Note: Descriptions are shown in the official language in which they were submitted.
CA 02599872 2007-08-30
SPECIFICATION
Novel Salts of Quinuclidine Derivative
Technical Field
The present invention relates to novel acid addition salts of
(-)-(3R)-quinuclidin-3-yl
(1 R)-1-phenyl-1,2,3,4-tetrahydroisoquinoline-2-carboxylate
(hereinafter referred to as Compound A) which are useful as drugs,
particularly as muscarine M3 receptor antagonists.
Background Art
It was made public that Compound A having the chemical formula
shown as below, since it displays an affinity and selectivity for muscarinic
M3 receptors, is useful as an M3 receptor antagonist in prophylaxis or
treatment of various M3 receptor-mediated diseases, particularly,
urologic diseases such as urinary incontinence or pollakisuria in neurotic
pollakisuria, neurogenic bladder, nocturnal enuresis, unstable bladder,
bladder spasm, chronic cystitis, etc., respiratory diseases such as
chronic obstructive pulmonary disease, chronic bronchitis, asthma and
rhinitis, or digestive tract diseases such as irritable bowel syndrome also
referred to as spastic colitis or diverticulitis (Patent document 1).
1
CA 02599872 2007-08-30
c z,, c N O.
O 0
N
Compound A
For the acid addition salt of Compound A, the above patent
document 1 discloses only one salt, the hydrochloride salt of Compound
A in Example 10, and no particular example is known regarding other
acid addition salts except the hydrochloride salt described in the above
patent document 1.
[Patent document 1 ] JP 3014457 B
Disclosure of Invention
The Compound A hydrochloride, only one known acid addition
salt of Compound A, can be obtained as a crystalline anhydride, but it
has been found that the hydrochloride is so hygroscopic as to deliquesce
in a conventional environment kept at a relative humidity of 70% (room
temperature) and accompanied by increase of impurities during
long-term storage.
In order to supply a safer pharmaceutical preparation or its drug
substance, accordingly, it has been desired that a lesser hygroscopic salt
of Compound A, particularly the salt highly stable to humidity, other
than the hydrochloride could be discovered.
The present inventors have investigated a variety of acid addition
salts of Compound A and found that particular acid addition salts of
Compound A are less hygroscopic and have higher stability to humidity
2
CA 02599872 2007-08-30
than the conventional hydrochloride salt. Thus, the invention was
completed. According to the invention, there is provided acid addition
salt of Compound A with an acid selected from the group S consisting of
(-)-(2S,3S)-tartaric acid, (+)-(2S,3S)-di-O-benzoyltartaric acid,
(+)-(2S,3S)-di-O-(4-methylbenzoyl)tartaric acid, (-)-L-phenylalanine,
benzenesulfonic acid, cyclohexanesulfamic acid, hydrobromic acid,
naphthalene-2-sulfonic acid, sebacic acid, (+)-camphor-l0-sulfonic
acid, p-toluenesulfonic acid, ethanesulfonic acid, methanesulfonic acid
and methyl phosphate.
Concretely, the invention provides an acid addition salt of
(-)-(3R)-quinuclidin-3-yl
(1 R)-1 -phenyl-1,2,3,4-tetrahydroisoquinoline-2-carboxylate, i.e.,
Compound A, selected from (-)-(3R)-quinuclidin-3-yl
(1 R)-1-phenyl-1,2,3,4-tetrahydroisoquinoline-2-carboxylate
(-)-(2S,3S)-tartrate, (-)-(3R)-quinuclidin-3-yl
(1 R)-1-phenyl-1,2,3,4-tetrahydroisoquinoline-2-carboxylate
(+)-(2S,3S)-di-O-benzoyltartrate, (-)-(3R)-quinuclidin-3-yl
(1 R)-1 -phenyl-1,2,3,4-tetrahydroisoquinoline-2-carboxylate
(+)-(2S,3S)-di-O-(4-methylbenzoyl)tartrate, (-)-(3R)-quinuclidin-3-yl
(1 R)-1-phenyl-1,2,3,4-tetrahydroisoquinoline-2-carboxylate
(-)-L-phenylaianinate, (-)-(3R)-quinuclidin-3-yl
(1 R)-1 -phenyl-1,2,3,4-tetrahydroisoquinoline-2-carboxylate
benzenesulfonate, (-)-(3R)-quinuclidin-3-yl
(1 R)-1-phenyl-1,2,3,4-tetrahydroisoquinoline-2-carboxylate
cyclohexanesulfamate, (-)-(3R)-quinuclidin-3-yl
3
CA 02599872 2007-08-30
(1 R)-1 -phenyl-1,2,3,4-tetrahydroisoquinoline-2-carboxylate
hydrobromide, (-)-(3R)-quinuclidin-3-yl
(1 R)-1-phenyl-1,2,3,4-tetrahydroisoquinoline-2-carboxylate
naphthalene-2-sulfonate, (-)-(3R)-quinuclidin-3-yl
(1 R)-.1-phenyl-1,2,3,4-tetrahydroisoquinoline-2-carboxylate sebacate,
(-)-(3R)-quinuclidin-3-yl
(1 R)-1-phenyl-1 ,2,3,4-tetrahydroisoquinoline-2-carboxylate
(+)-camphor-10-sulfonate, (-)-(3R)-quinuclidin-3-yl
(1 R)-1-phenyl-1,2,3,4-tetrahydroisoquinoline-2-carboxylate
p-toluenesulfonate, , (-)-(3R)-quinuclidin-3-yl
(1 R)-1-phenyl-1,2,3,4-tetrahydroisoquinoline-2-carboxylate
ethanesulfonate, (-)-(3R)-quinuclidin-3-yl
(1 R)-1-phenyl-1,2,3,4-tetrahydroisoquinoline-2-carboxylate
methanesulfonate, and (-)-(3R)-quinuclidin-3-yl
(1 R)-1-phenyl-1,2,3,4-tetrahydroisoquinoline-2-carboxylate methyl
phosphate.
