Note: Descriptions are shown in the official language in which they were submitted.
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
PHOSPHODIESTERASE 4D IN THE RYANODINE RECEPTOR COMPLEX
PROTECTS AGAINST HEART FAILURE
CROSS REFERENCE TO RELATED APPLICATIONS
[0001] This application claims the benefit of U.S. Provisional Application
Serial
No. 60/636,959, filed December 16, 2004; which is incorporated herein in its
entirety by
reference thereto.
STATEMENT OF GOVERNMENT INTEREST
[0002] This invention was made with government support under NIH Grant No.
NIH-ROl-HD20788. As such, the United States government may have certain rights
in
this invention.
FIELD OF THE INVENTION
[0003] This invention relates to novel compositions and methods to treat and
prevent disorders and diseases associated with the RyR receptors that regulate
calcium
channel functioning in cells.
BACKGROUND OF THE INVENTION
[0004] Througout this application, various publications are referenced in
parentheses by author and year. Full citations for these references are
provided at the end
of the specification immediately preceding the claims. The disclosures of
these
publications in their entireties are hereby incorporated by reference into
this application to
more fully describe the state of the art to which this invention pertains.
[0005] The sarcoplasmic reticulum (SR) is a structure in cells that functions,
among other things, as a specialized intracellular calcium (Ca2+) store.
Channels in the
SR called ryanodine receptors (RyRs) open and close to regulate the release of
Ca2+ from
the SR into the intracellular cytoplasm of the cell. Release of Ca2+ into the
cytoplasm
from the SR increases cytoplasmic Ca2+ concentration. Open probability (Po) of
the RyR
receptor refers to the likelihood that the RyR channel is open at any given
moment, and
therefore capable of releasing CaZ+ into the.cytoplasm from the SR.
[0006] There are three types of ryanodine receptors, all of which are highly-
related Ca2+ channels: RyRl, RyR2, and RyR3. RyRl is found predominantly in
skeletal
muscle as well as other tissues, RyR2 is found predominantly in the heart as
well as other
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
2
tissues, and RyR3 is found in the brain as well as other tissues. The RyR
channels are
formed by four RyR polypeptides in association with four FK506 binding
proteins
(FKBPs), specifically FKBP12 (calstabinl) and FKBP12.6 (calstabin2).
Calstabinl binds
to RyRI, calstabin2 binds to RyR2, and calstabinl binds to RyR3. The FKBP
proteins
(calstabinl and calstabin2) bind to the RyR channel (one molecule per RyR
subunit),
stabilize RyR-channel functioning, and facilitate coupled gating between
neighboring
RyR channels, thereby preventing abnormal activation of the channel during the
channel's closed state.
[0007] Besides the calstabin binding proteins, protein kinase A (PKA) also
binds
to the cytoplasmic surface of the RyR receptors. PKA phosphorylation of the
RyR
receptors causes partial dissociation of calstabins from RyRs. Dissociation of
calstabin
from RyR causes increased open probability of RyR, and therefore increased
CaZ+ release
from the SR into the intracellular cytoplasm.
[0008] CaZ+ release from the SR in skeletal muscle cells and heart cells is a
key
physiological mechanism that controls muscle performance, because increased
concentration of Ca2+ in the intracellular cytoplasm causes contraction of the
muscle.
[0009] Excitation-contraction (EC) coupling in skeletal muscles involves
electrical depolarization of the plasma membrane in the transverse tubule (T-
tubule),
which activates voltage-gated L-type Ca2+ channels (LTCCs). LTCCs trigger Ca2+
release from the SR through physical interaction with RyRl. The resulting
increase in
cytoplasmic Ca2+ concentration induces actin-myosin interaction and muscle
contraction.
To enable relaxation, intracellular Ca2+ is pumped back into the SR via SR
Ca2+-ATPase
pumps (SERCAs), which is regulated by phospholamban (PLB) depending on the
muscle
fiber type.
[0010] It has been shown that disease forms that result in sustained
activation of
the sympathetic nervous system and increased plasma catecholamine levels cause
maladaptive activation of intracellular stress pathways resulting in
destabilization of the
RyRl channel closed state and intracellular Ca2+ leak. SR Ca2+ leak via RyRl
channels
was found to deplete intracellular SR calcium stores, to increase compensatory
energy
consumption, and to result in significant acceleration of muscle fatigue. The
stress-
induced muscle defect permanently reduces isolated muscle and in vivo
performance
particularly in situations of increased demand.
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
3
[0011] It also has been shown that destabilization of the RyRl closed state
occurs
under pathologic conditions of increased sympathetic activation and involves
depletion of
the stabilizing calstabinl (FKBP12) channel subunit. Proof-of-principle
experiments
have shown that PKA. activation as an end effector of the sympathetic nervous
systems
increases RyRl PKA phosphorylation at Ser-2843 which decreases the binding
affinity of
calstabinl to RyRl and increases channel open probability.
100121 In cardiac striated muscle, RyR2 is the major Ca?+- release channel
required
for EC coupling and muscle contraction. During EC coupling, depolarization of
the
cardiac-muscle cell membrane during phase zero of the action potential
activates
voltage-gated Ca2+ channels. Ca?+ influx through the open voltage-gated
channels in turn
initiates Ca2+ release from the SR via RyR2. This process is known as Ca2}-
induced CaZ+
release. The RyR2-mediated, Ca2+-induced Ca2+ release then activates the
contractile
proteins in the cardiac cell, resulting in cardiac muscle contraction.
[0013] Phosphorylation of cardiac RyR2 by PKA is an important part of the
"fight
or flight" response that increases cardiac EC coupling gain by augmenting the
amount of
Ca2+ released for a given trigger. This signaling pathway provides a mechanism
by which
activation of the sympathetic nervous system, in response to stress, results
in increased
cardiac output. PKA phosphorylation of RyR2 increases the open probability of
the
channel by dissociating calstabin2 (FKBP 12.6) from the channel complex. This,
in turn,
increases the sensitivity of RyR2 to Caz+-dependent activation.
[0014] Despite advances in treatment, heart failure remains an important cause
of
mortality in Western countries. An important hallmark of heart failure is
reduced
myocardial contractility. In heart failure, contractile abnormalities result,
in part, from
alterations in the signaling pathway that allows the cardiac action potential
to trigger Ca2+
release via RyR2 channels and muscle contraction. In particular, in failing
hearts, the
amplitude of the whole-cell Ca2+ transient is decreased and the duration
prolonged.
[0015] Cardiac arrhythmia, a common feature of heart failure, results in many
of
the deaths associated with the disease. Atrial fibrillation (AF) is the most
common
cardiac arrhythmia in humans, and represents a major cause of morbidity and
mortality.
Structural and electrical remodeling - including shortening of atrial
refractoriness, loss of
rate-related adaptation of refractoriness, and shortening of the wavelength of
re-entrant
wavelets - accompany sustained tachycardia. This remodeling is likely
important in the
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
4
development, maintenance and progression of atrial fibrillation. Studies
suggest that
calcium handling plays a role in electrical remodeling in atrial fibrillation.
100161 Approximately 50% of all patients with heart disease die from fatal
cardiac
arrhythmias. In some cases, a ventricular arrhythmia in the heart is rapidly
fatal - a
phenomenon referred to as "sudden cardiac death" (SCD). Fatal ventricular
arrhythmias
and SCD also occur in young, otherwise-healthy individuals who are not known
to have
structural heart disease. In fact, ventricular arrhythmia is the most common
cause of
sudden death in otherwise-healthy individuals.
[0017] Catecholaminergic polymorphic ventricular tachycardia (CPVT) is an
inherited disorder in individuals with structurally normal hearts. It is
characterized by
stress-induced ventricular tachycardia - a lethal arrhythmia that causes SCD.
In subjects
with CPVT, physical exertion and/or stress induce bidirectional and/or
polymorphic
ventricular tachycardias that lead to SCD even in the absence of detectable
structural
heart disease. CPVT is predominantly inherited in an autosomal-dominant
fashion.
Individuals with CPVT have ventricular arrhythmias when subjected to exercise,
but do
not develop arrhythmias at rest. Studies have identified mutations in the
human RyR2
gene, on chromosome 1q42-q43, in individuals with CPVT.
[0018] Failing hearts (e.g., in patients with heart failure and in animal
models of
heart failure) are characterized by a maladaptive response that includes
chronic
hyperadrenergic stimulation. In heart failure, chronic beta-adrenergic
stimulation is
associated with the activation of beta-adrenergic receptors in the heart,
which, through
coupling with G-proteins, activate adenylyl cyclase and thereby increase
intracellular
cAMP concentration. cAMP activates cAMP-dependent PKA, which has been shown to
induce hyperphosphorylation of RyR2. Thus, chronic heart failure is a chronic
hyperadrenergic state which results in several pathologic consequences,
including PKA
hyperphosphorylation of RyR2.
[0019] The PKA hyperphosphorylation of RyR2 has been proposed as a factor
contributing to depressed contractile function and arrhythmogenesis in heart
failure.
Consistent with this hypothesis, PKA hyperphosphorylation of RyR2 in failing
hearts has
been demonstrated, in vivo, both in animal models and in patients with heart
failure
undergoing cardiac transplantation.
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
[0020] In failing hearts, the hyperphosphorylation of RyR2 by PKA induces the
dissociation of FKBP12.6 (calstabin2) from the RyR2 channel. This causes
marked
changes in the biophysical properties of the RyR2 channel, including increased
open
probability (Po) due to an increased sensitivity to Ca2-'-dependent
activation;
destabilization of the channel, resulting in subconductance states; and
impaired coupled
gating of the channels, resulting in defective EC coupling and cardiac
dysfunction. Thus,
PKA-hyperphosphorylated RyR2 is very sensitive to low-level Ca2+ stimulation,
and this
manifests itself as a diastolic SR Ca2+ leak through the PKA
hyperphosphorylated RyR2
channel.
[0021] The maladaptive response to stress in heart failure results in
depletion of
FKBP 12.6 from the channel macromolecular coinplex. This leads to a shift to
the left in
the sensitivity of RyR2 to Ca2+-induced Ca2+ release, resulting in channels
that are more
active at low-to-moderate Ca2+ concentrations. Over time, the increased "leak"
through
RyR2 results in resetting of the SR Ca2+ content to a lower level, which in
turn reduces
EC coupling gain and contributes to impaired systolic contractility.
[0022] Additionally, a subpopulation of RyR2 that are particularly "leaky" can
release SR Ca2+ during the resting phase of the cardiac cycle, diastole. This
results in
depolarizations of the cardiomyocyte membrane known as delayed after-
depolarizations
(DADs), which are known to trigger fatal ventricular cardiac arrhythmias.
[0023] In patients with CPVT mutations in their RyR2 and otherwise
structurally-
normal hearts, a similar phenomenon is at work. Specifically, it is known that
exercise
and stress induce the release of catecholamines that activate beta-adrenergic
receptors in
the heart. Activation of the beta-adrenergic receptors leads to PKA
hyperphosphorylation
of RyR2 channels. Evidence also suggests that the PKA hyperphosphorylation of
RyR2
resulting from beta-adrenergic-receptor activation renders mutated RyR2
channels more
likely to open in the relaxation phase of the cardiac cycle, increasing the
likelihood of
arrhythmias.
[0024] Cardiac arrhythmias are known to be associated with diastolic SR Ca2+
leaks in patients with CPVT mutations in their RyR2 and otherwise structurally-
normal
hearts. In these cases, the most common mechanism for induction and
maintenance of
ventricular tachycardia is abnormal automaticity. One form of abnormal
automaticity,
known as triggered arrhythmia, is associated with aberrant release of SR Ca2+,
which
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
6
initiates DADs. DADs are abnormal depolarizations in cardiomyocytes that occur
after
repolarization of a cardiac action potential. The molecular basis for the
abnormal SR
Ca2} release that results in DADs has not been fully elucidated. However, DADs
are
known to be blocked by ryanodine, providing evidence that RyR2 plays a key
role in
the pathogenesis of this aberrant Ca2+ release.
[0025] Co-pending application U.S. Serial No. 10/763,498 discusses RyR2 as a
target for treating and preventing heart failure and cardiac arrhythmias,
including atrial
fibrillation and cardiac arrhythmias that cause exercise-induced sudden
cardiac death
(SCD). RyR2 channels with 7 different CPVT mutations (e.g., S2246L, R2474S,
N4104K, R4497C, P2328S, Q4201R, V4653F) were found to have functional defects
that
resulted in channels that became leaky (i.e., a calcium leak) when stimulated
during
exercise. The mechanism for the VT in CPVT has been demonstrated to be the
same as
the mechanism for VT in heart failure.
[0026] It has been shown that exercise-induced arrhythmias and sudden death
(in
patients with CPVT) result from a reduced affinity of FKBP 12.6 (calstabin2)
for RyR2.
Additionally, it has been demonstrated that exercise activates RyR2 as a
result of
phosphorylation by adenosine 3', 5'-monophosphate (cAMP)-dependent protein
kinase
(PKA). Mutant RyR2 channels, which had normal function in planar lipid
bilayers under
basal conditions, were more sensitive to activation by PKA phosphorylation -
exhibiting
increased activity (open probability) and prolonged open states, as compared
with wild-
type channels. In addition, PKA-phosphorylated mutant RyR2 channels were
resistant to
inhibition by Mg2 a physiological inhibitor of the channel, and showed reduced
binding
to FKBP12.6 (aka calstabin2, which stabilizes the channel in the closed
state). These
findings indicate that, during exercise, when the RyR2 are PKA-phosphorylated,
the
mutant CPVT channels are more likely to open in the relaxation phase of the
cardiac
cycle (diastole), increasing the likelihood of arrhythmias triggered by SR
Ca2+ leak.
[0027] Additionally, co-pending U.S. Patent Application No. 09/288,606
discusses a method for regulating contraction of a subject's heart by
administering a
compound which regulates PKA phosphorylation of an RyR2 receptor and
specifically
decreases PKA phosphorylation. Co-pending U.S. Patent Application No.
10/608,723
also discusses a method for treating and prophylaxis for atrial
tachyarrhythmia and
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
7
exercise and stress-induced arrhytlunias by administration of an agent which
inhibits
PKA phosphorylation of RyR2.
[0028] Phosphodiesterases (PDEs) control the temporal and spatial dynamics of
the second messenger 3',5' cyclic adenosine monophosphate (cAMP), allowing for
highly
localized cAMP gradients in cells (Zaccolo and Pozzan, 2002). Localization of
PDEs in
close proximity to cAMP-dependent protein kinase A (PKA) is thought to control
access
of cAMP to the regulatory kinase subunit (Conti et al., 2003 and Houslay and
Adams,
2003). PKA phosphorylation of proteins mediates a wide variety of signals,
including
those generated during activation of the sympathetic nervous system (SNS) as
part of the
"fight or flight" response. On the other hand, chronic activation of the SNS
is a
characteristic finding in heart failure, and acute stimulation of the SNS has
been linked to
triggered arrhythmias associated with sudden cardiac death.
[0029] In the heart, phosphodiesterase 4 (PDE4) contributes to the regulation
of
cAMP levels in cardiac myocytes. In particular, PDE4 cAMP-hydrolyzing activity
has
been localized to the transverse (T) tubule/sarcoplasmic reticulum (SR)
junctional space
that is involved in excitation-contraction coupling (Mongillo et al., 2004 and
Zaccolo and
Pozzan, 2002). PDEs have been shown to be components of macromolecular
signaling
complexes via binding to targeting proteins including muscle A-kinase
anchoring proteins
(AKAPs) (Dodge et al., 2001). PDEs in cardiac muscle are complexed with
proteins that
mediate signals from SNS, including (3-adrenergic receptors and 0-arrestin
(Mongillo et
al., 2004, Perry et al., 2002 and Xiang et al., 2005).
[0030] The PDE superfamily is subgrouped into 11 families that include at
least
20 genes and 50 unique isoforms. Of these PDE families, only PDE4, PDE7, and
PDE8
are cAMP specific (Conti et al., 2003). Through alternative splicing and the
use of
multiple promotors, the PDE4D gene encodes nine variants (PDE4D1-9) with
identical
catalytic domains and carboxyl termini and unique amino termini important for
subcellular localization. For example, PDE4D3 binds to the targeting protein
mAKAP via
its unique N-terminal region, creating a mAKAP-PKA-PDE4D3 signaling module
(Dodge et al., 2001 and Tasken et al., 2001) in which PKA phosphorylation
increases
PDE4D3 activity approximately 2-fold (Carlisle Michel et al., 2004 and Sette
and Conti,
1996). Moreover, as the inventors have previously shown, mAKAP colocalizes
with the
ryanodine receptor (RyR2)/calcium-release channel in cardiac muscle (Ruehr et
al., 2003
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
8
and Yang et al., 1998), where it is part of the RyR2 macromolecular signaling
complex
(Marx et al., 2000 and Wehrens et al., 2003).
[0031] PKA-PDE signaling has been identified as a therapeutic target in
several
major diseases (Conti et al., 2003). Inhibitors of the PDE4 family are under
development
for asthma, chronic obstructive lung disease (COPD), cognitive disorders
including
Alzheimer's disease, and stroke (Gong et al., 2004, Gretarsdottir et al., 2003
and Vignola,
2004). However, nonspecific PDE inhibition with theophylline, commonly used to
treat
asthma and COPD, and trials using PDE3 inhibition to treat heart failure have
demonstrated increased mortality due to cardiac arrhythmias (Barnes, 2003 and
Packer et
al., 1991).
[0032] The inventors now, for the first time, definitively demonstrate that
PDE4D
deficiency in mice is associated with a cardiac phenotype comprised of a
progressive,
age-related cardiomyopathy and exercise-induced arrhythmias, despite normal
global
cAMP signaling. Furthermore, PDE4D3 was found to be an integral component of
the
RyR2 macromolecular signaling complex. RyR2 located on the sarcoplasmic
reticulum
(SR) is the major Ca2+-release channel required for excitation-contraction
coupling in
heart muscle. RyR2 channels were PKA hyperphosphorylated and exhibited a
"leaky"
phenotype in PDE4D-deficient mice, similar to RyR2 defects previously observed
by the
inventors in patients with heart failure and sudden cardiac death (SCD) (Marx
et al., 2000
and Wehrens et al., 2003). In failing huinan hearts, PDE4D31evels were reduced
in the
RyR2 complex. Moreover, mice with PDE4D deficiency exhibited accelerated
progression of heart failure following myocardial infarction associated with
RyR2
channels that were PKA hyperphosphorylated and exhibited a "leaky" phenotype.
