Note: Descriptions are shown in the official language in which they were submitted.
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
PHARMACEUTICAL COMPOSITIONS COMPRISING FENOFIBRATE AND
ATO RVASTATI N
Technical field
[0001] The present invention relates to compositions, particularly,
pharmaceutical
compositions in particulate form such as granulate or in solid dosage forms
comprising a combination of a fibrate and atorvastatin (also known as an HMG
CoA reductase inhibitor). More specifically, the invention relates to a solid
pharmaceutical composition comprising atorvastatin and a low dose, i.e. a
reduced
amount, of fenofibrate having improved bioavailability and/or improved
pharmacological response, i.e. improved effect. The composition may be in the
form of an immediate release formulation, a controlled release formulation or
a
combination thereof. The invention also relates to methods for making the
compositions in particulate form, i.e. as particles, and in solid dosage
forms.
Background art
[0002] Fibrates are drug substances that generally are poorly and variably
absorbed after oral administration. Normally they are prescribed to be taken
with
food in order to increase the bioavailability. There has been a number of
improvements in dosage form of the currently most used fibrate, fenofibrate,
in an
effort to increase the bioavailability of the drug and hence its efficacy.
Furthermore, clinical guidelines indicate that not only fibrate therapy but
also a
combination therapy with e.g. fenofibrate and a statin should be the most
effective
means of cholesterol and lipid management. In fact, treatment with fenofibrate
is
often prescribed together with a statin as clinicians seem to prefer the use
of e.g.
fenofibrate due to its triglyceride-lowering and HDL-C increasing effects
while a
statin is used for its positive effects on lowering LDL-C and raising HDL-C.
However, at present, such a combination therapy can only be achieved by the
use
of two separate products, i.e. the patient needs to take e.g. one fenofibrate
tablet
together with another tablet or capsule containing a statin.
[0003] Fenofibrate is chemically named 2-[4-(4-chlorobenzoyl]-2-methyl-
propanoic
acid, 1-methylethyl ester and has the following structural formula:
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
2
[0004]
c~+
J. [0005] Fenofibrate is a white solid. The compound is insoluble in water.
The
melting point is 79-82 C. Fenofibrate is metabolised to the active substance
fenofibric acid. Fenofibric acid has an elimination half-life of about 20
hours.
Measurement of the detected amount of fenofibric acid in the blood of a
patient
can reflect the efficacy of fenofibrate uptake. Fenofibric acid produces
reductions
in total cholesterol (total-C), LDL-C, apo-lipoprotein B, total triglycerides,
and
triglyceride rich lipoprotein (VLDL) in treated patients. In addition,
treatment with
fenofibrate results in increases in high density lipoprotein (HDL) and apo-
lipoprotein apoAl and apo All. Fenofibrate acts as a potent lipid regulating
agent
offering unique and clinical advantages over existing products in the fibrate
family
of drug substances. Fenofibrate produces substantial reduction in plasma
triglyceride levels in hypertriglyceridemic patients and in plasma cholesterol
and
LDL-C in hypercholesterolemic and mixed dyslipidemic patients.
[0006] Fenofibrate also reduces serum uric acid levels in hyperuricemic and
normal subjects by increasing the urinary excretion of uric acid.
[0007] Clinical studies have demonstrated that elevated levels of total
cholesterol,
low density lipoprotein cholesterol (LDL-C), and apo-lipoprotein B (apo B) are
associated with human atherosclerosis. Decreased levels of high density
lipoprotein cholesterol (HDL-C) and its transport complex, apolipoprotein A
(apo Al
and apo AII) are associated with the development of atherosclerosis.
[0008] Fenofibrate is also effective in the treatment of Diabetes Type II and
metabolic syndrome.
[0009] Further, the lipid improvements seen with fenofibrate therapy are
associated with reduced progression to microalbuminuria in patients with
Diabetes
Type II. A recent study shows that fenofibrate treatment for at least 3 years
is
effective in reducing the progression of renal disease in patients with
Diabetes
Type II without diabetic nephropathy (Am. J. Kidney Dis. 2005, vol. 45, p. 485-
493).
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
3
[0010] Fenofibrate is also indicated as adjunctive therapy to diet for
treatment of
adult patients with hypertriglyceridemia (Fredrickson Types IV and V
hyperlipedemia). Improving glycemic control in diabetic patients showing
fasting
chylomicronemia will usually reduce fasting triglycerides and eliminate
chylomicronemia and thereby obviating the need for pharmacologic intervention.
[0011] Fibrates are drug substances known to be are poorly and variably
absorbed
after oral administration. Normally they are prescribed to be taken with food
in
order to increase the bioavailability.
[0012] In general, it is known that the absorption and bioavailability of a
therapeutically active substance can be affected by a variety of factors when
administered orally. Such factors include the presence of food in the
gastrointestinal tract and, in general, the gastric residence time of a drug
substance is significantly longer in the presence of food than in the fasted
state. If
the bioavailability of a drug substance is affected beyond a certain point due
to the
presence of food in the gastrointestinal tract, the drug substance is said to
exhibit
a food effect. Food effects are important because there is a risk associated
with
administering the drug substance to a patient who has eaten recently. The risk
derives from the potential that absorption into the bloodstream may be
adversely
affected to the point that the patient risks insufficient absorption to remedy
the
condition for which the drug was administered. In the case of e.g. fenofibrate
the
situation is different in that food increases the uptake. Thus, lack of intake
of food
simultaneously with the drug substances may lead to insufficient absorption.
The
extent of absorption of a commercially available product Tricor (Lipanthyl )
containing fenofibrate (from Abbott Laboratories, IL, U.S.A.) is increased by
approximately 35% under fed as compared to fasting conditions.
[0013] Examples of commercially available fenofibrate drug products are: From
Abbott Laboratories: TriCor tablets 160 mg, 145 mg, 54 mg, 48 mg, Lipanthyl
capsules; from Reliant Pharmaceuticals Inc., NJ, U.S.A.: Antara capsules 130
mg, 43 mg. The fenofibrate present in these commercial products is in
micronized
form, i.e. crystalline fenofibrate in the form of fenofibrate particles as
such,
prepared by subjecting crystalline fenofibrate to a mechanical milling in
order to
reduce the particle size.
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
4
[0014] WO 04/041250 relates to nanoparticulate compositions of fenofibrate,
i.e.
fenofibrate particles having an effective average particle size of less than
about
2000 nm.
[0015] Atorvastatin is a synthetic reversible inhibitor of the microsomal
enzyme
HMG-CoA reductase. Atorvastatin is usually administered orally as the calcium
salt of the active hydroxy acid in a dosage range of 10-80 mg/day.
Atorvastatin
acid is converted to its lactone in vivo in humans, and these two forms appear
to
have approximately the same AUC.
[0016] Atorvastatin calcium is [R-(R*, R*)]-2-(4-fluorophenyl)-f3, 6-dihydroxy-
5-
[0017] (1 -methylethyl)-3-phenyl-4-[(phenylamino)carbonyl]-1 H-pyrrole-1 -
heptanoic
acid, calcium salt (2:1) trihydrate. The empirical formula of atorvastatin
calcium is
(C33H34FN205)2Ca3H2O and its molecular weight is 1209.42. The molecular
weight of atorvastatin as such is 546, pKa 4.46.
[0018] Atorvastatin calcium is a white to off-white crystalline or amorphous
powder
that is insoluble in aqueous solutions of pH 4 and below. Atorvastatin calcium
is
very slightly soluble in distilled water, pH 7.4 phosphate buffer, and
acetonitrile,
slightly soluble in ethanol, and freely soluble in methanol. The solubility of
the
calcium salt of atorvastatin is 1.23 mg/mL at pH 6.0; accordingly, it is
believed that
solubility in the intestinal lumen is not a limiting factor in vivo. The
calcium salt has
the following structural formula:
[0019]
OM OH D
1 t II
~ ~~PJF [;~~~ ~{ H ~ -IC:H,_ rC;H~ rL'~ C-~
~~ PJ c.H_, H c.~H. ~
= :?HatiJ
[0020] There exists a vast number of crystalline forms of atorvastatin calcium
(Forms I-XIV).
[0021] Atorvastatin magnesium is a crystalline or amorphous powder.
[0022] LipitorTM tablets (from Pfizer Inc.) for oral administration contain
10, 20, 40
or 80 mg atorvastatin and the following inactive ingredients: calcium
carbonate,
USP; candelilla wax, FCC; croscarmellose sodium, NF; hydroxypropyl cellulose,
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
NF; lactose monohydrate, NF; magnesium stearate, NF; microcrystalline
cellulose,
NF; Opadry White YS-1-7040 (hydroxypropyl-methylcellulose, polyethylene
glycol,
talc, titanium dioxide); polysorbate 80, NF; simethicone emulsion.
[0023] Atorvastatin is a synthetic lipid-lowering agent. Atorvastatin is a
selective,
competitive inhibitor of 3-hydroxy-3-methylglutaryl-coenzyme A(HMG-CoA)
reductase. This enzyme catalyzes the conversion of HMG-CoA to mevalonate, a
precursor of sterols, including cholesterol. Cholesterol and triglycerides
circulate in
the bloodstream as part of lipoprotein complexes. With ultracentrifugation,
these
complexes separate into HDL (high-density Iipoprotein), IDL (intermediate-
density
lipoprotein), LDL (low-density Iipoprotein), and VLDL (very-low-density
Iipoprotein)
fractions. Triglycerides (TG) and cholesterol in the liver are incorporated
into VLDL
and released into the plasma for delivery to peripheral tissues. LDL is formed
from
VLDL and is catabolized primarily through the high-affinity LDL receptor.
Clinical
and pathologic studies show that elevated plasma levels of total cholesterol
(total-
C), LDL-cholesterol (LDL-C), and apolipoprotein B (apo B) promote human
atherosclerosis and are risk factors for developing cardiovascular disease,
while
increased levels of HDL-C are associated with a decreased cardiovascular risk
[0024] Atorvastatin acid is highly soluble and permeable, and the drug is
completely and rapidly absorbed after oral administration; maximum plasma
concentrations occur within 1 to 2 hours. Extent of absorption increases in
proportion to the atorvastatin dose (for a dose below 40 mg). However,
atorvastatin acid is subject to extensive first-pass metabolism in the gut
wall and in
the liver, and the absolute bioavailability of atorvastatin acid (parent drug)
is
approximately 14% and the systemic availability of HMG-CoA reductase
inhibitory
activity is approximately 30%. The low systemic availability is attributed to
pre-
systemic clearance in gastrointestinal mucosa and/or hepatic first-pass
metabolism (oxidation, lactonization and glucuronidation; the metabolites are
eliminated by biliary secretion and direct secretion from blood to the
intestine).
Although food decreases the rate and extent of drug absorption by
approximately
25% and 9%, respectively, as assessed by Cmax and AUC, LDL-C reduction is
said to be similar whether atorvastatin is given with or without food. Plasma
atorvastatin concentrations are lower (approximately 30% for Cmax and AUC)
following evening drug administration compared with morning. However, LDL-C
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
6
reduction is said to be the same regardless of the time of day of drug
administration
[0025] WO 03013608 describes semi-solid pharmaceutical compositions
containing a fibrate and a statin prepared by melting the inactive substances,
adding the active substances and filling the melt into pharmaceutically
acceptable
capsules.
[0026] WO 03103640 discloses nanoparticulate compositions comprising statin
particles having an effective average particle size of less than about 2000
nm,
optionally in combination with other cholesterol lowering agents.
[0027] US-A-2003-0162827 discloses an atorvastatin composition with improved
bioavailability allegedly due to the presence of atorvastatin in a rapidly
dissolvable
and more solubilized state.
[0028] There is a need for developing a pharmaceutical composition that in a
single formulation, preferably in a single solid dosage form, contains a
fibrate and
atorvastatin as active substances, which composition is stable and provides
suitable and desirable biopharmaceutical properties of the active substances
(e.g.
for each of the active substances a suitable bioavailability, a suitable
pharmacological response, less dependency on food intake etc), and which
composition can be easily manufactured in large scale. Furthermore, there is a
need for developing formulations containing a fibrate and a statin, which
formulations can be further processed into pharmaceutical dosage forms with a
high degree of flexibility of choosing the particular kind of dosage form.
Within the
pharmaceutical field such flexibility can be obtained when the formulation is
in the
form of a solid product such as powder or particles.
[0029] Also, there is still a need for a composition that has a suitable or
even
improved bioavailability, that can substantially reduce or overcome the
differential
between the bioavailability of the drug in patients who are fasted versus the
bioavailability of the drug (in particular relevant for fenofibrate) in
patients who are
fed, and/or than can substantially reduce or overcome the intra- and/or inter-
individual variations observed with the current treatment. Furthermore, there
is
also a need for a composition that enables reduction in observed side effects
and
minimizes any possible drug-drug interactions.
Disclosure of the invention
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
7
Summary of the invention
[0030] The inventors have now successfully formulated a solid pharmaceutical
composition in particulate form comprising a combination of two active
substances, namely fenofibrate and atorvastatin or a pharmaceutically active
salt
thereof such as the calcium salt or the magnesium salt, where the fenofibrate
is at
least bioequivalent to the commercially available drug at present containing
the
lowest dose of fenofibrate (that is 130 mg, full dose).
[0031] The inventors have found that the bioavailability of the combination
drug
can be significantly enhanced by dissolving the active substance fenofibrate
in a
suitable vehicle and using the resulting composition for preparing a solid
dosage
form, i.e. a dosage form excluding material in liquid form. Fenofibrate is
known to
be insoluble in water and the present invention includes pharmaceutical
composi-
tions and formulations exhibiting release profiles which have significantly
increased in vivo bioavailability in patients in need thereof, especially
eliminating
the food effect of fenofibrate known from commercially available fenofibrate
tablets
(Tricor/Lipanthyl tablets or other drug products containing micronized
fenofibrate).
Especially, the inventors have succeeded in preparing a solid dosage form,
such
as a tablet, which ensures suitable bioavailability of the active substances
upon
oral administration. The advantages of a solid and stable dosage form useful
for
oral administration are well-known.
[0032] Further, the inventors have found that it is possible to obtain the
desired
pharmacological response in vivo (a reduction of the LDL-cholesterol level)
and at
the same time maintain the maximum obtainable increase in HDL-cholesterol by
administering a fenofibrate-atorvastatin combination composition comprising a
controlled release formulation of atorvastatin, preferably a delayed release
formulation, even a formulation with a reduced amount of atorvastatin is
contemplated, including a time-controlled coating or an enzyme controlled
coating
or a pressure controlled coating.
[0033] The compositions, i.e. the particulate composition and the solid dosage
forms, are manufactured without any need of addition of water or an aqueous
medium. As a result, the compositions of the invention have a very low content
of
moisture, i.e. less than about 2.5% w/w water, or less than about 2% w/w
water, or
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
8
less than about 1% w/w water are obtained, thereby ensuring suitable storage
stability, since both fenofibrate as well as atorvastatin is degradable by
water.
[0034] The solid pharmaceutical compositions in the form of particles and
solid
dosage forms of the present invention are useful for treatment of conditions
that
respond to fibrate and atorvastatin treatment, including hypercholesterolemia
and
hyperlipidemia.
[0035] Accordingly, in a first aspect the present invention provides a solid
pharmaceutical composition in particulate form, which composition comprises a
vehicle, an effective amount of atorvastatin or a pharmaceutically acceptable
salt
thereof, and an effective amount of fenofibrate exhibiting a bioavailability
which is
at least bioequivalent to a 130 mg Antara capsule.
[0036] Thus, the composition of the invention provides a combination drug
product
with a low dose of fenofibrate, i.e. a reduced amount of this active
substance,
while at the same time providing a pharmaceutical composition being
bioequivalent to commercially available fenofibrate-containing medicaments or,
alternatively, being even more efficient by exhibiting an increased
bioavailability
such as an AUCo-24 value for fenofibrate relative to the AUC 0-24 value for a
130 mg
Antara tablet of at least about 1.3.
[0037] In a preferred embodiment of the invention, the amount of fenofibrate
is less
than 130 mg. That is a low dosage, i.e. a reduced amount, as compared to the
commercially available medicaments providing various dosage forms typically
containing 160 mg, 145 mg or 130 mg of fenofibrate, usually micronized
fenofibrate.
[0038] In another preferred embodiment, the composition of the invention
comprises about 120 mg of fenofibrate. It is contemplated that the minimum
effective amount of fenofibrate is about 30 mg. The amount of atorvastatin in
the
composition may vary from about 5 mg to about 80 mg. Conventionally the amount
of fenofibrate present in the combination composition is higher that the
amount of
atorvastatin. However, effective co-formulations comprising a higher amount of
atorvastatin than of fenofibrate is contemplated. The relative amount of
atorvastatin to fenofibrate is at least 1:15.
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
9
[0039] Especially, essentially all of the fenofibrate present in the
composition is
dissolved in a suitable vehicle, which may be hydrophobic, hydrophilic or
water-
miscible.
[0040] In a second aspect, the invention relates to a solid oral dosage form
comprising the pharmaceutical composition. Useful solid dosage forms are in
the
form of tablets, beads, capsules, grains, pills, granulate, granules, powder,
pellets,
sachets or troches.
[0041] In a third aspect, the invention relates to a solid oral dosage form
comprising an immediate release formulation of fibrate, preferably
fenofibrate, and
a controlled release formulation of atorvastatin. In a preferred embodiment,
the
solid dosage form may be tablets prepared by compressing a mixture of fibrate
granulate and entero-coated atorvastatin granulate. In another preferred
embodiment, the solid dosage form may be fibrate granulate, fibrate granules,
fibrate grains, fibrate beads and/or fibrate pellets filled into capsules or
sachets
together with entero-coated atorvastatin granules, atorvastatin grains,
atorvastatin
beads and/or atorvastatin pellets.
[0042] In yet another aspect, the invention relates to a method of
manufacturing
the pharmaceutical compositions and the solid oral dosage forms of the
invention.
[0043] In further aspects, the invention relates to a method of treating
hyperlipid-
emia or hypercholesterolemia comprising administering to a human in need of
such treatment the pharmaceutical composition of this invention, and to use of
the
pharmaceutical composition or a solid dosage form of this invention for
manufac-
turing a medicament for treatment of hyperlipidemia or hypercholesterolemia in
mammals.
[0044] The pharmaceutical composition of the invention is advantageous by
being
in the form of particles, for example granulate, which can easily be further
processed into solid dosage forms, especially tablets or filled into capsules.
That
is, the pharmaceutical composition of the invention exhibits suitable
properties
such as for example being free-flowing, non-adherent and compressible.
[0045] Further aspects of the invention are evident from the following
description.
[0046] Comparison in vivo tests in dogs have shown, cf. the examples herein,
that
solid dosage forms and compositions of the invention exhibit significantly
enhanced bioavailability of fenofibrate compared to commercially available
solid
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
dosage forms containing the same active ingredient, i.e. to Tricor (Lipanthyl
)
tablets and Lipanthyl capsules (both from Abbott Laboratories, Illinois,
U.S.A.).
[0047] Further, it is believed that the present invention provides solid
dosage forms
and compositions of fenofibrate and atorvastatin capable of significantly
reducing
the intra- and/or inter-individual variation normally observed after oral
administration. Furthermore, the compositions and dosage forms according to
the
invention provide for a significant reduced food effect, i.e. the absorption
is
relatively independent on whether the patient takes the composition or dosage
form together with or without any meal. It is contemplated that a modified
release
formulation may reduce the number of gastro-intestinal related side effects.
Furthermore, it is contemplated that in comparison with commercially available
drug products, a significantly larger amount of fenofibrate is absorbed from
the
present composition and, accordingly, an equally less amount is excreted
unchanged via faeces. Finally, it is contemplated that the reduced amount of
fenofibrate in the composition of the invention significantly reduces any
negative
effects of possible drug-drug interactions (i.e. fenofibrate-atorvastatin).
[0048] As mentioned above, the present invention fulfils the need for
pharmaceutical compositions containing a combination of fenofibrate and
atorvastatin or a pharmaceutically acceptable salt thereof for oral use that
lead to
an improved treatment of conditions requiring lipid management (e.g.,
atherosclerosis, coronary heart diseases, diabetes management, obesity,
overweight, metabolic syndrome etc).
[0049] Furthermore, it is contemplated that the invention provides improved
bioavailability, especially of the fenofibrate component, and an improved
pharmacological response (LDL-cholesterol lowering and HDL-cholesterol
increase) of atorvastatin. Fenofibrate has a very poor solubility in water,
which
property is regarded as one of the major reasons for the poor bioavailability
of
fenofibrate. Accordingly, it is advantageous to provide a composition in which
the
fenofibrate is mainly in dissolved form. Improved bioavailability results in
improved
treatment. However, it may also be possible to obtain the same therapeutic
response with a decreased dose and/or a less frequent administration and less
variability in plasma levels and no food restrictions. Another way of
obtaining an
improved treatment of conditions where fenofibrate is indicated is by
balancing the
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
11
release of fenofibrate to the gastro-intestinal tract in such a manner that an
enhanced plasma concentration of fenofibrate is obtained initially or delayed
with
respect to the time of administration, i.e. by administering modified or
delayed
release compositions containing fenofibrate.
Drawings
[0050] Figure 1 shows the mean plasma concentration data of Lipanthyl 160 mg
fed state and Lipanthyl 160 mg fasted state (0-96 hours).
[0051] Figure 2 shows the mean plasma concentration data of invention
fenofibrate formulation (LCP-feno) 160 mg fed state and invention fenofibrate
formulation (LCP-feno) 160 mg fasted state (0-96 hours).
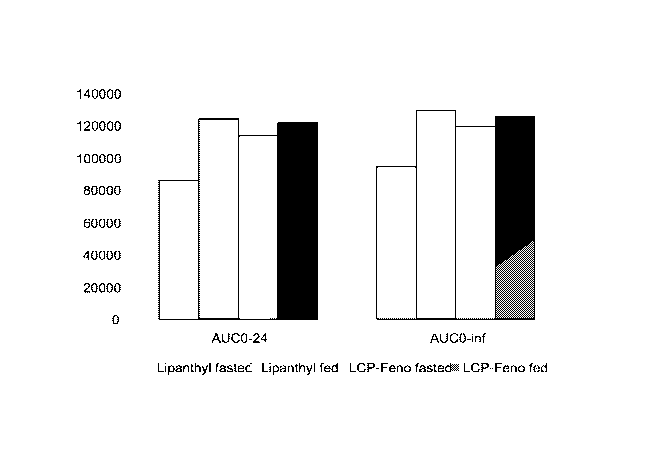
[0052] Figure 3 shows mean (average) AUCo-24 and mean (average) AUCo-inf for
each of Lipanthyl fasted state, Lipanthyl fed state, invention fenofibrate
formulation
(LCP-Feno) fasted state and invention fenofibrate formulation (LCP-Feno) fed
state.
Definitions
[0053] As used herein, the terms "active substance", "active pharmaceutical
substance", "active ingredient" and "active pharmaceutical ingredient" (API)
denote
any component that is intended to furnish pharmacological activity or other
direct
effect in the diagnosis, cure, mitigation, treatment, or prevention of
disease, or to
affect the structure or any function of the body of man or other animals. The
term
includes those components that may undergo chemical change in the manufacture
of the drug product and are present in the drug product in a modified form
intended to furnish the specified activity or effect.
[0054] In the present context, the term "hydrophilic" describes that something
'likes
water, i.e. a hydrophilic molecule or portion of a molecule is one that
typically is
electrically polarized and capable of forming hydrogen bonds with water
molecules, enabling it dissolve more readily in water than in oil or other
"non-polar"
solvents.
[0055] In the present context, the term "amphiphilic" describes a molecule (as
a
surfactant) having a polar water-soluble group attached to a water-insoluble
hydrocarbon chain. Thus, one end of the molecule is hydrophilic (polar) and
the
other is hydrophobic (non-polar).
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
12
[0056] In the present context, the term "hydrophobic" denotes a compound
tending
to be electrically neutral and non-polar, and thus preferring other neutral
and non-
polar solvents or molecular environments.
[0057] As used herein, the term "water-miscible" denotes a compound being
fully
or partly miscible with water. For example, certain polar lipids are partly
water-
miscible.
[0058] As used herein, the term "vehicle" means any solvent or carrier in a
pharmaceutical product that has no pharmacological role. For example, water is
the vehicle for xylocaine and propylene glycol is the vehicle for many
antibiotics.
[0059] In the present context, the term "solid dispersion" denotes a drug or
active
ingredient or substance dispersed on a particulate level in an inert vehicle,
carrier,
diluent or matrix in the solid state, i.e. usually a fine particulate
dispersion.
[0060] In the present context, the term "solid solution" denotes a drug or
active
ingredient or substance dissolved on a molecular level in an inert vehicle,
carrier,
diluent or matrix in the solid state.
[0061] In the present context, the term "interstitial crystalline solid
solution"
denotes a drug or active ingredient or substance dissolved on a molecular
level in
an inert vehicle, carrier, diluent or matrix in the solid state, where the
inert vehicle,
carrier, diluent or matrix forms a crystal lattice and the dissolved
fenofibrate
molecules occupy the interstitial spaces between the solvent molecules in the
crystal lattice, cf. the review article: Leuner C. and Dressman, J., European
Journal of Pharmaceutics and Biopharmaceutics 50 (2000) 47-60.
[0062] As used herein, the term "analog" means a chemical compound that is
structurally similar to another.
[0063] The term "drug" means a compound intended for use in diagnosis, cure,
mitigation, treatment, or prevention of disease in man or other animals.
[0064] In this context, the term "dosage form" means the form in which the
drug is
delivered to the patient. Examples of known dosage forms are parenteral,
topical,
oral (liquid or dissolved powder, tablet, capsule, sachet), suppository,
inhalation,
transdermal, etc.
[0065] As used herein, the term "bioavailability" denotes the degree to which
a
drug or other substance becomes available to the target tissue after
administration. In the present context, the term "suitable bioavailability" is
intended
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
13
to mean that administration of a composition according to the invention will
result
in a bioavailability that is improved compared to the bioavailability obtained
after
administration of the active substance(s) in a plain tablet; or the
bioavailability is at
least the same or improved compared to the bioavailability obtained after
administration of a commercially available product containing the same active
substance(s) in the same amounts. In particular it is desired to obtain
quicker and
larger and/or more complete uptake of the active compound, and thereby provide
for a reduction of the administered dosages or for a reduction in the number
of
daily administrations. Further, pharmaceutical compositions of the invention
may
also reduce or negate the need for the dosage form to be taken simultaneously
with intake of food (this is in particular relevant fenofibrate) thereby
allowing
patients more freedom to choose when to administer the drug.
[0066] As used herein, the term "bioequivalency" denotes a scientific basis on
which generic and brand name drugs are compared with one another. For
example, drugs are bioequivalent if they enter circulation at the same rate
when
given in similar doses under similar conditions. Parameters often used in
bioequivalence studies are tmax, cmax, AUCo-infniry, AUCo-t. Other relevant
parameters may be W50, W75 and/or MRT. Accordingly, at least one of these
parameters may be applied when determining whether bioequivalence is present.
In the present context, bioequivalency of two compositions is established by a
90% confidence interval of between 0.80 and 1.25 for AUC (either AUCo-infniry
or
AUCo-24). In addition, a 90% confidence interval of between 0.80 and about
1.40
for cmax is also required for bioequivalency. The combination composition of
the
invention, i.e. regarding the establishment of bioequivalency of the
fenofibrate
active ingredient, may be compared with standard commercial fenofibrate
formulations, for example 160 mg or 145 mg Tricor /Lipanthyl tablets or
capsules or 130 mg Antara capsules or similar, preferably 130 mg Antara
capsules.
[0067] In the present context "tmax denotes the time to reach the maximal
plasma
concentration (cmax) after administration; AUCo-infniry or AUC denotes the
area
under the plasma concentration versus time curve from time 0 to infinity; AUCo-
t
denotes the area under the plasma concentration versus time curve from time 0
to
time t, especially, AUCo-24 is the area under the plasma concentration versus
time
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
14
curve from time 0 to time 24 hr at steady state conditions; W5o denotes the
time
where the plasma concentration is 50% or more of Cmax; W75 denotes the time
where the plasma concentration is 75% or more of Cmax; and MRT denotes mean
residence time for each of the active pharmaceutical ingredients of the
compositions of the present invention.
[0068] In this context, the term "medicine" or "medicament" means a compound
used to treat disease, injury or pain. Medicine is designated "prophylactic,"
i.e. the
art of preserving health, and "therapeutic", i.e. the art of restoring health.
[0069] In the present context, the terms "controlled release" and "modified
release"
are intended to be equivalent terms covering any type of release of active
ingredient, e.g. fenofibrate or atorvastatin, from a composition of the
invention that
is appropriate to obtain a specific therapeutic or prophylactic response after
administration to a subject. A person skilled in the art knows how controlled
release/modified release differs from the release of plain tablets or
capsules. The
terms "release in a controlled manner" and "release in a modified manner' have
the same meaning as stated above. The terms include slow release (that results
in
a lower Cmax and later tmax, but the half-life remains unchanged), extended
release
(that results in a lower Cmax, later tmax, but apparent half-life is longer);
delayed
release (that result in an unchanged Cmax, but lag time and, accordingly, tmax
is
delayed, and the half-life remains unchanged) as well as pulsatile release,
burst
release, sustained release, prolonged release, chrono-optimized release, fast
release (to obtain an enhanced onset of action) etc. Included in the terms is
also
e.g. utilization of specific conditions within the body e.g., different
enzymes or pH
changes in order to control the release of the drug substance.
[0070] In this context, the term "erosion" or "eroding" means a gradual
breakdown
of the surface of a material or structure, for example of a tablet or the
coating of a
tablet.
The active drug substances
[0071 ] A first drug or active substance of the dosage forms and
pharmaceutical
compositions of this invention is a fibrate, usually fenofibrate as described
above
or an analog thereof. It should be understood that this invention includes
dosage
forms and compositions comprising a mixture of two, three or even four
different
fibrates and/or fibric acids. Examples of other useful fibrates are
bezafibrate,
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
ciprofibrate, clinofibrate, clofibrate, etofylline, clofibrate, gemfibrozil,
pirifibrate,
simfibrate and tocofibrate; particularly useful are gemfibrozil, fenofibrate,
bezafibrate, clofibrate, ciprofibrate and active metabolites and analogues
thereof
including any relevant fibric acid such as fenofibric acid.
[0072] A second drug or active substance of the dosage forms and
pharmaceutical
compositions of this invention is atorvastatin as described above or,
typically, a
pharmaceutically acceptable salt thereof such as the calcium salt or the
magnesium salt in either amorphous or crystalline form. The calcium salt
exists in
a number of crystalline forms (Forms I-XIV). However, it is contemplated that
any
type and physical form of atorvastatin is useful in the compositions and solid
dosage forms of the present invention.
[0073] The first and second active substance, i.e. fenofibrate and
atorvastatin or a
pharmaceutically acceptable salt thereof, is present in the composition or the
solid
dosage form of the invention in effective amounts together with a vehicle and
optionally further excipients or additives. It is believed that fenofibrate in
combination with atorvastatin may have an added effect; it has been shown that
use of the combination results in TG and LDL levels being more decreased while
HDL level is increased.
[0074] More specifically, the effective amount of fenofibrate is an amount
which is
at least bioequivalent to a 130 mg Antara capsule.
[0075] Alternatively, the amount of fenofibrate present in the composition of
the
invention exhibits an increased bioavailability as compared to 130 mg Antara
capsule by exhibiting a relative AUCo-24 value of 1.3 (AUC of fenofibrate of
the
invention relative to AUC of the 130 mg Antara capsule).
[0076] In one embodiment, fenofibrate is present in the composition of the
invention in an amount below about 130 mg.
[0077] In another embodiment, fenofibrate is present in the composition of the
invention in an amount of about 120 mg.
[0078] The fenofibrate of the solid composition or the solid dosage form of
this
invention provides, after oral administration, an AUCo-24value of fibric acid
(arithmetic mean) of at least 28,000 ng=h/mL, or at least of about 40,000
ng=h/mL,
or at least of about 79,000 ng=h/mL, or at least of about 118,000 ng=h/mL.
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
16
[0079] In the solid composition or the solid dosage form of this invention at
least
about 50% w/w, preferably at least about 75%w/w, of the total amount of active
substances or essentially all of the fenofibrate is dissolved in vehicle
selected from
the group consisting of a hydrophobic, a hydrophilic and a water-miscible
vehicle.
[0080] Normally, at least about 85% w/w, at least about 90% w/w, at least
about
95% w/w or at least about 98% w/w, or at least about 99% w/w, or at least
about
99.5% w/w, or 100% w/w of the fenofibrate is dissolved in the vehicle.
[0081] If those embodiments where the total amount of fenofibrate present in
the
composition or the solid dosage form of the invention is completely (100%)
dissolved in the vehicle, fenofibrate is present in the form of a solid
solution in the
particulate composition. The presence of a solid solution can be tested by a
DSC
test mentioned herein. It is contemplated that the fenofibrate forms an
interstitial
crystalline solid solution with the vehicle. The atorvastatin component may be
co-
dissolved or, at least when crystalline or semi-crystalline atorvastatin is
used,
dispersed homogeneously in the solid solution. However, it is contemplated
that
crystallization of a diminutive amount of any of the active substances
(notably
fenofibrate) from the solid solution may occur during storage of the solid
dosage
form of the invention, especially in tablets due to the possibility of
formation of
cavities in the tablet during manufacturing (tablet compression), which
cavities
may leave space for crystallization. Thus, the present invention includes
particulate material wherein the active substances, or at least the
fenofibrate, are
present in the form of a solid solution, but it is within the scope of the
present
invention that a minor or diminutive amount of the active substance(s) in
solid
solution may precipitate or crystallize upon storage.
[0082] In another embodiment of the invention, at least about 80% w/w,
preferably
100% w/w, of fenofibrate is dissolved in the vehicle, which is further
processed into
the particulate form as described herein. The solid particles, for examples
granulate, comprising the dissolved fenofibrate is then mixed or blended with
micronized atorvastatin, and the resulting composition is optionally subjected
to
conventional methods for preparing solid dosage forms, especially tablets.
Alternatively, the solid fenofibrate particles are mixed with entero-coated
atorvastatin particles, for example entero-coated granulate, and subjected to
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
17
conventional methods for preparing tablets or simply filled into capsules or
sachets.
[0083] As mentioned above, sufficient flowability is required of the
particulate
composition of the invention in order to obtain a suitable flexibility so that
different
dosage forms can be obtained. In a preferred embodiment, the solid composition
of the invention is free-flowing, i.e. has a suitable flowability as
determined
according to the method described in the European Pharmacopoeia (Ph.Eur.)
measuring the flow rate of the composition out of a funnel with a nozzle
diameter
of 10.0 mm.
[0084] In a specific embodiment, the concentration of fenofibrate in the
vehicle is
at least about 10% w/w, based on the total weight of the fibrate, the statin
and the
vehicle. In particular, the concentration of fenofibrate in the vehicle is at
least
about 15% w/w, or at least about 16% w/w, or at least about 17% w/w, or at
least
about 20% w/w, preferably at least 25% w/w, more preferably at least about 30%
w/w, especially at least about 35% w/w, based on the total weight of the
fibrate,
the statin and the vehicle.
[0085] The concentration of atorvastatin in the vehicle of the solid
composition or
solid dosage form according to the invention is at least about 1% w/w, based
on
the total weight of the fibrate, the statin and the vehicle. More
specifically, the
concentration of statin in the vehicle is at least about 1.5% w/w, or at least
about
2.5% w/w, or at least about 5% w/w, or at least about 7.5% w/w or at least
about
10% w/w, based on the total weight of the fibrate, the statin and the vehicle.
[0086] The present invention provides solid compositions and solid dosage
forms
for improved treatment of conditions that respond to fenofibrate and
atorvastatin
treatment, for example hyperlipidemia and hypercholesterolemia.
[0087] In a preferred embodiment of the invention, the fibrate is fenofibrate
present
in the solid dosage form or the pharmaceutical composition of this invention
in an
amount selected from the group consisting of 160 mg, 145 mg, 130 mg, 120 mg,
110 mg, 100 mg, 90 mg, 87 mg, 80 mg, 70 mg, 60 mg, 50 mg, 48 mg, 45 mg, 43
mg, 40 mg, 35 mg and 30 mg of fenofibrate. In a preferred embodiment, the
solid
dosage form comprises 145 mg of fenofibrate. In another preferred embodiment,
the solid dosage form comprises 130 mg of fenofibrate. In yet another
preferred
embodiment, the solid dosage form comprises 120 mg of fenofibrate. In yet
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
18
another preferred embodiment, the solid dosage form comprises 110 mg of
fenofibrate. In yet another preferred embodiment, the solid dosage form
comprises
50 mg of fenofibrate. In yet another preferred embodiment, the solid dosage
form
comprises 48 mg of fenofibrate. In yet another preferred embodiment, the solid
dosage form comprises 43 mg of fenofibrate. In yet another preferred
embodiment, the solid dosage form comprises 87 mg of fenofibrate.
[0088] Atorvastatin may be present (conveniently as atorvastatin calcium or as
atorvastatin magnesium, either in amorphous form, in semi-amorphous form, in
semi-crystalline form or in crystalline form) in an amount of from about 5 mg
to
about 80 mg, for example in an amount of about 5 mg or about 10 mg or about 15
mg or about 20 mg or about 25 mg or about 30 mg or about 35 mg or about 40 mg
or about 45 mg or about 50 mg or about 55 mg or about 60 mg or about 65 mg or
about 70 mg or about 75 mg or about 80 mg of atorvastatin or a
pharmaceutically
acceptable salt thereof, for example the calcium salt or the magnesium salt.
[0089] Examples of useful combinations are about 120 mg of fenofibrate and
about
mg of atorvastatin; about 120 mg of fenofibrate and about 20 mg of
atorvastatin; about 120 mg of fenofibrate and about 30 mg of atorvastatin;
about
120 mg of fenofibrate and about 40 mg of atorvastatin; about 120 mg of
fenofibrate
and about 10 mg of atorvastatin; about 110 mg of fenofibrate and about 10 mg
of
atorvastatin; about 110 mg of fenofibrate and about 20 mg of atorvastatin;
about
110 mg of fenofibrate and about 30 mg of atorvastatin; about 110 mg of
fenofibrate
and about 40 mg of atorvastatin.
Bioavailability
[0090] As described above, there remains a need for novel pharmaceutical
compositions comprising fenofibrate and atorvastatin exhibiting suitable
bioavailability and/or suitable pharmacological response of the active
substances
and/or reduced or eliminated food effect.
[0091] Clinical trial studies have shown, cf. the example herein, that the
fenofibrate
solid dosage forms and pharmaceutical compositions of the present invention
eliminate the food effect, i.e. may be administered in the fed or the fasted
state.
Accordingly, the present invention provides the patient the choice of taking
only
one tablet daily at any time over the commercially available fenofibrate-
containing
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
19
medicament which should be taken with food in order to achieve the desired
bioavailability of the active ingredient.
[0092] For atorvastatin, the liver is the primary site of action (first-pass
metabolism); accordingly, the pharmacological or therapeutic response is
correlated to the actual oral dose administered and not correlated to the
plasma
exposure. That is, plasma concentrations of atorvastatin acid and its
metabolites
do not correlate with LDL-cholesterol reduction at a given dose. Thus, the
efficacy
of atorvastatin may be better predicted by drug dose than by peak
concentration
(cmax). Without being bound to this theory, it is contemplated that the best
possible
total in vivo efficacy of orally administered atorvastation can be obtained by
providing the drug in a controlled release formulation or, alternatively, in a
delayed
release formulation, since atorvastatin is metabolized in vivo by cytochrome
P450
(CYP) 3A4 to two active metabolites (2-hydroxy-atorvastatin acid and 4-hydroxy-
atorvastatin acid), thus allowing the active substances to be released in
areas of
the intestine having a reduced CYP3A4 activity and optionally over an extended
period of time, resulting in a relatively larger amount of the administered
drug
actually reaching the liver. Accordingly, it is contemplated that the dose
level can
be reduced while maintaining the LDL-lowering effect. For atorvastatin, this
is
advantageous, since the effect of atorvastatin on the relative increase in HDL-
level
(desirable) seems reduced at increasing doses: A published study has shown a
5.7% increase in HDL-level (from baseline) at a dose of 10 mg/day; a 4.8%
increase in HDL-level at a dose of 20 mg/day, a 4.4% increase in HDL-level at
a
dose of 40 mg/day, and a 2.1 % increase in HDL-level at a dose of 80 mg/day.
Other statins typically show increasing HDL-levels with increasing statin
dose.
[0093] In one embodiment, the invention relates to a pharmaceutical
composition
in particulate form or solid dosage form comprising fenofibrate and
atorvastatin,
wherein the composition upon oral administration to a mammal in need thereof
exhibits an AUC/AUCcontrol value for fenofibrate of at least about 1.0, the
AUCcontrol
being determined using a commercially available product containing
fenofibrate,
and the AUC values being determined under similar conditions.
[0094] No absolute bioavailability data based on an injectable composition are
available e.g., for fenofibrate (most likely due to solubility problems in
aqueous
media). The commercially available compositions containing fenofibrate include
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
surface-active agents and/or e.g., a lipophilic medium. The surface-active
agents
may impart improved bioavailability and therefore, the bioavailability of such
a
composition may be sufficient already. However, there is still a need for
developing a flexible formulation technique that enables preparation of a
variety of
dosage forms. Accordingly, the requirement to such improved and/or more
flexible
compositions may be to obtain the same or better bioavailability than already
seen
from the commercially available products.
[0095] Accordingly, in further embodiments of the invention, the
AUC/AUCcontro1
value for fenofibrate obtained by administering the solid dosage form or
pharmaceutical composition of the invention is at least about 1.1 such as,
e.g., at
least about 1.2, at least about 1.3, at least about 1.4, at least about 1.5,
about 1.75
or more, about 1.8 or more, about 1.9 or more, about 2.0 or more, about 2.5 or
more, about 2.75 or more, about 3.0 or more, about 3.25 or more, about 3.5 or
more, about 3.75 or more, about 4.0 or more, about 4.25 or more, about 4.5 or
more, about 4.75 or more or about 5.0 or more, the AUC values being determined
under similar conditions.
[0096] Likewise, the cmaX value for fenofibrate obtained by administering the
solid
dosage form or pharmaceutical composition of the invention relative to the
cmax
value of commercially available Tricor (Lipanthyl ) tablets, or
alternatively to
commercially available Antara capsules, is at least about 1.1, or at least
about
1.2, or at least about 1.3, or at least about 1.4, or at least about 1.5, or
at least
about 1.6 or more, or at least about 2.0, or at least about 2.5, or at least
about 3.0,
the cmax values being determined under similar conditions.
[0097] Another object of the invention is to reduce or eliminate the food
effect.
Thus, in another aspect, the invention relates to a pharmaceutical composition
in
particulate form or solid dosage form comprising one or more fibrates,
especially
fenofibrate, wherein the composition or solid dosage form upon oral
administration
to a mammal in need thereof does not exhibit a significant adverse food effect
as
evidenced by a value of (AUCfea/AUCfaStea) of at least about 0.85 with a lower
90%
confidence limit of at least 0.75. In a specific embodiment, the
pharmaceutical
composition or solid dosage form of the invention has a value of
(AUCfea/AUCfaStea)
that is about 0.9 or more such as, e.g., about 0.95 or more, about 0.97 or
more or
about 1 or more.
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
21
[0098] In other words, the difference between a bioequivalence parameter
measured after oral administration to a mammal with and without food,
respectively, is less than about 25% such as, e.g., less than about 20%, less
than
about 15%, less than about 10% or less than about 5%.
[0099] In another aspect, the invention relates to a pharmaceutical
composition in
particulate form or solid dosage form comprising fenofibrate, wherein the
composition upon oral administration to a mammal in need thereof is
essentially
bioequivalent with a commercially available product containing fenofibrate
when
administered in the same or lower dose as the commercially available product
containing fenofibrate.
[00100] In specific embodiments thereof, the dose is at the most about 98%
w/w such as, e.g., at the most about 95% w/w, at the most about 90% w/w, at
the
most about 85% w/w, at the most about 80% w/w, at the most about 75% w/w, at
the most about 70% w/w, at the most about 65% w/w, at the most about 60% w/w,
at the most about 55% w/w or at the most about 50% w/w of the dose of
fenofibrate administered in the form of a commercially available product
containing
fenofibrate.
[00101] A major problem with treatment with fenofibrate is the large intra- or
inter-individual variation. Thus, in a further aspect the invention relates to
a
pharmaceutical composition in particulate form comprising fenofibrate, wherein
the
composition upon oral administration to a mammal in need thereof reduces inter-
and/or intra-individual variations compared to those of a commercially
available
product containing fenofibrate under the same conditions and in a dose that
provides an equivalent therapeutic effect.
[00102] In the comparison tests mentioned above, the commercially available
fenofibrate product is Tricor (Lipanthyl ) in the form of tablets or,
alternatively,
Tricor in the form of capsules, or Antara capsules.
[00103] A convenient method for determining whether a suitable amount of
fenofibrate has been absorbed may be to determine the content of unchanged
fibrate excreted via the faeces. Thus, in one embodiment the invention relates
to a
solid pharmaceutical composition or solid dosage form, wherein at most about
25% w/w such as, e.g., at the most about 20% w/w, at the most about 15% w/w,
at
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
22
the most about 10% w/w, at the most about 5% w/w of the fenofibrate contained
in
the composition is excreted in the faeces after oral administration.
The vehicle
[00104] Vehicles useful in the present context are vehicles, which are water-
miscible, hydrophilic or hydrophobic. Useful vehicles are non-aqueous
substances
which may be hydrophilic, lipophilic, hydrophobic and/or amphiphilic
materials. The
hydrophobic or hydrophilic or water-miscible vehicles will normally be liquid
at
ambient or elevated temperature. In the present context the term "a
hydrophobic
or a hydrophilic or water-miscible vehicle" is used in a very broad sense
including
oils, waxes, semi-solid materials and materials that normally are used as
solvents
(such as organic solvents) or co-solvents within the pharmaceutical industry,
and
the term also includes therapeutically and/or prophylactically active
substances
that are in liquid form at ambient temperature; furthermore the term includes
emulsions like e.g., micro-emulsions and nanoemulsions and suspensions.
[00105] The oils or oily materials that are suitable for use in the present
context are substances or materials, which have a melting point of at least
about
C and at the most about 250 C. In specific embodiments of the invention, the
oily material has a melting point of about 5 C or more such as, e.g., about 10
C or
more, about 15 C or more, about 20 C or more or about 25 C or more. In further
embodiments of the invention, the oily material has a melting point of at
least
about 25 C such as, e.g., at least about 30 C at least about 35 C or at least
about
40 C. For practical reasons, the melting point may normally not be too high,
thus
the oily material normally has a melting point of at the most about 250 C, at
the
most about 200 C, at the most about 150 C or at the most about 100 C. If the
melting point is higher a relatively high temperature may promote e.g.
oxidation or
other kind of degradation of an active substance in those cases where e.g. a
therapeutically and/or prophylactically active substance is included.
[00106] Typically, a suitable hydrophilic oil or oily material is selected
from
the group consisting of: polyether glycols such as, e.g., polyethylene
glycols,
polypropylene glycols; polyoxyethylenes; polyoxypropylenes; poloxamers and
mixtures thereof, or it may be selected from the group consisting of: xylitol,
sorbitol, potassium sodium tartrate, sucrose tribehenate, glucose, rhamnose,
lactitol, behenic acid, hydroquinon monomethyl ether, sodium acetate, ethyl
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
23
fumarate, myristic acid, citric acid, Sucro-ester 7, Sucro-ester 11, Sucro-
ester 15,
maltose, mannitol and mixtures thereof.
[00107] The pharmaceutical composition or a solid dosage form according to
the invention may have a concentration of oil or oily material in the
composition or
the dosage form of about 5% w/w or more such as, e.g., about 10% w/w or more,
about 15% w/w or more, about 20% w/w or more, about 25% w/w or more, about
30% w/w or more, about 35% w/w or more, about 40% w/w or more, about 45%
w/w or more, about 50 w/w or more, about 55% w/w or more, about 60% w/w or
more, about 65% w/w or more, about 70% w/w or more, about 75% w/w or more,
about 80% w/w or more, about 85% w/w or more, about 90% w/w or more or about
95% w/w or more.
[00108] In specific embodiments the concentration of the oily material in a
composition or solid dosage form of the invention is in a range from about 20%
to
about 80% w/w such as, e.g., from about 25% to about 75% w/w.
[00109] In general, the hydrophobic or hydrophilic or water-miscible vehicles
that are suitable for use in the present context are substances or materials
having
a melting point of at least about 0 C and at the most about 250 C.
[00110] Interesting hydrophobic or hydrophilic or water-miscible vehicles are
generally substances, which are used in the manufacture of pharmaceuticals as
so-called melt binders or solid solvents (in the form of solid dosage form),
or as co-
solvents or ingredients in pharmaceuticals for topical use.
[00111] It may be hydrophilic, hydrophobic and/or have surface-active
properties. In general hydrophilic and/or hydrophobic vehicles are suitable
for use
in the manufacture of a solid pharmaceutical composition in particulate form
or a
solid dosage form according to the invention. In a specific embodiment they
may
be used when the release of the active substance from the pharmaceutical
composition is designed to be immediate or non-modified or modified.
Hydrophobic vehicles are normally used in the manufacture of a modified
release
pharmaceutical composition. These considerations are simplified to illustrate
general principles, but there are many cases where other combinations of
vehicles
and other purposes are relevant and, therefore, the examples above should not
in
any way limit the invention.
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
24
[00112] Examples of hydrophobic vehicles useful in the present invention are
straight chain saturated hydrocarbons, paraffins; fats and oils such as cacao
butter, beef tallow, lard; higher fatty acid such as stearic acid, myristic
acid,
paimitic acid; hydrogenated tallow, substituted and/or unsubstituted
triglycerides,
yellow beeswax, white beeswax, carnauba wax, castor wax, Japan wax, and
mixtures thereof.
[0113] Examples of water-miscible vehicles useful in the present invention
are:
[0114] water-miscible polar lipids such as sorbitan esters, polyether glycol
esters;
higher alcohols such as cetanol, stearyl alcohol; glyceryl monooleate,
substituted
and/or unsubstituted monoglycerides, substituted and/or unsubstituted
diglycerides, and mixtures thereof.
[0115] In a more preferred embodiment, the vehicle is hydrophilic or water-
miscible. Preferably, the vehicle is selected from the group consisting of
polyethylene glycols, polyoxyethylene oxides, poloxamers, polyoxyethylene
stearates, poly-epsilon caprolactone and mixtures thereof.
[0116] Examples of useful hydrophilic or water-miscible vehicles are
polyvinylpyrrolidones, polyvinyl-polyvinylacetate copolymers (PVP-PVA),
polyvinyl
alcohol (PVA), PVP polymers, acrylic polymers, polymethacrylic polymers
(Eudragit RS; Eudragit RL, Eudragit NE, Eudragit E), myristyl alcohol,
cellulose
derivatives including hydroxypropyl methylcellulose (HPMC), hydroxypropyl
cellulose (HPC), methylcellulose, sodium carboxymethylcellulose, hydroxyethyl
cellulose, pectins, cyclodextrins, galactomannans, alginates, carragenates,
xanthan gums and mixtures thereof.
[0117] The vehicle is preferably a mixture of two or more substances.
[0118] The vehicle may also be an oily material as defined and described
below.
[0119] Preferably, the melting point of the vehicle is preferably in the range
of 10 C
to 250 C, preferably in the range of 30 C to 100 C, more preferably in the
range of
40 C to 75 C, especially in the range of 40 C to 70 C. In specific embodiments
of
the invention, the hydrophobic or hydrophilic or water-miscible vehicles have
a
melting point of about 5 C or more such as, e.g., about 10 C or more, about 15
C
or more, about 20 C or more or about 25 C or more. Normally, vehicles having
such a low melting point require addition of an oil-sorption material.
However, a
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
person skilled in the art will know when it is necessary to add such an oil-
sorption
material.
[0120] In the present context, melting points are determined by DSC
(Differential
Scanning Calorimetry). The melting point is determined as the temperature at
which the linear increase of the DSC curve intersects the temperature axis.
[0121] In a preferred embodiment of the invention, the vehicle is a
polyethylene
glycol having an average molecular weight in a range of from about 400 to
about
35,000 such as, e.g., from about 800 to about 35,000, from about 1,000 to
about
35,000 such as, e.g., polyethylene glycol 1,000, polyethylene glycol 2,000,
polyethylene glycol 3,000, polyethylene glycol 4,000, polyethylene glycol
5,000,
polyethylene glycol 6,000, polyethylene glycol 7,000, polyethylene glycol
8,000,
polyethylene glycol 9,000 polyethylene glycol 10,000, polyethylene glycol
15,000,
polyethylene glycol 20,000, or polyethylene glycol 35,000. In certain
situations
polyethylene glycol may be employed with a molecular weight from about 35,000
to about 100,000.
