Note: Descriptions are shown in the official language in which they were submitted.
CA 02608539 2007-11-14
WO 2006/122977 PCT/EP2006/062446
1
PROCESS FOR PREPARING 3a((3)-7a((3)-DIHYDROXY-6a((3)-ALKYL-5(3-
CHOLANIC ACID.
Description
FIELD OF THE INVENTION
The present invention concerns a process for the preparation of 3a-7a((3)-
dihydroxy-6a((3)-alkyl-5p-cholanic acids.
STATE OF THE ART
Farnesoid X receptors (FXR) are initially orphan nuclear receptors, identified
for
the first time from a cDNA library of rat liver (B.M Forman et al., Cell.
81:687-693
(1995)), they are members of the family of nuclear receptors of ligand-
activated
transcription factors, including the receptors of steroid, retinoid and
thyroid
hormones (D.J. Mangelsdorf , et al, Cell.83:841-850(1995)).
Several bile acids of a natural type bind together and activate FXR in
physiological
concentrations as described in W000/37077 and in particular chenodeoxycholic,
deoxycholic, litocholic acids and the relative conjugates with taurine and
glycine.
It is also believed that FXR are involved in the regulation of the homeostasis
of bile
acids and of cholesterol.
W002/072598 describes 3-a,7-a-dihydroxy-6a-alkyl-(allyl)-5(3-cholanic acids
with
general formula (A)
O
OH
COH OH
H
(A)
in which R' is ethyl, propyl or allyl which are also agonists of Farnesoid X
receptors.
CA 02608539 2007-11-14
WO 2006/122977 PCT/EP2006/062446
2
In particular the compound with formula (I) in which R' = ethyl is two
magnitude
order more powerful than chenodeoxycholic acid, the most powerful natural FXR
agonist
The compounds with general formula (A), used in particular to increase HDL
cholesterol, to lower triglycerides for the prevention and treatment of
hepatic
diseases of cholestatic origin, are prepared with a process comprising the
following stages:
i) reacting 3-a-hydroxy-7-keto-5p-cholanic acid of formula (II)
O
OH
HOO
H
(II)
with dihydro pyrane to obtain the corresponding 3-a-tetrahydropyranyloxy-7-
keto-
5(3-cholanic acid of formula (B)
O
OH
aod" O
(B)
ii) reacting the compound (B) with an alkyl bromide with formula R'Br in which
R'
has the meanings indicated above, to obtain the compound (C)
CA 02608539 2007-11-14
WO 2006/122977 PCT/EP2006/062446
3
O
OEt
HO" Or
H =
R'
(C)
iii) reducing the compound (C) with sodium borohydride to give (D),
O
OEt
HOC "'OH
H =
R'
(D)
iv) hydrolysing (D) to give the compounds (A).
Even though this process comprises only few stages, it presents a series of
drawbacks.
Firstly, in all stages the reaction products are purified on a chromatographic
column, namely a very expensive separation method that cannot be realised on
an
industrial scale.
Moreover the reaction yield in stage (ii) is extremely low (12-13%), with a
considerable decrease in the global yield, which is lower than 3.5%.
Moreover, still in this stage, hexamethylenphosphonamide is used as a
reactant,
which is a known cancerogenic agent
SUMMARY OF THE INVENTION
The Applicant has now found a process which makes it possible to obtain both
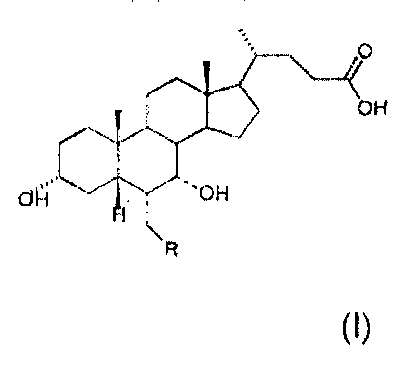
compounds with general formula (I)
CA 02608539 2007-11-14
WO 2006/122977 PCT/EP2006/062446
4
O
OH
"H C'OH
H
R
(I)
in which the dashed bond (----) in position 6 and 7 indicates that the
substituent
may be in position a or (3 chosen in the class consisting of:
i) 3-a,7-a-dihydroxy-6-a-alkyl-5R-cholanic acid with general formula (IA)
O
OH
OI-i O H
H
R
(IA)
ii) 3-a,7-a-dihydroxy-6-(3-alkyl-5(3-cholanic acid with general formula (IB)
O
OH
HO\" 4H0H
R
(IB)
CA 02608539 2007-11-14
WO 2006/122977 PCT/EP2006/062446
iii) 3-a,7-E3-dihydroxy-6-a-alkyl-5(3-cholanic acid with general formula (IC)
O
OH
HOOH
H =
R
5 (IC)
in which R is a linear or branched Cl-C5 alkyl, comprising the following
stages
a) esterifiying 3a-hydroxy-7-keto-5(3-cholanic acid (II)
O
OH
HO\\ Or
H
(II)
in methanol in an acidic environment to obtain methyl 3a-hydroxy-7-keto-5p-
cholanate (III),
O
OMe
HO\\ O
H
(III)
CA 02608539 2007-11-14
WO 2006/122977 PCT/EP2006/062446
6
b) Silylating methyl 3a-hydroxy-7-keto-5(3-cholanate (III) with
trimethylchlorosilane
to obtain the corresponding 3-a-trimethylsiloxy-7-keto-5(3-cholanate (IV),
O
OMe
SIC 3)3 O
(IV)
c) Silylating methyl 3a-tri m ethylsi loxy-7-keto-5 P-ch ola n ate (IV)
obtained in stage
(b) with trimethylchlorosilane in the presence of a strong base to obtain
methyl
3a-,7a-di-trimethylsiloxy-6-en-5(3-cholanate (V),
O
OMe
0~ OSI(CH3)3
(~SI(C 3)aH
(V)
d) Reacting methyl 3a-,7a-di-trimethylsiloxy-6-en-5(3-cholanate (V) with the
aidehyde R-CHO in which R has the meanings indicated above and a Lewis
acid, to obtain methyl 3a-hydroxy-6-aIkylidene-7-keto-5(3-cholanate (VI),
CA 02608539 2007-11-14
WO 2006/122977 PCT/EP2006/062446
7
O
OMe
OFi O
H I
(VI)
e) hydrolysis of methyl 3a-hydroxy-6-alkylidene-7-keto-5(3-cholanate to 3a-
hydroxy-6-alkylidene-7-keto-5(3-colanic acid (VII),
O
OH
OH O
H
R
(VII)
f) hydrogenating 3a-hydroxy-6-alkylidene-7-keto-5(3-cholanic in an aqueous
alkaline environment with Pd/C to 3a-hydroxy-6p-alkyl-7-keto-5(3-cholanic acid
O
OH
OFI
R
(VIII)
CA 02608539 2007-11-14
WO 2006/122977 PCT/EP2006/062446
8
g) optionally heat treating the intermediate (VIII) in an aqueous alkaline
environment to obtain the corresponding 3a-hydroxy-6a-alkyl-7-keto-5p-
cholanic (IX)
O
OH
dH O
R
(IX)
h) reducing the ketonic group in position (7) to 7-hydroxy group of the
intermediate (VIII) or (IX) according to one of the following alternative
operating
conditions:
h') reducing 3a-hydroxy-6a-alkyl-7-keto-5R-cholanic compound (IX) with
metallic
hydride to 3a-,7a-di-hydroxy-6a-alkyl -5(3-cholanic acid (IA),
h") reducing 3a-hydroxy-6a-alkyl-7-keto-5p-cholanic compound (IX) in the
presence of sodium and alcohol and obtaining 3a-,7(3-di-hydroxy-6a-alkyI-5(3-
colanic (IC);
h"') reducing 3a-hydroxy-6(3-alkyl-7-keto-5(3-cholanic (VIII) in the presence
of a
metallic hydride to 3a-,7a-di-hydroxy-6p-alkyl-5p-cholanic (IB).
The process according to the present invention in particular for obtaining 3a-
,7a-
di-hydroxy-6a-alkyl-5p-cholanic acids (IA) presents considerable advantages
with
respect to the known process described above. In fact, although it
contemplates a
larger number of stages, it allows the product with formula (I) to be obtained
with
decidedly satisfactory global yields (24.6%), in any case decidedly higher
than
those of the known process. Moreover, the intermediates do not need to be
purified by chromatography and the use of reagents, such as the highly toxic
hexamethylenphosphonamide is avoided.
CA 02608539 2007-11-14
WO 2006/122977 PCT/EP2006/062446
9
Lastly, with the process of the present invention and as shown above, it is
possible
to obtain the new compounds of formula (IB) and (IC) which may be used a
hepatoprotectors in particular for the treatment and prevention of hepatic
diseases
of cholestatic origin.
The present invention therefore concerns pharmaceutical compositions
containing
as active principle at least one of the acids (I-B) and (I-C) and the
respective
pharmaceutically acceptable salts in combination with suitable excipients
and/or
diluents.
DETAILED DESCRIPTION OF THE INVENTION
The esterifying reaction of the 3a-hydroxy-7-keto-5p-cholanic acid (II) in
stage (a)
of the process of the present invention is preferably carried out at a
temperature
between 30 and 60 C in an acid environment, in which the acid is preferably
methanesulphonic acid.
The silylating reaction of the hydroxy group in position 3a- of methyl 3a-
hydroxy-7-
keto-5p-cholanate contemplated in stage (b) of the process of the present
invention is preferably carried out in an apolar solvent, more preferably an
aromatic solvent, even more preferably toluene, in the presence of a hydrogen
ion
acceptor preferably consisting of a tertiary amine, of aliphatic, alicyclic or
heteroaromatic type, even more preferably said tertiary amine is
triethylamine.
According to a particularly preferred embodiment, before being used in stage
(c),
methyl 3-a-trimethylsiloxy-7-keto-5P-cholanate is not isolated and purified,
but on
the contrary in this stage the oily residue is used which is obtained after
evaporating the reaction solvent from which the salts have previously been
removed by water extraction.
The subsequent silylation of the ketonic group in position 7 contemplated in
stage
(c) of the process of the present invention is preferably carried out using as
the
strong base an alkaline amide obtained from ammonia or an alkaline amide of a
secondary aliphatic amine. According to a particularly preferred solution,
lithium
diisopropylamide is used as the strong base. This reaction is preferably
carried
out in a polar aprotic solvent, and even more preferably said solvent is
tetrahydrofuran.
CA 02608539 2007-11-14
WO 2006/122977 PCT/EP2006/062446
According to a preferred embodiment the product obtained in stage (c), before
being used in the following stage (d), is not isolated and purified, but on
the
contrary in this case too the oily residue is used obtained by evaporating the
reaction solvent from which the salts have been previously extracted with
water.
5 Stage (d) is preferably carried out in an apolar solvent, preferably chosen
from
alkyl halide, and even more preferably this solvent is methylene chloride.
Stage (d) is preferably carried out using boron trifluoride etherate as the
Lewis
acid at a temperature between -90 and -60 C for a period of 2 to 4 hours in
the
presence of the aldehyde R-CHO in which R has the desired meanings.
10 Subsequently, the reaction mixture is reacted at a temperature between 0
and
35 C for a period of 1 to 6 hours.
In this case too, before being used in the following stage (e), the product
obtained
in stage (d) is not isolated and purified, but the oily residue is used which
was
obtained after evaporating the reaction solvent, from which the salts and
water-
soluble components have been removed with water extraction.
Stage (e) is preferably carried out in an alcoholic solvent, preferably
methanol, in
the presence of an alkaline hydroxide, even more preferably said alkaline
hydroxide is an aqueous solution of 30% sodium hydroxide.
The temperature is preferably comprised between 20 and 60 C.
Stage (e) reaction product is preferably isolated after acidification, by
crystallisation with an organic solvent, preferably chosen from ethyl acetate
and
acetone, possibly in the presence of water.
The hydrogenation reaction contemplated in stage (f) is preferably carried out
in
an aqueous environment in the presence of an aqueous solution of sodium
hydroxide with pressure between 1 and 3 atmospheres. When the process of the
present invention contemplates stage (g), in particular when the compounds
with
general formula (IA) or (IC) have to be prepared, this stage is preferably
carried
out directly on the reaction mixture coming from hydrogenation reaction and is
preferably carried by heating said reaction at a temperature between 95 and
105 C for a few hours to allow the epimerization of the 6-p-ethyl group into 6-
a-
ethyl.
CA 02608539 2007-11-14
WO 2006/122977 PCT/EP2006/062446
11
The reaction product coming from stage (f) or possible stage (g) is isolated
from
the reaction mixture preferably using the following operating conditions.
1) adding the aqueous solution, from which the catalyst has been removed by
filtration is acidified preferably with 85% phosphoric acid,
2) ethyl acetate is added to the mixture obtained in stage (1) and the whole
is
heated at a temperature between 40 and 70 C.
3) this is then cooled to a temperature between 0 and 30 C and the precipitate
obtained is filtered and subsequently dried.
When the reduction of stage (h) is carried out according to the operative
conditions
contemplated in stage (h') to obtain the compound with formula (IA) or
according
to the operative conditions contemplated in stage (h"') to obtain the compound
with formula (IB) of the process of the present invention, the metallic
hydride is
preferably sodium borohydride and the reduction reaction is carried out in an
alkaline aqueous solution. The reaction is preferably carried out at a
temperature
between 70 and 105 C for 1 hour.
Instead, when the reduction of stage (h) is carried out according to the
operating
conditions contemplated in stage (h") it is preferably carried out in linear
or
branched Cl-C5 alcohol, even more preferably in sec-butyl alcohol, at the
solvent
reflux temperature. The product obtained from stage (h') or (h"') is
preferably
isolated according to the following operative conditions:
1') adding a water immiscible solvent to the reaction mixture, preferably an
apolar
solvent such as methylene chloride, and acidifying the mixture preferably with
phosphoric acid,
2') stirring and allowing to rest to the mixture obtained thereafter
eliminating the
aqueous phase,
3') extracting the product from the organic phase with water and ammonia,
4') adding phosphoric acid to the aqueous phase thus obtained and stirring the
whole for a few hours at a temperature between 20 and 50 C,
5') recovering and drying the precipitated product by filtration.
The process of the present invention is suitable in particular for the
preparation of
compounds with formula (I) in which R is preferably methyl.
CA 02608539 2007-11-14
WO 2006/122977 PCT/EP2006/062446
12
Some examples of preparation according to the process of the invention of the
compounds with formula (I) and in particular (IA), (IB) and (IC) in cui R =
methyl
are given for illustrative purposes, but not limitative.
EXAMPLE 1- PROCESS FOR PREPARING 3a-,7a-DIHYDROXY-6a-ETHYL-5(3-
CHOLANIC ACID of formula (IA) in which R =methyl
a) preparation of methyl 3-a-hydroxy-7-keto-5(3-6holanate (III).
17.0kg of 3-a-hydroxy-7-keto-5p-cholanic acid, 68kg of methanol and 0.17kg of
methansulphonic acid are charged into a reactor. The reaction mixture is then
heated to 30-60 C for 1 hour and 25.5kg of demineralised water are added. The
mixture obtained is then stirred, cooled to 20-25 C until a good precipitation
is
obtained, then cooled further to 0-15 C. The precipitate is filtered and
washed with
a mixture of water and methanol and further dried in a oven at about 40 C.
15kg
of methyl 3a-hyd roxy-7-keto-5 P-chol an ate (III) is thus obtained.
Stoichiometric
yield 85.2%.
b) preparation of methyl 3a-trimethylsiloxy-7-keto-5p-cholanate (IV),
15.0kg of methyl 3a-hydroxy-7-keto-5p-cholanate, 45kg of toluene, 7.5kg of
triethylamine, 7.5kg of trimethylchlorosilane are charged into a reactor.
The mixture is heated to 70-80 C and is kept under stirring at that
temperature for
about 1 hour, then 37.5kg of water are added and the mixture is stirred at 15-
20 C.
The lower aqueous phase is then separated and eliminated. The organic phase is
concentrated until an oily residue is obtained, which 15 kg of tetrahydrofuran
are
added to.
The solution thus obtained containing methyl 3a-trimethylsiloxy-7-keto-5(3-
cholanate (IV) is used in the following stage (c).
c) preparation of methyl 3a-, 7a-di-trimethylsililoxy-5~-cholanate (V)
30kg of tetrahydrofuran are loaded in a reaction, then the mixture is brought
to a
temperature between -90 and -60 C, 9.8kg of 100% lithium diisopropylamide
and 9.3kg of trimethylchlorosilane are added, and the whole solution of
tetrahydrofuran prepared in (b) and containing methyl 3-a-trimethylsiloxy-7-
keto-
5(3-cholanate is poured. The mixture is then stirred for about 1 hour at a
temperature between-60 and -90 C for 1 hour. A solution of 4.50kg of sodium
bicarbonate and 60 kg of water is then poured and the mixture is stirred at 0-
10 C,
CA 02608539 2007-11-14
WO 2006/122977 PCT/EP2006/062446
13
and the lower aqueous phase is separated and eliminated. The lower phase is
then concentrated until an oily residue is obtained, which 45.0Kg of methylene
chloride- are added to.
The solution of methyl 3a-,7a-di-trimethylsililoxy-5p-cholanate thus obtained
is
sent to the next stage (d).
d) preparation of methyl 3a-hydroxy-6-ethyliden-7-keto -5R-cholanate (VI) in
which
R = Methyl
The whole solution of methyl 3a,7-a-di-trimethylsililoxy-5(3-cholanate in
methylene
chloride coming from the preceding example in charged into a reactor and
cooled
to -90/-60 C 1.97kg of acetaldehyde and 5.5kg of boron trifluoride etherate
are
then added. The reaction mixture is kept under stirring at the above
temperature
for 2/4 hours. After that it is heated to 30-35 C and kept at that temperature
for
about 2/4 hours. Then 60kg of water are added. The mixture obtained is stirred
and the aqueous phase is separated. The solution thus obtained containing
methyl
3a-hydroxy-6-ethyliden-7-keto-5(3-cholanate is sent to the next stage.
e) preparation of 3a-hydroxy-6-ethyliden-7-keto-5p-cholanic (VII) acid in
which R
=CH3,
The solution of methyl 3-a-hydroxy-6-ethyliden-7-keto-5p-cholanate in
methylene
chloride obtained in the previous stage is charged into a reactor. The solvent
is
then removed by distillation until an oily residue is obtained, which 15 kg of
methanol are added to.
The reaction mixture is then heated to 45-50 C and 7.5kg of 30% sodium
hydroxide are poured , and the reaction mixture is kept at the above
temperature
for about 1 hour. Then 30kg of water are added. 45.0kg of methylene chloride
and
7.5kg of 85% phosphoric acid are subsequently added. The lower organic phase
is
separated and the aqueous phase is eliminated subsequently. The solvent is
removed from the organic phase by distillation until a pasty residue is
obtained.
About 37.5kg of ethyl acetate are added to the residue and the mixture is
heated
to 65-75 C, then cooled to 10-35 C. The precipitate obtained, filtered and
washed
with ethyl acetate, is dried.
CA 02608539 2007-11-14
WO 2006/122977 PCT/EP2006/062446
14
8.0kg of 3-a-hydroxy-6-ethyliden-7-keto-5p-cholanic acid are obtained, with a
stoichiometric yield of 51.8% calculated on methyl 3-a-hydroxy-7-keto-5(3-
cholanate.
preparation of 3-a-hydroxy-6-(3-ethyl-7-keto-5~-cholanic acid (IX) in which
R=CH3l
8.0 kg of 3-a-hydroxy-6-a-ethyliden-7-keto-5(3-cholanic acid, 48.0kg of water,
5.1kg of 30% sodium hydroxide, 0.80kg of 5% Palladium/Carbon are charged into
a reactor. The reaction mixture is hydrogenated at a pressure between 1 and 3
atmospheres, until the hydrogen absorption is no longer noted.
(g) preparation of 3a-hydroxy -6-a-ethyl-7-keto-5(3-cholanic acid (IX)
At the end of the reaction the mixture is heated to 95-105 C and is kept at
that
temperature for a few hours to allow the 3a-hydroxy-6-(3-ethyl-7-keto-5(3-
cholanic
acid (VIII) to convert into the corresponding epimer of the desired 3a-hydroxy
-6-
a-ethyl-7-keto-5(3-cholanic acid (IX).
The suspension is filtered, and the catalyst is recovered. 5.1 kg of 85%
phosphoric
acid 9.6 kg of ethyl acetate are added to the filtered solution, and the
reaction
mixture is heated to a temperature between 40 and 70 C. It is cooled to a
temperature between 0 and 30 C and the precipitate is recovered by filtration.
After washing with ethyl acetate, the precipitate is dried in a oven at 65 C.
5.0kg of
3a-hydroxy-6a-ethyl-7-keto-(3-cholanic acid are obtained. Stoichiometric
yield:
62.2%.
21
18 0
24
19 .17 23 OH
14
3 6 7
OH 0
H = 25
26
CA 02608539 2007-11-14
WO 2006/122977 PCT/EP2006/062446
Analysis
3-a-hydroxy-6-a-ethYl-7-keto-5p-cholanic (IX)
C26H4204 M.P. 185-188 C
The 1H-NMR analysis carried out with the instrument Bruker DRX-ADVANCE-
5 400Mhz, dissolving the specimen in CD3OD, gave the following results:
0.62 ppm (s, 3H of methyl C18); 0.76ppm, J=7.4Hz (t, 3H of methyl C26);
0.89ppm
J= 6.5Hz, (d, 3H, of methyl C21); 1.18ppm (s, 3H, of methyl C19), 2.21 ppm (m,
2H,
-CH2- of C23); 2.50ppm, J=11.17Hz (ypt, CH on C8); 2.85ppm J =13Hz and J=
5.4Hz (dd 1 H in C6), 3.50ppm (m, CH on C3).
10 The 13C NMR analysis carried out with the instrument Bruker DRX-ADVANCE-
200Mhz, dissolving the specimen to be analysed in a mixture of CD3OD and
CDCI3, gave the following results:
212.82ppm (C7); 179.44ppm (C24), 71.26ppm (C3), 54.77ppm (C17), 51.98ppm
(C14),18.84ppm (C21), 18.34ppm (C26), 12.09ppm (C18).
15 h') preparation of 3a,7a-dihydroxy-6a-ethyl-5j3-cholanic acid with formula
(I) in
which R= methyl.
5.0kg of 3a-hydroxy-6a-ethyl-7-keto-(3-cholanic acid, 5.0kg of water, 2.50 kg
of
sodium hydroxide are loaded in a reactor. The mixture -is then heated to 70-
105 C
and a mixture of sodium borohydride dissolved in 2.50 kg of water is poured,
the
mixture is then kept warm for 1 hour, cooled to room temperature, and 10.0kg
of
demineralised water, 15.0kg of methylene chloride and 3.00kg of 85% phosphoric
acid are added. The mixture is stirred, the lower organic phase is separated
and
the aqueous phase is removed.
Crystallization of the crude product is obtained by cooling the organic
solution.
This product is dissolved in 50kg of demineralised water and 1.10kg of 30%
ammonia. The mixture is then stirred until a complete solution is obtained,
and
keeping the mixture at 20-50 C, 1.50kg of phosphoric acid is poured. The
precipitated mixture is stirred, always at a temperature between 20 and 50 C,
then
the precipitate is recovered by filtration, washed with water and dried.
4.50 kg of 3a-,7a-di-hydroxy-6a-ethyl-5(3-cholanic acid of formula (I) are
obtained,
in which R= methyl. Stoichiometric yield: 89.6%.
CA 02608539 2007-11-14
WO 2006/122977 PCT/EP2006/062446
16
EXAMPLE 2 preparation of 3-a,7a-di-hydroxy-6p-ethyl-5R-cholanic acid of
formula
(IB) in which R = methyl.
The 3-a-hydroxy-6(3-ethyl-7keto-5p-cholanic acid of formula (VIII) prepared as
described in example 1 stages (a)-(f) and isolated as described in stage (g),
is
reduced using the same operating conditions described in example 1 stage (h').
3-
a,7a-di-hydroxy-6(3-ethyl-5p-cholanic acid of formula (IB) is then obtained in
which
R = methyl.
Analysis
21
18 ~/O
( 24
19 ~7 23 \O H
14
6 7
,,OH
H 25
26
3-a,7-a-di-hydroxy-6-(3-ethyl-5(3-cholanic (IB)
C26H4404 m.p. 115-118 C
The 1H-NMR analysis carried out with the instrument Bruker DRX-ADVANCE-
400MHz, dissolving the specimen in CD3OD, gave the following results:
0.70ppm (s, 3H of methyl C18); 0.95ppm (s, 3H, of methyl C19), 1.O0ppm,
J=7,65Hz
(t, 3H of methyl C26); 1.45ppm J= 3.5Hz, (d, 3H, of methyl C21);
2.25 ppm (m, 2H, -CH2- of C23); 3.40ppm (m, CH on C3), 3.62ppm (m, CH on C7).
The 13C NMR analysis carried out with the instrument Bruker DRX-ADVANCE-
200Mhz, dissolving the specimen to be analysed in a mixture of CD3OD and
CDCI3, gave the following results:
177.91 ppm (C24), 72.18ppm (C3), 71.68ppm (C7); 55.79ppm (C17), 50.83ppm
(C14),18.17ppm (C21), 14ppm (C26), 11.60ppm (C18).
EXAMPLE 3 preparation of 3-a,70-di-hydroxL-6a-ethyl-5p-cholanic acid with
formula (IC) in which R= methyl
According to the operative conditions described in example 1 stages (a)-(g),
the
CA 02608539 2007-11-14
WO 2006/122977 PCT/EP2006/062446
17
intermediate (IX) is prepared, to which is added until complete solution sec-
butyl
alcohol in which sodium has previously been dissolved in molar quantities with
respect to the compound (IX) between 3:1 and 3:2. 3-a,7P-di-hydroxy-6a-ethyl-
5R-
cholanic acid with formula (IC) is obtained in which R= methyl
Analysis
3-a,7-R-di-hydroxy-6-a-ethyl-5p-cholanic (IC)
C26H4404 m.p. 217-219 C
21
18 0
24
17 23 O H
&14
H :
OH H
= 25
26
The 1H-NMR analysis carried out with the instrument Bruker DRX-ADVANCE-
400MHz, dissolving the specimen in CD3OD, gave the following results:
0.56ppm (s, 3H of methyl C18) ; 0.73ppm, J=7.4Hz (t, 3H of methyl C26); 0.81
ppm
(s, 3H, of methyl C19), 0.82ppm J= 4.60Hz, (d, 3H, of methyl C21); 2.21ppm (m,
2H,
-CH2- of C23), 3.80ppm (br, 0-H of hydroxyl on C3, on C7 and of carboxyl C24);
3.10ppm (m, CH on C7); 3.44ppm (m, CH on C3).
The 13C NMR analysis carried out with the instrument Bruker DRX-ADVANCE-
200MHz, dissolving the specimen to be analysed in a mixture of CD3OD and
CDCI3, gave the following results:
179ppm (C24), 75.65ppm (C7), 71.87ppm (C3), 56ppm (C17), 55ppm (C14), 18.4ppm
(C21), 12.24ppm (C26), 11.20ppm (C18).