Pharmacological PDE4 inhibition was associated with exercise-induced cardiac
arrhythmias that were suppressed in mice harboring a mutation that prevents
PKA
phosphorylation of the RyR2 channel. These data indicate that PDE4D deficiency
contributes to heart failure and arrhythmias by promoting defective regulation
of the
RyR2 channel.
SUMMARY OF THE INVENTION
[0033] In view of the foregoing, there is a current need to identify new
methods
effective for treating and preventing disorders and diseases associated with
the RyR
recpetors that regulate calcium channel functioning in cells, including
skeletal muscular
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
9
disorders and diseases and especially cardiac disorders and diseases.
[0034] The inventors now show, for the first time that that PDE4D plays a
protective role in the heart against heart failure and arrhythmias. The
inventors also
demonstrate herein that PDE deficiency is associated with a severe cardiac
phenotype
consisting of heart failure and lethal cardiac arrhythmias. Further, the
inventors show
herein that PDE4D3 deficiency in the RyR2 complex contributes to PKA
hypersphosporylation of RyR2 in human and animal hearts, and that PDE4D3
activity
provides an important negative feedback mechanism to limit j3-AR-dependent PKA
phosphorylation of RyR2-Ser2808.
[0035] Accordingly, the present invention generally provides compositions
useful
for treating or preventing a ryanodine receptor associated disorder
comprising: a
phosphodiesterase (PDE)-associated agent; and optionally a pharmaceutically
acceptable
carrier. In a preferred embodiment of the invention, the PDE-associated agent
may be a
PDE protein, a nucleic acid encoding a PDE protein, a member of a PDE signal-
transduction pathway, and a modulator of a member of a PDE signal transduction
pathway. In one embodiment of the present invention, the PDE-associated agent
is
PDE4D protein or a nucleic acid encoding a PDE4D protein. The ryanodine
receptor
associated disorder may be a RyR2-l, RyR2 or RyR3 associated disorder.
[0036] The present invention also provides kits for use in delivering a PDE-
associated agent to a ryanodine receptor complex in a subject, comprising a
PDE-
associated agent, optionally with a pharmaceutically acceptable carrier and a
catheter.
[0037] Additionally, the invention provides methods for treating or preventing
a
ryanodine receptor associated disorder in a subject comprising augmenting PDE
in a
ryanodine receptor complex of the subject. In one embodiment of the invention,
PDE is
augmented in the ryanodine receptor complex by contacting the ryanodine
receptor
complex with a PDE-associated agent. In one embodiment of the invention, the
PDE-
associated agent is selected from the group consisting of a PDE protein, a
nucleic acid
encoding a PDE, a member of a PDE signal-transduction pathway, and a modulator
of a
member of a PDE signal transduction pathway. In a particular embodiment, the
nucleic
acid is operatively linked to an inducible promoter. In another embodiment of
the present
invention, the phosphodiesterase is PDE4D including PDE4D3. The subject of the
present invention may be any animal, including amphibians, birds, fish
mammals, and
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
marsupials, but is preferably a mammal, including but not necessarily limited
to a mouse,
rat, cat, dog, horse, monkey cow or pig. In an preferred embodiment, the
subject is
human.
[0038] In a particular embodiment of the invention, the ryanodine receptor
associated disorder is a RyR1 receptor associated disorder. In another
embodiment of the
present invention, the ryanodine receptor associated disorder is a RyR2
receptor
associated disorder. In a further embodiment, the ryanodine receptor
associated disorder
is an RyR3 receptor associated disorder.
[0039] In one embodiment of the present invention, the ryanodine receptor
associated disorder may be a cardiac disorder and/or disease including, but
not limited to,
an irregular heartbeat disorder or disease; exercise-induced irregular heart
beat disorder or
disease; sudden cardiac death; exercise-induced sudden cardiac death;
congestive heart
failure; chronic obstructive pulmonary disease; and high blood pressure. The
irregular
heartbeat disorders and diseases and exercise-induced irregular heartbeat
disorders and
diseases may include, but are not necessarily limited to, atrial and
ventricular arrhythmia;
atrial and ventricular fibrillation; atrial and ventricular tachyarrhythmia;
atrial and
ventricular tachycardia; catechlaminergic polymorphic ventricular tachycardia
(CPTV);
and exercise-induced variants thereof.
[0040] In another embodiment of the present invention, the ryanodine receptor
associated disorder is a skeletal muscular disorder and/or disease, including
but not
limited to skeletal muscle fatigue, exercise-induced skeletal muscle fatigue,
muscular
dystrophy, bladder disorders, and incontinence.
[0041] In an additional embodiment of the present invention, the ryanodine
receptor associated disorder is a cognitive disorder and/or disease including,
but not
necessarily limited to Alzheimer's Disease, dementia, forms of memory loss,
and age-
dependent memory loss. In a further embodiment of the present invention the
ryanodine
receptor associated disorder is a malignant hyperthermia, diabetes, or sudden
infant death
syndrome.
[0042] The present invention additionally provides methods for regulating PKA
phosphorylation of a ryanodine receptor comprising contacting the ryanodine
receptor
complex with an agent that modulates the level of PDE in the ryanodine
receptor
complex, wherein contacting the ryanodine receptor complex with an agent that
increases
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
11
the level of PDE in the complex results in a reduction of PKA phosporylation
of the
ryanodine receptor, and contacting the ryanodine receptor complex with an
agent that
decreases the level of PDE in the complex results in an increase of PKA
phosporylation
of the ryanodine receptor. In one embodiment of the invention, the agent is
selected from
the group consisting of a PDE protein, a nucleic acid encoding a PDE, a member
of a
PDE signal-transduction pathway, and a modulator of a member of a PDE signal
transduction pathway. In another embodiment of the present invention, the
phosphodiesterase is PDE4D or PDE4D3.
[0043] In a particular embodiment of the invention, the ryanodine receptor is
a
RyRl receptor. In another embodiment of the present invention, the ryanodine
receptor is
a RyR2 receptor. In a further embodiment, the ryanodine receptor is an RyR3
receptor.
100441 Also provided by the present invention are methods for decreasing PKA
phosphorylation of a ryanodine receptor by contacting the ryanodine receptor
complex
with an agent that increases the level of PDE in the ryanodine receptor
complex. In one
embodiment, the receptor is hyperphosphorylated prior to contacting the
ryanodine
receptor complex with the agent. In another embodiment, the agent is a PDE
protein, a
nucleic acid encoding a PDE, a member of a PDE signal-transduction pathway,
and a
modulator of a member of a PDE signal transduction pathway. In another
embodiment of
the present invention, the phosphodiesterase is PDE4D or PDE4D3.
[0045] In one embodiment of the invention, the ryanodine receptor is a RyRl
receptor. In another embodiment , the ryanodine receptor is a RyR2 associated
receptor.
In a further embodiment, the ryanodine receptor is a RyR3 associated receptor.
[0046] The present invention additionally provides methods for regulating Ca2}
release and reuptake in the sarcoplasmic reticulum of a cell comprising
contacting a
ryanodine receptor complex of the cell with an agent that modulates the level
of PDE,
wherein contacting the ryanodine receptor complex with an agent that increases
the level
of PDE results in a reduction of Ca2+ release from and reuptake into the
sarcoplasmic
reticulum and contacting the ryanodine receptor complex with an agent that
decreases the
level of PDE in the complex results in an increase of Ca2+ release from and
reuptake into
the sarcoplasmic reticulum.
[0047] In one embodiment of the invention, the agent is selected from the
group
consisting of a PDE protein, a nucleic acid encoding a PDE, a member of a PDE
signal-
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
12
transduction pathway, and a modulator of a member of a PDE signal transduction
pathway. In another embodiment of the present invention, the phosphodiesterase
is
PDE4D or PDE4D3.
[0048] In an embodiment of the invention, the ryanodine receptor is a RyR1
receptor. In another embodiment, the ryanodine receptor is a RyR2 receptor. In
a further
embodiment, the ryanodine receptor is a RyR3 receptor.
[0049] The present invention further provides methods for decreasing Ca2+
release
and reuptake in the sarcoplasmic reticulum of a cell comprising contacting a
ryanodine
receptor complex of the cell with an agent that increases the level of PDE. In
one
embodiment, the receptor is hyperphosphorylated prior to contacting the
ryanodine
receptor complex with the agent. In another embodiment, the agent is selected
from the
group consisting of a PDE protein, a nucleic acid encoding a PDE, a member of
a PDE
signal-transduction pathway, and a modulator of a member of a PDE signal
transduction
pathway. In a further embodiment, the phosphodiesterase is PDE4D, including
PDE4D3.
[0050] In an embodiment of the invention, the ryanodine receptor is a RyR1
receptor. In another embodiment, the ryanodine receptor is a RyR2 receptor. In
a further
embodiment, the ryanodine receptor is a RyR3 receptor.
BRIEF DESCRIPTION OF THE FIGURES
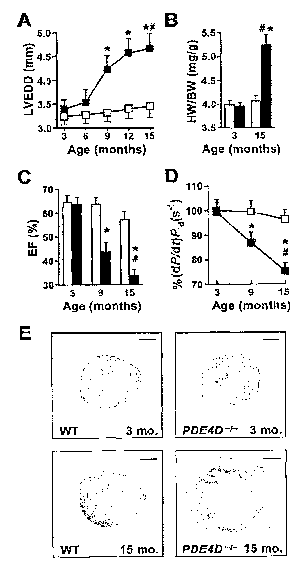
[0051] FIG. 1 PDE4D Deficiency Promotes Age-Related Cardiomyopathy. *p <
0.05 versus wt; #p < 0.05 versus PDE4D -'-. (A) Echocardiography at 3 month
intervals
showing progressive increase in left ventricular end-diastolic diameter
(LVEDD) in
PDE4D _' mice (open squares, wt; filled squares, PDE4D-' ; n= 12 each). Data
in (A)-
(D) are mean ~: SEM. (B) Age-dependent increase in heart-to-body-weight ratio
(HWBW) in PDE4D _'- mice (open bar, wt; filled bar, PDE4D-/-). (C) Age-
dependent
decrease in ejection fraction (EF) in PDE4D-/- mice (open bar, wt; filled bar,
PDE4D-'-).
(D) Reduced cardiac contractility (dP/dt/P;a) in PDE4D-/- mice at 3, 9, and 15
months of
age (open squares, wt; filled squares, PDE4D -/-). (E) Histology showing
dilated
cardiomyopathy in PDE4D-deficient mouse hearts.
[0052] FIG. 2 Normal cAMP and (3-Adrenergic-Receptor Levels in PDE4D-'-
Mouse Hearts and Increased cAMP Levels at the Z Lines Detected by FRET-PKA.
(A)
cAMP concentrations were not significantly increased in hearts of 3- to 6-
month-old
PDE4D-' mice (wt, n = 5; PDE4D-/-, n = 5; p= NS). Each heart was extracted and
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
13
assayed separately in quadruplicate experiments. Data in (A), (B), and (E) are
mean SD.
(B) 0-adrenergic-receptor density was unchanged in PDE4D-'- mice (wt, n = 5;
PDE4D -~, n = 5; p = NS). (C) Comparison of the B,,,aX values for (3-
adrenergic-receptor
density calculated separately for each of the five wt or five PDE4D -/-
knockout mice
investigated. (D) FRET-PKA showing increased cAMP-dependent signal over the Z
lines
(site of localization of RyR2) after low-dose isoproterenol stimulation in
PDE4D-/-
cardiomyocyte when compared to wild-type cardiomyocyte (white and black areas
represent sensing of highest and lowest cAMP concentrations, respectively).
(E) Bar
graph summarizes Z line intensity-profile analysis of 480 nrn/545 nm intensity
ratio from
five wt and six PDE4D -/- cells (*p < 0.05). Dimensions as indicated.
[0053] FIG. 3 shows Age-Dependent Alterations in RyR2-Channel Complex and
Function in PDE4D-' Murine Heart. (A) Immunoblots showing progressive increase
in
PKA phosphorylation of RyR2 at Ser2808 (second panel) but no change in CaMKII
phosphorylation at Ser2814 (third panel) and progressive decrease in
calstabin2 binding
to the RyR2 complex (fourth panel). PKA catalytic (-C) and regulatory (-RII)
subunits are
not changed during heart failure (HF) development (fifth and six panels). (B)
Quantification of age-dependent increase in RyR2 PKA phosphorylation (open
bars, wt;
filled bars, PDE4D-/-; *p < 0.05 versus wt, #p < 0.05 versus PDE4D -/-). Data
in (B)-(E)
and (G) are mean :L SD. (C) Quantification of age-dependent decrease in
calstabin2 in the
RyR2 complex (open bars, wt; filled bars, PDE4D-' ;*p < 0.05 versus wt, #p <
0.05
versus PDE4D-'-). (D) Quantification of CaMKII-specific RyR2 phosphorylation
showing no significant changes with aging. (E) Immunoprecipitation of RyR2
showing
complete absence of PDE activity in the RyR2 complex in PDE4D-'- mouse hearts.
*p <
0.05 versus wt; #p < 0.05 versus untreated wt. Rol, rolipram; Mil, milrinone.
(F) Single-
channel recordings of wt (left) and PDE4D -/- RyR2 (right) at 3 months of age
(top) and
15 months of age (bottom). Current trace from 3-month-old PDE4D-'- heart shows
slightly increased channel open probability (Po) with short openings
consistent with
increased PKA phosphorylation. At 15 months of age, RyR2 channels from PDE4D-/-
hearts showed significantly increased Po and subconductance states (indicated
by dotted
lines), as evidenced by current-amplitude histograms. Upper traces represent 5
s; lower
traces represent 500 ms. Channel openings are upward; full openings are 4 pA;
closed
state is indicated by "c." (G) Bar graphs summarizing Po, open frequency (Fo),
average
open time (To), and average closed time (Tc) at 15 months of age in wt and
PDE4D-/-
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
14
groups. *p < 0.001; n =10 each.
[0054] FIG. 4 PDE4D3 Is a Component of the RyR2 Ca2+-Release-Channel
Complex. (A) PAN-PDE4 antibody against the conserved UCR2 domain (a-4PAN, top
panel) and antibody raised against the N-terminal domain unique to PDE4D3 (a-
4D3,
bottom panel) were used to detect PDE4D isoforms in extracts of COS7 cells
overexpressing recombinant PDE4D splice variants 1 to 9. Samples were size
fractionated
on 6% SDS PAGE and blotted onto Immobilon-P membranes. (B) Immunoblotting with
splice-variant-specific anti-PDE4D3, anti-PDE4D$, and anti-PDE4D9 antibodies
shows
expression of all three major forms in the heart. However, immunoprecipitation
of RyR2
followed by splice-variant-specific immunoblot demonstrates that only PDE4D3
is
associated with RyR2. Reverse immunoprecipitation with a specific anti-PDE4D3
antibody confirms that PDE4D3 is physically associated with RyR2. (C)
Immunoblots of
cardiac lysates showing amounts of RyR2 and PDE4D3 in wt, PDE4D"-, and PDE4D-1-
mice. Bar graph shows a 37% reduction of PDE4D3 in the RyR2 complex in cardiac
lysates of PDE4D-" mice relative to wt. *p < 0.05, n = 3 for each genotype.
Data in (C)
and (D) are mean SD. (D) Coimmunoprecipitation using anti-RyR2 antibody,
showing
a 44% decrease of PDE4D3 bound to RyR2 in PDE4D+l- mouse hearts relative to
wt. *p
< 0.05, n= 3 for each genotype.
[0055] FIG. 5 Reduced PDE4D3 in the RyR2 Complex in Human Heart Failure.
(A) PDE4D3 was detected in human cardiac lysate and in the immunoprecipitated
RyR2
complex (IP:RyR2); IP:IgG, negative control. (B) RyR2 was immunoprecipitated
from
cardiac homogenates of normal human (N) and heart failure (HF) samples. PDE4D3
binding to RyR2 was significantly decreased in human HF. Increased PKA
phosphorylation was detected by a phosphoepitope-specific RyR2-Ser2808
antibody in
HF samples. (C) RyR2 bound PDE4D3 activity was significantly decreased in HF,
as
evidenced by close-proximity substrate cAMP catalysis (#p < 0.001). Data in
(C) and (D)
are mean SD. (D) Rolipram (R), a PDE4-specific inhibitor, but not milrinone
(M), a
PDE3-specific inhibitor, decreased RyR2-associated PDE activity submaximally;
C,
control (#p < 0.001 versus untreated sample).
[0056] FIG. 6 Cardiac Arrhythmias due to PDE4D3 Inhibition Are Suppressed in
Mice Harboring Mutant RyR2 that Cannot be PKA Phosphorylated. (A)
Susceptibility to
exercise-induced sustained ventricular arrhythmias (sVT) and nonsustained
ventricular
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
arrhythmias (nsVT) was significantly increased in PDE4D-'- compared to wt mice
(each
n = 6, *p < 0.05). (B) Rolipram (0.3 mg/kg body weight) maximally increased
RyR2-
Ser2808 PKA phosphorylation during exercise in vivo. Treatment as indicated on
top:
Rol, rolipram; Epi, epinephrine (0.1 mg/kg); white bars, no rolipram; black
bar, rolipram-
treated mice; *p < 0.05 between treatments in wt mice; #p < 0.001 wt versus
homozygous
RyR2-S2808A knockin (S2808A-"+) mice. Control mice were treated with placebo
(rolipram carrier, 0.5% DMSO). Data are mean ::[-- SD. (C) In rolipram-treated
mice,
susceptibility to exercise-induced sustained ventricular arrhythmias (sVT) and
nonsustained ventricular arrhythmias (nsVT) was significantly decreased in
homozygous
RyR2-S2808A knockin (S2808A+~) mice compared to wt mice (each n= 6, #p <
0.05).
(D) Mortality from sudden death was significantly increased in PDE4D-"- mice
24 and 72
hr after MI compared to wt mice. White bars, wt mice; black bars, PDE4D-"-
mice. *p <
0.01 between wt and PDE4D+/ groups.
[0057] FIG. 7 PDE4D3 Deficiency Promotes HF Progression. (A) LVEDD
increased in PDE4D+/- (black squares) compared to wt (open squares) mice
before
(control, CO) and 14 and 28 days after myocardial infarction (MI) (*p < 0.05
versus wt).
