Note: Descriptions are shown in the official language in which they were submitted.
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
1
2-Arylbenzothiazoles and uses thereof
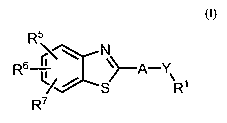
The present invention relates to 2-arylbenzothiazoles of the general formula
(I) or a salt or
a prodrug or a stereoisomer thereof. The compounds of the invention are
exceptionally
useful for the treatment of diseases associated with abnormal and
hyperproliferation of
cells in a mammal, especially humans. In particular, they are useful for the
treatment of all
forms of cancer.
Furthermore a process of preparing said 2-arylbenzothiazole derivatives is
disclosed.
Background of the invention
Protein kinases play a central role in the regulation of cellular functions.
This includes
processes like cell growth and division, cell differentiation and cell death,
but also many
other cellular activities. Protein kinases catalyze the transfer of phosphate
residues from
ATP on target proteins which as a consequence of this protein kinase mediated
phosphorylation change their three-dimensional structure and thereby their
physiological
function. Depending on the amino acid which is phosphorylated by a protein
kinase these
enzymes are grouped in two families, the so-called serine/threonine protein
kinases and the
tyrosine protein kinases.
Based on the human genome project it is known that in human beings there exist
518 DNA
sequences which eiicode for a protein kinase-like protein sequence. For
several of these
518 proteins it could be shown in the last about 20 years that modifications
in their related
gene sequences (e.g. point mutations, deletions or gene amplifications) result
in
pathological changes of the cellular activities of the corresponding protein
kinase. This is
in particular true for protein kinases which are involved in cell
proliferation and cell cycle
control, in survival of cells and cell death, in tumor angiogenesis, and in
formation of
tumor metastases.
Several so-called oncogenes are pathologically modified genes which in their
proto-
oncogenic form encode for protein kinases involved in normal, physiological
regulation of
cell growth and division.
Since protein kinases are key regulators of cell functions and since they can
show
dysregulated enzymatic activity in cells they are promising targets for the
development of
therapeutic agents. There are many ongoing drug discovery projects in the
pharmaceutical
industry with the goal to identify modulators of protein kinases. The major
focus is
currently on protein kinases involved in inflammation and cancer, but besides
this protein
kinases are currently discussed as promising targets in almost every area of
diseases.
In the field of tumors the first protein kinase inhibitors (Gleevec, Iressa)
have already
reached the market. In addition, a great number of protein kinase inhibitors
are currently in
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
2
various phases of clinical development. In most cases these compounds are
either targeting
subtypes of the EGF (Epidermal Growth Factor) receptor family or of the VEGF
(Vascular
Endothelial Growth Factor) receptor family. All these compounds have been
developed
with the goal to specifically inhibit one particular protein kinase, for which
there is
evidence that it interferes with one of the four major molecular processes of
tumor
progression. These four processes are (1) cell proliferation/cell cycle
control, (2) regulation
of programmed cell death (apoptosis) and cell survival, (3) tumor angiogenesis
and (4)
tumor metastasis.
The present invention relates to 2-arylbenzothiazole derivatives which may be
useful for
inhibition of protein kinases involved in diseases besides cancer, but which
are especially
useful as anti-tumor agents. This includes monospecific protein kinase
inhibitors, which
preferentially inhibit one protein kinase which is causatively involved in
tumor
progression, but also so-called multi-target protein kinase inhibitors, which
inhibit at least
two different protein kinases which either relate to the same or to two or
more different
molecular mechanism of tumor progression. As an example, such a compound could
be an
inhibitor of tumor angiogenesis and, in addition, also a stimulator of
apoptosis.
The concept of multi-target protein kinase inhibitors is a new approach
although the idea of
developing "multiplex protein kinase inhibitors" has already been described by
J. Adams et
al., Current Opinion in Chemical Biology 6, 486-492, 2002. Therein compounds
are
described, which, at the same time, inhibit several protein kinases, which
however all are
involved in one molecular mechanism of tumor progression, namely tumor
angiogenesis.
The object of the present invention is solved by the subject-matter of the
independent
claims. Further advantageous features, aspects and details of the invention
are evident from
the dependent claims, the description, the figures, and the examples of the
present
application.
Considering the lack of currently available treatment options for the majority
of the
conditions associated with protein kinases like ABL1, AKT1, AKT2, AKT3, ARK5,
Aurora-A, Aurora-B, Aurora-C, BRK, CDK1, CDK2, CDK3, CDK4, CDK5, CDK6,
CDK7, CDK9, CHK1, CK2, COT, CSK, DAPKI, EGF-R, EPHA1, EPHA2, EPHA4,
EPHBI, EPHB2, EPHB3, EPHB4, ERBB2, ERBB4, FAK, FGF-R1, FGF-R3, FGF-R4,
FGR, FLT3, GSK3-beta, IGF1-R, IKK-beta, IKK-epsilon, INS-R, IRAK4, ITK, JAK2,
JAK3, JNK3, KIT, LCK, LYN, MET, MST4, MUSK, NEK2, NEK6, NLK, PAK1, PAK2,
PAK4, PBK, PCTAIREI, PDGFR-alpha, PDGFR-beta, PDK1, PIM1, PIM2, PKC-alpha,
PKC-betal, PKC-beta2, PKC-delta, PKC-epsilon, PKC-eta, PKC-gamma, PKC-iota,
PKC-
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
3
mu, PKC-theta, PKC-zeta, PLK1, PRK1, RET, ROCK2, S6K, SAK, SGK1, SGK3, SNK,
SRC, SYK, TIE2, TSF1, TSK2, VEGF-R1, VEGF-R2, VEGF-R3, VRK1, WEE1, YES,
ZAP70 especially with protein kinases like EGF-R (cell proliferation), ERBB2
(cell
proliferation), PDGFR (cell proliferation), Aurora-A (cell cycle control),
Aurora-B (cell
cycle control), IGF1-R (apoptosis), VEGF-R2 (angiogenesis), VEGF-R3
(angiogenesis),
TIE2 (angiogenesis), EPHB4 (angiogenesis), and SRC kinase (metastasis), there
is still a
great need for new therapeutic agents that inhibit these protein targets.
2-Arylbenzothiazole derivatives described herein are a new group of protein
kinase
inhibitors which show differential inhibition of protein kinases, each of
which can be
assigned to one of the four molecular mechanisms of tumor development.
In WO 0149686 2-Phenyl-benzothiazoles substituted by an aminotriazine are
described as
UV-filters used as skin and hair sunscreens.
Similar compounds are described in WO 9825922, in EP 841341 and in JP
11060573, all of
them also substituted by a aminotriazine.
In EP 711818 2-Phenyl-benzothiazoles are claimed as liquid crystal
compositions.
The present invention relates to compounds of the general formula (1) or a
salt or a prodrug
or a stereoisomer thereof,
R5
R l ~A-Y'
6 'D:s N
/ R'
R7
formula (I)
wherein
Y independently represents a divalent linkage selected from S, 0, NR2, SO,
SO2;
A independently represents a divalent linkage selected from a five- or six-
membered aromatic carbocycle or heterocycle each of which is optionally
substituted by one to four substituents selected from R3 and R4, with the
proviso
that A-Y is not NR2 attached at the 2- or 4-position of a pyrimidine ring
represented
by A and A-Y is not NR2 attached to 2-halopyridine represented by A;
R2 independently represents H, alkyl, cycloalkyl, -COR", -SOR", -SO2R11, -CN,
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
4
hydroxyalkyl, haloalkyl, haloalkyloxy, or alkylamino;,
R3 independently represents H, -CORI l, -C02RI I, -SOR", -S02R' 1, -S03R11, -
NO2,
-CN, -CF3, -OCH3, -OCF3, alkyl, cycloalkyl, alkoxy, NH2, alkylamino,
-NR8COR' 1, halogen, -OH, -SH, alkylthio, hydroxyalkyl, haloalkyl, or
haloalkyloxy;
R4 independently represents H, -CORII, -C02R11, -SOR", -S02R", -S03R", -NO2,
-CN, -CF3, -OCH3, -OCF3, alkyl, cycloalkyl, alkoxy, -NH2, alkylamino,
-NRgCOR' 1, halogen, -OH, -SH, alkylthio, hydroxyalkyl, haloalkyl,
haloalkyloxy,
aryl or heteroaryl;
RS independently represents H, -COR", -C02R11, -SORII, -S02R", -S03R11, -NOz,,
-CN, -CF3, -OCH3, -OCF3, alkyl, cycloalkyl, alkoxy, -NH2, alkylamino,
-NR8COR", halogen, -OH, -SH, alkylthio, hydroxyalkyl, haloalkyl, haloalkyloxy,
aryl or heteroaryl;
R6 independently represents H, -COR11, -C02R11, -SOR", -S02R", -S03R11, -NO2,
-CN, -CF3, -OCH3, -OCF3, alkyl, cycloalkyl, alkoxy, -NH2, alkylamino,
-NR8COR11, halogen, -OH, -SH, alkylthio, hydroxyalkyl, haloalkyl,
haloalkyloxy,
aryl or heteroaryl;
R7 independently represents H, -COR", -C02R", -SOR' 1, -SOZRI l, -S03R11, -
NOZ,
-CN, -CF3, -OCH3, -OCF3, alkyl, cycloalkyl, alkoxy, -NH2, alkylamino,
-NR8COR", halogen, -OH, -SH, alkylthio, hydroxyalkyl, haloalkyl, haloalkyloxy,
aryl or heteroaryl;
R8 independently represents H, alkyl, cycloalkyl, -CORI l, -SOR", -SOZRI l,
hydroxyalkyl, haloalkyl, haloalkyloxy, aryl or heteroaryl;
R9 independently represents H, alkyl, cycloalkyl, hydroxyalkyl, haloalkyl,
haloalkyloxy, aryl or heteroaryl;
R" independently represents H, alkyl, cycloalkyl, -NR 8R9, -NR8 NR8R9, -
ONRgR9,
-NRgOR9, alkylamino, arylamino, aryl or heteroaryl;
R' independently represents one of the following groups:
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
* R13 * R13 * R13
R14 R14 N~ R14
i R17 i
12 / ~ 15 15 R17 N R15
R N R N R R16
R16 R16
R13
13
N ' R14 N R NN
~/ ~
~ ~ R15 R12a N R14 R12a R13
17
R R16a R14
* * *
R14 R15 R13 R13
~
~N l N
12 13a 12 / 14 12a 14
R N R R N R R R
R15
* * *
R13 R R 8 N ~ N
I N R18
~' , ~ R
R 13
N
N
12a 14 N R19 R14 N
R15 0 R$
* R8 R13
I
N N Z
jll~ ~ R13 i R14 i R13
14 N 15~ Z 15
R N R N R N
R14
R13
N/
R14
16 N ~N
R
R15
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
6
where * indicates the point of attachment
Z independently represents 0, NR8, or S;
R12 independently represents H, -NHR8; or one of the following groups:
R5
N'N N'5
or R
HN--..** HN~
where ** indicates the point of attachment.
R12a independently represents one of the following groups:
R5
i
N ~N'R5
N
or ~
HN~ HN~
where ** indicates the point of attachment.