[0122] In another interesting embodiment, the vehicle is polyethylene oxide
having
a molecular weight of from about 2,000 to about 7,000,000 such as, e.g. from
about 2,000 to about 100,000, from about 5,000 to about 75,000, from about
10,000 to about 60,000, from about 15,000 to about 50,000, from about 20,000
to
about 40,000, from about 100,000 to about 7,000,000 such as, e.g., from about
100,000 to about 1,000,000, from about 100,000 to about 600,000, from about
100,000 to about 400,000 or from about 100,000 to about 300,000.
[0123] In another embodiment, the vehicle is a poloxamer (PEO-PPO-PEO, a
polyethylene oxide-polypropylene oxide-polyethylene oxide tri-block polymer),
for
example Poloxamer 188, Poloxamer 237, Poloxamer 338 or Poloxamer 407 or
other block copolymers of ethylene oxide and propylene oxide such as the
Pluronic and/or Tetronic series from BASF. Suitable block copolymers of the
Pluronic series include polymers having a molecular weight of about 3,000 or
more such as, e.g. from about 4,000 to about 20,000 and/or a viscosity
(Brookfield) from about 200 to about 4,000 cps such as, e.g., from about 250
to
about 3,000 cps. Suitable examples include Pluronic F38, P65, P68LF, P75,
F77, P84, P85, F87, F88, F98, P103, P104, P105, F108, P123, F123, F127, 10R8,
17R8, 25R5, 25R8 etc. Suitable block copolymers of the Tetronic series
include
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
26
polymers having a molecular weight of about 8,000 or more such as, e.g., from
about 9,000 to about 35,000 and/or a viscosity (Brookfield) of from about 500
to
about 45,000 cps such as, e.g., from about 600 to about 40,000. The
viscosities
given above are determined at 60 C for substances that are pastes at room
temperature and at 77 C for substances that are solids at room temperature.
[0124] In a specific embodiment a particulate material according to the
invention
comprises as vehicle a mixture of a polyethylene glycol and a poloxamer in a
proportion (weight) of between about 1:3 and about10:1, preferably between
about 1:1 and about 5:1, more preferably between about 3:2and about 4:1,
especially between about 2:1 and about 3:1, in particular about 7:3.
[0125] In a preferred embodiment of the invention, the poloxamer is poloxamer
188.
[0126] In another preferred embodiment, polyethylene glycol is employed as a
vehicle, the PEG having an average molecular weight of about 6000 (PEG 6000).
[0127] The vehicle may also be a sorbitan ester such as, e.g., sorbitan di-
isostearate, sorbitan dioleate, sorbitan monolaurate, sorbitan
monoisostearate,
sorbitan monooleate, sorbitan monopaimitate, sorbitan monostearate, sorbitan
sesqui-isostearate, sorbitan sesquioleate, sorbitan sesquistearate, sorbitan
tri-
isostearate, sorbitan trioleate, sorbitan tristearate or mixtures thereof.
[0128] The vehicle may also comprise a mixture of different vehicles, for
example
a mixture of hydrophilic and/or hydrophobic materials.
[0129] Other suitable vehicles may be solvents or semi-solid excipients, for
example propylene glycol, complex fatty materials of plant origin including
theobroma oil, carnauba wax, vegetable oils like e.g. almond oil, coconut oil,
corn
oil, cottonseed oil, sesame oil, soy bean oil, olive oil, castor oil, palm
kernels oil,
peanut oil, rape oil, grape seed oil etc., hydrogenated vegetable oils such
as, e.g.
hydrogenated peanut oil, hydrogenated palm kernels oil, hydrogenated
cottonseed
oil, hydrogenated soy bean oil, hydrogenated castor oil, hydrogenated coconut
oil;
natural fatty materials of animal origin including beeswax, lanolin, fatty
alcohols
including cetyl, stearyl, lauric, myristic, paimitic, stearic fatty alcohols;
esters
including glycerol stearate, glycol stearate, ethyl oleate, isopropyl
myristate; liquid
interesterified semi-synthetic glycerides including Miglycol 810/812; amide or
fatty
acid alcolamides including stearamide ethanol, diethanolamide of fatty coconut
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
27
acids, acetic acid esters of mono and di-glycerides, citric acid esters of
mono and
di-glycerides, lactic acid esters of mono and diglycerides, mono and di-
glycerides,
poly-glycerol esters of fatty acids, poly-glycerol poly-ricinoleate, propylene
glycol
esters of fatty acids, sorbitan monostearates, sorbitan tristearates, sodium
stearoyl
lactylates, calcium stearoyl lactylates, diacetyl tartaric acid esters of mono
and di-
glycerides etc.
[0130] One of the advantages is that is it possible to incorporate a
relatively large
amount of vehicle and still have a material that is solid. Thus, it is
possible to
prepare solid compositions with a relatively high load of vehicle by use of an
oil
sorption material as mentioned above. Within the pharmaceutical field it is an
advantage to be able to incorporate a relatively large amount of a vehicle
(e.g.,
with oil or oily-like characteristics) in a solid composition especially in
those
situation where the active substance does not have suitable properties with
respect to water solubility (e.g., poor water solubility), stability in
aqueous medium
(i.e. degradation occurs in aqueous medium), oral bioavailability (e.g. low
bioavailability) etc., or in those situations where it is desired to modify
the release
of an active substance from a composition in order to obtain a controlled,
modified,
delayed, sustained and/or pulsed delivery of the active substance.
[0131] It is within the skills of the average practitioner to select a
suitable vehicle
being pharmaceutical acceptable, capable of dispersing, dissolving or at least
partly dissolving the active substances and having a melting point in the
desired
range using general knowledge and routine experimentation. Suitable vehicles
are
for example disclosed in WO 03/004001, which is incorporated herein by
reference.
[0132] The solid composition of the invention has a suitable flowability. In
order to
avoid any adherence to the manufacturing and/or filling equipment it is
important
that the particulate material is free-flowing. This characteristic is also
important in
those cases where it is desired to process the particulate material further,
for
example into solid dosage forms. When the particulate composition of the
invention is a free-flowing powder it can be immediately processed into e.g.
solid
dosage forms such as tablets, capsules or sachets. Normally, the particulate
composition has properties so as to allow manufacturing of tablets by direct
compression without addition of large amounts of further additives.
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
28
[0133] In some embodiments of the invention, the used vehicle is an oily
material
which may be present in a relatively high amount. In such cases it may be
necessary to include in the material a substance that has adsorbing or
absorbing
properties so that the final particulate material appears as a non-oily powder
and
not during storage release some of the vehicle that could result in a oily
surface.
Accordingly, the particulate material may contain one or more oil-sorption
materials, which - when tested as described herein - i) has an oil threshold
value
of 10% or more, when tested according to the Threshold Test herein, and at
least
one of ii) releases at least 30% of an oil, when tested according to the
Release
Test herein, and iii) in the form of a tablet, has a disintegration time of at
the most
1 hour, when tested according to Ph. Eur. Disintegration test, the tablet
containing
about 90% w/w or more of the oil-sorption material. In certain situations, it
has
been found that it is an advantage to incorporate a sorption material in the
composition in order e.g., to enable a high concentration of a vehicle has oil
or
oily-like character. In those cases where the vehicle has a melting point of
at the
most about 25 C, it may be especially suitable to incorporate a sorption
material.
Suitable examples of materials suitable as vehicles as well as sorption
materials
are given herein.
Pharmaceutically acceptable excipients and additives
[0134] In the present context the term "pharmaceutically acceptable
excipient(s)" is
intended to denote any material, which is inert in the sense that it
substantially
does not have any therapeutic and/or prophylactic effect per se. Such an
excipient
may be added with the purpose of making it possible to obtain a
pharmaceutical,
cosmetic and/or foodstuff composition, which have acceptable technical
properties. A pharmaceutical composition or a solid dosage form according to
the
invention may contain one or more pharmaceutically acceptable excipients.
[0135] Examples of suitable excipients for use in a composition or solid
dosage
form according to the invention include fillers, diluents, disintegrants,
binders,
lubricants etc. or mixtures thereof. As the composition or solid dosage form
according to the invention may be used for different purposes, the choice of
excipients is normally made taken such different uses into considerations.
Other
pharmaceutically acceptable excipients for suitable use are e.g. acidifying
agents,
alkalizing agents, preservatives, antioxidants, buffering agents, chelating
agents,
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
29
coloring agents, complexing agents, emulsifying and/or solubilizing agents,
flavors
and perfumes, humectants, sweetening agents, wetting agents etc.
[0136] Examples of suitable fillers, diluents and/or binders include lactose
(e.g.
spray-dried lactose, a-lactose, b-lactose, Tabletose , various grades of
Pharmatose , Microtose or Fast-Floc ), microcrystalline cellulose (various
grades of Avicel , Elcema , Vivacel , Ming Tai or Solka-Floc ),
hyd roxypropylcel I u lose, L-hyd roxypropylcel I u lose (low substituted),
hydroxypropyl
methylcellulose (HPMC) (e.g., Methocel E, F and K, Metolose SH of Shin-Etsu,
Ltd, such as, e.g. the 4,000 cps grades of Methocel E and Metolose 60 SH, the
4,000 cps grades of Methocel F and Metolose 65 SH, the 4,000, 15,000 and
100,000 cps grades of Methocel K; and the 4,000, 15,000, 39,000 and 100,000
grades of Metolose 90 SH), methylcellulose polymers (such as, e.g., Methocel
A,
Methocel A4C, Methocel A15C, Methocel A4M), hydroxyethylcellulose, sodium
carboxymethylcel I u lose, carboxymethylene, carboxymethyl hydroxyethylcel I u
lose
and other cellulose derivatives, sucrose, agarose, sorbitol, mannitol,
dextrins,
maltodextrins, starches or modified starches (including potato starch, maize
starch
and rice starch), calcium phosphate (e.g., basic calcium phosphate, calcium
hydrogen phosphate, dicalcium phosphate hydrate), calcium sulfate, calcium
carbonate, sodium alginate, collagen etc.
[0137] Specific examples of diluents are e.g., calcium carbonate, dibasic
calcium
phosphate, tribasic calcium phosphate, calcium sulfate, microcrystalline
cellulose,
powdered cellulose, dextrans, dextrin, dextrose, fructose, kaolin, lactose,
mannitol,
sorbitol, starch, pre-gelatinized starch, sucrose, sugar etc.
[0138] Specific examples of disintegrants are e.g. alginic acid or alginates,
microcrystalline cellulose, hydroxypropyl cellulose and other cellulose
derivatives,
croscarmellose sodium, crospovidone, polacrillin potassium, sodium starch
glycolate, starch, pregelatinized starch, carboxymethyl starch (e.g. Primogel
and
Explotab ) etc.
[0139] Specific examples of binders are e.g., acacia, alginic acid, agar,
calcium
carrageenan, sodium carboxymethylcellulose, microcrystalline cellulose,
dextrin,
ethylcellulose, gelatin, liquid glucose, guar gum, hydroxypropyl
methylcellulose,
methylcellulose, pectin, PEG, povidone, pregelatinized starch etc.
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
[0140] Glidants and lubricants may also be included in the second composition.
Examples include stearic acid, magnesium stearate, calcium stearate or other
metallic stearate, talc, waxes and glycerides, light mineral oil, PEG,
glyceryl
behenate, colloidal silica, hydrogenated vegetable oils, corn starch, sodium
stearyl
fumarate, polyethylene glycols, alkyl sulfates, sodium benzoate, sodium
acetate
etc.
[0141] Other excipients which may be included in a composition or solid dosage
form of the invention are e.g., flavoring agents, coloring agents, taste-
masking
agents, pH-adjusting agents, buffering agents, preservatives, stabilizing
agents,
anti-oxidants, wetting agents, humidity-adjusting agents, surface-active
agents,
suspending agents, absorption enhancing agents, agents for modified release
etc.
[0142] Other additives in a composition or a solid dosage form according to
the
invention may be antioxidants like e.g. ascorbic acid, ascorbyl paimitate,
butylated
hydroxyanisole, butylated hydroxytoluene, hypophosphorous acid,
monothioglycerol, potassium metabisulfite, propyl gallate, sodium
formaidehyide
sulfoxylate, sodium metabisulfite, sodium thiosulfate, sulfur dioxide,
tocopherol,
tocopherol acetate, tocopherol hemisuccinate, TPGS or other tocopherol
derivatives, etc. The carrier composition may also contain e.g., stabilising
agents.
The concentration of an antioxidant and/or a stabilizing agent in the carrier
composition is normally from about 0.1 % w/w to about 5% w/w.
[0143] A composition or solid dosage form according to the invention may also
include one or more surfactants or substances having surface-active
properties. It
is contemplated that such substances are involved in the wetting of the
slightly
soluble active substance and thus, contributes to improved solubility
characteristics of the active substance. Suitable surfactants for use in a
composition or a solid dosage form according to the invention are surfactants
such
as, e.g., hydrophobic and/or hydrophilic surfactants as those disclosed in WO
00/50007 in the name of Lipocine, Inc.
[0144] Specific examples of suitable surfactants are polyethoxylated fatty
acids
such as, e.g., fatty acid mono- or diesters of polyethylene glycol or mixtures
thereof such as, e.g., mono- or diesters of polyethylene glycol with lauric
acid,
oleic acid, stearic acid, myristic acid, ricinoleic acid, and the polyethylene
glycol
may be selected from PEG 4, PEG 5, PEG 6, PEG 7, PEG 8, PEG 9, PEG 10,
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
31
PEG 12, PEG 15, PEG 20, PEG 25, PEG 30, PEG 32, PEG 40, PEG 45, PEG 50,
PEG 55, PEG 100, PEG 200, PEG 400, PEG 600, PEG 800, PEG 1000, PEG
2000, PEG 3000, PEG 4000, PEG 5000, PEG 6000, PEG 7000, PEG 8000, PEG
9000, PEG 1000, PEG 10,000, PEG 15,000, PEG 20,000, PEG 35,000,
polyethylene glycol glycerol fatty acid esters, i.e. esters like the above-
mentioned
but in the form of glyceryl esters of the individual fatty acids; glycerol,
propylene
glycol, ethylene glycol, PEG or sorbitol esters with e.g., vegetable oils like
e.g.,
hydrogenated castor oil, almond oil, palm kernel oil, castor oil, apricot
kernel oil,
olive oil, peanut oil, hydrogenated palm kernel oil and the like,
polyglycerized fatty
acids like e.g., polyglycerol stearate, polyglycerol oleate, polyglycerol
ricinoleate,
polyglycerol linoleate, propylene glycol fatty acid esters such as, e.g.,
propylene
glycol monolaurate, propylene glycol ricinoleate and the like, mono- and
diglycerides like e.g. glyceryl monooleate, glyceryl dioleae, glyceryl mono-
and/or
dioleate, glyceryl caprylate, glyceryl caprate etc.; sterol and sterol
derivatives;
polyethylene glycol sorbitan fatty acid esters (PEG-sorbitan fatty acid
esters) such
as esters of PEG with the various molecular weights indicated above, and the
various Tween series (from ICI America, Inc.); polyethylene glycol alkyl
ethers
such as, e.g., PEG oleyl ether and PEG lauryl ether; sugar esters like e.g.
sucrose
monopaimitate and sucrose monolaurate; polyethylene glycol alkyl phenois like
e.g. the Triton X or N series (Union Carbide Chemicals & Plastics Technology
Corporation); polyoxyethylene-polyoxypropylene block copolymers such as, e.g.,
the Pluronic series from BASF Aktiengesellschaft, the Synperonic series from
ICI America, Inc., Emkalyx, Lutrol from BASF Aktiengesellschaft, Supronic
etc.
The generic term for these polymers is "poloxamers" and relevant examples in
the
present context are Poloxamer 105, 108, 122, 123, 124, 181, 182, 183, 184,
185,
188, 212, 215, 217, 231, 234, 235, 237, 238, 282, 284, 288, 331, 333, 334,
335,
338, 401, 402, 403 and 407; sorbitan fatty acid esters like the Span series
(from
ICI) or Arlacel series (from ICI) such as, e.g., sorbitan monolaurate,
sorbitan
monopaimitate, sorbitan monooleate, sorbitan monostearate etc.; lower alcohol
fatty acid esters like e.g., oleate, isopropyl myristate, isopropyl paimitate
etc.; ionic
surfactants including cationic, anionic and zwitterionic surfactants such as,
e.g.,
fatty acid salts, bile salts, phospholipids, phosphoric acid esters,
carboxylates,
sulfates and sulfonates etc.
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
32
[0145] When a surfactant or a mixture of surfactants is present in a
composition or
a solid dosage form of the invention, the concentration of the surfactant(s)
is
normally in a range of from about 0.1 - 80% w/w such as, e.g., from about 0.1
to
about 20% w/w, from about 0.1 to about 15% w/w, from about 0.5 to about 10%
w/w, or alternatively, from about 0.10 to about 80% w/w such as, e.g. from
about
to about 70% w/w, from about 20 to about 60% w/w or from about 30 to about
50% w/w.
[0146] In a specific aspect of the invention, the at least one of the one or
more
pharmaceutically acceptable excipient is selected from the group consisting of
silica acid or a derivative or salt thereof including silicates, silicon
dioxide and
polymers thereof; magnesium aluminosilicate and/or magnesium
aluminometasilicate, bentonite, kaolin, magnesium trisilicate, montmorillonite
and/or saponite.
Sorption materials
[0147] Materials such as those mentioned immediately above are especially
useful
as a sorption material for oily materials in pharmaceuticals, cosmetics and/or
foodstuff. In a specific embodiment, the material is used as a sorption
material for
oily materials in pharmaceuticals. The material that has the ability to
function as a
sorption material for oily materials is also denoted "oil sorption material".
[0148] Furthermore, in the present context the term "sorption" is used to
denote
"absorption" as well as "adsorption". It should be understood that whenever
one of
the terms is used it is intended to cover the phenomenon absorption as well as
adsorption. The terms "sorption material" and "oil sorption material" is
intended to
have the same meaning.
