Note: Descriptions are shown in the official language in which they were submitted.
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
6-11 Bridged Oxime Erythromycin Derivatives
RELATED APPLICATIONS
This application claims the benefit of U.S. Application No. 11/122,251, filed
on May 4, 2005 and U.S. Provisional Application No. 60/677,675, filed on May
4,
2005. The entire teachings of the above application(s) are incorporated herein
by
reference.
TECHNICAL FIELD
The present invention relates to novel semisynthetic macrolides having
antibacterial activity and useful in the treatment and prevention of bacterial
infections. More particularly, the invention relates to 6-11 bicyclic
macrolide,
ketolide, and anhydrolide derivatives, compositions containing such compounds
and
methods for using the same, as well as processes for making such compounds.
BACKGROUND OF THE INVENTION
Macrolide antibiotics play a therapeutically important role, particularly with
the emergence of new pathogens. Structural differences are related to the size
of the
lactone ring and to the number and nature (neutral or basic) of the sugars.
Macrolides are classified according to the size of the lactone ring (12, 14,
15 or 16
atoms). The macrolide antibiotic family (14-, 15- and 16-membered ring
derivatives) shows a wide range of characteristics (antibacterial spectrum,
side-
effects and bioavailability). Among the commonly used macrolides are
erythromycin, clarithromycin, and azithromycin. Macrolides possessing a 3-oxo
moiety in place of the 3-cladinose sugar are known as ketolides and have shown
enhanced activity towards gram-negative bacteria and macrolide resistant gram-
positive bacteria. Macrolides possessing a degree of unsaturation between
carbons 2
and 3 or between carbons 3 and 4 of the erythromycin macrocycle are known as
anhydrolides. The search for macrolide compounds which are active against MLSB-
resistant strains (MLSB = Macrolides-Lincosamides-type B Streptogramines) has
become a major goal, together with retaining the overall profile of the
macrolides in
terms of stability, tolerance and pharmacokinetics.
International Application WO 97/42205 of Elliott et al, published November
13, 1997, discloses 3-descladinose-2, 3-anhydroerythromycin derivatives having
a
1
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
cyclic carbamate and cyclic carbazate basic nuciear structure. rurtner aetaiis
were
also disclosed in J. Med Chein., 41, pp 1651-1659 (1998) and J. Med Chem., 41,
pp
1660-1670 (1998) by Elliott et al, and by Griesgraber et al, respectively.
United States Patent 5,444,051 discloses certain 6-0-substituted-3-
oxoerythromycin A derivatives. PCT application WO 97/10251, published March
20, 1997, discloses intermediates useful for preparation of 6-0-methyl3-
descladinose erythromycin derivatives. United States Patent 5,631,355
discloses
certain tricyclic 6-0-methyl3-oxo erythromycin derivatives. United States
Patent
5,527,780 discloses certain bicyclic 6-0-methyl-3-oxo erythromycin A
derivatives
(Agouridas, ROUSSEL) corresponding to EP application 596802, published May
11, 1994. United States Patents 5,866,549 and 6,075,011, and PCT application
WO
00/78773, published December 28, 2000, disclose certain 6-0-substituted
erythroinycin derivatives. United States Patent 6,124,269 and PCT application
WO
00/62783, published October 26, 2000, disclose certain 2-halo-6-0-substituted
ketolide derivatives. United States Patent 6,046,171 and PCT application WO
99/21864, published May 6, 1999, disclose certain 6,11-bridged erythromycin
derivatives.
PCT Application WO 03/095466 Al, published November 20, 2003 and
PCT Application WO 03/097659 Al, published November 27, 2003 disclose a
series of bicyclic erythromycin derivatives.
SUMMARY OF THE INVENTION
The present invention provides a novel class of C6-C 11 bridged oxime
erythromycin derivatives which possess antibacterial activity.
In one aspect of the present invention there are provided novel bridged
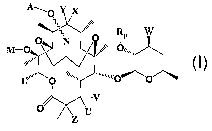
erythromycin compounds represented by the formulae as illustrated below:
A-, 0 Z'X
~ ;,..
O IN 0 p
.~' O
W
M-O ,,,,, \\\/
u,wuo
o
v
0
U
z
or their racemates, enantiomers, regioisomers, salts, esters or prodrugs
thereof, wherein
2
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
X and Y are independently selected from the group consisting of: hydrogen,
deuterium, halogen, Rl, ORI, S(O)nRj, -NR1C(O)R2, -NRIC(O)NR3R4, -
NR1S(O)õR2, -C(O)NR3R4, and -NR3R4;
Each of Rl and R2 is independently selected from the group consisting of:
hydrogen, acyl, silane, a substituted or unsubstituted, saturated or
unsaturated
aliphatic group, a substituted or unsubstituted, saturated or unsaturated
alicyclic
group, a substituted or unsubstituted aromatic group, a substituted or
unsubstituted
heteroaromatic group, or a substituted or unsubstituted heterocyclic group;
Each of R3 and R4 is independently selected from the group consisting of:
hydrogen, acyl, a substituted or unsubstituted, saturated or unsaturated
aliphatic
group, a substituted or unsubstituted, saturated or unsaturated alicyclic
group, a
substituted or unsubstituted aromatic group, a substituted or unsubstituted
heteroaromatic group, a substituted, or unsubstituted heterocyclic group; or
can be
taken together with the nitrogen atom to which they are attached to form a
substituted or unsubstituted heterocyclic or heteroaromatic ring;
or X and Y, taken together with the carbon atom to which they are attached,
are selected from the group consisting of: CO, C=CHR1, C=NR1, C=NC(O)Rl,
C=NOR1, C=NO(CH2)mRl, C=NNHR1, C=NNHCOR1, C=NNHCONR1R2,
C=NNHS(O)nRl, C=N-N=CHR1, C=N-N02, or C=N-ONO;
one of U or V is hydrogen and the other is independently selected from the
-0,,,,,", O
G
,O
I
group consisting of: Ri, ORI, OC(O)Rl, OC(O)NR3R4, S(O)õRl, Rt
or U and V, talcen together with the carbon atom to which they are attached,
are C=O;
one of J or G is hydrogen and the other is selected from: Rl, ORI, or NR3R4;
or, J and G, taken together with the carbon atom to which they are attached,
are selected from: C=O, C=NR1, C=NOR1, C=NO(CH2)mRl, C=NNHR1,
C=NNHCOR1, C=NNHCONR1R2, C=NNHS(O)õRl, or C=N-N=CHR1;
L is selected from the group consisting of: hydrogen, a substituted or
unsubstituted, saturated or unsaturated aliphatic group, a substituted or
unsubstituted, saturated or unsaturated alicyclic group, a substituted or
unsubstituted
3
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
aromatic group, a substituted or unsubstituted heteroaromatic group, or a
substituted
or unsubstituted heterocyclic group;
M is Rl;
W is NR3R4;
Z is hydrogen, alkyl or halogen;
Rp is hydrogen, hydroxy protecting group or hydroxy prodrug group;
m is an integer; and
n is 0, 1, or 2.
A is
Zj\ Y
X,
P ~ 11
Jv%fv
wherein:
Q' is N, CH or CF;
XI is 0, N, NRI, S, or CR5;
Yl is 0, N, NRI, S, CR5, or Se;
Zl is 0, N, NRI, S, or CR5;
R5 is independently selected from hydrogen, acyl, silane, a substituted or
unsubstituted, saturated or unsaturated aliphatic group, a substituted or
unsubstituted, saturated or unsaturated alicyclic group, a substituted or
unsubstituted
aromatic group, a substituted or unsubstituted heteroaromatic group, a
substituted or
unsubstituted heterocyclic group, NR3R4, OH, NHCORI or NHCONH2, and is
preferably, NH2 or NHR1.
With the proviso that a compound of Formula I is not selected from
compound having the following formula where A, Q, and Z as defined below in
the
table A.
4
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
A--_O Q
O N ON
\
H O HO
õ,nnp0
0
O 0
'o
Z
Table A
Compound A Q Z
~JIIL1 =
01 H N NAc H
=
02 NN NAc H
H
03 N~N I/ NH H
H
04 N ~
HZN--~ ~
s ~ NAc H
In one preferred embodiment, A is:
R5
N
wherein XI, and R5 is as defined previously.
5
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
In another preferred embodiment, A is:
X R5
1~
P11-1- N
.nrw
wherein Xi is 0, NH or S, and R5 is as defined previously.
In yet another embodiment, A is:
X1 R5
I
N
.n~'u
wherein Xi is 0, NH or S, and R5 is as defined previously.
In yet another embodiment, A is:
0 R5
N
,nnnr
wherein R5 is as defined previously.
In yet another embodiment, A is:
p NH2
I
N
,nrvv
In one preferred embodiment, A is selected from the compounds shown in
Table B.
6
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
Table B 12 F
N
Number A- HZN- S\ I
N
<v 13 F
01 S
HZN \S\~,~õM
N
02 HzN--/\/ S ~/ 14 H
N
N~
S-N
N H2N V~
03 15 0:0
HZN-N
1
04 S
HzN\ 16
-(N
H
17 N N\
05 N
N~ I /
06 p H2N
N~ \ 18 Np
HZN
07 HZN H2N
N/ 19 H N~
N
08 HZN~ H2N ~õrv
20 N
HZN--~~
09 HzN-,N I\ H
~~ /
S 21 N, \
S"N
" D /
HZN-~ ~
\ 22 rNN
'S/ N
11 N~ H
H I ZN O ~
s 23 0
7
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
24 ~ ~~ 30 N~
S N
/ 0 25 .N,4110
31 N
N N
26
e ~N ~ 32 N ~
'""~ N ~ ~
=' =
27 N
HN-~ /
U N /
33 0
HZN--<.,
N
28 0 ON:j
N 29 0
H2N--' O
N N
One preferred compound of the invention has the formula II:
A--_O Q
N
O N 0;~~' HOu,,,
HO ~~v' V =,.
O nm0 O
0 o
z II
Where in A, Q, and Z are as defined in Table C:
8
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
Table C
Number A Q Z
N Dol
Ac H
01 g N
HZN--~ '
02 NAc F
S-N
N x
~
03 NAc H
04 s
HZN--{~N NAc H
05 N I ~
NAc H
06 ,~
N ~ NAc H
HZN
07 HZN
N o NAc H
08 N /
HZN-~
NAc F
09 HZN~S c 0 H
N
HZN--~
NAc H
~
11 N
HZN--~~
O H
~
12 N-( / ~
HZNI ~
0 S , NAc H
9
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
13 F
HZN4S~ I NAc H
~
14 N H N ~~
HzN~ ~S ~
o , 0 H
15 F
H N--~ 0 H
z
S
H
16
N
N~ NAc H
~~~~
H2N
17 ~
HZN--~ ~
j
N NAc H
18 o
HZN--~ ~
N ~ 0 H
19 N /
AH~s ~ I NAc H
20 H
N~
N;Xi NAc H
HZN
21 N~
NAc F
HZN
'22 H
c F
N NA
A.,
~~~~
H2N
23 N
H2N_<
N
H NAc H
24 N
H2N-<
H 0 H
25 N, ~-
SeN /
NAc H
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
26 r "
C
s H NAc H
27 0
i
N NAc H
i
28 0
N I O H
29 ~
's " NAc H
30 clNmN
" NAc H
31 ~
i (/
j
" NAc H
32 HN-~ ~ /
//-j " NAc H
33 0
N =
,N NAc H
34
HZN4N ~ N NAc H
0
35 ~
~N~ I /
"~ NAc H
36 N ~
N~N ~ ~ NAc H
37 N
N , ,N ~ NAc H
0
38 HZN-<"N ~ I NC(O)OCH3 H
11
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
O /
39 HZN ~ ~ NH H
N
One preferred compound of the invention has the formula III:
O /
H2N--C ~ I
O I'
O RP W
IO
HO C\~ 1 / ==,
õO 0
0
O U
z III
wherein Rp, U, V, W, X, Y, L, and Z are as defined previously.
Another preferred compound of the invention has the formula IV:
o /
H-2N/ ~ I O
N-\
HO O~ O , OP,,, N
\~/
I 010 O
O
z
IV
wherein Z and Rp are as previously defined.
Yet another preferred compound of the invention has the formula V:
O
HZN-~
O 0
R
lP \ N
HO O~~/ V =
,,.õ~~'0 O
\'
O
O
z V
wherein Z and Rp are as defined previously.
12
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
In another aspect ot the invention, tnere are proviaeu nuvGi j-Q%,yliuv,
mlurvu
erythromycin compounds represented by the Formula VI:
B~O Y X
~~.
O ORp W
HO ~~~ II / O
p11111 O
O 0
O
'/
O
VI
or any racemates, enantiomers, regioisomers, salts, esters, or prodrugs
thereof
wherein X, Y, L, W, and Rp are as defined previously;
B is independently selected from hydrogen, acyl, silane, a substituted or
unsubstituted, saturated or unsaturated aliphatic group, a substituted or
unsubstituted, saturated or unsaturated alicyclic group, a substituted or
unsubstituted
aromatic group, a substituted or unsubstituted heteroaromatic group, or a
substituted
or unsubstituted heterocyclic group.
One preferred compound of Formula VI has the formula VII:
P-N / O
S N
H2N , 000 R N
O N P
HO O
O unnl0 0
O
I\'"
O
N
VII
wherein Rp is as previously defined.
DETAILED DESCRIPTION OF THE INVENTION
Definitions
Listed below are definitions of various terms used to describe this invention.
These definitions apply to the terms as they are used throughout this
specification
and claims, unless otherwise limited in specific instances, either
individually or as
part of a larger group.
13
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
An "aliphatic group" is non-aromatic moiety that may contain any
combination of carbon atoms, hydrogen atoms, halogen atoms, oxygen, nitrogen
or
other atoms, and optionally contain one or more units of unsaturation, e.g.,
double
and/or triple bonds. An aliphatic group may be straight chained, branched or
cyclic
and preferably contains between about 1 and about 24 carbon atoms, more
typically
between about 1 and about 12 carbon atoms. In addition to aliphatic
hydrocarbon
groups, aliphatic groups include, for example, polyalkoxyalkyls, such as
polyalkylene glycols, polyamines, and polyimines, for example. Such aliphatic
groups may be further substituted.
The terms "Cl-C3 alkyl," "C1-C6 alkyl," or "Cl-C12 alkyl," as used herein,
refer to saturated, straight- or branched-chain hydrocarbon radicals
containing
between one and three, one and twelve, or one and six carbon atoms,
respectively.
Examples of C1-C3 alkyl radicals include methyl, ethyl, propyl and isopropyl
radicals; examples of C1-C6 alkyl radicals include, but are not limited to,
methyl,
ethyl, propyl, propyl, butyl, pentyl, and hexyl radicals; and examples of C1-
C12 alkyl
radicals include, but are not limited to, ethyl, propyl, propyl, hexyl,
heptyl, octyl,
nonyl, decyl, undecyl, dodecyl radicals and the like.
The term "substituted alkyl," as used herein, refers to an alkyl, such as a C1-
C12 alkyl or C1-C6 alkyl group, substituted by one, two, three or more
aliphatic or
aromatic substituents.
Suitable aliphatic or aromatic substituents include, but are not limited to, -
F,
-Cl, -Br, -I, -OH, protected hydroxy, aliphatic ethers, aromatic ethers, oxo, -
NO2,
-CN, -CI-CIZ-alkyl optionally substituted with halogen (such as
perhaloalkyls), C2-
C12-alkenyl optionally substituted with halogen, -C2-C12-alkynyl optionally
substituted witli halogen, -NH2, protected amino, -NH -C1-C12-alkyl, -NH -C2-
C12-
alkenyl, -NH -C2-C12-allcynyl, -NH -C3-C12-cycloalkyl, -NH -aryl, -NH -
heteroaryl, -NH -heterocycloalkyl, -dialkylamino, -diarylamino,
-diheteroarylamino, -O-C1-C12-alkyl, -O-C2-C12-alkenyl, -O-Cz-C12-alkynyl, -O-
C3-C12-cycloalkyl, -0-aryl, -0-heteroaryl, -0-heterocycloalkyl, -C(O)- CI-C12-
alkyl, -C(O)- C2-C12-alkenyl, -C(O)- C2-C12-alkynyl, -C(O)-C3-C12-cycloalkyl,
-C(O)-aryl, -C(O)-heteroaryl, -C(O)-heterocycloalkyl, -CONH2, -CONH- C1-C12-
alkyl, -CONH- Cz-C12-alkenyl, -CONH- C2-C12-alkynyl, -CONH-C3-C12-
cycloalkyl, -CONH-aryl, -CONH-heteroaryl, -CONH-heterocycloalkyl, -C02- CI-
14
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
C12-alkyl, -C02- Ca-C12-alkenyl, -C02- C2-C12-alkynyl, -CO2-C3-Clz-cycloalkyl,
-C02-aryl, -C02-heteroaryl, -C02-heterocycloalkyl, -OC02- C1-C12-alkyl, -OC02-
C2-C12-alkenyl, -OC02- C2-C12-alkynyl, -OCO2-C3-CIZ-cycloalkyl, -OC02-aryl,
-OC02-heteroaryl, -OC02-heterocycloalkyl, -OCONH2, -OCONH- C1-C12-alkyl,
-OCONH- C2-C12-alkenyl, -OCONH- C2-C12-alkynyl, -OCONH- C3-C12-
cycloalkyl, -OCONH- aryl, -OCONH- heteroaryl, -OCONH- heterocycloalkyl,
-NHC(O)- C1-C12-a1ky1, -NHC(O)-C2-C12-alkenyl, -NHC(O)-CZ-Cla-alkynyl,
-NHC(O)-C3-C12-cycloalkyl, NHC(O)-aryl, -NHC(O)-heteroaryl, -NHC(O)-
heterocycloalkyl, -NHCO2- C1-C12-alkyl, -NHCO2- C2-C12-alkenyl, -NHCO2- C2-
C12-alkynyl, -NHCO2- C3-Cla-cycloalkyl, -NHC02- aryl, -NHCOZ- heteroaryl,
-NHCO2- heterocycloalkyl, NHC(O)NH2, NHC(O)NH- C1-C12-alkyl,
-NHC(O)NH-C2-C12-alkenyl, -NHC(O)NH-Ca-C12-alkynyl, NHC(O)NH-C3-C12-
cycloalkyl, -NHC(O)NH-aryl, -NHC(O)NH-heteroaryl, -NHC(O)NH-
heterocycloalkyl, NHC(S)NHZ, NHC(S)NH- Cl-C12-alkyl, -NHC(S)NH-C2-C12-
alkenyl, -NHC(S)NH-Ca-C12-alkynyl, -NHC(S)NH-C3-C12-cycloalkyl,
-NHC(S)NH-aryl, -NHC(S)NH-heteroaryl, -NHC(S)NH-heterocycloalkyl,
-NHC(NH)NH2, NHC(NH)NH- Cl-C12-alkyl, -NHC(NH)NH-C2-C12-alkenyl,
-NHC(NH)NH-C2-C12-alkynyl, -NHC(NH)NH-C3-C1z-cycloalkyl, -NHC(NH)NH-
aryl, -NHC(NH)NH-heteroaryl, -NHC(NH)NH-heterocycloalkyl, NHC(NH)-C1-
C12-alkyl, -NHC(NH)-C2-C12-alkenyl, NHC(NH)-C2-C12-alkynyl, -NHC(NH)-C3-
C12-cycloalkyl, -NHC(NH)-aryl, -NHC(NH)-heteroaryl, -NHC(NH)-
heterocycloalkyl, -C(NH)NH-C1-Cla-alkyl, -C(NH)NH-C2-C12-alkenyl,
-C(NH)NH-C2-Cla-alkynyl, -C(NH)NH-C3-Cia-cycloalkyl, -C(NH)NH-aryl,
-C(NH)NH-heteroaryl, -C(NH)NH-heterocycloalkyl, -S(O)-C1-C12-alkyl, - S(O)-
C2-C12-alkenyl, - S(O)-C2-C12-alkynyl, - S(O)-C3-C12-cycloalkyl, - S(O)-aryl, -
S(O)-heteroaryl, - S(O)-heterocycloalkyl -SOaNHa, -SO2NH- Cl-Cla-alkyl,
-SO2NH- C2-C12-alkenyl, -SO2NH- C2-C12-alkynyl, -SO2NH- C3-C12-cycloalkyl,
-SO2NH- aryl, -SO2NH- heteroaryl, -SO2NH- heterocycloalkyl, NHSO2-C1-C12-
alkyl, -NHSOa-C2-C1z-allcenyl, - NHSOz-Ca-Cla-alkynyl, -NHSO2-C3-C12-
cycloalkyl, -NHSOa-aryl, -NHSOa-heteroaryl, -NHSO2-heterocycloallcyl,
-CH2NH2, -CHaSOaCH3, -aryl, -arylalleyl, -heteroaryl, -heteroarylallcyl,
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
-heterocycloalkyl, -C3-C1z-cycloalkyl, polyalkoxyalkyl, polyalkoxy,
-methoxymethoxy, -methoxyethoxy, -SH, -S-CI-C12-alkyl, -S-C2-CI2-alkenyl, -S-
C2-C12-alkynyl, -S-C3-C12-cycloalkyl, -S-aryl, -S-heteroaryl, -S-
heterocycloalkyl,
or methylthiomethyl. It is understood that the aryls, heteroaryls, alkyls and
the like
can be further substituted.
The terms "C2-C12 alkenyl" or "C2-C6 alkenyl," as used herein, denote a
monovalent group derived from a hydrocarbon moiety containing from two to
twelve or two to six carbon atoms having at least one carbon-carbon double
bond by
the removal of a single hydrogen atom. Alkenyl groups include, but are not
limited
to, for example, ethenyl, propenyl, butenyl, 1-methyl-2-buten-1-yl, alkadienes
and
the like.
The term "substituted alkenyl," as used herein, refers to a"C2-C12 alkenyl"
or "C2-C6 alkenyl" group as previously defined, substituted by one, two, three
or
more aliphatic substituents.
The terms "C2-ClZ alkynyl" or "C2-C6 alkynyl," as used herein, denote a
monovalent group derived from a hydrocarbon moiety containing from two to
twelve or two to six carbon atoms having at least one carbon-carbon triple
bond by
the removal of a single hydrogen atom. Representative alkynyl groups include,
but
are not limited to, for example, ethynyl, 1-propynyl, 1-butynyl, and the like.
The term "substituted alkynyl," as used herein, refers to a"C2-C12 alkynyl"
or "C2-C6 alkynyl" group as previously defined, substituted by one, two, three
or
more aliphatic substituents.
The term "Cl-C6 alkoxy," as used herein, refers to a C1-C6 alkyl group, as
previously defined, attached to the parent molecular moiety through an oxygen
atom. Examples of C1-Cg-alkoxy include, but are not limited to, inethoxy,
ethoxy,
propoxy, isopropoxy, n-butoxy, sec-butoxy, tert-butoxy, n-pentoxy, neopentoxy
and
n-hexoxy.
The terms "halo" and "halogen," as used herein, refer to an atom selected
from fluorine, chlorine, bromine and iodine.
The terms "aryl" or "aromatic," as used herein, refer to a mono- or bicyclic
carbocyclic ring system having one or two aromatic rings including, but not
limited
to, phenyl, naphthyl, tetrahydronaphthyl, indanyl, idenyl and the like.
16
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
The terms "substituted aryl" or "substituted aromatic," as used herein, refer
to an aryl group, as previously defined, substituted by one, two, three or
more
aromatic substituents.
The term "arylalkyl," as used herein, refers to an aryl group attached to the
parent compound via a Cl-C3 alkyl or C1-C6 alkyl residue. Examples include,
but
are not limited to, benzyl, phenethyl and the like.
The term "substituted arylalkyl," as used herein, refers to an arylalkyl
group,
as previously defined, substituted by one, two, three or more aromatic
substituents.
The terms "heteroaryl" or "heteroaromatic," as used herein, refers to a mono-
, bi-, or tri-cyclic aromatic radical or ring having from five to ten ring
atoms of
which at least one ring atom is selected from S, 0 and N; zero, one, two,
three or
more ring atoms are additional heteroatoms independently selected from S, 0
and N;
and the remaining ring atoms are carbon, wherein any N or S contained within
the
ring may be optionally oxidized. Heteroaryl includes, but is not limited to,
pyridinyl, pyrazinyl, pyrimidinyl, pyrrolyl, pyrazolyl, imidazolyl, thiazolyl,
oxazolyl, isooxazolyl, thiadiazolyl, oxadiazolyl, thiophenyl, furanyl,
quinolinyl,
isoquinolinyl, benzimidazolyl, benzooxazolyl, quinoxalinyl, tetrazolyl and the
like.
The heteroaromatic ring may be bonded to the chemical structure through a
carbon
or hetero atom.
The terms "substituted heteroaryl" or "substituted heteroaromatic," as used
herein, refer to a heteroaryl group as previously defined, substituted by one,
two,
three or four aromatic substituents.
The term "alicyclic," as used herein, denotes a monovalent group derived
from a monocyclic or bicyclic saturated carbocyclic ring compound by the
removal
of a single hydrogen atom. Examples include, but not limited to, cyclopropyl,
cyclobutyl, cyclopentyl, cyclohexyl, bicyclo [2.2.1] heptyl, and bicyclo
[2.2.2] octyl.
The term "substituted alicyclic" group as previously defined, substituted by
one, two, three or more aliphatic substituents.
The terms "heterocyclic" as used herein, refers to a non-aromatic 5-, 6- or 7-
membered ring or a bi- or tri-cyclic group fused system, where (i) each ring
contains
between one and three heteroatoms independently selected from oxygen, sulfur
and
nitrogen, (ii) each 5-membered ring has 0 to 1 double bonds and each 6-
membered
ring has 0 to 2 double bonds, (iii) the nitrogen and sulfur heteroatoms may
17
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
optionally be oxidized, (iv) the nitrogen heteroatom may optionally be
quaternized,
(iv) any of the above rings may be fused to a benzene ring, and (v) the
remaining
ring atoms are carbon atoms which may be optionally oxo-substituted.
Representative heterocycloalkyl groups include, but are not limited to,
[1,3]dioxolane, pyrrolidinyl, pyrazolinyl, pyrazolidinyl, imidazolinyl,
imidazolidinyl, piperidinyl, piperazinyl, oxazolidinyl, isoxazolidinyl,
morpholinyl,
thiazolidinyl, isothiazolidinyl, quinoxalinyl, pyridazinonyl, tetrahydrofuryl,
and the
like.
The term "substituted heterocyclic," as used herein, refers to a heterocyclic
group, as previously defined, substituted by one, two, three or more aliphatic
substituents.
The term "heteroarylalkyl," as used herein, refers to a heteroaryl group
attached to the parent compound via a Cl-C3 alkyl or C1-C6 alkyl residue.
Examples include, but are not limited to, pyridinylmethyl, pyrimidinylethyl
and the
like.
The term "substituted heteroarylallcyl," as used herein, refers to a
heteroarylalkyl group, as previously defined, substituted by independent
replacement of one, two, or three or more aromatic substituents.
The term "C1-C3-alkylamino," as used herein, refers to one or two Ci-C3-
alkyl groups, as previously defined, attached to the parent molecular moiety
through
a nitrogen atom. Examples of C1-C3-alkylamino include, but are not limited to,
methylamino, dimethylamino, ethylamino, diethylamino, and propylamino.
The term "alkylamino" refers to a group having the structure -NH(C1-C12
allcyl) where C1-C12 alkyl is as previously defined.
The term "dialkylamino" refers to a group having the structure -N(C1-C12
alkyl) (C1-C12 alkyl), where CI-C12 alkyl is as previously defined. Examples
of
dialkylamino are, but not limited to, dimethylamino, diethylamino,
methylethylamino, piperidino, and the like.
The term "allcoxycarbonyl" represents an ester group, i.e., an alkoxy group,
attached to the parent molecular moiety through a carbonyl group such as
methoxycarbonyl, ethoxycarbonyl, and the like.
The term "carboxaldehyde," as used herein, refers to a group of formula -
CHO.
18
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
The term "carboxy," as used herein, refers to a group of formula -COOH.
The term "carboxamide," as used herein, refers to a group of formula -
C(O)NH(CI-C12 alkyl) or -C(O)N(C1-C12 alkyl) (CI-Cla alkyl), -C(O)NH2,
-NHC(O)(C1-C12 alkyl), -N(Cl-C1a alkyl)C(O)(C1-C12 alkyl) and the like.
The term "hydroxy protecting group," as used herein, refers to a labile
chemical moiety which is known in the art to protect a hydroxyl group against
undesired reactions during synthetic procedures. After said synthetic
procedure(s)
the hydroxy protecting group as described herein may be selectively removed.
Hydroxy protecting groups as known in the art are described generally in T.H.
Greene and P.G. M. Wuts, Protective Groups in Organic Synthesis, 3rd edition,
John
Wiley & Sons, New York (1999). Examples of hydroxyl protecting groups include
benzyloxycarbonyl, 4-nitrobenzyloxycarbonyl, 4-bromobenzyloxycarbonyl, 4-
methoxybenzyloxycarbonyl, methoxycarbonyl, tert-butoxycarbonyl,
isopropoxycarbonyl, diphenylmethoxycarbonyl, 2,2,2-trichloroethoxycarbonyl, 2-
(trimethylsilyl)ethoxycarbonyl, 2-furfuryloxycarbonyl, allyloxycarbonyl,
acetyl,
formyl, chloroacetyl, trifluoroacetyl, methoxyacetyl, phenoxyacetyl, benzoyl,
methyl, t-butyl, 2,2,2-trichloroethyl, 2-trimethylsilyl ethyl, 1, 1 -dimethyl-
2-propenyl,
3-inethyl- 3 -butenyl, allyl, benzyl, para-methoxybenzyldiphenylmethyl,
triphenylmethyl (trityl), tetrahydrofuryl, methoxymethyl, methylthiomethyl,
benzyloxymethyl, 2,2,2-triehloroethoxymethyl, 2-(trimethylsilyl)ethoxymethyl,
methanesulfonyl, para-toluenesulfonyl, trimethylsilyl, triethylsilyl,
triisopropylsilyl,
and the like. Preferred hydroxyl protecting groups for the present invention
are
acetyl (Ac or -C(O)CH3), benzoyl (Bz or -C(O)C6H5), and trimethylsilyl (TMS or-
Sl(CH3)3)=
The term "protected hydroxy," as used herein, refers to a hydroxy group
protected with a hydroxy protecting group, as defined above, including
benzoyl,
acetyl, trimethylsilyl, triethylsilyl, methoxymethyl groups, for example.
The term "hydroxy prodrug group", as used herein, refers to a promoiety
group which is known in the art to change the physicochemical, and hence the
biological properties of a parent drug in a transient manner by covering or
masking
the hydroxy group. After said synthetic procedure(s), the hydroxy prodrug
group as
described herein must be capable of reverting back to hydroxy group in vivo.
Hydroxy prodrug groups as known in the art are described generally in Kenneth
B.
19
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
Sloan, Prodrugs Topical and Ocular Drug Delivery, (Drugs and the
Pharmaceutical
Sciences; Volume 53), Marcel Dekker, Inc., New York (1992).
The term "amino protecting group," as used herein, refers to a labile
chemical moiety which is known in the art to protect an amino group against
undesired reactions during synthetic procedures. After said synthetic
procedure(s)
the amino protecting group as described herein may be selectively removed.
Amino
protecting groups as known in the are described generally in T.H. Greene and
P.G.
M. Wuts, Protective Groups in Or ag nic Synthesis, 3rd edition, John Wiley &
Sons,
New York (1999). Examples of amino protecting groups include, but are not
limited
to, t-butoxycarbonyl, 9-fluorenylmethoxycarbonyl, benzyloxycarbonyl, and the
like.
The term "protected amino," as used herein, refers to an amino group
protected with an amino protecting group as defined above.
The term "acyl" includes residues derived from acids, including but not
limited to carboxylic acids, carbamic acids, carbonic acids, sulfonic acids,
and
phosphorous acids. Examples include aliphatic carbonyls, aromatic carbonyls,
aliphatic sulfonyls, aromatic sulfinyls, aliphatic sulfinyls, aromatic
phosphates and
aliphatic phosphates.
The term "aprotic solvent," as used herein, refers to a solvent that is
relatively inert to proton activity, i.e., not acting as a proton-donor.
Examples
include, but are not limited to, hydrocarbons, such as hexane and toluene, for
example, halogenated hydrocarbons, such as, for example, methylene chloride,
ethylene chloride, chloroform, and the like, heterocyclic compounds, such as,
for
example, tetrahydrofuran and N-methylpyrrolidinone, and ethers such as diethyl
ether, bis-methoxymethyl ether. Such compounds are well lcnown to those
skilled in
the art, and it will be obvious to those skilled in the art that individual
solvents or
mixtures thereof may be preferred for specific compounds and reaction
conditions,
depending upon such factors as the solubility of reagents, reactivity of
reagents and
preferred temperature ranges, for example. Further discussions of aprotic
solvents
may be found in organic chemistry textbooks or in specialized monographs, for
example: Organic Solvents Physical Properties and Methods of Purification, 4th
ed.,
edited by John A. Riddick et al., Vol. II, in the Techniques of ChemistrY
Series,
John Wiley & Sons, NY, 1986.
The term "protogenic organic solvent" or "protic solvent," as used herein,
refers to a solvent that tends to provide protons, such as an alcohol, for
example,
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
methanol, ethanol, propanol, isopropanol, butanol, t-butanol, water and the
like.
Such solvents are well known to those skilled in the art, and it will be
obvious to
those skilled in the art that individual solvents or mixtures thereof may be
preferred
for specific compounds and reaction conditions, depending upon such factors as
the
solubility of reagents, reactivity of reagents and preferred temperature
ranges, for
exainple. Further discussions of protogenic solvents may be found in organic
chemistry textbooks or in specialized monographs, for example: Organic
Solvents
Physical Properties and Methods of Purification, 4th ed., edited by John A.
Riddick
et al., Vol. II, in the Techniques of ChemistrX Series, John Wiley & Sons, NY,
1986.
Combinations of substituents and variables envisioned by this invention are
only those that result in the formation of stable compounds. The term
"stable", as
used herein, refers to compounds which possess stability sufficient to allow
manufacture and which maintains the integrity of the compound for a sufficient
period of time to be useful for the purposes detailed herein (e.g.,
therapeutic or
prophylactic administration to a subject).
The synthesized compounds can be separated from a reaction mixture and
further purified by a method such as column chromatography, high pressure
liquid
chromatography, or recrystallization. As can be appreciated by the skilled
artisan,
further methods of synthesizing the compounds of the formulae herein will be
evident to those of ordinary skill in the art. Additionally, the various
synthetic steps
may be performed in an alternate sequence or order to give the desired
compounds.
Synthetic chemistry transformations and protecting group methodologies
(protection
and deprotection) useful in synthesizing the compounds described herein are
known
in the art and include, for example, those such as described in R. Larock,
Comprehensive Organic Transformations, VCH Publishers (1989); T.W. Greene and
P.G.M. Wuts, Protective Groups in Or ag nic Synthesis, 2d. Ed., John Wiley and
Sons
(1991); L. Fieser and M. Fieser, Fieser and Fieser's Reagents for Or ag nic S
nt~ hesis,
John Wiley and Sons (1994); and L. Paquette, ed., Encyclopedia of Reagents for
Organic S, nt~ hesis, John Wiley and Sons (1995), and subsequent editions
thereof.
The term "subject" as used herein refers to an animal. Preferably the animal
is a mammal. More preferably the mammal is a human. A subject also refers to,
for
example, dogs, cats, horses, cows, pigs, guinea pigs, fish, birds and the
like.
The compounds of this invention may be modified by appending appropriate
functionalities to enhance selective biological properties. Such modifications
are
21
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
lcnown in the art and may include those which increase biological penetration
into a
given biological system (e.g., blood, lymphatic system, central nervous
system),
increase oral availability, increase solubility to allow administration by
injection,
alter metabolism and alter rate of excretion.
The compounds described herein contain one or more asymmetric centers
and thus give rise to enantiomers, diastereomers, and other stereoisomeric
forms that
may be defined, in terms of absolute stereochemistry, as (R)- or (S)- , or as
(D)- or
(L)- for amino acids. The present invention is meant to include all such
possible
isomers, as well as their racemic and optically pure forins. Optical isomers
may be
prepared from their respective optically active precursors by the procedures
described above, or by resolving the racemic mixtures. The resolution can be
carried
out in the presence of a resolving agent, by chromatography or by repeated
crystallization or by some combination of these techniques which are known to
those skilled in the art. Further details regarding resolutions can be found
in Jacques,
et al., Enantiomers, Racemates, and Resolutions (John Wiley & Sons, 198 1).
When
the compounds described herein contain olefinic double bonds, other
unsaturation,
or other centers of geometric asymmetry, and unless specified otherwise, it is
intended that the compounds include both E and Z geometric isomers or cis- and
trans- isomers. Likewise, all tautomeric forms are also intended to be
included. The
configuration of any carbon-carbon double bond appearing herein is selected
for
convenience only and is not intended to designate a particular configuration
unless
the text so states; thus a carbon-carbon double bond or carbon-heteroatom
double
bond depicted arbitrarily herein as trans may be cis, trans, or a mixture of
the two in
any proportion.
As used herein, the term "pharmaceutically acceptable salt" refers to those
salts which are, within the scope of sound medical judgment, suitable for use
in
contact with the tissues of humans and lower animals without undue toxicity,
irritation, allergic response and the like, and are commensurate with a
reasonable
benefit/risk ratio. Pharmaceutically acceptable salts are well known in the
art. For
example, S. M. Berge, et al. describes pharmaceutically acceptable salts in
detail in
J. Pharmaceutical Sciences, 66: 1-19 (1977). The salts can be prepared in situ
during the final isolation and purification of the compounds of the invention,
or
separately by reacting the free base function with a suitable organic acid or
inorganic acid. Examples of pharmaceutically acceptable nontoxic acid addition
22
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
salts include, but are not limited to, salts of an amino group tormed witn
inorganic
acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric
acid
and perchloric acid or with organic acids such as acetic acid, maleic acid,
tartaric
acid, citric acid, succinic acid lactobionic acid or malonic acid or by using
other
methods used in the art such as ion exchange. Other pharmaceutically
acceptable
salts include, but are not limited to, adipate, alginate, ascorbate,
aspartate,
benzenesulfonate, benzoate, bisulfate, borate, butyrate, camphorate,
camphorsulfonate, citrate, cyclopentanepropionate, digluconate,
dodecylsulfate,
ethanesulfonate, formate, fumarate, glucoheptonate, glycerophosphate,
gluconate,
hemisulfate, heptanoate, hexanoate, hydroiodide, 2-hydroxy-ethanesulfonate,
lactobionate, lactate, laurate, lauryl sulfate, malate, maleate, malonate,
methanesulfonate, 2-naphthalenesulfonate, nicotinate, nitrate, oleate,
oxalate,
palmitate, pamoate, pectinate, persulfate, 3-phenylpropionate, phosphate,
picrate,
pivalate, propionate, stearate, succinate, sulfate, tartrate, thiocyanate, p-
toluenesulfonate, undecanoate, valerate salts, and the like. Representative
alkali or
alkaline earth metal salts include sodium, lithium, potassium, calcium,
magnesium,
and the like. Further pharmaceutically acceptable salts include, when
appropriate,
nontoxic ammonium, quaternary ammonium, and amine cations formed using
counterions such as halide, hydroxide, carboxylate, sulfate, phosphate,
nitrate, alkyl
having from 1 to 6 carbon atoms, sulfonate and aryl sulfonate.
As used herein, the term "pharmaceutically acceptable ester" refers to esters
which hydrolyze in vivo and include those that break down readily in the human
body to leave the parent compound or a salt thereof. Suitable ester groups
include,
for example, those derived from pharmaceutically acceptable aliphatic
carboxylic
acids, particularly alkanoic, alkenoic, cycloalkanoic and alkanedioic acids,
in which
each allcyl or alkenyl moiety advantageously has not more than 6 carbon atoms.
Exainples of particular esters include, but are not limited to, formates,
acetates,
propionates, butyrates, acrylates and ethylsuccinates.
The term "pharmaceutically acceptable prodrugs" as used herein refers to
those prodrugs of the compounds of the present invention which are, within the
scope of sound medical judgment, suitable for use in contact with the tissues
of
humans and lower animals with undue toxicity, irritation, allergic response,
and the
like, commensurate with a reasonable benefit/risk ratio, and effective for
their
intended use, as well as the zwitterionic forms, where possible, of the
compounds of
23
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
the present invention. "Prodrug", as used herein means a compound which is
convertible in vivo by metabolic means (e.g. by hydrolysis) to a compound of
the
invention. Various forms of prodrugs are known in the art, for example, as
discussed
in Bundgaard, (ed.), Design of Prodrugs, Elsevier (1985); Widder, et al.
(ed.),
Methods in Enzymology, vol. 4, Academic Press (1985); Krogsgaard-Larsen, et
al.,
(ed). "Design and Application of Prodrugs, Textbook of Drug Design and
Development, Chapter 5, 113-191 (1991); Bundgaard, et al., Journal of Drug
Deliver
Reviews, 8:1-38(1992); Bundgaard, J. of Pharmaceutical Sciences, 77:285 et
seq.
(1988); Higuchi and Stella (eds.) Prodrugs as Novel Drug Delivery Systems,
American Chemical Society (1975); and Bernard Testa & Joachim Mayer,
"Hydrolysis In Drug And Prodrug Metabolism: Chemistry, Biochemistry And
Enzymology," John Wiley and Sons, Ltd. (2002).
This invention also encompasses pharmaceutical compositions containing,
and methods of treating bacterial infections through administering,
pharmaceutically
acceptable prodrugs of compounds of the invention. For example, compounds of
the
invention having free amino, amido, hydroxy or carboxylic groups can be
converted
into prodrugs. Prodrugs include compounds wherein an amino acid residue, or a
polypeptide chain of two or more (e.g., two, three or four) amino acid
residues is
covalently joined through an amide or ester bond to a free amino, hydroxy or
carboxylic acid group of compounds of the invention. The amino acid residues
include but are not limited to the 20 naturally occurring amino acids commonly
designated by three letter symbols and also includes 4-hydroxyproline,
hydroxylysine, demosine, isodemosine, 3-methylhistidine, norvalin, beta-
alanine,
gamma-aminobutyric acid, citrulline homocysteine, homoserine, ornithine and
methionine sulfone. Additional types of prodrugs are also encompassed. For
instance, free carboxyl groups can be derivatized as amides or alkyl esters.
Free
hydroxy groups may be derivatized using groups including but not limited to
hemisuccinates, phosphate esters, dimethylaminoacetates, and
phosphoryloxymethyloxycarbonyls, as outlined in Advanced Drug Delivery
Reviews, 1996, 19, 115. Carbamate prodrugs of hydroxy and amino groups are
also
included, as are carbonate prodrugs, sulfonate esters and sulfate esters of
hydroxy
groups. Derivatization of hydroxy groups as (acyloxy)methyl and (acyloxy)ethyl
ethers wherein the acyl group may be an alkyl ester, optionally substituted
with
groups including but not limited to ether, amine and carboxylic acid
functionalities,
24
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
or where the acyl group is an amino acid ester as described above, are also
encompassed. Prodrugs of this type are described in J. Med. Chem. 1996, 39,
10.
Free amines can also be derivatized as amides, sulfonamides or phosphonamides.
All of these prodrug moieties may incorporate groups including but not limited
to
ether, amine and carboxylic acid functionalities.
As used herein, unless otherwise indicated, the term "bacterial infection(s)"
or "protozoa infections"; includes, but is not limited to, bacterial
infections and
protozoa infections that occur in mammals, fish and birds as well as disorders
related to bacterial infections and protozoa infections that may be treated or
prevented by administering antibiotics such as the compounds of the present
invention. Such bacterial infections and protozoa infections and disorders
related to
such infections include, but are not limited to, the following: pneumonia,
otitis
media, sinusitus, bronchitis, tonsillitis, cystic fibrosis (CF) and
mastoiditis related to
infection by Streptococcus pneumoniae, Haemophilus influenzae, Moraxella
catarrhalis, Staphylococcus aureus, Peptostreptococcus spp. or Pseudomonas
spp.;
pharynigitis, rheumatic fever, and glomerulonephritis related to infection by
Streptococcus pyogenes, Groups C and G streptococci, Clostridium diptheriae,
or
Actinobacillus haemolyticum; respiratory tract infections related to infection
by
Mycoplasma pneumoniae, Legionella pneumophila, Streptococcus pneumoniae,
Haemophilus influenzae, or Chlamydia pneumoniae; uncomplicated skin and soft
tissue infections, abscesses and osteomyelitis, and puerperal fever related to
infection by Staphylococcus aureus, coagulase-positive staphylococci (i.e., S.
epidermidis, S. hemolyticus, etc.), S. pyogenes, S. agalactiae, Streptococcal
groups
C-F (minute-colony streptococci), viridans streptococci, Corynebacterium spp.,
Clostridium spp., or Bartonella henselae; uncomplicated acute urinary tract
infections related to infection by S. saprophyticus or Enterococcus spp.;
uretliritis
and cervicitis; and sexually transmitted diseases related to infection by
Chlamydia
trachomatis, Haemophilus ducreyi, Treponema pallidum, Ureaplasma urealyticum,
or Nesseria gonorrheae; toxin diseases related to infection by S. aureus (food
poisoning and Toxic shock syndrome), or Groups A, S. and C streptococci;
ulcers
related to infection by Helicobacter pylori; systemic febrile syndromes
related to
infection by Borrelia recurrentis; Lyme disease related to infection by
Borrelia
burgdorferi; conjunctivitis, keratitis, and dacrocystitis related to infection
by C.
trachomatis, N. gonorrlioeae, S. aureus, S. pneumoniae, S. pyogenes, H.
influenzae,
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
or Listeria spp.; disseminated Mycobacterium avium complex (MAU) disease
related to infection by Mycobacterium avium, or Mycobacterium intracellulare;
tuberculosis disease related to infection by Mycobacterium tuberculosis;
gastroenteritis related to infection by Campylobacter jejuni; intestinal
protozoa
related to infection by Cryptosporidium spp. odontogenic infection related to
infection by viridans streptococci; persistent cough related to infection by
Bordetella
pertussis; gas gangrene related to infection by Clostridium perfringens or
Bacteroides spp.; Skin infection by S. aureus, Propionibacterium acne;
atherosclerosis related to infection by Helicobacter pylori or Chlamydia
pneumoniae; or the like.
Bacterial infections and protozoa infections and disorders related to such
infections that may be treated or prevented in animals include, but are not
limited to,
the following: bovine respiratory disease related to infection by P.
haemolytica., P.
multocida, Mycoplasma bovis, or Bordetella spp.; cow enteric disease related
to
infection by E. coli or protozoa (i.e., coccidia, cryptosporidia, etc.), dairy
cow
mastitis related to infection by S. aureus, S. uberis, S. agalactiae, S.
dysgalactiae,
Klebsiella spp., Corynebacterium, or Enterococcus spp.; swine respiratory
disease
related to infection by A. pleuropneumoniae., P. multocida, or Mycoplasma
spp.;
swine enteric disease related to infection by E. coli, Lawsonia
intracellularis,
Salmonella spp., or Serpulina hyodyisinteriae; cow footrot related to
infection by
Fusobacterium spp.; cow metritis related to infection by E. coli; cow hairy
warts
related to Infection by Fusobacterium necrophorum or Bacteroides nodosus; cow
pink-eye related to infection by Moraxella bovis, cow premature abortion
related to
infection by protozoa (i.e. neosporium); urinary tract infection in dogs and
cats
related to infection by E. coli; skin and soft tissue infections in dogs and
cats related
to infection by S. epidermidis, S. intermedius, coagulase neg. Staphylococcus
or P.
multocida; and dental or mouth infections in dogs and oats related to
infection by
Alcaligenes spp., Bacteroides spp., Clostridium spp., Enterobacter spp.,
Eubacterium spp., Peptostreptococcus spp., Porphfyromonas spp., Campylobacter
spp., Actinomyces spp., Erysipelothrix spp., Rhodococcus spp., Trypanosoma
spp.,
Plasmodium spp., Babesia spp., Toxoplasma spp., Pneumocystis spp., Leishmania
spp., and Trichomonas spp. or Prevotella spp. Other bacterial infections and
protozoa infections and disorders related to such infections that may be
treated or
prevented in accord with the method of the present invention are referred to
in J. P.
26
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
Sanford at al.,"The Sanford Guide To Antimicrobial Therapy," 26th Edition,
(Antimicrobial Therapy, Inc., 1996).
Antibacterial activity studies may be carried out using suitable assays as are
known in the art. Susceptibility tests can be used to quantitatively measure
the in
vitro activity of an antimicrobial agent against a given bacterial isolate.
Compounds
are tested for in vitro antibacterial activity by a micro-dilution method.
Minimal
Inhibitory Concentration (MIC) is determined in 96 well microtiter plates
utilizing
the appropriate Mueller Hinton Broth medium (CAMHB) for the observed bacterial
isolates. Antimicrobial agents are serially diluted (2-fold) in DMSO to
produce a
concentration range from about 64 g/ml to about 0.03 g/ml. The diluted
compounds (2 l/well) are then transferred into sterile, uninoculated CAIVIHB
(0.2
mL) by use of a 96 fixed tip-pipeting station. The inoculum for each bacterial
strain
is standardized to 5 x 105 CFU/inL by optical comparison to a 0.5 McFarland
turbidity standard. The plates are inoculated with 10 Uwell of adjusted
bacterial
inoculum. The 96 well plates are covered and incubated at 35 +/- 2 C for 24
hours in
ambient air environment. Following incubation, plate wells are visually
examined by
Optical Density measurement for the presence of growth (turbidity). The lowest
concentration of an antimicrobial agent at which no visible growth occurs is
defined
as the MIC. The compounds of the invention generally demonstrated an MIC in
the
range from about 64 g/ml to about 0.03 g/ml.
All in vitro testing follows the guidelines described in the Approved
Standards M7-A4 protocol, published by the National Committee for Clinical
Laboratory Standards (NCCLS).
The invention further provides compositions and methods of treating
subjects suffering from an inflammatory condition comprising administering to
a
subject in need thereof, a therapeutically effective amount of at least one
compound
of the invention. Specific examples of inflammatory conditions treatable
according
to the invention include, but are not limited to, scleritis; epi-scleritis;
allergic
conjunctivitis; pulmonary inflammatory diseases, particularly CF, asthma,
chronic
obstructive pulmonary disease (COPD), allergic bronchopulmonary aspergillosis
(ABPA), and sarcoidosis; procto-sigmoiditis; allergic rhinitis; arthritis;
tendonitis;
apthous stomatitis; and inflammatory bowel disease.
27
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
The invention further provides compositions and methods for i) prophylactic
treatment of those subjects susceptible to the symptoms CF including pulmonary
infection and inflammation associated with CF, ii) treatinent at the initial
onset of
symptoms of pulmonary infection and inflammation associated with CF, and iii)
treatment of ongoing or relapsing symptoms of infection and inflammation
associated with CF. In accordance with the invention a compound of the
invention,
is administered to a subject in need of treatment for CF, in amount sufficient
to
prevent, diminish or eradicate symptoms of CF including chronic pulmonary
inflammation and infection.
The pharmaceutical compositions of the present invention comprise a
therapeutically effective amount of a compound of the present invention
formulated
together with one or more pharmaceutically acceptable carriers or excipients.
As used herein, the term "pharmaceutically acceptable carrier or excipient"
means a non-toxic, inert solid, semi-solid or liquid filler, diluent,
encapsulating
material or formulation auxiliary of any type. Some examples of materials
which
can serve as pharmaceutically acceptable carriers are sugars such as lactose,
glucose
and sucrose; starches such as corn starch and potato starch; cellulose and its
derivatives such as sodium carboxymethyl cellulose, ethyl cellulose and
cellulose
acetate; powdered tragacanth; malt; gelatin; talc; excipients such as cocoa
butter and
suppository waxes; oils such as peanut oil, cottonseed oil, safflower oil,
sesame oil,
olive oil, corn oil and soybean oil; glycols such as propylene glycol; esters
such as
ethyl oleate and ethyl laurate; agar; buffering agents such as magnesium
hydroxide
and aluminun hydroxide; alginic acid; pyrogen-free water; isotonic saline;
Ringer's
solution; ethyl alcohol, and phosphate buffer solutions, as well as other non-
toxic
compatible lubricants such as sodium lauryl sulfate and magnesium stearate, as
well
as coloring agents, releasing agents, coating agents, sweetening, flavoring
and
perfuming agents, preservatives and antioxidants can also be present in the
composition, according to the judgment of the formulator.
The pharmaceutical compositions of this invention may be administered
orally, parenterally, by inhalation spray, topically, rectally, nasally,
buccally,
vaginally or via an implanted reservoir, preferably by oral administration or
administration by injection. The pharmaceutical compositions of this invention
may
contain any conventional non-toxic pharmaceutically-acceptable carriers,
adjuvants
or vehicles. In some cases, the pH of the forinulation may be adjusted with
28
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
pharmaceutically acceptable acids, bases or buffers to enhance the stability
of the
formulated compound or its delivery form. The term parenteral as used herein
includes subcutaneous, intracutaneous, intravenous, intramuscular,
intraarticular,
intraarterial, intrasynovial, intrasternal, intrathecal, intralesional and
intracranial
injection or infusion techniques.
Liquid dosage forms for oral administration include pharmaceutically
acceptable emulsions, microemulsions, solutions, suspensions, syrups and
elixirs. In
addition to the active compounds, the liquid dosage forms may contain inert
diluents
commonly used in the art such as, for example, water or other solvents,
solubilizing
agents and emulsifiers such as ethyl alcohol, isopropyl alcohol, ethyl
carbonate,
ethyl acetate, benzyl alcohol, benzyl benzoate, propylene glycol, 1,3-butylene
glycol, dimethylformamide, oils (in particular, cottonseed, groundnut, corn,
germ,
olive, castor, and sesame oils), glycerol, tetrahydrofurfuryl alcohol,
polyethylene
glycols and fatty acid esters of sorbitan, and mixtures thereof. Besides inert
diluents, the oral compositions can also include adjuvants such as wetting
agents,
emulsifying and suspending agents, sweetening, flavoring, and perfuming
agents.
Injectable preparations, for example, sterile injectable aqueous or oleaginous
suspensions, may be formulated according to the known art using suitable
dispersing
or wetting agents and suspending agents. The sterile injectable preparation
may also
be a sterile injectable solution, suspension or emulsion in a nontoxic
parenterally
acceptable diluent or solvent, for example, as a solution in 1,3-butanediol.
Among
the acceptable vehicles and solvents that may be employed are water, Ringer's
solution, U.S.P. and isotonic sodium chloride solution. In addition, sterile,
fixed oils
are conventionally employed as a solvent or suspending medium. For this
purpose
any bland fixed oil can be employed including synthetic mono- or diglycerides.
In
addition, fatty acids such as oleic acid are used in the preparation of
injectables.
The injectable formulations can be sterilized, for example, by filtration
through a bacterial-retaining filter, or by incorporating sterilizing agents
in the form
of sterile solid compositions which can be dissolved or dispersed in sterile
water or
other sterile injectable medium prior to use.
In order to prolong the effect of a drug, it is often desirable to slow the
absorption of the drug from subcutaneous or intramuscular injection. This may
be
accomplished by the use of a liquid suspension of crystalline or amorphous
material
with poor water solubility. The rate of absorption of the drug then depends
upon its
29
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
rate of dissolution, which, in turn, may depend upon crystal size anca
crystalline
form. Alternatively, delayed absorption of a parenterally administered drug
form is
accomplished by dissolving or suspending the drug in an oil vehicle.
Injectable
depot forms are made by forming microencapsule matrices of the drug in
biodegradable polymers such as polylactide-polyglycolide. Depending upon the
ratio of drug to polymer and the nature of the particular polymer employed,
the rate
of drug release can be controlled. Examples of other biodegradable polymers
include poly(orthoesters) and poly(anhydrides). Depot injectable formulations
are
also prepared by entrapping the drug in liposomes or microemulsions that are
compatible with body tissues.
Compositions for rectal or vaginal administration are preferably
suppositories which can be prepared by mixing the compounds of this invention
with suitable non-irritating excipients or carriers such as cocoa butter,
polyethylene
glycol or a suppository wax which are solid at ambient temperature but liquid
at
body temperature and therefore melt in the rectum or vaginal cavity and
release the
active compound.
Solid dosage forms for oral administration include capsules, tablets, pills,
powders, and granules. In such solid dosage forms, the active compound is
mixed
with at least one inert, pharmaceutically acceptable excipient or carrier such
as
sodium citrate or dicalciuin phosphate and/or: a) fillers or extenders such as
starches, lactose, sucrose, glucose, mannitol, and silicic acid, b) binders
such as, for
example, carboxymethylcellulose, alginates, gelatin, polyvinylpyrrolidinone,
sucrose, and acacia, c) humectants such as glycerol, d) disintegrating agents
such as
agar-agar, calcium carbonate, potato or tapioca starch, alginic acid, certain
silicates,
and sodium carbonate, e) solution retarding agents such as paraffin, f)
absorption
accelerators such as quaternary ammonium compounds, g) wetting agents such as,
for example, cetyl alcohol and glycerol monostearate, h) absorbents such as
kaolin
and bentonite clay, and i) lubricants such as talc, calcium stearate,
magnesium
stearate, solid polyethylene glycols, sodium lauryl sulfate, and mixtures
thereof. In
the case of capsules, tablets and pills, the dosage form may also comprise
buffering
agents.
Solid compositions of a similar type may also be employed as fillers in soft
and hard-filled gelatin capsules using such excipients as lactose or milk
sugar as
well as high molecular weight polyethylene glycols and the like.
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
The solid dosage forms of tablets, dragees, capsules, pills, and granules can
be prepared with coatings and shells such as enteric coatings and other
coatings well
known in the pharmaceutical formulating art. They may optionally contain
opacifying agents and can also be of a composition that they release the
active
ingredient(s) only, or preferentially, in a certain part of the intestinal
tract,
optionally, in a delayed manner. Examples of embedding compositions that can
be
used include polymeric substances and waxes.
Dosage forms for topical or transdermal administration of a compound of
this invention include ointments, pastes, creams, lotions, gels, powders,
solutions,
sprays, inhalants or patches. The active component is admixed under sterile
conditions with a pharmaceutically acceptable carrier and any needed
preservatives
or buffers as may be required. Ophthalmic formulation, ear drops, eye
ointments,
powders and solutions are also contemplated as being within the scope of this
invention.
The ointments, pastes, creams and gels may contain, in addition to an active
compound of this invention, excipients such as animal and vegetable fats,
oils,
waxes, paraffins, starch, tragacanth, cellulose derivatives, polyethylene
glycols,
silicones, bentonites, silicic acid, talc and zinc oxide, or mixtures thereof.
Powders and sprays can contain, in addition to the compounds of this
invention, excipients such as lactose, talc, silicic acid, aluminum hydroxide,
calcium
silicates and polyamide powder, or mixtures of these substances. Sprays can
additionally contain customary propellants such as chlorofluorohydrocarbons.
Transdermal patches have the added advantage of providing controlled
delivery of a compound to the body. Such dosage forms can be made by
dissolving
or dispensing the compound in the proper medium. Absorption enhancers can also
be used to increase the flux of the compound across the skin. The rate can be
controlled by either providing a rate controlling membrane or by dispersing
the
compound in a polymer matrix or gel.
For pulmonary delivery, a therapeutic composition of the invention is
formulated and administered to the subject in solid or liquid particulate form
by
direct administration e.g., inhalation into the respiratory system. Solid or
liquid
particulate forms of the active compound prepared for practicing the present
invention include particles of respirable size: that is, particles of a size
sufficiently
small to pass through the mouth and larynx upon inhalation and into the
bronchi and
31
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
alveoli of the lungs. Delivery of aerosolized therapeutics, particularly
aerosolized
antibiotics, is known in the art (see, for example U.S. Pat. No. 5,767,068 to
VanDevanter et al., U.S. Pat. No. 5,508,269 to Smith et al., and WO 98/43,650
by
Montgomery, all of which are incorporated herein by reference). A discussion
of
pulmonary delivery of antibiotics is also found in U.S. Pat. No. 6,014,969,
incorporated herein by reference.
According to the methods of treatment of the present invention, bacterial
infections, cystic fibrosis and inflammatory conditions are treated or
prevented in a
subject such as a human or another animal by administering to the subject a
therapeutically effective amount of a compound of the invention, in such
amounts
and for such time as is necessary to achieve the desired result.
By a "therapeutically effective amount" of a compound of the invention is
meant an amount of the compound which confers a therapeutic effect on the
treated
subject, at a reasonable benefit/risk ratio applicable to any medical
treatment. The
therapeutic effect may be objective (i.e., measurable by some test or marker)
or
subjective (i.e., subject gives an indication of or feels an effect). An
effective
amount of the compound described above may range from about 0.1 mg/Kg to about
500 mg/Kg, preferably from about 1 to about 50 mg/Kg. Effective doses will
also
vary depending on route of administration, as well as the possibility of co-
usage
with other agents. It will be understood, however, that the total daily usage
of the
compounds and compositions of the present invention will be decided by the
attending physician within the scope of sound medical judgment. The specific
therapeutically effective dose level for any particular subject will depend
upon a
variety of factors including the disorder being treated and the severity of
the
disorder; the activity of the specific compound employed; the specific
composition
employed; the age, body weight, general health, sex and diet of the subject;
the time
of administration, route of administration, and rate of excretion of the
specific
compound employed; the duration of the treatment; drugs used in combination or
contemporaneously with the specific compound employed; and lilee factors well
known in the medical arts.
The total daily dose of the compounds of this invention administered to a
human or other animal in single or in divided doses can be in amounts, for
example,
from 0.01 to 50 mg/kg body weight or more usually from 0.1 to 25 mg/kg body
weight. Single dose compositions may contain such amounts or submultiples
32
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
thereof to make up the daily dose. In general, treatment regimens according to
the
present invention comprise administration to a subject in need of such
treatment
from about 10 mg to about 1000 mg of the compound(s) of this invention per day
in
single or multiple doses.
The compounds of the formulae described herein can, for example, be
administered by injection, intravenously, intraarterially, subdermally,
intraperitoneally, intramuscularly, or subcutaneously; or orally, buccally,
nasally,
transmucosally, topically, in an ophthalmic preparation, or by inhalation,
with a
dosage ranging from about 0.1 to about 500 mg/kg of body weight, alternatively
dosages between 1 mg and 1000 mg/dose, every 4 to 120 hours, or according to
the
requirements of the particular drug. The methods herein contemplate
administration
of an effective amount of compound or compound composition to achieve the
desired or stated effect. Typically, the pharmaceutical compositions of this
invention will be administered from about 1 to about 6 times per day or
alternatively, as a continuous infusion. Such administration can be used as a
chronic
or acute therapy. The amount of active ingredient that may be combined with
pharmaceutically excipients or carriers to produce a single dosage form will
vary
depending upon the host treated and the particular mode of administration. A
typical
preparation will contain from about 5% to about 95% active compound (w/w).
Alternatively, such preparations may contain from about 20% to about 80%
active
compound.
Lower or higher doses than those recited above may be required. Specific
dosage and treatment regimens for any particular subject will depend upon a
variety
of factors, including the activity of the specific compound employed, the age,
body
weight, general health status, sex, diet, time of administration, rate of
excretion, drug
combination, the severity and course of the disease, condition or symptoms,
the
subject's disposition to the disease, condition or symptoms, and the judgment
of the
treating physician.
Upon improvement of a subject's condition, a maintenance dose of a
compound, composition or combination of this invention may be administered, if
necessary. Subsequently, the dosage or frequency of administration, or both,
may be
reduced, as a function of the symptoms, to a level at which the improved
condition is
retained when the symptoms have been alleviated to the desired level. Subjects
33
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
may, however, require intermittent treatment on a long-term basis upon any
recurrence of disease symptoms.
When the compositions of this invention comprise a combination of a
compound of the formulae described herein and one or more additional
therapeutic
or prophylactic agents, both the compound and the additional agent should be
present at dosage levels of between about 1 to 100%, and more preferably
between
about 5 to 95% of the dosage normally administered in a monotherapy regimen.
The
additional agents may be administered separately, as part of a multiple dose
regimen, from the compounds of this invention. Alternatively, those agents may
be
part of a single dosage form, mixed together with the compounds of this
invention in
a single composition.
The pharmaceutical compositions of this invention can be administered
orally to fish by blending said pharmaceutical compositions into fish feed or
said
pharmaceutical compositions may be dissolved in water in which infected fish
are
placed, a method commonly referred to as a medicated bath. The dosage for the
treatment of fish differs depending upon the purpose of administration
(prevention
or cure of disease) and type of administration, size and extent of infection
of the fish
to be treated. Generally, a dosage of 5 - 1000 mg, preferably 20 - 100 mg, per
kg of
body weight of fish may be administered per day, either at one time or divided
into
several times. It will be recognized that the above-specified dosage is only a
general
range which may be reduced or increased depending upon the age, body weight,
condition of disease, etc. of the fish.
Unless otherwise defined, all technical and scientific terms used herein are
accorded the meaning commonly known to one of ordinary skill in the art. All
publications, patents, published patent applications, and other references
mentioned
herein are hereby incorporated by reference in their entirety.
Abbreviations
Abbreviations which may be used in the descriptions of the scheme and the
examples that follow are:
Ac for acetyl;
AIBN for azobisisobutyronitrile;
Bu3SnH for tributyltin hydride;
CDI for carbonyldiimidazole;
dba for dibenzylidene acetone:
34
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
dppb for diphenylphosphino butane;
DBU for 1,8-diazabicyclo[5.4.0]undec-7-ene;
DEAD for diethylazodicarboxylate;
DMAP for dimethylaminopyridine;
DMF for dimethyl formamide;
DPPA for diphenylphosphoryl azide;
EtOAc for ethyl acetate;
EtOH for ethanol;
MeOH for methanol;
Ms for mesylate or O-S02-CF3;
NaN(TMS)2 for sodium bis(trimethylsilyl)amide;
NMMO for N-methylmorpholine N-oxide;
TEA for triethylamine;
THF for tetrahydrofuran;
TPP or PPh3 for triphenylphosphine;
MOM for methoxymethyl;
Boc for t - butoxycarbonyl;
Bz for benzoyl;
Bn for benzyl;
Ph for phenyl;
POPd for dihydrogen dichlorobis(di-tert-butylphosphinito-xP)palladate(II);
TBS for tert-butyl dimethylsilyl; or
TMS for trimethylsilyl.
Synthetic Methods
The compounds and processes of the present invention will be better
understood in connection with the following synthetic schemes that illustrate
the
methods by which the compounds of the invention may be prepared.
A preferred intermediate for the preparation of compounds represented by
formula I is a compound represented by formula VIII as illustrated below
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
Y, X
I
O ltP
W
O O
HO
L, .=.'o ,,,,, ,O
U
Z
VIII
wherein Rp, U, V, W, X, Y and Z are as previously defined.
Schemes 1- 2 describe processes for the preparation of compounds
according to the invention.
Compounds of formula VIII, which are useful as the starting materials for
the preparation of compounds of the present invention are prepared from
erythromycin using the procedures described in U.S. Patent No. 6,878,691 and
U.S.
Patent Application Publication No. 2004/0053861, incorporated herein by
reference.
Scheme 1
Y, X A-O I' X
O Rp W O_NH2 O ~ XI- O Rp W
0,,,,., HO ' 0
HO
~ 1 ~ .
....q p11 ,q~
O O O õnn O O
acid
,,~'V
O U O
Z Z
VIII (1-2)
Scheme 1 illustrates a process of preparing compounds of the present
invention by converting the bridged ketone of VIII into an oxime of formula (1-
2)
using the appropriate substituted hydroxylamine of the formula:
A" O-NH2
where A is as previously defined. This oxime formation can be accomplished,
using
the appropriate substituted hydroxylamine under either acidic or basic
conditions in
a variety of solvents. Representative acids include, but are not limited to,
hydrochloric acid, phosphoric acid, sulfuric acid, p-toluenesulfonic acid, and
pyridinium p-toluene sulfonate and the likes. Likewise, representative bases
include, but are not limited to, triethylamine, pyridine, diisopropylethyl
amine, 2,6-
36
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
lutidine, and the likes. Appropriate solvents include, but are not limited to,
methanol, ethanol, water, tetrahydrofuran, 1,2-dimethoxyethane, ethyl acetate
and
the likes. Preferably the reaction is carried out in ethanol using aqueous
hydrochloric
acid. Reaction temperature is generally, but not limited to, from -20 C to 40
C and
the reaction time is froml to 8 hours, preferably the reaction is carried out
at 0 C.
Scheme 2
A O YX A-O YX
N . .'.
O lP W
0 O O? w
HO O~ HO O~
tO 0
, \ ~f V ~ ~,,,,== \
I,,.,== Iõ==
"OH 4j"0-R
Z Z R = Rl, ORI, OC(O)Rl,
(2-1) (2-2) OC(O)NR3R4, S(O)nRl
Scheme 2 illustrates the procedure by which compounds of formula (2-1)
may be converted to compounds of formula (2-2) by treatment with isocyanates
of
formula Rl-NCO, acid chlorides of formula Rl-C(O)Cl or Alkyl isocyanates in
the
present of bases such as, but not limited to, sodium hydride, potassium
hydride,
potassium tert-butoxide, potassium hydroxide, KHMDS, and the like. The
reaction
is typically carried out in aprotic solvents such as, but not limited to, THF,
DMSO,
DMF, or dioxane and the likes. The temperature of the reaction is from 25 C
to 80
C. The preferred reaction time is from 5 to 20 hours.
Alternatively, some of the ester compounds of formula (2-2) can be prepared
by treating compounds of formula (2-1) with acids of formula R1-C(O)OH in the
presence of bases such as but not limited to Et3N, Pyridine, DMAP and coupling
agents such as but not limited to EDC, BOPCI, HATU, and the likes in aprotic
solvents such as but not limited to dichloromethane, ethylene chloride, THF,
DMF,
acetonitrile and the like at a temperature from 25 C to 80 C and the
reaction time
is from 2 to 24 hours.
Compounds of formula (2-1) also can be treated with with substituted tert-
butyl allyl carbonate in the presence of a palladium catalyst and a phosphine
additive to give allyl ethers.
37
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
Examples
The compounds and processes of the present invention will be better
understood in connection with the following examples, which are intended as an
illustration only and not limiting of the scope of the invention. Various
changes and
modifications to the disclosed embodiments will be apparent to those skilled
in the
art and such changes and modifications including, without limitation, those
relating
to the chemical structures, substituents, derivatives, formulations and/or
methods of
the invention may be made without departing from the spirit of the invention
and the
scope of the appended claims.
Example 1
~
H2N-~o ~
N ~ NAc
MN ,,~' "I N
HO~~O HO,,
' .
o ~~' ~O O
O O'
~ O
/,--NH2
Step 1 a: N
To a solution of 2-amino-4-methylphenol (157.6g, 1.28mo1) in 1500 ml of
EtOH at room temperature, under stirred condition, was added bromocyanogen
(135.OOg, 1.28mo1) in about 30 minutes. During the addition, the reaction
mixture
became warm and water bath was used to cool the reaction to room temperature.
After 5-6 hours, the reaction solvent was evaporated under reduced pressure.
The
residue was dissolved in about 1500 ml of EtOAc and washed with the saturated
NaHCO3 (1.5 L). The organic layer was separated and dried over anhydrous
MgSO4. Removal of solvent and dried by high vacuum gave about 150g of the
title
compound as pale brown color, which can be used in next step. ESI MS m/e: 149
(M+H)+.
0
0
s-N
N
Step lb: 0
A solution of succinic anhydride (141.1 g, 1.41 mol) and 70 g (about 0.47
mol) of compound of step la (70 g, 0.47 mol) in 2500 ml of anhydrous toluene
was
refluxed for overnight. After that, HATU (100g, 0.26 mol) and 4-
methylmorpholine
38
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
(41.36 ml, 0.376 mol) were added and the resulting mixture was refluxed for 2-
3
hours. TLC showed that the major spot was product (Rf = 0.35, acetone:hexane =
1:2). After reaction was completed, the solvent was evaporated and the residue
was
dissolved in about 2000 ml of CHZC12. The solution was washed with aqueous
NaHCO3. After pH was adjusted to 7-8 and washed with brine, organic phase was
separated and dried over MgSO4. Filtration and removal of solvent gave the
title
compound (98g) as fine white needle crystalline ESI MS m/e: 231 (M+H)+.
0
~
N
Br ~ /oN~
Step 1 c: o
To a solution of the compound of step lb (44.37g, 0.193mo1) in CC14 (1.5 L)
was added NBS (41.16g, 0.23mo1) and then the mixture were heated to reflux.
Benzoyl peroxide (0.75g) was added by three times. After refluxing for 24
hours, the
reaction was cooled to room temperature and the mixture was diluted with 1.5 L
of
CHZC12 , The organic phase was separated and washed with 4 L of saturated
NaHCO3 two times to adjust pH to 7-8. Dried over MgSO4 and removal of the
solvent under reduced pressure gave the title compound (57.3 g) as slight
yellow
solid which was used for next step without further purification. ESI MS m/e:
309/311(M+H)+.
o
o~
I~ N_ N N
/
Step 1 d: 0 0
To a solution of the compound of step 1c (57.3g, 0.185mo1) in 450 ml of
MeCN were added N-hydroxyphanthalimide (60.51g, 0.371mol) and 80 ml of
triethylamine. The reaction mixture was stirred at 50 C for 5 hours and
cooled to
room temperature. The reaction was added water (200 ml) and filtered. The pale
yellow solid was collected, washed with100ml of MeOH and ether in the 1:1
ratio.
Dried on vacuum to give the title compound (48 g) as pale solid. ESI MS m/e:
392
(M+H)+.
~ o
HZN-o I ~ N~NHZ
Step le:
A mixture of the compound of step ld (39.1g, 0.1 mol) in 500 ml of 2M
ammonia in methanol was stirred at room temperature for 16 hours and filtered.
The
39
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
filtrate was concentrated and purified on silica gel (2M NH3 in MeOH:CH2C12 =
5:95) to give the title compound (17g, 95%). ESI MS m/e: 180 (M+H)+.
O
H2N-~~ ~
N,Ac
0N AcN
HO . O''.
O "''O O
O O
Step lf:
To a solution of compound of step id (215 mg, 1.2 mmol) in 15 ml of
ethanol was added 1N HCI (2 ml). The mixture was cooled to 0 C and added
compound of formula VIII where X and Y taken together with the atom that they
are
attached is C=N-Ac and U and V taken together with the atom that they are
attached
is C=O, Z= H, Rp = Ac and W = NMe2 (711 mg, 1 mmol). The mixture was stirred
at 0 C for 1 hour and quenched with saturated NaHCO3 (50 ml). Extracted with
ethyl acetate (100 ml) and washed with brine (100 ml x 2). Dried over
anhydrous
Na2SO4 and concentrated to give crude title compound (828 mg, 95%, as a
mixture
of oximes, oximeE/Z = 4/1), which was used for next step reaction without
further
purification. ESI MS m/e: 872 (M+H)+.
H, N--~~
o ~ I
N ~ N AC
O~ o
O N ''O
HO HO,,,
"'
O /1,' 'O O
O O
Step l g:
A solution of compound of step lf (828 mg, 0.95 mmol) in 15 ml of
methanol was stirred at 60 C for 5 hours. The solvent was removed and the
residue
was purified on silica gel (2M NH3 in MeOH/ CH2C12 = 5/95) to give the title
compound (765mg, 97%, as a mixture of E/Z oximes E/Z - 4/1). The compound was
further separated on HPLC to give E-oxime isomer (430 mg) and Z-oxime isomer
(110mg).
E-oxime isomer: ESI MS m/e: 830 (M+H)+.
E-oxime isomer: 13C NMR (125 MHz, CDC13): 8 205.9, 191.4, 186.8, 184.7, 178.1,
167.8, 162.1, 153.3, 148.5, 143.0, 134.1, 125.6, 121.9, 116.8, 108.7, 103.0,
79.4,
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
76.4, 74.6, 70.5, 69.8, 66.1, 63.2, 62.9, 50.8, 40.5, 38.8, 31.2, 28.5, 25.3,
23.8, 21.5,
19.5, 17.8, 15.1, 14.1, 12.8.
Z-oxime isomer: ESI MS m/e: 830 (M+H)+.
Z-oxime isomer: 13C NMR (125 MHz, CDC13): S 206.2, 184.7, 176.9, 169.3, 163.0,
155.9, 148.5, 143.2, 133.6, 121.6, 116.7, 108.5, 103.0, 79.5, 79.0, 76.7,
76.2, 75.8,
70.5, 70.2, 69.7, 66.1, 58.2, 53.7, 51.0, 45.3, 40.5, 39.7, 39.0, 36.9, 28.5,
25.5, 23.4,
21.5, 20.3, 19.6, 17.3, 15.7, 14.5, 12.9, 12Ø
Example 2
O i
H2N4
O
O~,N N
HO OHO ,,
O ,''O O
O O
O
N' \
O O AHOO
O '0O
O O
Step 2a:
The title compound of step 2a was prepared according to experimental
procedure from U.S. Pat. No. 6,878,691 incorporated herein by reference.
0
O O\ Ac
O O 1 HO O
O '' O
O O
Step 2b:
To a solution of compound of step 2a (711 mg, 1 mmol) in 8 ml of
acetonitrile was added 1 N HCI (10 ml) at room temperature. The mixture was
stirred at room temperature for 4 hours and was quenched with saturated NaHCO3
(30 ml). Extracted with ethyl acetate (40 ml) and the organic phase was washed
with
brine (40 m1X2). After dried over anhydrous Na2SO4, the solvent was removed
and
the residue was purified on silica gel column (hexane: acetone = 1:1) to give
the title
compound (330 mg, 49%).
41
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
O /
HaN--~ ~ ~
N 0
z,N .,,
HOk/~/ 0 Ac N
~,.
0
'''v
0 ~''0 0
0 0
Step 2c:
To a solution of compound from step 2b (215 mg, 1.2 mmol) in 15 ml of
ethanol was added 1N HC1(2 ml). The mixture was cooled to 0 C and added
bridged ketone lcetolide, compound of step 2a (670 mg, 1 mmol). The mixture
was
stirred at 0 C for 1 hour and quenched with saturated NaHCO3 (50 ml).
Extracted
with ethyl acetate (100 ml) and washed with brine (100 ml X 2). Dried over
anhydrous Na2SO4 and concentrated to give crude title compound (764 mg, 92%,
E/Z - 1/1). ESI MS m/e: 831 (M+H)+.
0 i
HZN--~ ~ ~
N 0
0."
0 N O N
HO H
Y ~
I"~~'0 ~''0 0
0 O
Step 2d:
A solution of compound of step 2c (764mg, 0.92 mmol) in 15 ml of
methanol was stirred at 60 C for 6 hours. The solvent was removed and the
residue
was purified on silica gel (2M NH3 in MeOH/ CH2C12 = 5/95) to give the title
compound as a mixture of E/Z oxime isomers (690mg, 95%, E/Z - 1/1). The
compound was further separated on HPLC to give E-oxime isomer (280 mg) and Z-
oxime isomer (230mg).
E-oxime isomer: ESI MS m/e: 789 (M+H)+.
E-oxime isomer: 13C NMR (125 MHz, CDC13): 6 218.5, 205.6, 191.7, 168.0, 162.0,
152.8, 148.6, 143.0, 134.0, 122.1, 117.0, 108.7, 103.3, 79.3, 79.0, 76.5,
75.8, 74.5,
70.5, 69.8, 66.1, 63.0, 61.5, 51.0, 47.0, 46.2, 40.5, 39.5, 39.3, 28.5, 23.5,
21.4, 20.0,
18.6, 18.0, 14.6, 14.2, 12.6, 12.2.
Z-oxime isomer: ESI MS m/e: 789 (M+H)+.
Z-oxime isomer: 13C NMR (125 MHz, CDC13): 8 215.0, 205.7, 169.6, 162.2, 156.2,
149.0, 142.8, 131.4, 124.8, 116.3, 109.7, 102.5, 79.9, 78.7, 76.3, 76.2, 70.5,
69.7,
42
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
66.1, 59.2, 50.8, 46.3, 45.7, 40.5, 40.0, 39.1, 28.5, 23.1, 21.6, 19.7, 18.5,
17.2, 14.5,
13.1, 12.9, 11.8.
Example 3.
In accordance with Schemes 1 and 2, the following compounds of the
invention having the Formula II were prepared:
A---- O
'4
O N~ O
HO = N
~~ ~ / = ~
~,.Y s V HO,,,
~~. O ,... '',O
O
o = z II
wherein A, Q and Z are defined as in Table I.
TABLE I.
MS (ESI):
Example A- Q Z mlz 13C NMR (125 MHz,CDCI3):S
M+H +
NAc H 205.8,184.7,178.0,167.8, 154.3,153.8,153.0,
N ~ 136.0, 134.0,126.7,123.5, 121.5, 103.1, 79.3,
~ 79.2, 76.8, 75.8, 75.5, 74.7, 70.5, 69.7, 66.1,
Example 01 S 831 63.1, 62.9, 50.7, 46.2, 40.5, 38.8, 37.2, 31.2,
28.5, 25.3, 23.8, 21.5, 20.3, 19.5, 17.8, 15.1,
14.1
,~/~N NAc F
Example 02 H2N ~S 864
S-N NAc H
N~
Example 03 832
Example 04 ~S NAc H
HZN ~ 846
N
Example 05 NAc H 205.8, 184.7,178.0,167.8, 153.6,153.0,149.8,
/N ~ 140.3, 135.0,126.3,120.5, 110.8,103.0, 79.4,
815 76.8, 76.0, 74.6, 70.5, 69.7, 66.1, 63.1, 62.8,
O ~ 53.7, 50.7, 46.2, 40.5, 38.9, 28.5, 25.3, 23.8,
21.5,20.3,19.5,17.8,15.1,14.1,13.6,12.9
Example 06 ,O NAc H
N ~ 830
H N ~~~~
Example 07 H2N NAc H
N~ 830
0
43
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
Example 08 NAc F (selected)205.5, 205.3, 184.2, 165.1, 164.9,
162.4, 153.7,148.8, 142.7, 131.2,124.5,116.2,
109.3,104.1, 99.7, 98.1, 79.7, 76.5, 73.7, 70.6,
H2N- 848 69.8, 66.0, 63.1, 62.6, 41.2, 40.5, 39.0, 28.4,
o~ 25.3, 24.7, 24.5, 23.2, 21.4, 21.0,
17.5,15.0,14.3,12.6
Example 09 0 H 218.4, 205.6,168.0, 166.3,152.0, 132.1, 131.9,
N ~ 126.9, 121.5,119.1, 103.3, 79.3, 78.9, 76.4,
HZN--~~ 805 75.9, 74.6, 70.5, 69.8, 66.1, 13.0, 61.5, 50.9,
s 47.1, 46.2, 40.5, 39.5, 39.3, 28.5, 23.5, 21.5,
20.1, 18.6, 18.0, 14.6, 14.2, 12.6, 12.2.
Example 10 NAc H 205.9,184.7,178.0, 167.9, 162.2, 153.4, 148.9,
142.6,131.5,124.6,116.2,109.4, 103.1, 79.3,
HZN--N 830 76.8, 74.8, 70.5, 66.1, 63.2, 62.9, 50.8, 40.5,
o~ 38.8, 28.5, 25.4, 23.8, 21.5, 20.3, 19.6, 17.8,
15.1, 14.1, 12.8.
Example 11 0 H 216.3, 203.6,165.9, 160.1,150.8,146.8, 140.6,
129.1, 122.7,114.1, 107.5, 101.2, 77.2, 76.8,
N H2N--~ 789 74.7, 74.4, 73.8, 72.6, 68.4, 67.7, 63.9, 60.8,
59.4, 51.5, 48.9, 45.0, 44.0, 38.3, 37.4, 29.0,
26.3, 21.3, 19.3,18.0, 16.4, 15.9,12.6,12.1
Example 12 NAc H (selected): 205.8, 185.0,178.3,168.2, 161.6,
155.9, 153.4,148.9,133.0,131.6,126.6, 121.2,
N-{ ~ 1 889 120.0, 103.0, 79.4, 76.7, 76.2, 70.5, 69.7, 66.0,
HZN~ s~ 63.2, 50.8, 46.2, 40.5, 38.8, 28.5, 25.5, 23.7,
0 21.5, 20.2, 19,7, 18.0, 15.4, 14.1, 13.7, 12.7.
Example 13 NAc H (selected): 205.9,184.7,178.0,167.9, 167.4,
F 153.9, 153.7,152.0, 140.1,140.0,133.7, 132.9,
132.8, 116.4,112.4,112.3, 103.0, 79.3, 76.7,
HZN~N 864 75.6, 74.8, 70.5, 59.7, 66.0, 63.1, 62.9, 50.8,
s 46.3, 40.4, 38.8, 28.5, 25.3, 23.7, 21.5, 20.3,
19.6,17.8,15.1, 14.1, 13.7,12.8
Example 14 0 H (selected); 217.9, 205.6,168.9,162.0, 155.5,
NN ~ ~ 152.9, 131.8,126.8, 122.1,119.7, 102.8, 9.4,
HZN1 ~ 848 79.3, 76.5, 75.1, 70.5, 69.8, 66.1, 63.2, 50.9,
o 46.6, 46.3, 40.5, 39.4, 28.5, 23.2, 21.5, 18.2,
17.9,14.2, 14.0, 13.2,12.2.
Example 15 0 H (selected): 218.4, 205.7, 168.1,167.4,154.0,
153.1, 152.0,140.2,140.0, 133.8,133.7,132.8,
F 132.7, 116.7,112.7,112.5, 103.3, 79.3, 78.9,
823 76.8, 75.9, 75.8, 74.7, 70.5, 69.8, 66.0, 62.9,
H2N-~ ~ 61.5, 51.0, 47.2, 46.2, 40.4, 39.4, 39.3, 28.5,
s 23.5, 21.5, 20.1, 18.5,18.0,14.7, 14.2, 12.5,
12.2
Example 16 N, NAc H
N = ~ ~ 829
H N ~~~~
Example 17 o NAc H 205.9, 191.4,186.8,184.7, 178.1,167.8,162.1,
~ 153.3, 148.5,143.0, 134.1,125.6,121.9,116.8,
HZN N~ ~ 830 108.7, 103.0, 79.4, 76.4, 74.6, 70.5, 69.8, 66.1,
63.2, 62.9, 50.8, 40.5, 38.8, 31.2, 28.5, 25.3,
23.8, 21.5, 19.5, 17.8, 15.1, 14.1, 12.8.
Example 18 0 H 218.5, 205.6,191.7,168.0,162.0, 152.8, 148.6,
O 143.0, 134.0, 122.1, 117.0, 108.7, 103.3, 79.3,
H2N-~~ ~ 79.0, 76.5, 75.8, 74.5, 70.5, 69.8, 66.1, 63.0,
N~ 789 61.5, 51.0, 47.0, 46.2, 40.5, 39.5, 39.3, 28.5,
, 23.5, 21.4, 20.0, 18.6, 18.0, 14.6,14.2,12.6,
12.2.
44
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
Example 19 N/ NAc H
AH\s ~ ~ 888
Example 20 H N NAc H
N
N 830
H N
Example 21 0 ~ NAc F
N ~ ~ 848
HN
Example 22 N NAc F
, /
N ~ 847
HN
Example 23 N NAc H (selected); 206.2,184.8, 168.6,155.0, 153.8,
HZN-~ / 110.0, 103.4, 79.5, 76.7, 75.1, 70.5, 69.8, 66.0,
N~ 829 63.2, 63.0, 51.3, 40.5, 38.8, 31.2, 30.0, 28.4,
H 25.4, 23.7, 21.8, 20.6, 20.0,17.9, 15.1,14.1,
12.8.
Example 24 N 0 H (selected): 218.5, 206.0, 168.8, 155.1, 153.3,
HZN-~ ~ 129.8, 121.1, 103.4, 79.6, 79.1, 76.6, 75.4,
N~ 788 70.6, 69.8, 66.0, 62.9, 62.1, 53.7, 51.4, 47.6,
H 46.0, 40.5, 39.7, 39.4, 28.4, 23.4, 21.5, 20.4,
18.6, 18.2, 15.3, 14.4, 12.5, 12.2.
Example 25 NAc H 205.8,184.8,178.0,167.9,167.8,159.3,153.5,153
.3,146.3,143.8,143.7,135.0,133.9,1333.3,125.4,
123.7,121.9,120.2,111.1,103.0,79.4,79.2,76.5,7
Cs N ~ 897 6.4,74.8,70.5,69.8,66.1,63.2,62.9,50.8,46.2,40.
/~ 5,38.8,28.5,25.4,23.8,21.5,20.3,19.6,17.8,15.1,
14.1,13.7,12.9.
Example 26 NAc H 205.8, 184.7,178.0,167.8, 153.6,153.0, 149.8,
0 140.3,135.0,126.3, 120.5, 110.8,103.0, 78.4,
<\ ~ 815 76.0, 74.6, 70.5, 69.7, 66.2, 63.1, 62.9, 50.7,
46.2, 40.5, 38.8, 28.6, 25.4, 23.8, 21.5, 20.3,
19.5, 17.8, 15.1, 14.1, 13.6, 12.9
Example 27 0 H 218.4, 205.6,168.0,153.0, 149.8,140.3,134.9,
O/ 126.5,120.7,110.8,103.1, 79.3, 79.0, 76.1,
~ 775 75.8, 74.6, 70.5, 69.6, 66.2, 63.0, 61.5, 50.9,
N~ 47.0, 46.2, 40.5, 39.5, 39.3, 28.9, 23.5, 21.4,
20.0, 18.6,18.0,14.6, 14.3,12.6,12.2
Example 28 NAc H 205.8,184.7,178.0,167.8,157.5,155.1,153.7,150
.5,145.3,141.6,135.9,127.1,123.3,120.4,111.1,1
C ~~ ~ 898 03.0,79.4,76.8,75.8,74.6,70.5,69.7,66.1,63.1,62
s N / .9,50.7,40.5,38.8,29.9,29.5,28.5,25.4,23.8,21.5,
1 20.3,19.5,17.8,15.1,14.1,13.6,12.9.
Example 29 N~ NAc H
N N / 881
Example 30 NAc H
~ ~ 845
N
Example 31 NAc H (selected): 210.1, 184.7,177.9,170.0, 162.4,
153.7,148.5,143.1, 133.9,132.9,121.5,119.6,
o ~ 870 116.4, 108.7,102.4, 81.2, 79.7, 76.6, 76.3,
74.5, 70.6, 69.5, 66.2, 62.7, 60.8, 43.7, 40.7,
HN~N 40.5, 40.1, 39.3, 37.4, 29.0, 25.3, 22.7,21.6,
21.4, 20.6,19.8,18.3,16.2,14.8, 12.5.
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
Example 32 HO / NAc H (selected): 206.1,184.9, 178.0,168.1,153.8,
~ 806 150.1,120.4,113.7,103.0, 79.4, 76.7, 70.5,
H2N N 69.7, 66.0, 50.9, 40.5, 28.7, 25.4, 23.7, 21.5,
17.8, 17.2, 15.7, 15.1, 14.1, 12.8, 12Ø
Example 33 0 / NAc H
N ~N 816
ll I I
Example 34 NAc H (selected): 205.9,184.8,177.9,167.9,165.1,
157.6,153.8,152.9,140.5,115.5,114.9,102.9,
0 ~ 831 79.3, 76.8, 76.7, 75.1, 70.5, 69.6, 66.1, 63.1,
H2N--' ~ 50.9, 40.5, 38.8, 28.7, 25.6, 25.4, 23.8, 21.6,
N N 21.5, 20.4,19.6,17.8,17.2,15.2,14.1, 12.8,
12Ø
Example 35 NAc H (selected): 209.9,184.7,177.8,169.9,157.6,
153.9,144.3,133.6,133.3,132.8,131.0,121.1,
119.7,118.1,108.5,107.6, 102.4, 81.0, 79.7,
o ~ 910 76.5, 76.0, 74.4, 70.5, 69.5, 66.3, 62.7, 62.6,
" <, I i 60.7, 45.1, 43.7, 40.6, 39.3, 29.2, 25.3, 22.9,
21.5, 20.6,19.8,18.2,16.3,14.7, 12.7.
Example 36 N NAc H
815
Example 37 I NAc H
N /
N ~ 829
N
Example 38 O NC(0)0 H
HZN-~~ CH3
846 205.9,186.4,167.7,163.7,162.4,153.2,148.4,143
.0,134.1,121.8,116.6,108.7,102.9,79.5,78.9,77.
5,76.8,76.4,75.1,74.7,70.5,69.7,66.1,63.2,62.7,
54.1,53.1,50.7,40.5,38.7,32.0,29.5,28.6,23.8,21
.5,20.2,19.3,17.8,14.9,14.1,12.9,
Example 39 O NH H
HZN--~~
205.2,169.3,162.8,153.7,148.5,143.1,133.7,
788 121.6, 116.5, 108.8, 103.5, 80.4, 78.8, 78.6,
76.6, 75.9, 70.6, 69.8, 66.1, 64.7, 63.1, 51.6,
47.9, 41.4, 40.5, 38.3, 28.5, 23.0, 21.4, 21.1,
20.1, 17.4,15.8, 14.4, 14.0, 11.8
In accordance with Scheme 1, compounds of the invention having the Formula IX
were prepared:
Q O."A
,o'
O
~
YN HO
HO.,.
0
... '=~O
O
0 Z 0 IX
wherein A, Q, and Z are defined as in Table II.
46
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
Table II
Example A- Q Z MS (ESI): +sC NMR (125 MHz CDCI3):S
m/z (M+H)+ '
NAc H 815 205.8,184.6, 176.5,169.3,156.2, 153.0,
149.9, 140.3,134.8, 126.6, 120.8, 110.9,
Example 01 N> 102.6, 79.6, 79.0, 76.3, 76.2, 70.5, 69.7,
0 66.1, 58.4, 50.9, 45.4, 40.5, 29.9, 28.6, 25.5,
23.4, 21.6, 20.2, 19.7, 17.2, 15.6, 14.4, 12.9,
12.0
0 H 805 215.1, 205.7, 169.6, 166.8, 156.2, 152.1,
):s N'~--NHZ 132.0,126.6, 121.3, 118.9, 102.5, 80.0,
Example 02 78.7, 76.3, 76.1, 70.5, 69.7, 66.0, 50.8, 46.3,
45.7, 40.4, 40.0, 39.1, 28.5, 23.1, 21.6, 19.7,
18.5,17.3,14.5,13.1,11.8
0 H 789 215.0, 205.7, 169.6, 162.2, 156.2, 149.0,
):0 N'~--NHZ 142.8, 131.4, 124.8, 116.3, 109.7, 102.5,
Example 03 79.9, 78.7, 76.3, 76.2, 70.5, 69.7, 66.1, 59.2,
50.8, 46.3, 45.7, 40.5, 40.0, 39.1, 28.5, 23.1,
21.6,19.7,18.5,17.2,14.5,13.1, 12.9,11.8.
NAc H 864 (selected): 205.9, 184.6, 176.4, 169.4,
F 166.7, 156.4, 154.2, 152.2, 140.1, 140.0,
N 134.0, 133.9, 133.3, 133.2, 116.6, 116.5,
Example 04 '--NH2 112.6, 112.4, 102.6, 79.6, 79.0, 76.2, 75.8,
S 75.4, 70.5, 69.7, 66.1, 58.5, 50.9, 45.4, 40.5,
39.0, 28.5, 25.5, 23.3, 21.6, 20.2, 19.7, 17.2,
15.7,14.4,13.0,12Ø
0 H 848 (selected): 216.3, 205.4,169.6, 162.0,
N 155.8,149.0,131.9, 127.1,119.6, 102.3,
Example 05 ~ I ~"Y NH2 80.0, 79.0, 76.7, 76.3, 76.0, 70.5, 69.7, 66.0,
a 59.4, 50.7, 46.3, 45.7, 40.5, 40.1, 39.3, 29.9,
28.6, 23.1, 21.6, 19.8, 18.2,17.4, 14.5, 13.7,
13.0, 11.7.
F 0 H 823 (selected): 215.2, 205.7, 169.6, 167.2,
156.5,154.1,152.1, 140.2,140.1, 134.0,
N 133.9, 133.0, 132.9, 116.6, 112.5, 112.3,
Example 06 ~NHz 102.5, 76.3, 70.5, 69.7, 66.1, 59.1, 50.9,
46.3, 45.7, 40.5, 39.2, 28.5, 23.1, 21.6, 19.7,
18.5, 17.2, 14.5, 13.2, 12.8, 11.8
PCN ~ NAc H 830
Example 07 NHZ
In accordance with Scheme I, compounds of the invention having the
Formula X were prepared:
47
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
A "'A
HO O N O N
HO,,
o .. '''O
O
0 'oZ 0 x
wherein A, X and Z are defined as in Table III.
TABLE III
Example A- x z MS (ESI):
m/z (M+H)+
Example 01 HZN~ ~ ~ NH2 H 790
N ~
Example 4.
g NAc
H2N O 0 N 0 N
,,
HO HO5
011 O ~~''' O 0
O NI
Step 4a:
Br S
\ ~/OH
A solution of 5-bromo-2-thiophenecarboxaldehyde (13.08 g, 68.46 mmol) in
isopropanol (100 mL) was treated with NaBH4 (1.30 g, 34.23 mmol) at 0 C for
1.5
hours with stirring before HCI (1 M, 60 mL, 60 mmol) was charged. The mixture
was stirred for 0.5 hour before being partitioned (ethyl acetate and saturated
NaHCO3). The organics were washed with water, brine, and then dried (Na2SO4).
The volatile was removed by evaporation and dried in a vacuum to give the
title
compound (12.55 g, 95%).
48
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
Step 4b:
Bu3Sn S
JoH
Into a solution of the compound of Step 4a (5.02 g, 26.00 mmol) in THF (80
mL) was treated with NaH (95%, 730 mg, 28.9 mmol) at room temperature for 50
minutes with stirring. It was chilled to -78 C before n-BuLi (1.6 M in
hexanes, 20
mL, 32 mmol) was charged. The mixture was kept at -78 C for 1 hour before n-
Bu3SnC1(17.6 mL, 65 mmol) was introduced. The mixture was warmed up
naturally to room temperature and stirred over night. The volatile was
evaporated
off and the residue was partitioned (ethyl acetate and saturated NaHCO3). The
organics were washed with water, brine, and then dried (Na2SO4). Evaporation
followed by chromatography (silica, hexanes/ethyl acetate) gave the title
compound
(4.51 g, 43%).
Step 4c:
O
Br N
a
ON
1
Into a 250 mL round bottom flask was charged 2-amino-6-bromopyridine
(25.0 g, 0.144 mol) and phthalic anhydride (21.4g, 0.144 mol). The solid
mixture in
the open flask (with a slow flow of N2) was heated to 175 C and the
temperature
was kept there for lhour or until no vapor comes out. It was cooled down to
room
temperature and vacuumed for 10 hours to give the title compound as a tan
solid
(100% yield).
Step 4d: ~ S
N I
C:N OH A mixture of the compound from Step 4b (4.50 g, 11.16 mmol), the
compound from Step 4c (3.72 g, 12.28 mmol), and Pd(PPh3)4 (645 mg, 0.56 mmol)
in PhMe (50 mL) was degassed and heated at 100 C under N2 for 17 hours before
being cooled to 0 C. The insoluble was collected by filtration and washed with
PhMe to give the title compound (2.90 g). The filtrate and washings were
49
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
evaporated and the residue was chromatographed (silica, hexanes/ethyl acetate)
to
give another crop of the title compound (0.20 g).
ESIMS m/e: 337 (M+H)+.
Step 4e:
N \N I ~
CI
O
A suspension of the compound from Step 4d (3.10 g, 9.22 mmol) in
methylene chloride (50 mL) was treated with thionyl chloride (3.35 mL, 46.08
mmol) at 0 C. The mixture was warmed up naturally to room temperature and
stirred for 16 hours. The volatile was evaporated off. The residue was
partitioned
(CHaC12/saturated NaHCO3). The organics were washed with water, brine, and
then
dried (Na2SO4). The volatile was removed by evaporation and dried in vacuo to
give the title compound (3.253 g, 100%).
ESIMS m/e: 355/357 (M+H)+.
Step 4f:
o
N s
N
I / O-N
O
0
Into a solution of N-hydroxylphthalimide (2.40 g, 14.7 mmol) in DMF (20
mL) was added NaH (95%, 332 mg, 13.8 mmol) at 0 C. It was warmed up to room
temperature and stirred for 1 hour. It was added into a solution of the
compound
from Step 4e (3.25 g, 9.2 mmol) in DMF (25 mL). The mixture was stirred at 40
C
for 16 hours before being cooled to room temperature. It was diluted with
saturated
NaHCO3 and water. The insoluble was collected by filtration, washed with
saturated NaHCO3 and water, and dried to give the title compound (3.930 g,
89%).
ESIMS m/e: 482 (M+H)+.
Step 4g:
/
~N Is
/ 25 HZN 0-NHZ
A suspension of the compound from Step 4f (1.00 g, 2.08 mmol) in
methanolic ammonia (2M, 20 mL, 40 mmol) was heated at 55 C for 2 hours before
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
being cooled to room temperature. The insoluble was filtered off and washed
with
MeOH. The combined filtrate and washings were evaporated. The residue was
added CHZCl2 to dissolve the crude title compound (548 mg) after evaporation.
ESIMS m/e: 222 (M+H)+.
Step 4h:
N,Ac
O O Ac ~N~
HO 0
O "''O O
0 1"'O
NI
A mixture of compound of formula VIII where X and Y taken together with
the atom that they are attached is C=N-Ac, U = H, V = OH, Z = H, Rp = Ac and W
= NMe2 (356 mg, 0.50 mmol), 2-pyridylacetic acid hydrochloride (174 mg, 1.0
mmol), 1-(3-dimethylaminopropyl)-3-ethylcabodiimide hydrochloride (EDC-HCI,
192 mg, 1.0 mmol), triethylamine (0.28 mL, 2.0 mmol) and DMAP (10.0 mg) in
methylene chloride (5.0 mL) was stirred at room temperature for 22 hours
before
more 2-pyridylacetic acid hydrochloride (87 mg, 0.5 mmol) and EDC-HCI (192 mg,
1.0 mmol). It was stirred for another 3 hours before being partitioned (ethyl
acetate
and 10% K2C03). The organics were washed with water, brine, and then dried
(Na2SO4). The volatile was removed by evaporation and dried in a vacuum to
give a
yellow foam (450 mg) as the crude title compound.
ESIMS m/e: 832 (M+H)+.
Step 4i:
/ ~
_N g N,Ac
H2N O Ac "'N/
O O 1
HO 0~,,
'l"O O
O "'O
0 N /
Into a solution of the crude compound from Step 4g (166 mg, -0.62 mmol)
in ethanol (5.0 mL) and HCl (1 M, 2.5 mL) at-5 C was added the crude compound
from Step 4h (450 mg, 0.5 mmol at most). After being stirred for one hour,
more
51
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
crude compound from Step 4g (50 mg, -0.18 mmol) was added. The mixture was
stirred for another 1 hour before partition (ethyl acetate and saturated
NaHCO3).
The organics were washed with water, brine, and then dried (Na2SO4). It was
evaporated and the residue was chromatographed (silica, hexanes/acetone) to
give
the title compound (332 mg, 64%) as a 2:1 mixture.
ESIMS m/e: 1035 (M+H)+.
Step 4j:
/ \ / I
c
i_N g NAc
H2N O':~,~=~' \N/
~ HO~,,
HO Ory Iv
'" O O
O N
A solution of the compound from Step 4i (100 mg) in MeOH (3 mL) was
stirred at room temperature for 70 hours before evaporation to give the title
compound. The two bridged oxime isomers were separated by HPLC.
E-oxime isomer: ESIMS m/e: 993 (M+H)+. 13C NMR (CDC13, 125 MHz): 184.6,
178.0, 172.5, 170.4, 158.0, 153.9,153.2, 151.0, 149.1, 145.4, 141.8, 138.1,
136.6,
127.5, 124.4, 123.7, 122.3, 109.1, 106.7, 103.0, 82.3, 79.4, 78.5, 78.3, 76.4,
75.0,
70.8, 70.5, 69.1, 65.4, 63.1, 62.4, 43.8, 42.7, 40.4, 39.9, 38.3, 36.8, 35.8,
29.7, 29.2,
25.1, 23.2, 21.0, 19.9, 19.1, 17.5, 15.0, 14.3, 12.1, 9Ø
Z-oxime isomer: ESIMS m/e: 993 (M+H)+. 13C NMR (CDC13, 125 MHz): 184.4,
176.6, 173.7, 170.2, 158.0, 156.1, 153.9, 151.1, 149.1, 145.5, 141.2, 138.1,
136.6,
128.0, 124.4, 123.8, 122.3, 109.1, 106.7, 103.0, 83.3, 80.2, 79.0, 77.7, 77.5,
75.8,
70.8, 70.6, 70.5, 69.1, 65.4, 58.7, 43.2, 40.3, 39.1, 38.5, 36.5, 36.0, 29.7,
29.2, 25.2,
22.7, 21.1, 20.1, 19.6, 16.8, 15.4, 14.6, 11.3, 9.1.
Example 5. Compounds with improved antibacterial activities.
Table IV below provides MIC data of species from U.S. Patent No. 6,878,691 and
U.S. Patent Application Publication No. 2004/0053 86 1.
The values in the table are minimum inhibition concentration (MIC) and are
expressed as ug/mL.
Assays for MIC are described above.
52
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
Table IV
S. S. S. S. S. S. S. H.
Com- Structure aureus aureus aureus pneumoniae pneumoniae pyogenes pyogenes
influen
pound 29213 27660 33591 7701 700906 1323 2912 zae
33929
Al 0.25 0.13 >64 0.5 0.25 0.5 8 8
N N,Ac
O
HO~ 'N ,=~,"HO, N-
O
O
O A2 N 0.5 0.25 >64 0.5 0.25 1 16 16
N
N.Ac
0
HO~ 'N =,O HO N_
O 0
O O
A3 N N 0.25 0.25 >64 2 0.13 16 32 8
N N,Ac
O
HO~ 'N ,O ..HO N_
O 'O~
O O
A4 ~ ,N 8 64 64 >64
~ o
~N
HOV OHO
'O 0 O
O "0=.,
OAc
OMe
A5 >64 2 64 >64 >64
NH
O'~~=" N
O
HOV oHO,..
O ,,, Oo
O "9OH
A6 9' 1 1 16 1 8 2 8 >64
N)\
HO O HO.,,
O "O O
O 'O
O IN
53
CA 02605295 2007-10-16
WO 2006/119313 PCT/US2006/016882
Table V provides data for compounds of this invention demonstrating improved
microbiological activities against gram negative bacteria and resistant
organism. The
values in Table V are minimum inhibition concentration and are expressed as
ug/mL.
Table V
S. aureus S. aureus S. S. S. S. S. H.
Com- Structure 29213 27660 aureus pneumoniae pneumoniae pyogenes pyogen
influen
pound 33591 7701 700906 1323 es zae
BI H=N~N \ ~ <=0.06 <=0.06 >64 <=0.06 1 0.25 412 23929
S
0
H O \N t,o Hq N-
O O
0.125 <=0.06 >64 0.125 0.5 0.5 2 2
B2 H2N N,Ac
O
HO~ N O HO \N~
O O
O
O O
B3 H,N ,N ~ i <=0.06 <=0.06 32 0.125 0.25 0.5 4 2
0
0
\
NO~~~..0
H~ ~\[N_
O~
O O
0.125 0.125 32 0.125 0.125 0.5 1 4
B4 N N0 Ac
HO~~~ HO N
I :~ A
O O
The patent and scientific literature referred to herein establishes the
knowledge that is available to those with skill in the art. All United States
patents
and published or unpublished United States patent applications cited herein
are
incorporated by reference. All published foreign patents and patent
applications
cited herein are hereby incorporated by reference. All other published
references,
documents, manuscripts and scientific literature cited herein are hereby
incorporated
by reference.
While this invention has been particularly shown and described with
references to preferred embodiments thereof, it will be understood by those
skilled
in the art that various changes in form and details may be made therein
without
departing from the scope of the invention encompassed by the appended claims.
54