Note: Descriptions are shown in the official language in which they were submitted.
CA 02592388 2007-06-19
WO 2006/069193 PCT/US2005/046477
IMIDAZO[4,5-c]PYRIDINE COMPOUND AND METHOD
OF ANTIVIRAL TREATMENT
Field of the Invention
The present invention relates to compounds useful in the treatment or
prophylaxis of viral infections, particularly those by the Flaviviridae and
Picornaviridae families including hepatitis C virus (HCV).
Background of the Invention
The Flaviviridae family consists of 3 genera, the pestiviruses, the
flaviviruses and the hepaciviruses. It also contains the hepatitis G virus
(HGV/GBV-C), which has not yet been assigned to a genus. Pestiviruses such as
the Classical Swine Fever Virus (CSFV), the Bovine Viral Diarrhea Virus (BVDV)
and the Border Disease Virus (BDV) cause infections of domestic livestock
(respectively pigs, cattle and sheep) and are responsible for significant
economic
losses world-wide. BVDV, the prototypic representative of the pestivirus
genus, is
ubiquitous and causes a range of clinical manifestations, including abortion,
teratogenesis, respiratory problems, chronic wasting disease, immune system
dysfunction, and predisposition to secondary viral and bacterial infections.
Vaccines are used in some countries with varying degrees of success to
control pestivirus disease. In other countries, animal culling and slaughter
are
used to contain pestivirus disease outbreaks.
CA 02592388 2007-06-19
WO 2006/069193 PCT/US2005/046477
The World Health Organization estimates that world-wide 170 million
people (3% of the world's population) are chronically infected with HCV. These
chronic carriers are at risk of developing cirrhosis and/or liver cancer. In
studies
with a 10 to 20 year follow-up, cirrhosis developed in 20 - 30 % of the
patients, 1
to 5% of whom may develop liver cancer during the next ten years. The only
treatment option available today is the use of interferon a-2 (or its
pegylated
from) either alone or combined with ribavirin. However, sustained response is
only observed in about 40% of the patients and treatment is associated with
serious adverse effects. There is thus an urgent need for potent and selective
inhibitors of HCV.
The compound 3-[((2-dipropylamino)ethyl)thio]-5H-1,2,4-triazino[5,6-
b]indole has been reported to selectively inhibit the replication of BVDV and
other
pestiviruses (Baginski SG et al., Proc. Natl. Acad. Sci. U.S.A. 2000 Jul
5;97(14):7981-
6). Currently, no pharmaceutical strategy is available for controlling
pestivirus
infections.
Coxsackie viruses belong to the enteroviruses of the Picornaviridae family.
They cause a heterogeneous group of infections including herpangina, aseptic
meningitis, a common-cold-like syndrome, a non-paralytic poliomyelitis-like
syndrome, epidemic pleurodynia (an acute, febrile, infectious disease
generally
occurring in epidemics), hand-foot-mouth syndrome, pediatric and adult
pancreatitis and serious myocarditis.
Currently only pleconaril (3-13,5-dimethyl-4-[[3-methyl-5-
isoxazolyl)propyl]phenyl]-5-(trifluoromethyl-1,2,4-oxadiazole)) and enviroxime
(2-amino-l-(isopropylsulfonyl)-6-benzimidazole phenyl ketone oxime) have been
studied clinically for the treatment of infections with enteroviruses.
Pleconaril is a
so called "capsid function-inhibitor"; enviroxime prevents the formation of
the
RNA replicative intermediate. Enviroxime resulted in only modest clinical and
-2-
CA 02592388 2012-04-12
virological benefit in some studies and no benefits in others. Clinical
response
with pleconaril has been observed in some studies, but the compound has not
been approved by the Food and Drug Administration (hearing of March 18th,
2002).
Relevant disclosures include U.S. Patent Nos. 4,914,108; 4,988,707;
4,990,518; 5,137,896; 5,208,242; 5,227,384; 5,302,601; 5,374,638; 5,405,964;
5,438,063;
5,486,525; 6,479,508; and U.S. Patent Publication No. US2003/0108862 Al,
Canadian Patent No. 2423800 Al, German Patent Nos. 4211474 Al, 4236026,
4309969, 4318813, European Patent Nos. EP 0 138 552 A2, EP 0 706 795 A2,
EP 1 132 381 Al, Great Britain Patent No. 2158440 A, PCT Patent Publication
Nos.
WO 00/20416, WO 00/39127, WO 00/40583, WO 03/007945 Al, WO 03/010140
A2, WO 03/010141 A2, WO 93/02080, WO 93/14072, WO 96/11192, WO
96/12703, WO 99/27929, PCT-US2004/43112, PCT-BE2003/000117, PCT-
US2005/26606, Akamatsu, et al., "New Efficient Route for Solid-Phase Synthesis
of Benzimidazole Derivatives", 4:475-483, J. COMB. CHEM., 2002, Cleve et al.,
"Derivate des Imidazo[4.5-b]- and lmidazo[4.5-c]pyridins", 747:158-171, JUSTUS
LIEBIGS ANNALEN DER CHEMICA, 1971, Kiyama, et al., "Synthesis and
Evaluation of Novel Nonpeptide Angiotensin II Receptor Antagonists:
Imidazo[4,5-c]pyridine Derivatives with an Aromatic Substituent", 43(3):450-
60,
CHEM PHARM BULL, 1995, Mederski et al., "Synthesis and Structural
Assignment of Some N-substituted Imidazopyridine Derivatives", 48(48):10549-
58, TETRAHEDRON, 1992, Yutilov et al., 23(l):56-9, KHIMIKO-
FARMATSEVTICHESKII ZHURNAL, 1989.
A need exists for compounds having therapeutic properties, such as greater
oral bioavailability, reduced toxicity, optimal clearance, increased potency
and the
like against viruses belonging to the family of Flaviviridae including
hepatitis C
3
CA 02592388 2012-04-12
virus, and against viruses belonging to the family of Picornaviridae. These
and
other objects of this invention will be apparent to one skilled in the art
from
consideration of this specification as a whole.
Summary of the Invention
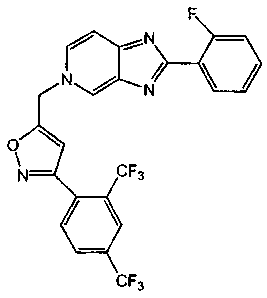
In accordance with the objects, the novel compound 5-((3-(2, 4-
trifluoromethyphenyl)isoxazol-5-yl)methyl)-2-(2-fluorophenyl)-5H-imidazo[4,5-
c]pyridine
F
:N
N / N
O
N CF3
CF3
or a salt thereof.
The invention also concerns a pharmaceutical composition comprising the
compound of this invention and at least one pharmaceutically acceptable
carrier.
The invention also concerns the use of the compound defined herein, or of
the pharmaceutical composition defined herein, for the preparation of a
medicament
for the treatment or prophylaxis of a flaviviral or picornaviral infection,
preferably
when the infection is a hepatitis C viral infection.
The invention also concerns the use of the compound defined herein, or of
the pharmaceutical composition defined herein, for the treatment or
prophylaxis of a
flaviviral or picornaviral infection, preferably wherein the infection is a
hepatitis C
viral infection.
4
CA 02592388 2012-04-12
The invention also concerns a veterinary composition comprising the
compound defined herein and at least one veterinary carrier, and its use for
the
treatment or prophylaxis of bovine viral diarrhoea virus BVDV.
Detailed Description of the Invention
The compound of this invention is employed for the treatment or
prophylaxis of flaviviral or picornaviral infections, in particular HCV and
BVDV.
