Note: Descriptions are shown in the official language in which they were submitted.
CA 02579732 2012-08-08
- 1 -
Tablets with site and time-controlled gastrointestinal release of active
ingredient
Description
Background of the invention
1. Field of the invention
The present invention describes a pharmaceutical dosage form with site-
and time-controlled gastrointestinal release of active ingredient.
2. Description of the Related Art
Release of non-steroidal anti-inflammatory drugs in the stomach frequently
causes ulcers of the gastric mucosa. This is why tablets with a gastro-
resistant coating are now employed almost exclusively. The disadvantage is
often that the active ingredient is released very quickly on entry into the
intestine. Thus, it is possible with this technology to achieve control only
of
the site, but not of the timing, of active ingredient release.
Absorption of some active ingredients is possible only in certain sections of
the gastrointestinal tract (absorption window). Active ingredient
entry/transfer into the plasma is often desired only when the pathological
state becomes particularly manifest at certain periods of the day (circadian
rhythm). This is the case for example with asthma or ischemies in the early
morning, joint pain in the morning, etc. On the other hand, the effect of some
medicaments is often desired only locally in the gastrointestinal tract, such
as for inflammations (e.g. in ulcerative colitis or Crohn's disease) or
infections in the gastrointestinal tract.
3o Coated tablets have been described frequently, especially with the aim
of
delayed release of active ingredient, in which case an initial phase without
release of the active ingredient (lag phase) is followed by the active
ingredient leaving the tablet.
CA 02579732 2007-03-07
WO 2006/027266
PCT/EP2005/009726
- 2 -
Thus, WO 02/072033 discloses that the amount of coating material applied
determines the lag phase. The coating consists of a swellable material
through the pores of which the active ingredient is then released. In this
case, the diffusion through the swellable matrix of the coating becomes the
release-determining factor. However, release through the pores often does
not take place spontaneously after the desired lag phase; on the contrary,
there is onset of more or less rapid release. In addition, the influences of
food on swelling and eroding coatings are very important.
US 5 464 633 describes a tablet for delayed release of an active substance.
The tablet consists of a core which comprises the active ingredient and a
polymer, and of a polymer-containing coating.
EP 0 463 877 describes a pharmaceutical preparation for controlled release
of an active ingredient, which comprises a core and a coating layer, where
the coating layer comprises a water-repellent salt and a copolymer.
A pharmaceutical preparation consisting of a core and of a multilayer coating
for release of the active ingredient in the lower part of the gastrointestinal
tract (colon) is known for example from EP 0 366 621. Film coatings which
are degraded only in the colon by bacteria present therein are, however,
unsuitable for releasing the active ingredient in upper sections of the
intestine.
WO 01/80824 (Eurand) describes a pharmaceutical form having a core
which, besides the active ingredient, also comprises a hydrophilic, swelling
polymer, and having a surrounding coating consisting of at least one water-
insoluble polymer.
EP 0 939 623 B1 and US 6 183 780 (Duphar) describe an oral dosage form
with delayed release consisting of a core and of a coating, where the coating
consists of one or more polymers, of a water-soluble plasticizer and of a
CA 02579732 2007-03-07
WO 2006/027266
PCT/EP2005/009726
- 3 -
substance which increases the brittleness of the coating. The disadvantages
of this form are, in particular, that influences of food are possible.
EP 1 067 910 (Bar-Shalom) describes an oral dosage form having at least
one erodable surface. EP 1 275 381 (Yamanouchi) likewise describes a core
tablet with coating, the latter consisting of a swellable hydrophilic polymer.
The effects of food in these cases are also great.
Administration of dilitiazern in the form of biologically inert pellets with a
plurality of layers is described in. US 6 620 439 (Elite Labs). In this case,
the
active ingredient is released some hours after intake to treat arterial
occlusions in the morning.
US Patent 5 792 476 describes a pharmaceutical composition for peroral
administration for rheumatoid arthritis, which comprises a glucocorticoid as
active ingredient and which leads to release in the small intestine. The
composition is a granulate which is laminated with an inner layer which is
resistant to a pH of 6.8, and with an outer layer which is resistant to a pH
of
1Ø
US Patent 6 488 960 describes a pharmaceutical dosage form for controlled
release of corticoids, reference being made to the formulations described in
US Patent 5 792 476.
WO 01/08421 describes a tablet having a core which is coated by at least
two layers, one of which completely encloses the other. The coating layers
can be produced by spray coating and/or pressing.
WO 01/68056 discloses a pharmaceutical preparation having a release
profile with a time delay, comprising a core and at least one hydrophilic or
lipophilic coating surrounding the core, where the coating is slowly swollen,
dissolved, eroded or changed in its structure in another way through the
water present in the release medium, so that the core or parts of the core
CA 02579732 2007-03-07
WO 2006/027266
PCT/EP2005/009726
- 4 -
become accessible to the release medium. The coating may be formed for
example as pressed coating.
