Note: Descriptions are shown in the official language in which they were submitted.
CA 02583054 2007-04-02
WO 2006/037160 PCT/AU2005/001511
1
POROUS POLYELECTROLYTE MATERIALS
Field of the Invention
The present invention relates to porous polyelectrolyte materials,
particularly nanoporous polyelectrolyte materials and to methods of making
such materials. In a preferred embodiment, the invention relates to nanoporous
polyelectrolyte spheres. In a preferred form of the invention, the materials
are
manufactured with the use of mesoporous silica spheres as templates. The
invention also relates to a method of manufacturing such materials, and in
particular, to a method of manufacturing such materials by a layer-by-layer
process.
Background of the Invention
Porous materials have been used as sacrificial host templates for the
synthesis of various materials. In a typical synthetic strategy the
constituent
materials are infiltrated into the pores of the porous material and subjected
to
conditions such that reactions occur leading to the formation of an
interconnected network within the pores. Removal of the template is then
carried out to leave the final product. Mesoporous silicas are porous
materials
with extremely high surface areas and homogenous pores in the range of 2-50
nm. Mesoporous silicas may have a number of different shapes however in one
known embodiment the mesoporous silica material is in the form of a particle
or
sphere. Silane grafting and in-situ synthesis doped with silane chemicals have
been employed to modify the siliceous surface with various functional groups
to
tailor the functional properties of the material.
Due to the unique pore structure of mesoporous silica materials, these
materials have been efficiently used as host porous supports or templates in
replication synthesis. A mesoporous silica material, in particular, provides a
confined space for controlled intra-pore inclusion of materials such as
metals,
metal oxides and carbons. These materials may be infiltrated into the pores,
followed by reduction, crosslinking or carbonization to obtain an
interconnected
network, followed by removal of the silica template (typically by dissolution)
to
form a porous material. Porous materials of this type, especially in
particulate
CA 02583054 2007-04-02
WO 2006/037160 PCT/AU2005/001511
2
form, are of interest in a diverse range of applications including controlled
drug
delivery, molecular separation technology and as hosts for chemical synthesis.
Accordingly it would be desirable to provide new materials of this type as
well as new methods of making such materials, as it would be expected that the
new materials may have a number of interesting properties.
The discussion of documents, acts, materials, devices, articles and the
like is included in this specification solely for the purpose of providing a
context
for the present invention. It is not suggested or represented that any or all
of
these matters formed part of the prior art base or were common general
knowledge in the field relevant to the present invention as it existed in
Australia
before the priority date of each claim of this application.
Summary of the Invention
The present invention aims to provide porous multilayer polyelectrolyte
materials, particularly nanoporous multilayer polyelectrolyte materials. In
one
embodiment, the nanoporous polyelectrolyte material is substantially spherical
and is produced by a layer-by-layer method utilizing a porous silica sphere,
preferably a mesoporous silica sphere, as a template. Preparation of
multilayer
films has the benefit of low cost production, simplicity and versatility and
has the
potential for preparation of materials with designed morphologies in the
presence of suitable templates.
Accordingly, in one embodiment of the invention, there is provided a
porous multilayer polyelectrolyte material including at least two layers of
polyelectrolyte material. In a particularly preferred embodiment the material
includes at least two layers of oppositely charged polyelectrolyte material.
In
another preferred embodiment the material is a nanoporous multilayer
polyelectrolyte material. The porous multilayer polyelectrolyte material is
preferably spherical or substantially spherical.
The pores in the material may be of a wide variety of sizes however the
material preferably includes pores with a pore size of from 5 to 50 nm, even
CA 02583054 2007-04-02
WO 2006/037160 PCT/AU2005/001511
3
more preferably 10 to 50 nm. In a particularly preferred embodiment the pores
are interconnecting to produce an interconnected porous network.
The material may include any suitable number of polyelectrolyte layers
with the number of layers being determined based on the desired properties of
the final material produced. Nevertheless it is preferred that there are from
two
to ten layers of polyelectrolyte material, more preferably from two to eight
layers
of polyelectrolyte material, even more preferably from 4 to 8 layers of
polyelectrolyte material. In one particularly preferred embodiment the
material
includes two layers of polyelectrolyte material. In one particularly preferred
embodiment each layer of polyelectrolyte material is oppositely charged to the
layer(s) of polyelectrolyte material adjacent to it. In another preferred
embodiment the material includes at least two adjacent layers having the same
charge. In a particularly preferred embodiment each layer of polyelectrolyte
material is cross-linked. In one preferred form of the invention the cross-
linking
is such that one or more of the layers of polyelectrolyte material is cross
linked
to an adjacent layer. In another preferred form each layer is internally cross-
linked. In a most preferred form of the invention the layers are both
internally
cross-linked and cross-linked to one or more adjacent layers.
The polyelectrolyte materials used to form the layers may be of any
suitable polyelectrolyte material however it is preferred that each layer
includes
a polyelectrolyte material independently selected from the group consisting of
polymers, biodegradable polymers, poly(amino acids), peptides, glycopeptides,
polypeptides. peptidoglycans, glycosaminoglycans, glycolipids, lipo-
polysaccharides, proteins, glycoproteins, polycarbohydrates; polynucleotides,
modified biopolymers; polysilanes, polysilanols, polyphosphazenes,
polysulfazenes, polysulfide, polyphosphates, nucleic acid polymers,
nucleotides, polynucleotides, RNA and DNA or a mixture thereof.
It is particularly preferred that each layer includes a polyelectrolyte
material independently selected from the group consisting of polyglycolic acid
(PGA), polylactic acid (PLA), poly-2-hydroxy butyrate (PHB), gelatins, (A, B)
polycaprolactone (PCL), poly (lactic-co-glycolic acid) (PLGA), carboxymethyl
CA 02583054 2007-04-02
WO 2006/037160 PCT/AU2005/001511
4
cellulose, carboxymethyl dextran, poly(allylamine hydrochloride), poly(acrylic
acid), poly(sodium 4-styrene sulphonate), poly (diallyldimethylammonium
chloride), poly(vinylsulfate), poly(L-glutamic acid) and poly(L-Iysine) or a
mixture
thereof.
It is particularly preferred that the polyelectrolyte material in at least one
layer contains an amine group. In another preferred embodiment the
polyelectrolyte material in at least one layer contains a carboxylic group. In
a
further preferred embodiment the material includes at least one layer of
poly(acrylic acid). In another preferred embodiment the material includes at
least one layer of poly(allylamine hydrochloride).
The molecular weight of the polyelectrolyte materials used to form the
layers may vary widely with the molecular weights being chosen to provide the
desired functionality to the finished product. It is preferred, however, that
each
polyelectrolyte material has a molecular weight of at least 100, more
preferably
at least 500. Accordingly each polyelectrolyte preferably has a molecular
weight that is from 100 to 1,000,000, even more preferably from 500 to
1,000,000, even more preferably from 500, to 500,000, yet even more
preferably 500 to 100,000, most preferably the polyelectrolyte material has a
molecular weight of from 1000 to 100,000.
As stated above, the materials of the invention may incorporate a wide
variety of polyelectrolyte materials depending upon the desired end use
application of the porous multilayer polyelectrolyte material. As such one can
select the surface or any of the layers of the material to impart the desired
functionality on the material. In one preferred embodiment the material used
to
form at least one polyelectrolyte layer is selected from the group consisting
of
peptides, glycopeptides, polypeptides. peptidoglycans, glycosaminoglycans,
glycolipids, lipopolysaccharides, proteins, glycoproteins and polynucleotides.
In
this embodiment it is preferred that at least one polyelectrolyte layer is a
protein
layer, preferably a protein layer wherein the protein has a molecular weight
of
from I to 500 kDa. In a particularly preferred embodiment the protein is
selected from the group consisting of lysosome, cytochrome C, catalase, bovine
CA 02583054 2007-04-02
WO 2006/037160 PCT/AU2005/001511
serum albumin, immunoglobulin G, protease, RNase A, trypsin, conalbumin,
lactoglobulin, myoglobin, ovalbumin, papain, penicillin acylase, and
subtilisin
Carlsberg.
5 As would be clear to a skilled addressee the polyelectrolyte materials of
the various layers may be chosen to impart a wide variety of functionality on
the
end product.
The invention also relates to methods for the production of materials of
this type which the applicants have found may be readily produced using the
developed techniques.
In yet a further embodiment there is provided a method of manufacturing
a porous muitilayer polyelectrolyte material including the steps of:
(i) providing a porous template;
(ii) depositing layer-by-layer polyelectrolyte material onto the porous
template; and
(iii) removing the template by exposure to a suitable solvent.
In one preferred embodiment the porous template is a mesoporous
template, such as a mesoporous silica template. In this embodiment the
polyelectrolyte material thus formed is a nanoporous material.
Any suitable template may be used in the method of the invention
however it is preferred that the template has an interconnected network of
pores. It is preferred that the template includes pores with a pore size in
the
range 2 to 50 nm, more preferably 10 to 50 nm. The template may be made of
any suitable material but is preferably a silica template. The template may be
any suitable shape but is preferably selected from the group consisting of
planar
supports, powder particles, fibres, tubes, films, membranes and spheres. A
particularly preferred shape for the template is spherical or substantially
spherical.
CA 02583054 2007-04-02
WO 2006/037160 PCT/AU2005/001511
6
In certain embodiments of the method of the invention the exposed
surface of the template is modified in order to facilitate bonding of the
polyelectrolyte material to the template. In a particularly preferred
embodiment
the exposed surface has been modified by grafting 3-aminopropyltriethoxysilane
(APTS) onto the exposed surface.
The steps of depositing the polyelectrolyte materials may be carried out
in a number of ways but the polyelectrolyte material is preferably deposited
in
layers of alternating charge. In general the step of depositing the
polyelectrolyte layers is carried out by contacting the template with a
solution
containing the polyelectrolyte material to be deposited. The solution may be
of
any suitable concentration but it will preferably have a concentration of
polyelectrolyte material of 0.001 to 100 mg mL-', more preferably a
concentration of polyelectrolyte material of 0.1 to 30 mg mL"1, more
preferably
concentration of polyelectrolyte material of 0.5 to 10 mg mL"1. In a
particularly
preferred embodiment the solution includes a salt. The salt preferably has a
concentration of from 0.001 to 5 M, more preferably a concentration of from
0.05 to 5 M, most preferably a concentration of from 0.1 to I M. Any suitable
salt may be used but it is preferred that the salt is sodium chloride.
The step of contacting the template with the solution may be carried out
for any period of time suitable to achieve the desired deposition of
polyelectrolyte. In one preferred embodiment the contacting is carried out for
from 15 minutes to 24 hours, more preferably the contacting is carried out for
from 2 hours to 20 hours, most preferably the contacting is carried out for
from 4
hours to 12 hours.
In order to facilitate the deposition the solution is preferably subjected to
ultrasound irradiation. It is preferred that following deposition each layer
of
polyelectrolyte material is cross-linked after being deposited and before
deposition of a further layer. The cross-linking may be carried out using any
technique well known in the art but is preferably cross-linked by heating at a
temperature of from 100 C to 250 C or by other chemical means. In another
preferred embodiment the polyelectrolyte layer is cross-linked using 1-ethyl-3-
CA 02583054 2007-04-02
WO 2006/037160 PCT/AU2005/001511
7
(3-dimethylaminopropyl) carbodiimide hydrochloride. The exact technique used
to cross-link a layer will depend upon the chemical structure of the
polyelectrolyte material used in that layer. Accordingly if cross-linking is
desired
this could readily be accomplished by a skilled addressee in the art.
In the methods of the invention the number of layers deposited may vary
depending upon the desired end use application. In a preferred embodiment a
plurality of layers are deposited. In one preferred embodiment two to ten
layers
are deposited, even more preferably from two to eight layers are deposited,
most preferably from four to 8 layers are deposited.
The polyelectrolyte materials used in the methods of the invention may
be chosen depending upon the desired end use application for the
polyelectrolyte material to be manufactured. It is preferred that the
polyelectrolyte material deposited to form each layer is independently
selected
from the group consisting of polymers, biodegradable polymers, poly(amino
acids), peptides, glycopeptides, polypeptides. peptidoglycans,
glycosaminoglycans, glycolipids, lipopolysaccharides, proteins, glycoproteins,
polycarbohydrates, modified biopolymers; polysilanes, polysilanols,
polyphosphazenes, polysulfazenes, polysulfide, polyphosphates, nucleic acid
polymers, nucleotides, polynucleotides, RNA and DNA.
In a particularly preferred embodiment the polyelectrolyte material
deposited to form each layer is independently selected from the group
consisting of polyglycolic acid (PGA), polylactic acid (PLA), poly-2-hydroxy
butyrate (PHB), gelatins, (A, B) polycaprolactone (PCL), poly (lactic-co-
glycolic
acid) (PLGA), carboxymethyl cellulose, carboxymethyl dextran, poly(allylamine
hydrochloride), poly(acrylic acid), poly(sodium 4-styrene sulphonate), poly
(diallyldimethylammonium chloride), poly(vinyisulfate), poly(L-glutamic acid)
and
poly(L-lysine) or a mixture thereof.
The molecular weight of the polyelectrolyte material used in the method
of the invention may be chosen to provide the desired properties to the final
product. It is preferred that each polyelectrolyte material has a molecular
CA 02583054 2007-04-02
WO 2006/037160 PCT/AU2005/001511
8
weight of at least 100, more preferably at least 500. Accordingly it is
preferred
that each polyelectrolyte material is chosen so that it has a molecular weight
of
500 to 1,000,000, even more preferably each polyelectrolyte material has a
molecular weight of from 500, to 500,000, yet even more preferably each
polyelectrolyte material has a molecular weight of from 500 to 100,000, most
preferably each polyelectrolyte material has a molecular weight of from 1000
to
100,000. _
In one embodiment it is preferred that the polyelectrolyte material
deposited to form at least one layer contains an amine group. In another
embodiment the polyelectrolyte material deposited to form at least one layer
contains a carboxylic group. In one particularly preferred embodiment the
polyelectrolyte material deposited to form at least one layer is poly(acrylic
acid).
In another preferred embodiment the polyelectrolyte material deposited to form
at least one layer is poly(allylamine hydrochloride).
The methods of the invention may be used to produce nanoporous
biomaterials. In order to produce biomaterials of this type the
polyelectrolyte
material deposited to form at least one layer is selected from the group
consisting of poly(amino acids), peptides, glycopeptides, polypeptides.
peptidoglycans, glycosaminoglycans, glycolipids, lipopolysaccharides,
proteins,
glycoproteins, polycarbohydrates; nucleic acid polymers, nucleotides,
polynucleotides, RNA and DNA, with proteins being particularly preferred. The
protein may have any suitable molecular weight but preferably has a molecular
weight of from 1 to 500 kDa, more preferably from 10 to 250 kDa. In a
particularly preferred embodiment of the methods of the invention the protein
is
selected from the group consisting of lysosome, cytochrome C, catalase, bovine
serum albumin, immunoglobulin G, protease, RNase A, trypsin, conalbumin,
lactoglobulin, myoglobin, ovalbumin, papain, penicillin acylase, and
subtilisin
Carlsberg.
The removal of the template is preferably carried out by exposure to
hydrofluoric acid. It is preferred that the hydrofiuoric acid has a
concentration of
from 0.01 to 10 M, more preferably from 1 to 10 M, most preferably about 5 M.
CA 02583054 2007-04-02
WO 2006/037160 PCT/AU2005/001511
9
In yet a further aspect the invention provides methods of delivering an
active agent to a target site the method including the steps of (I) adsorbing
the
active agent onto a multilayer polyelectrolyte material of the invention and
(ii)
delivering the polyelectrolyte material to the target site. The active agent
may
be adsorbed in any of a number of ways but us typically adsorbed by
suspending a polyelectrolyte material of the invention into a solution of the
active agent. The active agent is adsorbed onto the polyelectrolyte material
which can then be isolated from the solution. The polyelectrolyte material
with
the active agent adsorbed thereon may then be delivered to the target site
such
as by administration to the site so as to effectively deliver the active agent
to the
site. Any suitable active agent may be chosen for delivery such as therapeutic
agents including pharmaceuticals, veterinary chemicals and the like.
Alternatively the active agent may be a fragrance or a cleaning chemical which
is intended to be delivered to its site of action. The target site may be any
position or site that it would be desired for the active agent to be
administered.
In a further aspect the present invention provides the use of a multilayer
polyelectrolyte material of the invention as a micro reactor. It is found that
the
materials adsorb compounds and can thus be used to adsorb one or more
reactive species allowing them to be held proximal to each other to facilitate
reaction.
In yet an even further aspect the invention provides a method of
conducting a chemical reaction including contacting a solution containing one
or
more reactants with a polyelectrolyte material of the invention. The step of
contacting preferably involves addition of the polyelectrolyte material of the
invention to a solution containing the reactant(s) in question. The chemical
reaction may be carried out by the polyelectrolyte material acting as a micro
reactor for the chemical reactant(s) as discussed above or the polyelectrolyte
material may take an active part in the reaction. In a particularly preferred
embodiment the reaction is an enzymatic reaction, preferably an enzymatic
catalytic reaction of a reactant. In a most preferred embodiment the
polyelectrolyte material catalyses the reaction.
CA 02583054 2007-04-02
WO 2006/037160 PCT/AU2005/001511
As a result of their ability to adsorb chemical compounds the
polyelectrolyte materials of the invention may be used as adsorbents. In yet
an
even further aspect the invention provides methods of removing a compound
5 from solution including contacting the solution with a polyelectrolyte
material of
the invention, allowing sufficient time for the compound to be adsorbed by the
polyelectrolyte material and removing the polyelectrolyte material from the
solution. This method may be used to isolate drugs from solution or in the
purification of solutions containing trace amounts of compounds that it is
10 desired be removed from solution.
DESCRIPTION OF THE FIGURES
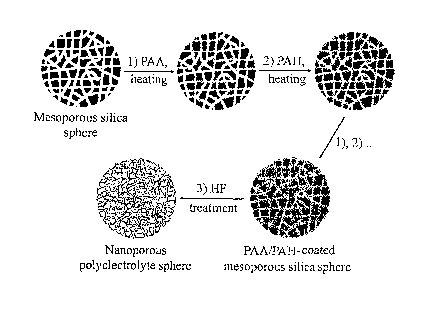
Figure 1 shows a schematic illustration showing the preparation of
nanoporous polyelectrolyte spheres (NPS). 3-aminopropyltriethoxysilane
(APTS) - modified spheres were layer-by-layer coated with polyelectrolytes of
opposite charge [poly(acrylic acid) (PAA) and poly(allylamine hydrochloride)
(PAH)] (steps I and 2, with the samples heated (160 C for 2 h) after
deposition
of each polyelectrolyte to partially cross link the layers. The spheres were
then
dissolved by exposure to hydrofluoric acid (HF) (step 3) yielding intact NPS.
Figure 2 (a) Fourier Transform Infrared (FTIR) spectra of the APTS-BMS
spheres before and after the alternate deposition of PAA and PAH layers. The
deposited layers were partially cross-linked by heating at 160 C for 2 h
prior to
recording each spectrum. The numbers correspond to the number of
polyelectrolyte layers deposited, commencing with PAA. APTS-BMS spheres
were used as the internal reference for measuring each spectrum. The spectra
are shifted in the vertical direction for clarity. (b) PAA amount deposited
onto
the APTS-BMS spheres as a function of PAA layer number, as determined by
FTIR at 1720 cm"1. The amount of PAA was calculated by using the
absorbance of APTS-BMS at 800 cm"' as a reference and assuming an average
APTS-BMS sphere size of 2.5 m and a density of 0.53 g mL"'.
Figure 3 TEM images of the NPS comprised of (a) (PAA/PAH)2/PAA
(NPS-5) and ultramicrotomed thin sections of the same spheres at (b) low and
CA 02583054 2007-04-02
WO 2006/037160 PCT/AU2005/001511
11
(c) higher magnification. The NPS-5 was partially cross-linked by heating at
160 C for 2 h after deposition of each polyelectrolyte layer. Images (b) and
(c)
clearly show the porosity of the polyelectrolyte spheres. The large difference
in
the diameters seen is a result of the ultramicrotoming process.
Figure 4 SEM images of (a, b, c) NPS-5 [(PAA/PAH)2/PAA] at different
magnifications, and (d) (PAA/PAH)Z/PAA capsules prepared when PAA and
PAH are deposited in the absence of added salt to the adsorption solution. The
NPS-5 and capsules were partially cross-linked by heating at 160 C for 2 h
after deposition of each poiyelectrolyte layer.
Figure 5 CLSM images of FITC-labelled lysozyme immobiiised in the
NPS-5 [(PAA/PAH)2/PAA] at (a) low and (b) higher magnification. The NPS-5
was partially cross-linked by heating at 160 C for 2 h after deposition of
each
polyelectrolyte layer.
Figure 6 Schematic illustration showing the preparation of nanoporous
protein particles (NPP). Protein is first loaded in the mesoporous silica
spheres
(step 1), after which the protein molecules are bridged by the infiltrated
polyelectrolyte (step 2). The mesoporous silica template is then dissolved by
exposure to HF/NH4F buffer (step 3), yielding intact NPP.
Figure 7 Nitrogen sorption isotherms of the native MS spheres
(diamonds), lysozyme-loaded MS (triangles), and the polyelectrolyte-connected
protein in the MS after PAA infiltration and subsequent cross-linking
(squares).
(The open symbols in the nitrogen sorption isotherms correspond to the
desorption branches.)
Figure 8 TEM images of the NPP-lys (a) and NPP-cyt (b) prepared using
8,000 Da PAA as the bridging molecule. TEM images of NPP-lys prepared
using PSS (70,000 Da) (c), and PAA (250,000 Da) (f) as the bridging molecules.
SEM images (d, e) of the NPP-lys prepared using 8,000 Da PAA as the bridging
molecule. The protein/PAA were cross-linked using EDC, followed by
dissolution of the MS template using HF/NH4F at pH -5.
CA 02583054 2007-04-02
WO 2006/037160 PCT/AU2005/001511
12
Figure 9 CLSM images of NPP-lys prepared using 8,000 Da (a) and
250,000 Da (b) PAA as the bridging molecules, respectively. Cross-linking of
lysozyme/PAA was accomplished using EDC, after which the MS template was
dissolved using HF/NH4F at pH -5. The samples were incubated in 0.1 mg mL-'
Rhodamine 6G solution for 60 min, washed with water four times, and then
dispersed in 50 mM PB.
Figure 10 TEM images of fibrous NPP-lys at different magnifications.
PAA with molecular weight of 8,000 Da was used as the bridging molecule.
Lysozyme/PAA was cross-linked using EDC as an initiator, followed by removal
of the MS template with HF/NH4F at pH -5.
DETAILED DESCRIPTION OF THE INVENTION
As used herein the terms "polyelectrolyte" or "polyelectrolyte material"
refers to a material that either has a plurality of charged moieties or has
the
ability to carry a plurality of charged moieties. A number of polyelectrolyte
materials are well known in the art and the polyelectrolyte may be a
positively
charged polyelectrolyte (or have the ability to be positively charged) or a
negatively charged polyelectrolyte (or have the ability to be negatively
charged)
or have a zero net charge. In addition the polyelectrolyte may be one where
the
charged moieties are relatively uniformly dispersed throughout the material
(such as a charged polymer e.g. PAA) or may be one where the charged
moieties are dispersed throughout the material. Proteins are an example of
polyelectrolytes where the charged moieties are dispersed throughout the
material as these molecules typically have areas of positive and negative
charge dispersed throughout the molecule. As would be appreciated by a
skilled worker, due to their ability to carry positive or negative charges,
the term
polyelectrolyte material therefore includes macromolecules which have the
ability to carry a plurality of charges, including bio-macromolecules such as
such as proteins, enzymes, polypeptides, peptides, polyoligonucleotides,
polysaccharides, polynucleotides, DNA, RNA and the like.
CA 02583054 2007-04-02
WO 2006/037160 PCT/AU2005/001511
13
Porous Multilayer Polyelectrolyte materials
As stated above the present invention provides a porous multilayer
polyelectrolyte material including at least two layers of polyelectrolyte
material.
In a preferred embodiment the material includes at least two layers of
oppositely
charged polyelectrolyte material.
The layers of polyelectrolyte material may be attracted to each other via
a number of mechanisms. Thus in one embodiment the layers are attracted via
electrostatic interactions and thus it is the differential net charge of the
adjacent
layers that lead to the layers being held together. In this circumstance it is
preferred that each layer is of alternating net charge such that each layer
has
an electrostatic attraction for the adjacent layer. In another embodiment the
layers may be attracted to each other via hydrogen bonding interactions such
that there is hydrogen bonding between the layers. Hydrogen bonding
interactions may occur with layers of opposite net charge or of the same
charge
and thus provides a mechanisms whereby layers with the same net charge can
be placed adjacent to each other.
Any suitable polyelectrolyte material may be used in each layer although
as stated above it is preferred that each layer is of alternating charge such
that
each layer has an attraction for the adjacent layers. Examples of preferred
polyelectrolyte materials for forming the layers include polymers,
biodegradable
polymers, poly(amino acids), peptides, glycopeptides, polypeptides.
peptidoglycans, glycosaminoglycans, glycolipids, lipopolysaccharides,
proteins,
glycoproteins, polycarbohydrates, modified biopolymers; polysilanes,
polysilanols, polyphosphazenes, polysulfazenes, polysulfide, polyphosphates,
nucleic acid polymers, nucleotides, polynucleotides, RNA and DNA.
Examples of materials that can be used as polyelectrolyte materials
include but are not limited to biodegradable polymers such as polyglycolic
acid
(PGA), polylactic acid (PLA), polyacrylic acid (PAA), polyamides, poly-2-
hydroxy
butyrate (PHB), gelatins, (A, B) polycaprolactone (PCL), poly (lactic-co-
glycolic
acid) (PLGA), flourescently labelled polymers, conducting polymers, liquid
crystal polymers, photoconducting polymers, photochromic polymers;
CA 02583054 2007-04-02
WO 2006/037160 PCT/AU2005/001511
14
poly(amino acids) including peptides and S-layer proteins; peptides,
glycopeptides, peptidoglycans, glycosaminoglycans, glycolipids,
lipopolysaccharides, proteins, glycoproteins, polypeptides, polycarbohydrates
such as dextrans, alginates, amyloses, pectins, glycogens, and chitins;
polynucleotides such as DNA, RNA and oligonucleotides; modified biopolymers
such as carboxymethyl celluiose, carboxymethyl dextran and lignin sulfonates;
polysilanes, polysilanols, poly phosphazenes, polysulfazenes, polysulfide and
polyphosphate. Preferred polymers include those with an amine group, for
example poly(allylamine hydrochloride) or a carboxylic acid group, for example
poly(acrylic acid). Other preferred polymers include poly(sodium 4-styrene
sulphonate), poly(diallyldimethylammonium chloride), poly(vinylsulfate), et
al.,
and the biocompatible polymers, such as poly(L-glutamic acid) and poly(L-
lysine) and mixtures thereof.
Preferred polyelectrolyte materials include materials (including polymers)
having a molecular weight of at least 100, more preferably at least 500.
Accordingly it is preferred that the polyelectrolyte material is chosen so
that the
material has a molecular weight of from 100 to 1,000,000, more preferably from
500 to 1,000,000, more preferably from 500, to 500,000, even more preferably
from 500 to 300,000, more preferably from 500 to 100,000, most preferably
from 1000 to 100,000. Preferred polyelectrolyte materials include materials
(such as polymers) with functional groups that can impart functionality on the
porous material. For example it is preferred that the polyelectrolytes used in
the
process contain functional groups that can be cross-linked under suitable
conditions to enable the various layers in the final material to *be
ultimately
cross-linked.
In one particularly preferred embodiment the poiyelectrolyte material in at
least one layer contains an amine group. In another preferred embodiment the
polyelectrolyte material in at least one layer contains a carboxylic group. It
is
particularly preferred that the material includes at least one layer of
poly(acrylic
acid). It is also particularly preferred that the material includes at least
one layer
of poly(allylamine hydrochloride).
CA 02583054 2007-04-02
WO 2006/037160 PCT/AU2005/001511
In a particularly preferred embodiment at least one polyelectrolyte layer
includes a material to impart a desired biological activity on the final
polyelectrolyte material. This is preferably achieved by ensuring that at
least
one polyelectrolyte layer is selected from the group consisting of peptides,
5 glycopeptides, polypeptides. peptidoglycans, glycosaminoglycans,
glycolipids,
lipopolysaccharides, proteins, glycoproteins and polynucleotides. If this is
done
it is preferred that the polyelectrolyte material is a protein, preferably a
protein
with a molecular weight of from 1 to 500 kDa. Examples of preferred proteins
include of lysosome, cytochrome C, catalase, bovine serum albumin,
10 immunoglobulin G, protease, RNase A, trypsin, conalbumin, lactoglobulin,
myoglobin, ovalbumin, papain, penicillin acylase, and subtilisin Carlsberg.
The material included to impart a desired biological activity on the final
polyelectrolyte material may be any layer of the final polyelectrolyte
material. It
15 may be an inner layer or it may be a surface layer.
The material should have at least two layers, but may in some
circumstances have a far greater number of layers. A preferred configuration
is
from two to ten layers, more preferably from two to eight layers, most
preferably
from four to eight layers. In one particularly preferred embodiment the
material
has two layers of polyelectrolyte material. In one preferred embodiment each
layer of polyelectrolyte material is oppositely charged to the layer(s) of
polyelectrolyte material adjacent to it. It is found that this is advantageous
as it
facilitates attraction between the layers and provides a more stable final
material. In an alternative embodiment the material includes at least two
adjacent layers of polyelectrolyte material with the same net charge.
In many embodiments of the invention the layers of polyelectrolyte
material are robust in that the attraction between molecules within a layer
and
between molecules of adjacent layers is such that the final multilayer
polyelectrolyte material are relatively stable to a variety of conditions. In
many
instances these molecules are resistant to leaching such that there is no loss
of
polyelectrolyte from the molecule in solution. In some instances however the
materials are not sufficiently stable to withstand the desired conditions of
use
CA 02583054 2007-04-02
WO 2006/037160 PCT/AU2005/001511
16
and steps are preferably taken to increase the stability of the materials such
as
by cross-linking the materials.
In one preferred form, each layer of polyelectrolyte material is cross-
linked in an intra-layer fashion. That is the layer is internally cross-linked
such
that molecules that make up the layer are linked to other molecules that make
up the layer such that the layers consist of a web of cross-linked
polyelectrolyte
molecules.
In another preferred embodiment there is cross-linking between one or
more adjacent layers of the multilayer polyelectrolyte material. In this
embodiment molecules in one layer are linked to molecules in an adjacent
layer. In this way networks of cross-linked molecules are created between
adjacent layers. Of course as would be clear to a skilled addressee in the art
a
combination of cross-linking strategies may be employed such that the final
multilayer polyelectrolyte. material may have cross linking both within one or
more layers and between one or more layers. It is found that cross-linking of
this type strengthens the final material and increases its rigidity.
The material preferably has pores with a pore size of from 1 to 100 nm,
more preferably 3 to 50 nm, even more preferably 5 to 50 nm, more preferably
from 10 to 50 nm. As such the materials are preferably nanoporous materials.
The pores are preferably interconnecting to produce an interconnected porous
network. The material may be of any suitable shape but is preferably
spherical.
The multilayer polyelectrolyte materials preferably have a particle size of
from 0.1 to 1000 m, more preferably from 0.1 to 100 m, even more preferably
0.1 to 20 m, most preferably from 0.4 to 5.0 m. In one preferred embodiment
the particles have a particle size of from 0.8 to 1.3 m. In another preferred
embodiment the particles have a size of from 1.4 to 2.1 m. In yet another
preferred embodiment the particles have a size of 1.6 to 2.4 m.
The polyelectrolyte materials - used in the layers of the multilayer
polyelectrolyte material are preferably chosen such that the porous
CA 02583054 2007-04-02
WO 2006/037160 PCT/AU2005/001511
17
polyelectrolyte materials are self-supporting in that the pores do not
collapse
under the weight of the material after template removal. This may be achieved
either by careful selection of the polyelectrolyte materials in the layers or
by
cross-linking of the layers to provide the required rigidity.
Method of Production of the Materials of the Invention
As stated above the porous electrolyte materials of the invention are
preferably produced using the layer-by-layer technique.
Accordingly the invention also provides a method of manufacturing a
porous multilayer polyelectrolyte material including the steps of:
(iv) providing a porous template;
(v) depositing layer-by-layer polyelectrolyte material onto the porous
template; and
(vi) removing the template by exposure to a suitable solvent.
The layer-by-layer technique typically exploits attractions between the
respective layers to form the final multi-layer material. For example it may
exploit the electrostatic attraction between oppositely charged species
deposited from solution. Alternatively if the layers are held together by
hydrogen bonding interactions it typically exploits the hydrogen bonding
interactions between the polyelectrolyte materials chosen. In one preferred
embodiment the polyelectrolyte material will be deposited in alternating
positive
and negative layers. In another preferred embodiment the polyelectrolyte
material is deposited such that there are at least two adjacent layers with
the
same net charge. The subsequent removal of the template leaves a porous
polyelectrolyte material, preferably a nanoporous polyelectrolyte material.
Figure 1 shows a schematic depiction of the preparation of a preferred
embodiment, namely a nanoporous polyelectrolyte sphere utilising PAA and
PAH. As depicted the process involves using a mesoporous silica sphere and
depositing a layer of PAA (stepl). This is followed by deposition of a layer
of
PAH (step 2) and then steps 1. and 2 are repeated the desired number of times
depending upon the number of layers desired in the final material. Once the
CA 02583054 2007-04-02
WO 2006/037160 PCT/AU2005/001511
18
desired number of layers has been deposited the process includes treatment of
the material to remove the template (step 3) to form the final porous
multilayer
polyelectrolyte material.
The porous template may be of any suitable type that provides an
interconnected network of pores. The pores may take a number of different
shapes and sizes however it is preferred that the porous template is a
mesoporous template. Mesoporous templates are templates in which there are
at least some pores, preferably a majority of pores having a pore size in the
range 2 to 50 nm. The mesoporous template may be made of a number of
suitable materials that allow for their subsequent removal although the
template
is preferably a mesoporous silica material. The template may take any suitable
form and may be for example in the form of planar supports, powder particles,
fibres, films, membranes or spheres. It is preferred that the template is
spherical or substantially spherical.
It is most preferred that the mesoporous material is a mesoporous silica
sphere in order to produce a spherical or substantially spherical nanoporous
polyelectrolyte material. It will be convenient to describe the invention in
terms
of a spherical material, but it shall be kept in mind that the porous
polyelectrolyte material produced by the process of the invention may be of
any
form, depending on the form of the template. Thus in general the final shape
of
the porous polyelectrolyte materials produced by the process of the invention
will take the general shape or form of the template used in their synthesis.
Thus
for example if the template is spherical then the final product will typically
be
spherical. If the template is a fibre then once again the final product will
typically be a fibre.
With many polyelectrolytes and templates the attraction between the
polyelectrolyte and the surface of the pores of the template is such that the
polyelectrolyte is naturally adsorbed onto the surface of the pores of the
template. In such cases the template can be used directly in the process
without any modification. In certain circumstances, however, the surface of
the
pores of the template and the polyelectrolyte may not have sufficient affinity
for
CA 02583054 2007-04-02
WO 2006/037160 PCT/AU2005/001511
19
the polyelectrolyte to be efficiently adsorbed onto the surface of the pores.
In
these cases it is preferred to modify the exposed surface of the pores of the
template prior to depositing the polyelectrolyte.
The surface of the pores of the template may be modified by addition of
functional moieties to enhance the adsorption of the polyelectrolyte onto the
pore surface. Any of a number of functional moieties can be added onto the
surface of the pores of the template with the choice of functional moiety
being
chosen to complement the polyelectrolyte being introduced as the first layer
during the process. A skilled worker in the area will generally have little
difficulty in choosing a functional moiety to introduce onto the surface of
the
template to complement the chosen polyelectrolyte. A particularly preferred
method of modifying the surface of a silica template for example is to graft a
moiety such as 3-aminopropyltriethoxysilane (APTS) onto the surface of the
silica. This introduces an amine surface functionality that can react with any
carboxyl groups on the polyelectrolyte to promote adsorption of the
electrolyte.
If it was desired to promote adsorption of a polyelectrolyte that contains
amino
moieties this could similarly be carried out by attaching carboxyl moieties to
the
exposed surface of the template.
As stated above it is preferred that the porous template is a mesoporous
silica material. In general, the mesoporous silica material may have a bimodal
pore structure, that is, having smaller pores of about 2-3 nm and larger pores
from about 10-40 nm.
The layers of polyelectrolyte material may be deposited in any of a
number of orders and the order of deposition of the layers will depend upon
the
desired final layer order in the final multilayer polyelectrolyte material. In
one
embodiment it is preferred that the polyelectrolyte layers are deposited in
layers
of alternating charge. In another preferred embodiment layers of the same net
charge may be deposited one after the other. As would be appreciated by a
skilled addressee a combination of these two embodiments may also be used.
CA 02583054 2007-04-02
WO 2006/037160 PCT/AU2005/001511
Each layer of polyelectrolyte material is typically deposited onto the
porous template by contacting the porous template with the polyelectrolyte.
The
contacting of the porous template with polyelectrolyte typically involves the
polyelectrolyte material being applied to the template such as a mesoporous
5 template in solution form in a suitable solvent. Generally, the
polyelectrolyte
material when applied in solution, will be in the form of an aqueous solution,
preferably an aqueous salt solution. The polyelectrolyte in solution typically
has
a concentration of from 0.001 to 100 mg mL"1, more preferably from about 0.1
to
mg mL-', most preferably from 0.5 to 10 mg mL"1.
If a salt solution is used the salt in solution preferably has a concentration
of from about 0.001 to 5 M, more preferably from 0.05 to 5 M, most preferably
from 0.1 to I M. It is preferred that the polyelectrolyte material is applied
in a
salt solution as, without wishing to be bound by theory, it is thought that in
the
presence of a salt solution, the polyelectrolyte material can be highly coiled
which assists in it penetrating into the mesopores of the template. In the
absence of salt, the polyelectrolyte will be mainly restricted to the outer
surface
assembly of the template as it will be presented with a long chain
configuration.
The salt may be of any suitable type but is typically selected from the group
consisting of potassium chloride, lithium chloride and sodium chloride with
sodium chloride being particularly preferred.
The polyelectrolyte material will generally infiltrate the pores within the
range of 10-40 nm. Preferred polyelectrolyte materials include any known
polymer material having a molecular weight of at least 100, more preferably
from 500 to 1,000,000, more preferably from 100 to 500,000, even more
preferably from 500 to 100,000, most preferably from 1000 to 100,000.
Preferred polyelectrolyte materials include polymers with functional groups
that
can impart functionality on the porous material. For example it is preferred
that
the polyelectrolytes used in the process contain functional groups that can be
cross-linked under suitable conditions to enable the various layers in the
final
material to be ultimately cross-linked.
CA 02583054 2007-04-02
WO 2006/037160 PCT/AU2005/001511
21
Any suitable polyelectrolyte material may be used in each layer although
it is preferred that each layer is of alternating charge such that each layer
has
an attraction for the adjacent layers. Examples of preferred polyelectrolyte
materials for forming the layers include polymers, biodegradable polymers,
poly(amino acids), peptides,. glycopeptides, polypeptides. peptidoglycans,
glycosaminoglycans, glycolipids, lipopolysaccharides, proteins, glycoproteins,
polycarbohydrates, modified biopolymers; polysilanes, polysilanols,
polyphosphazenes, polysulfazenes, polysulfide, polyphosphates, nucleic acid
polymers, nucleotides, polynucleotides, RNA and DNA.
Examples of materials that can be used as polyelectrolyte materials
include but are not limited to biodegradable polymers such as polyglycolic
acid
(PGA), polylactic acid (PLA), polyacrylic acid (PAA), polyamides, poly-2-
hydroxy
butyrate (PHB), gelatins, (A, B) polycaprolactone (PCL), poly (lactic-co-
glycolic
acid) (PLGA), flourescently labelled polymers, conducting polymers, liquid
crystal polymers, photoconducting polymers, photochromic polymers;
poly(amino acids) including peptides and S-layer proteins; peptides,
glycopeptides, peptidoglycans, glycosaminoglycans, glycolipids,
lipopolysaccharides, proteins, glycoproteins, polypeptides, polycarbohydrates
such as dextrans, alginates, amyloses, pectins, glycogens, and chitins;
polynucleotides such as DNA, RNA and oligonucleotides; modified biopolymers
such as carboxymethyl cellulose, carboxymethyl dextran and lignin sulfonates;
polysilanes, polysilanols, poly phosphazenes, polysulfazenes, polysulfide and
polyphosphate Preferred polymers include those with an amine group, for
example poly(allylamine hydrochloride) or a carboxylic acid group, for example
poly(acrylic acid). Other preferred polymers include poly(sodium 4-styrene
sulphonate), poly(diallyldimethylammonium chloride), poly(vinyisulfate), et
al.,
and the biocompatible polymers, such as poly(L-glutamic acid) and poly(L-
lysine) and mixtures thereof.
It is particularly preferred that at least one polyelectrolyte layer is
selected from the group consisting of poly(amino acids), peptides,
glycopeptides, polypeptides. peptidoglycans, glycosaminoglycans, glycolipids,
lipopolysaccharides, proteins, glycoproteins, polycarbohydrates; nucleic acid
CA 02583054 2007-04-02
WO 2006/037160 PCT/AU2005/001511
22
polymers, nucleotides, polynucleotides, RNA and DNA. If this is done it is
preferred that the polyelectrolyte material is a protein, preferably a protein
with a
molecular weight of from 1 to 500 kDa. Examples of preferred proteins include
lysosome, cytochrome C, catalase, bovine serum albumin, immunoglobulin G,
protease, RNase A, trypsin, conalbumin, lactoglobulin, myoglobin, ovalbumin,
papain, penicillin acylase, and subtilisin Carlsberg.
After addition of the solution of the polyelectrolyte material to the
template the mixture thus formed is typically agitated to allow the
polyelectrolyte
material to be adsorbed into the pores of the template. This can be done for
any suitable length of time but it is typically found that the solution is
agitated
from 15 minutes to 24 hours, more preferably from 2 hours to 20 hours, even
more preferably from 4 hours to 12 hours, most preferably about 6 hours.
Preferably ultrasound may be used during the depositing of the
polyelectrolyte on the mesoporous template to assist in allowing the
polyelectrolyte material to infiltrate the pores of the mesoporous template.
Accordingly after the solution of polyelectrolyte material has been mixed with
the template the mixture may be ultrasonicated. It has been found that with
the
application of ultrasound, together with agitation of the mixture of
polyelectrolyte
material and the mesoporous template, that higher molecular weight polymer
material can infiltrate the pores in an efficient manner.
Following the mixing as discussed above the template may be treated to
remove excess polyelectrolyte material that has not been adsorbed onto the
template. The treatment may involve centrifugation, washing or a mixture
thereof. This removes excess solution and increases the prospect that the next
layer will be able to be successfully added.
In a preferred process for the preparation of the nanoporous
polyelectrolyte material, the layers of polyelectrolyte are deposited layer-by-
layer with oppositely charged polyelectrolyte materials. That is, there will
be a
build-up of subsequent positive and negatively charged materials. The material
should have at least two layers, but may in some circumstances have a far
CA 02583054 2007-04-02
WO 2006/037160 PCT/AU2005/001511
23
greater number of layers. In principle the only limitation on the number of
layers
is the pore size of the porous template. Eventually as a plurality of layers
are
laid down they fill the pores completely thus stopping infiltration of any
further
polyelectrolyte material. A preferred configuration is from two to ten layers,
more preferably from four to eight layers.
In one preferred form, each layer of polyelectrolyte material is cross-
linked internally after being deposited and before deposition of a further
layer.
The layer may be cross-linked in any way well known in the art with the method
chosen being determined by the moiety on the polyelectrolyte material that
allows cross-linking.
In one preferred embodiment the polyelectrolyte layers are cross-linked
by subjecting them to heat. The cross-linking is generally performed by
heating
at a temperature of from about 100 C to 250 C, more preferably from 140 C
to 220 C, most preferably about 160 C. The amount of time taken to effect
cross-linking will vary depending on the nature of the cross-linking moieties
but
it typically takes from 30 minutes to 12 hours.
In another preferred embodiment the layer may be internally cross-linked
using other chemical means such as by the use of 1-ethyl-3-(3-
dimethylaminopropyl) carbodiimide hydrochloride. The exact means of
chemical cross-linking of the materials will depend upon the nature of the
polyelectrolyte materials chosen.
Using similar methodology the layers may also be cross-linked to each
other to provide further strength to the final multilayer material formed.
Cross-linking the polyelectrolyte material reinforces the strength of the
polyelectrolyte layer. The layers may also be cross linked in an inter layer
fashion such that one layer is cross- linked to the adjacent layers. This is
generally carried out by reaction of the layers with a chemical entity that is
able
to react with functional groups on each layer.
CA 02583054 2007-04-02
WO 2006/037160 PCT/AU2005/001511
24
In an alternative embodiment it is sometimes possible to cross-link the
layers after deposition of all layers of polyelectrolyte material.
Following the deposition of the desired number of layers the process
then involves removal of the template. The template may be removed by
exposure to a suitable solvent that is capable of dissolving the template. In
general the solvent will be chosen such that it is able to dissolve the
template
but such that it will not damage the polyelectrolyte layers. An example of a
suitable solvent is hydrofluoric acid or sodium hydroxide. It has been found
that
the silicone dioxide core of the mesoporous silica material can readily be
decomposed in hydrofluoric acid as it is converted to [SiF6]2" ion leaving the
polyelectrolyte layers. Preferably, the mixture containing the template is
shaken
when the template is exposed to the hydrofluoric acid. If hydrofluoric acid is
used it is found that the silica can be dissolved using a wide range of
concentrations of acid. The acid may be of any strength although it is
convenient to use an acid strength of from 1 to 10 M, more preferably about 5
M. Whereas hydrofluoric acid is preferred as a solvent, other suitable
solvents
would be well appreciated by the skilled practitioner. As such in principle
any
substance that can dissolve the template may be used as the solvent.
The main advantages of the layer-by-layer approach in the formation of a
nanoporous polyelectrolyte sphere is that it offers a facile route to
nanoporous
polyelectrolyte sphere production as it is based on self assembly of a
multilayer
species based on attractions between the layers such as electrostatic self-
assembly principles, thereby allowing the preparation of nanoporous
polyelectrolyte spheres of diverse composition. Further, it affords nanometer
level control of the deposited polyelectrolyte thickness and hence allowing
the
control of the functional groups of the mesoporous silica materials and
subsequently on the nanoporous polyelectrolyte sphere, depending on the
number of layers deposited.
Using the bimodal mesoporous silica spheres as a template has at least
the following benefits:
CA 02583054 2007-04-02
WO 2006/037160 PCT/AU2005/001511
(i) relatively regular spherical morphology makes it easier and
possible to follow the particle morphology variation after
polyelectrolyte deposition (it is difficult to do so with mesoporous
silica with fractal morphology);
5 (ii) bimodal mesoporous silica possesses large mesopores (10-40
nm) and very high pore volume (1.2 mL g-') for such pores, which
provides comparable size for the polyelectrolyte layer-by-layer
infiltration into the three-dimensional random pores in the bimodal
mesoporous silica templates. Self-standing nanoporous
10 polyelectrolyte spheres are yielded after removal of the bimodal
mesoporous silica templates.
Control of the functional groups within the siliceous mesopores can also
improve the material performance in the applications for which the nanoporous
15 polyelectrolyte sphere may be used. For example, depending on the
functional
group of the nanoporous polyelectrolyte sphere, the material may find use in
bio-molecule (i.e. protein) adsorption/separation, high efficient adsorbents
for
environmental protection (i.e. removal of heavy metal ions and toxic organic
molecules), enzyme immobilization and drug delivery. In addition the final
20 materials may be useful as adsorbents for dyes and could also be useful in
the
controlled release of fragrances in certain applications.
The porous polyelectrolyte materials of the invention may also be coated
using the layer-by-layer technique discussed herein to produce a shell on the
25 outer surface assembly of the porous polyelectrolyte material. This may be
useful in some applications such as where one desires to encapsulate any
entities adsorbed into the pores of the porous polyelectrolyte material.
Accordingly, one could treat the porous polyelectrolyte materials of the
invention
in solution to adsorb a material of interest such as an enzyme or
pharmaceutically active compound. Once the desired amount of material had
been absorbed into the pores, the porous polyelectrolyte material could then
be
surface coated using the methodology discussed above to produce an
encapsulated material. This could be used in applications such as sustained
drug delivery or the like by judicious selection of the coating layers.
CA 02583054 2007-04-02
WO 2006/037160 PCT/AU2005/001511
26
The invention will now be described with reference to the accompanying
examples.
Examples
Materials: Catalase (C-100), cytochrome C (C-2037), poly(acrylic acid)
(PAA, MW 8,000, and 250,000), poly(sodium 4-styrenesulfonate (PSS, MW
70,000), poly(L-glutamic acid) (PGA, Mw 1,500-3,000), 1-ethyl-3-(3-
dimethylaminopropyl) carbodiimide hydrochloride (EDC), hydrogen peroxide
(H202), hydrofluoric acid (HF), ammonium fluoride (NH4F), sodium metasilicate
(Na2SiO3) and cetyltrimethylammonium bromide (CTABr) were obtained from
Sigma-Aldrich and used as received. Lysozyme was purchased from Fluka
BioChemika. The mesoporous silica (MS) spheres were synthesised according
to a literature method (G. Schulz-Ekloff, J. Rathouskjr, A. Zukal, Int. J.
Inorg.
Mater. 1999, 1, 97). All PE solutions were of concentration 5 mg mL"'. The
solution used for dissolving the silica core was a mixture of 2 M HF and 8 M
NH4F at pH - 5. The water used in all experiments was prepared in a Millipore
Milli-Q purification system and had a resistivity higher than 18 MS2 cm.
Example I production of nanoporous polyelectrolyte spheres (NPS) using
Poly(acrylic acid) (PAA) and poly (allylamine hydrochloride) (PAH)
A mesoporous silica sphere was prepared in accordance with the
method described in a paper published in the International Journal of
Inorganic
Materials (1999) 97-102, titled Mesoporous Silica with Controlled Porous
Structure and Regular Morphology by Schulz-Ekloff et al. The molar ratio of
surfactant to silica (CTABr/Na2SiO3) used to prepare the material is much
higher (ca. two times) than that used to prepare conventional mesoporous
materials. Therefore, the produced particles contain domains with stable
silica
walls between the micelles as well as domains in which these walls are
unstable or even missing, which will form the larger mesopores after the
removal of surfactant micelles. The mesoporous silica sphere possesses a
bimodal mesoporous silica structure. That is, the bimodal mesoporous silica
template has a surface area of 630 m2 g1 and a pore volume of 1.72 mL g"1.
The material has a bimodal pore structure that is smaller pores in the range
of
CA 02583054 2007-04-02
WO 2006/037160 PCT/AU2005/001511
27
2-3 nm and larger pores in the range of from 10-40 nm with a volume of 1.28
mL g"1. The bimodal mesoporous silica spheres have a particle size
distribution
of 2-4 pm.
To modify the bimodal mesoporous silica surface with a layer of
functional groups (e.g. -NH2 groups), which will have specific adsorption with
subsequent polyelectrolyte deposition, a silanization method is applied
according to the literature method. In this process, the newly dried
mesoporous
silica powder was well dispersed in toluene by sonication for 20 min before
silane chemicals were added to the suspension. The molar ratio of the
mesoporous silica particles (calculated as Si02)/silane chemical/toluene was
fixed to be 5:1:500, and the suspension was refluxed for 24 h. The silane
grafted mesoporous silica particles were separated from the solution by
centrifugation, and washing in toluene and methanol twice, respectively.
Finally, the pellet was dried at 80 C for 12 h. The silane modification was
fulfilled through grafting 3-aminopropyltriethoxysilane (APTS) on the bimodal
mesoporous silica pore walls. The APTS grafted bimodal mesoporous silica
(denoted as APTS-BMS) has a surface area of 465 m2 g 1, and a pore volume
of 1.32 mL g'. Most of the pore volume in the APTS-BMS is contributed by the
mesopores ranging from 10-40 nm with a volume of about 1.0 mL g"1.
Poly(acrylic acid) (PAA) having a molecular weight of 2000 and poly
(ailylamine hydrochloride) (PAH) having a molecular weight of 15,000 were
used as the counter polyelectrolyte pairs for the layer-by-layer assembly in
the
mesoporous silica spheres. All polyelectrolyte solutions were of a
concentration
of 5 mg mL-' and contained 0.7 M NaCI. The adsorption of polyelectrolyte was
processed at ambient temperature for fifteen minutes in a sonication bath,
followed by shaking for six hours. The sample was separated following three
minutes by centrifugation (500g) and washed with 0.1 M NaCi solution four
times.
To reinforce the polyelectrolyte on the walls, cross-linking of the
polyelectrolyte after each layer of polyelectrolyte deposition was applied.
The
cross-linking was performed by heating of the sample at 160 C for two hours,
CA 02583054 2007-04-02
WO 2006/037160 PCT/AU2005/001511
28
according to a method described in the International Journal of Journal of the
American Chemical Society (1999) 1978-1979, titled Synthesis of Passivating,
Nylon-Like Coatings through Cross-Linking of Ultrathin Polyelectrolyte Films
by
Jeremy J. Harris et al. Under this treatment it was found that amide bonds are
formed by the -COOH groups (in PAA) and the -NH2 groups (in PAH).
It has been found that the polyelectrolytes assembled in the BMS
particles in the subsequent polyelectrolyte assembly step may dissolve and may
form aggregates on the particle surface and solution if the materials are
prepared without cross-linking. If cross-linking is applied, the surface of
the
polyelectrolyte assembled APTS-BMS particles is very smooth and the pore
structure still can be distinguished by transmission electron microscopy (TEM)
at high magnification in the APTS-BMS-polyelectrolyte samples and indicate
negligible aggregation on the APTS-BMS surface. This result means that
cross-linking can effectively stabilise the polyelectrolyte layers, and avoid
desorption of the previous polyelectrolyte layer in the subsequent
polyelectrolyte assembly.
The successful deposition of the PAA and PAH in the APTS-BMS
particles is further forcefully proved by FTIR. The FTIR spectra of the APTS-
BMS particles after different layer of PAA and PAH deposition are shown in
Figure 2(a). For the APTS-BMS spheres, the absorption band at 1635 cm', (i)
is assigned to the Si-OH vibrations and the N-H bending (scissoring)
vibrations
of APTS. The peaks at 1720 (ii), 1570 (iii) and 1400 (iv) cm-1 are attributed
to
the -COOH carbonyl and -COO' asymmetric and symmetric stretches,
respectively, of PAA. The intensities of the peaks at 1635 cm-1 (due to the N-
H
bending (scissoring) vibration of PAH for layer number ?2) and at 1720 cm-1
(corresponding to PAA deposition) increase with PAH and PAA layer number,
respectively, confirming the sequential deposition of PAA/PAH multilayers. The
following observations can be made from the spectra: (a) The presence of
-COOH (from PAA) after heating at 160 C indicates that only partial cross-
linking of the layers occurs. Only ca.10-15 % reduction in intensity of this
peak
was observed after heating the films. (b) The amide bonds formed as a result
of cross-linking (peak at -1670 cm"1) are not discernible, largely due to the
CA 02583054 2007-04-02
WO 2006/037160 PCT/AU2005/001511
29
relatively low cross-linking degree and masking from the peak at 1635 cm'1
(arising from the APTS-BMS substrate and PAH). (c) The total amount of PAA
deposited per APTS-BMS particle increases with PAA layer number, although
the amount adsorbed per layer decreases with increasing PAA layer number
(Figure 2b). This trend is attributed to increased blockage of the larger
mesopores in the APTS-BMS templates with increasing polyelectrolyte layer
number.
The PAAA deposition amount via the layer numbers is depicted in Figure
2b. With the first layer of PAA deposition, the sample weight (i.e. PAA
deposition amount) increased about 14 wt% of the original APTS-BMS
templates. After that, the deposition amount in each layer will gradually
decrease with the layer numbers increasing, which might be caused by partially
blocking of the smaller mesopores in the APTS-BMS templates.
To examine the influence of sphere porosity, the experiment used both
mesoporous silica spheres with only 2-3 nm pores and nonporous silica
spheres for comparison. No distinguishable peaks due to PAA and PAH in the
FTIR spectra of either of the PAA/PAH-coated silica spheres were observed,
even after deposition of seven layers (i.e., 3.5 PAA/PAH bilayers). This
indicates that polyelectrolyte deposition predominantly occurs in the larger
mesopores of the APTS-BMS particles, and that the contribution to the FTIR
intensities from polyelectrolyte adsorption on the outer surface of the
particles is
negligible.
The silicon dioxide skeleton was dissolved by hydrofluoric acid (10%
solution in water), while gently shaking the tubes for twelve hours. The
silicon
dioxide core can be decomposed in 1 M hydrofluoric acid within a few seconds
into [SiF6]2 ion, which can leave the polyelectrolyte layers during the
dissolution
without problems. Effective removal of the silicon dioxide wall is proved by
energy dispersive X-ray and FTIR spectra. Only a small amount of silicon
(0.8%) was detected after the template removal. The small amount of silicon is
most possibly caused by the silicon-alkyl groups (arising from APTS
modification), which is stable in the presence of hydrofluoric acid.
CA 02583054 2007-04-02
WO 2006/037160 PCT/AU2005/001511
Nitrogen adsorption measurements were also conducted to follow the
changes in the surface area of the APTS-BMS spheres after polyelectrolyte
deposition. The first layer of adsorbed PAA dramatically decreased the surface
5 area from 465 m2 g"1 (APTS-BMS template) to 284 m2 g"1. This is likely
caused
by the high PAA loading and blocking of some of the mesopores. Deposition of
subsequent PAH and PAA layers resulted in a surface area decrease per
polyelectrolyte adsorption step of approximately 20 m2 g'I. After deposition
of
seven layers, the surface area of the coated spheres was ca. 160 m2 g"1. These
10 data further confirm the stepwise deposition of polyeiectrolytes within the
APTS-
BMS spheres.
TEM was used to follow the nanoporous polyelectrolyte sphere
morphology and size with the layer number variation. The nanoporous
15 polyelectrolyte sphere prepared with different layer numbers of
polyelectrolyte is
denoted as NPS-n. It was found that the nanoporous polyelectrolyte materials
of the invention typically retain the original shape of the template and do
not
show any signs of collapse. The APTS-BMS-PAA sampie (one layer of PAA
deposition) totally dissolved in seconds after exposure to hydrofluoric acid
20 solution. Spherical morphology can be obtained for the sample (i.e. NPS-2
sample) with an additional layer of PAH deposition on the APTS-BMS-PAA
sample. The NPS-2 sample had a diameter of from 0.8 to 1.3 pm, representing
shrinkage of about 55% compared to the original APTS-BMS templates. With
more layers of polyelectrolyte, less shrinkage was found in the nanoporous
25 polyelectrolyte sphere products. For the NPS-7 sample, shrinkage of about
25% was found after the silica skeleton dissolution (diameter of approximately
1.4 to 2.1 pm). No obvious aggregation of the nanoporous polyelectrolyte
sphere was found from TEM low magnification images (Figure 3a). The inner
structure of the nanoporous polyelectrolyte sphere was examined by slicing the
30 spheres to a thickness of about 90 nm using the TEM microtome technique. It
was found that the inner part of the nanoporous polyelectrolyte sphere was
also
efficiently filled with polyelectrolytes (Figure 3b). At higher magnification
(Figure
3c) it can be observed that the porous structure was relatively homogeneous
with a pore size of from 5 to 50 nm.
CA 02583054 2007-04-02
WO 2006/037160 PCT/AU2005/001511
31
Scanning electron microscopy (SEM) was further used to observe the
morphology of the particles (Figure 4). No obvious aggregation of the
particles
is observed at low magnification (Figure 4a). With the magnification increase,
the roughness and porosity of the spheres becomes clear (Figure 4b). At high
magnification (Figure 4c), abundant and homogeneous pores in the range of
10-40 nm were found.
Figure 4d shows the collapsed capsule structure of the product prepared
by the same procedure except without salt in the polyelectrolyte solution.
This
indicates that, in the presence of salt, polyelectrolyte can be highly coiled
and
able to penetrate into the mesopores of the templates.
The porosity and adsorption ability of the nanoporous polyelectrolyte
sphere particles is also characterized through enzyme entrapment.
Approximately 10 mg of the nanoporous polyelectrolyte spheres were dispersed
in 15 mL of lysozyme (molecular weight 14.6 k Da) with a concentration of 1.0
mg mL"' in 50 mM phosphate buffer (pH 7.0) stock solution and shaken at room
temperature for twenty fours. The enzyme retained by the particles was
monitored by UV-vis spectroscopy, i.e., by monitoring the difference in
solution
between the protein absorbance at 280 nm before adsorption and after
separating the supernatants via centrifugation at a speed of 1000 g for five
minutes. For the NPS-5 particles, the weight will increase about 90% after
lysozyme immobilization, which means nearly half the weight in the enzyme
nanoporous polyelectrolyte sphere materials is contributed by the enzyme.