Both treatment with the 1,4-benzothiazepine JTV-519 (red line), which enhances
calstabin2 binding to RyR2, or crossing the PDE4D+/- mice with RyR2-S2808A
mice that
harbor RyR2 that cannot be PKA phosphorylated (green line) significantly
reduced the
remodeling of the left ventricle following MI (#p < 0.01 versus PDE4D+/-).
Data in (A)-
(C) are mean SEM. (B) Reduced cardiac EF in PDE4D+'- mice 28 days after MI.
wt,
open bars; PDE4D+/- mice, filled bars (*p < 0.05 versus CO same genotype).
Both
treatment with the 1,4-benzothiazepine JTV-519 (red bar), which enhances
calstabin2
binding to RyR2, or crossing the PDE4D+'- mice with RyR2-S2808A mice that
harbor
RyR2 that cannot be PKA phosphorylated (green bar) significantly improved left
ventricular EF following MI (#p < 0.01 versus PDE4D+'- MI). (C) Left
ventricular
contractility (dP/dt)/P;a normalized to 100% of control in wt (open bars) and
haploinsufficient PDE4D+/ mice (black bars) before (control, CO) and 28 days
after MI
(*p < 0.05 versus CO in same genotype). Treatment with JTV-519 (red bar) or
crossing
the PDE4D+/ mice with RyR2-S2808A mice (green bar) significantly improved
contractility following MI (#p < 0.01 versus PDE4D+'- MI). (D) Immunoblot
showing
levels of PDE4D3, calstabin2, and RyR2-Ser2808 PKA phosphorylation in the
immunoprecipitated RyR2-channel complex. JTV-519 treatment (red) or crossing
the
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
16
PDE4D+/- mice with RyR2-S2808A mice (green) significantly increased the
binding of
calstabin2 to RyR2 in the PDE4D-deficient mice. (E) Quantification of RyR2 PKA
phosphorylation in wt and PDE4D+/- mice before (CO) and 28 days after MI,
showing
significantly more RyR2 PKA hyperphosphorylation in PDE4D+/ mice (*p < 0.05
versus
wt at same time point). Treatment with JTV-519 (red bar) or crossing the
PDE4D+/ mice
with RyR2-S2808A mice (green bar) significantly reduced RyR2 PKA
phosphorylation
following MI (#p < 0.01 versus PDE4D+/ MI). Data in (E) and (F) are mean zL
SD. (F)
Quantification of RyR2-associated PDE activity showing significant decrease in
PDE4D+1- mice 28 days before (CO) and after MI compared to wt (*p < 0.05
versus wt).
Treatment with JTV-519 (red bar) or crossing the PDE4D+/ mice with RyR2-S2808A
mice (green bar) significantly increased RyR2-associated PDE activity
following MI (#p
< 0.01 versus PDE4D+/- MI).
DETAILED DESCRIPTION OF THE INVENTION
[0058] As used herein and in the appended claims the singular forms "a," "an,"
and "the" include plural references unless the content clearly dictates
otherwise. Thus,
for example, reference to "an agent" includes a plurality of such agents and
equivalents
thereof known to those skilled in the art, and so forth. All publications,
patent
applications, patents and other references mentioned herein are incorporated
by reference
in there entirety.
[0059] The inventors have previously shown in co-pending U.S. Application No.
10/7894,218 (incorporated in its entirety herein) that PDE4D3 is present in
the RyRl
receptor complex of skeletal muscle. Specifically, to determine whether mAKAP
and
PDE4D3 were present in the RyRl macromolecular complex, immunoprecipitation
with
anti-RyR-5029 followed by immunoblotting with antibodies that detect mAKAP and
PDE4D3 was used. The inventors found that both mAKAP and PDE4D3 co-
immunoprecipitate with RyRl and therefore are part of the RyRl macromolecular
complex. Moreover, the inventors found that compared to controls, the amount
of
PDE4D3 in the RyRl channel complex could contribute to a local increase in
cAMP and
increased PKA activity that likely explains PKA hyperphosphorylation of RyRl
in HF
skelatal muscle.
[0060] The inventors have also previously posited in co-pending U.S.
Application
No. 10/608,723 (incorporated in its entirety herein) that a chemical agent
that reduces
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
17
PKA phosphorylation of a RyR2 receptor could at via multifarious mechanisms,
including, inter alia, inhibiting PKA activity, or increasing the activity of
endogenous
phosphatases (PPl and PP2A have been shown to be present in the RyR2
macromolecular
complex), or increasing the activity of a phosphodiesterase which hydrolyzes
cAMP
(PDE4D3, which hydrolyzes cAMP, has also been shown to be present in the RyR2
macromolecular complex; Dodge et al., 2001).
[0061] The inventors demonstrate herein a novel function of PDE4D3 in the
regulation of intracellular Ca2+ release. PDE4D3 activity provides an
important negative
feedback mechanism to limit (3AR-dependent PKA phosphorylation of RyR2-Ser28o9
Under physiologic conditions, PDE4D3 regulates local PKA activity and channel
activation at RyR2-Ser 2809 and prevents excess accumulation of cAMP and
uncontrolled
PKA activation. In human heart failure, PDE4D3 deficiency contributes to RyR2
PKA
hyperphosphorylation, calstabin2 (FKBP12.6) depletion and hyperactive, "leaky"
RyR2
channels. Since global cAMP synthesis is decreased in HF (Feldman, 1987), a
reduced
capacity for cAMP hydrolysis by locally reduced PDE4D3 activity may be
causative for
RyR2 PKA hyperphosphorylation. Taken together, these data suggest that PDE4D3
plays
a protective role in the heart against HF and catecholaminergic arrhythmias.
On the other
hand, chronic pharmacologic inhibition of the PDE4 class of enzymes may
contribute to a
cardiac phenotype, particularly in individuals with underlying cardiac
disease. In addition
there are likely to be other signaling systems affected by reduced PDE4
activity, since 0-
arrestin targeting of PDE4D3 activity may be important for p2AR
desensitization28 and
PDE4D ablation differentially changes j3IAR versus (32AR signaling.
Iinportantly,
PDE4D3 deficiency was associated with stress-induced cardiac arrhythmias
indicating a
possible novel mechanism for drug-induced sudden death analogous to the drug-
induced
long QT syndrome caused by blockage of HERG potassium channels, suggesting
that
PDE4 inhibitors could increase the risk of sudden cardiac death via an RyR2-
mediated
mechanism.
[0062] In veiw of the foregoing, the present invention provides novel
compounds
and methods for treating and preventing disorders and diseases associated with
the RyR
receptors that regulate calcium channel functioning in cells.
[0063] Specifically, the present invention encompasses and provides
compositions
useful for treating or preventing a ryanodine receptor associated disorder
comprising a
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
18
phosphodiesterase (PDE)-associated agent; and optionally a pharmaceutically
acceptable
carrier. In a preferred embodiment of the invention, the PDE-associated agent
may be a
PDE protein, a nucleic acid encoding a PDE protein, a member of a PDE signal-
transduction pathway, and a modulator of a member of a PDE signal transduction
pathway. In one embodiment of the present invention, the PDE-associated agent
is
PDE4D protein or a nucleic acid encoding a PDE4D protein. The ryanodine
receptor
associated disorder may be a RyR2-l, RyR2 or RyR3 associated disorder.
[0064] As used herein the term "ryanodine receptor associated disorders" and
"disorders and diseases associated with the RyR receptors" means disorders and
diseases
that can be treated and/or prevented by modulating PDE in the RyR receptor
complex in
cells. "Ryanodine receptor associated disorders" and/or "Disorders and
diseases
associated with the RyR receptors" include, without limitation, cardiac
disorders and
diseases, skeletal muscular disorders and diseases, cognitive disorders and
diseases,
malignant hyperthermia, diabetes, and sudden infant death syndrome. Cardiac
disorder
and diseases include, but are not limited to, irregular heartbeat disorders
and diseases;
exercise-induced irregular heartbeat disorders and diseases; sudden cardiac
death;
exercise-induced sudden cardiac death; congestive heart failure; chronic
obstructive
pulmonary disease; and high blood pressure. Irregular heartbeat disorders and
diseases
include and exercise-induced irregular heartbeat disorders and diseases
include, but are
not limited to, atrial and ventricular arrhythmia; atrial and ventricular
fibrillation; atrial
and ventricular tachyarrhythmia; atrial and ventricular tachycardia;
catecholaminergic
polymorphic ventricular tachycardia (CPVT); and exercise-induced variants
thereof.
Skeletal muscular disorder and diseases include, but are not limited to,
skeletal muscle
fatigue, exercise-induced skeletal muscle fatigue, muscular dystrophy, bladder
disorders,
and incontinence. Cognitive disorders and diseases include, but are not
limited to,
Alzheimer's Disease, forms of memory loss, and age-dependent memory loss.
[0065] As used herein, "RyR" includes RyRl, RyR2, and RyR3, and also
includes an "RyR protein" and an "RyR analogue." An "RyR analogue" is a
functional
variant of the RyR protein, having RyR biological activity, that has 60% or
greater
amino-acid-sequence homology with the RyR protein. The RyR of the present
invention
are unphosphorylated, phosphorylated (e.g., by PKA), or hyperphosphorylated
(e.g., by
PKA). As further used herein, the term "RyR biological activity" refers to the
activity of
a protein or peptide that demonstrates an ability to associate physically
with, or bind with,
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
19
FKBP12 (calstabinl) in the case of RyRl and RyR3, and FKBP12.6 (calstabin2) in
the
case of RyR2 (i.e., binding of approximately two fold or, approximately five
fold, above
the background binding of a negative control), under the conditions of the
assays
described herein.
[0066] As used herein, "ryanodine receptor complex" or "RyR complex" refers to
the RyR macromolecular signaling complex as described herein.
[0067] Additionally, the present invention provides methods for treating or
preventing a ryanodine receptor associated disorder in a subject comprising
augmenting
PDE in a ryanodine receptor complex of the subject. In one embodiment of the
invention,
PDE is augmented in the ryanodine receptor complex by contacting the ryanodine
receptor complex with a PDE-associated agent. In one embodiment of the
invention, the
PDE-associated agent is selected from the group consisting of a PDE protein, a
nucleic
acid encoding a PDE, a member of a PDE signal-transduction pathway, and a
modulator
of a member of a PDE signal transduction pathway. In a particular embodiment,
the
nucleic acid is operatively linked to a.n inducible promoter. In another
embodiment of the
present invention, the phosphodiesterase is PDE4D including PDE4D3. The
subject of
the present invention may be an in vitro or in vivo system and any animal,
including
amphibians, birds, fish mammals, and marsupials, but is preferably a mammal,
including
but not necessarily limited to a mouse, rat, cat, dog, horse, monkey cow or
pig. In a
preferred embodiment of the invention, the subject is human.
[0068] In a particular embodiment of the invention, the ryanodine receptor
associated disorder is a RyRl receptor associated disorder. In another
embodiment of the
present invention, the ryanodine receptor associated disorder is a RyR2
receptor
associated disorder. In a further embodiment, the ryanodine receptor
associated disorder
is an RyR3 receptor associated disorder.
[0069] The cells of a subject include striated muscle cells. A striated muscle
is a
muscle in which the repeating units (sarcomeres) of the contractile myofibrils
are
arranged in registry througlZOut the cell, resulting in transverse or oblique
striations that
are observed at the level of a light microscope. Examples of striated muscle
cells include,
without limitation, voluntary (skeletal) muscle cells and cardiac muscle
cells. In one
embodiment, the cell used in the method of the present invention is a human
cardiac
muscle cell. As used herein, the term "cardiac muscle cell" includes cardiac
muscle
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
fibers, such as those found in the myocardium of the heart. Cardiac muscle
fibers are
composed of chains of contiguous heart-muscle cells, or cardiomyocytes, joined
end to
end at intercalated disks. These disks possess two kinds of cell junctions:
expanded
desmosomes extending along their transverse portions, and gap junctions, the
largest of
which lie along their longitudinal portions.
[0070] In one embodiment of the present invention, the ryanodine receptor
associated disorder may be a cardiac disorder and/or disease including, but
not limited to,
an irregular heartbeat disorder or disease; exercise-induced irregular heart
beat disorder or
disease; sudden cardiac death; exercise-induced sudden cardiac death;
congestive heart
failure; chronic obstructive pulmonary disease; and high blood pressure. The
irregular
heartbeat disorders and diseases and exercise-induced irregular heartbeat
disorders and
diseases may include, but are not necessarily limited to, atrial and
ventricular arrhythmia;
atrial and ventricular fibrillation; atrial and ventricular tachyarrhythmia;
atrial and
ventricular tachycardia; catechlaminergic polymorphic ventricular tachycardia
(CPTV);
and exercise-induced variants thereof.
[0071] In another embodiment of the present invention, the ryanodine receptor
associated disorder is a skeletal muscular disorder and/or disease, including
but not
limited to skeletal muscle fatigue, exercise-induced skeletal muscle fatigue,
muscular
dystrophy, bladder disorders, and incontinence.
[0072] In an additional embodiment of the present invention, the ryanodine
receptor associated disorder is a cognitive disorder and/or disease including,
but not
necessarily limited to Alzheimer's Disease, dementia, forms of memory loss,
and age-
dependent memory loss. In a further embodiment of the present invention the
ryanodine
receptor associated disorder is a malignant hyperthermia, diabetes, or sudden
infant death
syndrome.
[0073] As used herein, "effective amount" or "pharmaceutically effective
amount" refers to any amount of an agent which, when administered to a subject
suffering
from a disorder against which the agent is effective, causes reduction,
remission or
regression or prevents recurrence of the disorder. "Prophylactically effective
amount"
refers to any amount of an agent which, when administered to a subject prone
to suffer
from a disorder, inhibits the onset of the disorder.
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
21
[0074] The present invention additionally provides methods for regulating PKA
phosphorylation of a ryanodine receptor comprising contacting the ryanodine
receptor
complex with an agent that modulates the level of PDE in the ryanodine
receptor
complex, wherein contacting the ryanodine receptor complex with an agent that
increases
the level of PDE in the complex results in a reduction of PKA phosporylation
of the
ryanodine receptor, and contacting the ryanodine receptor complex with an
agent that
decreases the level of PDE in the complex results in an increase of PKA
phosporylation
of the ryanodine receptor. In one embodiment of the invention, the agent is
selected from
the group consisting of a PDE protein, a nucleic acid encoding a PDE, a member
of a
PDE signal-transduction pathway, and a modulator of a member of a PDE signal
transduction pathway. In preferred embodiment of the present invention, the
phosphodiesterase is PDE4D or PDE4D3.
[0075] In a particular embodiment of the invention, the ryanodine receptor is
a
RyRl receptor. In another embodiment of the present invention, the ryanodine
receptor is
a RyR2 receptor. In a further embodiment, the ryanodine receptor is an RyR3
receptor.
[0076] As used herein, "PKA phosphorylation" refers to a reaction in which a
phosphate group is substituted for a hydroxyl group by the enzyme protein
kinase A
(PKA).
[0077] The present invention also provides kits for use in delivering a PDE-
associated agent to a ryanodine receptor complex in a subject, comprising a
PDE-
associated agent, optionally with a pharmaceutically acceptable carrier and a
catheter.
[0078] Also provided by the present invention are methods for decreasing PKA
phosphorylation of a ryanodine receptor by contacting the ryanodine receptor
complex
with an agent that increases the level of PDE in the ryanodine receptor
complex. In one
embodiment, the receptor is hyperphosphorylated prior to contacting the
ryanodine
receptor complex with the agent. In another einbodiment, the agent is a PDE
protein, a
nucleic acid encoding a PDE, a member of a PDE signal-transduction pathway,
and a
modulator of a member of a PDE signal transduction pathway. In another
embodiment of
the present invention, the phosphodiesterase is PDE4D or PDE4D3.
[0079] In one embodiment of the invention, the ryanodine receptor is a RyRl
receptor. In another embodiment , the ryanodine receptor is a RyR2 associated
receptor.
In a fi.irther embodiment, the ryanodine receptor is a RyR3 associated
receptor.
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
22
[0080] The present invention additionally provides methods for regulating Ca2}
release and reuptake in the sarcoplasmic reticulum of a cell comprising
contacting a
ryanodine receptor complex of the cell with an agent that modulates the level
of PDE,
wherein contacting the ryanodine receptor complex with an agent that increases
the level
of PDE results in a reduction of Ca2+ release from and reuptake into the
sarcoplasmic
reticulum and contacting the ryanodine receptor complex with an agent that
decreases the
level of PDE in the complex results in an increase of CaZ+ release from and
reuptake into
the sarcoplasmic reticulum.
[0081] In one embodiment of the invention, the agent is selected from the
group
consisting of a PDE protein, a nucleic acid encoding a PDE, a member of a PDE
signal-
transduction pathway, and a modulator of a member of a PDE signal transduction
pathway. In a preferred embodiment of the present invention, the
phosphodiesterase is
PDE4D or PDE4D3.
[0082] In an embodiment of the invention, the ryanodine receptor is a RyRl
receptor. In another embodiment, the ryanodine receptor is a RyR2 receptor. In
a further
embodiment, the ryanodine receptor is a RyR3 receptor.
[0083] The present invention further provides methods for decreasing Ca2+
release
and reuptake in the sarcoplasmic reticulum of a cell comprising contacting a
ryanodine
receptor complex of the cell with an agent that increases the level of PDE. In
one
embodiment, the receptor is hyperphosphorylated prior to contacting the
ryanodine
receptor complex with the agent. In another embodiment, the agent is selected
from the
group consisting of a PDE protein, a nucleic acid encoding a PDE, a member of
a PDE
signal-transduction pathway, and a modulator of a member of a PDE signal
transduction
pathway. In a further embodiment, the phosphodiesterase is PDE4D, including
PDE4D3.
[0084] In an embodiment of the invention, the ryanodine receptor is a RyRl
receptor. In another embodiment, the ryanodine receptor is a RyR2 receptor. In
a further
embodiment, the ryanodine receptor is a RyR3 receptor.
[0085] PDEs are enzyme proteins, found in certain cells, which hydrolyze
phosphodiester bonds. PDEs regulate the local concentration of 3', 5' cyclic
adenosine
monophosphate (cAMP) within cells. In the heart, PDE4 contributes to the
regulation of
cAMP levels in cardiac myocytes. The PDE superfamily is subgrouped into 11
families
tht include at least 20 genes and 50 unique isoforms. The PDE4D gene encodes
nnine
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
23
variants (PDE4D1-9) with identical catalytic domains and carboxyl termini and
unique
amino termini important for subcellular localization. In a preferred
embodiment of the
present invention, the PDE is PDE4D3.
