Note: Descriptions are shown in the official language in which they were submitted.
DEMANDE OU BREVET VOLUMINEUX
LA PRESENTE PARTIE DE CETTE DEMANDE OU CE BREVET COMPREND
PLUS D'UN TOME.
CECI EST LE TOME 1 DE 2
CONTENANT LES PAGES 1 A 96
NOTE : Pour les tomes additionels, veuillez contacter le Bureau canadien des
brevets
JUMBO APPLICATIONS/PATENTS
THIS SECTION OF THE APPLICATION/PATENT CONTAINS MORE THAN ONE
VOLUME
THIS IS VOLUME 1 OF 2
CONTAINING PAGES 1 TO 96
NOTE: For additional volumes, please contact the Canadian Patent Office
NOM DU FICHIER / FILE NAME:
NOTE POUR LE TOME / VOLUME NOTE:
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
LEUCINE-RICH REPEAT (LRR) MOTIF CONTAINING PROTEINS
This invention relates to proteins, termed INSP168, INSP168-SV1, INSP149 and
INSP169, herein identified as leucine-rich repeat (LRR) motif containing
proteins,
and to the use of these proteins and nucleic acid sequences from the encoding
gene in
the diagnosis, prevention and treatment of disease.
All publications, patents and patent applications cited herein are
incorporated in full
by reference.
BACKGROUND
The process of drug discovery is presently undergoing a fundamental revolution
as the
era of functional genomics comes of age. The term "functional genomics"
applies to
an approach utilising bioinformatics tools to ascribe function to protein
sequences of
interest. Such tools are becoming increasingly necessary as the speed of
generation of
sequence data is rapidly outpacing the ability of research laboratories to
assign
functions to these protein sequences.
As bioinformatics tools increase in potency and in accuracy, these tools are
rapidly
replacing the conventional techniques of biochemical characterisation. Indeed,
the
advanced bioinformatics tools used in identifying the present invention are
now
capable of outputting results in which a high degree of confidence can be
placed.
Various institutions and commercial organisations are examining sequence data
as
they become available and significant discoveries are being made on an on-
going
basis. However, there remains a continuing need to identify and characterise
further
genes and the polypeptides that they encode, as targets for research and for
drug
discovery.
Introduction to Secreted Proteins
The ability of cells to make and secrete extracellular proteins is central to
many
biological processes. Enzymes, growth factors, extracellular matrix proteins
and
signalling molecules are all secreted by cells. This is through fusion of a
secretory
vesicle with the plasma membrane. In most cases, but not all, proteins are
directed to the
endoplasmic reticulum and into secretory vesicles by a signal peptide. Signal
peptides
are cis-acting sequences that affect the transport of polypeptide chains from
the
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
2
cytoplasm to a membrane bound compartrnent such as a secretory vesicle.
Polypeptides
that are targeted to the secretory vesicles are either secreted into the
extracellular matrix
or are retained in the plasma membrane. The polypeptides that are retained in
the plasma
membrane will have one or more transmembrane domains. Examples of signal
peptide
contaiiiing proteins that play a central role in the functioning of a cell are
cytokines,
hormones, extracellular matrix proteins, adhesion molecules, receptors,
proteases, and
growth and differentiation factors.
Introduction to PAL
The photoreceptor-associated leucine-rich repeat (LRR) protein (abbreviated to
PAL)
is a membrane glycoprotein that is specifically expressed in the photoreceptor
cells of
the retina (Gomi et al., J. Neuroscience, 2000, 20(9):3206-3213).
Sequencing of the PAL gene revealed that the PAL protein contains an LRR
motif, an
Ig C2-like domain and a fibronectin type III-like domain, all within its
extracellular
region. The LRR domain of PAL contains five contiguous LRRs. This combination
of
the three types of domain described above was identified as a new class of
transmembrane protein, although some previously known proteins contain two of
these three domains (e.g. Trk and NCAM) (Gomi et al., J. Neuroscience, 2000,
20(9):3206-3213).
The abundance of PAL mRNA was observed to increase over the time course of
development of the rat retina. Northern blotting experiments revealed that the
PAL
mRNA was specific to the photoreceptor cells within the retina. Western
blotting and
immunoprecipitation experiments with a PAL-specific polyclonal antibody showed
that PAL forms a strong homodimer structure that is resistant to SDS and high
temperature (Gomi et al., J. Neuroscience, 2000, 20(9):3206-3213).
A liuman homolog of PAL was also identified and was mapped to chromosome
10q23.2-23.3 by fluorescence in situ hybridisation (FISH). On the basis of
experiments carried out on rat retinal cells, it was postulated that PAL may
act as a
receptor for a certain trophic factor or for an adhesion molecule
participating in
morphogenesis. The human-, PAL protein was therefore considered to be a_
potential
candidate disease gene for inherited retinal disorders (Gomi et al., J.
Neuroscience,
2000, 20(9):3206-3213).
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
3
Other known retina-specific genes include rhodopsin, transducin, cGMP-gated
ion
channels, peripherin/rds and rom-1. Among these, a number of causative genes
for
inherited diseases of the retina have been identified. For example, mutations
in
rhodopsin or peripherin/rds contribute to autosomal dominant retinitis
pigmentosa.
Introduction to the fibronectin type 3 domains
Fibronectin Type III (FnIII) protein domains are formed by 80-100 amino acids
included
in several multimodular proteins, mostly associated to extracellular matrix
such as
tenascins (Joester A and Faissner A, Matrix Biol. 2001, 20:13-22), or Titins
(Skeie GO,
Cell Mol Life Sci. 2000, 57:1570-6) or to receptor proteins such as insulin
receptor
protein family (Marino-Buslje C et al., FEBS Lett. 1998, 441:331-6).
Despite remarkably similar tertiary structures, FnI1I modules share low
sequence
homology. Conversely, the sequence homology for the same FnIII module across
multiple species is notably higher, suggesting that sequence variability is
functionally
significant. Amongst the residues that are conserved, Prolines are of
particular
importance since prevent aggregation in multi-modular proteins containing this
domain
(Craig D et al., Structure 2004, 12:21-30; Craig D et al., Proc Natl Acad Sci
U S A.
2001, 98(10):5590-5; Cota E et al., J Mol Biol. 2001, 305:1185-94; Cota E et
al., J Mol
Biol. 2000, 302:713-25; Steward A et al., J Mol Biol. 2002, 318:935-40).
The paradigm of this protein module is human Fibronectin, a 2386-amino acid
glycoprotein of the extracellular matrix containing several protein modules,
usually
categorized in tllree types: FnI, FnII, and FnIII (or Fl, F2, or F3; Potts JR
and
Campbell ID, Matrix Biol. 1996, 15:313-20). Fibronectin circulates in a
soluble form
in the plasma and is also found in an insoluble, multimeric form within the
extracellular matrix at appropriate sites. The formation and the degradation
of these
insoluble fibrils is a dynamic, cell-dependent process that is mediated by a
series of
events involving the actin cytoskeleton and integrin receptors. Fibronectin
fibrils can
bind the surfaces of mammalian and bacterial cells and various molecules
including
collagen, fibrin, heparin, DNA, and actin. Fibronectin is involved in cell
adhesion/contractility/motility, opsonization, wound healing, and formation of
fibrotic
aggregates. For example, in certain chronic inflammatory diseases, including
asthma,
the loss of this regulation gives rise either to excess or to inappropriate
fibronectin
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
4
deposition that parallels the development of tissue fibrosis (Hocking DC,
Chest. 2002,
122(6 Suppl):275S-278S).
Finally, isolated fibronectin domains of extracellular matrix proteins, that
can be
generated physiologically by limited proteolysis or mechanical stress of the
fibronectin fibers, can modulate various biological and physiological
responses, such
as the neuronal regeneration, hippocampal learning and synaptic plasticity
(Meiners S
and Mercado ML, Mol Neurobiol. 2003, 27:177-96; Strekalova T et al., Mol Cell
Neurosci. 2002, 21:173-87), osteoblast adliesion, proliferation and
differentiation
(Kim TI et al., Biotechnol Lett. 2003, 25:2007-11), tissue degradation,
inflammation
and tumor progression (Labat-Robert J, Ageing Res Rev. 2004, 3:233-47).
Fragments
or splicing variants of FnIIl domain-containing proteins may become target for
antibodies and other proteins blocking them having important therapeutic or
diagnostic applications, such as cancer (Ebbinghaus C et al., Curr Pharm Des.
2004,
10:1537-49), inflammatory arthritis (Barilla ML and Carsons SE, Semin
Arthritis
Rheum. 2000, 29:252-65), or organ transplantation (Coito AJ et al., Dev
Immunol.
2000;7:239-48).
Introduction to the leucine-rich repeat (LRR) motif
The LRR motif is a relatively short motif of around 22-28 residues, and is
found in a
variety of cytoplasmic, membrane and extracellular proteins. Proteins
containing LRRs
are associated with a very wide range of biological functions, although all
are thought to
be involved in protein-protein interaction or cell adhesion. The LRR motif is
a repetitive
motif made up of several copies of the sequence LxxLxxLxLxxNxLxxL xxxxFxx.
LRRs
are often flanked by cysteine-rich repeat regions, an N-terminal LRR motif or
a leucine-
rich repeat C-terminal domain (LRRCT).
Introduction to Immunoglobulin Domains
The immunoglobulin (Ig) domain is a well characterised domain present in
hundreds
of proteins of varying functions. The basic Ig domain structure is a tetramer
of two
light chains and two heavy chains linked by disulphide bonds. Immunoglobulin
domain-containing cell surface recognition molecules have been shown to play a
role
in diverse physiological functions, many of which can play a role in disease
processes. Alteration of their activity is a means to alter the disease
phenotype and as
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
such identification of novel immunoglobulin domain-containing cell surface
recognition molecules is highly relevant as they may play a role in many
diseases,
particularly inflammatory disease, oncology, and cardiovascular disease.
Immunoglobulin domain-containing cell surface recognition molecules are
involved
5 in a range of biological processes, including: embryogenesis, maintenance of
tissue
integrity, leukocyte extravasation/inflammation, oncogenesis, angiogenesis,
bone
resorption, neurological dysfunction, thrombogenesis, and invasion/adherence
of
bacterial pathogens to the host cell.
The detailed characterisation of the structure and function of several
immunoglobulin-
domain containing cell surface recognition molecule families has led to active
programs by a number of pharmaceutical companies to develop modulators for use
in
the treatment of diseases involving inflamination, oncology, neurology,
immunology
and cardiovascular function. hninunoglobulin domain containing cell surface
recognition molecules are involved in virtually every aspect of biology from
embryogenesis to apoptosis. They are essential to the structural integrity and
homeostatic functioning of most tissues. It is therefore not surprising that
defects in
immunoglobulin domain containing cell surface recognition molecules cause
disease
and that many diseases involve modulation of immunoglobulin domain containing
cell surface recognition molecule function.
Immunoglobulin domain-containing cell surface recognition molecules are thus
known to play a role in diverse physiological functions, many of which can
play a role
in disease processes. Alteration of their activity is a means to alter the
disease
phenotype and as such identification of novel immunoglobulin domain-containing
cell
surface recognition molecules is highly relevant as they may play a role in
many
diseases, particularly immunology, inflainmatory disease, oncology,
cardiovascular
disease, central nervous system disorders and infection.
In summary, cell surface recognition molecules, including those containing
LRRs, Ig
domains or fibronectin type 3 domains, have been shown to play a role in
diverse
physiological functions, many of which can play a role in disease processes.
Alteration
of their activity is a means to alter the disease phenotype and as such
identification of
novel adhesion molecules is liighly relevant as they may play a role in many
diseases,
particularly inflammatory disease, oncology, cardiovascular disease and
bacterial
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
6
infection. The identification of further retina-specific cell surface
recognition
molecules, including paralogs of the human PAL protein (which will contain
LRRs, Ig
domains and fibronectin type 3 domains) is of great iinportance in the ongoing
investigation of retinal developments and retinal pathologies. Their
identification will
allow the development of new methods for the treatment and diagnosis of
retinal
diseases and disorders. Accordingly, there remains a need for the
identification of
such proteins to enable new drugs to be developed for the treatment and
prevention of
human disease.
THE INVENTION
The invention is based on the discovery that the INSP168, INSP168-SV1, INSP149
and INSP169 proteins are splice variants of a leucine-rich repeat (LRR) motif
containing sequence with similarity to PAL (SwissProt Acc. Code PALP HUMAN)
and to a nogo receptor homolog (SwissProt Acc. Code Q6X814).
In particular, the invention is based on the finding that polypeptides of the
present
invention are PAL-like and/or nogo-receptor like molecules.
In a first aspect of the invention, there is provided a polypeptide, which
polypeptide:
(i) comprises the amino acid sequence as recited in SEQ ID NO:2 (mature
INSP 168);
(ii) is a fragment thereof which functions as a biologically active
polypeptide and/or has an antigenic determinant in common with the
polypeptides of (i); or
(iii) is a functional equivalent of (i) or (ii).
A polypeptide according to part (i) of the first aspect of the invention may
comprise
the amino acid sequence as recited in SEQ ID NO:4 (mature INSP149), SEQ ID
NO:8
(mature INSP168-SV1) or SEQ ID NO:10 (mature INSP169).
Although the Applicant does not wish to be bound by this theory, it is
postulated that
the first 19 ainino acids of the INSP168, INSP168-SV1, INSP149 and INSP169
proteins forms a signal peptide, as shown in the scheinatic representation
below:
MHLFACLCIVLSFLEGVGCLCPSQCTCDYHGRNDGSGSR...
Thus, a polypeptide according to part (i) of the first aspect of the invention
may
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
7
comprise the amino acid sequence as recited in SEQ ID NO:30 (full length
INSP168),
SEQ ID NO:32 (full length INSP149), SEQ ID NO:34 (full length INSP168-SV1),
SEQ ID NO:36 (full lengtll INSP169) and SEQ ID NO:67 (INSP169 cloned
extracellular region).
A polypeptide according to part (i) of the first aspect of the invention may
comprise
the extracellular portion of the amino acid sequence as recited in SEQ ID NO:4
(mature INSP149) or SEQ ID NO:10 (mature INSP169). Thus, a polypeptide
according to part (i) of the first aspect of the invention may comprise the
amino acid
sequence as recited in SEQ ID NO:6 (INSP149 extracellular region) or SEQ ID
NO:12 (INSP 169 extracellular region). A polypeptide according to part (i) of
the first
aspect of the invention may comprise the amino acid sequence as recited in SEQ
ID
NO:67 (INSP 169 cloned extracellular region). The cloned extracellular region
of
INSP169 (SEQ ID NO:67) differs from the predicted extracellular region of
INSP169
at amino acid 524 (see Example 7 and Figure 8).
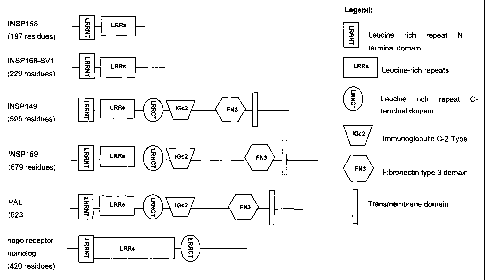
The INSP 168, INSP 168-SV l, INSP 149 and INSP 169 proteins each contain a
leucine-
rich repeat motif. The amino acid sequence of the leucine-rich repeat motif
present in
the INSP168, INSP168-SV1, INSP149 and INSP169 proteins is recited in SEQ ID
NO:14. Accordingly, preferred fragments of the INSP168, INSP168-SV1, INSP149
and INSP 169 proteins are fragments that comprise or consist of the amino acid
sequence as recited in SEQ ID NO:14 (LRR motif).
A polypeptide according to part (i) of the first aspect of the invention may
comprise
the amino acid sequence as recited in SEQ ID NO:2 (mature 1NSP 168), SEQ ID
NO:4
(mature INSP 149), SEQ ID NO:6 (INSP 149 extracellular region), SEQ ID NO:8
(mature INSP168-SV1), SEQ ID NO:10 (mature INSP169), SEQ ID NO:12
(INSP169 extracellular region), SEQ ID NO:14 (LRR motif), SEQ ID NO:30 (full
length INSP168), SEQ ID NO:32 (full length INSP149), SEQ ID NO:34 (full length
INSP168-SV1), SEQ ID NO:36 (full length INSP169) or SEQ ID NO:67 (INSP169
cloned extracellular region) and a histidine tag. Preferably the histidine tag
is located
at the C-terminus of the polypeptide. Preferably the histidine tag comprises
between 1
and 10 histidine residues (e.g. 1, 2, 3, 4, 5, 6, 7, 8, 9 or 10 residues).
More preferably,
the histidine tag comprises 6 residues. Thus, a polypeptide according to part
(i) of the
first aspect of the invention may comprise the amino acid sequence as recited
in SEQ
ID NO:16 (his tag mature INSP168), SEQ ID NO:18 (his tag mature INSP149), SEQ
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
8
ID NO:20 (his tag INSP 149 extracellular region), SEQ ID NO:22 (his tag mature
INSP168-SVl), SEQ ID NO:24 (his tag mature INSP169), SEQ ID NO:26 (his tag
INSP169 extracellular region), SEQ ID NO:28 (his tag LRR motif), SEQ ID NO:38
(his tag full length INSP 168), SEQ ID NO:40 (his tag full length INSP 149),
SEQ ID
NO:42 (his tag full length INSP 168-SV 1) or SEQ ID NO:44 (his tag full length
INSP 169).
Thus, the first aspect of the present invention provides a polypeptide which
comprises
or consists of the amino acid sequence as recited in SEQ ID NO:2 (mature
INSP168),
SEQ ID NO:4 (mature INSP149), SEQ ID NO:6 (INSP149 extracellular region), SEQ
ID NO:8 (mature INSP168-SV1), SEQ ID NO:10 (mature INSP169), SEQ ID NO:12
(INSP 169 extracellular region), SEQ ID NO:14 (LRR moti fl, SEQ ID NO:16 (his
tag
mature INSP168), SEQ ID NO:18 (his tag mature INSP149), SEQ ID NO:20 (his tag
INSP 149 extracellular region), SEQ ID NO:22 (his tag mature INSP 168-SV 1),
SEQ
ID NO:24 (his tag mature INSP169), SEQ ID NO:26 (his tag INSP 169
extracellular
region), SEQ ID NO:28 (his tag LRR motif), SEQ ID NO:30 (full length INSP168),
SEQ ID NO:32 (full length INSP149), SEQ ID NO:34 (full length INSP168-SV1),
SEQ ID NO:36 (fall length INSP169), SEQ ID NO:38 (his tag full length
INSP168),
SEQ ID NO:40 (his tag full length INSP149), SEQ ID NO:42 (his tag full length
INSP168-SVl), SEQ ID NO:44 (his tag full length INSP169), or SEQ ID NO:67
(INSP 169 cloned extracellular region). The first aspect of the present
invention also
provides a polypeptide which is a fragrnent of such a polypeptide and which
functions
as a biologically active polypeptide and/or has an antigenic determinant in
common
with such a polypeptide or which is a functional equivalent of such a
polypeptide.
The terms "the INSP 168 polypeptides" and "an INSP 168 polypeptide" as used
herein
include polypeptides comprising or consisting of the amino acid sequence as
recited
in SEQ ID NO:2 (mature INSP168), such as polypeptides comprising or consisting
of
the amino acid sequence as recited in SEQ ID NO:4 (mature INSP 149), SEQ ID
NO:6
(INSP149 extracellular region), SEQ ID NO:8 (mature INSP168-SV1), SEQ ID
NO:10 (mature INSP169), SEQ ID NO:12 (INSP169 extracellular region), SEQ ID
NO:16 (his tag mature INSP168), SEQ ID NO:18 (his tag mature INSP149), SEQ ID
NO:20 (his tag INSP149 extracellular region), SEQ ID NO:22 (his tag mature
INSP168-SV1), SEQ ID NO:24 (his tag mature INSP169), SEQ ID NO:26 (his tag
INSP169 extracellular region), SEQ ID NO:30 (full length INSP168), SEQ ID
NO:32
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
9
(full length INSP149), SEQ ID NO:34 (full length INSP168-SV1), SEQ ID NO:36
(full length INSP169), SEQ ID NO:38 (his tag full length INSP168), SEQ ID
NO:40
(his tag full length INSP149), SEQ ID NO:42 (his tag full length INSP168-SV1),
SEQ ID NO:44 (his tag full length INSP169) or SEQ ID NO:67 (INSP169 cloned
extracellular region).
INSP 149, INSP168 and INSP 169 were a set of predictions representing splice
variants of a leucine rich repeat-containing sequence with similarity to the
retina-
specific protein PAL.
INSP149 was a prediction for a 595 amino acid (1785 bp) ORF encoded in 5
exons.
INSP 168 was a prediction for a 197 amino acid (591 bp) ORF encoded in 3
exons.
INSP169 was a prediction for a 679 amino acid (2037 bp) ORF encoded in 4
exons.
INSP 149 and INSP 169 were predicted to be type I transmembrane proteins
comprising a leucine-rich repeat motif, an immunoglobulin domain and a
fibronectin
type 3 domain. The INSP 169 polypeptide is a splice variant of the INSP 149
polypeptide that is identical to INSP149 except for the longer final exon,
which
subsumes the final two exons of INSP149. The longer sequence encoded by
INSP169
has the same domain organisation as INSP149 but has a low complexity insert
between the Ig and FN III domains. INSP168 was essentially a truncated splice
variant of INSP149 and INSP169 and was predicted to represent a secreted
protein.
The INSP168 polypeptide is a splice variant of the INSP149 polypeptide that
comprises a stop codon at the start of its third exon. As a result of this
truncation in
the third exon, the INSP168 polypeptide lacks the transmembrane region found
in
exon 5 of INSP 149. A113 of the predictions contained 4 leucine rich repeat
regions in
the N-terminal portion (SEQ ID NO:14). INSP149 and INSP 169 also contained an
immunoglobulin domain and a fibronectin type III domain in the predicted
extracellular regions. An aligmnent of the 3 predicted amino acid sequences is
shown
in Figure 1. As noted above, a signal peptide was predicted spanning from
residues 1
to 19.
The open reading frame (ORF) of the INSP168 prediction has been cloned using a
pair of PCR primers (see Figures 2 and 3). The primer pair was tested on
selected
cDNA libraries derived from brain and retina and on brain and eye cDNA
templates
using Platinum Taq DNA Polymerase High Fidelity (HiFi). PCR products were
cloned and sequenced and a clone was identified, amplified from brain cDNA,
which
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
contained the expected INSP168 ORF. A second clone was identified, also
amplified
from brain cDNA, which contained a splice variant of the INSP168 ORF. This
clone
contained a 32 amino acid insertion towards the 3' end of the INSP168 ORF
which
represented a new exon 3. The insertion also caused a fraineshift such that
the new
5 exon 4 contained an extra 6 amino acids compared with the original INSP 168
exon 3.
This clone was called INSP168-SV1, and is also shown in the Figure 1
alignment.
The INSP 168 polypeptides are structurally related to the Retinal Specific
Protein PAL
(SwissProt Acc. Code PALP HUMAN) and to a nogo receptor homolog (SwissProt
Acc. Code Q6X814). An amino acid alignment between the INSP168, INSP168-SV1,
10 INSP 149 and INSP 169 polypeptides and PAL is shown in Figure 5, and the
schematic
representation of domains is shown in Figure 6.
As noted above, the cloned extracellular region of INSP 169 (SEQ ID NO:67)
differs
from the predicted extracellular region of INSP 169 at amino acid 524 (see
Example 7
and Figure 8).
PAL may be implicated in diseases of the retina, retinal pigment epithelium
(RPE),
and clloroids (see for example JP2001128686). These include ocular
neovascularization, ocular inflammation and retinal degenerations. Specific
examples
of these disease states include diabetic retinopathy, chronic glaucoma,
retinal
detachment, sickle cell retinopathy, senile macular degeneration, retinal
neovascularization, subretinal neovascularization; rubeosis iritis
inflammatory
diseases, chronic posterior and pan uveitis, neoplasms, retinoblastoma,
pseudoglioma,
neovascular glaucoma; neovascularization resulting following a combined
vitrectomy
and lensectomy, vascular diseases retinal ischemia, choroidal vascular
insufficiency,
choroidal thrombosis, neovascularization of the optic nerve, diabetic macular
edema,
cystoid macular edema, retinitis pigmentosa, retinal vein occlusion,
proliferative
vitreoretinopathy, angioid streak, and retinal artery occlusion, and,
neovascularization
due to penetration of the eye or ocular injury. Additional relevant diseases
include the
neuropathies, such as Leber's, idiopathic, drug-induced, optic, and ischemic
neropathies.
Nogo receptor-like proteins could be major inhibitors of CNS neuronal
regeneration
(Schwab ME. Curr Opin N6urobiol: 2004 Feb;14(l):118-24; Teng et al. J
Neurochem.
2004 May;89(4):801-6). Animals treated with antibodies targeted to Nogo-A
always
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
11
showed a higher degree of recovery in various behavioural tests (e.g. IN-1
Fab'
fragments or new purified IgGs against Nogo-A). In addition, a Nogo-66
antagonistic
peptide (NEP 1-40) effected significantly axon growth of the corticospinal
tract and
improved functional recovery in rats inflicted with mid-thoracic spinal cord
hemisections. Subcutaneous administration of NEP1-40 in spinal cord lesioned
animals resulted in extensive growth of corticospinal axons, sprouting of
serotonergic
fibres, synapse formation and enhanced locomotor recovery. Soluble Fc fusion
proteins of the Nogo receptor subunit NgR, which blocks Nogo, significantly
reduce
the inhibitory activity of myelin. Similar results were obtained after Nogo
gene
deletions and blockade of the downstream messengers Rho-A and ROCK in animal
models.
The leucine-rich repeat domain of SLIT proteins is sufficient for guiding both
axon
projection and neuronal migration in vitro (the LRR region of SLIT is
structurally
related to the LRR region of INSP168, INSP168-SV1, INSP149 and INSP169). As
such, the LRR region of INSP168, INSP168-SV1, INSP149 and INSP169 or
fragments containing the LRR region might be useful in the treatment of the
diseases
listed herein. SLIT-like proteins are thought to act as molecular guidance cue
in
cellular migration, and function appears to be mediated by interaction with
roundabaout homolog receptors (bind ROBOl and ROBO2 with high affinity).
During neural development, SLIT is involved in axonal navigation at the
ventral
midline of the neural tube and projection of axons to different regions. In
spinal chord
development, SLIT may play a role in guiding commissural axons once they
reached
the floor plate by modulating the response to netrin. SLIT may be implicated
in spinal
chord midline post-crossing axon repulsion. In the developing visual system,
SLIT
appears to function as repellent for retinal ganglion axons by providing a
repulsion
that directs these axons along their appropriate paths prior to, and after
passage
through, the optic chiasm. In vitro, SLIT collapses and repels retinal
ganglion cell
growth cones. SLIT seems to play a role in branching and arborization of CNS
sensory axons, and in neuronal cell migration. In vitro, Slit homolog 2
protein N-
product, but not Slit homolog 2 protein C-product, repells olfactory bulb (OB)
but not
dorsal root ganglia (DRG) axons, induces OB growth cones collapse and induces
branching of DRG axons. SLIT seems to be involved in regu ating leukocyte
migration.
