores in the device. Typically, the core and the band of soluble material are
coated
with a bioadhesive polymeric coating.
In a further example, the drug-eluting device is an osmotic delivery system.
Typically, the reservoir of such devices contains osmotic agents to draw water
across
a semi-permeable membrane and a swelling polymer to push drug out of the
device at
a controlled rate.
Preferably, drug-eluting devices of the invention release the drug contained
therein with zero-order kinetics.
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
EXEMPLIFICATION
Example 1
Fluoroscopy Study of Barium-Impregnated Trilayer Tablets with Bioadhesive
Polymer Outer Layers
Trilayer tablets were prepared by sequentially filling a 0.3287 X 0.8937 "00
capsule" die (Natoli Engineering) with 333 mg of either SpheromerTM I or
SpheromerTM III Bioadhesive polymer, followed by 233 mg of a blend of
hydroxypropylmethylcellulose (HPMC) 4000 cps and 100 mg of barium sulfate,
followed by an outer layer of 333 mg of either SpheromerTM I or III
bioadhesive
polymer. Trilayer tablets were prepared by direct compression at 2000 psi for
1
second using a Globepharma Manual Tablet Compaction Machine (MTCM-1). The
tablets were administered to feinale beagles that were fasted for 24 hrs. The
tablets
were also dosed to fasted beagles that had been fed with chow, 30 min before
dosing
(fed). Tablets were continuously imaged with fluoroscopy over the course of 6
hrs in
unrestrained dogs. Typical results are indicated below. Trilayer tablets with
SpheromerTM I or III in the bioadhesive layers remained in the stomach of
fasted dogs
for up to 3.5 hrs and resided in the stomach of fed dogs in excess of 6 hrs,
as shown in
FIGS. 2A-D. The tablets did not mix with food contents and remained in contact
with
stomach mucosa at the same location until they passed into the small
intestine.
Example 2
Fluoroscopy Study of Barium Impregnated Five-layer Tablets with SpheromerTM I
in
Outer Layers
Five-layer tablets were prepared by sequentially filling a 0.3287 X 0.8937 "00
capsule" die (Natoli Engineering) with the following mixtures:
46
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
Composition of 5 Layer Tablet
(1427 mg)
Position mg/tablet % w/w
250 x 2=500
Outer Bioadhesive Layer (two) mg
poly[fumaric-co-sebacic] acid
20:80 66.2
Eudragit RS PO 22.8
Sodium Chloride 10
Magnesiuin Stearate 1
Total 100%
100 x 2=200
Intermediate Contrast Layer (two) mg %w/w
Barium sulfate 100%
Central Core Layer (one) 727 mg % w/w
33% Itraconazole/Eudragit E100 46
Microcrystalline Cellulose
Granulation
Spray Dried Lactose 13.7
HPMC, 5 cps 30
HPMC, 100 cps 10
Magnesium Stearate 0.3
Total 100%
Five layer tablets, containing 100 mg of itraconazole, were prepared by direct
compression at 3000 psi for 5 second using a Globepharma Manual Tablet
Compaction Machine (MTCM-1). The tablets were administered to two female
beagles that were fasted for 24 hrs and then fed chow 30 min before dosing
(fed).
Tablets were continuously imaged with fluoroscopy over the course of 8 hrs in
unrestrained dogs. The tablets resided in the stomach of both fed dogs in
excess of 6
hrs and ultimately split apart and passed into the small intestine at 8 hrs
post-dosing.
The tablets did not mix with food contents and remained in contact with
stomach
mucosa at approximately the same location until they passed into the small
intestine.
Pharmacokinetic results are indicated below:
47
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
Plasma Itraconazole
Time Mean SD
(hr) (ng/ml) (ng/ml)
0 0 0
1 98.2 62
4 984 277.2
8 1545 7.1
12 858 200.8
16 719 207.2
18 633.5 173.2
24 433.58 72.8
28 370 38.2
32 336 38.2
48 307.5 77.1
AUC 27409.3 ng/m1*hr-
Cmax 1545 ng/ml
Tmax 8 hrs
By comparison, the iimovator drug Sporanox (Johnson & Johnson) is an
immediate release dosage form containing 100 mg of itraconazole. When tested
in the
same beagle model, an AUC of 22,000 ng/ml*hr-1, Cmax of 1200 ng/ml and T,,,ax
of 1.5
hrs were obtained. Itraconazole is a Biopharmaceutical Classification System
Class 2
drug that has negligible water solubility and good GI penneability. It is
slightly
soluble in water at pH <3.5, limiting the site of GI absorption to duodenum
and upper
jejunum.
Clearly, the gastroretentive bioadhesive formulation described in this example
delivered itraconazole for an extended period of time to the absorptive site
in
duodenum and upper jejunum. The 8 hr gastrointestinal residence time observed
by
fluoroscopy corresponds to the maximum itraconazole plasma concentration
achieved
at T max of 8 hrs.
Example 3
Sodium Valproate Tablets
Two different lots of sodium valproate bioadhesive tablet formulations, based
on the concentration gradient approach, were prepared. Tablets from the first
lot
utilized L-Dopa/BMA (SpheromerTM III) as the bioadhesive polymer while tablets
from the second lot were based on p(FA:SA) bioadhesive polymer. An additional
48
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
tablet lot using ethyl cellulose as a non-bioadhesive polymer was also
prepared. The
following granulation and blending steps were used to make the three lots:
Granulation
180.0 g of sodium valproate (Katwijk Chemie BV) were granulated using a
binder solution prepared previously by dissolving 10 g of ethyl cellulose (1 0-
FP, NF
Premium) and 10 g of polyvinylpyrrolidone, K-15 in 667 mL of ethanol. Binder
solution was applied onto the drug in a bench top fluidized-bed spray-coating
unit
(Vector Corp. model MFL.01). The following process parameters were used: fluid
bed N2 gas-flow = 60-140 LPM; spray-nozzle pressure = 15 psi; inlet
temperature =
50 C; exhaust temperature = 21-26 C; pump speed = 40 rpm; screen size ="I";
Wurster partition = medium; spray = bottom spray; and spray nozzle = medium.
The
granulation was dried and blended with 1% colloidal silicon dioxide. The
granulation
was stored in a 1-Liter glass jar containing DesiPak dessicant until used.
Blending
The granulation sodium valproate was blended with various excipients to
achieve the sodium valproate inner and outer layers compositions as shown
below.
The granulation was initially blended with ethyl cellulose or SpheromerTM I
(p(FA:SA)) or SpheromerTM III (DOPA grafted on BMA) in a blender for 5 minutes
followed by blending with magnesium stearate for additional 5 minutes.
Composition of Common Inner Layer Blend
Ingredients Weight (%)
Sodium Valproate Granulation 59.0
Ethyl Cellulose, 10FP 40.0
Magnesium Stearate 1.0
Total 100.0
49
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
Composition of Outer Layer Blends
Ingredients Weight (%)
Sodium Valproate Granulation 7.65
Spheromer I or Spheromer 91.35
III
Magnesium Stearate 1.0
Total 100.0
Trilayer Tablets
Trilayer tablets were compressed on the GlobePharma MCTM-1 manual tablet
press using 0.328" x 0.8937" capsule shaped, deep concave punches. First 200
mg of
outer blend was added to the die cavity and pre-compressed, then 987.2 mg of
inner
blend was added to the die cavity and pre-compressed again, and finally the
200 mg
outer blend was added and compressed at 3000 psi for 1s.
In vitro Dissolution Testing
Trilayer tablets were tested for dissolution testing in a USP I apparatus
using
pH 6.8 PBS buffer at 100 rpm and 37 C. The dissolution profiles for the lots
including SpheromerTM I and III are shown in FIGS. 4A and 4B, respectively.