[0086] As used herein, "PDE" includes both a "PDE protein" and a "PDE
analogue". Unless otherwise indicated, "protein" shall include a protein,
protein domain,
polypeptide, or peptide, and any fragment or variant thereof having protein
function. The
variants preferably have greater than about 75% homology with the naturally-
occurring
protein sequence, more preferably have greater than about 80% homology, even
more
preferably have greater than about 85% homology, and most preferably, have
greater than
about 90% homology with the protein sequence. In some embodiments, the
homology
may be as high as about 93-95%, 98%, or 99%. These variants may be
substitutional,
insertional, or deletional variants. The variants may also be chemically-
modified
derivatives: proteins which have been subjected to chemical modification, but
which
retain the biological characteristics of the naturally-occurring protein.
[0087] A "PDE analogue", as used herein, is a functional variant of the PDE
protein, having PDE biological activity, that has 60% or greater (preferably,
70% or
greater) amino-acid-sequence homology with the PDE protein. As further used
herein,
the term "PDE biological activity" refers to the activity of a protein or
peptide that
demonstrates an ability to hydrolyze cAMP, as described herein.
[0088] In accordance with methods described herein, PDE may be augmented or
increased in a cell or subcellular compartment, or more particularly, in a
ryanodine
receptor complex (RyR complex) of a cell by activating, facilitating,
inducing, or
stimulating one or more functions, activities, or effects (e.g., downstream
effects of the
PDE in the PDE signal transduction pathway) of PDE in a cell or in a RyR of a
cell,
particularly those that result in promotion of heart-tissue generation, or by
increasing the
amount, expression, or level of PDE in the cells. Furthermore, one or more PDE
functions, activities, effects, expression, and levels in a cell, subcellular
compartment or
RyR complex may be augmented by targeting PDE directly, or by targeting PDE
indirectly, via an enzyme or other endogenous molecule that regulates or
modulates the
functions, activities, effects, expression, and/or levels of PDE in the cell.
PDE expression
may also be augmented by engineering the PDE gene so that PDE is expressed on
an
inducible promoter. In such a case, PDE expression would be sustained in the
presence of
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
24
a suitable inducing agent, but would shut down once the supply of inducer was
depleted,
thereby bringing about a decrease in the amount or level of PDE in the cell.
PDE also
may be augmented in a cell or RyR complex by activating, facilitating,
inducing, or
stimulating the functions, activities, effects, expression, and levels of
endogenous PDE, or
by introduction of an exogenous PDE, particularly where the PDE is under the
control of
a strong promoter.
100891 The functions, activities, effects, expression, and/or levels of PDE
are
augmented in a cells or RyR complex by an amount effective to promote
hydrolyzation of
cAMP. This amount may be readily determined by the skilled artisan, based upon
known
procedures, including analysis of titration curves established in vivo,
methods disclosed
herein, and techniques known to one of skill in the art.
[0090] In the method of the present invention, the fi.inctions, activities,
effects,
expression, and/or levels of PDE in a cell, subeellular compartment or RyR
complex are
preferably augmented by contacting the cells (i.e., treating the cells) with a
PDE-
associated agent. As used herein, an "agent" shall include a protein,
polypeptide, peptide,
nucleic acid (including DNA, RNA, and an antisense oligonucleotide), antibody
(monoclonal and polyclonal), Fab fragment, F(ab')2 fragment, molecule,
compound,
antibiotic, drug, and any combinations thereof, and may be an agent reactive
with PDE or
a member of a PDE signal transduction pathway. The term "reactive", as used
herein,
means that the molecule or mimetic has affinity for, binds to, or is directed
against PDE
or a member of a PDE signal transduction pathway. A Fab fragment is a
univalent
antigen-binding fragment of an antibody, which is produced by papain
digestion. A
F(ab')2 fragment is a divalent antigen-binding fragment of an antibody, which
is produced
by pepsin digestion.
[0091] As further used herein, the term "PDE-associated agent" or "agent"
includes a PDE protein, including an exogenous PDE protein; a PDE nucleic acid
(i, e., a
nucleic acid encoding a PDE); a member of a PDE signal-transduction pathway
(including upstream and downstream effectors and activators, in either protein
or nucleic
acid form); and a modulator (e.g., inhibitor, activator, antagonist, or
agonist) of a member
of the PDE signal transduction pathway or system (i.e., a modulator which
affects the
expression, activity, function, and/or effect of a member of the PDE signal-
transduction
pathway), in either protein or nucleic acid form, including a modulator of PDE
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
expression. Additionally, as used herein, a "member of a PDE signal-
transduction
pathway" includes a downstream effector or an upstream regulator of PDE in
cells.
[0092] By way of example, activity of PDE in a cell, subcellular compartment
or
RyR complex may be augmented by contacting the cells or RyR complex with a
small
molecule or protein mimetic that stimulates PDE activity and/or that is
reactive with PDE
or a member of a PDE signal transduction pathway. Similarly, the level of PDE
in a cell
or RyR complex may be augmented by directly or indirectly causing, inducing,
or
stimulating the upregulation of PDE expression within a subject. Accordingly,
in one
embodiment of the present invention, activity of PDE is increased in a subject
by
administering to the subject a modulator of PDE expression.
[0093] In one embodiment of the present invention, the PDE-associated agent is
a
protein. Examples of proteins for use in the present invention include,
without limitation,
PDE proteins, members of the PDE signal-transduction pathway (including
upstream and
downstream effector and activator polypeptides), modulators (e.g., inhibitors,
activators,
antagonists, or agonists) of a member of the PDE signal-transduction
pathway/system,
PDE-associated antibodies (e.g., IgA, IgD, IgE, IgG, IgM, single-chain
antibodies, and
Fab' fragments, such as scFv) that are capable of binding and inhibiting a
negative
regulator of the PDE signal-transduction pathway, and PDE-associated ligands
(e.g., a
ligand for a member of the PDE signal-transduction pathway, and derivatives
thereof).
Preferably, the PDE-associated protein is PDE4D protein.
[0094] Where the protein of the present invention is an antibody, the protein
is
preferably a mammalian antibody (e.g., a human antibody) or a chimeric
antibody (e.g., a
humanized antibody). More preferably, the antibody is a human or humanized
antibody.
As used herein, the term "humanized antibody" refers to a genetically-
engineered
antibody in which the minimum portion of an animal antibody (e.g., an antibody
of a
mouse, rat, pig, goat, or chicken) that is generally essential for its
specific functions is
"fused" onto a human antibody. In general, a humanized antibody is 1-25%,
preferably 5-
10%, animal; the remainder is human. Humanized antibodies usually initiate
minimal or
no response in the human immune system. Methods for expressing fully human or
humanized antibodies in organisms other than human are well known in the art
(see e.g.,
U.S. Patent No. 6,150,584, Human antibodies derived from immunized xenomice;
U.S.
Patent No. 6,162,963, Generation of xenogenetic antibodies; and U.S. Patent
No.
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
26
6,479,284, Humanized antibody and uses thereof). In one embodiment of the
present
invention, the antibody is a single-chain antibody. In one embodiment, the
single-chain
antibody is a human or humanized single-chain antibody. In another embodiment
of the
present invention, the antibody is a murine antibody.
[0095] The PDE-associated agent of the present invention may also be a nucleic
acid. As used herein, a"nucleic acid" or "polynucleotide" includes a nucleic
acid, an
oligonucleotide, a nucleotide, a polynucleotide, and any fragment or variant
thereof. The
nucleic acid or polynucleotide may be double-stranded, single-stranded, or
triple-stranded
DNA or RNA (including cDNA), or a DNA-RNA hybrid of genetic or synthetic
origin,
wherein the nucleic acid contains any combination of deoxyribonucleotides and
ribonucleotides and any combination of bases, including, but not limited to,
adenine,
thymine, cytosine, guanine, uracil, inosine, and xanthine hypoxanthine. The
nucleic acid
or polynucleotide may be combined with a carbohydrate, lipid, protein, or
other materials.
In one embodiment of the present invention, the nucleic acid encodes PDE4D
protein.
[0096] The "complement" of a nucleic acid refers, herein, to a nucleic acid
molecule which is completely complementary to another nucleic acid, or which
will
hybridize to the other nucleic acid under conditions of high stringency. High-
stringency
conditions are known in the art (see e.g., Maniatis et al., Molecular Cloning:
A
Laboratory Manual, 2nd ed. (Cold Spring Harbor: Cold Spring Harbor Laboratory,
1989)
and Ausubel et al., eds., Current Protocols in Molecular Biology (New York,
NY: John
Wiley & Sons, Inc., 2001)). Stringent conditions are sequence-dependent, and
may vary
depending upon the circumstances. As used herein, the terin "cDNA" refers to
an isolated
DNA polynucleotide or nucleic acid molecule, or any fragment, derivative, or
complement thereof. It may be double-stranded, single-stranded, or triple-
stranded, it
may have originated recombinantly or synthetically, and it may represent
coding and/or
noneoding 5' and/or 3' sequences.
[0097] The nucleic acid agent of the present invention, for example, may be a
plasmid. Such a plasmid may comprise a nucleic acid sequence encoding PDE or
another
PDE-associated protein, although it is to be understood that other types of
nucleic acid
agents, such as recombinant viral vectors, may also be used for the purposes
of the
present invention. In one embodiment of the present invention, the nucleic
acid (e.g.,
plasmid) encodes at least one PDE-associated protein. In a preferred
embodiment, the
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
27
nucleic acid encodes PDE4D protein.
[0098] The term "plasmid", as used herein, refers generally to circular double-
stranded DNA, which is not bound to a chromosome. The DNA, for example, may be
a
chromosomal or episomal-derived plasmid. The plasmid of the present invention
may
optionally contain a terminator of transcription, a promoter, and/or a
discrete series of
restriction-endonuclease recognition sites, located between the promoter and
the
terminator. In the plasmid, a polynucleotide insert of interest (e.g., one
encoding a PDE-
associated protein) should be operatively linked to an appropriate promoter.
The
promoter may be its native promoter or a host-derived promoter. The promoter
may also
be a tissue-specific promoter, such as a cardiomyocyte-specific promoter or
other heart-
tissue-specific promoter. The promoter may further be a regulatable promoter,
which
may be turned off when the expression of the gene is no longer desired.
Examples of
promoters for use in the present invention include the actin promoter and
viral promoters.
Other suitable promoters will be known to the skilled artisan.
[0099] In another embodiment of the present invention, the nucleic acid (e.g.,
plasmid) encodes or comprises at least one gene-silencing cassette, wherein
the cassette is
capable of silencing the expression of genes that negatively affect the PDE
signal-
transduction pathway/system. It is well understood in the art that a gene may
be silenced
at a number of stages, including, without limitation, pre-transcription
silencing,
transcription silencing, translation silencing, post-transcription silencing,
and post-
translation silencing. In one embodiment of the present invention, the gene-
silencing
cassette encodes or comprises a post-transcription gene-silencing composition,
such as
antisense RNA or RNAi. Both antisense RNA and RNAi may be produced in vitro,
in
vivo, ex vivo, or in situ.
[00100] For example, the PDE-associated agent of the present invention may be
an
antisense RNA. Antisense RNA is an RNA molecule with a sequence complementary
to a
specific RNA transcript, or mRNA, whose binding prevents further processing of
the
transcript or translation of the mRNA. Antisense molecules may be generated,
synthetically or recombinantly, with a nucleic-acid vector expressing an
antisense gene-
silencing cassette. Such antisense molecules may be single-stranded RNAs or
DNAs,
with lengths as short as 15-20 bases or as long as a sequence complementary to
the entire
mRNA. RNA molecules are sensitive to nucleases. To afford protection against
nuclease
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
28
digestion, an antisense deoxyoligonucleotide may be synthesized as a
phosphorothioate,
in which one of the nonbridging oxygens surrounding the phosphate group of the
deoxynucleotide is replaced with a sulfur atom (Stein et al.,
Oligodeoxynucleotides as
inhibitors of gene expression: a review, Cancer Res., 48:2659-68, 1998).
[00101] Antisense molecules designed to bind to the entire mRNA may be made by
inserting cDNA into an expression plasmid in the opposite or antisense
orientation.
Antisense molecules may also function by preventing translation initiation
factors from
binding near the 5' cap site of the mRNA, or by interfering with interaction
of the mRNA
and ribosomes (see e.g., U.S. Patent No. 6,448,080, Antisense modulation of
WRN
expression; U.S. Patent Application No. 2003/0018993, Methods of gene
silencing using
inverted repeat sequences; U.S. Patent Application No., 2003/0017549, Methods
and
compositions for expressing polynucleotides specifically in smooth muscle
cells in vivo;
Tavian et al., Stable expression of antisense urokinase mRNA inhibits the
proliferation
and invasion of human hepatocellular carcinoma cells, Cancer Gene Ther.,
10:112-20,
2003; Maxwell and Rivera, Proline oxidase induces apoptosis in tumor cells and
its
expression is absent or reduced in renal carcinoma, J. Biol. Chem., 278:9784-
89, 2003;
Ghosh et al., Role of superoxide dismutase in survival of Leishmania within
the
macrophage, Biochem. J., 369:447-52, 2003; and Zhang et al., An anti-sense
construct of
full-length ATM cDNA imposes a radiosensitive phenotype on normal cells,
Oncogene,
17:811-8, 1998).
[00102] Oligonucleotides antisense to a member of the PDE signal-transduction
pathway/system may be designed based on the nucleotide sequence of the member
of
interest. For example, a partial sequence of the nucleotide sequence of
interest (generally,
15-20 base pairs), or a variation sequence thereof, may be selected for the
design of an
antisense oligonucleotide. This portion of the nucleotide sequence may be
within the 5'
domain. A nucleotide sequence complementary to the selected partial sequence
of the
gene of interest, or the selected variation sequence, then may be chemically
synthesized
using one of a variety of techniques known to those skilled in the art,
including, without
limitation, automated synthesis of oligonucleotides having sequences which
correspond to
a partial sequence of the nucleotide sequence of interest, or a variation
sequence thereof,
using commercially-available oligonucleotide synthesizers, such as the Applied
Biosystems Mode1392 DNA/RNA synthesizer.
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
29
[00103] Once the desired antisense oligonucleotide has been prepared, its
ability to
modulate PDE then may be assayed. For example, the antisense oligonucleotide
may be
contacted with a cells or RyR receptor complex, and the levels of PDE
expression or
activity in the cells or RyR complex may be determined using standard
techniques, such
as Western-blot analysis and immunostaining. Alternatively, the antisense
oligonucleotide may be delivered to a cell or RyR complex using a liposome
vehicle, then
the levels of PDE expression or activity in the cells may be determined using
standard
techniques, such as Western-blot analysis and inununostaining. Where the level
of PDE
expression in the cells is increased in the presence of the designed antisense
oligonucleotide, it may be concluded that the oligonucleotide could be an
appropriate
PDE-associated agent for use in modulating PDE in cells.
[00104] It is within the confmes of the present invention that
oligonucleotides
antisense to a member of the PDE signal-transduction pathway/system may be
linked to
another agent, such as a drug or a ribozyme, in order to increase the
effectiveness of
treatments using PDE-associated agents and/or to increase the efficacy of
targeting.
Moreover, antisense oligonucleotides may be prepared using modified bases
(e.g., a
phosphorothioate), as discussed above, to make the oligonucleotides more
stable and
better able to withstand degradation.
[00105] The PDE-associated agent of the present invention also may be an
interfering RNA, or RNAi, including PDE small interfering RNA (siRNA). As used
herein, "RNAi" refers to a double-stranded RNA (dsRNA) duplex of any length,
with or
without single-strand overhangs, wherein at least one strand, putatively the
antisense
strand, is homologous to the target mRNA to be degraded. As further used
herein, a
"double-stranded RNA" molecule includes any RNA molecule, fragment, or segment
containing two strands forming an RNA duplex, notwithstanding the presence of
single-
stranded overhangs of unpaired nucleotides. Additionally, as used herein, a
double-
stranded RNA molecule includes single-stranded RNA molecules forming
functional
stem-loop structures, such that they thereby form the structural equivalent of
an RNA
duplex with single-strand overhangs. The double-stranded RNA molecule of the
present
invention may be very large, comprising thousands of nucleotides; preferably,
however, it
is small, in the range of 21-25 nucleotides. In a preferred embodiment, the
RNAi of the
present invention comprises a double-stranded RNA duplex of at least 19
nucleotides.
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
[00106] In one embodiment of the present invention, RNAi is produced in vivo
by
an expression vector containing a gene-silencing cassette coding for RNAi.
(see e.g., U.S.
Patent No. 6,278,039, C. elegans deletion mutants; U.S. Patent Application No.
2002/0006664, Arrayed transfection method and uses related thereto; WO
99/32619,
Genetic inhibition by double-stranded RNA; WO 01/29058, RNA interference
pathway
genes as tools for targeted genetic interference; WO 01/68836, Methods and
compositions
for RNA interference; and WO 01/96584, Materials and methods for the control
of
nematodes). In another embodiment of the present invention, RNAi is produced
in vitro,
synthetically or recombinantly, and transferred into the microorganism using
standard
molecular-biology techniques. Methods of making and transferring RNAi are well
known
in the art (see e.g., Ashrafi et al., Genome-wide RNAi analysis of
Caenorhabditis elegans
fat regulatory genes, Nature, 421:268-72, 2003; Cottrell et al., Silence of
the strands:
RNA interference in eukaryotic pathogens, Trends Microbial., 11:37-43, 2003;
Nikolaev
et al., Parc: A Cytoplasmic Anchor for p53, Cell, 112:29-40, 2003; Wilda et
al., Killing of
leukemic cells with a BCR/ABL fusion gene RNA interference (RNAi), Oncogene,
21:5716-24,2002; Escobar et al., RNAi-mediated oncogene silencing confers
resistance to
crown gall tumorigenesis, Proc. Natl. Acad. Sci. USA, 98:13437-42, 2001; and
Billy et
al., Specific interference with gene expression induced by long, double-
stranded RNA in
mouse embryonal teratocarcinoma cell lines, Proc. Natl. Acad. Sci. USA,
98:14428-33,
2001).
[00107] In a further embodiment of the present invention, the plasmid is an
expression plasmid. The expression plasmid may contain sites for transcription
initiation,
termination, and, optionally, in the transcribed region, a ribosome-binding
site for
translation. The coding portions of the mature transcripts expressed by the
plasmid may
include a translationinitiating codon at the beginning, and a ter-mination
codon
appropriately positioned at the end of the polypeptide to be translated.
[00108] By way of example, the PDE-associated gene to be expressed from the
expression plasmid may be under the specific regulatory control of certain
types of
promoters. In one embodiment, these promoters are constitutive promoters.
Genes under
the control of these constitutive promoters will be expressed continually. In
another
embodiment, the promoters are inducible promoters. Genes under the control of
these
inducible promoters will be expressed only upon the presence of an inducer
molecule or
the absence of an inhibitor molecule, thereby providing a method to turn off
expression of
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
31
the gene when it is not desired. In yet another embodiment, the promoters are
cell-type-
specific promoters or tissue-specif c(e.g., heart-tissue-specific) promoters.
Genes under
the control of cell-type-specific promoters will be expressed only in certain
cell types,
preferably only in cardiomyocytes.
[00109] In another embodiment of the present invention, the PDE-associated
agent
is a modulator (e.g., inhibitor, activator, antagonist, or agonist) of PDE
expression/activity, including a modulator of a member of the PDE signal-
transduction
pathway/system. The modulator of the present invention may be a protein,
polypeptide,
peptide, nucleic acid (including DNA or RNA), antibody, Fab fragment, F(ab')2
fragment,
molecule, compound, antibiotic, or drug, including an agent reactive with PDE,
and an
agent that induces or upregulates PDE expression or activity.
[00110] Modulators of PDE or a member of the PDE signal-transduction pathway/
system may be identified using a simple screening assay. For example, to
screen for
candidate modulators of PDE, cells may be plated onto microtiter plates, then
contacted
with a library of drugs. Any resulting increase in, or upregulation of, PDE
expression
then may be detected directly or indirectly using a luminescence reporter,
nucleic acid
hybridization, and/or immunological techniques known in the art, including an
ELISA.
Additional modulators of PDE expression may be identified using screening
procedures
well known in the art or disclosed herein. It is within the confines of the
present
invention that the modulator of PDE expression may be linked to another agent,
or
administered in combination with another agent, such as a drug or a ribozyme,
in order to
increase the effectiveness of treatments using PDE-associated agents and/or
increase the
efficacy of targeting. Additional PDE-associated agents may be identified
using
screening procedures well known in the art, and methods described herein.
[00111] As discussed above, the present invention contemplates the use of
proteins
and protein analogues generated by synthesis of polypeptides in vitro, e.g.,
by chemical
means or in vitro translation of mRNA. For example, PDE may be synthesized by
methods commonly known to one skilled in the art (Modem Techniques of Peptide
and
Amino Acid Analysis (New York: John Wiley & Sons, 1981); Bodansky, M.,
Principles
of Peptide Synthesis (New York: Springer-Verlag New York, Inc., 1984)).
Examples of
methods that may be employed in the synthesis of the amino acid sequences, and
analogues of these sequences, include, but are not limited to, solid-phase
peptide
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
32
synthesis, solution-method peptide synthesis, and synthesis using any of the
commercially-available peptide synthesizers. The amino acid sequences of the
present
invention may contain coupling agents and protecting groups, which are used in
the
synthesis of protein sequences, and which are well known to one of skill in
the art.
[00112] In accordance with the method of the present invention, PDE in cells
may
be modulated, and cells may be contacted with a PDE-associated agent (e.g., by
introducing a PDE-associated agent directly into the cells) including stem
cells containing
a PDE-associated agent either in vitro, or in vivo in a subject. Where cells
are contacted
with a PDE-associated agent in vitro, the agent may be added directly to the
cell-culture
medium. Alternatively, a PDE-associated agent may be contacted with cells in
vivo in a
subject, by introducing the agent into the subject (e.g., by introducing the
agent directly
into cells of the subject) and/or administering the agent to the subject. The
subject may
be any animal, including amphibians, birds, fish, mammals, and marsupials, but
is
preferably a mammal (e.g., a human; a domestic animal, such as a cat, dog,
monkey,
mouse, and rat; or a commercial animal, such as a cow or pig). In a preferred
embodiment, the subject is a human.
[00113] The PDE-associated agent of the present invention may be contacted
with
a cell or RyR complex, either in vitro, or in vivo (including in situ) in a
subject, by known
techniques used for the introduction and administration of proteins, nucleic
acids, and
other drugs. Examples of methods for contacting the cells with (i.e., treating
the cells
with) a PDE-associated agent include, without limitation, absorption,
electroporation,
immersion, injection (including microinjection), introduction, liposome
delivery, stem
cell fusion (including embiyonic stem cell fusion), transduction,
transfection, transfusion,
vectors, and other protein-delivery and nucleic-acid-delivery vehicles and
methods.
[0100] When the cells are localized to a particular portion of a subject, it
may be
desirable to introduce the agent directly to the cells, by injection or by
some other means
(e.g., by introducing the agent into the blood or another body fluid).
Preferably, where
heart tissue cells are contacted with a PDE-associated agent in vivo in a
subject,
contacting is accomplished via a catheter inserted directly into the subject's
heart tissue.
A catheter would be useful in achieving targeted delivery of the agent to
heart tissue cells.
Targeted delivery is especially appropriate for cardiomyocytes, which are
joined by
intercalated disks. These disks should allow passage of the agent from one
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
33
cardiomyocyte to adjoining cardiomyocytes, thereby aiding in the distribution
of the
agent throughout the heart tissue.
[0101] Where a PDE-associated agent is a protein, it may be introduced into a
cell
or RyR complex directly, in accordance witli conventional techniques and
methods
disclosed herein. Additionally, a protein agent may be introduced into a cell
or RyR
complex indirectly, by introducing into the cells a nucleic acid encoding the
agent, in a
manner permitting expression of the protein agent. The PDE-associated agent
may be
introduced into cells, in vitro or in vivo, using conventional procedures
known in the art
,
including, without limitation, electroporation, DEAF dextran transfection,
calcium
phosphate transfection, monocationic liposome fusion, polycationic liposome
fusion,
protoplast fusion, creation of an in vivo electrical field, DNA-coated
microprojectile
bombardment, injection with recombinant replication-defective viruses,
homologous
recombination, in vivo gene therapy, ex vivo gene therapy, viral vectors, and
naked DNA
transfer, or any combination thereof. Recombinant viral vectors suitable for
gene therapy
include, but are not limited to, vectors derived from the genomes of such
viruses as
retrovirus, HSV, adenovirus, adeno-associated virus, Semilild Forest virus,
cytomegalovirus, lentivirus, and vaccinia virus.
[0102] The amount of nucleic acid to be used in the method of the present
invention is an amount sufficient to express an ainount of protein effective
to promote
PDE level. These amounts may be readily determined by the skilled artisan. It
is also
within the confines of the present invention to use an ex vivo approach,
wherein a nucleic
acid encoding a protein agent is introduced into suitable a cell or RyR
complex in vitro,
using conventional procedures, to achieve expression of the protein agent in
the cells.
Cells expressing protein agent are then introduced into a subject to promote
PDE activity
in vivo.
[0103] In accordance with the method of the present invention, a PDE-
associated
agent, including stem cells containing the agent, may be administered to a
human or
animal subject by known procedures, including, without limitation, oral
administration,
parenteral administration, transdermal administration, and by way of a
catheter. For
example, the agent may be administered parenterally, by intracranial,
intraspinal,
intrathecal, or subcutaneous injection. The agent of the present invention
also may be
administered to a subject in accordance with any of the above-described
methods for
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
34
effecting in vivo contact between a cell or RyR complex and PDE-associated
agents.
[0104] For oral administration, a formulation comprising a PDE-associated
agent
may be presented as capsules, tablets, powders, granules, or as a suspension.
The
formulation may have conventional additives, such as lactose, mannitol,
cornstarch, or
potato starch. The formulation also may be presented with binders, such as
crystalline
cellulose, cellulose derivatives, acacia, cornstarch, or gelatins.
Additionally, the
formulation may be presented with disintegrators, such as cornstarch, potato
starch, or
sodium carboxymethylcellulose. The formulation also may be presented with
dibasic
calcium phosphate anhydrous or sodium starch glycolate. Finally, the
formulation may
be presented with lubricants, such as talc or magnesium stearate.
[0105] For parenteral administration (i.e., administration by injection
through a
route other than the alimentary canal) or administration through a catheter, a
PDE-
associated agent may be combined with a sterile aqueous solution that is
preferably
isotonic with the blood of the subject. Such a formulation may be prepared by
dissolving
a solid active ingredient in water containing physiologically-compatible
substances, such
as sodium chloride, glycine, and the like, and having a buffered pH compatible
with
physiological conditions, so as to produce an aqueous solution, then rendering
said
solution sterile. The formulation may be presented in unit or multi-dose
containers, such
as sealed ampoules or vials. The formulation may be delivered by any mode of
injection,
including, without limitation, epifascial, intracapsular, intracranial,
intracutaneous,
intrathecal, intramuscular, intraorbital, intraperitoneal, intraspinal,
intrasternal,
intravascular, intravenous, parenchymatous, subcutaneous, or sublingual, or by
way of a
catheter.
[0106] For transdermal administration, an agent may be combined with skin
penetration enhancers, such as propylene glycol, polyethylene glycol,
isopropanol,
ethanol, oleic acid, N-methylpyrrolidone, and the like, which increase the
permeability of
the skin to the agent, and permit the agent to penetrate through the skin and
into the
bloodstream. The agent/enhancer composition also may be further combined with
a
polymeric substance, such as ethylcellulose, hydroxypropyl cellulose,
ethylene/vinylacetate, polyvinyl pyrrolidone, and the like, to provide the
composition in
gel form, which may be dissolved in solvent, such as methylene chloride,
evaporated to
the desired viscosity, and then applied to backing material to provide a
patch.
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
[0107] The method of the present invention may also be used either to treat a
RyR
receptor associated disorder or disease in vivo in a subject, or to prevent a
RyR receptor
associated disorder or disease in vivo in a subject. As the inventors have
demonstrated,
augmented PDE in a cell or RyR complex has the ability protect the heart
against heart
failure and catecholaminergic arrhythmias by regulating local PKA activity and
channel
activation at RyR2-Ser2809, and preventing excess accumulation of cAMP and
uncontrolled PKA activation.
[0108] The present invention also provides a therapeutic composition
comprising
a PDE-associated agent and, optionally, a pharmaceutically-acceptable carrier.
As
described above, the PDE-associated agent may include a PDE protein or nucleic
acid, a
PDE-associated protein, a PDE-associated nucleic acid, a member of the PDE
signal -
transduction pathway (including upstream and downstream effectors and
activators, in
protein or nucleic acid form), and a modulator (e.g., inhibitor, activator,
antagonist, or
agonist) of a member of the PDE signal-transduction pathway/system (i.e., a
modulator
which affects the expression and/or activity of PDE or a member of the PDE
signal
transduction pathway).
[0109] In accordance with the therapeutic composition of the present
invention,
the pharmaceutically-acceptable carrier must be "acceptable" in the sense of
being
compatible with the other ingredients of the composition, and not deleterious
to the
recipient thereof. The pharmaceutically-acceptable carrier employed herein is
selected
from various organic or inorganic materials that are used as materials for
pharmaceutical
formulations, and which may be incorporated as analgesic agents, buffers,
binders,
disintegrants, diluents, emulsifiers, excipients, extenders, glidants,
solubilizers,
stabilizers, suspending agents, tonicity agents, vehicles, and viscosity-
increasing agents.
If necessary, pharmaceutical additives, such as antioxidants, aromatics,
colorants, flavor-
improving agents, preservatives, and sweeteners, may also be added. Examples
of
acceptable pharmaceutical carriers include, without limitation, carboxymethyl
cellulose,
crystalline cellulose, glycerin, gum arabic, lactose, magnesium stearate,
methyl cellulose,
powders, saline, sodium alginate, sucrose, starch, talc, and water, among
others.
[0110] Formulations of the therapeutic composition of the present invention
may
be prepared by methods well-known in the pharmaceutical arts. For example, a
PDE-
associated agent may be brought into association with a carrier or diluent, as
a suspension
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
36
or solution. Optionally, one or more accessory ingredients (e.g., buffers,
flavoring agents,
surface active agents, and the like) also may be added. The choice of carrier
will depend
upon the route of administration. The PDE-associated agent is provided in an
amount
that is effective to hydrolyze cAMP in a cell or RyR complex in a subject to
whom the
therapeutic composition is administered. This amount may be readily determined
by the
skilled artisan.
[0111] In one embodiment of the present invention, the PDE-associated agent is
a
protein that is expressed in a target heart tissue cell using an expression
construct.
Expression of the protein may be controlled by methods known in the art,
including the
use of attenuators, downregulators, inhibitors, and other molecules known to
inhibit
protein expression. By way of example, where the therapeutic composition of
the present
invention is administered to a subject, such that the composition expresses a
PDE-
associated protein in the subject, this expression may be shut off in vivo by
subsequently
administering to the subject an attenuator, downregulator, inhibitor, or other
molecule that
will inhibit expression of the exogenous molecule. Control of expression of
the PDE-
associated protein is also advantageous, in that it allows one to turn off the
expression of
the protein when desired, thereby minimizing any harmful side-effects in a
subject to
whom the composition is administered. Continuous expression of such a protein,
beyond
an appropriate time limit, may harm the subject. For exainple, a significant
interference
with a PDE signal transduction pathway may cause neoplasia or apoptosis.
[0112] The therapeutic composition of the present invention may furtller
comprise
a vehicle for assisting in the delivery of the composition that target
specific cells,
including but not limited to heart tissue cells or skeletal muscle cells. A
variety of
biological delivery systems (e.g., antibodies, bacteria, liposomes, and viral
vectors)
currently exist for delivering drugs, genes, immunostimulators, pro-drug
converting
enzymes, radiochemicals, and otlzer therapeutic agents to the vicinity of
target cells (see
e.g., Ng et al., An anti-transferrin receptor-avidin fusion protein exhibits
both strong
proapoptotic activity and the ability to deliver various molecules into cancer
cells, Proc.
Natl. Acad. Sci. USA, 99:10706-11, 2002; Mastrobattista et al., Functional
characterization of an endosome-disruptive peptide and its application in
cytosolic
delivery of immunoliposome-entrapped proteins, J. Biol. Chem., 277:27135-43,
2002;
Fefer, "Special delivery" to cancer cells, Blood, 99:1503-04, 2002; Kwong et
al., The
suppression of colon cancer cell growth in nude mice by targeting 13-
catenin/TCF
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
37
pathway, Oncogene, 21:8340-46, 2002; Huser et al., Incorporation of decay-
accelerating
factor into the baculovirus envelope generates complement-resistant gene
transfer vectors,
Nat. Biotechnol., 19:451-55, 2001; Lu et al., Polymerizable Fab' antibody
fragments for
targeting of anticancer drugs, Nat. Biotechnol., 17:1101-04, 1999; and Chu et
al., Toward
highly efficient cell-typespecific gene transfer with retroviral vectors
displaying single-
chain antibodies, J. Virol. 71:720-25, 1997). For example, U.S. Patent No.
6,491,905
provides a prokaryotic cell stably carrying a vector that includes a DNA
sequence
encoding a purine nucleotide phosphorylase or hydrolase, and the use of such a
cell,
together with a purine pro-drug, to treat tumors.
[0113] In one embodiment of the present invention, the vehicle is a liposome.
Liposomal vesicles may be prepared by various methods known in the art, and
liposome
compositions may be prepared using any one of a variety of conventional
techniques for
liposome preparation known to those skilled in the art. Examples of such
methods and
techniques include, without limitation, chelate dialysis, extrusion (with or
without freeze-
thaw), French press, homogenization, microeinulsification; reverse phase
evaporation,
simple freeze-thaw, solvent dialysis, solvent infusion, solvent vaporization,
sonication,
and spontaneous formation. Preparation of the liposomes may be carried out in
a
solution, such as an aqueous saline solution, aqueous phosphate buffer
solution, or sterile
water. Liposome compositions also may be prepared by various processes
involving
shaking or vortexing.
[0114] The therapeutic composition of the present invention may be
incorporated
into the layers of a liposome, or enclosed within the interior of the
liposome. The
liposome containing the composition then may be fused cell, in accordance with
known
methods of fusion of liposomes to cell membranes, such that the coinposition
protein is
delivered into the membrane of the cell or into the interior of the cell, as
the case may be.
[0115] The present invention also provides a kit for use in delivering a PDE-
associated agent to a cells, cell subcompartment or RyR complex in a subject.
The kit
comprises a therapeutic composition and a catheter. As described above, the
therapeutic
composition may comprise a PDE-associated agent; optionally, a
pharmaceutically-
acceptable carrier; and, optionally, a liposome,=viral vector, or other
vehicle.
[0116] The present invention is described in the following Examples, which are
set forth to aid in the understanding of the invention, and should not be
construed to limit
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
38
in any way the scope of the invention as defined in the claims which follow
thereafter.
EXAMPLES
EXAMPLE 1- PDE4D-/- MICE AN RYR2-S2808A KNOCK IN MICE
[0117] Mouse genomic k-phage clones for segments of the murine ortholog of the
human RyR2 gene were isolated from a 129/SvEvTacfBR genomic library
(Stratagene,
La Jolla, CA). A 5.4kb Eco RI fragment containing exons 53 to 55 and the
flanking
intronic regions was isolated using a 250 bp 32P-labeled cDNA probe containing
serine
(S) 2808. The isolated 5.4kb fragment was subcloned into the Eco RI site
ofpBluescriptSK, and S2808 was mutated to alanine (A) using a Chameleon
Mutagenesis
Kit (Stratagene, La Jolla CA). In addition to the S2808A mutation, an extra
FSP I
restriction site was added to exon 55. The 5.4kb Eco RI fragment was then
excised and
cloned into the Eco RI site of pACN vector. The pACN plasmid was a backbone
vector
containing a cassette (ACN) with genes for neomycin resistance, Cre
recombinase and a
testes-specific promoter (tACE), flanked by loxP sites. The promoter tACE
initiates
expression of Cre recombinase only during spermatogenesis, resulting in
excision of the
ACN cassette. The 3' targeting arm, consisting of the 2463 bps upstream of the
Eco RI
site, was obtained by PCR of genomic mouse DNA. After adding Sal I sites to
botll ends
of this 2.4 kb fragment, it was cloned into the Sal I site of the pACN vector
containing the
mutated 5.4kb segment. The Kpn I linearized targeting vector was
electroporated into
MM13 mouse embryonic stem (ES) cells using established protocols. Gene-
targeted ES
cells were screened by Southern analysis using both 5' and 3' external probes
to confirm
homologous recombination, and injected into C57BI6 blastocysts. Founder mice
were
backcrossed to C57B16 mice. Germline offspring were identified by brown coat
color and
further verified by Southern blot analysis. Heterozygous males and females
were
intercrossed to obtain homozygous offspring.
[0118] PDE4D-'- mice were generated and genotyped as described (Jin et al.,
1999). RyR2-S2808A knockin mice, generated using homologous recombination as
described immediately above, exhibited normal cardiac structure and function,
and no
PKA phosphorylation of RyR2 was detected using a kinasing reaction with [y-32
P]ATP or
with a phosphoepitope-specific antibody that detects PKA-phosphorylated RyR2.
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
39
EXAMPLE 2- TRANSTHORACIC ECHOCARDIOGRAPHY AND IN VIVO
HEMODYNAMIC ANALYSES ON MICE
[0119] Transthoracic 2D echocardiography and in vivo hemodynamic analyses on
mice were performed as previously described. All animal studies were performed
according to protocols approved by the Institutional Animal Care and Use
Committee of
Columbia University and according to NIH guidelines. For transthoracic 2-D
echocardiography, mice were anesthetized with 1.0-1.5% isoflurane in Oz and
placed on a
37 C heating pad. Hearts were visualized parasternally along the short axis to
obtain 2-D
images and M-mode tracings of the anterior wall, left ventricular cavity, and
posterior
wall. Left ventricular dimensions and function were measured in triplicate
from different
cardiac cycles for the number of animals indicated.
[0120] Hemodynamic measurements were performed on PDE4D-~-, PDE4D+~- and
age-and litter-matched wild-type (WT) mice (3 to 15 months) anesthetized with
1.5%
isoflurane using a 1.4 F micromanometer-conductance catheter (SPR-839, Millar
Instruments) via the right carotid artery. Pressure-volume analysis was
performed using
hemodynamic analysis software (EMKA Technologies, VA) as described (van Rooij
et
al., 2004).
EXAMPLE 3- MYOCARDIAL INFARCT MODEL
[0121] PDE4D-1- and age-and-litter-matched wild-type mice (4 to 5 months old)
were anesthetized with 1.5% isoflurane and ventilated with a small-rodent
respirator
(Harvard Apparatus). A left thoracotomy was performed, and the left anterior
descending
artery (LAD) was ligated proximally with an 8-0 suture as described (Wehrens
et al.,
2005).
EXAMPLE 4- EXERCISE TESTING AND MOUSE ECG RECORDING
[0122] Ambulatory ECG recordings were performed using implantable
radiotelemetry transmitters (DSI) as described (Wehrens et al., 2004). For the
pharmacological experiments performed in wt and RyR2-S2808A knockin mice,
animals
were pretreated for 30 min by intraperitoneal injection with the PDE4
inliibitor rolipram
(0.3 mg/kg) or placebo (DMSO 0.5% as carrier) followed by the exercise
protocol
described above.
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
EXAMPLE 5 - (3-ADRENERGIC RECEPTOR MEASUREMENT
[0123] 0-adrenergic-receptor levels were assessed as previously described
(Reiken et al., 2003b). Aliquots of cardiac membrane preparations from 5 wild-
type
(WT) and 5 PDE4D"/- knockout mice were incubated for 2 hours in 0.5 mM Tris-
HCl
buffer, pH 7.4, containing increasing concentrations of [125I]-(-)-
cyanopindolol ([125I]-
CYP) before the reactions were filtered using GF/C microfiber filters from
Whatman. The
filters were washed three times with 3 ml of binding buffer, dried, and bound
radioligand
was measured in a y-radiation counter. [125I]- CYP binding was determined in
the
presence and absence of I M alprenolol to distinguish between specific and
nonspecific
(residual binding in the presence of 1 M alprenolol) binding.
EXAMPLE 6- IMMUNOPRECIPITATION AND IMMUNOBLOT ANALYSIS
[0124] RyR2 channels were immunoprecipitated and immunoblotted as
previously described (Marx et al., 2000). RyR2 channels were
immunoprecipitated from
100 g of human or murine cardiac homogenates using anti-RyR antibody
(Jayaraman et
al., 1992) in 0.5 ml of RIP A buffer (50 mM Tris-HCl buffer), pH 7.4, 0.9%
NaCI, 5.0
mM NaF, 1.0 mM Na3 V04, 0.25% Triton-XI00, and protease inhibitor mix (Roche)
overnight at 4 C. The use of human tissues was approved by the Institutional
Review
Board of Columbia-Presbyterian Medical Center. Normal and failing human heart
tissues
were obtained as previously described from patients undergoing cardiac
transplant (Marx
et al, 2000). Immunoprecipitates were separated by SDS-PAGE and the proteins
were
transferred onto nitrocellulose membranes overnight (Semi-Dry transfer blot,
Bio-Rad,
USA). Immunoblots were developed with an enhanced chemiluminescence system
using
primary antibodies against RyR (5029; 1:3,000) (Jayaraman et al., 1992), PDE
splice-
variant 4D3 (1: 1,000) (Reiken et al, 2003b) calstabin2 (I: 1,000) (Wehrens et
al., 2003),
PKA catalytic subunit (1:1,000), PPI (1:1,000) and PP2A (1:1,000)
(Transduction Labs,
Lexington, KY) (Marx et al., 2000). PKA phosphorylation of RyR2 was quantified
using
phospho-epitope specific RyR2-pSer2808 antibody (I :5,000) (Reiken et al.,
2003c).
Results were confirmed using a PKA back-phosphorylation assay as previously
described
(Reiken et al., 2003c). CaMKII phosphorylation of RyR2 was quantified using
phospho-
epitope specific antibody RyR2-pSer2814 (1:5,000) as described (Wehrens et
al., 2004).
Band densities were quantified using Quantity One software (Biorad, Hercules,
CA)
(Reiken et al., 2003a). Data presented represent > 4 individual experiments.
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
41
EXAMPLE 7- PHOSPHODIESTERASE ACTIVITY ASSAY
[0125] Phosphodiesterase (PDE) activity was measured using selective binding
of
5'-AMP to yttrium silicate beads with embedded scintillant. Immunoprecipitated
RyR2
complexes were incubated with 50 nM 3H-cyclic nucleotide (Amersham, 5-60
Ci/mM) in
50 mM Tris-HCl (pH 7.5), 8.3 mM MgC12, 1.7 mM EGTA, BSA 0.01% at 30 C for 30
min. The reaction was terminated by adding one-third of 5 mg/ml yttrium
silicate beads in
18 mM Zn acetate/Zn sulfate solution (3:1). After 30 min, hydrolysis was
quantified by a
scintillation counter (Wallac 1409, PerkinElmer).
EXAMPLE 8 - FRET-PKA ASSAY
[0126] Local intracellular cAMP concentrations were determined in murine
cardiomyocytes using fluorescence resonance energy transfer (FRET) between the
cyan
(CFP) and yellow (YFP) variants of green fluorescent protein as described
(Zaccolo and
Pozzan, 2002). Primary cultures of adult murine ventricular cardiac myocytes
from wild-
type and age-and litter-matched PDF4&hearts were isolated using a modified
Langendorffperfusion protocol with CaZ+free Tyrode solution followed by
collagenase
digestion (Type II, Worthington). Cardiomyocytes were infected with
recombinant
adenoviruses expressing CFP attached to the PKA regulatory subunit (RII-CFP)
and YFP
attached to the PKA catalytic subunit (C-YFP) as described previously (Warrier
et al.,
2005). Simultaneous infection of mouse cardiomyocytes occurred at a
multiplicity of
infection of 50 to 100 for each virus. Cells expressing approximately equal
amounts of
CFP and YFP as evidenced by fluorescence at 48-72 hrs, were used for
intracellular
cAMP imaging by FRET. Imaging was performed with an inverted microscope
(Olympus
IX70) equipped with a 40X water immersion objective (1.3 NA, Olympus) and a
CCD
camera (Hamamatsu, Orca ER) as described previously (Warrier et al., 2005).
Fluorescence images were acquired using 2x2 binning and analyzed using Simple
PCI
imaging software (Compix Inc.) and changes in cAMP concentrations at the Z-
line
containing RyR2 complexes were defined as the relative changes in the
intensity of CFP
and YFP measured at the Z-lines within a region of interest. Isoproterenol
bitartrate (Iso;
Sigma RBI) was prepared as stock solution and applied by rapid perfusion.
EXAMPLE 9 - BACK PHOSPHORYLATION OF PDE4D3
[0127] PKA phosphorylation of PDE4D3 was assessed using a kinasing reaction
on PDE4D3 immunoprecipitated from 100 g of human cardiac homogenates. PDE4D3
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
42
was immunoprecipitated from 100 g of human cardiac homogenates with anti-
PDE4D3
antibody (5 g/ml) in 0.5 ml of RIPA buffer overnight at 4 C. Samples were
incubated
with Protein A sepharose beads (Amersham Pharmacia Biotech, Piscataway, NJ) at
4 C
for 1 hour, beads were washed three times with 1 x kinase buffer (8 mM MgCl2,
10 mM
EGT A, and 50 mM Tris/piperazine-N,N'-bis(2ethanesulfonic acid), pH 6.8).
After
resuspending the beads in 10 l of 1.5 x kinase buffer containing PKA
catalytic subunit
(5 units, Sigma, St. Louis, MO), back phosphorylation of the
immunoprecipitated
PDE4D3 was initiated with 5 of 100 M Mg-ATP containing 10% [y-32P] A TP
(NEN
Life Sciences, Boston, MA). The reaction was terminated after 10 min at RT
with 5 l of
stop solution (4% SDS and 0.25 M DTT). Samples were heated to 95 C, size
fractionated
on 8% PAGE, and PDE4D3 radioactivity was quantified using a Molecular Dynamics
Phosphorimager and ImageQuant software (Ammersham Pharmacia).
EXAMPLE 10 - SINGLE CHANNEL RECORDINGS
[0128] RyR2 single channels were recorded in planar lipid bilayers as
previously
described (Marx et al., 2000). Symmetrical solutions used were (in mM) trans-
HEPES
250 and Ca(OH)2 53 (pH 7.35) and cis-HEPES 250, Tris 125, EGTA 1.0, and CaC12
0.5
(pH 7.35). Free Ca2+ concentrations were calculated by CHELATOR software. At
the
conclusion of each experiment, ryanodine (5 M) or ruthenium red (20 [tM) was
applied
to confirm RyR2-channel identity.
EXAMPLE 11- PKA PHOSPHORYLATION OF CARDIAC RYANODINE
RECEPTORS
[0129] PKA phosphorylation of RyR2 was assessed as previously described
(Marx et al., 2000). RyR2 was immunoprecipitated from 250 g of mouse heart
homogenate. PKA phosphorylation of RyR2 was initiated with 5 l of 100 M Mg-
ATP
(for autoradiography, the Mg-ATP contained 10% [32P]-yATP (NEN Life Sciences,
Boston, MA) in kinase buffer (8 M MgCI2, 10 mM EGTA, and 50 mM
Tris/piperazine-
N,N'-bis(2ethanesulfonic acid), pH 6.8). The reaction was terminated after 8
min at room
temperature with 5 l of stop solution (4% SDS and 0.25 M DTT). Samples were
heated
to 95 DC and size-fractionated on 6% SDS-PAGE.
EXAMPLE 12 - STATISTICAL ANALYSIS
[0130] Data are reported as mean SEM for in vivo experiments and mean SD
for biochemical studies. Differences between multiple experimental groups were
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
43
compared by analysis of variance (ANOVA) followed by Tukey's multiple
comparison
test. Analysis between two groups was performed by t test (paired or unpaired
as
appropriate). Serial studies were tested by repeated-measure ANOVA. A value of
p <
0.05 was considered significant.
EXAMPLE 13 - PDE4D GENE INACTIVATION CAUSES AGE-RELATED
CARDIOMYOPATHY
[0131] To explore the consequences of chronic PDE4D deficiency on cardiac
function, a mouse model of PDE4D gene inactivation (Jin et al., 1999) was
used.
Echocardiography of PDE4D-/ mice showed a progressive, age-dependent increase
in
left ventricular end-diastolic diameter (LVEDD), a hallmark of cardiac
dysfunction
(Figure IA; n = 12 each for wild-type [wt] and PDE4D-'-). PDE4D-'_ mice
exhibited
increased heart-weight-to-body-weight (HW/BW) ratios compared to wt controls
(Figure
1B). PDE4D _' mice had reduced ejection fractions (EF) and cardiac
contractility
(dP/dt)/P;d, documented by cardiac catheterization (Figures IC and 1D).
Histologic
examination of PDE4D-/- hearts confirmed that the 15-month-old hearts were
dilated,
with no other structural abnormalities (Figure JE). These data show that PDE4D
deficiency is associated with progressive cardiac dysfunction consistent with
a dilated
cardiomyopathy similar to that seen in patients with chronic heart failure.
EXAMPLE 14 - GLOBAL cAMP SIGNALING IS NORMAL IN
PDE4D-DEFICIENT MICE
[0132] It is well established that chronic hyperadrenergic signaling is
associated
with heart failure. Therefore, the inventors sought to determine whether the
mechanism
underlying the observed cardiac phenotype in PDE4D-/- mice was increased
global
cAMP signaling. However, there were no significant differences in global cAMP
levels
and (3-adrenergic receptors in hearts from PDE4D-/ mice (Figures 2A-2C). Total
cAMP-
hydrolyzing activity of PDE in the heart was only slightly decreased in PDE4D-
/- mice
(data not shown), consistent with PDE4D activity representing only a fraction
of total
cytosolic PDE cAMP-hydrolyzing activity in the heart (Mongillo et al., 2004
and Richter
et al., 2005). However, rolipram-sensitive PDE4 activity in PDE4D-/- heart was
reduced
by ~50% (data not shown). Whereas global cAMP signaling was not perturbed in
PDE4D-deficient mice, there was a significant increase in localized cAMP
levels at the
cardiomyocyte Z line (corresponding to the location of the RyR2 channel) in
cardiomyocytes isolated from PDE4D-/ mice following a low dose (1 nM) of
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
44
isoproterenol (Figures 2D and 2E). Thus, the abnormalities observed in cardiac
function
of PDE4D -/- mice must be explained by defects in localized cAMP-dependent
signaling.
EXAMPLE 15 - PKA PHOSPHORYLATION OF RYR2 IN PDE4D' MICE
[0133] While many proteins in the heart are regulated by PKA phosphorylation
and therefore can be affected by altered PDE4D gene expression, only a limited
number
of these PKA substrates are known to be dysregulated by PKA during heart
failure. For
example, it has been shown that PKA phosphorylation of phospholamban, a
regulator of
the SR Ca2+ uptake ptunp (SERCA2a), is decreased in heart failure. In
contrast, of the
other proteins known to be involved in regulating cardiac contractility, the
SR Ca2+-
release channel, RyR2, has been shown to be PKA hyperphosphorylated in heart
failure
(Antos et al., 2001, Marx et al., 2000, Reiken et al., 2003a, Reiken et al.,
2003b, Yano et
al., 2000 and Yano et al., 2003), although this finding has been challenged by
others
(Jiang et al., 2002).
[0134] Given that RyR2 PKA hyperphosphorylation has been linked to cardiac
dysfunction in humans and animal models and that cAMP concentrations are
increased in
the compartment of RyR2 Ca2+ release (Figures 2D and 2E), the inventors sought
to
determine whether RyR2 PKA hyperphosphorylation might play a role in the
observed
cardiac phenotype in PDE4D _' mice. Indeed, there was a progressive, age-
dependent
increase in PKA phosphorylation of RyR2 on Ser2808 (detected using a
phosphoepitope-
specific antibody) in PDE4D-'- mouse hearts (Figures 3A and 3B). PKA
hyperphosphorylation of RyR2 in PDE4D _'- mouse hearts was associated with
depletion
of the RyR2-stabilizing protein calstabin2 (FKBP12.6) that prevents Ca2+ leak
from the
SR into the cytosol through RyR2 during diastole in the heart (Figure 3C)
(Marx et al.,
2000 and Wehrens et al., 2003). Increased PKA phosphorylation of RyR2 was not
caused
by increased protein levels of PKA catalytic and regulatory subunits in the
RyR2 complex
(Figure 3A). Moreover, no changes in PP 1 or PP2A phosphatase levels in the
RyR2-
complex levels were detected (data not shown). Phosphorylation of RyR2 by
another
kinase that phosphorylates the channel, Ca2+/calmodulin-dependent protein
kinase A
(CaMKII) (detected using a phosphoepitope-specific antibody), was not altered
in
PDE4D-'- mouse hearts (Figures 3A and 3D).
[0135] In wt mice, PDE activity was associated with immunoprecipitated RyR2
channels (Figure 3E). RyR2-associated PDE activity was almost completely
inhibited by
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
the PDE4 antagonist rolipram but not by the PDE3 inhibitor milrinone.
Moreover, PDE
activity specifically associated with RyR2 channels was reduced to zero in the
PDE4D-/-
mice (Figure 3E). Taken together, these data suggested that one consequence of
PDE4D
deficiency is PKA hyperphosphorylation of RyR2, which has previously been
associated
with heart failure (Marx et al., 2000). Moreover, it appeared that the PDE
activity
associated with the RyR2 complex was encoded by the PDE4D gene.
EXAMPLE 16 - ABNORMAL RYR2 CHANNELS IN PDE4D-DEFICIENT HEARTS
[0136] To ascertain the functional consequences of PKA hyperphosphorylation of
RyR2 in hearts from PDE4D-deficient mice, the inventors examined the single-
channel
properties of RyR2 in planar lipid bilayers. Compared to channels from wt
mice, RyR2
from PDE4D-1- mice exhibited significantly increased open probability (Po) and
frequency of openings (Fo) and decreased mean open and closed times when
channels
were examined under conditions that mimic diastole in the heart (cytosolic
(cis) [Ca2+]
150 nM) (Figures 3F and 3G). Thus, PKA hyperphosphorylation of cardiac RyR2 in
PDE4D-deficient mice was associated with the same defects in RyR2-channel
function
("leaky" channels) previously linked to human heart failure (Marx et al.,
2000) and
genetically deterinined exercise-induced sudden cardiac death (Wehrens et al.,
2003).
EXAMPLE 17 - PDE4D3 IS A OF THE RYR2 MACROMOLECULAR COMPLEX
[0137] The finding that the PDE activity that coimmunoprecipitates with RyR2
was abrogated in channels from PDE4D -/- mouse hearts (Figure 3E) raised the
possibility
that a protein encoded by the PDE4D gene is an integral component of the RyR2
macromolecular signaling complex. Four genes constitute the type 4
phosphodiesterase
family (PDE4A, PDE4B, PDE4C, and PDE4D), and all are expressed as multiple
splice
variants (Conti et al., 2003 and Houslay and Adams, 2003). With the recent
discovery of
additional PDE4D variants (Gretarsdottir et al., 2003 and Wang et al., 2003),
a total of
nine PDE4D splice variants (PDE4D1-9) are known (Richter et al., 2005). The
inventors
generated an isoform-specific antibody against the unique N-terminal epitope
of PDE4D3
(Figure 4A). PDE4D splice variants were identified by RT-PCR in heart (data
not
shown), and PDE4D3, PDE4D8, and PDE4D9 expression in the heart was
demonstrated
using isoform-specific antibodies (Figure 4B), confirming that these are the
major
PDE4D isoforms expressed in heart muscle (Richter et al., 2005).
[0138] To examine the possibility that a specific PDE4D isoform is part of the
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
46
cardiac RyR2 channel complex, immunoprecipitations were performed using human
heart
extracts. RyR2 channels were immunoprecipitated with anti-RyR2 antibody and
assayed
for coimmunoprecipitation of PDE4D3, PDE4D8, or PDE4D9 with RyR2 by
immunoblotting. Using isoform-specific PDE4D antibodies, only PDE4D3 was
detected
in the RyR2 complex. In addition, anti-PDE4D3 antibody was used to
coimmunoprecipitate RyR2 (Figure 4B). The interaction between PDE4D3 and RyR2
was specific because PDE4D3 was excluded from immunoprecipitates using control
IgG
(Figure 4B). Moreover, the inventors used the PDE4D3-specific antibody to show
that
total PDE4D3 protein was decreased 37% in haploinsufficient PDE4D+/ mouse
hearts
and by 100% in homozygous PDE4D -/- hearts (Figure 4C). Finally, PDE4D3 in the
RyR2
macromolecular signaling complex was also decreased by 44% in PDE4D+l- mouse
hearts and by 100% in PDE4D-/ heart (Figure 4D). Taken together, these data
show that
PDE4D3 is an integral component of the RyR2 macromolecular complex in the
heart and
that PDE4D3 is the only PDE isoform in the RyR2 complex.
EXAMPLE 18 - PDE4D3 IS DECREASED IN THE RYR2 COMPLEX IN FAILING
HUMAN HEARTS
[0139] PDE4D3 was also associated with RyR2 from human hearts (Figure 5A).
In human heart failure (HF), PDE4D31evels in the RyR2 complex were decreased
by
43% (normal, n = 6 versus HF, n = 9; p < 0.001), and the RyR2 channels in the
human
HF samples were PKA hyperphosphorylated (Figure 5B). The inventors have
previously
shown that PKA hyperphosphorylation of RyR2 depletes calstabin2 from the RyR2
complex and significantly increases channel activity, consistent with a
diastolic SR Ca2}
leak in human heart failure (Marx et al., 2000 and Reiken et al., 2003a). The
cAMP-
hydrolyzing activity of RyR2 bound PDE4D3 was decreased by 42% in human HF
samples (n = 6, each experiment was performed in triplicate; p < 0.001),
providing a
possible explanation for chronic RyR2-Ser2808 PKA hyperphosphorylation
observed in
failing human hearts (Figure 5C). This reduction in PDE4D3 activity in the
human HF
RyR2 complex was comparable to that observed in the RyR2 complexes in PDE4D+/-
mice (Figure 4D). To explore the basis for the observed reduction in PDE4D3
amount
and activity in the HF RyR2 complex, the inventors examined PKA
phosphorylation of
PDE4D3, which has been shown to increase its activity (Sette and Conti, 1996)
and
binding to mAKAP (Carlisle Michel et al., 2004). The inventors observed a~40%
reduction in PKA phosphorylation of PDE4D3 in the human HF RyR2 complexes
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
47
compared to nonfailing controls (n = 3, p < 0.01), providing a possible
explanation for the
observed decrease in amount and activity of PDE4D3 in the HF RyR2 complexes
(data
not shown).
[0140] PDE4-specific inhibition with rolipram (10 M) significantly decreased
RyR2 bound PDE4D3 activity in normal human heart lysates (n = 3, p < 0.01),
whereas
the PDE3-specific inhibitor milrinone (10 ~LM) had no effect on RyR2-
associated PDE
activity (Figure 5D), confirming that the cAMP-hydrolyzing activity in the
RyR2
complex is due to PDE4. Thus, PDE4D3 is part of the human RyR2 signaling
complex,
and reduction of PDE4D3 activity in heart failure may contribute to RyR2 PKA
hyperphosphorylation and diastolic SR Caz+ leak observed in failing hearts
(Shannon et
al., 2003).
EXAMPLE 19 - CARDIAC ARRHYTHMIAS DUE TO PDE4 INHIBITION ARE
SUPPRESSED IN MICE HARBORING RYR2 THAT CANNOT BE PKA
PHOSPHORYLATED
[0141] As discussed above, the inventors have previously demonstrated a link
between PKA hyperphosphorylation of RyR2, "leaky" RyR2 channels, and exercise-
induced sudden cardiac death (Wehrens et al., 2003). Therefore, in the present
study, the
inventors sought to determine whether PDE4D-deficient mice, which exhibit PKA-
hyperphosphorylated RyR2, are more susceptible to exercise-induced cardiac
arrhythmias. Resting heart rate in PDE4D _/ mice at 3-4 months of age was
similar to wt,
consistent with unchanged baseline sympathetic activity (wt 584 ~: 22 bpm,
PDE4D _/-
603 32 bpm; p = NS). Since PDE inhibitors increase arrhythmogenic sudden
cardiac
death (Barnes, 2003 and Packer et al., 1991), the inventors tested the
susceptibility of
PDE4D-1- mice to cardiac arrhythmias during exercise followed by low-dose
epinephrine
injection (0.1 mg/kg) using a previously established protocol (Wehrens et al.,
2003 and
Wehrens et al., 2004). Exercise-induced sustained (sVT) and nonsustained
ventricular
arrhythmias (nsVT) were observed in 66% and 100% of PDE4D_1- mice,
respectively, but
in none of the wt mice (Figure 6A; each n= 6, p < 0.01).
[0142] In order to investigate whether a diastolic SR Ca2+ leak due to PKA
hyperphosphorylation of RyR2 directly contributes to a cardiac phenotype in
PDE4D-/
mice, the inventors treated wt mice with the PDE4 inhibitor rolipram. Rolipram
(0.3
mg/kg) inhibited cAMP-hydrolyzing PDE activity in the RyR2 complex in wt mice
(data
not shown) and resulted in significantly increased RyR2 PKA phosphorylation
during
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
48
exercise (Figure 6B). Following exercise and epinephrine injection (0.1
mg/kg),
ventricular arrhythmias or sudden death occurred in 100% of rolipram-treated
wt mice
(Figure 6C). Importantly, RyR2-S2808A knockin mice, which express a mutant
RyR2
that cannot be PKA phosphorylated (Figure 6B), were protected against rolipram-
induced
exercise-triggered arrhythmias (Figure 6C). These findings indicate that the
proarrhythmogenic effects of PDE4 inhibition are specifically due to PKA
hyperphosphorylation of RyR2 at Ser2808. Thus, PKA phosphorylation of RyR2 at
Ser2808 is necessary in order to generate triggered cardiac arrhythmias
associated with
PDE4 inhibition. Moreover, mortality due to sudden cardiac death at 24 and 72
hr
following myocardial infarction (MI, induced by ligation of the left anterior
descending
artery) was significantly increased in PDE4D+/ compared to wt mice (Figure
6D), fu.rther
suggesting that PDE deficiency in the RyR2-channel complex increases
susceptibility to
cardiac arrhythmias.
EXAMPLE 20 - EXACERBATION OF ACUTE HEART FAILURE ASSOCIATED
WITH PDE4D3 DEFICIENCY IS ATTENUATED IN MICE HARBORING RYR2
THAT CANNOT BE PKA PHOSPHORYLATED
[0143] Since PDE4D3 protein levels and cAMP-hydrolyzing activity in the RyR2
complex were reduced by 42% and 43% in human heart failure, respectively, the
inventors examined whether a partial reduction of PDE4D in haploinsufficient
PDE4D+/-
mice affects progression of HF. Heterozygous PDE4D+I- and wt control mice were
subjected to proximal left anterior descending (LAD) coronary artery ligation
to induce
myocardial infarction (MI), which results in progressive heart failure.
Similar to human
heart failure (Figure 5B), PDE4D+/ mice had a 44% reduction of PDE4D3 bound to
the
RyR2 complex as compared to control (Figure 4D) and developed significantly
worse
heart failure manifested by a larger increase in cardiac dimensions and more
depressed
cardiac contractility over a 28 day post-MI period (Figure 7). Cardiac
dimensions
(LVEDD) were 30% larger in PDE4D+/- hearts compared to wt 28 days post-MI
(Figure
7A), consistent with more severe cardiomyopathy. Infarct sizes were not
significantly
different in 4- to 5-month-old wt (35.8% ~: 3.1% LV, n=11) and PDE4D+I- mice
(37.2%
3.7% LV, n = 14). Cardiac function, measured by echocardiography and cardiac
catheterization, was reduced in haploinsufficient PDE4D+I- mice compared with
wt mice
following MI (Figures 7B and 7C). Accelerated HF progression in PDE4D+/- mice
was
associated with enhanced RyR2 PKA hyperphosphorylation and reduced PDE4D3
protein
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
49
levels in the RyR2 complex (Figures 7D and 7E) and significantly reduced
PDE4D3
enzymatic activity in the RyR2 complex (Figure 7F). In the RyR2 complex, PKA
catalytic and regulatory subunits, as well as the levels of the protein
phosphatases PP1
and PP2A, were not significantly different between wt and PDE4D+'- hearts
(data not
shown). Thus, reduction of PDE4D3 activity in the RyR2 complex in
haploinsufficient
PDE4D+/- mice to levels similar to those observed in RyR2 complexes from
failing
human hearts results in accelerated progression of heart failure.
[0144] Next, the inventors set out to determine whether the detrimental
effects of
PDE4D deficiency in the heart were dependent on dysfunction of the RyR2-
channel
complex due to PKA hyperphosphorylation and reduced binding of calstabin2.
Since
recent studies have demonstrated that the 1,4-benzothiazepine JTV-519
increases the
binding of calstabin2 to RyR2 in vivo, the inventors treated PDE4D+/ mice
subjected to
myocardial infarction with JTV-519. Treatment with JTV-519 (symbols)
significantly
increased the amount of calstabin2 bound to RyR2 (Figure 7D) and was
associated with
improved cardiac function following MI (Figures 7B and 7C). The inventors also
crossed
the PDE4D+/- mice with RyR2-S2808A mice to investigate the specific role of
PKA
hyperphosphorylation of RyR2 in the development of cardiac dysfunction in
PDE4D+/-
mice. RyR2-S2808A mice harbor RyR2 that cannot be PKA phosphorylated (Figure
7D).
Prevention of PKA hyperphosphorylation improved cardiac function in PDE4D+/
mice
subjected to MI (Figures 7A and 7C, green line and bars). Infarct sizes were
not
significantly different between PDE4D+/- mice, PDE4D+/ mice treated with JTV-
519
(38.6% 3.9% LV, n= 12), or PDE4D+/- mice crossed with RyR2-S2808A mice
(39.8%
4.3% LV, n= 11). These data demonstrate that normalizing RyR2 function, either
by
enhancing calstabin2 binding to RyR2 or by preventing PKA hyperphosphorylation
of
RyR2, improved cardiac function in PDE4D+'- mice following myocardial
infarction.
Taken together, these data suggest that the cardiac defects observed in PDE4D-
deficient
mice are due, at least in part, to defective RyR2 fiulction.
EXAMPLE 21- SUMMARY OF RESULTS
[0145] The present study shows that phosphodiesterase (PDE4D) deficiency is
associated with a severe cardiac phenotype consisting of heart failure and
lethal cardiac
arrhythmias. The importance of this cardiac phenotype in PDE4D-deficient mice
is
underscored by the fact that it is similar to that observed in humans with
heart failure:
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
decreased cardiac function and increased susceptibility to cardiac
arrhythmias. Moreover,
PDE inhibition has been associated with increased mortality in patients with
heart failure
(Packer et al., 1991), arrhythmias, and sudden cardiac death (Bittar and
Friedman, 1991
and Suissa et al., 1996), although the mechanism has been unknown. Finally,
since PDE4
inhibitors are being tested in clinical trials to treat common chronic
diseases including
Alzheimer's disease (Gong et al., 2004), asthma, and COPD (Giembycz, 2002), it
is
important to understand the consequences of long-term inhibition of PDE4
activity in the
heart, where PDE4 activity plays a major role in regulating cAMP-dependent
signals
(Perry et al., 2002, Verde et al., 1999 and Xiang et al., 2005).
[0146] Since the PDE4D deficiency caused no detectable alteration in global
cAMP levels or (3-adrenergic signaling in the heart, the cardiac phenotype in
PDE4D _'-
mice must be due to abnormalities in localized signaling, i.e., altered
microdomains of
cAMP, which is supported by the FRET imaging data showing increased cAMP
concentrations at the Z lines of PDE4D-deficient cardiomyocytes (where RyR2 is
present) after physiologic stimulation of 0-adrenergic receptors. Indeed, the
concept that
localized signaling regulates cAMP in the heart is supported by previous
findings
showing that PDE4 is a localized regulator of (3-adrenergic receptor (02-AR)
signaling in
cardiomyocytes (Baillie et a1.,.2003, Mongillo et al., 2004, Perry et al.,
2002 and Xiang et
al., 2005). Moreover, it has been shown that PDE4D3 can be targeted to
specific
compartments including the cardiomyocyte Z line via the targeting protein
mAKAP
(Carlisle Michel et al., 2004, Dodge et al., 2001, Sette and Conti, 1996 and
Yang et al.,
1998). These experiments were carefully designed to examine differences in
cAMP
levels at the Z line in cardiomyocytes from wt versus PDE4D-deficient mice
using low-
dose P-adrenergic stimulation (1 nM isoproterenol). Others have shown that
nonspecific
phannacologic PDE inhibition using maximal 0-adrenergic stimulation causes
cAMP
spillover into different compartments (Zaccolo and Pozzan, 2002).
[0147] There are likely many changes in cAMP-dependent signaling in the hearts
of PDE4D-deficient mice. For example, receptor-stimulated (3-arrestin-mediated
recruitment of PDE4 regulates G protein switching by the 02-AR in
cardiomyocytes
(Baillie et al., 2003). Moreover, PDE4D is an integral component of the 02-AR
signaling
complex (Xiang et al., 2005). Loss of other PDE4D isoforms (e.g., PDE4D8 and
PDE4D9) likely also contributes to localized alterations in cAMP levels in
PDE4D-'-
cardiomyocytes (Richter et al., 2005). This work shows that PKA
phosphorylation of
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
51
RyR2, which occurs at an early stage, before any structural or functional
abnormalities
were observed in PDE4D-/- mouse hearts (Figure 3), is a critical event since
crossing the
PDE4D-deficient mice with RyR2-S2808A mice inhibits the development of the
cardiac
phenotype (Figure 6 and Figure 7).
[01481 Indeed, there are at least two lines of evidence that strongly suggest
that
the cardiac phenotype in PDE4D-deficient mice is due to defects related to PKA
hyperphosphorylation of RyR2 and the resulting abnormal regulation of this
channel
required for EC coupling in the heart. First, the levels of PDE4D3 in RyR2-
channel
complexes from failing human hearts are reduced to the same degree as in RyR2
complexes in the hearts of PDE4D+'- mice, which develop accelerated heart
failure
following myocardial infarction. This suggests that PDE4D deficiency in the
RyR2
complex may play a role in PKA hyperphosphorylation of the channel and the
associated
cardiomyopathy. Second, and more importantly, sustained cardiac arrhythmias
associated
with pharmacologic PDE4 inhibition and accelerated progression of heart
failure
following MI were not observed in RyR2-S2808A mice harboring a RyR2 that
cannot be
PKA phosphorylated (Figure 6 and Figure 7). Thus, the present study indicates
that the
cardiac phenotype in PDE4D-deficient mice is directly related to defective
regulation of
RyR2 and provides support for the model whereby PKA hyperphosphorylation of
RyR2
causes a diastolic SR Ca2+ leak that (1) depletes SR Ca2+, contributing to
decreased
cardiac function (Marx et al., 2000), and (2) may provide a trigger for fatal
cardiac
arrhythmias (Wehrens et al., 2003).
[0149] Since the role of PKA hyperphosphorylation of RyR2 in heart failure
(Jiang et al., 2002) has been challenged, it is important to establish a
mechanism
underlying the PKA hyperphosphorylation of RyR2 and to show that this defect
can
specifically account for the observed cardiac phenotype. The inventors now
show that
PDE4D3 deficiency in the RyR2 complex contributes to PKA hyperphosphorylation
of
RyR2 in human and animal heart failure. Furthermore, a mutant RyR2 that cannot
be
PKA phosphorylated (RyR2-S2808A) protects against the cardiac effects of
PDE4D3
deficiency in the RyR2 complex in vivo. Indeed, the finding that PDE4D3 is
decreased in
RyR2 complexes in human heart failure helps address one of the controversial
issues in
this field, the mechanism whereby RyR2 become PKA hyperphosphorylated (Marx et
al.,
2000) despite decreases in global cAMP levels in failing human hearts (Regitz-
Zagrosek
et al., 1994).
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
52
[0150] The current findings suggest a novel function of PDE4D3 in the
regulation
of RyR2, the major intracellular Ca2+-release channel in the heart. PDE4D3
activity
provides an important negative-feedback mechanism to limit (3-AR-dependent PKA
phosphorylation of RyR2-Ser2808. Under physiologic conditions, PDE4D3 may
regulate
local PKA activity and channel activation via phosphorylation of RyR2-Ser2808,
thereby
preventing excess accumulation of cAMP (Zaccolo and Pozzan, 2002) and
uncontrolled
PKA-mediated activation of the channel. In human heart failure, loss of
negative
feedback due to PDE4D3 deficiency in the RyR2 complex likely contributes to
RyR2
PKA hyperphosphorylation; calstabin2 depletion; and hyperactive, "leaky" RyR2
channels (Marx et al., 2000 and Pieske et al., 1999). Taken together, these
data suggest
that PDE4D3 plays a protective role in the heart against heart failure and
arrhythmias.