R13 independently represents H, halogen, nitro, -CN, trifluoromethyl, alkyl,
aryl,
heteroaryl, -NR8R9, or -X2R18;
R13a independently represents H, nitro, -CN, trifluoromethyl, alkyl, aryl, or
heteroaryl;
R14 independently represents H, halogen, nitro, -CN, trifluoromethyl, alkyl,
aryl,
heteroaryl, -NR8R9, or -X2R18;
R15 independently represents H, halogen, nitro, -CN, trifluoromethyl, alkyl,
aryl,
heteroaryl, -NRgR9, or -X2R18;
R16 independently represents H, halogen, nitro, -CN, trifluoromethyl, alkyl,
aryl,
heteroaryl, -NRgR9, or -X2R18;
R16a independently represents H, halogen, nitro, -CN, trifluoromethyl, alkyl,
heteroaryl, -NR8R9, or -XZRIg;
Rl7 independently represents H, halogen, nitro, trifluoromethyl, alkyl, aryl,
heteroaryl, -NR8R9, or -X2R18;
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
7
X2 independently represents a direct bond, -0-, -CH2-, -OCO-, CO, -S-,
-SO-, -SO2-, -NRgCO-, -CONRg-, -SO2NRg-, -NRB- or -NR8SO2-;
Rlg independently represents H, alkyl, cycloalkyl, -COR", -SORII,
-SO2R11, -OCH3, -OCF3, hydroxyalkyl, haloalkyl, haloalkyloxy, or
one of the following groups:
# L,. N #
N N ~
~
N ~ ~N' R19 O
~
#/L , N #/L' #/~
0 aR19 N~ R 19
LN L ., Xs
O". R2o
m
N LN N
1000~
N '(IO L.__~ N O--R 20 N
2o 19
R .R
m m
#LN #/LN #LN
I (I...,
#,o,- L, N LN'o~, N LN
,O
R2o
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
8
#rL i
.N ~~N q
R20,O jj)_D ,
"O m .O
R20 R20
'k
OR20 )rOR19
O O
where # indicates the point of attachment
m independently represents an integer from 1-3;
L is absent or represents a divalent linkage group selected from alkylen,
cycloalkylen, heterocyclylen, arylen, or heteroarylen, wherein one or
more of the (-CH2-) groups may be replaced by an oxygen or a NRg,
and wherein one or more carbon atoms may be independently
substituted by one or two substituents selected from halogen, hydroxy,
alkoxy, haloalkyloxy, phoshonooxy, or phoshonooxyalkyl;
X3 independently represents -COOH, -COOalkyl, -CONR8R9, -OH,
-NR8R9, -SH, -SO3H, or -SO2NR8 R9;
R19 independently represents H, alkyl, cycloalkyl, alkylamino, or alkoxy;
R20 independently represents H, phosphonooxy, or phosphonooxyalkyl;
wherein
an alkyl group, if not stated otherwise, denotes a linear or branched C1-C6-
alkyl, preferably
a linear or branched chain of one to five carbon atoms, a linear or branched
C2-C6-alkenyl
or a linear or branched C2-C6-alkinyl group, which can be substituted by one
or more
substituents R';
the C1-C6-alkyl, C2-C6-alkenyl and C2-C6-alkinyl residue may be selected from
the group
comprising -CH3, -C2H5, -CH=CH2, -C=CH, -C3H7, -CH(CH3)2, -CH2-CH=CH2,
-C(CH3)=CH2, -CH=CH-CH3, -C=C-CH3, -CH2-C=CH, -C4H9, -CH2-CH(CH3)2,
-CH(CH3)-C2H5, -C(CH3)3, -C5Hii, -C6Hi3, -C(R')3, -C2(R')5, -CH2-C(R')3, -
C3(R')7,
-C2H4-C(R')3, -C2H4-CH=CH2, -CH=CH-C2H5, -CH=C(CH3)2, -CH2-CH=CH-CH3,
-CH=CH-CH=CH2, -CZH4-C-CH, -C=C-C2H5, -CH2-C=C-CH3, -C=C-CH=CH2,
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
9
-CH=CH-C=CH, -C=C-C=CH, -C2H4-CH(CH3)2, -CH(CH3)-C3H7, -CH2-CH(CH3)-C2H5,
-CH(CH3)-CH(CH3)2, -C(CH3)2-C2H5, -CH2-C(CH3)3, -C3H6-CH=CH2,
-CH=CH-C3H7, -C2H4-CH=CH-CH3, -CH2-CH=CH-C2H5, -CH2-CH=CH-CH=CH2,
-CH=CH-CH=CH-CH3, -CH=CH-CH2-CH=CH2, -C(CH3)=CH-CH=CH2,
-CH=C(CH3)-CH=CH2, -CH=CH-C(CH3)=CH2, -CH2-CH=C(CH3)2, C(CH3)=C(CH3)2,
-C3H6-C=CH, -C=C-C3H7, -C2H4-C=C-CH3, -CH2-C=C-CZH5, -CH2-C=C-CH=CH2, -CHZ-
CH=CH-C=CH, -CH2-C=C-C=CH, -C=C-CH=CH-CH3, -CH=CH-C=C-CH3,
-C=C-C=C-CH3, -C C-CH2-CH=CH2, -CH=CH-CH2-C=CH, -C=C-CH2-C=CH,
-C(CH3)=CH-CH=CH2, -CH=C(CH3)-CH=CH2, -CH=CH-C(CH3)=CH2, -C(CH3)=CH-
C=CH, -CH=C(CH3)-C=CH, -C=C-C(CH3)=CH2, -C3H6-CH(CH3)2, -C2H4-CH(CH3)-
C2H5, -CH(CH3)-C4H9, -CH2-CH(CH3)-C3H7, -CH(CH3)-CH2-CH(CH3)2, -CH(CH3)-
CH(CH3)-C2H5, -CH2-CH(CH3)-CH(CH3)2, -CH2-C(CH3)2-C2H5, -C(CH3)2-C3H7,
-C(CH3)2-CH(CH3)2, -C2H4-C(CH3)3, -CH(CH3)-C(CH3)3, -C4H8-CH=CH2, -CH=CH-
C4H9, -C3H6-CH=CH-CH3, -CH2-CH=CH-C3H7, -C2H4-CH=CH-C2H5, -CH2-
C(CH3)=C(CH3)2, -C2H4-CH=C(CH3)2, -C4Hg-C=CH, -C=C-C4H9, -C3H6-C=C-CH3,
-CH2-C=C-C3H7, -C2H4-C=C-C2H5;
R' independently represents H, -CO2R", -CONHR", -CR"O, -SO2NR", -NR"-CO-
haloalkyl, -NO2, -NR"-S02-haloalkyl, -NR"-S02-alkyl, -S02-alkyl, -NR"-CO-
alkyl, -CN,
alkyl, cycloalkyl, aminoalkyl, alkylamino, alkoxy, -OH, -SH, alkylthio,
hydroxyalkyl,
hydroxyalkylamino, halogen, haloalkyl, haloalkyloxy, aryl, arylalkyl or
heteroaryl;
R" independently represents H, haloalkyl, hydroxyalkyl, alkyl, cycloalkyl,
aryl, heteroaryl
or aminoalkyl;
an alkylene group denotes a divalent linear or branched C1-C6-alkylene,
preferably a linear
or branched chain of one to five carbon atoms, a linear or branched C2-C6-
alkenylene or a
linear or branched C2-C6-alkynylene group, which may be substituted by one or
more
substituents R';
a cycloalkylene group denotes a divalent non-aromatic ring system containing
three to
eight carbon atoms, preferably four to eight carbon atoms, wherein one or more
of the
carbon atoms in the ring may be substituted by a group E, E being 0, S, SO,
SOZ, N, or
NR", R" being as defined above;
a heterocyclylene group denotes a 3 to 8-membered divalent heterocyclic non-
aromatic
group which contains at least one heteroatom selected from 0, N, and S,
wherein the
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
heterocyclylene group may be fused to another non-aromatic ring and may be
substituted
by one or more substituents R', wherein R' is as defined above;
an arylene group denotes an aromatic divalent group having five to fifteen
carbon atoms,
5 which may be substituted by one or more substituents R', and may be fused to
another
aromatic ring, where R' is as defined above;
a heteroarylene group denotes a divalent 5- or 6-membered heterocyclic group
which
contains at least one heteroatom selected from 0, N, and S, wherein the
heterocyclylene
10 group may be fused to another aromatic ring and may be substituted by one
or more
substituents R', wherein R' is as defined above;
a cycloalkyl group denotes a non-aromatic ring system containing three to
eight carbon
atoms, preferably four to eight carbon atoms, wherein one or more of the
carbon atoms in
the ring can be substituted by a group E, E being 0, S, SO, SO2, N, or NR", R"
being as
defined above; the C3-Cg-cycloalkyl residue may be selected from the group
comprising
-cyclo-C3H5, -cyclo-C4H7, -cyclo-C5H9, -cyclo-C6H12, -cyclo-C7Hi3, -cyclo-
C8H15,
morpholine-4-yl, piperazinyl, 1-alkylpiperazine-4-yl;
an alkoxy group denotes an 0-alkyl group, the alkyl group being as defined
above; the
alkoxy group is preferably a methoxy, ethoxy, isopropoxy, t-butoxy or pentoxy
group;
an alkylthio group denotes an S-alkyl group, the alkyl group being as defmed
above;
an haloalkyl group denotes an alkyl group which is substituted by one to five
halogen
atoms, the alkyl group being as defined above; the haloalkyl group is
preferably a-C(R10)3,
io io io io io io io io io
-CR (R )2, -CR (R )R , -C2(R )5, -CH2-C(R )3, -CH2-CR (R )2, -CH2-
CRI0(R10')R10 , -C3(R10)7> or -C2H4-C(Rl0)3, wherein Rlo> Ri0 , Rlo"
represent F. Cl, Br or I
,
preferably F;
a hydroxyalkyl group denotes an HO-alkyl group, the alkyl group being as
defined above;
an haloalkyloxy group denotes an alkoxy group which is substituted by one to
five halogen
atoms, the alkyl group being as defined above; the haloalkyloxy group is
preferably a
-OC(R10)3, -0CRl0(R10')2, -OCRIO(Rlo')Rio , -OC2(Rl )5, -OCHZ-C(R10)3, -OCH2-
CRio(Rio )2, -OCH2-CR10(R10 )R10 , -OC3(Ri0)7 or -OC2H4-C(Ri0)3, wherein Rio,
Rio , Rio
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
11
represent F, Cl, Br or I, preferably F;
a hydroxyalkylamino group denotes an (HO-alkyl)2-N- group or HO-alkyl-NH-
group, the
alkyl group being as defined above;
an alkylamino group denotes an HN-alkyl or N-dialkyl group, the alkyl group
being as
defined above;
an arylamino group denotes an HN-aryl, or N-diaryl, or -N-aryl-alkyl group,
the alkyl and
aryl group being as defined above;
a halogen group is fluorine, chlorine, bromine, or iodine;
an aryl group denotes an aromatic group having five to fifteen carbon atoms,
which can be
substituted by one or more substituents R', where R' is as defined above; the
aryl group is
preferably a phenyl group, -o-C6H4- R', -m-C6H4- R', -p-C6H4- R', 1-naphthyl,
2-naphthyl,
1-anthracenyl or 2-anthracenyl;
a heteroaryl group denotes a 5- or 6-membered heterocyclic group which
contains at least
one heteroatom like 0, N, S. This heterocyclic group can be fused to another
aromatic ring.
For example, this group can be selected from a thiadiazole, thiazol-2-yl,
thiazol-4-yl,
thiazol-5-yl, isothiazol-3-yl, isothiazol-4-yl, isothiazol-5-yl, oxazol-2-yl,
oxazol-4-yl,
oxazol-5-yl, isooxazol-3-yl, isooxazol-4-yl, isooxazol-5-yl, 1,2,4-oxadiazol-3-
yl, 1,2,4-
oxadiazol-5-yl, 1,2,5-oxadiazol-3-yl, 1,2,5-oxadiazol-4-yl, 1,2,4-thiadiazol-3-
yl, 1,2,4-
thiadiazol-5-yl, isothiazol-3-yl, isothiazol-4-yl, isothiazol-5-yl, 1,2,5-
thiadiazol-3-yl, 1-
imidazolyl, 2-imidazolyl, 1,2,5-thiadiazol-4-yl, 4-imidazolyl, 1-pyrrolyl, 2-
pyrrolyl, 3-
pyrrolyl, 2-furanyl, 3-furanyl, 2-thienyl, 3-thienyl, 2-pyridyl, 3-pyridyl, 4-
pyridyl, 2-
pyranyl, 3-pyranyl, 4-pyranyl, 2-pyrimidinyl, 4-pyrimidinyl, 5-pyrimidinyl,
pyrid-2-yl,
pyrid-3-yl, pyrid-4-yl, pyrid-5-yl, pyrid-6-yl, 3-pyridazinyl, 4-pyridazinyl,
2-pyrazinyl, 1-
pyrazolyl, 3-pyrazolyl, 4-pyrazolyl, 1,2,3-triazol-4-yl, 1,2,3-triazol-5-yl,
1,2,4-triazol-3-yl,
1,2,4-triazol-5-yl, 1H-tetrazol-2-yl, 1H-tetrazol-3-yl, tetrazolyl, acridyl,
phenazinyl,
carbazolyl, phenoxazinyl, indolizine, 2-indolyl, 3-indolyl, 4-indolyl, 5-
indolyl, 6-indolyl,
7-indolyl, 1-isoindolyl, 3-isoindolyl, 4-isoindolyl, 5-isoindolyl, 6-
isoindolyl, 7-isoindolyl,
2-indolinyl, 3-indolinyl, 4-indolinyl, 5-indolinyl, 6-indolinyl, 7-indolinyl,
benzo[b]furanyl,
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
12
benzofurazane, benzothiofurazane, benzotriazol-1-yl, benzotriazol-4-yl,
benzotriazol-5-yl,
benzotriazol-6-yl, benzotriazol-7-yl, benzotriazine, benzo[b]thiophenyl,
benzimidazolyl,
benzothiazolyl, quinazolinyl, quinoxazolinyl, cinnoline, quinolinyl,
tetrahydroquinolinyl,
isoquinolinyl, or tetrahydroisoquinolinyl,purine, phthalazine, pteridine,
thiatetraazaindene,
thiatriazaindene, isothiazolopyrazine, isothiazolopyrimidine,
pyrazolotriazine,
pyrazolopyrimidine, imidazopyridazine, imidazopyrimidine, imidazopyridine,
imidazolotriazine, triazolotriazine, triazolopyridine, triazolopyrazine,
triazolopyrimidine,
triazolopyridazine group. This heterocyclic group can be substituted by one or
more
substituents R', wherein R' is as defined above;
a phosphonooxy group is -O-P(=O)(OH)2 or a salt thereof;
a phosphonooxyalkyl group denotes an -alkyl-O-P(=O)(OH)2 group or a salt
thereof, alkyl
being as defined above.
The invention also provides a pharmaceutical composition comprising a compound
of
formula (I), in free form, or in the form of a pharmaceutically acceptable
salt, or a prodrug
thereof, together with a pharmaceutically acceptable diluent or carrier
therefore.
The term "physiologically functional derivative" as used herein refers to
compounds which
are not pharmaceutically active themselves but which are transformed into
their
pharmaceutical active form in vivo, i.e. in the subject to which the compound
is
administered. Examples of physiologically functional derivatives are prodrugs
such as
those described below in the present application.
The term "prodrug" as used herein refers to compounds which are not
pharmaceutically
active themselves but which are transformed into their pharmaceutical active
form in vivo,
i.e. in the subject to which the compound is administered. Prodrugs of the
compounds of
the present invention include but are not limited to: esters, which are
transformed in vivo
into the corresponding active alcohol, esters, which are transformed in vivo
into the
corresponding active acid, imines, which are transformed in vivo into the
corresponding
amines, imines which are metabolized in vivo into the corresponding active
carbonyl
derivative (e.g. aldehyde or ketone), 1-carboxy-amines, which are
decarboxylated in vivo
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
13
into the active amine, phosphoryloxy-compounds, which are dephosporylated in
vivo by
phosphateases into the active alcohols, and amides which are metabolized into
the
corresponding active amine or acid respectively.
The term "stereoisomer" as used herein refers to compound with at least one
stereogenic
center, which can be R- or S-configurated. It has to be understood, that in
compounds with
more than one stereogenic center each of which independently from each other
can be R-
or S-configurated. The term "stereoisomer" as used herein also refers to salts
of the
compounds herein described with optically active acids or bases. The term
"stereoisomer"
also means cis/trans or E/Z isomerism. More particularly, the possible double
bond(s)
present in the various substituent of the compounds of the present invention
can be E or Z
configuration. These pure or impure geometrical isomers, alone or as a
mixture, form an
integral part of the compounds of the present invention. The term
"stereoisomer" includes
also all the isomeric forms, alone or as mixture, resulting from the presence
of one or more
axes and / or centers of symmetry in the molecules, and resulting in the
rotation of a beam
of polarized light. More particularly, it includes enatiomers and
diastereomers, in pure
form or as a mixture.
In addition, the present invention provides methods for preparing the
compounds of the
invention such as compounds of formula (I).
The compounds of formula (I) may be obtained via various methods. One
possibility for
the synthesis of compounds of formula (I) comprises the step of reacting a
compound of
formula (VII), wherein R5, R6, R7 , A, and Y are defined as above, with a
compound of
formula (VIII), wherein R' is as defined above and LG comprises a leaving
group such as
Cl, Br, or I. Either nucleophilic substitution or palladium-catalyzed cross-
coupling may be
applied. If Y= NRZ, R2 may be added before or after addition of R'.
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
14
R5
' N
R s >--A-Y' + LG-Rl
S H
R 7 formula (VIII)
formula (VII)
R5
s ' N
R ~ )-A-Y
R'
R7 '
formula (I)
Compounds of formula (VII) can be synthesized by reaction of a thioamine
according to
formula (X) with an carboxylic acid of the formula (IX).
R5
'~ NH2 + 0
s
R ~ ~A-Y\
SH HO H
R7
formula (X) formula (IX)
R5
s ' N
R ~ >--A-Y'
S H
R7
formula (VII)
Alternatively an activated acid derivative (e.g. acid chloride) may be used
instead of the
acid according to formula (TX).
Another way to synthesize compounds of the formula (VII) is the conversion of
compounds of formula (XI), a disulfide, into the benzothiazole of formula
(VII) by
reduction of the disulfide and subsequent condensation.
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
H ~Y~A
~O R5
R 5 NH 6 ~ N
R6 ~ R A_Y
~
L~/ S /~ S H
R7 R
formula (XI) 2 formula (VII)
In some cases in the synthesis of compounds of the formula (VII) and formula
(XI) Y may
be protected. This protection group has to be removed before converting them
into
compounds of the formula (I). Carboxylic acids of formula (IX) may arise from
nitrile
5 hydrolysis.
Compounds of the formula (XI) may be obtained by reaction of a disulfide
according to
formula (XII) with a compound of formula (IX) or alternatively an activated
carboxylic
acid derivative.
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
16
R5
s ' NH2 O
R S R5 + A-Y
R7 S ~/1 Rs HO H
,
7 formula (IX)
H2N R
formula (XII)
ilf
H --Y--,A
~O
R5
s NH
R ,
S
R7
formula (XI) 2
As far as compounds of the general formula (X) and formula (XII) could not be
purchased,
they were synthesized from the benzothiazoles, 2-aminobenzthiazoles or 2-
methyl-
benzothiazoles of formula (XIII) e.g. by hydrolysis under basic conditions.
5
R D"" N formula (X)
R~R21
and / or
s
R
formula (XII)
formula (XIII)
R21 = H, NH2, Me
A preferred embodiment of the invention are compounds of the formula (I)
wherein Y is
NR2or0.