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
12
INSP168, INSP168-SV1, fNSP149 and INSP169 and other INSP168 polypeptides
and/or fragments and functional equivalents thereof (e.g. fragments containing
the
LRR region) can be useful in the diagnosis and/or treatment of diseases for
which
other (e.g. above mentioned PAL- and Nogo receptor-like proteins) structurally
related proteins demonstrate therapeutic activity.
In particular, polypeptides of the invention consisting of and/or comprising
of the
mature (lacking a signal peptide) forms and/or cleaved forms of INSP168,
INSP168-
SV 1 and/or mature soluble forms of INSP 149 and/or INSP169, and/or agonists
thereof are useful for the diagnosis and/or treatment of diseases. Preferably,
the
soluble forms of INSP149 and/or INSP169 consist of the mature extracellular
part
and/or cleaved fragments of INSP149 and/or INSP 169. Antagonists of membrane
bound INSP149 and/or INSP169, for example antibodies, are useful for the
diagnosis
and/or treatment of diseases.
The INSP168, INSP168-SV1, INSP149 and INSP169 polypeptides maybe implicated
in diseases of the retina, spinal cord injuries (e.g. paraplegia) and
neurodegenerative
disorders. These include disorders of the central nervous system as well as
disorders
of the peripheral nervous system. Neurodegenerative disorders include, but are
not
limited to, brain injuries, cerebrovascular diseases and their consequences,
Parkinson's
disease, corticobasal degeneration, motor neuron disease (including
amyotrophic
lateral sclerosis, ALS), multiple sclerosis, traumatic brain injury, stroke,
post-stroke,
post- traumatic brain injury, and small-vessel cerebrovascular disease.
Dementias,
such as Alzheimer's disease, vascular dementia, dementia with Lewy bodies,
frontotemporal dementia and Parkinsonism, frontotemporal dementias (including
Pick's disease), progressive nuclear palsy, corticobasal degeneration,
Huntington's
disease, thalamic degeneration, Creutzfeld-Jakob dementia, HIV dementia,
schizophrenia with dementia, and Korsakoffs psychosis, as well as stroke and
trauma.
In particular, the INSP168, INSP168-SV1, INSP149 and INSP169 polypeptides may
be implicated in diseases of the retina, retinal pigment epithelium (RPE), and
choroids; ocular neovascularization, ocular inflammation and retinal
degenerations;
diabetic retinopathy, chronic glaucoma, retinal detachment, sickle cell
retinopathy,
senile macular degeneration, retinal neovascularization, subretinal
neovascularization;
rubeosis iritis inflammatory diseases, chronic posterior and pan uveitis,
neoplasms,
retinoblastoma, pseudoglioma, neovascular glaucoma; neovascularization
resulting
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
13
following a combined vitrectomy and lensectomy, vascular diseases retinal
ischemia,
choroidal vascular insufficiency, choroidal thrombosis, neovascularization of
the optic
nerve, diabetic macular edema, cystoid macular edema, retinitis pigmentosa,
retinal
vein occlusion, proliferative vitreoretinopathy, angioid streak, retinal
artery occlusion,
neovascularization due to penetration of the eye or ocular injury,
neuropathies;
Leber's, idiopathic, drug-induced, optic, and ischemic neropathies; spinal
cord
injuries, paraplegia, neurodegenerative disorders, disorders of the central
nervous
system, disorders of the peripheral nervous system, brain injuries,
cerebrovascular
diseases, Parkinson's disease, corticobasal degeneration, motor neuron
disease,
amyotrophic lateral sclerosis (ALS), multiple sclerosis, traumatic brain
injury, stroke,
post-stroke, post- traumatic brain injury, small-vessel cerebrovascular
disease,
dementias, Alzheimer's disease, vascular dementia, dementia with Lewy bodies,
frontotemporal dementia, Parkinsonism, frontotemporal dementias, Pick's
disease,
progressive nuclear palsy, corticobasal degeneration, Huntington's disease,
thalamic
degeneration, Creutzfeld-Jakob dementia, HIV dementia, schizophrenia with
dementia, Korsakoffs psychosis, stroke and trauma.
Neuro-inflammation is a common feature of several neurological diseases,
trauinatic
situations (at central or peripheral level), stroke (brain, heart, renal), or
infectious
diseases (mediated by viral agents such as HIV or bacterial agents such as
meningitis), leading to an excessive inflammatory response in central nervous
system.
Many stimuli, originated by neuronal or oligodendroglial cells suffering due
to these
various conditions, can trigger neuro-inflammation. In particular, astrocytes
can
secrete various chemokines and cytokines, inducing a recruitment of additional
leukocytes that in their turn will further stimulate astrocytes, leading to an
exacerbated response. In chronic neurodegenerative diseases such as multiple
sclerosis (MS), spinal muscular atrophies (SMA), Alzheimer's disease (AD),
Parkinson's disease (PD), Huntington's disease (HD), or amylotrophic lateral
sclerosis
(ALS), the presence of persistent neuro-inflammation is thought to be involved
in the
progression of the disease and in the case of AD in the secondary events such
as
micro-hemorrhagic events (Cacquevel M et al., Curr Drug Targets. 2004, 5: 529-
534;
Chavarria A et al., Autoimmun Rev. 2004, 3: 251-260; Ambrosini E and Aloisi F,
Neurochem Res. 2004, 29: 1017-103 8).
Limited axon regeneration and plasticity is central to the pathophysiology of
a range
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
14
of neurological disorders, including stroke, head trauma, multiple sclerosis,
and
neurodegenerative disease.
In addition to its role in the pathophysiology of neurological disorders as
well as in
loss of sight or blindness, nogo-like molecules are implicated in cancer.
Without
wishing to be bound to this theory, the polypeptides of the present invention
are
implicated in cancer through EGFR inhibition. Preferably, the cancer is lung
cancer.
The biological properties of the INSP168, INSP168-SV1, INSP149 and INSP169
polypeptides related to neuroprotection, maintenance of axonal integrity,
myelination
and re-/generation of myelin producing cells, can be tested in various assays
involving
cell lines. For example, the neuroimmunodulatory effects of a compound can be
evaluated in U373, a human astroglioma cell line in which the nuclear
translocation of
specific regulatory proteins involved in cytokine/chemokine expression can be
quantified (Le Roy E et al., J Virol. 1999, 73: 6582-9; Jin Y et al., J Infect
Dis. 1998,
177: 1629-1638; Acevedo-Duncan M et al., Cell Growth Differ. 1995, 6: 1353-
1365).
A series of assays has been performed, and have indicated that the addition of
a
culture medium containing INSP168 stiinulates Stat-2 nuclear translocation in
U373
cells. This first series of experiments thus revealed that INSP168 has the
capacity to
stimulate intracellular signalling. by inducing Stat-2 nuclear translocation
in U373
cells. Activation of Stat proteins signaling is known to be associated with
immunomodulation and eventually cell proliferation (Pfitzner E et al., Curr
Pharm
Des. 2004, 10: 2839-2850).
A polypeptide according to the first aspect of the invention may thus function
as an
activator of cell proliferation, as a neuromodulator (neuroimmunomodulator),
as a
modulator of the inflammatory response in the CNS, as a regulator of astrocyte
proliferation or as a regulator of axon projection, neuronal migration or
leukocyte
recruitment or migration.
Preferably, the activity of a polypeptide of the present invention can be
confirmed in
at least one of the following assays:
a) in the maintenance of neuronal cell survival, for example in the
regeneration
of injured adult neurons, or
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
b) in the modulation of neurite growth in animal models of spinal cord injury
(Fouad et al., Brain.Res.Rev. 2001, Vol. 36, pp.204-212; Bareyre et al.,
J.Neurosci. 2002, Vol.22, pp.7097-7110; GrandPre et al. Nature 2002,
Vol.417, pp.547-551; Li and Strittmatter, J.Neurosci. 2003, Vol.23, pp.4219-
5 4227; Liebscher et al. Ann.Neurol. 2005, Vol.58, pp.706-719), for example
in the modulation of myelin inhibition of neurite outgrowth, and other CNS
lesions such as cortical lesions or cerebral ischemia induced by middle
cerebral artery occlusion (Yu Hsuan Teng and Luen Tang, Journal of
Neurochemistry 2005, Vol.94, pp.865-874; Papadopoulos et al. Ann.Neurol.
10 2002, Vol.51, pp.433-441; Emerick et al. J.Neurosci. 2003, Vol.23, pp.4826-
4830), or
c) in the modulation of axonal growth of many neuron types, for example of
corticospinal tract (CST) axons, corticofugal, retinal, superior cervical
ganglion, spinal or hippocampal neurons, dorsal column axons, for example,
15 in the modulation of axonal plasticity of unlesioned cortical neurons (with
enhanced behavioural recovery), or
d) in the translocation of Stat2 as described in Example 6, or
e) in the up-regulation of growth factors or growth-related proteins, for
example
of Brain-derived neurotrophic factor (BDNF), Vascular Endothelial Growth
Factor (VEGF) and/or Growth-associated protein 43 (GAP-43), or
f) in the regeneration of nerve fibers, for example of raphespinal,
rubrospinal or
corticospinal fibers, or in the regeneration of injured optic nerve fibers
(for
example in an optic nerve crush model), or
g) in the improvement of locomotor function, or
h) in the regulation of secretases, for example of P-secretases, or
i) in the modulation of apoptosis, for example by modulating pro-apoptotic
proteins of the Bcl-2 family and/or mitochondrial proteins, or
j) in the modulation of Rho and/or Rho-associated kinase (ROCK), or
k) in the modulation of (3-secretases, for example of BACE1 activity, and/or
amyloid-(3 peptide generation and/or formation of amyloid plaques, or
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
16
1) in the regulation of ocular dominance (OD) plasticity, or
m) in the modulation of the phosphorylation of the epidermal growth factor
receptor (EGFR), or
n) in the stabilization of neural circuitry, or
o) in the modulation of dendritic plasticity.
As regards the Nogo receptor (NgR), the myelin inhibitory proteins Nogo,
myelin-
associated glycoprotein (MAG) and oligodendrocyte myelin glycoprotein (Omgp)
all
bind to the extracellular leucine-rich repeat (LRR) domain of NgR. Thus,
polypeptides of the invention comprising and/or consisting of the LRR domain
can at
least display activity in one of the above-mentioned assays.
Polypeptides of the invention consisting of and/or comprising of the mature
(lacking a
signal peptide) forins and/or cleaved forms of INSP168, INSP168-SV1 and/or
mature
soluble forms of INSP149 and/or INSP169, and/or agonists thereof are
preferably
used in the above-mentioned assays. Preferably, the soluble forms of INSP149
and/or
INSP169 consist of the mature extracellular part aiid/or cleaved fragments of
INSP 149 and/or INSP 169. Alternatively, antagonists of ineinbrane bound INSP
149
and/or INSP169, for example antibodies, can be used in the above-mentioned
assays.
Preferred epitopes of the polypeptides of the present invention can be
detected by
"affinity fingerprinting" as described in Schimmele and Pli.ickthun (Journal
of
Molecular Biology 2005, Vol.352, Issue 1, pp.229-241).
Polypeptides of the present invention may undergo cleavage by
metalloendopeptidase
and/or proprotein convertases such as zinc metalloproteinases, N-Arginine
dibasic
(NDR) convertase or subtilisin-like proprotein convertases. NDR cleavage sites
and
PCSK cleavage sites have been detected in the polypeptides of the present
invention.
The skilled artisan will appreciate that such resulting cleaved fraginents of
the
polypeptides of the present invention can be used for the diagnosis and/or
treatment of
diseases. Cleavage of membrane-bound INSP 149 and/or INSP 169 can yield
soluble
N- and C- terminal fragments useful on their own or as components of fusion
proteins
such as Fc fusion.
A NDR cleavage site has been detected in the full length polypeptides of the
present
invention at position 70-72 (RRI), located after the first LRR motif.
Surprisingly,
PCSK cleavage sites have been detected at position 207-209 (KRT) of full
length
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
17
INSP168-SV1 and at positions 439-441 (KRS) and 449-451 (KRN) of full length
membrane bound INSP169. Interestingly, the PCSK cleavage site in INSP168-SV1
is
located just after the LRR motifs and for INSP169 between the LRR motifs and
the
fibronectin domain. The polypeptides of the present invention also encompass
the
resulting cleaved N-fragments and/or C-fragments or mature forms thereof.
Preferably, the resulting cleaved fragments are soluble fragments. Preferably,
the
resulting fragments consist of:
(i) the first 208 amino acids of INSP168-SV1 or the mature form thereof, or
(ii) the first 440 amino acids of INSP 169 or the mature form thereof, or
(iii) the first 450 amino acids of INSP 169 or the mature form thereof.
This aspect of the invention also includes fusion proteins that incorporate
the
polypeptides of the first aspect of the invention.
An "antigenic determinant" of the present invention may be a part of a
polypeptide of
the present invention, which binds to an antibody-combining site or to a T-
cell
receptor (TCR). Alternatively, an "antigenic determinant" may be a site on the
surface
of a polypeptide of the present invention to which a single antibody molecule
binds.
Generally an antigen has several or many different antigenic determinants and
reacts
with antibodies of many different specificities. Preferably, the antibody is
immunospecific to a polypeptide of the invention. Preferably, the antibody is
immunospecific to a polypeptide of the invention, which is not part of a
fusion
protein. Preferably, the antibody is immunospecific to INSP168, INSP168-SV1,
INSP 149 or INSP 169 or a fragment thereof. Antigenic determinants usually
consist of
chemically active surface groupings of molecules, such as amino acids or sugar
side
chains, and can have specific three dimensional structural characteristics, as
well as
specific charge characteristics. Preferably, the "antigenic determinant"
refers to a
particular chemical group on a polypeptide of the present invention that is
antigenic,
i.e. that elicit a specific immune response.
The polypeptides AA104038 (SEQ ID NO:68), XP_853150 (SEQ ID NO:69) and
ENSCAFP00000016927 (SEQ ID NO:70), and their encoding nucleic acid sequences
are specifically excluded from the scope of this invention. .-
In a second aspect, the invention provides a purified nucleic acid molecule
which
encodes a polypeptide of the first aspect of the invention.
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
18
The term "purified nucleic acid molecule" preferably refers to a nucleic acid
molecule
of the invention that (1) has been separated from at least about 50 percent of
proteins,
lipids, carbohydrates, or other materials with which it is naturally found
when total
nucleic acid is isolated from the source cells, (2) is not linked to all or a
portion of a
polynucleotide to which the "purified nucleic acid molecule" is linked in
nature, (3) is
operably linked to a polynucleotide which it is not linked to in nature, or
(4) does not
occur in nature as part of a larger polynucleotide sequence. Preferably, the
isolated
nucleic acid molecule of the present invention is substantially free from any
other
contaminating nucleic acid molecule(s) or other contaminants that are found in
its
natural environment that would interfere with its use in polypeptide
production or its
therapeutic, diagnostic, prophylactic or research use. In a preferred
embodiment,
genomic DNA are specifically excluded from the scope of the invention.
Preferably,
genomic DNA larger than 10 kbp (kilo base pairs), 50 kbp, 100 kbp, 150 kbp,
200
kbp, 250 kbp or 300 kbp are specifically excluded from the scope of the
invention.
Preferably, the "purified nucleic acid molecule" consists of cDNA only.
Preferably, the purified nucleic acid molecule comprises or consists of the
nucleic
acid sequence as recited in the nucleic acid sequence as recited in SEQ ID NO:
1, SEQ
ID NO:3, SEQ ID NO:5, SEQ ID NO:7, SEQ ID NO: 9, SEQ ID NO:l l, SEQ ID
NO:13, SEQ ID NO:15, SEQ ID NO:17, SEQ ID NO:19, SEQ ID NO:21, SEQ ID
NO:23, SEQ ID NO:25, SEQ ID NO:27, SEQ ID NO:29, SEQ ID NO:31, SEQ ID
NO:33, SEQ ID NO:35, SEQ ID NO:37, SEQ ID NO:39, SEQ ID NO:41 , SEQ ID
NO: 43 and/or SEQ ID NO:66, or is a redundant equivalent or fragment of those
sequences.
Preferably, the purified nucleic acid molecule consists of the nucleic acid
sequence as
recited in SEQ ID NO:1, SEQ ID NO:3, SEQ ID NO:5, SEQ ID NO:7, SEQ ID NO:9,
SEQ ID NO:11, SEQ ID NO:13, SEQ ID NO:15, SEQ ID NO:17, SEQ ID NO:19,
SEQ ID NO:21, SEQ ID NO:23, SEQ ID NO:25, SEQ ID NO:27, SEQ ID NO:29,
SEQ ID NO:31, SEQ ID NO:33, SEQ ID NO:35, SEQ ID NO:37, SEQ ID NO:39,
SEQ ID NO:41, SEQ ID NO: 43 and/or SEQ ID NO:66, or is a redundant equivalent
or fragment of those sequences.
In a third aspect, the invention provides a purified nucleic acid molecule
which
hybridizes under high stringency conditions with a nucleic acid molecule of
the
second aspect of the invention. High stringency hybridisation conditions are
defined
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
19
as overnight incubation at 42 C in a solution comprising 50% formamide, 5XSSC
(150mM NaC1, 15mM trisodium citrate), 50mM sodium phosphate (pH7.6), 5x
Denhardts solution, 10% dextran sulphate, and 20 microgram/ml denatured,
sheared
salmon sperm DNA, followed by washing the filters in 0.1X SSC at approximately
65 C.
In a fourth aspect, the invention provides a vector, such as an expression
vector, that
contains a nucleic acid molecule of the second or third aspect of the
invention.
In a fifth aspect, the invention provides a host cell transformed with a
vector of the
fourth aspect of the invention.
In a sixth aspect, the invention provides a ligand which binds specifically
to, and
which preferably inhibits the activity of, a leucine-rich repeat motif
containing
polypeptide of the first aspect of the invention.
In a seventh aspect, the invention provides a compound that is effective to
alter the
expression of a natural gene which encodes a polypeptide of the first aspect
of the
invention or to regulate the activity of a polypeptide of the first aspect of
the
invention.
Such compounds may be identified using the assays and screening methods
disclosed
herein.
A compound of the seventh aspect of the invention may either increase
(agonise) or
decrease (antagonise) the level of expression of the gene or the activity of
the
polypeptide.
Importantly, the identification of the domain organisation and function of the
INSP168, INSP168-SV1, INSP149 and INSP169 polypeptides allows for the design
of screening methods capable of identifying coinpounds that are effective in
the
treatment and/or diagnosis of disease. Ligands and compounds according to the
sixth
and seventh aspects of the invention may be identified using such methods.
These
methods are included as aspects of the present invention. Using these methods,
it will
now be possible to identify inhibitors or antagonists of INSP168, INSP168-SV1,
INSP149 and INSP169, such as, for example, monoclonal antibodies, which may be
of use in modulating INSP168, INSP168-SV1, INSP149 and INSP169 activity in
vivo
in clinical applications. Such compounds are likely to be useful in
counteracting the
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
biological activity of the INSP 168, 1NSP 168-SV l, INSP 149 and INSP 169
polypeptides.
Another aspect of this invention resides in the use of an INSP168, INSP168-
SV1,
INSP149 or INSP169 gene or polypeptide as a target for the screening of
candidate
5 drug modulators, particularly candidate drugs active against leucine-rich
repeat (LRR)
motif containing protein related disorders.
A further aspect of this invention resides in methods of screening of
compounds for
therapy of leucine-rich repeat (LRR) motif containing protein related
disorders,
comprising determining the ability of a compound to bind to an INSP168,
INSP168-
10 SV1, INSPI49 or INSP169 gene or polypeptide, or a fragment thereof.
A further aspect of this invention resides in methods of screening of
compounds for
therapy of leucine-rich repeat (LRR) motif containing protein related
disorders,
comprising testing for modulation of the activity of an INSP168, INSP168-SV1,
INSP 149 or INSP 169 gene or polypeptide, or a fragment thereof.
15 In an eighth aspect, the invention provides a polypeptide of the first
aspect of the
invention, or a nucleic acid molecule of the second or third aspect of the
invention, or
a vector of the fourth aspect of the invention, or a host cell of the fifth
aspect of the
invention, or a ligand of the sixth aspect of the invention, or a compound of
the
seventh aspect of the invention, for use in therapy or diagnosis of diseases
in which
20 leucine-rich repeat motif containing proteins are implicated. Such diseases
include,
but are not limited to, diseases of the retina, retinal pigment epithelium
(RPE), and
choroids; ocular neovascularization, ocular inflammation and retinal
degenerations;
diabetic retinopathy, chronic glaucoma, retinal detachment, sickle cell
retinopathy,
senile macular degeneration, retinal neovascularization, subretinal
neovascularization;
rubeosis iritis inflammatory diseases, chronic posterior and pan uveitis,
neoplasms,
retinoblastoma, pseudoglioma, neovascular glaucoma; neovascularization
resulting
following a combined vitrectomy and lensectomy, vascular diseases retinal
ischemia,
choroidal vascular insufficiency, choroidal thrombosis, neovascularization of
the optic
nerve, diabetic macular edema, cystoid macular edema, retinitis piginentosa,
retinal
vein occlusion, proliferative vitreoretinopathy, angioid streak, retinal
artery occlusion,
neovascularization due to penetration of the eye or ocular injury,
neuropathies;
Leber's, idiopathic, drug-induced, optic, and ischemic neropathies; spinal
cord
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
21
injuries, paraplegia, neurodegenerative disorders, disorders of the central
nervous
system, disorders of the peripheral nervous system, brain injuries,
cerebrovascular
diseases, Parkinson's disease, corticobasal degeneration, motor neuron
disease,
amyotrophic lateral sclerosis (ALS), multiple sclerosis, traumatic brain
injury, stroke,
post-stroke, post- traumatic brain injury, small-vessel cerebrovascular
disease,
dementias, Alzheimer's disease, vascular dementia, dementia with Lewy bodies,
frontotemporal dementia, Parkinsonism, frontotemporal dementias, Pick's
disease,
progressive nuclear palsy, corticobasal degeneration, Huntington's disease,
thalamic
degeneration, Creutzfeld-Jakob dementia, HIV dementia, schizophrenia with
dementia, Korsakoffs psychosis, stroke and trauma.
The moieties of the first, second, third, fourth, fifth, sixth or seventh
aspect of the
invention may also be used in the manufacture of a medicament for the
treatment of
such diseases.
In a ninth aspect, the invention provides a method of diagnosing a disease in
a patient,
comprising assessing the level of expression of a natural gene encoding a
polypeptide
of the first aspect of the invention or the activity of a polypeptide of the
first aspect of
the invention in tissue from said patient and comparing said level of
expression or
activity to a control level, wherein a level that is different to said control
level is
indicative of disease. Such a method will preferably be carried out in vitro.
Similar
methods may be used for monitoring the therapeutic treatment of disease in a
patient,
wherein altering the level of expression or activity of a polypeptide or
nucleic acid
molecule over the period of time towards a control level is indicative of
regression of
disease.
A preferred method for detecting polypeptides of the first aspect of the
invention
comprises the steps of: (a) contacting a ligand, such as an antibody, of the
sixth aspect
of the invention with a biological sample under conditions suitable for the
formation
of a ligand-polypeptide complex; and (b) detecting said complex.
A number of different such methods according to the ninth aspect of the
invention
exist, as the skilled reader will be aware, such as metliods of nucleic acid
hybridization with short probes, point mutation analysis, polymerase chain
reaction
(PCR) amplification and methods using antibodies to detect aberrant protein
levels.
Similar methods may be used on a short or long term basis to allow therapeutic
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
22
treatment of a disease to be monitored in a patient. The invention also
provides kits
that are useful in these methods for diagnosing disease.
Preferably, the disease diagnosed by a method of the ninth aspect of the
invention is a
disease in which leucine-rich repeat motif containing polypeptides are
implicated, as
described above.
In a tenth aspect, the invention provides for the use of the polypeptide of
the first
aspect of the invention as an activator of cell proliferation, as a
neuromodulator
(neuroimmunomodulator), as a modulator of the inflammatory response in the
CNS,
as a regulator of astrocyte proliferation or as a regulator of axon
projection, neuronal
migration or leukocyte recruitment or migration. INSP168 and INSP168-SV1, or
truncated fonns of INSP 149 and INSP 169 (for example, lacking the
transmembrane
region) could be used as recombinant soluble antagonists of the endogenous
activity
of INSP149 and INSP169. Another suitable use of the INSP168 polypeptides is
use in
the screening of drug compounds that are effective against the diseases and
conditions
in which the INSP168 polypeptides are implicated.
In an eleventh aspect, the invention provides a pharmaceutical coinposition
comprising a polypeptide of the first aspect of the invention, or a nucleic
acid
molecule of the second or third aspect of the invention, or a vector of the
fourth aspect
of the invention, or a host cell of the fifth aspect of the invention, or a
ligand of the
sixth aspect of the invention, or a compound of the seventh aspect of the
invention, in
conjunction with a pharmaceutically-acceptable carrier.
In a twelfth aspect, the present invention provides a polypeptide of the first
aspect of
the invention, or a nucleic acid molecule of the second or third aspect of the
invention, or a vector of the fourth aspect of the invention, or a host cell
of the fifth
aspect of the invention, or a ligand of the sixth aspect of the invention, or
a compound
of the seventh aspect of the invention, for use in the manufacture of a
medicament for
the diagnosis or treatment of a disease in which leucine-rich repeat motif
containing
polypeptides are implicated. Such diseases include those described above in
connection with the eighth aspect of the invention.
In a thirteenth aspect, the invention provides a method of treating a disease
in a
patient comprising administering to the patient a polypeptide of the first
aspect of the
invention, or a nucleic acid molecule of the second or third aspect of the
invention, or
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
23
a vector of the fourth aspect of the invention, or a host cell of the fifth
aspect of the
invention, or a ligand of the sixth aspect of the invention, or a compound of
the
seventh aspect of the invention.
For diseases in which the expression of a natural gene encoding a polypeptide
of the
first aspect of the invention, or in which the activity of a polypeptide of
the first
aspect of the invention, is lower in a diseased patient when compared to the
level of
expression or activity in a healthy patient, the polypeptide, nucleic acid
molecule,
vector, host cell, ligand or compound administered to the patient should be an
agonist.
Conversely, for diseases in which the expression of the natural gene or
activity of the
polypeptide is higher in a diseased patient when compared to the level of
expression
or activity in a healthy patient, the polypeptide, nucleic acid molecule,
vector, host
cell, ligand or compound administered to the patient should be an antagonist.
Examples of such antagonists include antisense nucleic acid molecules,
ribozymes
and ligands, such as antibodies.
In a fourteenth aspect, the invention provides transgenic or knockout non-
human
animals that have been transformed to express higher, lower or absent levels
of a
polypeptide of the first aspect of the invention. Such transgenic animals are
very
useful models for the study of disease and may also be used in screening
regimes for
the identification of compounds that are effective in the treatment or
diagnosis of such
a disease.