[0149] A sorption material suitable for use according to the present invention
is a
solid pharmaceutically acceptable material, which - when tested as described
herein - i) has an oil threshold value of 10% or more, when tested according
to the
Threshold Test disclosed herein, and which material is used in a composition
of
the invention further fulfilling one or both of i) and ii): i) the composition
releases at
least 30% of the hydrophobic or a hydrophilic or water-miscible vehicle, when
tested according to the Release Test; ii) the composition contains, in the
form of a
tablet, at least about 90% w/w of the oil-sorption material, and exhibits a
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
33
disintegration time of at the most 60 minutes when tested according to the Ph.
Eur.
Disintegration Test.
[0150] The material is especially useful as a sorption material for oily
materials in
pharmaceuticals, cosmetics and/or foodstuff, especially in pharmaceuticals.
[0151] It is important that the oil sorption material fulfils at least two
tests. One of
the tests is mandatory, i.e. the Threshold Test must be met. This test gives a
measure for how much oily material the oil sorption material is able to absorb
while
retaining suitable flowability properties. It is important that an oil
sorption material
for use according to the invention (with or without oil absorbed) has a
suitable
flowability so that it easily can be admixed with other excipients and/or
further
processed into compositions without significant problems relating to e.g.
adherence to the apparatus involved. The test is described below in Materials
and
Methods and guidance is given for how the test is carried out. The Threshold
Test
involves the determination of the flowability of the solid material loaded
with
different amounts of oil.
[0152] From above it is seen that the oil threshold value normally must exceed
10% and often the oil sorption material has an oil threshold value of at least
about
15%, such as, e.g., at least about 20%, at least about 25%, at least about
30%, at
least about 35%, at least about 40%, or at least about 45%.
[0153] An especially suitable material for use according to the invention,
Aeroperl
300, has a very high oil threshold value of about 60%. Accordingly, materials
that
have an oil threshold value of at least about 50%, such as, e.g., at least
about
55% or at least about 60% are used in specific embodiments of the present
invention.
[0154] Furthermore, an oil sorption material for use according to the
invention must
fulfil at least one further test, namely a release test and/or a
disintegration test.
[0155] The release test gives a measure of the ability of an oil sorption
material to
release the oil that is absorbed to the material when contacted with water.
This
ability is very important especially in those situations where an active
substance is
contained in the oily material. If the oil sorption material is not capable of
releasing
the oil from the material then there is a major risk that the active substance
will
only to a minor degree be released from the material. Accordingly, it is
envisaged
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
34
that bioavailability problems relating to e.g., poor absorption etc. will
occur in such
situations.
[0156] The requirements for the release test are that the solid pharmaceutical
acceptable material, when tested as described herein, releases at least about
30%
such as, e.g., at least about 35%, at least about 40%, at least about 45%, at
least
about 50%, at least about 55% or at least about 60% of an oil. As it appears
from
the examples herein a suitable oil sorption material like Aeroperl 300 has a
much
higher release. Therefore, in a specific embodiment of the invention, the
solid
pharmaceutical acceptable material, when tested as described herein, releases
at
least about 65% such as, e.g., at least about 70%, at least about 75% or at
least
about 80% of an oil.
[0157] The disintegration test is not performed on the solid composition in
particulate form but on a tablet made of the solid material. A requirement
with
respect to disintegration is important in order to ensure that the solid
composition,
when included in solid dosage forms, does not impart unwanted properties to
the
dosage form e.g., leading to unwanted properties with respect to dissolution
and
bioavailability of the active substance contained in the dosage form. For some
of
the materials suitable for use according to the invention it is possible to
press
tablets containing 100% w/w of the solid material itself. If this is the case,
the test
is carried out on such tablets. However, it is envisaged that there may be
situations where it is rather difficult to prepare tablets from the solid
material alone.
In such cases it is possible to add pharmaceutically acceptable excipients
normally
used in the preparation of compressed tablets up to a concentration of 10% w/w
or
less. Examples of suitable pharmaceutically acceptable excipients include
fillers,
diluents, binders and lubricants. However, excipients, normally classified as
disintegrants, should be avoided.
[0158] Accordingly, the solid pharmaceutical acceptable material for use
according
to invention, when tested as described herein, in the form of a tablet should
have a
disintegration time of at the most 1 hour, when tested according to Ph. Eur.
Disintegration test, the tablet containing about 90% w/w or more, such as,
e.g.,
about 92.5% w/w or more, about 95% w/w or more, about 97.5% w/w or more or
about 100% of the pharmaceutically acceptable material.
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
[0159] In a further embodiment, the solid pharmaceutical acceptable material,
when tested as described herein, in the form of a tablet has a disintegration
time of
at the most about 50 min, such as, e.g., at the most about 40 min, at the most
about 30 min, at the most about 20 min, at the most about 10 min or at the
most
about 5 min, when tested according to Ph. Eur. Disintegration test, the tablet
containing about 90% w/w or more, such as, e.g., about 92.5% w/w or more,
about
95% w/w or more, about 97.5% w/w or more or about 100% of the
pharmaceutically acceptable material.
[0160] In a specific embodiment, the solid material used as a sorption
material
fulfils all three tests. Thus, the solid pharmaceutical acceptable material,
when
tested as described herein, i) has an oil threshold value of at least about
10%,
such as, e.g., at least about 15%, at least about 20%, at least about 25%, at
least
about 30%, at least about 35%, at least about 40%, at least about 45%, at
least
about 50%, at least about 55% or at least about 60%, ii) releases at least
about
30% such as, e.g., at least about 35%, at least about 40%, at least about 45%,
at
least about 50%, at least about 55%, at least about 60%, at least about 65%,
at
least about 70%, at least about 75% or at least about 80% of an oil, and iii)
in the
form of a tablet has a disintegration time of at the most 1 hour such as at
the most
about 50 min, at the most about 40 min, at the most about 30 min, at the most
about 20 min, at the most about 10 min or at the most about 5 min, when tested
according to Ph. Eur. Disintegration test, the tablet containing about 90% w/w
or
more, such as, e.g., about 92.5% w/w or more, about 95% w/w or more, about
97.5% w/w or more or about 100% of the pharmaceutically acceptable material.
[0161] Other specific embodiments of the invention are those, wherein the
solid
pharmaceutical material used as a sorption material in a composition of the
invention, when tested as described herein, i) has an oil threshold value of
at least
about 55%; the solid pharmaceutical material, when tested as described herein,
ii)
releases at least about 75% of an oil; and/or the solid pharmaceutical
material,
when tested as described herein, iii) in the form of a tablet has
disintegration time
of at the most about 10 min, when tested according to Ph. Eur. Disintegration
test,
the tablet containing about 97.5% w/w of the pharmaceutically acceptable
material.
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
36
[0162] The solid pharmaceutically acceptable material used as a sorption
material
in a composition according to the invention is normally a particulate material
in the
form of e.g. powders, particles, granules, granulates etc.
[0163] Such particulate material suitable for use as an oil sorption material
has
normally a bulk density of about 0.15 g/cm3 or more such as, e.g., at least
about
0.20 g/cm3 or at least about 0.25 g/cm3.
[0164] Furthermore, the oil sorption material normally has an oil absorption
value
of at least about 100 g oil/100 g such as, e.g., at least about 150 g oil/100
g, at
least about 200 g oil/100g, at least about 250 g oil/100 g, at least about 300
g
oil/100 g or at least about 400 g oil/100 g pharmaceutically acceptable
material.
The oil absorption value is determined as described in the experimental
section
herein.
[0165] The present inventors have found that a common feature of some of the
materials suitable for use as oil sorption material is that they have a
relatively large
surface area. Accordingly, pharmaceutically acceptable material for use as an
oil
sorption material according to the invention may have a BET surface area of at
least 5 m2/g such as, e.g., at least about 25 m2/g, at least about 50 m2/g, at
least
about 100 m2/g, at least about 150 m2/g, at least about 200 m2/g, at least
about
250 m2/g or at least about 275 m2/g.
[0166] As mentioned above one of the characteristic features of a
pharmaceutically acceptable material for use as an oil sorption material
according
to the invention is that it retains a good flowability even if it has been
loaded with
oily material. Thus, the flowability of the pharmaceutically acceptable
material
loaded with about 25% w/w or more such as, e.g. about 30% w/w or more, about
40% w/w or more, about 45% w/w or more, about 50% w/w or more, about 55%
w/w or more, about 60% w/w or more, about 65% w/w or more or about about 70%
w/w viscoleo will normally meet the Ph. Eur. requirements.
[0167] Notably, the oil sorption material may comprise a silica acid or a
derivative
or salt thereof such as, e.g., silicon dioxide or a polymer thereof as a
pharmaceutically acceptable excipient. However, dependent on the quality
employed a silicon dioxide may be a lubricant or it may be an oil sorption
material.
Qualities fulfilling the latter function seem to be most important.
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
37
[0168] In a specific embodiment, a composition or solid dosage form according
to
invention comprises a pharmaceutically acceptable excipient that is a silicon
dioxide product that has properties corresponding to Aeroperl 300.
[0169] Use of an oil sorption material in compositions or dosage forms
according
to the invention is very advantageous for the preparation of pharmaceutical,
cosmetic, nutritional and/or food compositions, wherein the composition
comprises
oily material. One of the advantages is that is it possible to incorporate a
relatively
large amount of and oily material and still have a material that is solid.
Thus, it is
possible to prepare solid compositions with a relatively high load of oily
materials
by use of an oil sorption material according to the invention. Within the
pharmaceutical field it is an advantage to be able to incorporate a relatively
large
amount of an oily material in a solid composition especially in those
situation
where the active substance does not have suitable properties with respect to
water
solubility (e.g. poor water solubility), stability in aqueous medium (i.e.
degradation
occurs in aqueous medium), oral bioavailability (e.g. low bioavailability)
etc., or in
those situations where it is desired to modify the release of an active
substance
from a composition in order to obtain a controlled, delayed, sustained and/or
pulsed delivery of the active substance. Thus, in a specific embodiment it is
used
in the preparation of pharmaceutical compositions.
[0170] The oil sorption material for use in the processing into solid
compositions
normally absorbs about 5% w/w or more, such as, e.g., about 10% w/w or more,
about 15% w/w or more, about 20% w/w or more, about 25% w/w or more, about
30% w/w or more, about 35% w/w or more, about 40% w/w or more, about 45%
w/w or more, about 50 w/w or more, about 55% w/w or more, about 60% w/w or
more, about 65% w/w or more, about 70% w/w or more, about 75% w/w or more,
about 80% w/w or more, about 85% w/w or more, about 90% w/w or more or about
95% w/w or more of an oil or an oily material and is still a solid material.
[0171] The oils and oily-like materials that can be absorbed are normally
liquid at
ambient or elevated temperature (for practical reasons the max. temperature is
about 250 C). They may be hydrophilic, lipophilic, hydrophobic and/or
amphiphilic
materials.
Method of manufacture
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
38
[0172] The particulate composition of the invention may be prepared by any
method which is suitable for incorporation of poorly water-soluble active
substances. The pharmaceutical compositions may be prepared by any
convenient method such as, e.g. granulation, mixing, spray drying etc. A
particularly useful method is the method disclosed in Applicants' co-pending
international application published as WO 03/004001, which describes a process
for preparation of particulate material by a controlled agglomeration method,
i.e. a
method, which enables a controlled growth in particle size. The method
involves
spraying a first composition comprising the active substance and a vehicle in
liquid
form onto a solid carrier. Normally, the vehicle has a melting point of at
least 5 C,
but the melting point must indeed be below the melting point of the active
substance. In the present invention, the melting point of the vehicle and
should not
exceed 250 C.
[0173] It is within the skills of the average practitioner to select a
suitable vehicle
being pharmaceutical acceptable, capable of dispersing or fully or at least
partly
dissolving the active substance and having a melting point in the desired
range
using general knowledge and routine experimentation. Suitable candidate for
carriers are described in WO 03/004001, which is herein incorporated by
reference.
[0174] In the present context, suitable vehicles are e.g., those mentioned as
vehicles or as oily materials as well as those disclosed in WO 03/004001. An
advantage of using the controlled agglomeration method described in WO
03/004001 is that it is possible to apply a relatively large amount of a
liquid system
to a particulate material without having an undesirable growth in particle
size.
Accordingly, in one embodiment of the invention, the particulate material of a
pharmaceutical composition has a geometric weight mean diameter dgw of _ 10
mm such as, e.g. _ 20 mm, from about 20 to about 2000, from about 30 to about
2000, from about 50 to about 2000, from about 60 to about 2000, from about 75
to
about 2000 such as, e.g. from about 100 to about 1500 mm, from about 100 to
about 1000 mm or from about 100 to about 700 mm, or at the most about 400 mm
or at the most 300 mm such as, e.g., from about 50 to about 400 mm such as,
e.g., from about 50 to about 350 mm, from about 50 to about 300 mm, from about
50 to about 250 mm or from about 100 to about 300 mm.
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
39
[0175] The compositions and dosage forms of the invention are preferably
formed
by spray drying techniques, controlled agglomeration, freeze-drying or coating
on
carrier particles or any other solvent removal process. The dried product
contains
the active substances present preferably in dissolved form either fully
dissolved as
a solid solution, for example forming an interstitial crystalline solid
solution, or
partly dissolved as a solid dispersion including a molecular dispersion and a
solid
solution.
[0176] However, the composition and dosage forms of the invention are
preferably
manufactured by a method comprising the steps of: i) bringing the vehicle in
liquid
form, i.e. melting the vehicle if solid at room temperature, ii) maintaining
the liquid
vehicle at a temperature below the melting point of the fibrate, iii)
dissolving the
desired amount of fibrate in the vehicle, iv) spraying the resulting solution
onto a
solid carrier having a temperature below the melting point of the vehicle, v)
mechanically working the resulting composition to obtain particles, i.e. a
particulate
material, and vi) optionally subjecting the particulate material to
conventional
methods for preparing solid dosage forms.
[0177] Alternatively, the solid oral dosage form of the invention may be
prepared
by a method comprising the steps of i) Bringing the vehicle in liquid form, if
applicable, ii) Maintaining the liquid vehicle at a temperature below the
melting
point of fenofibrate or a pharmaceutically acceptable salt thereof, iii)
Dissolving the
desired amount of fibrate in the vehicle, iv) Spraying the resulting solution
onto a
solid carrier having a temperature below the melting point of the vehicle, v)
Mechanically working the resulting composition to obtain particles, i.e. a
particulate
material containing fenofibrate, and, prior to or simultaneous with or after
applying
steps i) to v), vi) Bringing the vehicle in liquid form, if applicable, vii)
Maintaining
the liquid vehicle at a temperature below the melting point of atorvastatin or
a
pharmaceutically acceptable salt thereof, viii) Dissolving the desired amount
of
atorvastatin in the vehicle, ix) Spraying the resulting solution onto a solid
carrier
having a temperature below the melting point of the vehicle, x) Mechanically
working the resulting composition to obtain particles, i.e. a particulate
material
containing atorvastatin, followed by the steps of xi) Mixing the particulate
material
containing fenofibrate and the particulate material containing atorvastatin,
and xii)
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
Optionally subjecting the particulate material to conventional methods for
preparing solid dosage forms.
[0178] In yet another embodiment, the solid oral dosage form of the invention
is
prepared by a method comprising the steps of: i) bringing the vehicle in
liquid form,
if applicable, ii) maintaining the liquid vehicle at a temperature below the
melting
point of fenofibrate or a pharmaceutically acceptable salt thereof, iii)
dissolving the
desired amount of fenofibrate in the vehicle, iv) spraying the resulting
solution onto
a solid carrier having a temperature below the melting point of the vehicle,
v)
Mechanically working the resulting composition to obtain particles, i.e. a
particulate
material containing fenofibrate, and, prior to or simultaneous with or after
applying
steps i) to v), vi) micronizing atorvastatin or a pharmaceutically acceptable
salt
thereof, if applicable, followed by the steps of vii) mixing the particulate
material
containing fenofibrate and micronized atorvastatin, and viii) optionally
subjecting
the particulate material to conventional methods for preparing solid dosage
forms.
[0179] In yet another embodiment, the solid oral dosage form of the invention
is
prepared by a method comprising the steps of: i) Bringing the vehicle for
fibrate in
liquid form, if applicable, ii) Maintaining the liquid vehicle at a
temperature below
the melting point of the fibrate or a pharmaceutically acceptable salt
thereof, iii)
Dissolving the desired amount of fibrate in the vehicle, iv) Spraying the
resulting
solution onto a solid carrier having a temperature below the melting point of
the
vehicle, v) Mechanically working the resulting composition to obtain
particles, i.e. a
particulate material containing fibrate, and, prior to or simultaneous with or
after
applying steps i) to v), vi) Bringing the vehicle for atorvastatin in liquid
form, if
applicable, vii) dissolving or dispersing the desired amount of atorvastatin
in the
vehicle, viii) Spraying the resulting solution onto a solid carrier having a
temperature below the melting point of the vehicle, ix) Mechanically working
the
resulting composition to obtain particles, i.e. a particulate material
containing
atorvastatin, x) subjecting the particles to enteric coating, followed by the
steps of
xi) Mixing the particulate material containing fibrate and the entero-coated
particulate material containing atorvastatin, and xii) Optionally subjecting
the
particulate material to conventional methods for preparing solid dosage forms,
for
example compression into tablets of filling into capsules or sachets.
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
41
[0180] In an important embodiment of the invention, at least part of the
active
substances is present in the composition in the form of a solid dispersion
including
a molecular dispersion and a solid solution and an interstitial crystalline
solid
solution. Normally, about 10% or more such as, e.g., about 20% or more, about
30% or more, about 40% or more, about 50% or more, about 60% or more, about
70% or more, about 80% or more, about 90% or more such as, e.g., about 95% or
more or about 100% w/w of either the fenofibrate or the atorvastatin is
present in
the vehicle in the form of a solid dispersion, provided that at least about
80% w/w
of the total amount of active substances is dissolved in the vehicle.
[0181] The pharmaceutical compositions comprising the active substance at
least
partly in form of a solid dispersion or solution may in principle be prepared
using
any suitable procedure for preparing pharmaceutical compositions known within
the art.