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
17
A preferred embodiment of the invention are compounds of the formula (II)
R3
R5
~ ~ N Q'I'R,
R6 ~ "_Q
Q
R7 Q_I4
R
formula (H)
wherein
Q independently represents C, N, CH;
Y is as defined above; Rl, R3, R4, R5, R6and R7 are as defined above.
In a more preferred embodiment of the invention, in the compounds of formula
(II) are the
compounds where Q independently represents C or CH.
In a more preferred embodiment of the invention, in the compounds of formula
(II) are the
compounds where Y is attached at the 3- or 4-position of the cycle A.
A preferred embodiment of the invention are compounds of the formula (II)
wherein Y is
NR2orO.
A preferred embodiment of the invention are compounds of the formula (II)
wherein Y is
NR2 or 0 and where Y is attached at the 3- or 4-position of the cycle A.
A preferred embodiment of the invention are compounds of the formula (II)
wherein Q is
C or CH and R3 is F, Cl, or OCH3.
A preferred embodiment of the invention are compounds of the formula (II)
wherein Q is
C or CH and R3 is F, Cl, or OCH3 and where Y is attached at the 3- or 4-
position of the
cycle A.
A preferred embodiment of the invention are compounds of the formula (II)
wherein Y is
NR2 or 0 and wherein Q is C or CH and R3 is F, Cl, or OCH3.
A preferred embodiment of the invention are compounds of the formula (II)
wherein Y is
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
18
NRZ or 0 and wherein Q is C or CH and R3 is F, Cl, or OCH3 and where Y is
attached at
the 3- or 4-position of the cycle A.
In an even more preferred embodiment of the invention are compounds of the
formula (III)
R14
#13 R3 R15
R 5
N Q::QR6 Q R16a
~
~ S Q-I-Q NN
R R4
s
formula (III)
wherein
Q independently represents C, N, CH;
Y, R3, R4, R5, R6, R7 , R13, Rla, Rls, and R16a are as defmed above.
In a more preferred embodiment of the invention, in the compounds of formula
(III) are the
compounds where Y is attached at the 3- or 4-position of the cycle A.
A preferred embodiment of the invention are compounds of the formula (III)
wherein Y is
NRZ or O.
A preferred embodiment of the invention are compounds of the formula (III)
wherein Y is
NR2 or 0 and where Y is attached at the 3- or 4-position of the cycle A.
A preferred embodiment of the invention are compounds of the formula (III)
wherein Q is
C or CH and R3 is F, Cl, or OCH3.
A preferred embodiment of the invention are compounds of the formula (III)
wherein Q is
C or CH and R3 is F, Cl, or OCH3 and where Y is attached at the 3- or 4-
position of the
cycle A.
A preferred embodiment of the invention are compounds of the formula (III)
wherein Y is
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
19
NR2 or 0 and wherein Q is C or CH and R3 is F, Cl, or OCH3.
A preferred embodiment of the invention are compounds of the formula (III)
wherein Y is
NR2 or 0 and wherein Q is C or CH and R3 is F, Cl, or OCH3 and where Y is
attached at
the 3- or 4-position of the cycle A.
Another preferred embodiment of the invention are compounds of the formula
(IV)
R3
5
R
~ N Q:'%Y N R13
R6 Q y
//
i
L / Q. .Q N
R7 R I4
HN N
~ ~.
NH
formula (IV)
wherein
Q independently represents C, N, CH;
Y, R3, R4, R5, R6, R' and R13 are as defined above.
In a more preferred embodiment of the invention, in the compounds of formula
(IV) are the
compounds where Y is attached at the 3- or 4-position of the cycle A.
A preferred embodiment of the invention are compounds of the formula (IV)
wherein Y is
NR2or0.
A preferred embodiment of the invention are compounds of the formula (IV)
wherein Y is
NR2 or 0 and where Y is attached at the 3- or 4-position of the cycle A.
A preferred embodiment of the invention are compounds of the formula (IV)
wherein Q is
C or CH and R3 is F, Cl, or OCH3.
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
A preferred embodiment of the invention are compounds of the formula (IV)
wherein Q is
C or CH and R3 is F, Cl, or OCH3 and where Y is attached at the 3- or 4-
position of the
cycle A.
5 A preferred embodiment of the invention are compounds of the formula (IV)
wherein Y is
NRZ or 0 and wherein Q is C or CH and R3 is F, Cl, or OCH3.
A preferred embodiment of the invention are compounds of the formula (IV)
wherein Y is
NR2 or 0 and wherein Q is C or CH and R3 is F, Cl, or OCH3 and where Y is
attached at
10 the 3- or 4-position of the cycle A.
Exemplary compounds of formula (I) of the present invention include, but are
not limited
to, the following:
Name Compound
(4-Benzothiazol-2-yl-phenyl)-(4,6-dimethyl-pyrimidin-2-yl)-amine 1
(4-Benzothiazol-2-yl-2-methyl-phenyl)-(4,6-dimethyl-pyrimidin-2-yl)-amine 2
(3-Benzothiazol-2-yl-phenyl)-(4,6-dimethyl-pyrimidin-2-yl)-amine 3
(5-Benzothiazol-2-yl-2-chloro-phenyl)-(4,6-dimethyl-pyrimidin-2-yl)-amine 4
(4-Benzothiazol-2-yl-3-methoxy-phenyl)-(4,6-dimethyl-pyrimidin-2-yl)- 5
amine
2-Benzothiazol-2-yl-5-(4,6-dimethyl-pyrimidin-2-ylamino)-phenol 6
2-Benzothiazol-2-yl-4-(4,6-dimethyl-pyrimidin-2-ylamino)-phenol 7
(5-Benzothiazol-2-yl-2-methyl-phenyl)-(4,6-dimethyl-pyrimidin-2-yl)-amine 8
(4-Benzothiazol-2-yl-phenyl)-(6,7-dimethoxy-quinazolin-4-yl)-amine 9
(4-Benzothiazol-2-yl-phenyl)-[4-(4-methyl-piperazin-l-yl)-pyrimidin-2-yl]- 10
amine
2-(4-Benzothiazol-2-yl-phenyl)-N4-(5-methyl-lH-pyrazol-3-yl)-pyrimidine- 11
2,4-diamine
(3-Benzothiazol-2-yl-phenyl)-(6,7-dimethoxy-quinazolin-4-yl)-amine 12
2-Benzothiazol-2-yl-5-(6,7-dimethoxy-quinazolin-4-ylamino)-phenol 13
(4-Benzothiazol-2-yl-3-methoxy-phenyl)-(6,7-dimethoxy-quinazolin-4-yl)- 14
amine
(4-Benzothiazol-2-yl-phenyl)-(9H-purin-6-yl)-amine 15
(3-Benzothiazol-2-yl-phenyl)-(9H-purin-6-yl)-amine 16
(4-Benzothiazol-2-yl-2-methyl-phenyl)-(6,7-dimethoxy-quinazolin-4-yl)- 17
amine
(5-Benzothiazol-2-yl-2-chloro-phenyl)-(6,7-dimethoxy-quinazolin-4-yl)- 18
amine
(5-Benzothiazol-2-yl-2-methyl-phenyl)-(6,7-dimethoxy-quinazolin-4-yl)- 19
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
21
amine
2-Benzothiazol-2-yl-4-(6,7-dimethoxy-quinazolin-4-ylamino)-phenol 20
(3-Benzothiazol-2-yl-phenyl)-[4-(4-methyl-piperazin- 1 -yl)-pyrimidin-2-yl]-
21
amine
N2-(3-Benzothiazol-2-yl-phenyl)-N4-(5-methyl-lH-pyrazol-3-yl)-pyrimidine- 22
2,4-diamine
2-(4-Benzothiazol-2-yl-phenyl)-N4-methyl-pyrimidine-2,4-diamine 23
N4-(4-Benzothiazol-2-yl-phenyl)-N2-methyl-pyrimidine-2,4-diamine 24
4-(4-Benzothiazol-2-yl-phenyl)-6,7-dimethoxy-N2-methyl-quinazoline-2,4- 25
diamine
4-(4-Benzothiazol-2-yl-phenyl)-6,7-dimethoxy-N2-(5-methyl- 1 H-pyrazol-3- 26
yl)-quinazoline-2,4-diamine
4-(3-Benzothiazol-2-yl-phenyl)-6,7-dimethoxy-N2-methyl-quinazoline-2,4- 27
diamine
N2-(3-Benzothiazol-2-yl-phenyl)-N4-methyl-pyrimidine-2,4-diamine 28
N4-(3-Benzothiazol-2-yl-phenyl)-N2-methyl-pyrimidine-2,4-diamine 29
4-(3-Benzothiazol-2-yl-phenyl)-6,7-dimethoxy-N2-(5-methyl-lH-pyrazol-3- 30
yl)-quinazoline-2,4-diamine
(4-Benzothiazol-2-yl-phenyl)-{6-methoxy-7-[3-(4-methyl-piperazin-l-yl)- 31
propoxy] -quinazolin-4-yl } -amine
(4-Benzothiazol-2-yl-3-methoxy-phenyl)-{6-methoxy-7-[3-(4-methyl- 32
piperazin- 1 -yl)-propoxy] -quinazolin-4-yl }-amine
(4-Benzothiazol-2-yl-phenyl)-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-amine 33
(4-Benzothiazol-2-yl-3-methoxy-phenyl)-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)- 34
amine
(3-Benzothiazol-2-yl-phenyl)-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-amine 35
(4-Benzothiazol-2-yl-phenyl)-(2-methoxy-7H-pyrrolo[2,3-d]pyrimidin-4-yl)- 36
amine
(4-Benzothiazol-2-yl-3-methoxy-phenyl)-(2-methoxy-7H-pyrrolo[2,3- 37
d]pyrimidin-4-yl)-amine
(3-Benzothiazol-2-yl-phenyl)-(2-methoxy-7H-pyrrolo[2,3-d]pyrimidin-4-yl)- 38
amine
(4-Benzothiazol-2-yl=phenyl)-{7-methoxy-6-[3-(4-methyl-piperazin-l-yl)- 39
propoxy] -quinazolin-4-yl } -amine
(4-Benzothiazol-2-yl-3-methoxy-phenyl)-{7-methoxy-6-[3-(4-methyl- 40
piperazin-l-yl)-propoxy] -quinazolin-4-yl } -amine
(3-Benzothiazol-2-yl-phenyl)-{7-methoxy-6-[3-(4-methyl-piperazin-l-yl)- 41
propoxy] -quinazolin-4-yl } -amine
(3-Benzothiazol-2-yl-phenyl)-{6-methoxy-7-[3-(4-methyl-piperazin-l-yl)- 42
propoxy] -quinazolin-4-yl } -amine
(4-Benzothiazol-2-yl-3-methoxy-phenyl)-(7H-purin-6-yl)-amine 43
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
22
(4-Benzothiazol-2-yl-2-methyl-phenyl)-{6-methoxy-7-[3-(4-methyl-piperazin- 44
1-yl)-propoxy]-quinazolin-4-yl } -amine
2-Benzothiazol-2-yl-5-{6-methoxy-7-[3-(4-methyl-piperazin-l-yl)-propoxy]- 45
quinazolin-4-ylamino } -phenol
(5-Benzothiazol-2-yl-2-chloro-phenyl)-{6-methoxy-7-[3-(4-methyl-piperazin- 46
1-yl)-propoxy]-quinazolin-4-yl}-amine
(5-Benzothiazol-2-yl-2-methyl-phenyl)-{6-methoxy-7-[3-(4-methyl-piperazin- 47
1-yl)-propoxy]-quinolin-4-yl} -amine
(4-Benzothiazol-2-yl-2-trifluoromethoxy-phenyl)- {6-methoxy-7-[3-(4-methyl- 48
piperazin-l-yl)-propoxy]-quinazolin-4-yI } -amine
(4-Benzothiazol-2-yl-3-chloro-phenyl)- {6-methoxy-7-[3-(4-methyl-piperazin- 49
1-yl)-propoxy]-quinazolin-4-yl} -amine
(4-Benzothiazol-2-yl-3-fluoro-phenyl)-{6-methoxy-7-[3-(4-methyl-piperazin- 50
1-yl)-propoxy]-quinazolin-4-yl} -amine
(4-Benzothiazol-2-yl-2-methoxy-phenyl)- { 6-methoxy-7-[3 -(4-methyl- 51
piperazin-1-yl)-propoxy]-quinazolin-4-yl } -amine
(4-Benzothiazol-2-yl-2-fluoro-phenyl)-{6-methoxy-7-[3-(4-methyl-piperazin- 52
1-yl)-propoxy]-quinazolin-4-yl} -amine
2-Benzothiazol-2-yl-4-{6-methoxy-7-[3-(4-methyl-piperazin-1-yl)-propoxy]- 53
quinazolin-4-ylamino} -phenol
N2-(4-Benzothiazol-2-yl-3 -methoxy-phenyl)-N4-(5 -methyl-1 H-pyrazol-3 -yl)-
54
pyrimidine-2,4-diamine
N4-(4-Benzothiazol-2-yl-phenyl)-N2-(5-methyl-lH-pyrazol-3-yl)-pyridine- 55
2,4-diamine
4-(4-Benzothiazol-2-yl-phenylamino)-pyridine-2-carboxylic acid methylamide 56
(4-Benzothiazol-2-yl-phenyl)-pyridin-4-yl-amine 57
4-(4-Benzothiazol-2-yl-phenyl)-N2-methyl-pyridine-2,4-diamine 58
5-Benzothiazol-2-yl-2-{6-methoxy-7-[3-(4-methyl-piperazin-1-yl)-propoxy]- 59
quinazolin-4-ylamino } -phenol
{6-Methoxy-7-[3-(4-methyl-piperazin-l-yl)-propoxy]-quinazolin-4-yl}-[4-(5- 60
trifluoromethyl-benzothiazol-2-yi)-phenyl]-amine
(3-Benzothiazol-2-yl-4-chloro-phenyl)-{6-methoxy-7-[3-(4-methyl-piperazin- 61
1-yl)-propoxy]-quinazolin-4-yl} -amine
2-Benzothiazol-2-yl-6-{6-methoxy-7-[3-(4-methyl-piperazin-1-yl)-propoxy]- 62
quinazolin-4-ylamino} -phenol
(4-Benzothiazol-2-yl-3-methoxy-phenyl)- { 6-methoxy-7-[3 -(4-methyl- 63
piperazin-1-yl)-propoxy] -quinazolin-4-yl } -methyl-amine
(4-Benzothiazol-2-yl-phenyl)-(7-chloro-quinolin-4-yl)-amine 64
(4-Benzothiazol-2-yl-phenyl)-thieno[3,2-d]pyrimidin-4-yl-amine 65
(4-Benzothiazol-2-yl-3-methoxy-phenyl)-(7-chloro-quinolin-4-yl)-amine 66
(4-Benzothiazol-2-yl-3-methoxy-phenyl)-thieno[3,2-d]pyrimidin-4-yl-amine 67
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
23
(3-Benzothiazol-2-yl-phenyl)-(7-chloro-quinolin-4-yl)-amine 68
(3-Benzothiazol-2-yl-phenyl)-thieno[3,2-d]pyrimidin-4-yl-amine 69
[4-(5-Chloro-benzothiazol-2-yl)-phenyl]-{6-methoxy-7-[3-(4-methyl- 70
piperazin- 1 -yl)-propoxy]-quinazolin-4-yl } -amine
(4-Benzothiazol-2-yl-3 -methoxy-phenyl)-pyridin-4-yl-amine 71
(3-Benzothiazol-2-yl-phenyl)-pyridin-4-yl-amine 72
(4-Benzothiazol-2-yl-3-chloro-phenyl)-(7-chloro-quinolin-4-yl)-amine 73
(4-Benzothiazol-2-yl-3-chloro-phenyl)-thieno[3,2-d]pyrimidin-4-yl-amine 74
(4-Benzothiazol-2-yl-2-methyl-phenyl)-{7-methoxy-6-[3-(4-methyl-piperazin- 75
1-yl)-propoxy]-quinazolin-
2-Benzothiazol-2-yl-5-{7-methoxy-6-[3-(4-methyl-piperazin-1-yl)-propoxy]- 76
quinazolin-4-ylamino } -phenol
(4-Benzothiazol-2-yl-3-chloro-phenyl)- { 7-methoxy-6-[3-(4-methyl-piperazin-
77
1 -yl) -propoxy] -quinazo lin-4 -yl } -amine
(4-Benzothiazol-2-yl-3-fluoro-phenyl)-{7-methoxy-6-[3-(4-methyl-piperazin- 78
1 -yl)-propoxy]-quinazolin-4-yl } -amine
5-Benzothiazol-2-yl-2-{7-methoxy-6-[3-(4-methyl-piperazin-1-yl)-propoxy]- 79
quinazolin-4-ylamino } -phenol
(4-Benzothiazol-2-yl-2-methoxy-phenyl)-{7-methoxy-6-[3-(4-methyl- 80
piperazin-l-yl)-propoxy] -quinazolin-4-yl } -amine
(4-Benzothiazol-2-yl-2-fluoro-phenyl)-{7-methoxy-6-[3-(4-methyl-piperazin- 81
1-yl)-propoxy]-quinazolin-4-yl } -amine
{7-Methoxy-6-[3-(4-methyl-piperazin-l-yl)-propoxy]-quinazolin-4-yl}-[4-(5- 82
trifluoromethyl-benzothiazol-2-yl)-phenyl]-amine
[4-(5-Chloro-benzothiazol-2-yl)-phenyl]-{7-methoxy-6-[3-(4-methyl- 83
piperazin-1-yl)-propoxy]-quinazolin-4-yl } -amine
(4-Benzothiazol-2-yl-2-fluoro-phenyl)-(7-chloro-quinolin-4-yl)-amine 84
(4-Benzothiazol-2-yl-2-fluoro-phenyl)-thieno [3,2-d]pyrimidin-4-yl-amine 85
N2-(4-Benzothiazol-2-yl-phenyl)-N2-methyl-pyridine-2,4-diamine 86
(4-Benzothiazol-2-yl-phenyl)-(7-chloro-6-methoxy-quinazolin-4-yl)-amine 87
[4-(6-Fluoro-benzothiazol-2-yl)-phenyl]-{6-methoxy-7-[3-(4-methyl- 88
piperazin-l-yl)-propoxy]-quinazolin-4-yl} -amine
[4-(6-Chloro-benzothiazol-2-yl)-phenyl] - {6-methoxy-7- [3 -(4-methyl- 89
piperazin- 1 -yl)-propoxy] -quinazolin-4-yl} -amine
[4-(6-Chloro-benzothiazol-2-yl)-2-fluoro-phenyl] - { 6-methoxy-7-[3 -(4- 90
methyl-piperazin-1-yl)-propoxy] -quinazolin-4-yl } -amine
(4-Benzothiazol-2-yl-3-methyi-phenyl)-{6-methoxy-7-[3-(4-methyl-piperazin- 91
1-yl)-propoxy]-quinazolin-4-yl} -amine
[4-(6-Methoxy-benzothiazol-2-yl)-phenyl]-{6-methoxy-7-[3-(4-methyl- 92
piperazin- 1 -yl)-propoxy]-quinazolin-4-yl} -amine
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
24
4-(4-Benzothiazol-2-yl-phenoxy)-6-methoxy-7- [3 -(4-methyl-piperazin-l-yl)- 93
propoxy]-quinazoline
4-(4-Benzothiazol-2-yl-2-fluoro-phenoxy)-6-methoxy-7-[3-(4-methyl- 94
piperazin- 1 -yl)-propoxy]-quinazoline
2-(4-(6-methoxy-7-(3 -(pyrrolidin-l-yl)propoxy)quinazolin-4- 95
yloxy)phenyl)benzo [d]thiazole
2-(5-(6-methoxy-7-(3 -(pyrrolidin-1-yl)propoxy)quinazolin-4-yloxy)pyridin-2-
96
yl)benzo[d]thiazole
The compounds of the present invention can form salts with inorganic or
organic acids or
bases. Examples of pharmaceutically acceptable salts comprise without
limitation non-
toxic inorganic or organic salts such as acetate derived from acetic acid,
aconitate derived
from aconitic acid, ascorbate derived from ascorbic acid, benzoate derived
from benzoic
acid, cinnamate derived from cinnamic acid, citrate derived from citric acid,
embonate
derived from embonic acid, enantate derived from heptanoic acid, formiate
derived from
formic acid, fumarate derived from fumaric acid, glutamate derived from
glutamic acid,
glycolate derived from glycolic acid, chloride derived from hydrochloric acid,
bromide
derived from hydrobromic acid, lactate derived from lactic acid, maleate
derived from
maleic acid, malonate derived from malonic acid, mandelate derived from
mandelic acid,
methanesulfonate derived from methanesulfonic acid, naphtaline-2-sulfonate
derived from
naphtaline-2-sulfonic acid, nitrate derived from nitric acid, perchlorate
derived from
perchloric acid, phosphate derived from phosphoric acid, phthalate derived
from phthalic
acid, salicylate derived from salicylic acid, sorbate derived from sorbic
acid, stearate
derived from stearic acid, succinate derived from succinic acid, sulphate
derived from
sulphuric acid, tartrate derived from tartaric acid, toluene-p-sulfate derived
from p-
toluenesulfonic acid and others.
Salts of phosphonoxy- and phosphonoxyalkyl groups may be those formed with
alkali
metal ions e.g. sodium or potassium, or those formed with alkaline earth metal
ions e. g.
calcium or magnesium, or those formed with zinc ions.
Such salts of the compounds of the present invention may be anhydrous or
solvated. Such
salts can be produced by methods known to someone of skill in the art and
described in the
prior art.
Other salts like oxalate derived from oxalic acid, which is not considered as
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
pharmaceutically acceptable can be appropriate as intermediates for the
production of
compounds of the present invention or a pharmaceutically acceptable salt
thereof or a
prodrug or a stereoisomer thereof.
5 The compounds according to the invention and medicaments prepared therewith
are
generally useful for the treatment of cell proliferation disorders, for the
treatment or
prophylaxis of immunological diseases and conditions (as for instance
inflammatory
diseases, neuroimmunological diseases, autoimmune diseases or other).
The compounds of the present invention are useful for the treatment of
diseases which are
10 caused by malignant cell proliferation, such as all forms of solid tumors,
leukemias and
lymphomas. Therefore the compounds according to the invention and medicaments
prepared therewith are generally useful for regulating cell activation, cell
proliferation, cell
survival, cell differentiation, cell cycle, cell maturation and cell death or
to induce systemic
changes in metabolism such as changes in sugar, lipid or protein metabolism.
They can
15 also be used to support cell generation poiesis, including blood cell
growth and generation
(prohematopoietic effect) after depletion or destruction of cells, as caused
by, for example,
toxic agents, radiation, immunotherapy, growth defects, malnutrition,
malabsorption,
immune dysregulation, anemia and the like or to provide a therapeutic control
of tissue
generation and degradation, and therapeutic modification of cell and tissue
maintenance
20 and blood cell homeostasis.
These diseases and conditions include but are not limited to cancer as
hematological (e.g.
leukemia, myeloma), or lymphomas (e.g. Hodgkin's and non-Hodgekin's lymphoma),
or
solid tumors (for example breast, prostate, liver, bladder, lung, esophageal,
stomach,
25 colorectal, genitourinary, gastrointestinal, skin, pancreatic, brain,
uterine, colon, head and
neck, cervical, and ovarian, melanoma, astrocytoma, small cell lung cancer,
glioma, basal
and squameous cell carcinoma, sarcomas as Kaposi's sarcoma and osteosarcoma).
Other aspects of the present invention relate to 2-arylbenzothiazole
derivatives of the
present invention as new pharmaceutically active agents, especially for the
preparation of a
pharmaceutical composition for the treatment of diseases which are cured or
relieved by
the inhibition of one or several kinases and/or phosphatases.
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
26
In another more preferred embodiment of the invention the compounds of the
present
invention may be used for treating and/or preventing diseases by inhibition of
one or or
more kinases like: Aurora-A, Aurora-B, EGF-R, ERBB2, PDGFR, FLT3, IGFI-R, VEGF-
R2, VEGF-R3, EPHB4, TIE2, FAK and SRC.
The compounds according to the present invention or a salt or a prodrug or a
stereoisomer
thereof if desired with appropriate adjuvants and additives for the production
of a
medicament for the treatment or prevention of a disease characterized by
hyperproliferation of keratinocytes and/or T cells, especially inflammatory
disorders and
immune disorders, preferably selected from the group consisting of Addison's
disease,
alopecia areata, Ankylosing spondylitis, haemolytic anemia (anemia
haemolytica),
pernicious anemia (anemia perniciosa), aphthae, aphthous stomatitis,
arthritis,
arteriosclerotic disorders, osteoarthritis, rheumatoid arthritis,
aspermiogenese, asthma
bronchiale, auto-immune asthma, auto-immune hemolysis, Bechet's disease,
Boeck's
disease, inflammatory bowel disease, Burkitt's lymphoma, Crohn's disease,
chorioiditis,
colitis ulcerosa, Coeliac disease, cryoglobulinemia, dermatitis herpetiformis,
dermatomyositis, insulin-dependent type I diabetes, juvenile diabetes,
idiopathic diabetes
insipidus, insulin-dependent diabetes mellisis, autoimmune demyelinating
diseases,
Dupuytren's contracture, encephalomyelitis, encephalomyelitis allergica,
endophthalmia
phacoanaphylactica, enteritis allergica, autoimmune enteropathy syndrome,
erythema
nodosum leprosum, idiopathic facial paralysis, chronic fatigue syndrome,
febris
rheumatica, glomerulo nephritis, Goodpasture's syndrome, Graves' disease,
Harnman-
Rich's disease, Hashimoto's disease, Hashimoto's thyroiditis, sudden hearing
loss,
sensoneural hearing loss, hepatitis chronica, Hodgkin's disease,
haemoglobinuria
paroxysmatica, hypogonadism, ileitis regionalis, iritis, leucopenia, leucemia,
lupus
erythematosus disseminatus, systemic lupus erythematosus, cutaneous lupus
erythematosus, lymphogranuloma malignum, mononucleosis infectiosa, myasthenia
gravis,
traverse myelitis, primary idiopathic myxedema, nephrosis, ophthalmia
symphatica,
orchitis granulomatosa, pancreatitis, pemphigus, pemphigus vulgaris,
polyarteritis nodosa,
polyarthritis chronica primaria, polymyositis, polyradiculitis acuta,
psoriasis, purpura,
pyoderma gangrenosum, Quervain's thyreoiditis, Reiter's syndrome, sarcoidosis,
ataxic
sclerosis, progressive systemic sclerosis, scleritis, sclerodermia, multiple
sclerosis,
sclerosis disseminata, acquired spenic atrophy, infertility due to
antispermatozoan
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
27
antibodies, thrombocytopenia, idiopathic thrombocytopenia purpura, thymoma,
acute
anterior uveitis, vitiligo, AIDS, HIV, SCID and Epstein Barr virus associated
diseases such
as Sjorgren's syndrome, virus (AIDS or EBV) associated B cell lymphoma,
parasitic
diseases such as Leishmania, and immunesuppressed disease states such as viral
infections
following allograft transplantations, AIDS, cancer, chronic active hepatitis
diabetes, toxic
chock syndrome and food poisoning.
"Treatment" according to the present invention is intended to mean complete or
partial
healing of a disease, prevention of a disease, or alleviation of a disease, or
stop of
progression of a given disease.
The compounds of the present invention can further be used for diseases that
are caused by
protozoal infestations in humans and animals.
The compounds of the present invention can further be used for viral
infections or other
infections caused for instance by Pneumocystis carinii.
Furthermore, the invention relates to a method of treatment or prevention of
diseases which
comprises the administration of an effective amount of compounds of the
present invention
or a salt or prodrug or a stereoisomer thereof.
The compounds of the according invention and their pharmacologically
acceptable salts
can be administered to animals, preferably to mammals, and in particular to
humans, dogs
and chickens as therapeutics per se, as mixtures with one another or in the
form of
pharmaceutical preparations which allow enteral or parenteral use and which as
active
constituent contain an effective dose of at least one compound of the present
invention or a
salt thereof, in addition to customary pharmaceutically innocuous excipients
and additives.
The production of medicaments containing the compounds according to the
present
invention and their application can be performed according to well-known
pharmaceutical
methods.
While the compounds according to the present invention for use in therapy may
be
administered in the form of the raw chemical compound, it is preferred to
introduce the
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
28
active ingredient, optionally in the form of a physiologically acceptable salt
in a
pharmaceutical composition together with one or more adjuvants, excipients,
carriers,
buffers, diluents, and/or other customary pharmaceutical auxiliaries. Such
salts of the
compounds may be anhydrous or solvated.
In a preferred embodiment, the invention provides medicaments comprising
compounds
according to the present invention, or a salt or a prodrug or a stereoisomer
thereof, together
with one or more pharmaceutically acceptable carriers thereof, and,
optionally, other
therapeutic and/or prophylactic ingredients. The carrier(s) must be
"acceptable" in the
sense of being compatible with the other ingredients of the formulation and
not harmful to
the recipient thereof.
A medicament of the invention may be those suitable for oral, rectal,
bronchial, nasal,
topical, buccal, sub-lingual, transdermal, vaginal or parenteral (including
cutaneous,
subcutaneous, intramuscular, intraperitoneal, intravenous, intraarterial,
intracerebral,
intraocular injection or infusion) administration, or those in a form suitable
for
administration by inhalation or insufflation, including powders and liquid
aerosol
administration, or by sustained release systems. Suitable examples of
sustained release
systems include semipermeable matrices of solid hydrophobic polymers
containing the
compound of the invention, which matrices may be in form of shaped articles,
e.g. films or
microcapsules.
For preparing a medicament from a compounds of the present invention and
pharmaceutically acceptable carriers can be either solid or liquid. Solid form
preparations
include powders, tablets, pills, capsules, cachets, suppositories, and
dispersible granules. A
solid carrier can be one or more substances which may also act as diluents,
flavouring
agents, solubilizers, lubricants, suspending agents, binders, preservatives,
tablet
disintegrating agents, or an encapsulating material.
In powders, the carrier is a finely divided solid which is in a mixture with
the finely
divided active component. In tablets, the active component is mixed with the
carrier having
the necessary binding capacity in suitable proportions and compacted in the
shape and size
desired. Suitable carriers are magnesium carbonate, magnesium stearate, talc,
sugar,
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
29
lactose, pectin, dextrin, starch, gelatin, tragacanth, methylcellulose, sodium
carboxymethylcellulose, a low melting wax, cocoa butter, and the like. The
term
"preparation" is intended to include the formulation of the active compound
with
encapsulating material as carrier providing a capsule in which the active
component, with
or without carriers, is surrounded by a carrier, which is thus in association
with it.
Similarly, cachets and lozenges are included. Tablets, powders, capsules,
pills, cachets, and
lozenges can be used as solid forms suitable for oral administration.
For preparing suppositories, a low melting wax, such as a mixture of fatty
acid glyceride or
cocoa butter, is first melted and the active component is dispersed
homogeneously therein,
as by stirring. The molten homogenous mixture is then poured into convenient
sized
moulds, allowed to cool, and thereby to solidify. Compositions suitable for
vaginal
administration may be presented as pessaries, tampons, creams, gels, pastes,
foams or
sprays containing in addition to the active ingredient such carriers as are
known in the art
to be appropriate. Liquid preparations include solutions, suspensions, and
emulsions, for
example, water or water-propylene glycol solutions. For example, parenteral
injection
liquid preparations can be formulated as solutions in aqueous polyethylene
glycol solution.
The compounds according to the present invention may thus be formulated for
parenteral
administration (e.g. by injection, for example bolus injection or continuous
infusion) and
may be presented in unit dose form in ampoules, pre-filled syringes, small
volume infusion
or in multi-dose containers with an added preservative. The compositions may
take such
forms as suspensions, solutions, or emulsions in oily or aqueous vehicles, and
may contain
formulation agents such as suspending, stabilising and/or dispersing agents.
Alternatively,
the active ingredient may be in powder form, obtained by aseptic isolation of
sterile solid
or by lyophilization from solution, for constitution with a suitable vehicle,
e.g. sterile,
pyrogen-free water, before use.
Aqueous solutions suitable for oral use can be prepared by dissolving the
active component
in water and adding suitable colorants, flavours, stabilising and thickening
agents, as
desired. Aqueous suspensions suitable for oral use can be made by dispersing
the finely
divided active component in water with viscous material, such as natural or
synthetic
gums, resins, methylcellulose, sodium carboxymethylcellulose, or other well
known
suspending agents.
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
Also included are solid form preparations which are intended to be converted,
shortly
before use, to liquid form preparations for oral administration. Such liquid
forms include
solutions, suspensions, and emulsions. These preparations may contain, in
addition to the
5 active component, colorants, flavours, stabilisers, buffers, artificial and
natural sweeteners,
dispersants, thickeners, solubilizing agents, and the like.
In one embodiment of the present invention, the medicament is applied
topically or
systemically or via a combination of the two routes.
Preferably the medicament is prepared in form of an ointment, a gel, a
plaster, an
emulsion, a lotion, a foam, a cream of a mixed phase or amphiphilic emulsion
system
(oil/water-water/oil mixed phase), a liposome, a transfersome, a paste or a
powder.
Ointments and creams may, for example, be formulated with an aqueous or oily
base with
the addition of suitable thickening and/or gelling agents. Lotions may be
formulated with
an aqueous or oily base and will in general also contain one or more
emulsifying agents,
stabilising agents, dispersing agents, suspending agents, thickening agents,
or colouring
agents.
Compositions suitable for topical administration in the mouth include lozenges
comprising
the active agent in a flavoured base, usually sucrose and acacia or
tragacanth; pastilles
comprising the active ingredient in an inert base such as gelatin and
glycerine or sucrose
and acacia; and mouthwashes comprising the active ingredient in a suitable
liquid carrier.
Solutions or suspensions are applied directly to the nasal cavity by
conventional means, for
example with a dropper, pipette or spray. The compositions may be provided in
single or
multi-dose form. In the latter case of a dropper or pipette, this may be
achieved by the
patient administering an appropriate, predetermined volume of the solution or
suspension.
In the case of a spray, this may be achieved for example by means of a
metering atomising
spray pump.
Administration to the respiratory tract may also be achieved by means of an
aerosol
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
31
formulation in which the active ingredient is provided in a pressurised pack
with a suitable
propellant such as a chlorofluorocarbon (CFC) for example
dichlorodifluoromethane,
trichlorofluoromethane, or dichlorotetrafluoroethane, carbon dioxide, or other
suitable gas.
The aerosol may conveniently also contain a surfactant such as lecithin. The
dose of drug
may be controlled by provision of a metered valve.
Alternatively the active ingredients may be provided in the form of a dry
powder, for
example a powder mix of the compound in a suitable powder base such as
lactose, starch,
starch derivatives such as hydroxypropylmethyl cellulose and
polyvinylpyrrolidone (PVP).
Conveniently the powder carrier will form a gel in the nasal cavity The powder
composition may be presented in unit dose form for example in capsules or
cartridges of,
e.g., gelatin, or blister packs from which the powder may be administered by
means of an
inhaler.
In compositions intended for administration to the respiratory tract,
including intranasal
compositions, the compound will generally have a small particle size for
example of the
order of 5 microns or less. Such a particle size may be obtained by means
known in the art,
for example by micronization.
When desired, compositions adapted to give sustained release of the active
ingredient may
be employed.
The pharmaceutical preparations are preferably in unit dosage forms. In such
form, the
preparation is subdivided into unit doses containing appropriate quantities of
the active
component. The unit dosage form can be a packaged preparation, the package
containing
discrete quantities of preparation, such as packaged tablets, capsules, and
powders in vials
or ampoules. Also, the unit dosage form can be a capsule, tablet, cachet, or
lozenge itself,
or it can be the appropriate number of any of these in packaged form. Tablets
or capsules
for oral administration and liquids for intravenous administration and
continuous infusion
are preferred compositions.
Further details on techniques for formulation and administration may be found
in the latest
edition of Remington's Pharmaceutical Sciences (Maack Publishing Co. Easton,
Pa.).
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
32
Pharmaceutical compositions can also contain two or more compounds of the
present
invention or their pharmacologically acceptable salts and also other
therapeutically active
substances.
Thus, the compounds of the present invention can be used in the form of one
compound
alone or in combination with other active compounds - for example with
medicaments
already known for the treatment of the aforementioned diseases, whereby in the
latter case
a favorable additive, amplifying effect is noticed.
To prepare the pharmaceutical preparations, pharmaceutically inert inorganic
or organic
excipients can be used. To prepare pills, tablets, coated tablets and hard
gelatin capsules,
for example, lactose, corn starch or derivatives thereof, talc, stearic acid
or its salts, etc.
can be used. Excipients for soft gelatin capsules and suppositories are, for
example, fats,
waxes, semi-solid and liquid polyols, natural or hardened oils etc. Suitable
excipients for
the production of solutions and syrups are, for example, water, sucrose,
invert sugar,
glucose, polyols etc. Suitable excipients for the production of injection
solutions are, for
example, water, alcohols, glycerol, polyols or vegetable oils.
The dose can vary within wide limits and is to be suited to the individual
conditions in
each individual case. For the above uses the appropriate dosage will vary
depending on the
mode of administration, the particular condition to be treated and the effect
desired. In
general, however, satisfactory results are achieved at dosage rates of about 1
to 100 mg/kg
animal body weight preferably 1 to 50 mg/kg. Suitable dosage rates for larger
mammals,
for example humans, are of the order of from about 10 mg to 3 g/day,
conveniently
administered once, in divided doses 2 to 4 times a day, or in sustained
release form.
The following examples are included to demonstrate preferred embodiments of
the
invention. It should be appreciated by those of skill in the art that the
techniques disclosed
in the examples that follow represent techniques discovered by the inventors
to function
well in the practice of the invention, and thus can be considered preferred
modes for its
practice. However, those of skill in the art should, in light of the present
disclosure,
appreciate that many changes can be made in the specific embodiments that are
disclosed
without departing from the spirit and scope of the invention as set out in the
appended
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
33
claims. All references cited are incorporated herein by reference.
Examples
Abbreviations: min, minute(s); h, hour(s); r.t., room temperature; TLC, thin
layer
chromatography; XANTPHOS, 9,9-dimethyl-4,5-bis(diphenylphosphino)xanthene;
dba,
dibenzylideneacetone; DME, 1,2-dimethoxyethane; DIEA, N,N-
diisopropylethylamine;
EDC, 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide.
Preparative HPLC-MS: Waters 600 Multisolvent Delivery System with peparative
pump
heads. 2000 l or 5000 l Sample loop. Column, Waters X-Terra RP18, 7 m, 19 x
150
mm with X-Terra RP 18 guard cartridge 7 m, 19 x 10 mm; used at flow rate 20
ml/min or
YMC ODS-A, 120 A, 40 x 150 mm with X-Terra RP18 guard cartridge 7 m, 19 x 10
mm;
used at flow rate 50 ml/min. Make-up solvent: MeCN - H20 - HCO2H 80 : 20 :
0.05
(v:v:v). Eluent A, H20 + 0.1 % HCOZH; eluent B, MeCN. Different linear
gradients from 5
- 100% eluent B, adapted to sample. Injection volume: 500 l - 2000 l
depending on
sample.
Syntheses of Intermediates.
General procedure 1:
Syntheses of 4-chloroquinazolines with alkylamino sidechains: 4-Chloro-6-
methoxy-7-
[3-(4-methylpiperazin-1-yl)propoxy]quinazoline, 4-Chloro-7-methoxy-6-[3-(4-
methyl-
piperazin-l-yl)propoxy] quinazoline and 4-Chloro-6-methoxy-7-(3 -pyrrolidin-1-
yl-
propoxy)-quinazoline.
Step 1. To a solution of methyl vanillate or methyl isovanillate (7.29 g, 40
mmol) in
dimethylformamide (25 mL), potassium carbonate (8.29 g, 60 mmol) and benzyl
bromide
(5.26 mL, 44 mmol) were added. The mixture was heated to 100 C for 3 h. After
cooling
to r.t., water was added and the product was extracted several times with
ethyl acetate. The
combined organic phases were washed with water and brine. After drying over
Na2SO4,
the solvent was removed to yield methyl 4-benzyloxy-3-methoxybenzoate or
methyl 3-
benzyloxy-4-methoxybenzoate, respectively, quantitatively, which was used
without
further purification.
Step 2. Crude material of step 1 (40.0 mmol) was converted into methyl 4-
benzyloxy-5-
methoxy-2-nitrobenzoate or methyl 5-benzyloxy-4-methoxy-2-nitrobenzoate,
respectively,
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
34
in 91-94% yield as described in US 02/0026052 Al, page 51, reference example
15.
Step 3. In a 1 1 Schlenk flask filled with argon, product of step 2 (36.6
mmol) and
palladium on charcoal (1.17 g, 10% Pd, 1.1 mmol Pd) were combined and
tetrahydrofuran
(250 mL) was added. The argon was replaced with hydrogen (1 bar), and the
mixture was
vigorously stirred at r.t. until completion of the reaction. The palladium was
separated by
filtration through a pad of celite and the solvent was removed to obtain
methyl 2-amino-4-
hydroxy-5-methoxybenzoate or methyl 2-amino-5-hydroxy-4-methoxybenzoate,
respectively, quantitatively, which, again, was used without further
purification.
Step 4. A mixture of formamide (29 mL), ammonium formate (3.41 g, 54 mmol) and
crude
material of step 3 (36.0 mmol) was heated to 140 C for 4 h. After cooling to
r.t., water (75
mL) was added. After stirring for 1 h, the precipitated 7-hydroxy-6-methoxy-
3,4-
dihydroquinazolin-4-one or 6-hydroxy-7-methoxy-3,4-dihydroquinazolin-4-one,
respectively, was filtered off, washed with water and dried (76-85%).
Step 5. A mixture of product step 4 (30.5 mmol), acetic anhydride (21.5 mL,
229 mmol)
and pyridine (4.9 mL, 61 mmol) was heated to 100 C for 4 h. After cooling to
r.t., ice
water (200 mL) was added and the mixture was vigorously stirred for 1 h. The
precipitated
7-acetoxy-6-methoxy-3,4-dihydroquinazolin-4-one or 6-acetoxy-7-methoxy-3,4-
dihydroquinazolin-4-one, respectively, was filtered off, washed with water and
dried (93-
96%).
Step 6. Product step 5 (8.54 mmol) was converted into 4-chloro-7-hydroxy-6-
methoxyquinazoline or 4-chloro-6-hydroxy-7-methoxyquinazoline, respectively,
(58-95%)
by reacting them with thionylchloride (12 mL) and DMF (0.3 mL) at 85 C for
1.5 h.
Excess thionylchloride was removed by distillation. Traces of thionylchloride
were
removed by aceotropic distillation wit toluene (two times). Alternatively the
products step
5 can be converted into the chlorides by reacting them with a mixture of POC13
and PC15.
The acetyl groups were removed by hydrolysis with ammonium hydroxide (5 mL, 28-
30
wt%) in dioxane / water (100 mL / 20 mL) at 0 C to r.t.
Step 7.
General procedure 1:
Di-tert-butyl azodicarboxylate (0.478 g, 2.08 mmol) was added portionwise to a
mixture of
product step 6 (1.66 mmol), 3-(4-methylpiperazin-1-yl)-propan-l-ol (synthesis
described
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
below, 0.276 g, 1.74 mmol), and triphenylphosphine (0.544 g, 2.08 mmol) in
dichloromethane (20 mL) at r.t.. If necessary, further alcohol was added.
After stirring for
2 h, the solution was concentrated to 10 mL, mounted on silica and
chromatographed
(gradient, dichloromethane to dichloromethane : methanol = 3:2) to obtain the
desired
5 ethers (-73%).
Synthesis of 4-chloro-6-methoxy-7-[3-(4-methylpiperazin-1-
yl)propoxy]quinazoline:
The compound was synthesised according to general procedure 1 from 4-chloro-7-
hydroxy-6-methoxyquinazoline. LC/ESI-MS: m/z = 351 [M+H].
10 Synthesis of 4-chloro-7-methoxy-6-[3-(4-methylpiperazin-1-
yl)propoxy]quinazoline:
The compound was synthesised according to general procedure 1 from 4-chloro-6-
hydroxy-7-methoxyquinazoline. LC/ESI-MS: m/z = 351 [M+H].
Synthesis of 4-chloro-6-methoxy-7-(3-pyrrolidin-1-yl-propoxy)-quinazoline:
The compound was synthesised according to general procedure 1 from 4-chloro-7-
15 hydroxy-6-methoxyquinazoline. LC/ESI-MS: m/z = 322 [M+H].
Synthesis of 3-(4-methylpiperazin-1-yl)-propan-l-ol:
1-Methylpiperazine (6.99 mL, 63 mol) was dissolved in toluene (30 mL). 3-
Bromopro-
panol (2.62 mL, 30 mmol) was added slowly and the mixture was stirred
overnight at r.t..
After heating to 80 C for 2 h and cooling to r.t., the mixture was filtered
and the filter cake
20 was thoroughly washed with toluene. After removal of the solvent, the
residue was
subjected to Kugelrohr distillation (b.p., 180 C / 2 mbar) to obtain a
colourless oil (4.08 g,
25.8 mmol, 86%). LC/ESI-MS: m/z = 159 [M+H].
Synthesis of 3-morpholin-4-yl-propan-l-ol:
3-Morpholin-4-yl-propan=l-ol (b.p., 180 C / 1 mbar) was synthesized in analogy
to 3-(4-
25 methylpiperazin-1-yl)-propan-l-ol from 3-bromopropanol and morpholine.
LC/ESI-MS: m/z = 146 [M+H].
Synthesis of 3-Pyrrolidin-1-yl-propan-l-ol:
3-Pyrrolidin-1-yl-propan-l-ol (b.p., 230 C / 10 mbar) was synthesized in the
same manner
as 3-(4-methylpiperazin-1-yl)-propan-l-ol from 3-bromopropanol and
pyrrolidine.
30 LC/ESI-MS: m1z = 130 [M+H].
Synthesis of 2-(4-Methyl-piperazin-1-yl)-ethanol:
2-(4-Methyl-piperazin-1-yl)-ethanol (b.p., 115-135 C / 0.1 mbar) was
synthesized
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
36
according to the synthesis of 3-(4-methylpiperazin-1-yl)-propan-l-ol from 2-
bromo-
ethanol and 1-methylpiperazine. LC/ESI-MS: m/z = 145 [M+H].
Synthesis of 2-morpholin-4-yl-ethanol:
2-Morpholin-4-yl-ethanol (b.p., 130-145 C / 20 mbar) was synthesized according
to the
synthesis of 3-(4-methylpiperazin-1-yl)-propan-l-ol from 2-bromo-ethanol and
morpholine. LC/ESI-MS: m/z = 132 [M+H].
Synthesis of differently substituted 2- or 4-chloropyrimidines: 2-Chloro-4-(4-
methylpiperazinl -yl)pyrimidine, (2-chloropyrimidin-4-yl)-(5-methyl-lH-pyrazol-
3-yl)-
amine, (2-chloropyrimidin-4-yl)-methylamine, (4-chloropyrimidin-2-yl)-
methylamine.
Syntheses were performed in analogy to T. Kumagai et al., Bioorg. Med. Chem.
2001, 9,
1349-1355; S. F. Campbell et al., J. Med. Chem. 1987, 30, 1794-1798; US
2004/0132781
Al, [0851].
A mixture of 2,4-dichloropyrimidine (0.967 g, 6.49 mmol), the respective amine
1-
methylpiperazine (0.65 g, 6.49 mmol), 3-amino-5-methylpyrazole (0.63 g, 6.49
mmol) or
methylamine hydrochloride (0.44 g, 6.49 mmol), and ethyldiisopropylamine (2.83
mL,
16.22 mmol; 3.96 mL, 22.71 mmol in case of methylamine hydrochloride) in
ethanol (13
mL) was stirred at -10 C for 2 h and then at r.t. overnight. For weaker
nucleophiles like
aminopyrazoles, the mixture had to be stirred at 50 C for 4 h additionally to
get complete
conversion.
Mixtures were partitioned between Hz0/brine (3:1; 100 mL) and chloroform (3 x
70 mL).
Combined organic phases were washed once with brine (50 mL) and dried over
MgSO4.
2-Chloro-4-(4-methylpiperazinl -yl)pyrimidine:
Removal of solvent yielded a pale-beige solid, which was washed with ethyl
acetate /
ultrasound to give the desired product as a colourless powder, which was
further washed
with Et20. Additional product was obtained upon fractional crystallization of
the washing
solution. A total of 0.741 g (3.48 mmol, 54%) of 2-chloro-4-(4-
methylpiperazinl-yl)-
pyrimidine was obtained.
(2-Chloropyrimidin-4-yl)-(5-methylpyrazol-3 -yl)amine:
Upon fractional crystallization from chloroform/diethylether, (2-
chloropyrimidin-4-yl)-(5-
methylpyrazol-3-yl)amine (0.258 g, 1.23 mmol, 19%) was obtained as colourless
crystals.
(2-Chloropyrimidin-4-yl)-methylamine and (4-chloropyrimidin-2-yl)-methylamine:
Fractional crystallization from ethyl acetate/diethylether yielded (2-
chloropyrimidin-4-yl)-
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
37
methylamine as the major product within first fractions and enriched (4-
chloropyrimidin-2-
yl)-methylamine as minor product in the latter fractions, each as colorless
powder. A total
of 0.396 g (2.57 mmol, 40%) of (2-chloropyrimidin-4-yl)-methylamine and 0.118
g (0.822
mmol, 13%) of (4-chloropyrimidin-2-yl)-methylamine was attained.
General procedure 2:
Synthesis of 3- and 4-(benzothiazol-2-yl)-phenylamines and -phenols from 2-
aminothiophenols (according to I. Hutchinson et al., J. Med. Chem. 2001, 44,
1446-1455):
Differently substituted 2-aminothiophenols (1.1 equiv.) and 3- or 4-
aminobenzoic acids /
3- or 4-hydroxybenzoic acids (1.0 equiv.), respectively, were placed in a
flask and treated
with polyphosphoric acid (1.0 g per 1.0 mmol aminobenzoic acid) at 185 C for 5
h. For
aminobenzoic acids additionally substituted with free phenolic OH-groups, 3.0
equiv. of 2-
aminothiophenols had to be used.
Still hot, the mixture was neutralized by pouring it into icy NaOH solution (1
mmol NaOH
per 1.0 g polyphosphoric acid, dissolved in 10 mL H20 and mixed with ice). A
precipitation formed and still remaining polyphosphoric acid (brown-black
gummy slurry)
was dissolved by further addition of 5% aq. NaOH and ultrasonic treatment. The
precipitate was filtered off and washed thoroughly with 5% aq. NaOH and H20.
If
precipitate was too fine for filtering, a centrifuge was used for
sedimentation of the solid.
Product was next dissolved in 400 mL DMF/MeOH (1:1) at 60 C, cooled down to
r.t. and
precipitated by adding H20. Product was filtered off (frit 3 and subsequently,
frit 4) and
dried in high vacuum.
If direct precipitation did not succeed properly upon pouring into icy NaOH,
the aq. slurry
was extracted with CHC13/ethyl acetate 1:1 (3 x). Combined org. phases were
dried over
MgS04 and, if necessary, purified using chromatography on silica gel, solvent:
petroleum
ether / ethyl acetate 10:1 to 1:3 .
General procedure 3:
Synthesis of substituted 3- and 4-(benzothiazol-2-yl)-phenylamines from 2-
substituted
aminobenzothiazoles (according to I. Hutchinson et al., J. Med. Chem. 2001,
44, 1446-
1455):
Differently substituted 2-amino- or 2-methylbenzothiazoles (3 mmol) were added
to a
solution of potassium hydroxide (2.5 g) in water (5 mL). The resulting mixture
was heated
under reflux for 5 h, after which complete solution had occurred. After
cooling, the
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
38
reaction mixture was acidified (to pH 6) by the addition of acetic acid. Water
(50 mL) was
added, and the resulting mixture was stirred overnight. The solid precipitate
was collected
and purified by column chromatography (CH2C12) to give the corresponding bis(2-
aminoaryl)disulfide.
To a solution of the disulfide (1 mmol) in pyridine (5 mL), 4-nitrobenzoyl
chloride (2
mmol) or the corresponding derivative was added. The resulting mixture was
heated under
reflux for 30 min and then poured into water (15 mL). The precipitate was
collected,
washed with water (20 mL) and purified by column chromatography (CH2C12).
The precipitate was reductively cyclized as follows: To a solution of 10 M aq.
HCl (10
mL) and ethanol/H2O 10:1 (22 mL) was added the disulfide (1.6 mmol) and
tin(II) chloride
dihydrate (9.8 mmol). The reaction mixture was heated under reflux for 15 h,
cooled to 25
C, and poured into water (75 mL). Sodium hydroxide (2 g) was added slowly, and
the
mixture was stirred for 1 h. The precipitate was collected, washed with water
(10 mL) and
purified by column chromatography (CH2C12).
Synthesis of 2-(4-bromophenyl)-benzothiazole:
Preparation was performed according to the general procedure 2 described above
for 3-
and 4-(benzothiazol-2-yl)-phenylamines and -phenols from 2-aminothiophenols
and, in
this case, 4-bromobenzoic acid. LC/ESI-MS: m/z = 290 [M+H].
Synthesis of 4-benzothiazol-2-yl-phenylamine:
Preparation was performed according to the general procedure 2 described above
and, in
this case, 4-aminobenzoic acid. LC/ESI-MS: m/z = 227 [M+H].
Synthesis of 4-benzothiazol-2-yl-2-methyl-phenylamine:
Preparation was performed according to the general procedure 2 described above
and, in
this case, 4-amino-3-methyl-benzoic acid. LC/ESI-MS: m/z = 241 [M+H].
Synthesis of 3-benzothiazol-2-yl-phenylamine:
Preparation was performed according to the general procedure 2 described above
and, in
this case, 3-aminobenzoic acid. LC/ESI-MS: m/z = 227 [M+H].
Synthesis of 5-benzothiazol-2-yl-2-chloro-phenylamine:
Preparation was performed according to the general procedure 2 described above
and, in
this case, 3-amino-4-chloro-benzoic acid. LC/ESI-MS: m/z = 261 [M+H].
Synthesis of 4-benzothiazol-2-yl-3-methogy-phenylamine:
Preparation was performed according to the general procedure 2 described above
and, in
this case, 4-amino-2-methoxy-benzoic acid. LC/ESI-MS: m/z = 257 [M+H].
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
39
Synthesis of (4-benzothiazol-2-yl-3-methoxy-phenyl)-methyl-amine:
The compound was obtained as side product within the synthesis of 4-
benzothiazol-2-yl-3-
methoxy-phenylamine. LC/ESI-MS: m/z = 271 [M+H].
Synthesis of 5-amino-2-benzothiazol-2-yl-phenol:
Preparation was performed according to the general procedure 2 described above
and, in
this case, 4-amino-2-hydroxy-benzoic acid. LC/ESI-MS: m/z = 243 [M+H].
Synthesis of 4-amino-2-benzothiazol-2-yl-phenol:
Preparation was performed according to the general procedure 2 described above
and, in
this case, 5-amino-2-hydroxy-benzoic acid. LC/ESI-MS: m/z = 243 [M+H].
Synthesis of 5-benzothiazol-2-yl-2-methyl-phenylamine:
Preparation was performed according to the general procedure 2 described above
and, in
this case, 3-amino-4-methyl-benzoic acid. LC/ESI-MS: m/z = 241 [M+H].
Synthesis of 4-benzothiazol-2-yl-2-trifluoromethoxy-phenylamine:
Preparation was performed according to the general procedure 2 described above
and, in
this case, 4-amino-3-trifluoromethoxy-benzoic acid. LC/ESI-MS: m/z = 310
[M+H].
Synthesis of 4-benzothiazol-2-yl-3-chloro-phenylamine:
Preparation was performed according to the general procedure 2 described above
and, in
this case, 4-amino-2-chloro-benzoic acid. LC/ESI-MS: m/z = 261 [M+H].
Synthesis of 4-benzothiazol-2-yl-3-fluoro-phenylamine:
Preparation was performed according to the general procedure 2 described above
and, in
this case, 4-amino-2-fluoro-benzoic acid. LC/ESI-MS: m/z = 245 [M+H].
Synthesis of 4-benzothiazol-2-yl-2-methogy-phenylamine:
Preparation was performed according to the general procedure 2 described above
and, in
this case, 4-amino-3-methoxy-benzoic acid. LC/ESI-MS: m/z = 257 [M+H].
Synthesis of 4-benzothiazol-2-yl-2-fluoro-phenylamine:
Preparation was performed according to the general procedure 2 described above
and, in
this case, 4-amino-3-fluoro-benzoic acid. LC/ESI-MS: m/z = 245 [M+H].
Synthesis of 2-amino-5-benzothiazol-2-yl-phenol:
Preparation was performed according to the general procedure 2 described above
and, in
this case, 4-amino-3-hydroxy-benzoic acid. LC/ESI-MS: m/z = 243 [M+H].
Synthesis of 4-(5-trifluoromethyl-benzothiazol-2-yl)-phenylamine:
Preparation was performed according to the general procedure 2 described above
and, in
this case, 4-amino-benzoic acid and 2-amino-4-trifluoromethyl-benzenethiol.
LC/ESI-MS:
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
m/z = 295 [M+H].
Synthesis of 3-benzothiazol-2-yl-4-chloro-phenylamine:
Preparation was performed according to the general procedure 2 described above
and, in
this case, 5-amino-2-chloro-benzoic acid. LC/ESI-MS: m/z = 261 [M+H].
5 Synthesis of 2-amino-6-benzothiazol-2-yl-phenol:
Preparation was performed according to the general procedure 2 described above
and, in
this case, 3-amino-2-hydroxy-benzoic acid. LC/ESI-MS: m/z = 243 [M+H].
Synthesis of 4-(5-chloro-benzothiazol-2-yl)-phenylamine:
Preparation was performed according to the general procedure 2 described above
and, in
10 this case, 4-amino-benzoic acid and 2-amino-4-chloro-benzenethiol. LC/ESI-
MS: m/z =
261 [M+H].
Synthesis of 4-(6-fluoro-benzothiazol-2-yl)-phenylamine:
Preparation was performed according to the general procedure 3 described above
and, in
this case, starting from 6-fluoro-benzothiazol-2-ylamine. LC/ESI-MS: m/z = 245
[M+H].
15 Synthesis of 4-(6-chloro-benzothiazol-2-yl)-phenylamine:
Preparation was performed according to the general procedure 3 described above
and, in
this case, starting from 6-chloro-benzothiazol-2-ylamine. LC/ESI-MS: nilz =
261 [M+H].
Synthesis of 4-(6-chloro-benzothiazol-2-yl)-2-fluoro-phenylamine:
Preparation was performed according to the general procedure 3 described above
and, in
20 this case, starting from 6-chloro-benzothiazol-2-ylamine. LC/ESI-MS: m/z =
279 [M+H].
Synthesis of 4-benzothiazol-2-yl-3-methyl-phenylamine:
Preparation was performed according to the general procedure 2 described above
and, in
this case, 4-amino-2-methyl-benzoic acid. LC/ESI-MS: m/z = 241 [M+H].
Synthesis of 4-(6-methozy-benzothiazol-2-yl)-phenylamine:
25 Preparation was performed according to the general procedure 3 described
above and, in
this case, starting from 6-methoxy-benzothiazol-2-ylamine. LC/ESI-MS: m1z =
257
[M+H].
Synthesis of 2-fluoro-4-(6-fluoro-benzothiazol-2-yl)-phenylamine:
Preparation was performed according to the general procedure 3 described above
and, in
30 this case, starting from 2-amino-6-fluorobenzothiazole. LC/ESI-MS: m1z =
263 [M+H].
Synthesis of 4-(6-bromo-benzothiazol-2-yl)-phenylamine:
Preparation was performed according to the general procedure 3 described above
and, in
this case, starting from 6-bromo-benzothiazol-2-ylamine. LC/ESI-MS: m/z = 305
[M+H].
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
41
Synthesis of 4-(6-trifluoromethoxy-benzothiazol-2-yl)-phenylamine:
Preparation was performed according to the general procedure 3 described above
and, in
this case, starting from 6-trifluoromethoxy-benzothiazol-2-ylamine. LC/ESI-MS:
m/z =
311 [M+H].
Synthesis of 4-benzothiazol-2-yl-phenol:
Preparation was performed according to the general procedure 2 described above
and, in
this case, 4-hydroxy-benzoic acid. LC/ESI-MS: m/z = 228 [M+H].
Synthesis of 4-benzothiazol-2-yl-2-fluoro-phenol:
Preparation was performed according to the general procedure 2 described above
and, in
this case, 4-hydroxy-3-fluoro-benzoic acid. LC/ESI-MS: m/z = 246 [M+H].
General procedure 4:
Reaction of 4-chloroquinazolines without alkylamino sidechains with 3- and 4-
(benzothiazol-2-yl)-phenylamines (in analogy to T. Kumagai et al., Bioorg.