As used herein, "functional equivalent" refers to a protein or nucleic acid
molecule
that possesses functional or structural characteristics that are substantially
similar to a
polypeptide or nucleic acid molecule of the present invention. A functional
equivalent
of a protein may contain modifications depending on the necessity of such
modifications for the perforinance of a specific function. The term
"functional
equivalent" is intended to include the fragments, mutants, hybrids, variants,
analogs,
or chemical derivatives of a molecule.
Preferably, the "functional equivalent" may be a protein or nucleic acid
molecule that
exhibits any one or more of the functional activities of the polypeptides of
the present
invention.
Preferably, the "functional equivalent" may be a protein or nucleic acid
molecule that
displays substantially similar activity compared with INSP168, INSP168-SV1,
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
24
INSP149 or INSP169 or fragments thereof in a suitable assay for the
measurement of
biological activity or function. Preferably, the "functional equivalent" may
be a
protein or nucleic acid molecule that displays identical or higher activity
compared
with INSP168, INSP168-SV1, INSP149 or INSP169 or fragments thereof in a
suitable assay for the measurement of biological activity or function.
Preferably, the
"functional equivalent" may be a protein or nucleic acid molecule that
displays 50%,
60%, 70%, 80%, 90%, 95%, 98%, 99%, 100% or more activity compared with
INSP168, INSP168-SV1, INSP149 or INSP169 or fragments thereof in a suitable
assay for the measurement of biological activity or function.
Preferably, the "functional equivalent" may be a protein or polypeptide
capable of
exhibiting a substantially similar in vivo or in vitro activity as the
polypeptides of the
invention. Preferably, the "functional equivalent" may be a protein or
polypeptide
capable of interacting with other cellular or extracellular molecules in a
manner
substantially similar to the way in which the corresponding portion of the
polypeptides of the invention would. For example, a "functional equivalent"
would be
able, in an immunoassay, to diminish the binding of an antibody to the
corresponding
peptide (i.e., the peptide the amino acid sequence of which was modified to
achieve
the "functional equivalent") of the polypeptide of the invention, or to the
polypeptide
of the invention itself, where the antibody was raised against the
corresponding
peptide of the polypeptide of the invention. An equimolar concentration of the
functional equivalent will diminish the aforesaid binding of the corresponding
peptide
by at least about 5%, preferably between about 5% and 10%, more preferably
between about 10% and 25%, even more preferably between about 25% and 50%, and
most preferably between about 40% and 50%.
For example, functional equivalents can be fully functional or can lack
function in one
or more activities. Thus, in the present invention, variations can affect the
function,
for example, of the activities of the polypeptide that reflect its possession
of a leucine-
rich repeat (LRR) motif.
A summary of standard techniques and procedures which may be employed in order
to utilise the invention is given below. It will be understood that this
invention is not
limited to the particular methodology, protocols, cell lines, vectors and
reagents
described. It is also to be understood that the terminology used herein is for
the
purpose of describing particular embodiments only and it is not intended that
this
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
terminology should limit the scope of the present invention. The extent of the
invention is limited only by the terms of the appended claims.
Standard abbreviations for nucleotides and amino acids are used in this
specification.
The practice of the present invention will employ, unless otherwise indicated,
5 conventional techniques of molecular biology, microbiology, recombinant DNA
technology and immunology, which are within the skill of those working in the
art.
Such techniques are explained fully in the literature. Examples of
particularly suitable
texts for consultation include the following: Sambrook Molecular Cloning; A
Laboratory Manual, Second Edition (1989); DNA Cloning, Volumes I and II (D.N
10 Glover ed. 1985); Oligonucleotide Synthesis (M.J. Gait ed. 1984); Nucleic
Acid
Hybridization (B.D. Hames & S.J. Higgins eds. 1984); Transcription and
Translation
(B.D. Haines & S.J. Higgins eds. 1984); Animal Cell Culture (R.I. Freshney ed.
1986); Immobilized Cells and Enzymes (IRL Press, 1986); B. Perbal, A Practical
Guide to Molecular Cloning (1984); the Methods in Enzymology series (Academic
15 Press, Inc.), especially volumes 154 & 155; Gene Transfer Vectors for
Mammalian
Cells (J.H. Miller and M.P. Calos eds. 1987, Cold Spring Harbor Laboratory);
Immunochemical Methods in Cell and Molecular Biology (Mayer and Walker, eds.
1987, Academic Press, London); Scopes, (1987) Protein Purification: Principles
and
Practice, Second Edition (Springer Verlag, N.Y.); and Handbook of Experimental
20 Immunology, Volumes I-IV (D.M. Weir and C. C. Blackwell eds. 1986).
As used herein, the term "polypeptide" includes any peptide or protein
comprising
two or more amino acids joined to each other by peptide bonds or modified
peptide
bonds, i.e. peptide isosteres. This term refers both to short chains (peptides
and
oligopeptides) and to longer chains (proteins).
25 The polypeptide of the present invention may be in the form of a mature
protein or
may be a pre-, pro- or prepro- protein that can be activated by cleavage of
the pre-,
pro- or prepro- portion to produce an active mature polypeptide. In such
polypeptides,
the pre-, pro- or prepro- sequence may be a leader or secretory sequence or
may be a
sequence that is employed for purification of the mature polypeptide sequence.
As noted above, the polypeptide of the first aspect of the invention may form
part of a
fusion protein. For example, it is often advantageous to include one or more
additional amino acid sequences which may contain secretory or leader
sequences,
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
26
pro-sequences, sequences which aid in purification, or sequences that confer
higher
protein stability, for example during recombinant production. Alternatively or
additionally, the mature polypeptide may be fused with another compound, such
as a
compound to increase the half-life of the polypeptide (for exainple,
polyethylene
glycol).
Polypeptides of the invention are useful on their own, as components of fusion
proteins such as Fc fusion, and/or in combination with another agent.
Preferably, the
Fc fusion comprises the mature form of INSP168, the mature form of INSP168-SVl
or the mature form of the extracellular part of INSP 169 or INSP 149.
Preferably the agent is selected among interferon-beta, soluble NgR (e.g. Nogo-
66),
antibodies targeted to NgR, antibodies targeted to myelin inhibitors (e.g.
Nogo, MAG
or Omgp), CXCL10, agonists of serotonin receptors (e.g. 5-HT1A/2A/7), LIF,
EGFR
blockers such as Erlotinib, and/or methylprednisolone.
In a further preferred embodiment, a polypeptide of the invention, that may
comprise
a sequence having at least 85% of homology with INSP168, INSP168-SV1, INSP149
or INSP 169, is a fusion protein.
These fusion proteins can be obtained by cloning a polynucleotide encoding a
polypeptide comprising a sequence having at least 85% of homology with
INSP168,
INSP168-SVl, INSP149 or INSP169 in frame to the coding sequences for a
heterologous protein sequence.
The term "heterologous", when used herein, is intended to designate any
polypeptide
other than a human INSP168, INSP168-SV1, INSP149 or INSP169 polypeptide.
Examples of heterologous sequences, that can be comprised in the fusion
proteins
either at the N- or C-terminus, include: extracellular domains of membrane-
bound
protein, immunoglobulin constant regions (Fc regions), multimerization
domains,
domains of extracellular proteins, signal sequences, export sequences, and
sequences
allowing purification by affinity chromatography.
Many of these heterologous sequences are commercially available in expression
plasmids since these sequences are commonly included in fusion proteins in
order to
provide additional properties without significantly impairing the specific
biological
activity of the protein fused to them (Terpe K, 2003, Appl Microbiol
Biotechnol,
60:523-33). Examples of such additional properties are a longer lasting half-
life in
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
27
body fluids, the extracellular localization, or an easier purification
procedure as
allowed by the a stretch of Histidines forming the so-called "histidine tag"
(Gentz et
al. 1989, Proc Natl Acad Sci USA, 86:821-4) or by the "HA" tag, an epitope
derived
from the influenza hemagglutinin protein (Wilson et al. 1994, Cell, 37:767-
78). If
needed, the heterologous sequence can be eliminated by a proteolytic cleavage,
for
example by inserting a proteolytic cleavage site between the protein and the
heterologous sequence, and exposing the purified fusion protein to the
appropriate
protease. These features are of particular importance for the fusion proteins
since they
facilitate their production and use in the preparation of pharmaceutical
compositions.
For example, the INSP168, INSP168-SV1, INSP149 or INSP169 polypeptide may be
purified by means of a hexa-histidine peptide fused at the C-terminus of
INSP168,
INSP168-SV1, INSP149 or INSP169. When the fusion protein comprises an
immunoglobulin region, the fusion may be direct, or via a short linker peptide
wlzich
can be as short as 1 to 3 amino acid residues in lengtli or longer, for
example, 13
amino acid residues in length. Said linker may be a tripeptide of the sequence
E-F-M
(Glu-Phe-Met), for example, or a 13-amino acid linker sequence comprising Glu-
Phe-
Gly-Ala-Gly-Leu-Val-Leu-Gly-Gly-Gln-Phe-Met (SEQ ID NO:71) introduced
between the sequence of the substances 'of the invention and the
immunoglobulin
sequence. The resulting fusion protein has improved properties, such as an
extended
residence time in body fluids (i.e. an increased half-life), increased
specific activity,
increased expression level, or the purification of the fusion protein is
facilitated.
In a preferred embodiment, the protein is fused to the constant region of an
Ig
molecule. Preferably, it is fused to heavy chain regions, like the CH2 and CH3
domains of liuman IgG1, for example. Other isoforms of Ig molecules are also
suitable for the generation of fusion proteins according to the present
invention, such
as isofonns IgG2 or IgG4, or other Ig classes, like IgM or IgA, for example.
Fusion
proteins may be monomeric or multimeric, hetero- or homomultimeric.
In a further preferred embodiment, the functional derivative comprises at
least one
moiety attached to one or more functional groups, which occur as one or more
side
chains on the amino acid residues. Preferably, the moiety is a polyethylene
(PEG)
moiety. PEGylation may be carried out by known methods, such as the ones
described
in W099/55377, for example.
Polypeptides may contain amino acids other than the 20 gene-encoded amino
acids,
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
28
modified either by natural processes, such as by post-translational processing
or by
chemical modification techniques which are well known in the art. Among the
known
modifications which may commonly be present in polypeptides of the present
invention are glycosylation, lipid attachment, sulphation, gamma-
carboxylation, for
instance of glutamic acid residues, hydroxylation and ADP-ribosylation. Other
potential modifications include acetylation, acylation, amidation, covalent
attachment
of flavin, covalent attachment of a haeme moiety, covalent attachment of a
nucleotide
or nucleotide derivative, covalent attachment of a lipid derivative, covalent
attachment of phosphatidylinositol, cross-linking, cyclization, disulphide
bond
formation, demethylation, formation of covalent cross-links, formation of
cysteine,
formation of pyroglutamate, formylation, GPI anchor formation, iodination,
methylation, myristoylation, oxidation, proteolytic processing,
phosphorylation,
prenylation, racemization, selenoylation, transfer-RNA mediated addition of
amino
acids to proteins such as arginylation, and ubiquitination.
Modifications can occur anywhere in a polypeptide, including the peptide
backbone,
the ainino acid side-chains and the amino or carboxyl termini. In fact,
blockage of the
amino or carboxyl terminus in a polypeptide, or both, by a covalent
modification is
common in naturally-occurring and synthetic polypeptides and such
modifications
may be present in polypeptides of the present invention.
The modifications that occur in a polypeptide often will be a function of how
the
polypeptide is made. For polypeptides that are made recombinantly, the nature
and
extent of the modifications in large part will be determined by the post-
translational
modification capacity of the particular host cell and the modification signals
that are
present in the amino acid sequence of the polypeptide in question. For
instance,
glycosylation patterns vary between different types of host cell.
The polypeptides of the present invention can be prepared in any suitable
manner.
Such polypeptides include isolated naturally-occurring polypeptides (for
example
purified from cell culture), recombinantly-produced polypeptides (including
fusion
proteins), synthetically-produced polypeptides or polypeptides that are
produced by a
combination of these methods.
The functionally-equivalent polypeptides of the first aspect of the invention
may be
polypeptides that are homologous to the INSP 168 polypeptides. Two
polypeptides are
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
29
said to be "homologous", as the term is used herein, if the sequence of one of
the
polypeptides has a high enough degree of identity or similarity to the
sequence of the
other polypeptide. "Identity" indicates that at any particular position in the
aligned
sequences, the amino acid residue is identical between the sequences.
"Similarity"
indicates that, at any particular position in the aligned sequences, the amino
acid
residue is of a similar type between the sequences. Degrees of identity and
similarity
can be readily calculated (Computational Molecular Biology, Lesk, A.M., ed.,
Oxford
University Press, New York, 1988; Biocomputing. Informatics and Genome
Projects,
Smith, D.W., ed., Academic Press, New York, 1993; Computer Analysis of
Sequence
Data, Part 1, Griffin, A.M., and Griffin, H.G., eds., Humana Press, New
Jersey, 1994;
Sequence Analysis in Molecular Biology, von Heinje, G., Academic Press, 1987;
and
Sequence Analysis Primer, Gribskov, M. and Devereux, J., eds., M Stockton
Press,
New York, 1991). Preferably, percentage identity, as referred to herein, is as
determined using BLAST version 2.1.3 using the default parameters specified by
the
NCBI (the National Center for Biotechnology Information;
http://www.ncbi.nlm.nih.gov/) [Blosum 62 matrix; gap open penalty=ll and gap
extension penalty=l ].
Homologous polypeptides therefore include natural biological variants (for
example,
allelic variants or geographical variations within the species from which the
polypeptides are derived) and mutants (such as mutants containing amino acid
substitutions, insertions or deletions) of the INSP168 polypeptides. Such
mutants may
include polypeptides in which one or more of the amino acid residues are
substituted
with a conserved or non-conserved amino acid residue (preferably a conserved
amino
acid residue) and such substituted amino acid residue may or may not be one
encoded
by the genetic code. Typical such substitutions are among Ala, Val, Leu and
Ile;
among Ser and Thr; among the acidic residues Asp and Glu; among Asn and Gln;
among the basic residues Lys and Arg; or among the aromatic residues Phe and
Tyr.
Particularly preferred are variants in which several, i.e. between 5 and 10, 1
and 5, 1
and 3, 1 and 2 or just 1 amino acids are substituted, deleted or added in any
combination. Especially preferred are silent substitutions, additions and
deletions,
which do not alter the properties and activities of the protein. Also
especially
preferred in this regard are conservative substitutions. Such mutants also
include
polypeptides in which one or more of the amino acid residues includes a
substituent
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
group.
In accordance with the present invention, any substitution should be
preferably a
"conservative" or "safe" substitution, which is commonly defined a
substitution
introducing an amino acids having sufficiently similar chemical properties
(e.g. a
5 basic, positively charged amino acid should be replaced by another basic,
positively
charged amino acid), in order to preserve the structure and the biological
function of
the molecule.
The literature provide many models on which the selection of conservative
ainino
acids substitutions can be performed on the basis of statistical and physico-
chemical
10 studies on the sequence and/or the structure of proteins (Rogov SI and
Nekrasov AN,
2001). Protein design experiments have shown that the use of specific subsets
of
amino acids can produce foldable and active proteins, helping in the
classification of
ainino acid "synonyinous" substitutions which can be more easily accommodated
in
protein structure, and which can be used to detect functional and structural
homologs
15 and paralogs (Murphy LR et al., 2000). The groups of synonymous ainino
acids and
the groups of more preferred synonymous amino acids are shown in Table 1.
Specific, non-conservative mutations can be also introduced in the
polypeptides of the
invention with different purposes. Mutations reducing the affinity of the
protein may
increase its ability to be reused and recycled, potentially increasing its
therapeutic
20 potency (Robinson CR, 2002). Immunogenic epitopes eventually present in the
polypeptides of the invention can be exploited for developing vaccines
(Stevanovic S,
2002), or eliminated by modifying their sequence following known methods for
selecting mutations for increasing protein stability, and correcting them (van
den Burg
B and Eijsink V, 2002; WO 02/05146, WO 00/34317, WO 98/52976).
25 Preferred alternative, synonymous groups for amino acids derivatives
included in
peptide mimetics are those defined in Table 2. A non-exhaustive list of ainino
acid
derivatives also include aminoisobutyric acid (Aib), hydroxyproline (Hyp),
1,2,3,4-
tetrahydro-isoquinoline-3-COOH, indoline-2carboxylic acid, 4-difluoro-proline,
L-
thiazolidine-4-carboxylic acid, L-homoproline, 3,4-dehydro-proline, 3,4-
dihydroxy-
30 phenylalanine, cyclohexyl-glycine, and phenylglycine.
By "amino acid derivative" is intended an amino acid or amino acid-like
chemical
entity other than one of the 20 genetically encoded naturally occurring amino
acids. In
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
31
particular, the amino acid derivative may contain substituted or non-
substituted,
linear, branched, or cyclic alkyl moieties, and may include one or more
heteroatoms.
The amino acid derivatives can be made de novo or obtained from commercial
sources (Calbiochem-Novabiochem AG, Switzerland; Bachem, USA).
Various methodologies for incorporating unnatural amino acids derivatives into
proteins, using both in vitro and in vivo translation systems, to probe and/or
improve
protein structure and function are disclosed in the literature (Dougherty DA,
2000).
Techniques for the synthesis and the development of peptide mimetics, as well
as
non-peptide mimetics, are also well known in the art (Golebiowski A et al.,
2001;
Hruby VJ and Balse PM, 2000; Sawyer TK, in "Structure Based Drug Design",
edited
by Veerapandian P, Marcel Dekker Inc., pg. 557-663, 1997).
Such mutants also include polypeptides in which one or more of the amino acid
residues includes a substituent group.
Typically, greater than 80% identity between two polypeptides is considered to
be an
indication of functional equivalence. Preferably, functionally equivalent
polypeptides
of the first aspect of the invention have a degree of sequence identity with
the
INSP 168 polypeptides, or with active fragments thereof, of greater than 80%.
More
preferred polypeptides have degrees of identity of greater than 90%, 95%, 98%
or
99%, respectively.
The functionally-equivalent polypeptides of the first aspect of the invention
may also
be polypeptides which have been identified using one or more techniques of
structural
alignment. For example, the Inpharmatica Genome Threader technology that forms
one aspect of the search tools used to generate the Biopendium search database
may
be used (see PCT application published as WO 01/69507) to identify
polypeptides of
presently-unknown function which, while having low sequence identity as
compared
to the INSP168 polypeptides, are predicted to be cell surface recognition
molecules
by virtue of sharing significant structural homology with the INSP168
polypeptide
sequences. By "significant structural homology" is meant that the Inpharmatica
Genome Threader predicts two proteins to share structural homology with a
certainty
of 10% and above.
The polypeptides of the first aspect of the invention also include fragments
of the
INSP168 polypeptides and fragments of the functional equivalents of these
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
32
polypeptides, provided that those fragments retain the biological activity of
the
INSP 168 polypeptides, or have an antigenic determinant in common with these
polypeptides.
As used herein, the term "fragment" refers to a polypeptide having an amino
acid
sequence that is the same as part, but not all, of the amino acid sequence of
the
INSP168 polypeptides or one of their functional equivalents. The fragments
should
comprise at least n consecutive amino acids from the sequence and, depending
on the
particular sequence, n preferably is 7 or more (for example, 8, 10, 12, 14,
16, 18, 20
or more). Small fragments may form an antigenic determinant.
Nucleic acids according to the invention are preferably 10-2000 nucleotides in
length,
preferably 100-1750 nucleotides, preferably 500-1500, preferably 600-1200,
preferably 750-1000 nucleotides in length. Polypeptides according to the
invention
are preferably 10-700 amino acids in length, preferably 50-600, preferably 100-
500,
preferably 200-400, preferably 300-375 amino acids in length.
Fragments of the full length INSP168, INSP168-SVl, INSP149 and INSP169 exon
polypeptides and the INSP168, INSP168-SV1, INSP149 and INSP169 polypeptides
may consist of combinations of 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15,
16 or 17 of
neighbouring exon sequences in the INSP168, INSP168-SV1, 1NSP149 and INSP169
polypeptides, respectively.
Such fragments may be "free-standing", i.e. not part of or fused to other
amino acids
or polypeptides, or they may be comprised within a larger polypeptide of which
they
form a part or region. When coinprised within a larger polypeptide, the
fragment of
the invention most preferably forms a single continuous region. For instance,
certain
preferred embodiments relate to a fragment having a pre- and/or pro-
polypeptide
region fused to the amino terminus of the fragment and/or an additional region
fused
to the carboxyl terminus of the fragment. However, several fragments may be
comprised within a single larger polypeptide.
The polypeptides of the present invention or their immunogenic fragments
(comprising at least one antigenic determinant) can be used to generate
ligands, such
as polyclonal or monoclonal antibodies, that are immunospecific for the
polypeptides.
Such antibodies may be employed to isolate or to identify clones expressing
the
polypeptides of the invention or to purify the polypeptides by affinity
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
33
chromatography. The antibodies may also be employed as diagnostic or
therapeutic
aids, amongst other applications, as will be apparent to the skilled reader.
The term "immunospecific" means that the antibodies have substantially greater
affinity for the polypeptides of the invention than their affinity for other
related
polypeptides in the prior art. As used herein, the term "antibody" refers to
intact
molecules as well as to fragments thereof, such as Fab, F(ab')2 and Fv, which
are
capable of binding to the antigenic determinant in question. Such antibodies
thus bind
to the polypeptides of the first aspect of the invention.
By "substantially greater affinity" we mean that there is a measurable
increase in the
affinity for a polypeptide of the invention as compared with the affinity for
known
cell-surface receptors.
Preferably, the affinity is at least 1.5-fold, 2-fold, 5-fold 10-fold, 100-
fold, 103-fold,
104-fold, 105-fold, 106-fold or greater for a polypeptide of the invention
than for
known cell surface recognition molecules.
Preferably, there is a measurable increase in the affinity for a polypeptide
of the
invention as compared with known leucine-rich repeat (LRR) motif containing
proteins.
If polyclonal antibodies are desired, a selected mammal, such as a mouse,
rabbit, goat
or horse, may be immunised with a polypeptide of the first aspect of the
invention.
The polypeptide used to immunise the animal can be derived by recombinant DNA
technology or can be synthesized chemically. If desired, the polypeptide can
be
conjugated to a carrier protein. Commonly used carriers to which the
polypeptides
may be chemically coupled include bovine seruin albumin, thyroglobulin and
keyhole
limpet haemocyanin. The coupled polypeptide is then used to immunise the
animal.
Serum from the iinmunised animal is collected and treated according to known
procedures, for example by immunoaffinity chromatography.
Monoclonal antibodies to the polypeptides of the first aspect of the invention
can also
be readily produced by one skilled in the art. The general methodology for
making
monoclonal antibodies using hybridoma technology is well known (see, for
example,
Kohler,_ G. and Milstein, C., Nature 256: 495-497 (1975);_Kozbor et al.,
Immunology
Today 4: 72 (1983); Cole et al., 77-96 in Monoclonal Antibodies and Cancer
Therapy,
Alan R. Liss, Inc. (1985).
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
34
Panels of monoclonal antibodies produced against the polypeptides of the first
aspect
of the invention can be screened for various properties, i.e., for isotype,
epitope,
affinity, etc. Monoclonal antibodies are particularly useful in purification
of the
individual polypeptides against which they are directed. Alternatively, genes
encoding
the monoclonal antibodies of interest may be isolated from hybridomas, for
instance
by PCR techniques known in the art, and cloned and expressed in appropriate
vectors.
Chimeric antibodies, in which non-human variable regions are joined or fused
to
human constant regions (see, for example, Liu et al., Proc. Natl. Acad. Sci.
USA, 84,
3439 (1987)), may also be of use.
The antibody may be modified to make it less immunogenic in an individual, for
example by humanisation (see Jones et al., Nature, 321, 522 (1986); Verhoeyen
et al.,
Science, 239, 1534 (1988); Kabat et al., J. Immunol., 147, 1709 (1991); Queen
et al.,
Proc. Natl Acad. Sci. USA, 86, 10029 (1989); Gorman et al., Proc. Natl Acad.
Sci.
USA, 88, 34181 (1991); and Hodgson et al., Bio/Technology, 9, 421 (1991)). The
term "humanised antibody", as used herein, refers to antibody molecules in
which the
CDR amino acids and selected other amino acids in the variable domains of the
heavy
and/or light chains of a non-human donor antibody have been substituted in
place of
the equivalent amino acids in a human antibody. The humanised antibody thus
closely
resembles a human antibody but has the binding ability of the donor antibody.
In a further alternative, the antibody may be a "bispecific" antibody, that is
an
antibody having two different antigen-binding domains, each domain being
directed
against a different epitope.
Phage display technology may be utilised to select genes which encode
antibodies
with binding activities towards the polypeptides of the invention either from
repertoires of PCR amplified V-genes of lymphocytes from huinans screened for
possessing the relevant antibodies, or from naive libraries (McCafferty, J. et
al.,
(1990), Nature 348, 552-554; Marks, J. et al., (1992) Biotechnology 10, 779-
783).
The affinity of these antibodies can also be improved by chain shuffling
(Clackson, T.
et al., (1991) Nature 352, 624-628).
Antibodies generated by the above techniques, whether polyclonal or
monoclonal,
have additional utility in that they may be employed as reagents in
immunoassays,
radioimmunoassays (RIA) or enzyme-linked immunosorbent assays (ELISA). In
these
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
applications, the antibodies can be labelled with an analytically-detectable
reagent
such as a radioisotope, a fluorescent molecule or an enzyme.
Preferred nucleic acid molecules of the second and third aspects of the
invention are
those which encode a polypeptide sequence as recited in SEQ ID NO:2, SEQ ID
5 NO:4, SEQ ID NO:6, SEQ ID NO:8, SEQ ID NO:10, SEQ ID NO:12, SEQ ID
NO:14, SEQ ID NO:16, SEQ ID NO:18, SEQ ID NO:20, SEQ ID NO:22, SEQ ID
NO:24, SEQ ID NO:26, SEQ ID NO:28, SEQ ID NO:30, SEQ ID NO:32, SEQ ID
NO:34, SEQ ID NO:36, SEQ ID NO:38, SEQ ID NO:40, SEQ ID NO:42, SEQ ID
NO:44 and/or SEQ ID NO:67, and functionally equivalent polypeptides. These
10 nucleic acid molecules may be used in the methods and applications
described herein.
The nucleic acid molecules of the invention preferably comprise at least n
consecutive
nucleotides from the sequences disclosed herein where, depending on the
particular
sequence, n is 10 or more (for example, 12, 14, 15, 18, 20, 25, 30, 35, 40 or
more).
The nucleic acid molecules of the invention also include sequences that are
15 complementary to nucleic acid molecules described above (for example, for
antisense
or probing purposes).