These
dissolution profiles indicate that sodium valproate is released from the
tablet in two
phases, immediate release (outer layers) and sustained release (inner layer).
Water Uptake Studies
Sodium valproate trilayer tablet formulations (n=6) and a single-layer, matrix
tablet formulation (n=6) consisting of only the core layer of the trilayer
tablets were
incubated at 45 C and 60% relative huinidity for up to 56 hours, where weight
gain
was measured at regular intervals. The results are illustrated in FIG. 5. The
trilayer
tablet formulations had considerably less water uptake compared to the
unprotected
core layer tablet. Uptake of water by tablets containing L-DOPA-BMA
(SpheromerTM
III) and ethylcellulose in the outer layers was approximately the same,
whereas water
uptake by tablets with p[FA:SA] polymer (SpheromerTM I) in the outer layers
was
approximately 30% less than the other trilayer tablet formulations. Thus, the
use of
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
hydrophopic polymers in multilayer tablet formulations provides important
advantages for protection of hygroscopic drugs against water uptake.
Example 4
Carbidopa/Levodopa Tablets
Similar to the sodium valproate tablets described above, trilayer tablets
containing carbidopa and levodopa were prepared. The contents of the inner and
outer
layers are as follows:
Outer Layer Composition
Ingredients Portion of Outer Layer Portion of Tablet
Weight %) Weight (mg) Weight (mg)
Levodopa 10.0 20.0 40.0
Carbidopa, Monohydrate 2.7 5.40 10.8*
Citric Acid 2.0 4.0 8.0
Spheromer III 84.3 168.6 337.2
Magnesium Stearate 1.0 2.0 4.0
Total 100.0 200.0 400.0
*Equivalent to 10mg Carbidopa, anhydrous
Inner Layer Composition
Ingredients Weight (%) Weight (mg)
Levodopa 40.0 160.0
Carbidopa, Monohydrate 10.79 43.16
HPMC 100 cps 38.0 152.0
HPMC E5 4.71 18.84
Glutamic Acid 3.0 12.0
Corn Starch 3.0 12.0
Magnesium Stearate 0.5 2.0
Total 100.0 400.0
*Equivalent to 40mg Carbidopa, anhydrous
For the inner and outer layer compositions, all ingredients, except magnesium
stearate, were weighed and mixed thoroughly in a blender for five minutes. The
mixed ingredients were blended with magnesium stearate for additional five
minutes.
800 mg trilayer tablets were prepared as described in Example 3 using the
GlobePharma Manual Tablet Compaction Machine (MTCM-1) equipped with
51
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
standard 0.3287 x 0.8937" punches. The tablets were compressed at compression
force of 3000 psi for 1 s.
Bioadhesive tablets were tested for release profile in 0.1 N HCl at 37 0.5
C,
in the USP II dissolution apparatus at 50 rpm. The in vitro release profile of
levodopa
from the trilayer tablets is shown in FIG. 6, which confirm that there is an
immediate
release of levodopa (from the outer layers), followed by a sustained release
of
levodopa (from the inner layer).
Example 5
Comparative Performance of Acyclovir Trilayer Tablet and Zovirax Tablets
Acyclovir is categorized as a Class 3 drug according to the Biopharmaceutical
Classification system, because of its moderate water solubility and low
bioavailability
(10-20%). The drug is soluble only at acidic pH (pKa 2.27) limiting absorption
in the
gastrointestinal tract to duodenum and jejunum. There is no effect of food on
drug
absorption. Peak plasma levels are reached 3 to 4 hours following an oral
dose.
Bioavailability decreases with increasing drug dose. Elimination from plasma
has a
terminal half-life of 2.5 to 3.3 hours. Zovirax is normally dosed at either
200 mg
every 4 hrs or 400 mg every 12 hrs, depending on the antiviral indication.