Med. Chem.
2001, 9, 1349-1355):
A mixture of the respective 4-chloroquinazoline (0.221 mmol) and 3- or 4-
(benzothiazol-2-
yl)-phenylamine (0.221 mmol), respectively, in ethylene glycol (1.5 mL) was
heated to
100 C in a sealed vial for 3 h. In many cases, product precipitated upon
cooling of the
mixture to r.t. The resulting slurry was rinsed into a frit with ethanol, the
filter cake was
washed several times with the same solvent and fmally with some Et20. If
necessary,
product was further washed with CHC13/MeOH-ultrasound and Et20/CHC13-
ultrasound
and separated using a centrifuge.
General procedure 5:
Reaction of 4-chloroquinazolines with alkylamino sidechains with 3- and 4-
(benzothiazol-2-yl)-phenylamines
To a mixture of the respective 4-chloroquinazoline (0.130 mmol) and 3- or 4-
(benzothiazol-2-yl)-phenylamine (0.143 mmol), respectively, in ethylene glycol
(1.2 mL)
was added 4 M HCl in dioxane (2.0 equiv.), and the sealed vial was heated for
3 h to
110 C, or - in case of steric hindrance in the ortho-position of the amino
group of 3- or 4-
(benzothiazol-2-yl)-phenylamine - to 140 C.
The reaction mixture was partitioned between satd aq. NaHCO3/brine 1:3 (100
mL) and
CHC13 (100 mL, then 2x50 mL). Combined org. phases were re-extracted once
against 50
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
42
mL brine and dried over MgSO4. If necessary, product was purified by
preparative TLC (1
mm silica gel, CH2C12/MeOH 90:10 or prep. HPLC on reversed phase. Product was
crystallized from CHC13/Et20 or acetone/Et20.
General procedure 6:
Reaction of 4-chloroquinazolines with alkylamino sidechains with 3- and 4-
(benzothiazol-2-yl)-phenols (according to K. Kubo et al., J. Med. Chem. 2005,
48, 1359-
1366):
To a cooled solution (0 C) of substituted 3- or 4-(benzothiazol-2-yl)-phenol
(0.11 mmol)
in DMSO (1 mL) was added NaH (0.11 mmol) and the mixture stirred at r.t. for
10 min. 4-
Chloroquinazoline (0.11 mmol) was then added, and stirring continued at 130 C
for 12 h.
Water (5 mL) was added to the reaction mixture, which was further stirred for
5 min, and
the product was purified by preparative HPLC.
General procedure 7:
Reaction of 2- or 4-chloropyrimidines with 3- and 4-(benzothiazol-2-yl)-
phenylamines
(according to T. Kumagai et al., Bioorg. Med. Chem. 2001, 9, 1349-1355):
A mixture of the respective 2- or 4-chloropyrimidine (0.221 mmol; synthesized
as
described above) and 3- or 4-(benzothiazol-2-yl)-phenylamine (0.221 mmol),
respectively,
in ethylene glycol (1.5 mL) was heated to 160 C in a sealed vial for 3 h.
If product precipitated upon cooling of the mixture to r.t., the resulting
slurry was rinsed
into a frit with ethanol, the filter cake was washed several times with the
same solvent and
finally with some Et20. If necessary, product was further washed with
CHC13/acetone-
ultrasound and separated using a centrifuge.
If product stayed dissolved, mixtures were partitioned between satd aq.
NaHCO3/brine
(25+25 mL) and CHC13 (3x35 mL). Combined org. phases were dried over MgSO4. If
necessary, preparative TLC was performed (1 mm silica gel, petroleum
ether/CHZCl2/MeOH 30:90:20) or prep. HPLC on reversed phase. Product obtained
was
further washed with CHC13/MeOH-ultrasound and Et20/CHC13-ultrasound and
separated
using a centrifuge.
General procedure 8:
Palladium-catalyzed reaction of 2-chloro-4-(4-methyl-piperazin-1-yl)-
pyrimidine with
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
43
3- and 4-(benzothiazol-2-yl)-phenylamine (according to J. Yin et al., Org.
Lett. 2004, 4,
3481-3484):
An oven-dried G4 vial was charged subsequently with Pd2dba3 (8.10 mg, 0.009
mmol),
XANTPHOS ligand (15.3 mg, 0.027 mmol), pyrimidine (47.0 mg, 0,221 mmol), 3- or
4-
(benzothiazol-2-yl)-phenylamine (60.0 mg, 0.265 mmol) and K3PO4 (65.7 mg,
0.309
mmol). The tube was evacuated and purged with argon and dioxane (1.0 mL) was
added.
The vial was sealed and heated to 100 C for 21 h.
Mixtures were filtered through a pipette stuffed with cotton and then directly
mounted on a
prep. TLC plate (1 mm silica gel), reaction solvent was dried away in a
vigorous stream of
air, and separation was achieved using petroleum ether/CH2C12/MeOH 30:90:20.
Crude
product was crystallized from CHC13/Et20 to give products as beige powders in
20-38%
yield.
General procedure 9:
Reaction of 6-chloropurines with 3- and 4-(benzothiazol-2-yl)-phenylamine:
A mixture of the respective 6-chloropurine (0.221 mmol) and 3- or 4-
(benzothiazol-2-yl)-
phenylamine (0.243 mmol), respectively, in ethylene glycol (2.0 mL) was heated
to 110 C
in a sealed vial for 3 h. If reaction proceeded too sluggishly, 1.0 equiv 4 M
HCl in dioxane
was added and stirring continued for additional 3 h at 120 C. Product
precipitated upon
cooling of the mixture to r.t. Methanol/water 1:1 (40 mL) was added to the
mixtures, and
the resulting precipitate was filtered off and washed with 5% aq. HCl (3 x 10
mL) and
diethylether (2x10 mL). If necessary, product was further washed with
CHC13/MeOH-
ultrasound and Et20/CHC13-ultrasound and separated using a centrifuge.
General procedure 10:
Regioselective twofold 2,4-diamination of 2,4-dichloroquinazolines:
A mixture of 3- or 4-(benzothiazol-2-yl)-phenylamine (1.0 equiv.), 2,4-
dichloroquinazoline
(1.0 equiv.) and DIEA (1.0 equiv.) in n-BuOH was stirred at 120 C over night.
The respective amine (2.0 equiv.) was added and the mixture was overheated to
140 C in a
sealed vial for 3 h. If amine was used as a hydrochloride, additional DIEA had
to be added
(0.5 equiv.). For weakly nucleophilic amines like 3-amino-5-methylpyrazole,
NaI (1.0
equiv.) was used as additive.
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
44
Synthesis of (55) 1V4-(4-benzothiazol-2-yl-phenyl)1V2-(5-methyl-lH-pyrazol-3-
yl)-
pyridine-2,4-diamine:
Step 1(in analogy to S. L. Buchwald et al., J. Org. Chem. 2000, 65, 1158-
1174): An oven-
dried 624 vial was charged subsequently with Pd(OAc)2 (44.0 mg, 0.196 mmol),
biphenyl-
2-yl-di-tert-butyl-phosphine ligand (114 mg, 0.392 mmol), 2-chloro-pyridin-4-
ylamine
(252 mg, 1.96 mmol), 2-(4-bromophenyl)-benzothiazole ( 683 mg, 2.35 mmol) and
K3P04
(583 mg, 2.74 mmol). The tube was evacuated and purged with argon and DME (4.0
mL)
was added. The vial was sealed and heated to 100 C for 21 h.
The mixture was partitioned between half-saturated brine (50 mL, 1:1
H20/brine) and
CHC13 (2 x 35 mL). Combined org. phases were extracted once with half-
saturated brine
(50 mL, 1:1 H20/brine) and black fluffy precipitate formed was filtered off
and discarded.
Combined aq. phases were extracted again with CHC13 (2 x 35 mL). Combined org.
phases
were dried over MgSO4, mounted on silica gel and purified by chromatography on
using
PE/EE 20:1 to 1:2. Product fraction was further purified by prep. TLC (1 mm
silica gel,
PE/CH2C12/MeOH 70:70:15) and subsequent dissolving in acetone/MeOH and
crushing
out the product by adding H20. Crude product was treated with Et20 and
ultrasound, and
the suspension was layered with petroleum ether to give two crystal fractions,
10% total
yield of (4-benzothiazol-2-yl-phenyl)-(2-chloro-pyridin-4-yl)-amine.
Step 2: A mixture of product of step 1 (19.0 mg, 0.056 mmol) and 3-amino-5-
methylpyrazole (8.20 mg, 0.084 mmol) in ethylene glycol (0.5 mL) was treated
with 4 M
HCl in dioxane (14 L, 0.056 mmol) and heated to 160 C in a sealed G4 vial for
22 h.
The mixture was partitioned between satd aq. NaHCO3/brine 1:1 (50 mL) and
CHC13 (3 x
35 mL), combined org. phases were dried over MgSO4. Crude material was
purified by
prep. HPLC. Product fractions were partitioned between satd aq. NaHCO3 (75 mL
added to
HPLC phase upon removal of CH3CN) and CHC13 (3 x 50 mL), combined org. phases
were dried over MgSO4, and N4-(4-benzothiazol-2-yl-phenyl)-N2-(5-methyl-lH-
pyrazol-3-
yl)-pyridine-2,4-diamine was fmally crystallized from CHC13/Et20.
Synthesis of (58) N4-(4-Benzothiazol-2-yl-phenyl)-NZ-methyl-pyridine-2,4-
diamine
and (86) N2-(4-Benzothiazol-2-yl-phenyl)-N2-methyl-pyridine-2,4-diamine:
Step 1: A mixture of methylamine hydrochloride (149 mg, 2.20 mmol), 2-chloro-4-
aminopyridine (257 mg, 2.00 mmol), DIEA (174 L, 1.00 mmol) and NaI (300 mg,
2.00
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
mmol) in nBuOH (4 mL) was stirred at 120 C for 24 h.
Mixture was partitioned between NaHCO3/brine 1:1 (50 mL) and CHC13 (3 x 35
mL).
Combined org. phases were washed once with brine (50 mL) and dried over MgSO4.
The
aq. phase still contained some product along with high quantities of DIEA, so
water was
5 removed in vacuum and the remaining salt was washed with acetone/CHC13
several times.
Both organic fractions (first from extraction, second from salt) were purified
by prep. TLC
(1 mm silica gel each, CH2Cl2/MeOH 85:15). Product was crystallized from
CHC13/Et2O
and yielded 227 mg (92%) of a beige powder.
Step 2 (in analogy to S. L. Buchwald et al., J. Org. Chem. 2000, 65, 1158-
1174): An oven-
10 dried G4 vial was charged subsequently with Pd2dba3 (36.6 mg, 0.040 mmol),
biphenyl-2-
yl-di-tert-butyl-phosphine ligand (47.8 mg, 0.160 mmol), product step 1 (98.6
mg, 0.800
mmol, 2-(4-bromophenyl)-benzothiazole (155 mg, 0.880 mmol) and sodium tert-
pentoxide
(123 mg, 1.12 mmol). The tube was evacuated and purged with argon and toluene
(1.6 mL)
was added. The vial was sealed and heated to 110 C for 21 h.
15 The mixture was partitioned between half-saturated brine (50 mL, 1:1
H20/brine) and
CHCl3 (3 x 35 mL). Combined org. phases were dried over MgSO4 and purified by
prep
HPLC. Two product fractions were collected (constitutional isomers),
partitioned between
satd aq. NaHCO3 (20 mL) and CHC13 (3 x 30 mL) to remove any formic acid still
present
from HPLC and dried again over MgSO4. Both fractions were finally purified by
prep.
20 TLC (1 mm silica gel, CH2Cl2/MeOH 80:20), dissolved in a few drops CH2C12
and crushed
out with Et20 to give 1V4-(4-Benzothiazol-2-yl-phenyl)-N2-methyl-pyridine-2,4-
diamine
and N2-(4-Benzothiazol-2-yl-phenyl)-N2-methyl-pyridine-2,4-diamine,
respectively as
beige solids (yields around 1% each).
25 Synthesis of (56) 4-(4-benzothiazol-2-yl-phenylamino)-pyridine-2-carboxylic
acid
methylamide:
Step 1: A mixture of methylamine hydrochloride (544 mg, 8.05 mmol) and 4-
chloropicolinic acid (296 mg, 2.30 mmol) in DMF (5 mL) was treated with
EDC*HC1(661
mg, 3.45 mmol) and DIEA (2.0 mL, 11.5 mmol) at r.t. for 40 h.
30 was continued over night (LCMS: ST17921h).
Reaction mixture was partitioned between aq. satd NaHCO3 (50 mL) and CHC13 (3
x 35
mL). Combined org. phases were washed with aq. satd NH4C1 (2 x 50 mL) and
dried over
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
46
MgSO4. The organic phase was purified by chromatography on silica gel using
petroleum
ether/ethyl acetate 2:1 to pure ethyl acetate. Product was finally purified by
prep. HPLC.
Product fraction was again partitioned between satd aq. NaHCO3 (20 mL) and
CHC13 (3 x
30 mL) to remove any formic acid still present from HPLC, and dried over
MgSO4.
Resulting oil was dried only under reduced pressure, 25 mbar, as product is
volatile at high
vacuum.
Step 2: A mixture of 4-benzothiazol-2-yl-phenylamine (59.7 mg, 0.264 mmol),
product
step 1 (40.9 mg, 0.240 mmol) and 4.0 M HCl/dioxane (45 L, 0.180 mmol) in DMF
(0.5
mL) was heated to 160 C for 7 h.
Slurry was rinsed out into a frit (pore size 4) with EtOH, the filter cake was
washed several
times with the same solvent and finally with some Et20. Solid was treated with
CHC13-
ultrasound to remove impurities and 4-(4-benzothiazol-2-yl-phenylamino)-
pyridine-2-
carboxylic acid methylamide was separated using a centrifuge, resulting in
17.5 mg (20%)
of a beige powder.
Synthesis of (57) (4-benzothiazol-2-yl-phenyl)-pyridin-4-yl-amine:
When a mixture of 4-benzothiazol-2-yl-phenylamine (79.2 mg, 0.350 mmol) and
4-chloropicolinic acid (45.0 mg, 0.350 mmol) in DMF (0.5 mL) was heated to 160
C for
3 h, decarboxylation of product formed occurred immediately, but not of
starting picolinic
acid.
Mixture was filtered through a pipette stuffed with cotton wool and then
purified by prep.
TLC (1 mm silica gel), using PE/EE 1:1. (4-Benzothiazol-2-yl-phenyl)-pyridin-4-
yl-amine
was finally purified by prep. HPLC to give a pale yellow solid (9.10 mg, 8%).
General procedure 11:
Reaction of 4-chloropyridine with substituted 3- and 4-(benzothiazol-2-yl)-
phenylamines:
A mixture of 4-chloropyridine (40.0 mg, 0.350 mmol) and substituted 3- and 4-
(benzothiazol-2-yl)-phenylamines (0.350 mmol) in DMF (2.0 mL) was heated for 3
h at
160 C.
The mixtures were dissolved in DMSO (2 mL) and separated by prep. HPLC.
Product was
finally purified by additional prep. TLC (1 mm silica gel, CH2Cl2/MeOH,
90:10).
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
47
General procedure 12:
Reaction of 4,7-dichloroquinoline and 4-chloro-thieno[3,2-d]pyrimidine with
substituted 3- and 4-(benzothiazol-2-yl)-phenylamines:
A mixture of 4,7-dichloroquinoline or 4-chloro-thieno[3,2-d]pyrimidine (0.242
mmol) and
the respective 3- or 4-(benzothiazol-2-yl)-phenylamines (0.220 mmol) and 4.0 M
HCl/dioxane (65 L, 0.440 mmol) in ethylene glycol (1 mL) was heated to 110 C
for 6 h.
For reactions with 4,7-dichloroquinoline, addition of water resulted in
precipitation of
product, which was filtered off and washed with water, diethylether, and
petroleum ether.
For reactions with 4-chloro-thieno[3,2-d]pyrimidine, the mixtures were
separated by prep.
HPLC.
By following the methods described above, the compounds set out in the
following table
were prepared.
Com- LC/ESI-MS: General
Structure
pound [M+H] m/z = Procedure
N \ /
I S
I HN 333 7
NN
N \ /
I S
2 HN 347 7
N~N
~
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
48
N
S
3 333 7
NYNH
N
N \ /
S
4 CI 367 7
NYNH
N
HN r12
~ Oi 363 7
NN
N
I S
6 HN OH 349 7
NI ~- N
,
OH N
S
7 349 7
NYNH
N
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
49
N \ /
g 347 7
4111~
NYNH
N
S \ /
N--;7~N ~N
9 1 N I~ 415 4
H
O
S \ /
N 403 8
J N H
iNv
S
11 HN N I~ ~N 400 7
N/
N N N
H H
O
12 H S \/ 415 4
N N~N I S \ /
N";~ N , N
13 I N I~ OH 431 4
H
O
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
S \ /
~N
14 N N N I i Oi 445 4
~ ~ H
O
N
15 HN 345 9
N
IC N
H
N
I ~ S
16 345 9
HN N
N
N
.
\~-NH
N
~ ~ S
17 N N N 429 4
~ H
O
A
N
S
CI
18 ir N\ NH 449 4
N O
0 \ 1
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
51
N \ /
S
19 rN' NH 429 4
N
O
O\
Nr N N x
20 N 431 4
OH
0
O\ ~
HN \
/-\ N-{
21 -N N-~N N S 403 8
HN :I S
22 NN Lb 400 7
H N,N' N
H
S \ /
23 ~ I j ~N 334 7
N N N
H H
S \ /
24 N;-, ( I ~N 334 7
N N N
H
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
52
NH S \ /
N~N ~N
25 N I 444 10
~ H
O
"lO
-
HN, ~ -
N NH S \ /
ALI
26 N7" N (~ N 510 10
H
O (
~O
O O
S ~ /
27 N 444 10
HN N
N
NH
/
NYN
.
QN
2
8 S \ N oeNH 334 7
b
N N~YNN,
I ~ N
29 334 7
S N N
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
53
O O
S
N
30 HN N41N 510 10
NH
N
HN /
S \ /
N~N XYLN
31 H 541 5
Co
N~ ~
~N~
S \ /
N~N XN
~
32 H 0571 5
~
C'o
0"
,
S
OCN NH 33 NH 344 9
N O
S -
34 NH ~ 374 9
OCN
N~ ~ N H
N
H
~ \ / N
35 N HN /\N 344 9
ON
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
54
S
NH ~
N
36 N ~ NH 374 9
N
O
\
O
cIN)=1l 37 NH 404
\ N~
O
\
S NH
38 HN N 374 9
N=(
O
/
NH
/ S - O~
I O N
39 541 5
N N
\
-O -N N
~~ -N-\
N O
40 NH 571 5
N\_
S O
N
r'N"*-~O
~N,) H
N
41 N v N 541 5
S N N
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
O rN
H O,,,,,, N
N 1
42 I/ NvN 541 5
S ~N
\ /
-O
43 ~ NH N 375 9
OCN
N~NH
S
N / I
44 NH 555 5
N
O-'---" ON S OH
N
45 NH 557 5
N
N~
~N
, CI
N- \ I NH
46 S N\ \ OIN 575 5
~
L N
('~N\
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
56
i I
N~ ' N H
47 S N~ O~ 555 5
LN i O'*'-~N
---)
QS CF3
N / I O
~
48 NH 625 5
N ~ O~
N ~ O~~N
N
S ci
~
N /I
49 NH 575 5
N ~ O~
N ~ O"-~N
N
S F
N /I
:~
50 NH ON5559 5
~
N --,
O'*-~N
ON
S
N I O
51 NH 571 5
N ~
N ~ O'*~-ON
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
57
S
N ~ I F
~ NH
52 559 5
N ~ ~
N / O'---" O
HO
N~ NH
53 S N~ ~ O 557 5
N ~ O'~ON S
\ /
54 N 430 7
HN~~ XETN N N N N O
H H
S
55 ~ N N 399 see above
HN ~ ~ ~ ~ ,
N N N
H H
S \ /
56 H N' I I~ N 361 see above
~ N
O H
S \ /
57 ' I N 304 see above
N I%
H
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
58
S \ /
58 N~ I (~ \N 333 see above
~
N N
H H
\ / S
N , I OH
~
59 NH 557 5
N O( N O'*~-~ N~
~N
F 3 C S
~
N /I
60 ~ NH o 609 5
N ~
I
N O-~~N
ON
N~ SNH
CI ~
61 (~ 575 5
N ON,
NN.
N&NH SL(OH
62 557 5 N
O~"- 01.1
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
59
qs O~
N
63 585 5
N ooON
~N
N \
64 CI cHN /\ N 388 12
VJ ' ~
-
NN N \ ~
65 S 361 12
LS H
N \ ~
-
66 N ~ ~~ S 418 12
N ~
CI I~ H
N
67 N"- N S 391 12
S H O
Q_<N
S
68 N,\ / NH 388 12
\ /
CI
JZIIIIILN
N12
69 N
S H S 361
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
S
N / I
NH 575 5
N
I
N O~-' ON N
\ /
71 N\ S 334 11
N O
H I
N
72 I/ N I/ N 304 11
H S
N \ /
~ S
73 HN I/ CI 422 12
N / CI
N
74 S 395 12
N CI
S H
/ S
N
NH rN 555 5
N j 0N ,,/
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
61
Q\ / S OH
N I ~
76 ~ NH rN 557 5
N O
~
~
N O
S CI
N I
77 NH rN 575 5
N O
~N O
S F
~
N (~
78 NH rNI 559 5
N ~ ~ N ,/
~N ~ O
N OH
/
79 NH ~N 557 5
N ~
N ~ O
S
N O
80 NH rN~ 571 5
O~~ N ~N
N ao
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
62
N
N F
~ 4 N ~
/O - HN \ / S ~ /
81 O 559 5
~N
NJ
/
F3C \ ~ S
N
82 NH 609 5
N O~/~
'N O
CI \ ~ S
.
(~
83 N ~ NH rN 575 5
N ~ O~~NJ
'N ~ O
S
N /
84 NH 406 12
F C aCl
N S
N ~
85 NH 379 12
F N ~ S
N
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
63
S \ /
86 \ N I j \N 333 see above
H2N N
S \ /
N
N~N e
8
7 1 N 419 4
I~ H
Ci
F Q N
S
88 NH 559 5
N~ O
I ~
'N 0"~- ON N ,
CI q N
S
89 NH 575 5
N~ / O
~
'N ~ O'~-~N~
~N
CI \ ~ N
S I ~ F
90 NH 593 5
N
'~ O
~
N ~ ON,
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
64
N
S
NH
91 N 555 5
N')
O
~ N
I
S\
~
~
NH
92 - O 571 5
I ~
N
O-N--\
~
ON
S
N
~
\ I
93 0 542 6
N'
'
N ON
~
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
S
N F
94 O 560 6
N O
O~~ON N
95 O 513 6
N O\
IN
N ~ O~~ N
V
Qs
N' N
96 O 514 6
N 51 O~
Z~
N N
V
Materials and methods
In vitro Protein Kinase Assay
5 The effect of the 2-arylbenzothiazole derivatives was tested on recombinant,
human protein
kinases. All protein kinases were expressed in Sf9 insect cells as human
recombinant GST-
fusion proteins or as His-tagged proteins by means of the baculovirus
expression system.
Protein kinases were purified by affinity chromatography using either GSH-
agarose or Ni-
NTH-agarose. The purity and identity of each was checked by SDS-PAGE/silver
staining
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
66
and by western blot analysis with specific antibodies.
A proprietary protein kinase assay (33PanQinase Activity Assay) was used for
measuring
the kinase activity. All kinase assays were performed in 96-well F1ashPlatesTm
in a 50 1
reaction volume. The assay for all enzymes contained 60 mM HEPES-NaOH, pH 7.5,
3 mM MgC12, 3 mM MnC12, 3 M Na-orthovanadate, 1.2 mM DTT, 50 g/ml PEG20000
and 1 M [y-33P]-ATP (approx. 5x105 cpm per well).
The reaction cocktails were incubated at 30 C for 80 minutes. The reaction was
stopped
with 50 1 of 2% (v/v) H3PO4, plates were aspirated and washed two times with
200 l of
0.9% (w/v) NaC1. Incorporation of 33P; was determined with a microplate
scintillation
counter. All assays were performed with a BeckmanCoulter/ Sagian robotic
system.
Cellular Receptor Tyrosine Kinase Assay
The effect of 2-arylbenzothiazole derivatives was tested in cellular assays by
determining
the inhibition of the receptor tyrosine kinases (RTKs) of the growth factor
receptors EGF-
R, PDGF-R, TIE2, and VEGF-R2. To this end different cell lines expressing the
respective
growth factor receptor in appropriate amounts were used. 35,000 cells per well
were plated
in medium containing 10% fetal calf serum (FCS) in 48-well cell culture
dishes. After 24
hrs the FCS-containing medium was exchanged against medium without FCS, and
subsequently cells were starved in this medium overnight. On the next day test
compounds
at different concentrations in 100% DMSO were added to the cell culture medium
in a
1:100 dilution step resulting in a final DMSO assay concentration of 1%. After
90 min
preincubation with test compounds at 37 C, cells were stimulated at room
temperature for
several min with receptor-specific ligands. Receptor stimulation was followed
by cell lysis
using a lysis buffer complemented with standard protease and phosphatase
inhibitors.
The phosphorylation status of the various RTKs was quantified in 96-well
plates via a
sandwich ELISA using receptor-specific capture antibodies and a generic
biotinylated anti-
phosphotyrosine detection antibody. Finally optical density as measured at 450
nm after
addition of avidin-labelled horseradish peroxidase and Tetramethylbenzidine
(TMB) as a
substrate.
For each particular concentration of a test compound percental inhibition was
calculated
relative to maximal phosphorylation in stimulated, untreated cells ("high
control"). IC50
values were calculated based on sigmoidal inhibitor curves covering a
concentration range
of 9 concentrations of each test compound in half-logarithmic steps.
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
67
Cellular Aurora-B Kinase Assay
The effect of 2-arylbenzothiazole derivatives was tested in a cellular Aurora-
B assay by
measuring the effect of the test compounds on the endoreduplication of genomic
DNA.
Inhibition of Aurora-B results in endoreduplication of genomic DNA, which is
detectable
in cells as DNA-content higher then 4 n. Intercalation of fluorescent
Propidium Iodine (PI)
into DNA was used to quantify the DNA content by using a fluorescence
activated cell
sorter (FACS).
HT29 colon-carcinoma cells were seeded on day 1 of the experiment at 100,000
cells per
well in 6-well cell culture dishes in 3 ml of DMEM medium containing 10% FCS,
100
units/ml Pencillin, 100 mg/ml Streptomycin at 37 , 10% CO2. On day 2 test
compounds at
different concentrations in 100% DMSO were added to the medium in a 1:1000
dilution
step resulting in a final DMSO assay concentration of 0.1 %. Cells were
incubated with test
compounds for 3 days. On day 5 the cells were harvested by trypsinization,
combined with
corresponding supernatants, centrifuged, and resuspended in 80% methanol for
fixation
and permeabilization at 4 C overnight. On day 6 fixed cells were centrifuged,
rehydrated
in PBS/1% FCS for 1 h, and subsequently incubated with RNAse A and PI for 30
min at
room temperature.
Stained cells were analyzed for DNA-content by FACS as follows. For analyses
of the cell
cycle distribution of the cell population, 5000 single-cell-events of the
differently treated
cells were aquired by FACS . DNA-intercalated PI was detected by measuring
fluorescence emission using a 650 nm pass filter (FL3) upon excitation at 488
nm with an
argon laser. Single cell events were plotted in a histogram according to their
FL3-A signal.
Signal amplification for the first peak of the FL-3 amplitude (FL3-A) was set
to about 200
arbitrary units (AU). Using an untreated cell population, gates were defined
for each of the
different cell cycle phases. The area containing the gaussian-curve-shaped
first peak at 200
AU was defined as õcells in G1-phase" containing the double set of chromosomes
(2n).
The area around the peak at 400 AU was defined as õcells in G2/M-phase"
containing the
quadruple set of chromosomes (4n). Events inbetween GI and G2/M are defined as
õcells
in S-phase", those below G1 (subGl) as õapoptotic". Importantly, all events
beyond the
G2/M-gate were defined as õendoreduplicated cells" (EndoR). For each
concentration of a
test compound the percentage of EndoR-population as compared to the whole cell
population was determined. For estimation of IC50 values of Aurora-B
inhibition the
percentages of EndoR-populations were plotted versus compound concentrations.
CA 02615420 2008-01-15
WO 2007/009524 PCT/EP2006/004620
68
Cellular Aurora-B Kinase Histone H3 Phosphorylation Assay
The effect of compounds was tested in a cellular Aurora-B assay measuring
phosphory-
lation of the Aurora B-substrate protein Histone H3 at Serine 10 (HisH3-pS
10). Inhibition
of Aurora B results in reduction of HisH3-pSlO which was detected in a
specific immuno-
assay.
In the experiment, HT-29 colon-carcinoma cells were seeded on day 1 and on day
2 test
compounds at different concentrations were added. Cells were incubated with
test
compounds for 1 hour. Subsequently, Calyculin A was added for 30 min. For
DELFIA -
detection (PerkinElmer) of HisH3-pS 10, lysates were transferred to a
microtiterplate and
incubated with detecting antibody directed against HisH3-pS 10 and Europium-
labelled
secondary anti-IgG-antibody. Emission at 615 nm was measured upon excitation
at 340 nm
and the percentage of inhibition was calculated for each concentration of the
test
compoun&relative to controls without inhibitor. Mean values of HisH3-pS10
percentage
were plotted versus compound concentration for calculation of IC50-values.
Results
In vitro Protein Kinase Assay
The following examples show IC50 values lower than 500 nM on at least one
kinase
selected from Aurora-A, Aurora-B, EGF-R, ERBB2, PDGFR, IGF1-R, VEGF-R2, VEGF-
R3, EPHB4, TIE2, and SRC or display a beneficial activity profile by
inhibiting at least
two kinases from at least two different molecular mechanisms of tumor
progression with
IC50 values lower than 500 nM: 31, 32, 39, 40, 44, 45, 48, 49, 50, 51, 52, 54,
59, 60, 70,
75, 76, 77, 78, 79, 81, 83, 85, 87, 88, 89, 90, 91, 92, 93, 94, 95.
The following compounds show IC50 values lower than 10 M in the Cellular
Receptor
Tyrosine Kinase Assay and / or the Cellular Aurora-B Kinase Assay: 45, 51, 93,
94, 95.