Note: Descriptions are shown in the official language in which they were submitted.
CA 02620294 2008-02-14
WO 2007/022403 PCT/US2006/032249
OCULAR GENE THERAPY USING AVALANCHE-MEDIATED
TRANSFECTION
FIELD OF THE INVENTION
The present invention relates generally to medicine. More particularly, the
present invention
relates to a method of treating ocular diseases with gene therapy using
avalanche-mediated
transfection to genetically modify cells or tissue.
BACKGROUND
There are many ocular diseases that affect vision. Diseases of the conjunctiva
and cornea,
cataracts, uveal diseases, retinal diseases, loss of central acuity and visual
field abnormalities and
diseases of Bruch's membrane are a few examples. Age-related macular
degeneration is a
leading cause of vision loss in the aged population. In the less common but
more severe "wet"
form of age-related macular degeneration, choroidal neovascularization leads
to progressive
disease and vision loss.
Current therapeutics for treatment of many ocular conditions require the need
for frequent
intravitreal administration. Therapies involving delivery of proteins or
aptamers are examples of
such approaches, with the drawback that proteins and aptamers have short half-
lives and require
intravitreal administration every 4-6 weeks for life for maximal efficacy.
Gene therapy
approaches are potentially more long-term, with the possibility of lasting
many months or years.
Gene therapy can be in vivo, involving delivery of therapeutic genes directly
to the tissue of
interest, or can be ex vivo, where tissue selected for use is treated outside
the body prior to
implantation. The art has long sought gene therapy treatment methods that are
safe for the
patient and therapeutically viable.
1
CA 02620294 2008-02-14
WO 2007/022403 PCT/US2006/032249
One of the important factors in the efficacy and safety of gene therapy is the
method used to
introduce DNA into a cell. Viral vectors, such as retroviruses and
adenoviruses, enable high
expression of the introduced DNA but have safety concerns. Non-viral methods,
such as
liposomes, have low host iimnunogenicity but tend to suffer from inefficient
DNA delivery to
cells. Accordingly, there is a need in the art for new methods of introducing
DNA into cells and
tissues for the purpose of gene therapy.
2
CA 02620294 2008-02-14
WO 2007/022403 PCT/US2006/032249
SUMMARY OF THE INVENTION
The present invention provides a method of treating an ocular disease in a
subject. In a first step,
a nucleic acid is introduced into cells or a tissue. The nucleic acid is
introduced by electron
avalanche-mediated transfection. With this technique, a high electric field
induces a vapor
bubble and plasma discharge between an electrode and the surrounding medium.
The formation
of a vapor bubble generates mechanical stress. Plasma discharge through the
ionized vapor in the
bubble enables connectivity between the electrode and the surrounding medium,
so that the
mechanical stress and electric field are applied simultaneously, which results
in permeabilization
of the cells or tissue. This penneabilization in turn allows the nucleic acid
to enter the cell or
tissue. Cells or tissue containing the nucleic acid are then transplanted into
an ocular region of
the subject.
Cells and tissue according to the present invention are preferably autologous
(i.e. from the
subject), or allogeneic (i.e. from an individual of the same species). In the
case of cells, the cells
may be primary cells or cell lines. Preferred primary cells are conjunctival
fibroblasts,_scleral
cells, or epithelial cells. Preferred cell lines are fibroblast cell lines or
inuscle cell lines.
Preferred tissues are conjunctival tissue and scleral tissue. The cells or
tissue may be cultured
prior to transplantation. Alternatively, or in addition, the cells or tissue
may be placed in a cage,
such as a polymeric cage, or a scaffold or matrix to support the growth of the
cells.
In a preferred embodiment, the nucleic acid is DNA. The DNA may encode, for
example, a
therapeutic protein or an RNAi cassette, such as a short-hairpin RNA (shRNA).
Alternatively,
the DNA may be used for modifying an endogenous gene. For example, the DNA may
be an
oligonucleotide used for gene repair, or may be used for homologous
recombination with an
endogenous gene, for the purpose of modifying the gene. Modifications include,
for example,
modifying expression levels of the gene and/or replacing a mutant gene with a
wild-type copy of
3
CA 02620294 2008-02-14
WO 2007/022403 PCT/US2006/032249
the gene. In a particularly preferred einbodiment, the nucleic acid is part of
a plasmid. The
plasmid may, in addition to a therapeutic gene, contain a marker gene. In
order to obtain
genomic integration, the plasmid may contain integration elements, such as a
phiC31 attB site or
inverted repeats recognized by transposases such as Sleeping Beauty. In this
case, a source of
phiC31 integrase or a transposase would also be provided.
Genetically-modified cells or tissue may be transplanted into any ocular
region of the subject.
Preferred regions are the choroid, vitreous hutnor, retinal pigment
epithelium, near the macula,
and behind the sclera. In the case of a macular region, the ocular region may
be epiretinal to the
macula, subretinal to the macula, or intra-retinal to the macula. In the case
of the vitreous
huinor, the ocular region is preferably a region of the vitreous huinor near
the pars plana.
Any ocular disease may be treated according to the present invention. Examples
include, but are
not limited to, age-related macular degeneration, choroidal
neovascularization, retinal
degeneration, glaucoma, diabetic retinopathy, and retinal dystrophies.
Similarly, any subject
may be treated according to the present invention. Preferred subjects are
humans and non-
human mammals.
BRIEF DESCRIPTION OF THE FIGURES
The present invention together with its objectives and advantages will be
understood by reading
the following description in conjunction with the drawings, in which:
FIG. 1 shows the avalanche method according to the present invention.
FIG. 2 shows the use of the avalanche method according to the present
invention with
wire electrodes.
FIGS. 3-6 show examples of electrode geometries suitable for practicing the
avalanche
method according to the present invention.
4
CA 02620294 2008-02-14
WO 2007/022403 PCT/US2006/032249
FIG. 7 shows an example of a plasmid construct suitable for gene therapy of an
ocular
disease according to the present invention. The plasmid contains a nucleotide
sequence encoding for pigment epithelium-derived factor (PEDF) and a
nucleotide sequence encoding for enhanced green fluorescent protein (eGFP)
under control of a cytomegalovirus (CMV) proinoter, the two sequences linked
by
an internal ribosome entry site (IRES) coding sequence.
FIG. 8 shows ocular regions suitable for transplantation according to the
present
invention.
FIG. 9-10 show examples of electron avalanche-mediated transfection according
to the
present invention.
DETAILED DESCRIPTION OF THE INVENTION
Avalanche method
The present invention provides an ex vivo gene therapy method based on a novel
method of
introducing DNA into cells called the avalanche method. When sufficiently high
voltage is
applied to an electrode, a mechanical stress wave synchronized with a pulse of
electric current
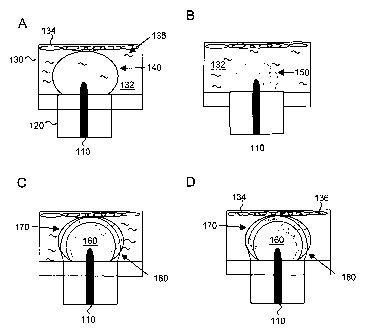
can be produced and applied to cells or tissue, as shown in FIG. 1. FIG. lA-C
shows three
stages that occur when a high voltage is applied to an electrode 110 covered
by insulation 120.
Electrode 110 is situated in tissue culture well 130, with conductive liquid
medium 132, cells
134, and nucleic acid 136. (While cells are pictured in this figure, tissue
could also be used).
When a voltage is first applied to electrode 110, (FIG. lA), an electric field
140 is generated
around the un-insulated portion of electrode 110. If the electric field in the
mediuin is
sufficiently high, generated Joule heat leads to rapid vaporization of liquid
medium 132 in the
areas adjacent to electrode 110, resulting in generation of a vapor bubble 150
(FIG. 1B). As
soon as vapor bubble 150 is formed, it disconnects the surface of electrode
110 from conductive
medium 132, so that the electric current stops flowing, and the electric field
on the target cells is
5
CA 02620294 2008-02-14
WO 2007/022403 PCT/US2006/032249
tenninated. To overcome this difficulty, the vapor in the bubble can be
ionized to forin ionized
vapor 160, which restores the electrical conductivity, as shown in FIG. 1C.
Ionized vapor 160,
also known as plasma, forms a kind of virtual electrode with electric field
170. During this
process, the formation of the vapor bubble, and its subsequent collapse,
causes a propagating
shock wave through the medium, exposing the cells or tissue to mechanical
stress 180. The
combination of the shock wave and the electric field leads to permeabilization
of cells 132, such
that nucleic acid 136 may enter cells 132 (FIG. 1D). Highlighting the role of
the plasma-
mediated electric discharge, the inventors have named this technique electron
avalanche-
mediated transfection, or, for siinplicity, the avalanche method.
The process described in FIG. 1 works when the electrode produces a relatively
uniform electric
field. Alternatively, electrodes with a very uneven electric field may be
used, so that the vapor
cavity formed at the apex does not cover the whole surface of the electrode
with a lower electric
field. This way the electric current to the medium will not be completely
disconnected. One
example of an electrode geometry with a non-uniform electric field is a
cylindrical probe, such
as a wire, with a sharp end. FIG. 2A shows an image of a wire electrode 210
producing a plasma
discharge 220. As can be seen from FIG. 2A, the plasma discharge is clearly
visible. It is also
clearly audible. FIG. 2B shows current 230 and voltage 240 versus time when a
voltage is
applied to a wire probe. In this particular example, the wire probe was 50 m
in diameter and
electrical pulses of up to 600 V were used to produce an electric field at the
tip of the wire of
about 30 kV/cm. However, these parameters may be varied. FIG. 2B shows that
when a voltage
is applied to such a probe, the initial 20 s of the waveform exhibits
reduction of the current due
to beginning of vaporization. This is followed by stabilization of
conductivity following
ionization of the vapor cavity. The ionized vapor cavity serves as a transient
electrode, which
can greatly exceed the size of the probe, as shown in FIG. 2A. As a result,
the distribution of the
6
CA 02620294 2008-02-14
WO 2007/022403 PCT/US2006/032249
electric field becomes inuch more uniform than the one generated initially on
the small wire
electrode, thus leading to more uniform electroporation of the target cells or
tissue.
FIG. 2C shows, for different diaineters of electrodes, the field strength
(kVhnin) along the
length of electrode 230 covered by insulator 240. The electrode diameter
indicated by the solid
line 250 is 10 m, the dotted line 260 is 25 m, and the dashed line 270 is 50
in. In this
particular experiment, 600 V was applied to the electrode. FIG. 2C shows that
for a cylindrical
electrode with a sharp tip, there is a rapid decrease in electric field as one
moves farther away
from the tip of the electrode. Thus, the strength of the electric field at the
apex of the electrode
can be varied by changing the electrode diameter.
Various types of probes may be used according to the present invention. FIG. 3
shows a version
of a probe in which active electrodes 310 are plated on a substrate 320. FIG.
3A shows a top
view and FIG. 3B shows a side view of the probe. In this probe, substrate 320
is surrounded by
return electrode 330. The pattern of active electrodes 310 on substrate 320
forms the necessary
proportion between electric field 340 and mechanical stress wave 350 due to
plasma discharge
352. The probe in FIG. 3 has a singularity of the electric field 340 at the
edges 312 of active
electrodes 310. Singularities serve as ignition points for plasma discharge
352 and generation of
mechanical stress wave 350. In FIG. 1, plasma occupies the whole volume of the
vapor cavity.
In contrast, in FIG. 3, the electric field at the edges of the thin electrode
is much higher than in
front of its flat part so vaporization and ionization will occur (or start)
primarily there. This
implementation is simple and inexpensive, but it does not provide the
flexibility to control
mechanical and electric pulse parameters separately.
Another probe iinplementation, which allows separate control of mechanical
stress wave 450 and
electric field 440, is shown in FIG. 4. (FIG. 4A is a top view, FIG. 4B is a
side view). In this
7
CA 02620294 2008-02-14
WO 2007/022403 PCT/US2006/032249
iinplementation, two types of active electrodes, 410 and 412, are patterned on
substrate 420, with
return electrode 430 surrounding substrate 420. Electrodes 412 may be driven
to generate an
electric field 440, while electrodes 410 may be driven to generate plasma 452
and mechanical
stress wave 450. (Plasma 454 also generates an accoinpanying electric field,
not shown).
Separate control of the ainplitude of stress wave and electric field might be
desirable for
optimization of penneabilization. Generating thein on the same electrode will
make these values
inutually dependent, while generation on two separate electrodes may provide
independent
control of these phenomena.
FIG. 5 shows an example of a transfection device suitable for molecular
delivery of nucleic acid
to adherent cells or tissue according to the present invention. In this
arrangement, cells 510 are
growing on an adherent surface 520 placed in a nonporous substrate 530, such
as a tissue culture
plate. Adherent surface 520 may be, for example, a tissue culture insert made
of porous material
such as polycarbonate. Cells could also be grown directly on nonporous
substrate 530. A
gelatinous matrix and/or feeder layer may also be present (not shown). A probe
540 with pillar
electrodes 542, return electrode 544, and connection 546 to a voltage source
(not shown) is
brought into a solution 550 containing nucleic acid 560. Pillar electrodes 542
are positioned a
finite distance from cells 510, e.g. about 1 mm. This finite distance is
preferably in the range of
about 0.5 mm to about 2 cm. In the embodiment shown, the return electrode 544
extends beyond
pillar electrodes 542 a distance equal to this finite distance such that the
finite distance is defined
when the return electrode 544 is touching adherent surface 520. However, this
distance could.be
defined by any substance. In addition, pillar electrodes 542 are preferably
about 0.5 mm to about
2 cm apart.
FIG. 6 shows an example of a transfection device suitable for molecular
delivery of nucleic acid
to cells or tissue in solution according to the present invention. In this
arrangement, cells or
8
CA 02620294 2008-02-14
WO 2007/022403 PCT/US2006/032249
tissue 610 are suspended in solution 620 with nucleic acids 630 in cuvette
640. Cuvette 640
contains return electrode 642, pillar electrodes 644, and connection 646 to a
voltage source (not
shown). In this design, pillar electrodes 644 are preferably between about 0.5
mm and about 2
em apart to provide adequate coverage of the solution voluine. In this
arrangeinent, the pillar
electrodes could be simultaneously or alternately active.
Regardless of probe design, to produce a strong stress wave, the electric
field on the electrode
surface should be sufficient for rapid vaporization of the liquid medium. In
addition, to maintain
connectivity, the electric field should be high enough to induce ionization of
the vapor. In this
way, both a mechanical stress wave and an electric field can be synchronized,
with maximal
intensity at the surface of the electrode. In addition to these concerns, the
plasma discharge must
be controlled in order to maximize transfection efficiency and minimize cell
death.
Several parameters may be varied to meet the above requirements, such as
electric field strength,
applied voltage, pulse duration, number of pulses, frequency, etc. The actual
values of these
parameters will depend on the specific electrode geometry. In general,
however, applied
voltages are preferably in the range of about 1 V to about 10 kV, more
preferably between about
100 V and about 1 W. Applied voltage preferably results in an electric field
between about 1 to
about 100 kV/cm, more preferably about 10 to about 50 kV/cm, and most
preferably about 30
kV/cm. Pulse duration is preferably in the range of about 1 ns to about 100
ms, more preferably
between about 100 ns and about 1 ms. Either monophasic or biphasic pulses are
suitable for the
purposes of the present invention. However, biphasic pulses are preferred as
they lead to less
gas formation, nerve and muscle response, and electrode erosion. Any nuinber
of pulses may be
used according to the present invention. The number of pulses is preferably
between about 1 and
100, more preferably between about 1 and 50. When multiple pulses are used,
the frequency of
9
CA 02620294 2008-02-14
WO 2007/022403 PCT/US2006/032249
pulses should be in the range of about 0.1 Hz to about 1 kHz. Preferably, the
frequency is less
than about 1 kHz to prevent heat accumulation.
Cells and Tissues
Any cell or tissue may be suitable for practicing the invention. Examples
include primary cells,
primary tissues, and cell lines. Preferred cells include conjunctival
fibroblasts, epithelial cells
and scleral cells. Preferred tissues include conjunctival tissue and scleral
tissue. Preferred cell
lines include fibroblast cell lines and muscle cell lines. The cells and
tissue are preferably
autologous or allogeneic.
In one einbodiinent, the method of the present invention involves obtaining
tissue from a subject
having or at risk of developing an undesirable eye condition. The condition
can range from a
minor or nuisance condition, such as dry eye, to a more serious disease with
possible vision loss,
such as age-related macular degeneration. Under the care of a skilled medical
provider, tissue
from the patent is harvested in an invasive, minimally invasive, or non-
invasive procedure, the
degree of invasiveness dictated, in part, by the tissue to be harvested.
Candidate tissues are
preferably those capable of transfection and production of a protein, and that
are capable of
survival in the transplanted environment.
In one aspect of this embodiment, tissue is harvested from the eye and it is
contemplated that any
tissue in the eye may be harvested in any feasible manner. For example,
conjunctival fibroblasts
can be excised from the eye by, for example, anesthetizing the conjunctiva
with a topical agent
such as propraracaine, cleansing and preparing the area with betadine or
another cidal agent, and
then taking a snip biopsy with a pair of toothed forceps and Wescott scissors.
Subconjunctival
anesthesia may be preferred by some surgeons or patients. The excised
conjunctiva or other
tissue is removed and then transfected either in the operating room or in an
adjacent area then
CA 02620294 2008-02-14
WO 2007/022403 PCT/US2006/032249
reimplanted in the appropriate location in the same session. Alternatively the
tissue can be
maintained under sterile conditions, taken to a sterile facility where
transfection and subsequent
subculture and testing can be perfonned, and reimplantation of the tissue
performed one to three
weeks later. A similar procedure can be performed on the sclera, except it may
be preferred to
use subconjunctival rather than topical anesthesia. In some instances
alternative tissue substrates
such as iris pigment epithelium may be substituted for conjunctiva or sclera.
Although a tissue
sainple of any size or dimension can be removed, typically a tissue sample of
approxiinately one
cubic millimeter of tissue or less is obtained. After removal of the tissue,
the site can sutured or
treated as needed.
In an alternative einbodiment, the tissue is harvested from a donor, rather
than the patient. In
this case, donor tissue would be isolated and transfected as described above
for autologous
transplantation. It may be transplanted after transfection in the same
session, or, alternatively the
tissue can be maintained under sterile conditions, taken to a sterile facility
where transfection and
subsequent subculture and testing can be performed, and reimplantation of the
tissue performed
one to three weeks later. In this case, donor tissue may be tested to
determine suitability of
transplantation, for example for viral or other pathogens or
irmnunocompatibility with recipient.
Nucleic Acids
Harvested cells or tissues, cell lines made from these cells or tissues, or
standard cell lines are
genetically modified according to the present invention with a nucleic acid as
described above.
The nucleic acid inay encode, for example, a therapeutic protein or an RNAi
cassette, such as a
shRNA. Alternatively, the nucleic acid may be used to repair or replace an
endogenous gene, for
example DNA used for homologous recombination, or an oligonucleotide used for
gene repair.
Modifications include, for exainple, modifying expression levels of the gene
and/or replacing a
mutant gene with a wild-type copy of the gene. The nucleic acid may be DNA or
RNA, but is
11
CA 02620294 2008-02-14
WO 2007/022403 PCT/US2006/032249
preferably DNA. Also preferably, the nucleic acid is a DNA construct, in
particular a cDNA or
synthetic DNA, and can be further modified to improve transcription and/or
translation in the
host cell, or to reduce or minunize gene silencing. The nucleic acid construct
may coinprise,
operably linlced, a promoter region, a nucleotide, and optionally, a
termination signal.
Preferably, this construct is part of a plasmid. Preferably, the cells or
tissue are stably transfected
so that the transplanted cells or tissue may act, for example, as a bio-
factory to produce a
therapeutic protein for a long period of time.
Multiple nucleic acid sequences can be introduced into the cells or tissue,
including multiple
copies of the saine nucleic acid sequence and/or multiple copies of differing
nucleic acid
sequences encoding for different therapeutic or marker proteins. In one
embodiment, each
nucleic acid sequence is present on a separate polynucleotide construct,
plasmid, or vector. In
another embodiment, both nucleic acid sequences are present on one
polynucleotide construct,
plasmid, or vector, with each sequence under the control of a separate
promoter. Alternatively,
and in yet another embodiment, both nucleic acid sequences are present on one
polynucleotide
construct, plasmid, or vector, with the polynucleotide structured so that it
is bicistronic and
where both nucleic acid sequences are under the control of a single promoter.
These various
embodiments are further described below.
With respect to the embodiments where two differing nucleic acid sequences are
present on one
polynucleotide construct, plasmid, or vector, each sequence can be under the
control of a
separate promoter or can be under the control of a single promoter. In
addition to a first nucleic
acid sequence encoding for a selected therapeutic protein, in this embodiment,
a second nucleic
acid sequence encoding, for example, a second therapeutic protein or a marker
is included in the
construct. Expression of this gene may be constitutive; in the case of a
selectable marker this
may be useful for selecting successfully transfected cells or for selecting
cells or transfected
12
CA 02620294 2008-02-14
WO 2007/022403 PCT/US2006/032249
populations of cells that are producing particularly high levels or optimal
therapeutic levels of
the protein. It will also be appreciated that a selectable marker may be used
to provide a means
for enriching for transfected cells or positively selecting for those cells
which have been
transfected, before reintroducing the cells into the patient, as will be
described below.
5-
Markers may include selectable drug resistance genes, metabolic enzyme genes,
fluorescent
proteins, bioluminescent proteins, or any other markers known in the art.
Exeinplary fluorescent
proteins include, but are not limited to: green fluorescent protein, cyan
fluorescent protein,
yellow fluorescent protein, DsRed fluorescent protein, AsRed fluorescent
protein, HcRed
fluorescent protein, and maxFP-green protein. When a marker gene is included
in the vector
construct, it will be appreciated that the marker can be used to quantify the
amount of
fluorescence after transfection and/or before transplantation and/or after
transplantation.
Quantitative determination of fluorescence can be undertaken after
transfection but before
transplanting the tissue using, for example, fluorescence microscopy, flow
cytometry, or
fluorescence-activated cell sorting (FACS) analysis, in order to quantify the
expression of
fluorescence markers ex vivo. After transplanting the tissue, in vivo
monitoring of the extent of
fluorescence, as a measure of production of the therapeutic protein, can be
done by examining
the patient with a fluorescent ophthalmoscope or a surgical microscope
equipped for
fluorescence imaging, and can be documented with a CCD camera. It will be
appreciated that
the marker gene can be used to indicate levels of transgene expression and can
be monitored by a
non-invasive or a minimally invasive procedure. If marker gene expression
decreases, another
tissue implant can be inserted into the patient to increase the level of
therapeutic protein. By
using a marker gene, diminished expression of the therapeutic protein can be
recognized early,
rather than waiting until decreased levels of the therapeutic gene lead to
disease progression.
13
CA 02620294 2008-02-14
WO 2007/022403 PCT/US2006/032249
It will be evident that for many gene therapy applications, selection for
expression of a marker
gene may not be possible or necessary. Also, it is possible that for in vivo
applications, vectors
without any internal promoters may be preferable. Single transcription unit
vectors, wliich may
be bi-cistronic or poly-cistronic, coding for one or two or more therapeutic
genes, may be
designed.
Where two or more genes are present and under transcriptional control of a
single promoter,
there may be an internal ribosome entry site (IRES), e.g. from picornaviral
RNA, to allow both
genes to be separately translated from a single transcript. Retroviruses
incorporating IRES
sequences are known in the art, for example in U.S. Patent No. 5,665,567.
Briefly, in bicistronic
or multicistronic vectors, the individual reading frames of the gene segments
encoding the
proteins lie on the transcription unit (expression unit). Expression of each
cistron is effected
using a single promoter, in conjunction with a specific nucleic acid sequence,
typically
untranslated regions of individual picorna viruses, e.g. poliovirus or
encephalomyocarditis virus,
or a cellular protein, e.g. BiP. In the picorna viruses, a short segment of
the 5' untranslated
region, the so-called IRES (internal ribosomal entry site) functions as an
initiator for translation
of reading frames.
By way of a specific example, and with reference to FIG. 7, the cells or
tissue can be transfected
with a plasmid having one promoter that drives the expression of a first
therapeutic protein, such
as pigment epithelium-derived factor (PEDF), and of a selectable marker, such
as a fluorescent
protein like enhanced green fluorescent protein (eGFP) under control of a
cytomegalovirus
(CMV) promoter. The CMV promoter is positioned at the 5' end of the construct.
Downstream
of the 3' end of the CMV promoter is the PEDF nucleotide sequence that encodes
for PEDF
protein. In the 3' direction of PEDF is an IRES site, which is designed to
allow translation of
14
CA 02620294 2008-02-14
WO 2007/022403 PCT/US2006/032249
inultiple genes on an inRNA transcript. Following the IRES site in the 3'
direction is the eGFP
coding sequence. The IRES will allow translation of eGFP as well as
translation of PEDF.
The promoter region of the construct can be chosen from ainong all promoter
regions that are
functional in maininalian cells, in particular human cells. The promoter can
be a strong or weak
promoter, a constitutive or a regulated/inducible promoter, a ubiquitous or
selective promoter.
The promoter can be of different origin such as cellular, viral, artificial,
and the like. Particular
types of promoters are house-keeping promoters, i.e., promoters from cellular
genes expressed in
inainmalian tissues or cells, or viral promoters (CMV, LTR, SV40, etc.).
Furthermore, the
promoter region can be modified artificially to include enhancer element(s),
inducibility
eleinent(s) and the like. The promoter, secretion and termination region
sequences can be
selected and adapted by the skilled artisan based on the polypeptide, the
pathology, the vector
used, etc. In this regard, the nucleic acid construct can be inserted into
various kinds of vectors
such as plasmids, episomes, artificial chromosomes and the like.
The nucleic acid construct can optionally include a secretion signal,
positioned between the
promoter and coding regions, which allows, or facilitates, the secretion of
the polypeptide
outside of the cells. The secretion signal may be homologous with respect to
the polypeptide
(i.e., from the same gene) or heterologous thereto (i.e., from any other gene
encoding a secreted
polypeptide, in particular a mammalian gene, or artificial). Examples of
secretion signals
include the signal peptide of vascular endothelial growth factor (VEGF), pre
pro nerve growth
sequence (NGS), and the like.
Various approaches may be used to achieve long-term expression of the nucleic
acid in the cells
or tissue. One approach involves a circular vector carrying a recoinbination
site and the
polynucleotide sequence encoding for the therapeutic protein, shRNA, etc., and
the transfection
CA 02620294 2008-02-14
WO 2007/022403 PCT/US2006/032249
is accompanied by introduction of a recoinbinase that facilitates
recombination between the
vector's recombination site and a second recombination site in the genome of
the cell being
transfected. Constructs carrying a recoinbination site, such as a phiC31 attB
site, are described,
for example, in U.S. Patent No. 6,632,672, which is incorporated by reference
herein. It will be
appreciated, however, that other means for long-tenn gene expression are
contemplated, such as
the other meinbers of the serine recoinbinase fainily, transposases (e.g.,
"Sleepifzg Beauty"),
DNA mini-circles, plasmids optimized for minimal gene silencing, or the_use of
a stable
extrachroinasoinal vector such as EBV. When using a phiC31 attB recombination
site, the
nucleic acid constructs are comprised of the phiC31 integrase system
(described in U.S. patents
6,632,672 and 6,808,925, which are incorporated by reference herein) to
achieve site-specific
integration into a target genome of interest.
Bacteriophage phi-.C31 integtrase recognizes pseudo-recombination sites
present in eukaryotic
cells. For genetic manipulation of a eukaryotic cell, phiC31 integrase and a
vector carrying a
phiC31 wild-type recombination site are placed into the cell. The wild-type
recombination
sequence aligns itself with a sequence in the eukaryotic cell genome and the
phiC31 integrase
facilitates a recombination that results in integration of a heterologous gene
into the eukaryotic
genome. It is contemplated that any attB site, any attP site, or any pseudo
att site is present on
any nucleotide sequence used to introduce genetic material into the genome of
the harvested or
cultured cells.
Accordingly, in one embodiment, the method of integrating a polynucleotide
sequence into a
genome of a cell comprises introducing into the cell (i) a circular targeting
construct, comprising
a first recoinbination site and a polynucleotide sequence of interest, and
(ii) a phiC31 integrase,
native or, imodified, wherein the genome of the cell coinprises a second
recombination site (i.e. a
16
CA 02620294 2008-02-14
WO 2007/022403 PCT/US2006/032249
pseudo att site) native to the human genome. Recoinbination between the first
and second
recoinbination sites is facilitated by the site-specific integrase.
The therapeutic gene and the attB sequence are preferably introduced into the
target cell as
circular plasmid DNA. The integrase may be introduced into the target cell (i)
as DNA encoding
the integrase on a second plasmid, (ii) mRNA encoding the integrase, or (iii)
in polypeptide
form. Once phiC31 is introduced into the cell, the cell is maintained under
conditions that allow
recoinbination between the first and second recombination sites and the
recombination is
mediated by the phiC31 integrase. The result of the recoinbination is site-
specific integration of
the polynucleotide sequence of interest in the genome of the cell.
By way of a specific example, and with reference again to FIG. 7, a plasmid is
constructed
having a cytomegalovirus (CMV) promoter that drives the expression of a
therapeutic protein,
pigment epithelium-derived factor (PEDF), and as a marker, enhanced green
fluorescent protein
(eGFP). In the 3' direction of the PEDF nucleotide sequence is an IRES site,
followed in the 3'
direction by the eGFP coding sequence. The IRES allows translation of eGFP as
well as
translation of PEDF. The plasmid, which also includes an attB nucleic acid
sequence, is detailed
in Example 1 and the plasmid sequence is identified herein as SEQ ID NO: 1.
Transfection of a wide variety of genes encoding for therapeutic proteins is
contemplated, and
preferred candidate genes include genes that encode for diffusible proteins
that act
extracellularly to have a therapeutic effect. In a preferred embodiment, a
nucleic acid sequence
coding for a protein with anti-angiogenic activity or with neurotrophic
activity is transfected into
human cells. Exemplary proteins include, but are not limited to, pigment
epithelium-derived
factor (PEDF), truncated soluble VEGF receptor sFlt-1, truncated soluble VEGF
receptor sFlk-1,
VEGFR-1, VEGFR-2, angiostatin, endostatin, tissue inhibitor of metalloprotease
3 (TIMP-3),
17
CA 02620294 2008-02-14
WO 2007/022403 PCT/US2006/032249
ExTek, ciliary neurotrophic factor (CNTF), brain-derived neurotrophic factor
(BDNF), bone
morphogenetic protein 4 (BMP4), alpha fibroblast growth factor (aFGF), beta
fibroblast growth
factor (bFGF), and any protein having activity on or within the compliment
factor H pathway.
Preferred biologically active polypeptides exhibit neurotrophic and/or anti-
angiogenic activity.
The most preferred biologically active polypeptides are autogenic and thus do
not invoke an
iimnune response in the subject or are known in the art not to invoke an
iininune response.
In a preferred embodiment, human cells are genetically modified to contain a
recombinant
nucleic acid construct that directs the cells to produce the therapeutic
protein encoded by the
recombinant nucleic acid. The cells can be immediately transplanted into the
subject or can be
cultured in vitro for a period of time. In a preferred embodiment, mammalian
cells modified
with a vector containing at least one nucleic acid sequence coding for a
therapeutic protein and
another nucleic acid sequence coding for a marker gene are prepared for
transplantation. When
the cells are cultured in vitro prior to transplantation, a selection step can
be performed in order
to isolate the cells that effectively contain the recombinant nucleic acid
construct and express the
polypeptide. The selection step will depend in part on the marker gene and can
involve
measuring fluorescence, screening for antibiotic resistance, or the like.
Cells expressing the
marker gene are selected for transplantation. In general, when the cells are
cultured for a period
of time after transfection, the treatment method is performed on a subject
over more than one
visit to the medical provider. In a first visit, the tissue is harvested. The
tissue cells are
transfected and cultured in vitro, during which time the level of expression
can be monitored and
stably-transfected cells from the tissue selected, by, for example,
quantifying expression of a
marker or of the desired protein by methods noted above for measuring marker
expression, for
transplantation. The subject returns to the medical provider for a second
visit during which the
transfected tissue is transplanted.
18
CA 02620294 2008-02-14
WO 2007/022403 PCT/US2006/032249
Alternatively, tissue can be obtained, transfected, and transplanted during a
single patient visit to
a medical provider. In this scenario, the level of expression of a marker or
the desired
therapeutic protein can be monitored in vivo, by methods mentioned above, such
as
ophthalmoscope or a surgical microscope.
In a preferred einbodiment, one or more nucleotide sequences coding for a
therapeutic protein
and one nucleotide sequence coding for a marker gene are present in the saine
polynucleotide
vector construct. The marker gene is coupled to the therapeutic gene by an
IRES sequence.
Quantification of the degree of fluorescence emitted from a cell or group of
clonal cells would
correlate with the amount of expression of the therapeutic protein, enabling
selection of stably
transfected cells or monitoring of protein expression after transplantation.
Transplantation
The transfected tissue or cells can be transplanted into the subject in any of
a number of different
iinplantation sites in or near the eye by a provider of medical care. FIG. 8
is a diagram showing
an eye 800 in cross-sectional view, and indicating some of the preferred sites
for placing
genetically modified cells or tissue into the patient. Identified anatomical
features are retina 830,
sclera 840, optic nerve 850, cornea 860, pupil 870 and iris 880. Sites in eye
800 preferred for
implanting the transfected cells or tissue include the vitreous humor 810,
near the pars plana 820,
near the posterior retina 832, or sub-sclerally 842. Other sites for
implanting tissue, which are
not specifically indicated in FIG. 8, include the choroid, retinal pigment
epithelium (RPE), and
near the macula epi-retinally, sub-retinally, or intra-retinally.
In a preferred embodiment, the transfected cells or tissue are implanted into
the subject in the
absence of an encapsulating member, such as a polymer capsule or a so-called
"cage". Especially
in the case where the method described herein employs autologous tissue or
cells, encapsulation
19
CA 02620294 2008-02-14
WO 2007/022403 PCT/US2006/032249
of the tissue or cells within a cage is not necessary for immunosuppression.
However,
encapsulation could be used to enhance graft survival and/or to reduce
possible splintering of
cells away from the graft to other sites in the eye. A number of cage designs
have been proposed
for ophthalmologic use for various purposes, as described in U.S. Patent Nos.
6,500,449 and
6,663,894. The cage would be able to house the tissue or cell transplant and
would have pores
large enough for proteins to diffuse out, but small enough so that cells could
not enter or leave.
The cage may contain a matrix or other materials to support cell survival and
cell anchoring to
prevent cell migration to other sites.
EXAMPLES
The following exainples are illustrative in nature and are in no way intended
to be limiting.
Example 1: Construction of a plasmid for transfection
The plasmid shown in FIG. 7 contains the sequence SEQ ID NO: 1. SEQ ID NO: 1
includes a
cytomegalovirus (CMV) promoter (1-589 bp), a nucleotide sequence encoding for
pigment
epithelium-derived factor (PEDF; 590-2131 bp), an internal ribosome entry site
(IRES) coding
sequence (b2151-2735 bp), and a nucleotide sequence encoding for enhanced
green fluorescent
protein (eGFP; bp 2739-3455), an sv4O polyA sequence (3612-3662 bp), a phi C31
attB site
(3952-4245 bp), a bacterial kan promoter (4541-4576 bp), SV40 origin and
promoter enhancer
(4653-4955 bp), neo for G418 selection (5004-5798 bp), and an pUC origin (6383-
7026 bp).
To make this plasmid, begin with vector pIRES-EGFP, commercially available
from Clontech.
Cut the vector with the restriction enzyme BsaI (New England Biolabs) to
linearize the vector,
make blunt ends (e.g., using DNA Polymerase I, Large (Klenow) Fragment, New
England
Biolabs), and treat with a phosphatase to remove the phosphate groups (e.g.,
using calf intestinal
phosphatase, New England Biolabs). Ligate this vector to the fragment
containing attB when
CA 02620294 2008-02-14
WO 2007/022403 PCT/US2006/032249
pTA-attB+ is cleaved with EcoRI and then its ends blunted, to fonn the plasmid
pIRES-EGFP-
attB.
In the second cloning step, use PCR ainplification with primers designed to
amplify the PEDF
gene from human cDNA. Cleave pIRES-EGFP-attB with the restriction enzyme
Srnal, which
linearizes the plasmid upstream of the IRES sequence and use a phosphatase to
remove the
phosphate groups. Ligate the PCR-ainplified fragment into the vector to forin
the plasmid
pPEDF-IRES-GFP-attB, shown in FIG. 7. '
Example 2: Transfection of conjunctival tissue with luciferase gene
A study was conducted in support of the inethod described herein, where a
luciferase marker
gene was transfected into conjunctiva tissue. Conjunctival tissue was
explanted from adult New
Zealand White rabbits and placed in tissue culture dishes. All samples were
placed in 1 mL
phosphate buffered saline solution with 100 micrograms of plasmid DNA encoding
the
luciferase gene under a CMV promoter. All samples were cultured in Dulbecco's
Modified
Eagle Medium (DMEM) plus 10% serum and antibiotic/antimicotic for 24 hours
after
transfection. Samples were then treated with luciferin substrate (150
micrograms luciferin per
ml medium) and imaged using the IVIS-200 system (Xenogen Corp.).
The conjunctival tissue, which contained conjunctival fibroblasts, was
transfected using electron-
avalanche mediated transfection with a luciferase inarker gene. A control
sample of tissue was
contacted with the luciferase gene in the absence of electron-avalanche
mediated transfection.
Twenty-four hours after transfection, bioluininescence was measured. As shown
in FIG. 9, the
tissue transfected with electron-avalanche mediated transfection emitted 2.2 x
105 photons/sec,
two orders of magnitude higher than the cells transfected in the absence of
the electron-
21
CA 02620294 2008-02-14
WO 2007/022403 PCT/US2006/032249
avalanche mediated transfection (4.6 x 103 photons/sec). Background emission
was measured at
3.7 x 103 photons/sec.
Example 3: Comparison of electron avalanclie versus traditional
electroporation in DNA
transfer
Because electroporation protocols vary for different tissues, experiments were
first conducted to
determine the optimal protocol for transfecting CAM from a developing chicken
egg using
traditional electroporation. CAM is a live, readily available, and inexpensive
tissue. Its epithelial
layer is unifonn and has high resistance, inalcing it a good model for
epithelial cell layers, such
as retinal pigment epithelium. In this model system, 100 g of pNBL2 plasmid
DNA encoding
the luciferase gene was pipetted onto the CAM, and pulses were applied.
Specifically, a 250- s,
150-V phase, followed by a 5-ms, 5-V phase in the same polarity was applied.
Optimal results
were achieved with 50 cycles applied at 1 Hz. The tissue was then cultured and
assayed for
luciferase bioluininescence. Luciferase expression using this method was about
104 photons/s.
For electron-avalanche transfection, a 50- m wire microelectrode 1 mm in
length was used to
apply a series of symmetric biphasic pulses, with each phase 250 s in
duration and 600 V in
amplitude. The microelectrode was scanned over a 4-mm2 area, and approximately
50 pulses
were applied. As shown in FIG. 10, the resultant luciferase expression was
about 109 photons/s,
1 0,000-fold higher than levels seen with conventional electroporation.
As one of ordinary skill in the art will appreciate, various changes,
substitutions, and alterations
could be made or otherwise impleinented without departing from the principles
of the present
invention. Accordingly, the scope of the invention should be determined by the
following claims
and their legal equivalents.
22
CA 02620294 2008-02-14
WO 2007/022403 PCT/US2006/032249
SEQUENCE LISTING
<110> Chalberg, Thomas W
Huie, Philip
Marmor, Michael F
Calos, Michele P
Blumenkranz, Mark S
Palanker, Daniel V
Vankov, Alexander B
<120> Ocular Gene Therapy Using Avalanche-Mediated Transfection
<130> S05-139
<150> 60/708,486
<151> 2005-08-15
<150> 11/360,984
<151> 2006-02-22
<160> 1
<170> PatentIn version 3.3
<210> 1
<211> 7166
<212> DNA
<213> Artificial
<220>
<223> Nucleotide sequence for the circular construct shown in FIG. 7
<400> 1
tagttattaa tagtaatcaa ttacggggtc attagttcat agcccatata tggagttccg 60
cgttacataa cttacggtaa atggcccgcc tggctgaccg cccaacgacc cccgcccatt 120
gacgtcaata atgacgtatg ttcccatagt aacgccaata gggactttcc attgacgtca 180
atgggtggag tatttacggt aaactgccca cttggcagta catcaagtgt atcatatgcc 240
aagtacgccc cctattgacg tcaatgacgg taaatggccc gcctggcatt atgcccagta 300
catgacctta tgggactttc ctacttggca gtacatctac gtattagtca tcgctattac 360
catggtgatg cggttttggc agtacatcaa tgggcgtgga tagcggtttg actcacgggg 420
CA 02620294 2008-02-14
WO 2007/022403 PCT/US2006/032249
atttccaagt ctccacccca ttgacgtcaa tgggagtttg ttttggcacc aaaatcaacg 480
ggactttcca aaatgtcgta acaactccgc cccattgacg caaatgggcg gtaggcgtgt 540
acggtgggag gtctatataa gcagagctgg tttagtgaac cgtcagatcc gctagcgcta 600
ccggactcag atctcgagct caagcttcga attctgcagt cgacggtacc gcgggcccgg 660
tcgctttaag aaaggagtag ctgtaatctg aagcctgctg gacgctggat tagaaggcag 720
caaaaaaagc tctgtgctgg ctggagcccc ctcagtgtgc aggcttagag ggactaggct 780
gggtgtggag ctgcagcgta tccacaggcc ccaggatgca ggccctggtg ctactcctct 840
gcattggagc cctcctcggg cacagcagct gccagaaccc tgccagcccc ccggaggagg 900
gctccccaga ccccgacagc acaggggcgc tggtggagga ggaggatcct ttcttcaaag 960
tccccgtgaa caagctggca gcggctgtct ccaacttcgg ctatgacctg taccgggtgc 1020
gatccagcac gagccccacg accaacgtgc tcctgtctcc tctcagtgtg gccacggccc 1080
tctcggccct ctcgctggga gcggagcagc gaacagaatc catcattcac cgggctctct 1140
actatgactt gatcagcagc ccagacatcc atggtaccta toaggagctc cttgacacgg 1200
tcactgcccc ccagaagaac ctcaagagtg cctcccggat cgtctttgag aagaagctgc 1260
gcataaaatc cagctttgtg gcacctctgg aaaagtcata tgggaccagg cccagagtcc 1320
tgacgggcaa ccctcgcttg gacctgcaag agatcaacaa ctgggtgcag gcgcagatga 1380
aagggaagct cgccaggtcc acaaaggaaa ttcccgatga gatcagcatt ctccttctcg 1440
gtgtggcgca cttcaagggg cagtgggtaa caaagtttga ctccagaaag acttccctcg 1500
aggatttcta cttggatgaa gagaggaccg tgagggtccc catgatgtcg gaccctaagg 1560
ctgttttacg ctatggcttg gattcagatc tcagctgcaa gattgcccag ctgcccttga 1620
ccggaagcat gagtatcatc ttcttcctgc ccctgaaagt gacccagaat ttgaccttga 1680
tagaggagag cctcacctcc gagttcattc atgacataga ccgagaactg aagaccgtgc 1740
aggcggtcct cactgtcccc aagctgaagc tgagttatga aggcgaagtc accaagtccc 1800
CA 02620294 2008-02-14
WO 2007/022403 PCT/US2006/032249
tgcaggagat gaagctgcaa tccttgtttg attcaccaga ctttagcaag atcacaggca 1860
aacccatcaa gctgactcag gtggaacacc gggctggctt tgagtggaac gaggatgggg 1920
cgggaaccac ccccagccca gggctgcagc ctgcccacct caccttcccg ctggactatc 1980
accttaacca gcctttcatc ttcgtactga gggacacaga cacaggggcc cttctcttca 2040
ttggcaagat tctggacccc aggggcccct aatatcccag tttaatattc caatacccta 2100
gaagaaaacc cgagggacag cagattccac aggacacgaa ggctgcccct gtaaggtttc 2160
aatgcataca ataaaagagc tttatcccta acttctgtta gggatccgcc cctctccctc 2220
ccccccccct aacgttactg gccgaagccg cttggaataa ggccggtgtg cgtttgtcta 2280
totgttattt tccaccatat tgccgtcttt tggcaatgtg agggcccgga aacctggccc 2340
tgtcttcttg acgagcattc ctaggggtct ttcccctctc gccaaaggaa tgcaaggtct 2400
gttgaatgtc gtgaaggaag cagttcctct ggaagcttct tgaagacaaa caacgtctgt 2460
agcgaccctt tgcaggcagc ggaacccccc acctggcgac aggtgcctct gcggccaaaa 2520
gccacgtgta taagatacac ctgcaaaggc ggcacaaccc cagtgccacg ttgtgagttg 2580
gatagttgtg gaaagagtca aatggctctc ctcaagcgta ttcaacaagg ggctgaagga 2640
tgcccagaag gtaccccatt gtatgggatc tgatctgggg cctcggtgca catgctttac 2700
atgtgtttag tcgaggttaa aaaaacgtct aggccccccg aaccacgggg acgtggtttt 2760
cctttgaaaa acacgatgat aatatggcca caaccatggt gagcaagggc gaggagctgt 2820
tcaccggggt ggtgcccatc ctggtcgagc tggacggcga cgtaaacggc cacaagttca 2880
gcgtgtccgg cgagggcgag ggcgatgcca cctacggcaa gctgaccctg aagttcatct 2940
gcaccaccgg caagctgccc gtgccctggc ccaccctcgt gaccaccctg acctacggcg 3000
tgcagtgctt cagccgctac cccgaccaca tgaagcagca cgacttcttc aagtccgcca 3060
tgcccgaagg ctacgtccag gagcgcacca tcttcttcaa ggacgacggc aactacaaga 3120
cccgcgccga ggtgaagttc gagggcgaca ccctggtgaa ccgcatcgag ctgaagggca 3180
CA 02620294 2008-02-14
WO 2007/022403 PCT/US2006/032249
tcgacttcaa ggaggacggc aacatcctgg ggcacaagct ggagtacaac tacaacagcc 3240
acaacgtcta tatcatggcc gacaagcaga agaacggcat caaggtgaac ttcaagatcc 3300
gccacaacat cgaggacggc agcgtgcagc tcgccgacca ctaccagcag aacaccccca 3360
tcggcgacgg ccccgtgctg ctgcccgaca accactocct gagcacccag tccgccctga 3420
gcaaagaccc caacgagaag cgcgatcaca tggtcctgct ggagttcgtg accgccgccg 3480
ggatcactct cggcatggac gagctgtaca agtaaagcgg ccgcgactct agatcataat 3540
cagccatacc acatttgtag aggttttact tgctttaaaa aacctcccac acctccccct 3600
gaacctgaaa cataaaatga atgcaattgt tgttgttaac ttgtttattg cagcttataa 3660
tggttacaaa taaagcaata gcatcacaaa tttcacaaat aaagcatttt tttcactgca 3720
ttctagttgt ggtttgtcca aactcatcaa tgtatcttaa ggcgtaaatt gtaagcgtta 3780
atattttgtt aaaattcgcg ttaaattttt gttaaatcag ctcatttttt aaccaatagg 3840
ccgaaatcgg caaaatccct tataaatcaa aagaatagac cgagataggg ttgagtgttg 3900
ttccagtttg gaacaagagt ccactattaa agaacgtgga ctccaacgtc aaagggcgaa 3960
aaaccgtcta tcagggcgat ggcccactac gtgaaccatc accctaatca agttttttgg 4020
ggtcgaggtg ccgtaaagca ctaaatcgga accctaaagg gagcccccga tttagagctt 4080
gacggggaaa gccggcgaac gtggcgagaa aggaagggaa gaaagcgaaa ggagcgggcg 4140
ctagggcgct ggcaagtgta gcggtcacgc tgcgcgtaac caccacaccc gccgcgctta 4200
atgcgccgct acagggcgcg tcaggtggca cttttcgggg aaatgtgcgc ggaaccccta 4260
tttgtttatt tttctaaata cattcaaata tgtatccgct catgagacaa taaccctgat 4320
aaatgcttca ataatattga aaaaggaaga gtcctgaggc ggaaagaacc agctgtggaa 4380
tgtgtgtcag ttagggtgtg gaaagtcccc aggctcccca gcaggcagaa gtatgcaaag 4440
catgcatctc aattagtcag caaccaggtg tggaaagtcc ccaggctccc cagcaggcag 4500
aagtatgcaa agcatgcatc tcaattagtc agcaaccata gtcccgcccc taactccgcc 4560
CA 02620294 2008-02-14
WO 2007/022403 PCT/US2006/032249
catcccgccc ctaactccgc ccagttccgc ccattctccg ccccatggct gactaatttt 4620
ttttatttat gcagaggccg aggccgcctc ggcctctgag ctattccaga agtagtgagg 4680
aggctttttt ggaggcctag gcttttgcaa agatcgatca agagacagga tgaggatcgt 4740
ttcgcatgat tgaacaagat ggattgcacg caggttctcc ggccgcttgg gtggagaggc 4800
tattcggcta tgactgggca caacagacaa tcggctgctc tgatgccgcc gtgttccggc 4860
tgtcagcgca ggggcgcccg gttctttttg tcaagaccga cctgtccggt gccctgaatg 4920
aactgcaaga cgaggcagcg cggctatcgt ggctggccac gacgggcgtt ccttgcgcag 4980
ctgtgctcga cgttgtcact gaagcgggaa gggactggct gctattgggc gaagtgccgg 5040
ggcaggatct cctgtcatct caccttgctc ctgccgagaa agtatccatc atggctgatg 5100
caatgcggcg gctgcatacg cttgatccgg ctacctgccc attcgaccoc caagcgaaac 5160
atcgcatcga gcgagcacgt actcggatgg aagccggtct tgtcgatcag gatgatctgg 5220
acgaagagca tcaggggctc gcgccagccg aactgttcgc caggctcaag gcgagcatgc 5280
ccgacggcga ggatctcgtc gtgacccatg gcgatgcctg cttgccgaat atcatggtgg 5340
aaaatggccg cttttctgga ttcatcgact gtggccggct gggtgtggcg gaccgctatc 5400
aggacatagc gttggctacc cgtgatattg ctgaagagct tggcggcgaa tgggctgacc 5460
gcttcctcgt gctttacggt atcgccgctc ccgattcgca gcgcatcgcc ttctatcgcc 5520
ttcttgacga gttcttctga gcgggactct ggggttcgaa atgaccgacc aagcgacgcc 5580
caacctgcca tcacgagatt tcgattccac cgccgccttc tatgaaaggt tgggcttcgg 5640
aatcgttttc cgggacgccg gctggatgat cctccagcgc ggggatctca tgctggagtt 5700
cttcgcccac cctaggggga ggctaactga aacacggaag gagacaatac cggaaggaac 5760
ccgcgctatg acggcaataa aaagacagaa taaaacgcac ggtgttgggt cgtttgttca 5820
taaacgcggg gttcggtccc agggctggca ctctgtcgat accccacaat tcggcttggc 5880
tgtcgacatg cccgccgtga ccgtcgagaa cccgctgacg ctgccccgcg tatccgcacc 5940
CA 02620294 2008-02-14
WO 2007/022403 PCT/US2006/032249
cgccgacgcc gtcgcacgtc ccgtgctcac cgtgaccacc gcgcccagcg gtttcgaggg 6000
cgagggcttc ccggtgcgcc gcgcgttcgc cgggatcaac taccgccacc tcgacccgtt 6060
catcatgatg gaccagatgg gtgaggtgga gtacgcgccc ggggagccca agggcacgcc 6120
ctggcacccg caccgcggct tcgagaccgt gacctacatc gtcgacggta cctggaattc 6180
caccgagacc ccattggggc caatacgccc gcgtttcttc cttttcccca ccccaccccc 6240
caagttcggg tgaaggccca gggctcgcag ccaacgtcgg ggcggcaggc cctgccatag 6300
cctcaggtta ctcatatata ctttagattg atttaaaact tcatttttaa tttaaaagga 6360
tctaggtgaa gatccttttt gataatctca tgaccaaaat cccttaacgt gagttttcgt 6420
tccactgagc gtcagacccc gtagaaaaga tcaaaggatc ttcttgagat cctttttttc 6480
tgcgcgtaat ctgctgcttg caaacaaaaa aaccaccgct accagcggtg gtttgtttgc 6540
cggatcaaga gctaccaact ctttttccga aggtaactgg cttcagcaga gcgcagatac 6600
caaatactgt ccttctagtg tagccgtagt taggccacca cttcaagaac tctgtagcac 6660
cgcctacata cctcgctctg ctaatcctgt taccagtggc tgctgccagt ggcgataagt 6720
cgtgtcttac cgggttggac tcaagacgat agttaccgga taaggcgcag cggtcgggct 6780
gaacgggggg ttcgtgcaca cagcccagct tggagcgaac gacctacacc gaactgagat 6840
acctacagcg tgagctatga gaaagcgcca cgcttcccga agggagaaag gcggacaggt 6900
atccggtaag cggcagggtc ggaacaggag agcgcacgag ggagcttcca gggggaaacg 6960
cctggtatct ttatagtcct gtcgggtttc gccacctctg acttgagcgt cgatttttgt 7020
gatgctcgtc aggggggcgg agcctatgga aaaacgccag caacgcggcc tttttacggt 7080
tcctggcctt ttgctggcct tttgctcaca tgttctttcc tgcgttatcc cctgattctg 7140
tggataaccg tattaccgcc atgcat 7166