Nucleic acid molecules of the present invention may be in the form of RNA,
such as
mRNA, or in the form of DNA, including, for instance cDNA, synthetic DNA or
genomic DNA. Such nucleic acid molecules may be obtained by cloning, by
chemical
20 synthetic techniques or by a combination tliereof. The nucleic acid
molecules can be
prepared, for example, by chemical synthesis using techniques such as solid
phase
phosphoramidite chemical synthesis, from genomic or cDNA libraries or by
separation from an organism. RNA molecules may generally be generated by the
in
vitro or in vivo transcription of DNA sequences.
25 The nucleic acid molecules may be double-stranded or single-stranded.
Single-
stranded DNA may be the coding strand, also known as the sense strand, or it
may be
the non-coding strand, also referred to as the anti-sense strand.
The term "nucleic acid molecule" also includes analogues of DNA and RNA, such
as
those containing modified backbones, and peptide nucleic acids (PNA). The term
30 "PNA", as used herein, refers to an antisense molecule or an anti-gene
agent which
comprises an oligonucleotide of at least five nucleotides in length linked to
a peptide
backbone of amino acid residues, which preferably ends in lysine. The terminal
lysine
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
36
confers solubility to the composition. PNAs may be pegylated to extend their
lifespan
in a cell, where they preferentially bind complementary single stranded DNA
and
RNA and stop transcript elongation (Nielsen, P.E. et al. (1993) Anticancer
Drug Des.
8:53-63).
A nucleic acid molecule which encodes the polypeptide of SEQ ID NO:2 may be
identical to the coding sequence of the nucleic acid molecule shown in SEQ ID
NO: 1.
A nucleic acid molecule which encodes the polypeptide of SEQ ID NO:4 may be
identical to the coding sequence of the nucleic acid molecule shown in SEQ ID
NO:3.
A nucleic acid molecule which encodes the polypeptide of SEQ ID NO:6 may be
identical to the coding sequence of the nucleic acid molecule shown in SEQ ID
NO:5.
A nucleic acid molecule which encodes the polypeptide of SEQ ID NO:8 may be
identical to the coding sequence of the nucleic acid molecule shown in SEQ ID
NO:7.
A nucleic acid molecule which encodes the polypeptide of SEQ ID NO:10 may be
identical to the coding sequence of the nucleic acid molecule shown in SEQ ID
NO:9.
A nucleic acid molecule which encodes the polypeptide of SEQ ID NO:12 may be
identical to the coding sequence of the nucleic acid molecule shown in SEQ ID
NO:11. A nucleic acid molecule which encodes the polypeptide of SEQ ID NO:14
may be identical to the coding sequence of the nucleic acid molecule shown in
SEQ
ID NO:13. A nucleic acid molecule wllich encodes the polypeptide of SEQ ID
NO:16
may be identical to the coding sequence of the nucleic acid molecule shown in
SEQ
ID NO:15. A nucleic acid molecule which encodes the polypeptide of SEQ ID
NO:18
may be identical to the coding sequence of the nucleic acid molecule shown in
SEQ
ID NO:17. A nucleic acid molecule which encodes the polypeptide of SEQ ID
NO:20
may be identical to the coding sequence of the nucleic acid molecule shown in
SEQ
ID NO: 19. A nucleic acid molecule which encodes the polypeptide of SEQ ID
NO:22
may be identical to the coding sequence of the nucleic acid molecule shown in
SEQ
ID NO:21. A nucleic acid molecule which encodes the polypeptide of SEQ ID
NO:24
may be identical to the coding sequence of the nucleic acid molecule shown in
SEQ
ID NO:23. A nucleic acid molecule which encodes the polypeptide of SEQ ID
NO:26
may be identical to the coding sequence of the nucleic acid molecule shown in
SEQ
ID NO:25. A nucleic acid molecule which encodes the polypeptide of SEQ ID
NO:28
may be identical to the coding sequence of the nucleic acid molecule shown in
SEQ
ID NO:27. A nucleic acid molecule which encodes the polypeptide of SEQ ID
NO:30
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
37
may be identical to the coding sequence of the nucleic acid molecule shown in
SEQ
ID NO:29. A nucleic acid molecule which encodes the polypeptide of SEQ ID
NO:32
may be identical to the coding sequence of the nucleic acid molecule shown in
SEQ
ID NO:3 1. A nucleic acid molecule which encodes the polypeptide of SEQ ID
NO:34
may be identical to the coding sequence of the nucleic acid molecule shown in
SEQ
ID NO:33. A nucleic acid molecule which encodes the polypeptide of SEQ ID
NO:36
may be identical to the coding sequence of the nucleic acid molecule shown in
SEQ
ID NO:35. A nucleic acid molecule which encodes the polypeptide of SEQ ID
NO:38
may be identical to the coding sequence of the nucleic acid molecule shown in
SEQ
ID NO:37. A nucleic acid molecule which encodes the polypeptide of SEQ ID
NO:40
may be identical to the coding sequence of the nucleic acid molecule shown in
SEQ
ID NO:39. A nucleic acid molecule which encodes the polypeptide of SEQ ID
NO:42
may be identical to the coding sequence of the nucleic acid molecule shown in
SEQ
ID NO:41. A nucleic acid molecule which encodes the polypeptide of SEQ ID
NO:44
may be identical to the coding sequence of the nucleic acid molecule shown in
SEQ
ID NO:43. A nucleic acid molecule which encodes the polypeptide of SEQ ID
NO:67
may be identical to the coding sequence of the nucleic acid molecule shown in
SEQ
ID NO:66.
These molecules also may have a different sequence which, as a result of the
degeneracy of the genetic code, encodes a polypeptide of SEQ ID NO:2, SEQ ID
NO:4, SEQ ID NO:6, SEQ ID NO:8, SEQ ID NO:10, SEQ ID NO:12, SEQ ID
NO:14, SEQ ID NO:16, SEQ ID NO:18, SEQ ID NO:20, SEQ ID NO:22, SEQ ID
NO:24, SEQ ID NO:26, SEQ ID NO:28, SEQ ID NO:30, SEQ ID NO:32, SEQ ID
NO:34, SEQ ID NO:36, SEQ ID NO:38, SEQ ID NO:40, SEQ ID NO:42, SEQ ID
NO:44 or SEQ ID NO:67. Such nucleic acid molecules may include, but are not
limited to, the coding sequence for the mature polypeptide by itself; the
coding
sequence for the mature polypeptide and additional coding sequences, such as
those
encoding a leader or secretory sequence, such as a pro-, pre- or prepro-
polypeptide
sequence; the coding sequence of the mature polypeptide, with or without the
aforementioned additional coding sequences, together with further additional,
non-
coding sequences, including non-coding 5' and 3' sequences, such as the
transcribed,
non-translated sequences that play a role in transcription (including
termination
signals), ribosome binding and mRNA stability. The nucleic acid molecules may
also
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
38
include additional sequences which encode additional amino acids, such as
those
which provide additional functionalities.
The nucleic acid molecules of the second and third aspects of the invention
may also
encode the fragments or the functional equivalents of the polypeptides and
fragments
of the first aspect of the invention. Such a nucleic acid molecule may be a
naturally-
occurring variant such as a naturally-occurring allelic variant, or the
molecule may be
a variant that is not known to occur naturally. Such non-naturally occurring
variants
of the nucleic acid molecule may be made by mutagenesis techniques, including
those
applied to nucleic acid molecules, cells or organisms.
Among variants in this regard are variants that differ from the aforementioned
nucleic
acid molecules by nucleotide substitutions, deletions or insertions. The
substitutions,
deletions or insertions may involve one or more nucleotides. The variants may
be
altered in coding or non-coding regions or both. Alterations in the coding
regions may
produce conservative or non-conservative amino acid substitutions, deletions
or
insertions.
The nucleic acid molecules of the invention can also be engineered, using
methods
generally known in the art, for a variety of reasons, including modifying the
cloning,
processing, and/or expression of the gene product (the polypeptide). DNA
shuffling
by random fragmentation and PCR reassembly of gene fragments and synthetic
oligonucleotides are included as techniques which may be used to engineer the
nucleotide sequences. Site-directed mutagenesis may be used to insert new
restriction
sites, alter glycosylation patterns, change codon preference, produce splice
variants,
introduce mutations and so fortlz.
Nucleic acid molecules which encode a polypeptide of the first aspect of the
invention
may be ligated to a heterologous sequence so that the combined nucleic acid
molecule
encodes a fusion protein. Such combined nucleic acid molecules are included
within
the second or third aspects of the invention. For example, to screen peptide
libraries
for inhibitors of the activity of the polypeptide, it may be useful to
express, using such
a combined nucleic acid molecule, a fusion protein that can be recognised by a
commercially-available antibody. A fusion protein may also be engineered to
contain
a cleavage site located between the sequence of the polypeptide of the
invention and
the sequence of a heterologous protein so that the polypeptide may be cleaved
and
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
39
purified away from the heterologous protein.
The nucleic acid molecules of the invention also include antisense molecules
that are
partially complementary to nucleic acid molecules encoding polypeptides of the
present invention and that therefore hybridize to the encoding nucleic acid
molecules
(hybridization). Such antisense molecules, such as oligonucleotides, can be
designed
to recognise, specifically bind to and prevent transcription of a target
nucleic acid
encoding a polypeptide of the invention, as will be known by those of ordinary
skill in
the art (see, for example, Cohen, J.S., Trends in Phann. Sci., 10, 435 (1989),
Okano,
J. Neurochem. 56, 560 (1991); O'Connor, J. Neurochem 56, 560 (1991); Lee et
al.,
Nucleic Acids Res 6, 3073 (1979); Cooney et al., Science 241, 456 (1988);
Dervan et
al., Science 251, 1360 (1991).
The term "hybridization" as used here refers to the association of two nucleic
acid
molecules with one another by hydrogen bonding. Typically, one molecule will
be
fixed to a solid support and the other will be free in solution. Then, the two
molecules
may be placed in contact with one another under conditions that favour
hydrogen
bonding. Factors that affect this bonding include: the type and volume of
solvent;
reaction temperature; time of hybridization; agitation; agents to block the
non-specific
attachment of the liquid phase molecule to the solid support (Denhardt's
reagent or
BLOTTO); the concentration of the molecules; use of compounds to increase the
rate
of association of molecules (dextran sulphate or polyethylene glycol); and the
stringency of the washing conditions following hybridization (see Sambrook et
al.
[supra]).
The inhibition of hybridization of a completely complementary molecule to a
target
molecule may be examined using a hybridization assay, as known in the art
(see, for
example, Sambrook et al [supra]). A substantially homologous molecule will
then
compete for and inllibit the binding of a completely homologous molecule to
the
target molecule under various conditions of stringency, as taught in Wahl,
G.M. and
S.L. Berger (1987; Methods Enzymol. 152:399-407) and Kimmel, A.R. (1987;
Methods Enzymol. 152:507-511).
"Stringency" refers to conditions in a hybridization reaction that favour the
association of very similar molecules over association of molecules that
differ. High
stringency hybridisation conditions are defined as overnight incubation at 42
C in a
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
solution comprising 50% formamide, 5XSSC (150mM NaC1, 15mM trisodium
citrate), 50mM sodium phosphate (pH7.6), 5x Denhardts solution, 10% dextran
sulphate, and 20 microgram/ml denatured, sheared salmon sperm DNA, followed by
washing the filters in 0.1X SSC at approximately 65 C. Low stringency
conditions
5 involve the hybridisation reaction being carried out at 35 C (see Sambrook
et al.
[supra]). Preferably, the conditions used for hybridization are those of high
stringency.
Preferred embodiments of this aspect of the invention are nucleic acid
molecules that
are at least 70% identical over their entire length to nucleic acid molecules
encoding
10 the INSP 168 polypeptides (such as SEQ ID NO:2, SEQ ID NO:4, SEQ ID NO:6,
SEQ ID NO:8, SEQ ID NO:10, SEQ ID NO:12, SEQ ID NO:16, SEQ ID NO:18,
SEQ ID NO:20, SEQ ID NO:22, SEQ ID NO:24, SEQ ID NO:26, SEQ ID NO:30,
SEQ ID NO:32, SEQ ID NO:34, SEQ ID NO:36, SEQ ID NO:38, SEQ ID NO:40,
SEQ ID NO:42, SEQ ID NO: 44, or SEQ ID NO:67) and nucleic acid molecules that
15 are substantially complementary to such nucleic acid molecules. Preferably,
a nucleic
acid molecule according to this aspect of the invention comprises a region
that is at
least 80% identical over its entire length to the nucleic acid molecules
having the
sequence recited in SEQ ID NO:1, SEQ ID NO:3, SEQ ID NO:5, SEQ ID NO:7, SEQ
ID NO:9, SEQ ID NO:11, SEQ ID NO:15, SEQ ID NO:17, SEQ ID NO:19, SEQ ID
20 NO:21, SEQ ID NO:23, SEQ ID NO:25, SEQ ID NO:29, SEQ ID NO:31, SEQ ID
NO:33, SEQ ID NO:35, SEQ ID NO:37, SEQ ID NO:39, SEQ ID NO:41, SEQ ID
NO:43 or SEQ ID NO:66 or a nucleic acid molecule that is compleinentary
thereto. In
this regard, nucleic acid molecules at least 90%, preferably at least 95%,
more
preferably at least 98% or 99% identical over their entire length to the same
are
25 particularly preferred. Preferred einbodiments in this respect are nucleic
acid
molecules that encode polypeptides which retain substantially the same
biological
function or activity as the INSP 168 polypeptides.
The invention also provides a process for detecting a nucleic acid molecule of
the
invention, comprising the steps of: (a) contacting a nucleic probe according
to the
30 invention with a biological sample under hybridizing conditions to form
duplexes;
and (b) detecting any such duplexes that are formed.
As discussed additionally below in connection with assays that may be utilised
according to the invention, a nucleic acid molecule as described above may be
used as
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
41
a hybridization probe for RNA, cDNA or genomic DNA, in order to isolate full-
length cDNAs and genomic clones encoding the INSP164 polypeptides and to
isolate
cDNA and genomic clones of homologous or orthologous genes that have a high
sequence similarity to the gene encoding this polypeptide.
In this regard, the following techniques, among others known in the art, may
be
utilised and are discussed below for purposes of illustration. Methods for DNA
sequencing and analysis are well known and are generally available in the art
and
may, indeed, be used to practice many of the embodiments of the invention
discussed
herein. Such methods may employ such enzymes as the Klenow fragment of DNA
polymerase I, Sequenase (US Biochemical Corp, Cleveland, OH), Taq polymerase
(Perkin Elmer), thermostable T7 polymerase (Amersham, Chicago, IL), or
combinations of polymerases and proof-reading exonucleases such as those found
in
the ELONGASE Amplification System marketed by Gibco/BRL (Gaithersburg, MD).
Preferably, the sequencing process may be automated using machines such as the
Hamilton Micro Lab 2200 (Hamilton, Reno, NV), the Peltier Thermal Cycler
(PTC200; MJ Research, Watertown, MA) and the ABI Catalyst and 373 and 377
DNA Sequencers (Perkin Elmer).
One method for isolating a nucleic acid molecule encoding a polypeptide with
an
equivalent function to that of the INSP168 polypeptides is to probe a genoinic
or
cDNA library with a natural or artificially-designed probe using standard
procedures
that are recognised in the art (see, for example, "Current Protocols in
Molecular
Biology", Ausubel et al. (eds). Greene Publishing Association and John Wiley
Interscience, New York, 1989,1992). Probes comprising at least 15, preferably
at least
30, and more preferably at least 50, contiguous bases that correspond to, or
are
coinpleinentary to, nucleic acid sequences from the appropriate encoding gene
(such
as SEQ ID NO:l, SEQ ID NO:3, SEQ ID NO:5, SEQ ID NO:7, SEQ ID NO:9, SEQ
ID NO:11, SEQ ID NO:15, SEQ ID NO:17, SEQ ID NO:19, SEQ ID NO:21, SEQ ID
NO:23, SEQ ID NO:25, SEQ ID NO:29, SEQ ID NO:31, SEQ ID NO:33, SEQ ID
NO:35, SEQ ID NO:37, SEQ ID NO:39, SEQ ID NO:41, SEQ ID NO:43 or SEQ ID
NO:66) are particularly useful probes. Such probes may be labelled with an
analytically-detectable reagent to facilitate their identification. Useful
reagents
include, but are not limited to, radioisotopes, fluorescent dyes and enzymes
that are
capable of catalysing the formation of a detectable product. Using these
probes, the
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
42
ordinarily skilled artisan will be capable of isolating complementary copies
of
genomic DNA, eDNA or RNA polynucleotides encoding proteins of interest from
human, mammalian or other animal sources and screening such sources for
related
sequences, for example, for additional members of the family, type and/or
subtype.
In many cases, isolated cDNA sequences will be incomplete, in that the region
encoding the polypeptide will be cut short, normally at the 5' end. Several
methods are
available to obtain full length cDNAs, or to extend short cDNAs. Such
sequences may
be extended utilising a partial nucleotide sequence and employing various
methods
known in the art to detect upstream sequences such as promoters and regulatory
elements. For example, one method which may be employed is based on the method
of Rapid Amplification of cDNA Ends (RACE; see, for example, Frohman et al.,
PNAS USA 85, 8998-9002, 1988). Recent modifications of this teclmique,
exemplified by the MarathonTM teclmology (Clontech Laboratories Inc.), for
example, have significantly simplified the search for longer cDNAs. A slightly
different technique, termed "restriction-site" PCR, uses universal primers to
retrieve
unknown nucleic acid sequence adjacent a known locus (Sarkar, G. (1993) PCR
Methods Applic. 2:318-322). Inverse PCR may also be used to amplify or to
extend
sequences using divergent primers based on a known region (Triglia, T. et al.
(1988)
Nucleic Acids Res. 16:8186). Another method whicll may be used is capture PCR
which involves PCR amplification of DNA fragments adjacent a known sequence in
human and yeast artificial chromosome DNA (Lagerstrom, M. et al. (1991) PCR
Methods Applic., 1, 111-119). Another method which may be used to retrieve
unknown sequences is that of Parker, J.D. et al. (1991); Nucleic Acids Res.
19:3055-
3060). Additionally, one may use PCR, nested primers, and PromoterFinderTM
libraries to walk genomic DNA (Clontech, Palo Alto, CA). This process avoids
the
need to screen libraries and is useful in finding intron/exon junctions.
When screening for full-length cDNAs, it is preferable to use libraries that
have been
size-selected to include larger cDNAs. Also, random-primed libraries are
preferable,
in that they will contain more sequences that contain the 5' regions of genes.
Use of a
randomly primed library may be especially preferable for situations in which
an oligo
d(T) library does not yield a full-length cDNA. Genomic libraries may be
useful for
extension of sequence into 5' non-transcribed regulatory regions.
In one embodiment of the invention, the nucleic acid molecules of the present
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
43
invention may be used for chromosome localisation. In this technique, a
nucleic acid
molecule is specifically targeted to, and can hybridize with, a particular
location on an
individual human chromosome. The mapping of relevant sequences to chromosomes
according to the present invention is an important step in the confirmatory
correlation
of those sequences with the gene-associated disease. Once a sequence has been
mapped to a precise chromosomal location, the physical position of the
sequence on
the chromosome can be correlated with genetic map data. Such data are found
in, for
example, V. McKusick, Mendelian Inheritance in Man (available on-line through
Johns Hopkins University Welch Medical Library). The relationships between
genes
and diseases that have been mapped to the same chromosomal region are then
identified through linkage analysis (coinheritance of physically adjacent
genes). This
provides valuable information to investigators searching for disease genes
using
positional cloning or other gene discovery techniques. Once the disease or
syndrome
has been crudely localised by genetic linkage to a particular genomic region,
any
sequences mapping to that area may represent associated or regulatory genes
for
further investigation. The nucleic acid molecule may also be used to detect
differences in the chromosomal location due to translocation, inversion, etc.
among
nonnal, carrier, or affected individuals.
The nucleic acid molecules of the present invention are also valuable for
tissue
localisation. Such techniques allow the determination of expression patterns
of the
polypeptide in tissues by detection of the mRNAs that encode them. These
techniques
include in situ hybridization techniques and nucleotide amplification
techniques, such
as PCR. Results from these studies provide an indication of the normal
functions of
the polypeptide in the organism. In addition, comparative studies of the
normal
expression pattern of mRNAs with that of mRNAs encoded by a mutant gene
provide
valuable insights into the role of mutant polypeptides in disease. Such
inappropriate
expression may be of a temporal, spatial or quantitative nature.
Gene silencing approaches may also be undertaken to down-regulate endogenous
expression of a gene encoding a polypeptide of the invention. RNA interference
(RNAi) (Elbashir, SM et al., Nature 2001, 411, 494-498) is one method of
sequence
specific post-transcriptional gene silencing that may be employed. Short dsRNA
oligonucleotides are synthesised in vitro and introduced into a cell. The
sequence
specific binding of these dsRNA oligonucleotides triggers the degradation of
target
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
44
mRNA, reducing or ablating target protein expression.
Efficacy of the gene silencing approaches assessed above may be assessed
through the
measurement of polypeptide expression (for example, by Western blotting), and
at the
RNA level using TaqMan-based methodologies.
The vectors of the present invention comprise nucleic acid molecules of the
invention
and may be cloning or expression vectors. The host cells of the invention,
which may
be transformed, transfected or transduced with the vectors of the invention
may be
prokaryotic or eukaryotic.
The polypeptides of the invention may be prepared in recombinant form by
expression of their encoding nucleic acid molecules in vectors contained
within a host
cell. Such expression methods are well known to those of skill in the art and
many are
described in detail by Sambrook et al (supra) and Fernandez & Hoeffler (1998,
eds.
"Gene expression systems. Using nature for the art of expression". Academic
Press,
San Diego, London, Boston, New York, Sydney, Tokyo, Toronto).
Generally, any system or vector that is suitable to maintain, propagate or
express
nucleic acid molecules to produce a polypeptide in the required host may be
used. The
appropriate nucleotide sequence may be inserted into an expression system by
any of
a variety of well-known and routine techniques, such as, for example, those
described
in Sambrook et al., (supra). Generally, the encoding gene can be placed under
the
control of a control element such as a promoter, ribosome binding site (for
bacterial
expression) and, optionally, an operator, so that the DNA sequence encoding
the
desired polypeptide is transcribed into RNA in the transformed host cell.
Examples of suitable expression systems include, for example, chromosomal,
episomal and virus-derived systems, including, for example, vectors derived
from:
bacterial plasmids, bacteriophage, transposons, yeast episomes, insertion
elements,
yeast chromosomal elements, viruses such as baculoviruses, papova viruses such
as
SV40, vaccinia viruses, adenoviruses, fowl pox viruses, pseudorabies viruses
and
retroviruses, or combinations thereof, such as those derived from plasmid and
bacteriophage genetic elements, including cosmids and phagemids. Human
artificial
chromosomes (HACs) may also be employed to deliver larger fragments of DNA
than
can be contained and expressed in a plasmid.
The pCR4-TOPO-INSP168, pCR4-TOPO-INSP168-SV1, pEAK12d INSP168-6HIS,
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
pENTR INSP168-6HIS, and pDEST12.2 INSP168-6HIS vectors are preferred
examples of suitable vectors for use in accordance with this invention.
Particularly suitable expression systems include microorganisms such as
bacteria
transformed with recombinant bacteriophage, plasmid or cosmid DNA expression
5 vectors; yeast transformed with yeast expression vectors; insect cell
systems infected
with virus expression vectors (for example, baculovirus); plant cell systems
transformed with virus expression vectors (for example, cauliflower mosaic
virus,
CaMV; tobacco mosaic virus, TMV) or with bacterial expression vectors (for
example, Ti or pBR322 plasmids); or animal cell systems. Cell-free translation
10 systems can also be employed to produce the polypeptides of the invention.
Introduction of nucleic acid molecules encoding a polypeptide of the present
invention into host cells can be effected by methods described in many
standard
laboratory manuals, such as Davis et al., Basic Methods in Molecular Biology
(1986)
and Sambrook et al.,[supra]. Particularly suitable methods include calcium
phosphate
15 transfection, DEAE-dextran mediated transfection, transvection,
microinjection,
cationic lipid-mediated transfection, electroporation, transduction, scrape
loading,
ballistic introduction or infection (see Sambrook et al., 1989 [supra];
Ausubel et al.,
1991 [supra]; Spector, Goldman & Leinwald, 1998). In eukaryotic cells,
expression
systems may either be transient (for example, episomal) or permanent
(chromosomal
20 integration) according to the needs of the system.
The encoding nucleic acid molecule may or may not include a sequence encoding
a
control sequence, such as a signal peptide or leader sequence, as desired, for
example,
for secretion of the translated polypeptide into the lumen of the endoplasmic
reticuluin, into the periplasmic space or into the extracellular environment.
These
25 signals may be endogenous to the polypeptide or they may be heterologous
signals.
Leader sequences can be removed by the bacterial host in post-translational
processing.
In addition to control sequences, it may be desirable to add regulatory
sequences that
allow for regulation of the expression of the polypeptide relative to the
growth of the
30 host cell. Examples of regulatory sequences are those which cause the
expression of a
gene to be increased or decreased in response to a chemical or physical
stimulus,
including the presence of a regulatory compound or to various temperature or
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
46
metabolic conditions. Regulatory sequences are those non-translated regions of
the
vector, such as enhancers, promoters and 5' and 3' untranslated regions. These
interact
with host cellular proteins to carry out transcription and translation. Such
regulatory
sequences may vary in their strength and specificity. Depending on the vector
system
and host utilised, any number of suitable transcription and translation
elements,
including constitutive and inducible promoters, may be used. For example, when
cloning in bacterial systems, inducible promoters such as the hybrid lacZ
promoter of
the Bluescript phagemid (Stratagene, LaJolla, CA) or pSportlTm plasmid (Gibco
BRL)
and the like may be used. The baculovirus polyhedrin promoter may be used in
insect
cells. Promoters or enhancers derived from the genomes of plant cells (for
example,
heat shock, RUBISCO and storage protein genes) or from plant viruses (for
example,
viral promoters or leader sequences) may be cloned into the vector. In
inammalian
cell systems, promoters from mammalian genes or from mammalian viruses are
preferable. If it is necessary to generate a cell line that contains multiple
copies of the
sequence, vectors based on SV40 or EBV may be used with an appropriate
selectable
marker.
An expression vector is constructed so that the particular nucleic acid coding
sequence is located in the vector with the appropriate regulatory sequences,
the
positioning and orientation of the coding sequence with respect to the
regulatory
sequences being such that the coding sequence is transcribed under the
"control" of
the regulatory sequences, i.e., RNA polymerase which binds to the DNA molecule
at
the control sequences transcribes the coding sequence. In some cases it may be
necessary to modify the sequence so that it may be attached to the control
sequences
with the appropriate orientation; i.e., to maintain the reading fraine.
The control sequences and other regulatory sequences may be ligated to the
nucleic
acid coding sequence prior to insertion into a vector. Alternatively, the
coding
sequence can be cloned directly into an expression vector that already
contains the
control sequences and an appropriate restriction site.