The therapeutic compound of this invention is administered to a subject
mammal (including a human) by any means well known in the art, i.e. orally,
intranasally, subcutaneously, intramuscularly, intradermally, intravenously,
intra-arterially, parenterally or by catheterization in a therapeutically
effective
amount, i.e., a flaviviral or picornaviral growth inhibiting amount or a
flaviviral
or picornaviral replication inhibiting amount. This amount is believed to be
an
CA 02592388 2007-06-19
WO 2006/069193 PCT/US2005/046477
amount that ensures a plasma level of between about 1pg/ml and 100 mg/ml,
optionally of 10 mg/ml. This optionally is achieved by administration of a
dosage
of in the range of 0.001 mg to 60 mg, preferably 0.01 mg to 10 mg, preferably
about 0.5 mg to 1.5 mg per day per kg bodyweight for humans. Dosages of 1, 3,
6,
10, 20, 30 and 60 mg/kg are suitable for conducting toxicity studies in dogs
and
for extrapolating to suitable doses in humans. The optimal dosage of the
compound of this invention will depend upon many factors known to the artisan,
including bioavailability of the compound, its metabolism and distribution in
the
subject, its toxicity and its potency, among others. Proper dosing typically
is
o determined in the preclinical and clinical settings, and is well within the
skill of
the ordinary artisan. The therapeutically effective amount of the compound of
this invention optionally is divided into several sub-units per day or is
administered daily or in more than one day intervals, depending upon the
nature
of the infection, the patient's general condition and the nature of the
compound of
this invention. Generally, the compound is administered daily.
The compound of this invention is employed in concert with other agents
effective against Picornaviral or Flaviviral infections. Such agents include,
for
instance, interferon alpha, ribavirin, and/or compounds falling within the
disclosures of EP1162196, WO 03/010141, WO 03/007945 WO 00/204425 and/or
WO 03/010140 (and other filings within their patent families). Such other
agents
are used in conventional amounts, although if the efficacy of the compound of
this invention and the other compound is additive then the amounts of each
active agent optionally are commensurately reduced, and more so if the agents
act
synergistically. In general, however, the agents are used in their ordinary
active
amounts in the compositions.
Co-administered agents generally are formulated into unitary
compositions with the compound of this invention so long as they are
chemically
compatible and are intended to be administered by the same route. If not, then
6 -
CA 02592388 2007-06-19
WO 2006/069193 PCT/US2005/046477
they optionally are provided in the form of a medical kit or package
containing
the two agents in separate repositories or compartments.
The present invention further provides veterinary compositions
comprising at least one compound of this invention together with a veterinary
carrier therefor, for example in the treatment of BVDV. Veterinary carriers
are
materials useful for the purpose of administering the composition and are
excipients which are otherwise inert or acceptable in the veterinary art and
are
compatible with the compound of this invention. These veterinary compositions
0 are administered orally, parenterally or by any other desired route.
The compound of this invention is provided as the free base or as a salt.
Salts typically are prepared by acid addition of certain organic and inorganic
acids to the free base. Examples include (1) inorganic acids such as
hydrohalogen
acids, e.g. hydrochloric or hydrobromic acid, sulfuric acid, nitric acid,
phosphoric
acid and sulfamic acids; or (2) organic acids such as acetic, propanoic,
hydroxyacetic, benzoic, 2-hydroxypropanoic, 2-oxopropanoic, lactic, fumaric,
tartaric, pyruvic, maleic, malonic, malic, salicylic (e.g. 2-hydroxybenzoic),
p-
aminosalicylic, isethionic, lactobionic, succinic, oxalic and citric acids;
organic
sulfonic acids, such as methanesulfonic, ethanesulfonic, benzenesulfonic, p-
toluenesulfonic, C1-C6 alkylsulfonic, benzenesulfonic, p-toluenesulfonic, and
cyclohexanesulfamic acids. Also included within the scope of this invention
are
the salts of the compound of this invention with one or more amino acids,
typically naturally-occuring amino acids such as one of the amino acids found
in
proteins. The acidic counterion desirably is physiologically innocuous and non-
toxic or otherwise pharmaceutically acceptable, unless the salt is being used
as an
intermediate in preparation of the compounds whereupon toxicity is not
relevant.
While the free base is preferred, suitable salts include mesylate
(methanesulfonic
acid) and HCI.
7 -
CA 02592388 2007-06-19
WO 2006/069193 PCT/US2005/046477
The compound of this invention includes the solvates formed with the
compound of this invention or their salts, such as for example hydrates,
alcoholates and the like.
The compound of this invention optionally is formulated with
conventional pharmaceutical carriers and excipients, which will be selected in
accord with ordinary practice. Tablets will contain excipients, glidants,
fillers,
binders and the like. Aqueous formulations are prepared in sterile form, and
when intended for delivery by other than oral administration generally will be
isotonic. Formulations optionally contain excipients such as those set forth
in the
"Handbook of Pharmaceutical Excipients" (1986) and include ascorbic acid and
other antioxidants, chelating agents such as EDTA, carbohydrates such as
dextrin,
hydroxyalkylcellulose, hydroxyalkylmethylcellulose and stearic acid.