[0182] A solid dispersion may be obtained in different ways e.g., by employing
organic solvents or by dispersing or dissolving the active substance in
another
suitable medium (e.g. an oily material that is in liquid form at room
temperature or
at elevated temperatures). Solid dispersions (solvent method) are prepared by
dissolving a physical mixture of the active substance (e.g. a drug substance)
and
the carrier in a common organic solvent, followed by evaporation of the
solvent.
The carrier is often a hydrophilic polymer. Suitable organic solvents include
pharmaceutical acceptable solvent in which the active substance is soluble
such
as methanol, ethanol, methylene chloride, chloroform, ethylacetate, acetone or
mixtures thereof.
[0183] Suitable water-soluble carriers include polymers such as polyethylene
glycol, poloxamers, polyoxyethylene stearates, poly-epsilon-caprolactone,
polyvinylpyrrolidone (PVP), polyvinylpyrrolidone-polyvinylacetate copolymer
PVP-
PVA (Kollidon VA64), poly-methacrylic polymers (Eudragit RS, Eudragit RL,
Eudragit NE, Eudragit E) and polyvinyl alcohol (PVA), hydroxypropyl cellulose
(HPC), hydroxypropyl methyl cellulose (HPMC), methyl cellulose, and
poly(ethylene oxide) (PEO).
[0184] Polymers containing acidic functional groups may be suitable for solid
dispersions, which release the active substance in a preferred pH range
providing
acceptable absorption in the intestines. Such polymers may be one ore more
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
42
selected from the group comprising hydroxypropyl methylcellulose phtalate
(HMPCP), polyvinyl acetate phtalate (PVAP), hydroxypropylmethylcellulose
acetate succinate (HPMCAS), alginate, carbomer, carboxymethylcellulose,
methacrylic acid copolymer (Eudragit L, Eudragit S), shellac, cellulose
acetate
phthalate (CAP), starch glycolate, polacrylin, methyl cellulose acetate
phtalate,
hydroxypropyulcellulose acetate phthalate, cellulose acetate terephtahalate,
cellulose acetate isophthalate and cellulose acetate trimellitate.
[0185] The weight ratio of active substance to polymer may be in a range of
from
about 3:1 to about 1:20. However, narrower ranges of from about 3:1 to about
1:5,
such as, e.g., from about 1:1 to about 1:3 or about may also be used.
[0186] Apart from using the organic solvent based method, solid dispersion or
solid
solutions of one or more fibrates may be also obtained by dispersing and/or
dissolving the active compound in the carrier composition used in the
controlled
agglomeration method. Stabilizing agents etc. may be added in order to ensure
the stability of the solid dispersion/solution.
[0187] There are a number of methods for combining fenofibrate and
atorvastatin
in the composition or solid dosage form of the invention:
1. In a first embodiment, a fenofibrate granulate is prepared as disclosed
in International Application PCT/DK2004/000667 and example 9 herein. The
fenofibrate granulate may be in the form of an immediate release formulation
or in
the form of a delayed release or even a controlled release formulation. A
atorvastatin granulate is prepared in the same manner as the fenofibrate
granulate, i.e. by dissolving or dispersing atorvastatin in a suitable vehicle
such as
the vehicle used for dissolving/dispersing fenofibrate and spraying the
dispersion
onto a suitable carrier to obtain a granulate. The two granulates are mixed
and
either compressed into tablets or filled into hard gelatine capsules. The
atorvastatin granulate is optionally subjected to entero-coating prior to
mixing, thus
providing a controlled release atorvastatin formulation. The atorvastatin
granulate
may also be in the form of a delayed release formulation.
2. In a second embodiment, a single granulate of fenofibrate and
atorvastatin is prepared by dissolving fenofibrate together with atorvastatin
in a
suitable vehicle as described herein, followed by spraying the solution (or
dispersion) on a a suitable carrier (as described herein), thereby obtaining a
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
43
particulate material, i.e. a single granulate, which may be compressed into
tablets
in a conventional manner or filled into hard gelatine capsules.
3. In a third embodiment, a single granulate of fenofibrate and
atorvastatin is prepared by dissolving fenofibrate in a suitable vehicle as
described
herein, followed by spraying the solution (or dispersion) on a mixture of a
suitable
carrier (as described herein) and the desired amount of atorvastatin, thereby
obtaining a particulate material, i.e. a single granulate, which may be
compressed
into tablets in a conventional manner or filled into hard gelatine capsules.
4. In a fourth embodiment, a fenofibrate granulate is prepared as disclosed in
International Application PCT/DK2004/000667 and example 9 herein. An
atorvastatin granulate corresponding to the granulate composition of Lipitor
tablets is prepared. The two granulates are mixed and either compressed into
tablets or filled into hard gelatine capsules.
5. In a fifth embodiment, a fenofibrate granulate is prepared as disclosed in
International Application PCT/DK2004/000667 and example 9 herein. Atorvastatin
is micronized and mixed with fenofibrate granulate and optionally conventional
excipients and/or additives such as glidants, fillers, binders or
disintegrators. The
mixture may be compressed into tablets or filled into hard gelatine capsules.
6. In a sixth embodiment, a fenofibrate granulate is prepared as disclosed
in International Application PCT/DK2004/000667 and example 9 herein. Granulate
is compressed into a tablet, and the tablet is coated with an aqueous
suspension
comprising a sufficient amount of atorvastatin including a film-forming
polymer and
stabilizers (antioxidants). The tablets might be sub-coated with a film-
forming
polymer before coating with the statin suspension below.
[0188] Examples of film polymers include water soluble agents such as
hydroxypropylmethylcellulose, Metolose (HPMC), hydroxypropylmethylcellulose,
Klucel (HPC), polyvinyl alcohol (PVA), polyvinylpyrrolidone (PVP) or
combinations of PVA and PVP (Kollicoat IR) and acid soluble acrylic polymer
(Eudragit E, soluble in gastric juice).
[0189] Examples of antioxidants include butylhydroxyanisol (BHA), ascorbyl
paimitate, ascorbic acid or combinations of BHA, ascorbyl paimitate and citric
acid.
[0190] Wetting and pH adjusting agent might be included in the coating
suspension.
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
44
[0191] The amount of atorvastatin in the coating suspension is between about
2%
w/w and about 40% w/w, such as for example between about 5% w/w and about
30% w/w. The skilled person will know how to determine the exact amount of
atorvastatin in the coating composition, when the desired amount of
atorvastatin in
the final composition and/or dosage form is known, for example 20 mg
atorvastatin
and 120 mg fenofibrate.
[0192] Coating of fenofibrate tablets is performed in conventional coating
equipment such as drum coater, perforated vessel or fluidized bed (Wurster
insert).
[0193] The atorvastatin coated fenofibrate tablets might be further coated
with a
suitable polymer to protect the atorvastatin from degradation.
Solid dosage forms
[0194] The pharmaceutical composition according to the invention is in solid,
particulate form and may be employed as such. However, in many cases it is
more
convenient to present the composition in the form of granules, pellets,
microspheres, nanoparticies and the like or in the form of solid dosage forms
including tablets, tablets, beads, capsules, grains, pills, granulates,
granules,
powder, pellets, sachets, lozenges, troches and the like.
[0195] A solid dosage form according to the invention may be a single unit
dosage
form or it may in the form of a poly-depot dosage form contain a multiplicity
of
individual units such as, e.g., pellets, beads and/or granules.
[0196] Usually, a pharmaceutical composition or a solid dosage form of the
invention is intended for administration via the oral, buccal or sublingual
administration route.
[0197] The dosage form of the invention is truly a solid, i.e. the dosage form
does
not comprise any liquid, semi-liquid or semi-solid material. Neither does the
solid
dosage form of the invention comprise a suspension, an emulsion or a micro-
emulsion.
[0198] The invention also relates to the above-mentioned presentation form.
Within
the scope of the invention are compositions/solid dosage forms that are
intended
to release the active substance in a fast release, a delayed release or
modified
release manner.
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
[0199] A solid dosage form according to the present invention comprises a
pharmaceutical composition in particulate form as described above. The details
and particulars disclosed under this main aspect of the invention apply
mutatis
mutandis to the other aspects of the invention. Accordingly, the properties
with
respect to increase in bioavailability, changes in bioavailability parameters,
reduction in adverse food effect as well as release of one or more fibrates
etc.
described and/or claimed herein for pharmaceutical compositions in particulate
form are analogues for a solid dosage form according to the present invention.
[0200] The solid dosage form of the invention, i.e. in unit dosage form,
comprises
comprises from about 30 to about 170 mg of fenofibrate and from about 5 to
about
80 mg of atorvastatin or a pharmaceutically acceptable salt thereof. In a
preferred
embodiment, the unit dosage form comprises about 160 mg of fenofibrate, or
about 145 mg of fenofibrate, or about 130 mg, or about 120 mg of fenofibrate,
or
about 110 mg of fenofibrate, and about 10 mg of atorvastatin, or about 15 mg
of
atorvastatin, or about 20 mg of atorvastatin, or about 25 mg of atorvastatin,
or
about 30 mg of atorvastatin, or about 40 mg of atorvastatin, or of a
pharmaceutically acceptable salt of atorvastatin. Preferably, the unit dosage
form
comprises fenofibrate and atorvastatin or pharmaceutically acceptable salt
thereof
in the (relative) weight ratio between fenofibrate and atorvastatin or a
pharmaceutically acceptable salt thereof from about 1:1 to about 40:1.
[0201] Usually, the concentration of the pharmaceutical composition in
particulate
form is in a range of from about 5 to 100% w/w such as, e.g., from about 10%
to
about 90% w/w, from about 15% to about 85% w/w, from about 20% to about 80%
w/w, from about 25% to about 80% w/w, from about 30% to about 80% w/w, from
about 35% to about 80% w/w, from about 40% to about 75% w/w, from about 45%
to about 75% w/w or from about 50% to about 70% w/w of the dosage form. In an
embodiment of the invention, the concentration of the pharmaceutical
composition
in particulate form is 50% w/w or more of the dosage form.
[0202] The solid dosage forms of the invention are stable. For example, the
fenofibrate is present in an amount of at least about 90%, or at least about
95%, or
at least about 99.3%, or at least about 100%, relative to the amount prior to
storage, when assayed after 3 months of storage at a temperature of about 40 C
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
46
and a relative humidity of about 75%. Also, the physical stability is high as
can be
seen from the Examples below.
[0203] The solid dosage form according to the invention is obtained by
processing
the particulate material according to the invention by means of techniques
well-
known to a person skilled in the art. Usually, this involves further addition
of one or
more of the pharmaceutically acceptable excipients mentioned herein.
[0204] The composition or solid dosage form according to the invention may be
designed to release fenofibrate and/or atorvastatin in any suitable manner
provided that the increase in bioavailability is maintained. Thus, the active
substance(s) may be released relatively fast in order to obtain an enhanced on-
set
of action, it may be released so as to follow zero or first order kinetics or
it may be
released in a controlled or modified manner in order to obtain a predetermined
pattern of release. Plain formulations are also within the scope of the
present
invention.
[0205] The composition or solid dosage form according to the invention may
also
be coated with a film coating, an enteric coating, a modified release coating,
a
protective coating, an anti-adhesive coating etc. In one embodiment of the
invention, a controlled release profile of atorvastatin is obtained by means
of
applying a time-controlled coating or en enzyme controlled coating or a
pressure
controlled coating.
[0206] A solid dosage form according to the invention may also be coated in
order
to obtain suitable properties e.g. with respect to release of the active
substance.
The coating may be applied on single unit dosage forms (e.g. tablets,
capsules) or
it may be applied on a poly-depot dosage form or on its individual units.
[0207] Suitable coating materials are e.g. methylcellulose,
hydroxypropylmethyl-
cellulose, hydroxypropylcellulose, acrylic polymers, ethylcellulose, cellulose
acetate phthalate, polyvinyl acetate phthalate, hydroxypropyl methylcellulose
phthalate, polyvinylalcohol, sodium carboxymethylcellulose, cellulose acetate,
cellulose acetate phthalate, gelatin, methacrylic acid copolymer, polyethylene
glycol, shellac, sucrose, titanium dioxide, carnauba wax, microcrystalline
wax,
zein.
[0208] Plasticizers and other ingredients may be added in the coating
material.
The same or different active substance may also be added in the coating
material.
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
47
[0209] The pharmaceutical composition or a solid dosage form according to the
invention is designed to release the fenofibrate in a suitable manner.
Specific
release patterns are disclosed in the appended claims to which reference is
made.
Herein is also given specific relevant absorption patterns. In specific
embodiments,
the compositions (i.e. particulate material or the solid dosage form) may
increase
the bioavailability of the fibrate and/or the atorvastatin after oral
administration.
The active substances may be released relatively fast in order to obtain an
enhanced on-set of action, it may be released so as to follow zero or first
order
kinetics or it may be released in a controlled or modified manner in order to
obtain
a predetermined pattern of release. Plain formulations are also within the
scope of
the present invention.
[0210] In a specific embodiment a solid dosage form of the invention results
in an
increased bioavailability of fenofibrate and/or atorvastatin relative to
existing
commercial fenofibrate and/or atorvastatin dosage forms when administered to a
mammal in need thereof.
[0211] With respect to fenofibrate a solid dosage form according to the
invention
may provide an AUCo-24 value of fibric acid relative to that of commercially
available Tricor (Lipanthyl ) tablets, or alternatively of commercially
available
Antara capsules, of at least about 1.1, or at least about 1.2, or at least
about 1.3,
or at least about 1.4, or at least about 1.5, or at least about 1.75 or more,
or at
least about 2.0, or at least about 2.5, or at least about 3.0, the AUCo-24
values
being determined under similar conditions. Moreover, a solid dosage form may
provide a cmax value relative to that of commercially available Tricor
(Lipanthyl )
tablets, or alternatively of commercially available Antara capsules, of at
least
about 1.1, or at least about 1.2, or at least about 1.3, or at least about
1.4, or at
least about 1.5, or at least about 1.6 or more, or at least about 2.0, or at
least
about 2.5, or at least about 3.0, the cmax values being determined under
similar
conditions.
[0212] With respect to atorvastatin, a solid dosage form according to the
invention
may provide an AUCo-24 value relative to that of commercially available
Lipitor
tablets of at least about 1.0, or at least about 1.1, or at least about 1.23,
or at least
about 1.3, or at least about 1.4, or at least about 1.75 or more, or at least
about
2.0, or at least about 2.5, or at least about 3.0, the AUCo-24 values being
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
48
determined under similar conditions. Moreover, a solid dosage form may provide
a
cmaX value relative to that of commercially available Lipitor tablets of at
least about
1.0, or at least about 1.1, or at least about 123, or at least about 1.3, or
at least
about 1.4, or at least about 1.5 or more, or at least about 2.0, or at least
about 2.5,
or at least about 3.0, the cmax values being determined under similar
conditions.
[0213] In a typical average blood plasma sample, the AUCo-24 of fenofibrate
resulting from the administration of 160 mg fenofibrate tablets are about
118,300
ng h/mL. However, wide individual variations in bioavailability are usually
observed.
Other aspects of the invention
[0214] A pharmaceutical composition or a solid dosage form according to the
invention is designed to release the fenofibrate in a suitable manner.
Specific
release patterns as well as specific absorption patterns are mentioned below.
[0215] In specific embodiments, the fenofibrate and/or the atorvastatin is
released
from the composition within about 2 hours such as, e.g., within about 1.5
hours or
within about 1 hour after oral administration, and/or about 50% w/w or more of
the
fibrate and/or the statin is released from the composition within about 30 min
after
oral administration, and/or about 50% w/w or more of the fibrate and/or the
statin
is released from the composition within about 20 min after oral
administration,
and/or about 60% w/w or more of the fibrate is released from the composition
within about 1.5 hours after oral administration, and/or about 60% w/w or more
of
the fibrate and/or the statin is released from the composition within about 1
hour
after oral administration, and/or about 70% w/w or more of the fibrate and/or
the
statin is released from the composition within about 1.5 hours after oral
administration, and/or about 70% w/w or more of the fibrate and/or the statin
is
released from the composition within about 1 hour after oral administration,
and/or
about 85% w/w or more of the fibrate and/or the statin is released from the
composition within about 45 min when tested in an in vitro dissolution test
according to USP dissolution test (paddle) employing water as dissolution
medium, 100 rpm and a temperature of about 37 C.
[0216] In another embodiment about 50% w/w or more of the fenofibrate and/or
the atorvastatin is released from the composition within about 20 min, 15 min
or
1 0min, and/or about 60% w/w or more of the fibrate and/or the statin is
released
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
49
from the composition within about 20 min or 15 min, and/or about 70% w/w or
more of the fibrate and/or the statin is released from the composition within
about
20 min or 15 min, when tested in an in vitro dissolution test according to USP
dissolution test (paddle) employing water as dissolution medium, 100 rpm and a
temperature of about 37 C.
[0217] In a still further embodiment about 50% w/w or more of the fenofibrate
and/or the atorvastatin contained in the composition is absorbed within about
8
hours, 7 hours, 6 hours or 5 hours, and/or about 60% w/w or more of the
fibrate
and/or statin contained in the composition is absorbed within about 8 hours or
7
hours after oral administration, and/or about 60% w/w or more of the fibrate
contained in the composition is absorbed within about 7 hours after oral
administration, and/or about 70% w/w or more of the fibrate contained in the
composition is absorbed within about 8 hours or 7 hours after oral
administration.
[0218] The details and particulars disclosed under this main aspect of the
invention
apply mutatis mutandis to the other aspects of the invention. Accordingly, the
properties with respect to increase in bioavailability, changes in
bioavailability
parameters, reduction in adverse food effect as well as release of one or more
fibrates etc. described and/or claimed herein for pharmaceutical compositions
in
particulate form are analogues for a solid dosage form according to the
present
invention.
Materials and methods
Materials
[00219] Fenofibrate (supplied by Sigma)
[00220] Lactose monohydrate 200 mesh (from DMV)
[00221] Granulated silicium oxide, Aeroperl 300, (Degussa)
[00222] Polyethylene glycol 6000, Pluracol E6000 (from BASF)
[00223] Poloxamer 188, Pluronic F-68 (from BASF)
[00224] Glyceryl monostearate, Rylo MD50, (from Danisco Cultor), Ph.Eur.
[00225] Avicel PH200 (microcrystalline cellulose) (from FMC)
[00226] Magnesium stearate
[00227] Tablets, capsules or granules might be enteric coated with different
types of polymers such as hydroxypropylmethylcellulose acetate succinate
(Aqoat), cellulose acetate phthalate CAP, hydroxypropylmethylcellulose
phtalate
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
HPMCP or methacrylic acid copolymers such as Eudragit L30D, Eudragit 100/S,
Eudragit 100/L.
TriCor /Lipanthyl tablet formulation
[00228] TRICOR (Lipanthyl ) tablets from Abbott Laboratories are
fenofibrate-containing tablets available for oral administration, either
containing 48
mg or 54 mg or 145 mg or 160 mg of fenofibrate per tablet.
[00229] The tablets contain the following inactive ingredients: colloidal
silicon
dioxide, crospovidone, lactose monohydrate, lecithin, microcrystalline
cellulose,
polyvinyl alcohol, povidone, sodium lauryl sulfate, sodium stearyl fumarate,
talc,
titanium dioxide, xanthan gum, colorant.
Equipment
[00230] Laboratory scale fluid bed equipment: Strea-1.
[00231] The melt feed unit is a prototype composed of separate units for
heating of air supplies for the atomizer, pressure tank and feeding tube.
Granulate
was sieved manually and mixed with extragranular excipients in a Turbula
mixer.
[00232] Tablet compression was performed on a single punch press, Diaf
TM20.
Methods
[0233] According to the method of the invention, the fenofibrate drug was
dissolved into the liquefied vehicle(s) and applied on the particulate
carrier(s) as
follows:
[0234] The vehicle(s) was melted in a beaker placed in a microwave oven. The
beaker was transferred to a temperature controlled heating plate supplied with
magnetic stirring. Fenofibrate was dissolved slowly in the liquefied vehicle
at a
temperature of 75 C under magnetic stirring. The hot solution was transferred
to
the pressure tank for melt spray application onto the carrier in the fluid
bed. The
granulate product was discharged from the fluid bed and sieved through sieve
0.7
mm or 1.0 mm manually. The sieved product was blended with magnesium
stearate for 0.5 min in a Turbula mixer. If an extragranular phase has to be
incorporated, the extragranular phase was premixed with granulate in 3 minutes
in
a Turbula mixer.
[0235] The tablet compression was performed on a single punch machine Diaf
TM20.
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
51
Threshold test
[0236] The test involves determination of flowability according to the method
described in Ph.Eur. by measuring the flow rate of the material out of a
funnel with
a nozzle diameter of 10.0 mm.
[0237] Viscoleo (medium chain triglycerides MCT; Miglyol 812 N from Condea)
was added to 100 g of the solid pharmaceutically acceptable material to be
tested
for use according to the invention and mixed manually. The mixture obtained
was
sieved through sieve 0.3 mm to assure a homogenous mixture. The oil was added
successively until a flow of 100 g of the mixture could not flow through the
nozzle.
If the material to be tested has a high bulk volume (e.g. like that of
Aeroperl 300)
only 50 g of the mixture is used when testing these blends. The maximal
concentration of oil where flow of material could be obtained is called the
Threshold Value (given as % w/w).
Release test
[0238] A fat-soluble colorant Sudan II (BDH Gur:f_~) obtained from BDH VWR
International 14.3 mg was dissolved in 50.0 g viscoleo (fractionated medium
chain
triglycerides).
[0239] 10 g of the oil was added to 10.0 g of the solid pharmaceutically
acceptable
material to be tested for use according to the present invention and mixed
until the
oil was fully absorbed in the solid material. The mixture was subsequently
sieved
through sieve 0.3 mm to achieve a homogeneous mixture.
[0240] 1.00 g of the mixture was transferred to a centrifugal tube and 3.00 ml
of
water was added. The suspension was mixed in a blood sample turner for 1 hour
and subsequently centrifuged for 10 minutes at 5000 rpm. The upper phase of
oil
and water was transferred carefully to a beaker and the water was evaporated
in
an oven at 80 C until constant weight. The amount of oil released from the
solid
material was calculated on basis of the weight of the remaining after
evaporation
of the water phase.
Disintegration test
[0241] The disintegration time was determined according to the method
described
in to Ph. Eur.
Dissolution test
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
52
[0242] The test was performed in accordance with Ph. Eur 2.9.3 using the
paddle
apparatus. The quantification was performed using HPLC with UV-detection.
[0243] Medium: 900 ml water with 0.75 % sodium lauryl sulfate (SLS)
[0244] Rotation speed: 50 rpm
[0245] Temperature: 37 C
[0246] Sampling time: 10, 20, 30, 45 and 60 minutes
[0247] Acceptance criteria: > 75 % at 45 minutes (for the stability study)
Determination of Bulk Density
[0248] The bulk density was measured by pouring 100 g of the powder in
question
in a 250 ml graduated cylinder. The bulk density is given as the tapped bulk
density in g/ml. The determination was performed according to Ph. Eur.
(apparent
volume).
Determination of Oil Absorption Value
[00249] The oil absorption value is determined by adding well-defined
amounts (a 10 g) of viscoleo to a well-defined amount of the pharmaceutically
acceptable material (100 g) to be tested. The oil absorption value (expressed
as g
viscoleo/100 g material) is reached when a further addition of 10 g oil
results in a
material that does not have suitable properties with respect to flowability,
i.e. the
material does not meet the meet the requirements when tested according to
Ph.Eur. (flowability test; see above under Threshold Test herein).
Determination of BET Surface Area
[0250] The apparatus applied was a Micromertics Gemini 2375. The method
applied was according to USP volumetric methods based on multiple point
determination.
Determination of Flowability
[0251] The flowability was determined according to the method described in
Ph.Eur. measuring the flow rate of the material out of a funnel with a nozzle
diameter of 10.0 mm.
Determination of weight variation
[0252] The tablets prepared in the Examples herein were subject to a test for
weight variation performed in accordance with Ph. Eur.
Determination of average tablet hardness
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
53
[0253] The tablets prepared in the Examples herein were subject to at test for
tablet hardness employing Schleuniger Model 6D apparatus and performed in
accordance with the general instructions for the apparatus.
Determination of solid solution
[0254] According to the present invention, the fenofibrate is dissolved in a
vehicle.
In order to substantiate this, a test involving differential scanning
calometry is
performed. The test is performed on the particulate composition, solid dosage
form
or mixture of vehicle and fibrate (after the solid solution is supposed to
form).
Standard DSC equipment connected to a PC is used.
[0255] Sample size: 10 mg in alu pans
[0256] Heating rate: 5 C /min from 27 C to 110 C
[0257] Evaluation: The fibrate and statin are considered to be in dissolved
state or non-crystalline if neither fibrate nor statin endoterm peaks are
observed
and if the melting intervals do not significantly shift compared with the
vehicle
alone.
Determination of geometric weight mean diameter dgw
[0258] The geometric weight mean diameter was determined by employment of a
method of laser diffraction dispersing the particulate material obtained (or
the
starting material) in air. The measurements were performed at 1 bar dispersive
pressure in Sympatec Helos equipment, which records the distribution of the
equivalent spherical diameter. This distribution is fitted to a log normal
volume-size
distribution.
[0259] When used herein, "geometric weight mean diameter" means the mean
diameter of the log normal volume-size distribution.
In vivo studies in Beagle dogs
[0260] In vivo studies with the purpose of determining the bioavailability of
the
compositions of the present invention relative to the bioavailability of the
commercially available fenofibrate tablet formulation, i.e. Tricor , was
performed
using Beagle dogs.
[0261] The experimental work was performed in Denmark using four male Beagle
dogs each having a body weight of 12-18 kg (starting weight). The studies were
conducted as open, non-randomised, cross-over studies. Each animal was its own
control. Oral doses of fenofibrate were administered according to the data
below.
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
54
[0262] The dogs were fasted overnight prior to dosing (water ad libitum) and
were
fed 5 hours after dosing (water ad libitum). Each dog was dosed with the
specified
dose of fenofibrate without taking the weight of the dog into consideration.
[0263] Blood samples were collected at vena jugularis externa at the following
points of time:
[0264] Pre-dose, 1, 1.5, 2, 3, 4, 6, 8, 12 and 24 hours after dosing. 4 ml of
blood
were collected, mixed with EDTA, and the samples were frozen (-80 C). The
blood
samples were analyzed using on-line extraction LC/MS and results were given in
mg/mL.
[0265] The determined full blood concentration profiles of fenofibrate were
treated
using the Pharmacokinetic software WinNonlin , (Pharsight, California;USA) to
calculate the pharmacokinetic parameters. All data are dose adjusted, when
necessary.
[0266] The following examples serve the purpose of illustration of the
invention
and are not intended to limiting the scope of the present invention.
Example 1
Immediate release tablet containing a fenofibrate and atorvastatin
[0267]
Table 1
Substance Ingredient mg
Drug Fenofibrate 130.00
Drug Atorvastatin calcium 10.00
Carrier Lactose 247.64
Vehicle PEG 6000 170.88
Vehicle Poloxamer 188 73.24
Excipient Magnesium stearate 2.69
Total 637.45
[0268] Fenofibrate and atorvastatin are mainly dissolved/dispersed in
polyethylene
glycol 6000 and poloxamer 188 (70:30 w/w ratio) at 70=C. The dispersion is
sprayed on 250 g lactose in a fluid bed Phast FB-1 00 with a Phast FS-1.7 melt-
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
spray unit. The particular material obtained is sieved through sieve 0.7 mm
and
blended with magnesium stearate for 0.5 min in a Turbula mixer.
[0269] The powder mixture is compressed into 13 mm tablets with strength of
130
mg fenofibrate and 10 mg atorvastatin in to a 637 mg tablet with compound cup
shaped.
[0270] Mean disintegration time: 20 min, Hardness: 45 N
Example 2
Immediate release tablet containing fenofibrate and atorvastatin
[0271]
Table 2
Substance Ingredient mg
Drug Fenofibrate 120.00
Drug Atorvastatin Mg 20.00
Carrier Lactose 261.00
Vehicle PEG 6000 171.00
Vehicle Poloxamer 188 73.00
Excipient Magnesium stearate 3.00
Total 648.00
[0272] Fenofibrate and atorvastatin are mainly dissolved/dispersed in
polyethylene
glycol 6000 and poloxamer 188 (70:30 w/w ratio) at 70 C. The dispersion is
sprayed on 261 g lactose in a fluid bed Phast FB-1 00 with a Phast FS-1.7 melt-
spray unit. The particular material obtained is sieved through sieve 0.7 mm
and
blended with magnesium stearate for 0.5 min in a Turbula mixer.
[0273] The powder mixture is compressed into 13 mm tablets with strength of
120
mg fenofibrate and 20 mg atorvastatin into a 648 mg tablet with compound cup
shaped.
[0274] Mean disintegration time: 25 min, Hardness: 47 N
Example 3
Immediate release tablet containing fenofibrate and atorvastatin
[0275]
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
56
Table 3
Substance Ingredient mg
Drug Fenofibrate 120.00
Drug Atorvastatin calcium 10.00
Carrier Lactose 241.00
Vehicle PEG 6000 171.00
Vehicle Poloxamer 188 73.00
Excipient Magnesium stearate 3.00
Total 618.00
[0276] Fenofibrate and atorvastatin are mainly dissolved/dispersed in
polyethylene
glycol 6000 and poloxamer 188 (70:30 w/w ratio) at 70 C. The dispersion is
sprayed on 250 g lactose in a fluid bed Phast FB-1 00 with a Phast FS-1.7 melt-
spray unit. The particulate material obtained is sieved through sieve 0.7 mm
and
blended with magnesium stearate for 0.5 min in a Turbula mixer.
[0277] The powder mixture is compressed into 12 mm tablets with strength of
120
mg fenofibrate and 10 mg atorvastatin into a 618 mg tablet with compound cup
shaped.
[0278] Mean disintegration time: 22 min, Hardness: 41 N
Example 4
Immediate release tablet containing fenofibrate and atorvastatin
[0279]
Table 4
Substance Ingredient mg
Drug Fenofibrate 120.00
Drug Atorvastatin (amorph.) 30.00
Carrier Lactose 266.00
Vehicle PEG 6000 171.00
Vehicle Poloxamer 188 73.00
Excipient Magnesium stearate 3.00
Total 673.00
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
57
[0280] Fenofibrate and atorvastatin are mainly dissolved/dispersed in
polyethylene
glycol 6000 and poloxamer 188 (70:30 w/w ratio) at 700C. The dispersion is
sprayed on 266 g lactose in a fluid bed Phast FB-1 00 with a Phast FS-1.7 melt-
spray unit. The particulate material is sieved through sieve 0.7 mm and
blended
with magnesium stearate for 0.5 min in a Turbula mixer.
[0281] The powder mixture is compressed into 13 mm tablets with strength of
120
mg fenofibrate and 30 mg atorvastatin into a 673 mg tablet with compound cup
shaped.
Example 5
Tablet based on lipophilic matrix of glyceryl monostearate
[0282]
Table 5
Substance Ingredient mg
Drug Fenofibrate 120.00
Drug Atorvastatin 10.00
Carrier Lactose 200 mesh 100.00
Vehicle Glycerylmonostearate 300.00
Excipient Magnesium stearate 2.00
532.00
[0283] Fenofibrate and atorvastatin are mainly dissolved/dispersed in glyceryl
monostearate at 70 C. The solution is sprayed on 200 g lactose in a fluid bed
Phast FB-1 00 with a Phast FS-1.7 melt-spray unit. The particulate material is
sieved through sieve 0.7 mm and blended with magnesium stearate for 0.5 min in
a Turbula mixer.
[0284] The powder mixture is compressed into 11 mm tablets with 532 mg tablet
with compound cup shape.
Example 6
Modified release poly-depot capsule based on swelling hydrocolloid matrix of
hyd roxyp ropylce l I u lose
[0285]
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
58
Table 6
Substance Ingredient mg
Drug Fenofibrate 120.00
Drug Atorvastatin calcium 20.00
Carrier HPMC 2910 3 cp 150.00
Carrier Lactose 200 mesh 50.00
Vehicle Glyceryl monostearate 300.00
Total 640.00
[0286] Fenofibrate and atorvastatin are mainly dissolved/dispersed in
glycerylmonostearate at 70 C. The solution is sprayed on a mixture of 50 g
lactose
and 150 g HPMC in a fluid bed Phast FB-1 00 with a Phast FS-1.7 melt-spray
unit.
The particulate material is sieved through sieve 0.7 mm and filled into hard
gelatine capsules (640 mg)
Example 7
Immediate release tablet
[0287]
Table 7
Substance Ingredient mg
Drug Fenofibrate 120.00
Drug Atorvastatin (amorph.) 40.00
Oil-sorption
material Aeroperl 300 95.00
Vehicle PEG 3000 195.00
Excipient Magnesium stearate 3.00
Total 463.00
[0288] Fenofibrate and atorvastatin are mainly dissolved/dispersed in
polyethylene
glycol 3000 at 70 C. The dispersion is sprayed on 95 g Aeroperl in a fluid bed
Phast FB-1 00 with a Phast FS-1.7 melt-spray unit. The particulate material is
sieved through sieve 0.7 mm and blended with magnesium stearate for 0.5 min in
a Turbula mixer.
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
59
[0289] The powder mixture is compressed into 11 mm tablets with strength of
120
mg fenofibrate and 40 mg atorvastatin into a 463 mg tablet with compound cup
shaped.
Example 8
Solid dosage forms according to the invention
[0290] The following compositions were prepared according to the method
described in Example 1 above.
[0291]
Table 8
Substance Ingredient B C D E F G
mg mg mg mg mg mg
Drug Fenofibrate 50 50 50.1 160 130 43
Drug Atorvastatin 10 10 10.0 40 20 10
Vehicle 1 PEG6000 171.1 124.3 - - 169 56
PEG4000 - - 244.6 - - -
GMS (Rylo) - - - 86.2 - -
Vehicle 2 Poloxamer188 73.3 53.3 - - 72 24
Carrier Lactose 231.9 - 232.0 163. 304 101
Aeroper1300 - 63.9 - 0 - -
Excipients Mg stearate 2.7 1.5 5.3 8.3 1.3 0.5
Avicel - - - 417. - -
Total 53 30 54 87 69 234
Hardness N 44 44 47 102
Disintegra minutes 14 30 48 >55
-tion time
Diameter mm 12 12 10 ObI. obl obl
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
Example 9 (A-E)
Methods of manufacturing fenofibrate -atorvastatin combinations
[0292] There are several useful methods for preparing combination products
according to this invention. The method is primarily selected from the desired
characteristics and performance of the composition or solid dosage form. In
examples 9A-9E is given a number of compositions and methods of production.
The methods shown are by no means intended to limit the scope of this
invention.
[0293] All granulates listed herein can either be filled into hard gelatin
capsules or
compressed into tablets.
[0294] The following fenofibrate granulate A is disclosed in international
application
PCT/DK2004/000667, granulate B is prepared in a similar manner:
[0295] Composition/table 9:
[0296]
Table 9
A B
Substance Ingredient mg mg
Drug Fenofibrate 160.0 120.0
Carrier Lactose 356.5 292.0
Vehicle PEG 6000 208.2 162.5
Vehicle Poloxamer 188 89.2 69.5
Excipient Magnesium stearate 4.1 6.0
818.0 650.0
[0297]
Example 9A
[00298] Al.
[0299] The fenofibrate granulate A (table 9) was used.
[0300] The fenofibrate granulate is mixed with another granulate containing
atorvastatin. This statin granulate is as follows:
[0301]
Table 10
Substance Ingredient mg
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
61
Drug Atorvastatin amorphous 10.00
Carrier Lactose 200 mesh 50.00
Vehicle PEG 6000 66.00
Vehicle Poloxamer 188 22.00
Excipient Magnesium stearate 2.00
150.00
[0302] The granulate obtained is sieved through sieve 0.7 mm and blended with
the fenofibrate granulate and magnesium stearate for 0.5 min in a Turbula
mixer.
[0303] The final granulate is compressed into 13.5 mm tablets with strength of
160
mg fenofibrate and 10 mg atorvastatin into a 970 mg tablet with compound cup
shaped.
[0304] Mean disintegration time: 24 min, Hardness: 49 N
[00305] A2.
[0306] The fenofibrate granulate B (table 9) was used, and tablets having the
following composition were prepared:
[0307] Tablet composition:
[0308]
Table 11
Substance Ingredient mg
Drug I Fenofibrate 120.0
Drug II Atorvastatin amorph. 10.0
Carrier Lactose monohydrate 332.0
Vehicle PEG 6000 (Macrogol) 163.0
Vehicle Poloxamer 188 70.0
Excipients Magnesium stearate 6.0
Avicel PH200
(microcryst. cellulose) 103.0
Calcium carbonate 33.0
Ac-d i-soI
(croscarmellose Na) 10.0
Klucel (hydroxypropyl-
cellulose) 3.0
Polysorbate 80 0.5
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
62
(Tween)
Total 850.5
[0309] Fenofibrate granulate was prepared as described in PCT/DK2004/000667.
[0310] Atorvastatin granulate was prepared in a conventional manner using wet
granulation, i.e. mixing atorvastatin, lactose and calcium carbonate, adding
the
appropriate amount of Klucel and Ac-di-sol, adding sterile water to the
mixture,
mixing and drying off the water, sifting the dried mixture and adding
magnesium
stearate and Avicel. The resulting tablets were oblong, white to slightly pale
yellow
tablets (7.0 mm x 18 mm).
Example 9B
[0311] A single granulate comprising fenofibrate and atorvastatin is made as
follows:
[0312]
Table 12
Substance Ingredient mg
Drug Fenofibrate 120.0
Drug Atorvastatin calcium 30.0
Carrier Lactose 329.0
Vehicle PEG 6000 188.0
Vehicle Poloxamer 188 81.0
Excipient Magnesium stearate 4.0
Total 752.0
[0313] Fenofibrate and atorvastatin are mainly dissolved/dispersed in
polyethylene
glycol 6000 and Poloxamer 188 (70:30 w/w ratios) at 70 C. The dispersion is
sprayed on 329 g lactose in a fluid bed Phast FB-1 00 with a Phast FS-1.7 melt-
spray unit. The particulate material obtained is sieved through sieve 0.7 mm
and
blended with magnesium stearate for 0.5 min in a Turbula mixer.
[0314] The granulate is compressed into 13.5 mm tablets with strength of 120
mg
fenofibrate and 30 mg atorvastatin into a 752 mg tablet with compound cup
shaped.
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
63
Example 9C
[0315] A single granulate comprising fenofibrate and atorvastatin is made as
follows:
[0316]
Table 13
Substance Ingredient mg
Drug Fenofibrate 120.00
Drug Atorvastatin amorph. 10.00
Carrier Lactose 349.00
Vehicle PEG 6000 208.00
Vehicle Poloxamer 188 89.00
Excipient Magnesium stearate 4.00
Total 780.00
[0317] Fenofibrate is dissolved in polyethylene glycol 6000 and Poloxamer 188
(70:30 w/w ratios) at 70=C. The dispersion is sprayed on a mixture of 349 g
lactose
and 10 g of atorvastatin in a fluid bed Phast FB-1 00 with a Phast FS-1.7 melt-
spray unit. The particulate material obtained is sieved through sieve 0.7 mm
and
blended with magnesium stearate for 0.5 min in a Turbula mixer.
[0318] The granulate is compressed into 13.5 mm tablets with strength of 120
mg
fenofibrate and 10 mg atorvastatin into a 780 mg tablet with compound cup
shaped.
Example 9D
[0319] A fenofibrate granulate A of table 9 was used.
[0320] The fenofibrate granulate is mixed with a granulate similar to the
granulate
composition of LipitorTM tablets of either 10, 20 or 40 mg of atorvastatin in
order to
obtain the same plasma profiles as those of LipitorTM .
[0321] LipitorTM based granulates may have the following composition(s):
[0322] 10 mg atorvastatin per 150 mg granulate:
[0323] Atorvastatin calcium trihydrate 10.9 mg
[0324] Microcrystalline cellulose 60.0 mg
[0325] Calcium carbonate 33.0 mg
[0326] Lactose monohydrate 32.8 mg
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
64
[0327] Croscarmellose sodium 9.0 mg
[0328] HPMC 3.0 mg
[0329] Polysorbate 80 0.6 mg
[0330] Magnesium stearate 0.7 mg
[0331] 20 mg atorvastatin per 300 mg granulate:
[0332] Atorvastatin calcium trihydrate 21.8 mg
[0333] Microcrystalline cellulose 120.0 mg
[0334] Calcium carbonate 66.0 mg
[0335] Lactose monohydrate 65.6 mg
[0336] Croscarmellose sodium 18.0 mg
[0337] HPMC 6.0 mg
[0338] Polysorbate 80 1.2 mg
[0339] Magnesium stearate 1.4 mg
[0340] 40 mg atorvastatin per 600 mg granulate:
[0341] Atorvastatin calcium trihydrate 43.4 mg
[0342] Microcrystalline cellulose 240.0 mg
[0343] Calcium carbonate 132.0 mg
[0344] Lactose monohydrate 131.2 mg
[0345] Croscarmellose sodium 36.0 mg
[0346] HPMC 12.0 mg
[0347] Polysorbate 80 2.4 mg
[0348] Magnesium stearate 3.0 mg
[0349] The fenofibrate granulate and the "Lipitor" granulate are mixed in a
turbula
mixer and the final granulate is then either filled into hard gelatin capsules
or
compressed into tablet with a suitable crushing strengths around 40-50 N.
Example 9E
[0350] A fenofibrate granulate B of table 9 was manufactured.
[0351] The fenofibrate granulate is mixed with micronized atorvastatin,
optionally
added conventional excipients or additives for tablet production like a
glidant, filler,
binder, or disintegrator.
[0352] The granulate is either filled into hard gelatin capsules or compressed
into
tablet with a suitable crushing strength.
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
Example 9F
[0353] A fenofibrate granulate B of table 9 was manufactured. The fenofibrate
granulate were compressed into oblong tablets 19.9 x 8 mm with a mean tablet
hardness of 80 N.
[0354] The combination product of fenofibrate and atorvastatin is prepared by
coating the fenofibrate tablets with a coating comprising atorvastatin, i.e.
an
aqueous suspension of atorvastatin including a film-forming polymer and
stabilizers (antioxidants).
[0355] The fenofibrate tablets may optionally be sub-coated with a film-
forming
polymer prior to coating with the atorvastatin suspension.
[0356] The aqueous suspension of atorvastatin has the following composition:
[0357]
Table 14
Substance %
Kollicoat 10
Atorvastatin 5
Ascorbic acid 0.03
BHA 0.01
Citric acid 0.75
Water 84.21
Total 100
[0358] The fenofibrate tablets are coated with the coating suspension in a
fluid bed
Phast FB 100 equipped with a coating insert (top-spray) using an inlet air
temperature of 50 C, a product temperature of about 40-45 C, a feed rate of 9
g/min and a tablet load of 700 g.
[0359] Each tablet is coated with approx 171 g coating suspension
corresponding
to 10 mg atorvastatin per tablet.
[0360] The atorvastatin-coated fenofibrate tablets may additionally be coated
with
a suitable polymer to protect atorvastatin from degradation.
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
66
Example 10
Formulations for in vivo studies in dogs
[0361] Compositions of the invention were investigated in in vivo studies in
dog. As
fenofibrate is a drug substance that has major bioavailability problems, the
study
was primarily to investigate whether an improved bioavailability could be
obtained.
Accordingly, no data with respect to the statin component is available.
[0362] Tablets of 50 mg and 160 mg strength with respect to fenofibrate,
respectively and having the following compositions were prepared as described
in
Example 1:
[0363]
Table 15
Substance Ingredient A B C D E
mg mg mg mg mg
Drug Fenofibrate 160.09 50.05 50.08 50.09 159.99
Vehicle 1 PEG6000 208.12 171.09 124.29 - -
PEG4000 - - - 244.57 -
GMS (Rylo) - - - - 86.15
Vehicle 2 Poloxamer18 89.19 73.33 53.27 - -
8
Carrier Lactose 356.51 231.87 - 232.02 163.01
Aeroper1300 - - 63.89 - -
Excipients Mg stearate 4.09 2.65 1.47 5.32 8.35
Avicel - - - - 417.50
Total 818.00 529.00 293.00 532.00 835.00
Hardness N 60 44 44 47 102
Disintegration Minutes 25 14 30 48 >55
time
Diameter Mm Oblong 12 12 10 Oblong
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
67
Example 11
Dissolution tests
[0364] The tablet formulation A from Example 10 was subjected to a dissolution
test as described in Methods with the following results:
[0365]
Table 16
Time (min) % dissolved
0 0
28
56
74
45 88
60 97
Example 12
Stability tests
[0366] Samples of the tablet formulation A from Example 10 was stored in PP
bottles under the following conditions, respectively, and subjected to a
dissolution
(stability) test as described in Methods after 1 month and 3 months of
storage; %
dissolved is the percentage of fenofibrate dissolved after 45 minutes:
[0367]
Table 17
Months % dissolved
25 C and 60% 30 C and 65% 40 C and 75%
RH RH RH
0 88 - -
1 99 88 90
3 90 97 90
[0368] Samples of the tablet formulation A was stored under the following
conditions, respectively, and subjected to a fibrate assay with the following
results:
[0369]
Table 18
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
68
Months mg fenofibrate
25 C and 60% 30 C and 65% 40 C and 75%
RH RH RH
0 163.8 - -
1 161.9 160.1 160.8
3 162.6 164.9 164.4
[0370] Samples of the inventive tablet formulation A was stored under the
following
conditions, respectively, and subjected to a degradation product test
according to
Ph. Eur. (Degradation products A, B, G and Unknown accumulated into Total
Degradation Product; HPLC method) with the following results:
[0371]
Table 19
Months Total Degradation Product, %w/w, impurity
25 C and 60% 30 C and 65% 40 C and 75%
RH RH RH
0 0.05 - -
1 0.05 0.05 0.05
3 0.05 0.05 0.05
Example 13
In vivo study in dogs
[0372] An in vivo study of formulation A from Example 10 160 mg in Beagle
dogs,
performed as described above under Methods, relative to Tricor , 160 mg
(Batch
no.: 098212E21), gave the following results:
[0373] Blood concentrations (mg/mL) (average of 4 dogs) after administration
of
formulation:
[0374]
Table 20
Time Formulation
(hr) Tricor Invention, A
(160mg) (160 mg)
0 n.a. n.a.
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
69
0.5 367.5 995.8
1.0 612.5 2209.3
1.5 722.0 2627.8
2.0 725.8 2097.3
3.0 443.8 1219.5
4.0 295.3 930.5
6.0 160.5 642.0
8.0 250.3 869.5
12.0 211.8 615.3
24.0 133.3 394.0
48.0 n.a. 164.5
[0375] Relative bioavailability based on AUC (invention, A/Tricor ): 306%.
[0376] Relative cmaX(invention, A/Tricor ): 356%.
Example 14
In vivo study in dogs
[0377] A second in vivo study of formulation A (Example 10), 160 mg in Beagle
dogs, performed as described above under Methods, relative to Tricor , 160 mg
(Batch no.: 098212E21), gave the following results:
[0378] Blood concentrations (mg/mL) (average of 4 dogs) after administration
of
formulation:
[0379]
Table 21
Time Formulation
(hr) Tricor (160mg) Invention, A
(160 mg)
0 0 0
0.5 339.3 3616.0
1.0 1318.8 3724.8
1.5 1313.3 2982.0
2.0 1390.0 2355.8
3.0 1361.3 1359.5
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
4.0 1019.3 1309.5
6.0 969.3 973.8
8.0 667.0 1113.0
12.0 390.3 768.5
24.0 183.3 295.0
48.0 85.0 302.0
[0380] Relative bioavailability based on AUC (invention, A/Tricor ): 198%.
[0381] Relative cmaX(invention, A/Tricor ): 238%.
Example 15
In vivo study in dogs
[0382] An in vivo study of the formulations B, C and D (Example 10), 2x50 mg
in
Beagle dogs, performed as described above under Methods, relative to
Lipanthyl 67M, 2x67 mg (Batch no.: 75641), gave the following results:
[0383] Blood concentrations (mg/mL) (average of 4 dogs) after administration
of
formulation:
[0384]
Table 22
Time Formulation
(hr) Lipanthyl 67M Invention, B Invention, C Invention, D
(2x67mg) (2x50 mg) (2x50 mg) (2x50 mg)
0 0 0 0 0
0.5 187.3 2769.5 227.3 546.0
1.0 669.5 3526.8 521.5 1381.5
1.5 960.3 3106.3 858.3 1615.5
2.0 895.3 2938.0 989.3 1566.8
3.0 433.0 2465.5 902.5 1503.3
4.0 240.0 1492.3 783.8 1719.0
6.0 77.8 809.5 655.8 1034.5
8.0 79.3 1202.8 409.0 1056.0
12.0 291.3 848.0 269.8 597.3
24.0 82.5 378.0 163.8 282.8
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
71
48.0 19.3 18.8 51.5 36.5
72.0 0 0 0 0
[0385] Relative bioavailability based on AUC (invention, B/ Lipanthyl 67M):
532%.
[0386] Relative cmaX(invention,BA/Lipanthyl 67M): 548%.
[0387] Relative bioavailability based on AUC (invention, C/ Lipanthyl 67M):
228%.
[0388] Relative cmaX(invention, C/Lipanthyl 67M): 161 %.
[0389] Relative bioavailability based on AUC (invention, D/ Lipanthyl 67M):
424%.
[0390] Relative cmaX(invention, D/Lipanthyl 67M): 329%.
Example 16
Clinical trial of fenofibrate formulation used in the fenofibrate and
atorvastatin
composition of the invention
[0391] A clinical trial study was carried out in order to determine the
pharmacokinetic profile of the fenofibrate formulation used in the combination
composition of this invention, 160 mg tablets taken with food and without food
in
comparison with Lipanthyl (Tricor ) 160 mg tablets taken with and without
food.
[0392] The study was conducted in Switzerland as a randomized, four-way cross-
over study including 24 healthy volunteers (aged 27-55 years; 21 males and 3
females; body weight > 65 kg); 23 subjects concluded the study, 1 subject
dropped
out after period 3 for personal reasons (missing period: Lipanthyl fasted).
[0393] The study was carried out as a combined PK and food-effect study
according to FDA guidelines.
[0394] The objective was to demonstrate that the fenofibrate formulation used
in
the combination composition of the present invention (administered in fed
state)
and Lipanthyl (administered in fed state) are bioequivalent and, further that
the
present fenofibrate formulation, when administered in fed state, is
bioequivalent to
the identical formulation administered in fasted state.
[0395] Conditions (fed state) were according to Guidance for Industry: Food-
effect
Bioavailability and Fed Bioequivalence Studies; CDER December 2002: An
overnight fast of the subjects of at least 10 hours; high-fat, high-calorie
breakfast
within 30 minutes or less; 800-1000 calories in total (150 from protein; 250
from
carbohydrate; 500-600 from fat); 240 ml plain water at study drug
administration.
CA 02582405 2007-04-02
WO 2006/037347 PCT/DK2005/050004
72
[0396] Conditions (fasted state) were according to Guidance for Industry: Food-
effect Bioavailability and Fed Bioequivalence Studies; CDER December 2002: An
overnight fast of the subjects of at least 10 hours; no breakfast and no food
intake
4 hours after drug administration; 240 ml plain water at study drug
administration.
[0397] The following results were found:
[0398] AUCo-24(invention fenofibrate formulation fed)/ AUCo-24 (invention
fenofibrate formulation fasted): 106.9% (Cl 101-114%).
[0399] AUCo-24(invention fenofibrate formulation fed)/ AUCo-24(Lipanthyl fed):
98.0% (Cl 93-103%).
[0400] AUCo-inf(invention fenofibrate formulation fed)/ AUCo-inf (invention
fenofibrate
formulation fasted): 104.9% (Cl 98-111 %).
[0401 ] AUCo-inf(invention fenofibrate formulation fed)/ AUCo-inf(Lipanthyl
fed):
97.1 % (Cl 92-102%).
[0402] AUCo-inf(Lipanthyl fed)/ AUCo-inf(Lipanthyl fed): 136%.
[0403] A product is considered bioequivalent with a reference product, when
AUCo-
t, AUCo-inf, Cmax is within 80-125% of the reference product, including the
90%
Confidence Intervals (CI).
[0404] The results shows a markedly increased bioavailability of LCP-Feno
fasted
compared to Lipanthyl fasted.
[0405] The results demonstrate bioequivalence of the present fenofibrate
formulation under fed conditions compared to Lipanthyl (AUCo-t,1 AUCo-inf ,
Cmax)
and of the present fenofibrate formulation fasted compared to the present
fenofibrate formulation fed (AUCo-t,AUCo-inf). It can thus be concluded that
the
fenofibrate formulation used in the combination product of the invention has
no
food effect.
[0406] This invention may be embodied in other forms or carried out in other
ways
without departing from the spirit or essential characteristics thereof. The
present
disclosure is therefore to be considered as in all aspects illustrate and not
restrictive, and all changes which come within the meaning and range of
equivalency are intended to be embraced therein.
[0407] Various references are cited throughout this Specification, each of
which is
incorporated herein by reference in its entirety.