Note: Descriptions are shown in the official language in which they were submitted.
CA 02613698 2012-05-09
SEPARATION OF CARBON DIOXIDE (CO2) FROM GAS MIXTURES BY
CALCIUM BASED REACTION SEPARATION (CaRS-CO2) PROCESS
Inventors: Liang-Shih Fan
Himanshu Gupta
Mahesh lyer
10
TECHNICAL FIELD OF THE INVENTION
[0002] The present invention relates to the application of chemical sorbents
for the separation of CO2 from gas mixtures.
BACKGROUND OF THE INVENTION
[0003] As used herein, the term "supersorbent" shall mean a sorbent as
taught in United States Patent No. 5,779,464 entitled "Calcium Carbonate
Sorbent
and Methods of Making and Using Same".
[0004] As used herein, the term "microporous" shall mean a pore size
distribution of less than 5 nanometers. As used herein, the term "mesoporous"
shall
mean a pore size distribution of from about 5 nanometers to about 20
nanometers.
[0005] Atmospheric CO2 concentration has been increasing steadily since the
industrial revolution. It has been widely accepted that the while the CO2
concentration was about 280 ppm before the industrial revolution, it has
increased
from 315 ppmv in 1959 to 370 ppmv in 2001 [Keeling, C.D. and T.P. Whorf. 2002.
1
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
Atmospheric CO2 records from sites in the SIO air sampling network. In Trends:
A
Compendium of Data on Global Change. Carbon Dioxide Information Analysis
Center, Oak Ridge National Laboratory, U.S. Department of Energy, Oak Ridge,
Tenn., U.S.A. This data is also available from
http://cdiac.esd.ornl.gov/ftp/maunaloa-
co2/maunaloa.co2].. Rising CO2 concentrations has been reported to account for
half
of the greenhouse effect that causes global warming [IPCC Working Group I.
IPCC
Climate Change 1995 - The Science of Climate Change: The Second Assessment
Report of the Intergovernmental Panel on Climate Change; Houghton, J. T.,
Meira
Filho, L.G., Callander, B.A., Harris, N., Kattenberg, A., Maskell K, Eds.;
Cambridge
University Press: Cambridge, U.K., 1996]. Although the anthropogenic CO2
emissions are small compared to the amount of CO2 exchanged in the natural
cycles, the discrepancy between the long life of CO2 in the atmosphere (50-200
years) and the slow rate of natural CO2 sequestration processes leads to CO2
build
up in the atmosphere. The IPCC (Intergovernmental Panel on Climate Change)
opines that "the balance of evidence suggests a discernible human influence on
the
global climate." Therefore, it is necessary to develop cost effective CO2
management schemes to curb its emission.
[0006] Many of the envisaged CO2 management schemes consist of three
parts - separation, transportation and sequestration of CO2 [FETC Carbon
Sequestration R&D Program Plan: FY 1999-2000. National Energy Technology
Laboratory, Department of Energy, Washington, DC, 1999]. The cost of
separation
and compression of CO2 to 110 bar (for transportation of CO2 in liquid state)
is
estimated at $30-50 per ton C02, and transportation and sequestration would
cost
about $1-3 per ton per 100 km and $1-3 per ton of CO2, respectively [Wallace,
D.
Capture and Storage of CO2. What Needs To Be Done. Presented at the 6th
2
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
Conference of the Parties, COP 6, to the United Nations Framework Convention
on
Climate Change; The Hague, The Netherlands, Nov 13-24, 2000;
www.iea.org/envissu/index.htm]. The capture of CO2 imposes severe energy
penalties thereby reducing the net electricity output by as much as 13-37%
[Herzog,
H.; Drake, E.; Adams, E. CO2 Capture, Reuse, and Storage Technologies for
Mitigating Global Climate Change. A White Paper; Final Report No. DE-AF22-
96PC01257, Jan 1997]. The dominating costs associated with the current CO2
separation technologies necessitate development of economical alternatives.
[0007] Historically, CO2 separation was motivated by enhanced oil recovery
[Kaplan, L. J. Cost-Saving Processes Recovers CO2 from Power-Plant Flue gas.
Chem.Eng. 1982, 89 (24), 30-31; Pauley, C. P.; Smiskey, P. L.; Haigh, S. N-ReN
Recovers CO2 from Flue Gas Economically. Oil Gas J. 1984, 82(20), 87-92].
Currently, industrial processes such as limestone calcination, synthesis of
ammonia
and hydrogen production require CO2 separation. Absorption processes employ
physical and chemical solvents such as Selexol and Rectisol, MEA and KS-2
[Reimer, P.; Audus, H.; Smith, A. Carbon Dioxide Capture from Power Stations.
IEA
Greenhouse R&D Programme, www.ieagreen.org.uk, 2001. ISBN 1 898373 15 9;
Blauwhoff, P.M.M.; Versteeg, G. F.; van Swaaij, W. P. M. A study on the
reaction
between CO2 and alkanoamines in aqueous solution. Chem. Eng. Sci.1984, 39(2),
207-225. Mimura, T.; Simayoshi, H.; Suda, T.; lijima, M.; Mitsuake, S.
Development
of Energy Saving Technology for Flue Gas Carbon Dioxide Recovery by Chemical
Absorption Method and Steam System in Power Plant. Energy Convers. Mgmt.
1997, 38, Suppl. P.S57-S62]. Adsorption systems capture CO2 on a bed of
adsorbent materials such as molecular sieves and activated carbon [Kikkinides,
E.S.;
Yang, R.T.; Cho, S.H. Concentration and Recovery of CO2 from flue gas by
pressure
3
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
swing adsorption. Ind. Eng. Chem. Res. 1993, 32, 2714-2720]. CO2 can also be
separated from the other gases by condensing it out at cryogenic temperatures.
Polymers, metals such as palladium, and molecular sieves are being evaluated
for
membrane based separation processes [Reimer, P.; Audus, H.; Smith, A. Carbon
Dioxide Capture from Power Stations. IEA Greenhouse R&D Programme,
www.ieagreen.org.uk, 2001. ISBN 1 898373 15 9].
[0008] Reaction based processes, as promulgated in this work, can be applied
to separate CO2 from gas mixtures. This process is based on a heterogeneous
gas-
solid non-catalytic carbonation reaction where gaseous CO2 reacts with solid
metal
oxide (represented by MO) to yield the metal carbonate (MCO3). The reaction
can be
represented by:
MO + CO2 - MCO3 (1)
Once the metal oxide has reached its ultimate conversion, it can be thermally
,regenerated to the metal oxide and CO2 by the calcination of the metal
carbonate
product. The calcination reaction can be represented by:
MCO3 - MO + CO2 (2)
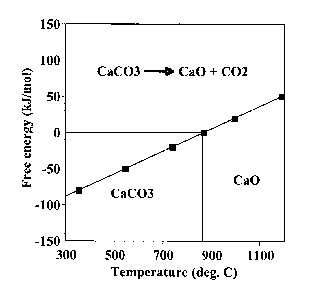
As an example of the above-mentioned scheme, Figure 1 shows the variation in
the
free energy of the carbonation reaction as a function of temperature for
calcium
oxide. From the figure, we can see that the carbonation reaction is
thermodynamically favored with a decrease in temperature (Gibbs free energy
declines with a decrease in temperature). However, at lower temperatures, the
carbonation reaction is kinetically slow. In fact, it takes geological time
scales for the
formation of CaCO3 by the reaction between CaO and atmospheric CO2 (at 280-360
ppm) at ambient temperatures. It should also be noted that the carbonation
reaction
would be favored as long as the free energy is negative. This creates an upper
4
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
bound of 890 C for carbonation to occur under a CO2 partial pressure of I
atm. The
equilibrium temperature for this reaction is a function of the partial
pressure of C02-
A reaction based CO2 separation process offers many advantages. Under ideal
conditions, MEA captures 60g C02/kg, silica gel adsorbs 13.2g C02/kg and
activated
carbon adsorbs 88g C02/kg. The sorption capacity of some metal oxides (such as
the modified CaO, presented in this study) is about 700g C02/kg of CaO. This
is
about an order of magnitude higher than the capacity of adsorbents/solvents
used in
other CO2 separation processes and would significantly reduce the size of the
reactors and the material handling associated with CO2 separation.
[0009] Numerous metal oxides exhibit the carbonation and calcination
reaction. The calcination temperature of a few metal carbonates (CaCO3 -750
C,
MgCO3 -385 C, ZnCO3 -340 C, PbCO3 -350 C, CuCO3 -225-290 C and MnCO3
-440 C) makes them viable candidates for this process. Apart from CaO, gas-
solid
carbonation of other metal oxides has not been widely studied. The carbonation
of
ZnO to ZnCO3 at 8-13 C was low when exposed to CO2 and H2O for over 100 days
(Sawada, Y.; Murakami, M.; Nishide, T. Thermal analysis of basic zinc
carbonate.
Part 1. Carbonation process of zinc oxide powders at 8 and 13 C. Thermochim.
Acta. 1996, 273, 95-102.). MnCO3 undergoes a more complex thermal degradation
phenomena. MnCO3 first decomposes to Mn02 at 300 C, which in turn changes to
Mn2O3 at 440 C. At higher temperatures (-900 C), the final thermal
decomposition
product was identified as Mn3O4 (Shaheen, W. M.; Selim, M. M. Effect of
thermal
treatment on physicochemical properties of pure and mixed manganese carbonate
and basic copper carbonate. Thermochim. Acta. 1998, 322(2), 117-128.).
Different
oxides of manganese provide the flexibility of exploiting the
carbonation/calcination
reaction over a wider temperature range. Aqueous phase MgO carbonation has
5
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
been studied for its suitability for mineral-based CO2 sequestration
(Fernandez, A.I.;
Chimenos, J.M.; Segarra, M.; Fernandez, M.A.; Espiell, F. Kinetic study of
carbonation of MgO slurries. Hydrometallurgy. 1999, 53, 155-167). The
carbonation
extent of Mg(OH)2 was about 10% between 387-400 C and 6% formation between
475-500 C (Butt, D.P.; Lackner, K.S.; Wendt, C.H.; Conzone, S.D.; Kung, H.;
Lu, Y-
C.; Bremser, J.K. Kinetics of Thermal Dehydroxylation and Carbonation of
Magnesium Hydroxide. J. Am. Ceram. Soc.1996, 79(7), 1892-1898). They
attributed
the low conversions to the formation of a non-porous carbonate product layer.
This
layer hinders the inward diffusion of CO2 and the outward diffusion of H2O (a
product
of the carbonation reaction) leading to low conversions. The carbonation of
PbO was
studied as a part of the chemical heat pump process (Kato, Y.; Saku, D.;
Harada, N.;
Yoshizawa, Y. Utilization of High Temperature Heat from Nuclear Reactor using
Inorganic Chemical Heat Pump. Progress in Nuclear Energy. 1998, 32(3-4), 563-
570. & Kato, Y.; Harada, N.; Yoshizawa, Y. Kinetic feasibility of a chemical
heat
pump for heat utilization from high temperature processes. Applied Thermal
Engineering. 1999, 19, 239-254). They reported 30% conversion in an hour under
100% CO2 atmosphere at 300 C. Furthermore, they found the reactivity of PbO
to
drop with the number of carbonation-calcination cycles.
[0010] Carbonation of calcium oxide has been widely studied. Related
applications of the CaO carbonation and calcination include the storage of
energy
(Barker, R. The Reversibility of the Reaction CaCO3 = CaO + CO2. J. App/.
Chem.
Biotechnol. 1973, 23, 733-742) and the zero emission coal alliance process,
consisting of hydrogasification of coal fueled by the heat of the carbonation
reaction
(Tinkler, M.J.; Cheh, C. Towards a Coal-capable Solid Oxide Fuel Cell System.
Proceedings of the 26th International Technical Conference on Coal Utilization
and
6
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
Fuel Systems; Clearwater, Florida, March 5-8, 2001; pp 569-570). The gas-solid
CaO-CO2 reaction proceeds through two rate-controlling regimes. The first
regime
involves a rapid, heterogeneous chemical reaction. In the second regime, the
reaction slows down due to the formation of an impervious layer of CaCO3. This
product layer prevents the exposure of unreacted CaO in the particle core to
CO2 for
further carbonation. The kinetics of the second regime is governed by the
diffusion of
ions through the CaCO3 product layer. The activation energy was estimated to
be 21
kcal/mol below 688 K and 43 kcal/mol above it for the product layer diffusion,
based
on the counter migration of C032" and 02- ions through the product layer
(Bhatia,
S.K.; and Perlmutter, D.D. Effect of the product layer on the kinetics of the
C02-Lime
Reaction. AIChE J. 1983, 29(1), 79-86).
[0011] The extent of the carbonation reaction reported in many studies has
also shown considerable variation. Stoichiometrically, 56g of CaO should react
with
44g of CO2 to form 100g of CaCO3. This translates to about 78.6-wt% capacity
for
CaO. However, the structural limitations prevent the attainment of theoretical
conversion. The extent of carbonation was only 23-wt% in 30 minutes at 600 C
(Dedman, A.J.; Owen, A.J. Calcium Cyanamide Synthesis, Part 4.- The reaction
CaO + CO2 = CaCO3. Trans. Faraday Soc.1962, 58, 2027-2035). A higher surface
area CaO sorbent provided 55-wt% CO2 sorption (Bhatia, S.K.; and Perlmutter,
D.D.
Effect of the product layer on the kinetics of the C02-Lime Reaction. AIChE J.
1983,
29(1), 79-86). 64-wt% CO2 sorption was achieved at 1050 C temperature and
11.74
atm CO2 pressure in 32 hours (Mess, D.; Sarofim, A.F.; Longwell, J.P. Product
Layer
Diffusion during the Reaction of Calcium Oxide with Carbon Dioxide. Energy and
Fuels. 1999, 13, 999-1005). However, the extent of carbonation at lower
temperature/pressure conditions that are more characteristic of CO2 containing
7
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
gaseous mixtures is absent in their work. The limitation in total conversion
stems
essentially from the nature of the initial pore size distribution of the CaO
sorbent.
Microporous sorbents (pore size < 2 nm) are very susceptible to pore blockage
and
plugging due to the formation of higher molar volume product (molar volume of
CaO:
17 cm3/mol; molar volume of CaCO3: 37 cm3/mol). CaO sorbents obtained from
naturally occurring precursors are usually microporous in nature. At the end
of the
kinetically controlled regime, diffusion processes through the product layer
control
the reaction rate. Similar structural limitations have prevented calcium-based
sorbents from attaining theoretical conversion for the sulfation reaction
between CaO
and sulfur dioxide (SO2) as well (Wei, S.-H.; Mahuli, S.K.; Agnihotri, R.;
Fan, L.-S.
High Surface Area Calcium Carbonate: Pore Structural Properties and Sulfation
Characteristics. Ind. Eng. Chem. Res. 1997, 36(6), 2141-2148). They suggested
that
a mesoporous structure, which maximizes porosity in the 5-20 nm pore size
range,
would be less susceptible to pore pluggage. This structure would also be able
to
provide sufficient surface area to ensure rapid kinetics. Their modified
precipitation
technique resulted in a mesoporous CaCO3 structure that also had a high BET
surface area determined by nitrogen (60 m2/g). A similar approach could also
enhance the reactivity of CaO sorbents towards the carbonation reaction, which
is
the focus of this study.
[0012] Lastly, it is important that the CaO sorbents maintain their reactivity
over many carbonation and calcination cycles. The conversion of CaO dropped
from
about 73% in the first carbonation cycle to 43% at the end of the 5th cycle at
866 C
(Barker, R. The Reversibility of the Reaction CaCO3 = CaO + CO2. J. App.'.
Chem.
Biotechnol. 1973, 23, 733-742 & Barker, R. The Reactivity of Calcium Oxide
Towards Carbon Dioxide and its use for Energy Storage. J. App!. Chem.
Biotechnol.
8
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
1974, 24, 221-227). Barker suggested that the CaCO3 layer is about 22 nm thick
and
his latter work showed repeated 93% conversion over 30 cycles at 629 C on 10
nm
CaO particles. In another study, cyclical studies conducted at a carbonation
temperature of 880 C and calcination at 860 C led to a drop in conversion
from
70% in the first carbonation to 38% in the 7t" carbonation step (Kato, Y.;
Harada, N.;
Yoshizawa, Y. Kinetic feasibility of a chemical heat pump for heat utilization
from
high temperature processes. Applied Thermal Engineering. 1999, 19, 239-254).