For long-term, high-yield production of a recombinant polypeptide, stable
expression
is preferred. For example, cell lines which stably express the polypeptide of
interest
may be transformed using expression vectors which may contain viral origins of
replication and/or endogenous expression elements and a selectable marker gene
on
the same or on a separate vector. Following the introduction of the vector,
cells may
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
47
be allowed to grow for 1-2 days in an enriched media before they are switched
to
selective media. The purpose of the selectable marker is to confer resistance
to
selection, and its presence allows growth and recovery of cells that
successfully
express the introduced sequences. Resistant clones of stably transformed cells
may be
proliferated using tissue culture techniques appropriate to the cell type.
Mammalian cell lines available as hosts for expression are known in the art
and
include many immortalised cell lines available from the American Type Culture
Collection (ATCC) including, but not limited to, Chinese hamster ovary (CHO),
HeLa, baby hamster kidney (BHK), monkey kidney (COS), C127, 3T3, BHK, HEK
293, Bowes melanoma and human hepatocellular carcinoma (for example Hep G2)
cells and a number of other cell lines.
In the baculovirus system, the materials for baculovirus/insect cell
expression systems
are commercially available in kit form from, inter alia, Invitrogen, San Diego
CA (the
"MaxBac" kit). These techniques are generally known to those skilled in the
art and
are described fully in Summers and Smith, Texas Agricultural Experiment
Station
Bulletin No. 1555 (1987). Particularly suitable host cells for use in this
system include
insect cells such as Drosophila S2 and Spodoptera Sf9 cells.
There are many plant cell culture and whole plant genetic expression systems
known
in the art. Examples of suitable plant cellular genetic expression systems
include those
described in US 5,693,506; US 5,659,122; and US 5,608,143. Additional examples
of
genetic expression in plant cell culture have been described by Zenk,
Phytochemistry
30, 3861-3863 (1991).
In particular, all plants from which protoplasts can be isolated and cultured
to give
whole regenerated plants can be utilised, so that whole plants are recovered
which
contain the transferred gene. Practically all plants can be regenerated from
cultured
cells or tissues, including but not limited to all major species of sugar
cane, sugar
beet, cotton, fruit and other trees, legumes and vegetables.
Examples of particularly preferred bacterial host cells include streptococci,
staphylococci, E. coli, Streptomyces and Bacillus subtilis cells.
Examples of particularly suitable host cells for fungal expression include
yeast cells
(for example, S. cerevisiae) and Aspergillus cells.
Any number of selection systems are known in the art that may be used to
recover
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
48
transformed cell lines. Examples include the herpes simplex virus thymidine
kinase
(Wigler, M. et al. (1977) Cell 11:223-32) and adenine
phosphoribosyltransferase
(Lowy, I. et al. (1980) Cell 22:817-23) genes that can be employed in tk- or
aprt
cells, respectively.
Also, antimetabolite, antibiotic or herbicide resistance can be used as the
basis for
selection; for example, dihydrofolate reductase (DHFR) that confers resistance
to
methotrexate (Wigler, M. et al. (1980) Proc. Natl. Acad. Sci. 77:3567-70);
npt, which
confers resistance to the aminoglycosides neomycin and G-418 (Colbere-Garapin,
F.
et al (1981) J. Mol. Biol. 150:1-14) and als or pat, which confer resistance
to
chlorsulfuron and phosphinotricin acetyltransferase, respectively. Additional
selectable genes have been described, examples of which will be clear to those
of skill
in the art.
Although the presence or absence of marker gene expression suggests that the
gene of
interest is also present, its presence and expression may need to be
confirmed. For
example, if the relevant sequence is inserted within a marker gene sequence,
transformed cells containing the appropriate sequences can be identified by
the
absence of marker gene fiuiction. Alternatively, a marker gene can be placed
in
tandem with a sequence encoding a polypeptide of the invention under the
control of a
single promoter. Expression of the marker gene in response to induction or
selection
usually indicates expression of the tandem gene as well.
Alternatively, host cells that contain a nucleic acid sequence encoding a
polypeptide
of the invention and which express said polypeptide may be identified by a
variety of
procedures known to those of skill in the art. These procedures include, but
are not
limited to, DNA-DNA or DNA-RNA hybridizations and protein bioassays, for
example, fluorescence activated cell sorting (FACS) or immunoassay techniques
(such as the enzyme-linked immunosorbent assay [ELISA] and radioimmunoassay
[RIA]), that include membrane, solution, or cliip based technologies for the
detection
and/or quantification of nucleic acid or protein (see Hampton, R. et al.
(1990)
Serological Methods, a Laboratory Manual, APS Press, St Paul, MN) and Maddox,
D.E. et al. (1983) J. Exp. Med, 158, 1211-1216).
A wide variety of labels and conjugation techniques are known by those skilled
in the
art and may be used in various nucleic acid and amino acid assays. Means for
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
49
producing labelled hybridization or PCR probes for detecting sequences related
to
nucleic acid molecules encoding polypeptides of the present invention include
oligolabelling, nick translation, end-labelling or PCR amplification using a
labelled
polynucleotide. Alternatively, the sequences encoding the polypeptide of the
invention may be cloned into a vector for the production of an mRNA probe.
Such
vectors are known in the art, are commercially available, and may be used to
synthesise RNA probes izz vitro by addition of an appropriate RNA polymerase
such
as T7, T3 or SP6 and labelled nucleotides. These procedures may be conducted
using
a variety of commercially available kits (Pharmacia & Upjohn, (Kalamazoo, MI);
Promega (Madison WI); and U.S. Biochemical Corp., Cleveland, OH)).
Suitable reporter molecules or labels, which may be used for ease of
detection,
include radionuclides, enzymes and fluorescent, chemiluminescent or
chromogenic
agents as well as substrates, cofactors, inhibitors, magnetic particles, and
the like.
Nucleic acid molecules according to the present invention may also be used to
create
transgenic animals, particularly rodent animals. Such transgenic animals form
a
further aspect of the present invention. This may be done locally by
modification of
somatic cells, or by germ line therapy to incorporate heritable modifications.
Such
transgenic animals may be particularly useful in the generation of animal
models for
drug molecules effective as modulators of the polypeptides of the present
invention.
The polypeptide can be recovered and purified from recombinant cell cultures
by
well-known methods including ammoniuin sulphate or ethanol precipitation, acid
extraction, anion or cation exchange chromatography, phosphocellulose
chromatography, hydrophobic interaction chromatography, affinity
chromatography,
hydroxylapatite chromatography and lectin chromatography. High performance
liquid
chromatography is particularly useful for purification. Well known techniques
for
refolding proteins may be employed to regenerate an active conformation wlien
the
polypeptide is denatured during isolation and or purification.
Specialised vector constructions may also be used to facilitate purification
of proteins,
as desired, by joining sequences encoding the polypeptides of the invention to
a
nucleotide sequence encoding a polypeptide domain that will facilitate
purification of
soluble proteins. Examples of such purification-facilitating domains include
metal
chelating peptides such as histidine-tryptophan modules that allow
purification on
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
immobilised metals, protein A domains that allow purification on immobilised
immunoglobulin, and the domain utilised in the FLAGS extension/affinity
purification system (Immunex Corp., Seattle, WA). The inclusion of cleavable
linker
sequences such as those specific for Factor XA or enterokinase (Invitrogen,
San
5 Diego, CA) between the purification domain and the polypeptide of the
invention may
be used to facilitate purification. One such expression vector provides for
expression
of a fusion protein containing the polypeptide of the invention fused to
several
histidine residues preceding a thioredoxin or an enterokinase cleavage site.
The
histidine residues facilitate purification by IMAC (immobilised metal ion
affinity
10 chromatography as described in Porath, J. et al. (1992), Prot. Exp. Purif.
3: 263-281)
while the thioredoxin or enterokinase cleavage site provides a means for
purifying the
polypeptide from the fusion protein. A discussion of vectors which contain
fusion
proteins is provided in Kroll, D.J. et al. (1993; DNA Cell Biol. 12:441-453).
If the polypeptide is to be expressed for use in screening assays, generally
it is
15 preferred that it be secreted into the culture medium of the host cell in
which it is
expressed. In this event, the polypeptides of the invention may be purified
from the
culture medium may be harvested prior to use in the screening assay, for
example
using standard protein purification techniques such as gel exclusion
chromatography,
ion-exchange chromatography or affinity chromatography. Examples of suitable
20 methods of protein purification are provided in the Examples herein. If
polypeptide is
produced intracellularly, the cells must first be lysed before the polypeptide
is
recovered.
Alternatively, it may be preferred that the polypeptides of the invention be
expressed
as cell-surface fusion proteins. In this event, the host cells may be
harvested prior to
25 use in the screening assay, for example using techniques such as
fluorescence
activated cell sorting (FACs) or immunoaffinity techniques.
As indicated above, the present invention also provides novel targets and
methods for
the screening of drug candidates or leads. These screening methods include
binding
assays and/or functional assays, and may be performed in vitro, in cell
systems or in
30 animals.
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
51
In this regard, a particular object of this invention resides in the use of an
INSP168
polypeptide as a target for screening candidate drugs for treating or
preventing
disorders in which leucine-rich repeat (LRR) motif containing proteins are
implicated.
Another object of this invention resides in methods of selecting biologically
active
compounds, said methods comprising contacting a candidate compound with a
INSP 168 gene or polypeptide, and selecting compounds that bind said gene or
polypeptide.
A further other object of this invention resides in methods of selecting
biologically
active compounds, said method comprising contacting a candidate compound with
recombinant host cell expressing a INSP 168 polypeptide with a candidate
compound,
and selecting compounds that bind said INSP168 polypeptide at the surface of
said
cells and/or that modulate the activity of the INSP 168 polypeptide.
A "biologically active" compound denotes any compound having biological
activity
in a subject, preferably therapeutic activity, more preferably a compound that
can be
used for treating disorders in which leucine-rich repeat (LRR) motif
containing
proteins are implicated, or as a lead to develop drugs for treating disorders
in which
leucine-rich repeat (LRR) motif containing proteins are implicated. A
"biologically
active" compound preferably is a compound that modulates the activity of a
INSP 168
polypeptide.
The above methods may be conducted in vitro, using various devices and
conditions,
including with immobilized reagents, and may further comprise asi additional
step of
assaying the activity of the selected compounds in a model of a disorder in
which
leucine-rich repeat (LRR) motif containing proteins are implicated, such as an
animal
model.
Binding to a target gene or polypeptide provides an indication as to the
ability of the
compound to modulate the activity of said target, and thus to affect a pathway
leading
to disorder in a subject. The determination of binding may be performed by
various
techniques, such as by labelling of the candidate compound, by competition
with a
labelled reference ligand, etc. For in vitro binding assays, the polypeptides
may be
used in essentially pure form, in suspension, immobilized on a support, or
expressed
in a membrane (intact cell, membrane preparation, liposome, etc.).
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
52
Modulation of activity includes, without limitation, stimulation of the
surface
expression of a receptor, and modulation of multimerization of said receptor
(e.g., the
formation of multimeric complexes with other sub-units), etc. The cells used
in the
assays may be any recombinant cell (i.e., any cell comprising a recombinant
nucleic
acid encoding a INSP168 polypeptide) or any cell that expresses an endogenous
INSP168 polypeptide. Examples of such cells include, without limitation,
prokaryotic
cells (such as bacteria) and eukaryotic cells (such as yeast cells, mammalian
cells,
insect cells, plant cells, etc.). Specific examples include E.coli, Pichia
pastoris,
Hansenula polymorpha, Schizosaccharomyces pombe, Kluyveromyces or
Saccharomyces yeasts, mammalian cell lines (e.g., Vero cells, CHO cells, 3T3
cells,
COS cells, etc.) as well as primary or established mammalian cell cultures
(e.g.,
produced from fibroblasts, einbryonic cells, epithelial cells, nervous cells,
adipocytes,
etc.).
Preferred selected compounds are agonists of INSP 168, i.e., compounds that
can bind
to INSP 168 and mimic the activity of an endogenous ligand thereof.
A further object of this invention resides in a method of selecting
biologically active
coinpounds, said method comprising contacting in vitro a test compound with a
INSP168 polypeptide according to the present invention and determining the
ability
of said test compound to modulate the activity of said INSP 168 polypeptide.
A further object of this invention resides in a method of selecting
biologically active
compounds, said method comprising contacting in vitro a test compound with a
1NSP 168 gene according to the present invention and determining the ability
of said
test compound to modulate the expression of said INSP168 gene, preferably to
stimulate expression thereof.
In another embodiment, this invention relates to a method of screening,
selecting or
identifying active compounds, the method comprising contacting a test compound
with a recombinant host cell comprising a reporter construct, said reporter
construct
comprising a reporter gene under the control of a INSP168 gene promoter, and
selecting the test compounds that modulate (e.g. stimulate or reduce,
preferably
stimulate) expression of the reporter gene.
The polypeptide of the invention can be used to screen libraries of compounds
in any
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
53
of a variety of drug screening techniques. Such compounds may activate
(agonise) or
inhibit (antagonise) the level of expression of the gene or the activity of
the
polypeptide of the invention and form a further aspect of the present
invention.
Preferred compounds are effective to alter the expression of a natural gene
which
encodes a polypeptide of the first aspect of the invention or to regulate the
activity of
a polypeptide of the first aspect of the invention.
Agonist or antagonist compounds may be isolated from, for example, cells, cell-
free
preparations, chemical libraries or natural product mixtures. These agonists
or
antagonists may be natural or modified substrates, ligands, enzymes, receptors
or
structural or functional mimetics. For a suitable review of such screening
techniques,
see Coligan et al., Current Protocols in Immunology 1(2):Chapter 5 (1991).
Compounds that are most likely to be good antagonists are molecules that bind
to the
polypeptide of the invention without inducing the biological effects of the
polypeptide
upon binding to it. Potential antagonists include small organic molecules,
peptides,
polypeptides and antibodies that bind to the polypeptide of the invention and
thereby
inhibit or extinguish its activity. In this fashion, binding of the
polypeptide to normal
cellular binding molecules may be inhibited, such that the normal biological
activity
of the polypeptide is prevented.
The polypeptide of the invention that is employed in such a screening
technique may
be free in solution, affixed to a solid support, borne on a cell surface or
located
intracellularly. In general, such screening procedures may involve using
appropriate
cells or cell membranes that express the polypeptide that are contacted with a
test
compound to observe binding, or stimulation or inhibition of a functional
response.
The functional response of the cells contacted with the test compound is then
compared with control cells that were not contacted witli the test compound.
Such an
assay may assess whether the test compound results in a signal generated by
activation of the polypeptide, using an appropriate detection system.
Inhibitors of
activation are generally assayed in the presence of a known agonist and the
effect on
activation by the agonist in the presence of the test compound is observed.
Methods for generating detectable signals in the types of assays described
herein will
be known to those of skill in the art. A particular example is cotransfecting
a construct
expressing a polypeptide according to the invention, or a fragment such as the
LBD,
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
54
in fusion with the GAL4 DNA binding domain, into a cell together with a
reporter
plasmid, an example of which is pFR-Luc (Stratagene Europe, Amsterdam, The
Netherlands). This particular plasmid contains a synthetic promoter with five
tandem
repeats of GAL4 binding sites that control the expression of the luciferase
gene. When
a potential ligand is added to the cells, it will bind the GAL4-polypeptide
fusion and
induce transcription of the luciferase gene. The level of the luciferase
expression can
be monitored by its activity using a luminescence reader (see, for example,
Lehman et
al. JBC 270, 12953, 1995; Pawar et al. JBC, 277, 39243, 2002).
A preferred method for identifying an agonist or antagonist compound of a
polypeptide of the present invention comprises:
(a) contacting a cell expressing on the surface thereof a polypeptide
according to the
first aspect of the invention, the polypeptide being associated with a second
component capable of providing a detectable signal in response to the binding
of a
compound to the polypeptide, with a compound to be screened under conditions
to
permit binding to the polypeptide; and
(b) determining whether the compound binds to and activates or inhibits the
polypeptide by measuring the level of a signal generated from the interaction
of the
compound with the polypeptide.
A further preferred method for identifying an agonist or antagonist of a
polypeptide of
the invention comprises:
(a) contacting a cell expressing on the surface thereof the polypeptide, the
polypeptide
being associated with a second component capable of providing a detectable
signal in
response to the binding of a compound to the polypeptide, with a compound to
be
screened under conditions to permit binding to the polypeptide; and
(b) determining whether the compound binds to and activates or inhibits the
polypeptide by comparing the level of a signal generated from the interaction
of the
compound with the polypeptide with the level of a signal in the absence of the
compound.
For example, a method such as FRET detection of a ligand bound to the
polypeptide
in the-presence of peptide co-activators (Norris et al., Science 285, 744,
1999) might
be used.
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
In further preferred embodiments, the general methods that are described above
may
further comprise conducting the identification of agonist or antagonist in the
presence
of labelled or unlabelled ligand for the polypeptide.
In another embodiment of the method for identifying agonist or antagonist of a
5 polypeptide of the present invention comprises:
determining the inhibition of binding of a ligand to cells which have a
polypeptide of
the invention on the surface thereof, or to cell membranes containing such a
polypeptide, in the presence of a candidate compound under conditions to
permit
binding to the polypeptide, and determining the ainount of ligand bound to the
10 polypeptide. A compound capable of causing reduction of binding of a ligand
is
considered to be an agonist or antagonist. Preferably the ligand is labelled.
More particularly, a method of screening for a polypeptide antagonist or
agonist
compound comprises the steps of
(a) incubating a labelled ligand with a whole cell expressing a polypeptide
according
15 to the invention on the cell surface, or a cell membrane containing a
polypeptide of
the invention,
(b) measuring the amount of labelled ligand bound to the whole cell or the
cell
membrane;
(c) adding a candidate compound to a mixture of labelled ligand and the whole
cell or
20 the cell membrane of step (a) and allowing the mixture to attain
equilibrium;
(d) measuring the amount of labelled ligand bound to the whole cell or the
cell
membrane after step (c); and
(e) comparing the difference in the labelled ligand bound in step (b) and (d),
such that
the compound which causes the reduction in binding in step (d) is considered
to be an
25 agonist or antagonist.
The polypeptides may be found to modulate a variety of physiological and
pathological processes in a dose-dependent manner in the above-described
assays.
Thus, the "functional equivalents" of the polypeptides of the invention
include
polypeptides that exhibit any of the same modulatory activities in the above-
described
30 assays in a dose-dependent manner. Although the degree of dose-dependent
activity
need not be identical to that of the polypeptides of the invention, preferably
the
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
56
"functional equivalents" will exhibit substantially similar dose-dependence in
a given
activity assay compared to the polypeptides of the invention.
In certain of the embodiments described above, simple binding assays may be
used, in
which the adherence of a test compound to a surface bearing the polypeptide is
detected by means of a label directly or indirectly associated with the test
compound
or in an assay involving competition with a labelled competitor. In another
embodiment, competitive drug screening assays may be used, in which
neutralising
antibodies that are capable of binding the polypeptide specifically compete
with a test
compound for binding. In this manner, the antibodies can be used to detect the
presence of any test compound that possesses specific binding affinity for the
polypeptide.
Persons skilled in the art will be able to devise assays for identifying
modulators of a
polypeptide of the invention. Of interest in this regard is Lokker NA et aL,
J. Biol.
Chem., 1997, Dec 26;272(52):33037-44, which reports an example of an assay to
identify antagonists (in this case neutralizing antibodies).
Assays may also be designed to detect the effect of added test compounds on
the
production of mRNA encoding the polypeptide in cells. For example, an ELISA
may
be constructed that measures secreted or cell-associated levels of polypeptide
using
monoclonal or polyclonal antibodies by standard methods known in the art, and
this
can be used to search for compounds that may inhibit or enhance the production
of the
polypeptide from suitably manipulated cells or tissues. The formation of
binding
complexes between the polypeptide and the compound being tested may then be
measured.
Another technique for drug screening which may be used provides for high
throughput screening of compounds having suitable binding affinity to the
polypeptide of interest (see International patent application W084/03564). In
this
method, large numbers of different small test compounds are synthesised on a
solid
substrate, which may then be reacted with the polypeptide of the invention and
washed. One way of immobilising the polypeptide is to use non-neutralising
antibodies. Bound polypeptide may then be detected using methods that are well
known in the art. Purified polypeptide can also be coated directly onto plates
for use
in the aforementioned drug screening techniques.
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
57
Assay methods that are also included within the terms of the present invention
are
those that involve the use of the genes and polypeptides of the invention in
overexpression or ablation assays. Such assays involve the manipulation of
levels of
these genes/polypeptides in cells and assessment of the impact of this
manipulation
event on the physiology of the manipulated cells. For example, such
experiments
reveal details of signalling and metabolic pathways in which the particular
genes/polypeptides are implicated, generate information regarding the
identities of
polypeptides with which the studied polypeptides interact and provide clues as
to
methods by which related genes and proteins are regulated.
Another technique for drug screening which may be used provides for high
throughput screening of compounds having suitable binding affinity to the
polypeptide of interest (see International patent application W084/03564). In
this
method, large numbers of different small test compounds are synthesised on a
solid
substrate, which may then be reacted with the polypeptide of the invention and
washed. One way of immobilising the polypeptide is to use non-neutralising
antibodies. Bound polypeptide may then be detected using methods that are well
known in the art. Purified polypeptide can also be coated directly onto plates
for use
in the aforementioned drug screening techniques.
The polypeptide of the invention may be used to identify membrane-bound or
soluble
receptors, through standard receptor binding techniques that are known in the
art, such
as ligand binding and crosslinking assays in which the polypeptide is labelled
with a
radioactive isotope, is chemically modified, or is fused to a peptide sequence
that
facilitates its detection or purification, and incubated with a source of the
putative
receptor (for example, a composition of cells, cell membranes, cell
supernatants,
tissue extracts, or bodily fluids). The efficacy of binding may be measured
using
biophysical techniques such as surface plasmon resonance (supplied by Biacore
AB,
Uppsala, Sweden) and spectroscopy. Binding assays may be used for the
purification
and cloning of the receptor, but may also identify agonists and antagonists of
the
polypeptide, that compete with the binding of the polypeptide to its receptor.
Standard
methods for conducting screening assays are well understood in the art.
In anotlier embodiment, this inventionrelates to the use of a INSP168, INSP168-
SV1,
INSP149 or INSP169 polypeptide or fragment thereof, whereby the fragment is
preferably a INSP168, INSP168-SV1, INSP149 or INSP169 gene-specific fragment,
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
58
for isolating or generating an agonist or stimulator of the INSP168, 1NSP168-
SV1,
INSP 149 or INSP 169 polypeptide for the treatment of a disorder, wherein said
agonist
or stimulator is selected from the group consisting of:
1. a specific antibody or fragment thereof including: a) a chimeric, b) a
humanized or c) a fully human antibody, as well as;
2. a bispecific or multispecific antibody,
3. a single chain (e.g. scFv) or
4. single domain antibody, or
5. a peptide- or non-peptide mimetic derived from said antibodies or
6. an antibody-mimetic such as a) an anticalin or b) a fibronectin-based
binding
molecule (e.g. trinectin or adnectin).
The generation of peptide- or non-peptide mimetics from antibodies is known in
the
art (Saragovi et al., 1991 and Saragovi et al., 1992).
Anticalins are also known in the art (Vogt et al., 2004). Fibronectin-based
binding
molecules are described in US6818418 and W02004029224.
Furthermore, the test compound may be of various origin, nature and
composition,
such as any small molecule, ilucleic acid, lipid, peptide, polypeptide
including an
antibody such as a chimeric, humanized or fully human antibody or an antibody
fragment, peptide- or non-peptide mimetic derived therefrom as well as a
bispecific or
multispecific antibody, a single chain (e.g. scFv) or single domain antibody
or an
antibody-mimetic such as an anticalin or fibronectin-based binding molecule
(e.g.
trinectin or adnectin), etc., in isolated form or in mixture or combinations.
The invention also includes a screening kit useful in the methods for
identifying
agonists, antagonists, ligands, receptors, substrates, enzymes, that are
described
above.
The invention includes the agonists, antagonists, ligands, receptors,
substrates and
enzymes, and other compounds which modulate the activity or antigenicity of
the
polypeptide of the invention discovered by the methods that are described
above.
As mentioned above, it is envisaged that the various moieties of the invention
(i.e. the
polypeptides of the first aspect of the invention, a nucleic acid molecule of
the second
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
59
or third aspect of the invention, a vector of the fourth aspect of the
invention, a host
cell of the fifth aspect of the invention, a ligand of the sixth aspect of the
invention, a
compound of the seventh aspect of the invention) may be useful in the therapy
or
diagnosis of diseases. To assess the utility of the moieties of the invention
for treating
or diagnosing a disease one or more of the following assays may be carried
out. Note
that although some of the following assays refer to the test compound as being
a
protein/polypeptide, a person skilled in the art will readily be able to adapt
the
following assays so that the other moieties of the invention may also be used
as the
"test compound".
The invention also provides pharmaceutical compositions comprising a
polypeptide,
nucleic acid, ligand or compound of the invention in combination with a
suitable
pharmaceutical carrier. These compositions may be suitable as therapeutic or
diagnostic reagents, as vaccines, or as other immunogenic compositions, as
outlined
in detail below.
According to the terminology used herein, a composition containing a
polypeptide,
nucleic acid, ligand or compound [X] is "substantially free of' impurities
[herein, Y]
when at least 85% by weight of the total X+Y in the composition is X.
Preferably, X
comprises at least about 90% by weight of the total of X+Y in the composition,
more
preferably at least about 95%, 98% or even 99% by weight.
The pharmaceutical compositions should preferably comprise a therapeutically
effective amount of the polypeptide, nucleic acid molecule, ligand, or
compound of
the invention. The term "therapeutically effective amount" as used herein
refers to an
amount of a therapeutic agent needed to treat, ameliorate, or prevent a
targeted
disease or condition, or to exhibit a detectable therapeutic or preventative
effect. For
any coinpound, the therapeutically effective dose can be estimated initially
either in
cell culture assays, for example, of neoplastic cells, or in animal models,
usually mice,
rabbits, dogs, or pigs. The animal model may also be used to determine the
appropriate concentration range and route of adininistration. Such information
can
then be used to determine useful doses and routes for administration in
humans.
The precise effective amount for a human subject will depend upon the severity
of the
disease state, general health of the subject, age, weight, and gender of the
subject,
diet, time and frequency of administration, drug combination(s), reaction
sensitivities,
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
and tolerance/response to therapy. This amount can be determined by routine
experimentation and is within the judgement of the clinician. Generally, an
effective
dose will be from 0.01 mg/kg to 50 mg/kg, preferably 0.05 mg/kg to 10 mg/kg.
Compositions may be administered individually to a patient or may be
administered in
5 combination with other agents, drugs or hormones.
A pharmaceutical coinposition may also contain a pharmaceutically acceptable
carrier, for administration of a therapeutic agent. Such carriers include
antibodies and
other polypeptides, genes and other therapeutic agents such as liposomes,
provided
that the carrier does not itself induce the production of antibodies harmful
to the
10 individual receiving the composition, and which may be administered without
undue
toxicity. Suitable carriers may be large, slowly metabolised macromolecules
such as
proteins, polysaccharides, polylactic acids, polyglycolic acids, polymeric
amino acids,
ainino acid copolymers and inactive virus particles.