The immobilization and distribution of enzyme in the nanoporous
polyelectrolyte sphere particles was further examined by confocal laser
scanning microscopy (CLSM) of a cross section of individual particles. Figure
5
shows the CLSM images of the NPS-5 spheres after incubating in fluorescein
isothiocyanate-labelled lysozyme (FITC-lysozyme) for one hour, followed by
washing with copious amounts of Milli-Q water. The bright spheres seen are
due to the homogenous distribution of FITC-lysozyme in the nanoporous
polyelectrolyte sphere particles with a relatively high enzyme amount (Figure
CA 02583054 2007-04-02
WO 2006/037160 PCT/AU2005/001511
32
5a). The fluorescence distribution in the nanoporous polyelectrolyte spheres
is
rather homogeneous, indicating effective immobilization of lysozyme molecule
in the particles. Excellent enzyme immobilization ability of the nanoporous
polyeiectrolyte sphere particles is mostly caused by the abundant pores in
nanoscale and high amount of functional chemical groups in the polyelectrolyte
network.
The results demonstrate the success of layer-by-layer assembly of
polyelectrolyte in mesoporous silica materials, to prepare nanoporous
polyelectrolyte materials. The functional chemical group types and amounts
can be controlled through layer-by-layer assembly. Compared with the
traditional silane modification technology, significantly higher amounts of
functional chemical groups (for example -NH2, -COOH etc.) are expected to be
grafted into the mesopores since the high molecular weight and long chain of
polyelectrolyte molecules which may coil in the pores (previous silane
modification is largely restricted to a single layer of functional group
modification
in the pore walls), hence influence the material adsorption properties and
application.
Nanoporous polyelectrolyte materials were obtained after removal of the
silicious skeleton. The size of the final nanoporous polyelectrolyte sphere
particles can be controlled by adjusting the polyelectrolyte deposition
layers.
Excellent adsorption (e.g. enzyme immobilization) ability of the nanoporous
polyelectrolyte spheres is found due to its abundant nanoscaled pore
structures
and high amount of functional groups in the polyelectrolyte networks. Electron
microscopy data show that the nanoporous polyelectrolyte spheres of the
invention have pores ranging from ca. 5-50 nm. The spheres show excellent
capacity for immobilization of enzymes (lysozyme). Since the method is
amenable to the deposition of diverse polyelectrolytes, the preparation of
nanoporous polyelectrolyte spheres of controlled composition and functionality
can be achieved by the present invention.
CA 02583054 2007-04-02
WO 2006/037160 PCT/AU2005/001511
33
Example 2
Production of Nanoporous Protein Particles (NPP with a protein as one of
the polyelectrolyfie layers
The general procedure was depicted in figure 6 and involves three main
steps. The first involves immobilizing protein in the MS spheres by solution
adsorption. Secondly, an oppositely charged polyelectrolyte (PE) is
infiltrated
into the protein-loaded mesopores, "bridging" the proteins. In the third step,
the
MS template is removed by exposure to a solution of hydrofluoric acid
(HF)/ammonium fluoride (NH4F), resulting in free-standing NPPs.
Particle Production
Several proteins with different molecular weight, size and isoelectric point
(pl) were chosen for investigation: lysozome (14.6 kDa, 3-4.5 nm, pl 11);
cytochrome C (12 kDa, 3 nm, pl 10.3); and catalase (250 kDa, 10.4 nm, pl 5.4).
The protein loading was performed by dispersing 10 mg of the MS particles in
the protein solution (20 mL of 0.5 mg mL"1 protein in 50 mM phosphate buffer
(PB) at pH 7) and mixing at 20 C for either 3 days (lysozyme or cytochrome C)
or 7 days (catalase). The amount immobilized was determined by monitoring
the difference in the protein absorbance in solution (lysozyme 280 nm;
cytochrome C 530 nm; catalase 405 nm) before and after adsorption. The
loadings for lysozyme, cytochrome C, and catalase are 400, 230, and 75 mg g"1
MS, respectively.
Following several washing cycles to remove loosely adsorbed protein, the
protein-loaded MS particles were dispersed in a 5'mg mL-1 poly(acrylic acid)
(PAA, M, 8 000) solution at pH 4.5, which contained 0.1 M NaCI. PAA was
allowed to infiltrate into the protein-loaded mesopores for 24 h at 20 C.
Excess
PAA was removed by two cycles of centrifugation and washing with 50 mM PB
at pH 7. To enhance the stability of the protein/PAA layers, cross-linking of
protein/PAA was performed by shaking the particles with 0.3 mL of EDC
solution (60 mg mL"1 in 50 mM PB) for 2 h at 20 C. The MS particle template
was then dissolved by adding 2 mL of a 2 M HF/8 M NH4F solution at 20 C to
the protein/PE loaded MS particles for 5 min, followed by two centrifugation
(1500 g for 5 min)/water washing cycles. The resulting NPPs were stored in 50
CA 02583054 2007-04-02
WO 2006/037160 PCT/AU2005/001511
34
mM PB at pH 7. The NPP materials composed of PE and lysozyme,
cytochrome C, or catalase are herein denoted as NPP-lys, NPP-cyt, and NPP-
cat, respectively.
Nitrogen Adsorption Measurements
Nitrogen adsorption measurements were conducted to follow the
variation in porosity of the MS spheres as a result of protein and PE
infiltration.
Figure 7 shows the nitrogen isotherms for the MS spheres before and after
lysozyme loading, and after PAA infiltration and cross-linking. The native MS
has a surface area of 630 m2 g 1 and a pore volume of 1.72 cm3 g 1. After
lysozyme immobilization, the surface area and pore volume of the particles
significantly decreased to 230 m2 g' and 0.88 cm3 g-1, respectively,
indicating
high amount of protein is loaded in the mesopores. Thermogravimetric analysis
(TGA) measurements showed the amount of lysozyme immobilized in the MS
spheres was 41 wt%, which is in close agreement with the loading determined
from UV- vis (40 wt%). The surface area and pore volume further decreased to
140 m2 g-' and 0.40 cm3 g-1, respectively, after PAA infiltration. TGA
experiments revealed that the amount of PAA infiltrated into the MS spheres
was 7.5 wt%.
Particle Stability
Structure stability was assessed by treatment of the lysozyme loaded MS
spheres in solution with different concentration of (NH4)2SO4. 1.5 mg of the
lysozyme loaded particles was added to 1 mL of (NH4)2SO4 solution with a salt
concentration of 0, 0.1, and 0.5 M, respectively, and incubated for 150 min at
20 C. The mixture was then centrifuged and the protein content in the
supernatant determined by UV-vis at 280 nm.
The stability of the PAA-connected catalase within the MS spheres was
examined by dispersing 2 mg particles in 2 mL PB solution (50 mM at pH 7.0) at
5 C for 14 days. The amount of protein desorbed during this time was
determined by measuring the supernatant activity after centrifugation of the
stock suspension.
CA 02583054 2007-04-02
WO 2006/037160 PCT/AU2005/001511
Results
PE bridging efficiency was assessed by treatment of the lysozyme loaded
MS spheres in solution with different concentration of (NH4)2SO4. About 9%,
25%, and 40% of the enzyme were desorbed from the lysozyme-loaded MS
5 particles after the treatment with 0, 0.1, and 0.5 M(NH4)2SO4, respectively.
After cross-linking the protein with PAA by EDC, no enzyme was detected in the
0 and 0.1 M(NH4)2SO4 solution, and only 0.18% lysozyme was desorbed in the
0.5 M(NH4)2SO4 solution. The stability of the PAA-connected catalase within
the MS spheres was examined by dispersing the samples in 50 mM PB solution
10 at 5 C for 14 days. The amount of protein desorbed during this time was
determined by measuring the supernatant activity after centrifugation of the
stock suspension. Catalase was selected because catalase decomposition of
H202 is very sensitive, allowing the measurement of trace amounts of protein
(one mole of catalase can decompose 5X10$ moles of H202 per min).
15 Experiments showed that 10% of the adsorbed protein desorbed from the
catalase-loaded MS particles during storage. After PAA infiltration, less than
7% of the immobilized protein desorbed. Protein loss was prevented by EDC
cross-linking the protein/PAA-infiltrated MS spheres; negligible protein was
detected in the supernatant. This indicates that the protein was firmly
20 immobilized in the mesopores, and that the immobilization-bridging strategy
provides an effective method for protein immobilization in porous materials.
Further, the cross-linked PAA/catalase-loaded MS spheres showed no loss of
activity over two weeks.
25 Particle Characterization
Transmission electron microscopy (TEM) samples were prepared by
placing a drop of a diluted capsule suspension (dispersed in water) onto a TEM
grid. A Philips CM 120 microscope operated at 120 kV was used for analysis. A
HP 8453 UV-vis spectrophotometer (Agilent, Palo Alto, CA) was used to
30 monitor the enzyme loading, activity and release amount. Confocal laser
scanning microscopy (CLSM) images were taken with an Olympus confocal
system equipped with a 60x oil immersion objective. Adsorption-desorption
measurements were conducted on a Micromeritics Tristar/surface area and
porosity analyser at 77 K using nitrogen as the adsorption gas. The surface
CA 02583054 2007-04-02
WO 2006/037160 PCT/AU2005/001511
36
areas were calculated by the Brunauer-Emmett-Teller (BET) method.
Thermogravimetric analysis (TGA) was performed on a Mettler Toledo/
TGA/SDTA851 e Module analyser.
Transmission electron microscopy (TEM) was used to examine the NPPs
obtained after silica removal. TEM shows that individual NPP-lys particles
were
produced, with no aggregation observed (Figure 8a). These particles retained
the original spherical shape of the MS templates, and did not show signs of
collapse, as is typically observed for PE capsules. The NPP-lys had diameters
ranging from 1.6-2.4 pm, some 20 % smaller than the MS template particles
(>90% of MS particles are within 2-3 pm). SEM also revealed the NPP-lys to be
individual particles (Figure 8d). At higher magnification, the surface
roughness
of the NPP-lys is apparent (Figure 8e). The highly efficient template role of
the
MS spheres for the preparation of the NPPs is attributed to the disordered
pore
structure of the larger mesopores (10-40 nm, 1.2 cm3 g"1) and the high surface
area (630 m2 g"1) of the MS particles. NPPs were not formed if the PAA
infiltration step was eliminated, even if EDC cross-linking was performed on
the
lysozyme-loaded MS spheres. This clearly indicates that PAA plays an
essential role in connecting the proteins. When using cytochrome C, which has
a similar size and pl as lysozome, the NPP-cyt formed (Figure 2b) were similar
in appearance to those shown in Figure 8a. However, for catalase, which has a
higher molecular weight and size (-10.4 nm) and lower pl (5.4), considerably
less protein loading was obtained in the MS spheres (-7.5 wt%). Both particles
similar to those shown in Figure 8a-c as well as non-spherical ("collapsed")
particles (-30%) were observed. The low protein loading most probably results
in collapse of some of the particles upon drying (data not shown). This
highlights the importance of high protein loadings in the MS spheres, which is
largely determined by the protein size and the protein pl, to generate stable
NPPs. Lowering the catalase deposition solution pH to ca. 5 did not yield
higher loadings because of the complex interplay of electrostatic and
secondary
interactions associated with protein adsorption.
Several other PEs were used to bridge the lysozyme-loaded MS spheres
to examine the effect of PE on the NPPs. When the polypeptide poly(L-
CA 02583054 2007-04-02
WO 2006/037160 PCT/AU2005/001511
37
glutamic acid) (PGA) was used and the PGA-lysozyme structure cross-linked
with EDC, NPP-lys were aiso formed. These NPPs are similar in appearance to
those obtained when PAA was used (Figure 8a). In both the PAA/lysozyme and
PGA/lysozyme systems, cross-linking was required to obtain spherical and
intact NPPs. In contrast, stable NPP-lys were prepared without the cross-
linking step if poly(sodium 4-styrenesulfonate) (PSS, M, 70 000, 5 mg mL"1 in
0.1 M NaCI) was used as the bridging PE (Figure 8c). This can be explained by
enhanced interactions between the lysozyme and PSS, compared with PAA or
PGA. The NPP-lys prepared using PSS as the bridging PE had diameters
ranging from 1.0-1.3 pm (Figure 8c), about 50% smaller than the MS template
particles. This shrinkage may be caused by the higher mobility of the non-
cross-linked protein. These results indicate that various PEs can be used as
bridging polymers and that the PE type can determine the final size of the
NPPs. The PE would also likely govern the porosity and stability of the
protein
spheres.
Experiments were conducted using linker PEs with different molecular
weights to provide evidence for the infiltration of PAA and subsequent
formation
of connected protein layers in the mesopores. The use of PAA with a much
high molecular weight (250 000 Da) than that used to generate the NPP-lys
shown in Figure 8a-c (70 000 Da), resulted in "collapsed capsule" structures
(Figure 8f). These data suggest that the high molecular weight PAA is too
large
to enter the protein-loaded mesopores, and therefore is mainly restricted to
the
outside surface of the particles. Hence, only a capsule-like complex of
lysozyme and PAA was formed. The presence of salt in the PE solution, which
affects the molecular size, also plays an essential role in the preparation of
NPPs. Only capsule-like materials (similar to those shown in Figure 8d) were
obtained when PSS (70 000 Da) solutions without sait were used. This is in
stark contrast to the NPP-lys prepared when salt (0.1 M NaCI) was used in the
PSS adsorption solution (see Figure 8c). In the absence of salt, PSS adopts a
more linear conformation, and is therefore also restricted to adsorption
mainly
on the outside particle surface.
CA 02583054 2007-04-02
WO 2006/037160 PCT/AU2005/001511
38
Confocal Laser Scanning Microscopy
Confocal laser scanning microscopy (CLSM) experiments were
conducted to investigate the inner structure of the NPPs. Figure 9 shows CLSM
images of the NPP-lys after incubation in Rhodamine 6G (MW 479 Da) solution
for 60 min, followed by washing with water. The bright spheres seen are due to
the homogeneous distribution of the dye in the particles (Figure 9a inset),
reflecting the non-hollow structure and the porous nature of the particles.
This
suggests that NPPs can be used as bioreactors and as drug loading vehicles.
The CLSM image also shows that NPPs are well separated in solution. For the
proteins linked by PAA of high molecular weight (250 000 Da), distinct
fluorescent rings are observed, indicating localization of most of the dye on
the
outside surface (Fig. 9b). In this case, a hollow-structured lysozyme-PAA
layer
is formed, which is in accordance with the capsule structure observed from TEM
(Figure 8d).
The NPP-lys has a lysozyme: PAA composition of -5:1 (weight to
weight), that is, about 83 wt% of the particles are protein. The protein
content is
significantly higher than that of the proteins adsorbed in preformed
nanoporous
PAA/PAH spheres (ca. 1:1) and in the MS spheres (ca. 0.4:1). Although PE is
used to bridge the protein molecules, the NPPs are composed mostly of protein:
the biomolecule content (mass:mass) is 12.5 times higher than protein loaded
in
MS spheres. Further the NPPs composed entirely of biocompatible materials
can be prepared by using, for example, polypeptides (i.e. PGA) as the bridging
molecule: The high protein content of such particles is of interest in drug
delivery, especially for improving drug efficacy and decreasing side effects.
Enzyme Activity Assay
The enzyme activity was determined spectrophotometrically using H202
as a substrate. The NPP-cafi (or free enzyme) was added to 11 mM H202 in 50
mM PB solution (pH 7.0) with rapid stirring. The decrease in absorbance at 240
nm (with an extinction coefficient of 0.041 mmol-1 cm 1) with time was
recorded
immediately after the enzyme was mixed into the above solution at 20 C. One
unit of catalase will decompose I pmol of H202 per minute at pH 7.0 and 20 C.
CA 02583054 2007-04-02
WO 2006/037160 PCT/AU2005/001511
39
The activity of catalase-loaded in the MS particles (-66% of the free
protein) is normalized as 100%. The activity slightly decreased to -91% after
PAA infiltration. After EDC cross-linking, the MS-immobilized and cross-linked
protein retains 75% of its activity (relative to catalase-loaded MS
particles),
which is significantly higher than that for catalase immobilized in MS spheres
and encapsulated with four PDDA/PSS bilayers (ca. 50%), and for catalase
cross-linked by glutaraidehyde in chitosan beads (-1 % activity). After silica
removal, the catalase activity increased from 75% to 86% (corresponding to
57% of the free protein in bulk solution), suggesting increased substrate
accessibility after removal of the MS template.
Example 3 Formation of Fibrous Particles
The method is also amendable to prepare nanoporous protein structures
with various morphologies. Nanoporous protein fibers (NPF) were prepared by
using MS templates with fiber morphologies using the procedures outlined in
examples 1 and 2. MS fibers have similar porosity to the MS spheres used,
with the exception of the worm-shaped morphology about 1 pm in diameter, and
are 10 to 30 pm in length. Figure 10 shows the NPF at different magnifications
after silica removal. The protein fibers have lengths of tens of micrometers
and
diameters of hundreds of nanometers, closely mimicking the sizes of the MS
fibers. Realization of these structures indicates that the PE-bridging
template
synthesis provides a facile method to control protein morphologies.
Industrial applicability
Layer-by-layer coating for nanoporous polyelectrolyte spheres may be
used in many applications, for example, in drug delivery with selectivity due
to
the control of surface functionality. The nanoporous polyelectrolyte particles
have high adsorption capacity and can be used in a number of applications, for
example drug delivery, separation of biomaterials such as enzymes or non-bio
materials such as separation of heavy metal ions or toxic organics molecules,
used as adsorbents for dyes and may also be useful in the controlled release
of
fragrances in certain applications.
CA 02583054 2007-04-02
WO 2006/037160 PCT/AU2005/001511
The nanoporous protein particles can be prepared with protein contents
as high as 83 wt% and from a range of polyelectrolytes/proteins, including
biocompatible bridging polymers such as polypeptides. The well retained
protein activities make such particles promising for functional protein drug
5 delivery
One particularly attractive use of the materials of the invention is in
methods of delivering an active agent to a target site the method including
the
steps of (I) adsorbing the active agent onto a multilayer polyelectrolyte
material
10 of the invention and (ii) delivering the polyelectrolyte material to the
target site.
The active agent may be adsorbed in any of a number of ways but us typically
adsorbed by suspending a polyelectrolyte material of the invention into a
solution of the active agent. The active agent is adsorbed onto the
polyelectrolyte material which can then be isolated from the solution. The
15 polyelectrolyte material with eth active agent adsorbed thereon may then be
delivered to the target site such as by administration to the site. Any
suitable
active agent may be chosen such as therapeutic agent including
pharmaceuticals, veterinary chemicals and the like. Alternatively the active
agent may be a fragrance or a cleaning chemical which is intended to be
20 delivered to its site of action.
The polyelectrolyte material of the invention also finds use as a micro
reactor. It is found that the materials adsorb compounds and can thus be used
to adsorb one or more reactive species allowing them to be held proximal to
25 each other to facilitate reaction.
Accordingly another application of the materials is in methods of
conducting a chemical reaction including contacting a solution containing one
or
more reactants with a polyelectrolyte material of the invention. The step of
30 contacting preferably involves addition of the polyelectrolyte material of
the
invention to a solution containing the reactant(s) in question. The chemical
reaction may be carried out by the polyelectrolyte material acting as a micro
reactor for the chemical reactant(s) as discussed above or it may actually
take
part in the reaction. In a particularly preferred embodiment the reaction is
an
CA 02583054 2007-04-02
WO 2006/037160 PCT/AU2005/001511
41
enzymatic reaction, preferably an enzymatic catalytic reaction of a reactant.
In
a most preferred embodiment the polyelectrolyte material catalyses the
reaction.
As a result of their ability to adsorb chemical compounds the
polyelectrolyte materials of the invention may be used as adsorbents.
Accordingly another use is in methods of removing a compound from solution
including contacting the solution with a polyelectrolyte material of the
invention,
allowing sufficient time for the compound to be adsorbed by the
polyelectrolyte
material and removing the polyelectrolyte material from the solution. This
method may be used to isolate drugs from solution or in the purification of
solutions containing trace amounts of compounds that it is desired be removed
from solution.
Finally, it is to be understood that various alterations, modifications
and/or additions may be introduced into the constructions and arrangements of
parts previously described without departing from the spirit or ambit of the
invention.
25