The
process described here leads to > 95% conversion due to the application of
novel
mesoporous CaO sorbents for C02 capture and maintains their reactivity over
repeated cycles of carbonation and calcination.
[0013] Part I (C02/SO2 combined reaction optimization)
[0014] INTRODUCTION
[0015] Carbon dioxide (C02) accounts for more than half of the enhanced
greenhouse effect, which is responsible for global warming.' The atmospheric
concentration of C02 has increased from 280 ppm before the Industrial
Revolution
to -365 ppm today. 2' 2,3 This is mainly due to the unabated emission of C02
as a
result of increasing consumption of fossil fuels such as coal, oil and natural
gas.
Point sources, such as electric utility plants that contribute to about one-
third of all
anthropogenic C02 emissions4, are ideal candidates for implementing C02
reduction
practices due to the relatively high concentration and quantity of C02 emitted
compared
to smaller, mobile sources. Coal consumption leads to high C02 emissions at
these
large point sources due to its dominant use in electricity generation (-52%)
and higher
energy specific C02 emission due to its high carbon to hydrogen content
compared to
other fossil fuels (g C02BTU).S Comprehensive C02 management scenarios
involve a three-step process that includes separation, transportation and safe
9
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
sequestration of C02. Economic analysis has,shown that C02 separation accounts
for 75-85% of the overall cost associated with carbon sequestration.6 Current
C02 separation technologies based on absorption, adsorption, membrane
separation, and cryogenic separation necessitate a low temperature and/or high
pressure of flue gas to enhance the C02 sorption capacity of the
sorbent/solvent
or the diffusion flux of C02 through the membrane. However, flue gas is
typically
characterized by sub-atmospheric pressure and high temperature. Metal oxides
are capable of reacting with C02 under existing flue gas conditions, thereby
reducing downstream process modifications. We have detailed elsewhere the
advantages of a high temperature reactive separation process based on the
carbonation and calcination reactions (CCR) of CaO to separate C02 from flue
gas.' The key advantage offered by this process is the enhanced C02 sorption
capacity (35-70 weight %) exhibited by the high reactivity CaO particles under
existing flue gas conditions over multiple cycles of CCRs.
[0016] Extensive screening of metal oxides has identified CaO as a potential
candidate for the CCR scheme. The carbonation reaction of CaO has been
studied for its role in chemical heat pumps8i9, energy storage systems10, zero
emission coal alliance processes", and in the enhanced production of hydrogen
from fossil fuels.' This reaction typically goes through a raid kinetic
controlled
regime, followed by a slower product-layer diffusion controlled regime.'
Naturally
occurring precursors (limestone and dolomite), are unable to achieve
stoichiometric
conversion in any carbonation step due to the predominant microporous
structure
which is susceptible to pore pluggage and pore mouth closure. In contrast,
mesoporosity, which dominates the pore structure of precipitated calcium
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
carbonate (PCC), synthesized under the influence of negatively charged
polyacrylate
ions yields greater than 90% carbonation conversion.7"4
[0017] For the viability of a CCR process, it is imperative that the CaO
sorbent
maintain high reactivity over multiple cycles. Previous studies in the
literature have
reported the performance of numerous CaO sorbents over multiple cycles.
Abanades
and co-workers summarized the CCR experimental data of previous studies on a
variety of CaO sorbents differing in their physical properties. They were able
to
develop a single correlation between the extent of carbonation as a function
of the
number of CCR cycles. 15,16 These sorbents experienced a similar loss in
reactivity
towards the carbonation reaction regardless of differences in particles size,
reactor types,
reaction conditions, sorbent characteristics and cycle times. They observed
that the
highest C02 sorption capacity retained by the sorbent was 24 wt% after 20
cycles.
[0018] Sulfur present in coal oxidizes to S02 during combustion. Calcium
based sorbents are widely used for the control of S02 emissions. The two
principal
calcium utilization processes are low temperature wet scrubbing and high
temperature furnace sorbent injection (FSI). In wet scrubbing, S02 capture
occurs
through ionic reactions in the aqueous phase. In high temperature (> 900 C)
FSI
systems, calcium oxide precursors (dolomite, Ca(OH)2 and limestone) and their
calcines reacts with S02 to form CaSO4 via the heterogeneous non-catalytic gas
solid reaction. Sulfation under these conditions has been extensively studied
and simulated using various models.","," PCC achieves a higher extent of
sulfation (-
70%) compared to naturally occurring limestone (-30%) at greater than 900 *C
within a
residence time of 700 milliseconds.
[0019] The flue gas generated by coal combustion typically contains 10-15%
C02, 3-4% 02, 5-7% H2O, 500-3000 ppm S02 and 150-500 ppm NOx in addition to
11
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
trace quantities of HCI, arsenic, mercury, and selenium. Separation of C02 by
its
absorption in monoethanolamine (MEA) is currently the most viable option for
commercial scale deployment. However, MEA forms thermally stable salts with
S02 and NOx, which do not decompose under the regeneration conditions employed
in the MEA process. It is necessary to lower S02 concentration to below 10 ppm
to
minimize the loss of the costly solvent. Economic analysis of this process,
based
on a parasitic consumption of MEA of 0.5-2 g MEA/kg C02 separated, show that
the cost associated with C02 separation lies in the $33-73/ton C02 avoided.' A
similar
hurdle is posed by S02 for a CaO based CCR process. CaO undergoes sulfation
with S02 forming CaS04, which cannot be thermally decomposed back to CaO
within
the operating temperature range of the proposed CCR process (400-800 C) as it
requires greater than 1100 C for its decomposition. Exposure of CaO to higher
temperatures leads to a loss in surface area and porosity due to excessive
.sintering, which drastically reduces its reactivity. Eventually, the CaSO4
buildup in
each cycle reduces the regenerative capacity of the CaO sorbent over
subsequent
cycles ultimately rendering it inactive. However, literature on the sulfation
of CaO
in the temperature range where CaCO3 is thermodynamically stable is scant.
Sulfation of calcium species in this temperature range is crucial for the
experiments covered in this paper because this study aims to investigate the
effect of
S02 on the carbonation of CaO.
[0020] The simultaneous hydration, carbonation and sulfation of reagent
grade 5 micron CaO particles have been previously investigated for an exposure
time of 2 hours in the 170-580 *C temperature range under differential
conditions.22
Low temperatures favor hydration over sulfation and carbonation. 380 'C marks
the termination of hydration and the onset of carbonation and sulfation.
12
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
Carbonation peaked at 520'C whereas sulfation dominated beyond 580 *C.
Furthermore, the sulfur species in the form of CaSO3 peaks at 24% at 300'C and
CaS04
is the only sulfur species above 585 'C. A high extent of sulfation has also
been
attained by 2mm sized macroporous (> 200 run) CaO particles synthesized by a
swelling technique involving water-acetic acid mixtures. 2311 The authors
attributed
the high sulfation extent to the increased access of S02 to the particle
surface due to
the macroporosity of the sorbent. Li et al. investigated combined carbonation
and
sulfation reactions on commercial grade calcium hydroxide .25 They carried out
these
reactions at 425-650 C by exposing the fines for 2 seconds under entrained
flow
conditions. The particles were then collected on a hot filter, maintained
between 450-
510 C and further exposed to the gas mixture for 2 hours. Their results
indicate an
increasing extent of direct sulfation of the carbonated product (CaC03) with
higher
residence time.2S The kinetic analysis and modeling of the reaction between
S02
and CaC03 in the temperature range where CaCO3 is thermodynamically stable,
was studied by Snow et al. and Hajaligol et al. 26,27 They exploited the
higher porosity
of calcium oxalate derived CaC03 to achieve about 90% sulfation in the 400-550
*C
temperature range. Tullin and Ljungstrom conducted thermogravimetric studies
on
the simultaneous carbonation and sulfation of CaO and CaCO3for a residence
time
of 10-180 minutes at 860 *C. The gas mixture consisted of 30-80% C021 3000 ppm
S02 and 3-4% oxygen 28,29 The initial increase in weight of the sorbent was
predominantly due to the carbonation reaction, which occurs to a higher extent
than
sulfation for the given inlet gas concentration levels. Further exposure of
the
sorbent to the reactant gas mixture results in the direct sulfation of the
CaCO3
so formed, and leads to a decrease in the overall extent of carbonation and an
increase in sulfation. In other experiments, they show that although both CaO
and
13
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
CaCO3 have similar reactivity towards the sulfation reaction, CaSO4 formed due
to
the direct carbonation of CaCO3 is more porous than the CaSO4product formed by
sulfation of CaO.
[0021] These literature studies indicate that carbonation occurs at faster
rate
compared to sulfation at temperatures around 700 *C due to higher
concentration of
C02. However, S02 will eventually react directly with CaCO3 leading to the
formation of
CaSO4. It is thus imperative to obtain experimental data on combined
carbonation and
sulfation reactions of CaO over multiple cycles to identify the process
conditions under
which the extent of carbonation can be maximized in the presence of S02.
Simultaneous
high temperature carbonation and sulfation experiments were performed in a
Thermogravimetric Analyzer (TGA). The study demonstrates the effect of solid
residence
time on the overall extent of simultaneous carbonation and sulfation.
Enhanced Hydrogen Production Integrated with CO2
Separation in a Single-Stage Reactor
[0022] There has been a global push towards the development of a hydrogen
economy. The main premise behind this drastic alteration in our energy usage
stems
from the fact that the use of hydrogen in portable and mobile applications
would be
the most environmentally beneficial process that leads only to the emission of
water.
However, the biggest issue that needs to be addressed for the success of the
hydrogen-based economy involves the source of hydrogen itself. While hydrogen
may be considered as the best "carrier" of energy, there is clearly no
hydrogen
"wells" on earth. The major processes for hydrogen production from fossil
fuels
consist of steam reforming of methane (SMR), coal gasification, catalytic
cracking of
natural gas, and partial oxidation of heavy oils. Other processes consist of
water
electrolysis, thermo chemical water decomposition, biological processes, etc.
14
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
(Rosen and Scott, 1998; Rosen, 1996). However, water electrolysis is not a
very
energy efficient process.
[0023] Water gas, a mixture of CO, C02, H2O and H2, is formed by the
gasification of coal by sub-stoichiometric air and/or steam. Irrespective of
the initial
concentration of these four gases, the reversible water gas shift (WGS)
reaction gets
initiated until the exact ratio of the concentration of these gases reaches a
particular
equilibrium constant KWGS that is a function of temperature. The WGS reaction
and
its equilibrium constant can be written as:
WGS Reaction: CO + H2O <=> CO2 + H2 AH = - 40.6 kJ/mol (1)
WGS equilibrium constant:
K WGS = [C02][H2] =812.9-6.628e+5+1.001e+8 (2)
[CO][H20] T T2
where T is in C. From equation (2), it can be observed that KWGS reduces with
increasing temperature. This means that processes aimed at converting coal-
derived
gas to hydrogen at high temperatures are thermodynamically restricted. While
catalysts aid in achieving this equilibrium, they cannot alter the value of K
to provide
a higher hydrogen yield. An effective technique to shift the reaction to the
right for
enhanced hydrogen generation has been to remove hydrogen from the reaction
mixture. This premise has lead to the development of hydrogen separation
membranes. However, membranes cannot completely remove hydrogen from the
mixture. Any remaining hydrogen would dilute CO2 after its utilization in
either a fuel
cell or gas turbine.
[0024] Another option for driving the WGS reaction forward is to remove CO2
from the reaction mixture by reacting it with CaO. The carbonation reaction
can be
written as:
Carbonation Reaction: CaO + CO2 -) CaCO3 (OH = - 183 kJ/mol) (3)
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
Under the appropriate reaction temperature, CO2 concentration can be lowered
down to ppm levels by reaction (3), thereby enabling the maximum production of
hydrogen from carbon via reaction (1). By conducting the reaction such that CO
is
the limiting reactant, we can ensure complete utilization of the fuel as well.
Besides
these advantages, CO2 is simultaneously removed from the gas mixture in the
form
of CaCO3, thereby improving the purity of the hydrogen stream (the other
contaminant being only water). The spent sorbent can then be calcined
separately to
yield pure CO2 stream, which is then amenable for compression and liquefaction
before its transportation to sequestration sites. Calcination reaction,
reverse of the
carbonation reaction can be written as:
Calcination Reaction: CaCO3 - CaO + CO2 (AH = + 183 kJ/mol) (4)
The resulting CaO sorbent is recycled to capture CO2 in the next cycle. This
cyclical
CCR process can be continued so long as the sorbent provides a satisfactory
CO2
capture.
[0025] To obtain high purity H2, the WGS reaction is generally carried out in
0
two stages for:.(1) high temperature shift (250-500 C) using iron catalysts
and (2)
0
low temperature shift (210-270 C) using copper-based catalysts (Gerhartz,
1993;
Bohlbro, 1969). Copper based catalysts are extremely intolerant to small
quantities
of sulfur (< 0.1 ppm) and hence the fuel gases need to be desulfurized
upstream of
the WGS reactor. Besides, to achieve satisfactory carbon monoxide conversion a
considerable quantity of high-pressure steam is required. For example, to
lower the
CO content of the typical fuel gas from 45 % (inlet) to 3% (outlet) a total
steam
addition of 1.18 kg/m3 of the gas is required, at a total pressure of 60 bar
and 410 OC
0
(Gerhartz, 1993). The steam to CO ratio at 550 C can be as high as 50 during a
16
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
single-stage operation or 7.5 for a more expensive dual-stage process to
obtain 99.5
% pure H2 (David, 1980). This is necessary due to the equilibrium limitation
inherent
in the WGS reaction. From the point of view of H2 production, even though
higher
temperatures lead to improved kinetics, WGS has poor equilibrium conditions at
the
higher temperatures. However, the continuous removal of the carbon dioxide
product
from the reaction chamber will incessantly drive the equilibrium-limited water-
gas
shift reaction forward. This will ensure a high yield and purity of hydrogen
with near
stoichiometric amounts of steam needed for the reaction. Besides, the reaction
can
now be carried out at higher temperatures leading to superior kinetics in the
forward
direction. Thus the major equilibrium related drawback in this process could
be
overcome. The continuous CO2 removal can be brought about by the carbonation
reaction of a metal oxide to give the corresponding metal carbonate. We have
identified a high reactivity, mesoporous calcium oxide as the potential
sorbent for the
in-situ CO2 capture given by eqn. 3.
[0026] The success of this process would effectively bridge coal gasification
to
fuel cell usage and chemical synthesis. Other side benefits of this process
involve
the potential for removal of sulfur and heavy metals such as arsenic and
selenium
from the fuel gas stream.
[0027] Recently, Harrison and co-workers reported a single-step sorption-
enhanced process to produce hydrogen from methane (Balasubramanian et al.,
1999; Lopez Ortiz and Harrison, 2001). They used the traditional concept of
SMR
with WGS using Ni-based catalyst to produce hydrogen, coupled with this novel
scheme of in-situ continuous CO2 capture using a calcium-based dolomite
sorbent.
They obtained high hydrogen yields with 97 % purity (dry basis).
17
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
[0028] However, they reported a low "calcium" conversion in the sorbent of
about 50% at the beginning of the breakthrough to about 83% at the end of the
test.
These conversion calculations are based on only the calcium portion of their
dolomite sorbent. Their total sorbent conversion will be much lower than these
values as dolomite does not entirely contain calcium based material. In fact,
dolomite
comprises of nearly 50wt.% calcium, which participates in the reaction to some
extent, and the remaining portion of the sorbent (mainly magnesium oxide)
stays
unreacted. Further, they attribute the incomplete conversions of the calcium
material
to the concept of pore filling and pluggage at the pore-mouths of these
sorbent
particles by CaCO3 product layer, preventing the access of CO2 in the gas to
unreacted CaO surface at the pore interiors.
[0029] Harrison and co-workers regenerated the dolomite sorbent in streams
of N2, 4%02 in N2 and pure CO2. They had to use high regeneration temperatures
of
0
800-950 C, especially while using pure CO2. Exposure of the reforming catalyst
to
an oxidizing atmosphere (viz. 02/N2 or C02) while regenerating the sorbent
used to
oxidize the Ni catalysts to NiO. Hence, the catalyst had to be reduced back to
Ni
before every cycle or the sorbent-catalyst mixture had to be separated after
every
run so that only the sorbent is subjected to the regeneration conditions.
Further, the
temperature of operation can be lowered by regeneration in a pure N2 stream.
