Note: Descriptions are shown in the official language in which they were submitted.
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
METHODS FOR NEUROPROTECTION
Cross Reference To Related Applications
This application claims the benefit of U.S. Provisional application Serial
Number 60/619,402 filed October 15, 2004 and U.S. Provisional application
Serial Number 60/698,403 filed July 12, 2005. These two Provisional
applications are hereby incorporated by reference.
BACKGROUND OF THE INVENTION
FIELD OF THE INVENTION
The present invention relates generally to the fields of pharmacology,
neurology and psychiatry and to methods of protecting the cells of a
mammalian central nervous system from damage or injury. More specifically,
this invention provides methods for the use of certain carbamate compounds
for neuroprotection.
DESCRIPTION OF THE RELATED ART
Injuries or trauma of various kinds to the central nervous system (CNS)
or the peripheral nervous system (PNS) can produce profound and long-lasting
neurological and/or psychiatric symptoms and disorders. One form that this
can take is the progressive death of neurons or other cells of the central
nervous system (CNS), i.e., neurodegeneration or neuronal degeneration.
Neuronal degeneration as a result of, for example; Alzheimer's disease,
multiple sclerosis, cerebral-vascular accidents (CVAs) stroke, traumatic brain
injury, spinal cord injuries, degeneration of the optic nerve, e.g., ischemic
optic
neuropathy or retinal degeneration and other central nervous system disorders
is an enormous medical and public health problem by virtue of both its high
incidence and the frequency of long- term sequelae. Animal studies and
clinical
trials have shown that amino acid transmitters (especially glutamate),
oxidative
stress and inflammatory reactions contribute strongly to cell death in these
conditions.
1
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
Upon injury or upon ischemic insult, damaged neurons release massive
amounts of the neurotransmitter glutamate, which is excitotoxic to the
surrounding neurons (Choi et al., (1988), Neuron 1: 623-634; Rothman et al.,
(1984), J. Neurosci. 4: 1884-1891; Choi end Rothman, (1990), Ann. Rev.
Neurosci. 13: 171-182; David et al., (1988), Exp. Eye Res. 46:657-662; Drejer
et al., (1985), J. Neurosci. 45:145-151. Glutamate is a negatively charged
amino acid that is an excitatory synaptic transmitter in the mammalian nervous
system. Although the concentration of glutamate can reach the millimolar range
in nerve terminals its extracellular concentration is maintained at a low
level to
prevent neurotoxicity. It has been noted that glutamate can be toxic to
neurons
if presented at a high concentration. The term "excitotoxicity" has been used
to
describe the cytotoxic effect that glutamate (and other such excitatory amino
acids) can have on neurons when applied at high dosages.
Physiologically, excessive release, inhibition of uptake, or both can
achieve high levels of glutamate. Normally, a low concentration of
extracellular
glutamate is maintained by both neurons and astrocytes. Neurons store
glutamate in intracellular stores and regulate its release. See, Reagan, R.F.,
Excitotoxicity and Central Nervous System Trauma, in The Neurobiology of
Central Nervous Trauma, New York, Oxford University Press, 1994, pp. 173-
181 (Salzman SK, Faden Al, eds). Astrocytes take up extracellular glutamate
by specific transporters and convert the glutamate into glutamine that is then
released for neuronal uptake. See, Robinson, M.B. & Dowd LA, Adv
Pharmacol, 1997; 37:69-115. In the process of excitotoxicity, glutamate is
released in a self-perpetuating manner by the neurons, resulting in excessive
or proionged activation of glutamate receptors.
The conjunction of such excessive glutamate stimulation on the energy-
depleted neurons taken with the compromised ability of the neurosupportive
astrocytes to sequester toxic levels of extracellular glutamate leads to
neuronal
death via necrosis and apoptosis. Various interventions are currently being
examined to reduce neuronal death associated with central nervous system
injuries and diseases. See, Kermer et al., Cell Tissue Res 298:383-395, 1999.
Such therapies include glutamate release inhibitors, glutamate receptor
2
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
antagonists, Ca2+ channel blockers, GABA receptor agonists, gangliosides,
neurotrophic factors, calpain inhibitors, caspase inhibitors, free radical
scavengers, immuno- and cell metabolism modulators.
For example, several studies have shown the involvement of glutamate
in the pathophysiology of:1) Huntington's disease (HD) (Coyle and Schwartz,
(1976), Nature 263: 244-246; 2) Alzheimer's disease (AD) (Maragos et al,
(1987), TINS 10: 65-68; 3) Epilepsy (Nadler et al, (1978), Nature 271: 676-
677); 4) Lathyrism (Spencer et al, (1986), Lancet 239: 1066- 1067; 5)
Amyotropic Lateral Sclerosis (ALS) and Parkinsonian dementia of Guam
(Caine et al, (1986), Lancet 2: 1067-1070) as well as in the neuropathology
associated with stroke, ischemia and reperfusion (See, Dykens et al, (1987),
J.
Neurochem. 49: 1222-1228).
Thus, injury to neurons may be caused by overstimulation of receptors
by excitatory amino acids including glutamate and aspartate (See, Lipton et
al.
(1994) New Engl. J. Med. 330:613 621). Indeed, the N-methyl-D-aspartate
(NMDA) subtype of glutamate receptor is suggested to have many important
roles in normal brain function, including synaptic transmission, iearning and
memory, and neuronal development (See, Lipston et al. (1994) supra; Meldrum
et al. (1990) Trends Pharm. Sci. 11:379-387). However, over-stimulation of the
NMDA subtype of glutamate receptor leads to increased free radical production
and neuronal cell death, which can be modulated by antioxidants (See, Herin
et al. (2001) J. Neurochem. 78:1307-1314; Rossato et al. (2002) Neurosci.
Lett. 318:137-140).
In addition, in many chronic neurodegenerative conditions, inflammation
and oxidative stress are key components of the pathology. These conditions
include Aizheimer's disease (AD). Alzheimer's disease (AD) is characterized by
the accumulation of neurofibrillary tangles and senile plaques, and a
widespread, progressive degeneration of neurons in the brain. Senile plaques
are rich in amyloid precursor protein (APP) that is encoded by the APP gene
located on chromosome 21. A commonly accepted hypothesis underlying
pathogenesis of AD is that abnormal proteolytic cleavage of APP leads to an
3
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
excess extracellular accumulation of beta-amyloid (AP) peptide that has been
shown to be toxic to neurons (See, Selkoe et al., (1996), J. Biol. Chem. 271:
487-498; Quinn et al., (2001), Exp. Neurol. 168: 203-212; Mattson et al.,
(1997), Alzheimer's Dis. Rev. 12: 1-14; Fakuyama et al., (1994), Brain Res.
667: 269-272).
Parkinson's disease (PD) is a progressive neurodegenerative disorder
characterized by a dysfunction of movement consisting of akinesia, rigidity,
tremor and postural abnormalities. This disease has been associated with the
loss of nigro-striatal dopaminergic neuronal integrity and functionality as
evidenced by substantial loss of dopaminergic neurons in substantia nigra pars
compacta (SNpc) (See, Pakkenberg et al. (1991) J. Neurol. Neurosurg.
Psychiat. 54:30-33), and a decrease in content, synaptic and vesicular
transporters of dopamine in the striatum (see, for example, Guttnan et al.
(1997) Neurology 48:1578-1583).
Death of neurons and supporting cells in the central (CNS) or peripheral
(PNS) nervous system of mammals including humans as a result of trauma,
injury of many kinds, ischemia, metabolic derangements, e.g., diabetes
hypoxia, toxins or surgical intervention causes both acute and chronic and
progressive loss of function and disability. Thus there is a need for the
development of methods and compounds that can protect the cells of the
mammalian nervous system from this degeneration, i.e., are neuroprotective.
SUMMARY OF THE INVENTION
The present invention relates in general to neuroprotective methods,
and more specifically to methods and compounds for prevention of damage to
cells of the mammalian central and peripheral nervous system resulting from
injury, trauma, surgery or acute or chronic disease processes.
This invention is based, in part, on the discovery that the administration
of one or more members of a family of carbamate compounds either alone or
in combination with one or more other neuroprotective medications provides a
neuroprotective effect on the mammalian nervous system.
4
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
L nm-. a.- ..._, ..._ . . .._ _
Neuroprotection provided by this invention includes protection from
damage resulting from neural injury or insult and from neurotoxicity,
including
excitotoxicity. Thus, neuroprotection provided by this invention will be
useful in
the treatment of acute and chronic neurodegenerative disorders that may
involve excitotoxicity, for example glutamate excitotoxicity, including
stroke/ischemia, surgical trauma, Traumatic Brain Injury (TBI), biunt, closed
or
penetrating head trauma, epilepsy, Huntington's disease, Amyotrophic Lateral
Sclerosis (ALS), diabetic neuropathy and hypoglycemic encephaiopathy.
Neuroprotection provided by this invention may be brought about upon
injured or diseased tissue or in a preventative fashion during or prior to
events
expected to lead to a neural insult.
The invention provides methods for providing neuroprotection; for
inhibiting cell degeneration or cell death; for treatment or prophylaxis of a
neurodegenerative disease; or for ameliorating the cytotoxic effect of a
compound (for example, a excitatory amino acid such as glutamate; a toxin; or
a prophylactic or therapeutic compound that exerts a cytotoxic side effect) in
a
subject in need thereof, by administering to the subject an effective amount
of
a compound of the invention, or it's pharmaceutically acceptable salt or ester
either alone or in combination with another medication along with a
pharmaceutically acceptabie excipient. In various embodiments, the methods
of the invention include protection against excitotoxicity, for example
glutamate
excitotoxicity.
In various embodiments, the subject, for example, a human, may be
suffering from neural insult or injury; or may be suffering from a condition
selected from substance abuse, trauma, stroke, ischemia, Huntington's
disease, Alzheimer's disease, Parkinson's disease, prion disease, variant
Creutzfeld-Jakob disease, amyotrophic or hypogiycemic encephalopathy; or
may be undergoing surgery or other intervention. The subject may have a pre-
existing condition that would benefit by neuroprotection or the patient may be
treated to reduce deleterious effects of a concomitant or subsequent neural
injury, such as may occur during surgery or other intervention.
5
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
Accordingly, the present invention provides methods for providing
neuroprotection comprising administering to a subject in need thereof a
therapeutically effective amount of a composition that comprises at least one
compound having Formula 1 or Formula 2:
Xl OH Rl
X2 p N
R2
O
X3 XS
X4
Formula 1
R3
N
R1
Xi O R4
O N
X2 R2
O
X3 X5
X4
Formula 2
wherein Ri, R2, R3, and R4 are, independently, hydrogen or C1-C4 alkyl; and
X1,
X2, X3, X4, and X5 are, independently, hydrogen, fluorine, chlorine, bromine
or
iodine. The said Ci-C4 alkyl group of Formula 1 or Formula 2 can be
substituted or unsubstituted. In one aspect of the present invention, the C1-
C4
alkyl group is substituted with a phenyl group. The phenyl group can be
unsubstituted or substituted. In certain embodiments, the phenyl group is
6
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
unsubstituted or substituted with halogen, C1-C4 alkyl, C1-C4 aikoxy, amino,
nitro, or cyano.
In the present invention, X1, X2, X3, X4, and X5 can be hydrogen,
fluorine, chlorine, bromine or iodine. In certain embodiments, X1, X2, X3, X4,
and X5 are, independently, hydrogen or chlorine. In a preferred embodiment of
the present invention, Xi is fluorine, chlorine, bromine or iodine. In one
aspect,
Xi is chlorine and X2, X3, X4, and X5 are, independentiy, hydrogen. In another
preferred embodiment, R1, R2, R3, and R4 are, independently, hydrogen.
The present invention provides enantiomers of Formula 1 or Formula 2
for providing neuroprotection in a subject. In certain embodiments, a
compound of Formula 1 or Formula 2 will be in the form of a single enantiomer
thereof. In other embodiments, a compound of Formula 1 or Formula 2 will be
in the form of an enantiomeric mixture in which one enantiomer predominates
with respect to another enantiomer. In one aspect, the enantiomer will
predominate to the extent of 90% or greater or to the extent of 98% or
greater.
The present invention also provides methods comprising administering
to a subject a neuroprotective amount of a composition that comprises at least
one compound having Formula 1 or Formula 2 wherein R1, R2, R3, and R4 are,
independently, hydrogen or C1-C4 alkyl; and Xi, X2, X3, X4, and X5 are,
independently, hydrogen, fluorine, chlorine, bromine or iodine. In one
embodiment, before administration of the composition to the subject, a
determination will be made as to whether or not the subject suffers from some
form of acute or chronic neurodegeneration or nervous system injury.
The present invention also provides methods comprising identifying a
patient at risk of deveioping acute or chronic neurodegeneration or nervous
system injury or a patient in need of treatment with a neuroprotective drug
(NPD), as defined below and administering a composition that comprises at
least one compound having Formula 1 or Formula 2 to the subject.
In certain embodiments of the present invention, a therapeutically
effective amount of a compound having Formula 1 or Formula 2 for providing
neuroprotection is from about 1.0 mg/Kg/dose to about 150 mg/Kg/dose. In a
70 kg human this would correspond to a daily dose of from about 70 mg/day to
about 10,500 mg/day.
7
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
In certain embodiments a therapeutically effective amount of
pharmaceutical composition for providing neuroprotection comprising one or
more of the enantiomers of this invention or a pharmaceutically acceptable
salt
or ester thereof and a pharmaceutically acceptable carrier or excipient is
administered to a subject or patient in need of treatment with a
neuroprotective
drug or NPD.
Pharmaceutical compositions comprising at least one compound having
Formula 1 or Formula 2 are administered to subjects in need thereof. In
certain embodiments, a subject or patient in need of treatment with a
neuroprotective drug or NPD may be one who has experienced some form of
acute trauma or injury to the cells of the central or peripheral nervous or
who
has some form of acute or chronic neurodegenerative disorder. In one aspect,
the subject or patient will be determined to be at risk for developing an
acute or
chronic neurodegenerative disorder at the time of administration, i.e., a
patient
in need of treatment with a neuroprotective drug. In other embodiments, a
subject in need thereof is one who has acute injury or trauma to the cells of
their nervous system at the time of administration.
BRIEF DESCRIPTION OF THE FIGURES
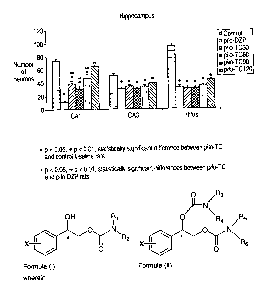
Figure 1: is a graph that shows the effects of increasing doses of TC on the
number of neurons in different areas of the hippocampus counted at 14 days
after li-pilo SE. Values are expressed as the number of neuronal cell bodies
in
each area of interest S.E.M.
Figure 2: is a graph that shows the effects of increasing doses of TC on the
number of neurons in different nuclei of the amygdala counted at 14 days after
li-pilo SE. Values are expressed as the number of neuronal cell bodies in each
area of interest S.E.M.
Figure 3: is a graph that shows the effects of increasing doses of TC on the
number of neurons in different nuclei of the thalamus counted at 14 days after
8
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
li-pilo SE. Values are expressed as the number of neuronal cell bodies in each
area of interest S.E.M.
Figure 4: is a graph that shows the effects of increasing doses of TC on the
number of neurons in different areas of the cortex counted at 14 days after Ii-
pilo SE. Values are expressed as the number of neuronal cell bodies in each
area of interest S.E.M.
Figure 5: is a graph that shows the effects of increasing doses of TC on the
latency to the first spontaneous seizure. Values are expressed as the mean
latency in days for each group S.E.M.
Figure 6: is a graph that shows the effects of increasing doses of TC on the
frequency of spontaneous seizures video-recorded over a 4 weeks period.
Values are expressed as the mean number of seizures S.E.M. The total
represents the total number of seizures observed during the 4 weeks of video-
recording and the mean represents the mean number of seizures per week.
The Anova test demonstrated an effect of the treatment on the total number of
seizures (p=0.045) and the mean number of seizures per week (p=0.045)
Figure 7: shows the total number of seizures video-recorded over four weeks
plotted according to the latency to the first spontaneous seizure (SL = short
latency, LL = long latency). Values are expressed as the mean number of
seizures for each subgroup S.E.M. The ANOVA test did not show any
significant effect of the treatment.
Figure 8: shows the correlation between the latency to the first spontaneous
seizure and the total number of seizures observed during the four following
weeks.
DETAILED DESCRIPTION OF THE INVENTION
The Carbamate Compounds of the Invention
9
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
The present invention provides methods of using certain 2-phenyl-1,2-
ethanediol monocarbomates and dicarbamates to provide neuroprotection to
mammals in need thereof.
Suitable methods for synthesizing and purifying carbamate compounds,
including carbamate enantiomers, used in the methods of the present invention
are well known to those skilled in the art. For example, pure enantiomeric
forms and enantiomeric mixtures of 2-phenyl-1, 2-ethanediol monocarbomates
and dicarbamates are described in United States Patent Numbers 5,854,283,
5,698,588, and 6,103,759, the disclosures of which are herein incorporated by
reference in their entirety.
Representative carbamate compounds according to the present
invention include those having Formula 1 or Formula 2:
Xl OH Ri
X2 O N
R2
X5
Xg
T
X4
Formula 1
/ R3
R1
X1 O R4
O N
X2 "--f R2
O
X3 X5
X4
Formula 2
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
wherein R1, R2, R3, and R4 are, independently, hydrogen or C1-C4 alkyl and Xi,
X2, X3, X4, and X5 are, independently, hydrogen, fluorine, chlorine, bromine
or
iodine.
"C1-C4 alkyl" as used herein refers to substituted or unsubstituted
aliphatic hydrocarbons having from 1 to 4 carbon atoms. Specifically included
within the definition of "alkyl" are those aliphatic hydrocarbons that are
optionally substituted. In a preferred embodiment of the present invention,
the
C1-C4 alkyl is either unsubstituted or substituted with phenyl.
The term "phenyl", as used herein, whether used alone or as part of
another group, is defined as a substituted or unsubstituted aromatic
hydrocarbon ring group having 6 carbon atoms. Specifically included within the
definition of "phenyl" are those phenyl groups that are optionally
substituted.
For example, in a preferred embodiment of the present invention, the, "phenyl"
group is either unsubstituted or substituted with halogen, C1-Cs4 aikyl, C1-C4
alkoxy, amino, nitro, or cyano.
In a preferred embodiment of the present invention, Xi is fluorine,
chlorine, bromine or iodine and X2, X3, X4, and X5 are hydrogen.
In another preferred embodiment of the present invention, X1, X2, X3, X4,
and X5 are, independently, chlorine or hydrogen.
In another preferred embodiment of the present invention, Ri, R2, R3,
and R4 are all hydrogen.
It is understood that substituents and substitution patterns on the
compounds of the present invention can be selected by one of ordinary skill in
the art to provide compounds that are chemically stable and that can be
readily
synthesized by techniques known in the art as well as the methods provided
herein.
Representative 2-phenyl-1, 2-ethanediol monocarbomates and
dicarbamates include, for example, the following compounds:
11
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
OH Ri
Xy
X2
~ R2
I
~
X3 X5
X4
Formula 3
OH R,
X1
X2 O
~ R2
I
/
Xg X5 O
X4
Formula 4
R3
O \ Ri
Xi R4
X2 ~ O N \ R2
O
X3 X5
4
Formula 5
12
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
,ve R3
/
X 0 R RIl
1 4
- N\
R2
X2 p qX5
O
X3 Formula 6
p1 OH
p NH2
O
Formula 7
NH2
O
ci
p '--f NH2
O
Formula 8
The present invention includes the use of isolated enantiomers of
Formula 1 or Formula 2. In one preferred embodiment, a pharmaceutical
composition comprising the isolated S-enantiomer of Formula 1 is used to
provide neuroprotection in a subject. In another preferred embodiment, a
13
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
pharmaceutical composition comprising the isolated R-enantiomer of Formula
2 is used to provide neuroprotection in a subject. In another embodiment, a
pharmaceutical composition comprising the isolated S-enantiomer of Formula
1 and the isolated R-enantiomer of Formula 2 can be used to provide
neuroprotection in a subject.
The present invention also inciudes the use of mixtures of enantiomers
of Formula 1 or Formula 2. In one aspect of the present invention, one
enantiomer will predominate. An enantiomer that predominates in the mixture
is one that is present in the mixture in an amount greater than any of the
other
enantiomers present in the mixture, e.g., in an amount greater than 50%. In
one aspect, one enantiomer will predominate to the extent of 90% or to the
extent of 91%, 92%, 93%, 94%, 95%, 96%, 97% or 98% or greater. In one
preferred embodiment, the enantiomer that predominates in a composition
comprising a compound of Formula 1 is the S-enantiomer of Formula 1. In
another preferred embodiment, the enantiomer that predominates in a
composition comprising a compound of Formula 2 is the R-enantiomer of
Formula 2.
In a preferred embodiment of the present invention, the enantiomer that
is present as the sole enantiomer or as the predominate enantiomer in a
composition of the present invention is represented by Formula 3 or Formula 5,
wherein Xi, X2, X3, X4, X5, Ri, R2, R3, and R4 are defined as above, or by
Formula 7 or Formula 8.
OH Rl
X1
X2 O N
R2
O
X3 X5
X4
Formula 3
14
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
R3
\ Ri
O R4
X1
X2
~ O N \ R2
O
Xg X5
4
Formula 5
pl OH
p NH2
O
Formula 7
NH2
CI
p NH2
O
Formula 8
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
The present invention provides methods of using enantiomers and
enantiomeric mixtures of compounds represented by Formula 1 and Formula
2. A carbamate enantiomer of Formula 1 or Formula 2 contains an asymmetric
chiral carbon at the benzylic position, which is the aliphatic carbon adjacent
to
the phenyl ring.
An enantiomer that is isolated is one that is substantially free of the
corresponding enantiomer. Thus, an isolated enantiomer refers to a
compound that is separated via separation techniques or prepared free of the
corresponding enantiomer. The term "substantially free," as used herein,
means that the compound is made up of a significantly greater proportion of
one enantiomer. In preferred embodiments, the compound includes at least
about 90% by weight of a preferred enantiomer. In other embodiments of the
invention, the compound includes at least about 99% by weight of a preferred
enantiomer. Preferred enantiomers can be isolated from racemic mixtures by
any method known to those skilled in the art, including high performance
liquid
chromatography (HPLC) and the formation and crystallization of chiral salts,
or
preferred enantiomers can be prepared by methods described herein.
Methods for the preparation of preferred enantiomers wouid be known
to one of skill in the art and are described, for example, in Jacques, et al.,
Enantiomers, Racemates and Resolutions (Wiley Interscience, New York,
1981); Wilen, S.H., et al., Tetrahedron 33:2725 (1977); Eliel, E.L.
Stereochemistry of Carbon Compounds (McGraw-Hill, NY, 1962); and Wilen,
S.H. Tables of Resolving Agents and Optical Resolutions p. 268 (E.L. Eliel,
Ed., Univ. of Notre Dame Press, Notre Dame, IN 1972).
Additionally, compounds of the present invention can be prepared as
described in United States Patent Number 3,265,728 (the disclosure of which
is herein incorporated by reference in its entirety and for all purposes),
3,313,692 (the disclosure of which is herein incorporated by reference in its
entirety and for all purposes), and the previously referenced United States
Patent Numbers 5,854,283, 5,698,588, and 6,103,759 ( the disclosures of
which are herein incorporated by reference in their entirety and for all
purposes).
The Nature of Neuroprotection
16
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
Patients with injury or damage of any kind to the central (CNS) or
peripheral (PNS) nervous system including the retina may benefit from these
neuroprotective methods. This nervous system injury may take the form of an
abrupt insult or an acute injury to the nervous system as in, for example,
acute
neurodegenerative disorders including, but not limited to; acute injury,
hypoxia-
ischemia or the combination thereof resulting in neuronal cell death or
compromise. Acute injury includes, but is not limited to, Traumatic Brain
Injury
(TBI) including, ciosed, blunt or penetrating brain trauma, focal brain
trauma,
diffuse brain damage, spinal cord injury, intracranial or intravertebral
lesions
(including, but not limited to, contusion, penetration, shear, compression or
laceration lesions of the spinal cord or whiplash shaken infant syndrome.
In addition, deprivation of oxygen or blood supply in general can cause
acute injury as in hypoxia and/or ischemia including, but is not limited to,
cerebrovascular insufficiency, cerebral ischemia or cerebral infarction
(including cerebral ischemia or infarctions originating from embolic occlusion
and thrombosis, retinal ischemia (diabetic or otherwise), glaucoma, retinal
degeneration, multiple sclerosis, toxic and ischemic optic neuropathy,
reperfusion following acute ischemia, perinatal hypoxic-ischemic injury,
cardiac
arrest or intracranial hemorrhage of any type (inciuding, but not limited to,
epidural, subdural, subarachnoid or intracerebral hemorrhage).
Trauma or injury to tissues of the nervous system may also take the
form of more chronic and progressive neurodegenerative disorders, such as
those associated with progressive neuronal cell death or compromise over a
period of time including, but not limited to, Alzheimer's disease, Pick's
disease,
diffuse Lewy body disease, progressive supranuclear palsy (Steel-Richardson
syndrome), multisystem degeneration (Shy-Drager syndrome), chronic epileptic
conditions associated with neurodegeneration, motor neuron diseases
(amyotrophic lateral sclerosis), multiple sclerosis, degenerative ataxias,
cortical
basal degeneration, ALS-Parkinson's-Dementia complex of Guam, subacute
sclerosing panencephalitis, Huntington's disease, Parkinson's disease,
synucleinopathies (including multiple system atrophy), primary progressive
aphasia, striatonigral degeneration, Machado-Joseph disease or
17
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
spinocerebellar ataxia type 3 and olivopontocerebellar degenerations, bulbar
and pseudobulbar palsy, spinal and spinobulbar muscular atrophy (Kennedy's
disease), primary lateral sclerosis, familial spastic paraplegia, Werdnig-
Hoffmann disease, Kugelberg-Welander disease, Tay-Sach's disease,
Sandhoff disease, familial spastic disease, Wohlfart-Kugelberg-Welander
disease, spastic paraparesis, progressive multifocal leukoencephalopathy,
familial dysautonomia (Riley-Day syndrome) or prion diseases (including, but
not iimited to Creutzfeld-Jakob disease, Gerstmann-Strussler-Scheinker
disease, Kuru disease or fatal familial insomnia).
In addition, trauma and progressive injury to the nervous system can
take place in various psychiatric disorders, including but not limited to,
progressive, deteriorating forms of Bipolar disorder or Schizoaffective
disorder
or Schizophrenia, Impulse Control disorders, Obsessive Compulsive disorder
(OCD), behavioral changes in Temporal Lobe Epilepsy and personality
disorders.
In one preferred embodiment the compounds of the invention would be
used to provide neuroprotection in disorders involving trauma and progressive
injury to the nervous system in various psychiatric disorders. These disorders
would be selected form the group consisting of; Schizoaffective disorder,
Schizophrenia, Impulse Control disorders, Obsessive Compulsive disorder
(OCD) and personality disorders.
In addition, trauma and injury make take the form of disorders
associated with overt and extensive memory loss including, but not limited to,
neurodegenerative disorders associated with age-related dementia, vascular
dementia, diffuse white matter disease (Binswanger's disease), dementia of
endocrine or metabolic origin, dementia of head trauma and diffuse brain
damage, dementia pugilistica or frontal lobe dementia, including but not
limited
to Pick's Disease.
Other disorders associated with neuronal injury include, but are not
limited to, disorders associated with chemical, toxic, infectious and
radiation
injury of the nervous system including the retina, injury during fetal
18
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
development, prematurity at time of birth, anoxic-ischemia, injury from
hepatic,
glycemic, uremic, electrolyte and endocrine origin, injury of psychiatric
origin
(including, but not limited to, psychopathology, depression or anxiety),
injury
from peripheral diseases and plexopathies (including plexus palsies) or injury
from neuropathy (including neuropathy selected from multifocal, sensory,
motor, sensory-motor, autonomic, sensory-autonomic or demyelinating
neuropathies (including, but not limited to Guillain-Barre syndrome or chronic
inflammatory demyelinating polyradiculoneuropathy) or those neuropathies
originating from infections, inflammation, immune disorders, drug abuse,
pharmacological treatments, toxins, trauma (including, but not limited to
compression, crush, laceration or segmentation traumas), metabolic disorders
(including, but not limited to, endocrine or paraneoplastic), Charcot-Marie-
Tooth disease (inciuding, but not limited to, type 1 a, 1 b, 2, 4a or 1-X
linked),
Friedreich's ataxia, metachromatic leukodystrophy, Refsum's disease,
adrenomyeloneuropathy, Ataxia-telangiectasia, Djerine-Sottas (including, but
not limited to, types A or B), Lambert-Eaton syndrome or disorders of the
cranial nerves).
Therefore, the term "neuroprotection" as used herein shall mean;
inhibiting, preventing, ameliorating or reducing the severity of the
dysfunction,
degeneration or death of nerve cells, axons or their supporting cells in the
central or peripheral nervous system of a mammal, including a human. This
includes the treatment or prophylaxis of a neurodegenerative disease;
protection against excitotoxicity or ameliorating the cytotoxic effect of a
compound (for example, a excitatory amino acid such as glutamate; a toxin; or
a prophylactic or therapeutic compound that exerts an immediate or delayed
cytotoxic side effect including but not limited to the immediate or delayed
induction of apoptosis) in a patient in need thereof.
Therefore, the term "a patient in need of treatment with a
neuroprotective drug (NPD)" as used herein will refer to any patient who
currently has or may develop any of the above syndromes or disorders, or any
disorder in which the patient's present clinical condition or prognosis could
19
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
benefit from providing neuroprotection to prevent the; development, extension,
worsening or increased resistance to treatment of any neurological or
psychiatric disorder.
The term "antiepileptic drug" (AED) will be used interchangeably with the
term "anticonvulsant agent," and as used herein, both terms refer to an agent
capable of inhibiting (e.g., preventing slowing, halting, or reversing)
seizure activity or ictogenesis when the agent is administered to a subject or
patient.
The term "treating" or "treatment" as used herein, refers to any indicia of
success in the prevention or amelioration of an injury, pathology or
condition,
including any objective or subjective parameter such as abatement; remission;
diminishing of symptoms or making the injury, pathology, or condition more
tolerable to the patient; slowing in the rate of degeneration or decline;
making
the final point of degeneration less debilitating; or improving a subject's
physical or mental well-being. The treatment or amelioration of symptoms can
be based on objective or subjective parameters; including the results of a
physical examination, neurological examination, and/or psychiatric
evaluations.
Accordingly, the term "treating" or "treatment" includes the administration of
the
compounds or agents of the present invention to provide neuroprotection. In
some instances, treatment with the compounds of the present invention will
done in combination with other neuroprotective compounds or AED's to
prevent, inhibit, or arrest the progression of neuronal death or damage or
brain
dysfunction or brain hyperexcitability.
The term "therapeutic effect" as used herein, refers to the effective
provision of neuroprotection effects to prevent or minimize the death or
damage or dysfunction of the cells of the patient's central or peripheral
nervous
system.
The term "a therapeutically effective amount" as used herein means a
sufficient amount of one or more of the compounds of the invention to produce
a therapeutic effect, as defined above, in a subject or patient in need of
such
neuroprotection treatment.
The terms "subject" or "patient" are used herein interchangeably and as
used herein mean any mammal including but not limited to human beings
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
including a human patient or subject to which the compositions of the
invention
can be administered. The term mammals include human patients and non-
human primates, as well as experimental animals such as rabbits, rats, and
mice, and other animals.