Among these salts, the preferred one is (-)-(3R)-quinuclidin-3-yl
(1 R)-1-phenyl-1,2,3,4-tetrahydroisoquinoline-2-carboxylate
(-)-(2S,3S)-tartrate; in another aspect, the preferred one is
(-)-(3R)-quinuclidin-3-yI
(1 R)-1-phenyl-1 ,2,3,4-tetrahydroisoquinoline-2-carboxylate
(+)-(2S,3S)-di-O-benzoyltartrate; in another aspect, the preferred one
is (-)-(3R)-quinuclidin-3-yl
(1 R)-1-phenyl-1,2,3,4-tetrahydroisoquinoline-2-carboxylate
(+)-(2S,3S)-di-O-(4-methylbenzoyl)tartrate; in another aspect, the
4
CA 02599872 2007-08-30
preferred one is (-)-(3R)-quinuclidin-3-yl
(1 R)-1-phenyl-1,2,3,4-tetrahydroisoquinoline-2-carboxylate
(-)-L-phenylalaninate; in another aspect, the preferred one is
(-)-(3R)-quinuclidin-3-yl
(1 R)-1-phenyl-1,2,3,4-tetrahydroisoquinoline-2-carboxylate
benzenesulfonate; in another aspect, the preferred one is
(-)-(3 R)-quinuclidin-3-yi
(1 R)-1-phenyl-1 ,2,3,4-tetrahydroisoquinoline-2-carboxylate
cyclohexanesulfamate; in another aspect, the preferred one is
(-)-(3R)-quinuclidin-3-yl
(1 R)-1-phenyl-1,2,3,4-tetrahydroisoquinoline-2-carboxylate
hydrobromide; in another aspect, the preferred one is
(-)-(3R)-quinuclidin-3-yl
(1 R)-1-phenyl-1,2,3,4-tetrahydroisoquinoline-2-carboxylate
naphthalene-2-sulfonate; in another aspect, the preferred one is
(-)-(3R)-quinuclidin-3-yi
(1 R)-1-phenyl-1 ,2,3,4-tetrahydroisoquinoline-2-carboxylate sebacate;
in another aspect, the preferred one is (-)-(3R)-quinuclidin-3-yl
(1 R)-1-phenyl-1,2,3,4-tetrahydroisoquinoline-2-carboxylate
(+)-camphor-1 0-sulfonate; in another aspect, the preferred one is
(-)-(3R)-quinuclidin-3-yl
(1 R)-1-phenyl-1,2,3,4-tetrahydroisoquinoline-2-carboxylate
p-toluenesulfonate; in another aspect, the preferred one is
(-)-(3R)-quinuclidin-3-yl
(1 R)-1-phenyl-1,2,3,4-tetrahydroisoquinoline-2-carboxylate
5
CA 02599872 2007-08-30
ethanesulfonate; in another aspect, the preferred one is
(-)-(3R)-quinuclidin-3-yI
(1 R)-1 -phenyl-1,2,3,4-tetrahydroisoquinoline-2-carboxylate
methanesulfonate; and in another aspect, the preferred one is
(-)-(3R)-quinuclidin-3-yI
(1 R)-1 -phenyl-1,2,3,4-tetrahydroisoquinoline-2-carboxylate methyl
phosphate.
Indeed, cyclohexanesulfamic acid is also called
cyclohexylsulfamic acid, sebacic acid is also called decandicarboxylic
acid, (+)-camphor-l0-sulfonic acid is also called
(+)-[(1 S,4R)-7,7-dimethyl-2-oxobicyclo[2.2.1 ]heptan-1-yl]methansulf
onic acid, and p-toluenesulfonic acid is also called
4-methylbenzenesulfonic acid.
In addition, according to the invention, a pharmaceutical
composition comprising as an active ingredient one or more acid
addition salts of Compound A, i.e., (-)-(3R)-quinuclidin-3-yl
(1 R)-1 -phenyl-1,2,3,4-tetrahydroisoquinoline-2-carboxylate,
particularly being a muscarine M3 receptor antagonist, is provided.
In the acid addition salts of the invention of Compound A with an
acid selected from the above-mentioned group S, the hygroscopicity is
improved and the stability to humidity is greatly enhanced in comparison
with the known Compound A hydrochloride. Thus, the salts of the
invention are very useful as drugs or their drug substances.
In particular, as commonly known, it is known that in
6
CA 02599872 2007-08-30
hygroscopically improved drugs or their drug substances, the problems
on storage at a humidity in storage conditions and on quality control are
reduced, and additionally the problem on weight variations of the active
ingredient in the pharmaceutical preparations during production of solid
preparations such as tablets or capsules is also reduced. That is, the
acid addition salts of Compound A of the invention are expected to show
stable shelf life and easiness of quality control since they have improved
hygroscopicity; thus, the salts may be considered to be easily handling
compounds in pharmaceutical preparation and contribute to provide
much better pharmaceutical preparations with high quality.
Brief Description of Drawings
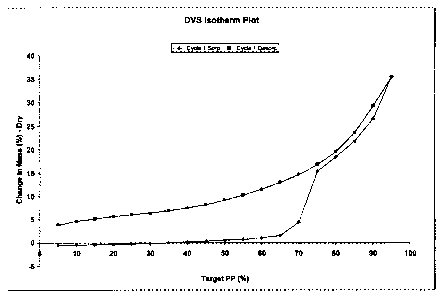
Fig. 1 shows an isothermal curve of water absorption and
desorption in the known Compound A hydrochloride.
Fig. 2 shows an isothermal curve of water absorption and
desorption in the compound of Example 1.
Fig. 3 shows an isothermal curve of water absorption and
desorption in the compound of Example 2.
Fig. 4 shows an isothermal curve of water absorption and
desorption in the compound of Example 3.
Fig. 5 shows an isothermal curve of water absorption and
desorption in the compound of Example 4.
Fig. 6 shows an isothermal curve of water absorption and
desorption in the compound of Example 5.
Fig. 7 shows an isothermal curve of water absorption and
7
CA 02599872 2007-08-30
desorption in the compound of Example 5-1.
Fig. 8 shows an isothermal curve of water absorption and
desorption in the compound of Example 6.
Fig. 9 shows an isothermal curve of water absorption and
desorption in the compound of Example 7.
Fig. 10 shows an isothermal curve of water absorption and
desorption in the compound of Example 8.
Fig. 11 shows an isothermal curve of water absorption and
desorption in the compound of Example 9.
Fig. 12 shows an isothermal curve of water absorption and
desorption in the compound of Example 10.
Fig. 13 shows an isothermal curve of water absorption and
desorption in the compound of Example 11.
Fig. 14 shows an isothermal curve of water absorption and
1 5 desorption in the compound of Example 12.