However, it would not solve the problem of CO2 separation due to the formation
of a
C02/N2 gas mixture. Calcination in a pure CO2 stream will result in higher
operating
temperatures due to the thermodynamic limitations of the calcination reaction
in
presence of the CO2 product. Higher temperatures and the presence of CO2
during
calcination would cause the sorbent to sinter. This is in agreement with the
results of
multiple carbonation-calcination cycle tests for dolomite by Harrison and co-
workers
18
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
(Lopez Ortiz and Harrison, 2001) in pure CO2 stream (800-950 C). They
observed a
decrease in "calcium" conversion from 83 % in the 1st cycle to about 69 % in
the 10th
cycle itself. However, a mesoporous high suface area calcium based sorbent
(precipitated calcium carbonate, PCC) developed at OSU has undergone 100 cycle
experiments. The PCC sorbent has shown 85% conversion in the 1st cycle 66.7%
in
the 10th cycle and 45.5% in the 100th cycle towards carbonation. These
experiments
were carried out in a TGA at 700 C in a 10% CO2 stream in the carbonation
cycle
and 100% N2 gas in the calcination cycle, with 30 minute residence times for
each
cycle. Therefore this project aims testing this PCC based sorbent towards
further
enhancing the WGSR and overcoming some of the problems faced by Harrison and
co-workers.
SUMMARY OF THE INVENTION
[0030] The present invention includes a calcium oxide, its usage for the
separation of CO2 from multicomponent gas mixtures and the optimum process
conditions necessary for enhancing the repeatability of the process.
[0031] A preferred method for separating carbon dioxide from a flow of gas
comprising carbon dioxide comprises the steps of: (1) directing the flow of
gas to a
gas-solid contact reactor, the gas-solid contact reactor contains at least one
sorbent
comprising at least one metal oxide; (2) reacting the carbon dioxide with the
at least
one sorbent so as to remove the carbon dioxide from said flow of gas, thereby
converting the at least one sorbent into spent sorbent; (3) calcining the
spent sorbent
so as to liberate the carbon dioxide from the spent sorbent, thereby
regenerating the
sorbent; and (4) repeating the aforementioned steps.
[0032] Although any metal oxide may be employed, it is preferred that the at
least one metal oxide is selected from the group consisting of: ZnO, MgO,
Mn02,
19
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
NiO, CuO, PbO, and CaO. Further, it is preferred that the spent sorbent is a
metal
carbonate.
[0033] It is preferred that the sorbent has a sorption capacity of at least
about
70 grams of carbon dioxide per kilogram of sorbent. However, it is even more
preferred that the sorbent has a sorption capacity of at least about 300 grams
of
carbon dioxide per kilogram of sorbent. Irrespective of the sorption capacity
of the
sorbent, it is preferred that the sorbent has substantially the same sorption
capacity
after calcining as the sorbent had prior to adsorbing the carbon dioxide.
[0034] Although any calcination method may be employed, it is preferred that
the calcining is performed under at least partial vacuum. It is also preferred
that the
calcining is performed by steam.
[0035] The present invention includes facilities practicing the aforementioned
method.
[0036] A method for separating carbon dioxide from a flow of gas comprising
carbon dioxide of the present invention comprises the steps of: (1) directing
the flow
of gas to a first gas-solid contact reactor, the first gas-solid contact
reactor containing
at least one sorbent, the sorbent comprising at least one metal oxide; (2)
reacting the
carbon dioxide in the flow of gas on the sorbent in the first gas-solid
contact reactor
so as to remove the carbon dioxide from the flow of gas; (3) directing the
flow of gas
to a second gas-solid contact reactor when the sorbent in the first gas-solid
contact
reactor is spent thereby forming spent sorbent, the second gas-solid contact
reactor
containing at least one sorbent, the sorbent comprising at least one metal
oxide; (4)
reacting the carbon dioxide in the flow of gas on the sorbent in the second
gas-solid
contact reactor so as to remove the carbon dioxide from the flow of gas; (5)
calcining
the spent sorbent from the first gas-solid contact reactor so as to generate
carbon
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
dioxide and to regenerate the sorbent; (6) directing the flow of gas to the
first gas-
solid contact reactor when the sorbent in the second gas-solid contact reactor
is
spent, thereby forming spent sorbent; and (7)calcining the spent sorbent from
the
second gas-solid contact reactor so as to generate carbon dioxide and to
regenerate
the sorbent.
[0037] Although any calcination method may be employed, it is preferred that
the calcining is performed under at least partial vacuum. It is also preferred
that the
calcining is performed by steam. This applies to both gas-solid contact
reactors.
[0038] Although any metal oxide may be utilized, it is preferred that the at
least one metal oxide is selected from the group consisting of: ZnO, MgO,
Mn02,
NiO, CuO, PbO, and CaO.
[0039] It is preferred that the sorbent has a sorption capacity of at least
about
70 grams of carbon dioxide per kilogram of sorbent. However, it is even more
preferred that the sorbent has a sorption capacity of at least about 300 grams
of
carbon dioxide per kilogram of sorbent. Irrespective of the sorption capacity
of the
sorbent, it is preferred that the sorbent has substantially the same sorption
capacity
after calcining as the sorbent had prior to adsorbing the carbon dioxide.
[0040] The present invention also includes facilities practicing the
aforementioned method
[0041] A method for regenerating a spent sorbent for carbon dioxide of the
present invention comprises the steps of: (1) providing a spent sorbent, the
spent
sorbent comprising metal carbonate; and (2) calcining the spent sorbent so as
to
liberate carbon dioxide gas and so as to regenerate the spent sorbent thereby
forming a sorbent comprising a metal oxide.
21
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
[0042] It is preferred that the spent sorbent is calcium carbonate. It is
further
preferred that the metal oxide is calcium oxide.
[0043] It is preferred that the sorbent has substantially the same sorption
capacity after calcining as the sorbent had prior to adsorbing the carbon
dioxide.
[0044] Although any calcination method may be employed, it is preferred that
the calcining is performed under at least partial vacuum. It is also preferred
that the
calcining is performed by steam. This applies to both gas-solid contact
reactors.
[0045] The present invention includes facilities practicing the aforementioned
method.
[0046] A method for producing a sorbent of the present invention comprises
the steps of: (1) obtaining a structurally altered high surface area calcium
carbonate
having a surface area of at least 25.0 m2/g, a pore volume of at least 0.05
cm3/g, and
a mesoporous pore size distribution; and (2) calcining the structurally
altered high
surface area calcium carbonate so as to produce a sorbent having a surface
area of
less than 22 m2/g, a pore volume of at least 0.005 cm3/g, and a mesoporous
pore
size distribution.
[0047] Although any calcination method may be employed, it is preferred that
the calcining is performed under at least partial vacuum. It is also preferred
that the
calcining is performed by steam. This applies to both gas-solid contact
reactors.
[0048] The present invention includes sorbents made according to the
aforementioned method.
[0049] A sorbent according to the present invention comprising calcium
oxide having a surface area of at least 12.0 m2/g and a pore volume of at
least 0.015
cm3/g, the calcium carbonate sorbent having sorption capacity of at least
about 70
grams of carbon dioxide per kilogram of sorbent.
22
CA 02613698 2012-05-09
[0049A] Various embodiments of this invention provide a method for separating
carbon dioxide and sulfur dioxide from a flow of gas comprising carbon dioxide
and
sulfur dioxide, said method comprising the steps of: (a) directing said flow
of gas to
a gas-solid contact reactor, said gas-solid contact reactor containing a
sorbent
comprising a metal oxide; (b) reacting said carbon dioxide and said sulfur
dioxide
with said sorbent at a temperature in the range of from about 600 to about 650
degrees Celsius so as to convert at least a portion of said sorbent to a metal
carbonate and at least a portion of said sorbent to a metal sulfate; (c)
directing at
least a portion of said metal carbonate to a calcinator; (d) calcining said
metal
carbonate so as to form said metal oxide and carbon dioxide; and (e)
replenishing
said sorbent in said gas-solid reactor with said metal oxide formed in said
calcinator. In some embodiments, the calcining may occur at a temperature in
the
range of from about 800 to about 850 degrees Celsius. The step of calcining
the
metal carbonate may be conducted under at least partial vacuum. The step of
calcining the metal carbonate may also be performed by steam. The metal oxide
may be selected from the group consisting of ZnO, MgO, MnO2, NiO, CuO, PbO,
and CaO. The metal oxide may be calcium oxide having a surface area of less
than 22 m2/g, a pore volume of at least 0.005 cm3/g, and a mesoporous pore
size
distribution. Also included is a facility practicing such methods of
separating carbon
dioxide and sulphur dioxide from a flow of gas, the facility comprising: a
source of
gas including carbon dioxide and sulfur dioxide; and a gas-solid reactor in
communication with a calcinatory.
22a
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
[0050] In addition to the novel features and advantages mentioned above,
other objects and advantages of the present invention will be readily apparent
from
the following descriptions of the drawing(s) and preferred embodiment(s).
BRIEF DESCRIPTION OF THE DRAWINGS
[0051] Figure 1 depicts the Gibbs Free Energy diagram for the carbonation
reaction, CaCO3->CaO + C02, as a function of temperature.
[0052] Figure 2 illustrates the performance of calcium oxide for the
carbonation reaction.
[0053] Figure 3 compares the XRD diffractograms of CaO derived from
various precursors.
[0054] Figure 4 is a schematic diagram of a carbonator reactor for the
synthesis of precipitated calcium carbonate.
[0055] Figure 5 shows the change in the pH of the slurry as a function of
Ca(OH)2 loading. (500 mL water, 0.0575% N40V dispersant, 4 scfh CO2).
[0056] Figure 6 depicts the effect of Ca(OH)2 loading on the morphology of
Precipitated Calcium Carbonate (PCC) (500 mL water, 0.0575% N40V dispersant,
4
scfh CO2).
(0057] Figure 7 compares the pore size distribution of four CaO precursors.
[0058] Figure 8 compares the conversion of four CaO sorbents under pure
CO2 at 650 C.
[0059] Figure 9 illustrates the effect of temperature on the carbonation of
PCC-CaO.
[0060] Figure 10 illustrates the carbonation-calcination cycles on Aldrich
CaCO3 and PCC at 700 C.
23
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
[0061] Figure 11 shows extended carbonation-calcination cycles on
precipitated calcium carbonate (PCC) powder at 700 C.
[0062] Figure 12 compares the effect of initial surface area of PCC-CaO to its
reactivity towards the carbonation reaction at 700 C.
[0063] Figure 13 depicts the effect of vacuum calcination on the reactivity of
PCC-CaO towards the carbonation reaction at 700 C.
[0064] Figure 14 provides a flow sheet depicting the integration of the
current
process in the overall coal-gasifier electric production facility.
[0065] Figure 15 illustrates thermodynamic data for predicting the temperature
zones for hydration and carbonation of CaO.
[0066] Figure 16 illustrates thermodynamic data for predicting the equilibrium
H2S concentration for CaO sulfidation with varying steam concentration (PTotal
-1
atm).
[0067] Figure 17 shows a modified reactor set-up with steam generating unit
for investigating WGS and carbonation reactions.
[0068] Figure 18 illustrates the set-up for combined vacuum/sweep gas
calcination experiments allowing the use of larger sorbent samples.
[0069] Figure 19 is a pore size distribution of the HTS and LTS obtained from
BET analysis.
[0070] Figure 20 shows the pore size distribution of various calcium oxide
precursors.
[0071] Figure 21 shows the effect of reaction temperature on the CO
conversion (0.5 g HTS catalyst, 3% CO, H20/CO ration = 3, total flow = 1.5
slpm).
[0072] Figure 22 shows the extent of reaction equilibrium as a function of
temperature for the WGS reaction.
24
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
[0073] Figure 23 is a breakthrough curve of CO conversion using a PCC-HTS
catalyst system (T=600C, 3% CO, 9% H2O, Total flow = 1.5 slpm).
[0074] Figure 24 is a breakthrough curve of CO conversion using a LH-HTS
catalyst system (T=600C, 3% CO, 9% H2O, total flow = 1.5 slpm).
[0075] Figure 25 provides a comparison of breakthrough curves for PCC-HTS
and LH-HTS systems (T=600C, 3% CO, 9% H2O, Total flow = 1.5 slpm).
[0076] Figure 26 depicts a typical steam generation scenario and use.
[0077] Figure 27 depicts one implementation of one embodiment of the
present invention.
[0078] Figure 28 depicts one implementation of one embodiment of the
present invention.
[0079] Figure 29 depicts one implementation of one embodiment of the
.present invention.
[0080] Figure 30 depicts one implementation of one embodiment of the
present invention.
[0081] Figure 31 depicts one implementation of one embodiment of the
present invention.
[0082] Figure 32 depicts one implementation of one embodiment of the
present invention.
[0083] Figure 33 depicts one implementation of one embodiment of the
present invention.
[0084] Figure 34 depicts one implementation of one embodiment of the
present invention.
[0085] Figure 35 depicts one implementation of one embodiment of the
present invention.
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
[0086] Figure 36 depicts one implementation of one embodiment of the
present invention.
[0087] Figure 37 depicts one implementation of one embodiment of the
present invention.
[0088] Figure 38 illustrates thermodynamic data for predicting the temperature
zones for sulfation of CaO as well as the direct sulfation of CaCO3.
(Sulfation was
considered at 1 atm total pressure, 4% 02 and 10% C02)
[0089] Figure 39 illustrates C02 capture capacity of various high temperature
sorbents over multiple carbonation-regeneration cycles.
[0090] Figure 40 provides a typical curve for combined carbonation and
sulfation of PCC-CaO for 3 cycles at 700 C for a residence time of 5 minutes
(3000
ppm Sot, 10% C02, 4% 02)
[0091] Figure 41 shows the effect of residence time on the extent of
carbonation (initial amount of CaO) of PCC-CaO for multiple cycles at 700 C
(3000
ppm S02, 10% C02, 4% 02)
[0092] Figure 42 shows the effect of residence time on the extent of sulfation
(initial amount of CaO) of PCC-CaO for multiple cycles at 700 C (3000 ppm S02,
10% C02, 4% 02)
[0093] Figure 43 shows the effect of residence time on the ratio of
carbonation
to sulfation of PCC-CaO for multiple cycles at 700 C (3000 ppm S02, 10% C02,
4%
02)
[0094] Figure 44 illustrates the extent of carbonation of PCC-CaO for multiple
cycles at 700 C (10% C02, 4% 02)
[0095] Figure 45 illustrates the extent of sulfation of PCC-CaO for multiple
cycles at 700 C (10%CO2, 4% 02)
26
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
[0096] Figure 46 shows the effect of residence time on the ratio of
carbonation
to sulfation of PCC-CaO for multiple cycles at 700 C for varying S02
concentrations
(3000 - 100 ppm So2, 10% Co2, 4% 02)
[0097] Figure 47 shows the effect of reaction temperature on the ratio of
carbonation to sulfation for increasing residence time (10% Co2, 3000 ppm S02)
[0098] Figure 48 illustrates the effect of reaction temperature on the extent
of
carbonation of PCC-CaO for increasing residence time (10% C02, 3000 ppm S02)
[0099] Figure 49 illustrates the effect of reaction temperature on the extent
of
sulfation of PCC-CaO for increasing residence time (10% C02, 3000 ppm S02.)
[00100] Figure 50 provides a flow sheet for the integration of the CCR process
in a coal fired utility.
[00101] Figure 51 illustrates the equilibrium partial pressure of C02 as
obtained
by thermodynamics (0 -1 atm)
[00102] Figure 52a illustrates a direct fired calcination configuration in
accordance with one embodiment.
[00103] Figure 52b illustrates an indirect fired calciner configuration in
accordance with one embodiment.
[00104] Figure 53 illustrates a schematic diagram of one embodiment of a
rotary calciner reactor set-up.