The term "pharmaceutically acceptable carrier" as used herein means any
material or substance with which the active ingredient is formulated in order
to
facilitate its preparation and/or its application or dissemination to the site
to be
treated. Suitable pharmaceutical carriers for use in the compositions of this
invention are well known to those skilled in the art. They include additives
such
as wetting agents, dispersing agents, adhesives, emulsifying agents, solvents,
glidants, coatings, antibacterial and antifungal agents (for example phenol,
sorbic
acid, chlorobutanol), isotonic agents (such as sugars or sodium chloride),
provided that the same are consistent with pharmaceutical practice, i.e. they
are
not toxic to mammals.
The pharmaceutical compositions of the present invention are prepared in
any known manner, for instance by homogeneously mixing, coating and/or
grinding the active ingredients in a one-step or multi-step procedure, with
the
selected carrier material and, where appropriate, other additives such as
surface-
active agents. Compositions containing the compound of this invention
-8-
CA 02592388 2007-06-19
WO 2006/069193 PCT/US2005/046477
formulated into microspheres (usually having a diameter of about 1 to 10 gm)
are
useful as controlled or sustained release formulations.
Suitable surface-active agents, also known as emulgents or emulsifiers, are
useful in the pharmaceutical compositions of the present invention. They are
non-
ionic, cationic and/or anionic materials having suitable emulsifying,
dispersing
and/or wetting properties. Suitable anionic surfactants include both water-
soluble soaps and water-soluble synthetic surface-active agents. Suitable
soaps are
alkaline or alkaline-earth metal salts, unsubstituted or substituted ammonium
1o salts of higher fatty acids (C,o-C,,), e.g. the sodium or potassium salts
of oleic or
stearic acid, or of natural fatty acid mixtures obtainable from coconut oil or
tallow
oil. Synthetic surfactants include sodium or calcium salts of polyacrylic
acids;
fatty sulphonates and sulphates; sulphonated benzimidazole derivatives and
alkylarylsulphonates. Fatty sulphonates or sulphates are usually in the form
of
alkaline or alkaline-earth metal salts, unsubstituted ammonium salts or
ammonium salts substituted with an alkyl or acyl radical having from 8 to 22
carbon atoms, e.g. the sodium or calcium salt of lignosulphonic acid or
dodecylsulphonic acid or a mixture of fatty alcohol sulphates obtained from
natural fatty acids, alkaline or alkaline-earth metal salts of sulphuric or
sulphonic
acid esters (such as sodium lauryl sulphate) and sulphonic acids of fatty
alcohol/ ethylene oxide adducts. Suitable sulphonated benzimidazole
derivatives
preferably contain 8 to 22 carbon atoms. Examples of alkylarylsulphonates are
the
sodium, calcium or alcoholamine salts of dodecylbenzene sulphonic acid or
dibutyl-naphthalenesulphonic acid or a naphthalene-sulphonic
acid/ formaldehyde condensation product. Also suitable are the corresponding
phosphates, e.g. salts of phosphoric acid ester and an adduct of p-nonylphenol
with ethylene and/or propylene oxide, or phospholipids. Suitable phospholipids
for this purpose are the natural (originating from animal or plant cells) or
synthetic phospholipids of the cephalin or lecithin type such as e.g.
phosphatidylethanolamine, phosphatidylserine, phosphatidylglycerine,
9 -
CA 02592388 2007-06-19
WO 2006/069193 PCT/US2005/046477
lysolecithin, cardiolipin, dioctanylphosphatidyl-choline,
dipalmitoylphoshatidyl -
choline and their mixtures.