In some embodiments the methods of the present invention will be
advantageously used to treat a patient who is not suffering or known to be
suffering from a condition that is known in the art to be effectively treated
with
carbamate compounds or presently known neuroprotective compounds or
AEDs. In these cases the decision to use the methods and compounds of the
present invention would be made on the basis of determining if the patient is
a
"patient in need of treatment with a neuroprotective drug (NPD)", as that term
is
defined above.
In some embodiments this invention provides methods of
neuroprotection. In certain embodiments, these methods comprise
administering a therapeutically effective amount of a carbamate compound of
the invention to a patient who has not yet developed overt, clinical signs or
symptoms of injury or damage to the cells of the nervous system but who may
be in a high risk group for the development of neuronal damage because of
injury or trauma to the nervous system or because of some known
predisposition either biochemical or genetic or the finding of a verified
biomarker of one or more of these disorders.
Thus, in some embodiments, the methods and compositions of the
present invention are directed toward neuroprotection in a subject who is at
risk
of developing neuronal damage but who has not yet developed clinical
evidence. This patient may simply be at "greater risk" as determined by the
recognition of any factor in a subject's, or their families, medical history,
physical exam or testing that is indicative of a greater than average risk for
developing neuronal damage. Therefore, this determination that a patient may
be at a "greater risk" by any available means can be used to determine
whether the patient should be treated with the methods of the present
invention.
Accordingly, in an exemplary embodiments, subjects who may benefit
from treatment by the methods and compounds of this invention can be
21
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
identified using accepted screening methods to determine risk factors for
neuronal damage. These screening methods include, for example,
conventional work-ups to determine risk factors including but not limited to:,
for
example, head trauma, either closed or penetrating, CNS infections, bacterial
or viral, cerebrovascular disease including but not limited to stroke, brain
tumors, brain edema, cysticercosis, porphyria, metabolic encephalopathy, drug
withdrawal including but not limited to sedative-hypnotic or alcohol
withdrawal,
abnormal perinatal history including anoxia at birth or birth injury of any
kind,
cerebral palsy, learning disabilities, hyperactivity, history of febrile
convulsions
as a child, history of status epilepticus, family history of epilepsy or any a
seizure related disorder, inflammatory disease of the brain including lupis,
drug intoxication either direct or by placental transfer, including but not
limited
to cocaine poisoning, parental consanguinity, and treatment with medications
that are toxic to the nervous system including psychotropic medications.
The determination of which patients may benefit from treatment with an
NPD in patients who have no clinical signs or symptoms may be based on a
variety of "surrogate markers" or "biomarkers".
As used herein, the terms "surrogate marker" and "biomarker" are used
interchangeably and refer to any anatomical, biochemical, structural,
electrical,
genetic or chemical indicator or marker that can be reliabiy correlated with
the
present existence or future development of neuronal damage. In some
instances, brain-imaging techniques, such as computer tomography (CT),
magnetic resonance imaging (MRI) or positron emission tomography (PET),
can be used to determine whether a subject is at risk for neuronal damage.
Suitable biomarkers for the methods of this invention inciude, but are not
limited to: the determination by MRI, CT or other imaging techniques, of
sclerosis, atrophy or volume loss in the hippocampus or overt mesial temporal
sclerosis (MTS) or similar relevant anatomical pathology; the detection in the
patient's blood, serum or tissues of a molecular species such as a protein or
other biochemical biomarker, e.g., elevated levels of ciliary neurotrophic
factor
(CNTF) or elevated serum levels of a neuronal degradation product; or other
evidence from surrogate markers or biomarkers that the patient is in need of
treatment with a neuroprotective drug.
22
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
It is expected that many more such biomarkers utilizing a wide variety of
detection techniques will be developed in the future. It is intended that any
such marker or indicator of the existence or possibie future development of
neuronal damage, as the latter term is used herein, may be used in the
methods of this invention for determining the need for treatment with the
compounds and methods of this invention.
A determination that a subject has, or may be at risk for developing,
neuronal damage would also include, for example, a medical evaluation that
includes a thorough history, a physical examination, and a series of relevant
bloods tests. It can also include an electroencephalogram (EEG), CT, MRI or
PET scan. A determination of an increased risk of developing neuronal
damage or injury may also be made by means of genetic testing, including
gene expression profiling or proteomic techniques. (See, Schmidt, D.
Rogawski, M. A. Epilepsy Research 50; 71-78 (2002), and Loscher, W,
Schmidt D. Epilepsy Research 50; 3-16 (2002))
For psychiatric disorders that may be stabilized or improved by a
neuroprotective drug, e.g., Bipolar Disorder, Schizoaffective disorder,
Schizophrenia, Impulse Control Disorders, etc. the above tests may also
include a present state exam and a detailed history of the course of the
patients symptoms such as mood disorder symptoms and psychotic symptoms
over time and in relation to other treatments the patient may have received
over time, e.g., a life chart. These and other specialized and routine methods
allow the clinician to select patients in need of therapy using the methods
and
formulations of this invention.
In some embodiments of the present invention carbamate compounds
suitable for use in the practice of this invention will be administered either
singly or concomitantly with at least one or more other compounds or
therapeutic agents, e.g., with other neuroprotective drugs or antiepileptic
drugs,
anticonvulsant drugs. In these embodiments, the present invention provides
methods to treat or prevent neuronal injury in a patient. The method includes
the step of; administering to a patient in need of treatment, an effective
amount
of one of the carbamate compounds disclosed herein in combination with an
effective amount of one or more other compounds or therapeutic agents that
23
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
have the ability to provide neuroprotection or to treat or prevent seizures or
epileptogenesis or the ability to augment the neuroprotective effects of the
compounds of the invention.
As used herein the term "concomitant administration" or "combination
administration" of a compound, therapeutic agent or known drug with a
compound of the present invention means administration of the drug and the
one or more compounds at such time that both the known drug and the
compound will have a therapeutic effect. In some cases this therapeutic effect
will be synergistic. Such concomitant administration can involve concurrent
(i.e. at the same time), prior, or subsequent administration of the drug with
respect to the administration of a compound of the present invention. A person
of ordinary skill in the art, would have no difficulty determining the
appropriate
timing, sequence and dosages of administration for particular drugs and
compounds of the present invention.
The said one or more other compounds or therapeutic agents may be
selected from compounds that have one or more of the following properties:
antioxidant activity; NMDA receptor antagonist activity, augmentation of
endogenous GABA inhibition; NO synthase inhibitor activity; iron binding
ability,
e.g., an iron chelator; calcium binding ability, e.g., a Ca (II) chelator;
zinc
binding ability, e.g., a Zn (II) chelator; the ability to effectively block
sodium or
calcium ion channels, or to open potassium or chloride ion channeis in the
CNS of a patient.
In some preferred embodiments, the one or more other compounds or
therapeutic agents would antagonize NMDA receptors by binding to the NMDA
receptors (e.g., by binding to the glycine binding site of the NMDA receptors)
and/or the agent would augment GABA inhibition by decreasing glial GABA
uptake.
In addition the said one or more other compounds or therapeutic agents
may be any agent known to suppress seizure activity even if that compound is
not known to provide neuroprotection. Such agents would include but not be
limited to any effective AED known to one of skill in the art or discovered in
the
future, for example suitable agents include, but are not limited to;
carbamazepine, clobazam, clonazepam, ethosuximide, felbamate, gabapentin,
24
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
lamotigine, levetiracetam, oxcarbazepine, phenobarbital, phenytoin,
pregabalin, primidone, retigabine, talampanel, tiagabine, topiramate,
valproate,
vigabatrin, zonisamide, benzodiazepines, barbiturates and sedative hypnotics
in general.
In addition, in some embodiments, the compounds of this invention will
be used, either alone or in combination with each other or in combination with
one or more other therapeutic medications as described above, or their salts
or
esters, for manufacturing a medicament for the purpose of providing
neuroprotection to a patient or subject in need thereof.
Carbamate Compounds as Pharmaceuticals:
The present invention provides enantiomeric mixtures and isolated
enantiomers of Formula 1 and/or Formula 2 as pharmaceuticals. The
carbamate compounds are formulated as pharmaceuticals to provide
neuroprotection in a subject.
In general, the carbamate compounds of the present invention can be
administered as pharmaceutical compositions by any method known in the art
for administering therapeutic drugs inciuding oral, buccal, topical, systemic
(e.g., transdermal, intranasal, or by suppository), or parenteral (e.g.,
intramuscular, subcutaneous, or intravenous injection.) Administration of the
compounds directly to the nervous system can include, for example,
administration to intracerebral, intraventricular, intacerebroventricular,
intrathecal, intracisternal, intraspinal or peri-spinal routes of
administration by
delivery via intracranial or intravertebral needles or catheters with or
without
pump devices.
In addition, in the case of diseases or disorders of the eye including but
not limited to; retinal ischemia (diabetic or otherwise), glaucoma, retinal
degeneration, macular degeneration, multiple sclerosis, toxic and ischemic
optic neuropathy the compounds of the present invention, including
combinations of compounds, can be administered by means of direct
exogenous application to the eye, i.e., to the sclera or otherwise, e.g., eye
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
drops or by ocular implant or other slow delivery device including
microspheres
including by direct injection into the vitreous humor etc..
Compositions can take the form of tablets, pills, capsules, semisolids,
powders, sustained release formulations, solutions, suspensions, emulsions,
syrups, elixirs, aerosols, or any other appropriate compositions; and comprise
at least one compound of this invention in combination with at least one
pharmaceutically acceptable excipient. Suitable excipients are well known to
persons of ordinary skill in the art, and they, and the methods of formulating
the compositions, can be found in such standard references as Alfonso AR:
Remington's Pharmaceutical Sciences, 17th ed., Mack Publishing Company,
Easton PA, 1985, the disclosure of which is incorporated herein by reference
in
its entirety and for all purposes. Suitable liquid carriers, especially for
injectable solutions, include water, aqueous saline solution, aqueous dextrose
soiution, and glycols.
The carbamate compounds can be provided as aqueous suspensions.
Aqueous suspensions of the invention can contain a carbamate compound in
admixture with excipients suitable for the manufacture of aqueous
suspensions. Such excipients can include, for example, a suspending agent,
such as sodium carboxymethylcellulose, methylcellulose,
hydroxypropylmethylcellulose, sodium alginate, polyvinylpyrrolidone, gum
tragacanth and gum acacia, and dispersing or wetting agents such as a
naturally occurring phosphatide (e.g., lecithin), a condensation product of an
alkylene oxide with a fatty acid (e.g., polyoxyethylene stearate), a
condensation
product of ethylene oxide with a long chain aliphatic alcohol (e.g.,
heptadecaethylene oxycetanol), a condensation product of ethylene oxide with
a partial ester derived from a fatty acid and a hexitol (e.g., polyoxyethylene
sorbitol mono-oleate), or a condensation product of ethylene oxide with a
partial ester derived from fatty acid and a hexitol anhydride (e.g.,
polyoxyethylene sorbitan mono-oleate).
The aqueous suspension can also contain one or more preservatives
such as ethyl or n-propyl p-hydroxybenzoate, one or more coloring agents, one
or more flavoring agents, and one or more sweetening agents, such as
sucrose, aspartame or saccharin. Formulations can be adjusted for osmolarity.
26
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
Oil suspensions for use in the present methods can be formulated by
suspending a carbamate compound in a vegetable oil, such as arachis oil, olive
oil, sesame oil or coconut oil, or in a mineral oil such as liquid paraffin;
or a
mixture of these. The oil suspensions can contain a thickening agent, such as
beeswax, hard paraffin or cetyl alcohol. Sweetening agents can be added to
provide a palatable oral preparation, such as glycerol, sorbitol or sucrose.
These formulations can be preserved by the addition of an antioxidant such as
ascorbic acid. As an example of an injectable oil vehicle, see Minto, J.
Pharmacol. Exp. Ther. 281:93-102, 1997. The pharmaceutical formulations of
the invention can also be in the form of oil-in-water emulsions. The oily
phase
can be a vegetable oil or a mineral oil, described above, or a mixture of
these.
Suitable emulsifying agents include naturally occurring gums, such as
gum acacia and gum tragacanth, naturally occurring phosphatides, such as
soybean lecithin, esters or partial esters derived from fatty acids and
hexitol
anhydrides, such as sorbitan mono-oleate, and condensation products of these
partial esters with ethylene oxide, such as polyoxyethylene sorbitan mono-
oleate. The emulsion can also contain sweetening agents and flavoring
agents, as in the formulation of syrups and elixirs. Such formulations can
also
contain a demulcent, a preservative, or a coloring agent.
The compound of choice, alone or in combination with other suitable
components, can be made into aerosol formulations (i.e., they can be
"nebulized") to be administered via inhalation. Aerosol formulations can be
placed into pressurized acceptable propellants, such as
dichlorodifluoromethane, propane, nitrogen, and the like.
Formulations of the present invention suitable for parenteral
administration, such as, for example, by intraarticular (in the joints),
intravenous, intramuscular, intradermal, intraperitoneal, and subcutaneous
routes, can include aqueous and non-aqueous, isotonic sterile injection
solutions, which can contain antioxidants, buffers, bacteriostats, and solutes
that render the formulation isotonic with the blood of the intended recipient,
and
aqueous and non-aqueous sterile suspensions that can include suspending
agents, solubilizers, thickening agents, stabilizers, and preservatives. Among
the acceptable vehicles and solvents that can be employed are water and
27
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
Ringer's solution, an isotonic sodium chloride. In addition, sterile fixed
oils can
conventionally be employed as a solvent or suspending medium. For this
purpose any bland fixed oil can be employed including synthetic mono- or
diglycerides. In addition, fatty acids such as oleic acid can likewise be used
in
the preparation of injectables. These solutions are sterile and generally free
of
undesirable matter.
Where the compounds are sufficiently soluble they can be dissolved
directly in normal saline with or without the use of suitable organic
solvents,
such as propylene giycol or polyethylene glycol. Dispersions of the finely
divided compounds can be made-up in aqueous starch or sodium
carboxymethyl cellulose solution, or in suitable oil, such as arachis oil.
These
formulations can be sterilized by conventional, well-known sterilization
techniques. The formulations can contain pharmaceutically acceptable
auxiliary substances as required to approximate physiological conditions such
as pH adjusting and buffering agents, toxicity adjusting agents, e.g., sodium
acetate, sodium chloride, potassium chloride, calcium chloride, sodium lactate
and the like.
The concentration of a carbamate compound in these formulations can
vary widely, and will be selected primarily based on fluid volumes,
viscosities,
body weight, and the like, in accordance with the particular mode of
administration selected and the patient's needs. For IV administration, the
formulation can be a sterile injectable preparation, such as a sterile
injectable
aqueous or oleaginous suspension. This suspension can be formulated
according to the known art using those suitable dispersing or wetting agents
and suspending agents. The sterile injectable preparation can also be a
sterile
injectable solution or suspension in a nontoxic parenterally acceptable
diluents
or solvent, such as a solution of 1,3-butanediol. The formulations of
commends can be presented in unit-dose or multi-dose sealed containers,
such as ampoules and vials. Injection solutions and suspensions can be
prepared from sterile powders, granules, and tablets of the kind previously
described.
A carbamate compound suitable for use in the practice of this invention
can be and is preferably administered orally. The amount of a compound of
28
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
the present invention in the composition can vary widely depending on the type
of composition, size of a unit dosage, kind of excipients, and other factors
well
known to those of ordinary skill in the art. In general, the final composition
can
comprise, for example, from 0.000001 percent by weight (% w) to 10 % w of
the carbamate compound, preferably 0.00001 % w to 1 % w, with the
remainder being the excipient or excipients.
Pharmaceutical formulations for oral administration can be formulated
using pharmaceutically acceptable carriers well known in the art in dosages
suitable for oral administration. Such carriers enable the pharmaceutical
formulations to be formulated in unit dosage forms as tablets, pills, powder,
dragees, capsules, liquids, lozenges, gels, syrups, slurries, suspensions,
etc.
suitable for ingestion by the patient.
Formulations suitable for oral administration can consist of (a)
liquid solutions, such as an effective amount of the pharmaceutical
formulation
suspended in a diluents, such as water, saline or PEG 400; (b) capsules,
sachets or tablets, each containing a predetermined amount of the active
ingredient, as liquids, solids, granules or gelatin; (c) suspensions in an
appropriate liquid; and (d) suitable emulsions.
Pharmaceutical preparations for oral use can be obtained through
combination of the compounds of the present invention with a solid excipient,
optionally grinding a resulting mixture, and processing the mixture of
granules,
after adding suitable additional compounds, if desired, to obtain tablets or
dragee cores. Suitable solid excipients are carbohydrate or protein fillers
and
include, but are not limited to sugars, including lactose, sucrose, mannitol,
or
sorbitol; starch from corn, wheat, rice, potato, or other plants; cellulose
such as
methyl cellulose, hydroxymethyl cellulose, hydroxypropylmethyl-cellulose or
sodium carboxymethylcellulose; and gums including arabic and tragacanth; as
well as proteins such as gelatin and collagen. If desired, disintegrating or
solubilizing agents can be added, such as the cross-linked polyvinyl
pyrrolidone, agar, alginic acid, or a salt thereof, such as sodium alginate.
Tablet forms can include one or more of lactose, sucrose, mannitol, sorbitol,
calcium phosphates, corn starch, potato starch, microcrystalline cellulose,
gelatin, colloidal silicon dioxide, talc, magnesium stearate, stearic acid,
and
29
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
other excipients, colorants, fillers, binders, diluents, buffering agents,
moistening agents, preservatives, flavoring agents, dyes, disintegrating
agents,
and pharmaceutically compatible carriers. Lozenge forms can comprise the
active ingredient in a flavor, e.g., sucrose, as well as pastilles comprising
the
active ingredient in an inert base, such as gelatin and glycerin or sucrose
and
acacia emulsions, gels, and the like containing, in addition to the active
ingredient, carriers known in the art.
The compounds of the present invention can also be administered in the
form of suppositories for rectal administration of the drug. These
formulations
can be prepared by mixing the drug with a suitable non-irritating excipient
that
is solid at ordinary temperatures but liquid at the rectal temperatures and
will
therefore melt in the rectum to release the drug. Such materials are cocoa
butter and polyethyiene glycols.
The compounds of the present invention can also be administered by
intranasal, intraocular, intravaginal, and intrarectal routes including
suppositories, insufflation, powders and aerosol formulations (for examples of
steroid inhalants, see Rohatagi, J. Clin. Pharmacol. 35:1187-1193, 1995; Tjwa,
Ann. Allergy Asthma Immunol. 75:107-111, 1995).
The compounds of the present invention can be delivered transdermally,
by a topical route, formulated as applicator sticks, solutions, suspensions,
emulsions, gels, creams, ointments, pastes, jellies, paints, powders, and
aerosols.
Encapsulating materials can also be employed with the compounds of
the present invention and the term "composition" can include the active
ingredient in combination with an encapsulating material as a formulation,
with
or without other carriers. For example, the compounds of the present invention
can also be delivered as microspheres for slow release in the body. In one
embodiment, microspheres can be administered via intradermal injection of
drug (e.g., mifepristone)-containing microspheres, which slowly release
subcutaneously (see Rao, J. Biomater Sci. Polym. Ed. 7:623-645, 1995; as
biodegradable and injectable gel formuiations (see, e.g., Gao, Pharm. Res.
12:857-863, 1995); or, as microspheres for oral administration (see, e.g.,
Eyles, J. Pharm. Pharmacol. 49:669-674, 1997). Both transdermal and
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
intradermal routes afford constant delivery for weeks or months. Cachets can
also be used in the delivery of the compounds of the present invention.
In another embodiment, the compounds of the present invention can be
delivered by the use of liposomes which fuse with the cellular membrane or are
endocytosed, i.e., by employing ligands attached to the liposome that bind to
surface membrane protein receptors of the cell resulting in endocytosis. By
using liposomes, particularly where the liposome surface carries ligands
specific for target cells, or are otherwise preferentially directed to a
specific
organ, one can focus the delivery of the carbamate compound into target cells
in vivo. (See, e.g., Al-Muhammed, J. Microencapsul. 13:293-306, 1996;
Chonn, Curr. Opin. Biotechnol. 6:698-708, 1995; Ostro, Am. J. Hosp. Pharm.
46:1576-1587, 1989).
The pharmaceutical formulations of the invention can be provided as a
salt and can be formed with many acids, including but not limited to
hydrochloric, sulfuric, acetic, lactic, tartaric, malic, succinic, etc. Salts
tend to
be more soluble in aqueous or other protonic solvents that are the
corresponding free base forms. In other cases, the preferred preparation can
be a lyophilized powder which can contain, for example, any or all of the
following: 1 mM-50 mM histidine, 0.1%-2% sucrose, 2%-7% mannitol, at a pH
range of 4.5 to 5.5, that is combined with buffer prior to use.
Pharmaceutically acceptabie salts and esters refers to salts and esters
that are pharmaceutically acceptable and have the desired pharmacological
properties. Such salts include salts that may be formed where acidic protons
present in the compounds are capable of reacting with inorganic or organic
bases. Suitable inorganic salts include those formed with the alkali metals,
e.g.
sodium and potassium, magnesium, calcium, and aluminum. Suitable organic
salts include those formed with organic bases such as the amine bases, e.g.
ethanolamine, diethanolamine, triethanolamine, tromethamine, N
methylglucamine, and the like. Pharmaceutically acceptable salts can also
include acid addition salts formed from the reaction of amine moieties in the
parent compound with inorganic acids (e.g. hydrochloric and hydrobromic
acids) and organic acids (e.g. acetic acid, citric acid, maleic acid, and the
alkane- and arene-sulfonic acids such as methanesulfonic acid and
31
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
benzenesulfonic acid). Pharmaceutically acceptable esters include esters
formed from carboxy, sulfonyloxy, and phosphonoxy groups present in the
compounds. When there are two acidic groups present, a pharmaceutically
acceptable salt or ester may be a mono-acid-mono-salt or ester or a di-salt or
ester; and similarly where there are more than two acidic groups present, some
or all of such groups can be salified or esterified.
Compounds named in this invention can be present in unsalified or
unesterified form, or in salified and/or esterified form, and the naming of
such
compounds is intended to include both the original (unsalified and
unesterified)
compound and its pharmaceutically acceptable salts and esters. The present
invention includes pharmaceutically acceptable salt and ester forms of Formula
1 and Formula 2. More than one crystal form of an enantiomer of Formula 1 or
Formula 2 can exist and as such are also included in the present invention.
A pharmaceutical composition of the invention can optionally contain, in
addition to a carbamate compound, at least one other therapeutic agent useful
in the treatment of a disease or condition associated with providing
neuroprotection.
Methods of formulating pharmaceutical compositions have been
described in numerous publications such as Pharmaceutical Dosage Forms:
Tablets. Second Edition. Revised and Expanded. Volumes 1-3, edited by
Lieberman et al; Pharmaceutical Dosage Forms: Parenteral Medications.
Volumes 1-2, edited by Avis et al; and Pharmaceutical Dosage Forms:
Disperse Systems. Volumes 1-2, edited by Lieberman et al; published by
Marcel Dekker, Inc, the disclosure of which are herein incorporated by
reference in their entireties and for all purposes.
The pharmaceutical compositions are generally formulated as sterile,
substantially isotonic and in full compliance with all Good Manufacturing
Practice (GMP) regulations of the U.S. Food and Drug Administration.
Dosage Regimens
The present invention provides methods of providing neuroprotection in
a mammal, including a human subject or patient, using the carbamate
compounds or compositions of the invention. The amount of the carbamate
32
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
compound necessary to provide neuroprotection is defined as a therapeutically
or a pharmaceutically effective dose. The dosage schedule and amounts
effective for this use, i.e., the dosing or dosage regimen will depend on a
variety of factors including the stage of the disease, the patient's physical
status, age and the like. In calculating the dosage regimen for a patient, the
mode of administration is also taken into account. In order to accomplish this
objective the compounds or compositions of this invention must be used in the
correct therapeutically effective amount or dose, as described below
The dosage schedule and amounts effective for this use, i.e., the dosing
or dosage regimen, will depend on a variety of factors including the precise
nature of the disease or injury, the patient's physical status, weight, age
and
the like. In calculating the dosage regimen for a patient, the mode of
administration is also taken into account.
The range of doses that are expected to be effective in producing a
neuroprotective effect in humans in the severe and acute clinical situations
that
are analogous to the lithium-pilocarpine rat model in Examples 1 and 2 and the
transient cerebral ischemia middle cerebral artery occlusion (MCAO) rat model
in Example 4 below are determined by comparing known effective doses and
blood levels in rats and humans.
Pharmacokinetics
In humans, it is known that the pharmacokinetics of one of the
compounds of the invention referred to herein as Test Compound (TC), i.e.,
Formula 7, are linear following single and repeated oral administration in
healthy adult men (see Example 5).
Blood levels in humans;
In toxicology studies in humans, oral administration of Test Compound
(TC) at various doses for 7 days produced the following C max's and AUC (0-
24):
1) At 100 mg. b.i.d. (200 mg in 24 hours or 2.85 mg/kg/day in a 70 kg.
human) C max was 3.6-micrograms/mL and AUC was 42.2 micrograms-
hour/mL;
33
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
2) At 250 mg. b.i.d (500 mg in 24 hours or 7.14 mg/kg/day in a 70 kg
human) C max was 8.2 micrograms/mL and AUC was 102.3 micrograms-
hour/mL;
3) At 500 mg. b.i.d. (1000 mg in 24 hours or 14.28 mg/kg/day in a 70 kg.
human) C max was 17.2-micrograms/mL and AUC was 204.1 micrograms-
hour/mL;
4) At 750 mg. b.i.d. (1500 mg in 24 hours or 21.4 mg/kg/day in a 70 kg
human) C max was 28.2-micrograms/mL and AUC was 322.7 micrograms-
hour/mL.
Blood levels in rats;
In toxicology studies in rats, oral administration of Test Compound (TC)
for 8 days produced the following C max and AUC:
1) At 30 mg/kg/day C max was 9.33 micrograms/ mL and AUC was
97.32 micrograms-hour/mL;
2) At 100 mg/kg/day C max was 20.63 micrograms/ mL and AUC was
230.33 micrograms-hour/mL
3) At 300 mg/kg/day C max was 70.34 micrograms/ mL and AUC was
525.95 micrograms-hour/mL
The doses tested in rats for anti-epileptogenic and neuroprotective
effects in Example 2 ranged from 30 mg/kg/day to 120 mg/kg/day. The lowest
dose tested in this Example, i.e., 30 mg/kg, produced some measurable
protective effects while the lowest dose tested in Example 1 was 10 mg/kg/day
and produced minimal protective effects (See Examples 1 and 2 below).
However, in the transient cerebral ischemia middle cerebral artery occlusion
(MCAO) rat model in Example 4 a dose of 10 mg/kg in rats showed moderate
but significant reduction in infarct size.
In rats, doses of 30 mg/kg/day of Test Compound (TC) would be
expected to produce blood levels of; C max of 9.33 micrograms/mL and an
AUC of 97.32 micrograms-hour/mL. In humans, these blood levels would be
expected from doses of about 500 mg/day to about 600 mg/day or from about
7.1 to about 8.6 mg/kg/day in a 70 kg human.
However, in Examples 1 and 2 relatively high doses and blood levels
were required because of the acute and very severe animal model that was
34
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
used and the need to produce the neuroprotective anti-epileptogenic effects
demonstrated. Also, in this severe acute animal model the compound was
given after the traumatic event or injury had occurred, i. e., the induction
of
status epilepticus by administration of Li-Pilocarpine. In the transient
cerebral
ischemia middle cerebral artery occlusion (MCAO) rat model in Example 4 the
compound was also administered 1 hour after the occlusion of the middle
cerebral artery. This type of post injury model is likely to correlate to
analogously acute and severe clinical situations in human patients such as not
starting the medication until after severe CNS injury has already occurred. In
such situations, it is expected that the dosages needed for a neuroprotective
and antiepileptogenic effect will be higher than what would likely be needed
in
less acute or severe circumstances or in chronic situations and especially
where the medication is used prophylactically.
In situation where the medication is used prophylactically in a primary
prevention or pre-treatment paradigm the required doses and blood levels
required to produce clinically important neuroprotective effects would be
expected to be somewhat lower than the human equivalent of the 10 mg/kg
dose used in Example 4 and significantly lower than the 30 mg/kg/day dose
found effective in Examples 2. As such, the doses expected to be
therapeutically effective in clinical practice in humans, in most cases, would
be
less than that identified in these severe animal models. The ED50 for Test
Compound for preventing seizures in rats is about 4 mg/kg to about 30 mg/kg
(depending on the time and experiment type) so a minimum effective dose of
10 mg/kg in the neuroprotection rat models is not unexpected. Based on this
data, an expected effective neuroprotective human dose would be similar to
the minimum dose required for anticonvulsant efficacy in humans. In a primary
prevention paradigm, where dosing is done well before any insult or
pathological process is initiated, the effective doses and blood levels in
humans would be expected to be somewhat lower than human equivalent of
the 10 mg/kg dose found minimally effective in the transient cerebral ischemia
middle cerebral artery occlusion (MCAO) rat model in Example 4.
In human patients, in a primary prevention paradigm where the
medication would begin prior to any injury or damage to the human nervous
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
system, the lower limits of a neuroprotective effective dose would be expected
to be about 100 mg/day to about 200 mg/day or about 1.43 mg/kg/day to about
2.85 mg/kg/day in a 70 kg. human, producing an expected C max (at 200
mg/day) of about 3.6-micrograms/mL and AUC of about 42.2 micrograms-
hour/mL.
In situations where the medication is started after the injury has been
sustained the dose range would be expected to be somewhat higher, for
example, from about 200 mg/day to about 400 mg/day or about 2.85 to about
5.71 mg/kg/day in a 70 kg human.
The compounds and compositions of the invention do not have a
theoretical upper end to their clinically effective dose range. Thus, the
upper
end of the therapeutically effective range would be determined by the
maximum amount that could be tolerated by the patient. However, the highest
dose tested in rats, i.e., 120 mg/kg, which showed very marked neuroprotective
and anti-epileptogenesis effects, would be expected, on the basis of the data
above, to have a C max and AUC similar or below those produced in humans
at a dose of 750 mg twice a day (1500 mg/day or approximately 21.4
mg/kg/day). A dose of 750 mg twice a day (total daily dose of 1500 mg) has
been used in humans and found to be easily tolerated. On this basis it would
be expected that the maximum tolerable dose would be considerably higher
than this for many patients. perhaps 2500 mg/day to 3000 mg/day or about
35.7 mg/kg/day to about 42.9 mg/kg/day in a 70 kg human.
Thus, for the purpose of providing neuroprotection to a human subject
or patient, the pharmaceutical compounds and compositions of the invention
may be administered at a dosage of from about 1.4 mg/kg/day to about 43.0
mg/kg/day (100 mg/day to 3000 mg/day in a 70 kg human), preferably from
about 2.9 mg/kg/day to about 35.7 mg/kg/day (200 mg/day to 2500 mg/day in a
70 kg human), more preferably from about 3.6 mg/kg/day to about 28.6
mg/kg/day (250 mg/day to 2000 mg/day in a 70 kg human), or even more
preferably from about 4.3 mg/kg/day to about 21.4 mg/kg/day (300 mg/day to
1500 mg/day in a 70 kg human) or most preferably from about 5.0 mg/kg/day
to about 17.1 mg/kg/day (350 mg/day to 1200 mg/day in a 70 kg human).
These dosages, however, may be varied depending the individual
36
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
characteristics and tolerances of the subject and the on the precise nature of
the condition being treated.
Based on this disclosure, a person of ordinary skill in the art will be able,
without undue experimentation, having regard to that skill, to determine a
therapeutically effective dose or amount of a particular substituted carbamate
compound of the invention for treating epilepsy and for producing a clinically
significant neuroprotective effect. (see, e.g., Lieberman, Pharmaceutical
Dosage Forms(Vols. 1-3, 1992); Lloyd, 1999, The art, Science and Technology
of Pharmaceutical Compounding; and Pickar, 1999, Dosage Calculations).
A therapeutically effective dose is also one in which any toxic or
detrimental side effects of the active agent is outweighed in clinical terms
by
therapeutically beneficial effects. It is to be further noted that for each
particular subject, specific dosage regimens should be evaluated and adjusted
over time according to the individual need and professional judgment of the
person administering or supervising the administration of the compounds. It is
also expected that the compositions of this invention could be initiated at a
low
or moderate dose and then increased to a fully therapeutically effective dose
and blood level over a period of time.
For treatment purposes, the compositions or compounds disclosed
herein can be administered to the subject in a single bolus delivery, via
continuous delivery over an extended time period, or in a repeated
administration protocol (e.g., by an hourly, daily or weekly, repeated
administration protocol). The pharmaceutical formulations of the present
invention can be administered, for example, one, two or more times daily, 3
times per week, or weekly. In one embodiment of the present invention, the
pharmaceutical formulations of the present invention are orally administered
once or twice daily.
In some embodiments, a treatment regimen with the compounds of the
present invention can commence, for example, after a subject suffers from a
brain damaging injury or other initial insult such as a stroke. In still other
embodiments, a treatment regimen with the compounds of the present
invention can commence before any damage or injury to the nervous system
has occurred but at a time when such damage or injury can expected or is
37
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
likely to occur. For example, such a treatment regimen can begin before a
subject undergoes a neurosurgical procedure or is likely to suffer other forms
or head or brain trauma, e.g., combat, violent sports or racing, recurrent
strokes, TIA's etc.
In certain embodiments, the carbamate compounds of the invention can
be administered daily for a set period of time (week, month, year) after
occurrence of the brain damaging injury or initial insult. An attendant
physician
will know how to determine that the carbamate compound has reached a
therapeutically effective level, e.g., clinical exam of a patient, or by
measuring
drug levels in the blood or cerebro-spinal fluid. One of skill in the art
would be
able to determine the maximum tolerable dose by means of a physical
examination to determine the presence and severity of side effects such as
slurred speech, lethargy or impaired coordination.
In this context, a therapeutically effective dosage of the biologically
active agent(s) can include repeated doses within a prolonged treatment
regimen that wili yield clinically significant results to provide a
neuroprotective
effect. Determination of effective dosages in this context is typically based
on
animal model studies followed up by human clinical trials and is guided by
determining effective dosages and administration protocols that significantly
reduce the occurrence or severity of targeted exposure symptoms or conditions
in the subject. Suitable models in this regard include, for example, murine,
rat,
porcine, feline, non-human primate, and other accepted animal model subjects
known in the art.
Alternatively, effective dosages can be determined using in vitro models
(e.g., immunologic and histopathologic assays). Using such models, only
ordinary calculations and adjustments are typically required to determine an
appropriate concentration and dose to administer a therapeutically effective
amount of the biologically active agent(s) (e.g., amounts that are
intranasally
effective, transdermally effective, intravenously effective, or
intramuscularly
effective to elicit a desired response).
In an exemplary embodiment of the present invention, unit dosage
forms of the compounds are prepared for standard administration regimens. In
this way, the composition can be subdivided readily into smaller doses at the
38
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
physician's direction. For example, unit dosages can be made up in packeted
powders, vials or ampoules and preferably in capsule or tablet form.
The active compound present in these unit dosage forms of the
composition can be present in an amount of, for example, from about 25 mg. to
about 800 mg or preferably in unit dosage amounts of about; 50, 100, 200 250,
400, 450, 500, and 600 mg of one or more of the active carbamate compounds
of the invention, for single or multiple daily administration, according to
the
particular need of the patient.
The methods of this invention also provide for kits for use in providing
neuroprotection. After a pharmaceutical composition comprising one or more
carbamate compounds of this invention, with the possible addition of one or
more other compounds of therapeutic benefit, has been formulated in a
suitable carrier, it can be placed in an appropriate container and labeled for
providing neuroprotection. Additionally, another pharmaceutical comprising at
least one other therapeutic agent useful in providing neuroprotection,
treatment
of epileptogenesis, epilepsy or another disorder or condition associated with
neuronal injury can be placed in the container as well and labeled for
treatment
of the indicated disease. Such labeling can include, for example, instructions
concerning the amount, frequency and method of administration of each
pharmaceutical.
Although the foregoing invention has been described in detail by way of
example for purposes of clarity of understanding, it will be apparent to the
artisan that certain changes and modifications are comprehended by the
disclosure and may be practiced without undue experimentation within the
scope of the appended claims, which are presented by way of illustration not
limitation. The following examples are provided to illustrate specific aspects
of
the invention and are not meant to be limitations.
EXPERIMENTAL EXAMPLES
The activities of a compound of Formula (I) and Formula (II) for use in
providing neuroprotection were evaluated in the following experimental
examples The activity of an isolated S-enantiomer of Formula 1 (e.g.,
Formula 7 shown above), herein referred to as the "Test Compound" or "TC"
39
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
or "test compound" was evaluated in the following experiments to determine
the efficacy of the compound for neuroprotection and in the treatment of
epileptogenesis in the model of temporal lobe epilepsy induced by lithium and
pilocarpine in the rat in Examples 1 and 2 and in other animal models useful
for the prediction of neuroprotective effects. These examples are intended to
be a way of illustrating various embodiments of the invention but not intended
to limit the invention in any way.
Example 1
The lithium-pilocarpine model of temporal lobe epilepsy
The model induced in rats by pilocarpine associated with lithium (Li-
Pilo) reproduces most of the clinical and neurophysiological features of human
temporal lobe epilepsy (Turski et al., 1989, Synapse 3:154-171; Cavalheiro,
1995, Ital J Neurol Sci 16:33-37). In adult rats, the systemic administration
of
pilocarpine leads to status epilepticus (SE). The lethality rate reaches 30-
50%
during the first days. In the surviving animals, neuronal damage
predominates within the hippocampal formation, the piriform and entorhinal
cortices, thalamus, amygdaloid complex, neocortex and substantia nigra. This
acute seizure period is followed by a "silent" seizure-free phase lasting for
a
mean duration of 14-25 days after which all animals exhibit spontaneous
recurrent convulsive seizures at the usual frequency of 2 to 5 per week
(Turski
et al., 1989, Synapse 3:154-171; Cavalheiro, 1995, Ital J Neurol Sci 16:33-37;
Dube et al., 2001, Exp Neurol 167:227-241).
Lithium-pilocarpine and treatments with the test compound
Male Wistar rats weighing 225-250 g, provided by Janvier Breeding Center
(Le Genest-St-Iste, France) were housed under controlled standard conditions
(light/dark cycle, 7.00 a.m.-7.00 p.m. lights on), with food and water
available ad
libitum. All animal experimentation was performed in accordance with the rules
of
the European Communities Council Directive of November 24, 1986 (86/609/EEC),
and the French Department of Agriculture (License N 67-97). For electrode
implantation, rats were anesthetized by an i.p. injection of 2.5 mg/kg
diazepam
(DZP, Valium, Roche, France) and 1 mg/kg ketamine chlorhydrate (Imalgene
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
1000, Rhone Merrieux, France). Four single-contact recording electrodes were
placed on the skull, over the parietal cortex, two on each side.
Status -epilepticus induction:
Treatment with the test compound and occurrence of spontaneous recurrent
seizures (SRS)
All rats received lithium chloride (3 meq/kg, i.p., Sigma, St Louis, Mo,
U.S.A.); about 20 h later, animals were placed into plexiglas boxes, in order
to record baseline cortical EEG. Methyiscopolamine bromide (1 mg/kg, s.c.,
Sigma) was administered to limit the peripheral effects of the convulsant. SE
was induced by injecting pilocarpine hydrochloride (25 mg/kg, s.c., Sigma) 30
min after methyl-scopolamine. The bilateral EEG cortical activity was
recorded during the whole duration of SE and behavioral changes were noted.
The effects of increasing doses of the Test Compound were studied on
3 groups of rats. The animals of the first group received 10 mg/kg of the test
compound, i.p., 1 h after the onset of SE (pilo-TC10) while the animals of
groups 2 and 3 received 30 and 60 mg/kg of the Test Compound (pilo-TC30
and pilo-TC60), respectively.
Another group was injected with 2 mg/kg diazepam (DZP, i.m.) at 1 h
after the onset of SE which are our standard treatment to improve animals
survival after SE (pilo-DZP). The control group received saline instead of
pilocarpine and the Test Compound (saline-saline). The pilo-Test Compound
rats surviving SE were then injected about 10 h after the first test compound
injection with a second i.p. injection of the same dose of the test
compound and were maintained under a twice daily treatment with the test
compound for 6 additional days. Pilo-DZP received a second injection of 1
mg/kg DZP on the day of SE at about 10 h after the first one. Thereafter, Pilo-
DZP and saline-saline rats received twice daily an equivalent volume of
saline.
The effects of the test compound on the EEG and on the latency to
occurrence of SRS were investigated by daily video recording of the animals
for 10 h per day and the recording of the electrographic activity twice a week
for 8 h.
Quantification of cell densities
41
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
Quantification of cell densities was performed at 6 days after SE on 8
pilo-DZP, 8 pilo-TC10, 7 pilo-TC30, 7 pilo-TC60, and 6 saline-saline rats. At
14
days after SE, animals were deeply anesthetized with 1.8 g/kg pentobarbital
(Dolethal , Vetoquinol, Lure, France. Brains were then removed and frozen.
Serial 20 pm slices were cut in a cryostat, air-dried during several days
before
thionine staining.
~ Quantification of cell densities was performed with a 10 x 10 boxes 1
cm2 microscopic grid on coronal sections according to the stereotaxic
coordinates of the rat brain atlas. (See Paxinos G, Watson C (1986) The
Rat Brain in Stereotaxic Coordinates, 2nd ed. Academic Press, San Diego)
~ Cell counts were performed twice in a blind manner and were the average
of at least 3 values from 2 adjacent sections in each individual animal.
Counts involved only cells larger than 10 pm, smaller ones being considered
as glial cells.
Timm staining
At 2 months after the onset of spontaneous recurrent seizures, mossy
fiber sprouting was examined on rats in the chronic period exposed to the test
compound or DZP and in 3 saiine-saline rats. Animals were deeply
anaesthetized and perfused transcardially with saline followed by 100 ml of
1.15% (w/v) Na2S in 0.1 M phosphate buffer, and 100 ml of 4% (v/v)
formaldehyde in 0.1 M phosphate buffer. Brains were removed from skull,
post-fixed in 4% formaldehyde during 3-5 h and 40 pm sections were cut on a
sliding vibratome and mounted on gelatin-coated slides.
The following day, sections were developed in the dark in a 26 C solution
of 50% (w/v) arabic gum (160 ml), sodium citrate buffer (30 ml), 5.7% (w/v)
hydroquinone (80 ml) and 10% (w/v) silver nitrate (2.5 ml) during 40-45 min.
The sections were then rinsed with tap water at 40 C during at least 45 min,
rinsed rapidly with distilled water and allowed to dry. They were dehydrated
in
ethanol and coverslipped.
Mossy fiber sprouting was evaluated according to criteria previously
described in dorsal hippocampus (Cavazos et al., 1991, J Neurosci 11:2795-
2803.), which are follows: 0 - no granules between the tips and crest of the
DG;
1 - sparse granules in the supragranular region in a patchy distribution
between
42
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
the tips and crest of DG; 2 - more numerous granules in a continuous
distribution between the tips and crest of DG; 3 - prominent granules in a
continuous pattern between tips and crest, with occasional patches of
confluent granules between tips and crest; 4 - prominent granules that form a
confluent dense laminar band between tips and crest and 5 - confluent dense
laminar band of granules that extends into the inner molecular layer.
Data analysis
For the comparison of the characteristics of SE in pilo-saline and pilo-
test compound animals, a non-paired Student's t-test was used. The
comparison between the number of rats seizing in both groups was performed
by means of a Chi square test. For neuronal damage, statistical analysis
between groups was performed using ANOVA followed by a Fisher's test for
multiple comparisons using the Statview software (Fisher RA, 1946a,
Statistical
Methods for Research Workers (10th edition) Oliver & Boyd, Edinburgh; Fisher
RA, 1946b, The Design of Experiments (4th edition) Oliver & Boyd, Edinburgh)
Behavioral and EEG characteristics of lithium-pilocarpine status epilepticus
A total number of Sprague-Dawley rats weighing 250-330 g were
subjected to Li-pilo induced SE. The behavioral characteristics of SE were
identical in both pilo-saline and pilo-test compound groups. Within 5 min
after
pilocarpine injection, rats developed diarrhea, piloerection and other signs
of
cholinergic stimulation. During the following 15-20 min, rats exhibited head
bobbing, scratching, chewing and exploratory behavior. Recurrent seizures
started around 15-20 min after pilocarpine administration. These seizures
which associated episodes of head and bilateral forelimb myoclonus with
rearing and falling progressed to SE at about 35-40 min after pilocarpine, as
previously described (Turski et al., 1983, Behav Brain Res 9:315-335.).
EEG patterns during SE
During the first hour of SE, in the absence of pharmacological treatment,
the amplitude of the EEG progressively increased while the frequency
decreased. Within 5 min after the injection of pilocarpine, the normal
background EEG activity was replaced with low voltage fast activity in the
cortex while theta rhythm (5-7 Hz) appeared in the hippocampus. By 15-20
43
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
min, high voltage fast activity superposed over the hippocampal theta rhythm
and isolated high voltage spikes were recorded only in the hippocampus while
the activity of the cortex did not substantially change.
By 35-40 min after pilocarpine injection, animals developed typical
electrographic seizures with high voltage fast activity present in both the
hippocampus and cortex which first occurred as bursts of activity preceding
seizures and were followed by continuous trains of high voltage spikes and
polyspikes lasting until the administration of DZP or the test compound. At
about 3-4 h of SE, the hippocampal EEG was characterized by periodic
electrographic discharges (PEDs, about one/sec) in the pilo-DZP and in the
pilo-10 group in both hippocampus and cortex. The amplitude of EEG
background activity was low in the pilo-TC60 animals. By 6-7 h of SE, spiking
activity was still present in the cortex and the hippocampus in the DZP-and
TC10-treated rats while the amplitude of the EEG decreased and came back
to baseline levels in the hippocampus of TC30 rats and in both structures
of TC60 treated rats. There was no difference between TC10, TC30, and
TC60 groups. By 9 h of SE, isolated spikes were still recorded in the
hippocampus of test compound-treated rats and occasionally in the cortex. In
both structures, the background activity was of very low amplitude at that
time.
Mortality induced by SE
During the first 48 h after SE, the degree of mortality was similar in
pilo-DZP rats (23%, 5/22), pilo-TC10 rats (26%, 6/23), and pilo-TC30 rats
(20%, 5/25), The mortality rate was largely reduced in pilo-TC60 rats in which
it
only reached 4% (1/23). The difference was statistically significant (p <
0.01).
EEG characteristics of the silent phase and occurrence of spontaneous
recurrent
seizures
The EEG patterns during the silent period were similar in pilo-DZP and
pilo-TC1 0, 30 or 60 rats. At 24 and 48 h days after SE, the baseline EEG was
still characterized by the occurrence of PEDs on which large waves or spikes
could be superimposed. Between 1 and 8 h after injection of the test
compound or vehicle injection, there was no change in the pilo-DZP or pilo-
TC10 groups. In TC30 and TC60 rats, the frequency and amplitude of PEDs
decreased as soon as 10 min after injection and were replaced by spikes of
44
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
large amplitude in the TC30 group and of low amplitude in the TC60 group.
By 4 h after injection the EEG had returned to baseline levels in the two
latter
groups. By 6 days after SE, the EEG was still of lower amplitude than before
pilocarpine injection and in most groups spikes could still be recorded,
occasionally in the pilo-DZP, -TC10 and -TC30 rats. In pilo-TC60 rats, the
frequency of large amplitude spikes was higher than in all other groups.
After the test compound or vehicle-injection, EEG recording was not
affected by the injection in the pilo-DZP and pilo-TC10 groups. In pilo-TC30
rats, the injection induced the occurrence of slow waves on the EEG of both
hippocampus and cortex and a decreased frequency of spikes in the pilo-
TC60 rats.
All the rats exposed to DZP, TC10 and TC30 and studied until the
chronic phase developed spontaneous recurrent seizures (SRS) with a similar
latency. The latency was 18.2 6.9 days (n = 9) in pilo-DZP rats, 15.4 5.1
days (n = 7) in pilo-TC10 rats, 18.9 9.0 days (n = 10) in pilo-TC30 rats. In
the group of rats subjected to TC60, a subgroup of rats became epileptic with
a latency similar to that of the other groups, i.e. 17.6 8.7 days (n= 7)
while
another group of rats became epileptic with a much longer delay ranging from
109 to 191 days post-SE (149.8 36.0 days, n = 4) and one rat did not
become epileptic in a delay of 9 months post-SE. The difference in the latency
to SRS between pilo-DZP, pilo-TC10, pilo-TC30 and the first subgroup of pilo-
TPM60 rats was not statistically significant. None of the saline-saline rats
(n = 5)
developed SRS.
To calculate the frequency of spontaneous recurrent seizures (SRS) in
pilocarpine-exposed rats, the seizure severity and distinguished stage III
(clonic
seizures of facial muscles and anterior limbs) and stage IV-V seizures
(rearing
and falling) was measured. The frequency of stage III SRS per week in pilo-
DZP and pilo-test compound rats was variable amongst the groups. It was low,
constant in the pilo-DZP and pilo-TC60 (with early SRS onset) groups during
the first 3 weeks and had disappeared during the 4th week in the pilo-DZP
group. The frequency of stage III SRS was higher in the pilo-TC10 group
where it was significantly increased over pilo-DZP values during weeks 3 and
4. The frequency of more severe stage IV-V SRS was highest during the first
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
week in most groups, except pilo-TC30 and TC60 with late seizure onset where
the SRS frequency was constant over the whole 4 weeks in TC30 group and
over the first two weeks in the pilo-TC60 group with late SRS onset in which
no
stage IV-V seizures where no seizures recorded after the second week. The
frequency of stage IV-V SRS was significantly reduced in the TC10, TC30 and
TC60 (with early SRS onset) groups (2.3-6,1 SRS per week) compared to the
pilo-DZP group (11.3 SRS per week) during the first week. During weeks 2-4v
the frequency of stage IV-V SRS was reduced in all groups compared to the
first week reaching values of 2-6 seizures per week, except in the pilo-TC60
group with early SRS onset where the frequency of seizures was significantly
reduced to 0.6-0.9 seizure per week compared to the pilo-DZP group in which
the frequency of SRS ranged from 3.3 to 5.8.
Cell densities in hippocampus, thalamus and cortex
In pilo-DZP rats compared to saline-saline rats, the number of cells was
massively decreased in the CAl region of the hippocampus (70% cell loss in
the pyramidal cell layer) while the CAS region was less extensively damaged
(54% cell loss in CA3a and 31% in CA3b). In the dentate gyrus, the pilo-DZP
rats experienced extensive cell loss in the hilus (73%) while the granule cell
layer did not show visible damage. Similar damage was observed in the
ventral hippocampus but cell counts were not performed in this region.
Extensive damage was also recorded in the lateral thalamic nucleus (91% cell
loss) while the mediodorsal thalamic nucleus was more moderately damaged
(56%). In the piriform cortex, cell loss was total in layers III-IV which was
no
longer visible and reached 53% in layer II in pilo-DZP rats. In the dorsal
entorhinal cortex, layers II and III-IV underwent slight damage (9 and 15%,
respectively). Layer II of the ventral entorhinal cortex was totally preserved
while layers III-IV underwent a 44% cell loss.
In the hippocampus of pilo-test compound animals, cell loss was
reduced compared to pilo-DZP rats in the CAl pyramidal layer in which the cell
loss reached 75% in pilo-DZP and 35 and 16% in the pilo-TC30 or pilo-TC60
animals, respectively. This difference was statistically significant at the
two test
compound doses. In the CAS pyramidal layer, the test compound did not
afford any protection in the CA3a area while the 60 mg/kg of the test
46
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
compound dose was significantly neuroprotective in CA3b. In the dentate
gyrus, the cell loss in the hilus was similar in pilo-test compound (69-72%)
and
pilo-DZP animals (73%). In the two thalamic nuclei, the 60 mg/kg dose was
also protective in reducing neuronal damage by 65 and 42% in the lateral and
mediodorsal nucleus, respectively. In the cerebral cortex, the treatment with
the test compound afforded neuronal protection compared to DZP only at the
highest dose, 60 mg/kg. At the two lowest doses, 10 and 30 mg/kg, the total
loss of cells and tissue disorganization observed in layers III-IV of the
piriform
cortex was identical in pilo-DZP rats and pilo-test compound rats and did not
allow any counting in any of the groups. In layers II and III-IV of the
piriform
cortex, the TC60 treatment reduced neuronal damage recorded in the pilo-DZP
rats by 41 and 44%, respectively. In the ventral entorhinal cortex,
neuroprotection was induced by TC60 administration in layers III-IV and
reached 31 % compared to pilo-DZP rats. In the entorhinal cortex, there was a
slight worsening of cell loss in pilo-TC10 rats compared with pilo-DZP rats in
layers III-IV of the dorsal entorhinal cortex (28% more damage) and layers III-
IV of the ventral entohinal cortex (35% more damage). At the other doses of
the test compound, cell loss in the entorhinal cortex was similar to the one
recorded in pilo-DZP rats.
Mossy fiber sproutinc1in hippocampus
All rats exhibiting SRS in pilo-DZP and pilo-TPM groups showed similar
intensity of Timm staining in the inner molecular layer of the dentate gyrus
(scores 2-4). Timm staining was present both on the upper and lower blades
of the dentate gyrus. The mean value of the Timm score in the upper blade
reached 2.8 0.8 in pilo-DZP rats (n = 9), 1.5 0.6 in pilo-TC10 rats (n =
7),
2.6 1.0 in pilo-TC30 rats (n = 10), and 1.5 0.7 in the whole group of pilo-
TC60 rats (n = 11). When the pilo-test compound at 60mg/kg.group was
subdivided according to the latency to SRS, the subgroup with early SRS
occurrence showed a Timm score of 1.8 0.6 (n = 6) and the subgroup of rats
with late occurrence or absence of SRS had a Timm score of 1.2 0.6 (n = 5).
The values recorded in the pilo-DZP rats were statistically significantly
different
from the values in the pilo-TC10 (p = 0.032) and the pilo-TC60 subgroup with
late or no seizures (p = 0.016).
47
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
Discussion and conclusions
The results of the present study show that a 7-day treatment with the
test compound starting at 1 h after the onset of SE is able to protect some
brain areas from neuronal damage, e.g., in the pyramidal cell layer of the CAl
and CA3b area, the mediodorsal thalamus, layers II and II MV of the piriform
cortex and layers III-IV of the ventral entorhinal cortex, but only at the
highest
dose the test compound, i.e. 60 mg/kg. The latter dose of the test compound
is also able to delay the occurrence of SRS, at least in a subgroup of animals
that became epileptic with a mean delay that was about 9-fold longer than in
the other groups of animals and one animal did not become epileptic in a delay
of 9 months after SE.
These results show that one compound with anti-ictal properties, which
are the classical properties of most antiepileptic-marketed drugs, is also
able to
delay epileptogenesis, i.e. to be antiepileptogenic. The data of the present
study show also that the test compound treatment, whatever the dose used,
decreases the severity of the epilepsy since it decreases the number of stage
IV-V seizures, mainly during the first week of occurrence and during the whole
period of 4-weeks observation with the test compound at 60mg/kg. treatment.
Moreover, in the TC10 group, there is a shift to an increase in the occurrence
of less severe stage III seizures that are more numerous than in the pilo-DZP
group.
EXAMPLE 2
The aim of this extended portion of the study was to pursue the study
reported in Example 1 above on the potential neuroprotective and
antiepileptogenic properties of the same Test Compound (TC) in the lithium-
pilocarpine (Li-Pilo) model of temporal lobe epilepsy. In the first study it
was
shown that TC was able to protect areas CAl and CA3 of the hippocampus,
piriform and ventral entorhinal cortex from neuronal damage induced by Li-Pilo
status epilepticus (SE). Most of these neuroprotective properties occurred at
the highest dose studied, 60 mg/kg and the treatment was able to delay the
occurrence of spontaneous seizures in 36% (4 out of 11) of the rats. In the
48
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
present example, the consequences of treatment by higher doses of TC on
neuronal damage and epileptogenesis is studied
The lithium-pilocarpine model of temporal lobe epilepsy
The model of epilepsy induced in rats by pilocarpine associated with
lithium (Li-Pilo) reproduces most of the clinical and neurophysiological
features
of human temporal lobe epilepsy (See, Turski L, lkonomidou C, Turski WA,
Bortolotto ZA, Cavalheiro EA (1989) Review: Cholinergic mechanisms and
epileptogenesis. The seizures induced by pilocarpine: a novel experimental
model of intractable epilepsy, Synapse 3:154-171; Cavalheiro EA (1995) The
pilocarpine model of epilepsy. Ital J Neurol Sci 16:33-37).
In adult rats, the systemic administration of pilocarpine leads to SE
which may last for up to 24 h. The lethality rate reaches 30-50% during the
first
days. In the surviving animals, neuronal damage predominates within the
hippocampal formation, the piriform and entorhinal cortices, thalamus,
amygdaloid complex, neocortex and substantia nigra. This acute seizure period
is followed by a "silent" seizure-free phase lasting for a mean duration of 14-
25
days after which all animals exhibit spontaneous recurrent convulsive seizures
at the usual frequency of 2 to 5 per week (See, Turski L, lkonomidou C, Turski
WA, Bortolotto ZA, Cavalheiro EA (1989) Review: Cholinergic mechanisms and
epileptogenesis, The seizures induced by pilocarpine: a novel experimental
model of intractable epilepsy Synapse 3:154-171; Cavalheiro EA (1995) The
pilocarpine model of epilepsy. Ital J Neurol Sci 16:33-37; Dube C, Boyet S,
Marescaux C, Nehlig A (2001) Relationship between neuronal loss and
interictal glucose metabolism during the chronic phase of the lithium-
pilocarpine model of epilepsy in the immature and adult rat. Exp Neurol
167:227-241)).
The current antiepileptic drugs (AED's) do not prevent epileptogenesis
and are only transiently efficient on recurrent seizures.
In our previous study, we studied the potential neuroprotective and
antiepileptogenic effects of increasing doses of Test Compound (TC) given in
monotherapy and compared to our standard diazepam (DZP) treatment mostly
given to prevent high mortality. These data show that a 7-day treatment with
10, 30 or 60 mg/kg TC starting at 1 h after the onset of SE is able to protect
49
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
some brain areas from neuronal damage. This effect is statistically
significant
in the pyramidal cell layer of the CAl and CA3b area, the mediodorsal
thalamus, layers II and III-IV of the piriform cortex and layers III-IV of the
ventral entorhinal cortex, but only at the highest dose of TC, i.e. 60 mg/kg.
Moreover, it appears that the latter dose of TC is also the only one that is
able
to delay the occurrence of spontaneous recurrent seizures (SRS), at least in a
subgroup of animals that became epileptic with a mean delay that was about 9-
fold longer than in the other groups of animals and one animal did not become
epileptic in a delay of 9 months after SE.
In the present study, the effects of different doses of Test Compound
(TC), i.e. 30, 60, 90 and 120 mg/kg (TC30, TC60, TC90 and TC120) were
tested using the same design as in the previous study. The treatment was
started one hour after the onset of SE and the animals were treated with a
second injection of the same dose of the drug. This early treatment of SE was
followed by a 6 days TC treatment. This report concerns the effects of the
four
different doses of TC on neuronal damage assessed in hippocampus,
parahippocampal cortices, thalamus and amygdala at 14 days after SE and on
the latency to and frequency of spontaneous epileptic seizures.
Methods
Animals
Adult male Sprague-Dawley rats provided by Janvier Breeding Center
(Le Genest-St-Isle, France) were housed under controlled, uncrowded
standard conditions at 20-22 C (light/dark cycle, 7.00 a.m.-7.00 p.m. lights
on),
with food and water available ad libitum. All animal experimentation was
performed in accordance with the rules of the European Communities Council
Directive of November 24, 1986 (86/609/EEC), and the French Department of
Agriculture (License N 67-97).
Status epilepticus induction, Test Compound (TC) treatment and occurrence of
SRS
All rats received lithium chloride (3 meq/kg, i.p., Sigma, St Louis, Mo,
U.S.A.) and about 20 h later, all animals received also methyiscopolamine
bromide (1 mg/kg, s.c., Sigma) that was administered to limit the peripheral
effects of the convulsant. SE was induced by injecting pilocarpine
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
hydrochloride (25 mg/kg, s.c., Sigma) 30 min after methyiscopolamine. The
effects of increasing doses of TC were studied in 5 groups of rats. The
animals
received either 2.5 mg/kg DZP, i.m., or 30, 60, 90 or 120 mg/kg TC (TC30 ,
TC60 , TC90 , TC120), i.p., at 1 h after the onset of SE. The control group
received vehicle instead of pilocarpine and TC. The rats surviving SE were
then injected about 10 h after the first TC injection with a second i.p.
injection
of 1.25 mg/kg DZP for the DZP group or of the same dose of TC as in the
morning and were maintained under a twice daiiy TC treatment (s.c.) for 6
additional days while DZP rats received a vehicle injection.
The effects of DZP and the 4 doses of TC on epileptogenesis were
investigated by daily video recording of the animals for 10 h per day. Video
recording was performed for 4 weeks during which the occurrence of the first
seizure was noted as well as the total number of seizures over the whole
period. Animals were then taken off the video recording system and kept for 4
additional weeks in our animal facilities before they were sacrificed after a
total
period of 8 weeks of epilepsy. The rats that did not exhibit seizures were
sacrificed after 5 months of video recording.
Quantification of cell densities
Quantification of cell densities was performed at two times after SE: a
first group was studied 14 days after SE and was composed by 7 DZP, 8 TC30
, 11 TC60 , 10 TC90 , 8 TC1 20 and 8 control rats not subjected to SE. A
second group used for the study of the latency to SRS was sacrificed either 8
weeks after the first SRS or at 5 months when no SRS could be seen in that
delay and was composed of 14 DZP, 8 TC30 , 10 TC60 , 11 TC90 , 9 TC120
rats. At the moment, neuronal counting is still in progress in the second
group
of animals studied for epileptogenesis and long-term counting and the data
concerning that part of the study will not be included in the present report.
For neuronal counting, animals were deeply anesthetized with 1.8 g/kg
pentobarbital (Dolethal , Vetoquinol, Lure, France). Brains were then removed
and frozen. Serial 20 m slices were cut in a cryostat, air-dried during
several
days before thionine staining. Quantification of cell densities was performed
with a 10 x 10 boxes 1 cm2 microscopic grid on coronal sections according to
the stereotaxic coordinates of the rat brain atlas (Paxinos G, Watson C(1986)
51
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
The Rat Brain in Stereotaxic Coordinates, 2nd ed. Academic Press, San
Diego). The grid of counting was placed on a well defined area of the cerebral
structure of interest and counting was carried out with a microscopic
enlargement of 200- or 400-fold defined for each single cerebral structure.
Cell
counts were performed twice on each side of three adjacent sections for each
region by a single observer unaware of the animal's treatment. The number of
cells obtained in the 12 counted fields in each cerebral structure was
averaged.
This procedure was used to minimize the potential errors that could result
from
double counting leading to overestimation of cell numbers. Neurons touching
the inferior and right edges of the grid were not counted. Counts involved
only
neurons with cell bodies larger than 10 pm. Cells with small cell bodies were
considered as glial cells and were not counted.
Data analysis
For neuronal damage and epileptogenesis, statistical analysis between
groups was performed by means of a one-way analysis of variance followed by
a post-hoc Dunnett or Fisher test using the Statistica software.
Results
Behavioral characteristics of lithium-pilocarpine status epilepticus
A total number of 143 Sprague-Dawley rats weighing 250-330 g were
subjected to lithium-pilocarpine (Li-pilo)-induced SE. In this number 10 did
not
develop SE while 133 rats developed a full characteristic Li-pilo SE. The
behavioral characteristics of SE were identical in both li-pilo-DZP and li-
pilo-TC
groups. Within 5 min after pilocarpine injection, rats developed diarrhea,
piloerection and other signs of cholinergic stimulation. During the following
15-
20 min, rats exhibited head bobbing, scratching, chewing and exploratory
behavior. Recurrent seizures started around 15-20 min after pilocarpine
administration. These seizures which associated episodes of head and bilateral
forelimb myocionus with rearing and falling progressed to SE at about 35-40
min after pilocarpine, as previously described (Turski L, lkonomidou C, Turski
WA, Bortolotto ZA, Cavalheiro EA (1989) Review: Cholinergic mechanisms and
epileptogenesis. The seizures induced by pilocarpine: a novel experimental
model of intractable epilepsy. Synapse 3:154-171; Dube C, Boyet S,
Marescaux C, Nehlig A (2001) Relationship between neuronal loss and
52
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
interictal glucose metabolism during the chronic phase of the lithium-
pilocarpine model of epilepsy in the immature and adult rat. Exp Neurol
167:227-241; Andre V, Rigoulot MA, Koning E, Ferrandon A, Nehlig A (2003)
Long-term pregabalin treatment protects basal cortices and delays the
occurrence of spontaneous seizures in the lithium-pilocarpine model in the
rat.
Epilepsia 44:893-903). The control group not subjected to SE and receiving
lithium and saline was composed of 20 rats.
In the group of 57 animals devoted to cell counting at 14 days after SE,
a total number of 13 rats died over the first 48 h after SE. The degree of
mortality varied with the treatment: 36% (4/11) of DZP rats, 33% (4/12) of
TC30
rats, 8% (1/12) of TC60 rats, 0% (0/10) of TC90 rats and 33% (4/12) of TC120
rats died. In the DZP group, the 4 rats died in the first 24 h after SE. In
the
group of TC30 rats, one rat died on the day of SE, one rat was dead by 24 h
after SE and two rats by 48 h. In the group of TC60 rats, one rat died at 48 h
after SE. In the group of TC1 20 rats, two rats were dead by 24 h and two by
48
h after SE.
In the group of 55 animals devoted to the study of the latency to SRS
and late cell counting, the degree of mortality over the first 48 h after SE
was
the following: 7% (1/14) of DZP rats, 27% (3/11) of TC30 rats, 0% (0/10) of
TC60 rats, 0% (0/11) of TC90 rats and 0% (0/9) of TC120 rats died. In the
group of DZP rats, one rat died during the first 24 h after SE. In the group
of
TC30 , two rats were dead by 24 h and one by 48 h after SE.
Cell densities in hippocampus and cortex in the early phase (14 days after SE)
In DZP rats compared to control rats, the number of neurons was
massively decreased in the CAl region of the hippocampus (85% drop out in
the pyramidal cell layer) while the CA3 region was less extensively damaged
(40% loss) (Table 1 and Figure 1). In the dentate gyrus, DZP rats experienced
extensive neuronal loss in the hilus (65%) while the granule cell layer did
not
show overt damage. The same distribution of damage was observed in the
ventral hippocampus but cell counts were not performed in this region.
In the thalamus, neuronal loss was moderate in the mediodorsal central
and lateral, the dorsolateral medial dorsal and in the central medial nuclei
(18,
24, 40 and 34% drop out, respectively), more marked in the mediodorsal
53
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
nucleus (49%) and major in the ventral lateral division of the dorsolateral
nucleus (90%) (Table 1 and Figure 2). In the amygdala, neuronal loss was
moderate in the medial ventral posterior nucleus (38%) and more marked in
the basolateral and medial dorsal anterior nuclei (73 and 53% drop out,
respectively). There was no neuronal damage in the central nucleus (Table 1
and Figure 3).
In the piriform cortex, neuronal loss was almost total in layer III (94%)
which was no longer really visible and reached 66 and 89% in dorsal and
ventral layer II, respectively in DZP rats compared to control saline-treated
rats.
In the dorsal entorhinal cortex, layers II and III-IV underwent slight damage
(18
and 24%, respectively) and in ventral layers II and III/IV, damage reached 22
and 74%, respectively (Table 1 below and Figure 4).
20
30
54
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
Table 1: Effects of increasing doses of Test Compound (TC) on the number of
neuronal cell bodies in the hippocampus, thalamus, amygdala and cerebral
cortex of rats subjected to li-pilo SE.
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
Control pilo-DZP pilo-TC30 pilo-TC60 pilo-TC90 pilo-TC120
(n=1 0) (n=7) (n=8) (n=1 1) (n=10) (n=8)
Hippocampus
74.8 10.9 39.3 31.9 65.5
CAl area 1.5 1.9** 4.4**00 4.4**0 47.7 6.6* 2.9
52.1 31.3 35.7 31.6 35.1 39.8
2.9** **
CA3 area 1.8** 1.4** 1.5**
2.7 2.9**
96.4 33.5 33.0 32.8 37.5 3.1 ** 44.8
Hilus 3.5 3.0** 3.2** 3.3** 2.9**
Thalamus
Mediodorsal 31.9 16.4 11.5 19.1 28.6
medial 0.9 1.9** 2.5** 2.6** 23.1 2.8 0.800
Mediodorsal 31.9 26.3 26.9 0.6* 24.1 1** 27.4 1.5 29.9 1.7
central 1.2 1.8**
Mediodorsal 25.9 19.6 20.5 18.9 24.4
lateral 0.6 0.8** 0.7** 0.6** 22 1.2* 1.100
Dorsolateral, 102.2 61 64.2 77.5 79.4 89.8
medial, dorsal 2.5 6.3** 9.3**00 3.9**00 3.1 ** 3.7*
Dorsolateral, 97.8 9.7 71.8 79.0
ventral lateral 1.7 2.5** + 8.8 2.8**
8.7** 5.300* 4.700
113.1 74.2 108.2
Central medial 5.9 7.4* 75.6 7.7* 83.7 9.6* 88.2 8.5 6.6
Amygdala
46.7 12.8 27.3 27.8 42.7
Basolateral 1.2 5.3** 4.9**0 4.3**0 40.7 1.6 1.300
Medial, dorsal 84.3 40.0 46.8 58.4 80.2
anterior 3.8 2.5** 5.0** 2.8**0 72.2 5. 2.600
Medial, ventral 35.1 21.8 22.3 26.2 30.7 34.7
posterior 1.7 2.4** 1.8** 2.9** 37 1.7
Cerebral
cortex
Piriform, layer 36.6 12.6 15.7 27.5 35.2
II, dorsal 0.8 4.2** 2.9** 2.8**0 32'4 1.100 1.100
Piriform, layer 33.0 3.6 13.7 30.5
II, ventral 0.8 0.7** 7=2 3.8** 4.2** 18.4 4.0 1.3
Piriform, layer 19.2 1.2
III 0.7 1.2** 1.8 1.8** 6.4 2.3** 9 3.0 15 2.2
Entorhinal, layer 23.5 23.4 23.9 26.3 0.9** 27.3
.9
29 0.6 0.6** 0.5** 0.5
II, dorsal 0.7**
Entorhinal, layer 26.8 21.7 23.3 25.4 1.1 25.1 0.6
11, ventral 0.7 1.3** 22.7 Q'9 0.8**
Entorhinal, layer 29.2 22.3 22.3 23.2 26.7 * 26.4
III/IV, dorsal 0.9 0.5** 0.5** 0.8** 0'8 0.700
Entorhinal, layer 28.7 7.7 13.2 16.5 23.7 1.5 24.5
III/IV, ventral 1.7 2.3** 1.9** 2.2** 1.400
* p < 0.05, ** p< 0.01, statistically significant difference between pilo-TC
and control li-saline rats
p < 0.05, p< 0.01, statistically significant differences between pilo-TC
and pilo-DZP rats
In the hippocampus of TC-treated animals, cell loss was significantly
reduced compared to DZP rats in CAl pyramidal cell layer. This reduction was
marked in TC30 , 60 or 90 rats (36-47% cell loss) and prominent in the TC120
56
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
group (12% cell loss). The differences were statistically significant at all
TC
doses (Table 1 and Figure 1). In the CA3 pyramidal layer, there was a
tendency to a slight neuroprotection induced by Test Compound, only at the
120 mg/kg dose but the difference with the DZP group was not significant. In
the dentate gyrus, the cell loss in the hilus was similar in the DZP and TC30
,
60 and 90 groups (61-66% drop out) and there was a slight tendency to
reduced damage in the TC120 group (53% neuronal loss) compared to DZP
animals (66% drop out). None of these differences was statistically
significant.
In the thalamus, neuronal loss was similar in DZP and TC30 and TC60
rats. TC was significantly protective at the 60 mg/kg dose in the dorsolateral
medial dorsal nucleus and at the two highest doses, 90 and 120 mg/kg in all
thalamic nuclei, although the difference did not reach significance in the
mediodorsal central and central medial nuclei in TC90 rats. In TC120 rats,
neuronal drop out was considerably reduced compared to DZP rats. It ranged
from 4-19% and the number of neurons was no longer significantly different
from control animals, except in the dorsolateral medial dorsal nucleus (Table
1
and Figure 2). In the amygdala, TC was significantly protective at the 30
mg/kg
dose in the basolateral nucleus and at the 60 mg dose, also in the medial
dorsal anterior nucleus. At the highest dose, TC was largely neuroprotective;
the number of neurons was no longer significantly different from the control
level and reached 86-99% of the control level in all amygdala nuclei (Table 1
and Figure 3).
In the cerebral cortex, the treatment with TC did not significantly protect
any cortical area compared to the DZP treatment at the dose of 30 mg/kg. At
60 mg/kg, TC significantly reduced neuronal loss only in layer II of the
dorsal
piriform cortex (25% drop out compared to 66% in the DZP group). At 90 and
120 mg/kg, TC significantly protected all three areas of the piriform cortex
compared to the DZP treatment and at the highest dose of TC, 120 mg/kg,
neuronal density reached 78-96% of control levels, even in piriform cortex,
dorsal layer II and layer III where the neuronal population was almost totally
depleted in the DZP group. In all layers of the dorsal and ventral entorhinal
cortex, the two lowest doses of TC, 30 and 60 mg/kg did not afford any
neuroprotection. The 90-mg/kg dose of TC significantly protected layers II and
57
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
III/IV of the ventral entorhinal cortex (4 and 17% damage remaining in layers
II
and II/IV of the dorsal part and in layer II of the ventral part compared to
19 and
73% in the DZP group). At the highest dose of TC, 120 mg/kg, all parts of the
entorhinal cortex, both dorsal and ventral were protected and the number of
neurons in these areas was no longer significantly different from the level in
controls (85-94% of neurons surviving compared to 27-81 % in the DZP group).
Latency to and frequency of recurrent seizures
The latency to spontaneous seizures reached a mean value of 15.5
2.3 days in the DZP group (14 rats) and was similar (11.6 2.5 days) in the
TC30 group (8 rats). At higher concentrations of TC, animals could be
subdivided in subgroups with short and long latencies. A short latency was
considered as any duration shorter than 40 days after SE. Some rats exhibited
a latency to the first spontaneous seizure that was similar to that recorded
in
the DZP and TC groups but the number of rats exhibiting this short latency
values progressively decreased with the increase in TC concentration. Thus at
30 mg/kg, 70% of the rats (7/10) had short latencies to seizures while at 90
and
120 mg/kg, this percentage reached 36% (4/11) and 11 %(1 /9), respectively
(Table 2 below and Figure 5).
58
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
Table 2: Effect of increasing doses of TC on the latency to spontaneous
seizures.
Treatment Number of Latency to the first spontaneous seizure
animals (days)
DZP 14 15.5 2.34
11.6 2.5
pilo-TC30 8
2 groups
pilo-TC60 10 Short latency (n=7) Long latency (n=3)
17.4 5.4 76.7f15.6**
3 groups
pilo-TC90 11 Short latency Long latency Non epileptic
(n=4) (n=2) (n=5)
14.8 5.7 52.0 1.0* 150** 00
3 groups
pilo-TC120 9 Short latency Long latency Non epileptic
(n=1) (n=4) (n=4)
13.0 84.5 16.7** 150** 00
** p < 0.01, * p < 0.05, statistically significant differences compared to the
pilo-DZP group
p < 0 01 p < 0.05, statistically significant differences compared to the
short latency group
In the TC60, 90 and 120 groups, the mean value of the rats with long
latencies was similar and ranged from 52 to 85 days. Finally, at the two
highest
doses of TC, we were able to identify a percentage of rats that did not
develop
any seizure over a duration of 150 days post-SE. The percentage of non-
epileptic rats reached 45% at both doses of TC.
The frequency of spontaneous seizures was similar over the four weeks
of recording. It showed a tendency to be higher in the DZP and TC30 groups
while it was lower in the TC60, 90 and 120 groups (Figure 6). These
differences did not reach statistical significance at the level of each
individual
weekly frequency but reached significance for the total or mean number of
seizures over the four weeks.
59
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
The number of seizures was also plotted according to the duration of the
latency to the first spontaneous seizure. Animals with a short latency showed
a
tendency to display 2-3 times more seizures over the four weeks of recording
than rats with a long latency period. No statistical analysis could be
performed
since the ANOVA did not show any significance, most likely because there was
only one animal in the short latency subgroup of the TC120 animals (Figure 7).
However, when all latency values were plotted against the number of seizures,
there was a significant inverse correlation leading to a straight line with a
correlation coefficient of - 0.4 (Figure 8).
To finalize this analysis, two more measurements will be performed. The
first one is cell counting on the animals that were video recorded and
followed
for 2 months after the first spontaneous seizure or sacrificed at 5 months to
study the potential correlation between the extent and location of brain
damage
and the occurrence of and/or latency to spontaneous seizures. The second
one will be to perform a one-year follow-up of seizure occurrence in a group
of
rats to study whether or not the animals that we declare "non epileptic" at 5
months will remain seizure free.
The results of the present study show that a treatment with TC starting
at 1 h after the onset of Li-pilo-induced SE has neuroprotective properties in
the CAl pyramidal cell layer of the hippocampus, and in all layers of the
ventral
and dorsal piriform and entorhinal cortex. TC protects also thalamus and
amygdala nuclei. However, TC is not protective at the dose of 30 mg/kg,
except in CA1, one thalamic and one amygdala nucleus. At the dose of 60
mg/kg, layer II of the dorsal piriform cortex and a second amygdala nucleus
are
also protected. At 90 and 120 mg/kg, the drug protects most cerebral regions
studied, except hippocampal CA3 and the hilus of the dentate gyrus. The latter
two structures plus the dorsolateral ventral dorsal thalamic nucleus are the
only
regions where the number of neurons remains significantly different from
controls at the dose of 120 mg/kg TC. From these data, the extremely powerful
neuroprotection properties of TC appear clearly. The molecule seems to
prevent neuronal death in most regions belonging to the circuit of limbic
epilepsy induced by Li-pilo, i.e., the hippocampus, thalamus, amygdala and
parahippocampal cortices. These are all the regions in which we have detected
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
MRI signal in the course of epileptogenesis in Li-pilo-treated rats (Roch C,
Leroy C, Nehlig A, Namer IJ (2002a) Contribution of magnetic resonance
imaging to the study of the lithium-pilocarpine model of temporal lobe
epilepsy
in adult rats. Epilepsia 43:325-335). The only two regions that are not
efficiently
protected by TC are CA3 pyramidal cell layer and the hilus of the dentate
gyrus. The latter region undergoes rapid and massive cell damage (Andre V,
Marescaux C, Nehlig A, Fritschy JM (2001) Alterations of the hippocampal
GABAergic system contribute to the development of spontaneous recurrent
seizures in the lithium-pilocarpine model of temporal lobe epilepsy.