[00105] Figure 54 shows the effect of temperature on LC calcination rate
(sample size: 500 mg; T: 700 - 750 C; Pvac: 25" Hg; FSG(N2) = 50 ml/min)
[00106] Figure 55 shows the effect of temperature on the rate of PCC
calcination (sample size: 500 mg; T: 700 - 750 C; Pvac: 25" Hg; FSG(N2) = 50
ml/min)
[00107] Figure 56 shows the effect of vacuum on PCC calcination (Sample
size: 500 mg; T: 750 C; Pvac: 10 - 25" Hg; FSG(N2) = 50 ml/min)
27
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
[00108] Figure 57 shows the effect of sweep gas flow (FSG) (Sample size: 1 Og,
T: 880 C; PvAC: 28" Hg; FSG(He) = 0 - 1000 mI/min)
[00109] Figure 58 shows the effect of diluent gas type (He, N2, Ar) (Sample
size: 10000 mg, T: 800 C; PvAC: 28" Hg; FSG(He, N2, Ar) = 120 ml/min)
DETAILED DESCRIPTION OF THE PREFERRED EMBODIMENT(S)
[00110] In accordance with the foregoing summary, the following presents a
detailed description of the preferred embodiment(s) of the invention that are
currently
considered to be the best mode.
Chemicals, Sorbents and Gases
[00111] Naturally occurring limestone (CaCO3) and hydrated lime (Ca(OH)2),
synthesized from it were obtained from Linwood Mining and Minerals. Dolomite
(CaCO3=MgCO3) was procured from the National Dolomite Company. The purity of
these ores was above 90%. High purity metal oxides such as ZnO, MgO, Mn02,
NiO,
CuO, PbO, CaO were obtained from Aldrich Chemical Company. Precipitated
calcium carbonate (PCC) was synthesized from Linwood hydrate by the procedure
described in a following section. N40V dispersant, a sodium salt of a
carboxylic
acid, used in the synthesis of PCC was obtained from Allied Colloid. The
synthesis
procedure is described in detail in a following section. N2 and CO2 used for
calcination and carbonation experiments were 99.999% and 99.9% pure,
respectively.
Sorbent Reactivity Testing and Structural Analysis
[00112] The reactivity testing of CaO sorbents for carbonation was carried out
in a Perkin Elmer Thermogravimetric Analyzer (TGA-7) apparatus. The balance
can
accurately measure up to 1 microgram. A small sample of the sorbent (5-20 mg)
is
placed in a quartz boat. The weight of the sample was recorded every second.
The
28
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
structural properties of CaO sorbents and their precursors were tested in a
NOVA
2200 analyzer (Quantachrome Company). The BET surface area, pore volume, and
pore size distribution were measured at -196 C using nitrogen as the
adsorbent.
Screening of Metal Oxides
[00113] Metal oxides such as ZnO, MgO, CuO, Mn02, NiO, PbO and CaO that
undergo the CCR scheme in the 800-200 C temperature range were analyzed for
their reactivity in a TGA. A powdered sample of these oxides was placed in a
quartz
pan and pure CO2 was passed over the sample metal oxide. The temperature was
then slowly raised and the weight of the sample was continuously monitored. An
increase in the weight of the sample is an indication of the formation of
metal
carbonate. Figure 2 provides experimental data for the carbonation of lime
(Ca(OH)2)
under flowing pure CO2 gas. With an increase in temperature, the weight of the
sample increases till the temperature reaches about 890 C. Calcination, which
is
thermodynamically favored above 890 C at 1 atm CO2 partial pressure, causes a
rapid decrease in weight until the sorbent converts completely to CaO. When
the
sample is reheated, the weight starts to increase again and the process is
repeated
once more. Besides proving that CaO is a viable candidate, the data also shows
recyclability of the sorbent.
XRD Analysis of CaO obtained from its precursors:
[00114] CaO was identified as a viable candidate for the carbonation-
calcination reactions. However, a variety of precursors can be calcined to
obtain the
CaO sorbents necessary for the carbonation reaction. Common and economical
precursors include calcium carbonate, calcium hydroxide and dolomite. The
other
important source of CaO is via the calcination of synthesized high surface
area
precipitated calcium carbonate. In order to compare the crystal structure of
the CaO
29
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
sorbents obtained from these sources, XRD patterns were obtained on all the
CaO
sorbents. Figure 3 depicts these diffractograms (a. Calcined Aldrich-CaO; b.
Dolomite-CaO; c. Ca(OH)2-CaO); d. PCC-CaO; e. Limestone-CaO; and f. Aldrich-
CaO). From this figure we can conclude that the crystal structure of the CaO
sorbents obtained from numerous sources is identical. Only the XRD pattern
corresponding to dolomite-derived CaO shows extra peaks due to the presence of
MgO in the calcined dolomite. Based on the similarity in all the CaO
structures, it can
be assumed that any difference in reactivity of CaO for carbonation is an
artifact of
the sorbent morphology and not due to the chemistry of the gas-solid reaction
that
occurs on the CaO surface.
Precipitated Calcium Carbonate (PCC) synthesis
[00115] Structurally altered high surface area CaO precursors were
synthesized based on the procedure outlined elsewhere (Fan, L.-S.; Ghosh-
Dastidar,
A.; Mahuli, S.; Calcium Carbonate Sorbent and Methods of Making the Same. US
Patent # 5,779,464 and Agnihotri, R.; Chauk, S.; Mahuli, S.; Fan, L.-S.
Influence of
Surface Modifiers on Structure of Precipitated Calcium Carbonate. Ind. Eng.
Chem.
Res. 1999, 38, 2283-2291). A schematic diagram of the slurry bubble column
used
for this purpose is shown in Figure 4. The carbonator 40 consists of a 2" OD
Pyrex
tube 40a. A porous frit 40d at the bottom, disposed over glass beads 40f,
provides
good distribution of CO2 40g through the slurry 40c. A K-type thermocouple 40h
inserted in the slurry continuously records the slurry temperature. A pH probe
40b
monitors the pH of the slurry as the reaction medium changes from a basic to
an
acidic solution as the reaction proceeds. First, 500 ml of distilled water is
poured into
the carbonator, followed by the addition of 0.0575g of N40V . 12.8g of Ca(OH)2
is
added to the solution to provide a loading of 2.56% by weight. This
corresponds to a
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
concentration of 16-sat (concentration of Ca(OH)2 is 16 times its saturation
solubility
limit). The solubility of Ca(OH)2 (- 0.16 g/1 00g water) leads to a pH of 12
at the start
of the experiment. The remaining Ca(OH)2 remains suspended in the solution.
The
ratio of N40V and Ca(OH)2 loading is chosen to create a surface charge of
zero on
the incipiently formed CaCO3 particles. The flow of CO2 40e into the
carbonator is
then started and the pH was continuously monitored. Figure 5 shows the change
in
pH with reaction time as a function of Ca(OH)2 loading. CO2 dissolved in water
provides carbonate ions that react with Ca" ions to form CaCO3 according to
the
reaction below:
Ca 2+ + C032" -3 CaCO3 (3)
CaCO3 has a much lower solubility in water (0.0012 g/1 00g water) compared to
Ca(OH)2 and thus precipitates out. As the reaction proceeds, Ca 2+ ions get
depleted,
but are continuously replenished by the suspended Ca(OH)2. Hence the pH
remains
12. As the reaction proceeds, Ca(OH)2 ultimately gets depleted and the
concentration of Ca 2+ ions cannot be maintained at its solubility limit. On
the other
hand, continued dissolution of CO2 gas leads to the accumulation of H+ ions
causing
the solution to become acidic. Eventually, the pH settles at about 6.0,
corresponding
to equilibrium solubility of CO2 in water at ambient temperature. This also
signals the
end of the carbonation of all Ca(OH)2. The slurry is then removed from the
precipitator, vacuum filtered and stored in a vacuum oven at 90-110 C for 20
hours
to completely remove the moisture. Higher Ca(OH)2 loading requires more
reaction
time as evident from Figure 5.
Effect of the ratio of Ca(OH)2 and dispersant on PCC morphology.
[00116] Precipitated calcium carbonate can be obtained by the reaction
between carbonate and calcium ions in solution. It is known that the CaCO3
nuclei
31
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
that precipitate out have positive surface charge on them that prevent
agglomeration
(Agnihotri, R.; Chauk, S.; Mahuli, S.; Fan, L.-S. Influence of Surface
Modifiers on
Structure of Precipitated Calcium Carbonate. Ind. Eng. Chem. Res. 1999, 38,
2283-
2291). The resulting structure is also microporous in nature. However, the
structural
properties of the synthesized PCC can be altered by the use of negatively
charged
dispersants that neutralize the surface charges. This makes the ratio between
the
Ca(OH)2 loading and the dispersant used very critical. Besides, the effect of
Ca(OH)2
loading in the slurry was studied to enhance the productivity of the
precipitation
process by synthesizing more PCC from the same slurry volume. 8-sat, 16-sat
and
24-sat were used as Ca(OH)2 loading levels, all other factors remaining
constant. It
can be seen from Figure 6 and Table 1 that at a concentration of 8-sat, there
is
proportionally more dispersant in the slurry causing the incipiently formed
CaCO3
particles to be negatively charged. The negative charge prevents the
agglomeration
of these nuclei eventually leading to the formation of microporous PCC as
shown in
Figure 6. Its surface area is also relatively lower. At a Ca(OH)2 loading
corresponding to 16-sat, the ratio of N40V and CaCO3 is balanced and the
surface
charge on the nuclei is zero. This allows optimal association of these nuclei
leading
to a predominantly mesoporous structure. The SA of PCC under these optimum
conditions is also the highest at 38.3 m2/g. As the loading of Ca(OH)2 is
raised to 24-
sat, there is not enough N40V dispersant to neutralize the surface charge on
all the
incipiently formed nuclei. There could possibly be some positively charged
particles.
This again creates non-optimum conditions leading to a loss in SA and PV
compared
to the 16-sat case. Another experiment was conducted to process a 32-sat
Ca(OH)2
slurry keeping the Ca(OH)2 to N40V ratio constant. The SA/PV of PCC
synthesized
from a 32-sat slurry was 37.07 m2/g and 0.139 cm3/g respectively; lending
support to
32
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
the fact that higher mass of PCC can be synthesized from the same amount of
slurry.
Ca(OH)2 loading Surface Area Pore Volume
weight % (m2/g) (cm3/g)
1.28 13.8 0.03
2.56 38.3 0.14
3.84 36.8 0.11
Table 1: Morphological properties of PCC as a function of N40V :Ca(OH)2
loading
ratio (500 ml water, 0.0575% N4OV dispersant, 4 scfh C02)-
Pore Structure of CaO sorbents
[00117] CaO sorbents were synthesized by calcining various CaO precursors
such as Linwood calcium carbonate (LC), dolomite (DL), Linwood calcium
hydroxide
(LH), and precipitated calcium carbonate (PCC). For convenience, the oxides
derived from these sources are termed as LC-CaO, FCD-CaO (for fully calcined
dolomite-CaO), LH-CaO, and PCC-CaO, respectively. The procedure involved
heating the precursor in flowing nitrogen beyond the calcination temperature
(800-
950 C) for an hour followed by its storage in a desiccator. Structural
properties such
as surface area (SA) and pore volume (PV) of these chemicals are listed in
Table 2
and their pore size distributions are shown in Figure 7. The SA of naturally
occurring
minerals, LC and dolomite was very low, 1.06 and 1.82 m2/g, respectively. LH
was
synthesized by first calcining the LC followed by its hydration. LH exhibited
a
considerably higher SA (13.19 m2/g) and PV compared to the LC. The SA of PCC
(38.3 m2/g), however, was the highest among all precursors. From Figure 5, we
can
infer that the structures of LC, DL and LH are predominantly microporous in
nature.
Most of the porosity lies in pores below 5 nm in diameter. In contrast, the
maximum
in PV occurs at 15 nm for PCC and most of its PV originates from mesopores in
the
5-25 nm range.
33
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
Sorbent Name Surface Area Pore Volume
(m2ig) (cm3/g)
LC 1.1 0.003
LC-CaO 17.8 0.078
Dolomite 1.8 0.004
FCD-CaO 29.8 0.08
LH 13.2 0.0453
LH-CaO 33.3 0.1
PCC 38.3 0.11
PCC-CaO 12.8 0.027
Table 2: Morphological properties (surface area and pore volume) of various
CaO
sorbents and their precursors.
Carbonation of CaO sorbents
[00118] The performance of these four CaO sorbents was tested in a TGA. The
experimental procedure consisted of placing 6-12 mg of the chosen CaO sorbent
in
a thin layer in a quartz pan to minimize external mass transfer resistances.
The
sorbent was then heated in flowing nitrogen (5.0 grade, 99.999% pure) to the
desired
temperature. The representative temperatures used in these experiments were
550
C, 600 C and 650 C. Once the desired temperature was reached, the flow was
switched to 100% CO2 stream. The increase in weight with time was recorded and
the conversion of CaO to CaCO3 was calculated from the increase in weight.
Only
the data obtained at 650 C is reported here. The performance of the four CaO
sorbents, LC-CaO, FCD-CaO, LH-CaO and PCC-CaO at 650 C is depicted in
Figure 8. Initially, CO2 diffuses into the pores of the LC-CaO and the
reaction takes
place on the CaO surface provided by the pores. The figure shows that there is
a
rapid increase in weight in the first 1-2 minutes. The conversion attained in
this
kinetically controlled regime depends on the initial surface area of the CaO
sorbent.
LC-CaO and FCD-CaO attained 40-45% conversion, while LH-CaO and PCC-CaO
34
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
attained about 60% and 54% conversion, respectively, in this regime. After
this
regime, conversion increases relatively slowly with time. The increase in
conversion
is only about 2-4% in the next hour for LC-CaO and FCD-CaO. This confirms the
susceptibility of micropores to pore filling and pore pluggage described
earlier due to
the formation of a higher volume product, CaCO3. The trend is not as dramatic
for
the case of LH-CaO because of its relatively higher initial surface area. The
conversion for LH-CaO increases by another 18% in the diffusion controlled
regime.
However, the increase in conversion for PCC-CaO is about 34-36% more in the
second regime. Since the PCC-CaO structure is mesoporous, the formation of
CaCO3 product layer is not able to plug all the pore mouths. This in turn
allows the
heterogeneous reaction to occur on a larger CaO surface. Once the kinetically
controlled regime is over, diffusion of ions occurs through a larger area,
ultimately
leading to a higher conversion of 88-90% for PCC-CaO. Figure 9 shows the
effect of
temperature on the carbonation of PCC-CaO. It can be seen that the extent of
conversion in the kinetic regime is different at different temperatures.
However,
unlike LC-CaO, the conversion at any temperature does not seem to taper off
and
given sufficient time, PCC-CaO is capable of attaining 90% or higher
conversion at
all of these temperatures.
Cyclic calcination and carbonation
[00119] One of the possible hurdles in the utilization of metal oxides for the
carbonation and calcination reaction scheme is its vulnerability to sintering
due to the
thermal cycling imposed by the cyclical nature of these reactions. Cyclical
studies
were carried out to quantify any loss in reactivity of these sorbents upon
multiple
cycles. The temperature chosen for cyclical studies was 700 C. This
temperature is
sufficient to achieve carbonation in the presence of pure CO2, and also to
calcine the
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
CaCO3 formed after the gas is switched from CO2 to N2. A variety of precursors
were first calcined in nitrogen at 700 C. The gas was then switched to pure
CO2 and
the weight gain continuously tracked. After reaching the ultimate conversion,
the gas
was switched back to N2. This process was repeated for 2-3 cycles. The data
obtained on Aldrich CaCO3 PCC undergoing this cyclical study is shown in
Figure 10. It can be seen that the reactivity of Aldrich CaCO3 a gradual
decrease even after the first cycle. In contrast, PCC completely regained its
mass
after the first calcination and carbonation cycle. At 700 C, we can deduce
that the
conversion is almost complete (>95%). The figure also shows that the
reactivity did
not decrease in the second cycle either. Under the reaction conditions chosen,
any
sintering did not seem to adversely affect the sorbent morphology. We
continued an
extended study of eleven calcination and carbonation cycles lasting over three
days
on PCC. The data is provided in Figure 11. It can be seen that the sorbent
reactivity
remained high and if enough reaction time is provided, the conversion could
reach
beyond 90% in every cycle. This is a positive result for the structural
viability of this
sorbent under multiple cycles.
Effect of Vacuum Calcination
[00120] The effect of initial surface area of CaO sorbents was studied. CaO
sorbents were synthesized from PCC under different calcination conditions. The
role
of surface area on the extent of carbonation is shown in Figure 12. Different
surface
area PCC-CaO sorbents were synthesized by the calcination of PCC at a range of
calcination temperature to induce varying degrees of sintering. It can be seen
that a
higher initial surface area (and its associated pore volume) leads to higher
reactivity
and conversion. Thus, it is necessary to identify calcination conditions that
optimize
the SA/PV and pore size distribution of PCC-CaO. It has been suggested in
literature
36
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
that CaO procured from the calcination of limestone under vacuum has a higher
reactivity. It was observed that under air calcination at 650 - 800 C, sharp
edges of
calcite powder were replaced by rounded surfaces and neck areas indicating
severe
sintering (Beruto, D., and Searcy, A.W., "Calcium oxides of high reactivity."