Suitable non-ionic surfactants include polyethoxylated and
polypropoxylated derivatives of alkylphenols, fatty alcohols, fatty acids,
aliphatic
amines or amides containing at least 12 carbon atoms in the molecule,
alkylarenesulphonates and dialkylsulphosuccinates, such as polyglycol ether
derivatives of aliphatic and cycloaliphatic alcohols, saturated and
unsaturated
fatty acids and alkylphenols, said derivatives preferably containing 3 to 10
glycol
1 o ether groups and 8 to 20 carbon atoms in the (aliphatic) hydrocarbon
moiety and 6
to 18 carbon atoms in the alkyl moiety of the alkylphenol. Further suitable
non-
ionic surfactants are water-soluble adducts of polyethylene oxide with
poylypropylene glycol, ethylenediaminopolypropylene glycol containing 1 to 10
carbon atoms in the alkyl chain, which adducts contain 20 to 250
ethyleneglycol
ether groups and/or 10 to 100 propyleneglycol ether groups. Such compounds
usually contain from I to 5 ethyleneglycol units per propyleneglycol unit.
Representative examples of non-ionic surfactants are nonylphenol -
polyethoxyethanol, castor oil polyglycolic ethers, polypropylene/ polyethylene
oxide adducts, tributylphenoxypolyethoxyethanol, polyethyleneglycol and
octylphenoxypolyethoxyethanol. Fatty acid esters of polyethylene sorbitan
(such
as polyoxyethylene sorbitan trioleate), glycerol, sorbitan, sucrose and
pentaerythritol are also suitable non-ionic surfactants.
Suitable cationic surfactants include quaternary ammonium salts,
particularly halides, having 4 hydrocarbon radicals optionally substituted
with
halo, phenyl, substituted phenyl or hydroxy; for instance quaternary ammonium
salts containing as N-substituent at least one C8C22 alkyl radical (e.g.
cetyl, lauryl,
palmityl, myristyl and oleyl) and, as further substituents, unsubstituted or
halogenated lower alkyl, benzyl and/or hydroxy-lower alkyl radicals.
- 10 -
CA 02592388 2007-06-19
WO 2006/069193 PCT/US2005/046477
A more detailed description of surface-active agents suitable for this
purpose is found in "McCutcheon's Detergents and Emulsifiers Annual" (MC
Publishing Crop., Ridgewood, New Jersey, 1981), "Tensid-Taschenbucw", 2nd ed.
(Hanser Verlag, Vienna, 1981) and "Encyclopaedia of Surfactants," (Chemical
Publishing Co., New York, 1981).
The compound of this invention is administered by any route appropriate
to the condition to be treated, such as oral, rectal, nasal, topical
(including ocular,
buccal and sublingual), vaginal and parenteral (including subcutaneous,
1 o intramuscular, intravenous, intradermal, intrathecal and epidural). The
preferred
route of administration may vary with for example the condition of the
recipient,
but is generally oral.
Formulations of the compound of this invention for oral administration
usually are presented as discrete units such as capsules, cachets or tablets
each
containing a predetermined amount of the active ingredient; as a powder or
granular form; as a solution or suspension in an aqueous liquid or a non-
aqueous
liquid; or as an oil-in-water liquid emulsion or a water-in-oil liquid
emulsion. The
compound of this invention optionally is presented as a bolus, electuary or
paste.
A tablet is made by compression or molding, optionally with one or more
accessory ingredients. Compressed tablets are prepared by compressing in a
suitable machine the compound of the invention in a free-flowing form such as
a
powder or granules, optionally mixed with a binder, lubricant, inert diluent,
preservative, surface active and/or dispersing agent. Molded tablets typically
are
made by molding in a suitable machine a mixture of the powdered compound
moistened with an inert liquid diluent. The tablets may optionally be coated
or
scored and may be formulated so as to provide slow or controlled release of
the
active ingredient therein.
- 11 -
CA 02592388 2007-06-19
WO 2006/069193 PCT/US2005/046477
For infections of the eye or other external tissues e.g. mouth and skin, the
formulations are optionally applied as a topical ointment or cream containing
the
active ingredient(s) in an amount of, for example, 0.075 to 20% w/w (including
active ingredient(s) in a range between 0.1% and 20% in increments of 0.1% w/w
such as 0.6% w/w, 0.7% w/w, etc), preferably 0.2 to 15% w/w and most
preferably 0.5 to 10% w/w. When formulated in an ointment, the compound is
employed with a paraffinic or a water-miscible ointment base. Alternatively,
the
compound is formulated in a cream with an oil-in-water cream base. If desired,
the aqueous phase of the cream base may include, for example, at least 30% w/w
of a polyhydric alcohol, i.e. an alcohol having two or more hydroxyl groups
such
as propylene glycol, butane 1,3-diol, mannitol, sorbitol, glycerol and
polyethylene
glycol (including PEG400) and mixtures thereof. The topical formulations may
desirably include a compound which enhances absorption or penetration of the
active ingredient through the skin or other affected areas. Examples of such
dermal penetration enhancers include dimethylsulfoxide and related analogs.