Hippocampus 11:452-468.; Roch C, Leroy C, Nehlig A, Namer IJ (2002a)
Contribution of magnetic resonance imaging to the study of the lithium-
pilocarpine model of temporal lobe epilepsy in adult rats. Epilepsia 43:325-
335)
and none of the neuroprotection that we used in previous studies have been
able to protect this structure. We have also on the basis of earlier studies
identified this structure as a key area in the initiation and maintenance of
epileptic seizures in the Li-pilo model. (Dube C, Marescaux C, Nehlig A (2000)
A metabolic and neuropathological approach to the understanding of plastic
changes occurring in the immature and adult rat brain during lithium-
pilocarpine
induced epileptogenesis. Epilepsia 41(Suppl 6):S36-S43)
Obviously, the present data demonstrate that epileptogenesis can be
prevented even though damage remains quite marked in this area. Long-term
cell counting on the group of animals that has been video recorded will be
able
to show whether or not the extent of damage in this region is critical for
epileptogenesis in this model.
The treatment did not affect the latency to the first spontaneous seizure at
the dose of 30 mg/kg. At the 3 higher doses, a percentage of animals
developed epilepsy as fast as the DZP or TC30 rats but the relative importance
of this subgroup was inversely related to the dose of TC used. Another
subgroup, constant in size (2-4 animals per group) developed epilepsy after a
4-6 times longer latency while at the two highest doses of the drug, 4-5 rats
had not become epileptic after 5 months, i.e. about 10 times the duration of
the
short latency and 2-3 times that of the long latency. This delay in the
occurrence of epilepsy might correlate with the number of neurons protected in
61
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
the basal cortices in the animals. This assumption is based on the fact that
we
noted some heterogeneity in the extent of neuroprotection in basal cortices of
the animals subjected the short term neuronal counting at 14 days after SE.
However, at the moment, we have not performed neuronal counting in the
animals used for the study of epileptogenesis and therefore, no conclusion can
be drawn on a potential relation between the number of neurons surviving in
basal cortices and the rate or even occurrence of epileptogenesis.
The data obtained in the present study are in line with the previous
study from this group reporting that the 60-mg/kg dose of Test compound (TC)
protected the hippocampus and the basal cortices from neuronal damage and
delayed the occurrence of recurrent seizures (see Example 1). They confirm
that the protection of the basal cortices could be a key factor in inducing a
disease modifying effect in the lithium-pilocarpine model of epilepsy. The key
role of the basal cortices as initiators of the epileptic process was
previously
demonstrated by our group in the lithium-pilocarpine model (Andre V, Rigoulot
MA, Koning E, Ferrandon A, Nehlig A (2003) Long-term pregabalin treatment
protects basal cortices and delays the occurrence of spontaneous seizures in
the lithium-pilocarpine model in the rat. Epilepsia 44:893-903; Roch C, Leroy
C,
Nehlig A, Namer IJ (2002a) Contribution of magnetic resonance imaging to the
study of the lithium-pilocarpine model of temporal lobe epilepsy in adult
rats.
Epilepsia 43:325-335; Roch C, Leroy C, Nehlig A, Namer IJ (2002b) Predictive
value of cortical injury for the development of temporal lobe epilepsy in P21-
day-old rats: a MRI approach using the lithium-pilocarpine model. Epilepsia
43:1129-1136.
References for Example 2
Andre V, Marescaux C, Nehlig A, Fritschy JM (2001) Alterations of the
hippocampal GABAergic system contribute to the development of spontaneous
recurrent seizures in the lithium-pilocarpine model of temporal lobe epilepsy.
Hippocampus 11:452-468.
~ Andre V, Rigoulot MA, Koning E, Ferrandon A, Nehlig A (2003) Long-term
pregabalin treatment protects basal cortices and delays the occurrence of
spontaneous seizures in the lithium-pilocarpine model in the rat. Epilepsia
44:893-903.
62
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
~ Cavalheiro EA (1995) The pilocarpine model of epilepsy. Ital J Neurol Sci
16:33-37.
~ Dube C, Marescaux C, Nehlig A (2000) A metabolic and neuropathological
approach to the understanding of plastic changes occurring in the immature
and adult rat brain during lithium-pilocarpine induced epileptogenesis.
Epilepsia 41(Suppl 6):S36-S43.
~ Dube C, Boyet S, Marescaux C, Nehlig A (2001) Relationship between
neuronal loss and interictal glucose metabolism during the chronic phase of
the lithium-pilocarpine model of epilepsy in the immature and adult rat. Exp
Neurol 167:227-241.
~ Paxinos G, Watson C (1986) The Rat Brain in Stereotaxic Coordinates, 2nd
ed. Academic Press, San Diego.
~ Roch C, Leroy C, Nehlig A, Namer IJ (2002a) Contribution of magnetic
resonance imaging to the study of the lithium-pilocarpine model of temporal
lobe epilepsy in adult rats. Epilepsia 43:325-335.
~ Roch C, Leroy C, Nehlig A, Namer IJ (2002b) Predictive value of cortical
injury for the development of temporal lobe epilepsy in P21-day-old rats: a
MRI approach using the lithium-pilocarpine model. Epilepsia 43:1129-1136.
~ Turski L, Ikonomidou C, Turski WA, Bortolotto ZA, Cavalheiro EA (1989)
Review: Cholinergic mechanisms and epileptogenesis. The seizures
induced by pilocarpine: a novel experimental model of intractable epilepsy.
Synapse 3:154-171.
Example 3
PC12 Cell Serum Withdrawal Model
Serum withdrawal is a cytotoxic environmental challenge that results in
cell death in cultured cell lines as well as in primary cells of various
tissue
origins, including nerve cells. In particular, pheochromocytoma (PC) 12 cells
have been widely employed as an in vitro neuronal cell model for a wide
variety
of neurodegenerative and cell death related disorders (Muriel, et al,
Mitochondrial free calcium levels (Rhod-2 fluorescence) and ultrastructural
63
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
alterations in neuronally differentiated PC12 cells during ceramide-dependent
cell death, J. Comp. Neurol., 2000, 426(2), 297-315; Dermitzaki, et al,
Opioids
transiently prevent activation of apoptotic mechanisms following short periods
of serum withdrawal, J. Neurochem., 2000, 74(3), 960-969; Carlile, et al,
Reduced apoptosis after nerve growth factor and serum withdrawal: conversion
of tetrameric glyceraidehyde-3-phosphate dehydrogenase to a dimer, Mol
Pharmacol., 2000, 57(1), 2-12). PC12 cells were cultured in sterile media
(RPMI 1640) supplemented with 10% heat-inactivated horse serum and 5%
fetal bovine serum (FBS). The culture medium also contained Penicillin-
Streptomycin-Neomycin antibiotic (50 µg, 50 µg, 100 µg,
respectively). Medium was exchanged every other day and the cells were
passed in log phase near confluence.
The control cells were cultured in regular media without any treatment.
An enantiomer of Formula 7 or Formula 8 (10 µM) was mixed well in the
medium and then applied to the cells. For the 2 day assay, an enantiomer of
Formula 7 or Formula 8 (10 µM) was only applied to the cells once at the
time of serum withdrawal. For the 7 day assay, an enantiomer of Formula 7or
Formula 8 (10. mu.M) was applied to the cells at the time of serum withdrawal
and every 48 hr thereafter when cells were changed with fresh new serum-free
medium. In the serum withdrawal group, the cells were cultured in serum-free
medium with no additional enantiomer of Formula 7 or Formula 8. Cell survival
was determined by the 3-(4, 5-dimethylthiazol-2-yl)-5-(3-carboxy- -
methoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium inner salt (MTS) assay at 2 or
7 days after serum withdrawal.
At the end of the experiment, cells were washed with fresh medium and
incubated with MTS solution in a humidified 37° C. with 5% C02
incubator for 1.5 hr. After the incubation period, the cells were immediately
analyzed using a Softmax program (Molecular Devices). MTS assay is a
calorimetric method for determining the number of viable cells in a given
experimental setting. The assay is based on the cellular conversion of the
tetrazolium salt, MTS, into a formazan that is soluble in tissue culture
medium
and measured directly at 490 nm in 96-well assay plates. The absorbance is
64
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
directly proportional to the number of living cells in culture. The arbitrary
absorbance reading in control cells is expressed as 100% survival rate.
Table 3 lists data demonstrating the effect on cell survival rate of the
orally administered enantiomer of Formula 7 and Formula 8 in the PC12 cell
serum withdrawal model.
TABLE 3 (% CELL SURVIVAL RATE)
2 Day 7 Day
Survival Rate Survival Rate
1% %
Control 100 100
Serum-free 49.6 2.6 23.8 2.6
Formula 7 69.4 1.7 79.9 4.0
Formula 8 66.4 5.4 85.2 0.6
Example 4
The Transient Cerebral lschemia Rat Model
The enantiomer of Formula 7 (test compound) was investigated in the
transient cerebral ischemia middle cerebral artery occlusion (MCAO) rat model
(as described in Nagasawa H. and Kogure K., Stroke, 1989, 20, 1037; and,
Zea Longa E., Weinstein P. R., Carlson S. and Cummins R., Stroke, 1989, 20,
84) using male Wistar rats at 10 and 100 mg/kg (i.v.). MK 801 (Dizocilpine
maleate; CAS Registry number 77086-22-7, a commercially available
neuroprotectant compound) was used as a positive control (3 mg/kg, i.p.).
Rats (n=12) were randomly allocated to one of four experimental groups
and were anesthetized. Blood flow from the internal carotid artery, anterior
cerebral artery and posterior cerebral artery into the middle cerebral artery
was
blocked by this procedure. One hour after blockage, animals were treated over
a 1 hour period with vehicie (administered i.v. over the one hour period),
control (administered as a single i.p. dose at the start of the one hour
period)
and two doses of the enantiomer of Formula 7 (administered i.v. over the one
hour period). Two hours after blockage, reperfusion was performed.
The animals were sacrificed and 20 mm-thick coronal sections of each brain
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
were prepared. One in every forty sections (i.e. every 800 nM) from the front
to
the occipital cortex was used to quantify the extent of the cerebral lesion.
Slides were prepared using sections stained (according to the Nissl procedure)
with cresyl violet and were examined under a light microscope.
Regional ischemic surface areas in the coronal sections of individual rats
were determined according to the presence of cells with morphological
changes. The areas of neuronal injury or infarction were measured and then
added. The cortex and striatum volume were calculated for each animal (total
ischemic surface area×0.8 mm (thickness)).
MCAO Model Analysis
The mean volumes (±S.E.M.) for each animal randomly assigned to
the four experimental groups were compared using one-way ANOVA (one way
ANOVA is a statistical method which compares 3 or more unmatched groups)
followed by Dunnett's t-test (both methods incorporated in Statview
512+software, BarinPower, Calabasas, Calif., USA).
As shown in Table 4 below, results were considered statistically
significant when the p value was < 0.05 compared to vehicle group (1p< 0.01;
2p< 0.05).
TABLE 4
Mean Infarct Volume (mm )
Treatment N S.E.M.
Cortex Striatum Total
Volume
Vehicle, 10 mUk 12 275.5 27.1 79.4 3.6 354.9 29.9
MK 801 @ 3 mg/kg 12 95.8 24.5 56.1 151.9
5.32 28.71
Formula 7@ 10 mg/kg 12 201.0 23.9 75.9 2.6 276.9 25.4
Formula7 @ 100 mg/kg 12 98.8 29.5 63.0 161.9
5.92 34.31
25
66
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
EXAMPLE 5
The Test Compound (TC) referred to in the example below is the
compound of Formula 7 and the same compound as in the other examples 1
and 2 above.
The purpose of this study was to assess the pharmacokinetics (PK) of
Test Compound (TC) following single and repeated oral administration in
healthy adult men at clinically relevant doses
METHODS:
Two single-center, placebo-controlled, double-blind, ascending-dose
studies were conducted in healthy men ?18 and <45 yrs. In study 1 (N=70),
subjects were randomly assigned to a single dose of Test Compound (TC) or
placebo. Escalated doses were received as 100, 250, 400, 750, 1000, 1250,
and 1500 mg. PK parameters were estimated from plasma and urine samples
collected up to 3 days post dose. Study 2 (N=53) evaluated the PK of repeated
doses of Test Compound (TC) in 4 dose groups (100, 250, 500, or 750 mg).
Within each group, 12 subjects were assigned to q12h treatment with drug or
placebo for 1 wk and were crossed over after a 14-day washout period. PK
parameters were estimated from plasma and urine samples on days 1 and 7.
RESULTS:
Single dose: Test Compound (TC) was rapidly absorbed following oral
administration. Cma,, and AUCo__ increased in proportion to dose over the
range
of 100-1500 mg. Mean tmax ranged from 1.3-2.7 h. Mean t1i2 (11.5 - 13.9 h),
CUF (2.87-3.67 Uh), and Vd/F (52.1-66.3 Uh) values were similar for all 7
dose groups.
Repeated doses: Plasma concentrations of Test Compound (TC)
reached steady state after 3-4 days as predicted from its single-dose half-
life.
Mean tmax occurred 1.3-1.8 h after dosing. The mean t1i2 (11.9-12.8 h) and
CUF (3.40-3.78 Uh) values at steady state were comparable to the PK
parameters following single-dose administration on day 1 and in study 1.
Steady-state Cmax and AUCO_12 increased in proportion to the dose.
As expected, there was a moderate degree of Test Compound (TC)
accumulation; Cmax and AUCO_12 were about two-fold higher on day 7 vs. day 1
(P<0.001). Mean CLR estimates for Test Compound (TC) were <5% of the
67
CA 02584854 2007-04-13
WO 2006/044472 PCT/US2005/036695
mean oral clearance, suggesting non-renal clearance as the primary
mechanism for Test Compound (TC) elimination.
CONCLUSIONS:
Test Compound (TC) exhibited linear PK after single (100-1500mg) and
repeated (100-750 mg bid) doses. It was rapidly absorbed and had a mean
elimination haif life of 11.5-13.9 h, allowing bid dosing. Following q12h
administration, Test Compound (TC) accumulated two-fold and was primarily
cleared by a non-renal pathway.
References cited
All references cited herein are incorporated herein by reference in their
entirety and for all purposes to the same extent as if each individual
publication
or patent or patent application was specifically and individually indicated to
be
incorporated by reference in its entirety for all purposes.
The discussion of references herein is intended merely to summarize
the assertions made by their authors and no admission is made that any
reference constitutes prior art. Applicants reserve the right to challenge the
accuracy and pertinence of the cited references.
The present invention is not to be limited in terms of the particular
embodiments described in this application, which are intended as single
illustrations of individual aspects of the invention. Many modifications and
variations of this invention can be made without departing from its spirit and
scope, as will be apparent to those skilled in the art. Functionally
equivalent
methods and apparatus within the scope of the invention, in addition to those
enumerated herein will be apparent to those skilled in the art from the
foregoing description and accompanying drawings. Such modifications and
variations are intended to fall within the scope of the appended claims. The
present invention is to be limited only by the terms of the appended claims,
along with the full scope of equivalents to which such claims are entitled.
68