Note: Descriptions are shown in the official language in which they were submitted.
CA 02587928 2007-05-18
WO 2006/061372 PCT/EP2005/056500
1
PHENYLPIPERAZINES WITH A COMBINATION OF AFFINITY FOR DOPAMINE -
D2 RECEPTORS AND SEROTONIN REUPTAKE SITES
The present invention relates to a group of novel phenylpiperazine derivatives
with a
dual mode of action: serotonin reuptake inhibition and affinity f or dopamine-
D2
receptors and to methods for the preparation of these compounds. The invention
also
relates to the use of a compound disclosed herein for the manufacture of a
medicament giving a beneficial effect. A beneficial effect is disclosed herein
or
apparent to a person skilled in the art from the specification and general
knowledge
in the art. The invention also relates to the use of a compound of the
invention for the
manufacture of a medicament for treating or preventing a disease or condition.
More
particularly, the invention relates to a new use for the treatment of a
disease or
condition disclosed herein or apparent to a person skilled in the art from the
specification and general knowledge in the art. In embodiments of the
invention
specific compounds disclosed herein are used for the manufacture of a
medicament
useful in the treatment of disorders in which dopamine-D2 receptors and
serotonin
reuptake sites are involved, or that can be treated via manipulation of those
targets.
Phenylpiperazine derivatives with a dual action as dopamine-D2 antagonists and
serotonin reuptake inhibitors are known from WO 01/014330. This combination is
useful for the treatment of schizophrenia and other psychotic disorders which
enables
a more complete treatment of all disease symptoms (e.g. positive symptoms and
negative symptoms).
In patent specification GB 1 378 080 (1974) oxime derivatives of halophenyl
piperazinyl-alkyl ketones have been disclosed that posses useful
pharmacological
activity, especially as analgesic agents, anti-inflammatory agents and
musculotropic
spasmolytic agents.
The goal of the present invention was to provide further compounds with a dual
action as dopamine-D2 antagonists and serotonin reuptake inhibitors.
CA 02587928 2007-05-18
WO 2006/061372 PCT/EP2005/056500
2
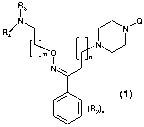
The invention relates to compounds of the general formula (1):
R3
I
N Q
R~
a
L m 0 n N
N
~ (1)
(R2)x
wherein:
- m and n independently are either 1, 2, 3, 4, 5, 6, 7 or 8,
- xis0, 1,2or3
- R2 is halogen, branched or unbranched alkyl(C,_6), phenyl, benzyl, branched
or
unbranched alkoxy(C,_s), trifluoromethyl or cyano
- R3 and R4 independently represent hydrogen, alkyl (C,_s), phenyl, benzyl or
acetyl
- group Q is chosen from structural fragments A-N
O OH O OH
O~ ~ O J I / O I
A H B C D
N N
~ 1 >~O 0 I~~O O~
0
1 ~
E F G N
17~ O CI O)
H J H K
(R1)
N /O N O
O~
L M N
wherein:
- yisl,2or3
- R, is halogen, branched or unbranched alkyl(C,_s), phenyl, benzyl, branched
or
unbranched alkoxy(C,_6), trifluoromethyl or cyano,
and tautomers, stereoisomers and N-oxides thereof, as well as
pharmacologically
acceptable salts, hydrates and solvates of said compounds of formula (1) and
its
tautomers, stereoisomers and N-oxides.
CA 02587928 2007-05-18
WO 2006/061372 PCT/EP2005/056500
3
Prodrugs of the compounds mentioned above are in the scope of the present
invention. Prodrugs are therapeutic agents which are inactive per se but are
transformed into one or more active metabolites. Prodrugs are bioreversible
derivatives of drug molecules used to overcome some barriers to the utility of
the
parent drug molecule. These barriers include, but are not limited to,
solubility,
permeability, stability, presystemic metabolism and targeting limitations
(Medicinal
Chemistry: Principles and Practice, 1994, ISBN 0-85186-494-5, Ed.: F. D. King,
p.
215; J. Stella, "Prodrugs as therapeutics", Expert Opin. Ther. Patents, 14(3),
277-
280, 2004; P. Ettmayer et al., "Lessons learned from marketed and
investigational
prodrugs", J.Med.Chem., 47, 2393-2404, 2004). Pro-drugs, i.e. compounds which
when administered to humans by any known route, are metabolised to compounds
having formula (1), belong to the invention. In particular this relates to
compounds
with primary or secondary amino or hydroxy groups. Such compounds can be
reacted with organic acids to yield compounds having formula (1) wherein an
additional group is present which is easily removed after administration, for
instance,
but not limited to amidine, enamine, a Mannich base, a hyd roxyl-methylene
derivative, an O-(acyloxymethylene carbamate) derivative, carbamate, ester,
amide
or enaminone.
N-oxides of the compounds mentioned above are in the scope of the present
invention. Tertiary amines may or may not give rise to N-oxide metabolites.
The
extend to what N-oxidation takes place varies from trace amounts to a near
quantitative conversion. N-oxides may be more active than their corresponding
tertiary amines or less active. Whilst N-oxides are easily reduced to their
corresponding tertiary amines by chemical means, in the human body this
happens
to varying degrees. Some N-oxides undergo nearly quantitative reductive
conversion
to the corresponding tertiary amines, in other cases the conversion is a mere
trace
reaction or even completely absent. (M.H. Bickel: "The pharmacology and
Biochemistry of N-oxides", Pharmaco-loaical Reviews, 21(4), 325 - 355, 1969).
Preferred compounds of the invention are compounds having formula (I) wherein
m
is 1, n is 2, 3, 4 or 5, x is 1, R2 is 4-fluoro or 4-trifluoromethyl, R3 and
R4
independently represent hydrogen or methyl, group Q is chosen from structural
fragments A, D, F or N, y is 1, and R, is branched or unbranched alkoxy(C,_3),
and
tautomers, stereoisomers a nd N -oxides thereof, as well as pharmacologically
acceptable salts, hydrates and solvates of said compounds of formula (1) and
its
tautomers, stereoisomers and N-oxides.
CA 02587928 2007-05-18
WO 2006/061372 PCT/EP2005/056500
4
It has been found that the compounds according to the invention show high
affinity fo r
both the dopamine D2 receptor and the serotonin reuptake site. The compounds
show
activity as antagonists at dopamine D2 receptors as they potentially
antagonize
apomorphine-induced climbing behaviour in mice. The compounds also show
activity
as inhibitors of serotonin reuptake, as they potentiate 5-HTP induced
behaviour in
mice. The compounds are active in therapeutic models sensitive to clinically
relevant
antipsychotics (e.g. the conditioned avoidance response; Van der Heyden &
Bradford,
Behav. Brain Res., 1988, 31:61-67) and antidepressants or anxiolytics (e.g.
suppression of stress-induced vocalization; van der Poel et al., Psycho-
pharmacology,
1989, 97: 147-148). In contrast to clinically relevant dopamine D2 receptor
antagonists
the described compounds have a low propensity to induce catalepsy in rodents
and as
such are likely to induce less extrapyramidal side effects than existing
antipsychotic
agents. The inhibitory activity of serotonin reuptake inherent in these
compounds may
be responsible for the therapeutic effects observed in behavioural models
sensitive to
either antidepressants or anxiolytics. The compounds can be used for the
treatment of
affections or diseases of the central nervous system caused by disturbances in
either
the dopaminergic or serotonergic systems, for example: aggression, anxiety
disorders,
autism, vertigo, depression, disturbances of cognition or memory, Parkinson's
disease,
and in particular schizophrenia and other psychotic disorders.
GENERAL ASPECTS OF SYNTHESES
The synthesis of all piperazine derivatives in this patent can be performed as
depicted in Scheme 1 for the preparation of compound 3.The starting phenyl
piperazines can be obtained as described in EP 0 189 612: Hartog, J et aL,
1985:
'New pharmaceutical compositions having a psychtropic activity'; Feenstra,
R.W.; de
Moes, J.P; Hofma, J.; Kling, H.; Kuipers, W; Long, S.K.; Tulp, M.T.M.; Van der
Heyden, J.A.M and Kruse, C.G.; 'New 1-aryl-4-(biarylmethylene)piperazines as
potential atypical antipsychotics sharing dopamine D2 receptor and serotonin
5HTIA
receptor affinities. Bioorg. & Med. Chem. Lett., 2001, 11, 2345-2349 and WO
01/14330. The alkylphenone derivatives 2 are commercially available.
CA 02587928 2007-05-18
WO 2006/061372 PCT/EP2005/056500
Scheme 1 a % ~ ~x
O
I \ J 1
a (NJ ~N)
O
O 2
5 (N)
H 3i 3ii 3
O /-N
CF3 CF3 O
a Reagents and conditions: (i) DIPEA, KI, CH 3CN, reflux; ~NH2
(ii) H2NO-CH2CH2NH2 = 2HCI, DIPEA, EtOH, reflux.
The selection of the particular synthetic procedures depends on factors known
to
those skilled in the art such as the compatibility of functional groups with
the reagents
used, the possibility to use protecting groups, catalysts, activating and
coupling
reagents and the ultimate structural features present in the final compound
being
prepared.
Pharmaceutically acceptable salts may be obtained using standard procedures
well
known in the art, for example by mixing a compound of the present invention
with a
suitable acid, for instance an inorganic acid such as hydrochloric acid, or
with an
organic acid.
PHARMACEUTICAL PREPARATIONS
The compounds of the invention can be brought into forms suitable for
administration
by means of usual processes using auxillary substances such as liquid or solid
carrier material. The pharmaceutical compositions of the invention may be
administered enterally, orally, parenterally (intramuscularly or
intravenously), rectally
or locally (topically). They can be administered in the form of solutions,
powders,
tablets, capsules (including microcapsuies), ointments (creams or gel) or
suppositories. Suitable excipients for such formulations are the
pharmaceutically
customary liquid or solid fillers and extenders, solvents, emulsifiers,
lubricants,
flavorings, colorings and/or buffer substances. Frequently used auxillary
substances
which may be mentioned are magnesium carbonate, titanium dioxide, lactose,
mannitol and other sugars, talc, lactoprotein, gelatin, starch, cellulose and
its
derivatives, animal and vegetable oils such as fish liver oil, sunflower,
groundnut or
sesame oil, polyethylene glycol and solvents such as, for example, sterile
water and
mono- or polyhydric alcohols such as glycerol.
CA 02587928 2007-05-18
WO 2006/061372 PCT/EP2005/056500
6
Compounds of the present invention are generally administered as
pharmaceutical compositions which are important and novel embodiments of the
invention because of the presence of the compounds, more particularly specific
compounds disclosed herein. Types of pharmaceutical compositions that may be
used include but are not limited to tablets, chewable tablets, capsules,
solutions,
parenteral solutions, suppositories, suspensions, and other types disclosed
herein or
apparent to a person skilled in the art from the specification and general
knowledge
in the art. In embodiments of the invention, a pharmaceutical pack or kit is
provided
comprising one or more containers filled with one or more of the ingredients
of a
pharmaceutical composition of the invention. Associated with such container(s)
can
be various written materials such as instructions for use, or a notice in the
form
prescribed by a governmental agency regulating the manufacture, use or sale of
pharmaceuticals products, which notice reflects approval by the agency of
manufacture, use, or sale for human or veterinary administration.
PHARMACOLOGICAL METHODS
In vitro affinity for dopamine-D2 receptors
Affinity of the compounds for dopamine -D2 receptor s was determined using the
receptor binding assay described by I. Creese, R. Schneider and S.H. Snyder:
"PH]-
Spiroperidol labels dopamine receptors in rat pituitary and brain",
Eur.J.Pharmacol.,
46, 377 - 381, 1977.
In vitro affinity for serotonin reuptake sites
Affinity of the compounds for serotonin reuptake sites was determined using
the
receptor binding assay described by E. Habert et al.,: "Characterisation of
(3H]-
paroxetine binding to rat cortical membranes", Eur.J.Pharmacol., 118, 107 -
114,
1985.
DOSAGES
The affinity of the compounds of the invention for dopamine-D2 receptors and
serotonine reuptake sites was determined as described above. From the binding
affinity measured for a given compound of formula (1), one can estimate a
theoretical
lowest effective dose. At a concentration of the compound equal to twice the
CA 02587928 2007-05-18
WO 2006/061372 PCT/EP2005/056500
7
measured Ki-value, 100% of the receptors likely will be occupied by the
compound.
Converting that concentration to mg of compound per kg of patient yields a
theoretical lowest effective dose, assuming ideal bioavailability.
Pharmacokinetic,
pharmacodynamic, and other considerations may alter the dose actually
administered to a higher or lower value. The dosage expediently administered
is
0.001 - 1000 mg/kg, preferably 0.1-100 mg/kg of patient's bodyweight.
TREATMENT
The term 'treatment' as used herein refers to any treatment of a mammalian,
preferably human condition or disease, and includes: (1) preventing the
disease or
condition from occurring in a subject which may be predisposed to the disease
but
has not yet been diagnosed as having it, (2) inhibiting the disease or
condition, i.e.,
arresting its development, (3) relievi ng the disease or condition, i.e.,
causing
regression of the condition, or (4) relieving the conditions caused by the
disease, i.e.,
stopping the symptoms of the disease.
The preparation of the compounds having formula (I) will now be described in
more
detail in the following Examples.
CA 02587928 2007-05-18
WO 2006/061372 PCT/EP2005/056500
8
EXAMPLES
EXAMPLE 1: MATERIALS AND METHODS
'H and 13C NMR spectra were recorded on a Bruker Avance DRX600 instrument
(600 MHz), Varian UN400 instrument (400 MHz) or on a Varian VXR200 instrument
(200 MHz) using DMSO-D6 or CDCI3 as solvents with tetramethylsilane as an
internal
standard. Chemical shifts are given in ppm (b scale) downfield from
tetramethylsilane. Coupling constants (J) are expressed in Hz. Flash
chromatography
was performed using silica gel 60 (0.040-0.063 mm, Merck). Column
chromatography was performed using silica gel 60 (0.063 -0.200 mm, Merck).
Melting
points were recorded on a Buchi B-545 melting point apparatus. Mass spectra
were
recorded on a Micromass QTOF-2 instrument with MassLynx application software
for
acquisition and reconstruction of the data. Exact mass measurement was done of
the
quasimolecular ion [M+H]+.
EXAMPLE 2: SYNTHESES OF SPECIFIC COMPOUNDS
The synthesis of compound 3 is a 2-step reaction starting from 4-(2,3 dihydro-
1,4 benzodioxin-5-yl)-1-piperazine (3i). 15 mmol of piperazine (3i) was
suspended
in 125 ml of acetonitril and 2 equivalents of diisopropylethyl-amine (DIPEA)
was
added. After 5 minutes stirring at room temperature, 1 equivalent (15 mmol) of
5-
chloro-1-(4-trifluoromethyl-phenyl)-pentane-1-one was added, followed by 1
equivalent of sodium iodide. This mixture was stirred at 80 C for 20 hours.
The
solvent was removed by evaporation and the residue dissolved in 100 ml of
dichloromethane.
The organic layer was washed with water and dried on magnesium sulphate before
evaporation. The residue was purified by column chromatography and this
yielded
6.4 mmol of the keto-derivative 3ii which was dissolved in 30 ml of methanol.
To this
solution 1 equivalent of O-(2-aminoethyl)-hydroxylamine di-HCI salt was added
and
this mixture was heated for 12 hours at 80 C. After evaporation of the
solvent, the
residue was dissolved in dichloromethane and washed with water. Drying of the
organic layer, using magnesium sulphate and evaporation of the solvent yielded
a
residue that was purified by column chromatography. The tri HCI -salt of
compound 3
was obtained after adding 3 equivalents of HCI in Ethanol to the purified
substance.
mp.156-60 C; overall yield 15%.
CA 02587928 2007-05-18
WO 2006/061372 PCT/EP2005/056500
9
The synthesis of compound 7 is a 2-step reaction starting from 2-isopropyloxy-
phenylpiperazine (7i). 4.2 mmol of phenyl piperazine 7i was suspended in 40 ml
acetonitril. Added were 2 eq. of DIPEA, 1 eq. of 4-chloro-1-(4-trifluoromethyl-
phenyl)-
butane-1-one and 1 eq. potassium iodide. This mixture was refluxed overnight
and
the solvent evaporated the next day. The residue was purified by column
chromatography, yielding 2.5 mmol of the pure keto-compound 7ii, which was
dissolved again in 30 ml of ethanol (100%). Added was 1 eq. of O-(2-
aminoethyl)
hydroxylamine di-HCI salt and 1 eq. of pyridine. This mixture was heated at 80
C for
4 hours. After solvent evaporation the residue was purified by column
chromatography and this yielded 2 mmol of an orange oil. The oily substance
was
dissolved in ethanol and added was 1 eq. of fumaric acid. The amorph fumaric
salt of
compound 7 was obtained after evaporation; overall yield 50%
The synthesis of compound 8 is a 2-step reaction starting from 4-(2,3 dihydro-
1,4 benzodioxin-5 yl)-1-piperazine (3i). 3.5 mmol of phenyl piperazine (3i)
was
suspended in 40 ml acetonitril. Added were 2 eq. of DIPEA, 1 eq. of 6-chloro-1-
(4-
trifluoromethyl-phenyl)-hexane-1-one and 1 eq. potassium iodide. This mixture
was
refluxed overnight and the solvent evaporated the next day. The residue was
purified
by column chromatography, yielding 1.7 mmol of the pure keto -compound 8ii,
which
was dissolved again in 10 ml of ethanol (100%). Added was 1 eq. of O-(2-
aminoethyl) hydroxylamine di HCI salt and this mixture was heated at 80 C for
4
hours. After solvent evaporation the residue was purified by column
chromatography
and this yielded 1.6 mmol of an yellow oil. The oily substance was dissolved
in
ethanol and added was 2 eq. of fumaric acid. The amorph fumaric salt of
compound
8 was obtained after evaporation; overall yield 45%.
The synthesis of compound 9 is a 2-step reaction starting from 4-(2,3 dihydro-
1,4 benzodioxin-5-yl)-1-piperazine. Compound 3ii (30 mmol) was dissolved in 20
ml of methanol. To this solution, 1 equivalent of O-(N-methyl-2-aminoethyl)-
hydroxylamine di-HCI salt was added and this mixture was heated for 5 hours at
80 C. After evaporation of the solvent, the residue was dissolved in
dichloromethane
and washed with sodium bicarbonate solution and followed by brine. The organic
layer was dried by using magnesium sulphate and evaporation of the solvent
yielded
a residue that was purified by column chromatography. The fumaric-sait of the
compound 9 was obtained after adding an ethanolic solution of 1 equivalents of
fumaric acid to the purified substance followed by evaporation of the solvent;
overall
yield 15%.
CA 02587928 2007-05-18
WO 2006/061372 PCT/EP2005/056500
The synthesis of compound 10 is a 2-step reaction starting from 4-(2,3 dihydro-
1,4 benzodioxin-5-yl)-1-piperazine (3i). 6 mmol of phenyl piperazine 3i was
suspended in 40 ml acetonitril. Added were 2 eq. of DIPEA, 1 eq. of 7-chloro-l-
(4-
5 trifluoromethyl-phenyl)-heptane-1-one and 1 eq. potassium iodide. This
mixture was
refluxed overnight and the solvent evaporated the next day. The residue was
purified
by column chromato-graphy, yielding 4.2 mmol of the pure keto-compound 1011,
which was dissolved again in 25 ml of ethanol (100%). Added was 1 eq. of O-(2-
aminoethyl) hydroxylamine di-HCI salt and this mixture was heated at 80 C for
4
10 hours. After solvent evaporation the residue was purified by column
chromatography
and this yielded 2 mmol of an yellowy oil. The oily substance was dissolved in
ethanol and added was 1.5 eq. of fumaric acid. The amorph fumaric salt of
compound 10 was obtained after evaporation; overall yield 35%.
Table 1. Compounds of the general formula (1)
R3
I
N Q
R~
a
N
n
m 0
11
N
~ (1)
(R2)x
wherein Q can be one of the structural fragments A-N
O, 0) O~OH
0 0
O J 0ON
A H B C D
II I ~ N~O ~
0 O
0
E
F G N
O
\O
O
(P70~ I O CI )
~ H K
H (R,)
N O N O
/
~ L ~ M N
CA 02587928 2007-05-18
WO 2006/061372 PCT/EP2005/056500
11
cpnd y R, Q x R2 R3 R4 n m mp ( C)
3 - - A 1 4-CF3 H H 3 1 156-160
4 1 2-OMe N 1 4-F H H 2 1 150-152
- - F 1 4-CF3 H H 3 1 158
6 - - D 1 4-CF3 H H 3 1 153-154
7 1 OCH(Me)2 N 1 4-CF3 H H 2 1 amorph
8 - - A 1 4-CF3 H H 4 1 amorph
9 - - A 1 4-CF3 Me H 3 1 amorph
- - A 1 4-CF3 H H 5 1 amorph
11 1 2-OMe N 1 4-CF3 H H 5 1 amorph
The specific compounds of which the synthesis is descri bed above are intended
to
further illustrate the invention in more detail, and therefore are not deemed
to restrict
5 the scope of the invention in any way. Other embodiments of the invention
will be
apparent to those skilled in the art from consideration of the specification
and
practice of the invention disclosed herein. It is thus intended that the
specification
and examples be considered as exemplary only, with a true scope and spirit of
the
invention being indicated by the claims.
EXAMPLE 3: FORMULATION OF COMP. 3 USED IN ANIMAL STUDIES
For oral (p.o.) administration : to the desired quantity (0.5-5 mg) of the
solid
compound 3 in a glass tube, some glass beads were added and the solid was
milled
by vortexing for 2 minutes. After addition of 1 ml of a solution of 1%
methylcellulose
in water and 2% (v/v) of Poloxamer 188 (Lutrol F68), the compound was
suspended
by vortexing for 10 minutes. The pH was adjusted to 7 with a few drops of
aqueous
NaOH (0.1N). Remaining particles in the suspension were further suspended by
using an ultrasonic bath.
For intraperitoneal (i.p.) administration: to the desired quantity (0.5-15 mg)
of the
solid compound 3 in a glass tube, some glass beads were added and the solid
was
milled by vortexing for 2 minutes. After addition of 1 ml of a solution of 1%
methylcellulose and 5% mannitol in water, the compound was suspended by
vortexing for 10 minutes. Finally the pH was adjusted to 7.
EXAMPLE 4: PHARMACOLOGICAL TESTRESULTS
Dopamine-D2 and serotonin reuptake receptor affinity data obtained according
to the
protocols given above are shown in the table below.
CA 02587928 2007-05-18
WO 2006/061372 PCT/EP2005/056500
12
Table 2. In vitro affinities of compounds of the invention
In vitro affinity
Dopamine-D2 5-HT reuptake
cpnd pK, pK,
3 8.3 8.2
4 8.6 7.3
8.2 8.3
6 6.6 8.3
7 8.1 7.0
8 8.3 8.0
9 8.4 7.5
8.4 8.5
11 8.3 8.3