WO 02/072034 discloses a pharmaceutical dosage form for delayed release,
having a core which comprises as active ingredient a glucocorticoid and a
material which brings about delayed release and includes at least one
natural or synthetic gum.
WO 2004/093843, the content of which is incorporated herein by reference,
discloses a tablet with a specific core geometry to release the active
ingredient in a specific delayed release manner.
Brief summary of the invention
The problem underlying the present invention was to provide a
pharmaceutical dosage form with site- and time-controlled release of active
ingredient, which makes reproducible in vivo release possible in the
particular desired sections of the intestine irrespective of the patient's
food
intake. It was further intended also for the active ingredient release process
itself to be controllable as optimally as possible depending on the relevant
medical indication.
This problem is solved by a pharmaceutical dosage form with site- and time-
controlled gastrointestinal release of active ingredient, comprising
(a) a core having at least one active ingredient and having at least one
swellable adjuvant such that the active ingredient is rapidly released
from the dosage form when the core is contacted with gastrointestinal
fluids; and
(b) an inert coating pressed onto the core, said coating being capable of
preventing substantial release of the active ingredient for a defined time
period following ingestion of the dosage form.
In another aspect, the present invention is directed to a method for the
CA 02579732 2007-03-07
WO 2006/027266
PCT/EP2005/009726
- 5 -
treatment of a patient in need of therapy with an active ingredient in a site-
and time-controlled dosage form, said method comprising administering to
said patient the pharmaceutical dosage form described herein.
In another aspect, the present invention is directed to a kit comprising at
least one unit dosage of a dosage form described herein with site- and time-
controlled gastrointestinal release of active ingredient. The kit optionally
contains instructional material for use of the unit dosage form.
In one aspect, the present invention relates to a method of producing a
tablet which releases a corticosteroid active ingredient at a predetermined
variable location in the GI tract, said method comprising:
determining the location in the GI tract at which it is desired to
deliver the corticosteroid;
forming a coated tablet having a core comprising the
corticosteroid and a swellable adjuvant, and an inert outer coating; and
compressing the coating of said tablet at a pressure chosen to
result in the release of the corticosteroid at said predetermined position.
In another aspect, the present invention relates to a coated tablet having a
core of a corticosteroid active ingredient and a coating, capable of releasing
the corticosteroid at a predetermined variable location the GI tract, the
coating being compressed to a degree which results in the release of the
corticosteroid at said predetermined location.
In another aspect, the present invention relates to a method of producing a
tablet which releases a corticosteroid active ingredient at a predetermined
variable location in the GI tract, said method comprising:
determining the location in the GI tract at which it is desired to
deliver the corticosteroid;
forming a coated tablet having a core comprising the
corticosteroid and a swellable adjuvant, and an inert outer coating;
CA 02579732 2007-03-07
WO 2006/027266
PCT/EP2005/009726
- 6 -
compressing the coating of said tablet at a pressure chosen to
result in the release of the corticosteroid at said predetermined position;
and
testing the in vitro release characteristics in a dissolution
apparatus in order to confirm release of the active ingredient at a specific
lag
time.
In another aspect, the present invention relates to a method for the
treatment of a local bowel disorder in the lower sections of the intestine,
which comprises administering to a patient in need thereof a coated tablet
having a core of a corticosteroid active ingredient and a coating, the coating
being compressed to a degree that results in the release of the
corticosteroid in the lower sections of the intestine.
Brief description of the drawings
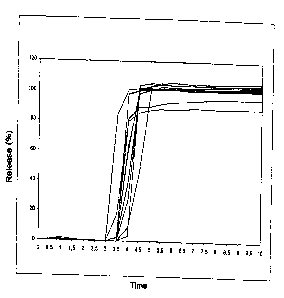
Figure 1 shows the in vitro release of the novel tablet containing 5 mg of
prednisone ("Prednisone TR") with a lag phase of about 4 h (500 ml of
water, paddle, USP)
Figure 2 shows the in vivo plasma level of prednisone after administration of
A) standard "Prednisone IR" (=Immediate release) tablet (intake 2 am) with
5 mg of prednisone,
B) Novel "Prednisone TR"tablet, "semi-fasted" (intake 8 pm) with 5 mg of
prednisone
C) Novel "Prednisone TR" tablet, fed-state (intake 8 pm) with 5 mg of
prednisone.
Figure 3 shows the in vivo plasma level of prednisolone after administration
of
A) standard "Prednisone IR" tablet (intake 2 am) with 5 mg of prednisone,
B) Novel "Prednisone TR" tablet, "semi-fasted" (intake 8 pm) with 5 mg of
prednisone
C) Novel "Prednisone TR" tablet, fed-state (intake 8 pm) with 5 mg of
CA 02579732 2007-03-07
WO 2006/027266
PCT/EP2005/009726
- 7 -
prednisone.
Figure 4 shows the in vitro release of a "Prednisone TR" tablet containing
mg of prednisone with a lag phase of 6 h (500 ml of water, paddle, USP)
5
Figure 5 shows an in vivo plasma level profile after administration of
prednisone tablets.
1) "Prednisone IR"standard tablet (intake 8 am)
2) "Prednisone IR" standard tablet (intake 2 am)
3) Novel "Prednisone TR" tablet with 6 h lag phase ("semi-fasted")(intake 8
Pm)
4) Novel "Prednisone TR" tablet with 6 h lag phase (fed state) (intake 8 pm)
Detailed description of the invention
It is possible for the site- and time-linked gastrointestinal release of
active
ingredient to differentiate two preferred embodiments:
1) Release in the upper sections of the intestine with the following
aims:
- avoidance of instabilities of the active ingredient in contact with
gastric juice,
- avoidance of side effects, such as ulcers, on release of the active
ingredient in the stomach,
- optimal site and timing of absorption of the active ingredient and its
entry into the plasma after release of the active ingredient in the
upper section of the small intestinal region,
- achievement of the systemic effect at the ideal time,
- display of a local effect in the upper sections of the intestine.
2) Release in the lower sections of the intestine with the following aims:
- local and targeted gastrointestinal release of active ingredients,
- avoidance of side effects by active ingredients after (unwanted)
absorption has taken place.
CA 02579732 2007-03-07
WO 2006/027266
PCT/EP2005/009726
- 8 -
It is common to both embodiments to increase markedly the medicament
efficacy and to reduce the side effects thereof.
A first preferred embodiment therefore provides a pharmaceutical dosage
form with a release of active ingredient in the upper sections of the
intestine
within a period of 2-6 hours. A second preferred embodiment provides a
pharmaceutical dosage form with a site- and time-controlled release of
active ingredient in the lower sections of the intestine within a period of
6-10 hours after intake.
The invention described herein relates to a novel timed-release ("TR")
dosage form which releases the active ingredient or the combination of
active ingredients, depending on the composition, the geometry and the
production conditions, at a particular site and/or at a particular time, and
thus makes it possible to ensure an optimal effect with reduced side effects.
Thus, experiments have already been carried out with prednisone as model
substance ("Prednisone TR") and can, because of the comparable
properties, also be applied to other active ingredients, e.g. corticosteroids.
The novel "TR" dosage form described herein differs from prior art
preparations. It surprisingly shows with a specific geometry of the press
coating with inert adjuvants and accurately adjusted production process
parameters a reproducible lag phase and subsequent rapid release (drug
release phase) of the active ingredient or the active ingredient combination.
The inert coating initially prevents release of the active ingredient or the
active ingredient combination over an exactly defined period, so that no
absorption can occur. The water present in the gastrointestinal tract
penetrates slowly in through the coating and, after a time which is previously
fixed by the pressure for compression, reaches the core. The coating
ingredients show neither swelling nor erosion of parts of the coating. When
CA 02579732 2012-08-08
- 9 -
the core is reached, the water penetrating in is very rapidly absorbed by the
hydrophilic ingredients of the core, so that the volume of the core increases
greatly and, as a consequence thereof, the coating completely bursts open,
and the active ingredient and the active ingredient combination respectively
is released very rapidly.
A particularly advantageous embodiment of this press-coated "TR" tablet is
achieved when a previously compressed core tablet is subsequently
compressed with a multilayer tablet press to a press-coated tablet.
=
The tablet coating typically consists of the following materials in order to
achieve a delayed release profile:
- polymer or copolymer of acrylic acid, methamlic acid etc. (e.g.
EudragitsTM or CarbopolTm),
- cellulose derivatives such as hydroxypropylmethylcellulose,
hydroxypropylcellulose, carboxymethylcellulose, ethylcellulose,
cellulose acetate,
- polyvinyl alcohol,
- polyethylene glycol,
- salts of higher fatty acids, esters of monohydric or polyhydric
alcohols with short-, medium- or long-chain, saturated or
unsaturated fatty acids. Specifically, stearic acid triglycerides (e.g.
DynersanTM) or glycerol behenate (e.g. CompritolTM) are used.
In addition, further adjuvants should also be added to these materials so that
the tablet coating can be compressed. Typically used here are fillers such as
lactose, various starches, celluloses and calcium hydrogen phosphate. The
glidant used is normally magnesium stearate, and in exceptional cases also
talc and glycerol behenate. A plasticizer is often also added to the coating
material, preferably from the group of polyethylene glycol, dibutyl phthalate,
Diethyl citrate or triacetin.
ln order to achieve an optimal release profile, the tablet core must also
fulfil
CA 02579732 2007-03-07
WO 2006/027266
PCT/EP2005/009726
- 10 -
certain tasks and exhibit certain properties. Thus, after the lag phase has
elapsed, a rapid release profile is achieved if typical disinteg rants are
added
to the inner core, which are derived for example from the group of the
following substances: cellulose derivatives, starch derivatives, crosslinked
polyvinylpyrrolidone. The use of a blowing agent, for example resulting from
a combination of a weak acid and a carbonate or bicarbonate, may also
promote rapid release. The tablet core typically consists additionally of
matrix or filling ingredients (e.g. lactose, cellulose derivatives, calcium
hydrogen phosphate or other substances known from the literature) and
lubricant or glidant (usually magnesium stearate, in exceptional cases also
talc and glycerol behenate).
The size of the core tablet preferably should not exceed 6 mm (preferably
5 mm) in diameter, because otherwise the press-coated tablet becomes too
large for convenient ingestion. As a result thereof, the dosages of the active
ingredients are in the range from 0.1 to 50 mg, very particularly between 1
and 20 mg.
The in vitro release profile of the "TR" dosage form according to the
invention is preferably such that less than 5% of the active ingredient is
released during the lag phase. After the release phase has started,
preferably N30%, particularly preferably 90%, of the active ingredient is
released within one hour. The in vitro release is preferably determined using
the USP paddle dissolution model in water.
The employed active ingredients are preferably derived from the group of
glucocorticoids and all show comparable physicochemical properties. Such
include cortisone, hydrocortisone, prednisone,
prednisolone,
methylprednisolone, budesonide, dexamethasone, fludrocortisone,
fiuocortolone, cloprednole, deflazacort, triamcinolone, and the corresponding
salts and esters thereof. This
applies in particular to prednisone,
prednisolone, methylprednisolone, budesonide,
dexamethasone,
fluocortolone, cloprednole, and defiazacort and the corresponding salts and
CA 02579732 2007-03-07
WO 2006/027266
PCT/EP2005/009726
-11 -
esters thereof.
In the present case of the "TR" tablet, the following combination of core
materials and coating materials has proved to be particularly suitable for
achieving a time- and site-controlled release with exclusion of pH and food
influences:
The coating preferably comprises:
- hydrophobic, waxy substances with an HLB value of less than about
5, preferably around 2. Carnauba wax, paraffins, cetyl ester waxes
are preferably employed therefor. Glycerol behenate has proved to
be particularly suitable. The use of about 20-60%, in particular about
30-50%, in the coating has proved to be very advantageous;
- non-fatty, hydrophobic filling materials such as calcium phosphate
salts, e.g. dibasic calcium phosphate. The use of about 25-75% of
these filling materials, in particular of about 40-60%, in the coating
has proved to be very advantageous here;
- in addition, the tablet coating preferably also consists of binders,
e.g. polyvinylpyrrolidone (PVP), typically in concentrations of about
4-12%, specifically about 7-10%, and glidants such as magnesium
stearate, in concentrations of about 0.1-2%, in the specific case of
about 0.5-1.5%. Colloidal silicon dioxide can for example be used as
flow regulator, normally in concentrations of about 0.25-1%. In
addition, to distinguish different dosages, a colorant can be added to
the tablet coating, preferably an iron oxide pigment in
concentrations of about 0.001-1%.
The core tablet preferably comprises:
- an active ingredient or an active ingredient combination from the
group of glucocorticoids, preferably prednisone, prednisolone,
= methylprednisolone, budesonide, dexamethasone, fludrocortisone,
fluocortolone, cloprednole, deflazacort, and triamcinolone, and the
corresponding salts and esters thereof. The dosages of the active
CA 02579732 2007-03-07
WO 2006/027266
PCT/EP2005/009726
- 12 -
ingredients are in the region of about 0.1-50 mg, very especially
between about 1 and 20 mg;
- in addition, the core tablet preferably comprises a filler such as,
for
example, lactose, starch derivatives or cellulose derivatives. Lactose
is preferably employed. The filler is typically present in
concentrations of about 50-90%, specifically of about 60-80%. A
disintegrant is additionally present and is typically crosslinked PVP
or sodium carboxymethylcellulose, typically in concentrations of
about 10-20%. It is additionally possible for a binder, e.g. PVP, to be
present, typically in concentrations of about 2-10%, specifically of
about 5.5-9%, and a lubricant such as magnesium stearate, in
concentrations of about 0.1-2%, in the specific case of about
0.5-1.5%. Colloidal silicon dioxide is normally used as flow
regulator, normally in concentrations of about 0.25-1%. It is
additionally possible, for visually distinguishing the core from the
coating, to add a colorant, preferably an iron oxide pigment in
concentrations of about 0.01-1%.
The pharmaceutical dosage form according to the invention is preferably
distinguished by the in vitro release and the in vivo release (on oral intake)
of
the active ingredient not differing by more than about one hour, particularly
preferably not more than about 30 minutes. It is further preferred for the in
vitro release to be substantially independent of the pH of the release
medium or/and of additions in the release medium which simulate high-fat
and low-fat food, i.e. to vary by preferably not more than about 20%. It is
further preferred for the in vivo release to be substantially independent of
food intake, with the time to reach the maximum plasma concentration (t.)
varying by not more than about 20%. The plasma level reached on in vivo
release is preferably independent of the gastrointestinal pH and of food
intake.
On in vivo release in the upper sections of the intestine, preferably
equivalent parameters, in particular a maximum plasma level (Cm) reached
CA 02579732 2007-03-07
WO 2006/027266
PCT/EP2005/009726
- 13 -
or/and an area reached under the plasma curve (AUC), as for a rapid-
release dosage form are achieved. It is particularly preferred for a Cmax of
at
least about 70%, preferably of at least about 80%, of the Cmõ of a rapid-
release dosage form, and an AUC which does not vary by more than about
25%, to be achieved. On release in lower sections of the intestine, the in
vivo plasma levels reached are much lower, this likewise being substantially
independent of the gastrointestinal pH and of food intake. The latter
embodiment of the invention is thus particularly suitable for the treatment of
local inflammatory bowel disease such as Crohn's disease or ulcerative
colitis, where a systemic effect is not desired. The first-mentioned
embodiment, with which absorption takes place in the upper sections of the
intestine, is by contrast suitable in particular for the treatment of
inflammatory diseases of the joints, associated with pain, such as, for
example, rheumatoid arthritis, allergies and nocturnal severe asthmatic
attacks, where a systemic effect is desired.
The process for producing the tablet takes place under usual conditions of
the pharmaceutical industry. Thus, standard technologies are used in the
production of the core tablet, such as weighing, sieving, mixing, aqueous
granulation in a high-speed mixer, fluidized-bed drying of the granules,
mixing and compression. Comparable methods are employed to produce the
coating, namely weighing, sieving, mixing, aqueous granulation in a high-
speed mixer, fluidized-bed drying of the granules, mixing and compression
to press-coated tablets.
The geometry of the press-coated tablet has, in addition to the composition,
a very great importance. It can be achieved only using a tablet machine for
producing press-coated tablets; spray coatings are unsuitable.
The ratio of the thickness of the press-coating on the sides of the tablets to
the upper side or lower side is preferably about 2.2-2.6 mm (for the side
edges):about 1.2-1.6 mm for the upper side of the tablet and about
1.0-1.4 mm (for the lower side of the tablet), particularly preferably about
CA 02579732 2007-03-07
WO 2006/027266
PCT/EP2005/009726
- 14 -
2.35-2.45 mm:about 1.35-1.45 mm (upper side of the tablet) and about
1.15-1.25 mm (lower side). This geometry results in the tablet remaining
sufficiently small to avoid problems with swallowing.
Tablets with a hardness of about 60-90 N, measured as specified in the
European Pharmacopoeia 4, 2.9.8, are thus achieved.
The timed-release ("TR") of active ingredient can be controlled by setting the
compressive forces during the application of the coating to the tablet core.
Thus, the compressive forces used for release in the upper sections of the
intestine are preferably up to about 600 kg, particularly preferably about
250-600 kg, whereas the compressive forces used for release of the active
ingredient in the lower sections of the intestine are preferably above about
600 kg, particularly preferably about 600-800 kg.
The pharmaceutical dosage form is particularly preferably in the form of a
tablet, but it is also possible to produce the dosage form as capsule.
The present invention is further illustrated by the following examples.
Examples
Example 1: Formulas
Core tablet consisting of:
Corticosteroidi 1 mg
Lactose 42.80 mg
Povidone 4 mg
Carboxymethylcellulose, Na 11 mg
Iron oxide, red 0.3 mg
Magnesium stearate 0.6 mg
Silicon dioxide 0.3 mg
or
CA 02579732 2007-03-07
WO 2006/027266
PCT/EP2005/009726
- 15 -
Corticosteroidl 5 mg
Lactose 38.80 mg
Povidone 4 mg
Carboxymethylcellulose, Na 11 mg
Iron oxide, red 0.3 mg
Magnesium stearate 0.6 mg
Silicon dioxide 0.3 mg
30 or
Corticosteroidl 10 mg
Lactose I 33.80 mg
Povidone 4 mg
Carboxymethylcellulose, Na 11 mg
Iron oxide, red 0.3 mg
Magnesium stearate 0.6 mg
Silicon dioxide 0.3 mg
Coating consisting of:
Calcium phosphate 50%
Glycerol behenate 40%
Povidone 8.4%
Iron oxide, yellow 0.1%
Magnesium stearate 1.0%
Silicon dioxide 0.5%
' Corticosteroid from the group of substances including cortisone,
hydrocortisone, prednisone, prednisolone, methylprednisolone, budesonide,
dexamethasone, fludrocortisone, fluocortolone, cloprednole, deflazacort,
triamcinolone and the corresponding salts and esters thereof.
The composition of the tablets ensures that the influences of food, pH and
motility of the gastrointestinal tract have no influence, and the active
ingredient escapes very rapidly from the tablet after completion of the lag
phase.
CA 02579732 2007-03-07
WO 2006/027266
PCT/EP2005/009726
- 16 -
Example 2: Production process and in vitro release
With a fixed tablet geometry, the lag phase of active ingredient release is
determined exclusively by the variably adjustable compressive force.
Prednisone was used as active ingredient in this case.
Thus, an average pressure of 400 kg for compression leads for example to a
lag phase of 4 hours. Table 1 summarizes the lag phases as a function of
the compressive force:
Table 1: Dependence of the lag phase [h] on the average
compressive force [kg]
Compressive force [kg] lag phase [h]
300 3
340 3.5
400 3.9
460 4.5
580 5
The lag phase is determined by means of the USP paddle dissolution model
with 100 rpm in water at a temperature of 37 C. Figure 1 shows typical
release behaviour (batch 0360).
Surprisingly, the lag phases and drug release phases in hours are
comparable in different release media for this formulation, with fixed
geometry and identical compressive force. This is evident from Table 2
(batch: G360).
Table 2: Lag phases and drug release phases [h] of the novel
"Prednisone TR" tablet with the active ingredient
prednisone in different release media, in vitro dissolution
release, 500 ml, paddle, USP
CA 02579732 2007-03-07
WO 2006/027266 PCT/EP2005/009726
- 17 -
Medium Average lag phase [h] Average drug release phase [h]
Water 4.1 0.7
pH 1.2 3.6 0.8
pH 4.5 3.8 0.9
pH 6.8 4.0 0.9
FaSSIF 4.2 0.8
FeSSIF 4.1 0.9
This surprising finding is very important because the aim which it is intended
to pursue is to achieve site- and time-controlled release without the
influence
of food.
Further experiments to correlate the compressive force with the lag phase
were undertaken with respect to 1 mg and 5 mg tablets containing
prednisone as the active ingredient. The results are summarized below:
Compressive Force Average lag phase Average lag phase
1 mg tablet 5 mg tablet
150 kg 2.2 2.2
200 kg 2.4 2.7
400 kg 3.4 3.9
600 kg 4.2 5.1
800 kg 4.8 5.6
1200 kg 6.0
Surprisingly, there are some differences in the required compression forces
between TR tablets of different strengths. Therefore, testing of the in-vitro
characteristics of each batch in a dissolution apparatus is preferred to
confirm release of the active at a specific lag-time. This can easily be
monitored by a color change of the dissolution medium. The color is
released from the colored core tablets.
Example 3: In vivo release
It was surprisingly possible to confirm in vivo exactly the delay, measured in
vitro, of prednisone release.
CA 02579732 2007-03-07
WO 2006/027266
PCT/EP2005/009726
- 18 -
It was possible to show in a pharmacokinetics study that with a delay of
4 hours in active ingredient release in vitro, the delay in vivo is exactly
the
same, and there is subsequently a very rapid rise in level. The resulting
plasma levels of prednisone after administration of the novel "Prednisone
TR" tablet are depicted in Figure 2. They agree very well in terms of time
with the in vitro release profile. It was additionally found that simultaneous
intake of food evidently likewise has no influence in vivo, and comparable
plasma levels are found as in the "semi-fasted" state. This is surprising
because food normally influences the motility of the gastrointestinal tract,
the
pH, the luminal metabolism, and normally interacts with the dose form. The
Guidance for Industry "Food Effect Bioavailability and Fed Bioequivalence
Studies" of the US FDA, Department of Health of December 2002 mentions
that a difference in reaching the trnax ought to be of no clinical relevance.
It is therefore very gratifying that the lag phase for the present "Prednisone
TR" tablet in vitro is 4 hours and this is also found in vivo with and without
food. In addition, food evidently has no influence on the maximum plasma
levels (C.) reached and the areas reached under the plasma curve (AUC)
either. The time until the maximum plasma concentration (t.) is reached is
likewise independent of the intake of food.
The difference in trnax between the tablet in the semi-fasted state compared
with simultaneous food intake is a maximum of 20% and is thus clinically
insignificant.
To demonstrate the influence of food on the release of the active ingredient
from the novel "Timed-Release" dosage form, the applicant has carried out a
pharmacokinetics study on 27 subjects. Three arms were compared:
administration in the evening (8 pm) of the novel "Prednisone TR" tablet with
standardized supper (fed state), administration in the evening (8 pm) of the
novel "Prednisone TR" tablet with light supper around 17.30 h (semi-fasted),
administration at night (2 am) of a standard Prednisone Immediate Release
tablet (Decortin, Merck, Germany). The study was carried out randomized, in
CA 02579732 2007-03-07
WO 2006/027266
PCT/EP2005/009726
- 19 -
cross-over design as single dose administration and thus complies with the
usual regulatory requirements.
The aim of the kinetics study was to achieve comparable plasma level
profiles in relation to Cma, and AUG for the novel tablet "Prednisone TR"
"semi-fasted" compared with "fed state" in relation to a standard "Prednisone
IR" tablet (with rapid release of active ingredient). The novel tablet
described
herein with the active ingredient prednisone showed that comparable
plasma level profiles can be achieved.
The plasma samples were taken at intervals of 0.5 and later of 1 hour.
The prednisone plasma levels found are depicted graphically in Figure 2,
and the principal pharmacokinetic characteristics are summarized in
Table 3.
Table 3: Pharmacokinetic parameters for prednisone after
administration of a single dose of 5 mg of prednisone as
"Prednisone IR" or "Prednisone TR" tablet in 27 healthy
male volunteers
Parameter Prednisone IR Prednisone Predisone
at 2 am TR;semi-fasted TR; fed state at p*
at 8 pm 8 pm
Cmax 20.9 20.3 22.0 0.54
(ng/mL) (19.2-22.7) (18.6-22.1) (20.1-23.9)
tmaõ (h) 2 (1.0-4.0) 6.0 (4.5-10.0) 6.5 (4.5-9.0) <0.0001
tag (h) 0.0 3.5 (2.0-5.5) 4.0 (3.5-5.0) <0.0001
(0.0-0.5)
AUCat 107 108 121 0.16
(h.ng/mL) (98.8-116) (99.1-117) (111-132)
AUC0.... 109 110 123 0.15
(h.ng/mL) (101-118) (102-119) (114-134)
t1,2 (h) 2.57 2.41 2.41 0.002
(2.51-2.63) (2.36-2.47) (2.36-2.47)
CA 02579732 2007-03-07
WO 2006/027266
PCT/EP2005/009726
- 20 -
tam and tag values are means (range). The other values are geometric
means (90% CI) obtained from ANOVA.
*. probability associated with the hypothesis, that there is no difference
between the formulations (ANOVA, except for traa, and tag: Friedman test)
It was also possible to confirm these results for prednisolone, a metabolite
of
prednisone, after administration of the novel "Prednisone TR" tablet.
Thus, it was also possible to show for prednisolone a comparability between
io Cam, and AUC of the novel "Prednisone TR" tablet "semi-fasted" with "fed
state". The plasma level profile of the metabolite prednisolone is therefore
also independent of food intake.
The plasma samples for determining prednisolone were taken at intervals of
0.5 and later of 1 hour.
The plasma levels found for prednisolone are depicted graphically in
Figure 3, and the principal pharmacokinetic characteristics are summarized
in Table 4.
Table 4: Pharmacokinetic parameters for prednisolone after
administration of a single dose of 5 mg of prednisone as
"Prednisone IR" or "Prednisone TR" tablet in 27 healthy
male volunteers
Parameter Prednisone IR Prednisone Prednisone TR;
at 2 am TR; semi-fased at fed state at 8 pm p*
8 pm
Cmax 135 113 132 0.036
(ng/mL) (124-147) (104-123) (121-143)
tm. (h) 1.0 5.0 (4.0-9.0) 5.5 (4.5-9.0) <0.0001
(0.5-3.0)
tag (h) 0.0 3.5 (2.0-5.5) 3.5 (3.0-5.0) <0.0001
(0.0-0.5)
CA 02579732 2007-03-07
WO 2006/027266 PCT/EP2005/009726
- 21 -
AUC04 614 561 647 0.081
(h.ng/mL) (571-661) (520-605) _ (599-698)
AUC0¨ 624 573 658 0.0076
(h.ng/mL) (582-670) (533-616) _ (612-707)
t112 (h) 2.66 2.66 2.71 0.11
(2.63-2.70) (2.62-2.69) _ (2.68-2.75)
tmax and tag values are means (range). The other values are geometric
means (90% Cl) obtained from ANOVA.
*: probability associated with the hypothesis, that there is no difference
between the formulations (ANOVA, except for trau and tag Friedman test)
Typical achieved Cmax values for 5 mg prednisone tablets after ingestion will
be in the range of from about 15 to about 25 ng/ml, and the AUC of
prednisone is from about 75 to about 150 h*ng/mL. The achieved C.
values for the prednisolone metabolite will be in the range of from about 100
to about 160 ng/ml, and the AUC of prednisolone is from about 500 to about
700 h*ng/mL.
It should additionally be mentioned that the coefficients of variation for
Cmõ,
tmõ and AUC for prednisone after administration of the standard tablet
"Prednisone IR" (at 2 am) and of the novel tablet "Prednisone TR" (at 8 pm)
with and without food are approximately comparable. This has not previously
been described for a tablet with modified release of active ingredient.
Table 5 summarizes the coefficients of variation for prednisone.
Table 5: Coefficients of variation for C., t., AUC for prednisone
plasma levels after administration of a standard tablet
"Prednisone IR", of the novel tablet "Prednisone TR"
"semi-fasted" and in "fed state"
Prednisone IR Prednisone prednisone
at 2 am TR; TR;
semi-fasted at fed state at
8 pm 8 pm
26 26 26
CA 02579732 2007-03-07
WO 2006/027266
PCT/EP2005/009726
- 22 -
,
CMaX Average _ 21.1 21.4 22.2 _
SD 3.56 5.65 3.66
(ng/ml) - 22.2 _
Median 20.8 21.4 ,
_ CV 16.9 26.4 16.4
tr.( (h) Average 2.06 6.21 6.5
-
SD 0.68 1.22 I 1.11
Median 2 6 6.5
CV 33.1 , 19.6 17.1
AUC,,..... Average 111 116 126
SD 17.5 31 , 24.3
(ngImrh) Median 106 122 130
_
CV (%) _ 15.8 26.6 19.2
50 The coefficients of variation of the pharnnacokinetic parameters for
the
metabolite prednisolone likewise differ negligibly when the standard tablet is
compared with the novel tablet.
Table 6: Coefficients of variation for C., t,õ.., AUC for prednisolone
plasma levels after administration of a standard tablet
55 "Prednisone IR", of the novel tablet "Prednisone TR"
"semi-fasted" and in fed state
_
Prednisone Prednisone
Prednisone TR semi-
TR; fed state
IR at 2 am fasted at 8 at 8 pm
Pm _
N 26 26 26
Cmax Average 138 121 -135_
SD 22.9 32.3 24.5
(ng/1111) Median 140 130 135
CV 16.6 26.8 18.2
-
tm" (h) Average 1.12 5.58 5.81
SD 0.67 1.2 1.16
Median 1 4 _5.5
CV 59.3 21.5 19.9
AUCo.... Average 638 611 _680
SD 112 178 142
(ng/ml*h)
Median 646 677 _713
CV (%) 17.7 29.2 20.9
The situation is quite different when a tablet with a longer lag phase (6
hours
CA 02579732 2007-03-07
WO 2006/027266
PCT/EP2005/009726
-23 -
in vitro) is administered. It is true that release after 6 hours is in this
case
also found in vitro. However, at the same time, the absorption is greatly
reduced because the release obviously takes place in lower sections of the
intestine, and absorption now takes place only to a smaller extent. This was
shown in a second pharmacokinetics study. Figure 5 shows a novel
"Prednisone TR" tablet with a lag phase of 6 hours, which can be produced
by means of a higher pressure for compression.
Typical achieved Cmax values for such 5 mg prednisone tablets after
ingestion will be in the range of less than 15 ng/ml, and the AUC of
prednisone is less than 75 h*ng/mL. The achieved Cmax values for the
prednisolone metabolite will be in the range of less than 100 ng/ml, and the
AUC of prednisolone is less than 500 h*ng/mL.
Very interesting novel therapeutic approaches derive therefrom, and this
invention relates thereto. Thus, the composition of the tablet, its specific
geometry and a compressive force which can be adjusted variably make it
possible for the coating of the tablet to release the active ingredient very
rapidly from the core tablet after an exactly fixed time. This is very
advantageous because the site of release can also be fixed accurately via
this precise presetting.
It is possible with a site of release on the one hand to treat local disorders
of
the gastrointestinal tract locally. For example, ulcerative colitis, an
inflammatory disorder of the bowel, may affect different sections of the
intestine. This novel timed-release ("TR") tablet is very advantageous
especially for disorders of lower sections of the intestine, because there is
now mainly local release of the active ingredient, but absorption thereof is
only negligible or very limited. It is possible thereby to avoid effects which
normally occur after uptake of the active ingredient into the blood.
However, with a precise controlled active ingredient release after an exactly
defined lag phase it is also possible to achieve the plasma level profiles
CA 02579732 2007-03-07
WO 2006/027266
PCT/EP2005/009726
-24 -
which correspond to those after administration of a rapid-release tablet.
However, the precondition for this is that the coating of the novel timed-
release ("TR") tablet exposes the active ingredient-containing core after less
than 6 hours, and the active ingredient can then be very rapidly dissolved
and absorbed. One application thereof is, for example, the administration of
corticoids for the treatment of inflammatory disorders to the joints, where
pro-
inflammatory cytokines are released in the early hours of the morning and
are thought to be responsible for the pain in the morning and the stiffness of
fingers in the morning. It is now possible through the novel tablet, with
intake
io at 10 pm, to enable release at 2 am, and thus to ensure the optimal
effect,
described by Arvidson et al. (Annals of Rheumatic Diseases 1997; 56:27-31)
of the cortisones on rheumatoid arthritis and, in addition, to contribute to a
crucial increase in patient compliance. Consequently, the tablets of the
present invention may be ingested once daily at bed-time, for example
between about 8 pm and about 12 am, and more preferably between about
9 pm and about 11 pm.
The present invention also provides a method for producing a tablet that
releases a corticosteroid active ingredient at a predetermined variable
location in the GI tract, said method comprising:
determining the location in the GI tract at which it is desired to
deliver the corticosteroid;
forming a coated tablet having a core comprising the
corticosteroid and a swellable adjuvant, and an inert outer coating;
compressing the coating of said tablet at a pressure chosen to
result in the release of the corticosteroid at said predetermined position;
and -
testing the in vitro release characteristics in a dissolution apparatus in
order to confirm release of the active ingredient at a specific lag time. The
in
vitro release characteristics can then be correlated to the suitable in vivo
release lag time.
In a preferred embodiment, the tablet core comprises a coloring material,
and the in vitro release of the active ingredient is determined by a color
change. The dissolution apparatus may be any standard apparatus in the
CA 02579732 2007-03-07
WO 2006/027266
PCT/EP2005/009726
- 25 -
industry, and preferably is in accordance with USP