Nature,
1976, 263, 221-222). The resulting CaO structure was highly crystalline as
well. In
contrast, the sharp edges of calcite were retained in the CaO obtained under
vacuum. The CaO however did not possess a high degree of crystallinity. The
latter
also showed high reactivity towards hydration. Vacuum calcination leads to the
formation of a metastable-nanocrystalline calcia structure while calcination
in helium
atmosphere lead to a stable microcrystalline calcia structure (Dash, S.,
Kamruddin,
M., Ajikumar, P.K., Tyagi, A.K., and Raj, B., "Nanocrystalline and metastable
phase
formation in vacuum thermal decomposition of calcium carbonate." Thermochimica
acta, 2000, 363, 129-135). Beruto et al., [1980] estimated the surface area
and pore
volume of limestone based CaO to be about 78-89 m2/g and 0.269 ml/g
respectively.
[00121] The effect of vacuum calcination was studied in this process. The
surface area of Linwood carbonate increased from 17.79 to 21.93 m2/g and pore
volume from 0.07815 to 0.1117 ml/g for calcination under nitrogen and under
vacuum, respectively. Similar enhancements were observed for PCC based CaO
sorbents as well. It has been observed that PCC-CaO is susceptible to high
degree
of sintering and the surface area of the sorbent falls off rapidly.
Calcination in
nitrogen resulted in surface areas below 13 m2/g repeatedly. However, vacuum
calcination lead to a surface area of 19.84 m2/g and 0.04089 ml/g pore volume.
The
carbonation characteristics are shown in Figure 13.
[00122] Vacuum calcination of PCC followed by the carbonation of PCC-CaO
was repeated over two cycles. PCC was first vacuum calcined to CaO-1 at 750
C.
37
CA 02613698 2011-01-31
Ca0-1 was carbonated to CC-2 at 700 C followed by its vacuum decomposition to
CaO-2 that is carbonated to CC-3. The values of surface area and pore volume
of
the sorbent at various stages are provided in Table 3 below:
Surface Area Pore Volume
(m2/g) (cc/g)
PCC 38.3 0.1416
CaO-1 12.63 0.02409
CC-2 6.5 0.0103
CaO-2 15.93 0.04008
CC-3 2.361 0.004483
Table 3: Structural properties of Calcium based sorbents undergoing vacuum
calcination at 750 C and carbonation at 700 C.
[00123] The data shows that PCC is susceptible to sintering because the CaO
obtained in the first cycle has a surface area of only 12.63 m2/g compared to
38.3
m2/g of PCC. As expected, pore filling leads to a drop in both properties when
CaO
1 carbonates. The extent of carbonation was beyond 90%. However, it can be
seen
that the SA of CaO obtained after the second vacuum calcination step, CaO 2,
is
15.93 m2/g, which is higher than the SA'of CaO 1. The pore volume of CaO 2 is
also
higher than that of CaO 1. These results prove that there is no systematic
decline in
SA and PV of sorbents with increasing calcination-carbonation cycles and that
this
combination is capable of providing a sustained conversion over many cycles.
[00124] See the article "Carbonation-Calcination Cycle Using High Reactivity
Calcium Oxide for Carbon Dioxide Separation from Flue Gas" by Himanshu Gupta
and Liang-S. Fan, published on the web July 11, 2002 by Ind. Eng. Chem. Res.
2002, 41, 4035-4042.
38
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
Enhanced Hydrogen Production Integrated with C02
Separation in a Single-Stage Reactor
[00125] A variety of chemical processes known to generate syngas include:
Steam Gasification: C + H2O -> CO + H2 (X)
Steam Methane Reforming: CH4 + H2O -> CO +3H2 (X)
Partial oxidation of Hydrocarbon: CXHy +02 -- CO + H2 (X)
[00126] The flow sheet shown in Figure 14 integrates the Calcium-based
reactive separation process under development in this project with a coal
gasifier
based electric power/chemical synthesis process plant 140. The main coal
gasifier
140a consists of a high pressure and high temperature unit that allows contact
between coal 140b, steam 140e and air/pure oxygen 140y in a variety of
schemes.
Boiler feed water 140d is preheated by passing it through gasifier 140a prior
to
steam tubine 140c. Waste from the gasifier is collected as slag 140z. Typical
fuel
gas compositions from various known coal gasifiers are shown in Table 4. Once
the
water gas mixture is formed at the exit of the gasifier 140a, CaO fines are
injected
140f into the gas duct that react with the CO2 present in the gas mixture
leading to
the formation of solid CaCO3. As the fuel gas flows past the WGS catalyst
monoliths
140g, the WGS reaction is effected forming more CO2 in the process. The
entrained
CaO particles react with the incipiently formed CO2 gas, thereby allowing
further
catalysis of the WGS reaction to occur. This process can be tailored to attain
as high
a H2 concentration as possible. At the exit of the WGS reactor, the reacted
CaCO3
particles are captured using a high temperature solids separator 140h (e.g., a
candle
filter or a high temperature ESP) and separated fuel gas stream. The spent
solids
are now sent to a rotary calciner 140k to thermally decompose the CaCO3 140j
back
to CaO 140f and pure CO2 140m. The high purity CO2 gas can now be economically
compressed 1401, cooled, liquefied and transported for its safe sequestration
140m.
39
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
The rotary calciner allows the calcium particles to remain segregated, which
is
crucial in maintaining a sorbent structure characterized by a higher porosity.
It was
previously observed in our studies that heaping of calcium sorbents leads to a
lower
porosity and consequently a lower reactivity over the next carbonation cycle.
The
calcination of the sorbent can also be effected under sub-atmospheric
conditions that
allow the removal of CO2 as soon as it is formed from the vicinity of the
calcining
sorbent, thereby aiding further calcination. This vacuum can be created by
means of
ejector systems that are widely used in maintaining vacuum in large vacuum
distillation units (VDU) in the petroleum refining industry. Lock and hopper
combinations and appropriate seals ensure that the sorbent can be effectively
separated from the CO2 stream and re-entrained in the fuel gas duct. The
hydrogen
enriched fuel gas 140i can now be used to generate electric power in a fuel
cell 140n
or used to make fuels and chemicals 140q without any low temperature clean up.
The fuel cell may receive a supply of air 140p and discharge steam 1400. The
hydrogen enriched fuel gas may be sent to gas turbine 140r used to drive
generator
140t to produce electricity and air compressor 140s to produce a stream of
compressed air. The stream of compressed air may be sent to air separator 140x
to
produce the air/oxygen of 140y. The discharge from gas turbine 140t may be
sent
through heat exchanger 140u prior to being discharged at stack 140v. The
absorbed
heat may be collected by steam turbine 140w to produce additional electricity.
Thermodynamic analysis
[00127] Primarily three important gas-solid reactions can occur when calcium
oxide (CaO) is exposed to a fuel gas mixture obtained from coal gasification.
CaO
can undergo hydration, carbonation and sulfidation reactions with H2O, CO2 and
H2S, respectively. These can be stoichiometrically represented as:
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
Hydration: CaO + H2O I Ca(OH)2 (5)
Carbonation: CaO + CO2 1 CaCO3 (6)
Sulfidation: CaO + H2S I CaS + H2O (7)
[00128] All these reactions are reversible and the extent of each of these
reactions depends on the concentrations of the respective gas species and the
reaction temperature. Detailed thermodynamic calculations were performed to
obtain
equilibrium curves for the partial pressures of H2O (PH2O), CO2 (PCO2) and H2S
(PH2S) as a function of temperature, for the hydration, carbonation, and
sulfidation
reactions using HSC Chemistry v 5.0 (Outokumpu Research Oy, Finland). The
equilibrium calculations were based on the fuel gas compositions that are
typical of
the different types of coal gasifiers. The details of the fuel gas mixtures
are illustrated
in Table 4.
Table 4: Typical fuel gas compositions obtained from different gasifiers.
(Stultz and Kitto, 1992)
Moving Moving Bed Fluidized Entrained Entrained
Bed, dry slagging Bed Flow, slurry Flow, dry
Oxidant air Oxygen Oxygen Oxygen Oxygen
Fuel Sub
tuminous Bituminous Lignite Bituminous Bituminous
B
Pressure 295 465 145 615 365
(psi)
CO 17.4 46 48.2 41 60.3
H2 23.3 26.4 30.6 29.8 30
CO2 14.8 2.9 8.2 10.2 1.6
H2O 16.3 9.1 17.1 2
N2 38.5 2.8 0.7 0.8 4.7
CH4+ 5.8 4.2 2.8 0.3
HCs
H2S + 0.2 1.1 0.4 1.1 1.3
COS
[00129] The relationship between the reaction temperature and the equilibrium
partial pressures of H2O and CO2 for the hydration and carbonation reactions
are
41
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
shown in Figure 15 (a). For a typical gasifier moisture composition ranging
from 12-
0
20 atm (PH2O) the hydration of CaO occurs for all temperatures below 550-575
C,
respectively. By operating above these temperatures, the CaO-hydration can be
prevented. Figure 15 (b) shows the typical equilibrium C02 partial pressures
(PCO2)
as a function of temperature. From the data in Table 4, it can be inferred
that the
typical PCO2 in the gasifiers ranges from 0.4-4.3 atm for entrained flow
(slurry) and
entrained flow (dry) gasifier systems respectively. The equilibrium
temperatures
0
corresponding to those PCO2 lie in the 830-1000 C range as shown in Figure
15(b).
Thus, by operating below these temperatures, we can effect the carbonation of
CaO.
For the reversible sulfidation of CaO (eqn 7) the thermodynamic calculations
depend
on the concentration of moisture in the system. Hence, Figure 16 depicts the
equilibrium H2S concentrations in ppm for varying moisture concentrations
(PH2O)
0
and 30 atm total pressure. For a typical operating temperature range of 800-
1000 C
the equilibrium H2S concentration is between 5700-1700 ppm respectively for 20
atm
0
PH2O. Consequently, at 800 C we need more than 5700 ppm H2S for the
sulfidation
0
of CaO to occur. This number changes to 570 ppm for a PH2O of 2 atm at 800 C.
Thus, by changing the moisture/steam concentration in the system we can
prevent
the sulfidation of CaO from occurring.
EXPERIMENTAL
Sorbent and Catalyst Characterization
[00130] The high and low temperature water gas shift (WGS) reaction catalysts
were procured from Sud-Chemie Inc., Louisville, KY. The high temperature shift
(HTS) catalysts comprises of iron (III) oxide supported on chromium oxide.
Precipitated calcium carbonate (PCC) was synthesized by bubbling C02 through a
42
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
slurry of hydrated lime. The neutralization of the positive surface charges on
the
CaCO3 nuclei by negatively charged N40V molecules forms CaCO3 particles
characterized by a higher surface area/pore volume and a predominantly
mesoporous structure. Details of this synthesis procedure have been reported
elsewhere (Agnihotri et al., 1999). Hydrated lime from a naturally occurring
limestone
(Linwood Hydrate, LH) and a naturally occurring limestone (Linwood Carbonate,
LC)
was obtained from Linwood Mining and Minerals Co.
[00131] The sorbents and catalyst were analyzed to determine their
morphologies using a BET analyzer. The BET surface areas, pore volumes, and
pore size distributions of the catalysts and sorbents were measured at -196 C
using
nitrogen as the adsorbent in a Nova 2200 Quantachrome BET analyzer. Special
care
was taken to ensure that all samples were vacuum degassed at 250 C for 5
hours
prior to BET analysis.
WGS Reactor Setup
[00132] A reactor setup was designed, underwent several iterations and was
assembled to carry out water gas shift reactions in the presence of CaO and
catalyst. The reactor design assembly used to carry out these experiments is
shown
in Figure 17. This setup enables us to carry out both the water gas shift
reaction in
the presence of CaO as well as the regeneration of the sorbent in flowing gas
such
as nitrogen and/or steam. The setup 170 consists of a tube furnace 170p, a
steel
tube reactor 170a, a steam generating unit 170c, a set of gas analyzers for
the
online monitoring of CO and CO2 concentrations 170n, a condenser 170m to
remove
water from the exit gas stream and a high pressure water syringe pump 170b.
[00133] All the reactant gases (H2, CO, C02, and NO are metered using
modified variable area flowmeters 170e - h respectively. The syringe pump is
used
43
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
to supply very accurate flow-rates of water into the heated zone of the steam-
generating unit in the 0.01-0.5 ml/min range. Once the steam is generated, it
is
picked up by the CO/N2 gas mixture 170i and enters the main reactor where the
sorbent/ catalyst mixture 1700 is loaded. All the lines connecting the steam-
generating unit to the main reactor are heated using heating tapes. The steam
generator is also packed with quartz wool 170d in order to distribute the
water drops
as they enter into the heating zone. The packing is utilized in order to
provide greater
surface for water evaporation and to dampen out fluctuations in steam
formation.
The main problem with a fluctuating steam supply is that the gas analyzers
used to
measure the exit CO and CO2 concentrations are sensitive to gas flow rates.
Even
though the steam is being condensed out before the gas is sent into the
analyzers,
surges in the steam supply still affect the overall gas flow rate, causing the
CO and
CO2 readings to fluctuate. The packing ultimately ensures a more continuous
and
constant overall gas flow rate into the main reactor and into the analyzers.
Thermocouple 170k is used to monitor the temperature inside reactor 170a. Any
extra gas inlets of reactor 170a are blocked 1701.
[00134] A steel tube reactor is used to hold the Ca-based sorbent and
catalyst,
and is kept heated using a tube furnace. The sorbent loading unit of the
reactor is
detachable which enables easy removal and loading of the sorbent and therefore
minimizes the sorbent loading time between runs. Also, the sorbent can be
changed
without having to cool down the entire reactor. The gas mixture 170j entering
the
reactor is preheated to the reaction temperature before contacting the
sorbent/
catalyst particles. The gases exiting the reactor first flow through a
condenser in
order to separate out the moisture and then to a set of gas analyzers.
Sub Atmospheric Calcination Reactor Setup
44
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
[00135] Once the Calcium based sorbent has reacted with the CO2 being
produced by the WGSR, the sorbent has to be regenerated for further use in
subsequent cycles. During the regeneration of the sorbent, carbon dioxide is
released from the sorbent. In order to minimize the necessity for further
treatment of
this released CO2 before sending it to sequestration sites, it is necessary to
regenerate the sorbent such that a pure stream of CO2 is released. Vacuum
calcination provides one method for ensuring that concentrated streams of CO2
are
release in the regeneration phase. The detailed setup is shown in Figure 18.
This
setup 180 was assembled to handle the regeneration of large quantities of
sorbent
(-10-20g per batch). The setup includes an alumina tube reactor 180b, which
would
hold the sorbent samples in a split tube furnace 180c that provides the heat
necessary to calcine the sorbent 180d, two Non Dispersive Infra Red (NDIR)
analyzers 180k -I to monitor the CO2 concentration (ranges 0-2500ppm and 0-
20%)
and two vacuum pumps 180f and 180i. 10g of sorbent yields about 2.4L of CO2 at
atmospheric pressure and temperature over the entire decomposition process:
This
gas needs to be diluted with air in order to ensure that the CO2 concentration
lies in
the detection range of the CO2 analyzers. Vacuum Pump 180f is a dry vacuum
pump procured from BOC Edwards capable of achieving vacuum levels as low as
50mtorr and gas flowrates of 6m3/hr. The C02 analyzers have their own inbuilt
pumps and are capable of drawing up to 2LPM from the header for online CO2
analysis. The second pump 180i is a smaller dry pump and is put in place to
ensure
that there is no pressure buildup in the 1/4" lines connecting the vacuum pump
to the
analyzers. Pump 180i discharges to vent 180j. The temperature of the furnace
is
controlled with a thermocouple inserted into the central zone of the furnace.
The
temperature of the reactor was also monitored using a second thermocouple
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
inserted into the center of the alumina tube. The setup is also capable of
combining
vacuum calcination with flow of sweep gas 180a. As it may not be feasible to
supply
very low vacuum levels for the calcination of the sorbent in industrial
settings, it may
be necessary to study the calcination process in combination with the addition
of
various sweep gases such as N2/ steam. Pressure gauges 180e, h and volumetric
flow meters are included to monitor the vacuum pressure in the reactor, the
pressure
in the 1 " lines and the flows of the sweep gases into the calciner and the
flow of the
air 180g used in the dilution of the exhaust CO2 before sending it to the
analyzers.
The analyzers are also connected to a data acquisition system 180m that can
record
analyzer readings every second.
[00136] INTRODUCTION:
[00137] CaO (s) + C02 (g) -> CaCO3 (s) (Carbonation) AH = - 178 kJ/mol
[00138] CaCO3 (s) --> CaO (s) + C02 (g) (Calcination) AH = + 178 kJ/mol
[00139] Thermodynamics of Calcination
[00140] The thermodynamics of calcination, evaluated by HSC chemistry
software, is represented in the form of equilibrium partial pressure of C02 as
a
function of temperature (Figure 1). It indicates that carbonation is favored
under
process conditions above the curve and calcination occurs at conditions below
the
equilibrium curve.
[00141] It has been amply demonstrated that the ultimate C02 capture capacity
(W) of most sorbents employed at high temperature monotonically falls with
increasing number of CCR cycles (Abanades and Alvarez, 2003, lyer et al.,
2004).
While numerous studies have been conducted on the carbonation reaction to
detail
the sorbent reactivity, kinetics, mechanism and its mathematical modeling,
sufficient
emphasis has not been placed on the calcination process, as it relates to this
C02
46
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
separation process. Current calciner designs primarily involve the combustion
of fuel
with air inside the rotating tube to supply the sensible heat and heat of
calcination
directly. The exiting gases, still dominated by nitrogen, are enriched in C021
which is
released from the calcining limestone. However, this design is not amenable to
generating a pure C02 stream. It is thus imperative that the calcination
designs and
methods be optimized to maintain the sorbent structure to maximize reactivity,
in a
way that the purity of the eventual C02 stream is not compromised.
[00142] Calcination Configurations
[00143] The CCR scheme can be carried out in two modes of operation viz.
temperature and pressure swing and any combination thereof. Calcination can be
induced by either increasing the temperature of the carbonated product or by
reducing the PCO2 in the calciner such that the process conditions fall below
the
thermodynamic equilibrium curve. Figure 2 shows various configurations of the
calciner operation which detail the mode of heat input to the calciner. The
usage of
air in Figure 2(a) represents direct calcination, which is representative of
the
commercial calcination process mentioned earlier. However, a similar reactor
design
can be implemented in the CCR scheme if pure oxygen is used in place of air.
The
fuel would then form only C02 and H2O due to its combustion. The released C02
from reacted product and the C02 from the fuel combustion. can now be further
purified by a simple condensation and removal of steam. Depending on the fuel
used
for the direct calcination, other trace gases such as SOx and NOx may be
emitted,
necessitating further control technologies. In particular, make-up calcium
would be
necessary to replace the sorbent consumed by S02 in the calciner (lyer et al.,
2004)
[00144] Alternatively, in the absence of pure oxygen, the heat of calcination
can
be supplied indirectly as shown in Figure 2(b). The addition of heat will
induce
47
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
calcination, which leads to C02 buildup in the calciner. However, the flow of
C02 out
of the reactor is possible only if the PCO2 becomes greater than I bar.
Thermodynamically, Pco2 becomes greater than 1 bar only above 890 C. It is
well
known that high temperatures cause sorbent sintering, which reduces its
porosity,
thereby leading to a drastic reduction in reactivity. Pressure swing mode of
operation
enables lowering of the calcination temperature to circumvent the sintering
problem.
A lower PCO2, required by pressure swing operation, is achieved by either
dilution of
evolved C02 or by an overall reduction in pressure of the calciner. For
example, a
reduction in PCO2 below 0.0272 bar would lower the calcination temperature to
below 700C. Lowering PC02 can be accomplished by flowing diluent gas through
the calciner. However, only steam is an acceptable diluent gas since any other
gas
such as air, nitrogen, etc. will mix with the evolved C02 defeating the
overall
objective of isolating a pure C02 stream. The reduction in overall calciner
pressure,
while maintaining 100% pure C02, can be achieved using a vacuum pump which
removes C02 as it evolves from calcination.
[00145] Literature Review
[00146] Past literature studies have shown that CaO resulting from the
calcination under vacuum has a higher reactivity. Beruto and Searcy (1976)
observed highly crystalline CaO structure characterized by rounded surfaces
and
neck areas indicative of severe sintering under air calcination at 650 - 800
0C. In
contrast, the sharp edges of calcite were retained in the CaO obtained under
vacuum. However, this CaO did not possess a high degree of crystallinity and
showed high reactivity towards hydration. Dash et al., (2000) also observed a
metastablenanocrystalline calcia under vacuum as opposed to the formation of
stable microcrystalline calcia under helium. Beruto et al., (1980) estimated
the
48
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
surface area and pore volume of limestone based CaO to be about 78-89 m2/g and
0.269 ml/g respectively. Ingram and Marrier (1963) reported that the rate of
reaction
varies linearly with the difference between equilibrium partial pressure and
the partial
pressure of C02 surrounding the solid. This further supports the need for sub-
atmospheric calcination.
[00147] Rao et al. (1989) used thermo-gravimetric reaction data aloni with a
grain model to arrive at the reaction rate constant expression of 1.18x103
exp(-
1.85x10 /RT). Samtani et al. (2002) investigated the kinetics of calcite
decomposition
under an atmosphere of N2 and determined an activation energy of 192.5kJ/mol
and
an I nA of 20.73 (where A is the pre-exponential value of the Arrhenius rate
law).
Calcite was determined to undergo a zero-order decomposition mechanism, and
further investigation into the effect of flow rate, heating rate and sample
size did not
yield any deviation in the kinetic parameters and mechanism of the process.
[00148] Steam enhances calcination due to better thermal properties compared
to air and possible catalysis of the reaction. Berger (1927) showed a 30%
increase in
calcination rate due to steam compared to air in the 600 - 1000 'C. Further,
the CaO
resulting from steam calcination exhibited higher rates of slaking, indicating
a higher
reactivity product compared the CaO obtained from air calcination. Maclntire
and
Stansel (1953) obtained complete calcination at 700 C as opposed to only 1.6%
in
air. The catalytic effect of steam, attributed to an activated calcium
bicarbonate
intermediate species, was responsible for a 20% increase in calcination rate
at 834
C (Terry and McGurk (1994)). Wang and Thompson (1995) hypothesized that the
catalysis by steam occurs by the surface adsorption of H2O that weakens the
CaO-
C02 bond. Dynamic XRD studies indicated a 35% increase in conversion due to an
addition of 0.77% steam over that in dry helium.
49
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
[00149] This study demonstrates the role of calcination temperature, level of
vacuum, thermal properties of diluent gas, effect of diluent flow, on the
kinetics of
calcination and the morphology of the resultant CaO sorbent. Previous studies
have
detailed the development of structurally altered precipitated calcium
carbonate
(PCC) sorbent which shows higher reactivity than CaO obtained from the
calcination
of naturally occurring limestone (Gupta and Fan, 2002).
[00150] EXPERIMENTAL
[00151] Reactor Design for Sub-atmospheric Calcination: Rotary Vacuum
Calciner
[00152] An important objective of this C02 separation technology is to yield a
pure/concentrated stream of C02 during the calcination process. Vacuum
calcination
provides one method for meeting this objective. An indirect electrically
heated rotary
calciner was designed to carry out the necessary calcination studies. The
calciner
design and setup have undergone a number of modifications however Figure 3
depicts the final setup which incorporates various aspects of prior
configurations.
The reactor setup as shown in Figure 3 was assembled to handle a wide range of
calcination conditions such as the calcination of 0.5-20g sorbent samples
under a
variety of vacuum, vacuum + sweep gas conditions and calcination temperatures
of
0
up to 950 C. Since early experiments had shown that sorbent heaping affects
both
the calcination kinetics as well as sorbent morphology, the reactor was
designed to
allow the calcination to take place under rotary motion which disperses the
sorbent
thereby minimizing sorbent heaping.
[00153] A quartz tube reactor was used to carry out calcination kinetic
studies.
The reactor tube was designed to have a conical shaped tapered central zone in
order to keep the particles from dispersing axially away from the heated zone.
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
Baffles were incorporated to ensure particle dispersion. The reactor was
placed on
two sets of rotary rollers, and was attached to a motor via a rubber belt
mechanism.
An electric split tube furnace was used to provide the necessary heat of
calcination.
Rotary seals enabled the rotation to take place while maintaining the desired
level of
vacuum level in the reactor tube. A vacuum level of -28 in Hg was achieved in
this
configuration. The gas exiting the calciner was further diluted with air in
order to
ensure that the C02 concentration fell in the detection range of the C02
analyzers.
A dry vacuum pump (Vacuum Pump 1) procured from BOC Edwards capable of
achieving vacuum levels as low as 50 millitorr and gas flowrates of 6m'/hr was
used
to supply the necessary reactor vacuum level.. The second pump in the setup is
a
smaller dry pump and was put in place to ensure that there was no pressure
buildup
in the '/+" lines connecting the vacuum pump to the analyzers. Two Non
Dispersive
Infra Red (NDIR) analyzers were used to monitor the C02 concentration (ranges
0-
2500ppm and 020%). These C02 analyzers have their own inbuilt pumps and are
capable of drawing upto 2LPM from the header for online C02 analysis. The
temperature of the furnace was controlled with a thermocouple inserted into
the
central zone of the furnace. The temperature of the reactor was monitored
using a
second thermocouple inserted into the center of the quartz / alumina tube.
Pressure
gauges and volumetric flow meters are included to monitor the vacuum pressure
in
the reactor, the pressure in the'/," lines and the flows of the sweep gases
into the
calciner and the flow of the air used in the dilution of the exhaust C02
before
sending it to the analyzers. The analyzers are also connected to a data
acquisition
system that can record analyzer readings every second.
[00154] The calcination studies were performed to investigate the role of
calcination temperature, level of vacuum, thermal properties of sweep gas and
effect
51
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
of gas flow on the kinetics of calcination and the morphology of the resultant
CaO
sorbent. Experiments were also performed to compare the calcination kinetics
of
PCC and LC and also to investigate the role of sorbent heaping as it relates
to scale-
up considerations of the calciner.
[00155] RESULTS AND DISCUSSION
[00156] Effect of Temperature
[00157] Sweep gas is necessary to aid calcination. Prior experiments carried
out under high vacuum conditions in the absence of sweep gas revealed a longer
time for calcination. Experiments were performed to determine the possibility
of
performing sub-atmospheric calcination in combination with the flow of gas.
PCC and
LC samples of 0.5g were calcined with a sweep N2 gas flow of 50m1/min under
25"Hg vacuum. Figures 4 and 5 show the conversion plots for LC and PCC at
0
temperature ranges of 700 C to 750 C. The resulting plots show that the
calcination
time for PCC is much lower than that for the naturally occurring LC. At 700 'C
PCC
takes about 2000 seconds to fully calcine whereas LC takes -3500 seconds.
Faster
calcination kinetics essentially translates to energy savings in the
calcination process
which is yet another advantage of PCC.
[00158] Effect of vacuum level
[00159] Further studies were performed to determine the effect of vacuum on
the kinetics of calcination. The kinetic conversion versus time plots curves
were
obtained for 0.5g PCC at vacuum levels of 10", 15", and 25" Hg vacuum with N2
sweep flows of 50ml/min. The results are plotted in Figure 6 and clearly show
that
higher vacuum levels translate to faster calcination times.
[00160] Effect of Diluent Flow rate
[00161] It can be observed from Figure 7 that the calcination time required
for
52
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
larger samples under pure vacuum conditions is too long. The heat of
calcination
under these conditions is predominantly supplied by radiative means. However,
the
addition of heat to the calcining sorbent through convective means also helps
accelerate calcination. This is accomplished through the use of a preheated
diluent
gas flow over the calcining sorbent. Figure 7 shows the effect of flow rate of
diluent
helium gas on the calcination behavior. It indicates that in the absence of
diluent
flow, we achieve only 78% calcination in 2000s. In contrast a steady diluent
flow of
120 ml/min attains -93% in 2000s. As the diluent flow is increased to 500
ml/min,
90% calcination occurs within 1200s. It is also useful to note that an
increase in
diluent flow to 1000 ml/min does not decrease the heat transfer resistance
significantly.
[00162] Effect of Diluent Type
[00163] Calcination experiments in the presence of different diluent gases
were
carried out to establish the influence of thermal properties (thermal
conductivity, heat
capacity) of gases on the calcination process. For example, at 1000K the
thermal
conductivity of He (0.354 W/m.K) is higher than that of N2 (0.0647 W/m.K)
(Perry
and Chilton, 1997), which could lead to a difference in the calcination rate
Figure 8
indicates the influence of helium, nitrogen and argon on the calcination of I
Og
samples of Linwood carbonate. It can be observed Figure 8 that helium indeed
causes a faster calcination. In a commercial operation, we cannot use these
particular gases because the gas mixture exiting the calciner will consist of
C02,
which is evolved from the calcination process and these diluent gases, thereby
defeating the overall purpose of isolating a pure C02 stream. However, these
experiments lay a foundation for the potential use of higher thermal
conductivity
gases such as steam (0.0978 W/m.K). Steam has the added advantage of ease of
53
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
separation from steam/C02 mixtures by its removal by condensation.
RESULTS AND DISCUSSIONS
Catalyst and Sorbent Characterization
[00164] The characterization of the high temperature shift (HTS) catalyst in a
2
BET analyzer revealed that the catalyst has a BET surface area of 85 m /g and
a
total pore volume of about 0.3 cc/g. The majority of the pores were found to
occur
around 180 A as evident from the maximum in its pore size distribution plot
shown in
Figure 19. In contrast, the low temperature shift (LTS) catalyst has a BET
surface
2
area of 52 m /g and a total pore volume of about 0.1 cc/g. The majority of
these
pores were found to occur around 37 A as evident from the maximum in its pore
size
distribution plot (Figure 19).
[00165] The surface area (SA) and pore volume (PV) of the three different CaO
precursors are provided in Table 5. Figure 20 shows the pore size distribution
(PSD)
of these precursor fines. It can be seen that LC fines do not have high SA/PV.
However, upon calcination and subsequent hydration, the SA/PV of the calcium
hydroxide (LH) fines increase as can be observed for the LH sample. The
porosity is
maximized in the microporous range (30-50 A range). In contrast, the SA/PV of
the
morphologically altered PCC are much higher. Further, most of the porosity
lies in
the 100-300 A range.
Table 5: Morphological properties of the natural and synthesized CaO
precursors and the HTS catalyst obtained from BET analyses.
Surface Pore
Sorbent Area Volume
(m2/g) (cc/g)
Linwood Carbonate (LC) 1.5 0.004
Linwood Hydrate (LH) 13.9 0.050
54
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
Precipitated Calcium Carbonate (PCC) 49.2 0.170
High Temperature Shift (HTS) catalyst 85 0.3
Water gas shift reaction (WGSR): Catalyst Testing and Analysis
[00166] The HTS catalyst was tested for its catalyst activity towards the WGS
reaction between 500-700 C. Blank runs (without any sorbent) were performed
in a
reaction mixture comprising of 3% CO and 9% H2O, the balance being 5.0 grade
N2. The total gas flow-rate was maintained at about 1.5 slpm and the steam/CO
ratio
was set at -3. Typically about 0.5 grams of the HTS catalyst was loaded in the
reactor prior to each run. The catalyst activity increases monotonically with
increasing reaction temperature. This is evident from Figure 21 below. The CO
conversion increases from 24.3% at 500 C to 69.3% at 550 C. It finally
reaches
about 80% at 600 C. Beyond 600 C the conversion does not change much but
remains steady at -78% at 700 C. This might be due to the equilibrium
limitations
governing the WGS reaction scheme is depicted in eqn (8) below:
CO + H2O --> C02 + H2 (8)
The data were further analyzed to check if the system was operating within the
domain of WGS equilibrium. The thermodynamic equilibrium constant (K) for any
temperature for this reaction was computed using the software "HSC Chemistry v
5.0" (Outokumpu Research Oy, Finland). The observed ratio was computed from
the
experimental data by obtaining the ratio of the partial pressures of the
products and
the reactants as per the eqn (9) below:
1 (PCO)(PHZo) (9)
~ Kobs = (PH2 X PCOZ )
Figure 22 illustrates the effect of temperature on the ratio of partial
pressures (Kobs)
obtained from the experimental data. This is compared with the thermodynamic
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
equilibrium values (Keq). From the figure it is evident that we are operating
in the
region that is below the thermodynamic equilibrium. At 500 C the Kobs is
0.028 while
the corresponding Keq is 4.77. Keq monotonically decreases with increasing
temperature. In contrast, Kobs increases with temperature for our operating
conditions. Thus, at 600 C the Kobs increases to 1.4 while the Keq moves down
to
2.5. This trend continues and it is clearly evident from the figure that the
system
moves closer to equilibrium as we progressively increase the temperature from
500
to 700 C.
Combined Carbonation and Water gas shift reaction:
Sorbent Testing and Analyses
[00167] The combined carbonation and WGS reaction was tested in the reactor
assembly used for the catalyst testing. The experimental conditions were
exactly
identical to that used for testing the catalyst. The runs were performed in a
reaction
mixture comprising of 3% CO and 9% H2O, the balance being 5.0 grade N2. The
total
gas flow-rate was maintained at about 1.5 slpm and the steam/CO ratio was set
at
-3. Typically about 0.5-1 g of the HTS catalyst was loaded in the reactor
prior to
each run. Different calcium oxide precursors were tested. Naturally occurring
limestone, Linwood Carbonate (LC) and the corresponding hydrated lime, Linwood
Hydroxide (LH) were obtained from Linwood Mining and Minerals Co. The
structurally modified calcium carbonate (PCC) was prepared in-house and the
details
are outlined below.
Sorbent testing without catalyst
[00168] The sorbents were initially tested for catalytic activity towards WGSR
and CO conversion without any HTS catalyst from 500-700 C. This would obviate
the need for any catalyst in the system. However, detailed investigation
resulted in
very miniscule activity and hence it was concluded that HTS catalyst was
required
56
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
for further combined reaction testing.
Combined reactions with PCC-HTS catalyst system
[00169] Typically about 0.5 g of HTS catalyst and 1.5 g of PCC were loaded in
the reactor and the temperature was ramped till 700 C in flowing N2. This
procedure
ensured the calcination of the calcium carbonate to calcium oxide and it was
monitored using CO2 analyzer. Subsequently, the reaction temperature was
lowered
to 600 C and the reaction gas mixture was allowed to flow through the system.
The
CO analyzer continuously monitored the CO flow through the system and the
breakthrough curve depicting the CO conversion with time is as shown in Figure
23
below. The system gives almost 100% conversion for first 240 seconds (4 min)
following which the initial reactivity of the sorbent slowly falls to give
about 90% CO
conversion at 1000 seconds (16.5 min). The sorbent gradually achieves its
maximum
loading capacity with time and finally at around 2500 seconds (42 min) the
sorbent
reaches its breakthrough loading. Beyond this the CO conversion of 81%
corresponds to that obtained with only the catalyst at 600 C. This can be
validated
from Figure 21.
[00170] The system was then switched to pure N2 flow and the reaction
temperature was increased to 700 C to drive the calcination of the CaCO3
formed
due to carbonation. Thus the reactions occurring in the system are:
Reaction phase:
WGSR: CO + H2O -> CO2 + H2 (7)
Carbonation: CaO + CO2 -> CaCO3 (8)
Regeneration phase:
Calcination: CaCO3 -> CaO + CO2 (9)
The termination of the calcination was ensured by monitoring the C02 released
57
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
using a CO2 analyzer. The reaction temperature was again lowered to 600 C and
the sorbent-catalyst system was subjected to the reaction mixture for a second
reaction cycle. The 2nd cycle CO breakthrough curve is also depicted in Figure
23. It
is evident from the figure that the CO conversion is not as superior as in the
1st
cycle. The CO conversion monotonically decreases to about 90% in 110 seconds,
80% in 240 seconds and gradually to about 50%. It is interesting to note that
at the
end of the breakthrough the sorbent-free catalytic CO conversion of 81% is not
achievable. This could be attributed to the loss in the catalytic activity
after the first
regeneration cycle. This is because the catalyst is subjected to CO2, an
oxidizing
atmosphere, during the calcination phase. Thus the deactivated catalyst is not
able
to augment the WGS reaction kinetics and hence we see a poor performance of
the
sorbent-catalyst system in the 2"d cycle. The solitary sorbent has been
subjected to
numerous carbonation calcination cycles and has shown satisfactory performance
(lyer et al, 2004).
Combined reactions with LH-HTS catalyst system
[00171] Typically about 1 g of the HTS catalyst and 1.3 g of LH were loaded in
the reactor and the temperature was ramped up slowly till 600 C in flowing
N2. This
procedure ensured the calcination of the calcium hydroxide to calcium oxide.
Calcium hydroxide decomposes above 400 C. Subsequently, the reaction gas
mixture was allowed to flow through the system. The CO analyzer continuously
monitored the CO flow through the system and the breakthrough curve depicting
the
CO conversion with time is as shown in Figure 24 below. The system gives
almost
100% conversion initially to give about 90% CO conversion at 900 seconds (15
min).
The sorbent gradually achieves its maximum loading capacity with time and
finally at
around 3000 seconds (50 min) the sorbent has achieved its breakthrough
loading.
58
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
Beyond this the CO conversion of 81 % corresponds to that obtained with only
the
catalyst at 600 C as was shown in Figure 21.
[00172] The system was then switched to pure N2 flow and the reaction
temperature was increased to 700 C to drive the calcination of the CaCO3
formed
due to carbonation. Subsequently, the reaction temperature was lowered to 600
C
and the LH-CaO/catalyst system was subjected to the reaction mixture for a
second
reaction cycle. The 2nd cycle CO breakthrough curve is also depicted in Figure
24. It
is evident from the figure that the CO conversion is not as superior as in the
1St cycle.
The CO conversion monotonically decreases to about 90% in 60 seconds, 80% in
180 seconds and gradually to about 30%. It is interesting to note that at the
end of
the breakthrough the sorbent-free catalytic CO conversion of 81 % is not
achievable.
This could be attributed to the loss in the catalytic activity after the first
regeneration
cycle. This is because the catalyst is subjected to C02, an oxidizing
atmosphere,
during the calcination phase. Thus the deactivated catalyst is not able to
augment
the WGS reaction kinetics and hence we see a poor performance of the sorbent-
catalyst system in the 2nd cycle. The solitary sorbent had been subjected to
numerous carbonation calcination cycles and has shown satisfactory performance
over few cycles.
Comparison of the PCC and LH sorbents
[00173] Figure 25 compares the CO conversion breakthrough curves for the
PCC and LH sorbent-catalyst systems. The curves are for the 1st reaction
cycle. The
CO conversion at any given time for PCC-CaO is always higher than that of LH-
CaO.
The PCC system gives almost 100% conversion for first 240 seconds (4 min)
while
the LH sorbent system sustains this conversion only in the initial few
seconds.
Subsequently, the PCC system gives about 90% CO conversion at 1000 seconds
59
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
(16.5 min) followed by 85% in 1600 seconds (27 min). In contrast, the LH
system
gradually gives about 90% CO conversion at 900 seconds (15 min) and followed
by
85% in 1200 seconds (20 min). Both the sorbent systems gradually achieve their
maximum loading capacity with time and finally at around 2500-3000 seconds
they
reach their breakthrough loading. Beyond this the CO conversion of 81%
corresponds to that obtained with only the catalyst at 600 C. Hence, it is
evident
from Figure 24 that the PCC-CaO performance dominates over that of LH-CaO at
any given time.
[00174] Figure 26 illustrates the generation 1 MWe of steam.
[00175] Figure 27 illustrates one embodiment of the present invention
providing
1.002 MWe total capacity.
[00176] Figure 28 illustrates a second embodiment of the present invention
providing 1 MWe total capacity.
j00177] Figure 29 illustrates another embodiment of the present invention
providing 1.33 MWe total capacity.
[00178] Figure 30 illustrates yet another embodiment of the present invention
providing 1.33 MWe total capacity
[00179] Figure 31 illustrates an alternative embodiment of the present
invention
providing 1.54 MWe total capacity.
[00180] Figure 32 illustrates yet another alternative embodiment of the
present
invention providing 1.07 MWe total capacity.
[00181] Figure 33 illustrates an alternative embodiment of the present
invention
providing 1 MWe total capacity.
[00182] Figure 34 illustrates an alternative embodiment of the present
invention
providing 1 MWe total capacity.
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
[00183] Figure 35 illustrates yet another embodiment of the present invention
providing 1.54 MWe total capacity.
[00184] Figure 36 illustrates an alternative embodiment of the present
invention
providing 1 MWe total capacity at 80% CO2 capture.
[00185] Figure 37 illustrates another embodiment of the present invention
providing 300 MWe total capacity at 90 CO2 capture.
C02/SO2 combined Reaction Optimization
[00186] EXPERIMENTAL
[00187] Chemicals, Sorbents and Gases
[00188] Naturally occurring limestone (CaCO3) was obtained from Linwood
Mining and Minerals Company (Linwood Carbonate, LC). Precipitated calcium
carbonate (PCC) was synthesized from Ca(OH)2, obtained from Fisher Scientific.
The pore structure of the synthesized PCC sorbent was tailored using an
anionic
dispersant, N40V , obtained from Ciba Specialty Chemicals, Corp. The details
of
the synthesis procedure is described elsewhere. 7' 7,14 This structurally
modified
PCC yields a predominantly mesoporous structure in the 5-20 nm range with a
surface area (SA) of 49.2 m /g and a pore volume (PV) of 0.17 cc/g obtained by
BET analysis. N2 and C02, obtained from Praxair, Inc were 99.999% and 99.9%
pure, respectively. Mixtures of 02 and S02 in N2 were also supplied by
Praxair, Inc.
The BET SA, PV, and pore size distribution (PSD) were measured at -196 *C
using nitrogen by a NOVA 2200 analyzer (Quantachrome Company).
[00189] Sorbent Reactivity Testing
[00190] The reactivity testing of the calcium-based sorbents was carried out
in
a Thermogravimetric Analyzer (TGA) procured from Perkin Elmer Corporation
(Model # TGA-7). A simplified schematic diagram of the experimental setup is
61
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
shown elsewhere (Iyer et al., 2004). The dome which houses the electronic
parts
of the balance was continuously flushed with a pure stream of N2 gas (TGA-N2)
to
ensure that corrosive gases do not adversely affect the equipment circuitry.
The sensitivity of the balance is 1 pg. In these experimental runs, the weight
of the
sample was recorded every 1-10 second intervals. The gas flows were accurately
maintained using variable area flow meters, obtained from Cole Parmer
Instrument Company. A small sample of the sorbent (about 10-12 mg) was
placed in a quartz sample holder and brought to 700 C under nitrogen flow. The
temperature of the TGA was then maintained at 700 C throughout the experiment
to
effect the calcination of PCC. After the calcination step, the valve was
switched to
allow the flow of reactant gas mixture over the calcined sorbent (PCC-CaO). An
automated multi-position valve (VICI Corporation, Model # EMTMA-CE) actuated
by a programmable electronic timer (VICI corporation, Model # DVSP4) was used
to switch between pure nitrogen stream and the reaction gas mixture at
programmed time intervals in order to effect the cyclical calcination and
carbonation and sulfation of the sorbent. The alternating flows are adjusted
to
minimize any variations in weight of the pan/sorbent system due to buoyancy
changes. The reactant gas mixture enters the TGA from the side port and gets
diluted by the TGA-N2 stream coming from the balance dome. The flow of the
reactant gas mixture causes an immediate increase in the weight of the sorbent
due to the formation of higher molecular weight products such as CaCO3 and
CaSO4. At the end of the set reaction residence time, the automated valve
toggles the flow back to the "calcination nitrogen". The sorbent weight starts
dropping immediately due to the calcination of the CaCO3 product that is
formed in
the previous reaction step. The raw data is then analyzed to obtain the
conversion plots.
62
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
[00191] RESULTS AND DISCUSSION
[00192] Thermodynamic analysis was carried out to understand the effect of
reaction temperature and gas concentration on the spontaneity of the various
reactions.
Thermodynamic analysis
[00193] Primarily four gas-solid reactions can occur when calcium oxide is
exposed to flue gas from coal combustion. CaO can undergo hydration,
carbonation
and sulfation reactions with H2O, C02 and S02, respectively. In addition, S02
can react with the CaCO3 formed due to the carbonation reaction, thereby
causing direct sulfation of the carbonate. These can be stoichiometrically
represented as:
[00194] Hydration: CaO + H2O -> Ca(OH)2 (1)
[00195] Carbonation: CaO + C02 --> CaCO3 (2)
[00196] Sulfation: CaO + S02 + 1/202 --> CaSO4 (3)
[00197] Sulfation: CaCO3 + S02 + 1/202 -> CaSO4 + C02 (4)
[00198] Thermodynamic calculations were performed to obtain equilibrium
curves for the partial pressures of H2O (PH20), C02 (Pco2) and S02 (Pso2) as a
function of temperature for the hydration, carbonation, sulfation and direct
sulfation
reactions using HSC Chemistry v 5.0 (Outokumpu Research Oy, Finland). The
equilibrium curves depicting the temperature dependent partial pressures of
H2O
and C02 for the hydration and carbonation reactions are shown in Figure 1 (a).
From
these equilibrium curves, we can predict that moisture does not react with CaO
beyond 350 *C in the 5-7% concentration range. At 10% C02, the equilibrium
temperature for CaO-CaCO3 system is 760 'C. Therefore, the temperature of the
carbonator needs to be kept below 760*C in order to effect the carbonation of
CaO in
a 10% C02 stream. A temperature of 700 C offers a reasonable rate of
carbonation
63
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
and calcination reactions and enabled us to carry out multiple CCR cycles
under
isothermal conditions. Thermodynamic data for the equilibrium temperature
versus
S02 concentration for the sulfation of CaO and direct sulfation of CaCO3 are
shown
in Figure 1(b). The S02 concentration for the sulfation of CaO system is
depicted in
terms of ppmv for a total system pressure of 1 bar at 4% 02. At 700 'C, the
equilibrium partial pressure of S02 is 1.84 and 5.72 ppt (parts per trillion)
for the
sulfation of CaO and the direct sulfation of CaCO3. Since S02 concentration in
the
inlet flue gas is in the 500-3000 ppm range, sulfation of CaO and the CaCO3
will
definitely occur until virtually all S02 is consumed. Table I summarizes the
temperature below which the three reactions are thermodynamically favored at
the
typical flue gas concentrations at 1 bar total pressure.
Reaction with CaO Hydration Carbonation Sulfation
Reactive component of the flue gas H2O C02 S02+02
Typical flue gas concentration 5-7% 10-15% 500 - 3000ppm
(vol%) S02,3-4%02
Equilibrium temperature below 330 - 350 760 - 790 1175-1245
which the reaction can proceed ( C)
[00199] Extended Cyclical Carbonation and Calcination Experiments
[00200] Earlier studies from our group have shown that PCC-CaO achieves
high conversions (>90%) towards carbonation as compared to 45-60% attained by
CaO derived from naturally occurring calcium sources.7 Life cycle testing on
PCC-
CaO, carried out in 100% C02 for an hour, did not show a significant drop in
reactivity for 2-3 CCR cycles. However, prior literature indicates a loss in
reactivity
over a higher number of CCR cycles. We carried out extended isothermal life
cycle
64
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
testing of LC and PCC sorbents at 700 *C. The carbonation was carried out in a
10%
C02 stream while pure N2 was used for calcination. Each of the CCR steps was
performed for 30 minutes. The sorption capacity of the sorbent is quantified
as wt%
C02 captured by the calcined sorbent. Theoretically, 56 grams of unsupported
CaO
sorbent should react with 44 grams of C02 corresponding to a maximum C02
sorption capacity of 78.6 wt% at 100% conversion. The wt% capacity, of the LC
based sorbent towards C02 capture reduces from 58% in the first cycle to 20%
at
the end of the 50 cycle. The microporous structure of LC, being susceptible to
pore
pluggage and pore mouth closure, does not attain high conversion. 7,24 This is
due to
the formation of CaCO3, whose molar volume (36.9 cc/mol) is higher than that
of the
reactant CaO (16.9 cc/mol). In contrast, we see that the conversion of PCC
based
sorbent over 100 cycles is distinctly higher. The capacity of PCC-CaO is 68
wt% in
the first cycle, which drops to 40 wt% by the 50th cycle and then slightly to
36 wt% by
the 100"' cycle (6000 minutes on stream). The high reactivity can be
attributed to the
predominant mesoporous structure of PCC, which allows the reactant gases to
access the entire surface of particle through the larger pores. The extent of
carbonation continues to rise significantly beyond the kinetic controlled
regime. This
fact was ascertained by extending the carbonation reaction time to 120 minutes
over
40 cycles, during which the sorbent retains 45 wt% capture after 40 cycles
(9600
minutes on stream). These results provide evidence that the reactivity of the
PCC-
CaO is governed solely by the reaction time provided and there is no
structural
limitation in attaining high conversion.
[00201] Figure 2 depicts graphically the wt% C02 capture attained by LC, PCC
and a host of other high temperature sorbents reported in the literature for
multiple
CCR cycles.30 While a variety of sorbents have been screened for this CCR
process,
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
a candidate sorbent that shows consistently high reactivity and sorption
capacity
over multiple cycles remains to be identified. The experimental conditions
used in the
studies referred to in Figure 5 are detailed in Table 2. This table highlights
important
process conditions such as carbonation and calcination temperatures, solid
residence times, number of cycles, sorption capacities (wt%), and the C02
concentration in the gas mixture during the reaction and regeneration steps.
PCC-
CaO attains 68 wt% increase in 30 minutes and 71.5 wt% after 120 min at the
end of
the first cycle. In contrast, earlier studies have shown a sorption capacity
of about 71
wt% (90% conversion) in a pure C02 stream after 60 min on stream at 650 C.
Hence, factors like C02 concentration, temperature and cycle time play a
significant
role in determining the sorption capacity for the same sorbent. The
experiments
conducted by Barker on 10 micron CaO powder demonstrate a drop in the sorption
capacity from 59 wt% in the first carbonation cycle to 8 wt% at the end of 25t
cycle.10
This work suggests that, due to the formation of a 22nni thick product layer,
particles
smaller than 22 nm in diameter should be able to achieve stoichiometric
conversion.
The author later proved this hypothesis by obtaining repeated 93% conversion
(73%
weight capture) of l0nm CaO particles over 30 cycles with a carbonation time
of 24
hours under 100% C02 at 577 C.31 In a PbO-CaO based chemical heat rump
process, PbO attained 3.6 wt% C02 capture in the first cycle, decreasing to
1.6 wt%
by the 6 cycle and CaO showed a drop in C02 capture from 53 wt% in the 1'4
cycle
to 27.5 wt% by the 5'h cycle.8 A lithium zirconate (Li2ZrO3) based sorbent
provided
20 wt% capacity over two cycles.32 In another study, researchers at Toshiba
Corp.
observed that the reactivity of lithium orthosilicate (Li4SiO4) was better
than that of
lithium zirconate. Extended cyclical studies performed on lithium
orthosilicate
samples attained 26.5 wt% sorption capacity over 50 cycles without any change
in
66
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
the reactivity.34 Harrison and co-workers developed an enhanced hydrogen
production process from the water gas shift reaction by removing C02 from the
gas
mixture through the carbonation of CaO. 12 Dolomitic limestone based CCR
process
yielded a 35 wt% capacity in the first cycle that fell to 11.4 wt% by the 148h
cycle
when the carbonation experiments were performed in pure C02 at 800 C and
calcination was conducted at 950 C. An explanation for the drop in capture
capacity
over multiple CCR cycles has been hypothesized by Abanades and Alvarez based
on the changing microporosity within the grains and the mesoporosity
surrounding
them due to sintering.16
[00202] Simultaneous Carbonation and Sulfation
[00203] The sulfation of CaO and the direct sulfation of the CaCO3 product
reduces the C02 sorption capacity of the CaO due to the formation of
"permanent"
CaSO4, thereby reducing its efficiency for the CCR process as discussed
earlier.
This part of the study involves the simultaneous carbonation and sulfation
reactions
followed by calcination over multiple cycles. The goals of this set of
experiments are:
[00204] (a) to identify the extent of carbonation (X o2) and sulfation (Xs,,,)
during
the simultaneous reactions
[00205] (b) to determine and optimize the trend in the ratio of carbonation to
sulfation as a function of residence time and reaction temperature
[00206] (c) to quantify the reduction in the ultimate carbonation capacity for
varying S02 concentrations (3000-100 ppm) over multiple CCR cycles
[00207] Figure 3 shows a sample plot of raw data typical for all the
experiments
conducted in this section. The x-axis represents the residence time and the y-
axis
shows the actual weight of the sorbent at any given instant. In all the
experiments,
calcination of PCC was carried out typically for 20-30 minutes. The residence
time
67
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
for the reaction step was maintained for 5 minutes in each of the three cycles
for this
specific run. From point A to point B, calcination of PCC occurs and the -56%
weight
remaining confirms that the sorbent at point B is pure CaO. The flow through
the
TGA is then switched to the reactant gas mixture causing the weight of the
sorbent
to increase from point B to point C due to the carbonation and sulfation
reactions. At
the end of the 5-minute reaction time, gas flow is switched back to N2 to
effect the
decomposition of the CaCO3 formed due to carbonation in the first reaction
cycle
causing the weight to drop from point C to point D. In contrast to CaCO3
decomposition, CaSO4 formed in the first reaction step remains intact. The
extent of
carbonation is calculated from the weight loss during the calcination step C
to D, and
the extent of sulfation can be estimated based on the difference between the
weight
at point D and that at the starting point B. Similarly, points D-E-F represent
the 20d
cycle and so forth. The trend observed from B to C to D is seen in every cycle
in
each experiment. The details of the conversion calculations are reported
elsewhere
(Iver et al. 2004).
[00208] Carbonation and sulfation occur as heterogeneous non-catalytic gas
solid reactions. Higher concentration of C02 (10% or 100,000 ppm) compared to
S02 (3000 ppm) could result in a higher conversion towards the carbonation
reaction. However, the higher free energy change associated with the sulfation
reaction thermodynamically favors it over the carbonation reaction. The
process
conditions employed can have a significant impact on the relative rates of
these two
reactions. The data obtained in all experiments has been presented in the form
Xco2, X502 and the ratio of carbonation to sulfation, R (Xco2/Xso2) as a
function of
reaction residence time
[00209] Effect of residence time
68
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
[00210] Figures 4-6 show the data obtained on the extent of carbonation and
sulfation as a result of simultaneous exposure of PCC-CaO to a gas mixture
containing 10% C02, 3000 ppm S02 and 4% 02. X-ray diffraction analysis of the
reacted sorbent revealed the presence of CaSO4, CaCO3 and CaO only. In the
first
cycle, XCO2 far exceeds the extent of sulfation (xso2) during the initial part
of the
reaction, thereby establishing the viability of a CCR process for C02
separation even
in the presence of S02. XCO2, which increased monotonically in the 0-10 minute
range, started to fall due to the direct sulfation of the CaCO3 formed,
consequently
leading to a higher XS02. From thermodynamic analysis presented earlier, it is
clear
that S02 concentration greater than 5.72 ppt will lead to direct sulfation at
700 C. In
fact, XS02 attained under simultaneous exposure to C02 and S02 is higher than
the
x502 obtained by either the pure sulfation of CaO or the direct sulfation of
CaC03
reaction, which are the only possible routes for sulfation. This indicates
that the
nascent CaC03, formed due to the parallel carbonation reaction, has a higher
reactivity for S02 than the CaC03, which forms a part of the stable crystal
structure
that characterizes the original PCC. After 10 minutes, XC02 starts dropping,
but it
continues to be higher than Xs02 until it reaches 40 minutes. Beyond 40
minutes, XCO2
starts dropping even below XS02 due to continued direct sulfation.
[00211] Figures 4 and 5 depict the effect of residence time over three CCR
cycles on xc02 and Xso2, respectively. PCC-CaO attains a maximum XC02 of -50
wt%
at 10 minute residence time. The data in Figures 4 and 5 show that xc02 and
XS02
decrease with increasing number of cycles for any residence time due to the
formation of CaSO4 which reduces the availability of CaO in the subsequent
cycle.
The primary reason for this observation is the fact that there is a loss in
the free CaO
due to the formation of non-regenerable CaSO4. Figure 5 shows that X502
remains
69
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
virtually the same in each of the three cycles until a residence time of 10
minutes. In
contrast, Xco2 shows a significant loss in reactivity over each subsequent
cycle in the
same duration. For a residence time of 60 minutes, XCO2, which was only 22.5%
in
the first cycle, reduced to almost zero in the second cycle, indicating a high
extent of
the direct sulfation reaction. In fact, the sorbent is completely spent at the
end of the
second cycle that it shows no reactivity to either gas in the third cycle. The
overall
XS02 for PCC-CaO at the end of three cycles was 88.2%
[00212] Figure 6 illustrates the ratio R obtained from XCO2 and XS02 attained
during simultaneous carbonation and sulfation. From Figure 6, we can observe
that
the magnitude of R is smaller than that derived from the "individual"
reactions and it
shifts to 5 minutes instead of 8 minutes seen earlier. This is probably due to
the fact
that the rate of sulfation is enhanced due to the simultaneous sulfation of
CaO and
the higher reactivity of nascent CaCO3 as explained earlier. From Figure 6, it
is
evident that the maximum in the ratio occurs at a reaction time of about 5 min
for all
the three cycles. The magnitude of the ratio falls with each subsequent cycle
and
longer residence time. This is due to the direct sulfation of the calcium
carbonate
product of carbonation reducing the xc02, increasing the xs02, and thereby
dropping the
ratio.
[00213] Effect of S02 concentration
[00214] Figures 7 and 8 below show the extent of carbonation and sulfation
respectively on PCC-CaO at 700 'C with varying SO2 concentration from 100 -
3000
ppm over multiple cycles. It is evident from the plots that the carbonation
conversions decrease with increasing cycles and S02 concentrations. The effect
of
sulfation is very drastic for 3000 ppm and not so severe with 100-300 ppm
range.
The extent of sulfation is also low in this range as can be observed from
Figure 8.
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
Figure 9 shows the ratio "R" for increasing CCR cycles with S02 concentrations
varying from 3000 to 100 ppm. It is evident from the plots that for each S02
concentration curve there exists a maximum in the ratio, which depends on the
residence time in the system. The ratio is maximum at 160 for a S02
concentration
of 100 ppm while it monotonically decreases and reaches a meager value of 5
for
3000 ppm S02 as seen earlier.
[00215] Effect of Temperature
[00216] Figures 10-12 depict the effect of reaction temperature (500-700 oC)
on
the ratio of carbonation to sulfation (R= XCO2I xso2), the extent of
carbonation
(XCO2) and the extent of sulfation (xS02) for increasing residence times (0-30
min).
The simultaneous experiments were conducted for a 3000 ppm S02, 10% C02 and
4% 02 stream. As observed in Figure 10, the ratio (R) decreases with
increasing_
residence time for all reaction temperatures (except for 700 0C). This is due
to the
onset of direct sulfation of CaCO3 product. It is interesting to note that for
any given
residence time the ratio for 650 C is the highest, which is followed by 600 C
and
subsequently by the values at 700 C (except at 2 min). The R values at 700 C
are
lower than that obtained at temperatures of both 600 and 650 C as sulfation
starts to
dominate at higher temperatures and the kinetics between these two competing
carbonation and sulfation reactions start to play a significant role in
determining their
0
ratios. Thus, 650 C seems to be the optimal temperature to operate with
minimal
sulfation effects for a 3000 ppm S02 and 10% C02 stream. Similarly, the
optimal
temperature for streams with varying S02 concentrations (3000-100 ppm) needs
to
be identified.
[00217] At 650 *C, the ratio starts with a value of 15 for a residence time
(RT) of
2 min and subsequently starts to monotonically decrease to about 9 for 5 min,
5 for
71
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
min and finally 2 for 30 min. As illustrated in Figure 11, the corresponding
xC02 is
34% for 2 min, which peaks to 45% at 20 min with a ratio of only 3. It is
evident that
the extent of carbonation is the highest for the temperature of 650 'C for any
0
residence time. The only exception is the XC02 of 52% at 700 C for a 10 min
5 residence time. However the R corresponding to this point is around 3.
Hence, 650
o C is still the preferred temperature of operation with optimal residence
times of 2-5
min giving XCO2 of 34-40% with corresponding ratios of 15-9 respectively.
Thus,
working at the optimum temperature where R as well as XC02 is the highest can
maximize the overall C02 capture capacity with minimal S02 effect.
10 [00218] A pilot scale plant, that integrates the CCR process with an actual
coal
fired combustor will be designed, installed and operated as part of this pilot
demonstration. B&W stoker boiler will be used in this process. A schematic of
the
process flow diagram is shown in Figure II.C-1. Please note that the process
flow
diagram could be altered based on future process modifications. It consists of
a coal
combustion unit that generates actual flue gas. The flue gas is then lowered
in its
S02 content by the injection of PCC. This FSI mode of sulfur capture has been
investigated in an earlier OCDO-OSU sponsored OSCAR project. Results from that
project will be factored in to use an optimal Ca/S ratio for S02 control. The
optimum
temperature for PCC injection is about 8001000C. The entrained fly ash and
partially sulfated solids (containing CaSO4 and unreacted CaO) are then
physically
removed from the flue gas through the use of a cyclone. The use of a single
cyclone
effectively separates >99% of all solids. This solid mixture is then safely
disposed.
[00219] The solids depleted flue gas is then subjected to C02 removal. This is
accomplished by injecting hot CaO powder through nozzles. The CaO injection
would occur in the 550-700C temperature range. The carbonator reaction, like
72
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
sulfation, is extremely fast, with the majority of reactions occurring in less
than one
second. This translates to an approximately 1.4 m long flue gas duct
requirement
under the process conditions assuming a residence time of 1 second. The
current
equipment under consideration provides ample opportunity for this residence
time.
The carbonation reaction releases tremendous quantity of heat causing a
significant
increase in flue gas temperature. However, adequate heat extraction surface
would
be provided and the actual heat transferred would be accurately quantified.
This is
one of the main outcomes of this project.
[00220] The carbonated sorbent is removed from the flue gas by an identical
cyclone, downstream of the carbonation reactor. Attempts would be made to
maintain the flue gas flowing between the two cyclones under isothermal
conditions.
This will be achieved by providing adequate heat transfer extraction to absorb
the
heat liberated during carbonation. Hence the flue gas temperature at both
cyclones
would be the same. This is advantageous as identical design can be used for
the two
cyclones. The use of identical cyclones enhances reliability of the CCR
process and
reduces design and testing costs associated with cyclones. The flue gas
leaving the
second "carbonation" cyclone is cooled and sent through another lower
temperature
particle capture device (PCD) and finally vented out of the building.
[00221] The carbonated hot solids are then sent to the calciner, which
provides
0
the heat required to raise the temperature of the solids to 770-830 C and
calcine the
carbonated portion of the solids. Prior laboratory data indicates that the
calcination
under sub-atmospheric conditions aids in maintaining a higher porosity CaO
sorbent,
which also exhibits higher reactivity. A water ejector will generate vacuum
for the
calcination. The water flowing through the throat of the eductor causes the
absolute
pressure to fall, allowing the suction of C02 out of the calciner. The two-
phase fluid is
73
CA 02613698 2007-12-28
WO 2007/002792 PCT/US2006/025266
then sent to a knockout drum, where water is separated from the "separated"
C02.
The water stored in the knockout drum is continuously recycled through the
water
pump for continuous vacuum building.
CONCLUSIONS
[00222] The enhanced water gas shift reaction for H2 production with in-situ
carbonation was studied at 600 C using HTS catalyst and calcium sorbents. A
naturally occurring calcium precursor (Linwood hydrate, LH) and a modified
mesoporous Precipitated Calcium Carbonate (PCC) were used for capturing CO2
for
two successive cycles. The PCC system gives almost 100% conversion for first 4
min followed by 90% at 16.5 min. In contrast, the LH sorbent system sustains
100%
conversion only in the initial few seconds and gradually gives about 90% CO
conversion at 15 min. Experimental evidence clearly shows that the PCC-CaO
performance dominates over that of LH-CaO at any given time.
74
CA 02613698 2012-05-09
[00223] While the invention has been described in connection with what is
presently considered to be the most practical and preferred embodiments, it is
to be
understood that the invention is not to be limited to the disclosed
embodiment(s), but
on the contrary, is intended to cover various modifications and equivalent
arrangements included within the scope of the appended claims,