[0151] These data further suggest that chronic pharmacologic PDE4 inhibition
could contribute to a cardiac phenotype including cardiac dysfunction and
arrhythmias,
particularly in individuals with underlying cardiac disease. In addition,
other signaling
systems may be affected by reduced PDE4D activity, e.g., (3-arrestin targeting
of PDE4D3
activity may be important for 02-AR desensitization (Perry et al., 2002).
Importantly,
PDE4D3 deficiency and pharmacologic PDE4 inhibition with rolipram was
associated
with stress-induced cardiac arrhythmias, which did not occur in mice lacking
the RyR2
PKA phosphorylation site at Ser2808. These findings suggest that PDE4
inhibitors could
increase the risk of cardiac arrhythmias due to "leaky" RyR2 channels as
observed in
individuals with genetic forms of sudden cardiac death linked to RyR2
mutations
(Lehnart et al., 2004 and Wehrens et al., 2003) and in patients with heart
failure. The
present study shows that phosphodiesterase (PDE4D) deficiency is associated
with a
severe cardiac phenotype consisting of heart failure and lethal cardiac
arrhythmias. The
importance of this cardiac phenotype in PDE4D-deficient mice is underscored by
the fact
that it is similar to that observed in humans with heart failure: decreased
cardiac function
and increased susceptibility to cardiac arrhythmias. Moreover, PDE inhibition
has been
associated with increased mortality in patients with heart failure (Packer et
al., 1991),
arrhythmias, and sudden cardiac death (Bittar and Friedinan, 1991 and Suissa
et al.,
1996), although the mechanism has been unknown. Finally, since PDE4 inhibitors
are
being tested in clinical trials to treat common chronic diseases including
Alzheimer's
disease (Gong et al., 2004), asthma, and COPD (Giembycz, 2002), it is
important to
understand the consequences of long-term inhibition of PDE4 activity in the
heart, where
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
53
PDE4 activity plays a major role in regulating cAMP-dependent signals (Perry
et al.,
2002, Verde et al., 1999 and Xiang et al., 2005).
[01521 Since the PDE4D deficiency caused no detectable alteration in global
cAMP levels or (3-adrenergic signaling in the heart, the cardiac phenotype in
PDE4D -/
mice must be due to abnormalities in localized signaling, i.e., altered
microdomains of
cAMP, which is supported by the FRET imaging data showing increased cAMP
concentrations at the Z lines of PDE4D-deficient cardiomyocytes (where RyR2 is
present) after physiologic stimulation of (3-adrenergic receptors. Indeed, the
concept that
localized signaling regulates cAMP in the heart is supported by previous
findings
showing that PDE4 is a localized regulator of (3-adrenergic receptor (02-AR)
signaling in
cardiomyocytes (Baillie et al., 2003, Mongillo et al., 2004, Perry et al.,
2002 and Xiang et
al., 2005). Moreover, it has been shown that PDE4D3 can be targeted to
specific
compartments including the cardiomyocyte Z line via the targeting protein
mAKAP
(Carlisle Michel et al., 2004, Dodge et al., 2001, Sette and Conti, 1996 and
Yang et al.,
1998). The inventors' experiments were carefully designed to examine
differences in
cAMP levels at the Z line in cardiomyocytes from wt versus PDE4D-deficient
mice using
low-dose 0-adrenergic stimulation (1 nM isoproterenol). Others have shown that
nonspecific pharmacologic PDE inhibition using maximal (3-adrenergic
stimulation
causes cAMP spillover into different compartments (Zaccolo and Pozzan, 2002).
[0153] There are likely many changes in cAMP-dependent signaling in the hearts
of PDE4D-deficient mice. For example, receptor-stimulated (3-arrestin-mediated
recruitment of PDE4 regulates G protein switching by the 02-AR in
cardiomyocytes
(Baillie et al., 2003). Moreover, PDE4D is an integral component of the [i2-AR
signaling
complex (Xiang et al., 2005). Loss of other PDE4D isoforms (e.g., PDE4D8 and
PDE4D9) likely also contributes to localized alterations in cAMP levels in
PDE4D -/
cardiomyocytes (Richter et al., 2005). The inventors' work shows that PKA
phosphorylation of RyR2, which occurs at an early stage, before any structural
or
functional abnormalities were observed in PDE4D-/- mouse hearts (Figure 3), is
a critical
event since crossing the PDE4D-deficient mice with RyR2-S2808A mice inhibits
the
development of the cardiac phenotype (Figure 6 and Figure 7).
[0154] Indeed, there are at least two lines of evidence that strongly suggest
that
the cardiac phenotype in PDE4D-deficient mice is due to defects related to PKA
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
54
hyperphosphorylation of RyR2 and the resulting abnormal regulation of this
channel
required for EC coupling in the heart. First, the levels of PDE4D3 in RyR2-
channel
complexes from failing human hearts are reduced to the same degree as in RyR2
complexes in the hearts of PDE4D+'- mice, which develop accelerated heart
failure
following myocardial infarction. This suggests that PDE4D deficiency in the
RyR2
complex may play a role in PKA hyperphosphorylation of the channel and the
associated
cardiomyopathy. Second, and more importantly, sustained cardiac arrhythmias
associated
with pharmacologic PDE4 inhibition and accelerated progression of heart
failure
following MI were not observed in RyR2-S2808A mice harboring a RyR2 that
cannot be
PKA phosphorylated (Figure 6 and Figure 7). Tllus, the present study indicates
that the
cardiac phenotype in PDE4D-deficient mice is directly related to defective
regulation of
RyR2 and provides support for the model whereby PKA hyperphosphorylation of
RyR2
causes a diastolic SR Ca2+ leak that (1) depletes SR Ca2+, contributing to
decreased
cardiac function (Marx et al., 2000), and (2) may provide a trigger for fatal
cardiac
arrhythmias (Wehrens et al., 2003).
[0155] Since the role of PKA hyperphosphorylation of RyR2 in heart failure
(Jiang et al., 2002) has been challenged, it is important to establish a
mechanism
underlying the PKA hyperphosphorylation of RyR2 and to show that this defect
can
specifically account for the observed cardiac phenotype. The inventors now
show that
PDE4D3 deficiency in the RyR2 complex contributes to PKA hyperphosphorylation
of
RyR2 in human and animal heart failure. Furtliermore, a mutant RyR2 that
camiot be
PKA phosphorylated (RyR2-S2808A) protects against the cardiac effects of
PDE4D3
deficiency in the RyR2 complex in vivo. Indeed, the finding that PDE4D3 is
decreased in
RyR2 complexes in human heart failure helps address one of the controversial
issues in
this field, the mechanism whereby RyR2 become PKA hyperphosphorylated (Marx et
al.,
2000) despite decreases in global cAMP levels in failing human hearts (Regitz-
Zagrosek
et al., 1994).
[0156] The current findings suggest a novel function of PDE4D3 in the
regulation
of RyR2, the major intracellular Ca2+-release channel in the heart. PDE4D3
activity
provides an important negative-feedback mechanism to limit (3-AR-dependent PKA
phosphorylation of RyR2-Ser2808. Under physiologic conditions, PDE4D3 may
regulate
local PKA activity and channel activation via phosphorylation of RyR2-Ser2808,
thereby
preventing excess accumulation of cAMP (Zaccolo and Pozzan, 2002) and
uncontrolled
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
PKA-mediated activation of the channel. In liuman heart failure, loss of
negative
feedback due to PDE4D3 deficiency in the RyR2 complex likely contributes to
RyR2
PKA hyperphosphorylation; calstabin2 depletion; and hyperactive, "leaky" RyR2
channels (Marx et al., 2000 and Pieske et al., 1999). Taken together, these
data suggest
that PDE4D3 plays a protective role in the heart against heart failure and
arrhythmias.
[0157] These data further suggest that chronic pharmacologic PDE4 inhibition
could contribute to a cardiac phenotype including cardiac dysfunction and
arrhythmias,
particularly in individuals with underlying cardiac disease. In addition,
other signaling
systems may be affected by reduced PDE4D activity, e.g., 0-arrestin targeting
of PDE4D3
activity may be important for 02-AR desensitization (Perry et al., 2002).
Importantly,
PDE4D3 deficiency and pharmacologic PDE4 inhibition with rolipram was
associated
with stress-induced cardiac arrhythmias, which did not occur in mice lacking
the RyR2
PKA phosphorylation site at Ser2808. These findings suggest that PDE4
inhibitors could
increase the risk of cardiac arrhythmias due to "leaky" RyR2 channels as
observed in
individuals with genetic forms of sudden cardiac death linked to RyR2
mutations
(Lehnart et al., 2004 and Wehrens et al., 2003) and in patients with heart
failure.
[0158] While the foregoing invention has been described in some detail for
purposes of clarity and understanding, it will be appreciated by one skilled
in the art,
from a reading of the disclosure, that various changes in form and detail can
be made
without departing from the true scope of the invention in the appended claims.
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
56
REFERENCES
Antos et al., Dilated cardiomyopathy and sudden death resulting from
constitutive
activation of protein kinase A, Circ. Res. 89, 997-1004 (2001).
Baille et al., beta-Arrestin-mediated PDE4 cAMP phosphodiesterase recruitment
regulates beta-adrenoceptor switching from Gs to Gi. Proc. Natl. Acada. Sci.
USA 100,
940-945 (2003).
Barnes, P.J., Theophylline: new perspectives for an old drug. Am. J. Respir.
Crit. Care
Med. 167, 813-8 (2003).
Bittar et al., The arrhythmogeneicity of theophylline. A multivariate analysis
of clinical
determinants. Chest 99, 1415-1420 (1991).
Bolger et al., Characterization of five different proteins produced by
alternatively spliced
mRNAs from the human cAMP-specific phosphodiesterase PDE4D gene. Biochem. J.
328 ( Pt 2), 539-48 (1997).
Bristow et al., Beta 1- and beta 2-adrenergic-receptor subpopulations in
nonfailing and
failing human ventricular myocardium: coupling of both receptor subtypes to
muscle
contraction and selective beta I-receptor down-regulation in heart failure.
Circ. Res. 59,
297-309 (1986).
Carlisle Michel et al., PKA-phosphorylation of PDE4D3 facilitates recruitment
of the
mAKAP signaling complex. Biochem. J. 381, 587-592 (2004).
Cohn et al., Plasma norepinephrine as a guide to prognosis in patients with
chronic
congestive heart failure. N. Engl. J. Med. 311, 819-23 (1984).
Conti et al., Cyclic AMP-specific PDE4 phosphodiesterases as critical
components of
cyclic AMP signaling. J. Biol. Chem. 278, 5493-6 (2003).
Dodge et al., mAKAP assembles a protein kinase A/PDE4 phosphodiesterase cAMP
signaling module. EMBO J. 20, 1921-30, (2001).
Feldman et al., Deficient production of cyclic AMP: pharmacologic evidence of
an
important cause of contractile dysfunction in patients with end-stage heart
failure.
Circulation 75, 331-9 (1987).
Franzini-Armstrong et al., Shape, size, and distribution of Ca(2+) release
units and
couplons in skeletal and cardiac muscles. Biophys. J. 77, 1528-39 (1999).
Giembycz, M.A., Development status of second generation PDE4 inhibitors for
asthma
and COPD: the story so far. Monaldi. Arch. Chest Dis. 57, 48-64 (2002).
Gong et al., Persistent improvement in synaptic and cognitive functions in an
Alzheimer
mouse model after rolipram treatment. J. Clin. Invest. 114, 1624-1634 (2004).
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
57
Gretarsdottir et al., The gene encoding phosphodiesterase 4D confers risk of
ischemic
stroke. Nat. Genet. 35, 131-8 (2003).
Houslay, et al., PDE4 cAMP phosphodiesterases: modular enzymes that
orchestrate
signaling cross-talk, desensitization and compartmentalization. Biochem. J.
370, 1-8
(2003).
Jiang et al., Abnormal Ca2+ release, but normal ryanodine receptors, in canine
and
human heart failure. Circ. Res. 91, 1015-1022 (2002).
Jayaraman et al., FK506 binding protein associated with the calcium release
channel
(ryanodine receptor). J. Biol. Chem. 267, 9474-7 (1992).
Jin et al., Impaired growth and fertility of cAMP-specific phosphodiesterase
PDE4D-
deficient mice. Proc. Natl. Acad. Sci. USA 96, 11998-2003 (1999).
Lellnart et al., Sudden death in familial polymorphic ventricular tachycardia
associated
with calcium release channel (ryanodine receptor) leak. Circulation 109, 3208-
14 (2004).
Marx et al., Phosphorylation-dependent regulation of iyanodine receptors: a
novel role for
leucine/isoleucine zippers. J. Cell. Biol. 153, 699-708 (2001).
Marx et al., PKA phosphorylation dissociates FKBP12.6 from the calcium release
channel (ryanodine receptor): defective regulation in failing hearts. Cell
101, 365-76
(2000).
Mongillo et al., Fluorescence resonance energy transfer-based analysis of cAMP
dynamics in live neonatal rat cardiac myocytes reveals distinct functions of
compartmentalized phosphodiesterases. Circ. Res., 95, 67-75 (2004).
Packer et al., Effect of oral milrinone on mortality in severe chronic heart
failure. The
PROMISE Study Research Group. N. Engl. J. Med. 325,1468-75 (1991).
Perry et al., Targeting of cyclic AMP degradation to beta 2-adrenergic
receptors by beta-
arrestins. Science 298, 834-6 (2002).
Pieske et al., Ca2+ handling and sarcoplasmic reticulum Ca2+ content in
isolated failing
and nonfailing human myocardium. Circ. Res. 85, 38-46 (1999).
Regitz-Zagrosek et al., Myocardial cyclic AMP and norepinephrine content in
human
heart failure. Eur. Heart J. 15 (Suppl. D) 7-13. (1994).
Reiken et al., Beta-blockers restore calcium release channel function and
improve cardiac
muscle performance in human heart failure. Circulation 107, 2459-66 (2003).
Reiken et al., Protein kinase A phosphorylation of the cardiac calcium release
channel
(ryanodine receptor) in normal and failing hearts. Role of phosphatases and
response to
isoproterenol. J. Biol. Chem. 278,444-53 (2003).
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
58
Richter et al., Splice variants of the cyclic nucleotide phosphodiesterase
PDE4D are
differentially expressed and regulated in rat tissue. Biochem. N. 388, 803-811
(2005).
Ruehr et al., Targeting the protein kinase A by muscle A kinase-anchoring
protein
(mAKAP) regulates phosphorylation and function of the skeletal muscle
ryanodine
receptor. J. Biol. Chem. 278, 24831-24836. (2003).
Sette et al., Phosphorylation and activation of a cAMP-specific
phosphodiesterase by the
cAMP-dependent protein kinase. Involvement of serine 54 in the enzyme
activation. J.
Biol. Chem. 271, 16526-34 (1996).
Sette et al., The ratPDE3/IVd phosphodiesterase gene codes for multiple
proteins
differentially activated by cAMP-dependent protein kinase. J. Biol. Chem. 269,
18271-4
(1994).
Shamion et al., Elevated sarcoplasmic reticulum Ca2+ leak in intact
ventricular myocytes
from rabbits in heart failure. Circ. Res. 93, 592-4 (2003).
Suissa et al., Bronchodilators and acute cardiac death. Am. J. Respir. Crit.
Care Med. 154,
1598-1602 (1996)
Tasken et al., Phosphodiesterase 4D and protein kinase a type II constitute a
signaling
unit in the centrosomal area. J. Biol. Chem. 276, 21999-2002 (2001).
van Rooij et al,. MCIPI overexpression suppresses left ventricular remodeling
and
sustains cardiac fiinction after myocardial infarction. Circ. Res. 94, e 18-26
(2004).
Verde et al., Characterization of the cyclic nucleotide phosphodiesterase
subtypes
involved in the regulation of the L-type Ca2+ current in rat ventricular
myocytes. Br. J.
Pharmacol. 127, 65-74 (1999).
Vignola, A.M., PDE4 inhibitors in COPD--a more selective approach to
treatment.
Respir. Med. 98,495-503 (2004).
Wang et al., Cloning and characterization of novel PDE4D isoforms PDE4D6 and
PDE4D7. Cell. Signal. 15, 883-891 (2003).
Wehrens et al,. FKBPI2.6 deficiency and defective calcium release channel
(ryanodine
receptor) function linked to exercise-induced sudden cardiac death. Cell 113,
829-40
(2003).
Welirens et al., Protection from cardiac arrhythmia through ryanodine receptor-
stabilizing
protein calstabin2. Science 304, 292-6 (2004).
Wehrens et al., Enhancing calstabin binding to ryanodine receptors improves
cardiac and
skeletal muscle function in heart failure. Proc. Natl. Acad. Sci. USA.102,
9607-12 (2005).
Wehrens et al., Intracellular Calcium Release Channels and Cardiac Disease.
Annu. Rev.
Physiol. (2004).
CA 02591924 2007-06-15
WO 2006/071603 PCT/US2005/045914
59
Wehrens et al., Ca2+/calmodulin-dependent protein kinase II phosphorylation
regulates
the cardiac ryanodine receptor. Circ. Res. 94, e61-70 (2004).
Xiang et al., Phosphodiesterase 4D in required for beta2 adrenoceptor subtype-
specific
signaling in cardiac cyocytes. Proc. Natl. Acad. Sci. USA 102, 909-914 (2005).
Yang et al., A-kinase anchoring protein 100 (AKAP 100) is localized in
multiple
subcellular compartments in the adult rat heart. J. Cell. Biol. 142, 511-22
(1998).
Yano et al., Altered stoichiometry of FKBP12.6 versus ryanodine receptor as a
cause of
abnormal Ca2+ leak through ryanodine receptor in heart failure. Circulation
102, 2131-36,
(2000).
Yano et al., FKBP12.6-Mediated Stablization of Calcium-Release Channel
(Ryanodine
Receptor) as a Novel Therapeutic Strategy Against Heart Failure. Circulation
107, 477-
484, (2003).
Zaccolo et al., Discrete micro domains with high concentration of cAMP in
stimulated rat
neonatal cardiac myocytes. Science 295, 1711-5 (2002).