The oily phase of the emulsions of this invention is constituted from known
ingredients in a known manner. While this phase may comprise merely an
emulsifier (otherwise known as an emulgent), it desirably comprises a mixture
of
at least one emulsifier with a fat or an oil or with both a fat and an oil.
Optionally,
a hydrophilic emulsifier is included together with a lipophilic emulsifier
which
acts as a stabilizer. It is also preferred to include both an oil and a fat.
Together,
the emulsifier(s) with or without stabilizer(s) make up the so-called
emulsifying
wax, and the wax together with the oil and fat make up the so-called
emulsifying
ointment base which forms the oily dispersed phase of the cream formulations.
The choice of suitable oils or fats for the formulation is based on achieving
the desired cosmetic properties. Thus the cream should optionally be a non-
greasy, non-staining and washable product with suitable consistency to avoid
leakage from tubes or other containers. Straight or branched chain, mono- or
12 -
CA 02592388 2007-06-19
WO 2006/069193 PCT/US2005/046477
dibasic alkyl esters such as di-isoadipate, isocetyl stearate, propylene
glycol
diester of coconut fatty acids, isopropyl myristate, decyl oleate, isopropyl
palmitate, butyl stearate, 2-ethylhexyl palmitate or a blend of branched chain
esters known as Crodamol CAP may be used, the last three being preferred
esters.
These may be used alone or in combination depending on the properties
required.
Alternatively, high melting point lipids such as white soft paraffin and/or
liquid
paraffin or other mineral oils can be used.
Formulations suitable for topical administration to the eye also include eye
1o drops wherein the active ingredient is dissolved or suspended in a suitable
carrier, especially an aqueous solvent for the active ingredient. The active
ingredient is optionally present in such formulations in a concentration of
0.5 to
20%, advantageously 0.5 to 10% particularly about 1.5% w/w.
Formulations suitable for topical administration in the mouth include
lozenges comprising the active ingredient in a flavored basis, usually sucrose
and
acacia or tragacanth; pastilles comprising the active ingredient in an inert
basis
such as gelatin and glycerin, or sucrose and acacia; and mouthwashes
comprising
the active ingredient in a suitable liquid carrier.
Formulations for rectal administration may be presented as a suppository
with a suitable base comprising for example cocoa butter or a salicylate.
Formulations suitable for nasal administration wherein the carrier is a solid
include a coarse powder having a particle size for example in the range 20 to
500
microns (including particle sizes in a range between 20 and 500 microns in
increments of 5 microns such as 30 microns, 35 microns, etc), which is
administered by aerosol or powder inhalers, of which numerous examples are
available. Suitable formulations wherein the carrier is a liquid, for
administration
as for example a nasal spray or as nasal drops, include aqueous or oily
solutions
of the active ingredient.
- 13 -
CA 02592388 2007-06-19
WO 2006/069193 PCT/US2005/046477
Formulations suitable for vaginal administration may be presented as
pessaries, tampons, creams, gels, pastes, foams or spray formulations
containing
in addition to the active ingredient such carriers as are known in the art to
be
appropriate.
Formulations suitable for parenteral administration include aqueous and
non-aqueous sterile injection solutions which may contain anti-oxidants,
buffers,
bacteriostats and solutes which render the formulation isotonic with the blood
of
the intended recipient; and aqueous and non-aqueous sterile suspensions which
may include suspending agents and thickening agents. The formulations are
presented in unit-dose or multi-dose containers, for example sealed ampoules
and
vials, and may be stored in a freeze-dried (lyophilized) condition requiring
only
the addition of the sterile liquid carrier, for example water for injections,
immediately prior to use. Extemporaneous injection solutions and suspensions
may be prepared from sterile powders, granules and tablets of the kind
previously described.
