Note: Descriptions are shown in the official language in which they were submitted.
CA 02610792 2007-12-03
WO 2006/130994 PCT/CA2006/000962
1
Synthesis of glycerolipid carbamates and dicarbamates and their use as
antitumor
compounds
PRIOR APPLICATION INFORMATION
This application claims the benefit of US Provisional Application
60/689,065, filed June 10, 2005.
BACKGROUND OF THE INVENTION
Prostate cancer (PCa) is the most common male cancer and the
second leading cause of cancer deaths in males (Jemal et al., 2003, Cancer
Statistics. CA Cancer J. Clin. 53, 5-26). Although androgen deprivation is
initially
effective, it does not cure the disease, and invariably the tumor recurs in an
androgen-independent form that is resistant to classical chemotherapy, and
usually metastasizes primarily to the bone (Saitoh et al., 1984, Cancer 54,
3078-
3084; Jacobs, 1983, Urology 21, 337-344). The molecular events underlying the
progression of the disease have yet to be elucidated; however, it is clear
that once
the disease progresses it does not respond to the current array of
chemotherapeutic agents (Koutsilieras and Tolis, 1985, Prostate 7, 31-39).
There
is, therefore, a clear need to develop new agents that kill hormone-
independent
prostate tumor cells and/or prevent their metastases.
A group of antitumor compounds collectively known as antitumor
ether lipids (AELs) act by perturbing intracellular signaling pathways,
leading to the
killing of the cells (Arthur and Bittman, 1998, Biochim. Biophys. Acta 1390,
85-102;
Bittman and Arthur, 1999, In Liposomes: Rational Design, A. S. Janoff, Ed.,
Marcel
Dekker, New York, pp. 125-144). These compounds are long-lived analogs of the
naturally occurring phospholipid, lysophosphatidylcholine (LPC). Insertion of
two
4ether bonds into LPC in place of the usual two ester bonds gives an analog
that is
highly resistant to metabolism at sites other than in the vicinity of the
phosphodiester linkage. AELs have the potential to deliver antitumor activity
without any mutagenicity because, unlike many other anticancer agents, they do
not interact directly with DNA. They possess cell-selective effects by
inhibiting the
proliferation and killing of cancer cells at concentrations that do not affect
normal
cells (Berdel, 1991, Br. J. Cancer 64, 208-211; Samadder and Arthur, 1999,
CA 02610792 2007-12-03
WO 2006/130994 PCT/CA2006/000962
2
Cancer Res. 59, 4808-4815). The prototype or gold standard AEL is known as 1-
O-octadecyl-2-O-methyl-glycerophosphocholine (ET-18-OCH3), which inhibits a
broad panel of tumor cell lines (Berdel et al., 1985, in Phospholipids and
Cellular
Regulation, Kuo, J. F., ed., 'Vol. 2, pp 41-73, CRC Press, Boca Raton, FL;
Lohmeyer and Bittman, 1994, Drugs Future 19, 1021-1037; Houlihan et al., 1995,
Med. Res. Rev. 15, 157-223; Mollinedo et al., 2004, Curr. Med. Chem. 2004, 11,
3163-3184) but it exhibits no known selectivity against specific cancer cell
types.
SUMMARY OF THE INVENTION
According to a first aspect of the invention, there is provided a
compound having a formula selected from the group consisting of:
0II
H.N Y Y.NJIN,H
~ '
~O ~ H XO
R'O R? P'O~~N'Mej R'O~R-P O~,
NMe3
O p
X=OorNH X=OorNH
Y=MeorOH Y=MeorOH
R' = C12-C2e alkyl or C1Z-CZO alkenyl R' = C12-C20 alkyl or C1Z-C20 alkenyl
R2=0orCHZ R2=0orCH2
0 0
Y N, Y N.H H.NP- NHY H, N~NHY
HO OH ~ OH ~ HO OH ~ OH ~ x HO~ ~/~OR' H O~ ~iOR1 HO~ OR' HHOp ~~OR~
G G G C,
X, Y, and R' as above X, Y, and R' as above X, Y, and R1 as above X, Y, and R'
as above
Z=OorCHZ Z=OorCHZ Z=OorCHZ Z=OorCHZ
G = OH, H, or NH2 G= OH, H, or NH2 G= OH, H, or NH2 G= OH, H, or NH2
MeZN /
O
15J O C14H2g-n
MeO~H ~ +
0-P-OCH2CH2NMe3
O
or
0
I ~ ~ C14H29
Me2N ~ ~ ~Ik
O O
MeHN~O~H O +
O-P-OCH2CH2NMe3
O
According to a second aspect of the invention, there is provided the
use of a compound having a formula selected from the group consisting of:
CA 02610792 2007-12-03
WO 2006/130994 PCT/CA2006/000962
3
0I
H.N Y Y.NII~N.H
R~O R? P'0----N+Me3 R1O,,-,~,R,P O,~NMe3
H
0 0
~ X~0
11 O p
X=OorNH X=OorNH
Y=MeorOH Y=MeorOH
R' = C12-C20 alkyl or C12-C20 alkenyl R' = C72-CZO alkyl or C12-C20 alkenyl
RZ=OorCHZ RZ=OorCHZ
0 0
Y.N.H Y, N,H H, NJLNHY H' N JL
NHY
HO OH O~X OH O~X HO OH O~X OH O 'X
HO~Z~iOR' HHOZOR1 HO~Z~iOR~ HH
G G G G
X, Y, and R' as above X, Y, and R' as above X, Y, and R' as above X, Y, and R'
as above
Z=0orCH2 Z=0orCH2 Z=0orCH2 Z=0orCHZ
G = OH, H, or NH2 G = OH, H, or NH2 G = OH, H, or NH2 G = OH, H, or NH2
M02N ~ N~
' 0
Moo H a~n
- qG
O-P-OCHyCH=NMe3
O
or
0
( ~ ~ Ct4H29
MeZN ~ ~
O O
MeHNO~H 0 +
O-P-OCH2CH2NMe3
O
in preparation of a medicament for treating cancer.
According to a third aspect of the invention, there is provided a
method of treating cancer in a patient comprising administering to a patient
in need
of such treatment an effect amount of a compound having a formula selected
from
the group consisting of:
CA 02610792 2007-12-03
WO 2006/130994 PCT/CA2006/000962
4
0II
H, N Y Y.NJIN.H
,-,, 0 H X~O
RlO R? P'O~~N.Me3 R'O~R\P O~,
NiMe3 11 0 O
X=OorNH X=OorNH
Y=MeorOH Y=MeorOH
RI = C12-C20 alkyl or C12-C20 alkenyl R' = Ctz-Czo alkyl or C1Z-C20 alkenyl
RZ = 0 or CH2 RZ = 0 or CHZ
0 0
Y N. Y, N.H H, Nlk NHY H, Nlk NHY
HO O~-X OX HO O~X OH O~X
HO O ZOR' H O O ZOR' HO O Z~iOR' HH-" Z~/OR'
G G G ~G ~
X, Y, and R' as above X, Y, and R' as above X, Y, and R' as above X, Y, and R'
as above
Z=0orCHZ Z=0orCH2 Z = 0 or CH2 Z = 0 or CH2
G = OH, H, or NH2 G OH, H, or NH2 G= OH, H, or NH2 G = OH, H, or NH2
MeZN / ~
O
O GUHaSn
Me0 H ~ +
p-OCHyCHqNMe3
O
or o
c,4HZ9
MezN JCI?
O
MeHN'O+ H 0
I, +
O-P_ OCH2CH2NMe3
O
BRIEF DESCRIPTION OF THE DRAWINGS
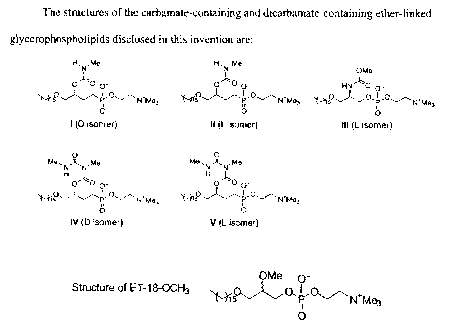
Figure 1 shows the structures of the carbamate lipids (compounds I,
II, and III) and dicarbamate lipids (compounds IV and V) of the invention. The
structure of the "gold standard"antitumor ether lipid ET-18-OCH3 is shown for
comparison.
Figure 2 is a graphical depiction of the results of an antiproliferative
evaluation of compound I against the following cancer cell lines: DU145, PC3,
BT549, HeLa, MDA-MB-231, A549, and MCF-7. Cells growing exponentially in 48-
well plates were incubated with different concentrations of compound I for 48
h.
The increases in cell numbers were determined by the CyQuant assay and are
expressed as a percentage of the increase in control wells with the vehicle
(0.1%
ethanol). The results are the average + SD from six different wells.
CA 02610792 2007-12-03
WO 2006/130994 PCT/CA2006/000962
Figure 3 is a graphical depiction of the results of an antiproliferative
evaluation of compound II against the following cancer cell lines: DU145, PC3,
BT549, HeLa, MDA-MB-231, A549, and MCF-7. Cells growing exponentially in 48-
well plates were incubated with different concentrations of compound II for 48
h.
5 The increases in cell numbers were determined by the CyQuant assay and are
expressed as a percentage of the increase in control wells with the vehicle
(0.1%
ethanol). The results are the average + SD from six different wells.
Figure 4 is a graphical depiction of the results of an antiproliferative
evaluation of compound III against the following cancer cell lines: DU145,
PC3,
BT549, HeLa, MDA-MB-231, A549, and MCF-7. Cells growing exponentially in 48-
well plates were incubated with different concentrations of compound III for
48 h.
The increases in cell numbers were determined by the CyQuant assay and are
expressed as a percentage of the increase in control wells with the vehicle
(0.1%
ethanol). The results are the average + SD from six different wells.
Figure 5 is a graphical depiction of the results of an antiproliferative
evaluation of compound IV against the following cancer cell lines: DU145, PC3,
BT549, HeLa, MDA-MB-231, A549, and MCF-7. Cells growing exponentially in 48-
well plates were incubated with different concentrations of compound IV for 48
h.
The increases in cell numbers were determined by the CyQuant assay and are
expressed as a percentage of the increase in control wells with the vehicle
(0.1%
ethanol). The results are the average + SD from six different wells.
Figure 6 is a graphical depiction of the results of an antiproliferative
evaluation of compound V against the following cancer cell lines: DU145, PC3,
BT549, HeLa, MDA-MB-231, A549, and MCF-7. Cells growing exponentially in 48-
well plates were incubated with different concentrations of compound V for 48
h.
The increases in cell numbers were determined by the CyQuant assay and are
expressed as a percentage of the increase in control wells with the vehicle
(0.1%
ethanol). The results are the average + SD from six different wells.
Figure 7 is a reaction scheme for the synthesis of carbamate
phosphonocholine (compound II) and dicarbamate phosphonocholine (compound
V) from 1, 3-O-benzylidine-1, 3,4-butanetriol.
Figure 8 is a reaction scheme for the synthesis of carbamate
phosphorylcholine (compound III) from 3-O-hexadecyl-sn-glycerol.
CA 02610792 2007-12-03
WO 2006/130994 PCT/CA2006/000962
6
Figure 9 shows the structure of 1-0-(7-N, N-dimethylamino-3-
pentadecanoyl-l-napthyl)-2-O-methyl-sn-glycero-3-phosphocholine.
Figure 10 shows the cytotoxic effect of HB40-6D on DU145 and PC3
cells. Cells were incubated with HB40-6D for 48 h in clear bottom black-coated
96-
well plates and the toxicity was determined by the Toxilight assay (Cambrex).
100% lysis of the cells at each concentration was obtained by adding the 100%
lysis reagent (Cambrex) to a parallel set of wells.
DESCRIPTION OF THE PREFERRED EMBODIMENTS
Unless defined otherwise, all technical and scientific terms used
herein have the same meaning as commonly understood by one of ordinary skill
in
the art to which the invention belongs. Although any methods and materials
similar
or equivalent to those described herein can be used in the practice or testing
of the
present invention, the preferred methods and materials are now described. All
publications mentioned hereunder are incorporated herein by reference.
We synthesized analogs of ET-18-OCH3 with a carbamate
(compounds I, II, and III) or a dicarbamate (compounds IV and V) moiety at the
C-
2 position instead of the OMe group in ET-18-OCH3 (shown in Figure 1). Four of
the analogs (compounds I, II, IV, and V) have a phosphonocholine head group at
the C-3 position instead of the phosphocholine as in ET-18-OCH3 and compound
III (Fig. 1). The phosphonate moiety was employed in order to resist the
action of
phospholipases, which are ubiquitous in cells and could cause the breakdown of
the compounds more rapidly than is desired. We also synthesized both the D
(compounds I and IV) and L (compounds li, 111, and V) stereoisomers of these
compounds.
Other substituents that can be made are a ureido analog with urea or
a hydroxureido analog with hydroxyurea at the C2 position. Additional analogs
are
carbamates in which the sn-3 phosphonocholine group is replaced with a C-
glycoside to yield glycosylated carbamates, and a carbamate with a naphthol
moiety at the C-1 position. A scheme that outlines the synthesis of these C-
glycosides and the naphthol analog appears herein. The figure below depicts
general structures for the compounds of the invention:
CA 02610792 2007-12-03
WO 2006/130994 PCT/CA2006/000962
7
0
H.N Y Y NAN.H
~0 H X~0
R'O R? P O'0 '/ NMe3 R'O,_,J-,,RP'O,/ NMey
0 ~
X=OorNH X=OorNH
Y=MeorOH Y=MeorOH
RI = C1y-Cyo alkyl or C12-C20 alkenyl R' = C12-C20 alkyl or C12-C20 alkenyl
RZ=OorCHp R2=0orCHz
0 0
Y, NH Y.N,H H,NJLNHY H'NxNHY
HO{ OH O~X OH O 20 OH O~X
HOC~Z~.jOR' HO0 Z,/'~i0R~ Z OR' HHO ~iORt
G G G G
X, Y, and R' as above X. Y, and R' as above X, Y, and R' as above X. Y, and R'
as above
Z=OorCHz Z=0orCHz Z=OorCHz Z=OorCH
z
G = OH, H, or NHZ G = OH, H, or NHZ G= OH, H, or NHZ G= OH, H, or NH2
Me.1N
~O
C,eHaa-
Me0 H ~
1
P-OCHzCIiZNMe3
0
or
0
C14H29
Me2N O
!OD?l
H O +
MeHN~O+O-P-OCH2CH2NMe3
O
We screened the compounds against a panel of human cancer cell
lines derived from a number of tissues. These were breast (MCF-7, BT549,
SKBr3, MDA-MB-231), lung (A549), cervix (HeLa), and prostate (DU145, PC3)
cancer cell lines. The results show that the compounds were very active
against
~the cell lines. In some cases, the IC50 values were much lower than values
reported for ET-18-OCH3, indicative of a greater degree of potency of these
new
compounds. The most striking observation is that the greatest inhibition of
cell
growth was found when the two prostate cell lines, DU145 and PC3, were treated
with these compounds. Compound I inhibited the proliferation of DU145 cells
with
an IC50 of 1 pM, and was toxic to the cells at a concentration of 2.5 pM.
Under
similar conditions, ET-18-OCH3 inhibited the proliferation of DU145 cells with
an
CA 02610792 2007-12-03
WO 2006/130994 PCT/CA2006/000962
8
IC50 value of 14 pM, and was not toxic even at a concentration of 30 pM
(Ashagbley, A. et al., 1966, Anticancer Res. 16, 1813-1818). (The IC50 value
refers
to the concentration at which the proliferation rate of the cells is inhibited
by 50%
while toxicity refers to the loss of viability of the cells.) When tested
against PC3
cells, another hormone-independent prostate cancer cell line derived from the
brain, compound I had an IC50 value of 1.2 pM, which is similar to that
observed in
DU145 cells (Fig. 2). These two prostate cancer cell lines are well-known
cellular
models of the hormone-insensitive stage of prostate cancer (Navone et al.,
1999,
Cancer Metastasis Rev. 17, 361-371; Sobel and Sadar, 2005, J. Urology 173, 342-
359), the most deadly form of the disease that does not respond to current
conventional chemotherapy. Because hormone-insensitive prostate cancer cells
are highly sensitive to the active compounds described herein, (compounds I,
II,
and III), we postulate that it may be possible to selectively kill prostate
cancer cells
with low concentrations without harming other cells in the body. The prostate
selectivity observed with compound I was also observed with compounds II and
III
(Table 1 and Figs. 3 and 4) and distinguishes these compounds from other
antitumor compounds.
In all of our studies, the carbamate- and dicarbamate-containing
phospholipid compounds were added to exponentially growing cells, and
incubation was for a period of only 48 h. The IC50 values of these compounds
are
impressive compared to the literature data in which established antitumor
compounds were added 24 h after cell seeding and drug incubations were for a
period of 72 h, with fresh addition of drug media every 24 h (Kreis et al.,
1997, Br.
J. Urol. 79, 196-202; Budman et al., 2002, Anti-Cancer Drugs 13, 1011-1016).
The results, which are displayed in Figures 2- 6 and Table 1, are:
The carbamate lipids (compounds I and II) were more active with
q
respect to blocking cancer cell growth than the corresponding dicarbamate
lipids
(compounds IV and V).
The D stereoisomers (compounds I and IV) were generally more
active than the corresponding L isomers (compound II and V) in blocking the
groinrth of the cancer cell lines tested.
Compound III showed the greatest prostate cancer cell selectivity,
followed by compounds I and II.
CA 02610792 2007-12-03
WO 2006/130994 PCT/CA2006/000962
9
The prostate selectivity was reduced when a second carbamate
moiety was introduced into the C2 position of the molecule (as in compounds IV
and V).
We have described procedures for the synthesis of ether glycerolipid
carbamates (compounds I, II and III) and dicarbamates (compounds IV and V).
We have determined that ether glycerolipid carbamates and
dicarbamates, compounds I, II, III, IV, and V, are useful in treating cancers
as they
possess both antiproliferative and cytotoxic effects.
We have determined that prostate cancer cell lines are particularly
sensitive to compounds I, II, and III relative to other cancer cell lines.
Therefore,
these compounds may provide a means for selectively treating prostate cancer
with minimal effects on other cells.
Similarly, 1-0-(7-N, N-dimethylamino-3-pentadecanoyl-l-napthyl)-2-
O-methyl-sn-glycero-3-phosphocholine.
Me2N
O
0 C14H29-n
MeO H 4 +
O-P-OCH2CH2NMe3
0
was added to proliferating cells at 0-20 pM for 48 hours, as shown in Table 2.
As
can be seen, this naphthol-alkyllysophospholipid analog is also highly
selective for
prostate cancer cells. Specifically, proliferating cells in 48-well plates
were
incubated with 0-20 pM of the compound for 48 hours. The cell numbers present
were determined by the CyQuantTM assay (Invitrogen), and the increase in
numbers were expressed relative to the controls receiving only the vehicle
(0.1%
ethanol).
The present invention also provides pharmaceutical compositions
comprising as active ingredients an effective amount of one or more of the
above-
CA 02610792 2007-12-03
WO 2006/130994 PCT/CA2006/000962
described carbamate lipids and dicarbamate lipids for use in treating cancer
in
general and selective treatment of prostate cancer. As used herein, an
"effective
amount" refers to an amount that is sufficient to inhibit proliferation of
cancerous
cells, for example, prostate cancer cells. As will be appreciated by one of
skill in
5 the art, suitable amounts and concentrations are described herein but will
of
course depend upon the mode of administration of the pharmaceutical
composition, the age, weight and general condition of the patient among other
factors.
In some embodiments, the carbamate lipids and dicarbamate lipids,
10 at concentrations or dosages discussed above, may be combined with a
pharmaceutically or pharmacologically acceptable carrier, excipient or
diluent,
either biodegradable or non-biodegradable. Exemplary examples of carriers
include, but are by no means limited to, for example, poly(ethylene-vinyl
acetate),
copolymers of lactic acid and glycolic acid, poly(lactic acid), gelatin,
collagen
matrices, polysaccharides, poly(D,L lactide), poly(malic acid),
poly(caprolactone),
celluloses, albumin, starch, casein, dextran, polyesters, ethanol,
methacrylate,
polyurethane, polyethylene, vinyl polymers, glycols, and mixtures thereof and
the
like. Standard excipients include gelatin, casein, lecithin, gum acacia,
cholesterol,
tragacanth, stearic acid, benzalkonium chloride, calcium stearate, glyceryl
monostearate, cetostearyl alcohol, cetomacrogol emulsifying wax, sorbitan
esters,
polyoxyethylene alkyl ethers, polyoxyethylene castor oil derivatives,
polyoxyethylene sorbitan fatty acid esters, polyethylene glycols,
polyoxyethylene
stearates, colloidol silicon dioxide, phosphates, sodium dodecylsulfate,
carboxymethylcellulose calcium, carboxymethylcellulose sodium,
methylcellulose,
hydroxyethylcellulose, hydroxypropylcellulose, hydroxypropylmethycellulose
phthalate, noncrystalline cellulose, magnesium aluminum silicate,
triethanolamine,
polyvinyl alcohol, polyvinylpyn-olidone, sugars and starches. See, for
example,
Remington: The Science and Practice of Pharmacy, 1995, Gennaro ed.
As will be apparent to those knowledgeable in the art, specific
carriers and carrier combinations known in the art may be selected based on
their
properties and release characteristics in view of the intended use.
Specifically, the
carrier may be pH-sensitive, thermo-sensitive, thermo-gelling, arranged for
sustained release or a quick burst. In some embodiments, carriers of different
CA 02610792 2007-12-03
WO 2006/130994 PCT/CA2006/000962
11
classes may be used in combination for multiple effects, for example, a quick
burst
followed by sustained release.
As discussed herein, the selectivity of the compounds for prostate
cancer lines is more surprising than the observed general antitumor
properties.
The latter is somewhat less surprising because the compound shares some
similarities to ET18-OCH3. Nevertheless the bulkiness of the moieties we have
inserted at the C-2 position makes the activity surprising since ET-18-OCH3-
like
compounds generally have a very small moiety at this position.
While not wishing to be bound to a specific hypothesis, the inventors
believe that the compounds described herein act to inhibit intracellular
pathways
that are essential for proliferation and/or activate pathways that lead to
cell death
via apoptosis or autophagy. The prostate selectivity may result from (A) a
greater
extent of accumulation of the carbamates, dicarbamates and ureido compounds as
a result of greater uptake and/or decreased metabolism in prostate cancer cell
lines relative to non prostate cells, (B) a high affinity of the carbamates,
dicarbamates, and ureido compounds for receptor-type molecules that are
preferentially found in prostate cancer cells, (C) a greater dependence of the
prostate cancer cells on pathways perturbed by the compounds for survival and
proliferation compared to non-prostate cancer lines."
The procedures for preparing these compounds and the spectral
data for characterizing their structures are as follows.
Procedures for the Syntheses of Carbamate Phosphonocholine (Compound II) and
Dicarbamate Phosphonocholine (Compound V)
As outlined in Figure 7, the preparation of carbamate
phosphonocholine (compound II) and dicarbamate phosphonocholine (compound
V) started with 3(S)-1,3-O-benzylidine-1,3,4-butanetrioi (2) (Yang et al.,
1999).
Alkylation of alcohol 2 with sodium hydride and 1-bromohexadecane under phase-
transfer conditions gave ether 3 in 91% yield. A benzylidine acetal of the 1,3-
dioxane type can undergo ring opening by treatment with N-bromosuccinimide
(NBS) to form a O-benzoyl bromohydrin (Hanessian and Plessas, 1969); thus
reaction of 3 with NBS in the presence of BaCO3 provided bromohydrin 4 in 87%
yield. Arbuzov reaction of bromohydrin 4 with P(OEt)3 at 150 C gave benzoyl
CA 02610792 2007-12-03
WO 2006/130994 PCT/CA2006/000962
12
phosphonate 5 in 79% yield. Base-induced hydrolysis of the benzoyl group of
phosphonate 5 provided hydroxyphosphonate 6 in 86% yield. During the
deprotection of the benzoate group by methanolysis the ethyl groups in
phosphonate 5 were converted to methyl groups by transesterification.
Hydroxyphosphonate 6 was reacted with methyl isocyanate, which was generated
in situ by the reaction of methyl iodide with KNCO in tetrahydrofuran/N,N-
dimethylformamide (THF/DMF) (10:1 v/v), furnishing a mixture of carbamate 7
and
carbamate 8 in 11 % and 53% yields, respectively. When the amount of DMF was
increased, the rate of carbamate formation was faster but the yield of
carbamate 7
decreased. The methyl groups in the phosphonate ester were removed by
treatment of 7 or 8 with trimethylsilyl bromide in CH2CI2, and a choline group
was
introduced by coupling of the resulting phosphonic acid with choline tosylate
promoted by trichloroacetonitrile in pyridine.
Procedures for the Syntheses of Carbamate Phosphonocholine (Compound I) and
Dicarbamate Phosphonocholine (Compound IV).
The enantiomer of compound II (compound I) and the enantiomer of
compound V (compound IV) were prepared starting from 3(R)-1,3-O-benzylidine-
1,3,4-butanetriol by using procedures similar to those described above.
Detailed Experimental Procedures for the Preparations of Compounds I, II, IV,
and
V.
General Information. 'H and 13C NMR spectra were recorded at 400
and 100 MHz, respectively, and were referenced to the residual chloroform at b
7.24 (1H) and 6 77.00 ppm (13C). Optical rotations were measured in a cell of
1-
dm pathiength on a digital polarimeter. TLC was carried out on aluminum-backed
silica gel GF plates (250-pm thickness), and the compounds were visualized by
charring with 10% sulfuric acid in ethanol and/or short wavelength UV light.
For
flash chromatography, silica gel 60 (230-400 ASTM mesh) was used. THF was
distilled from sodium and benzophenone before use. Pyridine, DMF, 1,2-
dichlorethane, EtN(Pr-/)z, and CH2CI2 were dried over CaHZ.
Preparation of (3S)-4-O-hexadecyl-1, 3-O-benzylidine-1,3,4-
butanetriol (3). To a suspension of 1.60 g (40.0 mmol) of NaH (60% in white
oil,
CA 02610792 2007-12-03
WO 2006/130994 PCT/CA2006/000962
13
washed twice with dry hexane) in 100 mL of dry THF was added 3.89 g (20.0
mmol) of (3S)-1,3-O-benzylidine-1,3,4-butanetriol (2) in 20 mL of THF at 0 C.
After the evolution of hydrogen had stopped, 6.2 mL (20.2 mmol) of 1-
bromohexadecane and 0.67 g (2.0 mmol) of n-Bu4NBr were added. The mixture
was stirred for 24 h at room temperature, and then the reaction was quenched
with
mL of MeOH. The volatile solvents were removed under reduced pressure, and
the residue was diluted with 200 mL of Et20 and washed with water. The organic
layer was dried over Na2SO4 and concentrated under reduced pressure to give a
residue. The residue was purified by column chromatography on silica gel
(elution
10 with hexane/EtOAc 25:1), affording 7.62 g(91 !0) of (3S)-4-O-hexadecyl-1,3-
0-
benzylidine-1,3,4-butanediol (3) (Yang et al., 1999, Org. Lett. 1, 2149-2151)
as a
white solid; mp 54-55 C; 'H NMR (CDCI3) b 0.88 (t, 3H, J = 6.6 Hz), 1.25 (br
s,
26H), 1.54-1.60 (m, 3H), 1.80-1.91 (m, 1 H), 3.44-3.52 (m, 3H), 3.62 (dd, 1H,
J=
5.8, 10.3 Hz), 3.95-4.01 (m, 1 H), 4.01-4.09 (m, 1 H), 4.29 (dd, 1 H, J = 4.1,
11.4
Hz), 5.53 (s, 1 H), 7.31-7.38 (m, 3H), 7.49 (dd, 2H, J = 1.6, 7.9 Hz); 13C NMR
(CDCI3) 6 14.1, 22.7, 26.1, 28.3, 29.3, 29.5, 29.6, 29.7, 30.9, 31.9, 66.9,
71.9,
73.7, 76.3, 101.2, 126.1, 128.2, 128.7, 129.0, 129.7, 138.6; MS (electrospray)
MH+
m/z calcd for C27H4703 419.35, found 419.2.
Preparation of 3(R)-4-O-hexadecyl-1,3-O-benzylidine-1,3,4-
butanetriol. The enantiomer of 3 was prepared in 90% yield from (3R)-1,3-0-
benzyfidine-1,3,4-butanetriol by the procedure described above.
Preparation of 3(S)-benzoyl-4-hexadecyloxy-l-bromobutane (4). A
mixture of 5.19 g (12.4 mmol) of (3S)-4-O-hexadecyl-1,3-O-benzylidine-1,3,4-
butanediol (3), 2.65 g (14.9 mmol) of NBS, and 1.10 g (5.57 mmol) of BaCO3 in
100 mL of CCI4 (or CICH2CH2CI) was heated at reflux for 4 h. The reaction
mixture
was passed through a pad of silica gel, which was rinsed with 100 mL of
hexane/EtOAc (10:1). The filtrate was concentrated to give a residue, which
was
purified by column chromatography on silica gel (elution with hexane/EtOAc
25:1)
to provide 5.36 g (87%) of bromide 4 as a pale yellow oil; [a]25p -24.9 (c
6.7,
CHCI3); 'H NMR (CDCI3) b 0.88 (t, 3H, J = 6.6 Hz), 1.26 (br s, 26H), 1.50-1.56
(m,
2H), 2.31-2.41 (m, 2H), 3.42-3.50 (m, 4H), 3.62-3.65 (m, 2H), 5.35-5.41 (m,
1H),
7.42-7.46 (m, 2H), 7.54-7.59 (m, 1 H), 8.05 (dd, 2H, J = 1.4, 8.4 Hz); 13C NMR
(CDCI3) 5 14.1, 22.6, 26.0, 28.7, 29.3, 29.4, 29.5, 29.6, 31.9, 34.5, 71.2,
71.67,
CA 02610792 2007-12-03
WO 2006/130994 PCT/CA2006/000962
14
71.72, 128.3, 129.6, 130.0, 133.0, 165.9; MS (electrospray) MH+ m/z calcd for
C27H46BrO3 497.26, found 497.2.
Preparation of 3(R)-benzoyl-4-hexadecyloxy-l-bromobutane. The
enantiomer of bromide 4 was prepared in 85% yield from 3(R)-4-O-hexadecyl-1,3-
O-benzylidine-1,3,4-butanediol by the procedure described above; [a]25p +20.5
(c
6.9, CHCI3).
Preparation of diethyl 3(S)-benzoyl-4-hexadecyloxy-l-
butanephosphonate (5). A solution of 4.98 g (10.0 mmol) of bromide 4 in 25 mL
(150 mmol) of triethyl phosphite [(EtO)3P] was heated at 150 C (oil bath
temperature) overnight. After the excess of (EtO)3P was removed by using a
stream of air, the residue was purified by column chromatography on silica gel
(elution with CHCI3/MeOH 25:1) to give 4.39 g (79%) of benzoyl phosphonate 5
as
a colorless oil; [a]25o -6.88 (c 5.8, CHCI3); 'H NMR (CDCI3) 6 0.88 (t, 3H, J
= 6.7
Hz), 1.26 (br s, 26H), 1.28-1.33 (m, 6H), 1.70-2.00 (m, 2H), 3.43-3.62 (m,
4H),
4.06-4.12 (m, 4H), 5.23-5.27 (m, 1 H), 7.42-7.46 (m, 2H), 7.54-7.59 (m, 1H),
8.04
(d, 2H, J = 7.0 Hz); 13C NMR (CDCI3) 6 14.1, 16.4 (d, J= 5.8 Hz), 22.2 (d, J =
143.2 Hz), 22.6, 24.3 (d, J = 4.0 Hz), 29.3, 29.4, 29.6, 29.7, 31.9, 61.6,
71.3, 71.7,
72.8 (d, J = 18.1 Hz), 127.7, 128.3, 129.6, 130.1, 133.0, 166.0; MS
(electrospray)
MH+ mlz calcd for C3jH5606P 555.39, found 555.3.
Preparation of diethyl 3(R)-benzoyl-4-hexadecyloxy-l-
butanephosphonate. The enantiomer of benzoyl phosphonate 5 was prepared in
80% yield from 3(R)-benzoyl-4-hexadecyloxy-l-bromobutane by the procedure
descried above; [a]25o +6.71 (c 6.0, CHCI3).
Preparation of dimethyl 4-hexadecyloxy-3(S)-hydroxy-l-
butanephosphonate (6). To 100 mL of dry MeOH was added 0.18 g (7.83 mmol)
of sodium metal. After the sodium metal had completely disappeared, a solution
of
3.91 g (7.05 mmol) of benzoyl phosphonate 5 in 10 mL of dry MeOH was added.
After the mixture was stirred overnight, the reaction was quenched by addition
of
500 NL (8.73 mmol) of AcOH and then concentrated under reduced pressure to
give a residue. The residue was purified by column chromatography on silica
gel
(elution with CHCI3IMeOH 10:1) to provide 2.56 g (86%) of hydroxy phosphonate
6
as a colorless oil; [a]25o -5.20 (c 5.0, C6H6); 'H NMR (CDCI3) 6 0.88 (t, 3H,
J = 6.8
Hz), 1.26 (br s, 26H), 1.54-1.58 (m, 2H), 1.70-1.90 (m, 4H), 2.78 (br s, 1H),
3.29
CA 02610792 2007-12-03
WO 2006/130994 PCT/CA2006/000962
(dd, 1H, J = 7.1, 9.4 Hz), 3.40-3.48 (m, 3H), 3.74 (d, 6H, J=10.8 Hz), 3.70-
3.85
(m, 1 H); 13C NMR (CDCI3) b 14.0, 20.7 (d, J = 142.1 Hz), 22.6, 26.0, 26.1 (d,
J =
4.7 Hz), 29.3, 29.4, 29.5, 29.6, 31.9, 52.3, 69.8 (d, J = 14.8 Hz), 71.6,
74.4; MS
(electrospray) MH+ m/z calcd for C22H4805P 423.3, found 423.3.
5 Preparation of dimethyl 4-hexadecyloxy-3(R)-hydroxy-1-
butanephosphonate. The enantiomer of hydroxy phosphonate 6 was prepared in
88% yield from diethyl 3(R)-benzoyl-4-hexadecyloxy-1-butanephosphonate by the
procedure described above; [a]25p +5.22 (C 5.1, C6H6).
Preparation of dimethyl 4-hexadecyloxy-3(S)-N-methylcarbamoyl-l-
10 butanephosphonate (7) and dimethyl 4-hexadecyloxy-3(S)-[N-(N'-
methylcarbamoyl)-N-methylcarbamoyl]-1-butanephosphonate (8). To a mixture of
8.11 g (100 mmol) of potassium cyanate, 3.22 g (10 mmol) of n-Bu4NBr, and 2.12
g (5.02 mmol) of hydroxy phosphonate 6 in 25 mL of 10:1 THF/DMF were added
3.2 mL (51.4 mmol) of methyl iodide and 1.8 mL (10.4 mmol) of (i-Pr)2NEt. The
15 mixture was stirred until hydroxy phosphonate 6 was completely consumed, as
indicated by TLC. The reaction mixture was diluted with CHCI3 and washed with
brine solution. The organic layer was dried over Na2SO4 and concentrated. The
product was purified by column chromatography on silica gel (elution with
CHCI3,
and then with 100:1 (v/v) CHC13/MeOH, 50:1 (v/v) CHCI31MeOH, 25:1 (v/v)
CHCI3/MeOH) to give 265 mg (11%) of carbamoyl phosphonate 7 and 1.43 g
(53%) of dicarbamoyl phosphonate 8 as colorless oils: Spectral data for
compound
7: [a]25p -5.82 (c 5.5, CHCI3); 'H NMR (CDCI3) b 0.88 (t, 3H, J = 6.4 Hz),
1.26 (br
s, 26H), 1.48-1.62 (m, 2H), 1.15-2.00 (m, 4H), 2.87 (d, 3H, J = 4.8 Hz), 3.25-
3.50
(m, 6H), 3.75 (d, 6H, J = 10.8 Hz), 4.90-5.00 (m, 1H), 7.60-7.70 (br s, 1 H);
13C
NMR (CDCI3) b 14.1, 20.7 (d, J = 151 Hz), 22.6, 24.0 (d, J = 5.0 Hz), 26.0,
26.1,
26.4, 29.3, 29.4, 29.5, 29.6, 31.9, 52.4, 67.8, 74.4 (d, J= 6.0 Hz), 153.7;
31P NMR
9
(CDCI3) b 35.5; MS (electrospray) MH+ m/z calcd for C24H51NO6P 480.3, found
480.3. Spectral data for compound 8: [a]25p -4.70 (c 5.6, CHCI3); 'H NMR
(CDCI3)
b 0.88 (t, 3H, J = 6.0 Hz), 1.25 (br s, 26H), 1.50-1.62 (m, 2H), 1.75-1.86 (m,
2H),
1.90-2.10 (m, 2H), 2.87 (d, 3H, J= 4.4 Hz), 3.22 (s, 3H), 3.42-3.52 (m, 4H),
3.51 (t,
2H, J = 5.2 Hz), 3.75 (d, 6H, J = 10.8 Hz), 4.90-5.00 (m, 1 H), 8.40-8.50 (br
s, 1 H);
13C NMR (CDCI3) b 14.1, 20.7 (d, J = 143 Hz), 22.4, 24.0 (d, J = 4.3 Hz),
26.0,
27.1, 29.3, 29.4, 29.5, 29.6, 30.7, 31.9, 52.5, 71.1, 71.7, 75.0 (d J = 17.5
Hz),
CA 02610792 2007-12-03
WO 2006/130994 PCT/CA2006/000962
16
155.2, 155.9; 31P NMR (CDCI3) 6 33.6; MS (electrospray) MH+ m/z calcd for
C26H54N207P 537.37, found 537.3.
Preparation of dimethyl 4-hexadecyloxy-3(R)-N-methylcarbamoyl-l-
butanephosphonate dimethyl and 4-hexadecyloxy-3(R)-[N-(N' methylcarbamoyl)-
N-methylcarbamoyl]-1-butanephosphonate. The enantiomers of carbamoyl
phosphonate 7 and dicarbamoyl phosphonate 8 were prepared in 12% and 50%
yields, respectively, from 4-hexadecyloxy-3(R)-hydroxy-1-butanephosphonate by
the procedure described above. Spectral data for the enantiomer of 7: [a]25p
+5.49 (c 5.6, CHCI3); MS (electrospray) MH+ mlz calcd for C24H51N06P 480.3,
found 480.3. Spectral data for the enantiomer of 8: [a]25o +4.55 (c 5.7,
CHCI3);
MS (electrospray) MH+ m/z calcd for C26H54N207P 537.37, found 537.3.
Preparation of 2'-(trimethylammonio)ethyl 4-hexadecyloxy-3(S)-N-
methylcarbamoyl-l-butane-phosphonate (Compound II). To a solution of 512 mg
(1.07 mmol) of carbamoyl phosphonate 7 in 25 mL of CH2CI2 was added 500 pL
(3.79 mmol) of trimethylsilyl bromide. After the mixture was allowed to stand
overnight at room temperature, the volatile materials were removed under
reduced
pressure to give a residue. To the residue was added 1.45 g (3.01 mmol) of
choline tosylate, and the mixture was dried overnight under high vacuum. After
the
dry mixture was dissolved in 50 mL of pyridine, 1.5 mL (15.0 mmol) of
trichloroacetonitrile was added, and the reaction mixture was heated for 48 h
at
50 C (oil bath temperature). On removal of most of the pyridine by rotary
evaporation, a brown residue was formed, which was dissolved in THF/H20 (10
mL, 9:1 v/v) and passed through a column of TMD-8 resin (previously
equilibrated
with the same solvent mixture). The product was purified by silica gel
chromatography (elution with CHCI3/MeOH/H20 65:25:4 vlvlv). The fractions
containing the product (as identified by TLC) were pooled and concentrated
under
reduced pressure. The residue was dissolved in CHCI3 (15-25 mL) and passed
through a Cameo filter three times to remove the suspended silica gel. The
filtrate
was concentrated to give a residue, which was lyophilized from benzene to
afford
395 mg (69%) of phosphonate 1a as a white powder; [a]25p -2.53 (c 0.21,
CHCI3/MeOH 1:1); 'H NMR (CDCI3/CD3OD) 6 0.89 (t, 3H, J = 6.4 Hz), 1.26 (br s,
26H), 1.32-1.35 (m, 2H), 1õ44-1.62 (m, 4H), 1.86-1.95 (m, 2H), 2.86 (d, 3H, J
= 4.8
Hz), 3.24 (s, 9H), 3.30-3.70 (m, 8H), 4.90-5.00 (m, 1H); 13C NMR (CDCI3) 6
14.1,
CA 02610792 2007-12-03
WO 2006/130994 PCT/CA2006/000962
17
20.7 (d, J = 143 Hz), 22.6, 25.1 (d, J = 4.1 Hz), 26.0, 26.1, 26.4, 29.3,
29.4, 29.5,
29.6, 31.9, 56.5 (d, J= 4.7 Hz), 65.8, 70.6 (d, J = 5.8 Hz), 75.4, 75.5, 74.9,
153.7;
MS (electrospray) M+ m/z calcd for C27H58N206P 537.37, found 537.4.
Preparation of 2'-(trimethylammonio)ethyl 4-hexadecyloxy-3(R)-N-
methylcarbamoyl-l-butanephosphonate (Compound I). The enantiomer of
compound li was prepared in 66% yield from dimethyl 4-hexadecyloxy-3(R)-N-
methylcarbamoyl-l-butanephosphonate by the procedure described above; [a]25p
.+2.44 (c 0.21, CHCI3/MeOH 1:1); MS (electrospray) M+ mlz calcd for
C27H58N206P 537.37, found 537.3.
Preparation of 2'-(tri methylam m on i o) ethyl 4-hexadecyloxy-3(S)-[N-
(N =methylcarbamoyl)-N-methylcarbamoyl]-1-butanephosphonate (Compound V).
Compound V was prepared in 71% yield from dimethyl 4-hexadecyloxy-3(S)-[N-
(N' methylcarbamoyl)-N-methylcarbamoyl]-1-butanephosphonate by the procedure
described above; [a]25o -2.07 (c 0.23, CHCI3/MeOH 1:1); ' H NMR
(CDCI3/CD3OD) 6 0.89 (t, 3H, J 6.4 Hz), 1.26 (br s, 26H), 1.44-1.62 (m, 4H),
1.86-1.95 (m, 2H), 2.87 (d, 3H, J 4.0 Hz), 3.24 (s, 3H), 3.26 (s, 9H), 3.35-
3.70
(m, 8H), 4.95-5.05 (m, 1H); 13C NMR (CDCI3) 5 14.1, 20.7 (d, J = 143 Hz),
22.6,
25.1 (d, J = 4.1 Hz), 26.0, 26.1, 26.4, 29.3, 29.4, 29.5, 29.6, 31.9, 56.5 (d,
J = 4.7
Hz), 65.8, 70.6 (d, J = 5.8 Hz), 75.4, 75.5, 74.8, 155.4, 156.1; MS
(electrospray)
M+ m/z calcd for C29H61 N307P 594.4, found 594.3.
Preparation of 2'-(trimethylammonio)ethyl 4-hexadecyloxy-3(R)-[N-
(N'-methylcarbamoyl)-N-methylcarbamoyl]-1-butanephosphonate (Compound IV).
The enantiomer of compound V was prepared in 68% yield from dimethyl 4-
hexadecyl oxy-3(R)-[N- (N ' methylcarbamoyl)-N-methylcarbamoyl]-1-
butanephosphonate by the procedure described above; [a]25p +1.98 (c 0.21,
CHCI31MeOH 1:1); MS (electrospray) M+ m/z calcd for C29H61 N307P 594.4, found
594.3.
References cited in the experimental procedures above:
1. Yang, G.; Franck, R. W.; Byun, H.-S.; Bittman, R.; Samadder, P.; Arthur, G.
Org.
Left. 1999, 1, 2149-2151.
2. Hanessian, S.; Plessas, N. R. J. Org. Chem. 1969, 34, 1035-1044.
CA 02610792 2007-12-03
WO 2006/130994 PCT/CA2006/000962
18
Procedures for the Synthesis of Carbamate Phosphorylcholine (Compound III)
As depicted in Figure 8, the preparation of carbamate
phosphorylcholine (compound III) started with 3-O-hexadecyl-sn-glycerol (2)
(van
Boeckel et al., 1982). Regioselective and stereospecific azidation of glycerol
2
with azidotrimethylsilane (Me3SiN3) under Mitsunobu conditions gave azido
alcohol
3 in 76% yield (He et al., 1999). Insertion of the phosphocholine head group
into
alcohol 3 provided 2-azidophosphorylcholine 5 in 69% overall yield by the
following
sequence of reactions: phosphorylation of alcohol 3 with 2-chloro-2-oxo-1,3,2-
dioxaphospholane, ring opening of phospholane intermediate 4 with
trimethylsilyl
bromide, and quaternization of the resulting ring-opened product with aqueous
trimethylamine. Finally, reduction of the azido group of 5 by catalytic
hydrogenation followed by reaction of amine 6 with methyl chloroformate
furnished
carbamate phosphorylcholine (compound III) (85% overall yield from 5).
Detailed Experimental Procedures for the Preparation of Compound 111.
2(S)-Azido-3-O-hexadecyl-1,3-propanediol (3). To a solution of 3.17 g (10.0
mmol) of 3-O-hexadecyl-sn-glycerol (2) and 3.42 g (13.0 mmol) of Ph3P in 180
mL
of CHZCI2 was added 3.2 mL (15 mmol) of diisopropyl azodicarboxylate (DIAD) at
0 C. After the mixture was stirred for 3 h under nitrogen, Me3SiN3 was added.
The mixture was stirred at the same temperature for 3 h, and then at room
temperature until glycerol 2 had reacted completely. The reaction mixture was
concentrated to give a yellow residue, which was dissolved in a minimum volume
of CH2CI2 and passed through a pad of silica gel in a sintered glass funnel.
The
pad was rinsed with hexane/EtOAc (50:1) until the excess yellow DIAD began to
elute. After concentration of the eluted silyloxy azide, the residue was
dissolved in
mL of THF and treated with 25 mL of a 1 M(n-Bu)~NF solution in THF. The
mixture was stirred at room temperature until all of the silyloxy azides were
consumed completely, and then was diluted with 250 mL of Et20 and washed with
water and brine. The organic layer was separated, dried over Na2SO4, and
30 concentrated The crude product was purified by column chromatography on
silica
gel (elution first 150 mL of 50:1 hexane/EtOAc and then with 6:1 hexane/EtOAc)
to
give 2.60 (76%) of azido alcohol 3 as a white solid: mp 37-39 C, (Ponpipom
and
Bugianesi, 1984). 38-29 C; [a]25o +14.5 (c 1.0, CHCI3), (Ponpipom and
Bugianesi
CA 02610792 2007-12-03
WO 2006/130994 PCT/CA2006/000962
19
1984. [a]Z'o +14.1 (c 1.0, CHCI3); ' H NMR (CDCI3) b 0.88 (t, 3H, J = 6.4
Hz), 1.26
(s, 26H), 1.52-1.62 (m, 2H), 2.19 (br s, 1 H), 3.47 (t, 2H, J = 6.8 Hz), 3.55-
3.63 (m,
4H), 3.65-3.80 (m, 1H);13C NMR (CDCI3) b 14.1, 22.7, 26.0, 29.3, 29.4, 29.5,
29.6,
29.7, 31.9, 62.3, 63.0, 70.9, 71.9.
Preparation of 2'-(trimethylammonio)ethyl 2(S)-azido-3-
hexadecyloxypropanephosphate (5).
To a solution of 342 mg (1.00 mmol) of 2(S)-azido-3-O-hexadecyl-1,3-
propanediol
(3) and 390 mg (3.02 mmol) of EtN(Pr-i)2 in 20 mL of CH2CI2 was added 250 NL
(2.72 mmol) of 2-chloro-1,3,2-dioxaphospholane-2-oxide at 0 C. After azido
alcohol 3 was consumed completely, Me3SiBr (1.0 mL, 7.6 mmol) was added at
0 C to carry out the ring-opening reaction of the phospholane. After the
mixture
was stirred for 2 h at room temperature, the volatile material was removed
under
reduced pressure to give a residue. The residue was dissolved in 40 mL of
0.9:1.5:1.5 (vlvlv) CHCI3/MeCN/2-PrOH and treated with 40 mL of 40% aqueous
Me3N solution for 2 days at room temperature. After concentration under
reduced
pressure, the residue was purified by column chromatography on silica gel
(elution
with CHCI3/MeOH/H20 65:25:4 v/v/v) to give 350 mg (69%) of
azidophosphorylcholine 5 as a white solid: [a]25p -4.3 (c 0.11, CHCI3),
[a]25D -4 5
(c 1.0, CHCI3); 'H NMR (CDCI3/CD3OD) 6 0.88 (t, 3H, J = 6.4 Hz), 1.26 (s,
26H),
1.50-1.60 (m, 2H), 3.30 (s, 9H), 3-43-3.65 (m, 4H), 3,25-3.50 (m, 3H), 3.90-
4.10
(m, 2H), 4.35-4.45 (m, 2H); 13C NMR (CDCI3/CD3OD) 6 13.6, 22.3, 25.6, 29.0,
29.1, 29.2, 29.3, 31.6, 44.4, 53.9, 59.5 (d, J= 5.0 Hz), 60.7 (d, J= 7.0 Hz),
65.7 (d,
J = 5.0 Hz), 69.6, 71.7; 31P NMR (CDCI3/CD3OD) 6 -2.03.
Preparation of 2'-(trimethylammonio)ethyl 2(S)-(N-methoxycarbonylamido)-3-
4
hexadecyloxypropanephosphate (III).
A mixture of 102 mg (0.20 mmol) of azidophosphorylcholine 5 and Pd/C (30 mg)
in
EtOH was stirred overnight under hydrogen atmosphere. After the catalyst was
removed by filtration, the filtrate was concentrated under reduced pressure to
give
crude amine 6. To a solution of vacuum-dried amine 6 in 10 mL of alcohol-free
CHCI3 were added 60 pL (0.44 mmol) of Et3N and 30 pL (0.39 mmol) of methyl
chloroformate at 0 C. The mixture was stirred overnight at room temperature
and
CA 02610792 2007-12-03
WO 2006/130994 PCT/CA2006/000962
then concentrated under reduced pressure to give a residue. The residue was
purified by column chromatography on silica gel (elution with CHCI3/MeOH/H20
65:25:4 v/v/v) to give 93 mg (85%) of carbamate phosphorylcholine III as a
white
solid: [a]25D -3.9 (c 0.10, CHCI3); 'H NMR (CDCI3) 6 0.88 (t, 3H, J = 6.4
Hz), 1.26
5 (s, 26H), 1.50-1.60 (m, 2H), 3.43 (s, 9H), 3-40-3.75 (m, 4H), 3.67 (s, 3H),
3,90-
4.30 (m, 5H), 4.60-4.80 (m, 2H), 6.51 (br s, 1 H); 31 P NMR (CDCI3) 6 -2.61.
References for the experimental procedures for preparation of compound III:
1. van Boeckel, C. A. A.; van del Marel, G. A.; Westerduin, P.; van Boom, J.
H.
10 Synthesis 1982, 399-402.
2. He, L.; Wanunu, M.; Byun, H.-S.; Bittman, R. J. Org. Chem. 1999, 64, 6049-
6055.
3. Ponpipom, M. M.; Bugianesi, R. L. Chem. Phys. Lipids 1984, 35, 29-37.
15 Synthesis of Ureido analog.
The ureido analog (VI) was made as follows: diethyl azodicarboxylate (DEAD)
was
added dropwise to a solution of the hydroxy phosphonocholine, PPh3, and HN3 in
toluene at 0 C, with stirring for 30 min. After hexane was added, the mixture
was filtered through a silica gel pad to yield the crude azide, which was
20 dissolved in Et20 and reduced to the amine with lithium aluminum
hydride at 0 C. After quenching of the reaction with water and filtration
through Celite, the crude amine was obtained. The amine was
dissolved in 2-propanol, and a solution of trimethylsilyl isocyanate in 2-
propanol was added at room temperature. After 4 h of stirring, the
mixture was concentrated under vacuum and the ureido-phosphonate
was obtained by silica gel chromatography.
7
H,N.Me
OH 0- NH2 0
I ab I HN O O
P'O~~N'Me3 '0--N'Me3
p O P' N=Me3
0
ureido analog of HB40-6D
Key: (a) HN3, DEAD, PPh3, toluene, 0 C; (b) LIAIHa, EtzO, 0 C; (c) TMSNCO, 2-
PrOH, rt.
CA 02610792 2007-12-03
WO 2006/130994 PCT/CA2006/000962
21
Schemes for the synthesis of ureido-C-galactoside (VII) and ureido-C-
aminogalactoside (VIII)
Ph Ph PhuPh
OBn PhuPh ~INI
Bn0 O + INI n-BuLi BnO OBn N_ 1. Tf20 BnO OBn OC18H33
BnOO OC18H33 THF, -78 C 0 / OC18H33 2. Et3SiH BnO O ~
OBn Bn0 OBn
per-0-benzyl galactosyl OBnOH
lactone
OBn H2N
BnO OC1eH33 1. TMSNCO HO OH
5%TFA 0 / O NH NHMe
THF/CH2CI2M20 Bo0 2. Mel HO OC18H33
OBn 3. H2, Pd/C OH
ureido-C-galactoside
Ph
Acccppp OAo PhI Ph AgOTf Ao0 OAc P h y5% TFA
~ N ~ N
AcO Br {Amolecularsieves Ac0 O~/OC+sHb THF/CH2CI=/HZ0
OAc HO~~OC+eHm CHiCl2 , rt, 18 h OAc
p e r-O-a cetylge la cto syI 2-N-(di p he nylm ethyle n e)-a m in o-
bromide t-O-hezadecyl glycerol
(from reaction ot 2-amino-l-O-hezadecyl
glycerol with Ph=C=NH)
Q Me,N-H
QA- TMSNCO AQ OAc HyI~NHTM B 1. n-au,NF HO OH Ol
Ac0 YH
yH
Ac0
O ~\'0~/~+eH33 2-PrOH, rt A.0 " -\ O~ /OC+sH, 2. Mel HO~O~'OCBHy
OAc n \OAc 3. hydrolysis OH
N-methylureldo 0-galactoside
Me.NH
Ac0 OAc same procedure as above HO OH
~ ~ O dH ureido 2-aminopalac[osida
AcO'Br Hp O O---~OC+eHa
NHAc NHZ
CA 02610792 2007-12-03
WO 2006/130994 PCT/CA2006/000962
22
Provided below is a reaction scheme for the synthesis of Naphthol
Carbamoyl Phosphonocholine derivative. 1-O-tert-Butyldimethylsilyl-2-(O-4'-
methoxybenzyl)-3-O-tert-butyldiphenylsilyl-sn-glycerol was converted to (S)-3-
iodo-
2-(O-4'-methoxybenzyl)-1-O-ten' butyldiphenylsilyl-propane-1,2-diol via
selective
desilylation of the tert-butyldimethylsilyl group, followed by tosylation of
the
liberated primary hydroxyl group and SN2 reaction with iodide ion. Coupling of
the
iodo glyceride with the naphthol derivative, compound 2 (which was synthesized
from 4-(dimethylamino)benzaldehyde as outlined in the accompanying scheme),
afforded compound 1. Compound 1 was converted to the naphthol phosphocholine
product by the following sequence of reactions: removal of the 4'-
methoxybenzyl
group, insertion of the carbamoyl moiety, desilylation, and installation of
the
phosphocholine moiety.
Synthesis of Naphthol Carbamoyl Phosphocholine Derivative
Me2N
OTMDS cpd.2
PMBO~H PMBO H
OTBDPS +OTBDPS KZC03 O compound 1
PMBO~
TBDPSO 0
CiaH2s
Synthesis of cpd. 2:
CHO \ C02Me C02Me O
CiaHzs
Me2N Me2N \ I~ COZNa Me N
2 Me2N
OR OH
compound 2
0 O
O
\ \
compound 1-- C14H29 _ I\ \ C14H29 I\ \ C14H29
Me2N0 Me2N Me2N i i
O
+OTBDPS MeHN MeHN O~H MeHN O~H O
LOH L 11 +
O-P-OCH2CH2NMe3
0
CA 02610792 2007-12-03
WO 2006/130994 PCT/CA2006/000962
23
Provided below is a reaction scheme for synthesis of naphthol
carbamoyl phosphonocholine derivative. The naphthol derivative of a carbamoyl
phosphonocholine was synthesized as outlined in the accompanying scheme. The
requisite four-carbon backbone of the phosphonolipid was prepared by using
malic
acid as the starting material. Arbuzov reaction of the primary bromide
afforded the
diethyl phosphonate ester. After installation of the carbamoyl moiety, the
reaction
sequence of desilylation, tosylation, and SN2 displacement with iodide ion
furnished iodobutyl phosphonate compound 1. Coupling with naphthol compound
2, followed by conversion of the phosphonate to the phosphonocholine, afforded
the napthol carbamoyl phosphonocholine product.
Synthesis of Naphthol Carbamoyl Phosphonocholine Derivative
CsHe CBH5
OO TBDPSCI ~ NBS,
NaH, O O BaCO3 OBz
P(OEt)3
HO~J --~ TBDPSO~J TBDPSO ~, THF, 24 hrt CICH2CH2CI, Br 150 C
from L-malic acid reflux
OR' OR2 Mel, H,N,Me H.N,Me
,
TBDPSO~~ I ORZ KNCO
n-Bu4NBr IJ-1 1. Bu4NF, THF ~
P, ~ -- O O OMe - _- O O OMe
O (i-Pr)2NEt, TBDPSO,_,I,_/.,P,OMe 2. TsCI, Et3N I~/. I OMe
~ R2 THF/DMF (10:1) õ 3. Nal P
NaOMe ~ R= Bz, R= Et O ~
MeOH R' = H, R2 = Me (insertion of the carbamoyl moiety) compound I
0 C14H29 H,N-Me
O ~
C14H29 1. compound 1, K2CO3 O O O O
MeZN i i ~p.O,-, N.Me3
2. TMSBr, CHZCIZ, rt O 11
OH 3. choline tosylate,
compound 2 CI3CCN, py, 50 C
NMe2
(from 4{dimethylemino)benzeldehyde)
The enantiomer can be synthesized from D-malic acid
CA 02610792 2007-12-03
WO 2006/130994 PCT/CA2006/000962
24
Therapeutic Methods and Uses
One method by which the compounds may have an antitumor effect
is by acting as an antiproliferative agent. One aspect the present invention
relates
to a method of inhibiting cell proliferation by administering an effective
amount of
the carbamate lipids and dicarbamate lipids to humans or other animals in
need.
Another method by which the carbamate, dicarbamate, and ureido lipids may have
an antitumor effect is by inducing death of the cancer cells. Thus another
aspect
of the invention provides a means of inducing cancer cell death by
administering
an effective amount of the carbamate and dicarbamate lipids.
The carbamate and dicarbamate lipids (compounds I-V) and uereido
and their C-glycoside derivatives ( compounds VI-VIII) in the invention can be
used
for treatment of all forms of cancer, malignant disease of hyperproliferative
diseases. These include but are not limited to breast cancer, leukemias,
lymphomas (Hodgkins and non-Hodgkins), plasmacytomas, histiocytomas,
melanomas, adenomas, sarcomas, carcinomas of solid tissues, hypoxic tumors,
squamous cell carcinomas, genitourinary cancers such as cervical, ovarian,
prostate, and bladder cancers, head and neck cancers, and nervous system
cancer.
Compounds I, II, and III may be particularly useful for the selective
treatment of prostate cancer, both the hormone-dependent and hormone-
independent forms, by administering lower doses with minimal effects on other
cells. They may be used in conjunction with hormone deprivation to eradicate
residual malignant prostate cells that may cause the recurrence of the hormone-
independent form of the disease.
The carbamate, dicarbamate, and ureido lipids described in this
invention may allow for the treatment of tumors resistant to chemotherapy
including multi-drug resistant varieties and may also allow for more effective
radiotherapy of tumors that currently respond poorly to radiotherapy such as
adenocarcinomas of the bowel and lung. The compounds described herein may
also be used in autologous bone marrow transfer, to purge the marrow stem
cells
of cancer cells prior to reintroducing the stem cells back into the patient.
CA 02610792 2007-12-03
WO 2006/130994 PCT/CA2006/000962
The carbamate, dicarbamate, and ureido lipids of the present
invention may also be used in combination with other antineoplastic drugs for
effective treatment of tumors. This includes known conventional drugs such as
antimetabolites, alkylating agents, antimicrobial antineoplastics,
antimicrotubular
5 agents, cisplatinum and its derivatives, and topoisomearase-interactive
agents.
Pharmaceutical Compositions.
The ether glycerolipid carbamates of the invention may be
incorporated into a pharmaceutical composition which may be useful for cancer
10 treatment. The pharmaceutical compositions of the invention can be prepared
by
known methods for the preparation of pharmaceutically acceptable compositions
which can be administered to patients so that effective quantities of the
active
compound is combined with an acceptable vehicle. The pharmaceutical
compositions of the invention can be for oral, topical, rectal, parenteral,
local,
15 intravenous, inhalant, or intracerebral. They may be solid or semisolid in
the form
of pills, tablets, creams, ointments, gelatin capsules, capsules, slow-release
capsules or pills, suppositories, soft gelatin capsules, gels, membranes,
tubelets,
and sprays. For parental and intracerebral use, those forms for intramuscular
or
subcutaneous administration can be used, or forms for infusion or intravenous
or
20 intracerebral injection can be used, and can therefore be prepared as
solutions of
the active compounds or as powders of the active. compound mixed with one or
more pharmaceutically acceptable excipients or diluents, suitable for use as
described above with the osmolarity compatible with the physiological fluid.
While the preferred embodiments of the invention have been
25 described above, it will be recognized and understood that various
modifications
may be made therein, and the appended claims are intended to cover all such
modifications which may fall within the spirit and scope of the invention.
CA 02610792 2007-12-03
WO 2006/130994 PCT/CA2006/000962
26
References
Arthur, G. and Bittman R. (1998). The inhibition of cell signaling pathways by
antitumor ether lipids. Biochim. Biophys. Acta 1390, 85-102.
Ashagbley, A., Samadder, P., Bittman, R., Erukulia, R. K., Byun, H.-S., and
Arthur,
G. (1996). Synthesis of ether linked analogs of lysophosphatidate and their
effect
on the proliferation of human epithelial cancer cells in vitro. Anticancer
Res. 16,
1813-1818.
Berdel, W. E. (1991). Membrane interactive lipids as experimental anticancer
drugs. Br. J. Cancer 64, 208-211.
Berdel, W. E., Andreesen, R., and Munder, P. G. (1985). In Phospholipids and
Cellular Regulation. Kuo, J. F., Ed., Vol. 2, pp. 41-73, CRC Press, Boca
Raton, FL.
Bittman, R. and Arthur, G. (1999). Antitumor ether lipids: biological and
biochemical effects. In Liposomes: Rational Design, Janoff, A. S., Ed., Marcel
Dekker, New York, pp. 125-144 (1999).
Budman, D. R., Calabro, A., and Kreis, W. (2002). Synergistic and antagonistic
combinations of drugs in human prostate cancer cell lines in vitro. Anti-
Cancer
Drugs 13, 1011-1016
Hanessian, S., and Plessas, N. R. (1969). J. Org. Chem. 1969, 34, 1035-1044.
He, L., Wanunu, M., Byun, H.-S., and Bittman, R. (1999). Regioselective and
stereospecific azidation of 1,2- and 1,3-diols by azidotrimethylsilane via a
Mitsunobu reaction. J. Org. Chem. 64, 6049-6055.
Houlihahn, W. J., Lohmeyer, M., Workman, P., and Cheon, S. H. (1995).
Phospholipid antitumor agents. Med. Res. Rev. 15, 157-223.
Jacobs, S. C. (1983). Spread of prostatic cancer to bone. Urology 21, 337-344.
Jemal, A., Murray, T., Samuels, A., Ghafoor, A., Ward, E., and Thun, M. J.
(2003).
Cancer Statistics. CA Cancer J. Clin. 53, 5-26.
Koutsilieras, M., and Tolis, G. (1985). Long term follow-up of patients with
advanced prostatic carcinoma treated with Buserelin (hoe 766) or orchiectomy:
classification of variables associated with disease outcome. Prostate 7, 31-
39.
Koutsilieris, M., Faure, N., Tolis, G., Larouche, B., Robert, G., and Ackman,
C. F.
(1986). Objective response and disease outcome in 59 patients with stage D2
prostatic cancer treated with either Buserilin or orchiectomy. Disease
aggressivity
and its association with response and outcome. Urology 27, 221-228.
Kreis, W., Budman, D. R., and Calabro, A. (1979). Unique synergism or
CA 02610792 2007-12-03
WO 2006/130994 PCT/CA2006/000962
27
antagonism of combinations of chemotherapeutic and hormonal agents in human
prostate cancer cell lines. Br. J. Urol. 79,196-202.
Lohmeyer, M., and Bittman, R. (1994) Antitumor ether lipids and
alkylphosphocholines. Drugs Future 19, 1021-1037.
Lu, X., Zhou, X., Kardash, D., and Arthur, G. (1993). Metabolism of
alkyllysophospholipid in epithelial cancer cell lines and inhibition of cell
growth.
Biochem. Cell Biol. 71, 122-126.
Mollinedo, F., Gajate, C., Martin-Santamaria, S., and Gago, F. (2004). ET-18-
OCH3 (edelfosine): a selective antitumour lipid targeting apoptosis through
intracellular activation of Fas/CD95 death receptor. Curr. Med. Chem.11, 3163-
3184.
Navone, N. M., Logothetis, C. J., von Eschenbach, A. C., and Troncoso, P.
(1999).
Model systems of prostate cancer: Uses and limitations. Cancer Metastasis Rev.
17, 361-371.
Ponpipom, M. M., and Bugianesi, R. L. (1984) Chem. Phys. Lipids, 35, 29-37.
Saitoh, H., Hilda, M., Shimbo, T., Nakamura, K., Yamagata, J., and Satoh, T.
(1984). Metastatic patterns of prostatic cancer. Correlation between sites and
number of organs involved. Cancer 54, 3078-3084.
Samadder, P., and Arthur, G. (1999). Decreased sensitivity to 1-O-octadecyl-2-
O-
methyl-glycerophosphocholine in MCF-7 cells adapted for serum-free growth
correlates with constitutive association of Raf-1 with cellular membranes.
Cancer
Res. 59, 4808-4815.
Samadder, P., Bittman, R., Byun, H.-S., and Arthur, G. (2004). Synthesis and
use
of novel ether phospholipids enantiomers to probe the molecular basis of the
enantiomer effects of alkyllysophospholipids: correlation of differential
activation of
c-Jun-NH2-terminal protein kinase with antiproliferative effects in neuronal
tumor
cells. J. Med. Chem. 47, 2710-2713.
Sobel, R. E., and Sadar, M. D. (2005). Cell lines used in prostate cancer
research:
a compendium of old and new lines - Part 1. J. Urol. 173, 342-359.
~
van Boeckel, C. A. A., van del Marel, G.A., Westerduin, P., and van Boom, J.
H.
(1982) Synthesis 1982, 399-402.
Yang, G., Franck, R. W., Byun, H.-S., Bittman, R., Samadder, P., and Arthur,
G.
(1999). Convergent C-glycolipid synthesis via the Ramberg-Backlund reaction:
active antiproliferative glycolipids. Org. Lett. 1,2149-2151.
CA 02610792 2007-12-03
WO 2006/130994 PCT/CA2006/000962
28
Table 1. Growth Inhibitory Properties of Carbamate and Dicarbamate Lipid
Compounds I-V on Human Tumor Cells [reported as IC50 ( M)]
Cell Line I II III IV V
DU145, prostate 1.2 2.1 2.4 4.8 8.2
PC3, prostate 1.2 2.5 1.9 2.8 4.1
BT549, breast 4.4 6.8 13.4 4.9 6.6
MDA-MB-231, 3.7 6.1 10.5 18.3 6.6
breast
MCF-7, breast 4 10.1 9.1 13.4 >20
HeLa, cervix 3.3 7.9 19.3 8.7 14.9
A549, lung 10.7 >20 >20 >20 >20
IC50 is the drug concentration ( M) required to inhibit the growth by 50%
after
incubation of exponentially growing cells with the drug for 48 hours.
CA 02610792 2007-12-03
WO 2006/130994 PCT/CA2006/000962
29
Table 2. Antiproliferative effect of 1-0(7-N, N-dimethylamino-3-pentadeconyl-l-
naphthyl)-2-O-methyl-sn-glycero-3-phosphocholine against human cancer cells
Cancer Cell Line IC50 NM
SK-N-SH (neuronal) < 20
SK-N-MC (neuronal) > 20
DU145 (prostate) 3.5
PC3 (prostate) 1.9
MCF-7 (breast) > 20
HepG2 (liver) > 20
A549 (lung) > 20
Proliferating cells in 48-well plates were incubated with 0-20 pM of the
compound
for 48 h. The cell numbers present were determined by the CyQuantTM assay
(Invitrogen), and the increase in numbers were expressed relative to the
controls
receiving only the vehicle (0.1 % ethanol).