Fig. 15 shows an isothermal curve of water absorption and
desorption in the compound of Example 1 3.
Fig. 16 shows an isothermal curve of water absorption and
desorption in the compound of Example 14.
Fig. 17 shows a powder X-ray diffraction pattern of the
compound of Example 1.
Fig. 18 shows a powder X-ray diffraction pattern of the
compound of Example 2.
Fig. 19 shows a powder X-ray diffraction pattern of the
compound of Example 3.
8
CA 02599872 2007-08-30
Fig. 20 shows a powder X-ray diffraction pattern of the
compound of Example 4.
Fig. 21 shows a powder X-ray diffraction pattern of the
compound of Example S.
Fig. 22 shows a powder X-ray diffraction pattern of the
compound of Example 5-1.
Fig. 23 shows a powder X-ray diffraction pattern of the
compound of Example 6.
Fig. 24 shows a powder X-ray diffraction pattern of the
compound of Example 7.
Fig. 25 shows a powder X-ray diffraction pattern of the
compound of Example 8.
Fig. 26 shows a powder X-ray diffraction pattern of the
compound of Example 9.
Fig. 27 shows a powder X-ray diffraction pattern of the
compound of Example 10.
Fig. 28 shows a powder X-ray diffraction pattern of the
compound of Example 11.
Fig. 29 shows a powder X-ray diffraction pattern of the
compound of Example 12.
Fig. 30 shows a powder X-ray diffraction pattern of the
compound of Example 13.
Fig. 31 shows a powder X-ray diffraction pattern of the
compound of Example 14.
9
CA 02599872 2007-08-30
Best Mode for Carrying Out the Invention
The acid addition salts of Compound A of the invention show a
stability to a sufficient degree for use in pharmaceutical preparations or
their drug substaces, have no hygroscopicity that affects the use as
drugs or their drug substances, and are expected to be chemically stable
or stable during storage. Therefore, all of the acid addition salts of the
invention are preferred as drugs or their drug substances, particularly as
drug substances for solid preparations.
(Manufacturing Method)
The acid addition salts of Compound A of the invention can be
produced according to the following manufacturing method.
That is, a solvent is added to a free base of Compound A at a ratio
of 1 mL/g - 100 mL/g to Compound A, and then an acid used in
formation of the salt or a solution containing the acid is added thereto in
the range of 0.5 to 2.0 equivalents to Compound A at room temperature.
When an insoluble material exists, the same solvent or a different solvent
is added, or the mixture is heated for dissolving the insoluble material to
give a solution, which is left with stirring or on standing at room
temperature or under cooling. When an insoluble material is still
remaining in spite of addition of solvent or heating, the mixture may be
filtered to remove it before crystallization of the salt. Thus, the
resulting crystals are collected by filtration and washed with a suitable
solvent to give the objective acid addition salt of Compound A. In this
operation for cooling to room temperature, it is sometimes effective to
cool the mixture more gradually or rapidly rather than merely standing
CA 02599872 2007-08-30
for cooling, in order to obtain better crystals.
The solvent/solvents which can be used in the above-mentioned
salt formation include water, acetic acid, acetone, anisole, 1 -butanol,
2-butanol, n-butyl acetate, t-butyl methyl ether, cumene,
dimethylsulfoxide, ethanol (EtOH), ethyl acetate (EtOAc), diethyl ether,
ethyl formate, formic acid, heptane, isobutyl acetate, isopropyl acetate
(iPrOAc), methyl acetate, 3-methyl-l-butanol, methyl ethyl ketone
(2-butanone), methyl isobutyl ketone, 2-methyl-l-propanol, pentane,
1-pentanol, 1-propanol, 2-propanol (2-PrOH), propyl acetate,
acetonitrile, chlorobenzene, chloroform, cyclohexane,
1,2-dichloroethene, dichloromethane, 1,2-dimethoxyethane, DMF, DMA,
1,4-dioxane, 2-ethoxyethanol, ethylene glycol, formamide, hexane,
methanol, 2-methoxyethanol, methyl butyl ketone, methylcyclohexane,
N-methylpyrrolidone, nitromethane, pyridine, sulfolane, THF, tetraline,
toluene, 1,1,2-trichloroethene, xylene, benzene, carbon tetrachloride,
1,2-dichloroethane, 1,1-dichloroethene, 1,1,1-trichloroethane,
diisopropyl ether, and the like.
Thus resulting crystals may be recrystallized in a conventional
manner as employed by a person skilled in the art to give much more
pure crystals.
A free base of Compound A which is a starting material in the
above-mentioned manufacturing method may be produced according to
the method as described in the above Patent document 1, i.e., European
Patent No. 0 801 067, or its corresponding or similar method, or a
method obviously employed by a person skilled in the art.
11
CA 02599872 2007-08-30
The acid addition salts of Compound A of the invention can be
used as drug substances in production of pharmaceutical preparations
by combining one or more of the acid addition salts of Compound A of
the invention with conventional pharmaceutical carriers or diluents
employed in this field. The pharmaceutical preparations may be
produced by a method usually employed in this field.
The pharmaceutical preparations containing the acid addition
salts of Compound A of the invention include orally administrable
preparations such as tablets, pills, capsules, granules, powders, liquids
and solutions, and the like; or parenteral preparations such as
intraarticular, intravenous, or intramuscular injections, suppositories,
percutaneous liquid preparations, ointments, transdermal stickers,
transmucosal liquid preparations, transmucosal stickers, inhalations,
and the like. Particularly, the oral preparations containing as a drug
substance an acid addition salt of Compound A, such as tablets, pills,
capsules, granules and powders, are advantageous as stable solid
preparations.
In the solid compositions for use in oral administration, one or
more of the active ingredients may be mixed with at least one inert
diluent, for example, lactose, mannitol, glucose, hydroxypropylcellulose,
fine crystal cellulose, starch, polyvinylpyrrolidone, magnesium
metasilicate aluminate, and the like. The compositions may contain
additives other than diluents in a conventional manner, for example,
lubricants such as magnesium stearate, disintegrating agents such as
fibrous calcium glycolate, stabilizers, or solubilizing agents. The
12
CA 02599872 2007-08-30
tablets or pills if required may be coated with sugar-coating or a gastric
or enteric coating film, such as sucrose, gelatin, hydroxypropylcellulose,
hydroxypropylmethylcellulose, and the like.
The liquid compositions for oral administration include
pharmaceutically acceptable emulsions, solutions, suspensions, syrups,
elixirs, and the like, which contain conventionally used diluents, for
example, purified water or ethanol. In addition to inert diluents, the
compositions may further contain auxiliary agents such as wetting agent
or suspending agent, sweetener, flavor, perfume, or preservative.
The injections for parenteral administration include sterile
aqueous or non-aqueous solutions, suspensions and emulsions. The
aqueous solutions and suspensions include, for example, distilled water
for injection and physiological saline. The non-aqueous solutions and
suspensions include, for example, propylene glycol, polyethylene glycol,
vegetable oils such as olive oil, alcohols such as EtOH, polysorbate 80,
and the like. Such a composition may further contain a preservative,
wetting agent, emulsifying agent, dispersant, stabilizer, solubilizing
agent, and the like. These may be sterilized for example by filtration
through a bacterium-impermeable filter, blending with a bactericide, or
irradiation. Alternatively, these may be made into a sterile solid
composition, which is dissolved in sterile water or sterile solvent for
injection just before use.
Since the pharmaceutical compositions of the present invention
comprise one or more of the acid addition salts of Compound A of the
invention, which are muscarinic M3 receptor antagonists, as the active
13
CA 02599872 2007-08-30
ingredient, the pharmaceutical compositions may be used for the
therapy or prophylaxis of a variety of diseases to which muscarinic M3
receptors contribute or may be 3mployed in diagnostic procedures.
That is, the pharmaceutical compositions of the invention, specifically,
are useful as regimen in the treatment of, for example, urinary urgency,
frequency/pollakisuria, urinary incontinence, nocturnal enuresis or
hyperreflexic bladder caused by urinary diseases, such as overactive
bladder, unstable bladder, neurogenic bladder, cystitis, etc.; in the
therapy or prophylaxis of bladder spasm caused by surgery or catheters;
in the treatment of respiratory diseases such as chronic obstructive
pulmonary disease, chronic bronchitis, asthma and rhinitis; in the
treatment of digestive diseases, such as irritable bowel syndrome; as the
relaxant used for examination of the digestive tract; as an agent to
ameliorate myopia or to promote mydriasis; or as an agent to treat or
prevent hyperhidrosis.
Examples
The invention will be explained specifically by the following
examples which are not intended as a limitation thereof and are not
intended to restrict the scope of the invention.
The thermal analysis and powder X-ray diffractometry were
performed according to the following methods.
(1) Thermal analysis
(Differential Scanning Calorimetry: DSC)
A sample (about 3 mg) was placed in a purpose-made aluminum
14
CA 02599872 2007-08-30
pan. The change of heat generated between the sample and a reference
(empty aluminum pan) was continuously measured and recorded under a
nitrogen atmosphere (50 mI/min) in the temperature range of room
temperature to 300oC at a rate of 10oC/min of ascending programmed
temperature. The apparatus including data processing was operated
according to the method and procedure directed in each device.
(Apparatus: Hi-Res DSC 2910, made by TA Instrument)
(Thermogravimetric apparatus: TGA)
A sample (about 3 mg) was placed in a purpose-made platinum
pan, and the sample weight was continuously measured and recorded
under a nitrogen atmosphere (100 mI/min) in the temperature range of
room temperature to 300oC at a rate of 10OC/min of ascending
programmed temperature. The apparatus including data processing
was operated according to the method and procedure directed in each
device. (Apparatus: Hi-Res TGA 2950, made by TA Instrument)
(2) Powder X-Ray Diffractometry
A sample (about 10 mg) was placed in a purpose-made sample
holder (5 mm wide, 18 mm long, 0.2 mm height), and the X-ray
diffraction pattern was carried out and the data recorded according to
the following condition. The apparatus including data processing was
operated according to the method and procedure directed in each device.
(Apparatus: MXP1 8TAHF22, made by MAC Science (Bruker at present))
(Condition)
X-ray radiation source: Cu; wavelength: 1.54056 angstrom; range of
measurement: 3.00 - 40.000; sampling interval: 0.020; scanning rate:
CA 02599872 2007-08-30
3.000/min; tube voltage: 40 kV; tube current: 200 mA; divergence slit:
1.000; scattering slit: 1.000; receiving slit: 0.1 5 mm
The values obtained from each spectrum sometimes in some
degree depend on the direction of crystal growth, particle size, and the
condition of measurement. These values should not be assessed
strictly, accordingly.
Reference Example 1: Preparation of Compound A in a free state
The title compound was prepared according to the method as
described in Japanese Patent No. 3014457.
Reference Example 2: Preparation of Compound A hydrochloride as a
reference compound
The title compound was prepared according to the method as
described in,Japanese Patent No. 3014457.
Example 1
Preparation of Compound A (-)-(2S,3S)-tartrate
To a solution of Compound A free base (26.0 g) in 260 mL of
EtOH was added 10.8 g of (-)-(2S,3S)-tartaric acid, and the mixture was
heated to be dissolved. After cooling to room temperature, the mixture
was stirred for 20 hours. The resulting crystals were collected by
filtration to give 30.6 g of white crystals. To a suspension of 1.00 g of
the crystals in 10 mL of EtOH was added 0.4 mL of water, and the mixture
was heated to give a solution. After cooling to room temperature, the
16
CA 02599872 2007-08-30
mixture was stirred for 6 hours. The resulting crystals were collected by
filtration to give 871 mg of the title compound as white crystals.
1 H-NMR(DMSO-d6, 2 5.9 C) :
1.40-1.98(4H,m),2.00-2.25(1 H,m),2.70-3.20(7H,m),3.33-3.53(2H,m),3
.83-3.94(1 H,m),3.99(2H,s),4.85(1 H,brs),6.25(1 H,brs),7.08-7.37(9H,m).
Peak top temperature of endothermia in DSC: 194 C
Fig. 17 shows a powder X-ray diffraction pattern of the
compound in Example 1.
Example 2
Preparation of Compound A (+)-(2S,3S)-di-O-benzoyltartrate
To a solution of Compound A free base (180 mg) in 1.8 mL of
EtOH was added 180 mg of (+)-(2S,3S)-di-O-benzoyltartaric acid, and
the mixture was stirred at room temperature for 12 hours. The
resulting crystals were collected by filtration, washed with EtOH and
dried under reduced pressure to give 284 mg of the title compound as
white crystals.