The compound of this invention is formulated into controlled release
compositions in which the release of the compound is controlled and regulated
to
allow less frequency dosing or to improve the pharmacokinetic or toxicity
profile
of the invention compound. Controlled release compositions are prepared in
accord with known methods, many of which involve formulating the active
compound with one or more polymer carriers such a polyester, polyamino acid,
polyvinyl pyrrolidone, ethylene-vinyl acetate copolymer, methylcellulose,
carboxymethylcellulose and/or protamine sulfate. The rate of drug release and
duration of action optionally is controlled by incorporating the active
ingredient
into particles, e.g. microcapsules, of a polymeric substance such as
hydrogels,
polylactic acid, hydroxymethylcellulose, polymethyl methacrylate and the other
above-described polymers. Also suitable are colloid drug delivery systems such
as
- 14 -
CA 02592388 2007-06-19
WO 2006/069193 PCT/US2005/046477
liposomes, microspheres, microemulsions, nanoparticles, nanocapsules and so
on.
Depending on the route of administration, the pharmaceutical composition,
e.g.,
tablets, may require protective coatings.
EXAMPLE 1
5-((3-(2, 4-trifluoromethyphenyl)isoxazol-5-yl)methyl)-2-(2-fluorophenyl)-5H-
imidazo [4,5-c]pyridine
2,4-(bis-trifluoromethyl)benzaldoxime
1o To aromatic aldehyde (0.021 mol) suspended in EtOH/H2O (1:2, 230 mL, 0.09
M)
was added hydroxylamine hydrochloride (1.58 g, 0.023 mol) and cooled to 4 C.
To
this solution was added aqueous NaOH 50% w/w (4.13 mL, 0.052 mol) dropwise.
After stirring for 1.5 h at room temperature, the reaction mixture was
acidified
with 2N aqueous HCI and extracted with CH,C12 (3 x 50 mL). The organic
solution was washed with saturated aqueous NaCl and dried over sodium sulfate.
Removal of solvent gave crude oxime (5.3 g, quant.) that was used directly in
the
next step.
3-(2 4-(bis-trifluoromethyl)phenyl)-5-(chloromethyl) isoxazole
2,4-(bis-trifluoromethyl)benzaldoxime (9.75 g, 0.038 mol) was suspended in
CH2C12 (45 mL, 0.85 M) and cooled to 4 C. Propargyl chloride (2.72 mL, 0.038
mol) was added to the reaction solution followed by dropwise addition of NaOCI
(10-13 % free chlorine, 37.6 mL, 0.061 mol). The reaction mixture was stirred
at
4 C for 15 min then heated to reflux for 3 h. After cooling to room
temperature,
the reaction was partitioned between CH2C12 and H2O. The organic layer was
separated, washed with saturated aqueous NaCl, and dried over sodium sulfate.
After removal of solvent, the crude product chloromethylisoxazole was purified
by column chromatography on silica (10% CHZC12/hexanes)(6.5 g,0.020 mol).
5-((3-(2 4-trifluoromethyphenyl)isoxazol-5-yl)methyl)-2-(2-fluorophenyl)-5H-
imidazo [4,5-clpyridine
- 15 -
CA 02592388 2007-06-19
WO 2006/069193 PCT/US2005/046477
To imidazopyridine (14.28 g, 0.067 mol) suspended in DMF (40 mL) was added
aqueous NaOH 10% w/w (32.2 mL, 0.080 mol) dropwise followed by addition of
the chloromethyl isoxazole from the previous step (26.3 g, 0.080 mol) in DMF
(16
mL). After stirring for 12 h at room temperature, solvents were evaporated to
give crude product as a tan solid. The crude solid was triturated with H20
(7x)
and crystallized (2x) from MeOH/H20 (2:1) to provide pure title product.
NMR; 300Mhz D6MSO
Chemical shift, multiplicity, # of protons:
6.1,s,2
7.0,s,1
7.3, t, 2
7.4-7.5, m, 1
7.8-7.9, d, 1
7.9-8.0, d, 1
8.2-8.4, m, 4
9.2,s,1
16 -