Pharmaceutically acceptable salts can be used therein, for example, mineral
acid salts
15 such as hydrochlorides, hydrobromides, phosphates, sulphates, and the like;
and the
salts of organic acids such as acetates, propionates, malonates, benzoates,
and the like.
A thorough discussion of pharmaceutically acceptable carriers is available in
Remington's Pharmaceutical Sciences (Mack Pub. Co., N.J. 1991).
Pharmaceutically acceptable carriers in therapeutic compositions may
additionally
20 contain liquids such as water, saline, glycerol and ethanol. Additionally,
auxiliary
substances, such as wetting or emulsifying agents, pH buffering substances,
and the
like, may be present in such compositions. Such carriers enable the
pharmaceutical
compositions to be formulated as tablets, pills, dragees, capsules, liquids,
gels, syrups,
slurries, suspensions, and the like, for ingestion by the patient.
25 Once formulated, the compositions of the invention can be administered
directly to
the subject. The subjects to be treated can be animals; in particular, human
subjects
can be treated.
The pharmaceutical compositions utilised in this invention may be administered
by
any number of routes including, but not limited to, oral, intravenous,
intramuscular,
30 intra-arterial, intramedullary, intrathecal, intraventricular, transdermal
or
transcutaneous applications (for example, see W098/20734), subcutaneous,
intraperitoneal, intranasal, enteral, topical, sublingual, intravaginal or
rectal means.
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
61
Gene guns or hyposprays may also be used to administer the pharmaceutical
compositions of the invention. Typically, the therapeutic compositions may be
prepared as injectables, either as liquid solutions or suspensions; solid
forms suitable
for solution in, or suspension in, liquid vehicles prior to injection may also
be
prepared.
Direct delivery of the compositions will generally be accomplished by
injection,
subcutaneously, intraperitoneally, intravenously or intramuscularly, or
delivered to
the interstitial space of a tissue. The compositions can also be administered
into a
lesion. Dosage treatinent may be a single dose schedule or a multiple dose
schedule.
If the activity of the polypeptide of the invention is in excess in a
particular disease
state, several approaches are available. One approach comprises administering
to a
subject an inhibitor compound (antagonist) as described above, along with a
pharmaceutically acceptable carrier in an amount effective to inhibit the
function of
the polypeptide, such as by blocking the binding of ligands, substrates,
enzymes,
receptors, or by inhibiting a second signal, and thereby alleviating the
abnormal
condition. Preferably, such antagonists are antibodies. Most preferably, such
antibodies are chimeric and/or humanised to minimise their immunogenicity, as
described previously.
In another approach, soluble forms of the polypeptide that retain binding
affinity for
the ligand, substrate, enzyme, receptor, in question, may be administered.
Typically,
the polypeptide may be administered in the form of fragments that retain the
relevant
portions.
In an alternative approach, expression of the gene encoding the polypeptide
can be
inhibited using expression blocking techniques, such as the use of antisense
nucleic
acid molecules (as described above), either internally generated or separately
administered. Modifications of gene expression can be obtained by designing
complementary sequences or antisense molecules (DNA, RNA, or PNA) to the
control, 5' or regulatory regions (signal sequence, promoters, enhancers and
introns)
of the gene encoding the polypeptide. Similarly, inhibition can be achieved
using
"triple helix" base-pairing methodology. Triple helix pairing is useful
because it
causes inhibition of the ability of the double helix to open sufficiently for
the binding
of polymerases, transcription factors, or regulatory molecules. Recent
therapeutic
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
62
advances using triplex DNA have been described in the literature (Gee, J.E. et
al.
(1994) In: Huber, B.E. and B.I. Carr, Molecular and Immunologic Approaches,
Futura
Publishing Co., Mt. Kisco, NY). The complementary sequence or antisense
molecule
may also be designed to block translation of mRNA by preventing the transcript
from
binding to ribosomes. Such oligonucleotides may be administered or may be
generated in situ from expression in vivo.
In addition, expression of the polypeptide of the invention may be prevented
by using
ribozymes specific to its encoding mRNA sequence. Ribozymes are catalytically
active RNAs that can be natural or synthetic (see for example Usman, N, et
al., Curr.
Opin. Struct. Biol (1996) 6(4), 527-33). Synthetic ribozymes can be designed
to
specifically cleave mRNAs at selected positions thereby preventing translation
of the
mRNAs into functional polypeptide. Ribozymes may be synthesised with a natural
ribose phosphate backbone and natural bases, as normally found in RNA
molecules.
Alternatively the ribozymes may be synthesised with non-natural backbones, for
exainple, 2'-O-methyl RNA, to provide protection from ribonuclease degradation
and
may contain modified bases.
RNA molecules may be modified to increase intracellular stability and half-
life.
Possible modifications include, but are not limited to, the addition of
flanking
sequences at the 5' and/or 3' ends of the molecule or the use of
phosphorothioate or 2'
0-methyl rather than phospliodiesterase linkages within the backbone of the
molecule. This concept is inherent in the production of PNAs and can be
extended in
all of these molecules by the inclusion of non-traditional bases such as
inosine,
queosine and butosine, as well as acetyl-, methyl-, thio- and similarly
modified forms
of adenine, cytidine, guanine, thymine and uridine which are not as easily
recognised
by endogenous endonucleases.
For treating abnormal conditions related to an under-expression of the
polypeptide of
the invention and its activity, several approaches are also available. One
approach
comprises administering to a subject a therapeutically effective amount of a
compound that activates the polypeptide, i.e., an agonist as described above,
to
alleviate the abnormal condition. Alternatively, a therapeutic amount of the
polypeptide in combination with a suitable pharmaceutical _carrier may be
administered to restore the relevant physiological balance of polypeptide.
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
63
Gene therapy may be employed to effect the endogenous production of the
polypeptide by the relevant cells in the subject. Gene therapy is used to
treat
permanently the inappropriate production of the polypeptide by replacing a
defective
gene with a corrected therapeutic gene.
Gene therapy of the present invention can occur in vivo or ex vivo. Ex vivo
gene
therapy requires the isolation and purification of patient cells, the
introduction of a
therapeutic gene and introduction of the genetically altered cells back into
the patient.
In contrast, in vivo gene therapy does not require isolation and purification
of a
patient's cells.
The tllerapeutic gene is typically "packaged" for administration to a patient.
Gene
delivery vehicles may be non-viral, such as liposomes, or replication-
deficient
viruses, such as adenovirus as described by Berkner, K.L., in Curr. Top.
Microbiol.
Immunol., 158, 39-66 (1992) or adeno-associated virus (AAV) vectors as
described
by Muzyczka, N., in Curr. Top. Microbiol. Imtnunol., 158, 97-129 (1992) and
U.S.
Patent No. 5,252,479. For example, a nucleic acid molecule encoding a
polypeptide of
the invention may be engineered for expression in a replication-defective
retroviral
vector. This expression construct may then be isolated and introduced into a
packaging cell transduced with a retroviral plasmid vector containing RNA
encoding
the polypeptide, such that the packaging cell now produces infectious viral
particles
containing the gene of interest. These producer cells may be administered to a
subject
for engineering cells in vivo and expression of the polypeptide in vivo (see
Chapter
20, Gene Therapy and other Molecular Genetic-based Therapeutic Approaches,
(and
references cited therein) in Human Molecular Genetics (1996), T Strachan and A
P
Read, BIOS Scientific Publishers Ltd).
Another approach is the administration of "naked DNA" in which the therapeutic
gene
is directly injected into the bloodstream or muscle tissue.
In situatioiis in which the polypeptides or nucleic acid molecules of the
invention are
disease-causing agents, the invention provides that they can be used in
vaccines to
raise antibodies against the disease causing agent.
Vaccines according to the invention may either be prophylactic (ie. to prevent
infection) or therapeutic (ie. to treat disease after infection). Such
vaccines comprise
immunising antigen(s), immunogen(s), polypeptide(s), protein(s) or nucleic
acid,
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
64
usually in combination with pharmaceutically-acceptable carriers as described
above,
which include any carrier that does not itself induce the production of
antibodies
harmful to the individual receiving the composition. Additionally, these
carriers may
function as immunostimulating agents ("adjuvants"). Furthermore, the antigen
or
iinmunogen may be conjugated to a bacterial toxoid, such as a toxoid from
diphtheria,
tetanus, cholera, H. pylori, and other pathogens.
Since polypeptides may be broken down in the stomach, vaccines comprising
polypeptides are preferably administered parenterally (for instance,
subcutaneous,
intramuscular, intravenous, or intradermal injection). Formulations suitable
for
parenteral adininistration include aqueous and non-aqueous sterile injection
solutions
which may contain anti-oxidants, buffers, bacteriostats and solutes which
render the
fonnulation isotonic with the blood of the recipient, and aqueous and non-
aqueous
sterile suspensions which may include suspending agents or thickening agents.
The vaccine formulations of the invention may be presented in unit-dose or
multi-
dose containers. For example, sealed ampoules and vials and may be stored in a
freeze-dried condition requiring only the addition of the sterile liquid
carrier
immediately prior to use. The dosage will depend on the specific activity of
the
vaccine and can be readily determined by routine experimentation.
Genetic delivery of antibodies that bind to polypeptides according to the
invention
may also be effected, for example, as described in International patent
application
W098/55607.
The technology referred to as jet injection (see, for example,
www.powderject.com)
may also be useful in the formulation of vaccine compositions.
A number of suitable methods for vaccination and vaccine delivery systems are
described in International patent application W000/29428.
This invention also relates to the use of nucleic acid molecules according to
the
present invention as diagnostic reagents. Detection of a mutated form of the
gene
characterised by the nucleic acid molecules of the invention which is
associated with
a dysfunction will provide a diagnostic tool that can add to, or define, a
diagnosis of a
disease, or susceptibility to adisease, which results from under-expression,
over-
expression or altered spatial or temporal expression of the gene. Individuals
carrying
mutations in the gene may be detected at the DNA level by a variety of
techniques.
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
Nucleic acid molecules for diagnosis may be obtained from a subject's cells,
such as
from blood, urine, saliva, tissue biopsy or autopsy material. The genomic DNA
may
be used directly for detection or may be amplified enzymatically by using PCR,
ligase
chain reaction (LCR), strand displacement amplification (SDA), or other
5 amplification techniques (see Saiki et al., Nature, 324, 163-166 (1986);
Bej, et al.,
Crit. Rev. Biochem. Molec. Biol., 26, 301-334 (1991); Birkenmeyer et al., J.
Virol.
Meth., 35, 117-126 (1991); Van Brunt, J., Bio/Technology, 8, 291-294 (1990))
prior
to analysis.
In one embodiment, this aspect of the invention provides a method of
diagnosing a
10 disease in a patient, comprising assessing the level of expression of a
natural gene
encoding a polypeptide according to the invention and comparing said level of
expression to a control level, wherein a level that is different to said
control level is
indicative of disease. The method may comprise the steps of:
a) contacting a sample of tissue from the patient with a nucleic acid probe
under
15 stringent conditions that allow the formation of a hybrid complex between a
nucleic acid molecule of the invention and the probe;
b) contacting a control sample with said probe under the same conditions used
in step
a);
c) and detecting the presence of hybrid complexes in said samples;
20 wherein detection of levels of the hybrid complex in the patient sample
that differ
from levels of the hybrid complex in the control sample is indicative of
disease.
A further aspect of the invention comprises a diagnostic method comprising the
steps
of:
a) obtaining a tissue sainple from a patient being tested for disease;
25 b) isolating a nucleic acid molecule according to the invention from said
tissue
sample; and
c) diagnosing the patient for disease by detecting the presence of a mutation
in the
nucleic acid molecule which is associated with disease.
To aid the detection of nucleic acid molecules in the- above-described
methods, an
30 amplification step, for example using PCR, may be included.
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
66
Deletions and insertions can be detected by a change in the size of the
amplified
product in comparison to the normal genotype. Point mutations can be
identified by
hybridizing amplified DNA to labelled RNA of the invention or alternatively,
labelled
antisense DNA sequences of the invention. Perfectly-matched sequences can be
distinguished from mismatched duplexes by RNase digestion or by assessing
differences in melting temperatures. The presence or absence of the mutation
in the
patient may be detected by contacting DNA with a nucleic acid probe that
hybridises
to the DNA under stringent conditions to fonn a hybrid double-stranded
molecule, the
hybrid double-stranded molecule having an unhybridised portion of the nucleic
acid
probe strand at any portion corresponding to a mutation associated with
disease; and
detecting the presence or absence of an unhybridised portion of the probe
strand as an
indication of the presence or absence of a disease-associated mutation in the
corresponding portion of the DNA strand.
Such diagnostics are particularly useful for prenatal and even neonatal
testing.
Point mutations and other sequence differences between the reference gene and
"mutant" genes can be identified by other well-known techniques, such as
direct DNA
sequencing or single-strand conformational polymorphism, (see Orita et al.,
Genomics, 5, 874-879 (1989)). For example, a sequencing primer may be used
with
double-stranded PCR product or a single-stranded template molecule generated
by a
modified PCR. The sequence determination is performed by conventional
procedures
with radiolabelled nucleotides or by automatic sequencing procedures with
fluorescent-tags. Cloned DNA segments may also be used as probes to detect
specific
DNA segments. The sensitivity of this method is greatly enhanced when combined
with PCR. Further, point inutations and other sequence variations, such as
polymorphisms, can be detected as described above, for example, through the
use of
allele-specific oligonucleotides for PCR amplification of sequences that
differ by
single nucleotides.
DNA sequence differences may also be detected by alterations in the
electrophoretic
mobility of DNA fragments in gels, with or without denaturing agents, or by
direct
DNA sequencing (for example, Myers et al., Science (1985) 230:1242). Sequence
changes at specific locations may also be revealed by nuclease protection
assays, such
as RNase and S1 protection or the chemical cleavage method (see Cotton et al.,
Proc.
Natl. Acad. Sci. USA (1985) 85: 4397-4401).
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
67
In addition to conventional gel electrophoresis and DNA sequencing, mutations
such
as microdeletions, aneuploidies, translocations, inversions, can also be
detected by in
situ analysis (see, for example, Keller et al., DNA Probes, 2nd Ed., Stockton
Press,
New York, N.Y., USA (1993)), that is, DNA or RNA sequences in cells can be
analysed for mutations without need for their isolation and/or immobilisation
onto a
membrane. Fluorescence in situ hybridization (FISH) is presently the most
coinmonly
applied method and numerous reviews of FISH have appeared (see, for example,
Trachuck et al., Science, 250, 559-562 (1990), and Trask et al., Trends,
Genet., 7,
149-154 (1991)).
In another embodiment of the invention, an array of oligonucleotide probes
comprising a nucleic acid molecule according to the invention can be
constructed to
conduct efficient screening of genetic variants, mutations and polymorphisms.
Array
technology methods are well known and have general applicability and can be
used to
address a variety of questions in molecular genetics including gene
expression,
genetic linkage, and genetic variability (see for example: M.Chee et al.,
Science
(1996), Vo1274, pp 610-613).
In one embodiment, the array is prepared and used according to the methods
described in PCT application W095/11995 (Chee et al); Lockhart, D. J. et al.
(1996)
Nat. Biotech. 14: 1675-1680); and Schena, M. et al. (1996) Proc. Natl. Acad.
Sci. 93:
10614-10619). Oligonucleotide pairs may range from two to over one million.
The
oligomers are synthesized at designated areas on a substrate using a light-
directed
chemical process. The substrate may be paper, nylon or other type of membrane,
filter, chip, glass slide or any other suitable solid support. In another
aspect, an
oligonucleotide may be synthesized on the surface of the substrate by using a
cheinical coupling procedure and an ink jet application apparatus, as
described in PCT
application W095/25116 (Baldeschweiler et al). In another aspect, a "gridded"
array
analogous to a dot (or slot) blot may be used to arrange and link cDNA
fragments or
oligonucleotides to the surface of a substrate using a vacuum system, thermal,
UV,
mechanical or chemical bonding procedures. An array, such as those described
above,
may be produced by hand or by using available devices (slot blot or dot blot
apparatus), materials (any suitable solid support), and machines (including
robotic
instruments), and may contain 8, 24, 96, 384, 1536 or 6144 oligonucleotides,
or any
other number between two and over one million which lends itself to the
efficient use
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
68
of commercially-available instrumentation.
In addition to the methods discussed above, diseases may be diagnosed by
methods
comprising determining, from a sample derived from a subject, an abnormally
decreased or increased level of polypeptide or mRNA. Decreased or increased
expression can be measured at the RNA level using any of the methods well
known in
the art for the quantitation of polynucleotides, such as, for example, nucleic
acid
amplification, for instance PCR, RT-PCR, RNase protection, Northern blotting
and
other hybridization methods.
Assay techniques that can be used to determine levels of a polypeptide of the
present
invention in a sample derived from a host are well-known to those of skill in
the art
and are discussed in some detail above (including radioimmunoassays,
competitive-
binding assays, Western Blot analysis and ELISA assays). This aspect of the
invention provides a diagnostic method which comprises the steps of: (a)
contacting a
ligand as described above with a biological sample under conditions suitable
for the
formation of a ligand-polypeptide coinplex; and (b) detecting said complex.
Protocols such as ELISA, RIA, and FACS for measuring polypeptide levels may
additionally provide a basis for diagnosing altered or abnormal levels of
polypeptide
expression. Normal or standard values for polypeptide expression are
established by
coinbining body fluids or cell extracts taken from normal mammalian subjects,
preferably humans, with antibody to the polypeptide under conditions suitable
for
complex formation The amount of standard complex formation may be quantified
by
various methods, such as by photometric means.
Antibodies which specifically bind to a polypeptide of the invention may be
used for
the diagnosis of conditions or diseases characterised by expression of the
polypeptide,
or in assays to monitor patients being treated with the polypeptides, nucleic
acid
molecules, ligands and other compounds of the invention. Antibodies useful for
diagnostic purposes may be prepared in the same maimer as those described
above for
therapeutics. Diagnostic assays for the polypeptide include methods that
utilise the
antibody and a label to detect the polypeptide in human body fluids or
extracts of cells
or tissues. The antibodies may be used with or without modification, and may
be
labelled by joining them, either covalently or non-covalently, with a reporter
molecule. A wide variety of reporter molecules known in the art may be used,
several
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
69
of which are described above.
Quantities of polypeptide expressed in subject, control and disease samples
from
biopsied tissues are compared with the standard values. Deviation between
standard
and subject values establishes the parameters for diagnosing disease.
Diagnostic
assays may be used to distinguish between absence, presence, and excess
expression
of polypeptide and to monitor regulation of polypeptide levels during
therapeutic
intervention. Such assays may also be used to evaluate the efficacy of a
particular
therapeutic treatment regimen in animal studies, in clinical trials or in
monitoring the
treatment of an individual patient.
A diagnostic kit of the present invention may comprise:
(a) a nucleic acid molecule of the present invention;
(b) a polypeptide of the present invention; or
(c) a ligand of the present invention.
In one aspect of the invention, a diagnostic kit may comprise a first
container
containing a nucleic acid probe that hybridises under stringent conditions
with a
nucleic acid molecule according to the invention; a second container
containing
primers useful for amplifying the nucleic acid molecule; and instructions for
using the
probe and primers for facilitating the diagnosis of disease. The kit may
further
comprise a third container holding an agent for digesting unhybridised RNA.
In an alternative aspect of the invention, a diagnostic kit may comprise an
array of
nucleic acid molecules, at least one of which may be a nucleic acid molecule
according to the invention.
To detect polypeptide according to the invention, a diagnostic kit may
comprise one
or more antibodies that bind to a polypeptide according to the invention; and
a reagent
useful for the detection of a binding reaction between the antibody and the
polypeptide.
Such kits will be of use in diagnosing a disease or susceptibility to disease,
particularly diseases in which leucine-rich repeat motif containing proteins
are
implicated. Such diseases include, but are not limited to, diseases of the
retina, retinal
pigment epithelium (RPE), and choroids; ocular neovascularization, ocular
inflammation and retinal degenerations; diabetic retinopathy, chronic
glaucoma,
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
retinal detachment, sickle cell retinopathy, senile macular degeneration,
retinal
neovascularization, subretinal neovascularization; rubeosis iritis
inflammatory
diseases, chronic posterior and pan uveitis, neoplasms, retinoblastoma,
pseudoglioma,
neovascular glaucoma; neovascularization resulting following a combined
vitrectomy
5 and lensectomy, vascular diseases retinal ischemia, choroidal vascular
insufficiency,
choroidal thrombosis, neovascularization of the optic nerve, diabetic macular
edema,
cystoid macular edema, retinitis pigmentosa, retinal vein occlusion,
proliferative
vitreoretinopathy, angioid streak, retinal artery occlusion,
neovascularization due to
penetration of the eye or ocular injury, neuropathies; Leber's, idiopathic,
drug-
10 induced, optic, and ischemic neropathies; spinal cord injuries, paraplegia,
neurodegenerative disorders, disorders of the central nervous system,
disorders of the
peripheral nervous system, brain injuries, cerebrovascular diseases,
Parkinson's
disease, corticobasal degeneration, motor neuron disease, amyotrophic lateral
sclerosis (ALS), multiple sclerosis, traumatic brain injury, stroke, post-
stroke, post-
15 traumatic brain injury, small-vessel cerebrovascular disease, dementias,
Alzheimer's
disease, vascular dementia, dementia with Lewy bodies, frontotemporal
dementia,
Parkinsonism, frontotemporal dementias, Pick's disease, progressive nuclear
palsy,
corticobasal degeneration, Huntington's disease, thalamic degeneration,
Creutzfeld-
Jakob deinentia, HIV dementia, schizophrenia with dementia, Korsakoffs
psychosis,
20 stroke and trauma.
Various aspects and embodiments of the present invention will now be described
in
more detail by way of example, with particular reference to INSP168, INSP168-
SV1,
INSP 149 and INSP 169.
It will be appreciated that modification of detail may be made without
departing from
25 the scope of the invention.
Brief description of the Figures
Figure 1: Amino acid aligmnent of INSP168, INSP168-SV1, INSP149 and INSP169
ORFs. The predicted transmembrane region is in bold. The predicted internal
LRR
region is boxed.
30 Fi ug re 2: Nucleotide sequence with translation of the PCR product INSP168
cloned
using primers INSP168-CP1 and INSP168-CP2. The predicted signal peptide is in
bold. The predicted internal LRR region is boxed. The position and sense of
the
SUBSTITUTE SHEET (RULE 26)
CA 02586486 2007-04-24
WO 2006/051333 71 PCT/GB2005/004390
primers are indicated by arrows.
Figure 3: Nucleotide sequence with translation of the PCR product INSP 168-SV
1
cloned using primers fNSP168-CP1 and INSP168-CP2. The predicted signal peptide
is in bold. The predicted internal LRR region is boxed. The position and sense
of the
primers are indicated by arrows.
Figure 4: Genomic organisation of the PCR product INSP168-SV1.
Figure 5: Amino acid alignment between INSP168, INSP168-SV1, INSP149 and
INSP 169 and Retinal Specific Protein PAL (SwissProt Acc. Code PALP HUMAN).
Fi ug re 6: Schematic domain representation of INSP168, INSP168-SV1, INSP149,
INSP 169, Retinal Specific Protein PAL (SwissProt Acc. Code PALP HUMAN) and
nogo receptor homolog (SwissProt Acc. Code Q6X814).
Fi ug re 7: Effect of INSP168 on Stat-2 nuclear translocation in U373 in two
distinct
experiments. The two left hand columns illustrate the effect of addition of
medium
only, the two middle columns illustrate the effect of addition of the positive
control
IFN-beta, and the two right hand columns illustrate the effect of addition of
INSP168
to the medium.
Fi , ru cDNA coding sequence and deduced peptide sequence of the cloned
INSP 169 extracellular domain. The position and orientation of primers used
for
cloning and sequencing are indicated by arrows. The sequence in bold
corresponds to
the centrally located BamHI site used in the assembly of the full length
clone.
Fi ug re 9: Nucleotide sequence of the cloned INSP169 extracellular domain.
Figure 10: Peptide sequence of the cloned cDNA from the N teriuinus to the TM
domain. Signal sequence is in italic; the leucine rich repeat region is in
bold; the
immunoglubulin C-2 domain is underlined; the fibronectin type3 domain is in
bold
Z5 italic.
SUBSTITUTE SHEET (RULE 26)
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
72
TABLE 1
Amino Acid Synonymous Groups More Preferred Synonymous Groups
Ser Gly, Ala, Ser, Thr, Pro Thr, Ser
Arg Asn, Lys, Gln, Arg, His Arg, Lys, His
Leu Phe, Ile, Val, Leu, Met Ile, Val, Leu, Met
Pro Gly, Ala, Ser, Thr, Pro Pro
Thr Gly, Ala, Ser, Thr, Pro Thr, Ser
Ala Gly, Thr, Pro, Ala, Ser Gly, Ala
Val Met, Phe, Ile, Leu, Val Met, Ile, Val, Leu
Gly Ala, Thr, Pro, Ser, Gly Gly, Ala
Ile Phe, Ile, Val, Leu, Met Ile, Val, Leu, Met
Phe Trp, Phe,Tyr Tyr, Phe
Tyr Trp, Phe,Tyr Phe, Tyr
Cys Ser, Thr, Cys Cys
His Asn, Lys, Gln, Arg, His Arg, Lys, His
Gln Glu, Asn, Asp, Gln Asn, Gln
Asn Glu, Asn, Asp, Gln Asn, Gln
Lys Asn, Lys, Gln, Arg, His, Arg, Lys, His
Asp Glu, Asn, Asp, Gln Asp, Glu
Glu Glu, Asn, Asp, Gln Asp, Glu
Met Phe, Ile, Val, Leu, Met Ile, Val, Leu, Met
Trp Trp, Phe,Tyr Trp
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
73
TABLE 2
Amino Acid Synonymous Groups
Ser D-Ser, Thr, D-Thr, allo-Thr, Met, D-Met, Met(O), D-Met(O), L-
Cys, D-Cys
Arg D-Arg, Lys, D-Lys, homo-Arg, D-homo-Arg, Met, Ile, D-.Met, D-
Ile, Orn, D-Orn
Leu D-Leu, Val, D-Val, AdaA, AdaG, Leu, D-Leu, Met, D-Met
Pro D-Pro, L-I-thioazolidine-4-carboxylic acid, D-or L-1-oxazolidine-
4-carboxylic acid
Thr D-Thr, Ser, D-Ser, allo-Thr, Met,D-Met, Met(O), D-Met(O), Val,
D-Val
Ala D-Ala, Gly, Aib, B-Ala, Acp, L-Cys, D-Cys
Val D-Val, Leu, D-Leu, Ile, D-Ile, Met, D-Met, AdaA, AdaG
Gly Ala, D-Ala, Pro, D-Pro, Aib, .beta.-Ala, Acp
Ile D-Ile, Val, D-Val, AdaA, AdaG, Leu, D-Leu, Met, D-Met
Phe D-Phe, Tyr, D-Thr, L-Dopa, His, D-His, Trp, D-Trp, Trans-3,4, or
5-phenylproline, AdaA, AdaG, cis-3,4, or 5-phenylproline, Bpa,
D-Bpa
Tyr D-Tyr, Phe, D-Phe, L-Dopa, His, D-His
Cys D-Cys, S--Me--Cys, Met, D-Met, Thr, D-Thr
Gln D-Gln, Asn, D-Asn, Glu, D-Glu, Asp, D-Asp
Asn D-Asn, Asp, D-Asp, Glu, D-Glu, Gln, D-Gln
Lys D-Lys, Arg, D-Arg, homo-Arg, D-homo-Arg, Met, D-Met, Ile, D-
Ile, Orn, D-Orn
Asp D-Asp, D-Asn, Asn, Glu, D-Glu, Gln, D-Gln
Glu D-Glu, D-Asp, Asp, Asn, D-Asn, Gln, D-Gln
Met D-Met, S--Me--Cys, Ile, D-Ile, Leu, D-Leu, Val, D-Val
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
74
Examples
Example 1: Cloning of INSP 168 and INSP 168-SV 1
Preparation of human cDNA templates
First strand cDNA was prepared from a variety of normal human tissue total RNA
sainples (Clontech, Stratagene, Ambion, Biochain Institute and in-house
preparations)
using Superscript II RNase H- Reverse Transcriptase (Invitrogen) according to
the
manufacturer's protocol. Oligo (dT)15 primer (1 l at 500 g/ml) (Promega), 2
g
human total RNA, 1 l 10 mM dNTP mix (10 mM each of dATP, dGTP, dCTP and
dTTP at neutral pH) and sterile distilled water to a final volume of 12 l
were
combined in a 1.5 ml Eppendorf tube, heated to 65 C for 5 min and then
chilled on
ice. The contents were collected by brief centrifugation and 4 l of 5X First-
Strand
Buffer, 2 l 0.1 M DTT, and 1 l RnaseOUT Recombinant Ribonuclease Inhibitor
(40 units/ l, Invitrogen) were added. The contents of the tube were mixed
gently and
incubated at 42 C for 2 min; then 1 1 (200 units) of SuperScript II enzyme
was
added and mixed gently by pipetting. The mixture was incubated at 42 C for 50
min
and then inactivated by heating at 70 C for 15 min. To remove RNA
complementary
to the cDNA, 1 l (2 units) of E. coli RNase H (Invitrogen) was added and the
reaction
mixture incubated at 37 C for 20 min. The final 21 l reaction mix was
diluted by
adding 179 l sterile water to give a total volume of 200 l. The cDNA
templates
used for the amplification of INSP 168 were derived from brain and eye RNA.
cDNA libraries
Human cDNA libraries (in bacteriophage lambda (k) vectors) were purchased from
Stratagene or Clontech or prepared at the Serono Pharmaceutical Research
Institute in
k ZAP or k GT10 vectors according to the manufacturer's protocol (Stratagene).
Bacteriophage k DNA was prepared from small scale cultures of infected E. coli
host
strain using the Wizard Lambda Preps DNA purification system according to the
manufacturer's instructions (Promega, Corporation, Madison WI.). cDNA library
templates used for the amplification of INSP168 were derived from brain, fetal
brain,
retina, and a mixed brain-lung-testis library.
Gene specific cloning primers for PCR
A pair of PCR primers having a length of between 18 and 25 bases was designed
for
amplifying the predicted coding sequence of the virtual cDNA using Primer
Designer
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
Software (Scientific & Educational Software, PO Box 72045, Durham, NC 27722-
2045, USA). PCR primers were optimized to have a Tm close to 55 + 10 C and a
GC
content of 40-60%. Primers were selected which had high selectivity for the
target
sequence (INSP 168) with little or no none specific priming.
5 PCR amplification of INSP168 f~~om human cDNA templates
Gene-specific cloning primers (INSP168-CP1 and INSP168-CP2, Figure 2, Figure 3
and Table 3) were designed to amplify a cDNA fragment of 614 bp covering the
entire of the predicted INSP 168 cds. The primer pair was used with the human
cDNA
samples and cDNA libraries listed above as PCR templates. PCR was performed in
a
10 final volume of 50 l containing 1X Platinum Taq High Fidelity (HiFi)
buffer, 2
mM MgSO4, 200 M dNTPs, 0.2 M of each cloning primer, 1 unit of Platinum
Taq DNA Polymerase High Fidelity (HiFi) (Invitrogen), and approximately 20 ng
of
template cDNA. Cycling was performed using an MJ Research DNA Engine,
programmed as follows: 94 C, 2 inin; 40 cycles of 94 C, 30 sec, 55 C, 30
sec, and
15 68 C, 1 min; followed by 1 cycle at 68 C for 7 min and a holding cycle at
4 C.
30 l of each ainplification product was visualized on a 0.8 % agarose gel in
1 X TAE
buffer (Invitrogen). Products of approximately the expected molecular weight
were
seen in the PCR products amplified from brain and eye cDNA templates. These
products was purified from the gel using the Promega Wizard PCR Preps DNA
20 Purification System, eluted in 50 l of water and subcloned directly.
Subcloning of PCR Products
The PCR products were subcloned into the topoisomerase I modified cloning
vector
(pCR4-TOPO) using the TA cloning kit purchased from the Invitrogen Corporation
using the conditions specified by the manufacturer. Briefly, 4 l of gel
purified PCR
25 product was incubated for 15 min at room temperature with 1 l of TOPO
vector and
1 l salt solution. The reaction mixture was then transformed into E. coli
strain
TOP10 (Invitrogen) as follows: a 50 l aliquot of One Shot TOP10 cells was
thawed
on ice and 2 l of TOPO reaction was added. The mixture was incubated for 15
min
on ice and then heat shocked by incubation at 42 C for exactly 30 s. Samples
were
30 returned to ice and 250 l of warm (room temperature) SOC media was added.
Samples were incubated with shaking (220 rpm) for 1 h at 37 C. The
transformation
mixture was then plated on L-broth (LB) plates containing ampicillin (100
g/ml) and
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
76
incubated overnight at 37 C.
Colony PCR
Colonies were inoculated into 50 l sterile water using a sterile toothpick. A
10 l
aliquot of the inoculum was then subjected to PCR in a total reaction volume
of 20 l
containing 1X AmpliTaqTM buffer, 200 M dNTPs, 20 pmoles of T7 primer, 20
pmoles of T3 primer, 1 unit of AmpliTaqTM (Applied Biosystems) using an MJ
Research DNA Engine. The cycling conditions were as follows: 94 C, 2 min; 30
cycles of 94 C, 30 sec, 48 C, 30 sec and 72 C for 1 min. Samples were
maintained
at 4 C (holding cycle) before further analysis.
PCR reaction products were analyzed on 1 % agarose gels in 1 X TAE buffer.
Colonies which gave PCR products of approximately the expected molecular
weight
(614 bp or 267 bp + 105 bp due to the multiple cloning site (MCS)) were grown
up
overnight at 37 C in 5 ml L-Broth (LB) containing ampicillin (100 g /ml),
with
shaking at 220 rpm.
Plasmid DNA preparation and sequencing
Miniprep plasmid DNA was prepared from the 5 ml culture using a Biorobot 8000
robotic system (Qiagen) or Wizard Plus SV Minipreps kit (Promega cat. no.
1460)
according to the manufacturer's instructions. Plasmid DNA was eluted in 80 l
of
sterile water. The DNA concentration was measured using a Spectramax 190
photometer (Molecular Devices). Plasmid DNA (200-500 ng) was subjected to DNA
sequencing with the T7 and T3 primers using the BigDye Terminator system
(Applied
Biosystems cat. no. 4390246) according to the manufacturer's instructions. The
primer sequences are shown in Table 3. Sequencing reactions were purified
using
Dye-Ex columns (Qiagen) or Montage SEQ 96 cleanup plates (Millipore cat. no.
LSKS09624) then analyzed on an Applied Biosystems 3700 sequencer.
Sequence analysis identified a clone, amplified from brain cDNA, which matched
the
expected INSP168 sequence. The plasmid map of the cloned PCR product is pCR4-
TOPO-INSP168. The nucleotide sequence with translation of the PCR product
INSP168 is shown in Figure 2.
'30 A second clone was identified, also amplified from brain, which contained
the
INSP168 cds with a 77 bp insertion towards the 3' end of the sequence. This
led to an
insertion of 32 amino acids and a frameshift such that an ORF of 229 amino
acids was
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
77
produced. The insertion represented an additional exon, giving an ORF encoded
in 4
exons. A stop codon was not identified but one present 2 bp downstream of the
3' end
of the new sequence in genomic DNA was assumed to be functional. The
nucleotide
sequence with translation of the PCR product INSP168-SV1 is shown in Figure 3.
This clone was named pCR4-TOPO-INSP168-SV1. The genomic organisation of the
INSP168-SV1 cds is shown in Figure 4. The plasmid map of the cloned PCR
product
is pCR4-TOPO-INSP 168-SV 1. The position of the INSP 168-CP 1 amplification
primer meant that the final base of the cds was missing - this base was added
during
transfer into the Gateway entry vector pDONR 221 (see below).
Table 3: INSP168 cloning and sequencing primers
Primer Sequence (5'-3')
INSP168-CPl GAG CAA TGC ATC TCT TTG CAT GTC
INSP168-CP2 AGC CGT AAC GTT CTA TCA GC
INSP168-EX1 GCA GGC TTC GCC ACC ATG CAT CTC TTT GCA TGT CT
INSP168-EX2 TG ATG GTG ATG GTG GCC AAG AAT AAT CCT GCT TGG
GCP Forward G GGG ACA AGT TTG TAC AAA AAA GCA GGC TTC GCC ACC
GGG GAC CAC TTT GTA CAA GAA AGC TGG GTT TCA ATG GTG ATG
GCP Reverse
GTG ATG GTG
pEAK12F GCC AGC TTG GCA CTT GAT GT
pEAK12R GAT GGA GGT GGA CGT GTC AG
21M13 TGT AAA ACG ACG GCC AGT
M13REV CAG GAA ACA GCT ATG ACC
T7 TAA TAC GAC TCA CTA TAG G
T3 ATT AAC CCT CAC TAA AGG
Underlined sequence = Kozak sequence
Bold = Stop codon
Italic sequence = His tag
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
78
Example 2: Construction of Mammalian Cell Expression Vectors for INSP 168
pCR4-TOPO-INSP168 was used as PCR template to generate pEAK12d and
pDEST12.2 expression clones containing the INSP168 ORF sequence with a 3'
sequence encoding a 6HIS tag using the Gatewaym cloning methodology
(Invitrogen).
Generation of Gateway compatible INSP168 ORF fused to an in fi=afne 6HIS tag
sequence
The first stage of the Gateway cloning process involves a two step PCR
reaction which
generates the ORF of INSP 168 flanked at the 5' end by an attBl recombination
site and
Kozak sequence, and flanked at the 3' end by a sequence encoding an in frame 6
histidine (6HIS) tag, a stop codon and the attB2 recoinbination site (Gateway
compatible
cDNA). The first PCR reaction (in a final volume of 50 l) contains
respectively: 1 l
(40 ng) of plasmid pCR4-TOPO-INSP168, 1.5 gi dNTPs (10 mM), 10 g1 of lOX Pfx
polymerase buffer, 1 l MgSO4 (50 mM), 0.5 l each of gene specific primer
(100 M)
(INSP168-EXI and INSP168-EX2), and 0.5 l Platinum Pfx DNA polymerase
(Invitrogen). The PCR reaction was performed using an initial denaturing step
of 95 C
for 2 min, followed by 12 cycles of 94 C for 15 s; 55 C for 30 s and 68 C
for 2 min;
and a holding cycle of 4 C. The amplification product was directly purified
using the
Wizard PCR Preps DNA Purification System (Promega) and recovered in 50 1
sterile
water according to the manufacturer's instructions.
The second PCR reaction (in a final volume of 50 gl) contained 10 l purified
PCR 1
product, 1.5 gl dNTPs (10 mM), 5 l of lOX Pfx polymerase buffer, 1 g1 MgSO4
(50
mM), 0.5 l of each Gateway conversion primer (100 M) (GCP forward and GCP
reverse) and 0.5 1 of Platinuin Pfx DNA polymerase. The conditions for the
2nd PCR
reaction were: 95 C for 1 min; 4 cycles of 94 C, 15 sec; 50 C, 30 sec and
68 C for 2
min; 25 cycles of 94 C, 15 sec; 55 C , 30 sec and 68 C, 2 min; followed by
a holding
cycle of 4 C. PCR product was visualized on 0.8 % agarose gel in 1 X TAE
buffer
(Invitrogen) and the band migrating at the predicted molecular mass (661 bp)
was
purified from the gel using the Wizard PCR Preps DNA Purification System
(Promega)
and recovered in 50 l sterile water according to the manufacturer's
instructions.
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
79
Subcloning of Gateway compatible INSPI68 ORF into Gateway entry vector
pDONR221 and expression vectors pEAK12d and pDEST12. 2
The second stage of the Gateway cloning process involves subcloning of the
Gateway
modified PCR products into the Gateway entry vector pDONR221 (Invitrogen) as
follows: 5 l of purified product from PCR2 were incubated with 1.5 l
pDONR221
vector (0.1 g/ l), 2 l BP buffer and 1.5 l of BP clonase enzyme mix
(Invitrogen) in a
final volume of 10 l at RT for 1 h. The reaction was stopped by addition of
proteinase
K 1 l (2 g/ l) and incubated at 37 C for a further 10 min. An aliquot of
this reaction
(1 l) was used to transform E. coli DH10B cells by electroporation as
follows: a 25 1
aliquot of DH10B electrocompetent cells (Invitrogen) was thawed on ice and I
g1 of the
BP reaction mix was added. The mixture was transferred to a chilled 0.1 cm
electroporation cuvette and the cells electroporated using a BioRad Gene-
PulserTM
according to the manufacturer's recommended protocol. SOC media (0.5 ml) which
had
been pre-warmed to room temperature was added immediately after
electroporation. The
mixture was transferred to a 15 ml snap-cap tube and incubated, with shaking
(220 rpm)
for 1 h at 37 C. Aliquots of the transformation mixture (10 l and 50 g1)
were then
plated on L-broth (LB) plates containing kanamycin (40 g/ml) and incubated
overnight
at 37 C.
Plasmid mini-prep DNA was prepared from 5 ml cultures from 6 of the resultant
colonies using a Qiaprep BioRobot 8000 system (Qiagen). Plasmid DNA (150-200
ng)
was subjected to DNA sequencing with 21M13 and M13Rev primers using the
BigDyeTerminator system (Applied Biosystems cat. no. 4336919) according to the
manufacturer's instructions. The primer sequences are shown in Table 3.
Sequencing
reactions were purified using Montage SEQ 96 cleanup plates (Millipore cat.
no.
LSKS09624) then analyzed on an Applied Biosystems 3700 sequencer.
Plasmid eluate (2 1 or approx. 150 ng) from one of the clones which contained
the
correct sequence (pENTR INSP 168-6HIS) was then used in a recombination
reaction
containing 1.5 1 of either pEAK12d vector or pDESTl2.2 vector (0.1 g / l),
2 l LR
buffer and 1.5 l of LR clonase (Invitrogen) in a final volume of 10 1. The
mixture was
incubated at RT for 1 h, stopped by addition of proteinase K (2 g) and
incubated at 37
C for a further 10 min. An aliquot of this reaction (1 ul) was used to
transform E. coli
DH10B cells by electroporation as follows: a 25 l aliquot of DH10B
electrocompetent
cells (Invitrogen) was thawed on ice and 1 l of the LR reaction mix was
added. The
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
mixture was transferred to a chilled 0.1 cm electroporation cuvette and the
cells
electroporated using a BioRad Gene-PulserTM according to the manufacturer's
recommended protocol. SOC media (0.5 ml) which had been pre-warmed to room
temperature was added immediately after electroporation. The mixture was
transferred to
5 a 15 mi snap-cap tube and incubated, with shaking (220 rpm) for 1 h at 37
C. Aliquots
of the transformation mixture (10 l and 50 l) were then plated on L-broth
(LB) plates
containing ampicillin (100 g/ml) and incubated overnight at 37 C.
Plasmid mini-prep DNA was prepared from 5 ml cultures from 6 of the resultant
colonies subcloned in eacll vector using a Qiaprep Bio Robot 8000 (Qiagen).
Plasmid
10 DNA (200-500 ng) in the pEAK12d vector was subjected to DNA sequencing
witli
pEAK12F and pEAK12R primers as described above. Plasmid DNA (200-500 ng) in
the
pDEST12.2 vector was subjected to DNA sequencing with 21M13 and M13Rev primers
as described above. Primer sequences are shown in Table 3.
CsCl gradient purified maxi-prep DNA was prepared from a 500 ml culture of the
15 sequence verified clone (pEAK12d INSP168-6HIS) using the method described
by
Sambrook J. et al., 1989 (in Molecular Cloning, a Laboratory Manual, 2nd
edition, Cold
Spring Harbor Laboratory Press). Plasmid DNA was resuspended at a
concentration of 1
g/ l in sterile water (or 10 mM Tris-HCl pH 8.5) and stored at -20 C.
Endotoxin-free maxi-prep DNA was prepared from a 500 ml culture of the
sequence
20 verified clone (pDEST12.2 INSP168-6HIS) using the EndoFree Plasmid Mega kit
(Qiagen) according to the manufacturer's instructions. Purified plasmid DNA
was
resuspended in endotoxin free TE buffer at a final concentration of at least 3
g/ l and
stored at -20 C.
Example 3: Tissue Distribution and Expression Levels of INSP168
25 Further experiments may now be performed to determine the tissue
distribution and
expression levels of the 1NSP168 polypeptide in vivo, on the basis of the
nucleotide
and amino acid sequence disclosed herein.
The presence of the transcripts for 1NSP168 may be investigated by PCR of cDNA
from different human tissues. The INSP 168 transcripts may be present at very
low
30 levels in the samples tested. Therefore, extreme- care is needed in the
design of
experiments to establish the presence of a transcript in various human tissues
as a
small amount of genomic contamination in the RNA preparation will provide a
false
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
81
positive result. Thus, all RNA should be treated with DNAse prior to use for
reverse
transcription. In addition, for each tissue a control reaction may be set up
in which
reverse transcription was not undertaken (a -ve RT control).
For example, 1 g of total RNA from each tissue may be used to generate cDNA
using
Multiscript reverse transcriptase (ABI) and random hexamer primers. For each
tissue,
a control reaction is set up in which all the constituents are added except
the reverse
transcriptase (-ve RT control). PCR reactions are set up for each tissue on
the reverse
transcribed RNA samples and the minus RT controls. INSP168-specific primers
may
readily be designed on the basis of the sequence information provided herein.
The
presence of a product of the correct molecular weight in the reverse
transcribed
sample together with the absence of a product in the minus RT control may be
taken
as evidence for the presence of a transcript in that tissue. Any suitable cDNA
libraries
may be used to screen for the INSP 168 transcripts, not only those generated
as
described above.
The tissue distribution pattern of the INSP 168 polypeptides will provide
further useful
information in relation to the function of those polypeptides.
Example 4: Expression and Purification of INSP168
Further experiments may now be performed using expression vectors.
Transfection of
mammalian cell lines with these vectors may enable the high level expression
of the
INSP 168 polypeptides and thus enable the continued investigation of the
functional
characteristics of the INSP168 polypeptides. The following material and
methods are
an example of those suitable in such experiments.
Cell Culture
Human Embryonic 1,,'-idney 293 cells expressing the Epstein-Barr virus Nuclear
Antigen (HEK293-EBNA, Invitrogen) are maintained in suspension in Ex-cell VPRO
serum-free medium (seed stock, maintenance medium, JRH). Sixteen to 20 hours
prior to transfection (Day-1), cells are seeded in 2x T225 flasks (50m1 per
flask in
DMEM / F12 (1:1) containing 2% FBS seeding medium (JRH) at a density of 2x105
cells/ml). The next day (transfection day 0) transfection takes place using
the
JetPEITM reagent (2 1/ g of plasmid DNA, PolyPlus-transfection). For each
flask,
plasmid DNA is co-transfected with GFP (fluorescent reporter gene) DNA. The
transfection mix is then added to the 2xT225 flasks and incubated at 37 C
(5%CO2)
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
82
for 6 days. Confirmation of positive transfection may be carried out by
qualitative
fluorescence examination at day 1 and day 6 (Axiovert 10 Zeiss).
On day 6 (harvest day), supernatants from the two flasks are pooled and
centrifuged
(e.g. 4 C, 400g) and placed into a pot bearing a unique identifier. One
aliquot (500 1)
is kept for QC of the 6His-tagged protein (internal bioprocessing QC).
Scale-up batches may be produced by following the protocol called "PEI
transfection
of suspension cells", referenced BP/PEI/HH/02/04, with PolyEthylenelmine from
Polysciences as transfection agent.
Purification pyocess
The culture medium sample containing the recombinant protein with a C-terminal
6His tag is diluted with cold buffer A (50mM NaH2PO4; 600mM NaCI; 8.7 % (w/v)
glycerol, pH 7.5). The sample is filtered then through a sterile filter
(Millipore) and
kept at 4 C in a sterile square media bottle (Nalgene).
The purification is performed at 4 C on the VISION workstation (Applied
Biosystems) connected to an automatic sample loader (Labomatic). The
purification
procedure is composed of two sequential steps, metal affinity chromatography
on a
Poros 20 MC (Applied Biosystems) column charged with Ni ions (4.6 x 50 mm,
0.83m1), followed by gel filtration on a Sephadex G-25 medium (Amersham
Pharmacia) column (1,0 x 10cm).
For the first chromatography step the metal affinity column is regenerated
with 30
column volumes of EDTA solution (100mM EDTA; 1M NaCl; pH 8.0), recharged
with Ni ions througli washing with 15 column volumes of a 100mM NiSO4
solution,
washed with 10 column volumes of buffer A, followed by 7 column volumes of
buffer B (50mM NaH2PO4; 600mM NaCl; 8.7 % (w/v) glycerol, 400mM; imidazole,
pH 7.5), and finally equilibrated with 15 column volumes of buffer A
containing
15mM imidazole. The sample is transferred, by the Labomatic sample loader,
into a
200ml sample loop and subsequently charged onto the Ni metal affinity column
at a
flow rate of 10m1/min. The column is washed with 12 column volumes of buffer
A,
followed by 28 column volumes of buffer A containing 20mM imidazole. During
the
20mM imidazole wash loosely attached contaminating proteins are eluted from
the
column. The recombinant His-tagged protein is finally eluted with 10 column
volumes of buffer B at a flow rate of 2ml/min, and the eluted protein is
collected.
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
83
For the second chromatography step, the Sephadex G-25 gel-filtration colunm is
regenerated with 2m1 of buffer D(1.137M NaCI; 2.7mM KC1; 1.5mM KH2PO4; 8mM
Nk,HPO4i pH 7.2), and subsequently equilibrated with 4 column volumes of
buffer C
(137mM NaCI; 2.7mM KCI; 1.5mM KH2PO4i 8mM Na2HPO4; 20% (w/v) glycerol;
pH 7.4). The peak fraction eluted from the Ni-column is automatically loaded
onto the
Sephadex G-25 column through the integrated sample loader on the VISION and
the
protein is eluted with buffer C at a flow rate of 2 ml/min. The fraction was
filtered
through a sterile centrifugation filter (Millipore), frozen and stored at -80
C. An
aliquot of the sainple is analyzed on SDS-PAGE (4-12% NuPAGE gel; Novex)
Western blot with anti-His antibodies. The NuPAGE gel may be stained in a 0.1
%
Coomassie blue R250 staining solution (30% methanol, 10% acetic acid) at room
temperature for lh and subsequently destained in 20% methanol, 7.5% acetic
acid
until the background is clear and the protein bands clearly visible.
Following the electrophoresis the proteins are electrotransferred from the gel
to a
nitrocellulose membrane. The membrane is blocked with 5% milk powder in buffer
E
(137mM NaCI; 2.7mM KCI; 1.5mM KH2PO4; 8mM Na2HPO4; 0.1 % Tween 20, pH
7.4) for lh at room temperature, and subsequently incubated with a mixture of
2
rabbit polyclonal anti-His antibodies (G-18 and H-15, 0.2gghnl each; Santa
Cruz) in
2.5% milk powder in buffer E overnight at 4 C. After a further 1 hour
incubation at
room temperature, the membrane is washed with buffer E (3 x 10min), and then
incubated with a secondary HRP-conjugated anti-rabbit antibody (DAKO, HRP
0399)
diluted 1/3000 in buffer E containing 2.5% milk powder for 2 hours at room
temperature. After washing with buffer E(3 x 10 minutes), the membrane is
developed with the ECL kit (Amersham Pharmacia) for 1 min. The membrane is
subsequently exposed to a Hyperfilm (Amersham Pharmacia), the film developed
and
the western blot image visually analysed.
For samples that showed detectable protein bands by Coomassie staining, the
protein
concentration may be determined using the BCA protein assay kit (Pierce) with
bovine serum albumin as standard.
Furthermore, overexpression or knock-down of the expression of the
polypeptides in
ce111ines_may be used to determine the effect on transcriptional activation of
the host
cell genome. Dimerisation partners, co-activators and co-repressors of the
INSP168
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
84
polypeptides may be identified by immunoprecipitation combined with Western
blotting and immunoprecipitation combined with mass spectroscopy.
Example 5: Biological Significance of INSP168 INSP168-SV1, INSP149 and
INSP 169
As explained above, INSP168, INSP168-SV1, INSP149 and INSP169 are structurally
related to the Retinal Specific Protein PAL (SwissProt Acc. Code PALP HUMAN)
and to a nogo receptor homolog (SwissProt Acc. Code Q6X814). An amino acid
alignment between INSP168, INSP168-SV1, INSP149 and INSP169 and PAL is
shown in Figure 5, and the schematic representation of domains is shown in
Figure 6.
PAL may be implicated in diseases of the retina, retinal piginent epithelium
(RPE),
and choroids (see for example JP2001128686). These include ocular
neovascularization, ocular inflammation and retinal degenerations. Specific
examples
of these disease states include diabetic retinopathy, chronic glaucoma,
retinal
detachment, sickle cell retinopathy, senile macular degeneration, retinal
neovascularization, subretinal neovascularization; rubeosis iritis
inflammatory
diseases, chronic posterior and pan uveitis, neoplasms, retinoblastoma,
pseudoglioma,
neovascular glaucoma; neovascularization resulting following a combined
vitrectomy
and lensectomy, vascular diseases retinal ischemia, choroidal vascular
insufficiency,
choroidal thrombosis, neovascularization of the optic nerve, diabetic macular
edema,
cystoid macular edema, retinitis pigmentosa, retinal vein occlusion,
proliferative
vitreoretinopathy, angioid streak, and retinal artery occlusion, and,
neovascularization
due to penetration of the eye or ocular injury. Additional relevant disease
include the
neuropathies, such as Leber's, idiopathic, drug-induced, optic, and ischemic
neropathies.