A trilayer tablet controlled release (CR) forinulation includes an inner core
and
an outer bioadhesive coating. The controlled-release inner core blend contains
400 mg
of acyclovir blended with glutamic acid, functioning as an acidulant, and
Ethocel. The
outer bioadhesive coating contains SpheromerTM III and excipients. The inner
core
blend is sandwiched between outer bioadhesive layers and direct compressed to
create
a bioadhesive, trilayer tablet.
The trilayer tablet is designed to reside in the stomach for greater than 6
hrs in
the fed state and release acyclovir downstream, in a controlled manner, to the
duodenum and upper jejunum, the main absorptive sites.
Cohorts of six, female beagle dogs (10-12 kg) were dosed with either Zovirax
or the trilayer tablet 30 min after a standard meal. 200 mg capsules of
Zovirax were
dosed four times, every 6 hrs and compared to the trilayer tablets, containing
400 mg
of acyclovir, dosed twice, every 12 hrs. The total drug dose in both cases was
800 mg
administered over 24 hrs. 1 ml blood samples were collected at appropriate
intervals
52
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
extending to 48 hours. Plasma was collected after centrifugation for 10 min at
3,000
rpm and 4 C. Samples were stored frozen at -20 C until analyzed.
Serum acyclovir was determined by LC/MS/MS. Turbulent flow
chromatography using a 2300 HTLCTM system (Cohesive Technologies, Franklin,
MA) was coupled to tandem-mass spectrometry (MS/1VIS) performed on a triple
stage quadropole from Perkin Elmer SCIEX API 365 (Sciex, Concord, Ontario,
Canada) with an atmospheric pressure ionization (API) chamber. The limit of
detection of acyclovir in dog plasma was 10 ng/ml.
For each dog, the following pharmacokinetic parameters were calculated for the
parent drug, acyclovir: maximum observed concentration (Cmax), time at which
Cmax was observed (tmax), and area under the plasma concentration versus time
curve (AUC) carried out to 48 hrs (AUCO-t).
The effect of repeat dosing on plasma drug levels is shown in FIG. 7. The
AUC's of the iminediate release Zovirax capsules and the trilayer tablets were
nearly
identical (167 ug/ml*hr for Zovirax compared to 164 ug/ml*hr for the trilayer
tablets, n=6 dogs/study).
These data demonstrate that two doses of the trilayer tablet maintained plasma
levels of acyclovir essentially as effectively as two doses of the Zovirax
capsules.
Thus, the trilayer tablets do not need to be administered as frequently as the
Zorivax
capsules.
Example 6
Phannacokinetics of Bioadhesive Itraconazole Tablets Containing Bioadhesive
Excipient
Bioadhesive, trilayer tablets containing 100 mg itraconazole in the central
core
layer were compressed using 0.3287 X 0.8937" capsule-shaped dies (Natoli
Engineering) at 3000 psi for 3 seconds in a GlobePharma Manual Tablet
Compaction
Machine (MTCM-1), as described above. The composition of the tablet was as
follows:
53
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
Drug Layer Composition
mg per
Component Function tablet %w/w
30% Itraconazole/
Eudragit E100 Layered Drug/Polymer
onto Microcrystalline Complex 334 46.0
Cellulose (Emcoce190
M)
H romellose 100 c s Rate-Controlling 73 10.0
~ p Polymer
Hypromellose 5 cps Rate-Controlling 218 30.0
Polymer
Spray-dried Lactose Compressible binder 100 13.7
(Fast Flo 316)
Magnesium Stearate Lubricant 2 0.3
Total 727 100.0
Bioadhesive Layer Composition
%w/w
Component Function tablet
Poly (Fumaric-co-
sebacic)anhydride 20:80 Bioadhesive Polymer 331 66.2
polymer (SpheromerTM I)
Fumaric anhydride
oligomer (FAO) Bioadhesive Excipient 50 10.0
(SpheromerTM II)
Eudragit RSPO Binder 114 22.8
Magnesium Stearate Lubricant 5 1.0
Total 500 100.0
The trilayer tablets were tested for dissolution release profile in 900 mL of
simulated gastric fluid (SGF), pH 1.2 in a USP II apparatus at 100 rpm. The
results
are shown in FIG. 8. Approximately 50% of the itraconazole was released within
8
hrs and 85% was released within 16 hrs. In contrast, dissolution of Sporanox
(approx. 85% drug release) occurred within 60 minutes.
Sporanox capsules and the bioadhesive trilayer tablets, each containing
100mg of itraconazole, were administered to cohorts of six beagle dogs in the
fed
state and plasma levels of itraconazole were measured using LC/MS/MS.
Pharmacokinetic profiles of two formulations in FIG. 9.
54
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
The area under the plasma itraconazole vs time curve (AUC), maximum
concentration (Cmax) and time required to achieve Cmax (Tmax) were calculated
and
the results are indicated in the table below.
AUC Cmax Tmax
Formulation (ng/mL*hr) (ng/mL) (hr)
Sporanox 22741 1382.5 2.0
Trilayer Tablets 21292 1162.8 8.0
The bioadhesive trilayer tablets were able to achieve an AUC similar to that
of
the Sporanox capsules. As per the Biopharmaceutical Classification System,
itraconazole is a Class II drug, known to be absorbed only in the upper small
intestine.
The longer Tmax of the bioadhesive Spherazole formulation, compared to the
Sporanox capsule, is characteristic of a controlled release formulation, as
well as
indicative of retention in the gastrointestinal tract.
Example 7
Bioadhesion of Coated Tablets
The bioadhesion of SpheromerTM II, SpheromerTM III, Gantrez AN-119 BF
(2,5-Furandione and methoxyethene) and hydrated Carbopol 934P NF (cross-
linked
polyacrylic acid homopolymer) films, prepared by dip-coating on nylon
supports, was
tested using a Texture Analyzer TA XT II tensile tester, with pig intestine as
the
biological substrate. The parameters measured were fracture strength (peak
force of
detachment normalized for cross-sectional surface area) and tensile work (area
under
the deformation versus load curve).
Polymer films on supports were prepared by dip-coating in concentrated
polymer solution and drying. Twenty percent (w/v) solutions were made for all
test
materials except for Carbopol 934P, which was a 2% (w/v) solution in water.
SpheromerTM II was dissolved with an equal amount of Eudragit RL 100 in
dichlorometliane. The films on supports were air-dried for 24 hrs after
dipping and
lyophilized overnight to remove residual solvents.
Pig intestine was cut into at least 1 in 2 sections, mounted into a
perforated,
plastic holder with the mucus side up and submerged in phosphate buffered
saline
(PBS, pH 6.8). A fresh piece of tissue was used for each test. A polymer-
coated
CA 02578845 2007-02-27
WO 2006/031420 PCT/US2005/030651
support was mounted on the Texture Analyzer, and brought into contact with the
pig
intestine sample. An uncoated support was used as the control. After 7
minutes, the
support was lifted away from the sample tissue and the load versus deformation
curve
was plotted. Instrumental settings are listed in the table below:
Texture Analyzer Settings
CAPTION VALUE UNITS
Pre-Test Speed 0.50 mm/sec
Test Speed 0.50 mm/sec
Post-Test Speed 0.50 mm/sec
Force 5.0 g
Time 420.00 sec
Trigger Type Button -
Trigger Force 5.0 g
Trigger Distance 0.000 mm
The results of the assay are shown in FIGS. 10 and 11.
While this invention has been particularly shown and described with
references to preferred embodiments thereof, it will be understood by those
skilled in
the art that various changes in form and details may be made therein without
departing from the scope of the invention encompassed by the appended claims.
56