I H-NMR(DMSO-d6: 70 C) :
1 .52-1 .90(4H,m),2.16(1 H,brs),2.76-3.1 6(7H,m),3.37-3.56(2H,m),3.89(
1 H,dt,J=1 3.2,5.4Hz),4.85-4.92(1 H,m),5.68(2H,s),6.23(1 H,s),7.1 1-7.33(
9H,m),7.43-7.55(4H,m),7.57-7.63(2H,m),7.90-7.96(4H,m).
Peak top temperature of endothermia in DSC: 159 C
Fig. 18 shows a powder X-ray diffraction pattern of the
compound in Example 2.
17
CA 02599872 2007-08-30
Example 3
Preparation of Compound A (+)-(2S,3S)-di-O-(4methylbenzoyl)tartrate
To a solution of Compound A free base (1.00 g) in 20 mL of EtOH
was added 1.12 g of (+)-(2S,3S)-di-O-(4-methylbenzoyl)tartaric acid,
and the mixture was stirred at room temperature for 22 hours. The
resulting precipitates were collected by filtration to give 1 .60 g of the
title compound as white crystals.
I H-NMR(DMSO-d6: 70 0C) :
1.53-1.88(4H,m),2.1 5(1 H,brs),2.32-2.38(6H,m),2.76-3.16(7H,m),3.42(
1 H,ddd,.)=13.6,8.8,5.2Hz),3.50(1 H,dd,.J=14.4,8.8Hz),3.90(1 H,dt,.J=13.2
,5.2Hz),4.88(1 H,dtj=8.8,4.4Hz),5.64(2H,s),6.23(1 H,s),7.1 1-7.34(1 3H,
m),7.77-7.84(4H,m).
Peak top temperature of endothermia in DSC: 1600C
Fig. 19 shows a powder X-ray diffraction pattern of the
compound in Example 3.
Example 4
Preparation of Compound A (-)-L-phenylaianinate
To a solution of Compound A free base (1.13 g) in 11.25 mL of
EtOH were added 515 mg of (-)-L-phenylalanine and 4.5 mL of water,
and the mixture was heated to be dissolved. The mixture was then
stirred at room temperature for 10 hours. The resulting crystals were
collected by filtration, washed with a mixture of water-EtOH and dried
under reduced pressure to give 1.12 g of the title compound as white
crystals.
18
CA 02599872 2007-08-30
~ H-NMR(DMSO-d6: 70.0 C) :
1.20-1.36(1 H,m),l .41-1.70(3H,m),1.86-1.95(1 H,m),2.50-2.95(8H,m),3
.03-3.17(2H,m),3.33-3.47(2H,m),3.84-3.95(1 H,m),4.60-4.71(1 H,m),6.
23(1 H,s),7.1 1-7.33(14H,m).
Peak top temperature of endothermia in DSC: 1 18 C and 235 C
Fig. 20 shows a powder X-ray diffraction pattern of the
compound in Example 4.
Example 5
Preparation of Compound A benzenesulfonate (1)
To a solution of Compound A free base (2.69 g) in 40 mL of EtOAc
was added 1.31 g of benzenesulfonic acid monohydrate was added, and
the mixture was stirred at room temperature for 1 hour. The resulting
precipitates were collected by filtration. To a suspension of the
precipitates in 30 mL of 2-butanone was added 0.35 mL of water. The
resulting mixture was heated to be dissolved. After cooling to room
temperature, the mixture was stirred for 60 hours. The resulting
crystals were collected by filtration to give 2.49 g of the title compound
as white crystals.
1 H-NMR(DMSO-d6: 70 C) :
1.65-1.99(4H,m),2.23(1 H,brs),2.77-2.96(2H,m),3.06-3.32(5H,m),3.44(
1 H,ddd,J=1 3.6,8.4,5.2Hz),3.66(1 H,dd,J=1 3.6,8.4Hz),3.91(1 H,dt,J=12.8
,5.6Hz),4.97(1 H,dt,J=8.4,4.4Hz),6.25(1 H,s),7.1 1-7.35(12H,m),7.59-7.6
4(2H,m),9.39(1 H,brs).
Peak top temperature of endothermia in DSC: 178 C
19
CA 02599872 2007-08-30
Fig. 21 shows a powder X-ray diffraction pattern of the
compound in Example 5.
Example 5-1
Preparation of Compound A benzenesulfonate (2)
To a solution of Compound A free base (7.00 g) in 70 mL of
acetone were added 3.40 g of benzenesulfonic acid monohydrate and 70
mL of tert-butyl methyl ether, and the mixture was stirred using a
mechanical stirrer at room temperature for 9 hours. The resulting
precipitates were collected by filtration to give 8.10 g of the title
compound as white crystals.
The 1H-NMR spectrum of the product was identical with that of
Example 5, but the DSC and the powder X-ray diffraction pattern
suggested that the products of Example 5 and Example 5-1 showed
crystal polymorphism.
Peak top temperature of endothermia in DSC: 170I)C
Fig. 22 shows a powder X-ray diffraction pattern of the
compound in Example 5-1.
Example 6
Preparation of Compound A cyclohexanesulfamate
To a solution of Compound A free base (500 mg) in 5 mL of
2-PrOH was added 494 mg of cyclohexanesulfamic acid, and the mixture
was stirred at room temperature for 13 hours. The resulting
precipitates were collected by filtration to give 550 mg of the title
CA 02599872 2007-08-30
compound as white crystals.
I H-NMR(DMSO-d6: 70 C) :
1.00-1 .35(1 OH,m),1.46-2.08(14H,m),2.23(1 H,brs),2.77-3.30(1 1 H,m),3
.44(1 H,ddd,J=1 3.6,8.8,5.2Hz),3.64(1 H,dd,J=1 3.8,8.8Hz),3.91(1 H,dt,j=
12.8,5.6Hz),4.96(1 H,dtj=8.4,4.4Hz),6.25(1 H,s),7.1 0-7.36(9H,m).
Peak top temperature of endothermia in DSC: 127 C and 1 70 C
Fig. 23 shows a powder X-ray diffraction pattern of the
compound in Example 6.
Example 7
Preparation of Compound A hydrobromide
To a solution of Compound A free base (200 mg) in 1.0 mL of
EtOH was added 95 mg of 47% hydrobromic acid. To the reaction
mixture, 1.1 mL of diisopropyl ether was added with stirring, and the
resulting mixture was stirred at 5 C for 18 hours. The obtaining
precipitates were collected by filtration to give 165 mg of the title
compound as white crystals.
1 H-NMR(DMSO-d6: 70 C) :
1 .65-1.98(4H,m),2.24(1 H,brs),2.77-2.97(2H,m),3.05-3.35(5H,m),3.45(
1 H,ddd,J=13.6,8.8,5.2Hz),3.65(1 H,ddj=13.2,8.4Hz),3.91(1 H,dt,J=12.8
,5.6Hz),4.97(1 H,dtj=8.8,4.4Hz),6.26(1 H,s),7.1 1-7.35(9H,m),9.68(1 H,b
rs).
Elemental Analysis: (calculated for C23H26N202.HBr) C, 62.31, H, 6.14, N,
6.32, Br, 18.02. (found) C, 62.04, H, 6.10, N, 6.09, Br, 1 7.73.
Peak top temperature of endothermia in DSC: 199 C
21
CA 02599872 2007-08-30
Fig. 24 shows a powder X-ray diffraction pattern of the
compound in Example 7.
Example 8
Preparation of Compound A naphthalene-2-sulfonate
To a solution of Compound A free base (100 mg) in 1.0 mL of
EtOH was added 65 mg of naphthalene-2-sulfonic acid hydrate, and the
mixture was stirred for 26 hours. The resulting crystals were collected
by filtration to give 79 mg of the title compound as slightly grey crystals.
1 H-NMR(DMSO-d6: 70 OC) :
1.65-2.00(4H,m),2.24(1 H,brs),2.77-2.97(2H,m),3.05-3.32(5H,m),3.45(
1 H,ddd,J=14.0,9.2,5.2Hz),3.65(1 H,dd,J=14.0,8.8Hz),3.90(1 H,dt,J=12.8
,5.6Hz),4.97(1 H,dtj=8.0,4.4Hz),6.25(1 H,s),7.1 2-7.34(9H,m),7.49(2H,d
t,J=10.4,4.0Hz),7.74(1 H,dd,J=8.4,1.6Hz),7.82(1 H,dj=8.OHz),7.85-7.95
(2H,m),8.14(1 H,s),9.35(1 H,brs).
Peak top temperature of endothermia in DSC: 1 780C
Fig. 25 shows a powder X-ray diffraction pattern of the
compound in Example 8.
Example 9
Preparation of Compound A sebacate
To a solution of Compound A free base (300 mg) in 1.0 mL of
EtOH was added 171 mg of sebacic acid, and the mixture was stirred for
3 hours. The resulting crystals were collected by filtration and washed
with ethanol to give 165 mg of the title compound as white crystals.
22
CA 02599872 2007-08-30
'H-NMR(DMSO-d6: 26.1 OC) :
1.16-2.00(17H,m),2.17(4H,t,J=7.2),2.50-2.97(7H,m),3.02-3.08(1 H,m),
3.28-3.50(1 H,m),3.78-3.98(1 H,m),4.65(1 H,brs),6.24(1 H,brs),7.12-7.26
(l OH,m).
Peak top temperature of endothermia in DSC: 127oC
Fig. 26 shows a powder X-ray diffraction pattern of the
compound in Example 9.
Example 10
Preparation of Compound A(+)-camphor-l0-sulfonate
To a solution of Compound A free base (200 mg) in 2 mL of
acetone was added 138 mg of (+)-camphor-l0-sulfonic acid, and the
mixture was stirred at room temperature for 5 hours. The resulting
precipitates were collected by filtration to give 191 mg of the title
compound as white crystals.
1H-NMR(DMSO-d6: 70OC) :
0.76(3H,s),1 .08(3H,s),1 .20-1.33(2H,m),1 .65-1 .98(7H,m),2.18-2.28(2H,
m),2.37-2.42(1 H,m),2.65-2.97(4H,m),3.05-3.31(5H,m),3.44(1 H,ddd,.J=
1 3.6,8.8,4.8Hz),3.65(1 H,ddj=1 3.6,8.4Hz),3.91(1 H,dt,J=1 3.2,5.6Hz),4.
97(1 H,dt,)=8.8,4.4Hz),6.25(1 H,s),7.1 1-7.35(9H,m),9.44(1 H,brs).
Peak top temperature of endothermia in DSC: 1980C
Fig. 27 shows a powder X-ray diffraction pattern of the
compound in Example 10.
Example 11
23
CA 02599872 2007-08-30
Preparation of Compound A p-toluenesulfonate
To a solution of Compound A free base (200 mg) in a mixture of
1.5 mL of acetone and 0.5 mL of tert-butyl methyl ether was added 105
mg of p-toluenesulfonic acid monohydrate, and the mixture was stirred
at room temperature for 17 hours. The resulting precipitates were
collected by filtration to give 83 mg of the title compound as white
crystals.
1 H-NMR(DMSO-d6:70 C) :
1.65-2.00(4H,m),2.24(1 H,brs),2.28(3H,s),2.76-2.96(2H,m),3.05-3.30(5
H,m),3.44(1 H,ddd,J=1 3.6,8.0,5.OHz),3.65(1 H,dd,J=13.6,8.OHz),3.91(1 H
,dt,J=12.8,5.6Hz),4.97(1 H,dtj=8.4,4.4Hz),6.25(1 H,s),7.09(2H,dj=7.6H
z),7.1 1-7.35(9H,m),7.47-7.52(2H,m),9.38(1 H,brs).
Peak top temperature of endothermia in DSC: 1 50 C
Fig. 28 shows a powder X-ray diffraction pattern of the
compound in Example 11.
Example 12
Preparation of Compound A ethanesulfonate
To a solution of Compound A free base (100 mg) in 1.0 mL of
2-butanone was added 31 mg of ethanesulfonic acid, and the mixture
was stirred at room temperature for 6.5 hours. The resulting
precipitates were collected by filtration to give 95 mg of the title
compound as white crystals.
1 H-NMR(DMSO-d6:70 C) :
1.07(3H,t,J=7.6Hz),1.62-2.10(4H,m),2.24(1 H,brs),2.39(2H,q,J=7.6Hz),2
24
CA 02599872 2007-08-30
.76-2.96(2H,m),3.08-3.32(SH,m),3.44(1 H,dddj=13.6,8.8,4.8Hz),3.65(
1 H,ddj=13.6,8.8Hz),3.91(1 H,dtj=12.4,5.2Hz),4.92-5.02(1 H,m),6.25(1
H,s),7.10-7.35(9H,m),9.51(1 H, brs).
Peak top temperature of endothermia in DSC: 2330C
Fig. 29 shows a powder X-ray diffraction pattern of the
compound in Example 12.
Example 13
Preparation of Compound A methanesulfonate
To a solution of Compound A free base (200 mg) in 1.0 mL of
2-butanone was added a solution of 53 mg of methanesulfonic acid in
1.0 mL of iPrOAc, and the mixture was stirred at room temperature for
0.5 hours. The resulting precipitates were collected by filtration to give
187 mg of the title compound as white crystals.
1 H-NHR(DMSO-d6 : 700C) :
1.62-2.02(4H,m),2.24(1 H,brs),2.30(3H,s),2.76-2.96(2H,m),3.00-3.34(5
H,m),3.44(1 H,ddd,,J=13.6,8.8,4.8Hz),3.65(1 H,dd,j=13.6,8.6Hz),3.91(1 H
,dt,J=1 3.2,5.2Hz),4.90-5.40(1 H,m),6.25(1 H,s),7.08-7.36(9H,m),9.37(1
H,brs).
Peak top temperature of endothermia in DSC: 178 OC
Fig. 30 shows a powder X-ray diffraction pattern of the
compound in Example 13.
Example 14
Preparation of Compound A methyl phosphate
CA 02599872 2007-08-30
To a solution of Compound A free base (200 mg) in 2.0 mL of
EtOAc and 0.5 mL of 2-butanone was added 98 mg of methyl phosphate,
and the mixture was stirred at room temperature for 22 hours. The
resulting precipitates were collected by filtration to give 124 mg of the
title compound as white crystals.
IH-NHR(DMSO-d6: 70 C) :
1 .43-1.54(1 H,m),l .57-1.81(3H,m),2.01-2.10(1 H,m),2.77-2.99(7H,m),3
.29-3.46(2H,m),3.42(3H,d,)=10.8Hz),3.90(1 H,dt,J=13.2,5.6Hz),4.76-4.
84(1 H,m),6.24(1 H,s),7.1 2-7.33(9H,m).
Peak top temperature of endothermia in DSC: 195 C
Fig. 31 shows a powder X-ray diffraction pattern of the
compound in Example 14.
The effect of the acid addition salt of Compound A of the
invention was confirmed by the following Test Examples.
Test Example 1: Evaluation of hygroscopicity
A sample (about 5 mg) was placed in a purpose-made quartz
holder, and the sample weight at the respective humidity was
continuously measured and recorded in the following conditions. The
apparatus including data processing was operated according to the
method and procedure directed in each device. (Apparatus: dynamic
steam adsorption measuring apparatus DVS Advantage, made by SMS)
(Condition)
Measuring temperature: 25 C; drying before measurement: not done;
humidity at the beginning: 5% RH; maximum humidity: 95% RH; humidity
26
CA 02599872 2007-08-30
at the end: 5% RH; step: 5% RH; equilibrium standard: 0.03 wt% in 5 min;
maximum equilibrated time: 180 min; sampling interval: 20 sec; data
recording interval: 1 min
The charts obtained in these tests are shown in Fig. 1 to Fig. 14.
The weight change within the range of measuring condition is shown in
Table 1.
Table 1
Compound Weight Change (%) Compound Weight Change (%)
tested tested
Example 1 <1 Example 8 <4
Example 2 <1 Example 9 <1
Example 3 <1 Example 10 <2
Example 4 <2 Example 11 <6
Example 5 <1 Example 12 <5
Example 5-1 <1 Example 13 <25
Example 6 <3 Example 14 <11
Example 7 <4 Reference >35 (deliquescent)
Example 2
As shown in Fig. 1 and Table 1, Compound A hydrochloride which
is a known compound began to rapidly take up moisture at about 65%
relative humidity and the absorption of moisture was recognized as a
weight gain of 1 5% or more.at 75% or more relative humidity. The
weight change exceeded 35% within the range of measuring condition,
i.e., 5 - 95% relative humidity, and this change was accompanied by
deliquescence. On the other hand, as shown in Fig. 2 to Fig. 16 and
Table 1, the acid addition salts of Compound A of the invention was
confirmed to have improved hydgroscopicity in comparison with the
known Compound A hydrochloride and have much better properties as
drugs or their drug substances.
27
CA 02599872 2007-08-30
Test Example 2: Evaluation of Stability (1)
A sample (about 0.5 mg) was weighed in a glass vial and applied
to a forced degradation test in the following storage conditions.
Condition 1: 1200C - tightly closed - 24 hours
Condition 2: 250C - relative humidity 93% - open - 5 days
Condition 3: 250C - NUV (near ultraviolet ray) irradiated - tightly closed -
5 days
A sample after storage was dissolved in 1 mL of MeOH and used
as a sample solution. The sample solution was diluted 100 times with
MeOH and used as a standard solution. Impurities contained in the
sample solution was quantitatively analyzed using the standard solution.
In this determination, the apparatus including data processing was
operated according to the method and procedure directed in each device.
(Apparatus: LC-MSD 1 100 series, made by Agilent)
The test results are shown in Table 2.
28
CA 02599872 2007-08-30
Table 2
Change from Impurity Amt. before
Impurities before Storage (%)
Storage (%) Condition 1 Condition 2 Condition 3
Example 1 1.5 0.2 -0.1 -0.3
Example 2 0.0 0.2 0.1 0.0
Example 3 0.0 2.3 0.1 0.0
Example 4 1.9 50.8 -0.3 -0.3
Example 5 1.6 0.3 0.1 -0.1
Example 5-1 1.8 -0.1 0.0 -0.2
Example 6 1.4 8.1 0.0 0.1
Example 7 0.5 -0.2 0.2 0.3
Example 8 2.0 1.3 0.2 2.6
Example 9 3.7 -0.3 0.2 -0.8
Example 10 0.9 0.0 0.0 0.0
Example 11 0.5 -0.1 0.1 0.0
Example 12 1.4 -0.2 -0.1 -0.1
Reference
0.1 0.2 3.8 2.0
Example 2
Test Example 3: Evaluation of Stability (2)
A sample (about 5 mg) was weighed in a glass mess-flask of 10
mL and applied to a stress testing in the following storage conditions.
Condition 1: 5 G - shading - tightly closed
Condition 2: 40 C - relative humidity 75% - open
Condition 3: 60 C - shading - tightly closed
Condition 4: 80 C - shading - tightly closed
Condition 5: 25 C - D65 (1000 lux) - tightly closed
Chemical stability: After storage, MeOH was placed in the
mess-flask containing the sample up to the level of the marked line, and
the resulting solution was used as a sample solution. The sample
solution was diluted 100 times with MeOH to prepare a standard solution,
which was used in determination of impurities contained in the sample
29
CA 02599872 2007-08-30
solution. Detection of the impurities was performed by means of UV at
210 nm. The apparatus including data processing was operated
according to the method and procedure directed in each device.
(Apparatus: LC-MSD 1 100 series, made by Agilent)
The test results are shown in Table 3.
Table 3
(%)
Test Condition Term Impurities
Example 1 Reference Example 2
2 weeks 0.00 0.25
Condition 1 1 month 0.00 0.21
2 months 0.00 0.18
2 weeks 0.00 8.72
Condition 2 1 month 0.00 8.19
2 months 0.00 9.20
2 weeks 0.00 0.34
Condition 3 1 month 0.00 0.22
2 months 0.00 0.24
2 weeks 0.00 0.43
Condition 4 1 month 0.00 0.39
2 months 0.00 0.47
2 weeks 0.00 1.73
Condition 5 1 month 0.00 2.38
2 months 0.00 3.68
As shown in Table 2 and Table 3, it was found that impurities
increased during storage in Compound A hydrochloride which was a
known compound. In particular, it became clear that Compound A
hydrochloride is not so stable to humidity (Condition 2 in Test Example 2
and Condition 2 in Test Example 3) and light (Condition 3 in Test
Example 2 and Condition 5 in Test Example 3). On the other hand, as
shown in Table 2 and Table 3, it was confirmed that the acid addition
salts of the invention showed almost no increase of impurities, indicating
CA 02599872 2007-08-30
it being chemically and physically highly stable in comparison with the
known Compound A hydrochloride and have much better properties as
drugs or their drug substances. In this connection, the reason of the
increase of impurities recognized in the compounds of Example 4 and
Example 6 was considered that they have somewhat low melting points
that they melted under Condition 1 of Test Example 2 (stability test to
heat) or the preserved condition was near the melting point.
Test Example 4: Functional affinity estimate for muscarinic M3 receptors
in bladders
According to a method as described by Ikeda et al., (2002,
Naunyn-Schmiedeberg's Archives of Pharmacology, Vol.366, p.97-103),
functional affinity estimates of test compound for muscarinic M3
receptors were determined using changes in intracellular Ca2+. Below,
the method is described in brief.
Smooth muscle cells were isolated from guinea pig bladders from
which the epidermis had been removed, loaded with a calcium-sensitive
fluorescent dye Fura2 and suspended in phenol red-free Hanks' buffer
solution supplemented with 20mM HEPES (pH 7.4) and 0.1% bovine
serum albumin. An aliquot of cell suspension (490 L) was continuously
stirred, kept at 28oC and monitored for the ratio of fluorescence (500
nm) at 340 nm excitation to that at 380 nm excitation. To each aliquot,
5 L of test compound and carbachol solutions were serially added with a
2 minute interval, and the peak increase in the ratio over the level just
before stimulation was used in data analysis. The concentration
31
CA 02599872 2007-08-30
required for 50% stimulation or 50% inhibition, that is, EC5o or IC5o, was
obtained by sigmoidal curve fitting, then IC50values of test compound
were converted into Ki values based on the EC5o value of carbachol using
the Cheng-Prusoff equation.
Table 4 shows the results.
Table 4
Compound Tested IC5o (ng/mL) Ki (nM)
Example 1 2.4 0.75 0.68 0.21
Example 2 3.2 0.97 0.62 0.16
Example 3 3.8 1.2 0.69 0.19
Example 4 2.5 0.044 0.66 0.046
Example 5 1.8 0.79 0.47 0.12
Example 6 4.3 1.2 0.84 0.10
Example 7 1.8 0.49 0.66 0.28
Example 8 2.3 0.87 0.55 0.12
Example 9 2.2 0.33 0.56 0.048
Example 10 2.6 0.42 0.65 0.12
Example 1 1 2.2 0.87 0.52 0.13
Example 12 1.9 0.30 0.54 0.05 5
As shown above, the acid addition salts of Compound A of the
present invention proved to have the affinity for muscarinic M3 receptors
that is sufficient for their use as a medicine.
Test Example 5: Inhibitory effects on carbachol-evoked bladder
contraction in anesthetized mice
The method for determination of inhibitory effects of test
compounds on bladder contraction is described as follows.
Female mice, weighing 30 to 35 g, were anesthetized with a
sub-lethal dose (75 mg/kg, i.v.) of sodium pentobarbital and placed
supine on a heating pad to maintain the body temperature. A
32
CA 02599872 2007-08-30
polyethylene catheter (PE1 0) was cannulated to the bladder via the
urethra and secured by purse-string suture at the external urethral
opening. Another catheter was inserted into the femoral vein for
injections of drug solutions at a 3 mL volume. The bladder catheter was
connected to a pressure transducer through a three-way stopcock valve.
The bladder was emptied by drainage of urine through the catheter, and
then distended with about 100 L of physiological saline, and the
intravesical pressure was measured.
After intravesical pressure stabilization, the muscarinic agonist
carbachol (10 g/kg) was administered repeatedly with an interval of 15
minutes or longer. By this means, bladder contractions occurred in a
reproducible manner over 2 hours without deterioration of physical
conditions. After three responses to carbachol were obtained, a test
compound injection was followed by carbachol administration 10
minutes later and this procedure was repeated with increasing doses of
the test compound. In four or five mice per test compound, the percent
inhibition of means of prior drug responses was obtained and the dose
of test compound required for 50% inhibition (IDso) was estimated by the
linear regression analysis. Mice showing rhythmic bladder constriction
were not used for the data analysis.
Following IDso values were obtained: 0.079 mg/kg for Example 1,
0.090 mg/kg for Example 4, 0.059 mg/kg for Example 5, 0.050 mg/kg
for Example 7, and 0.057 mg/kg for Example 8.
The result clearly suggest that the acid addition salts of
Compound A of the present invention have inhibitory effects on
33
CA 02599872 2007-08-30
contraction of the bladder induced by the muscarinic agonist carbachol.
34