Nogo receptor-like proteins could be major inliibitors, of CNS neuronal
regeneration
(Schwab ME. Curr Opin Neurobiol. 2004 Feb;14(1):l 18-24; Teng et al. J
Neurochem.
2004 May;89(4):801-6). Animals treated with antibodies targeted to Nogo-A
always
showed a higher degree of recovery in various behavioural tests (e.g. IN-1
Fab'
fragments or new purified IgGs against Nogo-A). In addition, a Nogo-66
antagonistic
peptide (NEP1-40) effected significantly axon growth of the corticospinal
tract and
improved functional recovery in rats inflicted with mid-thoracic spinal cord
hemisections. Subcutaneous administration of NEP1-40 in spinal cord lesioned
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
animals resulted in extensive growth of corticospinal axons, sprouting of
serotonergic
fibres, synapse formation and enhanced locomotor recovery. Soluble Fc fusion
proteins of the Nogo receptor subunit NgR, which blocks Nogo, significantly
reduce
the inhibitory activity of myelin. Similar results were obtained after Nogo
gene
5 deletions and blockade of the downstream messengers Rho-A and ROCK in animal
models.
The leucine-rich repeat domain of SLIT proteins is sufficient for guiding both
axon
projection and neuronal migration in vitro (the LRR region of SLIT is
structurally
related to the LRR region of INSP168, INSP168-SV1, INSP149 and INSP169). SLIT-
10 like proteins are thought to act as molecular guidance cue in cellular
migration, and
function appears to be mediated by interaction with roundabaout homolog
receptors
(bind ROBO1 and ROBO2 with high affinity). During neural development, SLIT are
involved in axonal navigation at the ventral midline of the neural tube and
projection
of axons to different regions. In spinal chord development, SLIT may play a
role in
15 guiding commissural axons once they reached the floor plate by modulating
the
response to netrin. SLIT may be implicated in spinal chord midline post-
crossing
axon repulsion. In the developing visual system appears to function as
repellent for
retinal ganglion axons by providing a repulsion that directs these axons along
their
appropriate paths prior to, and after passage through, the optic chiasm. In
vitro, SLIT
20 collapses and repels retinal ganglion cell growth cones. SLIT seems to play
a role in
branching and arborization of CNS sensory axons, and in neuronal cell
migration. In
vitro, Slit homolog 2 protein N-product, but not Slit homolog 2 protein C-
product,
repells olfactory bulb (OB) but not dorsal root ganglia (DRG) axons, induces
OB
growth cones collapse and induces branching of DRG axons. SLIT seeins to be
25 involved in regulating leukocyte migration.
INSP168, INSP168-SVl, INSP149 and INSP169 and/or fragments thereof (e.g.
fragments containing the LRR region) can be useful in the diagnosis and/or
treatment
of diseases for which other (e.g. above mentioned PAL- and Nogo receptor-like
proteins) structurally related proteins demonstrate therapeutic activity.
30 As such, INSP168, INSP168-SV1, INSP149 and INSP169 may be implicated in
diseases of the retina, spinal cord injuries (e.g. paraplegia) and
neurodegenerative
disorders. These include disorders of the central nervous system as well as
disorders
of the peripheral nervous system. Neurodegenerative disorders include, but are
not
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
86
limited to, brain injuries, cerebrovascular diseases and their consequences,
Parkinson's
disease, corticobasal degeneration, motor neuron disease (including
amyotrophic
lateral sclerosis, ALS), multiple sclerosis, traumatic brain injury, stroke,
post-stroke,
post- traumatic brain injury, and small-vessel cerebrovascular disease.
Dementias,
such as Alzheimer's disease, vascular dementia, dementia with Lewy bodies,
frontotemporal dementia and Parkinsonism, frontotemporal dementias (including
Pick's disease), progressive nuclear palsy, corticobasal degeneration,
Huntington's
disease, thalamic degeneration, Creutzfeld-Jakob dementia, HIV dementia,
schizophrenia with dementia, and Korsakoffs psychosis, as well as stroke and
trauma.
Thus, INSPI49, INSP168, INSP168-SV1 and INSP169 may be implicated in diseases
of the retina, retinal piginent epithelium (RPE), and choroids; ocular
neovascularization, ocular inflammation and retinal degenerations; diabetic
retinopathy, chronic glaucoma, retinal detaclunent, sickle cell retinopathy,
senile
macular degeneration, retinal neovascularization, subretinal
neovascularization;
rubeosis iritis inflammatory diseases, chronic posterior and pan uveitis,
neoplasms,
retinoblastoma, pseudoglioma, neovascular glaucoma; neovascularization
resulting
following a combined vitrectomy and lensectomy, vascular diseases retinal
ischemia,
choroidal vascular insufficiency, choroidal throinbosis, neovascularization of
the optic
nerve, diabetic macular edema, cystoid macular edema, retinitis pigmentosa,
retinal
vein occlusion, proliferative vitreoretinopathy, angioid streak, retinal
artery occlusion,
neovascularization due to penetration of the eye or ocular injury,
neuropathies;
Leber's, idiopathic, drug-induced, optic, and ischemic neropathies; spinal
cord
injuries, paraplegia, neurodegenerative disorders, disorders of the central
nervous
systein, disorders of the peripheral nervous system, brain injuries,
cerebrovascular
diseases, Parkinson's disease, corticobasal degeneration, motor neuron
disease,
amyotrophic lateral sclerosis (ALS), multiple sclerosis, traumatic brain
injury, stroke,
post-stroke, post- traumatic brain injury, small-vessel cerebrovascular
disease,
dementias, Alzheimer's disease, vascular dementia, dementia with Lewy bodies,
frontotemporal dementia, Parkinsonism, frontotemporal dementias, Pick's
disease,
progressive nuclear palsy, corticobasal degeneration, Huntington's disease,
thalamic
degeneration, Creutzfeld-Jakob dementia, HIV dementia, schizophrenia with
dementia, Korsakoffs psychosis, stroke and trauma.
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
87
Example 6: Neuroprotective activities of INSP 168
Neuro-inflammation is a common feature of several neurological diseases,
traumatic
situations (at central or peripheral level), stroke (brain, heart, renal), or
infectious
diseases (mediated by viral agents such as HIV or bacterial agents such as
meningitis), leading to an excessive inflammatory response in central nervous
system.
Many stimuli, originated by neuronal or oligodendroglial cells suffering due
to these
various conditions, can trigger neuro-inflammation. In particular, astrocytes
can
secrete various chemokines and cytokines, inducing a recruitment of additional
leukocytes that in their turn will further stimulate astrocytes, leading to an
exacerbated response. In chronic neurodegenerative diseases such as multiple
sclerosis (MS), spinal muscular atrophies (SMA), Alzheimer's disease (AD),
Parkinson's disease (PD), Huntington's disease (HD), or amylotrophic lateral
sclerosis
(ALS), the presence of persistent neuro-inflammation is thought to be involved
in the
progression of the disease and in the case of AD in the secondary events such
as
micro-hemorrhagic events (Cacquevel M et al., Curr Drug Targets. 2004, 5: 529-
534;
Chavarria A et al., Autoimmun Rev. 2004, 3: 251-260; Ambrosini E and Aloisi F,
Neurochem Res. 2004, 29: 1017-1038).
The biological properties of INSP168 related to neuroprotection, maintenance
of
axonal integrity, myelination and re-/generation of myelin producing cells,
can be
tested in various assays involving cell lines. For example, the
neuroimmunodulatory
effects of a compound can be evaluated in U373, a human astroglioma cell line
in
which the nuclear translocation of specific regulatory proteins involved in
cytokine/chemokine expression can be quantified (Le Roy E et al., J Virol.
1999, 73:
6582-9; Jin Y et al., J Infect Dis. 1998, 177: 1629-1638; Acevedo-Duncan M et
aL,
Cell Growth Differ. 1995, 6: 1353-1365).
A series of assays was performed on the human astroglioma cell line U373 to
check
whether INSP 168 can affect the translocation of transcription factors such as
Stat-2
(Signal transducer and activator of transcription-2, a transcription factor
induced by
cytokines and modulating IFNbeta response; Banninger G and Reich NC, J Biol
Chem. 2004, 279: 39199-39206; Leonard WJ, Int J Hematol. 2001, 73: 271-277)
from
the cytoplasm to the nucleus.
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
88
U373 cells (ECACC ref no: 89081403) were seeded at the density of 4000
cells/well
in 96-well-plates (Packard ViewPlate-96, black; Cat. No. 6005225) in 80 l of
DMEM containing 10% FCS (Fetak Calf Serum) and left overnight at 37 C in a
humidified 5% C02 incubator. The following day, 20 l of culture medium alone,
or
containing the protein to test (medium added with 1000 IU/ml of recombinant
IFNbeta, or medium from His-tagged INSP168 expressing cells) was added to the
cells. Thirty minutes after, the medium was removed and cells were fixed with
3.7%
formaldehyde (Sigma; Cat. No. 25,254-9) and processed for immunostaining using
commercial kits (for c-Jun immunostaining, Cellomics c-Jun activation HitKit,
Cat.
No. K01-0003-1; for Stat-2, Cellomics Stat-2 activation HitKit, Cat. No. K01-
0005-1)
according to the manufacturer's instructions. After staining, plates were read
on an
image analysis system (ArrayScan II HCS System; Cellomics).
Results were expressed as "nuclear translocation units". The nuclear
translocation unit
is the measure of the fluorescence intensity of the target in the nuclear
region minus
that of the cytoplasm region, reported as an average for all analyzed cells in
the well
(approx. 100 cells/well). In order to compare several experiments, results
were also
expressed as the percentage of maximal stimulation calculated with the
positive
control (IFNbeta). Statistics were performed using Student's T test or measure
analysis of variance (ANOVA) and one-way ANOVA, followed by Dunnett's test
depending of the number of groups per experiments. The level of significance
was set
at p < 0.05. The results were expressed as mean :L standard error of the mean
(s.e.m.).
The addition of a culture medium containing INSP 168 was found to stimulate
Stat-2
nuclear translocation in U373 cells, as indicated by the statistically
significant
increase of fluorescence intensity in the nuclei. The response corresponds to
20-30%
of the maximal level achieved with IFNbeta, the positive control (Fig. 7; **
means
p<0.005, *** means p<0.0005).
This first series of experiments revealed that INSP168 has the capacity to
stimulate
intracellular signaling by inducing Stat-2 nuclear translocation in U373
cells.
Activation of Stat proteins signaling is known to be associated with
immunomodulation and eventually cell proliferation (Pfitzner E et al., Curr
Pharm
Des. 2004, 10: 2839-2850).
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
89
Example 7: Cloning of the INSP 169 extracellular domain (INSP 169ec)
INSP169 is a re-prediction of INSP149 which encodes a protein of 679 ainino
acids
spanning 4 coding exons. A transmembrane (TM) domain is predicted near the C-
terminus. The N-terminal extracellular domain extends over 580 amino acids and
includes a cluster of 4 Leucine rich repeats (LRR) flanked by Cys-rich
domains, an
IgC-2 domain and a fibronectin type 3 domain. The extracellular domain of this
prediction has been cloned.
The cloning strategy used was to prepare an initial pool of RNAs from a wide
variety
of human tissues (see below) and from this to make a single preparation of
multi-
tissue polyA+ mRNA as template for reverse transcription. Gene specific cDNA
primers were designed for a small set of the predictions (typically 5-10
sequences),
and aliquots of the resulting cDNA mix provided templates for separate PCR
reactions using primers designed to obtain the corresponding coding region.
Amplified fragments were then purified by gel electorphoresis and cloned into
the
Bluescript cloning vector by virtue of specific restriction sites added to the
ends of the
PCR primers.
In the case of INSP169ec, a two step strategy was employed which makes use of
a
unique BamHI restriction site in the central part of this sequence. A first RT-
PCR was
performed to obtain the 1063nt sequence between the TM domain to the BamH1
site;
a second RT-PCR was performed to obtain the remaining 683nt sequence upstream
of
the BamHI site as far as the initiator methionine codon. These two fragments
were
then assembled following BamHI digestion and ligation.
The gene specific priiners used are described below and in Table 4 below. A
cDNA of
1740 nucleotides was obtained and sequence analysis of this insert revealed
the
predicted cDNA sequence for the complete extracellular domain of INSP169. The
coding region and position of the oligonucleotide primers used in the cloning
of
INSP169ec are shown in Figure S.
7.1 Preparation of a human multi-tissue cDNA template
A preparation of human RNA was prepared by mixing approximately 10 g total
RNA from each of the following sources:
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
Brain (Clontech), Heart (Clontech), Kidney (Clontech), Liver (Clontech), Lung
(Clontech), Placenta (Clontech), Skeletal Muscle (Clontech), Small Intestine
(Clontech), Spleen (Clontech), Thymus (Clontech), Uterus (Clontech) Bone
Marrow
(Clontech) Thyroid (Clontech), Ovary (Ambion), Testis (Ambion), Prostate
5 (Ambion), Skin (Resgen), Pancreas (Clontech), Salivary gland (BD
Biosciences),
Adrenal gland (BD Biosciences), Breast (Ambion), Pituitary gland (BioChain
Institut), Stomach (Ambion), Mammary gland (Clontech), Lymph Node (BioChain
Institut), Adipose tissue (BioChain Institut), Bladder (BioChain Institut),
Appendix
(BioChain Institut), Artery (BioChain Institut), Throat (BioChain Institut),
Universal
10 Human Reference (Stratagene), Foetal Kidney (Stratagene), Foetal Brain
(BioChain
Institut), Foetal Spleen (BioChain Institut), Foetal Liver (BioChain
Institut), Foetal
Heart (BioChain Institut), Foetal Lung (BioChain Institut), Peripheral blood
monocytes (prepared in-house from buffy coat).
The resulting pool of total RNA was fractionated by chromatography on a pre-
packed
15 oligo-dT column (Stratagene) according to the protocol supplied by the
manufacturer.
Approximately 400 g total RNA yielded 16 g polyA+ mRNA which was aliquotted
and frozen at -80 C.
7.2 RT-PCR cloning of INSP169ec
7.2.1 Stage 1: Cloning of nucleotides 678-1740
20 7.2.1.1 cDNA synthesis
In the first stage of cloning, a gene specific cDNA primer for INSP169 (AS502)
located in the TM domain, was pooled with gene specific cDNA primers for 9
other
predictions, each at a final concentration of 1pM. The pooled cDNA primer set
was
diluted 10 fold into 50 l of a mixutre containing 1 x RT buffer, 500 M each
dNTPs,
25 l0U/ l RNAguard (Pharmacia) and 1 g denatured polyA+ RNA prepared as
described above. cDNA synthesis was initiated by addition of l0U Omniscript
reverse
transcriptase (Qiagen) and allowed to proceed for 1h at 37 C. At the end of
the
reaction, 5 1 of the cDNA mix was used for PCR amplification as described
below.
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
91
7.2.1.2 PCR afnplification fof I1NSP169 nucleotides 678-1740
Top strand (AS503) and bottom strand (AS504) PCR primers (see Table 4) were
designed to span the predicted coding sequence between an internal BamHI site
and
the TM domain. EcoRI restrictions sites were added at the 5' end of each
primer. A
reaction mixture was set up containing 1 x PCR buffer, 0.2mM each dNTP, 0.5 M
each PCR primer, 5 1 cDNA above, and the PCR reaction was initiated by
addition of
5U PfuTurbo (Stratagene). Cycling conditions for 'touchdown' PCR were: 94 C 2
min (I cycle); 94 C 30 sec, 64 C (decreasing by 1 C each cycle) 30 sec, 72
C 90
sec (14 cycles); 94 C 30 sec, 50 C 30 sec, 72 C 90 see (25 cycles); 72 C 7
min (1
cycle). An aliquot of the PCR reaction was analysed by electrophoresis in a
0.8%
agarose gel and the remainder was purified using the Wizard PCR Cleanup System
(Promega) as recommended by the manufacturer, prior to subcloning of the PCR
products.
7.2.1.3 Subcloning PCR products
An aliquot of the purified PCR reation was digested with EcoRI (New England
Biolabs) for 2h at 37 C using the enzyme buffer supplied by the manufacturer.
In parallel, an appropriate amount of the Bluescript BSK- cloning vector was
digested
with EcoRI and dephosphorylated using calf intestinal alkaline phosphatase
(Roche
Diagnostics) according to the supplier's recommendations. The full length
linearized
and dephosphorylated cloning vector was separated on a 0.8% agarose gel,
excised
and purified using the Wizard Cleanup System (Promega) according to the
protocol
provided by the manufacturer. The purified vector DNA and PCR products were
mixed in a molar ratio of 1:3 and precipitated overnight at -20 C in the
presence of
2.5 volumes ethanol. The precipitated DNA was recovered by centrifugation,
washed
in 70% ethanol, dried under vacuum and ligated in a final volume of l0 1 using
the
Rapid Ligation Kit (Roche Diagnostics) according to the protocol supplied by
the
manufacturers.
The ligation mixture was then used to transform E. coli strain JM101 as
follows: 50 l
aliquots of competent JM101 cells were thawed on ice and 1 1 or 5 1 of the
ligation
mixture was added. The cells were incubated for 40 min on ice and then heat
shocked
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
92
by incubation at 42 C for 2min. lml of warm (room temperature) L-Broth (LB)
was
added and samples were incubated for a further 1 h at 37 C. The
transformation
mixture was then plated on LB plates containing ampicillin (100 g/ml) IPTG
(0.1 M) and X-gal (50 g/ml) and incubated overnight at 37 C. Single white
colonies
were chosen for plasmid isolation.
7.2.1.4 Plasmid DNA preparation, restriction digestion and sequence analysis.
Miniprep plasmid DNA was prepared from 5 ml cultures using a Biorobot 8000
robotic system (Qiagen) according to the manufacturer's instructions. Plasmid
DNA
was eluted in 80 l sterile water. The DNA concentration was measured using an
Eppendorf BO photometer or Spectramax 190 photometer (Molecular Devices).
Aliquots of miniprep plasmid DNAs (100-200ng) were digested with EcoRI for 2h
at
37 C and analysed by electrophoresis in 0.8% agarose gels. Plasmids with
inserts of
the expected size of about 1.lkb were selected for DNA sequence analysis.
Inserts
were sequenced from either end by mixing 200-500 ng plasmid DNA with the
either
the T7 or T3 sequencing primers (see Table 4). Sequencing reactions were
processed
using the BigDye Terminator system (Applied Biosystems cat. no. 4390246)
according to the manufacturer's instructions. Products were purified using Dye-
Ex
columns (Qiagen) or Montage SEQ 96 cleanup plates (Millipore cat. no.
LSKS09624)
and analyzed on an Applied Biosystems 3700 sequencer.
7.2.1.5 Results of cloning and sequence analysis of nucleotides 678-1740.
The predicted mRNA coding sequence for the region spanning the internal BainHI
site to the TM domain was confirmed. The DNA miniprep #14 was taken as a
representative clone for further work.
7.2.2 Stage 2: Cloning of nucleotides 1-683
7.2.2.1 cDNA synthesis
In the second stage of cloning, a gene specific cDNA primer for INSP169
(AS515)
located immediately downstream of the internal BamHI site, was pooled with
gene
specific cDNA primers for 6 other predictions, each at a final concentration
of 1pM.
The pooled cDNA primer set was diluted 10 fold into 40 1 of a mixutre
containing 1
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
93
x RT buffer, 500 M each dNTPs, 10U/ l RNAguard (Pharmacia) and 1 g denatured
polyA+ RNA prepared as described above. cDNA synthesis was initiated by
addition
of l0U Omniscript reverse transcriptase (Qiagen) and allowed to proceed for lh
at 37
C. At the end of the reaction, 5 1 of the cDNA mix was used for PCR
amplification
as described below.
7.2.2.2 PCR amplification for INSP169 nucleotides 1-683
Top strand (AS516) and bottom strand (AS517) PCR primers (see Table 4) were
designed to span the predicted coding sequence between the initiator
methionine and
the internal BamHI site. A BamH1 restriction site was added at the 5' end of
AS516.
A reaction mixture was set up containing 1 x PCR buffer, 0.2mM each dNTP, 0.5
M
each PCR primer, 5 l cDNA above, and the PCR reaction was initiated by
addition of
5U PfuTurbo (Stratagene). Cycling conditions for 'touchdown' PCR were: 94 C 2
min (I cycle); 94 C 30 sec, 64 C (decreasing by 1 C each cycle) 30 sec, 72
C 80
sec (14 cycles); 94 C 30 sec, 50 C 30 sec, 72 C 80 sec (25 cycles); 72 C 7
min (1
cycle). An aliquot of the PCR reaction was analysed by electrophoresis in a
0.8%
agarose gel and the remainder was purified using the Wizard PCR Cleanup System
(Promega) as recommended by the manufacturer, prior to subcloning of the PCR
products.
7.2.2.3 Subcloning PCR products
An aliquot of the purified PCR reation was digested with BamHI (New England
Biolabs) for 2h at 37 C using the enzyme buffer supplied by the manufacturer.
In parallel, an appropriate amount of the Bluescript BSK- cloning vector was
digested
with BamH1 and dephosphorylated using calf intestinal alkaline phosphatase
(Roche
Diagnostics) according to the supplier's recommendations. The full length
linearized
and dephosphorylated cloning vector was separated on a 0.8% agarose gel,
excised
and purified using the Wizard Cleanup System (Promega) according to the
protocol
provided by the manufacturer. The purified vector DNA and PCR products were
mixed in a molar ratio of 1:3 and precipitated overnight at -20 C in the
presence of
2.5 volumes ethanol. The precipitated DNA was recovered by centrifugation,
washed
in 70% ethanol, dried under vacuum and ligated in a final volume of 10 1 using
the
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
94
Rapid Ligation Kit (Roche Diagnostics) according to the protocol supplied by
the
manufacturers.
The ligation mixture was then used to transform E. coli strain JM101 as
follows: 50 l
aliquots of competent JM101 cells were thawed on ice and l l or 5 l of the
ligation
mixture was added. The cells were incubated for 40 min on ice and then heat
shocked
by incubation at 42 C for 2min. lml of warm (room temperature) L-Broth (LB)
was
added and samples were incubated for a further 1 h at 37 C. The
transformation
mixture was then plated on LB plates containing ampicillin (100 g/ml) IPTG
(0.1 M) and X-gal (50 g/ml) and incubated overnight at 37 C. Single white
colonies
were chosen for plasmid isolation.
7.2.2.4 Plasfnid DNA preparation, restriction digestion and sequence analysis.
Miniprep plasmid DNA was prepared from 5 ml cultures using a Biorobot 8000
robotic system (Qiagen) according to the manufacturer's instructions. Plasmid
DNA
was eluted in 80 1 sterile water. The DNA concentration was measured using an
Eppendorf BO photometer or Spectramax 190 photometer (Molecular Devices).
Aliquots of miniprep plasmid DNAs (100-200ng) were digested with BamHI for 2h
at
37 C and analysed by electrophoresis in 0.8% agarose gels. Plasmids with
inserts of
the expected size of about 0.7kb were selected for DNA sequence analysis.
Inserts
were sequenced from either end by mixing 200-500 ng plasmid DNA with the
either
the T7 or T3 sequencing primers (see Table 4). Sequencing reactions were
processed
using the BigDye Terminator system (Applied Biosystems cat. no. 4390246)
according to the manufacturer's instructions. Products were purified using Dye-
Ex
columns (Qiagen) or Montage SEQ 96 cleanup plates (Millipore cat. no.
LSKS09624)
and analyzed on an Applied Biosystems 3700 sequencer.
7.2.2.5 Results of cloning and sequence analysis of nucleotides 1-683
The predicted mRNA coding sequence for the region between the initiator
methionine
and the internal BamHI site was confirmed. The DNA miniprep #15 was taken as a
representative clone for further work.
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
7.3 Assembly of t/te complete INSP169 extracellular clomain
Aliquots of the DNA miniprep #14 from stage 1 and DNA miniprep#15 from stage 2
were digested with BamHI (New England Biolabs) for 2h at 37 C using the
enzyme
buffer supplied by the manufacturer. The full-length, linearized plasmid from
5 miniprep#14 and the 0.7kb excised fragment from miniprep#15 were separated
on
0.8% agarose gel, excised and purified using the Wizard Cleanup System
(Promega)
according to the protocol provided by the manufacturer. The two purified DNAs
were
mixed in a molar ratio of 1:3 respectively, and precipitated overnight at -20
C in the
presence of 2.5 volumes ethanol. The precipitated DNA was recovered by
10 centrifugation, washed in 70% ethanol, dried under vacuum and ligated in a
final
volume of 10 1 using the Rapid Ligation Kit (Roche Diagnostics) according to
the
protocol supplied by the manufacturers.
Transformation of competent JM101with aliquots of the ligation mix, plasmid
isolation and sequence analysis were performed as described in sections
7.2.1.3 and
15 7.2.1.4 above.
7.4 Results of sequence analysis of full length INSPI69ec
Sequence analysis of 12 miniprep DNAs allowed identification of clones in
which the
fragment corresponding to nucleotides 1-683 was inserted in the correct
orientation
upstream of the sequence correponding to nucleotides 678-1740 and in which the
20 junction between the two fraginents is the correctly religated BamHI site
(position
678-683).
DNA miniprep #11 was selected and the complete sequence was verified using
sequencing primers T3, T7, AS515 and AS599 (see Table 4). Compared to the
prediction, a single SNP was found, G524A, which results in a codon change
from
25 Ser>Asn (underlined in Fig 8).
CA 02586486 2007-04-24
WO 2006/051333 PCT/GB2005/004390
96
Table 4: Primers used for cloning and sequencing of INSP169ec
Oligonucleotide Description DNA Sequence (5'-3')
AS502 RT primer (in TM) caccacgagaaggagagacc
AS503 5' PCR internal primer gcgaattcgctatagtgcttctggatcc
AS504 3' PCR primer gcgaattctcattgagaatcatctccttcaac
AS515 internal RT primer gaggcgctcaggttcactgc
AS516 5' PCR primer gcggatccatgcatctctttgcatgtctg
AS517 3' PCR internal primer catcagtggatccagaagcac
AS599 internal sequencing primer caatccttggttctgtgactg
T7 seq primer taatacgactcactatagg
T3 seq primer attaaccctcactaaagg
DEMANDE OU BREVET VOLUMINEUX
LA PRESENTE PARTIE DE CETTE DEMANDE OU CE BREVET COMPREND
PLUS D'UN TOME.
CECI EST LE TOME 1 DE 2
CONTENANT LES PAGES 1 A 96
NOTE : Pour les tomes additionels, veuillez contacter le Bureau canadien des
brevets
JUMBO APPLICATIONS/PATENTS
THIS SECTION OF THE APPLICATION/PATENT CONTAINS MORE THAN ONE
VOLUME
THIS IS VOLUME 1 OF 2
CONTAINING PAGES 1 TO 96
NOTE: For additional volumes, please contact the Canadian Patent Office
NOM DU FICHIER / FILE NAME:
NOTE POUR LE TOME / VOLUME NOTE: