Note: Descriptions are shown in the official language in which they were submitted.
DEMANDE OU BREVET VOLUMINEUX
LA PRESENTE PARTIE DE CETTE DEMANDE OU CE BREVET COMPREND
PLUS D'UN TOME.
CECI EST LE TOME 1 DE 2
CONTENANT LES PAGES 1 A 129
NOTE : Pour les tomes additionels, veuillez contacter le Bureau canadien des
brevets
JUMBO APPLICATIONS/PATENTS
THIS SECTION OF THE APPLICATION/PATENT CONTAINS MORE THAN ONE
VOLUME
THIS IS VOLUME 1 OF 2
CONTAINING PAGES 1 TO 129
NOTE: For additional volumes, please contact the Canadian Patent Office
NOM DU FICHIER / FILE NAME:
NOTE POUR LE TOME / VOLUME NOTE:
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
MODULATION OF CELL BARRIER DYSFUNCTION
The invention was made with U.S. Government support under contract nos.
DE12322, DE00470, R01-GM-62344-01 and DE015830 awarded by the National
Institutes
of Health. The U.S. Government has certain rights to this invention.
Cross Reference to Related Application
This application claims the benefit of Provisional U.S. Patent Application
Nos.
60/687,568, filed June 3, 2005, 60/731,009, filed October 28, 2005, and
60/760,851, filed
January 20, 2006. The application also claims the priority benefit of
international (PCT)
patent application PCT/US06/07892, which designates the United States. Each of
the above-
identified patent applications is hereby expressly incorporated herein by
reference in its
entirety.
Field of the Invention
The invention generally relates to the field of prophylactic and therapeutic
use
of opioid receptor antagonists in modulating cell barrier dysfunction
characteristic of a
disorder or disease afflicting vertebrates (e.g., mammals) such as.humans.
Background
Vertebrate (e.g., mammalian) cell barrier dysfunction results in a change in
permeability of a cell barrier contributing to the internal
compartmentalization of a
multicellular organism and/or to the segregation of internal and external
environments of
such an organism. Typically, cell barrier dysfunctions are revealed as an
increase in the
permeability of a particular cell layer, such as the layer of endothelial
cells found in the
vasculature of higher eukaryotes or the layer of epithelial cells found in
tissues exposed to the
external environment, including the skin, lung and gut. A variety of disorders
and diseases
afflicting vertebrates such as humans can involve cell barrier dysfunction.
Collectively, these
maladies affect the quality of life of humans and other animals (e.g.,
domesticated animal,
zoo or exotic animals, pets) while contributing to the increasingly burdensome
cost of health
care. In the following description, a particular cell type, such as
endothelial cells or epithelial
cells, will be used for ease of exposition, with the understanding that cell
barrier dysfunction
applies to a variety of cell types, including the aforementioned endothelial
and epithelial
cells.
1
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
For example, endothelial cells provide a semi-selective barrier between the
blood and underlying vasculature. Disruption of this barrier results in
increased vascular
permeability and organ dysfunction. For example, the inflammatory process
increases
macromolecular transport by decreasing cell-cell and cell-matrix adhesion and
by increasing
centripetally directed tension, resulting in the formation of intercellular
gaps. Agents that
enhance endothelial cell barrier function provide a desirable therapeutic
strategy for a variety
of inflammatory diseases, atherosclerosis and acute lung injury.
Cell barrier dysfunction can be caused or exacerbated by a variety of factors,
including microbial pathogens, and by a variety of agents, including thrombin,
ionomycin,
LPS, and the like. Microbial pathogens such as P. aeruginosa can express
various peptides
and virulence factors that can disrupt barrier function. Microbiologists have
long recognized
that many bacteria activate their virulence genes in response to ambient
environmental cues.
In general such physico-chemical cues signal environmental stress or
adversity, such as
changes in redox status, pH, osmolality, and the like. For example, P.
aeYuginosa and other
bacteria can express a lectin/adhesin PA-I. The distribution of PA-I in
bacteria can be either
primarily intracytoplasmic or extracellular, depend#ig on its environment.
When bacteria are
grown in ideal growth conditions, about 85% of PA-I is located intracellularly
with small, but
significant, amounts located within the cytoplasmic membrane, on the outer
membrane, and
in the periplasmic space. In sharp contrast, within the intestinal tract of a
stressed host, PA-I
abundance is increased and localizes to the outer membrane, facilitating the
adherence of P.
aeruginosa to the intestinal epithelium. In addition, there is evidence that
free PA-I is shed
into the extracellular milieu and can be detected at concentrations as high as
25 g/ml in both
culture supernatants and sputum from P. aeruginosa infected lungs. This
finding is of
considerable importance, as treatment of cultured epithelial cells (e.g. T-84,
Caco-2bbe,
MDCK, airway epithelial cells) with 25 g/ml purified PA-I causes a profound
permeability
defect. This effect is also seen in the intestinal tract in vivo. These
effects are of clinical
significance because P. aeruginosa is the most common gram-negative bacterium
isolated
among cases of nosocomial infection and carries the highest reported fatality
rate of all
hospital acquired infections. The mere presence of this pathogen within the
intestinal tract of
a critically ill patient is associated with a four-fold increase in mortality,
independent of its
dissemination to remote organs.
Although there has been very little work on specific membrane sensors that
activate virulence gene expression in P. aeruginosa, two sensor proteins
located within the
2
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
cell membrane of P. aeruginosa, termed CyaB, GacS have been shown to respond
to three
known external signals, host cell contact, low calcium, and beet seed extract.
CyaB (via
cAMP) and GacS4 (via phosphorylation), activate the transcriptional regulators
Vfr and GacA
respectively, which, along with the cell density sensitive PcrA, exert global
regulatory
influences on two central systems for virulence gene regulation in P.
aeruginosa, the QS and
RpoS signaling systems. Mutant strains defective in CyaB and GacS have
attenuated
lethality in mice following lung instillation.
Host cellular elements such as seed extract and cell contact, activate the
membrane biosensors CyaB and GacS. These two component transmembrane alarm
systems
then activate two main global regulators of virulence, Vfr and GacA. Vfr is
involved in the
activation of LasRI which in turn promotes the activation of the Rh1RI system
of QS. GacA
induces the transcription of lasR and YhIR genes, and is also implicated in
the expression of
rpoS. Finally a third system, PQS, induces expression of both RhIR and RpoS.
Thus,
activation of any of the membrane biosensors could lead to the expression of
PA-I with the
involvement of a number of different pathways.
Opioids comprise a large group of compounds that are distributed in virtually
every tissue of the body and are abundantly released in response to various
stress conditions;
for example dynorphin and (3-endorphin appear to be the predominantly released
endogenous
opioids following stress (S. Yoshida, et al., Surg Endosc 14, 137 (2000), C.
Stemini, S.
Patiemo, I. S. Selmer and A. Kirchgessner, Neurogastroenterol Motil 16 Suppl
2, 3 (2004)).
Morphine and morphine derivatives (opiates) as well as morphine-like compounds
(opioids)
are among the most widely used analgesic drugs in the world and are often
administered at
high doses even at continuous dosing intervals in post-operative care, chronic
pain
management, and in critically ill patients such as patients with advanced
cancer or AIDS.
Intravenously applied morphine has been demonstrated to accumulate at tissues
sites of
bacterial infection such as the intestinal mucosa, at concentrations as high
as 100 gM (P.
Dechelotte, A. Sabouraud, P. Sandouk, I. Hackbarth and M. Schwenk, Drug Metab
Dispos
21, 13 (1993)) and has been shown to readily cross the intestinal wall into
the lumen (M. M.
Doherty and K. S. Pang, Pharm Res 17, 291 (2000)). Therefore it is likely that
an
opportunistic pathogen such as P. aeruginosa, which is present in greater than
50% of the
intestines of critically ill patients within 3 days of hospitalization, is
exposed to both
endogenously released and exogenously applied opioid compounds. Clinical data
suggest
3
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
that bacterial transmigration across the gut may lead to increased rates of
sepsis in burn or
ICU patients who have diminished gut motility.
The association of opioids and infection is well established (Risdahl, et al.,
J
Neuroimmunol 83:4 (1998)), including evidence that opioids enhance HIV
infection of
human macrophages by upregulating CCR5 receptor. Ho et al., J. Pharm. And Exp.
Ther.
307:1158-1162 (2003). Nonetheless, most of the work in this area has focused
on the
suppressive effects of opioids on the immune system (Eisenstein, et al., Adv
Exp Med Biol
493, 169 (2001)). Although opioids have been shown to suppress a variety of
immune cells
resulting in impaired clearance of bacteria and enhanced mortality in animals
(Wang, et al., J
Leukoc Biol 71, 782 (2002)), it has not been previously considered that opioid
compounds
might also directly activate the virulence of bacteria.
Opioids and opioid antagonists such as morphine and DAMGO (([D-Ala2, N-
MePhe4, Gly5-ol], a mu opioid enkephalin) bind to the mOP-R present in the
central nervous
system (CNS) and peripheral tissue. The mOP-R is expressed in a variety of
cell types
including endothelial cells and epithelial cells. The mOP-R is a G protein-
coupled receptor
with multiple isoforms resulting from alternative splicing of mRNA encoded
from a single
gene. Most mu opioid receptor antagonists, including naloxone, exist in an
uncharged state
and readily pass through the blood-brain barrier (BBB) to reverse CNS-
dependent analgesic
effects. MNTX, however, is a charged molecule that is known to be unable to
penetrate the
BBB. The effects of MNTX and other quaternary derivatives of noroxymorphone
(QDNM)
on cell barrier regulation have not been reported.
Several receptors have been implicated in cell (e.g., endothelial cell)
barrier
function. One important receptor family is the sphingosine-l-phosphate (SIP)
receptors (also
called Edg receptors, endothelial differentiation gene). S1P binds to the
plasma membrane G
protein-coupled S1P receptors 1(Edgl), 2 (Edg5), 3.(Edg3), 4 (Edg6) and 5
(Edg8)
expressed in a variety of cell types including endothelium. Human endothelial
cells exhibit
high expression of S 1P 1 and S 1P3 with S 1P 1 signaling coupled to the Gi
pathway and Rac 1
activation, whereas S1P3 signaling couples to the Gi, Gq/ll and G12/13
pathways and
activates RhoA to a much greater extent than Rac 1. S 1 P 1 receptor-dependent
activation of
Rac1 has been shown to promote vascular integrity. In contrast, S1P3 receptor-
dependent
activation of RhoA can potentially regulate endothelial cell barrier
disruption.
4
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
Src (pp60Src, c-Src tyrosine kinase) is a non-receptor tyrosine kinase that
contains an amino-terminal myristoylation site, Src Homology (SH) sites (i.e.,
SH2 and
SH3), a tyrosine kinase catalytic domain and regulatory tyrosine
phosphorylation sites.
Activation of Src promotes endothelial cell barrier disruption and endothelial
cell contraction.
Inhibition of Src attenuates edema and tissue injury after myocardial
infarction.
Protein tyrosine phosphatases (PTPs) are a diverse superfamily encoded by
over 100 genes that regulate a myriad of cellular events. One PTP highly
expressed in lung
endothelium is the receptor-like protein tyrosine phosphatase mu (RPTP ).
Structurally,
RPTP is composed of extracellular MAM (Meprin-A5-protein-M-type-RPTP (RPTP ),
Immunoglobulin (Ig)-like and Fibronectin type 3 (FN3)-like domains and
intracellular PTP
catalytic domains. RPTP is localized at endothelial cell junctions and
regulates vascular
integrity.
While in vitro assays have been enormously useful and continue to provide
important information on the mechanisms of bacterial pathogenesis, they cannot
accurately
reproduce all aspects of the host pathogen interaction, as a pathogen will
encounter several
radically different environments in the host at various points during
infection. Consequently,
a gene that seems important in in vitro studies, may not be important in vivo,
and genes that
appear unimportant in an in vitro assay may play a critical role during a
natural infection.
Furthermore, it has recently been shown that bacteria growing on the surface
of solid agar
have a markedly different physiology from those in broth, as judged by
differential regulation
of nearly one-third of their functional genome. Therefore, experiments must
now be
designed that control for the variables of the growth environment and host
environment,
while at the same'time allowing for measurements of gene expression patterns
and phenotype
analysis which are not possible in more traditional models, such as stressed
mice.
Severe sepsis continues to be the number one cause of mortality among
critically ill patients. Interventions to attenuate regulatory arms of the
systemic immune
response have resulted in clinical failure. Alternatively, newer and more
powerful antibiotics
have resulted in the emergence of highly resistant stains of bacteria for
which there is no
foreseeable therapy other than de-escalating their use. P. aeruginosa is now
on the
international list of emerging resistant pathogens posing a real and present
danger to the
public.
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
Thus, a need continues to exist in the art for methods of preventing,
mitigating
or treating cell barrier dysfunction, including endothelial cell barrier
dysfunction and
epithelial cell barrier dysfunction. Further, the need for compositions and
methods to
alleviate a symptom associated with a cell barrier dysfunction condition has
not been
satisfied.
Summary
The invention satisfies at least one of the foregoing needs in the art in
providing compositions and methods for preventing or treating cell barrier
dysfunction by
administering an effective amount of an opioid receptor antagonist (OP-RA).
The invention
is directed in important embodiments to preventing or treating an endothelial
or epithelial cell
barrier dysfunction. Specifically, the invention relates to the cell barrier
dysfunction
inhibitory effect of opioid receptor antagonists, including peripherally
restricted antagonists
(e.g., polar or charged antagonists typified by methylnaltrexone) as well as
centrally acting
antagonists. The methods are effective in preventing or treating the barrier
dysfunction and
attendant conditions and symptoms arising therefrom, associated with a variety
of diseases
and disorders, such as inflammation, atherosclerosis, and microbial
pathogenesis. As
particular nonlimiting examples, the conditions with which the cell barrier
dysfunction occurs
may be gut-derived sepsis, a bum injury, a chemical contact injury, acute lung
injury,
neonatal necrotizing enterocolitis, severe neutropenia, toxic colitis,
inflammatory bowel
disease, Crohn's disease, enteropathy, transplant rejection, pouchitis, pig-
bel, uremic
pericardial effusion, leakage in the vitreous of the eye, macular
degeneration, retinal
dysfunction, and infection (e.g., viral infection, bacterial infection,
opportunistic bacterial
infection, Clostridium dificile infection, Pseudomonas aeruginosa infection,
Pseudomonas-
mediated ophthalmologic infection, Pseudomonas-mediated otologic infection and
Pseudomonas-mediated cutaneous infection).
The opioid receptor antagonists useful in the inventions described herein are
set forth more comprehensively in the detailed description below, which
description is
incorporated into this summary by reference. Examples of suitable opioid
receptor
antagonists include heterocyclic amine compounds that belong to several
classes of
compounds. One class is the tertiary derivatives of morphinan and, in
particular, the tertiary
derivatives of noroxymorphone. In one embodiment, the tertiary derivative of
noroxymorphone, e.g., naloxone or naltrexone, is contemplated. Another class
is the
6
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
quatemary derivatives of morphinan and, in particular, the quaternary
derivatives of
noroxyrnorphone. Another class is the N-substituted piperidines. Another class
is the
quaternary derivatives of benzomorphans. In particular embodiments, the opioid
receptor
antagonist is a peripheral p.-opioid receptor antagonist, such as N-
methylnaltrexone,
alvimopan, ADL 08-0011, a piperidine-N-alkylcarboxylate, a quaternary
morphinan, an
opium alkaloid derivative or a quatemary benzomorphan coinpound. Further, the
quaternary
morphinan compound may be a quaternary salt of N-methylnaltrexone, N-
methylnaloxone,
N-methylnalorphine, N-diallylnormorphine, N-allyllevallorphan or N-
methylnalmefene. In
some embodiments, the quaternary benzomorphan compound is 2'-hydroxy-5,9-
dimethyl-2,2-
diallyl-6,7-benzomorphanium-bromide; 2'-hydroxy-5,9-dimethyl-2-n-propyl-6,7-
benzomorphan; 2'-hydroxy-5,9-dimethyl-2-allyl-6,7-benzomorphan; 2'-hydroxy-5,9-
dimethyl-2-n-propyl-2-allyl-6,7-benzomorphanium bromide; 2'-hydroxy-5,9-
dimethyl-2-n-
propyl-2-propargyl-6,7-benzomorphanium bromide ; or 2'-acetoxy-5,9-dimethyl-2-
n-propyl-
2-allyl-6,7-benzomorphanium bromide. In some embodiments, the method further
comprises
administration of a high molecular weight polyethylene glycol-like compound
having an
average molecular weight of at least 15 kilodaltons.
In preferred embodiments, the antagonist is a mu opioid receptor antagonist.
In some embodiments, the antagonist is a peripheral opioid receptor
antagonist, e.g., MNTX,
which may also inhibit VEGF, platelet-derived growth factor (PDGF),
sphingosine-l-
phosphate (S1P) and/or hepatocyte growth factor (HGF)-stimulated or induced
cell barrier
dysfunction.
As mentioned, in some embodiments of the invention, the opioid receptor
antagonist is a mu opioid receptor antagonist. In other embodiments, the
opioid receptor
antagonist is a kappa opioid receptor antagonist. The invention also
encompasses
administration of more than one opioid receptor antagonist, including
combinations of mu
opioid receptor antagonists, combinations of kappa opioid receptor antagonists
and
combinations of mu and kappa opioid receptor antagonists, for example, a
combination of
methylnaltrexone and alvimopan (or ADL 08-0011), or a combination of
naltrexone and
methylnaltrexone.
The invention described herein involves the prevention and/or treatment of
cell barrier dysfunction in vertebrates, and more preferably mammals.
hnportant subjects or
"patients" to be treated are farm animals (e.g., horses, goats, cows, sheep,
pigs, fish and
chickens), domestic animals (dogs and cats) and humans.
7
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
The invention described herein involves prevention or treatment of cell
barrier
dysfunction. Prevention as used herein means administration of an opioid
receptor antagonist
to a patient at risk of a cell barrier dysfunction in an amount effective to
inhibit the
appearance of, to lessen the development of or to prevent altogether the
appearance of a
sSnilptom or adverse medical condition arising from the cell barrier
dysfunction. Treatment
as used herein means administration of an opioid receptor antagonist to a
patient having or
believed to have a condition or symptom associated with a cell barrier
dysfunction in an
amount effective to inhibit, to halt the further development of, to lessen or
to eliminate
altogether a symptom or adverse medical condition arising from the cell
barrier dysfunction.
An opioid receptor antagonist, such as a mu opioid receptor antagonist (mOP-
RA) like methylnaltrexone (MNTX), inhibits cell barrier dysfunction. For
example, mu
opioid receptor antagonists, including MNTX, inhibit opiate-, thrombin- and
LPS-induced
endothelial cell barrier dysfunction by mu opioid receptor (mOP-R)-dependent,
and -
independent, mechanisms. The mOP-R-independent mechanisms of mOP-RA (e.g.,
MNTX)-
induced endothelial cell barrier regulation include activation of receptor-
like protein tyrosine
phosphatase mu (R.PTP ) and inhibition of thrombin- and LPS-induced, Src-
dependent, SIP3
receptor transactivation (tyrosine phosphorylation). Thus, mOP-RAs such as
MNTX are
useful as cell barrier protective agents.
The invention described herein provides methods for enhancing cell barrier
function (e.g., endothelial and/or epithelial cell barrier function),
comprising administering to
a patient in need of such treatment a composition comprising an effective
amount of one or
more opioid receptor antagonists. For example, cell barrier function can be
disrupted in
certain inflammatory syndromes. Thus, the invention provides a method of
preventing or
treating inflammatory syndromes, e.g., acute lung injury, as well as
atherosclerosis and
microbial pathogenesis (e.g., infection), which are characterized by a cell
barrier dysfunction,
typically an epithelial or endothelial cell barrier dysfunction. The methods
described herein
also involve treating or preventing a symptom arising from cell barrier
dysfunction associated
with any of these diseases.
In connection with all aspects of the inventions described herein, the patient
preferably is a human. In some embodiments, the human patient is free of
cancer, and/or is
not in a methadone maintenance program, and/or is not immunosuppressed. In
some
embodiments, the patient is not experiencing post operative bowel dysfunction.
The patient
may be, or may not be, on concurrent opioid therapy, depending on the
particular disorder the
8
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
patient has, the severity of the disorder, and the need the patient has for
pain management. In
some embodiments, the patient is taking concurrent opioid therapy. In some
embodiments,
the patient is not taking concurrent opioid therapy. In some embodiments, the
patient is
taking concurrent chronic opioid therapy. In some embodiments, the patient is
not taking
concurrent chronic opioid therapy. In some embodiments, the patient is
receiving a dose of
an opioid antagonist that is independent of any dose of opioid therapy
concurrently
administered.
In some embodiments, the effective amount is such that the patient has
effective circulating blood plasma levels of the opioid antagonist
continuously for at least 1
week, at least 2 weeks, at least three weeks and, even at least 4 weeks. In
one embodiment,
the opioid antagonists are used peri-operatively. By peri-operatively, it is
meant before ( e.g.,
in preparation for), during, and/or immediately after a surgical procedure
(i.e., up to three or
even up to five days). The opioid antagonists.act to attenuate, preserve, or
maintain the cell
barrier function, thereby inhibiting inflammation, inhibiting infection
including opportunistic
infection, and inhibiting recurrence of and/or the metastasis of a tumor in
the case of a
surgical procedure involving.removal of a tumor-and particularly a tumor that
is not an
endothelial cell tumor.
The invention also includes the co-administration of the opioid antagonists
with agents that are not opioid antagonists, but which are nonetheless useful
in treating a
disorder, condition or symptom associated with a cell barrier dysfunction.
Examples of such
agents include anti-cancer agents, anti-neovascularization agents (for
example, anti-VEGF
monoclonal antibody), anti-infective agents (e.g., antibacterial agents and
anti-viral agents),
anti-inflammatory agents, anti-atherosclerotic agents, anti-thrombotic agents,
and the like.
An aspect of the invention provides a method of treating a disorder
characterized by a cell barrier dysfunction comprising administering to a
subject free of an
opioid-induced side effect an effective amount of a g-opioid receptor
antagonist. The opioid-
induced side effects include opioid-induced constipation, irritable bowel
syndrome, post-
operative ileus or bowel dysfunction, opioid-induced nausea, opioid-induced
vomiting,
urinary retention, delayed gastrointestinal tract emptying, reduced
gastrointestinal tract
motility and opioid-induced suppression of the immune system. In some
embodiments, the
cell barrier dysfunction may be attributable to endothelial cells, epithelial
cells, or both types
of cells.
9
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
Another aspect of the invention provides a method of reducing the risk of
developing a disorder characterized by a cell barrier dysfunction comprising
administering to
a subject at risk of developing the disorder a prophylactically effective
amount of an opioid
receptor antagonist.
Another aspect of the invention provides a method of reducing a symptom
associated with a cell barrier disorder, comprising administering to a subject
in need thereof
an opioid receptor antagonist, wherein the compound is administered in an
amount effective
to reduce at least one symptom of the disorder.
Another aspect of the invention is a method of preventing tumor cell
metastasis comprising peri-operatively administering an effective amount of an
opioid
receptor antagonist to a patient having a tumor amenable to surgical
intervention. In some
embodiments the tumor cell is not an endothelial cell tumor.
Another aspect of the invention provides a method for preventing an infection
or for lowering the risk of an infection by administering to a patient in need
of such treatment
an effective amount of an opioid receptor antagonist. In some embodiments, the
patient has a
traumatic injury, such as an internal injury, a surgery, an acute lung injury,
or a burn. In
other embodiments, the patient is subjected to high levels of stress. In some
enibodiments the
infection is from an opportunistic infectious agent. In some embodiments the
infection is a
bacterial infection. In some embodiments the infectious agent is Clostridium
dificile, or
another bacterium capable of developing a virulent phenotype, such as
Pseudomonas
aeruginosa.
Another aspect of the invention provides a method of inhibiting the expression
of a bacterial PA-I lectin/adhesin by a bacterium in a patient comprising
administering an
effective amount of an opioid receptor antagonist to a subject at risk of
developing or
suffering from bacterial pathogenesis. Any known bacterial pathogen, such as
Clostridium
dificile, or bacterium capable of developing a virulent phenotype, such as
Pseudomonas
aeruginosa, that is further capable of expressing a PA-I lectin/adhesin
ortholog is
contemplated.
Another aspect of the invention provides a method for modulating the activity
of a bacterial MvfR protein comprising administering an effective amount of an
opioid
receptor antagonist to a subject at risk of developing or suffering from
bacterial pathogenesis.
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
Another aspect of the invention provides a method of decreasing the
permeability of, or preventing the increase in permeability of, an epithelium
to a bacterial
toxin comprising administering to a subject an amount of an opioid receptor
antagonist
effective in reducing, or inhibiting an increase in, transepithelial cell
electrical resistance.
Another aspect of the invention provides a method for preventing or treating
sepsis by administering to a patient in need of such treatment an effective
amount of an
opioid receptor antagonist.
Another aspect of the invention provides a method for preventing or treating
inflammation by administering to a patient in need of such treatment an
effective amount of
an opioid receptor antagonist. In some embodiments, the patient has
inflammation from a
traumatic injury, such as an internal injury, a surgery, an acute lung injury,
or a bum. In
other embodiments, the patient has inflammation from an infection. In some
embodiments
the infection is a bacterial infection. In some embodiments the infectious
agent is
Clostridium dificile, or another bacterium capable of developing a virulent
phenotype, such as
Pseudomonas aeruginosa.
Another aspect of the invention provides a method of mitigating a cell barrier
dysfunction free of -opioid receptor-dependent effects, comprising
administering to a
subject free of an opioid-induced side effect an effective amount of a
peripheral -opioid
receptor antagonist. In some embodiments, the peripheral -opioid receptor
antagonist is N-
methylnaltrexone. Also in some embodiments, the cell barrier dysfunction is
induced by an
inducing agent selected from the group consisting of thrombin and bacterial
lipopolysaccharide. This aspect of the invention also extends to methods
wherein a protein
phosphatase is activated in the cell, such as methods in which an S IP3
receptor
phosphorylation is reduced. In some embodiments of this method, a protein
tyrosine
phosphatase, such as a receptor protein tyrosine phosphatase , is activated.
Yet another aspect of the invention is a method of mitigating a cell barrier
dysfunction induced by transactivation of a S1P3 receptor, comprising
administering to a
subject free of an opioid-induced side effect an effective amount of a
peripheral g-opioid
receptor antagonist. In some embodiments, the peripheral -opioid receptor
antagonist is N-
methylnaltrexone.
Still another aspect according to the invention is a method of using an opioid
receptor antagonist in the preparation of a medicament for treating,
ameliorating, or
11
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
preventing a disorder or a symptom of a disorder selected from the group
consisting of
inflammation, atherosclerosis, acute lung injury, gut-derived sepsis, a bum
injury, a chemical
contact injury, neonatal necrotizing enterocolitis, severe neutropenia, toxic
colitis,
inflammatory bowel disease, Crohn's disease, enteropathy, transplant
rejection, pouchitis,
pig-bel, uremic pericardial effusion, leakage in the vitreous of the eye,
macular degeneration,
retinal dysfunction, infection (e.g., viral infection, bacterial infection,
opportunistic bacterial
infection, Clostridium dificile infection, Pseudonaonas aeYuginosa infection,
Pseudomonas-
mediated ophthalmologic infection, Pseudomonas-mediated otologic infection and
Pseudomonas-mediated cutaneous infection).
Using a combination of in vivo and molecular methods, surgical stress has
been shown to cause the release of host cell-derived Bacterial Signaling
Compounds (host
stress-derived BSCs) into the intestinal lumen that directly activate the
virulence machinery
of P. aeruginosa. The release of such host-derived BSCs, which include
morphine, K and S
opioid receptor agonists, and Interferon gamma (IFN-y), can shift the
phenotype of P.
aeruginosa, or other members of the normal intestinal flora, from that of
indolent colonizer to
lethal pathogen. Exposure of P. aeruginosa to host stress-derived BSCs induces
the
expression of the PA-I lectin/adhesin (PA-I), a key virulence gene involved in
lethal gut-
derived sepsis in mice. In at least some instances, induction of PA-I
expression is mediated
by a transcriptional regulator of virulence gene expression, MvfR. PA-I
induces an epithelial
permeability defect to at least two potent cytotoxins of this organism,
exotoxin A and
elastase, causing lethal gut-derived sepsis and other disorders characterized
by an epithelial
cell barrier dysfunction.. The data provide evidence for a model in which
opportunistic
pathogens sense host stress and vulnerability by perceiving key mediators
released by the
host into the intestinal tract during stress, such as the stress resulting
from surgery. These
host stress-derived compounds directly activate critical genes in P.
aeruginosa leading to
enhanced virulence.
Opioids, released in increased amount during physiological.stress, directly
induce the expression of several quorum sensing-dependent virulence factors in
P.
aeruginosa, such as pyocyanin, biofilm, and the lectin/adhesin PA-I.
Specifically, U-50,488
(bremazocine, i.e., trans-3,4-dichloro-N-methyl-N[2-(1-
pyrolidinyl)cyclohexyl]benzeneacetamide methanesulfonate, an exemplary x-
opioid receptor
agonist, induces pyocyanin production in P. aeruginosa via the global
virulence
transcriptional regulator MvfR. U-50,488 also induces pyocyanin at cell
densities below
12
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
those that would normally produce pyocyanin. These findings indicate that
opioids, whether
exogenous or endogenous, function as host stress-derived bacterial signaling
molecules
capable of activating a virulence response in P. aeruginosa. One aspect
according to the
invention provides a method of treating a disorder characterized by a barrier
dysfunetion
(e.g., an epithelial cell or an endothelial cell) comprising administering, to
a subject receiving
at least one opiate or experiencing release of at least one endogenous opioid
(e.g., an
endorphin) but not experiencing an opioid-induced side effect, an effective
amount of a -
opioid receptor antagonist. An opioid-induced side effect includes an opioid-
induced
constipation, irritable bowel syndrome, post-operative ileus or bowel
dysfunction, opioid-
induced nausea, opioid-induced vomiting, urinary retention, delayed
gastrointestinal tract
emptying, reduced gastrointestinal tract motility and opioid-induced
suppression of the
immune system. In some embodiments, the patient will not be undergoing
treatment for
cancer or methadone treatment for drug addiction. In some embodiments, the
subject will not
be receiving or experiencing an exogenous or an endogenous opioid.
In an aspect, the invention thus provides a method of reducing the risk of
developing a disorder characterized by a cell barrier dysfunction (e.g., an
epithelial cell or an
endothelial cell) comprising administering to a subject at risk of developing
the disorder a
propliylactically effective amount of a -opioid receptor antagonist. Another
aspect of the
invention is drawn to a method of reducing a symptom associated with a cell
barrier disorder
(e.g., an epithelial or endothelial cell barrier disorder), comprising
administering to a subject
in need thereof a -opioid receptor antagonist, wherein the compound is
administered in an
amount effective to reduce at least one symptom of the disorder. Another
aspect of the
invention is a method of inhibiting the expression of a bacterial PA-I
lectin/adhesin
comprising administering an effective amount of a -opioid receptor antagonist
to a subject at
risk of developing or suffering from bacterial pathogenesis. In some
embodiments of this
method, the bacterial PA-I lectin/adhesin is found in a bacterium residing in
a mammalian
intestine. In some embodiments of this aspect, the bacterial PA-I
lectin/adhesin is a
Pseudomonad PA-I lectin/adhesin. An important Pseudomonad is Pseudom.onas
aeruginosa.
Another aspect of the invention is directed to a method of modulating the
activity of a
bacterial MvfR protein comprising administering an effective amount of a g-
opioid receptor
antagonist to a subject at risk of developing or suffering from bacterial
pathogenesis. In some
embodiments, the bacterial MvfR protein is found in a bacterium residing in a
mammalian
intestine. Also in some embodiments, the bacterial MvfR protein is a
Pseudomonad MvfR
13
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
protein, preferably a Pseudomonas aeruginosa MvfR protein. In another
enumerated aspect,
the invention provides a method of decreasing the permeability of, or
preventing the increase
in permeability of, an epithelium to a bacterial toxin comprising
administering to a subject an
amount of a -opioid receptor antagonist effective in reducing, or inhibiting
an increase in,
transepithelial cell electrical resistance (i.e., transcellular electrical
resistance of an
epithelium). An epithelium in the context of this aspect of the invention
comprises at least
two epithelial cells. In some embodiments, the epithelial cells are intestinal
epithelial cells.
Also contemplated in this aspect of the invention is a subject that comprises
a microbial
pathogen, such as Pseudomonas aeruginosa or Clostridium dificile.
In all of the aspects of the invention, any mode of administering the opioid
receptor antagonist that is known in the art is contemplated, and in
particular, delivery by
parenteral, oral, subcutaneous, transcutaneous, subcutaneous implantation,
intramuscular,
intravenous, intrathecal, intraocular, intravitreous, ophthalmologic,
intraspinal, intrasynovial,
topical, rectal, transepithelial including transdermal, buccal, sublingual,
intramuscular,
intracavity, and aural routes, as well as by nasal inhalation including via
insufflation and
aerosol. Microbial pathogens, such as P. aeruginosa, not only inhabit the
intestinal tract,
these patliogens are also capable of ophthalmologic, otologic and cutaneous
infection of
subjects (e.g., humans). Thus, the invention comprehends administering the
opioid receptor
antagonist by direct routes, e.g., as by topical delivery, cutaneous delivery,
intravitreous
delivery, and intracerebroventricular delivery, to achieve localized,
therapeutically useful
concentrations of the antagonist. In addition, the invention comprehends
treatment of any
disorder caused, at least in part, by a microbial pathogen such as P.
aeruginosa, which
includes Pseudomonas-mediated ophthalmologic, Pseudomonas-mediated otologic or
Pseudomonas-mediated cutaneous disorders, by administering an opioid receptor
antagonist
through conventional systemic routes, including intravitreously,
intracerebroventricularly,
and topically (e.g., ophthalmologically, otologically, cutaneously), at levels
sufficient to
achieve therapeutically useful systemic levels of the antagonist.
Other features and advantages of the present invention will be better
understood by reference to the following detailed description, including the
drawing and the
examples.
Brief Description of the Drawing
14
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
Figure 1 is a panel of graphs, bar graphs and immunoblots showing that IFN-y
induces the expression of the PA-I lectin in P. aeruginosa.
Figure 2 is a panel of bar graphs showing that the presence of rlall and rhlR,
core quorum sensing signaling elements in P. aeruginosa, are required for the
PA-I
expression and pyocyanin production in response to IFN-y.
Figure 3 is a panel of graphs, an epimicrograph, immunoblots and MS/MS
spectra showing the identification of the IFN-y binding site to solubilized
membrane fractions
of P. aeruginosa (PAO1).
Figure 4 is a panel of bar charts and graphs showing the binding
characteristics of the IFN-y to membrane fractions of P. aeruginosa (PAO 1).
Figure 5 is a panel of graphs, bar charts and immunoblots showing that IFN-y
binds to OprF and induces PA-I expression.
Figure 6 is a panel of bar graphs and graphs showing that MvfR plays a key
role in the effect of U-50,488 and C4-HSL on PCN production.
Figure 7 is a bar graph showing the inhibition of morphine-induced PA-I
lectin/adhesin expression in the separate presences of -opioid receptor
antagonists
methylnaltrexone (MNTX) and naloxone (NAL).
Figure 8 is a panel of graphs and bar graphs showing the effects of adenosine
and inosine on PA-I expression.
Figure 9 is a panel of graphs and bar graphs showing the effects of
methylnaltrexone (MNTX) and DAMGO on human endothelial cell barrier
regulation.
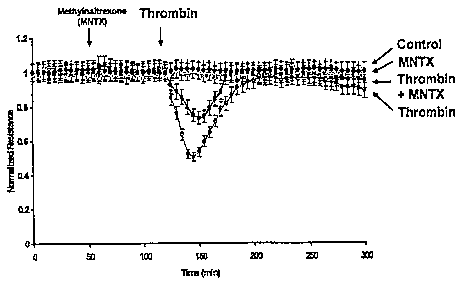
Figure 10 is a panel of graphs showing the effects of MNTX effects on non-
opioid agonist-induced human endothelial cell barrier regulation.
Figure 11 is a bar graph showing the differential effects of MNTX and
naloxone on agonist-induced human endothelial cell barrier disruption.
Figure 12 is a panel of bar graphs and immunoblots showing the effects of
silencing Mu opioid receptor, S 1 P1 receptor or S IP3 receptor on MNTX-
induced protection
from human endothelial cell barrier disruption.
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
Figure 13 is a panel of immunoblots and bar graphs showing the effects of
MNTX, naloxone and Src on SiP3 receptor transactivation (tyrosine
phosphorylation).
Figure 14 is a panel of bar graphs showing the analysis of agonist-induced
total cellular tyrosine phosphatase activity in human endothelial cells.
Figure 15 is a panel of graphs and immunoblots showing the effects of S1P3
receptor transactivation and endothelial cell barrier function by receptor
tyrosine phosphatase
mu (RPTP ).
Figure 16 is a panel of bar graphs showing the regulation of agonist-induced
total cellular tyrosine phosphatase activity and MNTX-induced protection from
human
endothelial cell barrier disruption by RPTP .
Figure 17 is a panel of immunhistochemical stains and bar graphs showing the
effect of MNTX on LPS-induced pulmonary vascular hyper-permeability in vivo.
Figure 18 is a panel of bar graphs showing the effects of silencing mu opioid
receptor expression using siRNA on agonist-induced barrier function.
Tigure 19 is a scheinatic illustration of pathways relevant to cell barrier
function, and dysfunction.
Detailed Description
A wide variety of inflammatory disorders, tumor metastasis, and a variety of
other diseases and disorders are characterized by a cell barrier dysfunction
manifested as an
increased cell barrier permeability or loss of selective permeability and
concomitant
exudation of cells, cellular contents, fluid or protein across the barrier.
For example, an
endothelial cell barrier dysfunction can lead to increased vascular
permeability and a
resulting extravasation of protein and fluids, characteristic of inflammatory
processes.
McVerry et al., Cell. Signal. 17:131-139 (2005). Analogously, a cell barrier
dysfunction can
become permissive for tumor cell metastasis. An epithelial cell barrier
dysfunction arising in
the context of, e.g., microbial pathogenesis of the mammalian intestine, can
lead to a variety
of illnesses, including gut-derived sepsis. Microbial pathogenesis, moreover,
can be the
product of infection by a pathogen (e.g., Clostridium dificile) or by the
phenotypic shift of a
normally benign member of the normal flora associated with an organism (e.g.,
intestinal
flora) to a pathogenic or virulent state (e.g., Pseudomonas aeruginosa).
Beyond these
16
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
illustrative examples, multi-cellular organisms such as vertebrates (e.g.,
mammals, including
humans) generally exhibit supracellular compartmentalization resulting in
discrete spacings
for tissues, organs, and organ systems. Chief contributors to this necessary
compartmentalization are the several kinds of cell barriers. Exemplified in
terms of
endothelial and epithelial cell barriers, there are cell barriers associated
with most tissues,
organs, and organ systems, e.g., brain (e.g., cerebral endothelial
lining/blood brain barrier),
spleen, liver, eye, lung, vasculature (blood and lymph), kidney, bladder,
ureter, urethra,
alimentary canal, including the small and large intestines, lung, and the
like. The invention
provides methods for preventing, reducing or eliminating a cell barrier
dysfunction associated
with a disease or disorder that is capable of lowering the quality of life or
that deleteriously
impacts the health of a subject or patient that has the disease or disorder.
Identification of host stress signaling compounds and the membrane receptors,
to which they bind, such as receptors on host cells (e.g., epithelial and
endothelial cells) as
well as receptors on pathogenic microbes such as infectious bacteria, will
lead to the
discovery of therapeutic targets that will allow for prevention or treatment
in a variety of cell
barrier diseases and disorders, including the infection, at its most proximate
point.
Furthermore, the identification of conserved receptors, e.g., bacterial
receptors common to
other microbial species, will then lead to the development of receptor
antagonists or decoys.
Such an approach of rendering recipient cells (e.g., colonizing pathogens)
insensate to host
stress activators has the potential to provide efficacious and cost-effective
treatment for a
wide variety of diseases and disorders characterized,by cell barrier
dysfunction.
An "abnormal condition" is broadly defined to include mammalian diseases,
mammalian disorders and any abnormal state of mammalian health that is
characterized by a
cell barrier dysfunction. Exemplary cells that may exhibit a cell barrier
dysfixnction, or be at
risk of developing such a dysfunction, include endothelial cells and
epithelial cells. The
abnormal conditions may be found in humans, non-human mammals, or any mammal.
"Bum injury" means (i) damage to mammalian tissue resulting from exposure
of the tissue to heat, for example in the form of an open flame, steam, hot
fluid, and a hot
surface.
A "chemical contact injury" refers to an injury caused by direct contact with
a
chemical and can involve a chemical burn or other injury.
17
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
"Severe neutropenia" is given its ordinary and accustomed meaning of a
marked decrease in the number of circulating neutrophils.
"Administering" is given its ordinary and accustomed meaning of delivery of a
therapeutic to an organism in need by any suitable means recognized in the
art. Exemplary
fonns of administering include delivery by parenteral, oral, subcutaneous,
transcutaneous,
subcutaneous implantation, intramuscular, intravenous, intrathecal,
intraocular, intravitreous,
ophthalmologic, intraspinal, topical, rectal, transdermal, sublingual,
intramuscular,
intracavity, and aural routes, as well as by nasal inhalation (e.g.,
nebulizing spray). The
mechanism of delivery may be direct puncture or injection, or gel or fluid
application to an
eye, ear, nose, mouth, anus or urethral opening, as well as cannulation.
An "effective dose" is that amount of a substance that provides a beneficial
effect on the organism receiving the dose and may vary depending upon the
purpose of
administering the dose, the size and condition of the organism receiving the
dose, and other
variables recognized in the art as relevant to a determination of an effective
dose. The
process of detennining an effective dose involves routine optimization
procedures that are
within the skill in the art.
An "animal" is given its conventional meaning of a non-plant, non-protist
living being. A preferred animal is a mammal, such as a human.
In the context of the present disclosure, a "need" is an organismal, organ,
tissue, or cellular state that could benefit from administration of an
effective dose to an
organism characterized by that state. For example, a human at risk of
developing gut-derived
sepsis, or presenting a symptom thereof, is an organism in need of an
effective dose of a
product, such as a pharmaceutical composition, according to the present
invention.
"Average molecular weight" is given its ordinary and accustomed meaning of
the arithmetic mean of the molecular weights of the components (e.g.,
molecules) of a
composition, regardless of the accuracy of the determination of that mean. For
example,
polyethylene glycol, or PEG, having an average molecular weight of 3.5
kilodaltons may
contain PEG molecules of varying molecular weight, provided that the
arithmetic mean of
those molecular weights is determined to be 3.5 kilodaltons at some level of
accuracy, which
may reflect an estimate of the arithmetic mean, as would be understood in the
art.
Analogously, PEG 15-20 means PEG whose molecular weights yield an arithmetic
mean
18
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
between 15 and 20 kilodaltons, with that arithmetic mean subject to the
caveats noted above.
These PEG molecules include, but are not limited to, simple PEG polymers. For
example, a
plurality of relatively smaller PEG molecules (e.g., 7,000 to 10,000 daltons)
may be joined,
optionally with a linker molecule such as a phenol, into a single molecule
having a higher
average molecular weight (e.g., 15,000 to 20,000 daltons).
"PA-I," or "PA-I lectin/adhesin," or "PA-IL" expression means the production
or generation of an activity characteristic of PA-I lectin/adhesin. Typically,
PA-I
lectin/adhesin expression involves translation of a PA-I lectin/adhesin-
encoding mRNA to
yield a PA-I lectin/adhesin polypeptide having at least one activity
characteristic of PA-I
lectin/adhesin. Optionally, PA-I lectin/adhesin further includes transcription
of a PA-I
lectin/adhesin-encoding DNA to yield the aforementioned mRNA.
"Intestinal pathogen" means a microbial pathogen capable of causing, in
whole or part, gut-derived sepsis in an animal such as a human. Intestinal
pathogens known
in the art are embraced by this definition, including gram negative bacilli
such as the
Pseudomonads (e.g., Pseudomonas aeruginosa).
"Pathogenic quorum" means aggregation or association of a sufficient number
of pathogenic organisms (e.g., P. aeruginosa) to initiate or maintain a quorum
sensing signal
or communication that a threshold concentration, or number, of organisms
(e.g., intestinal
pathogens) are present, as would be known in the art.
"Transcellular Electrical Resistance," or TER, is given the meaning this
phrase
has acquired in the art, which refers to a measurement of electrical
resistance across cells of a
given type (e.g., epithelial or endothelial cells), which is non-exclusively
useful in assessing
the status of tight junctions between such cells. A related term "TEER," is
used herein to
refer to "transepithelial cell electrical resistance," or "transendothelial
cell electrical
resistance," and the particular usage will be apparent from context.
"Pharmaceutical composition" means a formulation of compounds suitable for
therapeutic administration, to a living animal, such as a human patient.
Preferred
pharmaceutical compositions according to the invention are described in the
copending U.S.
Patent Publication No. 20040266806 the contents of which are herein
incorporated herein by
reference in their entireties. The pharmaceutical compositions of the
invention may comprise
a solution balanced in viscosity, electrolyte profile and osmolality,
comprising an electrolyte,
19
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
dextran-coated L-glutamine, dextran-coated inulin, lactulase, D-galactose, N-
acetyl D-
galactosamine and 5-20% PEG (15,000-20,000). The compounds are preferably
combined
with a pharmaceutical carrier selected on the basis of the chosen route of
administration and
standard pharmaceutical practice as described, for example, in Remington's
Pharmaceutical
Sciences (Mack Pub. Co., Easton, Pa., 1980), the disclosures of which are
hereby
incorporated herein by reference, in their entireties.
"Adjuvants," "carriers," or "diluents" are each given the meanings those terms
have acquired in the art. An adjuvant is one or more substances that serve to
prolong the
immunogenicity of a co-administered immunogen. A carrier is one or more
substances that
facilitate the manipulation, such as by translocation of a substance being
carried. A diluent is
one or more substances that reduce the concentration of, or dilute, a given
substance exposed
to the diluent.
"Alkyl" refers to an aliphatic hydrocarbon group which is saturated and which
may be straight, branched or cyclic and has from 1 to about 10 carbon atoms in
the chain, as
well as all combinations and subcombinations of chains therein. Exemplary
alkyl groups
include methyl, ethyl, n-propyl, isopropyl, butyl, isobutyl, sec-butyl, tert-
butyl, pentyl, hexyl,
heptyl, octyl, nonyl and decyl.
"Lower alkyl" refers to an alkyl group having 1 to about 6 carbon atoms.
"Alkenyl" refers to an aliphatic hydrocarbon group containing at least one
carbon-carbon double bond and having from 2 to about 10 carbon atoms in the
chain, as well
as all combinations and sub- combinations of chains therein. Exemplary alkenyl
groups
include vinyl, propenyl, butenyl, pentenyl, hexenyl, heptenyl, octenyl,
nonenyl and decenyl
groups.
"Alkynyl" refers to an aliphatic hydrocarbon group containing at least one
carbon-carbon triple bond and having from 2 to about 10 carbon atoms in the
chain, as well
as combinations and sub-combinations of chains therein. Exemplary alkynyl
groups include
ethynyl, propynyl, butynyl, pentynyl, hexynyl, heptynyl, octynyl, nonynyl and
decynyl
groups.
"Alkylene" refers to a bivalent aliphatic hydrocarbon group having from 1 to
about 6 carbon atoms, and all combinations and subcombinations of chains
therein. The
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
alkylene group may be straight, branched or cyclic. Optionally, there may be
inserted within
the alkylene group one or more oxygen, sulfur or optionally substituted
nitrogen atoms,
wherein the nitrogen substituent is an alkyl group, as described previously.
"Alkenylene" refers to an alkylene group containing at least one carbon-
carbon double bond. Exemplary alkenylene groups include ethenylene (-CH=CH-)
and
propenylene (-CH=CHCH2-).
"Cycloalkyl" refers to any stable monocyclic or bicyclic ring having from
about 3 to about 10 carbons, and all combinations and subcombinations of rings
therein.
Optionally, the cycloalkyl group may be substituted with one or more
cycloalkyl-group
substituents. Exemplary cycloalkyl groups include cyclopropyl, cyclobutyl,
cyclopentyl,
cyclohexyl, cycloheptyl and cyclooctyl groups.
"Cycloalkyl-substituted alkyl" refers to a linear alkyl group, preferably a
lower
alkyl group, substituted at a terminal carbon with a cycloalkyl group,
preferably a C3-C8
cycloalkyl group. Exemplary cycloalkyl-substituted alkyl groups include
cyclohexylmethyl,
cyclohexylethyl, cyclopentylethyl, cyclopentylpropyl, cyclopropylmethyl and
the like.
"Cycloalkenyl" refers to an olefinically unsaturated cycloaliphatic group
having from about 4 to about 10 carbons, and all combinations and
subcombinations of rings
therein.
"Alkoxy" refers to an alkyl substituted hydroxyl, or alkyl-O, group, where
alkyl is as previously described. Exemplary alkoxy groups include, for
example, methoxy,
ethoxy, propoxy, butoxy and heptoxy.
"Alkoxy-alkyl" refers to a di-alkyl ether, or alkyl-O-alkyl, group, where
alkyl
is as previously described.
"Acyl" means an alkyl-CO group wherein alkyl is as previously described.
Exemplary acyl groups include acetyl, propanoyl, 2-methylpropanoyl, butanoyl
and
palmitoyl.
"Aryl" refers to an aromatic carbocyclic group containing from about 6 to
about 10 carbons, and all combinations and subcombinations of rings therein.
Optionally, the
21
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
aryl group may be substituted with one or two or more aryl group substituents.
Exemplary
aryl groups include phenyl and naphthyl.
"Aryl-substituted alkyl" refers to a linear alkyl group, preferably a lower
alkyl
group, substituted at a terminal carbon with an optionally substituted aryl
group, preferably
an optionally substituted phenyl ring. Exemplary aryl-substituted alkyl groups
include, for
example, phenylmethyl, phenylethyl and 3-(4-methylphenyl)propyl.
"Heterocyclic" refers to a monocyclic or multicyclic ring system carbocyclic
group or radical containing from about 4 to about 10 members, and all
combinations and
subcombinations of rings therein, wherein one or more of the members of the
ring is an
element other than carbon, for example, nitrogen, oxygen or sulfur. The
heterocyclic group
may be aromatic or nonaromatic. Exemplary heterocyclic groups include, for
example,
pyrrole and piperidine groups.
"Halo" refers to fluoro, chloro, bromo or iodo. '
"Opium alkaloid derivative" refers to mu opioid receptor antagonists (e.g.,
peripheral antagonists) that are synthetic or semi-synthetic derivatives or
analogs of opium
alkaloids.
"Substantially no agonist activity," in connection with the opium alkaloid
derivatives, means that, at a concentration of 1 M, the maximal measured
physiological
response of a receptor, e.g., electrically stimulated guinea pig ileum, is
about 60% or less
relative to morphine.
"HMW PEG-like compounds" refer to relatively high molecular weight PEG
compounds, defined as having an average molecular weight greater than 3.5
kilodaltons (kD).
Preferably, HMW PEG has an average molecular weight greater than 5 kilodaltons
and, in
particular embodiments, HMW PEG has an average molecular weight at least 8
kilodaltons,
more than 12 kilodaltons, at least 15 kilodaltons, and between 15 and 20
kilodaltons.
Additionally, "HMW PEG-like compounds includes HMW PEG derivatives wherein
each
such derivative is an HMW PEG containing at least one additional functional
group.
Preferred HMW PEG derivatives are cationic polymers. Exemplary functional
groups
include any of the alkoxy series, preferably C1-C10, any of the aryloxy
series, phenyl and
substituted phenyl groups. Such functional groups may be attached at any point
to an HMW
22
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
PEG molecule, including at either terminus or in the middle; also included are
functional
groups, e.g., phenyl and its substituents, that serve to link to snialler PEG
molecules or
derivative thereof into a single HMW PEG-like compound. Further, the HMW PEG-
like
molecules having an additional functional group may have one such group or
more than one
such group; each molecule may also have a mixture of additional functional
groups, provided
such molecules are useful in stabilizing at least one therapeutic during
delivery thereof or in
treating, ameliorating or preventing a disease, disorder or condition of an
epithelial cell.
"Media" and "medium" are used to refer to cell culture medium and to cell
culture media throughout the application. The singular or plural number of the
nouns will be
apparent from context in each usage.
The term "peripheral" opioid receptor antagonist designates an opioid receptor
antagonist, including a -opioid receptor antagonist, that acts primarily on
physiological
systems and components external to the central nervous system, i.e., the
antagonist does not
readily cross the blood-brain barrier. In some embodiments, the peripheral
opioid receptor
antagonists employed in the methods of the invention exhibit high levels of
activity with
respect to gastrointestinal tissue, while exhibiting reduced, and preferably
substantially no,
central nervous system (CNS) activity. The term "substantially no CNS
activity," as used
herein, means that less than 20% of the pharmacological activity of the
peripheral opioid
receptor antagonists exhibited outside the CNS is exhibited inside the CNS. In
preferred
embodiments, the peripheral opioid receptor antagonists employed in the
inventive methods
exhibit less than 15% of their pharmacological activity in the CNS, witli less
than about 10%
being more preferred. In even more preferred embodiments, the peripheral
opioid receptor
antagonists employed in the methods of the invention exhibit less than 5% of
their
pharmacological activity in the CNS, with about 0% (i.e., no CNS activity)
also being more
preferred. Preferred peripheral opioid receptor antagonists of the invention
are quaternary
derivatives of noroxymorphone, such as R-methylnaltrexone.
In general terms, a model of lethal sepsis in mice has been developed which
provides unique insight into the process by which microbial pathogens can
cause lethal sepsis
syndrome from within the intestinal tract of a physiologically stressed host.
Three
physiologic "hits" result in mortality, e.g., surgical stress (30%
hepatectomy), starvation (48
hour of water only) and the introduction of P. aeruginosa into the distal
intestinal tract
(cecum). This model results in 100% mortality, whereas elimination of any one
of the three
23
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
factors results in complete survival. A single virulence determinant has been
identified in
Pseudornonas aeruginosa, PA-I, that is expressed in vivo in response to
locally released
compounds unique to the intestinal tract of a physiologically stressed host.
That PA-I plays a
role in lethal gut-derived sepsis, such as in mice, was demonstrated by
experiments in which
mutanized strains of P. aeruginosa, void of PA-I yet capable of secreting
exotoxin A, had
markedly attenuated effects on the barrier function of cultured epithelial
cells and were
completely apathogenic in the mouse model of lethal gut-derived sepsis. PA-I
lectin/adhesin
plays a key role in the lethal effect of this organism by creating a
permeability defect to
potent and lethal cytotoxins of P. aeruginosa, such as exotoxin A and
elastase. The lethal
effect of intestinal P. aeruginosa appears to occur completely independent of
its
extraintestinal dissemination (translocation). Surprisingly, systemic
injection (intravenous,
intraperitoneal) of an equal dose of P. aeruginosa in this model produces no
mortality and no
systemic inflammation. Taken together, the data provide strong evidence that
sepsis can be
generated by a microbial pathogen whose virulence is activated locally by host
stress-derived
bacterial signaling compounds (BSC) generated during surgical stress.
Observation that P. aeruginosa is much more virulent and lethal when present
on an epithelial surface than when bloodborne is supported by a lung model of
sepsis.
Intravenous injection of a highly cytotoxic strain of P. aey-uginosa, PA103,
resulted in no
systemic cytokine release and no mortality in rabbits, whereas lung
instillation of an equal
dose (approximately 108 cfu/ml) resulted in significant systemic cytokine
release (TNFa, IL-
8) and 100% mortality. An extensive number of studies have now demonstrated
that the most
virulent and lethal strains of P. aeruginosa causing sepsis following lung
instillation are not
those that display the most invasive (translocating/disseminating) phenotype,
but rather are
those strains that are most disruptive of cellular integrity and epithelial
permselectivity to its
locally released cytotoxins. These observations, coupled with the findings
that P. aeruginosa
produces a 25-fold increase in its extracellular virulence factors (i.e.,
elastase, alkaline
protease) when cultured in the presence of epithelial cells, suggests that the
lethality of this
pathogen is governed by its interaction with, and activation by, the
epithelium itself.
Experimental data show that both soluble and contact-mediated elements of the
intestinal
epithelium exposed to stress (e.g., surgery, hypoxia, heat shock), enhance the
capacity of P.
aeruginosa to express PA-I, which is capable of causing a profound disruption
in the cellular
integrity of both intestinal and lung epithelial cells.
24
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
The main mechanism of action by which P. aeruginosa induces mortality from
within the intestinal tract of a stressed host is via a PA-I-induced
permeability defect to its
lethal cytotoxins, exotoxin A and elastase. Instillation of a combination of
purified PA-I with
either exotoxin A or elastase into the cecum of surgically stressed and sham-
operated control
mice induced significant mortality, whereas injection of either compound alone
had no effect.
The clinical role of PA-I was examined by screening fecal samples of patients
with severe
sepsis for whom no source could be identified. Among strains of P. aer=uginosa
isolated from
the feces of critically ill patients, as well as among numerous laboratory and
environmental
strains, the PA-I genotype has been found to be highly prevalent. There is now
convincing
evidence that the intestinal tract environment is a unique niche in which key
virulence
determinants in highly lethal pathogens (i.e., Vibrio cholera) are activated
by yet-unknown
"cues" that are present only during active infection.
The gene encoding PA-I (the lecA gene) is an ideal biological "read-out" and
reporter gene in which to examine overall virulence gene expression in P.
aeruginosa in
response to host stress-derived BSCs.
The precise host cell elements that activate bacterial biosensors are not
known.
Because PA-I expression is'both QS and RpoS dependent, GFP-PA-I reporter
strains
(described herein) provide a unique opportunity to screen for host cell-
derived bacterial
signaling compounds released during stress that activate membrane sensors,
leading to PA-I
expression.
Various opioid receptor agonists, including endogenous morphine alkaloids,
are released and maintained at sustained concentrations during severe stress.
Opioids are
highly conserved compounds and various bacteria and fungi, including P.
aeruginosa,
synthesize and metabolize morphine. Similarly, as shown herein, elements of
the immune
system, such as IFN-y, can also serve as potent host stress-derived-BSCs. P.
aeruginosa is
able to sense the presence of the IFN-y and respond by expressing two quorum
sensing
dependent virulence factors, PA-I and pyocyanin. From the perspective of P.
aeYuginosa, the
ability to sense and respond to host immune activation, in particular to IFN-y
whose function
is directed at bacterial clearance, provides this organism with a
countermeasure against host
immune activation. In particular, Interferon-y is shown below to bind to an
outer membrane
protein in P. aeruginosa, OprF, resulting in the expression of a quorum
sensing-dependent
virulence determinant, the PA-I lectin. IFN- y also bound E. coli membranes.
These
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
observations provide details of the mechanisms by which prokaryotic organisms
are directly
signaled by immune activation in their eukaryotic host.
Exposure of P. aeruginosa to opioids leads to the expression of several
quoruin sensing-dependent virulence factors in P. aeruginosa. That the QS
system might be
activated by opioids is a significant finding given that QS controls the
expression of hundreds
of virulence genes in P. aeruginosa (M. Schuster, M. L. Urbanowski and E. P.
Greenberg,
Proc Natl Acad Sci U S A 101, 15833 (2004)).
Data disclosed herein provide evidence that MvfR is required for PCN
production in response to U-50,488. In addition, data from the present study
suggest that
PCN production in response to U-50,488 also requires the synthesis of
Pseudomonas
quinolone signal (PQS), since methyl anthranilate attenuated the U-50,488-
mediated effect on
PCN production. That C4-HSL also requires intact MvfR to produce PCN, coupled
with the
finding of highly up-regulated PCN production in strains harboring multiple
nzvfR genes, is
consistent with quorum sensing activation relying not only on the binding of
QS signaling
molecules to their core QS transcriptional regulators ( i.e., RhIR, LasR), but
also having QS
signals activating proximal transcriptional regulators.
The data disclosed herein establish that opioid compounds may vary in their
ability to induce a particular virulence phenotype in P. aeruginosa. It is
contemplated that
there are multiple host-stress-derived bacterial signaling compounds that are
able to influence
the state of virulence in P. aeruginosa. Norepinephrine can also affect the QS-
dependent
virulence factor PA-IL in P. aeruginosa (J. Alverdy, et al., Ann Surg 232, 480
(2000)) and
soluble compounds released into the media by hypoxic intestinal epithelial
cells also induce
PA-IL expression. Consistent with these disclosures is the disclosure that
norepinephrine
directly affects QS circuitry in E. coli (V. Sperandio, A. G. Torres, B.
Jarvis, J. P. Nataro and
J. B. Kaper, Proc Natl Acad Sci U S A 100, 8951 (2003)).
The invention provides methods of screening for modulators of the signaling
induced by one or more BSCs, including such modulators as opioid receptor
agonists,
morphine, and interferon gamma. These therapeutics are delivered to an
organism, such as a
human patient, in need thereof. Dosage levels and delivery routes and
schedules will vary
depending upon circumstances readily identified and accommodated by those
skilled in the
art using routine procedures.
26
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
The therapeutics according to the invention may further comprise a HMW
PEG-like compound, which may be administered by any means suitable for the
condition or
disorder to be treated. The compound(s) may be delivered orally, such as in
the form of
tablets, capsules, granules, powders, or with liquid formulations including
syrups; by
sublingual; buccal; or transdermal delivery; by injection or infusion
parenterally,
subcutaneously, transcutaneously, subcutaneous implantation, intravenously,
intramuscularly,
intrathecally, intraocularly, ophthalmologically, intraspinally, topically, or
intrastemally (e.g.,
as sterile injectable aqueous or non-aqueous solutions or suspensions);
orally, nasally, such as
by inhalation spray; aurally, rectally such as in the form of suppositories;
vaginally or
urethrally via suppository or infusion, e.g., via cannulation, or liposomally,
and intracavity
delivery. Dosage unit formulations containing non-toxic, pharmaceutically
acceptable
vehicles or diluents may be administered. The compounds may be administered in
a form
suitable for imniediate release or extended release. Immediate release or
extended release
may be achieved with suitable pharmaceutical compositions known in the art.
Exemplary compositions for oral administration include suspensions which
may contain, for example, microcrystalline cellulose for imparting bulk,
alginic acid or
sodium alginate as a suspending agent, methylcellulose as a viscosity
enhancer, sweeteners or
flavoring agents such as those known in the art; and immediate release tablets
which may
contain, for example, microcrystalline cellulose, dicalcium phosphate, starch,
magnesium
stearate and/or lactose and/or other excipients, binders, extenders,
disintegrants, diluents and
lubricants, such as those known in the art. The inventive compounds may be
orally delivered
by sublingual and/or buccal administration, e.g., with molded, compressed, or
freeze-dried
tablets. Exemplary compositions may include fast-dissolving diluents such as
mannitol,
lactose, sucrose, and/or cyclodextrins. Also included in such formulations may
be excipients
such as a relatively high molecular weight cellulose (AVICEL ) or a
polyethylene glycol
(PEG; GoLytely , 3.34 kD); an excipient to aid mucosal adhesion such as
hydroxypropyl
cellulose (HPC), hydroxypropyl methyl cellulose (HPMC), sodium carboxymethyl
cellulose
(SCMC), and/or maleic anhydride copolymer (e.g., GANTREZ ). Lubricants,
glidants,
flavors, coloring agents and stabilizers may also be added for ease of
fabrication and use.
Exemplary compositions for nasal aerosol or inhalation administration include
solutions wllich may contain, for example, benzyl alcohol or other suitable
preservatives,
27
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
absorption promoters to enhance absorption and/or bioavailability, and/or
other solubilizing
or dispersing agents such as those known in the art.
Exemplary compositions for intestinal administration include solutions or
suspensions which may contain, for example, suitable non-toxic diluents or
solvents, such as
mannitol, 1,3-butanediol, water, Ringer's solution, an isotonic sodium
chloride solution, or
other suitable dispersing or wetting and suspending agents, including
synthetic mono- or
diglycerides and fatty acids, including oleic acid. Contemplated in this
context are
suppositories which may contain, for example, suitable non-irritating
excipients, such as
cocoa butter, synthetic glyceride esters or polyethylene glycols (e.g.,
GoLytely ).
The effective amount of a compound of the present invention may be
determined by one of ordinary skill in the art. The specific dose level and
frequency of
dosage for any particular subject may vary and will depend upon a variety of
factors,
including the activity of the specific compound employed, the metabolic
stability and length
of action of that compound, the species, age, body weight, general health, sex
and diet of the
subject, the mode and time of administration, rate of excretion, drug
combination, and
severity of the particular condition. Preferred subjects for treatment include
animals, most
preferably mammalian species such as humans, and domestic animals such as
dogs, cats,
horses, and the like, at risk of developing a microbe-mediated epithelial
condition or disease,
such as gut-derived sepsis, or at risk of developing an inflammatory disorder,
e.g., acute lung
injury, characterized by cell barrier dysfunction. Generally, the peripheral
opioid receptor
antagonists of the invention are administered in an effective amount, e.g.,
from 10-6 M to 10-9
M. Patient drug plasma levels may be measured using routine HPLC methods known
to
those of skill in the art.
The invention provides methods of administering opioid receptor antagonists
to treat, prevent, or alleviate a symptom associated with, a disease or
disorder
characteristically exhibiting a cell barrier dysfunction. The opioid receptor
antagonist may be
a mu opioid antagonist, or the antagonist may be a kappa opioid antagonist.
The invention
also encompasses administration of more than one opioid antagonist, including
combinations
of mu antagonists, combinations of kappa antagonists, and combinations of at
least one mu
antagonist and at least one kappa antagonist; the invention further
comprehends
administration of combinations of at least one centrally acting opioid
receptor antagonist and
at least one peripherally restricted opioid receptor antagonist. For example,
a combination of
28
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
methylnaltrexone and either alvimopan or its metabolite ADL 08-0011, or a
combination of
naltrexone and methylnaltrexone, may be administered.
As described below in the examples, and in particular Example 26, it has also
been found that both morphine and DAMGO induce cell barrier dysfunction, such
as
pulmonary microvascular endothelial cell barrier disruption. Communication
between blood
and tissue occurs through the delivery of molecules and circulating substances
across the
endothelial barrier by directed transport either through or between cells.
Certain
inflammatory syndromes, for example, acute lung injury and sepsis, reduce
barrier function.
Such barrier disruption results in increased vascular permeability and organ
dysfunction.
Disclosed below are data establishing that a peripheral opioid receptor
antagonist in
accordance with the invention enhanced endothelial cell barrier function.
Specifically, the
cell barrier disruption is blocked by pretreatment with a peripheral opioid
receptor antagonist.
For example, pretreatment with a peripheral opioid receptor antagonist (e.g.,
MNTX) protects
against cell barrier dysfunction arising from either opioid receptor-
dependent or opioid
receptor-independent effects. Of course, the peripheral opioid receptor
antagonist is also
useful in protecting against cell barrier dysfunction arising from both
opioid receptor-
dependent effects, e.g., effects of opioid receptor agonist (e.g., morphine)
binding, and
opioid receptor-independent effects, e.g., effects realized without a
contribution from a
opioid receptor, such as thrombin- and/or lipopolysaccharide (LPS)-dependent
cell barrier
dysfunction or disruption, such as in endothelial cells. Thus, opioid
receptor antagonists,
e.g., peripheral opioid receptor antagonists, are useful in the prevention
or treatment of
inflammatory syndromes, e.g., acute lung injury, atherosclerosis, and other
diseases
characterized by a cell barrier dysfunction. Thus, the methods of the
invention have
therapeutic value in the treatment of those syndromes characterized by barrier
dysfunction or
disruption, e.g., atherosclerosis, acute lung injury, microbial infection, and
the like. It is,
therefore, contemplated that the invention includes methods of reducing cell
barrier
disruption by administering to the cells an effective amount of a cell barrier
enhancement
protective agent, e.g., MNTX.
The methods of the invention also encompass treating patients who are
undergoing treatment with opioid receptor agonists, although in some
embodiments, the
patients are not chronic recipients of any opioid receptor agonist. The opioid
receptor
agonists may be exogenously or endogenously supplied, and the agonist may be a
naturally
occurring opioid or a non-naturally occurring synthetic compound. As but one
example of a
29
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
method of treating a patient undergoing treatment with an opioid receptor
agonist, cancer
patients frequently receive morphine to manage pain associated with advanced
stages of the
disease and, while the opioid receptor antagonists are useful in this
context in providing
beneficial effects on cell barrier dysfunction without undermining efforts to
manage pain,
these opioid receptor antagonists also find use in treating cancer at a much
earlier stage. In
particular, the opioid receptor antagonists are beneficially administered to
cancer patients
having pre-metastatic stage tumors, e.g., peri-operatively, where pain
management may not
dictate the need for a g opioid receptor agonist such as morphine. At this
relatively early
stage in the progression of many cancers, a opioid receptor antagonist
provides therapeutic
support of normal cell barrier function, facilitating resistance to the
metastatic processes (i.e.,
tumor cell seeding) that exploit cell barrier dysfunction. Consequently, g
opioid receptor
antagonists have a particular application in pre-metastatic cancer patient
populations, which
are populations typically free of chronic recipients of opioid receptor
agonists like morphine.
In a particular embodiment of this aspect of the invention, a opioid
receptor antagonist, e.g.,
a peripheral opioid receptor antagonist such as MNTX, is administered intra-
or peri-
operatively during cancer surgery. It is expected that any type of cancer
amenable to surgery
will be amenable to peri-operative administration of a opioid receptor
antagonist. Without
wishing to be bound by theory, the surgical intervention creates a host stress
that may signal
cells, such as endothelial and/or epithelial cells of a wide variety of
tissues, organs and organ
systems (e.g., lung, gut, vasculature, eye) in a manner that leads to a cell
barrier dysfunction
that facilitates cancer cell mobilization or metastasis. Indirect evidence in
support of this
non-binding theory is available in a retrospective study of breast cancer
patients undergoing
surgery. Exploration of "surgical stress" led to a comparative study of
regional anesthesia, in
the form of paravertebral anesthesia (levobupivacaine), versus post-operative
morphine
analgesia for the surgical patients. The results showed a substantial
reduction in tumor
recurrence and metastasis when regional anesthesia was administered rather
than post-
operative morphine. The results are consistent with the view that the
difference in outcomes
was attributable to the deleterious effect of morphine rather than the
beneficial effect of
regional anesthetics. Thus, any agent, such as opioid receptor antagonists,
including
peripheral opioid receptor antagonists, that counteracts the effects of
morphine would be
beneficial in the peri-operative environment of cancer surgery, regardless of
whether an
opioid agonist such as morphine were contemplated as part of the surgical
treatment or post-
operative care protocol.
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
Opioid receptor agonists include, but are not limited to, morphine, methadone,
codeine, meperidine, fentidine, fentanil, sufentanil, alfentanil and the like.
Opioid receptor
agonists are classified by their ability to agonize one type of receptor an
order of magnitude
more effectively than another. For example, the relative affinity of morphine
for the mu
receptor is 200 times greater than for the kappa receptor, and it is therefore
classified as a mu
opioid receptor agonist. Some opioid compounds may act as agonists towards one
receptor
type and as antagonists toward another receptor type; such and are classified
as
agonist/antagonists, (also known as mixed or partial agonists).,
"Agonist/antagonist," "partial
agonist," and "mixed agonist" are used interchangeably herein. These opioids
include, but
are not limited to, pentazocine, butorphanol, nalorphine, nalbufine,
buprenorphine,
bremazocine, and bezocine. Many of the agonist/antagonist group of opioids are
agonists of
the kappa receptors and antagonists of the mu receptors. Further, it is
envisioned that the
active metabolites of opioid receptor agonists will also be active in the
methods of the
invention. For example, the metabolites of morphine, morphine 3-glucuronide
and morphine
6-glucuronide, are expected to be active in preventing, reducing or
eliminating cell barrier
dysfunction.
The ability to selectively antagonize peripheral opioid receptors to avoid,
e.g.,
unacceptable interference with patient pain management indicates that
peripheral opioid
receptor antagonists will be useful in addressing cell barrier dysfunction-
related diseases and
disorders. The peripheral opioid receptor antagonists form a class of
compounds that can
vary in structure while maintaining the restriction to peripheral receptor
interaction. These
compounds include tertiary and quaternary morphinans, in particular
noroxymorphone
derivatives, N-substituted piperidines, and in particular, piperidine-N-
alkylcarboxylates, and
tertiary and quaternary benzomorphans. Peripherally restricted antagonists,
while varied in
structure, are typically charged, polar and/or of high molecular weight, each
of which
impedes crossing of the blood-brain barrier.
Examples of opioid receptor antagonists that cross the blood-brain barrier and
are centrally (and peripherally) active include, e.g., naloxone, naltrexone
(each of which is
commercially available from Baxter Pharmaceutical Products, Inc.) and
nalmefene (available,
e.g., from DuPont Pharma). These may be of value in attenuating cell barrier
dysfunction in
certain patients, such as those not being treated for pain management or other
opiate
treatment.
31
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
In certain embodiments, the present methods involve the administration to a
patient of a peripheral -opioid receptor antagonist that is a piperidine-N-
alkylcarboxylate
compound. Piperidine-N-alkylcarboxylate opioid antagonists include, for
example, the
compounds disclosed in U.S. Patent Nos. 5,250,542; 5,159,081; 5,270,328; and
5,434,171,
the disclosures of which are hereby incorporated herein by reference, in their
entireties. A
class of piperidine-N-alkylcarboxylate opioid antagonists include those having
the following
formula (I):
1fiIO---~
A
Xf
wherein:
RI is hydrogen or alkyl;
R2 is hydrogen, alkyl or alkenyl;
R3 is hydrogen, alkyl, alkenyl, aryl; cycloalkyl, cycloalkenyl, cycloalkyl-
substituted alkyl,
cycloalkenyl-substituted alkyl or aryl-substituted alkyl;
R4 is hydrogen, alkyl or alkenyl;
A is QR5 or NR6 R7; wherein:
R5 is hydrogen, alkyl, alkenyl, cycloalkyl, cycloalkenyl, cycloalkyl-
substituted alkyl,
cycloalkenyl-substituted alkyl, or aryl-substituted alkyl;
R6 is hydrogen or alkyl;
32
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
R7 is hydrogen, alkyl, alkenyl, cycloalkyl, aryl, cycloalkyl-substituted
alkyl, cycloalkenyl,
cycloalkenyl-substituted alkyl, aryl-substituted alkyl, aryl-substituted
alkyl, or alkylene
substituted B or, together with the nitrogen atom to which they are attached,
R6 and R7 form
a heterocyclic ring; B is
C(=O)W or NR8 R9; wherein;
R8 is hydrogen or alkyl;
R9 is hydrogen, alkyl, alkenyl, cycloalkyl-substituted alkyl, cycloalkyl,
cycloalkenyl,
cycloalkenyl-substituted alkyl, aryl or aryl-substituted alkyl or, together
with the nitrogen
atom to which they are attached, R8 and R9 form a heterocyclic ring;
W is OR10, NRl l R12, or OE; wherein
R10 is hydrogen, alkyl, alkenyl, cycloalkyl, cycloalkenyl, cycloalkyl-
substituted alkyl,
cycloalkenyl-substituted alkyl, or aryl-substituted alkyl;
Rl 1 is hydrogen or alkyl;
R12 is hydrogen, alkyl, alkenyl, aryl, cycloalkyl, cycloalkenyl, cycloalkyl-
substituted alkyl,
cycloalkenyl-substituted alkyl, aryl-substituted alkyl or alkylene substituted
C(=O)Y or,
together with the nitrogen atom to which they are attached, Rl l and R12 form
a heterocyclic
ring;
E is
o==~ ~ .
33
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
alkylene substituted (C=O)D, or --Rl 30C(=O)R14;
wherein
R13 is alkyl substituted alkylene;
R14 is alkyl;
D is OR15 or NR16 R17;
wherein:
R15 is hydrogen, alkyl, alkenyl, cycloalkyl, cycloalkenyl, cycloalkyl-
substituted alkyl,
cycloalkenyl-substituted alkyl, or aryl-substituted alkyl;
R16 is hydrogen, alkyl, alkenyl, aryl, aryl-substituted alkyl, cycloalkyl,
cycloalkenyl,
cycloalkyl-substituted alkyl or cycloalkenyl-substituted alkyl;
Rl7 is hydrogen or alkyl or, together with the nitrogen atom to which they are
attached, R16
and R17 form a heterocyclic ring;
YisORl8orNR19R20;
wherein:
R18 is hydrogen, alkyl, alkenyl, cycloalkyl, cycloalkenyl, cycloalkyl-
substituted alkyl,
cycloalkenyl-substituted alkyl, or aryl-substituted alkyl;
R19 is hydrogen or alkyl;
R20 is hydrogen, alkyl, alkenyl, aryl, cycloalkyl, cycloalkenyl, cycloalkyl-
substituted alkyl,
cycloalkenyl-substituted alkyl, or aryl-substituted alkyl or, together with
the nitrogen atom to
which they are attached, R19 and R20 form a heterocyclic ring;
34
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
R21 is hydrogen or alkyl; and
n is 0 to about 4;
or a stereoisomer, prodrug, or pharmaceutically acceptable salt, hydrate or N-
oxide thereof
In the above formula (I), Rl is hydrogen or alkyl. In some embodiments, Rl
is hydrogen or Cl -C5 alkyl. In important embodiments, Rl is hydrogen.
In the above formula (I), R2 is hydrogen, alkyl or alkenyl. In some
embodiments, R2 is hydrogen, Cl -C5 alkyl or C2 -C6 alkenyl. In some
embodiments, R2 is
alkyl, with C1 -C3 alkyl being more preferred.
In the above formula (I), R3 is hydrogen; alkyl, alkenyl, aryl, cycloalkyl,
cycloalkenyl, cycloalkyl-substituted alkyl, cycloalkenyl-substituted alkyl or
aryl-substituted
alkyl. In some embodiments, R3 is hydrogen, Cl -C10 alkyl, C3 -C10 alkenyl,
phenyl,
cycloalkyl, C5 -C8 cycloalkenyl, cycloalkyl-substituted Cl -C3 alkyl, C5 -C8
cycloalkyl-
substituted Cl -C3 alkyl or phenyl-substituted Cl -C3 alkyl. In some
embodiments, R3 is
benzyl, phenyl, cyclohexyl, or cyclohexylmethyl.
In the above formula (I), R4 is hydrogen, alkyl or alkenyl. In some
embodiments, R4 is hydrogen, Cl -C5 alkyl or C2 -C6 alkenyl. In other
embodiments, R4 is
C 1-C3 alkyl, with methyl being more preferred.
In the above formula (I), A is OR5 or NR6 R7.
In the above formula (I), R5 is hydrogen, alkyl, alkenyl, cycloalkyl,
cycloalkenyl, cycloalkyl-
substituted alkyl, cycloalkenyl-substituted alkyl, or aryl-substituted alkyl.
In some
embodiments, R5 is hydrogen, Cl -ClO alkyl, C2 -ClO alkenyl, cycloalkyl, C5 -
C8
cycloalkenyl, cycloalkyl-substituted Cl -C3 alkyl, C5 -C8 cycloalkenyl-
substituted Cl -C3
alkyl, or phenyl-substituted Cl -C3 alkyl. Also in some embodiments, R5 is
hydrogen or
alkyl, with C 1-C3 alkyl being more preferred.
In the above formula (I), R6 is hydrogen or alkyl. In some embodiments, R6 is
hydrogen or
Cl -C3 alkyl. In some embodiments, R6 is hydrogen.
In the above formula (I), R7 is hydrogen, alkyl, alkenyl, cycloalkyl, aryl,
cycloalkyl-
substituted alkyl, cycloalkenyl, cycloalkenyl-substituted alkyl, aryl-
substituted alkyl, aryl-
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
aubstituted alkyl or alkylene substituted B. In some embodiments, R7 is
hydrogen, C1 -C10
alkyl, C3 -C10 alkenyl, phenyl, cycloalkyl, cycloalkyl-substituted C1 -C3
alkyl, C5 -C8
cycloalkenyl, C5 -C8 cycloalkenyl-substituted Cl -C3 alkyl, phenyl-substituted
Cl -C3 alkyl
or (CH2)q -B. In some embodiments, R7 is (CH2)q -B.
In certain alternative embodiments, in the above formula (I), R6 and R7 form,
together with
the nitrogen atom to which they are attached, a heterocyclic ring.
The group B in the definition of R7 is
~ j ~~.. -R~N
C(=0)W or NR8 R9. In some embodiments, B is C(=O)W.
The group R8 in the definition of B is hydrogen or alkyl. In some embodiments,
R8 is
hydrogen or Cl -C3 alkyl.
The group R9 in the definition of B is hydrogen, alkyl, alkenyl, cycloalkyl-
substituted alkyl,
cycloalkyl, cycloalkenyl, cycloalkenyl-substituted alkyl, aryl or aryl-
substituted alkyl. In
some embodiments, R9 is hydrogen, Cl -ClO alkyl, C3 -C10 alkenyl, cycloalkyl-
substituted
Cl -C3 alkyl, cycloalkyl, C5 -C8 cycloalkenyl, C5 -C8 cycloalkenyl-substituted
Cl -C3
alkyl, phenyl or phenyl-substituted C1 -C3 alkyl.
In certain alternative embodiments, in the definition of B, R8 and R9 form,
together with the
nitrogen atom to which they are attached, a heterocyclic ring.
The group W in the definition of B is OR10, NRl 1 R12 or OE.
The group R10 in the definition of W is hydrogen, alkyl, alkenyl, cycloalkyl,
cycloalkenyl,
cycloalkyl-substituted alkyl, cycloalkenyl-substituted alkyl, or aryl-
substituted alkyl. In some
embodiments, R10 is hydrogen, C1 -C10 alkyl, C2 -C10 alkenyl, cycloalkyl, C5 -
C8
cycloalkenyl, cycloalkyl-substituted Cl -C3 alkyl, C5 -C8 cycloalkenyl-
substituted Cl -C3
alkyl, or phenyl-substituted Cl -C3 alkyl. Also in some embodiments, R10 is
hydrogen,
alkyl, Cl -C5 alkyl, phenyl-substitutecl alkyl, phenyl-substituted C1 -C2
alkyl, cycloalkyl or
cycloalkyl-substituted alkyl, C5 -C6 cycloalkyl-substituted C1 -C3 alkyl.
The group R11 in the definition of W is hydrogen or alkyl. In some
embodiments, Rl 1 is
hydrogen or Cl -C3 alkyl.
The group R12 in the definition of W is hydrogen, alkyl, alkenyl, aryl,
cycloalkyl,
cycloalkenyl, cycloalkyl-substituted alkyl, cycloalkenyl-substituted alkyl,
aryl-substituted
36
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
alkyl or alkylene-substituted C(=O)Y. In some embodiments, R12 is hydrogen, Cl
-C10
alkyl, C3 -C10 alkenyl, phenyl, cycloalkyl, C5 -C8 cycloalkenyl, cycloalkyl-
substituted Cl -
C3 alkyl, C5 -C8 cycloalkenyl-substituted C1 -C3 alkyl, phenyl-substituted Cl -
C3 alkyl, or
alkylene-substituted C(=O)Y. Also in some embodiments, R12 is hydrogen, alkyl,
some C1 -
C3 alkyl or (CH2)m C(O)Y, where m is 1 to 4.
The group Y in the definition of R12 is OR18 or NRl9 R20.
In certain alternative embodiments, in the definition of W, R12 and R13 form,
together with
the nitrogen atom to which they are attached, a heterocyclic ring.
The group E in the definition of W is:
alkylene substituted (C=O)D, or --R13 OC(=O)R14. In some embodiments, E is:
(CH2)m (C=O)D (where m is as defined above), or --R13 OC(=O)R14.
The group R13 in the definition of E is alkyl substituted alkylene. In some
embodiments,
R13 is Cl -C3 alkyl substituted methylene. In some embodiments, R13 is --
CH(CH3)-- or --
CH(CH2 CH3)--.
The group R14 in the definition of E is alkyl. In some embodiments, Rl4 is Cl -
C10 alkyl.
The group D in the definition of E is D is OR15 or NR16 R17.
The group R15 in the definition of D is hydrogen, alkyl, alkenyl, cycloalkyl,
cycloalkenyl,
cycloalkyl-substituted alkyl, cycloalkenyl-substituted alkyl, or aryl-
substituted alkyl. In some
embodiments, Rl5 is hydrogen, C1 -Cl0 alkyl, C2 -C10 alkenyl, cycloalkyl, C5 -
C8
cycloalkenyl, cycloalkyl-substituted Cl -C3 alkyl, C5 -C8 cycloalkenyl-
substituted Cl -C3
alkyl, or phenyl-substituted Cl -C3 alkyl. Also in some embodiments, R15 is
hydrogen or
alkyl, with C1 -C3 alkyl being more preferred.
The group R16 in the definition of D is hydrogen, alkyl, alkenyl, aryl, aryl-
substituted alkyl,
cycloalkyl, cycloalkenyl, cycloalkyl-substituted alkyl or cycloalkenyl-
substituted alkyl. In
37
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
some embodiments, R16 is hydrogen, Cl -C10 alkyl, C3 -Cl0 alkenyl, phenyl,
phenyl-
substituted Cl -C3 alkyl, cycloalkyl, C5 -C8 cycloalkenyl, cycloalkyl-
substituted Cl -C3
alkyl, C5 -C8 cycloalkenyl-substituted Cl -C3 alkyl. In some embodiments, R16
is methyl
or benzyl.
The group Rl 7 in the definition of D is hydrogen or alkyl. In some
embodiments, Rl7 is
hydrogen or C1 -C3 alkyl. In even more some embodiments, R17 is hydrogen.
In certain alternative embodiments, in the definition of D, R16 and R17 form,
together with
the nitrogen atom to which they are attached, a heterocyclic ring.
The group R18 in the definition of Y is hydrogen, alkyl, alkenyl, cycloalkyl,
cycloalkenyl,
cycloalkyl-substituted alkyl, cycloalkenyl-substituted alkyl, or aryl-
substituted alkyl. In some
embodiments, R18 is hydrogen, C 1-C 10 alkyl, C2 -C 10 alkenyl, cycloalkyl, C5
-C8
cycloalkenyl, cycloalkyl-substituted Cl -C3 alkyl, C5 -C8 cycloalkenyl-
substituted Cl -C3
alkyl, or phenyl-substituted Cl -C3 alkyl. In some embodiments, Rl8 is
hydrogen or C1 -C3
alkyl.
The group R19 in the definition of Y is hydrogen or alkyl. In some
embodiments, R19 is
hydrogen or Cl -C3 alkyl.
The group R20 in the definition of Y is hydrogen, alkyl, alkenyl, aryl,
cycloalkyl,
cycloalkenyl, cycloalkyl-substituted alkyl, cycloalkenyl-substituted alkyl, or
aryl-substituted
alkyl. In some embodiments, R20 is hydrogen, CI -C10 alkyl, C3 -C10 alkenyl,
phenyl,
cycloalkyl, C5 -C8 cycloalkenyl, cycloalkyl-substituted Cl -C3 alkyl, C5 -C
gcycloalkenyl-
substituted C1 -C3 alkyl, or phenyl-substituted Cl -C3 alkyl. In some
embodiments, R20 is
hydrogen or Cl -C3 alkyl.
In certain alternative embodiments, in the definition of Y, R19 and R20 form,
together with
the nitrogen atom to which they are attached, a heterocyclic ring.
The group R21 in the definition of B is hydrogen or alkyl. In some
embodiments, R21 is
hydrogen or Cl -C3 alkyl. In some embodiments, R21 is hydrogen.
In the above formula (I), n is 0 to about 4. In some embodiments, n is about 1
or 2.
In the above definition of R7, q is about 1 to about 4. In some embodiments, q
is about 1 to
about 3.
In the above definition of E, m is about 1 to about 4. In some embodiments, m
is about 1 to
about 3.
The compounds of formula (I) can occur as the trans and cis stereochemical
isomers by virtue
of the substituents at the 3- and 4-positions of the piperidine ring, and such
stereochemical
isomers are within the scope of the claims. The term "trans", as used herein,
refers to R2 in
38
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
position 3 being on the opposite side from the methyl group in position 4,
whereas in the
"cis" isomer R2 and the 4-methyl are on the same side of the ring. In the
methods of the
present invention, the compounds employed may be the individual stereoisomers,
as well as
mixtures of stereoisomers. In some embodiments, the methods of the present
invention
involve compounds of formula (I) wherein the group R2 at the 3-position is
situated on the
opposite side of the ring, i.e., trans to the methyl group in the 4-position
and on the same side
of the ring. These trans isomers can exist as the 3R,4R-isomer, or the 3S,4S-
isomer.
The terms "R" and "S" are used herein as cominonly used in organic chemistry
to denote specific configuration of a chiral center. The term "R" refers to
"right" and refers
that configuration of a chiral center with a clockwise relationship of group
priorities (highest
to second lowest) when viewed along the bond toward the lowest priority group.
The term
"S" or "left" refers to that configuration of a chiral center with a
counterclockwise
relationship of group priorities (highest to second lowest) when viewed along
the bond
toward the lowest priority group. The priority of groups is based upon their
atomic number
(heaviest isotope first). A partial list of priorities and a discussion,of
stereochemistry is
contained in the book: The Vocabulary of Organic Chemistry, Orchin, et al.,
John Wiley and
Sons Inc., page 126 (1980), which is incorporated herein by reference in its
entirety.
Piperidine-N-alkylcarboxylate compounds for use in the methods of the
present invention are those of formula (I) in which the configuration of
substituents on the
piperidine ring is 3R and 4R.
When R3 is not hydrogen, the carbon atom to which R3 is attached is
asymmetric. As such, this class of compounds can further exist as the
individual R or S
stereoisomers at this chiral center, or as mixtures of stereoisomers, and all
are contemplated
within the scope of the present invention. A substantially pure stereoisomer
of the
compounds of this invention can be used, i.e., an isomer in which the
configuration at the
chiral center to which R3 is attached is R or S, i.e., those compounds in
which the
configuration at the three chiral centers is 3R, 4R, S or 3R, 4R, R.
Furthermore, other asymmetric carbons can be introduced into the molecule
depending on the structure of A. As such, these classes of coinpounds can
exist as the
individual R or S stereoisomers at these chiral centers, or as mixtures of
stereoisomers, and
all are contemplated as being within the scope of methods of the present
invention.
39
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
Certain piperidine-N-alkylcarboxylate compounds for use in the methods of
the present invention include the following:
U--OCH2 CH3; U--OH; G--OH; U--NHCH2 C(O)NHCH3; U--NHCH2 C(O)NH2; G--
NHCH2 C(O)NHCH3; U--NHCH2 C(O)NHCH2 CH3; G--NH(CH2)3 C(O)OCH2 CH3; G--
NHCH2 C(O)OH; M--NHCH2 C(O)NH2; M--NH(CH2)2 C(O)OCH2 (C6 H5); X--OCH2
CH3; X--OH; X--NH(CH2)2 CH3; Z--NH(CH2)3 C(O)OCH2 CH3; X--NHCH2 C(O)OH;
Z--NH(CH2)2 N(CH3)2; Z--NH(CH2)2 C(O)NHCH2 CH3; X--OCH2 (C6 H5); X--
N(CH3)2; Z--NH(CH2)3 C(O)NHCH3; Z--NH(CH2)3 C(O)NH2; Z--NH(CH2)3
C(O)NHCH2 CH3; X--OCH2 C(O)OCH3; X--OCH2 C(O)NHCH3; and X--N(CH3)CH2
C(O)CH2 CH3; in which:
L?
t3 =
1.4
t:'r rcg'ri~~ flis
'F P
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
:~ ,rc~rrt.c xit~
t3
X rt~rt 4 ti>
vvhcrcin ciz
itnr<s-~
yr $ 4:
4F~
INI
Important piperidine-N-alkylcarboxylate compounds for use in the methods of
the present
invention include the following:
Z--OH; Z--NH(CH2)2 C(O)OH; G--NH(CH2)2 C(O)NH2; G--NH(CH2)2 C(O)NHCH3; G--
NHCH2 C(O)NH2; G--NHCH2 C(O)NHCH2 CH3; G--NH(CH2)3 C(O)NHCH3; G--
NH(CH2)2 C(O)OH; G--NH(CH2)3 C(O)OH; X--NH2; X--NHCH(CH3)2; X--OCH2
CH(CH3)2; X--OCH2 C6 H5; X--OH; X--O(CH2)4 CH3; X--O--(4-methoxycyclohexyl); X-
-OCH(CH3)OC(O)CH3; X--OCH2 C(O)NHCH2 (C6 H5); M--NHCH2 C(O)OH; M--
NH(CH2)2 C(O)OH; M--NH(CH2)2 C(O)NH2; U-NHCH2 C(O)OCH2 CH3; and U-
NHCH2 C(O)OH; wherein Z, G, X, M and U are as defined above.
Stated another way, in accordance with some embodiments of the invention,
the compound of formula (I) has the formula Q--CH2 CH(CH2 (C6 H5))C(O)OH, Q--
CH2
CH2 CH(C6 H5)C(O)NHCH2 C(O)OCH2 CH2, Q--CH2 CH2 CH(C6 H5)C(O)NHCH2
C(O)OH, Q--CH2 CH2 CH(C6 H5)C(O)NHCH2 C(O)NHCH3, Q--CH2 CH2 CH(C6
H5)C(O)NHCH2 C(O)NHCH2 CH3, G--NH(CH2)2 C(O)NH2, G--NH(CH2)2
C(O)NHCH3, G--NHCH2 C(O)NH2, G--NHCH2 C(O)NHCH3, G--NHCH3 C(O)NHCH2
CH3, G--NH(CH2)3 C(O)OCH2 CH3, G--NH(CH2)3 C(O)NHCH3, G--NH(CH2)2
C(O)OH, G--NH(CH2)3 C(O)OH, Q--CH2 CH(CH2 (C6 H11))C(O)NHCH2 C(O)OH, Q--
CH2 CH(CH2 (C6 H11))C(O)NH(CH2)2 C(O)OH, Q--CH2 CH(CH2 (C6
H11))C(O)NH(CH2)2 C(O)NH2, Z--NHCH2 C(O)OCH2 CH3, Z--NHCH2 C(O)OH, Z--
NHCH2 C(O)NH2, Z--NHCH2 C(O)N(CH3)2, Z--NHCH2 C(O)NHCH(CH3)2, Z--NHCH2
C(O)OCH2 CH(CH3)2, Z--NH(CH2)2 C(O)OCH2 (C6 H5), Z--NH(CH2 C(O)OH, Z--
41
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
NH(CH2)2 C(O)NHCH2 CH3, Z--NH(CH2)3 C(O)NHCH3, Z--NHCH2 C(O)NHCH2
C(O)OH, Z--NHCH2 C(O)OCH2 C(O)OCH3, Z--NHCH2 C(O)O(CH2)4 CH3, Z--NHCH2
C(O)OCH2 C(O)NHCH3, Z--NHCH2 C(O)O--(4-methoxycyclohexyl), Z--NHCH2
C(O)OCH2 C(O)NHCH2 (C6 H5) or Z--NHCH2 C(O)OCH(CH3)OC(O)CH3; wherein Q, G
and Z are as defined above.
In some embodiments, the compound of formula (I) has the formula
(3R,4R,S)--Z--NHCH2 C(O)OCH2 CH(CH3)2, (+)--Z--NHCH2 C(O)OH, (-)--Z--NHCH2
C(O)OH, (3R,4R,R)--Z--NHCH2 C(O)--OCH2 CH(CH3)2, (3S,4S,S)--Z--NHCH2
C(O)OCH2 CH(CH3)2, (3 S,4S,R)--Z--NHCH2 C(O)OCH2 CH(CH3)2, (3R,4R)--Z--
NHCH2 C(O)NHCH2 (C6 H5) or (3R,4R)--G--NH(CH2)3 C(O)OH, where Z and G are as
defined above. In some embodiments, the compound of formula (I) has the
formula (+)--Z--
NHCH2 C(O)OH or (-)--Z--NHCH2 C(O)OH where Z is as defined above.
Compounds of formula (I) that act locally, such as on the gut, have high
potency and are orally active. An embodiment of the present invention is the
compound (+)-
-Z--NHCH2 C(O)OH, i.e., the compound of the following formula (II).
Cn~
J25
~ ~;C GO
The compound of formula (II) has low solubility in water except at low or high
pH
conditions. Zwitterionic character may be inherent to the compound, and may
impart
desirable properties such as poor systemic absorption and sustained local
affect on the gut
following oral administration.
In an altenlate embodiment, the methods of the present invention may involve
administering to a patient a peripheral mu-opioid receptor antagonist that is
a quatemary
morphinan compound. Examples of quaternary morphinan compounds that may be
suitable
for use in the methods of the present invention include, for example,
quaternary salts of N-
methylnaltrexone, N-methylnaloxone, N-methylnalorphine, N-diallylnormorphine,
N-
allyllevallorphan and N-methylnalmefene.
42
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
In yet another alternate embodiment, the methods of the present invention may
involve administering to a patient a peripheral mu-opioid receptor antagonist
in the form of
an opium alkaloid derivative. The term "opium alkaloid derivative", as used
herein, refers to
peripheral mu-opioid receptor antagonists that are synthetic or semi-synthetic
derivatives or
analogs of opium alkaloids. In preferred form, the opium alkaloid derivatives
employed in
the methods of the present invention exhibit high levels of morphine
antagonism, while
exhibiting reduced, and preferably substantially no, agonist activity. The
term "substantially
no agonist activity", as used herein in connection with the opium alkaloid
derivatives, means
that the maximal response with respect to electrically stimulated guinea pig
ileum, at a
concentration of 1 M, is about 60% or less relative to morphine. In some
embodiments, the
opium alkaloid derivatives employed in the present methods have a maximal
response with
respect to guinea pig ileum, at a concentration of 1 M, of about 50% or less
relative to
morphine, with a maximal response of about 40% or less being more preferred.
In some
embodiments, the opium alkaloid derivatives employed in the present methods
have a
maximal response with respect to guinea pig ileum, at a concentration of I M,
of about 30%
or less relative to morphine, with a maximal response of about 20% or less. In
still other
embodiments, the opium alkaloid derivatives employed in the present methods
have a
maximal response with respect to guinea pig ileum, at a concentration of 1 M,
of about 10%
or less relative to morphine. In certain embodiments, the opium alkaloid
derivatives have a
maximal response with respect to guinea pig ileum, at a concentration of 1 M,
of about 0%
(i.e., no response).
Suitable methods for determining maximal response of opium alkaloid
derivatives with respect to electrically stimulated guinea pig ileum are
described, for
example, in U.S. Pat. Nos. 4,730,048 and 4,806,556, the disclosures of which
are hereby
incorporated herein by reference, in their entireties.
In some embodiments, the opium alkaloid derivatives employed in the
methods of the present invention have the following formulas (III) or (IV):
43
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
z
tit) ~lI It'
tzt
III
IV
wherein:
R is alkyl, cycloalkyl-substituted alkyl, aryl, aryl-substituted alkyl or
alkenyl;
Z is hydrogen or OH;
R' is X'-J(L)(T), wherein:
J is alkylene or alkenylene;
L is hydrogen, amino, or alkyl optionally substituted with CO2 H, OH or
phenyl; and
T is CO2 H, S03 H, amino or guanidino;
X' is a direct bond or C(=O); and
R" is NH--J(L)(T) or guanidino; or a stereoisomer, prodrug, or
pharmaceutically acceptable
salt, llydrate or N-oxide thereof.
In the compounds of formulas (III) and (IV) above, R is alkyl, cycloalkyl-
substituted alkyl,
aryl, aryl-substituted alkyl or alkenyl. In some embodiments, R is Cl -C5
alkyl, C3 -C6
cycloakyl-substituted alkyl, aryl, arylalkyl or trans-C2 -C5 alkenyl. In some
embodiments, R
is C1 -C3 alkyl, allyl or cyclopropylmethyl, with cyclopropylmethyl being even
more
preferred.
In the compounds of formulas (III) and (IV) above, Z is hydrogen or OH. In
some
embodiments, Z is OH.
In the compounds of formulas (III) and (IV), R' is X--J(L)(T) and R" is NH--
J(L)(T) or
guanidino.
44
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
In the definitions of R' and R", G is alkylene or alkenylene. In some
embodiments, J is C 1-
C5 alkylene, C2 -C6 alkylene interrupted by an oxygen atom, or C2 -C5
alkenylene.
In the definitions of R' and R", L is hydrogen, amino, or alkyl optionally
substituted with
C02 H, OH or phenyl. In some embodiments, L is hydrogen, amino, or Cl -C5
alkyl
optionally substituted with CO2 H, OH or phenyl. In some einbodiments, L is
hydrogen or
amino.
In the definitions of R' and R", T is CO2 H, S03 H, amino or guanidino. In
some
embodiments, T is C02 H or guanidino.
In the definition of R', X is a direct bond or C(=O).
Important opioid alkaloid derivatives that may be employed in the methods of
the present
invention include compounds of formula (III) wherein R is cyclopropylmethyl, Z
is OH, and
R' is selected from C(=O)(CH2)2 CO2 H, C(=O)(CH2)3 C02 H, C(=0)CH--CHCO2 H,
C(=O)CH2 OCH2 CO2 H, C(=O)CH(NH2)(CH2)3 NHC(--NH)NH2 or C(=O)CH(NH2)CH2
CO2 H. Also important are opioid alkaloid derivatives of formula (III) wherein
R is
cyclopropylmethyl, Z is OH, and R' is CH2CO2H. In other embodiments, the
opioid alkaloid
derivatives that may be employed in the methods of the present invention
include compounds
of formula (IV) wherein R is cyclopropylmethyl, Z is OH, and R" is NHCH2CO2H.
For
example, N-methylnaltrexone (or methylnaltrexone, MNTX) has the following
formula (V):
V
iH2~ X
N CH3
OH
/ \
O
O
HO O
Methods for synthesis, formulating and manufacturing MNTX have been
described in a co-pending U.S. Patent Application (nuinber not yet assigned)
titled
"SYNTHESIS OF (R)-N-METHYLNALTREXONE", Attorney Docket No.
P0453.70119US01, filed on May 25, 2006, and hereby incorporated by reference
in its
entirety.
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
Other opioid alkaloid derivatives that may be employed in the methods of the
present invention are described, for example, in U.S. Pat. Nos. 4,730,048 and
4,806,556, the
disclosures of which are hereby incorporated herein by reference, in their
entireties.
In still other embodiments, the methods of the present invention may involve
administering to a patient a peripheral mu-opioid receptor antagonist compound
in the form
of a quaternary benzomorphan compound. In some embodiments, the quatemary
benzomorphan compounds employed in the methods of the present invention
exhibit high
levels of morphine antagonism, while exhibiting reduced, and preferably
substantially no,
agonist activity. The term "substantially no agonist activity", as used herein
in connection
with the quaternary benzomorphan compounds, means that the maximal response
with .
respect to electrically stimulated guinea pig ileum, at a concentration of 1
M, is about 60%
or less relative to morphine. In some embodiments, the quatemary benzomorphan
compounds einployed in the present methods have a maximal response with
respect to guinea
pig ileum, at a concentration of 1 M, of about 50% or less relative to
morphine, with a
maximal response of about 40% or less being more preferred. In some
embodiments, the
quaternary benzomorphan compounds employed in the present methods have a
maximal
response with respect to guinea pig ileum, at a concentration of 1 M, of
about 30% or less
relative to morphine, with a maximal response of about 20% or less being. In
some
embodiments, the quaternary benzomorphan compounds employed in the present
methods
have a maximal response with respect to guinea pig ileum, at a concentration
of 1 M, of
about 10% or less relative to morphine. In certain embodiments, the quaternary
benzomorphan compounds have a maximal response with respect to guinea pig
ileum, at a
concentration of 1 jiM, of about 0% (i.e., no response).
In some embodiments, the quaternary benzomorphan compounds employed in
the methods of the present invention have the following formula (VI):
VI
46
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
where:
R24 is hydrogen or acyl; and
R25 is alkyl or alkenyl;
or a stereoisomer, prodrug, or pharmaceutically acceptable salt, hydrate or N-
oxide thereof.
In the above formula (VI), R24 is hydrogen or acyl. In some embodiments, R24
is hydrogen
or Cl -C6 acyl. In some embodiments, R24 is hydrogen or Cl -C2 acyl. In some
embodiments, R24 is hydrogen or acetoxy, with hydrogen being still more
preferred.
In the above formula (VI), R25 is alkyl or alkenyl. In some embodiments,
R25 is Cl -C6 alkyl or C2 -C6 alkenyl. In some embodiments, R25 is Cl -C3
alkyl or C2 -
C3 alkenyl. In some embodiments, R25 is propyl or allyl.
Important quaternary benzomorphan compounds that may be employed in the
methods of the present invention include the following compounds of formula
(VI): 2'-
hydroxy-5,9-dimethyl-2,2-diallyl-6,7-benzomorphanium-bromide; 2'-hydroxy-5,9-
dimethyl-
2-n-propyl-6,7-benzomorphan; 2'-hydroxy-5,9-dimethyl-2-allyl-6,7-benzomorphan;
2'-
hydroxy-5,9-dimethyl-2-n-propyl-2-allyl-6,7-benzomorphanium-bromide; 2'-
hydroxy-5,9-
dimethyl-2-n-propyl-2-propargyl-6,7-benzomorphanium-bromide; and 2'-acetoxy-
5,9-
dimethyl-2-n-propyl-2-allyl-6,7-benzomorphanium-bromide.
Other quaternary benzomorphan compounds that may be employed in the
methods of the present invention are described, for example, in U.S. Pat. No.
3,723,440, the
disclosures of which are hereby incorporated herein by reference, in their
entirety.
Other mu opioid receptor antagonists which may be employed in the methods
and compositions of the present invention, in addition to those exemplified
above, would be
readily apparent to one of ordinary skill in the art, once armed with the
teachings of the
present disclosure.
The compounds employed in the methods of the present invention may exist in
prodrug form. As used herein, "prodrug" is intended to include any covalently
bonded
47
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
carriers which release the active parent drug, for example, as according to
formulas (I) or (II)
or other formulas or compounds employed in the methods of the present
invention in vivo
when such prodrug is administered to a mammalian subject. Since prodrugs are
known to
enhance numerous desirable qualities of pharmaceuticals (e.g., solubility,
bioavailability,
manufacturing, etc.) the compounds employed in the present methods may, if
desired, be
delivered in prodrug form. Thus, the present invention contemplates methods of
delivering
prodrugs. Prodrugs of the compounds employed in the present invention, for
example
formula (I), may be prepared by modifying functional groups present in the
compound in
such a way that the modifications are cleaved, either in routine manipulation
or in vivo, to
yield the phannacologically active moiety.
Accordingly, prodrugs include, for example, compounds described herein in
which a hydroxy, amino, or carboxy group is bonded to any group that, when the
prodrug is
administered to a mammalian subject, cleaves to fonn a free hydroxyl, free
amino, or
carboxylic acid, respectively. Examples include, but are not limited to,
acetate, formate and
benzoate derivatives of alcohol and amine functional groups; and alkyl,
carbocyclic, aryl, and
alkylaryl esters such as methyl, ethyl, propyl, iso-propyl, butyl, isobutyl,
sec-butyl, tert-butyl,
cyclopropyl, phenyl, benzyl, and phenethyl esters, and the like.
The compounds employed in the methods of the present invention may be
prepared in a number of ways well known to those skilled in the art. The
compounds can be
synthesized, for example, by the methods described below, or variations
thereon as '
appreciated by the skilled artisan. All processes disclosed in association
with the present
invention are contemplated to be practiced on any scale, including milligram,
gram,
multigram, kilogram, multikilogram or commercial industrial scale.
Compounds employed in the present methods may contain one,or more
asymmetrically substituted carbon atoms, and may be isolated in optically
active or racemic
fonns. Thus, all chiral, diastereomeric, racemic forms and all geometric
isomeric forms of a
structure are intended, unless the specific stereochemistry or isomeric form
is specifically
indicated. It is well known in the art how to prepare and isolate such
optically active forms.
For example, mixtures of stereoisomers may be separated by standard techniques
including,
but not limited to, resolution of racemic forms, normal, reverse-phase, and
chiral
chromatography, preferential salt formation, recrystallization, and the like,
or by chiral
synthesis either from chiral starting materials or by deliberate synthesis of
target chiral
centers.
48
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
As will be readily understood, functional groups present may contain
protecting groups during the course of synthesis. Protecting groups are known
per se as
chemical functional groups that can be selectively appended to and removed
from
functionalities, such as hydroxyl groups and carboxyl groups. These groups are
present in a
chemical compound to render such functionality inert to chemical reaction
conditions to
which the compound is exposed. Any of a variety of protecting groups may be
employed
with the present invention. Protecting groups include the benzyloxycarbonyl
group and the
tert-butyloxycarbonyl group. Other protecting groups that may be employed in
accordance
with the present invention may be described in Greene, T. W. and Wuts, P. G.
M., Protective
Groups in Organic Synthesis 2d. Ed., Wiley & Sons, 1991.
Piperidine-N-alkylcarboxylate compounds according to the present invention
may be synthesized employing methods taught, for example, in U.S. Pat. Nos.
5,250,542,
5,434,171, 5,159,081, and 5,270,328, the disclosures of which are hereby
incorporated herein
by reference in their entireties. For example, the 3-substituted-4-methyl-4-(3-
hydroxy- or
alkanoyloxyphenyl)piperidine derivatives employed as starting materials in the
synthesis of
the present compounds may be prepared by the general procedure taught in U.S.
Pat. No.
4,115,400 and U.S. Pat. No. 4,891,379, the disclosures of which are hereby
incorporated
herein by reference in their entireties. The starting material for the
synthesis of compounds
described herein, (3R,4R)-4-(3-hydroxypheny)-3,4-dimethylpiperidine, may be
prepared by
the procedures described in U.S. Pat. No. 4,581,456, the disclosures of which
are hereby
incorporated herein by reference, in their entirety, but adjusted as described
such that the
.beta.-stereochemistry is preferred.
The first step of the process may involves the formation of the 3-
alkoxyphenyllithium reagent by reacting 3-alkoxybromobenzene with an
alkyllithiuin
reagent. This reaction may be performed under inert conditions and in the
presence of a
suitable non-reactive solvent such as dry diethyl ether or preferably dry
tetrahydrofuran.
Preferred alkyllithium reagents used in this process are n-butyllithium, and
especially sec-
butyllithium. Generally, approximately an equimolar to slight excess of
alkyllithium reagent
may be added to the reaction mixture. The reaction may be conducted at a
temperature of
from about -20 C and about -100 C, more preferably from about -50 C. to about -
55 C.
Once the 3-alkoxyphenyllithium reagent has formed, approximately an
equimolar quantity of a 1-alkyl-4-piperidone may be added to the mixture while
maintaining
the temperature between -20 C and -100 C. The reaction is typically complete
after about 1
49
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
to 24 hours. At this point, the reaction mixture may be allowed to gradually
warm to room
temperature. The product may be isolated by the addition to the reaction
mixture of a
saturated sodium chloride solution to quench any residual lithium reagent. The
organic layer
may be separated and further purified if desired to provide the appropriate 1-
alkyl-4-(3-
alkoxyphenyl)piperidinol derivative.
The dehydration of the 4-phenylpiperidinol prepared above may be
accomplished with a strong acid according to well known procedures. While
dehydration
occurs in various amounts with any one of several strong acids such as
hydrochloric acid,
hydrobromic acid, and the like, dehydration is preferably conducted with
phosphoric acid, or
especially p-toluenesulfonic acid in toluene or benzene. This reaction may be
typically
conducted under reflux conditions, more generally from about 50 C and 150 C.
The product
thus formed may be isolated by basifying an acidic aqueous solution of the
salt form of the
product and extracting the aqueous solution with a suitable water immiscible
solvent. The
resulting residue following evaporation can then be further purified if
desired.
The 1-alkyl-4-methyl-4-(3-alkoxyphenyl)tetrahydropyridine derivatives may
be prepared by a metalloenamine alkylation. This reaction is preferably
conducted with n-
butyllithium in tetrahydrofuran (THF) under an inert atmosphere, such as
nitrogen or argon.
Generally, a slight excess of n-butyllithium may be added to a stirring
solution of the 1-alkyl-
4-(3-alkoxyphenyl)-tetrahydropyridine in THF cooled to a temperature in the
range of from
about is -50 C to about 0 C, more preferably from about -20 C to -10 C. This
mixture may
be stirred for approximately 10 to 30 minutes followed by the addition of
approximately from
1.0 to 1.5 equivalents of methyl halide to the solution while maintaining the
temperature of
the reaction mixture below 0 C. After about 5 to 60 minutes, water may be
added to the
reaction mixture and the organic phase may be collected. The product can be
purified
according to standard procedures, but the crude product is preferably purified
by either
distilling it under vacuum or slurrying it in a mixture of hexane:ethyl
acetate (65:35, v:v) and
silica gel for about two hours. According to the latter procedure, the product
may be then
isolated by filtration followed by evaporating the filtrate under reduced
pressure.
The next step in the process may involve the application of the Mannich
reaction of aminomethylation to non-conjugated, endocyclic enamines. This
reaction is
preferably carried out by combining from about 1.2 to 2.0 equivalents of
aqueous
formaldehyde and about 1.3 to 2.0 equivalents of a suitable secondary amine in
a suitable
solvent. While water may be the preferred solvent, other non-nucleophilic
solvents, such as
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
acetone and acetonitrile can also be employed in this reaction. The pH of this
solution may
be adjusted to approximately 3.0 to 4.0 with an acid that provides a non-
nucleophilic anion.
Examples of such acids include sulfuric acid, the sulfonic acids such as
methanesulfonic acid
and p-toluenesulfonic acid, phosphoric acid, and tetrafluoroboric acid, with
sulfuric acid
being preferred. To this solution may be added one equivalent of a 1-alkyl-4-
methyl-4-(3-
alkoxyphenyl)tetrahydropyridine, typically dissolved in aqueous sulfuric acid,
and the pH of
the solution may be readjusted with the non-nucleophilic acid or a suitable
secondary amine.
The pH is preferably maintained in the range of from about 1.0 to 5.0, with a
pH of about 3.0
to 3.5 being more preferred during the reaction. The reaction is substantially
complete after
about 1 to 4 hours, more typically about 2 hours, when conducted at a
temperature in the
range of from about 50 C to about 80 C, more preferably about 70 C. The
reaction may then
be cooled.to approximately 30 C, and added to a sodium hydroxide solution.
This solution
may then be extracted with a water immiscible organic solvent, such as hexane
or ethyl
acetate, and the organic phase, following thorough washing with water to
remove any
residual formaldehyde, may be evaporated to dryness under reduced pressure.
The next step of the process may involve the catalytic hydrogenation of the
prepared 1-alkyl-4-methyl-4-(3-alkoxyphenyl)-3-tetrahydropyridinemethanamine
to the
corresponding trans-l-alkyl-3,4-dimethyl-4-(3-alkoxyphenyl)piperidine. This
reaction
actually occurs in two steps. The first step is the hydrogenolysis reaction
wherein the exo C--
N bond is reductively cleaved to generate the 3-methyltetrahydropyridine. In
the second step,
the 2,3-double bond in the tetrahydropyridine ring is reduced to afford the
desired piperidine
ring.
Reduction of the enamine double bond introduced the crucial relative
stereochemistry at the 3 and 4 carbon atoms of the piperidine ring. The
reduction generally
does not occur with complete stereoselectivity. The catalysts employed in the
process may
be chosen from among the various palladium and preferably platinum catalysts.
The catalytic hydrogenation step of the process is preferably conducted in an
acidic reaction medium. Suitable solvents for use in the process include the
alcohols, such as
methanol or ethanol, as well as ethyl acetate, tetrahydrofuran, toluene,
hexane, and the like.
Proper stereochemical outcome may be dependent on the quantity of catalyst
employed. The quantity of catalyst required to produce the desired
stereochemical result may
51
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
be dependent upon the purity of the starting materials in regard to the
presence or absence of
various catalyst poisons.
The hydrogen pressure in the reaction vessel may not be critical but can be in
the range of from about 5 to 200 psi. Concentration of the starting material
by volume is
preferably around 20 mL of liquid per gram of starting material, although an
increased or
decreased concentration of the starting material can also be employed. Under
the conditions
specified herein, the length of time for the catalytic hydrogenation may not
be critical
because of the inability for over-reduction of the molecule. While the
reaction can continue
for up to 24 hours or longer, it may not be necessary to continue the
reduction conditions
after the uptake of the theoretical two moles of hydrogen. The product may
then be isolated
by filtering the reaction mixture for example through infusorial earth, and
evaporating the
filtrate to dryness under reduced pressure. Further purification of the
product thus isolated
may not be necessary and preferably the diastereomeric mixture may be carried
directly on to
the following reaction.
The alkyl substituent may be removed from the 1-position of the piperidine
ring by standard dealkylation procedures. Preferably, a chloroformate
derivative, especially
the vinyl or phenyl derivatives, may be employed and removed with acid. Next,
the prepared
alkoxy compound may be dealkylated to the corresponding phenol. This reaction
may be
generally carried out by reacting the compound in a 48% aqueous hydrobromic
acid solution.
This reaction may be substantially complete after about 30 minutes to 24 hours
when
conducted at a temperature of from about 50 C to about 150 C, more preferably
at the reflux
temperature of the reaction mixture. The mixture may then be worked up by
cooling the
solution, followed by neutralization with base to an approximate pH of 8. This
aqueous
solution may be extracted with a water immiscible organic solvent. The residue
following
evaporation of the organic phase may then be used directly in the following
step.
The compounds employed as starting materials to the compounds of the
invention can also be prepared by brominating the 1-alkyl-4-methyl-4-(3-
alkoxyphenyl)-3-
tetrahydropyridinemetlianamine at the 3-position, lithiating the bromo
compound thus
prepared, and reacting the lithiated intermediate with a methylhalide, such as
methyl bromide
to provide the corresponding 1-alkyl-3,4-dimethyl-4-(3-
alkoxyphenyl)tetrahydropyridinemethanamine. This compound may then be reduced
and
converted to the starting material as indicated above.
52
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
The compounds of the present invention can exist as the individual
stereoisomers. Preferably reaction conditions are adjusted as disclosed in
U.S. Pat. No.
4,581,456 or as set forth in Example 1 of U.S. Pat. No. 5,250,542 to be
substantially
stereoselective and provide a racemic mixture of essentially two enantiomers.
These
enantiomers may then be resolved. A procedure which may be employed to prepare
the
resolved starting materials used in the synthesis of these compounds includes
treating a'
racemic mixture of alkyl-3,4-dimethyl-4-(3-alkoxyphenyl)piperidine with either
(+)- or (-)-
ditoluoyl tartaric acid to provide the resolved intermediate. This compound
may then be
dealkylated at the 1-position with vinyl chloroformate and finally converted
to the desired 4-
(3-hydroxyphenyl)piperidine isomer.
As will be understood by those skilled in the art, the individual enantiomers
of
the invention can also be isolated with either (+) or (-) dibenzoyl tartaric
acid, as desired,
from the corresponding racemic mixture of the compounds of the invention.
Preferably the
(+)-trans enantiomer is obtained.
Although the (+)trans-3,4 stereoisomer is preferred, all of the possible
stereoiosmers of the compounds described herein are within the contemplated
scope of the
present invention. Racemic mixtures of the stereoisomers as well as the
substantially pure
stereoisomers are within the scope of the invention. The term "substantially
pure", as used
herein, refers to at least about 90 mole percent, more preferably at least
about 95 mole
percent and most preferably at least about 98 mole percent of the desired
stereoisomer' is
present relative to other possible stereoisomers.
Intermediates can be prepared by reacting a 3,4-alkyl-substituted-4-(3-
hydroxyphenyl)piperidine with a compound of the formula LCH2 (CH2),C1 CHR3
C(O)E
where L is a leaving group such as chlorine, bromine or iodine, E is a
carboxylic acid, ester
or amide, and R3 and n are as defined hereinabove. Preferably L may be
chlorine and the
reaction is carried out in the presence of a base to alkylate the piperidine
nitrogen. For
example 4-chloro-2-cyclohexylbutanoic acid, ethyl ester can be contacted with
(3R,4R)-4-(3-
hydroxyphenyl)-3,4-dimethylpiperidine to provide 4-[(3R,4R)-4-(3-
hydroxyphenyl)-3,4-
dimethyl-l-piperidine]butanoic acid, ethyl ester. Although the ester of the
carboxylic acid
may be preferred, the free acid itself or an amide of the carboxylic acid may
be used.
In alternative synthesis, the substituted piperidine can be contacted with a
methylene alkyl ester to alkylate the piperidine nitrogen. For example, 2-
methylene-3-
53
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
phenylpropanoic acid, ethyl ester can be contacted with a desired piperidine
to provide 2-
benzyl-3-piperidinepropanoic acid ethyl ester.
Another synthetic route can involve the reaction of a substituted piperidine
with a haloalkylnitrile. The nitrile group of the resulting piperidine
alkylnitrile can be
hydrolyzed to the corresponding carboxylic acid.
With each of the synthetic routes, the resulting ester or carboxylic acid can
be
reacted with an amine or alcohol to provide modified chemical structures. In
the preparation
of amides, the piperidine-carboxylic acid or -carboxylic acid ester may be
reacted with an
amine in the presence of a coupling agent such as dicyclohexylcarbodiimide,
boric acid,
borane-trimethylamine, and the like. Esters can be prepared by contacting the
piperidine-
carboxylic acid with the appropriate alcohol in the presence of a coupling
agent such as p-
toluenesulfonic acid, boron trifluoride etherate or N,N'-carbonyldiimidazole.
Alternatively,
the piperidine-carboxylic acid chloride can be prepared using a reagent such
as thionyl
chloride, phosphorus trichloride, phosphorus pentachloride and the like. This
acyl chloride
can be reacted with the appropriate amine or alcohol to provide the
corresponding amide or
ester.
Opium alkaloid derivatives according to the present invention may be
synthesized employing methods taught, for example, in U.S. Pat. Nos. 4,730,048
and
4,806,556, the disclosures of which are hereby incorporated herein by
reference in their
entireties. For example, opium alkaloid derivatives of formula (III) may be
prepared by
attaching hydrophilic, ionizable moieties R' and R" to the 6-amino group of
naltrexamine
(formula (III) where R is (cyclopropyl)methyl, Z is OH and R! is H) or
oxymorphamine
(formula (III) where R is CH3, Z is OH and R! is H). The opium alkaloid
derivatives of
formula IV may be prepared by converting the 6-keto-group of oxymorphone
(formula (VII)
where R is CH3 and Z is OH) or naltrexone (formula (VII) where R is
(cyclopropyl)methyl
and Z is OH; see also formula V) to the ionizable, hydrophilic group (R"N=) by
a Schiff base
reaction with a suitable amino-compound.
VII
54
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
'~----:t
= r.
~tr~ o r~
In a similar fashion, deoxy-opiates of formulae (III) and (IV) wherein Z is
hydrogen may be
prepared from readily available starting materials.
The compounds of formula (VII) may be synthesized employing methods
taught, for example, in U.S. Pat. No. 3,723,440, the disclosures of which are
hereby
incorporated herein by reference in their entirety.
The antagonist may be orally administered, for example, with an inert diluent
or with an assimilable edible carrier, or it may be enclosed in hard or soft
shell gelatin
capsules, or it may be compressed into tablets, or it may be incorporated
directly with the
food of the diet. For oral therapeutic administration, the antagonist may be
incorporated with
excipient and used in the form of ingestible tablets, buccal tablets, troches,
capsules, elixirs,
suspensions, syrups, wafers, and the like. The amount of antagonist in such
therapeutically
useful compositions is adjusted to achieve suitable dosages using routine
techniques within
the skill in the art. An exemplary dosage for an antagonist is an oral dosage
unit form
containing from about 0.1 to about 1000 mg of antagonist.
The tablets, troches, pills, capsules and the like may also contain one or
more
of the following: a binder, such as gum tragacanth, acacia, corn starch or
gelatin; an
excipient, such as dicalcium phosphate; a disintegrating agent, such as corn
starch, potato
starch, alginic acid and the like; a lubricant, such as magnesium'stearate; a
sweetening agent
such as sucrose, lactose, saccharin, and/or a flavoring agent, such as
peppermint, oil of
wintergreen or cherry flavoring. When the unit dosage form is a capsule, it
may contain, in
addition to materials of the above type, a liquid carrier. Various other
materials may be
present as coatings or to otherwise modify the physical form of the dosage
unit. For instance,
tablets, pills, or capsules may be coated with shellac, sugar or both. A syrup
or elixir may
contain the active compound, sucrose as a sweetening agent, methyl and
propylparabens as
preservatives, a dye, and/or flavoring, such as cherry or orange flavor. Of
course, any
material used in preparing any unit dosage form is preferably pharmaceutically
pure and
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
substantially non-toxic in the amount employed. In addition, the active
compound may be
incorporated into sustained-release preparations and formulations.
The antagonist may also be administered parenterally or intraperitoneally.
Solutions of the antagonists in unmodified form or as pharmacologically
acceptable salts are
contemplated and can be prepared in water suitably mixed with a surfactant,
such as
hydroxypropylcellulose. A dispersion can also be prepared in glycerol, liquid
polyethylene
glycols, preferably a high molecular weight polyethylene glycol of average
molecular weight
at least 15 kDa, mixtures thereof and in oils. In addition, any route of
administration
disclosed herein or known in the art may be used.
Pharmacologically and pharmaceutically acceptable salts for inclusion in
administrable compositions include, but are not limited to, those prepared
from the following
acids: hydrochloric, hydrobromic, sulfuric, nitric, phosphoric, maleic,
acetic, salicylic, p-
toluenesulfonic, tartaric, citric, methanesulfonic, formic, succinic,
naphthalene-2-sulfonic,
palmoic, 3-hydroxy-2-naphthalenecarboxylic, and benzene sulfonic. Suitable
buffering
agents include, but are not limited to, acetic acid and salts thereof (1-2%
WN); citric acid and
salts thereof (1-3% WN); boric acid and salts thereof (0.5-2.5% WN); and
phosphoric acid
and salts thereof (0.8-2% WN). Suitable preservatives include, but are not
limited to,
benzalkonium chloride (0.003-0.03% WN); 5 chlorobutanol (0.3-0.9% WIN);
parabens (0.01-
0.25% WN) and thimerosal (0.004-0.02% WN). For ease of administration, a
pharmaceutical
composition of the peripheral opioid antagonist may also contain one or more
pharmaceutically acceptable excipients, such as lubricants, diluents, binders,
carriers, and
disintegrants. Other auxiliary agents may include, e.g., stabilizers, wetting
agents,
emulsifiers, salts for influencing osmotic pressure, coloring, flavoring
and/or aromatic active
compounds.
A pharmaceutically acceptable carrier or excipient refers to a non-toxic
solid,
semi-solid or liquid filler, diluent, encapsulating material or formulation
auxiliary of any
type. For example, suitable pharmaceutically acceptable carriers, diluents,
solvents or
vehicles include, but are not limited to, water, salt (buffer) solutions,
alcohols, gum arabic,
mineral and vegetable oils, benzyl alcohols, polyethylene glycols, gelatin,
carbohydrates such
as lactose, amylose or starch, magnesium stearate, talc, silicic acid, viscous
paraffin,
vegetable oils, fatty acid monoglycerides and diglycerides, pentaerythritol
fatty acid esters,
hydroxy methylcellulose, polyvinyl pyrrolidone, and the like. Proper fluidity
may be
maintained, for example, by the use of coating materials such as lecithin, by
the maintenance
56
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
of the required particle size in the case of dispersions and by the use of
surfactants.
Prevention of the action of microorganism may be ensured by the inclusion of
various
antibacterial and antifungal agents such as paraben, chlorobutanol, phenol,
sorbic acid and
the like.
If a pharmaceutically acceptable solid carrier is used, the dosage form of the
antagonist(s) may be tablets, capsules, powders, suppositories, or lozenges.
If a liquid carrier
is used, soft gelatin capsules, transdermal patches, aerosol sprays, topical
cream, syrups or
liquid suspensions, emulsions or solutions may be the dosage form.
For parental application, particularly suitable are injectable, sterile
solutions,
preferably non-aqueous or aqueous solutions, as well as dispersions,
suspensions, emulsions,
or implants, including suppositories. Ainpoules are convenient forms in which
to administer
unit dosages.
The pharmaceutical forms suitable for injectable use include, for example,
sterile aqueous solutions or dispersions and sterile powders for the
extemporaneous
preparation of sterile injectable solutions or dispersions. In all cases, the
form is preferably
sterile; for administration via injection, the form is preferably sufficiently
non-viscous to
provide acceptable syringeability according to norms established in the art.
The antagonist
forms are preferably stable under the conditions of manufacture and storage
and are
preferably resistant to untoward contamination. The carrier may be a solvent
or dispersion
medium containing, for example, water, ethanol, polyol (for example, glycerol,
propylene
glycol, liquid polyethylene glycol, and the like), suitable mixtures thereof,
and vegetable oils.
The proper fluidity can be maintained, for example, by the use of a coating,
such as lecithin,
by the maintenance of the required particle size in the case of a dispersion,
and by the use of
surfactants. In many cases, it will be preferable to include isotonic agents,
for example,
sugars or sodium chloride. Prolonged absorption of the injectable compositions
may be
achieved by the use of agents delaying absorption, for example, aluminum
monostearate and
gelatin.
Sterile injectable solutions may be prepared by incorporating the active
compounds in the required amounts, in the appropriate solvent, with various of
the otlier
ingredients disclosed above, as required, followed by filter sterilization or
sterilization via
irradiation. Generally, dispersions may be prepared by incorporating the
sterilized active
ingredient into a sterile vehicle which contains the basic dispersion medium
and the required
57
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
other ingredients from those disclosed above. In the case of sterile powders
for the
preparation of sterile injectable solutions, the preferred methods of
preparation may include
vacuum drying and/or a freeze drying technique which yields a powder of the
active
ingredient, plus any additional desired ingredient from the previously
sterilized solution
thereof.
An injectable depot form may also be suitable and may be made by forming a
microcapsule matrix of the drug in a biodegradable polymer such as polylactide-
polyglycolide, poly(orthoesters) and poly(anhydrides). Depending upon the
ratio of drug to
polymer and the nature of the particular polymer employed, the rate of drug
release can be
controlled. Depot injectable formulations are also prepared by entrapping the
drug in
liposomes or microemulsions which are compatible with body tissues. The
injectable
formulations may be sterilized, for example, by filtration through a bacterial-
retaining filter
or by incorporating sterilizing agents in the form of sterile solid
compositions which can be
dissolved or dispersed in sterile water or other sterile injectable media just
prior to use.
For enteral application, particularly suitable are tablets, dragees, liquids,
drops,
suppositories, or capsules such as soft gelatin capsules. A syrup, elixir, or
the like can be
used wherein a sweetened vehicle is employed.
Another delivery system may include a time-release, delayed-release or
sustained-release (extended release) delivery system. Such a system can avoid
repeated
administrations of a compound of the invention, increasing convenience to the
patient and the
physician and maintaining sustained plasma levels of compounds where desired.
Many types
of controlled-release delivery systems are available and known to those of
ordinary skill in
the art. Sustained- or controlled-release compositions can be formulated,
e.g., as liposomes
or by protecting the active compound with differentially degradable coatings,
such as by
microencapsulation, multiple coatings, and the like.
For example, compounds of the invention may be combined with
pharmaceutically acceptable sustained-release matrices, such as biodegradable
polymers, to
form therapeutic compositions. A sustained-release matrix, as used herein, is
a matrix
typically composed of one or more polymers that are degradable by enzymatic or
acid-base
hydrolysis or by dissolution. Once inserted into the body, the matrix is acted
upon by
enzymes and body fluids. A sustained-release matrix may be desirably chosen
from
biocompatible materials such as liposomes, polymer-based systems such as
polylactides
58
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
(polylactic acid), polyglycolide (polymer of glycolic acid), polylactide co-
glycolide
(copolymers of lactic acid and glycolic acid), polyanhydrides,
poly(ortho)esters,
polysaccharides, polyamino acids, hyaluronic acid, collagen, chondroitin
sulfate,
polynucleotides, polyvinyl propylene, polyvinylpyrrolidone, and silicone;
nonpolymer
systems are composed of chemical components such as carboxylic acids, fatty
acids,
phospholipids, amino acids, lipids such as sterols, hydrogel release systems,
silastic systems,
peptide-based systems, implants, and the like. Specific examples include, but
are not limited
to: (a) erosional systems in which the polysaccharide is contained in a form
within a matrix,
as disclosed in U.S. Pat. Nos. 4,452,775, 4,675,189, and 5,736,152 (herein
incorporated by
reference in their entireties), and (b) diffusional systems in which an active
component
permeates, at a controlled rate, from a polymer such as described in U.S. Pat.
Nos. 3,854,480,
5,133,974 and 5,407,686 (herein incorporated by reference in their
entireties). In addition,
pump-based hard-wired delivery systems can be used, some of which are adapted
for
implantation. Suitable enteric coatings are described in PCT publication No.
WO 98125613
and U.S. Pat. No. 6,274,591, both incorporated herein by reference.
Use of a long-term sustained-release implant may be particularly suitable for
treatment of chronic conditions. "Long-terni" release, as used herein, means
that the implant
is constructed and arranged to deliver therapeutic,levels of the active
ingredient for at least 7
days, and suitably 30 to 60 days. Long-term sustained-release implants are
well-known to
those of ordinary skill in the art and include some of the release system
described above.
For topical application, one embodiment employs, as a nonsprayable form, a
viscous to semi-solid or solid form coinprising a carrier compatible with
topical application
and having a dynamic viscosity preferably greater than water. Suitable
formulations include,
but are not limited to, solutions, suspensions, emulsions, cream, ointments,
powders,
liniments, salves, aerosols, and the like, which are optionally sterilized or
mixed with
auxiliary agents, e.g., preservatives, and the like.
Transdermal or iontophoretic delivery of pharmaceutical compositions of the
peripheral opioid antagonists is also contemplated.
The therapeutic compounds of this invention may be administered to a patient
alone or in combination with a pharmaceutically acceptable carrier. As noted
above, the
relative proportions of active ingredient and carrier may be determined, for
example, by the
59
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
solubility and chemical nature of the compounds, chosen route of
administration and standard
pharmaceutical practice.
The dosage of the compounds of the present invention that will be most
suitable for prophylaxis or treatment will vary with the form of
administration, the particular
antagonist chosen, and the physiological characteristics of the particular
patient under
treatment. Typically, a daily dosage may range from about 0.001 to about 100
milligrams of
the peripheral -opioid receptor antagonist (and all combinations and
subcombinations of
ranges therein), per kilogram of patient body weight. Preferably, the a daily
dosage may be
about 0.01 to about 10 milligrams of the peripheral -opioid receptor
antagonist per kilogram
of patient body weight. Also preferred is a daily dosage of about 0.1
milligrams of the
peripheral -opioid receptor antagonist per kilogram of patient body weight.
With regard to a
typical dosage form, for example in tablet form, the peripheral -opioid
receptor antagonist is
present in an amount of about 0.1 to about 4 milligrams.
In one embodiment of this invention the product is orally administered
wherein an antagonist is enteric coated. By enteric coating an antagonist, it
is possible to
control its release into the gastrointestinal tract such that the antagonist
is not released in the
stomach, but rather is released in the intestine. Another embodiment of this
invention where
oral administration is desired provides for a combination product wherein one
of the
products, e.g., a -opioid receptor antagonist, is coated with a sustained-
release material
which effects a sustained-release throughout the gastrointestinal tract and
also serves to
minimize physical contact between the -opioid receptor antagonist and any
other compound
in the product. Furthermore, the sustained-released component can be
additionally enteric
coated such that the release of this component occurs only in the intestine.
Still another
approach involves the formulation of a combination product in which the one
component is
coated with a sustained and/or enteric release polymer, and the other
component is also
coated with a polymer such as a low-viscosity grade of hydroxypropyl
methylcellulose
(HPMC) or other appropriate material as known in the art, in order to further
separate the
active components. The polymer coating serves to form an additional barrier to
interaction
with the other component.
In some embodiments, compounds of the invention are administered in a
dosing regimen that provides a continuous dose of the compound to a subject,
i.e., a regimen
that eliminates the variation in internal drug levels found with conventional
regimens.
Suitably, a continuous dose may be achieved by administering the compound to a
subject on
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
a daily basis using any of the delivery methods disclosed herein. In one
embodiment, the
continuous dose may be achieved using continuous infusion to the subject, or
via a
mechanism that facilitates the release of the compound over time, for example,
a transdermal
patch, or a sustained release formulation. Suitably, compounds of the
invention are
continuously released to the subject in amounts sufficient to maintain a
concentration of the
compound in the plasma of the subject effective to inhibit or reduce cell
barrier dysfunction.
Compounds in accordance with the invention, whether provided alone or in
combination with
other therapeutic agents, are provided in an effective amount to prevent,
reduce or eliminate a
cell barrier dysfunction. It will be understood, however, that the total daily
usage of the
compounds and compositions of the present invention will be decided by the
attending
physician within the scope of sound medical judgment. The specific effective
dose level for
any particular patient will depend upon a variety of factors including the
disorder being
treated and the severity of the disorder; activity of the specific compound
employed; the
specific composition employed; the age, body weight, general health, sex and
diet of the
patient; the time of administration; the route of administration; the rate of
excretion of the
specific compound employed; the duration of the treatment; drugs used in
combination or
coincidental with the specific compound employed and like factors well known
in the
medical arts. For example, it is well within the level of ordinary skill in
the art to start doses
of the compound at levels lower than those required to achieve the desired
therapeutic effect
and to gradually increase the dosage until the desired effect is achieved.
If desired, the effective daily dose may be divided into multiple doses for
purposes of administration. Consequently, single-dose compositions may contain
such
amounts or submultiples thereof to make up the daily dose. As noted, those of
ordinary skill
in the art will readily optimize effective doses and co-administration
regimens as determined
by good medical practice and the clinical condition of the individual patient.
Generally, oral doses of the opioid receptor antagonists, particularly
peripheral
receptor antagonists, will range from about 1 to about 80 mg/kg body weight
per day. It is
expected that oral doses in the range from 2 to 20 mg/kg body weight will
yield beneficial
results. Generally, parenteral administration, including intravenous and
subcutaneous
administration, will range from about 0.001 to 5 mg/kg body weight. It is
expected that doses
ranging from 0.05 to 0.5 mg/kg body weight will yield the desired results.
Dosage may be
adjusted appropriately to achieve desired drug levels, local or systemic,
depending on the
mode of administration. For example, it is expected that the dosage for oral
administration of
61
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
the opioid antagonists in an enterically-coated formulation would be from 10
to 30% of the
non-coated oral dose. In the event that the response in a patient is
insufficient to such doses,
even higher doses (or effectively higher dosages by a different, more
localized, delivery
route) may be employed to the extent that patient tolerance permits. Multiple
doses per day
are contemplated to achieve appropriate systemic levels of compounds.
Appropriate system
levels can be determined by, for example, measurement of the patient's plasma
level for the
drug using routine HPLC methods known to those of skill in the art.
In some embodiments of the invention, the opioid receptor antagonists are co-
administered with an opioid compound. The term "co-administration" is meant to
refer to a
combination therapy by any administration route in which two or more agents
are
administered to a patient or subject. Co-administration of agents may also be
referred to as
combination therapy or combination treatment. The agents may be in the same
dosage
formulations or separate formulations. For combination treatment with more
than one active
agent, where the active agents are in separate dosage formulations, the active
agents can be
administered concurrently, or they each can be administered at separate times.
The agents
may be administered simultaneously or sequentially (i.e., one agent may
directly follow
administration of the other or the agents may be given episodically, i.e., one
can be given at
one time followed by the other at a later time, e.g., within a week), as long
as they are given
in a manner sufficient to allow both agents to achieve effective
concentrations in the body.
The agents may also be administered by different routes, e.g., one agent may
be administered
intravenously while a second agent is administered intramuscularly,
intravenously or orally.
In other words, the co-administration of the opioid receptor antagonist
compound with an
opioid compound is suitably considered a combined pharmaceutical preparation
which
contains an opioid receptor antagonist and an opioid compound or agent, the
preparation
being adapted for the administration of the opioid receptor antagonist on a
daily or
intermittent basis, and the administration of the opioid agent on a daily or
intermittent basis.
Thus, the opioid receptor antagonists may be administered prior to,
concomitant with, or after
administration of the opioids.
Co-administrable agents also may be formulated as an admixture as, for
example, in a single formulation or single tablet. These formulations may be
parenteral or
oral, such as the formulations described in, e.g., U.S. Pat. Nos. 6,277,384;
6,261,599;
5,958,452 and PCT Publication No. WO 98125613, each hereby incorporated by
reference.
62
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
In addition, any mode of administration disclosed herein or known in the art
to be compatible
with the contemplated co-administration is a suitable mode of administration.
In yet another aspect of the invention, the peripheral opioid receptor
antagonist
may be co-administered with an opioid or opioid receptor agonist, and another
therapeutic
agent that is not an opioid or opioid receptor agonist. The opioids and
peripheral opioid
receptor agonists are described above. Suitable therapeutic agents include
anti-biotics and
anti-inflammatory agents. The formulations may be prepared using standard
formulation
methods known to those of skill in the art.
Antibiotics include: Acedapsone; Acetosulfone Sodium; Alamecin; Alexidine;
Amdinocillin; Amdinocillin Pivoxil; Amicycline; Amifloxacin; Amifloxacin
Mesylate;
Amikacin; Amikacin Sulfate; Aminosalicylic acid; Aminosalicylate sodium;
Amoxicillin;
Amphomycin; Anipicillin; Ampicillin Sodium; Apalcillin Sodium; Apramycin;
Aspartocin;
Astromicin Sulfate; Avilamycin; Avoparcin; Azithromycin; Azlocillin;
Azlocillin Sodium;
Bacampicillin Hydrochloride; Bacitracin; Bacitracin Methylene Disalicylate;
Bacitracin Zinc;
Bambermycins; Benzoylpas Calcium; Berythromycin; Betamicin Sulfate; Biapenem;
Biniramycin; Biphenamine Hydrochloride; Bispyrithione Magsulfex; Butikacin;
Butirosin
Sulfate; Capreomycin Sulfate; Carbadox; Carbenicillin Disodium; Carbenicillin
Indanyl
Sodium; Carbenicillin Phenyl Sodium; Carbenicillin Potassium; Carumonam
Sodium;
Cefaclor; Cefadroxil; Cefamandole; Cefamandole Nafate; Cefamandole Sodium;
Cefaparole;
Cefatrizine; Cefazaflur Sodium; Cefazolin; Cefazolin Sodium; Cefbuperazone;
Cefdinir;
Cefepime; Cefepime Hydrochloride; Cefetecol; Cefixime; Cefinenoxime
Hydrochloride;
Cefinetazole; Cefinetazole Sodium; Cefonicid Monosodium; Cefonicid Sodium;
Cefoperazone Sodium; Ceforanide; Cefotaxime Sodium; Cefotetan; Cefotetan
Disodium;
Cefotiam Hydrochloride; Cefoxitin; Cefoxitin Sodium; Cefpimizole; Cefpimizole
Sodium;
Cefpiramide; Cefpiramide Sodium; Cefpirome Sulfate; Cefpodoxime Proxetil;
Cefprozil;
Cefroxadine; Cefsulodin Sodium; Ceftazidime; Ceftibuten; Ceftizoxime Sodium;
Ceftriaxone
Sodium; Cefuroxime; Cefuroxime Axetil; Cefuroxime Pivoxetil; Cefuroxime
Sodium;
Cephacetrile Sodium; Cephalexin; Cephalexin Hydrochloride; Cephaloglycin;
Cephaloridine;
Cephalothin Sodium; Cephapirin Sodium; Cephradine; Cetocycline Hydrochloride;
Cetophenicol; Chloramphenicol; Chloramphenicol Palmitate; Chloramphenicol
Pantothenate
Complex; Chloramphenicol Sodium Succinate; Chlorhexidine Phosphanilate;
Chloroxylenol;
Chlortetracycline Bisulfate; Chlortetracycline Hydrochloride; Cinoxacin;
Ciprofloxacin;
Ciprofloxacin Hydrochloride; Cirolemycin; Clarithromycin; Clinafloxacin
Hydrochloride;
63
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
Clindamycin; Clindamycin Hydrochloride; Clindamycin Palmitate Hydrochloride;
Clindamycin Phosphate; Clofazimine; Cloxacillin Benzathine; Cloxacillin
Sodium;
Cloxyquin; Colistimethate Sodium; Colistin Sulfate; Coumermycin; Coumermycin
Sodium;
Cyclacillin; Cycloserine; Dalfopristin; Dapsone; Daptomycin; Demeclocycline;
Demeclocycline Hydrochloride; Demecycline; Denofingin; Diaveridine;
Dicloxacillin;
Dicloxacillin Sodium; Dihydrostreptomycin Sulfate; Dipyrithione;
Dirithromycin;
Doxycycline; Doxycycline Calcium; Doxycycline Fosfatex; Doxycycline Hyclate;
Droxacin
Sodium; Enoxacin; Epicillin; Epitetracycline Hydrochloride; Erythromycin;
Erythromycin
Acistrate; Erythromycin Estolate; Erythromycin Ethylsuccinate; Erythromycin
Gluceptate;
Erythromycin Lactobionate; Erythromycin Propionate; Erythromycin Stearate;
Ethambutol
Hydrochloride; Ethionamide; Fleroxacin; Floxacillin; Fludalanine; Flumequine;
Fosfomycin;
Fosfomycin Tromethamine; Fumoxicillin; Furazolium Chloride; Furazolium
Tartrate;
Fusidate Sodium; Fusidic Acid; Gentamicin Sulfate; Gloximonam; Gramicidin;
Haloprogin;
Hetacillin; Hetacillin Potassium; Hexedine; Ibafloxacin; Imipenem;
Isoconazole; Isepamicin;
Isoniazid; Josamycin; Kanamycin Sulfate; Kitasamycin; Levofuraltadone;
Levopropylcillin
Potassium; Lexithromycin; Lincomycin; Lincomycin Hydrochloride; Lomefloxacin;
Lomefloxacin Hydrochloride; Lomefloxacin Mesylate; Loracarbef; Mafenide;
Meclocycline;
Meclocycline Sulfosalicylate; Megalomicin Potassium Phosphate; Mequidox;
Meropenem;
Methacycline; Methacycline Hydrochloride; Methenamine; Methenamine Hippurate;
Methenamine Mandelate; Methicillin Sodium; Metioprim; Metronidazole
Hydrochloride;
Metronidazole Phosphate; Mezlocillin; Mezlocillin Sodium; Minocycline;
Minocycline
Hydrochloride; Mirincamycin Hydrochloride; Monensin; Monensin Sodium;
Nafcillin
Sodium; Nalidixate Sodium; Nalidixic Acid; Natamycin; Nebramycin; Neomycin
Palmitate;
Neomycin Sulfate; Neomycin Undecylenate; Netilmicin Sulfate; Neutramycin;
Nifuradene;
Nifuraldezone; Nifuratel; Nifuratrone; Nifurdazil; Nifurimide; Nifurpirinol;
Nifurquinazol;
Nifurthiazole; Nitrocycline; Nitrofurantoin; Nitromide; Norfloxacin;
Novobiocin Sodium;
Ofloxacin; Ormetoprim; Oxacillin Sodium; Oximonam; Oximonam Sodium; Oxolinic
Acid;
Oxytetracycline; Oxytetracycline Calcium; Oxytetracycline Hydrochloride;
Paldimycin;
Parachlorophenol; Paulomycin; Pefloxacin; Pefloxacin Mesylate; Penamecillin;
Penicillin G
Benzathine; Penicillin G Potassium; Penicillin G Procaine; Penicillin G
Sodium; Penicillin V;
Penicillin V Benzathine; Penicillin V Hydrabamine: Penicillin V Potassium;
Pentizidone
Sodium; Phenyl Aminosalicylate; Piperacillin Sodium; Pirbenicillin Sodium;
Piridicillin
Sodium; Pirlimycin Hydrochloride; Pivampicillin Hydrochloride; Pivampicillin
Pamoate;
Pivampicillin Probenate; Polymyxin B Sulfate; Porfiromycin; Propikacin;
Pyrazinamide;
64
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
Pyrithione Zinc; Quindecamine Acetate; Quinupristin; Racephenicol; Ramoplanin;
Ranimycin; Relomycin; Repromicin; Rifabutin; Rifametane; Rifamexil; Rifamide;
Rifampin;
Rifapentine; Rifaximin; Rolitetracycline; Rolitetracycline Nitrate:
Rosaramicin; Rosaramicin
Butyrate; Rosaramicin Propionate; Rosaramicin Sodium Phosphate; Rosaramicin
Stearate;
Rosoxacin; Roxarsone; Roxithromycin; Sancycline; Sanfetrinem Sodium;
Sarmoxicillin;
Sarpicillin; Scopafingin; Sisomicin; Sisomicin Sulfate; Spariloxacin;
Spectinomycin
Hydrochloride; Spiramycin; Stallimycin Hydrochloride; Steffimycin;
Streptomycin Sulfate;
Streptonicozid; Sulfabenz; Sulfabenzamide; Sulfacetamide; Sulfacetamide
Sodium;
Sulfacytine; Sulfadiazine; Sulfadiazine Sodium; Sulfadoxine; Sulfalene;
Sulfamerazine;
Sulfameter; Sulfamethazine; Sulfamethizole; Sulfamethoxazole;
Sulfamonomethoxine;
Sulfamoxole; Sulfanilate Zinc; Sulfanitran; Sulfasalazine; Sulfasomizole;
Sulfathiazole;
Sulfazamet; Sulfisoxazole; Sulfisoxazole Acetyl; Sulfisoxazole Diolamine;
Sulfomyxin;
Sulopenem; Sultamicillin; Suncillin Sodium; Talampicillin Hydrochloride;
Teicoplanin;
Temafloxacin Hydrochloride; Temocillin; Tetracycline; Tetracycline
Hydrochloride;
Tetracycline Phosphate Complex; Tetroxoprim; Thiamphenicol; Thiphencillin
Potassium;
Ticarcillin Cresyl Sodium; Ticarcillin Disodium; Ticarcillin Monosodium;
Ticlatone;
Tiodonium Chloride; Tobramycin; Tobramycin Sulfate; Tosufloxacin;
Trimethoprim;
Trimethoprim Sulfate; Trisulfapyrimidines; Troleandomycin; Trospectomycin
Sulfate;
Tyrothricin: Vancomycin; VancomycinHydrochloride; Virginiamycin; or
Zorbamycin.
Antiviral agents include: Acemannan; Acyclovir; Acyclovir Sodium;
Adefovir; Alovudine; Alvircept Sudotox; Amantadine Hydrochloride; Aranotin;
Arildone;
Atevirdine Mesylate; Avridine; Cidofovir; Cipamfylline; Cytarabine
Hydrochloride; -
Delavirdine Mesylate; Desciclovir; Didanosine; Disoxaril; Edoxudine;
Enviradene;
Enviroxime; Famciclovir; Famotine Hydrochloride; Fiacitabine; Fialuridine;
Fosarilate;
Foscamet Sodium; Fosfonet Sodium; Ganciclovir; Ganciclovir Sodium;
Idoxuridine;
Kethoxal; Lamivudine; Lobucavir; Memotine Hydrochloride; Methisazone;
Nevirapine;
Penciclovir; Pirodavir; Ribavirin; Rimantadine Hydrochloride; Saquinavir
Mesylate;
Somantadine Hydrochloride; Sorivudine; Statolon; Stavudine; Tilorone
Hydrochloride;
Trifluridine; Valacyclovir Hydrochloride; Vidarabine; Vidarabine Phosphate;
Vidarabine
Sodium Phosphate; Viroxime; Zalcitabine; Zidovudine; Zinviroxime.
Antifiuigal agents include: Acrisorcin; Ambruticin; Amphotericin B;
Azaconazole; Azaserine; Basifungin; Bifonazole; Biphenamine Hydrochloride;
Bispyrithione
Magsulfex; Butoconazole Nitrate; Calcium Undecylenate; Candicidin; Carbol-
Fuchsin;
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
Chlordantoin; Ciclopirox; Ciclopirox Olamine; Cilofungin; Cisconazole;
Clotrimazole;
Cuprimyxin; Denofungin; Dipyrithione; Doconazole; Econazole; Econazole
Nitrate;
Enilconazole; Ethonam Nitrate; Fenticonazole Nitrate; Filipin; Fluconazole;
Flucytosine;
Fungimycin; Griseofulvin; Hamycin; Isoconazole; Itraconazole; Kalafungin;
Ketoconazole;
Lomofungin; Lydimycin; Mepartricin; Miconazole; Miconazole Nitrate; Monensin;
Monensin Sodium; Naftifine Hydrochloride; Neomycin Undecylenate; Nifuratel;
Nifurmerone; Nitralamine Hydrochloride; Nystatin; Octanoic Acid; Orconazole
Nitrate;
Oxiconazole Nitrate; Oxifungin Hydrochloride; Parconazole Hydrochloride;
Partricin;
Potassium Iodide; Proclonol; Pyrithione Zinc; Pyrrolnitrin; Rutamycin;
Sanguinarium
Chloride; Saperconazole: Scopafungin; Selenium Sulfide; Sinefingin;
Sulconazole Nitrate;
Terbinafine; Terconazole; Thiram; Ticlatone; Tioconazole; Tolciclate;
Tolindate; Tolnaftate;
Triacetin; Triafungin; Undecylenic Acid; Viridofulvin; Zinc Undecylenate; or
Zinoconazole
Hydrochloride.
Anti-inflammatory agents include: Alclofenac; Alclometasone Dipropionate;
Algestone Acetonide; Alpha Amylase; Amcinafal; Amcinafide; Amfenac Sodium;
Amiprilose Hydrochloride; Anakinra; Anirolac; Anitrazafen; Apazone;
Balsalazide
Disodium; Bendazac; Benoxaprofen; Benzydamine Hydrochloride; Bromelains;
Broperamole; Budesonide; Carprofen; Cicloprofen; Cintazone; Cliprofen;
Clobetasol
Propionate; Clobetasone Butyrate; Clopirac; Cloticasone Propionate;
Cormethasone Acetate;
Cortodoxone; Deflazacort; Desonide; Desoximetasone; Dexamethasone
Dipropionate;
Diclofenac Potassium; Diclofenac Sodium; Diflorasone Diacetate; Diflumidone
Sodium;
Diflunisal; Difluprednate; Diftalone; Dimethyl Sulfoxide; Drocinonide;
Endrysone;
Enlimomab; Enolicam Sodium; Epirizole; Etodolac; Etofenamate; Felbinac;
Fenamole;.
Fenbufen; Fenclofenac; Fenclorac; Fendosal; Fenpipalone; Fentiazac; Flazalone;
Fluazacort;
Flufenamic Acid; Flumizole; Flunisolide Acetate; Flunixin; Flunixin Meglumine;
Fluocortin
Butyl; Fluorometholone Acetate; Fluquazone; Flurbiprofen; Fluretofen;
Fluticasone
Propionate; Furaprofen; Furobufen; Halcinonide; Halobetasol Propionate;
Halopredone
Acetate; Ibufenac; lbuprofen; Ibuprofen Aluminum; Ibuprofen Piconol; Ilonidap;
Indomethacin; Indomethacin Sodium; Indoprofen; Indoxole; Intrazole;
Isoflupredone
Acetate; Isoxepac; Isoxicam; Ketoprofen; Lofemizole Hydrochloride; Lornoxicam;
Loteprednol Etabonate; Meclofenamate Sodium; Meclofenamic Acid; Meclorisone
Dibutyrate; Mefenamic Acid; Mesalamine; Meseclazone; Methylprednisolone
Suleptanate;
Momiflumate; Nabumetone; Naproxen; Naproxen Sodium; Naproxol; Nimazone;
Olsalazine
66
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
Sodium; Orgotein; Orpanoxin; Oxaprozin; Oxyphenbutazone; Paranyline
Hydrochloride;
Pentosan Polysulfate Sodium; Phenbutazone Sodium Glycerate; Pirfenidone;
Piroxicam;
Piroxicam Cinnamate; Piroxicam Olamine; Pirprofen; Prednazate; Prifelone;
Prodolic Acid;
Proquazone; Proxazole; Proxazole Citrate; Rimexolone; Romazarit; Salcolex;
Salnacedin;
Salsalate; Sanguinarium Chloride; Seclazone; Sermetacin; Sudoxicam; Sulindac;
Suprofen;
Talmetacin; Talniflumate; Talosalate; Tebufelone; Tenidap; Tenidap Sodium;
Tenoxicam;
Tesicam; Tesimide; Tetrydamine; Tiopinac; Tixocortol Pivalate; Tolmetin;
Tolmetin Sodium;
Triclonide; Triflumidate; Zidometacin; Glucocorticoids or Zomepirac Sodium.
Examples
Example I
Construction of GFP-PA I reporter strains
A plasmid containing the GFP-PA-I fusion construct was constructed using
conventional
recombinant DNA techniques. The EGFP gene encoding green fluorescent protein
was
amplified using the pBI-EGFP plasmid (Clontech) as a template. Xbal and Pstl
restriction
sites were introduced using primers TCTAGAACTAGTGGATCCCCGCGGATG (SEQ ID
NO: 1) and GCAGACTAGGTCGACAAGCTTGATATC (SEQ ID NO: 2). The PCR
product was cloned directly into the pCR 2.1 vector using a TA-cloning kit
(Invitrogen),
followed by transformation of the pCR2.1/EGFP construct into E.coli DH5a. The
EGFP
gene was excised from this construct by digestion with Xbal and Pstl and the
fragment
containing the excised gene was cloned into the E.coli-P. aeruginosa shuttle
vector pUCP24,
which had been digested with the same restriction enzymes. The resulting
construct (i.e.,
pUCP24/EGFP), containing the EGFP gene in the shuttle vector, was typically
electroporated
at 25 F and 2500 V into P. aeruginosa electrocompetent cells. Cells
containing
pUCP24/EGFP were selected by gentamicin (Gm)challenge, typically at 100 g/ml.
A
derivative of pUCP24/EGFP was generated that placed the PA-I l.ectin/adhesin
gene in close
proximity to the EGFP gene, effectively linking the genes genetically. In
addition to
incorporating the structural lecA gene, the construct contained the QS lux box
and RpoS
consensus sequences in the 5' non-coding region of ZecA, along with rRNA
sequence. The
derivative construct was termed pUCP24/PLL-EGFP. One of skill would understand
how to
make and use the above-described construct, as well as other suitable
constructs for providing
lecA, alone or in physical proximity to a marker gene such as EGFP, using any
of a variety'of
techniques.
67
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
Example 2
Location of PA-I
PA-I lectin/adhesin was localized to a previously undescribed structural
appendage on the outer surface of P. aeruginosa, using conventional
techniques.
Example 3
Correlation of in vitro and in vivo observations
C. elegans is suitable as an in vivo model system for BSC signaling and its
role in the production of PA-I. C. elegans is accepted as a highly accurate
and predictable
model in which to study the host response to P. aeruginosa. C. elegans worms
feed on lawns
of P. aeruginosa growing on solid agar and, thus, provides an ideal system in
which to study
microbial pathogenesis, especially in regard to gut-derived sepsis, since the
mode of
infectivity is via the digestive tract. These nematodes readily feed on
bacteria such as E. coli
growing on solid agar plates, yet when fed specific strains of P. aeruginosa,
mortality rates
exceed 50% within 72 hours. Mortality rates with this model have been shown to
be
dependent on both the agar envirorunent as well as the strain of P.
aeruginosa. Certain
strains are highly lethal in this model (e.g., PA14), whereas other strains
(PAOl) show
intermediate kill rates. The ability to feed C. elegans on lawns of the
completely sequenced
P. aeruginosa strain PAOl, and selected transposon mutants, while enriching
agar plates with
various host stress-derived BSCs screened for their ability to express PA-I,
makes available a
rapid screening system for genes that actively participate in in vivo
virulence against the
intestinal epithelium. With this approach, the virulence phenotype observed in
vitro has been
transferred to an in vivo model, with the expectation that results obtained
with such a model
will prove much more reliable in accurately characterizing the virulence
phenotype observed
in human patients suffering from an epithelial cell barrier dysfunction.
Example 4
In vitro recapitulation of the in vivo "cues " released duning surgical stress
In vitro studies demonstrated that pH, osmolality, and norepinephrine did not
change PA-I expression, while opioids, interferon-gamma, C4-HSL, and media
from hypoxic
and hyperthermic intestinal epithelial cells induced PA-I expression. PA-I was
functionally
expressed in epithelial cell assays in the presence of the PA-I-inducing
compounds.
Example 5
68
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
Toxin flux across epitlaelia
Exotoxin A was labeled with AlexaFluor 594, and its transepithelial flux was
measured at varying levels of decrease of transepithelial resistance (TEER) of
MDCK
monolayers that was achieved by apical application of MDCK cells to different
concentrations of pure PA-I protein. A five-fold increase in exotoxin A flux
across MDCK
cells was found when transepithelial resistance was decreased below 50% of
control.
Purified PA-I decreased the TEER of epithelial cells to the same degree as P.
aeruginosa.
PA-I null mutants of P. aeruginosa had a significantly attenuated effect on
the transepithelial
resistance of MDCK cells. Techniques used in conducting the experiments are
described in
Example 23, below, or are conventional in the art.
Example 6
Response of epithelia to purified PA-I
The degree of cell polarity (i.e. degree of cell confluency and tight
junctional
apposition) has been shown to dictate the degree of response to purified PA-I
protein. Cells
that were loosely confluent had a more profound fall in TEER in response to PA-
I compared
to "tighter" and more differentiated cell monolayers. In addition, wounded
monolayers
exposed dense areas of PA-I binding. Cell culturing was performed as described
in Example
24, below; relative confluency was assessed using conventional techniques as
would be
known in the art.
Example 7
Soluble host factors induce expression of PA-I lectin/adhesin
GFP-reporter strains permit demonstrations that virulence gene expression in
P. aeruginosa is expressed in vivo within the intestinal tract of a stressed
(30% hepatectomy)
host. EGPF reporter constructs were specifically designed to contain known
upstream
regulatory regions involved in PA-I expression (e.g., lux box (QS promoter
elements) and
RpoS). The EGFP-PA-I reporter strain, termed PLL-EGFP, was then injected into
the cecum
of sham-operated (control) mice and mice undergoing surgical hepatectomy.
Twenty-four
hours later, feces and washed cecal mucosa were then assayed for the presence
of fluorescent
bacteria. Both within the cecal lumen and in response to contact with the
intestinal
epithelium, PA-I was expressed in vivo (three- to six-fold over control
levels) in response to
elements of the local intestinal microenvironment (cecum) of mice subjected to
catabolic
(surgical) stress. These findings were verified in the non-reporter strain,
PA27853, using an
69
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
assay in which bacterial RNA is extracted from fresh feces using an RNA
protection system.
Reiterative studies were performed in which PA27853 was introduced into the
cecum of
control and hepatectomized mice and then bacterial RNA recovered from fresh
feces 24
hours later for quantitative RT-PCR (QRT-PCR) of both PA-I and exotoxin A
(about 600%
and 800% respectively). This assay provides a precise molecular "snapshot" of
the effect of
the in situ cecal environment on P. aeruginosa virulence gene expression.
Results
demonstrated that the cecal microenvironment of a stressed host induced PA-I
and exotoxin
A virulence gene expression. Next, in order to determine whether these
findings were due to
soluble factors released into the intestinal lumen, particulate-free filtrates
were prepared from
cecal luminal contents from control and hepatectomized mice and added to fresh
cultures of
the reporter strain PLL-EGFP. Results demonstrated that when PA-I GFP reporter
strains
were exposed to filtered cecal contents from mice exposed to surgical
hepatectomy, a 248%
12 increase in fluorescence was observed compared to 112% 15 for filtered
cecal contents
from sham-operated mice (P<0.001). These results indicated that a soluble
factor is present
in the intestinal lumen following surgical stress that activates PA-I
expression. Two
remaining issues included, first, whether the soluble PA-I-inducing components
are generated
from within the intestinal tract itself or from the systemic compartment and,
second, whether
the soluble PA-I-inducing components are specific to hepatectomy-induced
stress. To
address these issues an animal model of segmental intestinal ischemia was
developed in
which an isolated loop of intestine (6 cm, proximal ileum) was luminally
cannulated and
timed aliquots of luininal perfusates were collected following 10 minutes of
ischemia
followed by 10 minutes of reperfusion. Blood was then obtained at the end of
the experiment
in order to determine the effect of systemic factors on PA-I expression. The
results indicated
that 1) intestinal ischemia, similar to hepatectomy, can release soluble
factors into the
intestinal lumen capable of signaling P. aeruginosa to express PA-I; 2) these
factors may
originate from the intestinal tract itself, since during ischemia the
intestine is isolated from
systemic factors; 3) blood components do not induce PA-I expression; and 4)
the presence of
the normal flora, virtually absent in flushed small bowel segments, appears to
play no role in
this response. To rule out the possibility that the in vivo expression of PA-I
was due to
secondary effects of surgical stress on physico-chemical changes in the local
microenvironment, P. aeruginosa strain PA-27853 and reporter strains (PLL-
EGFP) were
exposed to ambient hypoxia (0.3% 02), pH changes (6-8), and 80% CO2. None of
these
conditions induced PA-I expression. In addition, reporter strains exposed to
the blood or
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
liver tissue of mice following sham-operation or hepatectomy, did not display
enhanced
fluorescence. These studies suggest that bacterial signaling components
released in response
to surgical and ischemic stress are highly concentrated in the intestinal
tract and are generated
by host-cell derived factors that can be isolated from, and detected within,
the intestinal
lumen. Based on these results, it is expected that any form of stress (e.g.,
surgery, injury such
as traumatic injury, illness, heat, starvation, hypoxia, and the like) to
epithelial cells, such as
intestinal epithelial cells, will typically lead to a change in the level of
at least one soluble
factor involved in bacterial signaling, i.e., at least one soluble BSC.
Example 8
Bacterial Signaling Compounds (BSCs) inducing PA-I lectin/adhesin expression
are found in
epithelial cells
Using Caco-2 intestinal epithelial cells, the issue of whether components of
intestinal epithelial cells themselves played a role in triggering the
expression of PA-I was
addressed. Strain PA27853 was exposed to media (apical and basolateral) and
Caco-2 cell
fractions (cytosolic, nuclear, membrane) at various time intervals. PA-I mRNA
was
measured in PA27853 in response to the various Caco-2 cell media fractions in
the presence
and absence of Ga1Nac, a sugar that binds specifically to.PA-I and prevents P.
aeruginosa
adherence to Caco-2 cells. Media alone from Caco-2 cells grown in transwells
(apical or
basolateral) had no effect on PA-I expression. However, Caco-2 cell membrane
fractions
triggered the accumulation of a very high abundance of PA-I mRNA (>10 fold
increase)-an
effect that was attenuated in the presence of Ga1Nac. These in vitro findings
are in agreement
with the above mouse studies showing that PA-I can be activated in response to
contact with
the intestinal epithelium, yet in the unstressed Caco-2 cell system, luminal
contents (apical
media) had no effect, similar to the control mice. Experiments in which
PA27853 were
inoculated onto the apical surface of Caco-2 cells and allowed to densely
adhere (extended
culture), demonstrated an increase in PA-I inRNA, which was nearly completely
abolished in
the presence of Ga1Nac. Thus, PA-I expression is influenced by both membrane-
bound and
soluble factors, and it is contemplated that modulators of the bacterial
signaling process
include, but are not limited to, effectors (i.e., enhancers, activators, and
inhibitors) of a
soluble factor, a membrane-bound factor, or both.
Example 9
71
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
Stressed Caco-2 cells release soluble factors that induce PA-I lectin/adhesin
expression
In order to recapitulate the type of stress that the intestinal epithelium is
exposed to under conditions of surgical injury, a confluent monolayer of Caco-
2 cells was
subjected to hypoxic stress (1 hour 0.3 % hypoxia + 30 minutes normoxic
recovery). A PA-I
GFP reporter strain, PLL-EGFP, was then exposed to the apical media from
stressed and non-
stressed cells. The results demonstrated a rapid and significant increase in
PA-I promoter
activity in these strains based on relative fluorescence units (RFU's) of PLL-
EGFP. Results
were confirmed by Northern blot analysis. Analysis of the spatial and temporal
dynamics of
these experiments was carried out using fluorescent microscopy. In hypoxic
cells, contact-
induced expression of PA-I promoter activity was observed and demonstrated
preferential
adherence of bacteria to the tri-cellular junctions of Caco-2 cells (Fig 8B).
Reiterative
experiments exposing Caco-2 cells to heat shock stress (42 C 1 h + 2, h
recovery)
demonstrated similar findings to hypoxia. A near ten-fold increase in
fluorescence was
observed in the PA-I GFP reporter strain exposed to apical media from heat
shock stressed
Caco-2 cells. Membrane fractions from both hypoxic and heat shock stressed
Caco-2 cells
induced extremely high PA-I expression (approximately 100 fold) that could not
be
quantifiably distinguished between groups.
Media from hypoxic and heat shock stressed Caco-2 cells were next
fractionated into 5 molecular weight fractions (<3, 3-10, 10-20, 20-30, >30
kDa) using
centricones, to determine if a specific MW fraction could be identified that
induces PA-I
expression. In addition, to determine if the bacterial signaling compound (s)
was a protein,
fractions were treated with heat inactivation and the protein inhibitor,
proteinase K. For the
hypoxic media the identified fraction was 10-30 kD and for the heat shock
fraction the
identified fraction was 30-50 W. Both fractions were inactivated, consistent
with the BSC
being proteins. Data from these experiments strongly suggest that there are
two distinct
bacterial signaling compounds released into the apical media in response to
hypoxic and heat
shock stress in Caco-2 cells that are proteins (peptides). These findings are
significant
because 1) the fractionated compounds are soluble and can be mass produced in
unlimited
supply by growing large sheets of Caco-2 cells, and 2) the compounds are
proteins and
therefore can be easily characterized by mass spectrometry and identified.
Although more
highly purified and characterized factors will facilitate technological
development, screens
for inodulators of the activity (e.g., bacterial signaling activity) of such
factors are presently
72
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
available, with variations on a given screening methodology apparent to one of
ordinary skill
using no more than routine procedures.
Stirnulated imnaune cells release factors that induce PA-I lectin/adhesin
expression
Immune elements released at the mucosal epitllelial surface, the primary site
of colonization for P. aeruginosa, were considered to be suitable candidates
to serve as host
stress-derived bacterial signaling compounds. As a physiologically relevant in
vitro system
to determine whether immune factors can activate P. aeruginosa virulence,
supernatants from
antigen-stimulated T cells were evaluated for their ability to increase PA-I
expression in the
P. aeruginosa strain PLL-EGFP/27853, which carries a PA-I-GFP reporter
construct. P.
aeruginosa cells were incubated with supernatants from stimulated T-cells and
PA-I
expression was assessed by GFP expression levels (fluorescence). Media from
activated T
cells, which release a comprehensive array of cytokines (D. J.
Schwartzentruber, S. L.
Topalian, M. Mancini, S. A. Rosenberg, Jlmmunol 146, 3674 (May 15, 1991)),
induced PA-I
expression as assessed by enhancement of fluorescence in the PA-I-GFP fusion
reporter
strain (L. Wu et al., Gastroenterology 126, 488 (Feb, 2004)) (Fig. lA).
To determine whether this effect was due to cytokines, the reporter strain was
exposed to various cytokines (lluman IL-2, IL-4, IL-6, IL-8, IL-10, IL-12,
Interferon gamma
(IFN-y) and tumor necrosis factor alpha (TNF-a) with only IFN-y showing a
significant
increase in PA-I expression beginning at early stationary phase of growth
(Fig. 1C). None of
the cytokines tested had any significant effect on bacterial growth (Fig. 1B).
To test whether
IFN-y was required in the media of activated T-cells to enhance PA-I
expression, we depleted
IFN-y from the culture media of activated T cells using specific antibody.
Immunodepletion
of the media of IFN- y resulted in the complete loss of its PA-I inducing
capacity (Fig. lA),
suggesting that IFN-y is essential for PA-I expression in this system. To
further confirm the
role of IFN-y as a host stress-derived bacterial signaling compound, we
exposed the
completely genomically sequenced strain of P. aeruginosa, PAO 1 (C. K. Stover
et al.,lVature
406, 959 (Aug 31, 2000)), to human recombinant IFN-y, TNF-a, and various other
cytokines
(IL-2, IL-4, IL-8, IL-10) and measured lecA (encoding for PA-I) mRNA by
Northern blot.
IFN-y, but not TNF-a or other cytokines, induced lecA mRNA (Fig. 1D). These
data
indicated that human IFN-y functions as a host cell-derived bacterial
signaling molecule to
which P. aeruginosa responds with enhanced virulence.
Exam 1~e10
73
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
Identification of host stress-derived BSCs by screening candidate agents: the
role of
cytokines
As a method to rapidly identify host BSCs, P. aeruginosa strains were
exposed to media containing adenosine (released by Caco-2 cells in response to
hypoxia)
TNFa, IL-2, IL-6 IL-8 (released by epithelia in response to bacterial
invasion/ischemia), and
IFNy (released by intraepithelial lymphocytes in response to bacterial
invasion/ischemia). In
addition, strains were exposed to apical media from Caco-2 cells basolaterally
exposed to
single and combinations of the various epithelial-derived cytokines. Dr.
Jerrold Turner, has
demonstrated that basolateral exposure of Caco-2 cells to the combination of
IFN y and TNF
a activates cellular signaling proteins that dramatically alter tight
junctional proteins and
function. Media from Caco-2 cells exposed to various combinations of these
cytokines had
no effect on PA-I expression. However, IFN-y alone induced a direct effect on
PA-I
expression while none of the other compounds alone had any effect. Another
issue was
whether IFN 7 binding to P. aeruginosa could be demonstrated for strain
PA27853. Using
both ELISA, immunofluorescence microscopy, and flow cytometry, the binding
characteristics of IFN7 were determined for both whole bacteria and membrane
fractions of
P. aeruginosa. Results demonstrated that IFN-y showed high binding affinity to
whole
bacterial cells of PA27853. These effects were also observed with strain PA01.
Next,
solubilized and separated membrane proteins of P. aeruginosa (PA27853) were
solubilized
and separated, which showed that IFN-y avidly binds to a single 30 kDa protein
band. It has
been difficult to immunoprecipitate a significant quantity of this protein
from PA27853, but it
has been determined that this protein can also be immunoprecipitated from E.
coli. Next,
IFN-y binding specificity, to whole bacterial cells, was determined, using
reiterative binding
studies in the presence of various gram-negative bacterial strains, including
P. aeruginosa.
Multiple strains of bacteria displayed IFN-y binding by ELISA binding assays
suggesting that
an IFN-y binding site may be conserved across a wide variety of procaryotic
cells. Finally, in
order to detennine if PA-I was functionally expressed in PA27853 in the
presence IFN-y,
PA27853 was inoculated onto Caco-2 cell monolayers in the presence of IFN-y
and the effect
on barrier dysregulating dynamics of PA27853 against this cell line were
assessed to
determine if IFN-y shifted the dynamics. IFN-y enhanced the barrier
dysregulating effect of
PA27853 against the intestinal epithelium after five hours of incubation by
about 20%. Thus,
cytokines such as IFN- y are embraced by the invention as effective modulators
of bacterial
signaling and, ultimately, of eukaryotic (e.g., epithelial) cell barrier
function.
74
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
The expression of virulence in P. aerugiyrosa is highly regulated by the
quorum sensing signaling system (QS), a hierarchical system of virulence gene
regulation
that is dependent on bacterial cell density and hence growth phase (M.
Whiteley, K. M. Lee,
E. P. Greenberg, Proc Natl Acad Sci U S A 96, 13904 (Nov 23, 1999)) (S. P.
Diggle, K.
Winzer, A. Lazdunski, P. Williams, M. Camara, JBacteriol 184, 2576 (May,
2002)).
Therefore in order to determine the effect of growth phase on the response of
P. aeruginosa
to IFN-y, bacteria were harvested at various growth phases following exposure
to IFN-y, and
PA-I mRNA and protein measured by Northern blot and immunoblot respectively.
Both PA-
I mRNA and protein were increased in response to IFN-y at early stationary
phase of growth
(Fig lE, 1F). PA-I protein expression in PAO1 was also dose dependent, with
the greatest
increase seen with 100 ng/ml (Fig. 1 G). Taken together these results
suggested the exposure
of P. aeruginosa to IFN-y enhanced PA-I expression but was not able to shift
its expression
to an earlier phase of growth.
To determine whether IFN-y induced PA-I via activation of the quorum
sensing signaling system, we measured rhlI gene expression in PAOl in response
to IFN-y by
Northern blot. IFN -y induced rhlI transcription in PAOl (Fig. 2A, 2B). RhII
is the gene
required for the synthesis of C4-HSL (C4-homoserine lactone), a core quorum
sensing
signaling molecule that plays a key role in the expression of PA-I (M. R.
Parsek, E. P.
Greenberg, Proc Natl Acad Sci (USA) 97, 8789 (Aug 1, 2000)). We next
determined if
exposure of P: aeruginosa to IFN-y would lead to the synthesis of C4-HSL. PAO
1 was
exposed to 100 ng/ml of IFN-,y and C4-HSL measured in bacterial supernatants.
C4-HSL
synthesis was increased in PAOl exposed to IFN-7 (Fig 2C). To verify that
activation of the
QS system by IFN-y led to the production of other QS-dependent virulence
products, we
measured pyocyanin production, a redox active compound, in PAO1 at various
phases of
growth following exposure to IFN-y and showed that IFN-y increased pyocyanin
production
in PAOl (Fig 2D). Finally, to determine whether rhll and rhlR are required for
the
production of pyocyanin (PCN) and PA-I expression in response to IFN-y, an
rhll mutant P.
aeruginosa strain and, independently, an rhlR- mutant P. aeruginosa strain
were exposed to
IFN-y. PCN production arid PA-I expression induced by IFN-y were abolished in
these
mutant strains (Fig 2E, 2F). These data suggest that the QS system plays a key
role in the
response of P. aeraiginosa to IFN-y.
Exam lpell
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
Interferon-y bincls to the surface of P. aeruginosa
IFN-y direct binding to a protein on the surface of P. aerugifaosa, in the
course
of virulence activation, was also investigated. ELISA binding assays were
performed by first
coating microtiter plates with P. aeruginosa (strain PAO1), then adding
recombinant human
IFN-y (rH IFN-y), followed by biotin-labeled anti-IFN-y antibody. IFN-y avidly
bound to
whole fixed cells of P. aeruginosa in a dose-dependent manner (Fig. 3A). The
ELISA data
were confirmed by the results of invmunofluorescent imaging of bacterial cells
exposed to
IFN-y followed by biotin-labeled anti- IFN-y antibody and Alexa 594-labeled
streptavidin.
The vast majority of bacterial cells (73% 3.2% vs. 8.5% 2.5%) bound IFN-y
(Fig. 3B).
The binding capacity of the IFN-y to the P. aeruginosa was affected by
bacterial growth
phase (Fig. 4A). In order to localize the binding site of IFN-y to P.
aeruginosa (PAO1),
equal protein concentrations of membrane and cytosol fractions of P.
aeruginosa were
prepared and coated onto ELISA microtiter plates. ELISA binding assays showed
that IFN-y
preferentially bound to membrane fractions of P. aeruginosa (Fig. 4B). To
determine if the
observed membrane binding by IFN-y was protein dependent, membrane fractions
were
treated with proteinase K for 3 hours and IFN-y binding assessed. Binding by
IFN-y to P.
aeruginosa membranes after treatment with proteinase K was decreased (Fig. 4C)
suggested
that IFN-y binds to protein on the bacterial cell membrane. We next determined
if other
cytokines similarly would bind to P. aeruginosa cell membranes by performing
reiterative
binding studies with human TNF-a, IL-2, IL-4, IL-10, EGF, and TGF-0. No
binding was
observed with any of these cytokines (Fig. 4D). Taken together these data
indicate IFN-y
bound to membrane protein on P. aeruginosa.
To isolate the putative protein to which IFN-y binds on the cell membrane of
P. aeruginosa, membrane proteins solubilized with mild detergents were
initially shown to
retain their binding capacity to IFN-y by ELISA (Fig. 3C). Prior to isolation
of the putative
binding protein of IFN-y, we sought to determine whether IFN-y bound to single
or multiple
membrane proteins. Membrane proteins were then separated by non-denaturing gel
electrophoresis, transferred to PVDF membranes and hybridized with IFN-y
followed by
biotin-labeled anti-IFN-y antibody. Results demonstrated a single
immunoreactive band of
about 35 kD. Immunoreactivity was IFN-y dose-dependent (Fig. 3D). In order to
identify the
putative binding protein, membrane protein was extracted from 4L of freshly
grown P.
aeruginosa and fractionated by molecular weight between 10-160 kD. Solubilized
protein
was then immunoprecipitated using-IFN-y and anti-IFN-y antibody. BSA was used
as a
76
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
control. Immunoprecipitation resulted in the appearance of a distinct protein
with a
molecular weight of about 35 kD. To further confirm that the protein isolated
by
immunoprecipitation was dependent on the presence of IFN-y, equally divided
solubilized
membrane protein fractions were mixed with and without IFN-y and then
immunoprecipitated with anti-IFN-y antibody. The 35 kD band appeared only in
the
solubilized membrane protein mixed with IFN-y (Fig. 3E). The IFN-y-dependent
band was
identified by ESI-TRAP-Electrospray LC-MSMS Ion Trap as the P. aeruginosa
outer
membrane porin OprF (Fig. 3F). These data established that IFN-y binds to the
P.
aeruginosa outer membrane protein OprF (A. O. Azghani, S. Idell, M. Bains, R.
E. Hancock,
Microb Pathog 33, 109 (Sep, 2002)).
To verify that OprF represented the major binding site for IFN-y in P.
aeruginosa strain PAO1, solubilized membrane proteins from OprF knockout
strains of P.
aeruginosa strain PAO1 (M. A. Jacobs et al., Proc Natl Acad Sci USA 100, 14339
(Nov 25,
2003)) were tested for their ability to bind IFN-y in comparison to the wild-
type strain using
the established ELISA and immunoprecipitation technique. ELISA binding assays
of
solubilized membrane proteins demonstrated reduced binding of IFN-y in OprF-
strains (Fig.
5A). Immunoprecipitation of solubilized membrane protein using IFN-y and
specific
antibody confirmed the role of OprF by showing complete loss of the
approxiinately 35 kD
band in the OprF mutant strain (Fig. 5B). To verify the functional role of
OprF on the
responsiveness of P. aeruginosa to IFN-y, we examined the expression of the PA-
I protein in
wild-type and OprF mutant strains exposed to 100 ng/ml of IFN-y. Results
demonstrate that
mutant strains failed to increase the expression of the PA-I protein in
response to an effective
stimulating dose of IFN-y as compared to the wild-type strain (Fig. 5C). The
results from
reporter gene fusion of wild-type and OprF mutant strains also demonstrated
that IFN-y
activated PA-I expression through OprF (Fig. 5I)). To further verify the role
of OprF, OprF
was reconstituted in mutant P. aeruginosa strain 31899 using the plasmid
pUCP24/OprF.
Reconstituted strains demonstrated recovery of their responsiveness to IFN-y
with an increase
in PA-I protein expression (Fig. 5E). Finally, we verified the binding between
OprF and
IFN-y by showing that purified OprF directly binds human IFN-y (Fig. 5F) in a
dose-
dependent manner.
Exam lpe 12
77
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
Identification of host stress-derived BSCs by screening candidate agents: the
role of
endogenous opioids
Although it was known that the counter-regulatory hormone, norepinephrine,
increased the binding of P. aeruginosa to human 0 erythrocytes, there has been
no
information relating to the involvement of PA-I in the process_ Accordingly,
an assay to
detect the presence of extracellular PA-I was performed. It was possible that
norepinephrine
would function as a host BSC for P. aeruginosa and, thus, affect human 0
erthyrocytes in a
manner similar to the way it affected E. coli. Despite extensive analyses, PA-
I expression
was not affected by this compound. The screening of other catecholamines, all
without
positive results, led to the expectation that opioids, particularly morphine
alkaloids, would
activate PA-I. Endogenous morphine has been documented to be released in
direct
proportion to the magnitude of surgical stress/injury in both animals and
humans. Initially,
morphine was assessed for its effects. Interestingly, exposure of Pseudomonas
strain
PA27853 to physiologic concentrations of morphine (13 [,M) resulted in a four-
fold increase
in PA-I expression (in comparison, in the same assay C4-HSL induced about a 16-
fold
increase in PA-I expression). As morphine is considered to be a non-selective
opioid,
specific endogenoizs opioid agonists with high selective affinity to , x and
8 receptors were
tested for their abilities to induce PA-I lectin/adhesin expression in strains
PA27853 and
PAO1. Also tested were two pure peptide agonists, endomorphine-1 (E1) (Tyr-
Pro-Trp-
Phe-NH2; SEQ ID N0:24) and endomorphine-2 (E2) (Tyr-Tyr-Pro-Phe-Phe-NH2; SEQ
ID
N0:25), the potent x opioid non-peptide agonist U-50488, and the potent 8
opioid non-
peptide agonist BW373U86 for their respective abilities to induce PA-I
expression in the
reporter strain P. aeruginosa PA27853/PLL-EGFP. Results demonstrated that
agonists
targeting the x and S receptors had the greatest effect on PA-I expression as
judged by
increased fluorescence of the GFP reporter strain. In order to determine if PA-
I was
functionally expressed when exposed to the various opioid agonists, the
agonists were tested
for their abilities to shift the barrier-dysregulating dynamics of PA27853 in
MDCK cells.
Results show that all three of the opioids that induced PA-I expression
(morphine, x and 8
agonists), shifted the virulence of PA27853 as judged by a more profound
decrease in the
TEER of MDCK cells following apical exposure (about 15%, 20%, and 25%
additional
TEER decrease, respectively).
In order to determine if morphine could shift the in vivo virulence of P.
aeruginosa, mice were implanted with slow release morphine pellets that
release a.daily dose
78
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
of morphine that is similar to that used clinically (pellets obtained from the
National Institute
on Drug Abuse (NIDA). Control mice were implanted with a placebo pellet. Mice
drank
infant formula spiked with a daily inoculum of lx l Og cfulml of PA27853. All
the morphine
treated mice developed severe sepsis (4/4) and significant mortality while
none of the control
mice appeared septic and all survived. Finally, agonists were tested for their
ability to induce
biofilm in PA27853, a quorum sensing dependent phenotype. Biofilm production
by P.
aer uginosa and other organisms has been established to be a major phenotype
indicative of
enhanced virulence. The opioid ic and S agonists significantly increased
biofilm production
in strains PA27853, about 150% and 180% of PA27853 induction respectively.
Taken
together, these studies demonstrate that opioid agonists can directly
influence the virulence,
and potential lethality, of P. aeruginosa. It is expected that opioid agonists
and antagonists,
whether found endogenously or not, and whether purified from a natural source,
chemically
synthesized, or produced by a combination thereof, are contemplated by the
invention as
usef-ul modulators of the bacterial signaling affecting microbial pathogenesis
generally, and
eukaryotic (e.g., epithelial or endothelial) cell barrier function more
specifically.
Example 13
Role of K-opioids in P. aeruginosa virulence expression
Opioid compounds, known to accumulate in tissues such as the lung and
intestine following stress, directly activate the virulence of P. aeruginosa
as judged by
pyocyanin production, biofilm formation, and the expression of the PA-IL
protein.
Specifically, pyocyanin production was enhanced in the presence of the
selective ic-opioid
receptor agonist, U-50,488, and the naturally occurring endogenous peptide
dynorphin, also a
selective ic-opioid receptor agonist. To understand the regulatory pathway(s)
involved in
opioid-induced virulence gene expression in P. aeruginosa, the effect of U-
50,488 on
multiple mutant P. aeruginosa strains defective in key elements involved in
pyocyanin
production was examined. Results demonstrated that the global transcriptional
regulator,
MvfR, plays a key role in pyocyanin production in response to U-50,488. Intact
MvfR was
also shown to be required for P. aeruginosa to respond to C4-HSL, a key quorum
sensing
signaling molecule known to activate hundreds of virulence genes. Taken
together, these
studies indicate that opioid compounds serve as host-derived signaling
molecules that can be
perceived by bacteria during host stress for the purposes of activating their
virulence
phenotype.
79
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
Bacterial strains and culture conditions. P. aeruginosa strains PAOl and
27853, and their derivative strains (Table 1) were routinely grown in tryptic
soy broth (TSB)
supplemented when necessary with tetracycline (Tc), 60 gg/ml, and/or
gentamicin (Gm), 100
g/ml. Alkaloid opiates morphine, a preferable -opioid receptor agonist (A.
Shahbazian, et
al., Br J Pharmacol 135, 741 (2002)), U-50,488, a specific x-opioid receptor
agonist (J. '
Szmuszkovicz, Prog Drug Res 53, 1 (1999)), and BW373U86, a specific 8- opioid
receptor
agonist (S. F. Sezen, V. A. Kenigs and D. R. Kapusta, J Pharmacol Exp Ther
287, 238
(1998)), along with the peptide opioid dynorphin, a specific x-opioid receptor
agonist (Y.
Zhang, E. R. Butelman, S. D. Schlussman, A. Ho and M. J. Kreek,
Psychopharmacology
(Berl) 172, 422 (2004)), and specific x-opioid-receptor antagonist nor-
binaltorphimine (A.
Shahbazian, et al., Br J Pharmacol 135, 741 (2002)) were used in the
experiments. Morphine
was purchased from Abbott Laboratories, U-50,488, BW373U86, dynorphin, nor-
binaltorphimine, and methyl anthranilate from Sigma-Aldrich, and C4-HSL from
Fluka.
Complementation of MvfR mutant with mvfR gene. Amplified mvfR was
directly cloned in pCR2.1 (Invitrogen), digested with XbaI-HinDIII restriction
endonucleases
and subcloned into pUCP24 under the Plac promoter to create pUCP24/mvfR. The
plasmids
pUCP24 (blank control) and pUCP24/mvfR were electroporated in strain 13375,
defective in
MvfR production, to create the P. aeruginosa strain 13375/MvfR (Tables 1, 2).
Complementation of GacA mutant with gacA gene. The gacA gene, a
member of a two-component signaling method involved in the elaboration of
virulence in
many gram-negative bacteria, was amplified and directly cloned into pCR2.1
(Invitrogen).
The gene was then excised with Xbal-HinDIII restriction endonucleases and
subcloned into
pUCP24 under the Plac promoter to create pUCP24/gacA. The plasmids pUCP24
(blank
control) and pUCP24/gacA were electroporated in P. aeruginosa strain PA06281,
defective
in GacA production, to create the P. aeruginosa strain PA06281/ GacA (Tables
1, 2).
Truncation of MvfR. PCR products of truncated mvfR genes amplified from
pUCP24/MvfR and their respective primers (Tables 1, 2) were purified using a
Geneclean kit
(Qbiogene), digested with XbaI-HinDII1 restriction endonucleases, and ligated
into pUCP24
followed by electroporation into P. aeruginosa strain 13375.
Pyocyanin assay. Bacteria were grown in TSB at 37 C under shaking
conditions at 220 rpm, with opioid compounds added at the early exponential
phase of
bacterial growth (OD600,,,,, of about 0.15-0.2). After incubation, pyocyanin
was extracted
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
from culture media in 6 chloroform followed by re-extraction in 0.2 M HCl, and
measured at
OD520 nm as described (D. W. Essar, L. Eberly, A. Hadero and I. P. Crawford, J
Bacteriol
172, 884 (1990)).
PA-IL assays. Irnmunoblotting and fluorescence of the GFP-PA-IL reporter
strain were used to determine the effect of opioids on PA-IL expression. For
irnmunoblotting, P. aeYuginosa PAO1 was grown in TSB media with or without 100
M U-
50,488, and cells were collected at the late exponential phase of growth
(OD600 nm = 1.8).
Equal amounts of protein from each saznple were separated by 15% SDS-PAGE,
transferred
to a PDF membrane, and probed with affinity-purified rabbit polyclonal anti-PA-
IL
antibodies. The probed membranes were treated with anti-rabbit horseradish
peroxidase-
conjugated IgG, and developed using SuperSignal West Femto chemoluminescent
substrate
(Pierce). For PA-IL expression detected by fluorescence, a bacterial culture
of the GFP-PA-
IL reporter strain 27853/PLL-EGFP (L. Wu, et al., Gastroenterology 126, 488
(2004)) was
plated at a final concentration of 108 CFU/ml in 96-well fluorometry plates
(Costar) in
conventioizal media, i.e., HDMEM media containing 10% FBS and HEPES buffer
with or
without 60 gM of U-50,488. Incubation was performed at 37 C, 100 rpm, and
fluorescence
reading was performed hourly with a 96-well fluorometry Plate Reader (Synergy
HT, Biotec
Inc.) at excitation/emission of 485/528 nm. Fluorescence intensity was
normalized to cell
density measured at 600 nm.
Biofilm formation assay. Bacterial cells were plated in quadruplicate in 96-
well U-bottom plates (Falcon) at a concentration of 107 CFU/ml in M63S media
(13.6 g
KH2PO4 1-1, 2.0 g(NIH4)2SO41-1, 0.5 mg FeSO4x7H2O 1-1), supplemented with 0.5%
casamino acids, 1mM MgSO4x7H2O and 0.2% glucose, and incubated overniglit at
37 C
under static conditions. U-50,488 was added at the inoculation point. After
inoculation, the
wells were rinsed thoroughly with water and the attached material was stained
with 0.1%
crystal violet, washed with water, and solubilized in ethanol. Solubilized
fractions were
collected and absorbance measured at 590 nm as described (G. A. O'Toole and R.
Kolter,
Mol Microbiol 28, 449 (1998)) with a Plate Reader.
ic-opioid receptor agonists U-50,488 and dynorphin stimulate pyocyanin
production in P. aerugisiosa.
P. aeruginosa harvested from the intestine of surgically stress mice appeared
intensely green compared to P. aeYugifzosa from the intestines of sham-
operated control mice.
81
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
Thus, P. aeruginosa might be responding to a signal to produce increased
amounts of
pyocyanin (PCN) in response to environmental cues unique to the intestinal
tract of stressed
mice. Pyocyanin, a redox active compound that increases intracellular oxidant
stress, has
been shown to play a key role in the virulence of P. aeruginosa in animal
models mediating
tissue damage and necrosis during lung infection (G. W. Lau, H. Ran, F. Kong,
D. J. Hassett
and D. Mavrodi, Infect Immun 72, 4275 (2004)). P. aeruginosa PAOl was exposed
to
peptide opioids and alkaloid opiates representing groups of g-, x-, and S-
opioid receptor
agonists. Results indicated that following overnight exposure, the alkaloid
opiate U-50,488, a
specific x-opioid receptor agonist, induced an intensely bright green color in
P. aes uginosa
PAOI, while no such effect was observed with any of the remaining compounds.
To verify
that the color change was due to PCN production, pyocyanin was measured at
OD520 nm (D.
W. Essar, L. Eberly, A. Hadero and I. P. Crawford, J Bacteriol 172, 884
(1990)). Results
demonstrated that U-50,488 induced a dose-dependent effect on PCN production
that was
observed with P. aeruginosa strains PAO1 and 27853. Exposure of P. aeruginosa
to
dynorphin, a naturally occurring specific x-opioid receptor peptide agonist,
also enhanced
PCN production in a dose-dependent manner. Reiterative experiments performed
in the
presence of the specific x-opioid receptor antagonist norbinaltorpliimine
(NOR),
demonstrated that NOR attenuates enhanced PCN production in PAOI following
exposure to
U-50,488 in a dose-dependent manner and completely inhibits enhanced PCN
production at a
dose of 200gM.
The x-opioid-receptor agonist U-50,488 shifts pyocyanin production at
lower cell densities in P. aeruginosa.
We assessed the dynamics of PCN production in response to U-50,488 at
varying cells densities, since the expression of QS-dependent genes is known
to occur at high
bacterial cell densities when QS signaling molecules reach their threshold
concentrations. As
a positive control, bacteria were exposed to C4-homoserine lactone (C4-HSL), a
QS signaling
molecule involved in PCN regulation (M. R. Parsek and E. P. Greenberg, Proc
Natl Acad Sci
U S A 97, 8789 (2000)). We found that exposure of PAO 1 to U-50,488 had a
similar effect
to exposure of cells to C4-HSL, resulting in a shift in the production of PCN
at lower cell
densities. Neither compound had an effect on bacterial growth in TSB media.
The x-opioid-receptor agonist U-50,488 exerts its inducing effect on
pyocyanin production via elements of the quorum sensing system in Pseudomonas
aerugifzosa.
82
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
The pathways of PCN regulation and biosynthesis have been described in '
detail (D. V. Mavrodi, et al., J Bacteriol 183, 6454 (2001), E. Deziel, et
al., Proc Natl Acad
Sci U S A 101, 1339 (2004), T. R. de Kievit, Y. Kakai, J. K. Register, E. C.
Pesci and B. H.
Iglewski, FEMS Microbiol Lett 212, 101 (2002), S. L. McKnight, B. H. Iglewski
and E. C.
Pesci, J Bacteriol 182, 2702 (2000)). In order to define potential pathways by
which U-
50,488 induces PCN production, mutant strains defective in key genes involved
in PCN
production were exposed to U-50,488 and the effect on pyocyanin production was
measured.
First, mutants defective in genes encoding core elements of the QS system (J.
P. Pearson, E.
C. Pesci and B. H. Iglewski, J Bacteriol 179, 5756 (1997)) (lasR, lasl, rizlI,
rhlR) were
analyzed and the results demonstrated that exposure to U-50,488 did not
restore PCN
production (relative to non-mutant strains) in any of these mutants. The roles
of the global
virulence regulators GacA and MvfR on PCN production were then investigated.
Both GacA
(C. Reimmann, et al., Mol Microbiol 24, 309 (1997)) and MvfR (E. Deziel, et
al., Proc Natl
Acad Sci U S A 101, 1339 (2004)) have been slzown to play a major role in PCN
production
in P. aeruginosa. Neither AGacA nor AMvfR produced PCN, as expected, and
exposure to
U-50,488 could not restore PCN production. C4-HSL was also unable to restore
PCN
production in the gacA and mvfR mutants. The finding that C4-HSL did not
restore PCN
production in the GacA mutant is consistent with the finding that the
analogous QS molecule,
N-hexanoyl-HSL (C6-HSL), did not restore phenazine production in a AGacA
mutant of P.
aureofaciens (S. T. Chancey, D. W. Wood and L. S. Pierson, 3rd, Appl Environ
Microbiol 65,
2294 (1999)). Seven additional nzvfR mutants from the comprehensive transposon
library
(M. A. Jacobs, et al., Proc Nat1 Acad Sci.U S A 100, 14339 (2003)) (i.e.,
numbers 8902,
47418, 35448, 51955, 21170, 47853, and 47198) were exposed to C4-HSL in order
to
confirm this finding. Results demonstrated that none of these mutants produced
PCN in the
presence of 1 mM C4-HSL.
MvfR is involved in the ability of U-50,488 and C4-HSL to enhance PCN
production in PAO1.
In order to define the possible role of MvfR and GacA in the U-50,488-
mediated upregulation of PCN synthesis, we complemented AMvfR and AGacA with
their
respective genes on the multicopy plasmid pUCP24 (S. E. West, H. P. Schweizer,
C. Dall, A.
K. Sample and L. J. Runyen-Janecky, Gene 148, 81 (1994)). Both complemented
mutants
produced significantly higher amounts of PCN (Fig. 6A,B). The addition of C4-
HSL and U-
50,488 further increased the already elevated PCN production in AMvfR /mvfR
(Fig. 6C). In
83
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
contrast, PCN production in AGacA/gacA was decreased, albeit minimally, when
exposed
overnight to either 1 mM U-50,488 or 100 gM C4-HSL (Fig. 6D). Dynamic tracking
of PCN
production in the complemented mutant OMvfR/mvfR exposed to U-50,488 and C4-
HSL
demonstrated a shift in PCN production at lower cell densities (Fig. 6E),
similar to that
observed in the parental strain PAO 1. The gacA complemented mutant,
AGacA/gacA, itself
produced PCN at lower cell densities than those observed with the parental
strain PAO1.
Exposure of AGacA/gacA to C4-HSL had no effect on the dynamics of PCN
production
whereas exposure to U-50,488 delayed PCN production. (Fig. 6F). These results
indicate
that MvfR is involved in the up-regulation of PCN production by exogenously
applied U-
50,488 and C4-HSL.
Intact substrate-binding and DNA-binding domains of MvfR are required
for U-50,488 to enhance PCN production in PAO1.
MvfR belongs to a family of prokaryotic LysR transcriptional regulators that
possess a helix-turn-helix DNA-binding motif at the N terminus and a substrate
binding
domain at the C terminus. A NCBI Conserved Domain Search revealed similar
domains in
MvfR: a LysR DNA-binding domain located at 6-64 aa, and a LysR substrate
binding domain
located at 156-293 amino acids. Therefore PAO1 mutants were constructed
producing N-
and C-terminus-truncated MvfR to determine if specific domains could be
identified that play
a functional role in mediating the -K-opioid receptor agonist effect on PCN
production.
Results indicated that the mutant lacking amino acids 121-332, defective in
the DNA-binding
domain, did not produce any PCN, and did not respond to U-50,488 or C4-HSL.
Mutants
lacking either amino acids 1-299 or 1-293, truncated at their C termini
without affecting the
substrate binding domain, produced PCN and responded to U-50,488 and C4-HSL
with
enhanced PCN production. Further deletions, however, including amino acids
Arg293,
Leu292, and Phe284, did affect the substrate binding domain in mutants 1-292,
1-291, and 1-
283. All three mutants failed to produce PCN and did not respond to U-50,488
and C4-HSL.
These results confirm a key functional role for MvfR in mediating enhanced PCN
production
in P. aerugifaosa in response to U-50,488 and C4-HSL.
The effect of U-50,488 on PCN production is dependent on MvfR
regulated synthesis of Pseudomonas Quinolone Signal (PQS).
MvfR might play a critical role in PCN production via positive transcriptional
regulation of the phnAB and PQS ABCDE operons that encode two 12 precursors of
PQS,
84'
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
anthranilic acid (AA) and 4-hydroxy-2-heptylquinolone (HHQ) (E. Deziel, et
al., Proc Natl
Acad Sci U S A 101, 1339 (2004)). Therefore the mutants APhnA and APqsA were
examined for their ability to produce PCN in the presence of U-50,488. Neither
mutant
produced PCN. Exposure of each mutant to U-50,488 resulted in a slight
increase in PCN
production, although the increase was much less than that observed with the
wild-type strain
PAO 1. These data suggested that MvfR-regulated PQS synthesis may be important
for the
ability of U-50,488 to enhance PCN production. Finally, reiterative
experiments were
performed with a P. aef uginosa mutant defective in the phzAl gene, which is
part of the
operon that contains the core genes for PCN biosynthesis and that is directly
preceded by the
lux box (D. V. Mavrodi, et al., J Bacteriol 183, 6454 (2001)). OPhzAl produced
no PCN
even when exposed to U-50,488.
To confirm that PQS plays a role in the pathway by which U-50,488 enhances
PCN production, U-50,488 was applied to P. aeruginosa incubated with 2 mM
methyl
anthranilate (MA), a compound previously shown to inhibit PQS synthesis in P.
aeruginosa
(S. P. Diggle, et al., Mol Microbiol 50, 29 (2003), M. W. Calfee, J. P.
Coleman and E. C.
Pesci, Proc Natl Acad Sci U S A 98, 11633 (2001)). Results demonstrated that
MA inhibited
the ability of U-50,488 to enhance PCN production in PAO1. These findings
indicate that U-
50,488 triggers PCN production in P. aeruginosa via a signal transduction
pathway that
includes the activation of transcriptional regulator MvfR and the synthesis of
the MvfR-
regulated molecule, PQS.
U-50,488 stimulates other QS-regulated virulence determinants in P.
aeruginosa including biofilm formation and PA-IL production.
To determine if other QS-dependent phenotypes could be expressed in
response to U-50,488, we measured biofilm production (T. R. De Kievit, R.
Gillis, S. Marx,
C. Brown and B. H. Iglewski, Appl Environ Microbio167, 1865 (2001)) and PA-IL
lectin
expression (K. Winzer, et al., J Bacteriol 182, 6401 (2000), M. Schuster, M.
L. Urbanowski
and E. P. Greenberg, Proc Natl Acad Sci U S A 101, 15833 (2004)) in P.
aeruginosa exposed
to this opiate. U-50,488 enhanced biofilm formation in PAO1 in a concentration-
dependent
manner. PA-IL expression was dynamically tracked in response to U-50,488 using
the green
fluorescent PA-IL reporter strain P. aeruginosa 27853/PLL-EGFP (L. Wu, et al.,
Gastroenterology 126, 488 (2004)). Marked fluorescence was observed in this
strain
following 9 hours of growth in HDMEM media. Results were confirmed in strain
PAO1 by
immunoblotting using rabbit polyclonal antibody against PA-IL .
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
The effect of U-50,488 on PCN production in P. aerugitaosa can be
inhibited by the anti-infective high molecular weight polymer PEG 15-20.
A high molecular weight polymer, PEG 15-20, protects mice against lethal
sepsis due to P. aeruginosa by interfering with the ability of both host
elements (epithelial
cell contact) and the QS signaling molecule C4-HSL to enhance P. aeruginosa
virulence
without affecting bacterial growth (L. Wu, et al., Gastroenterology 126, 488
(2004)). The
capacity of PEG 15-20 to interfere with the U-50, 488 effect on P. aeruginosa
was assessed
by measuring PCN production in the media of P. aeruginosa PAOl incubated in
the presence
of 5% PEG 15-20 and 0.5 mM U-50,488 or 0.2 mM C4-HSL. Results demonstrated
that
PEG 15-20 had a strong inhibitory effect on both U-50, 488- and C4-HSL-
mediated up-
regulation of PCN production.
86
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
Table 1. Bacterial strains
P. aeruginosa strains Relevant genotype
PA27853 Wild type
PAOI Wild type
PAO-JP-1 OLasI (lasl::Tc~
PAO-RI ALasR (lasR::Tc~
PDOIOO ARh1I rhlI::Tn501
PAO-MWI ARh1IALasI (rlilI::Tn501 lasl::tetA)
PA044488 ARhIR (rhIR:: ISphoA/hah)
PA06281 AGacA ( acA::S r /Sm~
PA06281/pUCP24/GacA AGacA complemented with gacA
on pUCP24
PA06281/pUCP24 AGacA transformed with blank
UCP24
PA08902 AMvfR (mvfR:: ISlacZ/hah)
PA047418 AMvfR (mv :: ISphoAlhah)
PA035448 AMvfR (mv :: ISphoAlhah)
PA051955 AMvfR (mv :: IS hoAlhah)
PAO21170 AMvfR (mv :: ISIacZ/hah)
PA047853 OMvfR (mv :: ISphoA/hah)
PA047198 AMvfR (mv :: ISphoAlhah)
PA013375 AMvfR (mvfR:: ISlacZ/hah)
PA013375/pUCP24/MvfR AMvfR complemented with mvfR
on pUCP24
PA013375/pUCP24 AMvfR transformed with blank
pUCP24
PA053589 APqsA qsA::ISphoA/hah)
PA037309 APhzA (phzA:: IS hoAlhah)
PA047305 APhzAl hzAl::ISphoA/hah)
PA03375/pUCP24/MvfR. 1- AMvfR complemented with pUCP24
299 harboring mvfR truncated with 33 aa
at C terminus
PA013375/pUCP24/MvfR AMvfR complemented with pUCP24
1-293 harboring mifR truncated with 39 aa
at C terminus
PA013375/pUCP24/MvfR AMvfR complemented with pUCP24
1-292 harboring rni?fR. truncated with 40 aa
at C terminus
PA013375/pUCP24/MvfR AMvfR complemented with pUCP24
1-291 harboring mvfR truncated with 41 aa
at C terminus
PA013375/pUCP24/MvfR AMvfR complemented with pUCP24
1-283 harboring mvfR truncated with 49 aa
at C terminus
PA013375/pUCP24/MvfR AMvfR complemented with pUCP24
121-332 harboring tnvfR truncated with 120 aa
at N terminus
27853/PLL-EGFP Green fluorescent PA-IL reporter
strain
87
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
Table 2. Pr=imers designed for conaplementation and truncation
Strain Template Primers
13375/MvfR PAOI DNA forward 5'-AAGGAATAAGGGATGCCTATTCA-3' SEQ ID NO: 3
reversed 5'-CTACTCTGGTGCGGCGCGCTGGC-3' SEQ ID NO: 4
PA0281/GacA PAOI DNA forward 5'-CGACGAGGTGCAGCGTGATTAAGGT-3' SEQ ID NO: 5
reversed 5'-CTAGCTGGCGGCATCGACCATGC-3' SEQ ID NO: 6
13375/1-299 pUCP24/mvfR MvfrXbal 5'-GCTCTAGAAAGGAATAAGGGATGCCTAT-3 'SEQ ID
NO: 7
C33HindIII 5'-CCCAAGCTTCTAACGCTGGCGGCCGAGTTC 3'
SEQ ID NO: 8
13375/1-293 pUCP241mvfR MvfrXbaI 5'-GCTCTAGAAAGGAATAAGGGATGCCTAT-3 'SEQ ID
NO: 7
C3 9HindIII 5' -CCCAAGCTTCTAGCGCAGGCGCTGGCGGGC-3'
SEQID NO:9
13375/1-292 pUCP24/rnvfR MvfrXbaI 5'-GCTCTAGAAAGGAATAAGGGATGCCTAT-3 'SEQ ID
NO: 7
C40HindIII 5'-CCCAAGCTTCTACAGGCGCTGGCGGGCGCT-3'
SEQIDNO:10
13375/1-291 pUCP24/mvjR MvfrXbal 5'-GCTCTAGAAAGGAATAAGGGATGCCTAT-3 'SEQ ID
NO: 7
C41 HindIII 5' -CCCAAGCTTCTAGCGCTGGCGGGCGCTTTC-3'
SEQ ID NO: 11
13375/121-232 pUCP24/mvfR N120Xbal
5'-GCTCTAGAAAGGAATAAGGGATGGTCAGCCTGATACGC-3
'SEQ ID NO: 12
MvfRHindIIl
5'-CCCAAGCTTCTACTCTGGTGCGGCGCGCTGGC-3'] SEQ ID
NO: 13
Example 13 A
P. aeruginosa PAOI expresses abundant PA I and alters MDCK naonolayer
permeability in a
PA-I-dependent manner
In order to verify that the sequenced P. aeruginosa strain, PAO 1, expressed
PA-I, and to verify that strains altered the TEER of MDCK cells in a PA-I-
dependent
manner, both wild type and PA-I mutant strains deleted of the PA-I gene (lecA)
were assayed
for PA-I protein expression and their abilities to decrease MDCK monolayer
TEER. PA-I
protein expression is highly abundant and responds to varying doses of C4-HSL,
its cognate
quorum sensing signaling molecule. In addition, in this strain, the ability of
P. aeruginosa to
decrease MDCK monolayer integrity (TEER) is highly dependent on the expression
of PA-I.
Also, it was determined that the PA-I induced permeability defect in MDCK
cells was of
sufficient magnitude to permit the apical to basolateral flux of exotoxin A
across the
monolayers, with a PA-I-induced TEER decrease of over 50% resulting in a five-
fold
increase in exotoxin A flux. Finally PA-I protein has been shown to be
abundantly expressed
in PAO1 when strains were exposed to the various opioid agonists. For PA-I
protein, the S
88
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
agonist (BW373U86) induced a response equal to C4-HSL. The data establish that
PA-I
expression affects eukaryotic cell barrier function. Thus, it is expected that
modulators of
PA-I expression, as well as modulators of PA-I activity, will be useful in
affecting the
virulence phenotype of microbial pathogens and will be useful in affecting the
eukaryotic
(e.g., epithelial) cell barrier dysfunction associated with that phenotype.
Example 14
Host cell-derived bacterial signaling conzponents enhance the barrier-
dysregulating
properties of P. aeruginosa agaiiast epithelial cells
In order to demonstrate that host stress BSCs could shift the barrier-
dysregulating dynamics of P. aeruginosa against the epithelium, media and cell
membrane
fractions from Caco-2 cells exposed to hypoxia were added to the apical wells
of MDCK
cells apically inoculated with PA27853. TEER was measured over time. C4-HSL
was also
added to serve as a positive control for PA-I expression. Both media and cell
membranes
enhanced the barrier-dysregulating properties of P. aeruginosa (PA27853)
against MDCK
cells at four hours, at levels comparable to the level resulting from C4-HSL
exposure. None
of the host cell derived bacterial signaling compounds alone had any effect on
MDCK TEER.
The results demonstrate that the microbial pathogen (e.g., P. aeruginosa) is
necessary to alter
the barrier function of host cells.
Exam lp e 15
PA-I is expressed in vivo within the digestive tube of Caenorhabditis elegans
The PA-I-GFP reporter plasmid was introduced into P. aeruginosa strain
PA14, a strain highly lethal to C. elegans, by electroporation. Worms were
then fed GFP-
tagged PA14 and PA27853 and examined for fluorescent bacteria. Worms feeding
on lawns
of PA14 and PA27853 displayed fluorescent bacteria within the digestive tube,
whereas no
fluorescence was seen within the surrounding media, indicating that PA-I
promoter activity is
activated by local factors within the worm digestive tube. Finally the killing
dynamics of
strain PA- 14, a highly lethal strain in this model, was compared to the
dynami'cs associated
with the completely sequenced PAO1 strain. The strain of E. coli (OP50) upon
which worms
normally feed, resulted in 100% survival, whereas, PA-14 displayed fast
killing dynamics, as
predicted. The PAO1 strain displayed slow killing with only a 50% mortality
rate at 80
hours. Thus PAO1 exhibits killing dynamics that will allow assessments of
whether host
stress-derived BSCs shift the killing curve to that of a more virulent strain.
It is expected that
BSCs, whether soluble or membrane-bound, will shift the killing dynamics of
relatively
89
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
quiescent, or benign, microbes towards the dynamics exhibited by lethal
microbial strains.
Stated in the alternative, it is expected that a BSC will shift the phenotype
of a microbe
towards a vinilent phenotype. Modulators of such activities are expected to be
useful in
preventing and treating disorders associated with the display of a virulence
phenotype by any
such microbe and in particular by P. aeruginosa. Such modulators are also
expected to be
used in methods for ameliorating a symptom of such a disorder.
Example 16
P. aeruginosa genes involved in BSC-induced PA-I lectin/adhesin gene
expression
The data demonstrate that i) morphine, the potent opioid agonists U-50488 and
BW373U86, which target x and F receptors, respectively, and IFN-y, induce a
robust
response in P. aeruginosa strains PA27853 and PAOI to express PA-I; ii) PA-I
expression is
dependent on multiple elements of the virulence gene regulatory circuitry in
P. aeruginosa,
including the quorum sensing signaling system (QS) and RpoS. The data will
show the genes
that are required for opioids and IFN-y to elicit a PA-I response in P.
aeruginosa and will
facilitate a determination of whether these host stress-derived BSCs use
common genes and
membrane receptor proteins to activate PA-I expression.
A. Genes required for P. aeruginosa PA-I expression responsive to moYphine,
Kand J
opioid agonists, and IFN-y
At least two techniques are contemplated for use in gene identification: 1)
perform transcriptome analysis on P. aeruginosa strain PAO1 exposed to
morphine, x and S
opioid receptor agonists, and IFN-y, and 2) establish a functional role for
candidate genes
identified in the transcriptome analysis by screening the corresponding
transposon mutants
for their ability to up-regulate PA-I protein expression in response to
opioids and IFN-y.
Transcniptonae analysis
Genes in strain PAO1 whose expression is increased in the presence of opioids
and/or IFN-y will constitute the initial focus. Transcriptome analyses is
performed using
Affymetrix GeneChip genome arrays in strain PAO1 to identify the genes that
respond to the
host cell elements such as morphine (non-selective opioid receptor agonist), U-
50488 (K
receptor agonist) , BW373U86 (8 opioid receptor agonist), and IFN-y. Time and
dose
variables for the following experiments are based on data for PA-I expression
(mRNA) in
strain PAO1.
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
Briefly, bacteria are grown in TSB overnight and diluted 1:100 in TSB
containing either morphine (20 p.M), x agonist (80 M), S agonist (80 M), or
IFN-y (10
g/ml). Bacteria are then grown to an OD600 of 0.5, 1.0, and 2.0, representing
three stages of
growth: exponential phase, late exponential phase, and stationary phase,
respectively. These
three time points will permit the capture of genes that are expressed early in
the PA-I
signaling pathway as well as during time points of high cell density. For
transcriptome
analysis, RNA is isolated from bacterial cells (treated and non-treated with
morphine, x and 6
opioid receptor agonists, and IFN-y) at the three designated points in the
growth phase.
cDNA synthesis, fragmentation, labeling, and hybridization, as well as P.
aeruginosa
GeneChip genome array processing, are performed as described herein or as
known in the art.
Each experiment is preferably performed in triplicate.
Functiotaal analysis of candidate genes
Genes showing at least a 2.5-fold change in expression resulting from
exposure to morphine, x and 8 opioid receptor agonists, and/or IFN-,y, are
individually tested
for their specific role in PA-I protein expression by screening mutant strains
from a PAO1
transposon library (University of Washington Genome Center, see below) using
dot blot
analysis. Briefly, strains are grown in sequential runs using 384-well
inicrotiter plates at 2
separate bacterial cell densities (OD600 of 1.0 and 2.0) predetermined to
respond to the
inducing compound (opioids, IFN-y). Dose-response curves are generated with
varying
doses of the PA-I inducing compounds at different bacterial cell densities in
wild-type strains
and in several mutant strains to determine the optimal conditions for
screening. Experiments
are performed separately for morphine, U-50488, BW373U86, and IFN-y. Briefly,
either
morphine, U-50488, BW373U86, or IFN-y are added to the wells containing mutant
strains at
the predetermined dose. All runs are performed with the wild-type strain as a
control. The
PA-I-inducing compound is added to the well for a predetermined time. Next,
the supernatant
is removed and the bacterial cell pellet is lysed by the addition of lysis
solution directly into
the well. The entire 384-well plate is then spun down (4000g) and the
supernatant transferred
to an Immobilon P-PDF membrane using a 384 replicator. Membranes are then
treated with
anti-PA-I primary and secondary antibodies. Dot blot analysis allows for rapid
identification
of all of the mutant strains that do not up-regulate PA-I in the presence of
host stress-derived
bacterial signaling compounds, thereby identifying genes that are required for
PA-I
expression. All assays are preferably performed in triplicate (3 cell
densities x 5 groups (4
experimental + 1 control) x triplicate (3) assays = 45 gene arrays).
91
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
It is expected that many of the genes that have already been established to
play
a role in PA-I expression, including genes in the QS and RpoS regulon, will be
identified.
However, it is expected that new and unanticipated functions for known genes
will also be
identified. Further, if CyaB or GacS transcripts are increased in response to
opioids or IFN-y,
and if Cya B and GacS transposon knockouts do not respond to either opioids or
IFN-y with
an increase in PA-I, then the role of these established biosensors as two-
component regulators
of opioids or IFN-y signaling to P. aeruginosa will be confirmed. Combining
the results of
the transcriptome analyses with the functional analyses of the transposon
library will allow us
to determine whether opioids and IFN-y activate common membrane biosensors and
common
downstream genes involved in PA-I expression. It is possible that one or more
of the non-
peptide opioids diffuses directly into the bacterial cell cytoplasm where it
initiates gene
activation downstream of the two-component membrane biosensors. If this is the
case, then
all of the transposon knockout strains encoding membrane proteins are expected
to respond
with an increase in PA-I and microarray data will demonstrate that levels of
transcripts
encoding membrane proteins will be unaltered by either opioids or IFN-y.
However, it is
possible that membrane biosensors are constitutively expressed and therefore
gene expression
will not change in response to opioids or IFN-y. If this is the case, then the
entire transposon
library will be screened for PA-I expression in response to opioids or IFN-y,
approaches that
are feasible given the high-throughput nature of the dot-blot technique. Of
note here is that
gene expressions can be triggered at different times during culturing and can
respond to an
exogenous compound(s) to varying degrees depending on the concentration of
compound.
The genomically sequenced strain PAO1 makes abundant PA-I and the anti-PA-I
lectin/adhesin antibodies are highly specific.
The data demonstrate that opioid receptor agonists and IFN-,y signal P.
aeruginosa to express PA-I mRNA and protein. In addition, these PA-I signaling
compounds
induce P. aeruginosa to express a more virulent phenotype against the
epithelium. The genes
that control PA-I expression are dependent on two key global regulatory
systems that activate
hundreds of virulence genes in P. aeruginosa. The activation of these
interconnected systems
of virulence gene regulation are directly influenced by membrane biosensors
that recognize
elements of host cells and include, but are not limited to, CyaB and GacS, via
a hierarchical
cascade involving the transcriptional regulators Vfr and Gac A. Genes that are
differentially
expressed in response to opioids and IFN- y will be identified using an
unbiased
transcriptome analysis approach. This approach was chosen instead of pursuing
individual
92
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
candidate genes involved in known pathways of PA-I expression because all
previous studies
have been performed only at high cell densities and in the absence of any host
cell elements.
Accordingly, previously described gene expression patterns may not be
applicable in the
physiologic models. The goal of this study is to identify and functionally
validate the genes
that are involved in PA-I expression in response to morphine, x and b opioid
receptor
agonists, and IFN-y.
B. Identify the receptors in P. aeruginosa that bind morphine and IFN-y
The data show that a single solubilized membrane protein from P. aeruginosa
can be isolated that avidly binds IFN-y. In addition, morphine also binds to
membrane
protein fractions. Because antibody is available that specifically recognizes
each of IFN-y
and morphine, initial studies are examining the effect of these two BSCs.
Using the
commercial antibodies, the membrane proteins that bind IFN-y and/or morphine
are
identified, and optionally purified. This protein-based approach provides data
which
complements the experiments described above.
Two approaches available for use in identifying membrane proteins that bind
IFN-y and/or morphine are now described. First, membrane proteins of P.
aeYuginosa strain
PAOI are solubilized using mild detergents. The binding capacity of
solubilized protein
fractions for IFN-y or morphine is then determined using simple ELISA binding
assays.
Protein fractions are then immunoprecipitated using the respective antibody
and proteins are
identified, e.g., by Maldi-MS.
Confirmation of the identity of a binding protein(s) is achieved by
determining
that a transposon knockout of the gene encoding the candidate protein(s) does
not respond to
IFN-y or morphine with an increase in PA-I, using the techniques described
herein. In order
to confirm the function of candidate proteins showing fidelity in these two
analyses,
candidate proteins are re-expressed in the corresponding transposon knockout
to verify that
the PA-I response is re-established. Additionally, receptor antagonists may
also be
developed.
The data indicate that membrane receptors for morphine and IFN- y can be
identified by identifying proteins from solubilized membranes. A potential
limitation using
this technique is that morphine could diffuse directly into the bacterial
cytoplasm and interact
with a downstream target and not a membrane protein. This possibility is
consistent with
results demonstrating that morphine does not change the transcript profiles of
any genes
93
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
encoding membrane proteins, but the data for IFN-y disclosed herein is
inconsistent with this
interpretation. In addition, morphine binding to a solubilized bacterial
membrane protein was
demonstrated using fluorescent imaging and analysis. Also, there is the
possibility that
transmembrane proteins or proteins that bind host stress-derived BSCs could be
secreted into
the culture medium and not be present within bacterial membranes. An example
of such
proteins are the bacterial iron binding proteins (enterochelin), which are
released by bacteria
into the culture medium and then re-enter the bacterial cells. Under such
circumstances, the
screening of cytosolic fractions and inner and outer membrane preparations are
contemplated,
along with iterative experiments probing for binding proteins with specific
antibodies. Any
discordance between the transposon mutant experiments and the proteins
purified from
bacterial membranes will be reconciled by analyzing IFN-y- membrane protein or
morphine-
membrane protein interactions directly using surface plasmon resonance and
mass
spectrometry.
Example 17
The impact of host signaling on microbial virulence states
The data demonstrate that PA-I knockout strains (lecA-) do not decrease the
TEER of cultured epithelial cells. The lethality of strains of P. aeruginosa
exposed to opioid
agonists and IFN-y can be defined in vivo using the well-characterized
invertebrate,
Caenorhabditis elegans, and the established model of gut-derived sepsis in
mice.
A. The defect in epitlaelial barrier function induced by P. aerugiiaosa
exposed to
opioid agonists and IFN-y and the role of PA-I in this response
One issue is whether opioids or IFN-y can activate P. aeruginosa to express a
lethal phenotype against an epithelium, as judged by an increase in exotoxin A
flux across
epithelial cell monolayers, through the action of its PA-I lectin/adhesin.
To address that issue, MDCK cells are grown to confluence to maintain a
stable TEER in transwells. Cells are apically inoculated with P. aeruginosa
strain PAO 1 (107
cfu/ml) in the presence and absence of varying doses of morphine (about 20
M), x agonist
(about 80 gM), 6 agonist (about 80 M), or IFN-y (about 10 gg/ml). To optimize
the effect
of opioids and IFN-y on the barrier-dysregulating effect of P. aeruginosa
against epithelial
cells, dose and time response curves are generated. TEER is measured using
chopstick
electrodes hourly for 8 hours. The apical to basolateral flux of exotoxin A
using Alexa-594-
labeled exotoxin A is determined in iterative experiments performed at each
hourly time
94
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
point in order to correlate the decrease in TEER to exotoxin A flux for each
condition. In
selected experiments in which a significant permeability defect to exotoxin A
is established,
the specific role of PA-I is defined by performing iterative experiments in
the presence and
absence of 0.3% Ga1NAc (N-acetylgalactoside) and 0.6% mellibiose, two
oligosaccharides
that specifically bind to PA-I78. Irrelevant sugars (heparin/mannose) are used
as negative
controls. Iterative studies are also performed using the PA-I transposon
knockout (lecA-)
mutant to define the specific role of PA-I in strains exposed to opioids and
IFN-y. It is
expected that PA-I will be expressed and localized to the microbial pathogen
cell surface,
where it will be situated in position to interact with host epithelial cells,
thereby influencing,
at a minimum, the cell barrier properties of the epithelial cells.
It is expected that opioids and IFN-y will decrease the TEER of MDCK cells.
Exotoxin A flux that is increased in cell monolayers with a low TEER will
suggest that the
opioids and IFN-,y alone can induce a lethal phenotype in P. aeruginosa. If
the Ga1NAc,
mellibiose inhibition studies, or the PA-I lectin/adhesin knockout strains,
prevent P.
aeruginosa from altering TEER and exotoxin A flux across the cell monolayers,
then this will
indicate that the observed response is PA-I-mediated. If the PA-I knockout
mutant strains
alter TEER and exotoxin A flux in response to opioids or IFN-y, then this will
indicate that
PA-I alone may not be responsible for the virulence of P. aerx-cginosa against
the intestinal
epithelium. Data from these studies are directly compared and correlated to
worm and mouse
lethality studies (see below) to determine if these in vitro assays accurately
predict a lethal
phenotype in vivo, as expected.
Example 18
The roles of opioid agonists and IFN-y on gut-derived sepsis due to P.
aeruginosa as
revealed using Caenorhabditis elegans and surgically stressed naice
The data provide strong evidence that opioid agonists and IFN-y enhance the
virulence of P. aeruginosa in vitro through the action of PA-I. Yet the degree
to which
opioid agonists and IFN-y influence the in vivo lethality of P. aeYuginasa is
unknown. Thus,
the ability of opioids and 1FN-y to enhance the in vivo lethality of P.
aeruginosa is assessed,
e.g., in two complementary animal models.
Wild-type N2 Caenorhabditis elegans worms are grown to the L4 larval stage
on normal growth medium (NGM) with E. coli OP50 as a nutrient source.
Specialized agar
plates are prepared onto which the PA-I-inducing compounds (vehicle (negative
control)),
opioids (morphine, x and S agonist), IFN-7, and C4-HSL (positive control))
will be added
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
and adsorbed into the agar as described for ethanol. The ability to embed
bioactive
compounds into the G. elegans growth agar is well described. Lawns of P.
aeruginosa (wild
type PAO1 and PA-I knockout PAO1 (lecA-)) are then grown on solid at agar
plates by
adding cultures of P. aeruginosa previously grown overnight in liquid media.
Worms from
the NGM medium are transferred onto the prepared culture dishes and killing
dynamics
assessed over time at temperature conditions of 25 C. Experiments are
perfonned at different
doses and re-dosing schedules to establish the optimum conditions under which
a killing
effect for each of the PA-I-inducing compounds can be demonstrated.
To test the ability of PA-I inducing compounds to enllance the lethality of P.
aeruginosa in the established mouse model of gut-derived sepsis, mice are
fasted for 24 hours
and are subjected to general anesthesia, a 30% surgical hepatectomy, and cecal
instillation of
106 efu/ml of wild-type PAOl or PAO1 (lecA-) via direct puncture. Dose-
response curves
for P. aeruginosa in this model have been established and show that 106 cfu/ml
of P.
aeruginosa induces a 50% mortality rate at 48 hours. In order to demonstrate
that opioid
agonists or IFN-y enhance the lethality of P. aeruginosa in this model,
varying doses of each,
are suspended in 1 ml of 0.9% NaC1 and injected retrograde into the ileum in
order to provide
a constant supply of the PA-I-inducing compound for 24 hours. Normal saline
alone is used
for controls. This maneuver is known to be efficacious in delivering a
continuous supply of
an exogenous compound to the cecum in this model. Mice are fed water only for
the next 24-
48 hours and mortality recorded. Mice that appear moribund are sacrificed and
the cecal
mucosa, liver, and blood are cultured for P. aeruginosa growth on Pseudomonas
isolation
agar (PIA) in order to quantify bacterial adherence and dissemination
patterns. The mice
used in the study include two strains (wild-type + PA-I knockout) and, with 6
groups of 10
mice per group, a total of 120 mice is suitable.
Increased mortality in worms feeding on lawns of P. aeruginosa in the
presence of opioids and/or IFN-y demonstrates the ability of these compounds
to induce a
lethal phenotype in this organism against the intestinal epithelium. The
demonstration of
enhanced killing of worms in these experiments also serves to establish the
feasibility and
applicability of this model. As disclosed herein, in the absence of PA-I-
inducing compounds,
C. elegans displays a 50% mortality rate at 80 hours. In testing opioids
and/or IFN-y, or in
screening for modulators of PA-I lectin/adhesin activity in general, it should
be noted that,
following 48 hours of growth and reproduction, worms can reproduce and progeny
worms
can be indistinguishable from the parent worms and overgrow the plates. If
killing dynamics
96
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
in response to PA-I-inducing compounds are such that observations extend past
48 hours,
then use of a temperature sensitive mutant, e.g., C. elegans GLP4 (which does
not reproduce
at 25 C), is preferred. Complementary experiments in mice will verify results
obtained with
worms.
The use of mouse studies to conf rm results obtained with C. elegans
preferably includes verification that luminally delivered PA-I-inducing
compounds are
efficacious in up-regulating PA-I as a general measure of enhanced virulence.
To control for
this possibility, experiments are performed to show that the PA-I-inducing
compounds =
injected into the small bowel enhance PA-I expression in the mouse cecum. One
approach
involves the use of quantitative RT-PCR for PA-I and exotoxin A on freshly
isolated RNA
from cecal contents 24 hours following cecal instillation of P. aeruginosa. An
alternative
approach to delivering opioids and IFN-y directly into the cecum is to
engineer non-
pathogenic E. coli strains that produce both morphine and IFN-y. The
feasibility of making
recombinant morphine and IFN-y in E. coli is well documented. Mice subjected
to a surgical
stress (e.g., hepatectomy) are then co-inoculated directly into the cecum with
the LD50 dose
of P. aeruginosa (approximately 106) and the morphine- and/or IFN-y-producing
E. coli
strain. In this manner, P. aeruginosa would be directly exposed to a constant
supply of the
PA-I-inducing compound such as might naturally occur in vivo. Relevant here is
the
knowledge in the art that numerous microbial strains (E. coli, Pseudomonas,
Candida)
naturally produce opioids, especially morphine. In addition, the "microbial
soup" typical of a
critically ill patient consists of highly pathogenic and resistant strains of
bacteria that compete
for nutrients in a highly adverse environment. Therefore, not only will the
use of morphine-
and/or IFN-y-producing E. coli constitute a feasible alternative approach to
obtaining in vivo
mouse data, it may also recapitulate actual events in vivo. Finally, C.
elegans normally feed
on E. coli strains that do not induce mortality. The availability of morphine-
and/or IFN-y-
producing E: coli strains could also be used in the C. elegans assays. Others
have shown the
feasibility of this approach is feasible in mice, as shown by delivering IL-10
into the
intestinal mucosa of mice using direct intestinal instillation of bacteria
that produce
recombinant IL-10. The use of the C. elegans assay is expected to result in
the rapid
identification of therapeutics and prophylactics that modulate expression of a
virulence
phenotype by microbial pathogens in contact with, or proximity to, a mammal.
The virulence
phenotype is amenable to assessment using a variety of measures, many of them
indirect,
e.g., measurement of epithelial cell barrier function.
97
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
Example 19
Opioids and/or IFN-y r-elease into the intestinal lumen Yesulting from host
stress
Endogenous morphine concentrations in the blood of humans and animals
increase in direct response to the degree of surgical stress. The neural
network of the
mammalian intestine contains the most abundant concentration of opioid
receptors in the
body. Morphine has been recently shown to enhance the release of nitric oxide
in the
mammalian gastrointestinal tract via the 3 opiate receptor subtype. In
addition, it has been
shown that the nematode, AscaYis suum, produces and liberates morphine in the
gut.
Similarly, IFN-y has been shown to be released by the gut from intestinal
intraepithelial
lymphocytes in response to a variety of stressors, including bacterial
challenge and
ischemia/reperfusion injury (I/R).
To demonstrate that C. elegans produces or releases morphine, worms are
grown penmissively at 20 C in massive cultures in liquid medium to 1 x 106
worms using
conventional culturing techniques. Stock cultures are treated with antibiotics
24 hours prior
to the imposition of stress conditions. Worms are separated from any remaining
bacteria by
sedimentation and sucrose flotation as known in the art. Worms are then
exposed to either
heat stress (35 C for 1 hour) followed by 2 hours of recovery, or hypoxic
stress (0.3% 02 for
45 minutes) followed by 1 hour of normoxic recovery, as described. Control
worms are
maintained at 20 C and 21 % 02. Both the growth medium and the supernatant of
homogenized C. elegans are preferably assayed for morphine by HPLC/ GC/MS
using
conventional techniques. To determine whether morphine and IFN-y are produced
by, or
released into, the mouse intestine following surgical stress, groups of mice
(n=10/group) are
subjected to a 30% hepatectomy or segmental mesenteric ischemia as described
below.
Surgical stress involving the hepatectomy model consists of performing a 30%
surgical
hepatectomy or sham laparotomy for controls and 24 hours later by harvesting
the cecal
tissue, the cecal luminal contents, and blood for morphine and IFN-,y assays.
The ischemia
reperfusion model (I/R) involves isolation of a 10 cm segment of distal ileum
that is
luminally cannulated and subjected to 10 minutes of ischemia (segmental artery
clamp)
followed by 10 minutes of reperfusion. Luminal perfusion with 2 ml of Ringers
solution is
performed to collect the luminal contents before and after I/R. Luminal
contents, the
homogenized intestinal segment, and blood are assayed for morphine by HPLC and
GC/MS;
IFN-y is assayed by ELISA using a specific anti-IFN-y antibody. A suitable
number of mice
for such assays is 30-50 mice.
98
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
Release of significant amounts of morphine and/or IFN-y into the gut
following surgical stress confirms that P. aeYuginosa has been exposed to
highly active
compounds capable of activating or enhancing its virulence phenotype during
host stress. In
addition, a better understanding of the precise concentration of morphine
and/or IFN-y to
which P. aeyuginosa are exposed in vivo can be determined by these
experiments. Whether
morphine is released in high concentration in the lumen versus within the
intestinal tissues is
amenable to experimental determination. If luminal levels of morphine are
elevated in
hepatectomy versus controls, mice can be decontaminated with antibiotics
(e.g.,
ciprofloxacin, metronidazole). Following such decontamination, the extent to
which the
luminal flora contribute to the opioid level can be determined using
conventional techniques.
It should be noted that, in addition to, e.g., morphine, other opioids and
cytokines may be
released from microbial pathogens such as P. aeruginosa that actively
participate as host
stress-derived BSCs. It is also possible that both opioids and IFN-y are
enzymatically
degraded in the intestinal lumen. An alternative approach would be to use
quantitative
immuno-fluorescence of stained tissues to assess morphine and IFN-y presence
in tissues as
antibodies specifically recognizing these compounds are readily available.
Notwithstanding
the preceding observations, these compounds have been measured by others from
luminal
contents without difficulty.
Example 20
Use of knockout mice to confirm the role of BSCs on PA-I lectin/adhesin
activity
IFN-y is a key immune element that actively participates in both the local and
systemic clearance of bacteria during acute infection. Animal models have
shown that IFN-y
knockout mice have higher mortality rates following infectious challenge at
local tissue sites
(lung) compared to IFN-y-sufficient mice in association with diminished
ability to clear
bacteria. Virtually all of the studies that have assessed the role of IFN-y on
P. aeruginosa
infection in vivo have been performed in non-stressed mice where the
infectious challenge
has been instilled into the lung, and not in stressed mice, such as surgically
stressed mice.
The lethality of intestinal P. aeruginosa is tested in IFN-y knockout mice and
wild-type controls (n=10 each group) in an established model of gut-derived
sepsis. Mice
fasted for 24 hours undergo 30% surgical hepatectomies followed by
instillation of 106
cfu/ml of wild type PAO1 into each cecum via direct puncture. Mice are then
allowed water
only for the remainder of the experiment and mortality is followed for 48
hours. Mice that
appear moribund are sacrificed and the cecal mucosa, liver, and blood is
quantitatively
99
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
cultured on Pseudoinonas isolation agar (PIA) to determine the rates of
bacterial adherence
and dissemination. To determine if PA-I expression in P. aeruginosa is
attenuated in IFN-,y,
a GFP PA-I reporter strain is injected directly into the cecum of mice
subjected to a 30%
hepatectomy and bacterial strains are recovered 24 hours later to determine
fluorescence.
The results of these experiments guide the performance of complementary
studies using the
segmental mesenteric ischemia model. Briefly, the lumena of 10 cm ileal
segments subjected
to sham ischemia (no clamp), 10 minutes of ischemia, and 10 minutes of
reperfusion is
perfused with Ringers solution and the timed aliquots of the perfusates is
collected from both
IFN-y knockout mice and their wild-type cohorts. Use of the GFP-PA-I reporter
strains
facilitates the determination of the extent to which each perfusate induces PA-
I promoter
activity. A suitable number of mice for such studies is 50 mice, divided into
five groups with
ten mice in each group.
The display of attenuated lethality by P. aeruginosa in IFN-y knockout mice is
consistent with IFN-y playing a role as a host stress-derived bacterial
signaling compound, or
protein, during stress (e.g., surgical stress). IFN-y may be only one of many
signals
necessary to orchestrate a fully lethal virulence repertoire for P. aeruginosa
under the
circumstances of catabolic stress, however. It is noted that IFN-y knockout
mice subjected to
hepatectomy may develop an overcompensated and excessive inflammatory response
to
intestinal P. aeruginosa, resulting in increased mortality that is based more
on immune
response than enhanced microbial virulence. Tissue and blood culture results
from these
studies are used to determine whether mortality is due, in part, to such
overcompensation. An
alternative approach to distinguish between these possibilities is to perform
studies in IFN-,y
knockout mice and their matched wild-type cohorts (with and without surgical
hepatectomy)
to determine if there is a mortality difference when groups of mice are
systemically
inoculated (e.g., intraperitoneal, intravenous, lung instillation) with P.
aeruginosa.
Example 21
Scf-eens for stress-induced bacterial signaling conapounds
The data disclosed herein establishes that i) filtered luminal contents from
the
cecum of mice subjected to hepatectomy, or from the small bowel lumen of
intestinal
segments subjected to mesenteric arterial occlusion, induce a strong signal in
P. aeruginosa
to express PA-I; and ii) media and membrane preparations from hypoxic or heat-
shocked
Caco-2 cells induce PA-I expression.
100
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
A. Stress-derived BSCs that are present in the naedia of Caco-2 cells exposed
to ischernia
and heat shock stress and that induce PA-I expression in P. aeruginosa
Intestinal epithelial hypoxia is a common consequence of critical illness
following surgical stress and is often an inadvertent consequence of its
treatment. In
addition, hypertherinia often develops during the acute stress response to
injury and infection.
Disclosed herein are data demonstrating that hypoxic or hyperthermic stress to
cultured
intestinal epithelial cells (Caco-2) causes the release of soluble PA-I-
inducing compounds
into the cell culture medium. This example discloses the isolation and
identification of PA-I-
inducing compounds that are released by Caco-2 cells exposed to hypoxia and
hyperthermic
stress.
Two sets of experiments are preferably performed. In the first set of
experiments, Caco-2 cells grown to confluence in cell culture plates (150 cm)
are exposed to
either normoxia (21% Oa) or hypoxia (0.3% 02 for 2 hours followed by 1 hour of
normoxic
recovery). In the second set of experiments Caco-2 cells are exposed to
normothermic (37 C)
or hyperthermic (immersed in water bath at 42 C for 23 minutes followed by 3
hours
recovery) conditions. Paired samples from each set of experiments are then
processed to
identify the specific host stress-derived bacterial signaling compound(s)
using GFP-PA-I
reporter strains as a detection system. Media from Caco-2 cells is collected,
filtered through
a 0.22 m filter (Millipore) and separated by molecular weight using
centricones with a MW
cutoff of 3, 10, 30, 50, 100 KDa (<3, 3-10, 10-30, 30-50, 50-100, >100 KDa).
All fractions
are preferably tested in 96 well plates to determine fractions that activate
PA-I expression
using PA-I GFP reporter strains. Two preferred approaches are contemplated for
use in
identifying the proteins that activate PA-I in the stress-conditioned media
(hypoxia,
hyperthermia). The first approach subjects bioactive fractions (i.e those that
induce PA-I),
and their molecular weight-matched control fractions (non-PA-I-inducing), to
Maldi-Mass
Spectrometry (MS) analysis. Spectra from the control media fractions are
compared to the
fractions of stress-conditioned media to determine the appearance of possible
protein
molecular ions present only in the samples that induce PA-I. This will allow
us to subtract
proteins that are present in both non-PA-I-inducing and PA-I-inducing
fractions. In order to
separate the molecular ion protein peaks that are present only in the PA-I-
inducing fractions,
bioactive fractions are loaded onto an HPLC equipped with a Vydac C4 column.
Eluted
samples are collected as fractions and individual fractions are tested for the
ability to induce
PA-I expression using the GPF-PA-I reporter strain. Proteins are then further
separated,
101
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
preferably by MW, hydrophobicity, and charge using stepwise well-controlled
physico-
chemical separation techniques in the HPLC system. Samples pre-fractionated in
this manner
should simplify the observed mass spectra and increase the likelihood of
observing any
putative protein(s) that induce PA-I expression. For any such proteins,
identification using
bottom-up proteomics techniques is performed.
An alternative to the use of molecular ion spectra, suitable in studies
presenting highly complex spectra, is the classical approach for protein
purification using
conventional techniques such as ion exchange, hydrophobic, size exclusion,
and/or affinity
chromatography. Purification of host stress-derived BSCs is preferably
assessed using the
GFP-PA-I reporter strain.
For protein identification, protein-containing fractions are digested by using
trypsin and digested fractions are analyzed with a LC/MSD XCT ion trap mass
spectrometer
system (Agilent Technologies, Santa Clara, CA). Data analysis for the
data,from the mass
spectrometer is carried out using the SpectrumMill software platform (Agilent
Technologies,
Santa Clara, CA). Confirmation of the ability of identified proteins to induce
PA-I
expression is conveniently achieved in the PA-I:EGFP reporter strain by
measuring
fluorescence, and in P. aerugiyaosa strain PAO1 by immunoblot analysis.
Two protein fractions from Caco-2 cells that induce PA-I expression have
been identified. Identification of specific active proteins (i.e., epithelial
cell-derived PA-I
signaling proteins) within the fraction(s) is achieved using any known
technique, and
preferably using a proteomics facility such as the University of Chicago
proteomics facility.
Many of these proteins may originate from the cell membranes themselves, since
the most
potent induction of PA-I expression occurs following contact with an
epithelial cell
membrane. In addition to protein identification, antibodies specifically
recognizing such
proteins are contemplated for such uses as cellular (e.g., Caco 2)
localization studies.
Although there are more classical approaches to protein identification, mass
spectrometry is
the most cost effective and rapid approach. For non-proteinaceous PA-I
inducing
compounds, lipid assays are contemplated that involve adjusting fraction pH to
3.5, followed
by HPLC using, e.g., a Sep-Pak C18 column. Eluted samples are trapped on a
fraction
collector, evaporated to dryness, and re-suspended in PBS for PA-I reporter
assays. The
structure of the active compound is preferably identified with IT/LC/MS/MS.
For bacterial
signaling compounds that are neither protein nor lipid, relevant fractions are
resolved by
IT/LC/MS/MS using a C18 column and a quadrapole-time of flight mass
spectrometer and
102
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
NMR. Individual compounds are determined by their mass-fragmentation spectra,
isolated,
and tested for PA-I inducing activity using GFP reporter strains. Alternative
approaches,
such as 2D-SDS-PAGE electrophoresis for protein separation and TLC for non-
protein
separation, are also contemplated. Proteins separated by 2D-SDS-PAGE are
typically
transferred to a polyvinylidene difluoride transfer protein membrane for
automated Edman
degradation N-terminal sequence determination using an ABI 477A protein
sequencer
(Applied Biosystems). Protein identification is further facilitated by
sequence comparison to
database(s).
In addition to the foregoing screens for modulators, the invention
contemplates any assay for a modulator of the expression of a virulence
phenotype by a
microbe in association with, or proximity to, a mammal such as a human. In
particular, the
invention comprehends a wide variety of assays for modulators of, e.g.,
eukaryotic cell
barrier function, such as epithelial cell barrier function (e.g., epithelial
cells of the intestine,
lung, and the like). The invention further comprehends numerous assays for
modulators of
PA-I lectin/adhesin activity, whether due to a modulation of the specific
activity of PA-I or a
modulation of the expression of PA-I of constant specific activity, or both.
In general, the
invention contemplates any assay known in the art as useful for identifying
compounds
and/or compositions having at least one of the above-described
characteristics.
Example 22
Miscellaneous Methods
A. Screens for PA-I nzodulatoYs using a PA-I reporter corastYuct
Media from Caco-2 cells exposed to either hypoxia or heat shock stress
induced PA-I expression in P. aeruginosa. Candidate PA-I inducer compounds
that are
released into the extracellular milieu following epithelial stress include
ATP, lactate, cAMP,
cytokines, and heat shock proteins.
The aforementioned candidate modulators, and other candidate modulators
found in properly conditioned media, are identified using screening methods
that constitute
another aspect of the invention. Screens for such modulators are conveniently
conducted in
96-well plates that contain the GFP-PA-I reporter strain PA27853/PLL-EGFP (see
Example
24, below). The reporter strain is exposed to varying concentrations of
candidate host stress
BSCs including, but not limited to, heat shock proteins (HSP 25, 72, 90, 110),
extracellular
nucleosides and nucleotides (adenosine, ATP, cAMP) and cytokines (IL-1-18).
Agents are
103
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
added to the wells and dynamic assessment of bacterial fluorescence is carried
out over 12
hours. Positive results are preferably verified by Western blot analysis of PA-
I expression.
For proteins that induce a PA-I response, the invention further comprehends
assays to
identify the receptors on P. aeruginosa to which such proteins bind. In one
embodiment of
this aspect of the invention, the identified protein inducer of PA-I activity
is used as a probe
to screen, e.g., a comprehensive library of P. aef-uginosa by dot blot
analysis. Confirmation
of the screen results is available by assaying the protein-binding capacity of
a lysate from a
corresponding clone from a P. aeruginosa transposon library in which the
relevant coding
region has been disrupted by insertional inactivation.
Identified modulators are then subjected to additional in vitro and in vivo
virulence assays to 'refine the understanding of the role in virulence
expression played by
such modulators.
B. Caco-2 and MDCK cell culture, measurement of TEER and exotoxin A flux.
Caco-2 cells and MDCK cells are well-differentiated epithelial cell lines that
maintain a stable TEER when grown in confluent monolayer. Apical to
basolateral exotoxin
A flux across monolayers is assessed with Alexa 594-labeled exotoxin A using
standard flux
measurements.
C. Bacterial Strains
P. aeruginosa strain PAO1 was obtained from the University of Washington
Genome Center and is preferably used in the procedures disclosed herein, where
appropriate.
D. Caenorhabditis elegans Assays.
Use of the nematode to assay for the lethality of P. aeruginosa is
accomplished using standard protocols, as described herein.
E. Antibodies.
Antibodies to PA-I are generated using conventional techniques. Preferably,
such antibodies are purified by affinity chromatography. I.FN-y and morphine
antibodies are
commercially available.
F. Dot Blot assays for menabNatae binding.
ImmunoDot Blot assays for the detection of bacterial proteins in large matrix
systems are known in the art. The technique has been validated as highly
sensitive and
accurate.
104
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
G. Trcznscriptome analysis of bacterial strain PAOl.
RNA is isolated from bacterial cultures exposed to opioids and/or IFN-y as
described herein at optical densities of 0.5, 1.0, 2Ø Between 1 x 109 and 2
x 109 cells are
then mixed with RNA Protect Bacteria reagent (Qiagen) and treated as
recommended by the
manufacturer's mechanical disruption and lysis protocol. RNA is purified by
using RNeasy
mini columns (Qiagen), including the on-column DNase I digestion described by
the
manufacturer. In addition, the eluted RNA is preferably treated for 1 hour at
37 C with
DNase I(0.1 U per g of RNA). DNase I is then removed by using DNA-Free
(Ambion) or
by RNeasy column purification. RNA integrity is monitored by agarose gel
electrophoresis
of glyoxylated samples. Further sample preparation and processing of the P.
aeruginosa
GeneChip genome arrays are then done as described by the manufacturer
(Affymetrix). For
cDNA synthesis 12 g of purified RNA is preferably combined with semirandom
hexamer
primers with an average G+C content of 75%, and Superscript II reverse
transcriptase (Life
Technologies). Control RNAs from yeast, Arabidopsis, and Bacillus subtilis
genes are added
to the reaction mixtures to monitor assay performance. Probes for these
transcripts are tiled
on the GeneChip arrays. RNA is removed from the PCR mixtures by alkaline
hydrolysis.
The cDNA synthesis products are purified and fragmented by brief incubation
with DNase I,
and the 3' termini of the fragmentation products are labeled with biotin-
ddUTP. Fragmented
and labeled cDNA is hybridized to an array by overnight incubation at 50 C.
Washing,
staining, and scanning of microarrays is performed with an Affymetrix fluidic
station.
H. Expression profiling.
The Affymetrix Microarray Software suite (MAS) (version 5.0) is a suitable
software choice for determining transcript levels and whether there are
differences in
transcript levels when different samples are compared. Affymetrix scaling is
used to
normalize data from different arrays. A scale factor is derived from the mean
signal of all of
the probe sets on an array and a user-defined target signal. The signal from
each individual
probe set is multiplied by this scale factor. For any given array, between 18
and 28% of the
mRNAs are considered absent by MAS, indicating that the corresponding genes
are not
expressed at levels above background levels. Furthermore, it is known in the
art that the
average changes in control transcript intensities are less than twofold for
any comparison of
array data. This indicates that the efficiency of cDNA synthesis and labeling
is similar from
sample to sample. For comparative analyses, the log2 ratio for absolute
transcript signals
obtained from a given pair of arrays will be calculated by using MAS. A
statistical algorithm
105
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
of the software is also assigned a change call for each transcript pair, which
indicates whether
the level of a transcript is significantly increased, decreased, or not
changed compared to the
level for the baseline sample. The baseline samples are those derived from
cultures ofP.
aef-uginosa PAO-1 without any added opioids or IFN-y. Graphical analyses of
the signal log
ratios from each experiment (anypair of arrays) is performed to display a
normal distribution
with a mean very close to zero (no change). Among the transcripts with
significant increases
or decreases compared to the baseline in one or more samples, those that
showed at least a
2.5-fold change are subjected to further analysis. For cluster analyses and
transcript pattern
analyses, GeneSpring software (Silicon Genetics, Redwood City, Calif.) is
contemplated as a
suitable choice. The fold change values for each gene will be normalized
independentlyby
defining the half-maximal value for the gene as 1 and representing all other
values as a ratio
that includes that value. This scaling procedure will allow direct visual
comparison of gene
expression patterns within an experiment, as well as between experiments.
GeneSpring is
also contemplated for use in sorting genes according to the P. aeruginosa
genome project.
I. Solubilization of non-denatured and denatured membrane pYoteins fnactions
from P.
aeruginosa.
P. aeruginosa cells are washed with PBS and re-suspended in PBS containing
a protein inhibitor cocktail. For preparation of membrane fractions, P.
aeruginosa cells are
disrupted by French pressure and centrifuged at 10000g x30 minutes to
eliminate debris. The
supematant is recentrifuged at 50000gx60 minutes. The pellet is solubilized in
4% CHAPS
at 37 C for 3 hours. After being recentrifuged at 50000gX60 minutes, the
supematant is spun
through a 100K centricone and dialyzed against PBS. The binding capacity of
the solubilized
protein to y-IFN is confirmed by ELISA binding assay.
J. Statistical Analysis and Protein-Protein Interactions.
For statistical analysis, all data are preferably loaded into the SigmaStat
platform software and appropriate tests applied. Protein-protein interaction
studies are
performed using conventional protocols, as would be known in the art.
K. Maldi-MS analysis.
Samples (0.5 L) are mixed with an equal volume of a 5 mg/mL solution of a-
cyanohydroxycinnamic acid in 30% acetonitrile in water with 0.1 % TFA and are
then
manually spotted onto a 192 spot target plate (Applied Biosystems, Foster
City, CA). The
plate is inserted into a 4700 MALDI TOF/TOF (Applied Biosystems, Foster City,
CA)
106
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
operated in linear mode. Samples are desorbed by a 200 Hz YAG laser. The
acquisition
program is set to acquire a summed spectrum (200-1000) shots across the range
5000 to
100000 Thompsons.
L. Digestion of a protein containing fraction by using trypsin to prepare for
protein
identification.
The protein extract sample is diluted in 50 mM ammonium carbonate buffer,
pH 8.5, containing 0.1 % Rapigest SF acid labile detergent (Waters Corp,
Miliford, MA).
The sample is heated to 100 C for 10 minutes to completely denature the
proteins. Ten L of
mM TCEP is added to reduce disulfide bonds and the sample is incubated for 10
minutes
at 37 C. The pooled sample is digested with Lys-C (12.5 ng/ L) at a mass ratio
of 1:100 for
3 hours at 37 C and then digested with trypsin (12.5 ng/ L) at a mass ratio of
1:50
(trypsin:protein) for 3 hours at 37 C. Digestion is halted by adding PMSF to
final
concentration of 1 mM. After digestion, 10 L of TFA is added to the solution
and the
sample is incubated for 45 minutes at 37 C to destroy the acid labile Rapigest
detergent.
M. LC/MSD XCT ion trap mass spectrometry analysis.
A digested protein sample is injected (10 L) onto an HPLC (Agilent
Technologies 1100) containing a Cl 8 trapping column (Agilent Technologies,
Santa Clara,
CA) containing Zorbax 300SB-C18 (5 x 0.3 mm). The column valve is switched to
its
secondary position 5 minutes after injection and the trapped peptides are then
eluted onto a
75 m id Zorbax Stablebond (300 A pore) colunm and chromatographed using a
binary
solvent system consisting of A: 0.1% formic acid and 5% acetonitrile and B:
0.1% formic
acid and 100% acetonitrile at a flow rate of 300 nL/minute. A gradient is run
from 15% B to
55% B over 60 minutes on a reversed-phase column (75 m id Zorbax Stablebond
(300 A
pore), and the eluting peptides are sprayed into a LC/MSD XCT ion trap mass
spectrometer
system (Agilent Technologies, Santa Clara, CA), equipped with an orthogonal
nanospray ESI
interface. The mass spectrometer is operated in positive ion mode with the
trap set to data
dependent auto-MS/MS acquisition mode. Source conditions are: Vcap -4500V,
drying gas
flow 8 L/minute, drying gas temperature 230 C and CapEx 65V. The instrument is
set to
complete a mass scan from 400-2200 Thompsons in one second. Peaks eluting from
the LC
column that have ions above 100,000 arbitrary intensity units trigger the ion
trap to isolate the
ion and perform an MS/MS experiment scan after the MS full scan. The
instrument's
dynamic ion exclusion filter is set to allow the instrument to record up to 2
MS/MS spectra
107
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
for each detected ion to maximize the acquisition of qualitative data from
peptides (by
preventing high abundance peptides from dominating the subsequent MS/MS
experiments)
and the excitation energy is set to "smart frag" mode to insure the generation
of useful
product ion spectra from all peptides detected. Data files that result are
then transferred to a
file server for subsequent data reduction.
N. The mass spectrometer data analysis with the SpectruinMill software
platform.
SpectrumMill is derived from the MS-Tag software package and is
contemplated as a suitable software platform. Raw data is extracted from the
MS data files
using the data extractor module and the data is then subjected to protein
library search and de
Novo spectral interpretation by the Sherenga module. SpectrumMill is designed
to minimize
spurious identifications obtained from the MS/MS spectra of peptides by
careful filtering and
grouping of related MS and MS/MS data during extraction from the raw da.ta
file. The
library searching and de Novo interpretation identify the detected proteins
form the individual
peptides. The results for all proteins detected are collected and listed by
protein name,
detected peptide sequence(s), and search score. The reports are exported to an
Excel
spreadsheet file for inclusion in a result database. hi addition, data
extracted from the raw
data files from the ion trap are preferably submitted to the Mascot
(MatrixScience Inc,
London, UK) search program and searched against both the NCBI non-redundant
protein
database and the SWISSPROT protein database. The identifications from these
two systems
are correlated to arrive at a final consensus list of identified proteins.
O. Separation of lipid fractions on HPLC system.
Fractions are pH adjusted to 3.5, and run across a Sep-Pak C18 column on a
HPLC system (Millipore corp., Milford, MA). The columns are washed with ddH2O,
and
compounds are eluted with increasingly polar mobile phases (hexane-methyl
formate-
methanol). Fractions are concentrated under a stream of nitrogen and
reconstituted in either 1
ml PBS (for PA-I reporter assay) or 100 ul of methanol (for UV/HPLC). Active
fractions
from Sep-Pak are preferably further resolved by a C18 reversed-phase HPLC
column (150
mm x 5 mm, Phenomenex, Torrance, CA) with acidified (0.1% acetic acid)
MetOH:H20
(60:40 vol/vol) at 1 ml/minute on a 1050 series HPLC using ChemStationTM
software
(Hewlett Packard, Palo Alto, CA).
ExMle 23
108
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
The separate effects of both tertiary and peripheral -opioid receptor
antagonists on nlorphine-induced PA-I lectin/adhesin expression in Pseudomonas
aeruginosa
were investigated. The P. aeruginosa strain used for the study was the PA-I
lectin/adhesin
reported strain 27853/PLL-EGFP, described above. PA-I lectin/adhesin assays
were
performed as described herein except where specifically indicated. The
reporter strain was
incubated in wells of a 96-well plate, and fluorescence and cell density were
measured using
conventional techniques. Results presented in Fig. 7 represent fluorescence
data normalized
to cell densities after 20 hours of incubation. Bars represent median of
twelve values stdv.
Apparent from Fig. 7 is the effect of 20 gM morphine on PA-IL expression, as
well as the
separate inhibitory effects of each of 20 M methylnaltrexone and 20 M
naloxone on the
morphine-induced expression of PA-I lectin/adhesin.
As shown in Fig. 7, these opioid-induced increases in PA-I lectin/adhesin are
significantly attenuated by either of the g-opioid receptor antagonists,
naloxone or
methylnaltrexone. The effects on opiate-mediated virulence may be mediated
through
classical mu opioid receptors or in subtypes of opioid receptors or splice
variants. Without
wishing to be bound by theory, this effect may be mediated by MAPK/ERR
phosphorylation
similar to or related to VEGF. The data establish that both tertiary -opioid
receptor
antagonists, e.g., naloxone, and peripheral g-opioid receptor antagonists,
e.g.,
methylnaltrexone, are useful compounds, both prophylactically and
therapeutically, in
addressing the clinical effects of microbial pathogens on host organisms.
Exa=le 24
Hypoxia-induced PA- lectira adhesin expression
The aim of the study described in this Example was to determine whether
intestinal epithelial hypoxia, a common response to surgical stress, could
activate PA-I
expression. Because splanchnic vasosconstriction and intestinal epithelial
hypoxia are a
common consequence of surgical injury, the aim of the experiments described
herein was to
determine the specific role of the intestinal epithelium in signaling to P.
aeYuginosa by
examining the effect of epithelial cell hypoxia and reoxygenation on PA-I
expression. A
fusion construct was generated to express green flixorescent protein
downstream of the PA-I
gene, serving as a stable reporter strain for PA-I expression in P.
aeruginosa, as described
herein. Polarized Caco-2 monolayers were exposed to ambient hypoxia (0.1-0.3%
02) for 1
hour, with or without a recovery period of normoxia (21% 02) for 2 hours, and
then
109
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
inoculated with P. aeruginosa containing the PA-I reporter construct. Hypoxic
Caco-2
monolayers caused a significant increase in PA-I promoter activity relative to
normoxic
monolayers (165% at 1 h; P < 0.001). Similar activation of PA-I was also
induced by cell-
free apical, but not basal, media from hypoxic Caco-2 monolayers. PA-I
promoter activation
was preferentially enhanced in bacterial cells that physically interacted with
hypoxic
epithelia. As shown below, the virulence circuitry of P. aeruginosa is
activated by both
soluble and contact-mediated elements of the intestinal epithelium during
hypoxia and
normoxic recovery.
Human epithelial cells.
Caco-2BBe cells expressing SGLT1 were maintained in DMEM with 25 mM
glucose (high-glucose DMEM) with 10% fetal calf serum, 15 mM HEPES, pH 7.4,
and 0.25
mg/ml geneticin, as previously described (Turner JR et al., Am J Physio1273:
C1378-1385,
1997). Caco-2 cells were plated on 0.33-cm2 collagen-coated, 0.4- m pore size
polycarbonate
membrane Transwell supports (Coming-Costar, Acton, MA) for 20 days, and media
were
replaced with identical media without geneticin at least 24 h before bacterial
inoculation.
GFP fusion constructs of wilcl-type P. aeruginosa.
P. aeruginosa (ATCC-27853, American Type Culture Collection, Manassas,
VA) was transformed with the plasmid pUCP24/PLL-EGFP. This construct harbors a
PA27853 chromosomal DNA fragment containing an upstream regulatory region of
PA-I
followed by the entire PA-I gene fused at the COOH terminal with an enhanced
green
fluorescent protein (EGFP) gene excised from the pBI-EGFP plasmid (Clontech,
Palo Alto,
CA). Expression of the PA-I lectin was detected by fluorescence microscopy and
fluorimetry
of this reporter strain as previously described (Wu L. et al., Ann Surg. 238,
754-764, 2003).
Dynamic f uorimetr-y.
Caco-2 cells were grown to confluence on collagen-coated 96-well fluorimetry
plates (Becton Dickinson Labware, Bedford, MA) and maintained in a 37 C
incubator with
5% CO2 and 21% 02. The day before experiments, media were removed and replaced
with
150 l of antibiotic-free media. Three experimental conditions were created
using a
modification of the methods previously described by Xu et al. J Trauina 46:280-
285, 1999).
In control conditions, Caco-2 cells were maintained in a 5% C02-21% 02
incubator for 2 h.
Hypoxic conditions were achieved by placing Caco-2 cells in a humidified
hypoxia chamber
110
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
at 37 C with 5% CO-95% N2 for 2 h. Measured 02 in the chambers varied between
0.1 and
0.3%. To simulate a reperfusion or reoxygenation state (normoxic recovery),
after 2 h of
Caco-2 cell hypoxia, hypoxic media were completely replaced with fresh,
normoxic
HDMEM media, and the cells were allowed to recover under normoxia (37 C, 5%
C02-21 l0
02) for 2 h before bacterial inoculation. The fluorescent reporter strain
PA27853/PLL-EGFP
was next added to the three groups of Caco-2 cells. Bacteria were cultured
overnight in Luria-
Bertani broth containing 20 g/ml gentamicin at 37 C under shaking conditions.
After - 12 h
of growth, 50 1 of the bacterial suspension were added to the 96-well plates
of Caco-2 cells.
Care was taken to ensure that all bacterial samples were cultured for
identical periods of time
and that wells contained equal cell densities. Fluorescence was tracked
immediately
following bacterial inoculation and then hourly thereafter up to 3 h using a
96-well
microplate fluorimeter (Synergy HT, Biotek, Winooski, VT). Plates were
maintained in
standard incubators at 37 C with 5% C02-21% 02 between all measurements.
Fluorescence
values were calculated as follows: %control = 100 x(RFUxt-, - RFUxt--
o)/(RFUcrõ -
.RFUct--o), where RFUx refers to the hypoxic or normoxic recovery groups and
RFUc refers to
the control at time n.
Exposure of bacteria to filtered media from Caco-2 cells and potential PA-I-
inducing
candidate molecules.
In this set of experiments, reiterative conditions of control, hypoxia, and
normoxic recovery (i.e., reperfusion/reoxygenation) were created in 96-well
plates containing
confluent Caco-2 cells. Media from all wells were then collected and passed
through a 0.22-
gm filter and stored on ice. Ninety-six-well fluorimetry plates without Caco-2
cells (Costar
3631, Coming, Corning, NY) were then prepared by adding a 20- 1 bacterial
suspension
containing overnight growing cultures of PA27853/ PLL-EGFP. Media from the
three
experimental groups were then added to the wells, and fluorescence was
assessed over 5 h,
with plates maintained at 37 C with continuous orbital shaking (100 rpm)
between
measurements. To screen for potential PA-I-inducing compounds that might be
present in the
media of hypoxic Caco-2 cell media, purified adenosine, D-lactate, and L-
lactate (Sigma-
Aldrich, St. Louis, MO) were added to wells containing HDMEM across a range of
physiologically relevant dosages, which were then tested as described above.
Fluorescent microscopy.
111
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
To visually correlate results from the above experiments to the spatiotemporal
effects of PA27853/PLLEGFP on hypoxic Caco-2 cells, cells were grown to
confluence on
Bioptechs dishes (Bioptechs, Butler, PA) and exposed to 2 h of hypoxia
followed by
inoculation with PA27853/PLL-EGFP. Experiments were perfornied on a 37 C
microscopy
stage and visualized using an inverted fluorescence microscope (Axiovert 100,
Carl Zeiss,
Thornwood, NY). Z-stacks were collected every 30 min for 3 h. Images were
analyzed for
bacterial distribution using ImageJ graphics analysis software (Version 1.31,
National
Institutes of Health, Bethesda, MD).
Caco-2 cell barriei- function during hypoxia and noYmoxic recovery in the
presence of P.
aeruginosa or purified PA-I.
Caco-2 monolayer transepithelial electrical resistance (TER), a measure of
barrier function, was assessed using agar bridges and Ag-AgCl-calomel
electrodes and a
voltage clamp (University of Iowa Bioengineering, Iowa City, IA). TER was
calculated
using Ohm's law. Fluid resistance was subtracted from all values. Two
microliters of
overnight cultures of PA27853 normalized to cell density or 50 g of purified
PA-I (Sigma-
Aldrich) were added to the apical chamber of the Caco-2 cell transwells
following exposure
to hypoxia and normoxic recovery as detailed above. Caco-2 cell TER was
assessed every
hour, and cells were maintained at 37 C with 5% C02-21% 02 throughout the
experiment. To
determine the effect of PA27853 on the barrier function of Caco-2 cells under
conditions of
sustained hypoxia, reiterative experiments were performed under continuous
hypoxia (37 C,
5% CO2-95% N2), in which TER measurements were made every hour for 7 h within
the
hypoxic chamber using an EVOM resistance measurement apparatus (World
Precision
Instruments, Sarasota, FL).
Northern blot analysis.
In selected experiments, PA-I expression was confirmed using Northern blot
analyses.
Statistical analysis.
Data were analyzed, and statistical significance was determined using Prism
4.0 (GraphPad Software, San Diego, CA). Statistical significance was defined
as P < 0.05 by
Student's t-test or two-way ANOVA, as appropriate.
RESULTS
112
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
PA27853/PLL-EGFP P. aeruginosa respond to the environment of Caco-2 cell
hypoxia and
nornYoxic recovery Wltll enlZancedfluorescence.
To determine whether the green fluorescent protein (GFP) reporter strain
PA27853/PLL-EGFP would display increased PA-I proinoter activity when added to
Caco-2
cells exposed to hypoxia (2 h at <0.3% 02) and normoxic recovery (hypoxia
followed by 2 h
of recovery in normoxic conditions), reporter strains were added to the media
of Caco-2 cells
exposed to the two conditions. GFP reporter strains demonstrated significantly
more PA-I
promoter activity, as measured by fluorescence, within 1 h of incubation with
Caco-2 cells
exposed to either hypoxia or normoxic recovery. The media pH in all
experimental
conditions was measured at all time points and demonstrated no significant
difference among
control, hypoxia, and normoxic recovery groups because all media were buffered
(data not
shown). However, to show that the pH of media did not influence fluorescence
in
PA27853/PLL-EGFP, strains were incubated in media at pH 6.5, 7.4, and 7.7. The
percent
change in fluorescence was not different among groups (6.5 = 106 10, 7.4 =
100 12, 7.7 =
112 12; P = not significant). Similarly, to rule out an effect of
hypercarbia or hypoxia alone
on PA-I promoter activity in our reporter strains, strains were subjected to
hypoxia (0.1 % for
2 h) and hypercarbia (80% CO2 for 2 h). No difference in fluorescence was
observed between
groups (data not shown). Taken together, these findings demonstrate that media
from Caco-2
cells exposed to hypoxia with or without normoxic recovery activate PA-I
promoter activity.
Fluorescence imaging of GFP reporter strains in the Caco-2 cell environment.
To determine whether epithelial cell contact contributes to the expression of
GFP in our PA-I reporter strain, Caco-2 cells were imaged by fluorescent
microscopy
following exposure to hypoxia and apical inoculation with PA27853/PLL-EGFP.
Fluorescence imaging demonstrated that PA27853/PLL-EGFP exposed to hypoxic
Caco-2
monolayers appeared niarkedly more fluorescent than bacteria exposed to
normoxic
monolayers at the 120-min time point. Multiple images of the bacterial/Caco-2
cell coculture
demonstrated that more bacteria were located near or within the plane of the
cell monolayers
exposed to hypoxia than in nonhypoxic cells. Quantitative analysis of multiple
microscopy
images revealed an average of 658 :h 78 bacteria/high-powered field at the
level of the
surface of hypoxic epithelia, whereas no bacteria were seen in plane-matched
controls (P <
0.001).
113
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
PA27853/PLL-EGFP reporter strains respond to a paracrine factor present in
znedia fronz
Caco-2 cells exposed to hypoxia and nornzoxic recovery.
To determine whether soluble compounds released into the media in response
to Caco-2 cell hypoxia are capable of activating PA-I expression independent
of bacterial
contact with the epithelium, we tested the ability of media from hypoxic Caco-
2 cell cultures
to enhance fluorescence in our reporter strain. PA27853/PLL-EGFP bacteria
exposed to
filtered media from Caco-2 cells exposed to hypoxia and normoxic recovery
developed a
significant enhancement of fluorescence that appeared greatest at the 5-h time
point (Fig. 40;
control: 3.7% + SD 3.9; hypoxia: 12.6% SD 5.8; normoxic recovery: 13.1 %
SD 3.9; P <
0.001 by 2-way repeated measures ANOVA). Results were confirmed by Northern
blot
analysis. To determine whether this paracrine factor was isolated to the
apical or basolateral
compartments, we performed reiterative experiments in which isolated media
from the
basolateral and apical compartments of hypoxic monolayers, as well as mixtures
of apical and
basolateral media, were added to wells containing the GFP-PA-I reporter strain
PA27853/PLL-EFGP. Only those bacteria exposed to hypoxic media from the apical
chamber
or liypoxic mixed media showed a statistically significant increase over
controls (>125%
change, normalized to initial value; P < 0.05).
Adenosine alone induces PA I expression in P. aeruginosa.
To deterniine whether candidate compounds specifically released by hypoxic
Caco-2 cells could induce the expression of PA-I, we tested the effect of D-
lactate, L-lactate,
and adenosine in our GFP-PA-I reporter strains. D_ and Llactate had no effect
on PA-I
promoter activity (data not shown); however, PLL/PA27853 responded with
enhanced
fluorescence to 10 mM adenosine, raising the possibility that adenosine
released by hypoxic
Caco-2 cells could be the putative mediator of the increased PA-I response
observed in the
above studies. However, the time required for upregulation of PA-I expression
was longer
than that observed in response to hypoxic cell media, suggesting that other
factors may be
involved in the signaling pathway.
Caeo-2 cells exposed to hypoxia and nornzoxic recovery resist the barrier-
dysregulating
effect of puried PA-Z
To determine whether conditions of hypoxia and nornloxic recovery enhance
or attenuate the barrier-dysregulating properties of PA27853 against Caco-2
cells, TER was
114
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
measured in Caco-2 cells apically inoculated with either PA27853 or purified
PA-I following
exposure to hypoxia and norrnoxic recovery. Despite the ability of media from
hypoxic and
reoxygenated Caco-2 cells to increase the expression of PA-I in P. aeruginosa,
the TER of
Caco-2 cells exposed to these conditions were unchanged in response to a P.
aeruginosa
inoculated with purified PA-I exhibited an attenuated drop in TER compared
with normoxic
cells.
Caco-2 cells exposed to sustained hypoxia conzpletely resist tlze barrier
dysregulating effect
of PA27853.
To determine whether Caco-2 cells exposed to sustained hypoxia could resist
the barrier-dysregulating effect of PA27853, the TER of Caco-2 cells apically
inoculated with
PA27853 in an environment of sustained hypoxia was measured. Caco-2 cells
maintained
TER equal to hypoxic Caco-2 cells without bacteria and completely resisted the
predicted
decrease in TER at the 7-h time point. That Caco-2 cells partially resist the
barrier-
dysregulating effect of strains of PA27853 despite increased PA-I expression
could be
explained by previous observations suggesting that epithelial cells normally
respond to
hypoxia with an enhancement of local mucosal defense proteins and barrier
function.
Soluble factors present in the media of hypoxic Caco-2 cells induce incYeased
barrier
resistance in normoxic cells.
To determine whether the normoxic Caco-2 cells could be induced to increase
their resistance to barrier dysregulation by P. aeruginosa through signals
present in hypoxic
cell media, we exchanged the apical and basolateral media of normoxic Caco-2
cells with
filtered media from the apical and basolateral compartments of hypoxic Caco-2
cells and
tested the barrier function of these cells when apically inoculated with P.
aeruginosa.
Normoxic Caco-2 cells exposed to media from hypoxicepithelia displayed a
prolonged
resistance to barrier dysregulation induced by P. aeruginosa, suggesting that
normoxic
epithelia may be activated to enhance their barrier function in the presence
of soluble
mediators produced during hypoxia.
Although P. aeruginosa is not considered to be an intestinal pathogen in the
classic sense, it induces one of the most rapid and profound decreases in
intestinal epithelial
TER of any bacteria reported to date. We have previously reported, in both
Caco-2 and T-84
cells, that P. aeruginosa (PA27853) can induce an 80% decrease in TER within 4
h following
its apical inoculation. If defined by this criterion alone, P. aeruginosa is
among the most
115
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
pathogenic organisms to the intestinal epithelium yet described. The
observation that as many
as 5% of the normal population harbor this pathogen within their intestinal
tracts, coupled
with our animal studies demonstrating that control mice do not develop any
symptoms of
infection following the direct introduction of large quantities of P.
aeruginosa into the
cecum, suggest that this organism behaves like a clas'sic opportunist,
switching virulence
genes on and off in response to selected environmental cues. Although it is
well established
that environmental cues such as pH, redox state, and nutrient composition can
activate
virulence gene expression in bacteria through a variety of membrane- bound
biosensor
kinases, there are no previous reports suggesting that bacterial signaling
compounds are
released by host cells following physiological or ischemic stress. From the
standpoint of the
evolutionary fitness of the microbe, however, it is logical that a pathogen
might recognize the
biochemistry of host cell stress, because possessing a system that recognizes
host
susceptibility would allow for a more accurate assessment of the costs versus
benefits of host
invasion. Yet, whereas it is well established that intestinal pathogens can
communicate
directly with the cells to which they adhere, that such a molecular dialogue
might be
bidirectional is poorly described.
To demonstrate that bacteria sense and respond directly to host cells, we used
the PA-I lectin/adhesin of P. aeruginosa as a reporter gene. The PA-I lectin
is under tight
regulatory control of two key systems of virulence gene regulation in P.
aeruginosa: the
quorum-sensing signaling system and the alternative sigma factor RpoS. The
quoruin-sensing
signaling system and RpoS are interconnected systems of virulence gene
regulation in P.
aeruginosa that control the expression of hundreds of virulence genes in this
pathogen.
Because PA-I expression is dependent on the function of both quorum sensing
and RpoS, it
serves as a relevant biological readout for generalized virulence gene
activation in P. -
aeruginosa. The finding that soluble elements of intestinal epithelial cells
and, in particular,
adenosine can activate PA-I expression, suggests that specific host cell-
derived compounds
may be released that signal colonizing pathogens such as P. aeruginosa to a
weak and
susceptible host. That adenosine alone can activate PA-I expression is an
important finding
given that adenosine is released. and can accumulate in the extracellular
milieu of hypoxic
tissues at high concentrations. During active intestinal inflammation, 5 -AMP
derived from
migrating polymorphonuclear leukocytes is converted to adenosine by the apical
surface
epithelium of the intestine. Strohmeier et al. (14) have demonstrated that
under nornal
conditions, the human intestinal epithelial cell line T-84 can convert
substantial amounts of
116
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
5'-AMP that accumulate to as much as 5 mM adenosine in the apical media within
30 min.
Although in the present study, activation of PA-I promoter activity in P.
aeruginosa required
what appeared to be an unphysiological dose of adenosine, the precise
concentration of
adenosine to which P. aeruginosa might be exposed within the intestinal tract
during
prolonged hypoxia and reoxygenation is unknown. In addition, adenosine
exposure required
6 h before PA-I promoter activity was observed, whereas with hypoxic media PA-
I promoter
activity was observed at 4 h. As a matter of speculation, an opportunistic
organism like P.
aeruginosa may require an inordinately potent and prolonged host-derived
signal for it to
invest the resources and energy required to mount a toxic offensive against
the intestinal
epithelium. Under such circumstances, P. aeruginosa might "sense" that the
host on which its
survival depends is subjected to an extreme degree of inflammation and
vulnerability and
hence represents a liability to its survival.
Given that PA-I expression was increased in response to Caco-2 cell hypoxia
and normoxic recovery, we expected to see a more profound decrease in TER when
P.
aeruginosa was apically inoculated onto Caco-2 cells exposed to these
conditions. That
enhanced PA-I expression in P. aeruginosa did not decrease Caco-2 cell TER
during hypoxia
could be explained by the enhancing effect of hypoxia itself on Caco-2 cell
barrier function.
This possibility is supported by the finding that hypoxic media transferred to
nonnoxic Caco-
2 cells enhanced their resistance to P. aeruginosa. This notion is further
supported by the
finding that hypoxic Caco-2 cells resist the barrier-dysregulating property of
purified PA-I,
again suggesting that hypoxia enhanced epithelial barrier function to the
barrier-dysregulating
effects of the PA-I protein of P. aeruginosa. These findings are also in
agreement with the
known enhancing effect of hypoxia on intestinal epithelial barrier function.
Furuta and
colleagues have demonstrated that exposure of Caco-2 cells to hypoxia
increases the
expression of both mucin and trefoil peptides, and they have also observed TER
to be
preserved or even increased in Caco-2 cells during hypoxia: This response
makes
physiological sense given that under such circumstances, the intestinal
epithelial surface will
be vulnerable to a potentially hostile flora. However, during reperfusion,
which here we have
termed normoxic recovery, Caco-2 cells eventually succumb to the potent
barrier-
dysregulating effect of P. aeruginosa. This is consistent with both clinical
and animal studies
where the greatest alteration in intestinal permeability and systemic
proinflammatory
activation occurs during the reperfusion phase following ischemic injury to
the intestine.
117
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
In summary, herein we demonstrate that P. aeruginosa is capable of sensing
and responding to local elements of host cell stress. Host-derived bacterial
signaling
compounds appear to be released by intestinal epithelial cells in response to
hypoxia and
normoxic recovery, which are often present during critical illness and its
treatment. Further
elucidation of the precise host compounds or signals that are sensed by
colonizing
nosocomial pathogens, such as P. aer-uginosa, could lead to a better
understanding of how
infection continues to complicate the course of the most critically ill
patients.
Example 25
This study was designed to determine whether the intestinal tract of a
stressed
host is a unique environment in which the virulence of P. aeruginosa is
enhanced in vivo. In
order to further investigate this question, the inventors created a reporter
strain of P.
aeruginosa with GFP inserted downstream of the PA-I gene and the quorum
sensing and
RpoS promoters as described herein. To further understand how surgical stress
and intestinal
hypoxia might play a role in activating the virulence of P. aeruginosa, the
inventors
investigated whether HIF-1-a may play a central role in this response. It is
well known that
hypoxia results in the accumulation of HIF-1-a in intestinal epithelial cells.
Given the
increasingly important role of HIF-1- a activation in intestinal epithelial
homeostasis, the
investigators sought to determine if HIF-1-a activation mediates the release
of soluble
compounds that activate P. aeruginosa virulence as judged by expression of the
PA-I
lectin/adhesin.
To accomplish this an established Caco 2 cell line that has been stably
transfected with HIF-1-a and its parental cell line were used. Briefly, both
cell lines were
grown to confluence. The media was collected and filtered through 0.22u
filters to remove
any potential cellular components. Media was then added to microtiter wells
containing a
fixed bacterial cell population of the GFP/PA-I reporter strain described
above. Fluorescence
was dynamically tracked over time and was calculated according to the
following formula:
RFU HIF t=n RFU HIF t=0
OD HIF t=n OD HIF t--0
%ofcontrol
R-FU Control t=n R-FU Conrtrol t=0
OD Control t=n OD Control t=0
118
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
The results demonstrated that there is a time-dependent induction of PA-I
expression
observed in GFP/PA-I reporter strains exposed to HIF-1 a media compared to
control
(>400% and >600% change in PA-I expression in comparison to control at 7 and 8
hours,
respectively, following inoculation). This finding were confirmed by Western
blot analysis
in reiterative experiments. '
In order to identify the potential compounds that activate PA-I, the media
from
three groups of Caco-2 cells were examined, namely, control cells, Caco2 cells
exposed to
hypoxia, and Caco2 cells with forced expression of HIF-1 a. Media fractions
were separated
into 4 molecular weight fractions which were added to the microtiter plates
containing the
PA-UGFP reporter strains and evaluated by dynamic fluorimetry.
Results from these experiments demonstrated that media fractions with MW of
<3kDa induced PA-I expression significantly (>800% and >700% increase in HIF-1-
a and
hypoxic media, respectively, at.7 hours following incubation).
Further studies were performed to show that HIF and hypoxic conditions have
similar effects.
Because of the MW of the potential inducing compound, the inventors examined
the known
genes that are expressed in response to HIF-1- a activation. Within this MW
range we
identified potential candidate compounds related to nucleotide metabolisi'n.
In particular, we
were interested in adenosine since it has been shown to be released in high
concentrations
following intestinal epithelial hypoxia and HIF-1- a activation. Adenosine
accumulates in
the media of intestinal epithelial cells exposed to hypoxia and / orHIF-1 a
activation, through
a mechanism that involves upregulation of 5'-nucleosidase (CD73) activity.
Therefore media fractions were examined by HPLC/MS/MS for adenosine by
comparing 3kDa centricon filtered media from control Caco-2 cells, hypoxic
cells (0.1-0.3%
02 for 2 hrs, and HIF-1- a overexpressing cells. Adenosine was greatly
elevated in HIF-1-a
activated and hypoxic cell media (>8000% increase).
When the effect of effect of adenosine on PA-I expression in the above-
described reporter strain, it was seen that PA-I expression was increased in
the presence
adenosine that was both dose- and time-dependent (Fig. 8A). Results were
confirmed by
Western blot (inset in Fig. 8A). For completeness the effect of ATP, ADP, and
AMP at
similar concentrations was tested and revealed no evident inducing effect.
In order to determine if adenosine was the putative component within the
media of HIF-1- a-activated Caco-2 cells that induces the expression of PA-I,
adenosine
119
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
deaminase was added to deplete the media of adenosine. Surprisingly, these
experiments
resulted in an even greater increase in PA-I expression, raising the
possibility the metabolite
of adenosine, namely inosine, plays a role in PA-I expression (Fig. 8).
Adenosine deaminase
is predicted to be present in P. aeYugifaosa based on its DNA sequence. In a
related study
inosine induced PA-I expression at a concentration 10-fold less than adenosine
(Fig. 8C).
Reiterative experiments to directly compare the change in PA-I expression
over time between inosine and adenosine demonstrate that not only is the
effect of inosine
greater, but it occurs at an earlier time point. Further studies showed that
inosine induces PA-
I expression at an earlier time point and at lower cell densities (OD)
compared to adenosine.
In conclusion, the present example demonstrates that hypoxia or Forced
expression of HIF-1-a in Caco-2 cells results in the extracellular release of
soluble
compounds that activate the virulence circuitry of P. aeruginosa. Further, the
data presented
herein show that adenosine and inosine may play an important role in this
response.
Example 26
This Example provides data establishing that a mu opioid receptor antagonist
in the form of MNTX inhibits opiate-, thronibin- and LPS-induced endothelial
cell barrier
disruption by mu opioid receptor (mOP-R)-dependent, and independent,
mechanisms. The
mOP-R-independent mechanisms of MNTX-induced endothelial cell barrier
regulation
include activation of receptor-like protein tyrosine phosphatase mu (RPTPg)
and inhibition of
thrombin- and LPS-induced, Src-dependent, S1P3 receptor transactivation
(tyrosine
phosphorylation). The results indicate that MNTX is useful as a cell barrier
protective agent,
such as an endothelial cell barrier protective agent. Although the data
disclosed in this
Example relate to pulmonary microvascular endothelial cells, the behavior of
these cells
exemplifies the behavior of any endothelial (or epithelial) cell towards
opioid receptor
agonists and antagonists. The data were generated using the following
materials and
methods.
Materials and Methods
Cell Culture and Reageyats - Human pulmonary microvascular endothelial cell
were obtained from Canibrex (Walkersville, MD) and cultured as previously
described (2) in
EBM-2 complete medium (Cambrex) at 37 C in a humidified atmosphere of 5% COZ,
95%
air, with passages 6-10 used for experimentation. Unless otherwise specified,
reagents were
obtained from Sigma (St. Louis, MO). Morphine sulfate was purchased from
Baxter
120
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
(Deerfield, IL). Reagents for SDS-PAGE electrophoresis were purchased from Bio-
Rad
(Richmond, CA), Immobilon-P transfer membranes were from Millipore (Millipore
Corp.,
Bedford, MA), and gold microelectrodes were from Applied Biophysics (Troy,
NY). Rabbit
anti-mu opioid receptor antibody was purchased from Abcam (Cambridge, MA).
Rabbit anti-
S1P1 receptor antibody was purchased from Affinity Bioreagents (Golden, CO).
Mouse anti-
S 1P3 receptor antibody was purchased from Exalpha Biologicals (Watertown,
MA). Mouse
anti-RPTP antibody was purchased from Cell Signaling Technologies (Danvers,
MA).
Mouse anti-phospho-tyrosine antibody, mouse anti-pp60src antibody and
recombinant active
Src were purchased from Upstate Biotechnologies (Lake Placid, NY). PP2 was
purchased
from Calbiochem (San Diego, CA). Mouse anti-(3-actin antibody, rabbit anti-
phospho-
tyrosine (418) Src antibody, naloxone, DAMGO, thrombin, LPS and ionomycin were
purchased from Sigma (St. Louis, MO). Secondary horseradish peroxidase (HRP)-
labeled
antibodies were purchased from Amersham Biosciences (Piscataway, NJ).
Immunoprecipitation and Immunoblotting - Cellular materials from treated or
untreated HPMVEC were incubated with IP buffer (50 mM HEPES (pH 7.5), 150 mM
NaCl,
20 mM MgC12, 1% Nonidet P-40 (NP-40), 0.4 mM Na3VO4, 40 mM NaF, 50 M okadaic
acid, 0.2 mM phenylmethylsulfonyl fluoride, 1:250 dilution of Calbiochem
protease inhibitor
mixture 3). The samples were then immunoprecipitated with anti- S1P3 receptor
IgG
followed by SDS-PAGE in 4-15% polyacrylainide gels, transferred onto
ImmobilonTM
membranes, and developed with specific primary and secondary antibodies.
Visualization of
immunoreactive bands was achieved using enhanced chemiluminescence (Amersham
Biosciences).
Construction and Transfection ofsiRNA against nau opioid receptor, SIPI,
SIP3, RPTP,u - The siRNA sequence(s) targeting human mOP-R, S1P1, S1P3, RPTPg
were
generated using mRNA sequences from Gen-BankTM (gi:56549104, gi:87196352,
gi:38788192, and gi:18860903, respectively). For each mRNA (or scramble), two
targets
were identified. Specifically, mOP-R target sequence 1 (5'-
AACGCCAGCAATTGCACTGAT-3'; SEQ ID NO:14), mOP-R target sequence 2(5'-
AATGTCAGATGCTCAGCTCGG-3'; SEQ ID NO:15), S1P1 target sequence 1(5'-
AAGCTACACAAAAAGCCTGGA-3'; SEQ ID NO:16), S 1 P1 target sequence 2(5'-
AA.AAAGCCTGGATCACTCATC-3'; SEQ ID NO:17), SiP3 target sequence 1(5'-
AACAGGGACTCAGGGACCAGA-3'; SEQ ID NO:18), S 1 P3 target sequence 2 (5'-
AAATGAATGTTCCTGGGGCGC-3'; SEQ ID NO:19), RPTP target sequence 1(5-
121
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
AATCTGAAGGTGATGACTTCA-3'; SEQ ID NO:20), RPTP target sequence 2(5'-
AACACCTTGACTAAACCGACT-3'; SEQ ID NO:21), scrambled sequence 1(5'-
AAGAGAAATCGAAACCGAA.AA-3'; SEQ ID NO:22) and scramble sequence 2 (5'-
AAGAACCCAATTAAGCGCAAG-3'; SEQ ID NO:23) were utilized. Sense and antisense
oligonucleotides were provided by the Johns Hopkins University DNA Analysis
Facility or
were purchased from Integrated DNA Technologies (Coralville, IA). For
construction of the
siRNA, a transcription-based kit from Ambion was used (SilencerTM siRNA
construction kit).
Human lung endothelial cells were then transfected with siRNA using
siPORTamineTM as the
transfection reagent (Ambion, TX) according to the protocol provided by
Ambion. Cells
(about 40% confluent) were serum-starved for 1 hour followed by incubated with
3 M (1.5
RM of each siRNA) of target siRNA (or scramble siRNA or no siRNA) for 6 hours
in serum-
free medium. The serum-containing medium was then added (1 % serum final
concentration)
for 42 hours before biochemical experiments and/or functional assays were
conducted.
DeteNnaination of tyYosine phosphorylation of the SIP3 Receptor - Solubilized
proteins in IP buffer were immunoprecipitated with mouse anti-S1P3 receptor
antibody
followed by SDS-PAGE in 4-15% polyacrylamide gels and transfer onto
ImmobilonTm
membranes (Millipore Corp., Bedford, MA). After blocking nonspecific sites
with 5%
bovine serum albumin, the blots were incubated with either mouse anti-S 1 P3
antibody or
mouse anti-phospho-tyrosine antibody followed by incubation with horseradish
peroxidase
(HRP)-labeled goat anti-rabbit or goat anti-mouse IgG. Visualization of
immunoreactive
bands was achieved using enhanced chemiluminescence (Amersham Biosciences).
Tyrosine Phosphatase Activity Assay - Treated or untreated HPAEC lysates
and/or immunoprecipitated RPTP were analyzed for tyrosine phosphatase
activity using the
fluorometric RediplateTM 96 EnzChek Tyrosine Phosphatase Assay Kit (Invitrogen
(Molecular Probes), Eugene, OR). Briefly, cellular materials were incubated in
reaction
buffer at 30 C and then added to a 96-well plate coated with 6,8-difluoro-4-
methylumbelliferyl phosphate (DiFMUP). Tyrosine phosphatase activity cleaves
DiFMUP
into DiFMU with excitation/emission maxima of 358/452 nm.
In vitro SIP3 Receptor Phosphofylation/Dephosphorylation - The S 1P3
receptor phosphorylation/dephosphorylation reaction was carried out in 50 l
of the reaction
mixture containing 40 mM Tris-HCl (pH 7.5), 2 mM EDTA, 1 mM dithiothreitol, 7
mM
MgCl2, 0.1% CHAPS, 100 M ATP, purified enzymes (i.e. 100 ng of recombinant
active Src
and/or immunoprecipitated RPTP obtained from MNTX-treated (1 hour)
endothelial cells)
122
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
and immunoprecipitated S I P3 receptor obtained from human pulmonary
endothelial cells that
were serum-starved for one hour. After incubation for 30 minutes at 30 C, the
reaction
mixtures were boiled in SDS sample buffer and subjected to SDS-PAGE.
Immunoblots were
performed using mouse anti-phospho-tyrosine, mouse anti-pp60src, mouse anti-
RPTP or
mouse anti-S1P3 antibody followed by incubation with horseradish peroxidase
(HRP)-labeled
goat anti-mouse IgG. Visualization of immunoreactive bands was achieved using
enhanced
chemiluminescence (Amersham Biosciences).
Measurement of Endotlzelial Cell Electrical Resistance - Cell barrier
properties were measured using a highly sensitive biophysical assay with an
electrical cell-
substrate impedance sensing system (Applied Biophysics Inc., Troy, NY), as
described
previously in Garcia et al., Am. J. Physiol. 273:LI 72-L184 (1997); J. Appl.
Physiol. 89:2333-
2343 (2000); J. Clin. Invest. 108:689-701 (2000). The cells were cultured to
confluence in
polycarbonate wells containing evaporated small gold microelectrodes (10"4 cm)
and culture
medium was used as electrolyte. The total electrical resistance was measured
dynamically
across the monolayer and was determined by the combined resistance between the
basal
surface of the cell and the electrode, reflective of focal adhesion, and the
resistance between
the cells. As cells adhered and spread out on the microelectrode, TER
increased (maximal at
confluence), whereas cell retraction, rounding, or loss of adhesion was
reflected by a decrease
in TER. The small gold electrode and the larger counter electrodes (1 cm2)
were connected
to a phase-sensitive Ion-in amplifier with a built-in differential
preamplifier (Applied
Biophysics). A I-V, 4000-Hz alternating current signal was supplied through a
MQ resistor
to approximate a constant-current source. Voltage and phase data were stored
and computer
processed using conventional techniques. Experiments were conducted only on
cells that
achieved >1000Q (10 microelectrodes per well) of steady-state resistance.
Resistance was
expressed by the in-phase voltage (proportional to the resistance), which was
normalized to
the initial voltage and expressed as a fraction of the nonnalized resistance
value, as
previously described (Garcia et al., (1997)). These measurements provided a
sensitive
biophysical assay that indicates the state of cell shape and focal adhesion
reflective of
changes in para-cellular permeability. TER values from each microelectrode
were pooled at
discrete time points and plotted versus time as the mean S.E.
Animal Preparation and Treatment - Male C57BL/6J mice (8-10 weeks,
Jackson Laboratories, Bar Harbor, ME) were anesthetized with intraperitoneal
ketamine (150
mg/kg) and acetylpromazine (15 mg/kg) before exposure of the right internal
jugular vein via
123
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
neck incision. LPS (2.5 mg/kg) or water (control) were instilled intravenously
through the
internal jugular vein. Four hours later, mice received methylnaltrex.one
(MNTX, 10 mg/kg)
or water control through the internal jugular vein. The animals were allowed
to recover for
24 hours after LPS before bronchoalveolar lavage protein analysis and/or lung
immunohistochemistry.
Mouse Lung Immunohistochemistny - To characterize the expression of
proteins in mouse lung vascular endothelial cells, lungs from control
(untreated) mice were
fonnalin-fixed, 5 micron paraffin sections were obtained, hydrated and epitope
retrieval was
performed (DakoCytomation Target Retrieval Solution, pH=6.0, DakoCytomation,
Carpinteria, CA). The sections were then histologically evaluated by either
anti-mu opioid
receptor, anti-RPTP or anti-S1P3 receptor antibody and secondary HRP-labeled
polymer
with DAB staining (Dako EnVisionTM + System, HRP (DAB) (DakoCytomation,
Carpinteria,
CA)), followed by hematoxylin QS counterstaining (Vector Laboratories,
Burlingame, CA).
Negative controls for immunohistochemical analysis were done by the same
method as above
but without primary antibody. Immunostained sections were photographed (100x)
using a
Leica Axioscope (Bannockbum, IL).
DeteYmination of Bronchoalveolan Lavage Protein - Bronchoalveolar lavage
(BAL) was performed by an intratracheal injection of 1 cc of Hank's balanced
salt solution
followed by gentle aspiration. The recovered fluid was processed for protein
concentration
(BCA Protein Assay Kit; Pierce Chemical Co., Rockford, IL).
Statistical Analysis - Student's t test was used to compare the means of data
from two or more different experimental groups. Results are expressed as means
S.E.
Results
The role of inethylnaltrexone (MNTX) in agonist-induced endothelial cell
barrier disruption. Endothelial cell barrier disruption is a causative factor
in a variety of
pathologies, including atherosclerosis and acute lung injury. The effects of
methylnaltrexone
(MNTX), a charged peripheral mu opioid receptor (mOP-R) antagonist, on
pulmonary
microvascular endothelial cell integrity was examined using transendothelial
resistance
(TER). Figure 9-A,B indicate that ligands for the mOP-R (i.e., morphine
sulfate (MS) and
DAMGO) induced endothelial cell barrier disruption in a dose-dependent manner.
These
barrier disruptive effects were blocked by pre-treatment with a
physiologically relevant dose
of MNTX (0.1 M)). Decreasing the dose of MNTX below 0.1 M attenuated its
barrier
124
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
protective effects while increasing the dose of MNTX beyond 0.1 M did not
significantly
alter its actions (Figure 9-C).
Next, the effects of MNTX on non-mOP-R-dependent agonist-induced
endothelial cell barrier regulation were investigated. Thrombin induced a
rapid transient
decrease in endothelial cell barrier function (Figure 10-A).
Lipopolysaccharide (LPS)
induced a delayed (about 4-hour) endothelial cell barrier-disruptive response
(Figure 10-B).
MNTX (0.1 M) attenuated endothelial cell barrier disruption from thrombin
(Figure 10-A)
and LPS (Figure 48-B) but not from the Ca2+ ionophore, ionomycin (Figure 10-
D). These
results indicated selectivity in MNTX-mediated endothelial cell barrier
protection. The
protective effects of MNTX were not limited to barrier-disrupting agents, as
MNTX
increased the sustained endothelial cell barrier-enhancing effect of
sphingosine- 1 -phosphate
(S1P) (Figure 10-C).
Methylnaltrexone is a charged molecule that cannot cross the blood-brain
barrier (BBB). This property allows MNTX to selectively block peripheral mOP-R
activity.
The effects of another mOP-R antagonist, naloxone, which is uncharged and
promotes both
peripheral and CNS mOP-R inhibition, on agonist-induced endothelial cell
barrier regulation
were examined. Both MNTX and naloxone (0.1 M) blocked MS- and DAMGO-induced
endothelial cell barrier disruption. However, naloxone did not display the
same endothelial
cell barrier-protective effects as MNTX with thrombin and LPS challenge
(Figure 11).
The role of S1P3 receptor transactivation in agonist-induced endothelial cell
barrier dysfunction. Considering the actions of MNTX on opiate and SIP-induced
endothelial cell barrier regulation, the effects of silencing (siRNA) mOP-R or
S 1 P receptor
subtypes on MNTX-regulated endothelial cell integrity were investigated
(Figures 12 and
18). Silencing mOP-R expression had little effect on MNTX protection from
thrombin- and
LPS-induced'endothelial cell barrier disruption indicating potential mOP-R-
independent
effects of MNTX. Endothelial cells express both S1P1 and S1P3 receptors with
S1P1 receptor
activating Rac 1-mediated signaling, while S 1 P3 receptor activates RhoA-
mediated signaling.
The silencing of S1P1 receptor had previously been shown to completely
eliminate the
barrier-protective effects of S1P (1 M). At higher concentrations (10 to 30
M), S1P-
induced barrier disruption is likely due to S1P3 receptor activation. In
contrast to S1P1
receptor, silencing S1P3 receptor inhibited thrombin- and LPS-induced, and
MNTX
protection from, endothelial cell barrier disruption (Figure 12-B,C; Figure
18).
125
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
It is known that S i P, receptor transactivation is important in agonist-
induced
endothelial cell barrier enhancement. Considering the results in Figure 12, it
was expected
that S1P3 receptor transactivation would be an important regulatory mechanism
in endothelial
cell barrier disruption. Figure 13 provides data indicating that barrier
disrupting, but not
barrier enhancing (i.e. S 1 P at 1[,M), agents promoted Src activation and Src
family kinase-
mediated S1P3 receptor transactivation (tyrosine phosphorylation). Further,
inhibition of Src
family kinases by PP2 blocked agonist-induced barrier disruption but did not
affect S1P-
mediated endothelial cell barrier enhancement. Finally, pre-treatment with
MNTX
completely blocked agonist-induced S 1 P3 receptor transactivation. In
contrast, naloxone pre-
treatment blocked the effects of morphine and DAMGO, but not thrombin or LPS,
on S1P3
receptor transactivation.
The role of receptor protein tyrosine phosphatase mu (RPTP ) in MNTX-
mediated protection from agonist-induced endothelial cell barrier disruption.
The results in
Figure 13 indicated that MNTX blocked agonist-induced S 1 P3 receptor
transactivation
(tyrosine phosphorylation). One mechanism of attenuating S1P3 receptor
tyrosine
phosphorylation is through regulation of tyrosine phosphatase activity. The
results indicated
that MNTX (but not naloxone, morphine, DAMGO or S1P) increased total
endothelial cell
tyrosine phosphatase activity (Figure 14).
An important tyrosine phosphatase implicated in regulating human pulmonary
endothelial cell-cell contacts is the receptor tyrosine phosphatase mu (RPTP
). MNTX, but
not naloxone, treatment of human pulmonary microvascular endothelial cells
(HPMVEC)
enhanced RPTP tyrosine phosphatase activity (Figure 15-A). Further, silencing
RPTP
prolonged thrombin-induced S1P3 receptor tyrosine phosphorylation (Figure 15-
B). In vitro
analysis of isolated S1P3 receptor indicated that MNTX-stimulated RPTP
blocked Src
tyrosine phosphorylation of the S 1P3 receptor (Figure 15-C). In addition,
silencing RPTP
(but not mOP-R or S1P3 receptor) protein expression significantly attenuated
the MNTX-
mediated increase of total endothelial cell tyrosine phosphatase activity
(Figure 16-A).
Finally, silencing =RPTP inhibited the protective effects of MNTX of, and
enhanced the
thrombin- and LPS-induced effects on, endothelial cell barrier disruption
(Figure 16-B,C).
The role of MNTX in LPS-induced pulmonazy vascular hyperpermeability in
vivo. Similar to the results from human pulmonary microvascular endothelial
cells,
immunohistochemistry revealed that endothelial cells in mouse lung vasculature
expressed
mOP-R, RPTP and S1P3 receptor (Figure 17-A). Next, the effect of MNTX on LPS-
126
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
induced endothelial cell barrier dysfunction in vivo was examined. Intravenous
injection of
LPS-induced endothelial cell-mediated vascular leakiness in mouse lung was
measured by
the protein concentration in bronchoalveolar lavage (BAL) fluid (Figure 17-8).
MNTX (10
mg/kg) alone did not affect pulmonary vascular permeability. However,
intravenous
injection of MNTX four hours after LPS delivery attenuated mouse pulmonary
hyper-
permeability (Figure 17-B).
In this Example, data is presented that shows that methylnaltrexone (MNTX),
a selective peripheral mu opioid receptor (mOP-R) antagonist, provided
protection from
agonist-induced endothelial cell barrier disruption through mOP-R-dependent,
and -
independent, mechanisms. The results indicate that S1P3 receptor
transactivation is an
important regulator of agonist-induced endothelial cell barrier disruption.
MNTX stimulated
mOP-R-independent receptor tyrosine phosphate mu (RPTP ) activity, which is
important in
inhibiting agonist-induced SIP3 receptor transactivation (Src-mediated
tyrosine
phosphorylation). MNTX exhibited clinical utility for the treatment of
diseases that involve
cell barrier disruption, such as diseases associated with endothelial cell
barrier dysfunction
like atherosclerosis and acute lung injury.
The mu opioid receptor antagonist, naloxone, is fairly lipid-soluble and
crosses the blood-brain barrier easily). Despite numerous attempts at
regulating doses, mOP-
R antagonists have proven unsuitable for patients receiving opiates for pain
management
because of analgesia reversal and breakthrough pain. MNTX is a quaternary
derivative of the
pure narcotic antagonist naltrexone. The addition of the methyl group to
naltrexone at the
amine in the ring forms the compound N-methylnaltrexone with greater polarity
and lower
lipid solubility. MNTX does not cross the blood-brain barrier and thus could
play a
therapeutic role in reversing the peripheral effects of opiates in palliative
care, especially for
patients taking high doses of opiates for analgesia. MNTX is expected to have
a clinical role
in the perioperative period, in the ICU (e.g., patients with burns), or with
advanced medical
illness. Because this population is most at risk for defects in cell barrier
function, particularly
pulmonary dysfiuzction, these work disclosed herein focused on MNTX rather
than the
tertiary opiate antagonists.
In previous studies of opiates and MNTX, the plasma concentrations of drugs
appeared to be well within the range of the effects disclosed in the in vitro
study. Peak
plasma concentrations of intravenous or intramuscular morphine in normal
therapeutic doses
are 80 ng/ml. In one comprehensive review, analgesia in cancer patients was
associated with
127
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
steady-state concentrations of morphine in plasma ranging from 6 to 364 ng/ml.
A meta-
analysis of dose-adjusted peak plasma concentrations of morphine revealed a
Cma, of 1-10
nM/L per mg of morphine, although there were some differences between single-
and
multiple-dosing and populations. Taken as a whole, the plasma concentration of
morphine
and MNTX in patients after parenteral or oral administration is consistent
with the levels that
regulated endothelial cell barrier function in the in vitro model. Similarly,
the concentrations
of MNTX in the in vitro study were similar to those achieved in clinical
trials of the drug. In
methadone maintenance patients who received mean doses of 0.1 mg/kg MNTX
intravenously, the mean plasma levels of MNTX were 162 ng/ml. After repeated
IV doses of
MNTX in volunteers, levels of MNTX in plasma were maintained well above the
range in
which we observed an effect on endothelial cell barrier function.
MNTX, but not naloxone, provided protection from both thrombin- and LPS-
induced endothelial cell barrier disruption. Thrombin induced rapid, transient
endothelial cell
barrier disruption through activation of PAR (Protease-Activated Receptors),
with consequent
Ca2+, RhoA and Ras/lMAP kinase signaling. In contrast, LPS induced a delayed
endothelial
cell barrier-disruptive response by activating a receptor complex of TLR4,
CD14 and MD2,
with consequent NF-xB activation and cytokine production. Considering the
contrasting
mechanisms of these agonists, MNTX is expected to provide cell barrier
protection
(including endothelial cell barrier protection) from a wide range of
disrupting agents.
It is known that SIP1 receptor transactivation (AKT-mediated threonine
phosphorylation) is a key component in agonist-induced endothelial cell
barrier enhancement.
In this Example, these findings have been extended to show that
transactivation (Src-
mediated tyrosine phosphorylation) of the S1P3 receptor played an important
role in agonist-
induced endothelial cell barrier disruption. S1P3 receptor signaling activated
the small G-
protein, RhoA, which is involved in actin cytoskeletal reorganization.
In agreement with these results, researchers have reported that inhibition of
Src protected from endothelial cell barrier disruption. Src regulates
endothelial cell
contraction and vascular permeability. Inhibition of Src stabilized a VEGF
receptor
2/cadheriri complex and reduced edema after myocardial infarction.
RPTP was established herein as playing an important role in regulating
endothelial cell barrier integrity. RPTPg is highly expressed in the lung
vasculature, where it
is localized to endothelial cell-cell junctions. Consistent with the results
disclosed herein,
128
CA 02609985 2007-11-27
WO 2007/053194 PCT/US2006/021604
researchers have shown that silencing RPTP expression in HPMVEC inhibited
barrier
function. RPTP can associate with various cell surface receptors, including
VE-cadherin,
N-cadherin, c-Met and the VEGF receptor. These findings were extended to show
that
RPTP regulated S1P3 receptor transactivation. RPTP further interacted with
signaling
molecules including IQGAPl, cdc42, RACKl, a-catenin, 0-catenin and PKCS.
The in vivo model of pulmonary vascular permeability demonstrated that
MNTX alone does not affect basal vascular integrity. However, MNTX attenuated
LPS-
induced vascular barrier disruption. These results are in agreement with the
protective effects
of MNTX on LPS-induced HPMVEC barrier disruption in vitro. Therefore, MTNX is
expected to be a useful therapeutic treatment (including preventative and
ameliorative
treatments) for diseases involving cell barrier disruption or dysfunction,
such as endothelial
cell barrier dysfunction.
Having thus described at least one embodiment of each of several aspects of
the
invention, it is to be appreciated that various alterations, modifications,
and improvements
will readily occur to those skilled in the art. Such alterations,
modifications, and
improvements are intended to be part of this disclosure, and are intended to
be within the
spirit and scope of the invention. Accordingly, the foregoing description and
drawings are by
way of example only.
129
DEMANDE OU BREVET VOLUMINEUX
LA PRESENTE PARTIE DE CETTE DEMANDE OU CE BREVET COMPREND
PLUS D'UN TOME.
CECI EST LE TOME 1 DE 2
CONTENANT LES PAGES 1 A 129
NOTE : Pour les tomes additionels, veuillez contacter le Bureau canadien des
brevets
JUMBO APPLICATIONS/PATENTS
THIS SECTION OF THE APPLICATION/PATENT CONTAINS MORE THAN ONE
VOLUME
THIS IS VOLUME 1 OF 2
CONTAINING PAGES 1 TO 129
NOTE: For additional volumes, please contact the Canadian Patent Office
NOM DU FICHIER / FILE NAME:
NOTE POUR LE TOME / VOLUME NOTE: