Note: Descriptions are shown in the official language in which they were submitted.
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
TRANSGENIC PLANT-BASED METHODS FOR PLANT PESTS USING
RNAi
FIELD OF THE INVENTION
The present invention relates generally to genetic control of pest
infestations. More
specifically, the present invention relates to double-stranded RNA recombinant
technologies
for repressing or inhibiting expression of a target coding sequence in a pest.
INTRODUCTION
The environment is replete with pests and numerous methods have attempted to
control pests infestations of plants.
Compositions for controlling microscopic pest
infestations have been provided in the form of antibiotic, antiviral , and
antifungal
compositions. Methods for controlling infestations by larger pests, such as
nematodes, have
typically been in the form of chemical compositions that are applied to the
surfaces on which
pests reside, or administered to infested animals in the form of pellets,
powders, tablets,
pastes, or capsules.
Commercial crops are often the targets of insect attack. Substantial progress
has been
made in the last a few decades towards developing more efficient methods and
compositions
for controlling insect infestations in plants. Chemical pesticides have been
very effective in
eradicating pest infestations. However, there are several disadvantages to
using chemical
pesticidal agents. Not
only are chemical pesticides potentially detrimental to the
environment, but chemical pesticides are not selective and are harmful to
various crops and
non-target fauna. Chemical pesticides persist in the environment and generally
are slow to be
metabolized, if at all. They accumulate in the food chain, and particularly in
the higher
predator species. Accumulation of these chemical pesticidal agents results in
the
development of resistance to the agents and in species higher up the
evolutionary ladder, can
act as mutagens and/or carcinogens to cause irreversible and deleterious
genetic
modifications.
Because of the dangers associated with chemical pesticides, molecular
approaches
have been developed for controlling pest infestations on plants. For example,
Bacillus
thuringiensis (B.t.) bacteria have been commercially available and used as
environmentally
safe and acceptable insecticides for more than thirty years. The decrease in
application of
chemical pesticidal agents has resulted in cleaner soils and cleaner waters
running off of the
soils into the surrounding streams, rivers, ponds and lakes. In addition to
these
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
environmental benefits, there has been a noticeable increase in the numbers of
beneficial
insects in crop fields in which transgenic insect resistant crops are grown
because of the
decrease in the use of chemical insecticidal agents.
RNA Interference (RNAi) provides a potentially powerful tool for controlling
gene
expression because of its specificity of target selection dnd remarkably high
efficiency in
target mRNA suppression. RNAi refers to the process of sequence-specific post-
transcriptional gene silencing mediated by short interfering RNAs (siRNAs)
(Zamore, P. et
al., Cell 101:25-33 (2000); Fire, A. et al., Nature 391:806 (1998); Hamilton
et al., Science
286, 950-951 (1999); Lin et al., Nature 402:128-129 (1999)). While the
mechanics
underlying RNAi are not fully characterized, it is thought that the presence
of dsRNA in a
cell triggers RNAi by activating the ribonuclease III enzyme Dicer ( Zamore,
P. et al., (2000);
Hammond et al., Nature 404, 293 (2000)). Dicer processes the dsRNA into short
pieces
called short interfering RNAs (siRNAs), which are about 21 to about 23
nucleotides long and
comprise about 19 base pair duplexes (Zamore et al., (2000); Elbashir et al.,
Genes Dev., 15,
188 (2001)). Following delivery into cells, the siRNA molecules associate
with an
endonuclease complex, commonly referred to as an RNA-induced silencing complex
(RISC),
which brings together the antisense strand of the siRNA and the cellular mRNA
gene target.
RISC cleaves the mRNA, which is then released and degraded. Importantly, RISC
is then
capable of degrading additional copies of the target mRNA.
Accordingly, the present invention provides methods and compositions for
controlling
pest infestation by repressing, delaying, or otherwise reducing gene
expression within a
particular pest.
SUMMARY OF THE INVENTION
In one aspect, the invention provides an isolated nucleotide sequence
comprising a
nucleic acid sequence set forth in SEQ ID NOs: 1, 3, 5, 7, 9, 11, 13, 15, 17,
19, 21, 23, 49 -
158, 159, 160-163, 168, 173, 178, 183, 188, 193, 198, 203, 208, 215, 220, 225,
230, 240-247,
249, 251, 253, 255, 257, 259, 275-472, 473, 478, 483, 488, 493, 498, 503, 508-
513, 515, 517,
519, 521, 533 - 575, 576, 581, 586, 591, 596, 601, 603, 605, 607, 609, 621 -
767, 768, 773,
778, 783, 788, 793, 795, 797, 799, 801, 813 - 862, 863, 868, 873, 878, 883,
888, 890, 892,
894, 896, 908- 1040, 1041, 1046, 1051, 1056, 1061, 1066-1071, 1073, 1075,
1077, 1079,
1081, 1083, 1085, 1087, 1089, 1091, 1093, 1095, 1097, 1099, 1101, 1103, 1105,
1107, 1109,
1111, 1113, 1161 - 1571, 1572, 1577, 1582, 1587, 1592, 1597, 1602, 1607, 1612,
1617, 1622,
2
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
1627, 1632, 1637, 1642, 1647, 1652, 1657, 1662, 1667, 1672, 1677, 1682, 1684,
1686, 1688,
1690, 1692, 1694, 1696, 1698, 1700, 1702, 1704, 1730 - 2039, 2040, 2045, 2050,
2055,
2060, 2065, 2070, 2075, 2080, 2085, 2090, 2095, 2100, 2102, 2104, 2106, 2108,
2120 -
2338, 2339, 2344, 2349, 2354, 2359, 2364, 2366, 2368, 2370, 2372, 2384 - 2460,
2461,
2466, 2471, 2476, and 2481. In one embodiment, there is provided a double
stranded
ribonucleotide sequence produced from the expression of a polynucleotide
sequence, wherein
ingestion of said ribonucleotide sequence by a plant pest inhibits the growth
of said pest. In a
further embodiment, ingestion of said sequence inhibits expression of a
nucleotide sequence
substantially complementary to said sequence. In another embodiment, a cell
transformed
with the polynucleotide. In yet another embodiment, a plant or plant cell is
transformed with
the polynucleotide. In a a further embodiment, a seed or product is produced
from the
transformed plant. In a still further embodiment, the product is selected from
the group
consisting of food, feed, fiber, paper, meal, protein, starch, flour, silage,
coffee, tea, and oil.
In another aspect, the invention provides a nucleotide sequence having at
least 70%
sequence identity to a nucleic acid sequence set forth in any of SEQ ID NOs:
1, 3, 5, 7, 9, 11,
13, 15, 17, 19, 21, 23, 49- 158, 159, 160-163, 168, 173, 178, 183, 188, 193,
198, 203, 208,
215, 220, 225, 230, 240-247, 249, 251, 253, 255, 257, 259, 275-472, 473, 478,
483, 488, 493,
498, 503, 508-513, 515, 517, 519, 521, 533 - 575, 576, 581, 586, 591, 596,
601, 603, 605,
607, 609, 621 - 767, 768, 773, 778, 783, 788, 793, 795, 797, 799, 801, 813 -
862, 863, 868,
873, 878, 883, 888, 890, 892, 894, 896, 908- 1040, 1041, 1046, 1051, 1056,
1061, 1066-
1071, 1073, 1075, 1077, 1079, 1081, 1083, 1085, 1087, 1089, 1091, 1093, 1095,
1097, 1099,
1101, 1103, 1105, 1107, 1109, 1111, 1113, 1161 - 1571, 1572, 1577, 1582, 1587,
1592, 1597,
1602, 1607, 1612, 1617, 1622, 1627, 1632, 1637, 1642, 1647, 1652, 1657, 1662,
1667, 1672,
1677, 1682, 1684, 1686, 1688, 1690, 1692, 1694, 1696, 1698, 1700, 1702, 1704,
1730 -
2039, 2040, 2045, 2050, 2055, 2060, 2065, 2070, 2075, 2080, 2085, 2090, 2095,
2100, 2102,
2104, 2106, 2108, 2120 - 2338, 2339, 2344, 2349, 2354, 2359, 2364, 2366, 2368,
2370,
2372, 2384 - 2460, 2461, 2466, 2471, 2476, and 2481. In one embodiment, there
is provided
a double stranded ribonucleotide sequence produced from the expression of a
polynucleotide
sequence, wherein ingestion of said ribonucleotide sequence by a plant pest
inhibits the
growth of said pest. In a further embodiment, ingestion of said sequence
inhibits expression
of a nucleotide sequence substantially complementary to said sequence. In
another
embodiment, a cell transformed with the polynucleotide. In yet another
embodiment, a plant
or plant cell is transformed with the polynucleotide. In a a further
embodiment, a seed or
3
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
product is produced from the transformed plant. In a still further embodiment,
the product is
selected from the group consisting of food, feed, fiber, paper, meal, protein,
starch, flour,
silage, coffee, tea, and oil.
In another aspect, the invention provides an ortholog of a gene comprising at
least 17
contiguous nucleotides of any of SEQ ID NOs: 1, 3, 5, 7, 9, 11, 13, 15, 17,
19, 21, 23, 49 -
158, 159, 160, 163, 168, 173, 178, 183, 188, 193, 198, 203, 208, 215, 220,
225, 230, 247,
249, 251, 253, 255, 257, 259, 275-472, 473, 478, 483, 488, 493, 498, 503, 513,
515, 517, 519,
521, 533 - 575, 576, 581, 586, 591, 596, 601, 603, 605, 607, 609, 621 - 767,
768, 773, 778,
783, 788, 793, 795, 797, 799, 801, 813 - 862, 863, 868, 873, 878, 883, 888,
890, 892, 894,
896, 908- 1040, 1041, 1046, 1051, 1056, 1061, 1071, 1073, 1075, 1077, 1079,
1081, 1083,
1085, 1087, 1089, 1091, 1093, 1095, 1097, 1099, 1101, 1103, 1105, 1107, 1109,
1111, 1113,
1161 - 1571, 1572, 1577, 1582, 1587, 1592, 1597, 1602, 1607, 1612, 1617, 1622,
1627, 1632,
1637, 1642, 1647, 1652, 1657, 1662, 1667, 1672, 1677, 1682, 1684, 1686, 1688,
1690, 1692,
1694, 1696, 1698, 1700, 1702, 1704, 1730 - 2039, 2040, 2045, 2050, 2055, 2060,
2065,
2070, 2075, 2080, 2085, 2090, 2095, 2100, 2102, 2104, 2106, 2108, 2120 - 2338,
2339,
2344, 2349, 2354, 2359, 2364, 2366, 2368, 2370, 2372, 2384 - 2460, 2461, 2466,
2471,
2476, and 2481, or a complement thereof. In one embodiment, there is provided
a double
stranded ribonucleotide sequence produced from the expression of a
polynucleotide
sequence, wherein ingestion of said ribonucleotide sequence by a plant pest
inhibits the
growth of said pest. In a further embodiment, ingestion of said sequence
inhibits expression
of a nucleotide sequence substantially complementary to said sequence. In
another
embodiment, a cell transformed with the polynucleotide. In yet another
embodiment, a plant
or plant cell is transformed with the polynucleotide. In a a further
embodiment, a seed or
product is produced from the transformed plant. In a still further embodiment,
the product is
selected from the group consisting of food, feed, fiber, paper, meal, protein,
starch, flour,
silage, coffee, tea, and oil.
In another aspect, the invention provides a plant comprising a double stranded
ribonucleic acid sequence derived from a pest species. In one embodiment, the
pest is
selected from a group consisting of insects, arachnids, crustaceans, fungi,
bacteria, viruses,
nematodes, flatworms, roundworms, pinworms, hookworms, tapeworms,
trypanosomes,
schistosomes, botflies, fleas, ticks, mites, and lice. In another embodiment,
the plant is
cytoplasmic male steril. In another embodiment, the sequence inhibits a pest
biological
4
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
activity. In another embodiment, the sequence inhibits expression of a target
sequence. In a
further embodiment, the target sequence is an insect, nematode, bacteria, or
fungi sequence.
In another aspect, the invention provides a method for controlling pest
infestation,
comprising providing a pest with plant material comprising a polynucleotide
sequence that
inhibits a pest biological activity. In one embodiment, the polynucleotide
sequence is set
forth in any of SEQ ID NOs: 1, 3, 5, 7, 9, 11, 13, 15, 17, 19, 21, 23, 49 -
158, 159, 160-163,
168, 173, 178, 183, 188, 193, 198, 203, 208, 215, 220, 225, 230, 240-247, 249,
251, 253, 255,
257, 259, 275-472, 473, 478, 483, 488, 493, 498, 503, 508-513, 515, 517, 519,
521, 533 -
575, 576, 581, 586, 591, 596, 601, 603, 605, 607, 609, 621 - 767, 768, 773,
778, 783, 788,
793, 795, 797, 799, 801, 813 - 862, 863, 868, 873, 878, 883, 888, 890, 892,
894, 896, 908-
1040, 1041, 1046, 1051, 1056, 1061, 1066-1071, 1073, 1075, 1077, 1079, 1081,
1083, 1085,
. 1087, 1089, 1091, 1093, 1095, 1097, 1099, 1101, 1103, 1105, 1107, 1109,
1111, 1113, 1161 -
1571, 1572, 1577, 1582, 1587, 1592, 1597, 1602, 1607, 1612, 1617, 162, 1627,
1632, 1637,
1642, 1647, 1652, 1657, 1662, 1667, 1672, 1677, 1682, 1684, 1686, 1688, 1690,
1692, 1694,
1696, 1698, 1700, 1702, 1704, 1730 - 2039, 2040, 2045, 2050, 2055, 2060, 2065,
2070,
2075, 2080, 2085, 2090, 2095, 2100, 2102, 2104, 2106, 2108, 2120 - 2338, 2339,
2344,
2349, 2354, 2359, 2364, 2366, 2368, 2370, 2372, 2384 - 2460, 2461, 2466, 2471,
2476, and
2481, or a complement thereof.
In another aspect, the invention provides a pesticide comprising a plant
expressing a
target polynucleotide sequence.
In another aspect, the invention provides a method for controlling pest
infestation,
comprising: (a) identifying a target sequence in a pest; (b) introducing said
sequence into a
plant; and (c) providing said plant, or portion thereof, to said pest.
In another aspect, the invention provides a method for controlling pest
infestation,
comprising: (a) identifying a target sequence in a first pest species; (b)
searching for an
orthologous target sequence in a second pest species; (c) introducing said
orthologous
sequence into a plant; and (d) providing said plant, or portion thereof, to
said second pest. In
another embodiment, the target is a gene from L. decemlineata and said plant
is selected from
the group consisting of acacia, alfalfa, apple, apricot, artichoke, ash tree,
asparagus, avocado,
banana, barley, beans, beet, birch, beech, blackberry, blueberry, broccoli,
brussels sprouts,
cabbage, canola, cantaloupe, carrot, cassava, cauliflower, cedar, a cereal,
celery, chestnut,
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
cherry, Chinese cabbage, citrus, Clementine, clover, coffee, cotton, cowpea,
cucumber,
cypress, eggplant, elm, endive, eucalyptus, fennel, figs, fir, geranium,
grape, grapefruit,
groundnuts, ground cherry, gum hemlock, hickory, kale, kiwifruit, kohlrabi,
larch, lettuce,
leek, lemon, lime, locust, pine, maidenhair, maize, mango, maple, melon,
millet, mushroom,
mustard, nuts, oak, oats, okra, onion, orange, an ornamental plant or flower
or tree, papaya,
palm, parsley, parsnip, pea, peach, peanut, pear, peat, pepper, persimmon,
pigeon pea, pine,
pineapple, plantain, plum, pomegranate, potato, pumpkin, radicchio, radish,
rapeseed,
raspberry, rice, rye, sorghum,sallow, spinach, spruce, squash, strawberry,
sugarbeet,
sugarcane, sunflower, sweet potato, sweet corn, tangerine, tea, tobacco,
tomato, trees,
triticale, turf grasses, turnips, a vine, walnut, watercress, watermelon,
wheat, yams, yew, and
zucchini.
In another aspect, the invention provides a method for improving crop yield,
comprising:(a) introducing a polynucleotide into a plant; and (b) cultivating
said plant to
allow polynucleotide expression, wherein said expression inhibits feeding by a
pest and loss
of yield due to pest infestation. In one embodiment, the pest is selected from
the group
consisting of insects, nematodes, and fungi. In another embodiment,
polynucleotide
expression produces an RNA molecule that suppresses a target gene in an insect
pest that has
ingested a portion of said crop plant, wherein said target gene performs at
least one essential
function selected from the group consisting of feeding by the pest, viability
of the pest, pest
cell apoptosis, differentiation and development of the pest or any pest cell,
sexual
reproduction by the pest, muscle formation, muscle twitching, muscle
contraction, juvenile
hormone formation and/or reduction, juvenile hormone regulation, ion
regulation and
transport, maintenance of cell membrane potential, amino acid biosynthesis,
amino acid
degradation, sperm formation, pheromone synthesis, pheromone sensing, antennae
formation,
wing formation, leg formation, egg formation, larval maturation, digestive
enzyme formation,
haemolymph synthesis, haemolymph maintenance, neurotransmission, larval stage
transition,
pupation, emergence from pupation, cell division, energy metabolism,
respiration,
cytoskeletal structure synthesis and maintenance, nucleotide metabolism,
nitrogen
metabolism, water use, water retention, and sensory perception.
In another aspect, the invention provides a method for producing a commodity
product, comprising: (a) identifying a target sequence in a pest; (b)
introducing said sequence
into a plant cell; (c) growing said plant cell under conditions suitable for
generating a plant;
6
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
and (d) producing a commodity product from said plant or part thereof. In
another
embodiment, the target is a gene from L. decendineata and said plant is
selected from the
group consisting of acacia, alfalfa, apple, apricot, artichoke, ash tree,
asparagus, avocado,
banana, barley, beans, beet, birch, beech, blackberry, blueberry, broccoli,
brussels sprouts,
cabbage, canola, cantaloupe, carrot, cassava, cauliflower, cedar, a cereal,
celery, chestnut,
cherry, Chinese cabbage, citrus, Clementine, clover, coffee, cotton, cowpea,
cucumber,
cypress, eggplant, elm, endive, eucalyptus, fennel, figs, fir, geranium,
grape, grapefruit,
groundnuts, ground cherry, gum hemlock, hickory, kale, kiwifruit, kohlrabi,
larch, lettuce,
leek, lemon, lime, locust, pine, maidenhair, maize, mango, maple, melon,
millet, mushroom,
mustard, nuts, oak, oats, okra, onion, orange, an ornamental plant or flower
or tree, papaya,
palm, parsley, parsnip, pea, peach, peanut, pear, peat, pepper, persimmon,
pigeon pea, pine,
pineapple, plantain, plum, pomegranate, potato, pumpkin, radicchio, radish,
rapeseed,
raspberry, rice, rye, sorghum,sallow, spinach, spruce, squash, strawberry,
sugarbeet,
sugarcane, sunflower, sweet potato, sweet corn, tangerine, tea, tobacco,
tomato, trees,
triticale, turf grasses, turnips, a vine, walnut, watercress, watermelon,
wheat, yams, yew, and
zucchini.
In another embodiment, the target is a gene from P. cochleariae and the plant
is
selected from the group consisting of acacia, alfalfa, apple, apricot,
artichoke, ash tree,
asparagus, avocado, banana, barley, beans, beet, birch, beech, blackberry,
blueberry, broccoli,
brussels sprouts, cabbage, canola, cantaloupe, carrot, cassava, cauliflower,
cedar, a cereal,
celery, chestnut, cherry, Chinese cabbage, citrus, clementine, clover, coffee,
cotton, cowpea,
cucumber, cypress, eggplant, elm, endive, eucalyptus, fennel, figs, fir,
geranium, grape,
grapefruit, groundnuts, ground cherry, gum hemlock, hickory, kale, kiwifruit,
kohlrabi, larch,
lettuce, leek, lemon, lime, locust, pine, maidenhair, maize, mango, maple,
melon, millet,
mushroom, mustard, nuts, oak, oats, okra, onion, orange, an ornamental plant
or flower or
tree, papaya, palm, parsley, parsnip, pea, peach, peanut, pear, peat, pepper,
persimmon,
pigeon pea, pine, pineapple, plantain, plum, pomegranate, potato, pumpkin,
radicchio, radish,
rapeseed, raspberry, rice, rye, sorghum,sallow, spinach, spruce, squash,
strawberry,
sugarbeet, sugarcane, sunflower, sweet potato, sweet corn, tangerine, tea,
tobacco, tomato,
trees, triticale, turf grasses, turnips, a vine, walnut, watercress,
watermelon, wheat, yams,
yew, and zucchini.
7
CA 02622660 2008-03-14
WO 2007/074405
PCT/1B2006/004003
In another embodiment, said target is a gene from E. varivetis and the plant
is selected
from the group consisting of acacia, alfalfa, apple, apricot, artichoke, ash
tree, asparagus,
avocado, banana, barley, beans, beet, birch, beech, blackberry, blueberry,
broccoli, brussels
sprouts, cabbage, canola, cantaloupe, carrot, cassava, cauliflower, cedar, a
cereal, celery,
chestnut, cherry, Chinese cabbage, citrus, clementine, clover, coffee, cotton,
cowpea,
cucumber, cypress, eggplant, elm, endive, eucalyptus, fennel, figes, fir,
geranium, grape,
grapefruit, groundnuts, ground cherry, gum hemlock, hickory, kale, kiwifruit,
kohlrabi, larch,
lettuce, leek, lemon, lime, locust, pine, maidenhair, maize, mango, maple,
melon, millet,
mushroom, mustard, nuts, oak, oats, okra, onion, orange, an ornamental plant
or flower or
tree, papaya, palm, parsley, parsnip, pea, peach, peanut, pear, peat, pepper,
persimmon,
pigeon pea, pine, pineapple, plantain, plum, pomegranate, potato, pumpkin,
radicchio, radish,
rapeseed, raspberry, rice, rye, sorghum, sallow, spinach, spruce, squash,
strawberry,
sugarbeet, sugarcane, sunflower, sweet potato, sweet corn, tangerine, tea,
tobacco, tomato,
trees, triticale, turf grasses, turnips, a vine, walnut, watercress,
watermelon, wheat, yams,
yew, and zucchini.
In another embodiment, the target is a gene from A. grandis and the plant is
selected
from the group consisting of acacia, alfalfa, apple, apricot, artichoke, ash
tree, asparagus,
avocado, banana, barley, beans, beet, birch, beech, blackberry, blueberry,
broccoli, brussels
sprouts, cabbage, canola, cantaloupe, carrot, cassava, cauliflower, cedar, a
cereal, celery,
chestnut, cherry, Chinese cabbage, citrus, clementine, clover, coffee, cotton,
cowpea,
cucumber, cypress, eggplant, elm, endive, eucalyptus, fennel, figs, fir,
geranium, grape,
grapefruit, groundnuts, ground cherry, gum hemlock, hickory, kale, kiwifruit,
kohlrabi, larch,
= lettuce, leek, lemon, lime, locust, pine, maidenhair, maize, mango,
maple, melon, millet,
mushroom, mustard, nuts, oak, oats, okra, onion, orange, an ornamental plant
or flower or
tree, papaya, palm, parsley, parsnip, pea, peach, peanut, pear, peat, pepper,
persimmon,
pigeon pea, pine, pineapple, plantain, plum, pomegranate, potato, pumpkin,
radicchio, radish,
rapeseed, raspberry, rice, rye, sorghum, sallow, spinach, spruce, squash,
strawberry,
sugarbeet, sugarcane, sunflower, sweet potato, sweet corn, tangerine, tea,
tobacco, tomato,
trees, triticale, turf grasses, turnips, a vine, walnut, watercress,
watermelon, wheat, yams,
yew, and zucchini.
In another embodiment, the target is a gene from T castaneum and the plant is
selected from the group consisting of acacia, alfalfa, apple, apricot,
artichoke, ash tree,
8
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
asparagus, avocado, banana, barley, beans, beet, birch, beech, blackberry,
blueberry, broccoli,
brussels sprouts, cabbage, canola, cantaloupe, carrot, cassava, cauliflower,
cedar, a cereal,
celery, chestnut, cherry, Chinese cabbage, citrus, clementine, clover, coffee,
cotton, cowpea,
cucumber, cypress, eggplant, elm, endive, eucalyptus, fennel, figs, fir,
geranium, grape,
grapefruit, groundnuts, ground cherry, gum hemlock, hickory, kale, kiwifruit,
kohlrabi, larch,
lettuce, leek, lemon, lime, locust, pine, maidenhair, maize, mango, maple,
melon, millet,
mushroom, mustard, nuts, oak, oats, okra, onion, orange, an ornamental plant
or flower or
tree, papaya, palm, parsley, parsnip, pea, peach, peanut, pear, peat, pepper,
persimmon,
pigeon pea, pine, pineapple, plantain, plum, pomegranate, potato, pumpkin,
radicchio, radish,
rapeseed, raspberry, rice, rye, sorghum, sallow, spinach, spruce, squash,
strawberry,
sugarbeet, sugarcane, sunflower, sweet potato, sweet corn, tangerine, tea,
tobacco, tomato,
trees, triticale, turf grasses, turnips, a vine, walnut, watercress,
watermelon, wheat, yams,
yew, and zucchini.
In another embodimentt, the target is a gene from M persicae and the plant is
selected
from the group consisting of acacia, alfalfa, apple, apricot, artichoke, ash
tree, asparagus,
avocado, banana, barley, beans, beet, birch, beech, blackberry, blueberry,
broccoli, brussels
sprouts, cabbage, canola, cantaloupe, carrot, cassava, cauliflower, cedar, a
cereal, celery,
chestnut, cherry, Chinese cabbage, citrus, Clementine, clover, coffee, cotton,
cowpea,
cucumber, cypress, eggplant, elm, endive, eucalyptus, fennel, figs, fir,
geranium, gape,
grapefruit, groundnuts, ground cherry, gum hemlock, hickory, kale, kiwifruit,
kohlrabi, larch,
lettuce, leek, lemon, lime, locust, pine, maidenhair, maize, mango, maple,
melon, millet,
mushroom, mustard, nuts, oak, oats, okra, onion, orange, an ornamental plant
or flower or
tree, papaya, palm, parsley, parsnip, pea, peach, peanut, pear, peat, pepper,
persimmon,
pigeon pea, pine, pineapple, plantain, plum, pomegranate, potato, pumpkin,
radicchio, radish,
rapeseed, raspberry, rice, rye, sorghum, sallow, spinach, spruce, squash,
strawberry,
sugarbeet, sugarcane, sunflower, sweet potato, sweet corn, tangerine, tea,
tobacco, tomato,
trees, triticale, turf grasses, turnips, a vine, walnut, watercress,
watermelon, wheat, yams,
yew, and zucchini.
In another embodiment, the target is a gene from N. lugens and the plant is
selected
from the group consisting of acacia, alfalfa, apple, apricot, artichoke, ash
tree, asparagus,
avocado, banana, barley, beans, beet, birch, beech, blackberry, blueberry,
broccoli, brussels
sprouts, cabbage, canola, cantaloupe, carrot, cassava, cauliflower, cedar, a
cereal, celery,
9
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
chestnut, cherry, Chinese cabbage, citrus, clementine, clover, coffee, cotton,
cowpea,
cucumber, cypress, eggplant, elm, endive, eucalyptus, fennel, figs, fir,
geranium, grape,
grapefruit, groundnuts, ground cherry, gum hemlock, hickory, kale, kiwifruit,
kohlrabi, larch,
lettuce, leek, lemon, lime, locust, pine, maidenhair, maize, mango, maple,
melon, millet,
mushroom, mustard, nuts, oak, oats, okra, onion, orange, an ornamental plant
or flower or
tree, papaya, palm, parsley, parsnip, pea, peach, peanut, pear, peat, pepper,
persimmon,
pigeon pea, pine, pineapple, plantain, plum, pomegranate, potato, pumpkin,
radicchio, radish,
rapeseed, raspberry, rice, rye, sorghum, sallow, spinach, spruce, squash,
strawberry,
sugarbeet, sugarcane, sunflower, sweet potato, sweet corn, tangerine, tea,
tobacco, tomato,
trees, triticale, turf grasses, turnips, a vine, walnut, watercress,
watermelon, wheat, yams,
yew, and zucchini.
In another embodiment, the target is a gene from C. suppressalis and the plant
is
selected from the group consisting of acacia, alfalfa, apple, apricot,
artichoke, ash tree,
asparagus, avocado, banana, barley, beans, beet, birch, beech, blackberry,
blueberry, broccoli,
brussels sprouts, cabbage, canola, cantaloupe, carrot, cassava, cauliflower,
cedar, a cereal,
celery, chestnut, cherry, Chinese cabbage, citrus, clementine, clover, coffee,
cotton, cowpea,
cucumber, cypress, eggplant, elm, endive, eucalyptus, fennel, figs, fir,
geranium, grape,
grapefruit, groundnuts, ground cherry, gum hemlock, hickory, kale, kiwifruit,
kohlrabi, larch,
lettuce, leek, lemon, lime, locust, pine, maidenhair, maize, mango, maple,
melon, millet,
mushroom, mustard, nuts, oak, oats, okra, onion, orange, an ornamental plant
or flower or
tree, papaya, palm, parsley, parsnip, pea, peach, peanut, pear, peat, pepper,
persimmon,
pigeon pea, pine, pineapple, plantain, plum, pomegranate, potato, pumpkin,
radicchio, radish,
rapeseed, raspberry, rice, rye, sorghum,sallow, spinach, spruce, squash,
strawberry,
sugarbeet, sugarcane, sunflower, sweet potato, sweet corn, tangerine, tea,
tobacco, tomato,
trees, triticale, turf grasses, turnips, a vine, walnut, watercress,
watermelon, wheat, yams,
yew, and zucchini.
In another embodiment, the target is a gene from P. xylostella and the plant
is selected
from the group consisting of acacia, alfalfa, apple, apricot, artichoke, ash
tree, asparagus,
avocado, banana, barley, beans, beet, birch, beech, blackberry, blueberry,
broccoli, brussels
sprouts, cabbage, canola, cantaloupe, carrot, cassava, cauliflower, cedar, a
cereal, celery,
chestnut, cherry, Chinese cabbage, citrus, lementine, clover, coffee, cotton,
cowpea,
cucumber, cypress, eggplant, elm, endive, eucalyptus, fennel, figes, fir,
geranium, gape,
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
grapefruit, groundnuts, ground cherry, gum hemlock, hickory, kale, kiwifruit,
kohlrabi, larch,
lettuce, leek, lemon, lime, locust, pine, maidenhair, maize, mango, maple,
melon, millet,
mushroom, mustard, nuts, oak, oats, okra, onion, orange, an ornamental plant
or flower or
tree, papaya, palm, parsley, parsnip, pea, peach, peanut, pear, peat, pepper,
persimmon,
pigeon pea, pine, pineapple, plantain, plum, pomegranate, potato, pumpkin,
radicchio, radish,
rapeseed, raspberry, rice, rye, sorghum, sallow, spinach, spruce, squash,
strawberry,
sugarbeet, sugarcane, sunflower, sweet potato, sweet corn, tangerine, tea,
tobacco, tomato,
trees, triticale, turf grasses, turnips, a vine, walnut, watercress,
watermelon, wheat, yams,
yew, and zucchini.
In another embodiment, the target is a gene from A. domesticus and the plant
is
selected from the group consisting of acacia, alfalfa, apple, apricot,
artichoke, ash tree,
asparagus, avocado, banana, barley, beans, beet, birch, beech, blackberry,
blueberry, broccoli,
brussels sprouts, cabbage, canola, cantaloupe, carrot, cassava, cauliflower,
cedar, a cereal,
celery, chestnut, cherry, Chinese cabbage, citrus, clementine, clover, coffee,
cotton, cowpea,
cucumber, cypress, eggplant, elm, endive, eucalyptus, fennel, figes, fir,
geranium, grape,
grapefruit, groundnuts, ground cherry, gum hemlock, hickory, kale, kiwifruit,
kohlrabi, larch,
lettuce, leek, lemon, lime, locust, pine, maidenhair, maize, mango, maple,
melon, millet,
mushroom, mustard, nuts, oak, oats, okra, onion, orange, an ornamental plant
or flower or
tree, papaya, palm, parsley, parsnip, pea, peach, peanut, pear, peat, pepper,
persimmon,
pigeon pea, pine, pineapple, plantain, plum, pomegranate, potato, pumpkin,
radicchio, radish,
rapeseed, raspberry, rice, rye, sorghum, sallow, spinach, spruce, squash,
strawberry,
sugarbeet, sugarcane, sunflower, sweet potato, sweet corn, tangerine, tea,
tobacco, tomato,
trees, triticale, turf grasses, turnips, a vine, walnut, watercress,
watermelon, wheat, yams,
yew, and zucchini.
In another embodiment, the target is a gene from a fungus and said plant is
selected
from the group consisting of acacia, alfalfa, apple, apricot, artichoke, ash
tree, asparagus,
avocado, banana, barley, beans, beet, birch, beech, blackberry, blueberry,
broccoli, brussels
sprouts, cabbage, canola, cantaloupe, carrot, cassava, cauliflower, cedar, a
cereal, celery,
chestnut, cherry, Chinese cabbage, citrus, clementine, clover, coffee, cotton,
cowpea,
cucumber, cypress, eggplant, elm, endive, eucalyptus, fennel, figes, fir,
geranium, grape,
grapefruit, groundnuts, ground cherry, gum hemlock, hickory, kale, kiwifruit,
kohlrabi, larch,
lettuce, leek, lemon, lime, locust, pine, maidenhair, maize, mango, maple,
melon, millet,
11
CA 02622660 2014-09-26
mushroom, mustard, nuts, oak, oats, okra, onion, orange, an ornamental plant
or flower or
tree, papaya, palm, parsley, parsnip, pea, peach, peanut, pear, peat, pepper,
persimmon,
pigeon pea, pine, pineapple, plantain, plum, pomegranate, potato, pumpkin,
radicchio, radish,
rapeseed, raspberry, rice, rye, sorghum, sallow, spinach, spruce, squash,
strawberry,
sugarbeet, sugarcane, sunflower, sweet potato, sweet corn, tangerine, tea,
tobacco, tomato,
trees, triticale, turf grasses, turnips, a vine, walnut, watercress,
watermelon, wheat, yams,
yew, and zucchini.
In another embodiment, the invention provides the use of an isolated
nucleotide
sequence, a double stranded ribonucleotide sequence, a cell, a plant, or a
product, for treating
insect infestation of plants.
In another embodiment, the invention provides the use of an isolated
nucleotide
sequence, a double stranded ribonucleotide sequence, a cell, a plant, or a
product, for treating
nematode infestation of plants.
In another embodiment, the invention provides the use of an isolated
nucleotide
sequence, a double stranded ribonucleotide sequence, a cell, a plant, or a
product, for treating
fungal infestation of plants.
In another aspect, the present invention relates to an isolated double
stranded RNA
molecule comprising annealed complementary strands, wherein at least one of
the strands
comprises a polyribonucleotide which is: (i) a polyribonucleotide
complementary to at least
21 contiguous nucleotides of a target gene, the target gene comprising the
nucleotide
sequence of any one of SEQ ID NOs: 3, 795, 890, 1073, 1684, and 2366; (ii) a
polyribonucleotide complementary to at least 21 contiguous nucleotides of a
target gene, the
target gene encoding the amino acid sequence of any one of SEQ ID NOs: 4, 796,
891,
1074, 1685, and 2367; and (iii) a polyribonucleotide having at least 85%
sequence identity
with the polyribonucleotide of (i) or (ii).
In another aspect, the present invention relates to an isolated polynucleotide
encoding the double stranded RNA as defined above. In another aspect, the
present
invention relates to a vector comprising the polynucleotide as defined above.
In another
aspect, the present invention relates to a cell comprising the double stranded
RNA
molecule, the polynucleotide molecule, or the vector as defined above.
12
CA 02622660 2014-09-26
In another aspect, the present invention relates to a product comprising the
double
stranded RNA molecule, the polynucleotide molecule, or the vector as defined
above,
wherein the product is: food, feed, fiber, paper, meal, protein, starch,
flour, silage, coffee,
tea, oil, crushed seed or grain, or silage.
In another aspect, the present invention relates to a method for controlling
pest
infestation, the method comprising providing a pest with plant material
comprising the
double stranded RNA molecule as defined above.
In another aspect, the present invention relates to a method for improving
crop yield,
the method comprising: (a) introducing the polynucleotide molecule as defined
above, or
the vector as defined above; and (b) cultivating the plant to allow expression
of the
polynucleotide molecule or vector, wherein the expression inhibits feeding by
a pest and
loss of crop yield due to pest infestation.
In another aspect, the present invention relates to the use of: (a) the
isolated double
stranded RNA molecule as defined above; (b) the isolated polynucleotide as
defined above;
(c) the vector as defined above; (d) the cell as defined above; or (e) the
product as defined
above, for treating insect infestation of plants or for the manufacture of a
pesticide for same.
In another aspect, the present invention relates to the use of: (a) the
isolated double
stranded RNA molecule as defined above; (b) the isolated polynucleotide as
defined above;
(c) the vector as defined above; (d) the cell as defined above; or (e) the
product as defined
above, for treating nematode infestation of plants or for the manufacture of a
pesticide for
same.
In another aspect, the present invention relates to the use of: (a) the
isolated double
stranded RNA molecule as defined above; (b) the isolated polynucleotide as
defined above;
(c) the vector as defined above; (d) the cell as defined above; or (e) the
product as defined
above, for treating fungal infestation of plants or the for the manufacture of
a pesticide for
same.
In another aspect, the present invention relates to a pesticide comprising:
(a) the
isolated double stranded RNA molecule as defined above; (b) the isolated
polynucleotide as
12a
CA 02622660 2014-09-26
defined above; (c) the vector as defined above; (d) the cell as defined above;
or (e) the
product as defined above, for use in treating insect, nematode, or fungal
infestation of
plants.
BRIEF DESCRIPTION OF THE DRAWINGS
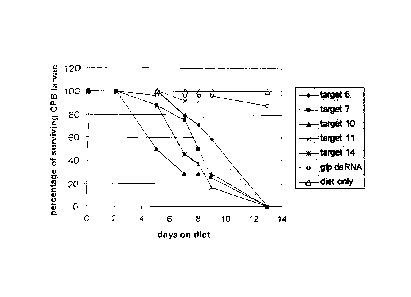
Figure 1-LD: Survival of L. decemlineata on artificial diet treated with
dsRNA.
Insects of the second larval stage were fed diet treated with 50 IA of
topically-applied solution
of dsRNA (targets or gfp control). Diet was replaced with fresh diet
containing topically-
applied dsRNA after 7 days. The number of surviving insects were assessed at
days 2, 5, 7, 8,
9, & 13. The percentage of surviving larvae was calculated relative to day 0
(start of assay).
Target LD006: (SEQ ID NO: 178); Target LD007 (SEQ ID NO: 183); Target LD010
(SEQ
ID NO: 188); Target LD011 (SEQ ID NO: 193); Target LD014 (SEQ ID NO: 198); gfp
dsRNA (SEQ ID NO: 235).
Figure 2-LD: Survival of L. decemlineata on artificial diet treated with
dsRNA.
Insects of the second larval stage were fed diet treated with 50 .1 of
topically-applied solution
of dsRNA (targets or gfp control). Diet was replaced with fresh diet only
after 7 days. The
number of surviving insects was assessed at days 2, 5, 6, 7, 8, 9, 12, & 14.
The percentage of
surviving larvae was calculated relative to day 0 (start of assay). Target
LD001 (SEQ ID NO:
12b
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
163); Target LD002 (SEQ ID NO: 168); Target LD003 (SEQ ID NO: 173); Target
LD015
(SEQ ID NO: 215); Target LD016 (SEQ ID NO: 220); gfp dsRNA (SEQ ID NO: 235).
Figure 3-LD: Average weight of L. decemlineata larvae on potato leaf discs
treated
with dsRNA. Insects of the second larval stage were fed leaf discs treated
with 20 jtl of a
topically-applied solution (10 ng/ 1) of dsRNA (target LD002 or gfp). After
two days the
insects were transferred on to untreated leaves every day.
Figure 4-LD: Survival of L. decemlineata on artificial diet treated with
shorter
versions of target LD014 dsRNA and concatemer dsRNA. Insects of the second
larval stage
were fed diet treated with 50 1 of topically-applied solution of dsRNA (gfp
or targets). The
number of surviving insects were assessed at days 3, 4, 5, 6, & 7. The
percentage of surviving
larvae were calculated relative to day 0 (start of assay).
Figure 5-LD: Survival of L. decemlineata larvae on artificial diet treated
with
different concentrations of dsRNA of target LD002 (a), target LD007 (b),
target LD010 (c),
target LD011 (d), target LD014 (e), target LD015 (f), LD016 (g) and target
LD027 (h).
Insects of the second larval stage were fed diet treated with 50 1 of
topically-applied solution
of dsRNA. Diet was replaced with fresh diet containing topically-applied dsRNA
after 7
days. The number of surviving insects were assessed at regular intervals. The
percentage of
surviving larvae were calculated relative to day 0 (start of assay).
Figure 6-LD. Effects of E. coli strains expressing dsRNA target LD010 on
survival
of larvae of the Colorado potato beetle, Leptinotarsa decemlineata, over time.
The two
bacterial strains were tested in separate artificial diet-based bioassays: (a)
AB309-105; data
points for pGBNJ003 and pGN29 represent average mortality values from 5
different
bacterial clones, (b) BL21(DE3); data points for pGBNJ003 and pGN29 represent
average
mortality values from 5 different and one single bacterial clones,
respectively. Error bars
represent standard deviations.
Figure 7-LD. Effects of different clones of E. coli strains (a) AB309-105 and
(b)
BL21(DE3) expressing dsRNA target LD010 on survival of larvae of the Colorado
potato
beetle, Leptinotarsa decemlineata, 12 days post infestation. Data points are
average mortality
values for each clone for pGN29 and pGBNJ003. Clone 1 of AB309-105 harbouring
plasmid
pGBNJ003 showed 100% mortality towards CPB at this timepoint. Error bars
represent
standard deviations.
13
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
Figure 8-LD. Effects of different clones of E. coli strains (a) AB309-105 and
(b)
BL21(DE3) expressing dsRNA target LD010 on growth and development of larval
survivors
of the Colorado potato beetle, Leptinotarsa decemlineata, 7 days post
infestation. Data points
are % average larval weight values for each clone (one clone for pGN29 and
five clones for
pGBNJ003) based on the data of Table 10. Diet only treatment represents 100%
normal
larval weight.
Figure 9-LD. Survival of larvae of the Colorado potato beetle, Leptinotarsa
decemlineata, on potato plants sprayed by double-stranded RNA-producing
bacteria 7 days
post infestation. Number of larval survivors were counted and expressed in
terms of %
mortality. The bacterial host strain used was the RNaseIII-deficient strain
AB309-105. Insect
gene target was LD010.
Figure 10-LD. Growth/developmental delay of larval survivors of the Colorado
potato beetle, Leptinotarsa decemlineata, fed on potato plants sprayed with
dsRNA-
producing bacteria 11 days post infestation. The bacterial host strain used
was the RNaseIII-
deficient strain AB309-105. Data figures represented as percentage of normal
larval weight;
100 % of normal larval weight given for diet only treatment. Insect gene
target was LD010.
Error bars represent standard deviations.
Figure 11-LD. Resistance to potato damage caused by larvae of the Colorado
potato
beetle, Leptinotarsa decemlineata, by double-stranded RNA-producing bacteria 7
days post
infestation. Left, plant sprayed with 7 units of bacteria AB309-105 containing
the pGN29
plasmid; right, plant sprayed with 7 units of bacteria Ab309-105 containing
the pGBNJ003
plasmid. One unit is defined as the equivalent of 1 ml of a bacterial
suspension at OD value
of 1 at 600 nrn. Insect gene target was LD010.
Figure 12-LD. Survival of L. decemlineata adults on potato leaf discs treated
with
dsRNA. Young adult insects were fed double-stranded-RNA-treated leaf discs for
the first
two days and were then placed on untreated potato foliage. The number of
surviving insects
were assessed regularly; mobile insects were recorded as insects which were
alive and
appeared to move normally; moribund insects were recorded as insects which
were alive but
appeared sick and slow moving ¨ these insects were not able to right
themselves once placed
on their backs. Target LD002 (SEQ ID NO: 168); Target LD010 (SEQ ID NO: 188);
Target
LD014 (SEQ ID NO: 198); Target LD016 (SEQ ID NO: 220); gfp dsRNA (SEQ ID NO:
235).
14
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
Figure 13-LD. Effects of bacterial produced target double-stranded RNA against
larvae of L. decemlineata. Fifty ill of an OD 1 suspension of heat-treated
bacteria expressing
dsRNA (SEQ ID NO: 188) was applied topically onto the solid artificial diet in
each well of a
48-well plate. CPB larvae at L2 stage were placed in each well. At day 7, a
picture was taken
of the CPB larvae in a plate containing (a) diet with bacteria expressing
target 10 double-
stranded RNA, (b) diet with bacteria harbouring the empty vector pGN29, and,
(c) diet only.
Figure 14-LD Effects on CPB larval survival and growth of different amounts of
inactivated E. coil AB309-105 strain harbouring plasmid pGBNJ003 topically
applied to
potato foliage prior to insect infestation. Ten Li larvae were fed treated
potato for 7 days.
Amount of bacterial suspension sprayed on plants: 0.25 U, 0.08 U, 0.025 U,
0.008 U of target
and 0.25 U of pGN29 (negative control; also included is Milli-Q water). One
unit (U) is
defined as the equivalent bacterial amount present in 1 ml of culture with an
optical density
value of 1 at 600nm. A total volume of 1.6 ml was sprayed on to each plant.
Insect gene
target was LD010.
Figure 15-LD Resistance to potato damage caused by CPB larvae by inactivated
E.
coil AB309-105 strain harbouring plasmid pGBNJ003 seven days post infestation.
(a) water,
(b) 0.25 U E. coil AB309-105 harbouring pGN29, (c) 0.025 U E. coil AB309-105
harbouring
pGBNJ003, (d) 0.008 U E. coil AB309-105 harbouring pGBNJ003. One unit (U) is
defined
as the equivalent bacterial amount present in 1 ml of culture with an optical
density value of 1
at 600nm. A total volume of 1.6 ml was sprayed on to each plant. Insect gene
target was
LD010.
Figure 1-PC: Effects of ingested target dsRNAs on survival and growth of P.
cochleariae larvae. Neonate larvae were fed oilseed rape leaf discs treated
with 25 1.d. of
topically-applied solution of 0.1 g/ 1 dsRNA (targets or gfp control). Afer 2
days, the
insects were transferred onto fresh dsRNA-treated leaf discs. At day 4, larvae
from one
replicate for every treatment were collected and placed in a Petri dish
containing fresh
untreated oilseed rape foliage. The insects were assessed at days 2, 4, 7, 9 &
11. (a) Survival
of E. varivestis larvae on oilseed rape leaf discs treated with dsRNA. The
percentage of
surviving larvae was calculated relative to day 0 (start of assay). (b)
Average weights of P.
cochleariae larvae on oilseed rape leaf discs treated with dsRNA. Insects from
each replicate
were weighed together and the average weight per larva determined. Error bars
represent
standard deviations. Target 1: SEQ ID NO: 473; target 3: SEQ ID NO: 478;
target 5: SEQ ID
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
NO: 483 --; target 10: SEQ ID NO: 488; target 14: SEQ ID NO: 493; target 16:
SEQ ID NO:
= 498; target 27: SEQ ID NO: 503; gfp dsRNA: SEQ ID NO: 235.
Figure 2-PC: Survival of P. cochleariae on oilseed rape leaf discs treated
with
different concentrations of dsRNA of (a) target PC010 and (b) target PCO27.
Neonate larvae
were placed on leaf discs treated with 25 W. of topically-applied solution of
dsRNA. Insects
were transferred to fresh treated leaf discs at day 2. At day 4 for target
PC010 and day 5 for
target PCO27, the insects were transferred to untreated leaves. The number of
surviving
insects were assessed at days 2, 4, 7, 8, 9 & 11 for PC010 and 2, 5, 8, 9 & 12
for PCO27. The
percentage of surviving larvae was calculated relative to day 0 (start of
assay).
Figure 3-PC: Effects of E. coli strain AB309-105 expressing dsRNA target PC010
on
survival of larvae of the mustard leaf beetle, P. cochleariae, over time. Data
points for each
treatment represent average mortality values from 3 different replicates.
Error bars represent
standard deviations. Target 10: SEQ ID NO: 488
Figure 1-EV: Survival of E. varivestis larvae on bean leaf discs treated with
dsRNA.
Neonate larvae were fed bean leaf discs treated with 25 ,1 of topically-
applied solution of 1
410 dsRNA (targets or gfp control). Afer 2 days, the insects were transferred
onto fresh
dsRNA-treated leaf discs. At day 4, larvae from one treatment were collected
and placed in a
plastic box containing fresh untreated bean foliage. The insects were assessed
for mortality at
days 2, 4, 6, 8 & 10. The percentage of surviving larvae was calculated
relative to day 0 (start
of assay). Target 5: SEQ ID NO: 576; target 10: SEQ ID NO: 586; target 15: SEQ
ID NO:
591; target 16: SEQ ID NO: 596; gfp dsRNA: SEQ ID NO: 235.
Figure 2-EV: Effects of ingested target dsRNAs on surival of E. varivestis
adults and
resistance to snap bean foliar insect damage. (a) Surivival of E. varivestis
adults on bean leaf
treated with dsRNA. Adults were fed bean leaf discs treated with 75 IA of
topically-applied
solution of 0.1 g/u1 dsRNA (targets or gfp control). After 24 hours, the
insects were
transferred onto fresh dsRNA-treated leaf discs. After a further 24 hours,
adults from one
treatment were collected and placed in a plastic box containing potted fresh
untreated whole
bean plants. The insects were assessed for mortality at days 4, 5, 6, 7, 8, &
11. The
percentage of surviving adults was calculated relative to day 0 (start of
assay). Target 10:
SEQ ID NO: 586; target 15: SEQ ID NO: 591; target 16: SEQ ID NO: 596; gfp
dsRNA: SEQ
ID NO: 235. (b) Resistance to bean foliar damage caused by adults of the E.
varivestis by
16
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
dsRNA. Whole plants containing insects from one treatment (see (a)) were
checked visually
for foliar damage on day 9. (i) target 10; (ii) target 15; (iii) target 16;
(iv) gfp dsRNA; (v)
untreated.
Figure 1-TC: Survival of T. castaneum larvae on artificial diet treated with
dsRNA of
target 14. Neonate larvae were fed diet based on a flour/milk mix with 1 mg
dsRNA target
14. Control was water (without dsRNA) in diet. Four replicates of 10 first
instar larvae per
replicate were performed for each treatment. The insects were assessed for
survival as
average percentage means at days 6, 17, 31, 45 and 60. The percentage of
surviving larvae
was calculated relative to day 0 (start of assay). Error bars represent
standard deviations.
Target TC014: SEQ ID NO: 878.
Figure 1-MP: Effect of ingested target 27 dsRNA on the survival ofMyzus
persicae
nymphs. First instars were placed in feeding chambers containing 50 I of
liquid diet with 2
iug/ 1 dsRNA (target 27 or gfp dsRNA control). Per treatment, 5 feeding
chambers were set
up with 10 instars in each feeding chamber. Number of survivors were assessed
at 8 days post
start of bioassay. Error bars represent standard deviations. Target MP027: SEQ
ID NO: 1061;
gfp dsRNA: SEQ ID NO: 235.
Figure 1-NL: Survival of Nilaparvata lugens on liquid artificial diet treated
with
dsRNA. Nymphs of the first to second larval stage were fed diet supplemented
with 2 mg/ml
solution of dsRNA targets in separate bioassays: (a) NL002, NL003, NL005,
NL010; (b)
NL009, NL016; (c) NL014, NL018; (d) NL013, NL015, NL021. Insect survival on
targets
were compared to diet only and diet with gfp dsRNA control at same
concentration. Diet was
replaced with fresh diet containing dsRNA every two days. The number of
surviving insects
were assessed every day
Figure 2-NL: Survival of Nilaparvata lugens on liquid artificial diet treated
with
different concentrations of target dsRNA NL002. Nymphs of the first to second
larval stage
were fed diet supplemented with 1, 0.2, 0.08, and 0.04 mg/ml (final
concentration) of NL002.
Diet was replaced with fresh diet containing dsRNA every two days. The numbers
of
surviving insects were assessed every day.
DETAILED DESCRIPTION OF THE INVENTION
The present invention provides a means for controlling pest infestations by
administering to a pest a target coding sequence that post-transcriptionally
represses or
17
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
inhibits a requisite biological function in the pest. In one aspect, the
invention contemplates
feeding a pest with one or more double stranded or small interfering
ribonucleic acid (RNA)
molecules transcribed from all or a portion of a target coding sequence that
is essential for the
pest's sustenance and survival. Therefore, the present invention relates to
sequence-specific
inhibition of coding sequences using double-stranded RNA (dsRNA), including
small
interfering RNA (siRNA), as a means for pest control.
Until now, it has been impractical to provide dsRNA molecules in the diet of
most
pest species because RNA molecules are easily degraded by nucleases in the
environment
and were thought unstable in mildly alkaline or acidic environments, such as
those found in
the digestive tracts of most invertebrate pests. Therefore, there has existed
a need for
improved methods of modulating gene expression by repressing, delaying, or
otherwise
reducing gene expression within a particular pest for the purpose of
controlling pest
infestation or to introduce novel phenotypic traits.
The inventors herein have identified means for controlling pest infestation by
providing a dsRNA molecules in the diet of said pest. The sequence of the
dsRNA
corresponds to part or whole of an essential pest gene and causes
downregrulation of the pest
target via RNA interference (RNAi). As a result of the downregulation of mRNA,
the
dsRNA prevents expression of the target pest protein and results in one or
more of (but not
limited to) the following attributes: reduction in feeding by the pest,
reduction in viability of
the pest, death of the pest, inhibition of differentiation and development of
the pest, absence
of or reduced capacity for sexual reproduction by the pest, muscle formation,
juvenile
hormone formation, juvenile hormone regulation, ion regulation and transport,
maintenance
of cell membrane potential, amino acid biosynthesis, amino acid degradation,
sperm
formation, pheromone synthesis, pheromone sensing, antennae formation, wing
formation,
leg formation, development and differentiation, egg formation, larval
maturation, digestive
enzyme formation, haemolymph synthesis, haemolymph maintenance,
neurotransmission,
cell division, energy metabolism, respiration, apoptosis, and any component of
a eukaryotic
cells' cytoskeletal structure, such as, for example, actins and tubulins. Any
one or any
combination of these attributes can result in effective inhibition of pest
infestation, and in the
case of a plant pest, inhibition of plant infestation.
All technical terms employed in this specification are commonly used in
biochemistry, molecular biology and agriculture; hence, they are understood by
those skilled
18
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
in the field to which this invention belongs. Those technical terms can be
found, for example
in: MOLECULAR CLONING: A LABORATORY MANUAL, 3rd ed., vol. 1-3, ed. Sambrook
and
Russel, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y., 2001;
CURRENT
PROTOCOLS IN MOLECULAR BIOLOGY, ed. Ausubel et al., Greene Publishing
Associates and
Wiley-Interscience, New York, 1988 (with periodic updates); SHORT PROTOCOLS IN
MOLECULAR BIOLOGY: A COMPENDIUM OF METHODS FROM CURRENT PROTOCOLS IN
MOLECULAR BIOLOGY, 5th ed., vol. 1-2, ed. Ausubel et al., John Wiley & sons,
Inc., 2002;
GENOME ANALYSIS: A LABORATORY MANUAL, vol. 1-2, ed. Green et al., Cold Spring
Harbor
Laboratory Press, Cold Spring Harbor, N.Y., 1997.
Methodology involving plant biology techniques are described here and also are
described in detail in treatises such as METHODS IN PLANT MOLEULAR BIOLOGY: A
LABORATORY COURSE MANUAL, ed. Maliga et al., Cold Spring Harbor Laboratory
Press,
Cold Spring Harbor, N.Y., 1995. Various techniques using PCR are described,
for example,
in Innis et al., PCR PROTOCOLS: A GUIDE TO METHODS AND APPLICATIONS, Academic
Press,
San Diego, 1990 and in Dieffenbach and Dveksler, PCR PRIMER: A LABORATORY
MANUAL,
2nd e
a Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y., 2003. PCR-
primer
pairs can be derived from known sequences by known techniques such as using
computer
programs intended for that purpose, e.g., Primer, Version 0.5, 1991, Whitehead
Institute for
Biomedical Research, Cambridge, MA. Methods for chemical synthesis of nucleic
acids are
discussed, for example, in Beaucage & Caruthers, Tetra. Letts. 22: 1859-62
(1981), and
Matteucci & Caruthers, J. Am. Chem. Soc. 103: 3185 (1981).
Restriction enzyme digestions, phosphorylations, ligations, and
transformations were
done as described in Sambrook et al., MOLECULAR CLONING: A LABORATORY MANUAL,
2nd
ed. (1989), Cold Spring Harbor Laboratory Press. All reagents and materials
used for the
growth and maintenance of bacterial cells were obtained from Aldrich Chemicals
(Milwaukee, Wis.), DIFCO Laboratories (Detroit, Mich.), Invitrogen
(Gaithersburg, Md.), or
Sigma Chemical Company (St. Louis, Mo.) unless otherwise specified.
Agrobacterium or bacterial transformation: as is well known in the field,
Agrobacteria that are used for transforming plant cells are disarmed and
virulent derivatives
of, usually, Agrobacterium tumefaciens, Agrobacterium rhizo genes, that
contain a vector.
The vector typically contains a desired polynucleotide that is located between
the borders of a
T-DNA. However, any bacteria capable of transforming a plant cell may be used,
such as,
19
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
Rhizobium trifolii, Rhizobium leguminosarum, Phyllobacterium myrsinacearum,
SinoRhizobium meliloti, and MesoRhizobium loti.
Angiosperm: vascular plants having seeds enclosed in an ovary. Angiosperms are
seed plants that produce flowers that bear fruits. Angiosperms are divided
into
dicotyledonous and monocotyledonous plants.
Biological activity refers to the biological behavior and effects of a protein
or peptide
and its manifestations on a pest. For example, an inventive RNAi may prevent
translation of
a particular mRNA, thereby inhibiting the biological activity of the protein
encoded by the
mRNA or other biological activity of the pest.
In the present description, an RNAi molecule may inhibit a biological activity
in a
pest, resulting in one or more of (but not limited to) the following
attributes: reduction in
feeding by the pest, reduction in viability of the pest, death of the pest,
inhibition of
differentiation and development of the pest, absence of or reduced capacity
for sexual
reproduction by the pest, muscle formation, juvenile hormone formation,
juvenile hormone
regulation, ion regulation and transport, maintenance of cell membrane
potential, amino acid
biosynthesis, amino acid degradation, sperm formation, pheromone synthesis,
pheromone
sensing, antennae formation, wing formation, leg formation, development and
differentiation,
egg formation, larval maturation, digestive enzyme formation, haemolymph
synthesis,
haemolymph maintenance, neurotransmission, cell division, energy metabolism,
respiration,
apoptosis, and any component of a eukaryotic cells' cytoskeletal structure,
such as, for
example, actins and tubulins.
Commodity product encompasses any product made or otherwise derived from a
plant, including but not limited to food, feed, fiber, paper, meal, protein,
starch, flour, silage,
coffee, tea, and oil.
Complementary DNA (cDNA) refers to single-stranded DNA synthesized from a
mature mRNA template. Though there are several methods, cDNA is most often
synthesized
from mature (fully spliced) mRNA using the enzyme reverse transcriptase. This
enzyme
operates on a single strand of mRNA, generating its complementary DNA based on
the
pairing of RNA base pairs (A, U, G, C) to their DNA complements (T, A, C, G).
Two
nucleic acid strands are substantially complementary when at least 85% of
their bases pair.
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
Desired Polynucleotide: a desired polynucleotide of the present invention is a
genetic element, such as a promoter, enhancer, or terminator, or gene or
polynucleotide that is
to be transcribed and/or translated in a transformed cell that comprises the
desired
polynucleotide in its genome. If the desired polynucleotide comprises a
sequence encoding a
protein product, the coding region may be operably linked to regulatory
elements, such as to
a promoter and a terminator, that bring about expression of an associated
messenger RNA
transcript and/or a protein product encoded by the desired polynucleotide.
Thus, a "desired
polynucleotide" may comprise a gene that is operably linked in the 5'- to 3'-
orientation, a
promoter, a gene that encodes a protein, and a terminator. Alternatively, the
desired
polynucleotide may comprise a gene or fragment thereof, in a "sense" and/or
"antisense"
orientation, the transcription of which produces nucleic acids that may affect
expression of an
endogenous gene in the plant cell. A desired polynucleotide may also yield
upon transcription
a double-stranded RNA product upon that initiates RNA interference of a gene
to which the
desired polynucleotide is associated. A desired polynucleotide of the present
invention may be
positioned within a vector, such that the left and right border sequences
flank or are on either
side of the desired polynucleotide. The present invention envisions the stable
integration of one
or more desired polynucleotides into the genome of at least one host cell. A
desired
polynucleotide may be mutated or a variant of its wild-type sequence. It is
understood that all
or part of the desired polynucleotide can be integrated into the genome of a
host. It also is
understood that the term "desired polynucleotide" encompasses one or more of
such
polynucleotides. Thus, a vector of the present invention may comprise one,
two, three, four,
five, six, seven, eight, nine, ten, ormore desired polynucleotides.
Dicotyledonous plant (dicot) is a flowering plant whose embryos have two seed
halves or cotyledons, branching leaf veins, and flower parts in multiples of
four or five.
Examples of dicots include but are not limited to, Eucalyptus, Populus,
Liquidamber, Acacia,
teak, mahogany, cotton, tobacco, Arabidopsis, tomato, potato, sugar beet,
broccoli, cassava,
sweet potato, pepper, poinsettia, bean, alfalfa, soybean, carrot, strawberry,
lettuce, oak,
maple, walnut, rose, mint, squash, daisy, geranium, avocado, and cactus.
Foreign, with respect to a nucleic acid, means that that nucleic acid is
derived from
non-host organisms. According to the present invention, foreign DNA or RNA
represents
nucleic acids that are naturally occurring in the genetic makeup of fungi,
bacteria, viruses,
mammals, fish or birds, but are not naturally occurring in the host that is to
be transformed.
Thus, a foreign nucleic acid is one that encodes, for instance, a polypeptide
that is not
21
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
naturally produced by the transformed host. A foreign nucleic acid does not
have to encode a
protein product.
Fungi or fungal cell(s) as used herein refers to any cell present within or
derived
from an organism belonging to the Kingdom Fungi. The methods of the invention
are
applicable to all fungi and fungal cells that are susceptible to gene
silencing by RNA
interference and that are capable of internalising double-stranded RNA from
their immediate
environment.
In one embodiment of the invention, the fungus may be a mould, or more
particularly
a filamentous fungus. In other embodiments of the invention, the fungus may be
a yeast.
In one embodiment the fungus may be an ascomycetes fungus, i.e. a fungus
belonging to the Phylum Ascomycota.
In preferred, but non-limiting, embodiments and methods of the invention the
fungal
cell is chosen from the group consisting of:
a fungal cell of, or a cell derived from a plant pathogenic fungus, such as
but not
limited to Acremoniella spp., Allomyces spp., Alternaria spp. (e.g. Alternaria
brassicola or
Alternaria solani), Amoiphothec spp., Ascochyta spp. (e.g. Ascochyta pisi),
Aspergillius spp.,
Aureobasidium spp., Blastocladiella spp., Botrytis spp. (e.g. Botrytis cinerea
or Botiyotinia
fuckeliana), Candida spp., Cladosporium spp., Cercospora spp. (e.g. Cercospora
kikuchii or
Cercospora zaea-maydis), Chaetomium spp., Cladosporium spp. (e.g. Cladosporium
fulvum),
Colletotrichum spp. (e.g. Colletotrichum lindemuthianum), Coccidioides spp.,
Conidiobolus
spp., Coprinopsis spp., Colynascus spp., Cryphonectria spp., Cryptococcus
spp.,
Cunninghamella spp., Curvularia spp., Debarymyces spp., Diplodia spp. (e.g.
Diplodia
maydis), Emericella ssp., Encephalitozoon spp., Eremothecium spp., Erysiphe
spp. (e.g.
Erysiphe graminis fsp. graminis, Etysiphe graminis fsp. hordei or Erysiphe
pisi), Erwinia
armylovora, Fusarium spp. (e.g. Fusarium nivale, Fusarium sporotrichioides,
Fusarium
oxysporum, Fusarium graminearum, Fusarium germinearum, Fusarium culmorum,
Fusarium
solani, Fusarium moniliforme or Fusarium roseum), Gaeumanomyces spp. (e.g.
Gaeumanomyces graminis fsp. tritici), Geomyces spp., Gibberella spp. (e.g.
Gibberella
zeae), Gloeophyllum spp., Glomus spp., Helminthosporium spp. (e.g.
Helminthosporium
turcicum, Helminthosporium carbonum, Helminthosporium mavdis or
Helminthosporium
sigmoideum), Hypocrea spp., Kluyveromyces spp., Lentinula spp., Leptosphaeria
salvinii,
Leucosporidium spp., Macrophomina spp. (e.g. Macrophomina phaseolina),
Magnaportha
spp. (e.g. Magnaporthe oiyzae), Metharhizium spp., Mucor spp., Mycosphaerella
spp.,
Neurospora spp., Nectria spp. (e.g. Nectria heamatococca), Ophiostoma spp.,
22
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
Paracocidioides spp, Peronospora spp. (e.g. Peronospora manshurica or
Peronospora
tabacina), Phoma spp. (e.g. Phoma betae), Phaeopsheria spp., Phanerochaete
spp.,
Phakopsora spp. (e.g. Phakopsora pachyrhizi), Phymatotrichum spp. (e.g.
Phymatotrichum
omnivorum), Phytophthora spp. (e.g. Phytophthora cinnamomi, Phytophthora
cactorum,
Phytophthora phaseoli, Phytophthora parasitica, Phytophthora citrophthora,
Phytophthora
megasperma fsp. soiae or Phytophthora infestans), Plasmopara spp. (e.g.
Plasmopara
viticola), Pneumocystis spp., Podosphaera spp. (e.g. Podosphaera leucotricha),
Puccinia
spp. (e.g. Puccinia sorghi, Puccinia striiformis, Puccinia graminis fsp.
tritici, Puccinia
asparagi, Puccinia recondita or Puccinia arachidis), Pythium spp. (e.g.
Pythium
aphanidermatum), Pyronema spp., Pyrenophora spp. (e.g. Pyrenophora tritici-
repentens or
Pyrenophora teres), Pyricularia spp. (e.g. Pyricularia oryzae), Pythium spp.
(e.g. Pythium
ultimum), Rhincosporium secalis, Rhizoctonia spp. (e.g. Rhizoctonia solani,
Rhizoctonia
oryzae or Rhizoctonia cerealis), Rhizopus spp. (e.g. Rhizopus chinensid),
Saccharomyces
spp., Scerotium spp. (e.g. Scerotium rolfsii), Sclerotinia spp. (e.g.
Sclerotinia sclerotiorum),
Septoria spp. (e.g. Septoria lycopersici, Septoria glycines, Septoria nodorum
or Septoria
triticz), Spizellomyces spp., Thermomyces spp., Thielaviopsis spp. (e.g.
Thielaviopsis
basicola), Tilletia spp., Trametes spp., Trichoderma spp. (e.g. Trichoderma
virde),
Trichophyton spp., Uncinula spp. (e.g. Uncinula necator), Ustilago maydis
(e.g. corn smut),
Venturia spp. (e.g. Venturia inaequalis or Venturia pirina) Yarrwia spp. or
Verticillium spp.
(e.g. Verticillium dahliae or Verticillium albo-atrum);
Gene refers to a polynucleotide sequence that comprises control and coding
sequences necessary for the production of a polypeptide or precursor. The
polypeptide can
be encoded by a full length coding sequence or by any portion of the coding
sequence. A
gene may constitute an uninterrupted coding sequence or it may include one or
more introns,
bound by the appropriate splice junctions. Moreover, a gene may contain one or
more
modifications in either the coding or the untranslated regions that could
affect the biological
activity or the chemical structure of the expression product, the rate of
expression, or the
manner of expression control. Such modifications include, but are not limited
to, mutations,
insertions, deletions, and substitutions of one or more nucleotides. In this
regard, such
modified genes may be referred to as "variants" of the "native" gene.
Genetic element is any discreet nucleotide sequence such as, but not limited
to, a
promoter, gene, terminator, intron, enhancer, spacer, 5'-untranslated region,
3'-untranslated
region, or recombinase recognition site.
23
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
Genetic modification refers to the stable introduction of DNA into the genome
of
certain organisms by applying methods in molecular and cell biology.
"Gene suppression" or "down-regulation of gene expression" or "inhibition of
gene
expression" are used interchangeably and refer to a measurable or observable
reduction in
gene expression or a complete abolition of detectable gene expression, at the
level of protein
product and/or mRNA product from the target gene. Down-regulation or
inhibition of gene
expression is "specific" when down-regulation or inhibition of the target gene
occurs without
manifest effects on other genes of the pest.
Depending on the nature of the target gene, down-regulation or inhibition of
gene
expression in cells of a pest can be confirmed by phenotypic analysis of the
cell or the whole
pest or by measurement of mRNA or protein expression using molecular
techniques such as
RNA solution hybridization, nuclease protection, Northern hybridization,
reverse
transcription, gene expression monitoring with a microarray, antibody binding,
enzyme-
linked immunosorbent assay (ELISA), Western blotting, radioimmunoassay (RIA),
other
immunoassays, or fluorescence-activated cell analysis (FACS).
Gymnosperm, as used herein, refers to a seed plant that bears seed without
ovaries.
Examples of gymnosperms include conifers, cycads, ginkgos, and ephedras.
Homology, as used herein relates to sequences; Protein, or nucleotide
sequences are
likely to be homologous if they show a "significant" level of sequence
similarity or more
preferably sequence identity. Trudy homologous sequences are related by
divergence from a
common ancestor gene. Sequence homologs can be of two types:(i) where homologs
exist in
different species they are known as orthologs. e.g. the a-globin genes in
mouse and human
are orthologs; (ii) paralogues are homologous genes within a single species.
e.g. the a- and 0-
globin genes in mouse are paralogs.
Host cell refers to a microorganism, a prokaryotic cell, a eukaryotic cell, or
cell line
cultured as a unicellular entity that may be, or has been, used as a recipient
for a recombinant
vector or other transfer of polynucleotides, and includes the progeny of the
original cell that
has been transfected. The progeny of a single cell may not necessarily be
completely
identical in morphology or in genomic or total DNA complement as the original
parent due to
natural, accidental, or deliberate mutation.
Insect as used herein can be any insect, meaning any organism belonging to the
Kingdom Animals, more specific to the Phylum Arthropoda, and to the Class
Insecta or the
Class Arachnida. The methods of the invention are applicable to all insects
and that are
24
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
susceptible to gene silencing by RNA interference and that are capable of
internalising
double-stranded RNA from their immediate environment.
In one embodiment of the invention, the insect may belong to the following
orders:
Acari, Araneae, Anoplura, Coleoptera, Collembola, Dermaptera, Dictyoptera,
Diplura,
Diptera, Embioptera, Ephemeroptera, Grylloblatodea, Hemiptera, Homoptera,
Hymenoptera,
Isoptera, Lepidoptera, Mallophaga, Mecoptera, Neuroptera, Odonata, Orthoptera,
Phasmida,
Plecoptera, Protura, Psocoptera, Siphonaptera, Siphunculata, Thysanura,
Strepsiptera,
Thysanoptera, Trichoptera, and Zoraptera.
In preferred, but non-limiting, embodiments and methods of the invention the
insect is
chosen from the group consisting of:
an insect which is a plant pest, such as but not limited to Nilaparvata spp.
(e.g. N.
lugens (brown planthopper)); Laodelphax spp. (e.g. L. striatellus (small brown
planthopper));
Nephotettix spp. (e.g. N virescens or N cincticeps (green leafhopper), or
Nnigropictus (rice
leafhopper)); Sogatella spp. (e.g. S. furcifera (white-backed planthopper));
Blissus spp. (e.g.
B. leucopterus leucopterus (chinch bug)); Scotinophora spp. (e.g. S.
vermidulate (rice
blackbug)); Acrosternum spp. (e.g. A. hilare (green stink bug)); Parnara spp.
(e.g. P. guttata
(rice skipper)); Chilo spp. (e.g. C. suppressalis (rice striped stem borer),
C. auricilius (gold-
fringed stem borer), or C. polychrysus (dark-headed stem borer)); Chilotraea
spp. (e.g. C.
polychrysa (rice stalk borer)); Sesamia spp. (e.g. S. inferens (pink rice
borer)); Trypolyza
spp. (e.g. T. innotata (white rice borer), or T. incertulas (yellow rice
borer)); Cnaphalocrocis
spp. (e.g. C. medinalis (rice leafroller)); Agromyza spp. (e.g. A. oryzae
(leafminer), or A.
parvicornis (corn blot leafminer)); Diatraea spp. (e.g. D. saccharalis
(sugarcane borer), or D.
grandiosella = (southwestern corn borer)); Narnaga spp. (e.g. N aenescens
(green rice
caterpillar)); Xanthodes spp. (e.g. X transversa (green caterpillar));
Spodoptera spp. (e.g. S.
frugiperda (fall armyworm), S. exigua (beet armyworm), S. littoralis (climbing
cutworm) or
S. praefica (western yellowstriped armyworm)); Mythimna spp. (e.g. Mythmna
(Pseudaletia)
seperata (armyworm)); Helicoveipa spp. (e.g. H. zea (corn earworm)); Colaspis
spp. (e.g. C.
brunnea (grape colaspis)); Lissorhoptrus spp. (e.g. L. oryzophilus (rice water
weevil));
Echinocnemus spp. (e.g. E. squamos (rice plant weevil)); Diclodispa spp. (e.g.
D. armigera
(rice hispa)); Oulema spp. (e.g. 0. oryzae (leaf beetle); Sitophilus spp.
(e.g. S. oryzae (rice
weevil)); Pachydiplosis spp. (e.g. P. oryzae (rice gall midge)); Hydrellia
spp. (e.g. H.
griseola (small rice leafrniner), or H. sasakii (rice stem maggot)); Ch/orops
spp. (e.g. C.
oryzae (stem maggot)); Ostrinia spp. (e.g. 0. nubilalis (European corn
borer)); Agrotis spp.
(e.g. A.ipsilon (black cutworm)); Elasmopalpus spp. (e.g. E. lignosellus
(lesser cornstalk
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
borer)); Melanotus spp. (wireworms); Cyclocephala spp. (e.g. C. borealis
(northern masked
chafer), or C. immaculata (southern masked chafer)); Popillia spp. (e.g. P.
japonica
(Japanese beetle)); Chaetocnema spp. (e.g. C. pulicaria (corn flea beetle));
Sphenophorus
spp. (e.g. S. maidis (maize billbug)); Rhopalosiphum spp. (e.g. R. maidis
(corn leaf aphid));
Anuraphis spp. (e.g. A. maidiradicis (corn root aphid)); Melanoplus spp. (e.g.
M
femurrubrum (redlegged grasshopper) M differentialis (differential
grasshopper) or M
sanguinipes (migratory grasshopper)); Hylemya spp. (e.g. H. platura (seedcorn
maggot));
Anaphothrips spp. (e.g. A. obscrurus (grass thrips)); Solenopsis spp. (e.g. S.
milesta (thief
ant)); or spp. (e.g. T. urticae (twospotted spider mite), T. cinnabarinus
(carmine spider mite);
Helicoverpa spp. (e.g. H. zea (cotton bollworm), or H. armigera (American
bollworm));
Pectinophora spp. (e.g. P. gossypiella (pink bollworm)); Earias spp. (e.g. E.
vittella (spotted
bollworm)); Heliothis spp. (e.g. H. virescens (tobacco budworm)); Anthonomus
spy). (e.g. A.
grandis (boll weevil)); Pseudatomoscelis spp. (e.g. P. seriatus (cotton
fleahopper));
Trialeurodes spp. (e.g. T. abutiloneus (banded-winged whitefly) T.
vaporariorum
(greenhouse whitefly)); Bemisia spp. (e.g. B. argentifolii (silverleaf
whitefly)); Aphis spp.
(e.g. A. gossypii (cotton aphid), A. mellifera); Lygus spp. (e.g. L.
lineolaris (tarnished plant
bug) or L. hesperus (western tarnished plant bug)); Euschistus spp. (e.g. E.
conspersus
(consperse stink bug)); Chlorochroa spp. (e.g. C. sayi (Say stinkbug)); Nezara
spp. (e.g. N
viridula (southern green stinkbug)); Thrips spp. (e.g. T. tabaci (onion
thrips)); Frankliniella
spp. (e.g. F. fusca (tobacco thrips), or F. occidentalis (western flower
thrips)); Leptinotarsa
spp. (e.g. L. decemlineata (Colorado potato beetle), L. juncta (false potato
beetle), or L.
texana (Texan false potato beetle)); Lema spp. (e.g. L. trilineata (three-
lined potato beetle));
Epitrix spp. (e.g. E. cucumeris (potato flea beetle), E. hirtipennis (flea
beetle), or E. tuberis
(tuber flea beetle)); Epicauta spp. (e.g. E. vittata (striped blister
beetle)); Empoasca spp. (e.g.
E. fabae (potato leafhopper)); Myzus spp. (e.g. M. persicae (green peach
aphid)); Paratrioza
spp. (e.g. P. cockerelli (psyllid)); Conoderus spp. (e.g. C. falli (southern
potato wireworm), or
C. vespertinus (tobacco wireworm)); Phthorimaea spp. (e.g. P. operculella
(potato
tuberworm)); Macrosiphum spp. (e.g. M euphorbiae (potato aphid)); Thyanta spp.
(e.g. T.
pallidovirens (redshouldered stinkbug)); Phthorimaea spp. (e.g. P. operculella
(potato
tuberworm)); Helicoverpa spp. (e.g. H. zea (tomato fruitworm); Keiferia spp.
(e.g. K
lycopersicella (tomato pinworm)); Limonius spp. (wireworms); Manduca spp,
(e.g. M sexta
(tobacco hornworm), or M. quinquemaculata (tomato hornworm)); Liriomyza spp.
(e.g. L.
sativae, L. trifolli or L. huidobrensis (leafminer)); Drosophilla spp. (e.g.
D. melanogaster, D.
yakuba, D. pseudoobscura or D. simulans); Carabus spp. (e.g. C. granulatus);
Chironomus
26
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
spp. (e.g. C. tentanus); Ctenocephalides spp. (e.g. C. fells (cat flea));
Diaprepes spp. (e.g. D.
abbreviatus (root weevil)); Ips spp. (e.g. I. pini (pine engraver)); Tribolium
spp. (e.g. T.
castaneum (red floor beetle)); Glossina spp. (e.g. G. morsitans (tsetse fly));
Anopheles spp.
(e.g. A. gambiae (malaria mosquito)); Helicoverpa spp. (e.g. H armigera
(African
Bollworm)); Acyrthosiphon spp. (e.g. A. pisum (pea aphid)); Apis spp. (e.g. A.
melifera
(honey bee)); Homalodisca spp. (e.g. H. coagulate (glassy-winged
sharpshooter)); Aedes spp.
(e.g. Ae. aegypti (yellow fever mosquito)); Bombyx spp. (e.g. B. mori
(silkworm), B.
mandarina); Locusta spp. (e.g. L. migratoria (migratory locust)); Boophilus
spp. (e.g. B.
microplus (cattle tick)); Acanthoscurria spp. (e.g. A. gomesiana (red-haired
chololate bird
eater)); Diploptera spp. (e.g. D. punctata (pacific beetle cockroach));
Heliconius spp. (e.g. H.
erato (red passion flower butterfly) or H melpomene (postman butterfly));
Curculio spp. (e.g.
C. glandium (acorn weevil)); Plutella spp. (e.g. P. xylostella (diamontback
moth));
Amblyomma spp. (e.g. A. variegatum (cattle tick)); Anteraea spp. (e.g. A.
yamamai
(silkmoth)); Belgica spp. (e.g. B. antartica), Bemisa spp. (e.g. B. tabaci),
Bicyclus spp.,
Biphillus spp., Callosobruchus spp., Choristoneura spp., Cicindela spp., Culex
spp.,Culicoides spp., Diaphorina spp., Diaprepes spp., Euclidia spp., Glossina
spp., Gryllus
spp., Hydropsyche spp., Ju/odis spp., Lonomia spp., Lutzomyia spp., Lysiphebus
spp,
Me/adema spp, Mycetophagus spp., Nasonia spp., Oncometopia spp., Papilio spp.,
Pedicu/us
spp., Plodia spp., Rhynchosciara spp., Sphaerius spp., Toxoptera spp.,
Trichoplusa spp., and
Armigeres spp. (e.g. A. subalbatus);
"Pest control agent" or "gene suppression agent" refers to a particular RNA
molecule comprising a first RNA segment and a second RNA segment, wherein the
complementarity between the first and the second RNA segments results in the
ability of the
two segments to hybridize in vivo and in vitro to form a double stranded
molecule. It may
generally be preferable to include a third RNA segment linking and stabilizing
the first and
second sequences such that a stem can be formed linked together at one end of
each of the
first and second segments by the third segment to forms a loop, so that the
entire structure
forms into a stem and loop structure, or even more tightly hybridizing
structures may form
into a stem-loop knotted structure. Alternatively, a symmetrical hairpin could
be formed
without a third segment in which there is no designed loop, but for steric
reasons a hairpin
would create its own loop when the stem is long enough to stabilize itself.
The first and the
second RNA segments will generally lie within the length of the RNA molecule
and be
substantially inverted repeats of each other and linked together by the third
RNA segment.
The first and the second segments correspond invariably and not respectively
to a sense and
27
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
an antisense sequence with respect to the target RNA transcribed from the
target gene in the
target insect pest that is suppressed by the ingestion of the dsRNA molecule.
The pest control agent can also be a substantially purified (or isolated)
nucleic acid
molecule and more specifically nucleic acid molecules or nucleic acid fragment
molecules
thereof from a genomic DNA (gDNA) or cDNA library. Alternatively, the
fragments may
comprise smaller oligonucleotides having from about 15 to about 250 nucleotide
residues,
and more preferably, about 15 to about 30 nucleotide residues.
Introduction, as used herein, refers to the insertion of a nucleic acid
sequence into a
cell, by methods including infection, transfection, transformation, or
transduction.
Monocotyledonous plant (monocot) is a flowering plant having embryos with one
cotyledon or seed leaf, parallel leaf veins, and flower parts in multiples of
three. Examples of
monocots include, but are not limited to turfgrass, maize, rice, oat, wheat,
barley, sorghum,
orchid, iris, lily, onion, and palm.
Nematodes, or roundworms, are one of the most common phyla of animals, with
over
20,000 different described species (over 15,000 are parasitic). They are
ubiquitous in
freshwater, marine, and terrestrial environments, where they often outnumber
other animals
in both individual and species counts, and are found in locations as diverse
as Antarctica and
oceanic trenches. Further, there are a great many parasitic forms, including
pathogens in
most plants and animals.
The methods of the invention are applicable to all nematodes and that are
susceptible to
gene silencing by RNA interference and that are capable of internalising
double-stranded RNA
from their immediate environment.
In one embodiment of the invention, the nematode may belong to the family of
the
Heteroderidae, encompassing the genera Heterodera and Globodera.
In preferred, but non-limiting, embodiments and methods of the invention the
insect is
chosen from the group coprisingbut not limited to: Meloidogyne spp. (e.g. M.
incognita, M.
javanica, M graminicola, M arenaria, M chitwoodi, M. hapla or M paranaensis);
Heterodera spp. (e.g. H oiyzae, H. glycines, H zeae or H. schachtii);
Globodera spp. (e.g. G.
pallida or G. rostochiensis); Rotylenchulus spp. (e.g. R. reniformis);
Pratylenchus spp. (e.g. P.
coffeae, P. Zeae or P. goodeyi); Radopholus spp. (e.g. R. similis);
Hirschmaniella spp. (e.g. H.
oryzae); Ancylostoma spp. (e.g. A. caninum, A. ceylanicum, A. duodenale or A.
tubaeforme);
Anisakid; Aphelenchoides spp. (e.g. A. Besseyi); Ascarids; Ascaris spp., (e.g.
A. suum or A.
lumbridoides); Belonolaimus spp.; Brugia spp. (e.g. B. malayi or B. pahangi);
Bursaphelenchus spp.; Caenorhabditis spp. (e.g. C. elegans, C briggsae or C.
remanei);
28
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
Clostridium spp. (e.g. C. acetobutylicum); Cooperia spp. (e.g. C. oncopizora);
Criconemoides
spp.; Cyathostomum spp. (e.g. C. catinatum, C. coronatum or C. pateratum);
Cylicocyclus spp.
(e.g. C. insigne, C. nassatus or C. radiatus); Cylicostephanus spp. (e.g. C.
goldi or C.
longibursatus); Diphyllobothrium; Dirofilaria spp. (e.g. D. immitis);
Ditylenchus spp. (e.g. D.
dipsaci, D. destructor or D. Angustus); Enterobius spp. (e.g. E.
vermicularis); Haemonchus
spp. (e.g. H. contortus); Helicotyknchus spp.; Hoplolaimus spp.; Litomosoides
spp. (e.g. L.
sigmodontis); Longidorus spp. (e.g. L. macrosoma); Necator spp. (e.g. N.
americanus);
Nippostrongylus spp. (e.g. N. brasiliensis); Onchocerca spp. (e.g. 0.
volvulus); Ostertagia
spp. (e.g. 0. ostertagi); Parastrongyloides spp. (e.g. P. trichosuri);
Paratrichodorus spp. (e.g.
P. minor or P. teres); Parelaphostrongylus spp. (e.g. P. tenuis); Radophulus
spp.;
Scutellonerna. spp.; Strongyloides spp. (e.g. S. Ratti or S. stercoralis);
Teladorsagia spp. (e.g.
T circumcincta); Toxascaris spp. (e.g. T. leonina); Toxocara spp. (e.g. T.
canis or T. cati);
Trichinella spp. (e.g. T. britovi, T. spiralis or T. spirae); Trichodorus spp.
(e.g. T.
similis);Trichuris spp. (e.g. T. muris, T. vulpis or T. trichiura);
Tylenchulus spp.;
Tylenchorhynchus spp.; Uncinaria spp. (e.g. U stenocephala); Wuchereria spp.
(e.g. W.
bancrofti); Xiphinema spp. (e.g. X Index or X americanum).
Plant parasitic nematodes cause severe crop losses. The most common genera
are:
Aphelenchoides (foliar nematodes), Meloidogyne (root-knot nematodes),
Heterodera,
Globodera (cyst nematodes) such as the potato root nematode, Nacobbus,
Pratylenchus
(lesion nematodes), Ditylenchus , Xi phinema, Longidorus , Trichodorus . Other
nematodes
attack bark and forest trees. The most important representative of this group
is
Bursaphelenchus xylophilus, the pine wood nematode, present in Asia and
America and
recently discovered in Europe.
Normal cell refers to a cell of an untransforrned phenotype or exhibiting a
morphology of a non-transformed cell of the tissue type being examined.
Operably linked means combining two or more molecules in such a fashion that
in
combination they function properly in a plant cell. For instance, a promoter
is operably
linked to a structural gene when the promoter controls transcription of the
structural gene.
Orthologs are genes that are related by vertical descent from a common
ancestor and
encode proteins with the same function in different species Due to their
separation following
a speciation event, orthologs may diverge, but usually have similarity at the
seqence and
structure levels. Two genes that are derived from a common ancestor and encode
proteins
with similar function are refered to as orthologous. Identification of
orthologs is critical for
reliable predictions of gene function in newly sequenced genomes.
29
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
Pest or target pest includes but not limited to insects, arachnids,
crustaceans, fungi,
bacteria, viruses, nematodes, flatworms, roundworms, pinworms, hookworms,
tapeworms,
trypanosomes, schistosomes, botflies, fleas, ticks, mites, and lice that are
pervasive in the
human environment and damage plants. A pest may ingest or contact one or more
cells,
tissues, or products produced by a plant transformed with a double stranded
gene suppression
agent.
Pesticide refers to any substance or mixture of substances intended for
preventing,
destroying, repelling, or mitigating any pest. A pesticide may be a chemical
substance or
biological agent, such as a transgenic plant, used against pests including
insects, plant
pathogens, weeds, nematodes, and microbes that compete with humans for food,
destroy
property, spread disease, or are a nuisance.
Phenotype is a distinguishing feature or characteristic of a plant, which may
be
altered according to the present invention by integrating one or more "desired
polynucleotides" and/or screenable/selectable markers into the genome of at
least one plant cell
of a transformed plant. The "desired polymcleotide(s)" and/or markers may
confer a change in
the phenotype of a transformed plant, by modifying any one of a number of
genetic, molecular,
biochemical, physiological, morphological, or agronomic characteristics or
properties of the
transformed plant cell or plant as a whole. Thus, expression of one or more,
stably integrated
desired polynucleotide(s) in a plant genome, may yield a phenotype selected
from the group
consisting of, but not limited to, increased disease tolerance, increased
insect tolerance,
increased drought tolerance, enhanced cold and frost tolerance, improved
vigor, enhanced
color, enhanced health and nutritional characteristics, improved storage,
enhanced yield,
enhanced salt tolerance, enhanced heavy metal toleranceõ increased water-
stress tolerance,
enhanced sweetness, improved vigor, improved taste, improved texture,
decreased phosphate
content, increased germination, increased micronutrient uptake, improved
starch composition,
and improved flower longevity.
Plant tissue: a "plant" is any of various photosynthetic, eukaryotic,
multicellular
organisms of the kingdom Plantae characteristically producing embryos,
containing
chloroplasts, and having cellulose cell walls. A part of a plant, i.e., a
"plant tissue" may be
treated according to the methods of the present invention to produce a
transgenic plant.
Many suitable plant tissues can be transformed according to the present
invention and
include, but are not limited to, somatic embryos, pollen, leaves, stems,
calli, stolons,
microtubers, and shoots. Thus, the present invention envisions the
transformation of
angiosperm and gymnosperm plants such as acacia, alfalfa, apple, apricot,
artichoke, ash tree,
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
asparagus, avocado, banana, barley, beans, beet, birch, beech, blackberry,
blueberry, broccoli,
brussels sprouts, cabbage, canola, cantaloupe, carrot, cassava, cauliflower,
cedar, a cereal,
celery, chestnut, cherry, chinese cabbage, citrus, clemintine, clover, coffee,
corn, cotton,
cowpea, cucumber, cypress, eggplant, elm, endive, eucalyptus, fennel, figes,
fir, geranium,
grape, grapefruit, groundnuts, ground cherry, gum hemlock, hickory, kale,
kiwifruit, kohlrabi,
larch, lettuce, leek, lemon, lime, locust, pine, maidenhair, maize, mango,
maple, melon,
millet, mushroom, mustard, nuts, oak, oats, okra, onion, orange, an ornamental
plant or
flower or tree, papaya, palm, parsley, parsnip, pea, peach, peanut, pear,
peat, pepper,
persimmon, pigeon pea, pine, pineapple, plantain, plum, pomegranate, potato,
pumpkin,
radicchio, radish, rapeseed, raspberry, rice, rye, sorghumõ sallow, soybean,
spinach, spruce,
squash, strawberry, sugarbeet, sugarcane, sunflower, sweet potato, sweet corn,
tangerine, tea,
tobacco, tomato, trees, triticale, turf grasses, turnips, a vine, walnut,
watercress, watermelon,
wheat, yams, yew, and zucchini.
According to the present invention "plant tissue" also encompasses plant
cells. Plant
cells include suspension cultures, callus, embryos, meristematic regions,
callus tissue, leaves,
roots, shoots, gametophytes, sporophytes, pollen, seeds and microspores. Plant
tissues may
be at various stages of maturity and may be grown in liquid or solid culture,
or in soil or
suitable media in pots, greenhouses or fields. A plant tissue also refers to
any clone of such a
plant, seed, progeny, propagule whether generated sexually or asexually, and
descendents of
any of these, such as cuttings or seed.
Plant transformation and cell culture: broadly refers to the process by which
plant
cells are genetically modified and transferred to an appropriate plant culture
medium for
maintenance, further growth, and/or further development. Such methods are well
known to
the skilled artisan.
Progeny: a "progeny" of the present invention, such as the progeny of a
transgenic
plant, is one that is born of, begotten by, or derived from a plant or the
transgenic plant.
Thus, a "progeny" plant, i.e., an "Fl" generation plant is an offspring or a
descendant of the
transgenic plant produced by the inventive methods. A progeny of a transgenic
plant may
contain in at least one, some, or all of its cell genomes, the desired
polynucleotide that was
integrated into a cell of the parent transgenic plant by the methods described
herein. Thus, the
desired polynucleotide is "transmitted" or "inherited" by the progeny plant.
The desired
polynucleotide that is so inherited in the progeny plant may reside within a T-
DNA construct,
which also is inherited by the progeny plant from its parent. The term
"progeny" as used
herein, also may be considered to be the offspring or descendants of a group
of plants.
31
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
Promoter: promoter is intended to mean a nucleic acid, preferably DNA that
binds _ _
RNA polymerase and/or other transcription regulatory elements. As with any
promoter, the
promoters of the current invention will facilitate or control the
transcription of DNA or RNA
to generate an mRNA molecule from a nucleic acid molecule that is operably
linked to the
promoter. As stated earlier, the RNA generated may code for a protein or
polypeptide or may
code for an RNA interfering, or antisense molecule.
A plant promoter is a promoter capable of initiating transcription in plant
cells
whether or not its origin is a plant cell. Exemplary plant promoters include,
but are not
limited to, those that are obtained from plants, plant viruses, and bacteria
such as
Agrobacterium or Rhizobium which comprise genes expressed in plant cells.
Examples of
promoters under developmental control include promoters that preferentially
initiate
transcription in certain tissues, such as xylem, leaves, roots, or seeds. Such
promoters are
referred to as tissue-preferred promoters. Promoters which initiate
transcription only in
certain tissues are referred to as tissue-specific promoters. A cell type-
specific promoter
primarily drives expression in certain cell types in one or more organs, for
example, vascular
cells in roots or leaves, e.g. a root-specific promoter. An inducible or
repressible promoter
is a promoter which is under environmental control. Examples of environmental
conditions
that may effect transcription by inducible promoters include anaerobic
conditions or the
presence of light. Tissue specific, tissue preferred, cell type specific, and
inducible promoters
constitute the class of non-constitutive promoters. A constitutive promoter is
a promoter
which is active under most environmental conditions, and in most plant parts.
Polynucleotide is a nucleotide sequence, comprising a gene coding sequence or
a
fragment thereof, a promoter, an intron, an enhancer region, a polyadenylation
site, a
translation initiation site, 5' or 3' untranslated regions, a reporter gene, a
selectable marker or
the like. The polynucleotide may comprise single stranded or double stranded
DNA or RNA.
The polynucleotide may comprise modified bases or a modified backbone. The
polynucleotide may be genomic, an RNA transcript (such as an mRNA) or a
processed
nucleotide sequence (such as a cDNA). The polynucleotide may comprise a
sequence in
either sense or antisense orientations.
An isolated polynucleotide is a polynucleotide sequence that is not in its
native state,
e.g., the polynucleotide is comprised of a nucleotide sequence not found in
nature or the
polynucleotide is separated from nucleotide sequences with which it typically
is in proximity
or is next to nucleotide sequences with which it typically is not in
proximity.
32
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
Recombinant nucleotide sequence refers to a nucleic acid molecule that
contains a
genetically engineered modification through manipulation via mutagenesis,
restriction
enzymes, and the like.
RNA interference (RNAi) refers to sequence-specific or gene-specific
suppression of
gene expression (protein synthesis) that is mediated by short interfering RNA
(siRNA).
Seed: a "seed" may be regarded as a ripened plant ovule containing an embryo,
and a
propagative part of a plant, as a tuber or spore. A seed may be incubated
prior to
microorganism-mediated transformation, in the dark, for instance, to
facilitate germination.
Seed also may be sterilized prior to incubation, such as by brief treatment
with bleach. The
resultant seedling can then be exposed to a desired bacterium for
transformation
Selectable/screenable marker: a gene that, if expressed in plants or plant
tissues,
makes it possible to distinguish them from other plants or plant tissues that
do not express
that gene. Screening procedures may require assays for expression of proteins
encoded by
the screenable marker gene. Examples of selectable markers include the
neomycin
phosphotransferase (NPTI1) gene encoding kanamycin and geneticin resistance,
the
hygromycin phosphotransferase (HPT or APHIV) gene encoding resistance to
hygromycin, or
other similar genes known in the art.
Sequence identity: as used herein, "sequence identity" or "identity" in the
context of
two nucleic acid sequences includes reference to the residues in the two
sequences which are
the same when aligned for maximum correspondence over a specified region.
Where
sequences differ in conservative substitutions, the percent sequence identity
may be adjusted
upwards to correct for the conservative nature of the substitution. Sequences
which differ by
such conservative substitutions are said to have "sequence similarity" or
"similarity." Means
for making this adjustment are well-known to those of skill in the art.
As used herein, percentage of sequence identity means the value determined by
comparing two optimally aligned sequences over a comparison window, wherein
the portion
of the polynucleotide sequence in the comparison window may comprise additions
or
deletions (i.e., gaps) as compared to the reference sequence (which does not
comprise
additions or deletions) for optimal alignment of the two sequences. The
percentage is
calculated by determining the number of positions at which the identical
nucleic acid base
occurs in both sequences to yield the number of matched positions, dividing
the number of
33
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
matched positions by the total number of positions in the window of comparison
and
multiplying the result by 100 to yield the percentage of sequence identity.
"Sequence identity" has an art-recognized meaning and can be calculated using
published techniques. See COMPUTATIONAL MOLECULAR BIOLOGY, Lesk, ed. (Oxford
University Press, 1988), BIOCOMPUTING: INFORMATICS AND GENOME PROJECTS, Smith,
ed.
(Academic Press, 1993), COMPUTER ANALYSIS OF SEQUENCE DATA, PART I, Griffin &
Griffin, eds., (Humana Press, 1994), SEQUENCE ANALYSIS IN MOLECULAR BIOLOGY,
Von
Heinje ed., Academic Press (1987), SEQUENCE ANALYSIS PRIMER, Gribskov &
Devereux,
eds. (Macmillan Stockton Press, 1991), and Carillo & Lipton, SIAM J. Applied
Math. 48:
1073 (1988). Methods commonly employed to determine identity or similarity
between two
sequences include but are not limited to those disclosed in GUIDE To HUGE
COMPUTERS,
Bishop, ed., (Academic Press, 1994) and Carillo & Lipton, supra. Methods to
determine
identity and similarity are codified in computer programs. Preferred computer
program
methods to determine identity and similarity between two sequences include but
are not
limited to the GCG program package (Devereux et alõ Nucleic Acids Research 12:
387
(1984)), BLASTN, FASTA (Atschul et al., I Mol. Biol. 215: 403 (1990)), and
FASTDB
(Brutlag et al., Comp. App. Biosci. 6: 237 (1990)).
Short hairpin RNA (shRNA) are short single-stranded RNAs having a high degree
of secondary structure such that a portion of the RNA strand forms a hairpin
loop.
Short interfering RNA (siRNA) refers to double-stranded RNA molecules from
about 10 to about 30 nucleotides long that are named for their ability to
specifically interfere
with gene protein expression.
Target sequence refers to a nucleotide sequence in a pest that is selected for
suppression or inhibition by double stranded RNA technology. A target sequence
encodes an
essential feature or biological activity within a pest.
Transcriptional terminators: The expression DNA constructs of the present
invention typically have a transcriptional termination region at the opposite
end from the
transcription initiation regulatory region. The transcriptional termination
region may be
selected, for stability of the mRNA to enhance expression and/or for the
addition of
polyadenylation tails added to the gene transcription product. Translation of
a nascent
34
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
polypeptide undergoes termination when any of the three chain-termination
codons enters the
A site on the ribosome. Translation termination codons are UAA, UAG, and UGA.
Transfer DNA (T-DNA): an bacterial T-DNA is a genetic element that is well-
known as an element capable of integrating a nucleotide sequence contained
within its
borders into another genome. In this respect, a T-DNA is flanked, typically,
by two "border"
sequences. A desired polynucleotide of the present invention and a selectable
marker may be
positioned between the left border-like sequence and the right border-like
sequence of a T-
DNA. The desired polynucleotide and selectable marker contained within the T-
DNA may be
operably linked to a variety of different, plant-specific (i.e., native), or
foreign nucleic acids,
like promoter and terminator regulatory elements that facilitate its
expression, i.e., transcription
and/or translation of the DNA sequence encoded by the desired polynucleotide
or selectable
marker.
Transformation of plant cells: A process by which a nucleic acid is stably
inserted
into the genome of a plant cell. Transformation may occur under natural or
artificial
conditions using various methods well known in the art. Transformation may
rely on any
known method for the insertion of nucleic acid sequences into a prokaryotic or
eukaryotic
host cell, including Agrobacterium-mediated transformation protocols such as
'refined
transformation' or 'precise breeding', viral infection, whiskers,
electroporation,
microinjection, polyethylene glycol-treatment, heat shock, lipofection and
particle
bombardment.
Transgenic plant: a transgenic plant of the present invention is one that
comprises at
least one cell genome in which an exogenous nucleic acid has been stably
integrated.
According to the present invention, a transgenic plant is a plant that
comprises only one
genetically modified cell and cell genome, or is a plant that comprises some
genetically
modified cells, or is a plant in which all of the cells are genetically
modified. A transgenic
plant of the present invention may be one that comprises expression of the
desired
polynucleotide, i.e., the exogenous nucleic acid, in only certain parts of the
plant. Thus, a
transgenic plant may contain only genetically modified cells in certain parts
of its structure.
Variant: a "variant," as used herein, is understood to mean a nucleotide
sequence
that deviates from the standard, or given, nucleotide sequence of a particular
gene. The
terms, "isoform," "isotype," and "analog" also refer to "variant" forms of a
nucleotide
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
sequence. An nucleotide sequence that is altered by the addition, removal or
substitution of
one or more nucleotides, may be considered a "variant" sequence. "Variant" may
also refer
to a "shuffled gene" such as those described in Maxygen-assigned patents.
It is understood that the present invention is not limited to the particular
methodology,
protocols, vectors, and reagents, etc., described herein, as these may vary.
It is also to be
understood that the terminology used herein is used for the purpose of
describing particular
embodiments only, and is not intended to limit the scope of the present
invention. It must be
noted that as used herein and in the appended claims, the singular forms "a,"
"an," and "the"
include plural reference unless the context clearly dictates otherwise. Thus,
for example, a
reference to "a gene" is a reference to one or more genes and includes
equivalents thereof
known to those skilled in the art and so forth.
I. Target Pests
The present invention provides methodology and constructs for controlling pest
infestations by administering to a pest a target coding sequence that post-
transcriptionally
represses or inhibits a requisite biological function in the pest. As used
herein, the term
"pest" refers to insects, arachnids, crustaceans, fungi, bacteria, viruses,
nematodes,
flatworms, roundworms, pinworms, hookworms, tapeworms, trypanosomes,
schistosomes,
botflies, fleas, ticks, mites, and lice and the like that are pervasive in
humans, animals, and
plants. A pest may ingest or contact one or more cells, tissues, or products
produced by a
plant transformed with a double stranded gene suppression agent.
A "pest resistance" trait is a characteristic of a transgenic plant host that
causes the
plant to be resistant to attack from a pest that typically is capable of
inflicting damage or loss
to the plant. Such pest resistance can arise from a natural mutation or more
typically from
incorporation of recombinant DNA that confers pest resistance. To impart
insect resistance
to a transgenic plant, a recombinant DNA can, for example, be transcribed into
a RNA
molecule that forms a dsRNA molecule within the tissues or fluids of the
recombinant plant.
The dsRNA molecule is comprised in part of a segment of RNA that is identical
to a
corresponding RNA segment encoded from a DNA sequence within an insect pest
that
prefers to feed on the recombinant plant. Expression of the gene within the
target insect pest
is suppressed by the dsRNA, and the suppression of expression of the gene in
the target insect
pest results in the plant being insect resistant.
36
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
Suitable pests include any herbivore that causes damage to a plant or portion
thereof.
The invention contemplates insect, nematode, and fungal pests in particular.
Insect pests are of particular interest and include but are not limited to:
from the order
Lepidoptera, for example, Acleris spp Adoxophyes spp Aegeria spp Agrotis spp.,
Alabama
argillaceae, Amylois spp., Anticarsia gemmatalis, Archips spp, Argyrotaenia
spp.,
Autographa spp., Busseola fusca, Cadra cautella, Carposina nipponensis, Chilo
spp.,
Choristoneura spp., Clysia ambiguella, Cnaphalocrocis spp., Cnephasia spp.,
Cochylis spp.,
Coleophora spp.,. Crocidolomia binotalis, Cryptophlebia leucotreta, Cydia
spp., Diatraea
spp., Diparopsis castanea, Earias spp., Ephestia spp., Eucosma spp.,
Eupoecilia ambiguella,
Euproctis spp., Euxoa spp., Grapholita spp., Hedya nubiferana, Heliothis spp.,
Hellula
undalis, Hyphantria cunea, Keiferia lycopersicella, Leucoptera scitella,
Lithocollethis spp.,
Lobesia botrana, Lymantria spp., Lyonetia spp., Malacosoma spp., Mamestra
brassicae,
Manduca sexta, Operophtera spp., Ostrinia Nubilalis, Pammene spp., Pandemis
spp., Panolis
flammea, Pectinophora gossypiella, Phthorimaea opercuklla, Pieris rapae,
Pieris spp.,
Plutella xylostella, Prays spp., Scirpophaga spp., Sesamia spp., Sparganothis
spp.,
Spodoptera spp., Synanthedon spp., Thaumetopoea spp., Tortrix spp.,
Trichoplusia ni and
Yponomeuta spp.;
from the order Coleoptera, for example, Agriotes spp., Anthonomus spp.,
Atomaria
linearis, Chaetocnema tibialis, Cosmopolites spp., Curculio spp., Dermestes
spp., Epilachna
spp., Eremnus spp., Leptinotarsa decemlineata, Lissorhoptrus spp., Melolontha
spp.,
Orycaephilus spp., Otiorhynchus spp., Phlyctinus spp., Popillia spp.,
Psylliodes spp.,
Rhizopertha spp., Scarabeidae, Sitophilus spp., Sitotroga spp., Tenebrio spp.,
Tribolium spp.
and Trogoderma spp.;
from the order Orthoptera, for example, Blatta spp., Blattella spp.,
Gtyllotalpa spp.,
Leucophaea maderae, Locusta spp., Periplaneta ssp., and Schistocerca spp.;
from the order Isoptera, for example,Reticulitemes ssp;
from the order Psocoptera, for example,Liposcelis spp.;
from the order Anoplura, for example,Haematopinus spp., Linognathus spp.,
Pediculus spp., Pemphigus spp. and Phylloxera spp.;
from the order Mallophaga, for example,Damalinea spp. and Trichodectes spp.;
from the order Thysanoptera, for example,Franklinella spp., Hercinothrips
spp.,
Taeniothrips spp., Thrips palmi, Thrips tabaci and Scirtothrips aurantii;
from the order Heteroptera, for example, Cimex spp., Distantiella theobroma,
Dysdercus spp., Euchistus spp., Eurygaster spp., Leptocorisa spp., Nezara
spp., Piesma spp.,
37
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
Rhodnius spp., Sahlbergella singularis, Scotinophara spp., Triatoma spp.,
Miridae family
spp. such as Lygus hesperus and Lygus lineoloris, Lygaeidae family spp. such
as Blissus
leucopterus, and Pentatomidae family spp.;
from the order Homoptera, for example, Aleurothrixus floccosus, Aleyrodes
brassicae, Aonidiella spp., Aphididae, Aphis spp., Aspidiotus spp., Bemisia
tabaci,
Ceroplaster spp., Clzrysomphalus aonidium, Cluysomphalus dictyospermi, Coccus
hesperidum, Empoasca spp., Eriosoma larigerum, Erythroneura spp., Gascardia
spp.,
Laodelphax spp., Lacanium corni, Lepidosaphes spp., Macrosiphus spp., Myzus
spp.,
Nehotettix spp., Nilaparvata spp., Paratoria spp., Pemphigus spp., Planococcus
spp.,
Pseudaulacaspis spp., Pseudococcus spp., Psylla ssp., Pulvinaria aethiopica,
Quadraspidiotus spp., Rhopalosz:phum spp., Saissetia spp., Scaphoideus spp.,
Schizaphis spp.,
Sitobion spp., Trialeurodes vaporariorum, Trioza erytreae and Unaspis citri;
from the order Hymenoptera, for example, Acromyrmex, Atta spp., Cephus spp.,
Diprion spp., Diprionidae, Gilpinia polytoma, Hoplocampa spp., Lasius sppp.,
Monomorium
pharaonis , Neodiprion spp, Solenopsis spp. and Vespa ssp.;
from the order Diptera, for example, Aedes spp., Antherigona soccata, Bibio
hortulanus, Calliphora erythrocephala, Ceratitis spp., Chrysomyia spp., Culex
spp.,
Cuterebra spp., Dacus spp., Drosophila melanogaster, Fannia spp., Gastrophilus
spp.,
Glossina spp., Hypoderma spp., Hyppobosca spp., Liriomysa spp., Lucilia spp.,
Melanagromyza spp., Musca ssp., Oestrus spp., Orseolia spp., Oscinella fit,
Pegomyia
hyoscyami, Phorbia spp., Rhagoletis pomonella, Sciara spp., Stomoxys spp.,
Tabanus spp.,
Tannia spp. and Tipula spp.,
from the order Siphonaptera, for example,Ceratophyllus spp. und Xenopsylla
cheopis
and
from the order Thysanura, for example,Lepisma saccharina.
Nematode pests of a particular interest include, for example, A. caninum, A.
ceylancium, H. contortus, 0. ostertagi, C. elegans, C. briggsae, P. pacificus,
S. stercoralis, S.
ratti, P. trichosuri, M. arenaria, M chitwoodi, M hapla, M incognita, M
javanica, M
paraensis, G. rostochiensis, G. pallida, H. glycines, H. schattii, P.
penetrans, P. vulnus, R.
similis, Z. punctata, A. suum, T canis, B. malayi, D. immitis, 0. volvulus, T.
vulpis, T
spiralis, X index. A. duodenale, A. lumbricoides, as well as species from the
following
genera: Aphelenchoides, Nacobbus, Ditylenchus, Longidorus, Trichodorus, and
Bursaphelenchus
38
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
Fungal pests of particular interest include but are not limited to
Acremoniella spp.,
Alternaria spp. (e.g. Alternaria brassicola or Alternaria solani), Ascochyta
spp. (e.g.
Ascochyta pin), Botzytis spp. (e.g. Botrytis cinerea or Botryotinia
fuckeliana), Cladosporium
spp., Cercospora spp. (e.g. Cercospora kikuchii or Cercospora zaea-maydis),
Cladosporium
spp. (e.g. Cladosporium fulvum), Colletotrichum spp. (e.g. Colletotrichum
lindemuthianum),
Curvularia spp., Diplodia spp. (e.g. Diplodia maydis), Erysiphe spp. (e.g.
Erysiphe graminis
fsp. graminis, Erysiphe graminis fsp. hordei or Erysiphe pis , Erwinia
armylovora,
Fusarium spp. (e.g. Fusarium nivale, Fusarium sporotrichioides, Fusarium
axysporum,
Fusarium graminearum, Fusarium germinearum, Fusarium culmorum, Fusarium
solani,
Fusarium moniliforme or Fusarium roseum), Gaeumanomyces spp. (e.g.
Gaeumanomyces
graminis fsp. triticz), Gibberella spp. (e.g. Gibberella zeae),
Helminthosporium spp. (e.g.
Helminthosporium turcicum, Helminthosporium carbonum, Helminthosporium mavdis
or
Helminthosporium sigmoideum), Leptosphaeria salvinii, Macrophomina spp. (e.g.
Macrophomina phaseolina), Magnaportha spp. (e.g. Magnaporthe oryzae),
Mycosphaerella
spp., Nectria spp. (e.g. Nectria heamatococca), Peronospora spp. (e.g.
Peronospora
manshurica or Peronospora tabacina), Phoma spp. (e.g. Phoma betae), Phakopsora
spp.
(e.g. Phakopsora pachyrhizi), Phymatotrichum spp. (e.g. Phymatotrichum
omnivorum),
Phytophthora spp. (e.g. Phytophthora cinnamomi, Phytophthora cactorum,
Phytophthora
phaseoli, Phytophthora parasitica, Phytophthora citrophthora, Phytophthora
megasperma
fsp. soiae or Phytophthora infestans), Plasmopara spp. (e.g. Plasmopara
viticola),
Podosphaera spp. (e.g. Podosphaera leucotricha), Puccinia spp. (e.g. Puccinia
sorghi,
Puccinia striiformis, Puccinia graminis fsp. tritici, Puccinia asparagi,
Puccinia recondita or
Puccinia arachidis), Pythium spp. (e.g. Pythium aphanidermatum), Pyrenophora
spp. (e.g.
Pyrenophora tritici-repentens or Pyrenophora teres), Pyricularia spp. (e.g.
Pyricularia
ozyzae), Pythium spp. (e.g. Pythium ultimum), Rhincosporium secalis,
Rhizoctonia spp. (e.g.
Rhizoctonia solani, Rhizoctonia oryzae or Rhizoctonia cerealis), Rhizopus spp.
(e.g. Rhizopus
chinensid), Scerotium spp. (e.g. Scerotium
Sclerotinia spp. (e.g. Sclerotinia
sclerotiorum), Septoria spp. (e.g. Septoria lycopersici, Septoria glycines,
Septoria nodorum
or Septoria triticz), Thielaviopsis spp. (e.g. Thielaviopsis basicola),
Tilletia spp., Trichoderma
spp. (e.g. Trichoderma virde), Uncinuia spp. (e.g. Uncinula necator), Ustilago
maydis (e.g.
corn smut), Venturia spp. (e.g. Venturia inaequalis or Venturia pirina) or
Verticillium spp.
(e.g. Verticillium dahliae or Verticillium albo-atrum);
39
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
II. Identification of Target Sequences
The present invention provides a method for identifying and obtaining a
nucleic acid
comprising a nucleotide sequence for producing a dsRNA or siRNA. For example,
such a
method comprises: (a) probing a cDNA or genomic DNA library with a
hybridization probe
comprising all or a portion of a nucleotide sequence or a homolog thereof from
a targeted
insect; (b) identifying a DNA clone that hybridizes with the hybridization
probe; (c) isolating
the DNA clone identified in step (b); and (d) sequencing the cDNA or genomic
DNA
fragment that comprises the clone isolated in step (c) wherein the sequenced
nucleic acid
molecule transcribes all or a substantial portion of the RNA nucleotide acid
sequence or a
homolog thereof.
Additionally, the present invention contemplates a method for obtaining a
nucleic acid
fragment comprising a nucleotide sequence for producing a substantial portion
of a dsRNA or
siRNA comprising: (a) synthesizing first and a second oligonucleotide primers
corresponding
to a portion of one of the nucleotide sequences from a targeted pest; and (b)
amplifying a
cDNA or genomic DNA template in a cloning vector using the first and second
oligonucleotide primers of step (a) wherein the amplified nucleic acid
molecule transcribes a
substantial portion of a dsRNA or siRNA of the present invention.
In practicing the present invention, a target gene may be derived from any
pest that
causes damage to crop plants and subsequent yield losses. Several criteria may
be employed
in the selection of preferred target genes. The gene is one whose protein
product has a rapid
turnover rate, so that dsRNA inhibition will result in a rapid decrease in
protein levels. In
certain embodiments it is advantageous to select a gene for which a small drop
in expression
level results in deleterious effects for the recipient pest. If it is desired
to target a broad range
of insect species, for example, a gene is selected that is highly conserved
across these species.
Conversely, for the purpose of conferring specificity, in certain embodiments
of the
invention, a gene is selected that contains regions that are poorly conserved
between
individual insect species, or between insects and other organisms. In certain
embodiments it
may be desirable to select a gene that has no known homologs in other
organisms.
As used herein, the term "derived from" refers to a specified nucleotide
sequence that
may be obtained from a particular specified source or species, albeit not
necessarily directly
from that specified source or species.
In one embodiment, a gene is selected that is expressed in the insect gut.
Targeting
genes expressed in the gut avoids the requirement for the dsRNA to spread
within the insect.
Target genes for use in the present invention may include, for example, those
that share
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
substantial homologies to the nucleotide sequences of known gut-expressed
genes that
encode protein components of the plasma membrane proton V-ATPase (Dow et al.,
1997,;
Dow, 1999), for instance the V-ATPase B or E subunit. This protein complex is
the sole
energizer of epithelial ion transport and is responsible for alkalinization of
the midgut lumen.
The V-ATPase is also expressed in the Malpighian tubule, an outgrowth of the
insect hindgut
that functions in fluid balance and detoxification of foreign compounds in a
manner
analogous to a kidney organ of a mammal.
In another embodiment, a gene is selected that is essentially involved in the
growth,
development, and reproduction of an insect. Exemplary genes include but are
not limited to
the structural subunits of ribosomal proteins and a beta-coatamer gene, CHD3
gene.
Ribosomal proteins such as S4 (RpS4) and S9(RpS9) are structural constituents
of the
ribosome involved in protein biosynthesis and which are components of the
cytosolic small
ribosomal subunit, the ribosomal proteins such as L9 and L19 are structural
constituent of
ribosome involved in protein biosynthesis which is localised to the ribosome.
The beta-
coatamer gene in C. elegans encodes a protein which is a subunit of a
multimeric complex
that forms a membrane vesicle coat Similar sequences have been found in
diverse organisms
such as Arabidopsis thaliana, Drosophila melanogaster, and =Saccharomyces
cerevisiae.
Related sequences are found in diverse organisms such as Leptinotarsa
decemlineata,
Phaedon cochleariae, Epilachna varivetis, Anthonomus grandis, Tribolium
castaneum,
Myzus persicae, Nilaparvata lugens, Chilo suppressalis, Plutella xylostella
and Acheta
domesticus. Other target genes for use in the present invention may include,
for example,
those that play important roles in viability, growth, development,
reproduction, and
infectivity. These target genes include, for example, house keeping genes,
transcription
factors, and insect specific genes or lethal knockout mutations in
Caenorhabditis or
Drosophila. The target genes for use in the present invention may also be
those that are from
other organisms, e.g., from a nematode (e.g., Meloidogyne spp. or Heterodera
spp.), other
insects or arachnidae (e.g. Leptinotarsa spp., Phaedon spp., Epilachna spp.,
Anthonomus
spp., Tribolium spp., Myzus spp., Nilaparvata spp., Chilo spp., Plutella spp.,
or Acheta spp.,.
Additionally, the nucleotide sequences for use as a target sequence in the
present invention
may also be derived from viral, bacterial, fungal, insect or fungal genes
whose functions have
been established from literature and the nucleotide sequences of which share
substantial
similarity with the target genes in the genome of an insect.
For many of the insects that are potential targets for control by the present
invention,
there may be limited information regarding the sequences of most genes or the
phenotype
41
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
resulting from mutation of particular genes. Therefore, genes may be selected
based on
information available concerning corresponding genes in a model organism, such
as
Caenorhabditis or Drosophila, or in some other insect species. Genes may also
be selected
based on available sequence information for other species, such as nematode or
fungal
species, in which the genes have been characterized. In some cases it will be
possible to
obtain the sequence of a corresponding gene from a target insect by searching
databases, such
as GenBank, using either the name of the gene or the gene sequence. Once the
sequence is
obtained, PCR may be used to amplify an appropriately selected segment of the
gene in the
insect for use in the present invention.
In order to obtain a DNA segment from the corresponding gene in an insect
species,
for example, PCR primers may be designed based on the sequence as found in C.
elegans or
Drosophila, or an insect from which the gene has already been cloned. The
primers are
designed to amplify a DNA segment of sufficient length for use in the present
invention.
Amplification conditions are selected so that amplification will occur even if
the primers do
not exactly match the target sequence. Alternately, the gene, or a portion
thereof, may be
cloned from a genomic DNA or cDNA library prepared from the insect pest
species, using a
known insect gene as a probe. Techniques for performing PCR and cloning from
libraries are
known. Further details of the process by which DNA segments from target insect
pest
species may be isolated based on the sequence of genes previously cloned from
an insect
species are provided in the Examples. One of ordinary skill in the art will
recognize that a
variety of techniques may be used to isolate gene segments from insect pest
species that
correspond to genes previously isolated from other species.
III. Methods for inhibiting or suppressing a target gene
The present invention provides methods for inhibiting gene expression of one
or
multiple target genes in a target pest using dsRNA methods. The invention is
particularly
useful in the modulation of eukaryotic gene expression, in particular the
modulation of
expression of genes present in pests that exhibit a digestive system pH level
that is from
about 4.5 to about 9.5, more preferably from about 5.0 to about 8.0, and even
more preferably
from about 6.5 to about 7.5. For plant pests with a digestive system that
exhibits pH levels
outside of these ranges, delivery methods may be desired for use that do not
require ingestion
of dsRNA molecules.
The methods of the invention encompass the simultaneous or sequential
provision of
two or more different double-stranded RNAs or RNA constructs to the same
insect, so as to
42
CA 02622660 2013-06-13
achieve down-regulation or inhibition of multiple target genes or to achieve a
more potent
inhibition of a single target gene.
Alternatively, multiple targets are hit by the provision of one double-
stranded RNA
that hits multiple target sequences, and a single target is more efficiently
inhibited by the
presence of more than one copy of the double stranded RNA fragment
corresponding to the
target gene. Thus, in one embodiment of the invention, the double-stranded RNA
construct
comprises multiple dsRNA regions, at least one strand of each dsRNA region
comprising a
nucleotide sequence that is complementary to at least part of a target
nucleotide sequence of
an insect target gene. According to the invention, the dsRNA regions in the
RNA construct
may be complementary to the same or to different target genes and/or the dsRNA
regions
may be complementary to targets from the same or from different insect
species. Use of such
dsRNA constructs in a plant host cell, thus establishes a more potent
resistance to a single or
to multiple insect species in the plant. In one embodiment, the double
stranded RNA region
comprises multiple copies of the nucleotide sequence that is complementary to
the target
gene. Alternatively, the dsRNA hits more than one target sequence of the same
target gene.
The invention thus encompasses isolated double stranded RNA constructs
comprising at least
two copies of said nucleotide sequence complementary to at least part of a
nucleotide
sequence of an insect target. DsRNA that hits more than one of the above-
mentioned targets,
or a combination of different dsRNA against different of the above mentioned
targets are
developed and used in the methods of the present invention. Suitable dsRNA
nucleotides and
dsRNA constructs are described in W02006/046148 by applicant.
The terms "hit", "hits", and "hitting" are alternative wordings to indicate
that at least
one of the strands of the dsRNA is complementary to, and as such may bind to,
the target
gene or nucleotide sequence.
The modulatory effect of dsRNA is applicable to a variety of genes expressed
in the
pests including, for example, endogenous genes responsible for cellular
metabolism or
cellular transformation, including house keeping genes, transcription factors,
and other genes
which encode polypeptides involved in cellular metabolism.
As used herein, the phrase "inhibition of gene expression" or "inhibiting
expression of
a target gene in the cell of an pest" refers to the absence (or observable
decrease) in the level
of protein and/or mRNA product from the target gene. Specificity refers to the
ability to
inhibit the target gene without manifest effects on other genes of the cell
and without any
effects on any gene within the cell that is producing the dsRNA molecule. The
inhibition of
43
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
gene expression of the target gene in the insect pest may result in novel
phenotypic traits in
the insect pest.
"Gene suppression" refers to any of the well-known methods for reducing the
levels
of gene transcription to mRNA and/or subsequent translation of the mRNA. Gene
suppression is also intended to mean the reduction of protein expression from
a gene or a
coding sequence including posttranscriptional gene suppression and
transcriptional
suppression. Posttranscriptional gene suppression is mediated by the homology
between of
all or a part of a mRNA transcribed from a gene or coding sequence targeted
for suppression
and the corresponding double stranded RNA used for suppression, and refers to
the
substantial and measurable reduction of the amount of available mRNA available
in the cell
for binding by ribosomes. The transcribed RNA can be in the sense orientation
to effect what
is called co-suppression, in the anti-sense orientation to effect what is
called anti-sense
suppression, or in both orientations producing a dsRNA to effect what is
called RNA
interference (RNAi).
Transcriptional suppression is mediated by the presence in the cell of a dsRNA
gene
suppression agent exhibiting substantial sequence identity to a promoter DNA
sequence or
the complement thereof to effect what is referred to as promoter trans
suppression. Gene
suppression may be effective against a native plant gene associated with a
trait, e.g., to
provide plants with reduced levels of a protein encoded by the native gene or
with enhanced
or reduced levels of an affected metabolite. Gene suppression can also be
effective against
target genes in plant pests that may ingest or contact plant material
containing gene
suppression agents, specifically designed to inhibit or suppress the
expression of one or more
homologous or complementary sequences in the cells of the pest. Post-
transcriptional gene
suppression by anti-sense or sense oriented RNA to regulate gene expression in
plant cells is
disclosed in U.S. Pat. Nos. 5,107,065, 5,759,829, 5,283,184, and 5,231,020.
The use of
dsRNA to suppress genes in plants is disclosed in WO 99/53050, WO 99/49029,
U.S. Patent
Application Publication No. 2003/0175965, and 2003/0061626, U.S. Patent
Application
No.10/465,800, and U.S. Patent Nos. 6,506,559, and 6,326,193.
A beneficial method of post transcriptional gene suppression in plants employs
both
sense-oriented and anti-sense-oriented, transcribed RNA which is stabilized,
e.g., as a hairpin
and stem and loop structure. A preferred DNA construct for effecting post
transcriptional
gene suppression is one in which a first segment encodes an RNA exhibiting an
anti-sense
orientation exhibiting substantial identity to a segment of a gene targeted
for suppression,
which is linked to a second segment in sense orientation encoding an RNA
exhibiting
44
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
substantial complementarity to the first segment. Such a construct forms a
stem and loop
structure by hybridization of the first segment with the second segment and a
loop structure
from the nucleotide sequences linking the two segments (see W094/01550,
W098/05770,
US 2002/0048814, and US 2003/0018993).
According to one embodiment of the present invention, there is provided a
nucleotide
sequence, for which in vitro expression results in transcription of a dsRNA
sequence that is
substantially homologous to an RNA molecule of a targeted gene in a pest that
comprises an
RNA sequence encoded by a nucleotide sequence within the genome of the pest.
Thus, after
the pest ingests, or otherwise uptakes, the dsRNA sequence incorporated in a
diet or sprayed
on a plant surface, a down-regulation of the nucleotide sequence corresponding
to the target
gene in the cells of a target pest is affected.
Inhibition of a target gene using the dsRNA technology of the present
invention is
sequence-specific in that nucleotide sequences corresponding to the duplex
region of the
RNA are targeted for genetic inhibition. RNA containing a nucleotide sequences
identical to
a portion of the target gene is preferred for inhibition. RNA sequences with
insertions,
deletions, and single point mutations relative to the target sequence have
also been found to
be effective for inhibition. In performance of the present invention, it is
preferred that the
inhibitory dsRNA and the portion of the target gene share at least from about
80% sequence
identity, or from about 85% sequence identity, or from about 90% sequence
identity, or from
about 95% sequence identity, or from about 99% sequence identity, or even
about 100%
sequence identity. Alternatively, the duplex region of the RNA may be defined
functionally
as a nucleotide sequence that is capable of hybridizing with a portion of the
target gene
transcript. A less than full length sequence exhibiting a greater homology
compensates for a
longer less homologous sequence. The length of the identical nucleotide
sequences may be at
least about 25, 50, 100, 200, 300, 400, 500 or at least about 1000 bases.
Normally, a
sequence of greater than 20-100 nucleotides should be used, though a sequence
of greater
than about 200-300 nucleotides would be preferred, and a sequence of greater
than about
500-1000 nucleotides would be especially preferred depending on the size of
the target gene.
The invention has the advantage of being able to tolerate sequence variations
that might be
expected due to genetic mutation, strain polymorphism, or evolutionary
divergence. The
introduced nucleic acid molecule may not need to be absolute homology, may not
need to be
full length, relative to either the primary transcription product or fully
processed mRNA of
the target gene. Therefore, those skilled in the art need to realize that, as
disclosed herein,
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
100% sequence identity between the RNA and the target gene is not required to
practice the
present invention.
IV. Methods for preparing dsRNA
dsRNA molecules may be synthesized either in vivo or in vitro. The dsRNA may
be
formed by a single self-complementary RNA strand or from two complementary RNA
strands. Endogenous RNA polymerase of the cell may mediate transcription in
vivo, or
cloned RNA polymerase can be used for transcription in vivo or in vitro.
Inhibition may be
targeted by specific transcription in an organ, tissue, or cell type;
stimulation of an
environmental condition (e.g., infection, stress, temperature, chemical
inducers); and/or
engineering transcription at a developmental stage or age. The RNA strands may
or may not
be polyadenylated; the RNA strands may or may not be capable of being
translated into a
polypeptide by a cell's translational apparatus.
A RNA, dsRNA, siRNA, or miRNA of the present invention may be produced
chemically or enzymatically by one skilled in the art through manual or
automated reactions
or in vivo in another organism. RNA may also be produced by partial or total
organic
synthesis; any modified ribonucleotide can be introduced by in vitro enzymatic
or organic
synthesis. The RNA may be synthesized by a cellular RNA polymerase or a
bacteriophage
RNA polymerase (e.g., T3, T7, SP6). The use and production of an expression
construct are
known in the art (see, for example, WO 97/32016; U.S. Pat. No's. 5,593, 874,
5,698,425,
5,712,135, 5,789,214, and 5,804,693). If synthesized chemically or by in vitro
enzymatic
synthesis, the RNA may be purified prior to introduction into the cell. For
example, RNA
can be purified from a mixture by extraction with a solvent or resin,
precipitation,
electrophoresis, chromatography, or a combination thereof. Alternatively, the
RNA may be
used with no or a minimum of purification to avoid losses due to sample
processing. The
RNA may be dried for storage or dissolved in an aqueous solution. The solution
may contain
buffers or salts to promote annealing, and/or stabilization of the duplex
strands.
V. Polynucleotide Sequences
Provided according to the invention are nucleotide sequences, the expression
of which
results in an RNA sequence which is substantially homologous to an RNA
molecule of a
targeted gene in a pest that comprises an RNA sequence encoded by a nucleotide
sequence
within the genome of the pest. Thus, after ingestion of the dsRNA sequence
down-regulation
of the nucleotide sequence of the target gene in the cells of the pest may be
obtained resulting
in a deleterious effect on the maintenance, viability, proliferation,
reproduction, and
infestation of the pest.
46
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
Each "nucleotide sequence" set forth herein is presented as a sequence of
deoxyribonucleotides (abbreviated A, G, C and T). However, by "nucleotide
sequence" of a
nucleic acid molecule or polynucleotide is intended, for a DNA molecule or
polynucleotide, a
sequence of deoxyribonucleotides, and for an RNA molecule or polynucleotide,
the
corresponding sequence of ribonucleotides (A, G, C and U) where each thymidine
deoxynucleotide (T) in the specified deoxynucleotide sequence in is replaced
by the
ribonucleotide uridine (U).
As used herein, "nucleic acid" refers to a single or double-stranded polymer
of
deoxyribonucleotide or ribonucleotide bases read from the 5' to the 3' end. A
nucleic acid
may also optionally contain non-naturally occurring or altered nucleotide
bases that permit
correct read through by a polymerase and do not reduce expression of a
polypeptide encoded
by that nucleic acid. "Nucleotide sequence" or "nucleic acid sequence" refers
to both the
sense and antisense strands of a nucleic acid as either individual single
strands or in the
duplex.
The term "ribonucleic acid" (RNA) is inclusive of RNAi (inhibitory RNA), dsRNA
(double stranded RNA), siRNA (small interfering RNA), mRNA (messenger RNA),
miRNA
(micro-RNA), tRNA (transfer RNA, whether charged or discharged with a
corresponding
acylated amino acid), and cRNA (complementary RNA) and the term
"deoxyribonucleic
acid" (DNA) is inclusive of cDNA and genomic DNA and DNA-RNA hybrids.
The words "nucleic acid segment", "nucleotide sequence segment", or more
generally
"segment" will be understood by those in the art as a functional term that
includes both
genomic sequences, ribosomal RNA sequences, transfer RNA sequences, messenger
RNA
sequences, operon sequences and smaller engineered nucleotide sequences that
express or
may be adapted to express, proteins, polypeptides or peptides.
Accordingly, the present invention relates to an isolated nucleic molecule
comprising
a polynucleotide having a sequence selected from the group consisting of any
of the
polynucleotide sequences of SEQ ID NOs: 1, 3, 5,7, 9, 11, 13, 15, 17, 19, 21,
23, 49 - 158,
159, 160, 163, 168, 173, 178, 183, 188, 193, 198, 203, 208, 215, 220, 225,
230, 247, 249,
251, 253, 255, 257, 259, 275-472, 473, 478, 483, 488, 493, 498, 503, 513, 515,
517, 519, 521,
533 - 575, 576, 581, 586, 591, 596, 601, 603, 605, 607, 609, 621 - 767, 768,
773, 778, 783,
788, 793, 795, 797, 799, 801, 813 - 862, 863, 868, 873, 878, 883, 888, 890,
892, 894, 896,
908- 1040, 1041, 1046, 1051, 1056, 1061, 1071, 1073, 1075, 1077, 1079, 1081,
1083, 1085,
1087, 1089, 1091, 1093, 1095, 1097, 1099, 1101, 1103, 1105, 1107, 1109, 1111,
1113, 1161 -
1571, 1572, 1577, 1582, 1587, 1592, 1597, 1602, 1607, 1612, 1617, 1622, 1627,
1632, 1637,
47
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
1642, 1647, 1652, 1657, 1662, 1667, 1672, 1677, 1682, 1684, 1686, 1688, 1690,
1692, 1694,
1696, 1698, 1700, 1702, 1704, 1730 - 2039, 2040, 2045, 2050, 2055, 2060, 2065,
2070,
2075, 2080, 2085, 2090, 2095, 2100, 2102, 2104, 2106, 2108, 2120 - 2338, 2339,
2344,
2349, 2354, 2359, 2364, 2366, 2368, 2370, 2372, 2384 - 2460, 2461, 2466, 2471,
2476 and
2481. The invention also provides functional fragments of the polynucleotide
sequences of
SEQ ID NOs: 1, 3, 5, 7, 9, 11, 13, 15, 17, 19, 21, 23, 49- 158, 159, 160, 163,
168, 173, 178,
183, 188, 193, 198, 203, 208, 215, 220, 225, 230, 247, 249, 251, 253, 255,
257, 259, 275-472,
473, 478, 483, 488, 493, 498, 503, 513, 515, 517, 519, 521, 533 - 575, 576,
581, 586, 591,
596, 601, 603, 605, 607, 609, 621 - 767, 768, 773, 778, 783, 788, 793, 795,
797, 799, 801,
813 - 862, 863, 868, 873, 878, 883, 888, 890, 892, 894, 896, 908- 1040, 1041,
1046, 1051,
1056, 1061, 1071, 1073, 1075, 1077, 1079, 1081, 1083, 1085, 1087, 1089, 1091,
1093, 1095,
1097, 1099, 1101, 1103, 1105, 1107, 1109, 1111, 1113, 1161 - 1571, 1572, 1577,
1582, 1587,
1592, 1597, 1602, 1607, 1612, 1617, 1622, 1627, 1632, 1637, 1642, 1647, 1652,
1657, 1662,
1667, 1672, 1677, 1682, 1684, 1686, 1688, 1690, 1692, 1694, 1696, 1698, 1700,
1702, 1704,
1730 - 2039, 2040, 2045, 2050, 2055, 2060, 2065, 2070, 2075, 2080, 2085, 2090,
2095,
2100, 2102, 2104, 2106, 2108, 2120 - 2338, 2339, 2344, 2349, 2354, 2359, 2364,
2366,
2368, 2370, 2372, 2384 - 2460, 2461, 2466, 2471, 2476 and 2481. The invention
further
provides complementary nucleic acids, or fragments thereof, to any of the
polynucleotide
sequences of SEQ ID NOs: 1, 3,5, 7, 9, 11, 13, 15, 17, 19, 21, 23, 49 - 158,
159, 160, 163,
168, 173, 178, 183, 188, 193, 198, 203, 208, 215, 220, 225, 230, 247, 249,
251, 253, 255,
257, 259, 275-472, 473, 478, 483, 488, 493, 498, 503, 513, 515, 517, 519, 521,
533 - 575,
576, 581, 586, 591, 596, 601, 603, 605, 607, 609, 621 -767, 768, 773, 778,
783, 788, 793,
795, 797, 799, 801, 813 - 862, 863, 868, 873, 878, 883, 888, 890, 892, 894,
896, 908- 1040,
1041, 1046, 1051, 1056, 1061, 1071, 1073, 1075, 1077, 1079, 1081, 1083, 1085,
1087, 1089,
1091, 1093, 1095, 1097, 1099, 1101, 1103, 1105, 1107, 1109, 1111, 1113, 1161 -
1571, 1572,
1577, 1582, 1587, 1592, 1597, 1602, 1607, 1612, 1617, 1622, 1627, 1632, 1637,
1642, 1647,
1652, 1657, 1662, 1667, 1672, 1677, 1682, 1684, 1686, 1688, 1690, 1692, 1694,
1696, 1698,
1700, 1702, 1704, 1730 - 2039, 2040, 2045, 2050, 2055, 2060, 2065, 2070, 2075,
2080,
2085, 2090, 2095, 2100, 2102, 2104, 2106, 2108, 2120 - 2338, 2339, 2344, 2349,
2354,
2359, 2364, 2366, 2368, 2370, 2372, 2384 - 2460, 2461, 2466, 2471, 2476 and
2481, as well
as a nucleic acid, comprising at least 15 contiguous bases, which hybridizes
to any of the
polynucleotide sequences of SEQ ID NOs: 1, 3, 5, 7, 9, 11, 13, 15, 17, 19, 21,
23, 49 - 158,
159, 160, 163, 168, 173, 178, 183, 188, 193, 198, 203, 208, 215, 220, 225,
230, 247, 249,
251, 253, 255, 257, 259, 275-472, 473, 478, 483, 488, 493, 498, 503, 513, 515,
517, 519, 521,
48
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
533 - 575, 576, 581, 586, 591, 596, 601, 603, 605, 607, 609, 621 - 767, 768,
773, 778, 783,
788, 793, 795, 797, 799, 801, 813 - 862, 863, 868, 873, 878, 883, 888, 890,
892, 894, 896,
908- 1040, 1041, 1046, 1051, 1056, 1061, 1071, 1073, 1075, 1077, 1079, 1081,
1083, 1085,
1087, 1089, 1091, 1093, 1095, 1097, 1099, 1101, 1103, 1105, 1107, 1109, 1111,
1113, 1161 -
1571, 1572, 1577, 1582, 1587, 1592, 1597, 1602, 1607, 1612, 1617, 1622, 1627,
1632, 1637,
1642, 1647, 1652, 1657, 1662, 1667, 1672, 1677, 1682, 1684, 1686, 1688, 1690,
1692, 1694,
1696, 1698, 1700, 1702, 1704, 1730 - 2039, 2040, 2045, 2050, 2055, 2060, 2065,
2070,
2075, 2080, 2085, 2090, 2095, 2100, 2102, 2104, 2106, 2108, 2120 - 2338, 2339,
2344,
2349, 2354, 2359, 2364, 2366, 2368, 2370, 2372, 2384 - 2460, 2461, 2466, 2471,
2476 and
2481.
The present invention also provides orthologous sequences, and complements and
fragments thereof, of the polynucleotide sequences of SEQ ID NOs: 1, 3, 5, 7,
9, 11, 13, 15,
17, 19, 21, 23, 49 - 158, 159, 160, 163, 168, 173, 178, 183, 188, 193, 198,
203, 208, 215,
220, 225, 230, 247, 249, 251, 253, 255, 257, 259, 275-472, 473, 478, 483, 488,
493, 498, 503,
513, 515, 517, 519, 521, 533 - 575, 576, 581, 586, 591, 596, 601, 603, 605,
607, 609, 621 -
767, 768, 773, 778, 783, 788, 793, 795, 797, 799, 801, 813 - 862, 863, 868,
873, 878, 883,
888, 890, 892, 894, 896, 908- 1040, 1041, 1046, 1051, 1056, 1061, 1071, 1073,
1075, 1077,
1079, 1081, 1083, 1085, 1087, 1089, 1091, 1093, 1095, 1097, 1099, 1101, 1103,
1105, 1107,
1109, 1111, 1113, 1161 - 1571, 1572, 1577, 1582, 1587, 1592, 1597, 1602, 1607,
1612, 1617,
1622, 1627, 1632, 1637, 1642, 1647, 1652, 1657, 1662, 1667, 1672, 1677, 1682,
1684, 1686,
1688, 1690, 1692, 1694, 1696, 1698, 1700, 1702, 1704, 1730 - 2039, 2040, 2045,
2050,
2055, 2060, 2065, 2070, 2075, 2080, 2085, 2090, 2095, 2100, 2102, 2104, 2106,
2108, 2120
- 2338, 2339, 2344, 2349, 2354, 2359, 2364, 2366, 2368, 2370, 2372, 2384 -
2460, 2461,
2466, 2471, 2476 and 2481 of the invention. Accordingly, the invention
encompasses target
genes which are insect orthologs of a gene comprising a nucleotide sequence as
represented
in any of SEQ ID NOs: 1, 3, 5, 7, 9, 11, 13, 15, 17, 19, 21, 23, 49 - 158,
159, 160, 163, 168,
173, 178, 183, 188, 193, 198, 203, 208, 215, 220, 225, 230, 247, 249, 251,
253, 255, 257,
259, 275-472, 473, 478, 483, 488, 493, 498, 503, 513, 515, 517, 519, 521, 533 -
575, 576,
581, 586, 591, 596, 601, 603, 605, 607, 609, 621 - 767, 768, 773, 778, 783,
788, 793, 795,
797, 799, 801, 813 - 862, 863, 868, 873, 878, 883, 888, 890, 892, 894, 896,
908- 1040, 1041,
1046, 1051, 1056, 1061, 1071, 1073, 1075, 1077, 1079, 1081, 1083, 1085, 1087,
1089, 1091,
1093, 1095, 1097, 1099, 1101, 1103, 1105, 1107, 1109, 1111, 1113, 1161 -1571,
1572, 1577,
1582, 1587, 1592, 1597, 1602, 1607, 1612, 1617, 1622, 1627, 1632, 1637, 1642,
1647, 1652,
1657, 1662, 1667, 1672, 1677, 1682, 1684, 1686, 1688, 1690, 1692, 1694, 1696,
1698, 1700,
49
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
1702, 1704, 1730 - 2039, 2040, 2045, 2050, 2055, 2060, 2065, 2070, 2075, 2080,
2085,
2090, 2095, 2100, 2102, 2104, 2106, 2108, 2120 - 2338, 2339, 2344, 2349, 2354,
2359,
2364, 2366, 2368, 2370, 2372, 2384 - 2460, 2461, 2466, 2471, 2476 and 2481. By
way of
example, insect orthologues may comprise a nucleotide sequence as represented
in any of
SEQ ID NOs: 49-123, 275-434, 533-562, 621-738, 813-852, 908-1010, 1161-1437,
1730-
1987, 2120-2290, 2384-2438, or a fragment thereof of at least 15, 16, 17, 18,
19, 20, 21, 22,
23, 24, 25, 26 or 27 nucleotides . A non-limiting list of insect or arachnida
orthologs genes or
sequences comprising at least a fragment of 15, preferably at least 17 bp of
one of the
sequences of the invention is given in Tables 4.
The invention also encompasses target genes which are nematode orthologs of a
gene
comprising a nucleotide sequence as represented in any of SEQ ID NOs: 1, 3, 5,
7, 9, 11, 13,
15, 17, 19, 21, 23, 49 - 158, 159, 160, 163, 168, 173, 178, 183, 188, 193,
198, 203, 208, 215,
220, 225, 230, 247, 249, 251, 253, 255, 257, 259, 275-472, 473, 478, 483, 488,
493, 498, 503,
513, 515, 517, 519, 521, 533 - 575, 576, 581, 586, 591, 596, 601, 603, 605,
607, 609, 621 -
767, 768, 773, 778, 783, 788, 793, 795, 797, 799, 801, 813 - 862, 863, 868,
873, 878, 883,
888, 890, 892, 894, 896, 908- 1040, 1041, 1046, 1051, 1056, 1061, 1071, 1073,
1075, 1077,
1079, 1081, 1083, 1085, 1087, 1089, 1091, 1093, 1095, 1097, 1099, 1101, 1103,
1105, 1107,
1109, 1111, 1113, 1161 -1571, 1572,1577, 1582, 1587, 1592, 1597, 1602, 1607,
1612, 1617,
1622, 1627, 1632, 1637, 1642, 1647, 1652, 1657, 1662, 1667, 1672, 1677, 1682,
1684, 1686,
1688, 1690, 1692, 1694, 1696, 1698, 1700, 1702, 1704, 1730 - 2039, 2040, 2045,
2050,
2055, 2060, 2065, 2070, 2075, 2080, 2085, 2090, 2095, 2100, 2102, 2104, 2106,
2108, 2120
- 2338, 2339, 2344, 2349, 2354, 2359, 2364, 2366, 2368, 2370, 2372, 2384 -
2460, 2461,
2466, 2471, 2476 and 2481 of the invention. By way of example, nematode
orthologs may
comprise a nucleotide sequence as represented in any of SEQ ID NOs: 124-135,
435-446,
563, 564, 739-751, 853, 854, 1011-1025, 1438-1473, 1988-2001, 2291-2298, 2439-
2440 of the
invention, or a fragment of at least 15, 16, 17, 18, 19, 20 or 21 nucleotides
thereof. According
to another aspect, the invention thus encompasses any of the methods described
herein for
controlling nematode growth in an organism, or for preventing nematode
infestation of an
organism susceptible to nemataode infection, comprising contacting nematode
cells with a
double-stranded RNA, wherein the double-stranded RNA comprises annealed
complementary strands, one of which has a nucleotide sequence which is
complementary to
at least part of the nucleotide sequence of a target gene comprising a
fragment of at least 17,
18, 19, 20 or 21 nucleotides of any of the sequences as represented in SEQ ID
NOs: 1, 3, 5, 7,
9, 11, 13, 15, 17, 19, 21, 23, 49- 158, 159, 160, 163, 168, 173, 178, 183,
188, 193, 198, 203,
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
208, 215, 220, 225, 230, 247, 249, 251, 253, 255, 257, 259, 275-472, 473, 478,
483, 488, 493,
498, 503, 513, 515, 517, 519, 521, 533 - 575, 576, 581, 586, 591, 596, 601,
603, 605, 607,
609, 621 - 767, 768, 773, 778, 783, 788, 793, 795, 797, 799, 801, 813 - 862,
863, 868, 873,
878, 883, 888, 890, 892, 894, 896, 908- 1040, 1041, 1046, 1051, 1056, 1061,
1071, 1073,
1075, 1077, 1079, 1081, 1083, 1085, 1087, 1089, 1091, 1093, 1095, 1097, 1099,
1101, 1103,
1105, 1107, 1109, 1111, 1113, 1161 - 1571, 1572, 1577, 1582, 1587, 1592, 1597,
1602, 1607,
1612, 1617, 1622, 1627, 1632, 1637, 1642, 1647, 1652, 1657, 1662, 1667, 1672,
1677, 1682,
1684, 1686, 1688, 1690, 1692, 1694, 1696, 1698, 1700, 1702, 1704, 1730 - 2039,
2040,
2045, 2050, 2055, 2060, 2065, 2070, 2075, 2080, 2085, 2090, 2095, 2100, 2102,
2104, 2106,
2108, 2120 - 2338, 2339, 2344, 2349, 2354, 2359, 2364, 2366, 2368, 2370, 2372,
2384
2460, 2461, 2466, 2471, 2476 and 2481, whereby the double-stranded RNA is
taken up by
the fungus and thereby controls growth or prevents infestation. The invention
also relates to
nematode-resistant transgenic plants comprising a fragment of at least 17, 18,
19, 20 or 21
nucleotides of any of the sequences as represented in SEQ ID NOs: 1, 3, 5, 7,
9, 11, 13, 15,
17, 19, 21, 23, 49 - 158, 159, 160, 163, 168, 173, 178, 183, 188, 193, 198,
203, 208, 215,
220, 225, 230, 247, 249, 251, 253, 255, 257, 259, 275-472, 473, 478, 483, 488,
493, 498, 503,
513, 515, 517, 519, 521, 533 - 575, 576, 581, 586, 591, 596, 601, 603, 605,
607, 609, 621 -
767, 768, 773, 778, 783, 788, 793, 795, 797, 799, 801, 813 - 862, 863, 868,
873, 878, 883,
888, 890, 892, 894, 896, 908- 1040, 1041, 1046, 1051, 1056, 1061, 1071, 1073,
1075, 1077,
1079, 1081, 1083, 1085, 1087, 1089, 1091, 1093, 1095, 1097, 1099, 1101, 1103,
1105, 1107,
1109, 1111, 1113, 1161 - 1571, 1572, 1577, 1582, 1587, 1592, 1597, 1602, 1607,
1612, 1617,
1622, 1627, 1632, 1637, 1642, 1647, 1652, 1657, 1662, 1667, 1672, 1677, 1682,
1684, 1686,
1688, 1690, 1692, 1694, 1696, 1698, 1700, 1702, 1704, 1730 - 2039, 2040, 2045,
2050,
2055, 2060, 2065, 2070, 2075, 2080, 2085, 2090, 2095, 2100, 2102, 2104, 2106,
2108, 2120
- 2338, 2339, 2344, 2349, 2354, 2359, 2364, 2366, 2368, 2370, 2372, 2384 -
2460, 2461,
2466, 2471, 2476 and 2481. A non-limiting list of nematode orthologs genes or
sequences
comprising at least a fragment of 15, preferably at least 17 bp of one of the
sequences of the
invention is given in Tables 5.
According to another embodiment, the invention encompasses target genes which
are
fungal orthologs of a gene comprising a nucleotide sequence as represented in
any of SEQ ID
NO:s 1, 3, 5, 7, 9, 11, 13, 15, 17, 19, 21, 23, 49 - 158, 159, 160, 163, 168,
173, 178, 183, 188,
193, 198, 203, 208, 215, 220, 225, 230, 247, 249, 251, 253, 255, 257, 259, 275-
472, 473, 478,
483, 488, 493, 498, 503, 513, 515, 517, 519, 521, 533 - 575, 576, 581, 586,
591, 596, 601,
603, 605, 607, 609, 621 - 767, 768, 773, 778, 783, 788, 793, 795, 797, 799,
801, 813 - 862,
51
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
863, 868, 873, 878, 883, 888, 890, 892, 894, 896, 908- 1040, 1041, 1046, 1051,
1056, 1061,
1071, 1073, 1075, 1077, 1079, 1081, 1083, 1085, 1087, 1089, 1091, 1093, 1095,
1097, 1099,
1101, 1103, 1105, 1107, 1109, 1111, 1113, 1161 -1571, 1572, 1577, 1582, 1587,
1592, 1597,
1602, 1607, 1612, 1617, 1622, 1627, 1632, 1637, 1642, 1647, 1652, 1657, 1662,
1667, 1672,
1677, 1682, 1684, 1686, 1688, 1690, 1692, 1694, 1696, 1698, 1700, 1702, 1704,
1730 -
2039, 2040, 2045, 2050, 2055, 2060, 2065, 2070, 2075, 2080, 2085, 2090, 2095,
2100, 2102,
2104, 2106, 2108, 2120 - 2338, 2339, 2344, 2349, 2354, 2359, 2364, 2366, 2368,
2370,
2372, 2384 - 2460, 2461, 2466, 2471, 2476 and 2481 of the invention. By way of
example,
fungal orthologs may comprise a nucleotide sequence as represented in any of
SEQ ID
NOs:136-158, 447-472, 565-575, 752-767, 855-862, 1026-1040, 1474-1571, 2002-
2039,
2299-2338, 2441-2460, or a fragment of at least 17, 18, 19, 20, 21, 22, 23,
24, 25, 26 or 27
nucleotides thereof.. According to another aspect, the invention thus
encompasses any of the
methods described herein for controlling fungal growth on a cell or an
organism, or for
preventing fungal infestation of a cell or an organism susceptible to fungal
infection,
comprising contacting fungal cells with a double-stranded RNA, wherein the
double-stranded
RNA comprises annealed complementary strands, one of which has a nucleotide
sequence
which is complementary to at least part of the nucleotide sequence of a target
gene
comprising a fragment of at least 17, 18, 19, 20 or 21 nucleotides of any of
the sequences as
represented in SEQ ID NOs: 1, 3, 5, 7, 9, 11, 13, 15, 17, 19, 21, 23, 49- 158,
159, 160, 163,
168, 173, 178, 183, 188, 193, 198, 203, 208, 215, 220, 225, 230, 247, 249,
251, 253, 255,
257, 259, 275-472, 473, 478, 483, 488, 493, 498, 503, 513, 515, 517, 519, 521,
533 - 575,
576, 581, 586, 591, 596, 601, 603, 605, 607, 609, 621 - 767, 768, 773, 778,
783, 788, 793,
795, 797, 799, 801, 813 - 862, 863, 868, 873, 878, 883, 888, 890, 892, 894,
896, 908- 1040,
1041, 1046, 1051, 1056, 1061, 1071, 1073, .1075, 1077, 1079, 1081, 1083, 1085,
1087, 1089,
1091, 1093, 1095, 1097, 1099, 1101, 1103, 1105, 1107, 1109, 1111, 1113, 1161 -
1571, 1572,
1577, 1582, 1587, 1592, 1597, 1602, 1607, 1612, 1617, 1622, 1627, 1632, 1637,
1642, 1647,
1652, 1657, 1662, 1667, 1672, 1677, 1682, 1684, 1686, 1688, 1690, 1692, 1694,
1696, 1698,
1700, 1702, 1704, 1730 - 2039, 2040, 2045, 2050, 2055, 2060, 2065, 2070, 2075,
2080,
2085, 2090, 2095, 2100, 2102, 2104, 2106, 2108, 2120 - 2338, 2339, 2344, 2349,
2354,
2359, 2364, 2366, 2368, 2370, 2372, 2384 - 2460, 2461, 2466, 2471, 2476 and
2481,
whereby the double-stranded RNA is taken up by the fungus and thereby controls
growth or
prevents infestation. The invention also relates to fungal-resistant
transgenic plants
comprising a fragment of at least 17, 18, 19, 20 or 21 of any of the sequences
as represented
in SEQ ID NOs: 1, 3, 5, 7, 9, 11, 13, 15, 17, 19, 21, 23, 49- 158, 159, 160,
163, 168, 173,
52
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
178, 183, 188, 193, 198, 203, 208, 215, 220, 225, 230, 247, 249, 251, 253,
255, 257, 259,
275-472, 473, 478, 483, 488, 493, 498, 503, 513, 515, 517, 519, 521, 533 -
575, 576, 581,
586, 591, 596, 601, 603, 605, 607, 609, 621 - 767, 768, 773, 778, 783, 788,
793, 795, 797,
, 799, 801, 813 - 862, 863, 868, 873, 878, 883, 888, 890, 892, 894, 896, 908-
1040, 1041,
1046, 1051, 1056, 1061, 1071, 1073, 1075, 1077, 1079, 1081, 1083, 1085, 1087,
1089, 1091,
1093, 1095, 1097, 1099, 1101, 1103, 1105, 1107, 1109, 1111, 1113, 1161 - 1571,
1572, 1577,
1582, 1587, 1592, 1597, 1602, 1607, 1612, 1617, 1622, 1627, 1632, 1637, 1642,
1647, 1652,
1657, 1662, 1667, 1672, 1677, 1682, 1684, 1686, 1688, 1690, 1692, 1694, 1696,
1698, 1700,
1702, 1704, 1730 - 2039, 2040, 2045, 2050, 2055, 2060, 2065, 2070, 2075, 2080,
2085,
2090, 2095, 2100, 2102, 2104, 2106, 2108, 2120 - 2338, 2339, 2344, 2349, 2354,
2359,
2364, 2366, 2368, 2370, 2372, 2384 - 2460, 2461, 2466, 2471, 2476 and 2481. A
non-
limiting list of fungal orthologs genes or sequences comprising at least a
fragment of 15,
preferably at least 17 bp of one of the sequences of the invention is given in
Tables 6.
In a further embodiment, a dsRNA molecule of the invention comprises any of
SEQ
ID NOs: 1,3, 5, 7,9, 11, 13, 15, 17, 19, 21, 23, 49 - 158, 159, 160, 163, 168,
173, 178, 183,
188, 193, 198, 203, 208, 215, 220, 225, 230, 247, 249, 251, 253, 255, 257,
259, 275-472, 473,
478, 483, 488, 493, 498, 503, 513, 515, 517, 519, 521, 533 - 575, 576, 581,
586, 591, 596,
601, 603, 605, 607, 609, 621 - 767, 768, 773, 778, 783, 788, 793, 795, 797,
799, 801, 813 -
862, 863, 868, 873, 878, 883, 888, 890, 892, 894, 896, 908- 1040, 1041, 1046,
1051, 1056,
1061, 1071, 1073, 1075, 1077, 1079, 1081, 1083, 1085, 1087, 1089, 1091, 1093,
1095, 1097,
1099, 1101, 1103, 1105, 1107, 1109, 1111, 1113, 1161 - 1571, 1572, 1577, 1582,
1587, 1592,
1597, 1602, 1607, 1612, 1617, 1622, 1627, 1632, 1637, 1642, 1647, 1652, 1657,
1662, 1667,
1672, 1677, 1682, 1684, 1686, 1688, 1690, 1692, 1694, 1696, 1698, 1700, 1702,
1704, 1730
- 2039, 2040, 2045, 2050, 2055, 2060, 2065, 2070, 2075, 2080, 2085, 2090,
2095, 2100,
2102, 2104, 2106, 2108, 2120 - 2338, 2339, 2344, 2349, 2354, 2359, 2364, 2366,
2368,
2370, 2372, 2384 - 2460, 2461, 2466, 2471, 2476 and 2481, though the sequences
set forth in
SEQ ID NOs: 1, 3, 5, 7, 9, 11, 13, 15, 17, 19, 21, 23, 49- 158, 159, 160, 163,
168, 173, 178,
183, 188, 193, 198, 203, 208, 215, 220, 225, 230, 247, 249, 251, 253, 255,
257, 259, 275-472,
473, 478, 483, 488, 493, 498, 503, 513, 515, 517, 519, 521, 533 - 575, 576,
581, 586, 591,
596, 601, 603, 605, 607, 609, 621 - 767, 768, 773, 778, 783, 788, 793, 795,
797, 799, 801,
813 - 862, 863, 868, 873, 878, 883, 888, 890, 892, 894, 896, 908- 1040, 1041,
1046, 1051,
1056, 1061, 1071, 1073, 1075, 1077, 1079, 1081, 1083, 1085, 1087, 1089, 1091,
1093, 1095,
1097, 1099, 1101, 1103, 1105, 1107, 1109, 1111, 1113, 1161 - 1571, 1572, 1577,
1582, 1587,
1592, 1597, 1602, 1607, 1612, 1617, 1622, 1627, 1632, 1637, 1642, 1647, 1652,
1657, 1662,
53
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
1667, 1672, 1677, 1682, 1684, 1686, 1688, 1690, 1692, 1694, 1696, 1698, 1700,
1702, 1704,
1730 - 2039, 2040, 2045, 2050, 2055, 2060, 2065, 2070, 2075, 2080, 2085, 2090,
2095,
2100, 2102, 2104, 2106, 2108, 2120 - 2338, 2339, 2344, 2349, 2354, 2359, 2364,
2366,
2368, 2370, 2372, 2384 - 2460, 2461, 2466, 2471, 2476 and 2481 are not
limiting. A dsRNA
molecule of the invention can comprise any contiguous target gene from a pest
species (e.g.,
about 15 to about 25 or more, or about 15, 16, 17, 18, 19, 20, 21, 22, 23, 24,
or 25 or more
contiguous nucleotides).
By "isolated" nucleic acid molecule(s) is intended a nucleic acid molecule,
DNA or
RNA, which has been removed from its native environment. For example,
recombinant DNA
molecules contained in a DNA construct are considered isolated for the
purposes of the
present invention. Further examples of isolated DNA molecules include
recombinant DNA
molecules maintained in heterologous host cells or purified (partially or
substantially) DNA
molecules in solution. Isolated RNA molecules include in vitro RNA transcripts
of the DNA
molecules of the present invention. Isolated nucleic acid molecules, according
to the present
invention, further include such molecules produced synthetically.
Nucleic acid molecules of the present invention may be in the form of RNA,
such as
mRNA, or in the form of DNA, including, for instance, cDNA and genomic DNA
obtained
by cloning or produced synthetically. The DNA or RNA may be double-stranded or
single-
stranded. Single-stranded DNA may be the coding strand, also known as the
sense strand, or
it may be the non-coding strand, also referred to as the anti-sense strand.
VI. Sequence Analysis
Unless otherwise indicated, all nucleotide sequences determined by sequencing
a
DNA molecule herein were determined using an automated DNA sequencer (such as
the
Model 373 from Applied Biosystems, Inc.). Therefore, as is known in the art
for any DNA
sequence determined by this automated approach, any nucleotide sequence
determined herein
may contain some errors. Nucleotide sequences determined by automation are
typically at
least about 95% identical, more typically at least about 96% to at least about
99.9% identical
to the actual nucleotide sequence of the sequenced DNA molecule. The actual
sequence can
be more precisely determined by other approaches including manual DNA
sequencing
methods well known in the art. As is also known in the art, a single insertion
or deletion in a
determined nucleotide sequence compared to the actual sequence will cause a
frame shift in
translation of the nucleotide sequence such that the predicted amino acid
sequence encoded
by a determined nucleotide sequence may be completely different from the amino
acid
54
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
sequence actually encoded by the sequenced DNA molecule, beginning at the
point of such
an insertion or deletion.
In another aspect, the invention provides an isolated nucleic acid molecule
comprising
a polynucleotide which hybridizes under stringent hybridization conditions to
a portion of the
polynucleotide in a nucleic acid molecule of the invention described above. By
a
polynucleotide which hybridizes to a "portion" of a polynucleotide is intended
a
polynucleotide (either DNA or RNA) hybridizing to at least about 15
nucleotides, and more
preferably at least about 20 nucleotides, and still more preferably at least
about 30
nucleotides, and even more preferably more than 30 nucleotides of the
reference
polynucleotide. These fragments that hybridize to the reference fragments are
useful as
diagnostic probes and primers. For the purpose of the invention, two sequences
hybridize
when they form a double-stranded complex in a hybridization solution of 6X
SSC, 0.5% SDS,
5X Denhardt's solution and 100 jig of non-specific carrier DNA. See Ausubel et
al., section 2.9,
supplement 27 (1994). Sequences may hybridize at "moderate stringency," which
is defined as
a temperature of 60 C in a hybridization solution of 6X SSC, 0.5% SDS, 5X
Denhardt's
solution and 1001..ig of non-specific carrier DNA. For "high stringency"
hybridization, the
temperature is increased to 68 C. Following the moderate stringency
hybridization reaction, the
nucleotides are washed in a solution of 2X SSC plus 0.05% SDS for five times
at room
temperature, with subsequent washes with 0.1X SSC plus 0.1% SDS at 60 C for
lh. For high
stringency, the wash temperature is increased to 68 C. For the purpose of the
invention,
hybridized nucleotides are those that are detected using 1 ng of a
radiolabeled probe having a
specific radioactivity of 10,000 cpm/ng, where the hybridized nucleotides are
clearly visible
following exposure to X-ray film at -70 C for no more than 72 hours.
The present application is directed to such nucleic acid molecules which are
at least
60 %, 65 %, 70 %, 75 %, 80 %, 85 %, 90%, 95%, 96%, 97%, 98%, 99% or 100%
identical to
a nucleic acid sequence described in any of SEQ ID NOs: 1, 3, 5, 7, 9, 11, 13,
15, 17, 19, 21,
23, 49- 158, 159, 160, 163, 168, 173, 178, 183, 188, 193, 198, 203, 208, 215,
220, 225, 230,
247, 249, 251, 253, 255, 257, 259, 275-472, 473, 478, 483, 488, 493, 498, 503,
513, 515, 517,
519, 521, 533 - 575, 576, 581, 586, 591, 596, 601, 603, 605, 607, 609, 621 -
767, 768, 773,
778, 783, 788, 793, 795, 797, 799, 801, 813 - 862, 863, 868, 873, 878, 883,
888, 890, 892,
894, 896, 908- 1040, 1041, 1046, 1051, 1056, 1061, 1071, 1073, 1075, 1077,
1079, 1081,
1083, 1085, 1087, 1089, 1091, 1093, 1095, 1097, 1099, 1101, 1103, 1105, 1107,
1109, 1111,
1113, 1161 - 1571, 1572, 1577, 1582, 1587, 1592, 1597, 1602, 1607, 1612, 1617,
1622, 1627,
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
1632, 1637, 1642, 1647, 1652, 1657, 1662, 1667, 1672, 1677, 1682, 1684, 1686,
1688, 1690,
1692, 1694, 1696, 1698, 1700, 1702, 1704, 1730 - 2039, 2040, 2045, 2050, 2055,
2060,
2065, 2070, 2075, 2080, 2085, 2090, 2095, 2100, 2102, 2104, 2106, 2108, 2120 -
2338,
2339, 2344, 2349, 2354, 2359, 2364, 2366, 2368, 2370, 2372, 2384 - 2460, 2461,
2466,
2471, 2476 and 2481. Preferred, however, are nucleic acid molecules which are
at least 95%,
96%, 97%, 98%, 99% or 100% identical to the nucleic acid sequence shown in of
SEQ ID
NOs: 1, 3, 5, 7, 9, 11, 13, 15, 17, 19, 21, 23, 49 - 158, 159, 160, 163, 168,
173, 178, 183, 188,
193, 198, 203, 208, 215, 220, 225, 230, 247, 249, 251, 253, 255, 257, 259, 275-
472, 473, 478,
483, 488, 493, 498, 503, 513, 515, 517, 519, 521, 533 - 575, 576, 581, 586,
591, 596, 601,
603, 605, 607, 609, 621 - 767, 768, 773, 778, 783, 788, 793, 795, 797, 799,
801, 813 - 862,
863, 868, 873, 878, 883, 888, 890, 892, 894, 896, 908- 1040, 1041, 1046, 1051,
1056, 1061,
1071, 1073, 1075, 1077, 1079, 1081, 1083, 1085, 1087, 1089, 1091, 1093, 1095,
1097, 1099,
1101, 1103, 1105, 1107, 1109, 1111, 1113, 1161 - 1571, 1572, 1577, 1582, 1587,
1592, 1597,
1602, 1607, 1612, 1617, 1622, 1627, 1632, 1637, 1642, 1647, 1652, 1657, 1662,
1667, 1672,
1677, 1682, 1684, 1686, 1688, 1690, 1692, 1694, 1696, 1698, 1700, 1702, 1704,
1730 -
2039, 2040, 2045, 2050, 2055, 2060, 2065, 2070, 2075, 2080, 2085, 2090, 2095,
2100, 2102,
2104, 2106, 2108, 2120 - 2338, 2339, 2344, 2349, 2354, 2359, 2364, 2366, 2368,
2370,
2372, 2384 -2460, 2461, 2466, 2471, 2476 and 2481. Differences between two
nucleic acid
sequences may occur at the 5' or 3' terminal positions of the reference
nucleotide sequence or
anywhere between those terminal positions, interspersed either individually
among
nucleotides in the reference sequence or in one or more contiguous groups
within the
reference sequence.
As a practical matter, whether any particular nucleic acid molecule is at
least 95%,
96%, 97%, 98% or 99% identical to a reference nucleotide sequence refers to a
comparison
made between two molecules using standard algorithms well known in the art and
can be
determined conventionally using publicly available computer programs such as
the BLASTN
algorithm. See Altschul et al., Nucleic Acids Res. 25:3389-3402 (1997).
In one embodiment of the invention, a nucleic acid comprises an antisense
strand
having about 15 to about 30 (e.g., about 15, 16, 17, 18, 19, 20, 21, 22, 23,
24, 25, 26, 27, 28,
29, or 30) nucleotides, wherein the antisense strand is complementary to a RNA
sequence or
a portion thereof encoding a protein that controls cell cycle or homologous
recombination,
and wherein said siNA further comprises a sense strand having about 15 to
about 30 (e.g.,
about 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, or 30)
nucleotides, and wherein
56
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
said sense strand and said antisense strand are distinct nucleotide sequences
where at least
about 15 nucleotides in each strand are complementary to the other strand.
In one embodiment, the present invention provides double-stranded nucleic acid
molecules of that mediate RNA interference gene silencing. In another
embodiment, the
siNA molecules of the invention consist of duplex nucleic acid molecules
containing about
15 to about 30 base pairs between oligonucleotides comprising about 15 to
about 30 (e.g.,
about 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, or 30)
nucleotides. In yet
another embodiment, siNA molecules of the invention comprise duplex nucleic
acid
molecules with overhanging ends of about 1 to about 32 (e.g., about 1, 2, or
3) nucleotides,
for example, about 21-nucleotide duplexes with about 19 base pairs and 3'-
terminal
mononucleotide, dinucleotide, or trinucleotide overhangs. In yet another
embodiment, siNA
molecules of the invention comprise duplex nucleic acid molecules with blunt
ends, where
both ends are blunt, or alternatively, where one of the ends is blunt.
An siNA molecule of the present invention may comprise modified nucleotides
while
maintaining the ability to mediate RNAi. The modified nucleotides can be used
to improve in
vitro or in vivo characteristics such as stability, activity, and/or
bioavailability. For example,
a siNA molecule of the invention can comprise modified nucleotides as a
percentage of the
total number of nucleotides present in the siNA molecule. As such, a siNA
molecule of the
invention can generally comprise about 5% to about 100% modified nucleotides
(e.g., about
5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%,
85%, 90%, 95% or 100% modified nucleotides). The actual percentage of modified
nucleotides present in a given siNA molecule will depend on the total number
of nucleotides
present in the siNA. If the siNA molecule is single stranded, the percent
modification can be
based upon the total number of nucleotides present in the single stranded siNA
molecules.
Likewise, if the siNA molecule is double stranded, the percent modification
can be based
upon the total number of nucleotides present in the sense strand, antisense
strand, or both the
sense and antisense strands.
VII. Nucleic Acid Constructs
A recombinant nucleic acid vector may, for example, be a linear or a closed
circular
plasmid. The vector system may be a single vector or plasmid or two or more
vectors or
plasmids that together contain the total nucleic acid to be introduced into
the genome of the
bacterial host. In addition, a bacterial vector may be an expression vector.
Nucleic acid
molecules as set forth in SEQ ID NOs: 1, 3, 5, 7, 9, 11, 13, 15, 17, 19, 21,
23, 49- 158, 159,
57
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
160, 161, 162, 163, 168, 173, 178, 183, 188, 193, 198, 203, 208, 215, 220,
225, 230, 240 -
246, 247, 249, 251, 253, 255, 257, 259, 275-472, 473, 478, 483, 488, 493, 498,
503, 508-512,
513, 515, 517, 519, 521, 533 - 575, 576, 581, 586, 591, 596, 601, 603, 605,
607, 609, 621 -
767, 768, 773, 778, 783, 788, 793, 795, 797, 799, 801, 813 -862, 863, 868,
873, 878, 883,
888, 890, 892, 894, 896, 908- 1040, 1041, 1046, 1051, 1056, 1061, 1066-1070,
1071, 1073,
1075, 1077, 1079, 1081, 1083, 1085, 1087, 1089, 1091, 1093, 1095, 1097, 1099,
1101, 1103,
1105, 1107, 1109, 1111, 1113, 1161 - 1571, 1572, 1577, 1582, 1587, 1592, 1597,
1602, 1607,
1612, 1617, 1622, 1627, 1632, 1637, 1642, 1647, 1652, 1657, 1662, 1667, 1672,
1677, 1682,
1684, 1686, 1688, 1690, 1692, 1694, 1696, 1698, 1700, 1702, 1704, 1730 - 2039,
2040,
2045, 2050, 2055, 2060, 2065, 2070, 2075, 2080, 2085, 2090, 2095, 2100, 2102,
2104, 2106,
2108, 2120 - 2338, 2339, 2344, 2349, 2354, 2359, 2364, 2366, 2368, 2370, 2372,
2384 -
2460, 2461, 2466, 2471, 2476 and 2481, or fragments thereof can, for example,
be suitably
inserted into a vector under the control of a suitable promoter that functions
in one or more
microbial hosts to drive expression of a linked coding sequence or other DNA
sequence.
Many vectors are available for this purpose, and selection of the appropriate
vector will
depend mainly on the size of the nucleic acid to be inserted into the vector
and the particular
host cell to be transformed with the vector. Each vector contains various
components
depending on its function (amplification of DNA or expression of DNA) and the
particular
host cell with which it is compatible. The vector components for bacterial
transformation
generally include, but are not limited to, one or more of the following: a
signal sequence, an
origin of replication, one or more selectable marker genes, and an inducible
promoter
allowing the expression of exogenous DNA.
Promoters
"Operably linked", as used in reference to a regulatory sequence and a
structural
nucleotide sequence, means that the regulatory sequence causes regulated
expression of the
linked structural nucleotide sequence. "Regulatory sequences" or "control
elements" refer to
nucleotide sequences located upstream (5' noncoding sequences), within, or
downstream (3'
non-translated sequences) of a structural nucleotide sequence, and which
influence the timing
and level or amount of transcription, RNA processing or stability, or
translation of the
associated structural nucleotide sequence. Regulatory sequences may include
promoters,
translation leader sequences, introns, enhancers, stem-loop structures,
repressor binding
sequences, and polyadenylation recognition sequences and the like.
An expression vector for producing a mRNA can also contain an inducible
promoter
that is recognized by the host bacterial organism and is operably linked to
the nucleic acid
58
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
encoding, for example, the nucleic acid molecule coding the D. v. virgifera
mRNA or
fragment thereof of interest. Inducible promoters suitable for use with
bacterial hosts include
g-lactamase promoter, E. coli phage PL and PR promoters, and E. coli galactose
promoter,
arabinose promoter, alkaline phosphatase promoter, tryptophan (trp) promoter,
and the
lactose operon promoter and variations thereof and hybrid promoters such as
the tac
promoter. However, other known bacterial inducible promoters. are suitable.
The invention contemplates promoters that function in different plant species.
Promoters useful for expression of polypeptides in plants include those that
are inducible,
viral, synthetic, or constitutive as described in Odell et al. (1985), and/or
promoters that are
temporally regulated, spatially regulated, and spatio-temporally regulated.
Preferred
promoters include the enhanced CaMV35S promoters, and the FMV35S promoter. For
the
purpose of the present invention, e.g., for optimum control of species that
feed on roots, it
may be preferable to achieve the highest levels of expression of these genes
within the roots
of plants. A number of root-enhanced promoters have been identified and are
known in the
art (Lu et al., 2000; U.S. Patent No. 5,837,848 and 6,489,542).
In one embodiment the plant transformation vector comprises an isolated and
purified
DNA molecule comprising a promoter operatively linked to one or more
nucleotide
sequences of the present invention. The nucleotide sequence is selected from
the group
consisting of SEQ ID NOs: 1, 3, 5, 7,9, 11, 13, 15, 17, 19, 21, 23, 49 - 158,
159, 160, 161,
162, 163, 168, 173, 178, 183, 188, 193, 198, 203, 208, 215, 220, 225, 230, 240
- 246, 247,
249, 251, 253, 255, 257, 259, 275-472, 473, 478, 483, 488, 493, 498, 503, 508-
512, 513, 515,
517, 519, 521, 533 - 575, 576, 581, 586, 591, 596, 601, 603, 605, 607, 609,
621 - 767, 768,
773, 778, 783, 788, 793, 795, 797, 799, 801, 813 - 862, 863, 868, 873, 878,
883, 888, 890,
892, 894, 896, 908- 1040, 1041, 1046, 1051, 1056, 1061, 1066-1070, 1.071,
1073, 1075,
1077, 1079, 1081, 1083, 1085, 1087, 1089, 1091, 1093, 1095, 1097, 1099, 1101,
1103, 1105,
1107, 1109, 1111, 1113, 1161 -1571, 1572, 1577, 1582, 1587, 1592, 1597, 1602,
1607, 1612,
1617, 1622, 1627, 1632, 1637, 1642, 1647, 1652, 1657, 1662, 1667, 1672, 1677,
1682, 1684,
1686, 1688, 1690, 1692, 1694, 1696, 1698, 1700, 1702, 1704, 1730 - 2039, 2040,
2045,
2050, 2055, 2060, 2065, 2070, 2075, 2080, 2085, 2090, 2095, 2100, 2102, 2104,
2106, 2108,
2120 - 2338, 2339, 2344, 2349, 2354, 2359, 2364, 2366, 2368, 2370, 2372, 2384 -
2460,
2461, 2466, 2471, 2476 and 2481. The nucleotide sequence includes a segment
coding all or
part of an RNA present within a targeted pest RNA transcript and may comprise
inverted
repeats of all or a part of a targeted pest RNA. The DNA molecule comprising
the expression
vector may also contain a functional intron sequence positioned either
upstream of the coding
59
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
sequence or even within the coding sequence, and may also contain a five prime
(5')
untranslated leader sequence (i.e., a UTR or 5'-UTR) positioned between the
promoter and
the point of translation initiation.
Selectable marker genes
A recombinant DNA vector or construct of the present invention will typically
comprise a selectable marker that confers a selectable phenotype on plant
cells. Selectable
markers may also be used to select for plants or plant cells that contain the
exogenous nucleic
acids encoding polypeptides or proteins of the present invention. The marker
may encode
biocide resistance, antibiotic resistance (e.g., kanamycin, G418 bleomycin,
hygromycin, etc.),
or herbicide resistance (e.g., glyphosate, etc.). Examples of selectable
markers include, but
are not limited to, a neo gene which codes for kanamycin resistance and can be
selected for
using kanamycin, G418, etc., a bar gene which codes for bialaphos resistance;
a mutant EPSP
synthase gene which encodes glyphosate resistance; a nitrilase gene which
confers resistance
to bromoxynil; a mutant acetolactate synthase gene (ALS) which confers
imidazolinone or
sulfonylurea resistance; and a methotrexate resistant DHFR gene. Examples of
such
selectable markers are illustrated in U.S. Patents 5,550,318; 5,633,435;
5,780,708 and
6,118,047.
A recombinant vector or construct of the present invention may also include a
screenable marker. Screenable markers may be used to monitor expression.
Exemplary
screenable markers include a 13-glucuronidase or uidA gene (GUS) which encodes
an enzyme
for which various chromogenic substrates are known (Jefferson, 1987; Jefferson
et al., 1987);
an R-locus gene, which encodes a product that regulates the production of
anthocyanin
pigments (red color) in plant tissues (Dellaporta et al., 1988); a p-lactamase
gene (Sutcliffe et
al., 1978), a gene which encodes an enzyme for which various chromogenic
substrates are
known (e.g., PADAC, a chromogenic cephalosporin); a luciferase gene (Ow et
al., 1986) a
xylE gene (Zukowsky et al., 1983) which encodes a catechol dioxygenase that
can convert
chromogenic catechols; an a-amylase gene (Ikatu et al., 1990); a tyrosinase
gene (Katz et al.,
1983) which encodes an enzyme capable of oxidizing tyrosine to DOPA and
dopaquinone
which in turn condenses to melanin; an a -galactosidase, which catalyzes a
chromogenic a-
gal actose substrate.
Preferred plant transformation vectors include those derived from a Ti plasmid
of
Agrobacterium tumefaciens (e.g. U.S. Patent Nos. 4,536,475, 4,693,977,
4,886,937,
5,501,967 and EP 0 122 791). Agrobacterium rhizogenes plasmids (or "Ri") are
also useful
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
and known in the art. Other preferred plant transformation vectors include
those disclosed,
e.g., by Herrera-Estrella (1983); Bevan (1983), Klee (1985) and EP 0 120 516.
In general it is preferred to introduce a functional recombinant DNA at a non-
specific
location in a plant genome. In special cases it may be useful to insert a
recombinant DNA
construct by site-specific integration. Several site-specific recombination
systems exist
which are known to function implants include cre-lox as disclosed in U.S.
Patent 4,959,317
and FLP-FRT as disclosed in U.S. Patent 5,527,695.
A transformation vector can be readily prepared using methods available in the
art.
The transformation vector comprises one or more nucleotide sequences that
is/are capable of
being transcribed to an RNA molecule and that is/are substantially homologous
and/or
complementary to one or more nucleotide sequences encoded by the genome of the
insect,
such that upon uptake of the RNA there is down-regulation of expression of at
least one of
the respective nucleotide sequences of the genome of the insect.
A plant transformation vector may contain sequences from more than one gene,
thus
allowing production of more than one dsRNA for inhibiting expression of two or
more genes
in cells of a target pest. One skilled in the art will readily appreciate that
segments of DNA
whose sequence corresponds to that present in different genes can be combined
into a single
composite DNA segment for expression in a transgenic plant. Alternatively, a
plasmid of the
present invention already containing at least one DNA segment can be modified
by the
sequential insertion of additional DNA segments between the enhancer and
promoter and
terminator sequences. In the insect control agent of the present invention
designed for the
inhibition of multiple genes, the genes to be inhibited can be obtained from
the same insect
species in order to enhance the effectiveness of the insect control agent. In
certain
embodiments, the genes can be derived from different insects in order to
broaden the range of
insects against which the agent is effective. When multiple genes are targeted
for
suppression or a combination of expression and suppression, a polycistronic
DNA element
can be fabricated as illustrated and disclosed in Fillatti, Application
Publication No. US
2004-0029283.
The transformation vector may be termed a dsDNA construct and may also be
defined
as a recombinant molecule, an insect control agent, a genetic molecule or a
chimeric genetic
construct. A chimeric genetic construct of the present invention may comprise,
for example,
nucleotide sequences encoding one or more antisense transcripts, one or more
sense
transcripts, one or more of each of the aforementioned, wherein all or part of
a transcript
61
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
therefrom is homologous to all or part of an RNA molecule comprising an RNA
sequence
encoded by a nucleotide sequence within the genome of an insect.
VIII. Plants for Genetic Engineering
A "plant" is any of various photosynthetic, eukaryotic, multicellular
organisms of the
kingdom Plantae characteristically producing embryos, containing chloroplasts,
and having
cellulose cell walls. A part of a plant, i.e., a "plant tissue" may be treated
according to the
methods of the present invention to produce a transgenic plant. Many suitable
plant tissues
can be transformed according to the present invention and include, but are not
limited to,
somatic embryos, pollen, leaves, stems, calli, stolons, microtubers, and
shoots.
Thus, the present invention envisions the transformation of angiosperm and
gymnosperm plants such as acacia, alfalfa, apple, apricot, artichoke, ash
tree, asparagus,
avocado, banana, barley, beans, beet, birch, beech, blackberry, blueberry,
broccoli, brussels
sprouts, cabbage, canola, cantaloupe, carrot, cassava, cauliflower, cedar, a
cereal, celery,
chestnut, cherry, chinese cabbage, citrus, clemintine, clover, coffee, corn,
cotton, cowpea,
cucumber, cypress, eggplant, elm, endive, eucalyptus, fennel, figes, fir,
geranium, grape,
grapefruit, groundnuts, ground cherry, gum hemlock, hickory, kale, kiwifruit,
kohlrabi, larch,
lettuce, leek, lemon, lime, locust, pine, maidenhair, maize, mango, maple,
melon, millet,
mushroom, mustard, nuts, oak, oats, okra, onion, orange, an ornamental plant
or flower or
tree, papaya, palm, parsley, parsnip, pea, peach, peanut, pear, peat, pepper,
persimmon,
pigeon pea, pine, pineapple, plantain, plum, pomegranate, potato, pumpkin,
radicchio, radish,
rapeseed, raspberry, rice, rye, sorghumõ sallow, soybean, spinach, spruce,
squash,
strawberry, sugarbeet, sugarcane, sunflower, sweet potato, sweet corn,
tangerine, tea,
tobacco, tomato, trees, triticale, turf grasses, turnips, a vine, walnut,
watercress, watermelon,
wheat, yams, yew, and zucchini.
According to the present invention "plant tissue" also encompasses plant
cells. Plant
cells include suspension cultures, callus, embryos, meristematic regions,
callus tissue, leaves,
roots, shoots, gametophytes, sporophytes, pollen, seeds and microspores. Plant
tissues may
be at various stages of maturity and may be grown in liquid or solid culture,
or in soil or
suitable media in pots, greenhouses or fields. A plant tissue also refers to
any clone of such a
plant, seed, progeny, propagule whether generated sexually or asexually, and
descendents of
any of these, such as cuttings or seed.
62
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
"Progeny" of the present invention, such as the progeny of a transgenic plant,
is one
that is born of, begotten by, or derived from a plant or the transgenic plant.
Thus, a
"progeny" plant, i.e., an "Fl" generation plant is an offspring or a
descendant of the
transgenic plant produced by the inventive methods. A progeny of a transgenic
plant may
contain in at least one, some, or all of its cell genomes, the desired
polynucleotide that was
integrated into a cell of the parent transgenic plant by the methods described
herein. Thus, the
desired polynucleotide is "transmitted" or "inherited" by the progeny plant.
The desired
polynucleotide that is so inherited in the progeny plant may reside within a T-
DNA construct,
which also is inherited by the progeny plant from its parent. The term
"progeny" as used
herein, also may be considered to be the offspring or descendants of a group
of plants.
A "seed" may be regarded as a ripened plant ovule containing an embryo, and a
propagative part of a plant, as a tuber or spore. Seed may be incubated prior
to
Agrobacterium-mediated transformation, in the dark, for instance, to
facilitate germination.
Seed also may be sterilized prior to incubation, such as by brief treatment
with bleach. The
resultant seedling can then be exposed to a desired strain of Agrobacterium or
other suitable
bacterium for transformation.
The present invention extends to methods as described herein, wherein the
insect is
Leptinotarsa decemlineata (Colorado potato beetle) and the plant is potato,
eggplant, tomato,
pepper, tobacco, ground cherry or rice, corn or cotton.
The present invention extends to methods as described herein, wherein the
insect is
Phaedon cochleariae (mustard leaf beetle) and the plant is mustard, chinese
cabbage, turnip
greens, collard greens or bok choy.
The present invention extends to methods as described herein, wherein the
insect is
Epilachna varivetis (Mexican bean beetle) and the plant is beans, field beans,
garden beans,
snap beans, lima beans, mung beans, string beans, black-eyed beans, velvet
bean, soybeans,
cowpea, pigeon pea, clover or alfalfa.
The present invention extends to methods as described herein, wherein the
insect is
Anthonomus grandis (cotton boll weevil) and the plant is cotton.
The present invention extends to methods as described herein, wherein the
insect is
Tribolium castaneum (red flour beetle) and the plant is in the form of stored
grain products
such as flour, cereals, meal, crackers, beans, spices, pasta, cake mix, dried
pet food, dried
flowers, chocolate, nuts, seeds, and even dried museum specimens
63
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
The present invention extends to methods as described herein, wherein the
insect is
Myzus persicae (green peach aphid) and the plant is a tree such as Prunus,
particularly peach,
apricot and plum; a vegetable crop of the families Solanaceae, Chenopodiaceae,
Compositae,
Cruciferae, and Cucurbitaceae, including but not limited to, artichoke,
asparagus, bean,
beets, broccoli, Brussels sprouts, cabbage, carrot, cauliflower, cantaloupe,
celery, corn,
cucumber, fennel, kale, kohlrabi, turnip, eggplant, lettuce, mustard, okra,
parsley, parsnip,
pea, pepper, potato, radish, spinach, squash, tomato, turnip, watercress, and
watermelon; a
field crops such as, but not limited to, tobacco, sugar beet, and sunflower; a
flower crop or
other ornamental plant.
The present invention extends to methods as described herein, wherein the
insect is
Nilaparvata lugens and the plant is a rice species n
The present invention extends to methods as described herein, wherein the
insect is
Chilo suppressalis (rice striped stem borer) and the plant is a rice plant,
bareley, sorghum,
maize, wheat or a grass.
The present invention extends to methods as described herein, wherein the
insect is
Plutella xylostella (Diamondback moth) and the plant is a Brassica species
such as, but not
limited to cabbage, chinese cabbage, Brussels sprouts, kale, rapeseed,
broccoli, cauliflower,
turnip, mustard or radish.
The present invention extends to methods as described herein, wherein the
insect is
Acheta domesticus (house cricket) and the plant is any plant as described
herein or any
organic matter.
IX. Methods for Genetic Engineering
The present invention contemplates introduction of a nucleotide sequence into
a plant
to achieve pest inhibitory levels of expression of one or more dsRNA
molecules. The
inventive pol3mucleotides and polypeptides may be introduced into a host plant
cell by standard
procedures known in the art for introducing recombinant sequences into a
target host cell. Such
procedures include, but are not limited to, transfection, infection,
transformation, natural
uptake, calcium phosphate, electroporation, microinjection biolistics and
microorganism-
mediated transformation protocols. See, for example, Miki et al., 1993,
"Procedure for
Introducing Foreign DNA into Plants", In: Methods in Plant Molecular Biology
and
Biotechnology, Glick and Thompson, eds., CRC Press, Inc., Boca Raton, pages 67-
88. The
methods chosen vary with the host plant.
64
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
Microorganism-mediated gene transfer refers to the use of a microorganism for
introducing a foreign gene into a host plant. While Agrobacterium (Horsch et
al., Science
227:1229-31, 1985) has been widely use for transferring genes into a plant, it
is not the only
bacteria capable of transforming plants. For example, it has been shown that
several species of
bacteria outside the Agrobacterium genus can be modified to mediate gene
transfer to diverse
plants. Bacteria from two families, and three genera, Rhizobium sp. NGR234,
Sinorhizobium
meliloti and Mesorhizobium loti, were made competent for gene transfer by
acquisition of
both a disarmed Ti plasmid and a binary vector. Broothaerts, W. et al. Nature
Feb 10, 433
(7026):629-633 (2005). Stable transformation of three plant species, tobacco,
rice and
Arabidopsis, was achieved with these non-Agrobacterium species using leaf
disk, scutellum-
derived callus or floral dip. Id. Thus, diverse plant-associated bacteria,
when harboring a
disarmed Ti plasmid and binary vector (or presumably a co-integrate or whole
Ti plasmid),
are readily able to transfer T-DNA to plants and may be used in accordance
with the present
invention.
A transgenic plant of the present invention is one that comprises at least one
cell
genome in which an exogenous nucleic acid has been stably integrated.
According to the
present invention, a transgenic plant is a plant that comprises only one
genetically modified
cell and cell genome, or is a plant that comprises some genetically modified
cells, or is a
plant in which all of the cells are genetically modified. A transgenic plant
of the present
invention may be one that comprises expression of the desired polynucleotide,
i.e., the
exogenous nucleic acid, in only certain parts of the plant. Thus, a transgenic
plant may
contain only genetically modified cells in certain parts of its structure.
Methods for the creation of transgenic plants and expression of heterologous
nucleic
acids in plants in particular are known and may be used with the nucleic acids
provided
herein to prepare transgenic plants that exhibit reduced susceptibility to
feeding by a target
pest organism. Plant transformation vectors can be prepared, for example, by
inserting the
dsRNA producing nucleic acids disclosed herein into plant transformation
vectors and
introducing these into plants. One known vector system has been derived by
modifying the
natural gene transfer system of Agrobacteriurn tumefaciens. The natural system
comprises
large Ti (tumor-inducing)-plasmids containing a large segment, known as T-DNA,
which is
transferred to transformed plants. Another segment of the Ti plasmid, the vir
region, is
responsible for T-DNA transfer. The T-DNA region is bordered by terminal
repeats. In the
modified binary vectors the tumor-inducing genes have been deleted and the
functions of the
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
vir region are utilized to transfer foreign DNA bordered by the T-DNA border
sequences.
The T-region may also contain a selectable marker for efficient recovery of
transgenic plants
and cells, and a multiple cloning site for inserting sequences for transfer
such as a dsRNA
encoding nucleic acid.
A transgenic plant formed using Agrobacterium or other microorganism-mediated
transformation methods typically contains a recombinant nucleotide sequence
inserted into
one chromosome and is referred to as a transgenic event. Such transgenic
plants can be
referred to as being heterozygous for the inserted exogenous sequence. A
transgenic plant
homozygous with respect to a transgene can be obtained by selfing an
independent segregant
transgenic plant to produce Fl seed. One fourth of the Fl seed produced will
be homozygous
with respect to the transgene. Germinating Fl seed results in plants that can
be tested for
heterozygosity or homozygosity, typically using a SNP assay or a thermal
amplification assay
that allows for the distinction between heterozygotes and homozygotes (i.e., a
zygosity
assay).
Accordingly, the present invention also provides plants or plant cells,
comprising the
polynucleotides or polypeptides of the current invention. In one embodiment,
the plants are
angiosperms or gymnosperms. Beyond the ordinary meaning of plant, the term
"plants" is also
intended to mean the fruit, seeds, flower, strobilus etc. of the plant. The
plant of the current
invention may be a direct transfectant, meaning that the vector was introduced
directly into the
plant, such as through Agrobacterium, or the plant may be the progeny of a
transfected plant.
The progeny may also be obtained by asexual reproduction of a transfected
plant. The second
or subsequent generation plant may or may not be produced by sexual
reproduction, i.e.,
fertilization. Furthermore, the plant can be a gametophyte (haploid stage) or
a sporophyte
(diploid stage).
X. Conventional breeding/crosses
In addition to direct transformation of a plant with a recombinant nucleic
acid
construct, transgenic plants can be prepared by crossing a first plant having
a recombinant
nucleic acid construct with a second plant lacking the construct. For example,
recombinant
nucleic acid for gene suppression can be introduced into first plant line that
is amenable to
transformation to produce a transgenic plant that can be crossed with a second
plant line to
introgress the recombinant nucleic acid for gene suppression into the second
plant line.
66
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
It may be advantageous to express a recombinant nucleic acid construct in a
male-
sterile plant, for example, as a means for reducing concern about transgene
flow to
neighboring plants.
The present invention can be, in practice, combined with other insect control
traits in
a plant to achieve desired traits for enhanced control of insect infestation.
Combining insect
control traits that employ distinct modes-of-action can provide insect-
protected transgenic
plants with superior durability over plants harboring a single insect control
trait because of
the reduced probability that resistance will develop in the field.
The combination of certain dsRNA constructs with one or more pest control
protein
genes may result in synergies that enhance the pest control phenotype of a
transgenic plant.
Pest bioassays employing artificial diet- or whole plant tissue can be used to
define dose-
responses for larval mortality, for example, or growth inhibition using both
dsRNAs and pest
control proteins. One skilled in the art can test mixtures of dsRNA molecules
and pest
control proteins in bioassay to identify combinations of actives that are
synergistic and
desirable for deployment in pest-protected plants (Tabashnik, 1992). Synergy
in killing pests
has been reported between different insect control proteins (for review, see
Schnepf et al.,
1998). It is anticipated that synergies will exist between certain dsRNAs and
between certain
dsRNAs and certain insect control proteins.
XI. Quantifying inhibition of target gene expression
Inhibition of target gene expression may be quantified by measuring either the
endogenous target RNA or the protein produced by translation of the target RNA
and the
consequences of inhibition can be confirmed by examination of the outward
properties of the
cell or organism. Techniques for quantifying RNA and proteins are well known
to one of
ordinary skill in the art. Multiple selectable markers are available that
confer resistance to
ampicillin, bleomycin, chloramphenicol, gentamycin, hygromycin, kanamycin,
lincomycin,
methotrexate, phosphinothricin, puromycin, spectinomycin, rifampicin, and
tetracyclin, and
the like.
In certain embodiments gene expression is inhibited by at least 10%,
preferably by at
least 33%, more preferably by at least 50%, and yet more preferably by at
least 80%. In
particularly preferred embodiments of the invention gene expression is
inhibited by at least
80%, more preferably by at least 90%, more preferably by at least 95%, or by
at least 99%
within cells in the pest so a significant inhibition takes place. Significant
inhibition is
intended to refer to sufficient inhibition that results in a detectable
phenotype (e.g., cessation
67
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
of larval growth, paralysis or mortality, etc.) or a detectable decrease in
RNA and/or protein
corresponding to the target gene being inhibited. Although in certain
embodiments of the
invention inhibition occurs in substantially all cells of the insect, in other
preferred
embodiments inhibition occurs in only a subset of cells expressing the gene.
For example, if
the gene to be inhibited plays an essential role in cells in the insect
alimentary tract, inhibition
of the gene within these cells is sufficient to exert a deleterious effect on
the insect.
XII. Products
The invention also provides commodity products containing one or more of the
sequences of the present invention, and produced from a recombinant plant or
seed
containing one or more of the inventive nucleotide sequences. A commodity
product
containing one or more of the sequences of the present invention is intended
to include, but
not be limited to, meals, oils, crushed or whole grains or seeds of a plant,
any food product
comprising any meal, oil, or crushed or whole grain of a recombinant plant or
seed, or any
silage, fiber, paper, or other product derived from an inventive plant
containing one or more
of the sequences of the present invention. The detection of an inventive
sequence is a
= commodity product provides de facto evidence that the commodity comprises
a transgenic
plant, or portion thereof, expressing an inventive sequence for controlling
pest infestation
using dsRNA mediated gene suppression methods.
********************************
Specific examples are presented below of methods for identifying target
sequences
comprising at least of one or more double stranded RNA molecules exemplified
herein
intended to suppress an essential feature or function within the pest., as
well as for
introducing the target sequences into plants. They are meant to be exemplary
and not as
limitations on the present invention.
Example 1: Silencing C.elegans target genes in C. elegans in High Throughput
Screening
A C. elegans genome wide library was prepared in the pGN9A vector (WO
01/88121)
between two identical T7-promoters and terminators, driving its expression in
the sense and
antisense direction upon expression of the T7 polymerase, which was induced by
IPTG.
This library was transformed into the bacterial strain AB301-105 (DE3) in 96
well
plate format. For the genome wide screening, these bacterial cells were fed to
the nuclease
deficient C. elegans nuc-1(e1392) strain.
68
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
Feeding the dsRNA produced in the bacterial strain AB301-105 (DE3), to C.
elegans
nuc-1 (e1392) worms, was performed in a 96 well plate format as follows: nuc-1
eggs were
transferred to a separate plate and allowed to hatch simultaneously at 20 C
for
synchronization of the Li generation. 96 well plates were filled with 100 !IL
liquid growth
medium comprising IPTG and with 10 pit bacterial cell culture of 0D6001 AB301-
105 (DE3)
of the C. elegans dsRNA library carrying each a vector with a C. elegans
genomic fragment
for expression of the dsRNA. To each well, 4 of the synchronized Li worms were
added and
were incubated at 25 C for at least 4 to 5 days. These experiments were
performed in
quadruplicate. In the screen 6 controls were used:
- pGN29 = negative control, wild type
- pGZ1 = unc-22 = twitcher phenotype
- pGZ18 = chitin synthase = embryonic lethal
- pGZ25 = pos-1 = embryonic lethal
- pGZ59 = bli-4D = acute lethal
- ACC = acetyl co-enzyrn A carboxylase = acute lethal
After 5 days, the phenotype of the C. elegans nuc-1 (e1392) worms fed with the
bacteria producing dsRNA were compared to the phenotype of worms fed with the
empty
vector (pGN29) and the other controls. The worms that were fed with the dsRNA
were
screened for lethality (acute or larval) lethality for the parent (Po)
generation, (embryonic)
lethality for the first filial (F1) generation, or for growth retardation of
Po as follows: (i)
Acute lethality of Po: Li 's have not developed and are dead, this phenotype
never gives
progeny and the well looks quite empty; (ii) (Larval) lethality of Po: Po
died in a later stage
than Li, this phenotype also never gives progeny. Dead larvae or dead adult
worms are found
in the wells; (iii) Lethality for Fl: Ll 's have developed until adult stage
and are still alive.
This phenotype has no progeny. This can be due to sterility, embryonic
lethality (dead eggs
on the bottom of well), embryonic arrest or larval arrest (eventually ends up
being lethal): (iv)
Arrested in growth and growth retardation/delay: Compared to a well with
normal
development and normal # of progeny.
For the target sequences presented in Table 1A, it was concluded that dsRNA
mediated
silencing of the C. elegans target gene in nematodes, such as C. elegans, had
a fatal effect on
the growth and viability of the worm.
Subsequent to the above dsRNA silencing experiment, a more detailed
phenotyping
experiment was conducted in C. elegans in a high throughput format on 24 well
plates. The
dsRNA library produced in bacterial strain AB301-105 (DE3), as described
above, was fed
69
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
to C. elegans nuc-1 (e1392) worms on 24 well plates as follows: nuc-1 eggs
were transferred
to a separate plate and allowed to hatch simultaneously at 20 C for
synchronization of the Li
generation. Subsequently 100 of the synchronized Li worms were soaked in a
mixture of
500 jtL S-complete fed medium, comprising 5 p.g/mL cholesterol, 4 L/mL PEG
and 1mM
IPTG, and 500 pL of bacterial cell culture of 0D6001 AB301-105 (DE3) of the C.
elegans
dsRNA library carrying each a vector with a C. elegans genomic fragment for
expression of
the dsRNA. The soaked Li worms were rolled for 2 hours at 25 C.
After centrifugation and removal of 950 IAL of the supernatant, 5 L of the
remaining
and resuspended pellet (comprising about 10 to 15 worms) was transferred in
the middle of
each well of a 24 well plate, filled with a layer of agar LB broth. The
inoculated plate was
incubated at 25 C for 2 days. At the adult stage, 1 adult worm was singled and
incubated at
25 C for 2 days for inspection of its progeny. The other adult worms are
inspected in situ on
the original 24 well plate. These experiments were performed in quadruplicate.
This detailed phenotypic screen was repeated with a second batch of worms, the
only
difference being that the worms of the second batch were incubated at 20 C for
3 days.
The phenotype of the worms fed with C. elegans dsRNA was compared to the
phenotype of C. elegans nuc-1 (e1392) worms fed with the empty vector.
Based on this experiment, it was concluded that silencing the C. elegans
target genes as
represented in Table lA had a fatal effect on the growth and viability of the
worm and that
the target gene is essential to the viability of nematodes. Therefore these
genes are good
target genes to control (kill or prevent from growing) nematodes via dsRNA
mediated gene
silencing. Accordingly, the present invention encompasses the use of nematode
orthologs of
the above C. elegans target gene, to control nematode infestation, such as
nematode
infestation of plants.
Example 2: Identification of D. melanogaster orthologs
As described above in Example 1, numerous C. elegans lethal sequenes were
identified and can be used for identifying orthologs in other species and
genera. For
example, the C. elegans lethal sequences can be used to identify orthologous
D.
melanogasters sequences. That is, each C. elegans sequence can be querried
against a public
database, such as GenBank, for orthologous sequences in D. melanogaster.
Potential D.
melanogaster orthologs were selected that share a high degree of sequence
homology (E
value preferably less than or equal to 1E-30) and the sequences are blast
reciprocal best hits,
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
the latter means that the sequences from different organisms (e.g. C. elegans
and D.
melanogaster) are each other's top blast hits. For example, sequence C from C.
elegans is
compared against sequences in D. melanogaster using BLAST. If sequence C has
the D.
melanogaster sequence D as best hit and when D is compared to all the
sequences of C.
elegans, also turns out to be sequence C, then D and C are reciprocal best
hits. This criterium
is often used to define orthology, meaning similar sequences of different
species, having
similar function. The D. melanogaster sequence identifiers are represented in
Table 1A.
Example 3: Leptinotarsa decemlineata (Colorado potato beetle)
A. Cloning partial gene sequences from Leptinotarsa
decemlineata
High quality, intact RNA was isolated from 4 different larval stages of
Leptinotarsa
decemlineata (Colorado potato beetle; source: Jeroen van Schaik, Entocare CV
Biologische
Gewasbeschertning, Postbus 162, 6700 AD Wageningen, the Netherlands) using
TRIzol
Reagent (Cat. Nr. 15596-026/15596-018, Invitrogen, Rockville, Maryland, USA)
following
the manufacturer's instructions. Genomic DNA present in the RNA preparation
was
removed by DNase treatment following the manufacturer's instructions (Cat. Nr.
1700,
Promega). cDNA was generated using a commercially available kit (SuperScript
TM III
Reverse Transcriptase, Cat. Nr. 18080044, Invitrogen, Rockville, Maryland,
USA) following
the manufacturer's instructions.
To isolate cDNA sequences comprising a portion of the LD001, LD002, LD003,
LD006, LD007, LD010, LD011, LD014, LD015, LD016 and LD018 genes, a series of
PCR
reactions with degenerate primers were performed using Amplitaq Gold (Cat. Nr.
N8080240,
Applied Biosystems) following the manufacturer's instructions.
The sequences of the degenerate primers used for amplification of each of the
genes
are given in Table 2-LD, which displays Leptintarsa decemlineata target genes
including primer
sequences and cDNA sequences obtained. These primers were used in respective
PCR reactions
with the following conditions: 10 minutes at 95 C, followed by 40 cycles of 30
seconds at
95 C, 1 minute at 55 C and 1 minute at 72 C, followed by 10 minutes at 72 C.
The resulting
PCR fragments were analyzed on agarose gel, purified (QIAquick Gel Extraction
kit, Cat. Nr.
28706, Qiagen), cloned into the pCR8/GW/topo vector (Cat. Nr. K2500 20,
Invitrogen), and
sequenced. The sequences of the resulting PCR products are represented by the
respectiveSEQ ID NOs as given in Table 2-LD and are referred to as the partial
sequences.
71
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
The corresponding partial amino acid sequence are represented by the
respective SEQ ID
NOs as given in Table 3-LD, where the start of the reading frame is indicated
in brackets.
B. dsRNA production of the Leptinotarsa decemlineata genes
dsRNA was synthesized in milligram amounts using the commercially available
kit
T7 RibomaxTM Express RNAi System (Cat. Nr. P1700, Promega). First two separate
single
5' T7 RNA polymerase promoter templates were generated in two separate PCR
reactions,
each reaction containing the target sequence in a different orientation
relative to the T7
promoter.
For each of the target genes, the sense T7 template was generated using
specific T7
forward and specific reverse primers. The sequences of the respective primers
for amplifying
the sense template for each of the target genes are given in Table 8-LD. The
conditions in
the PCR reactions were as follows: 4 minutes at 95 C, followed by 35 cycles of
30 seconds at
95 C, 30 seconds at 55 C and 1 minute at 72 C, followed by 10 minutes at 72 C.
The anti-
sense T7 template was generated using specific forward and specific T7 reverse
primers in a
PCR reaction with the same conditions as described above. The sequences of the
respective
primers for amplifying the anti-sense template for each of the target genes
are given in Table
8-LD. The resulting PCR products were analyzed on agarose gel and purified by
PCR
purification kit (Qiaquick PCR Purification Kit, Cat. Nr. 28106, Qiagen) and
NaC104
precipitation. The generated T7 forward and reverse templates were mixed to be
transcribed
and the resulting RNA strands were annealed, DNase and RNase treated, and
purified by
sodium acetate, following the manufacturer's instructions. The sense strand of
the resulting
dsRNA for each of the target genes is given in Table 8-LD. Table 8-LD displays
sequences
for preparing ds RNA fragments of Leptinotarsa decemlineata target sequences
and
concatemer sequences, including primer sequences.
C. Cloning Leptinotarsa decemlineata genes into plant vector
pK7GWIWG2D(II)
Since the mechanism of RNA interference operates through dsRNA fragments, the
target nucleotide sequences of the target genes, as selected above, were
cloned in anti-sense
and sense orientation, separated by the intron - CmR - intron, whereby CmR is
the
chloramphenicol resistance marker, to form a dsRNA hairpin construct. These
hairpin
constructs were generated using the LR recombination reaction between an attL-
containing
72
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
entry clone (see Example 1) and an attR-containing destination vector (=
pK7GWIWG2D(II)). The plant vector pK7GWIWG2D(II) was obtained from the
VIB/Plant
Systems Biology with a Material Transfer Agreement. LR recombination reaction
was
performed by using LR ClonaseTm II enzyme mix (Cat. Nr. 11791-020, Invitrogen)
following
the manufacturer's instructions. These cloning experiments resulted in a
hairpin construct for
each of the LD002, LD006, LD007, LD010, LD011, LD014 and LD016 genes, having
either
the promoter - sense - intron - CmR - intron - antisense orientation, or
promoter - antisense -
intron - CmR - intron ¨ sense orientation, and wherein the promoter is the
plant operable 35S
promoter. The binary vector pK7GWIWG2D(II) with the 35S promoter is suitable
for
transformation into A. tumefaciens.
For LD002, a double digest with restriction enzymes BsoBI & PvuI was done on
LD002 cloned into pCR8/GW/topo (see Example 3A). For LD006, a digest with
restriction
enzyme BsoBI was done on LD006 cloned into pCR8/GW/topo (see Example 3A). For
LD007, a digest with restriction enzyme BsoBI was done on LD007 cloned into
pCR8/GW/topo (see Example 3A). For LD007, a digest with restriction enzyme
BsoBI was
done on LD007 cloned into pCR8/GW/topo (see Example 3A). For LD010, a double
digest
with restriction enzymes PvuI & Pvull was done on LD010 cloned into
pCR8/GW/topo (see
Example 3A). For LD014, a digest with restriction enzyme BsoBI was done on
LD014
cloned into pCR8/GW/topo (see Example 1). For LD016, a digest with restriction
enzyme
BsoBI was done on LD016 cloned into pCR8/GW/topo (see Example 3A). The band
containing the gene of interest flanked by the attL sites using Qiaquick Gel
Extraction Kit
(Cat. Nr. 28706, Qiagen) was purified. An amount of 150 ng of purified
fragment and 150 ng
pK7GWIWG2D(II) was added together with the LR clonase II enzyme and incubated
for at
least lh at 25 C . After proteinase K solution treatment (10 min at 37 C), the
whole
recombination mix was transformed into Top 10 chemically competent cells.
Positive clones
were selected by restriction digest analysis. The complete sequence of the
hairpin construct
for:
- LD002 (antisense - intron - CmR - intron - sense) is set forth inSEQ ID NO::
240;
- LD006 (sense - intron - CmR - intron - antisense) is set forth in SEQ ID
NO::
241;
- LD007 sense - intron - CmR - intron - antisense) is set forth in SEQ ID
NO::
242;
73
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
- LD010 (sense - intron - CmR - intron - antisense) is set forth in SEQ ID
NO::
243;
- LD011 (antisense - intron - CmR - intron - sense) is set forth in SEQ ID
NO::
244;
- LD014 (sense - intron - CmR - intron - antisense) is set forth in SEQ ID
NO::
245;
- LD016 (antisense - intron - CmR - intron - sense) is recited in SEQ m
NO:: 246;
Table 9-LD provides complete sequences for each hairpin construct.
D. Screening dsRNA targets using artificial diet for activity
against
Leptinotarsa decemlineata
Artificial diet for the Colorado potato beetle was prepared as follows
(adapted from
Gelman et al., 2001, J. Ins. Sc., vol. 1, no. 7, 1-10): water and agar were
autoclaved, and the
remaining ingredients (shown in Table 2 below) were added when the temperature
'dropped to
55 C. At this temperature, the ingredients were mixed well before the diet
was aliquoted
into 24-well plates (Nunc) with a quantity of 1 ml of diet per well. The
artificial diet was
allowed to solidify by cooling at room temperature. Diet was stored at 4 C
for up to three
weeks.
Table 2: Ingredients for Artificial diet
Ingredients Volume for 1 L
water 768m1
agar 14g
rolled oats 40g
Torula yeast 60g
lactalbumin 30g
hydrolysate
casein lOg
fructose 20g
Wesson salt mixture 4g
tomato fruit powder 12.5g
74
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
potato leaf powder 25g
b-sitosterol lg
sorbic acid 0.8g
methyl paraben 0.8g
Vanderzant vitamin 12g
mix
neomycin sulfate 0.2g
aureomycin 0.130g
rifampicin 0.130g
chloramphenicol 0.130g
nystatin 0.050g
soybean oil 2m1
wheat germ oil 2m1
Fifty 1 of a solution of dsRNA at a concentration of 1 mg/ml was applied
topically
onto the solid artificial diet in the wells of the multiwell plate. The diet
was dried in a
laminair flow cabin. Per treatment, twenty-four Colorado potato beetle larvae
(2nd stage),
with two insects per well, were tested. The plates were stored in the insect
rearing chamber at
25 2 C, 60 % relative humidity, with a 16:8 hours light:dark photoperiod.
The beetles were
assessed as live or dead every 1, 2 or 3 days. After seven days, for targets
LD006, LD007,
LD010, LD011, and LD014, the diet was replaced with fresh diet with topically
applied
dsRNA at the same concentration (1 mg/ml); for targets LD001, LD002, LD003,
LD015, and
LD016, the diet was replaced with fresh diet only. The dsRNA targets were
compared to diet
only or diet with topically applied dsRNA corresponding to a fragment of the
GFP (green
fluorescent protein) coding sequence (SEQ ID NO: 235).
Feeding artificial diet containing intact naked dsRNAs to L. decemlineata
larvae
resulted in significant increases in larval mortalities as indicated in two
separate bioassays
(Figures 1LD-2LD).
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
All dsRNAs tested resulted ultimately in 100 % mortality after 7 to 14 days.
Diet with
or without GFP dsRNA sustained the insects throughout the bioassays with very
little or no
mortality.
Typically, in all assays observed, CPB second-stage larvae fed normally on
diet with
or without dsRNA for 2 days and molted to the third larval stage. At this new
larval stage the
CPB were observed to reduce significantly or stop altogether their feeding,
with an increase
in mortality as a result.
E. Bioassay of dsRNA targets using potato leaf discs for
activity
against the Leptinotarsa decemlineata
An alternative bioassay method was employed using potato leaf material rather
than
artificial diet as food source for CPB. Discs of approximately 1.1 cm in
diameter (or 0.95
cm2) were cut out off leaves of 2 to 3-week old potato plants using a suitably-
sized cork
borer. Treated leaf discs were prepared by applying 20 pi of a 10 ng/til
solution of target
LD002 dsRNA or control gfp dsRNA on the adaxial leaf surface. The leaf discs
were allowed
to dry and placed individually in 24 wells of a 24-well multiplate (Nunc). A
single second-
larval stage CPB was placed into each well, which was then covered with tissue
paper and a
multiwell plastic lid. The plate containing the insects and leaf discs were
kept in an insect
chamber at 28 C with a photoperiod of 16h light/8h dark. The insects were
allowed to feed
on the leaf discs for 2 days after which the insects were transferred to a new
plate containing
fresh treated leaf discs. Thereafter, the insects were transferred to a plate
containing untreated
leaf discs every day until day 7. Insect mortality and weight scores were
recorded.
Feeding potato leaf discs with surface-applied intact naked dsRNA of target
LD002 to
L. decemlineata larvae resulted in a significant increase in larval
mortalities (i.e. at day 7 all
insects were dead; 100 % mortality) whereas control gfp dsRNA had no effect on
CPB
survival. Target LD002 dsRNA severely affected the growth of the larvae after
2 to 3 days
whereas the larvae fed with gfp dsRNA at the same concentration developed as
normal
(Figure 3-LD).
F. Screening shorter versions of dsRNAs using artificial diet
for
activity against Leptinotarsa decemlineata
76
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
This example exemplifies the finding that shorter (60 or 100bp) dsRNA
fragments on
their own or as concatemer constructs are sufficient in causing toxicity
towards the Colorado
potato beetle.
LD014, a target known to induce lethality in Colorado potato beetle, was
selected for
this example. This gene encodes a V-ATPase subunit E (SEQ ID NO:: 15).
A 100 base pair fragment, LD014_F1, at position 195-294 on SEQ ID NO:: 15 (SEQ
=
ID NO: 159) and a 60 base pair fragment, LD014_F2, at position 235-294 on SEQ
ID NO::
15 (SEQ ID NO: 160) were further selected. See also Table 7-LD.
Two concatemers of 300 base pairs, LD014_Cl and LD014_C2, were designed (SEQ
ID NO:: 161 and SEQ ID NO:: 162). LD014_Cl contained 3 repeats of the 100 base
pair
fragment described above (SEQ ID NO:: 159) and LD014_C2 contained 5 repeats of
the 60
base pair fragment described above (SEQ ID NO:: 160). See also Table 7-LD.
The fragments LD014_F1 and LD014_F2 were synthesized as sense and antisense
primers. These primers were annealed to create the double strands DNA
molecules prior to
cloning. Xbal and Xmal restrictions sites were included at the 5' and 3' ends
of the primers,
respectively, to facilitate the cloning.
The concatemers were made as 300 base pairs synthetic genes. Xbal and Xmal
restrictions sites were included at the 5' and 3' ends of the synthetic DNA
fragments,
respectively, to facilite the cloning.
The 4 DNA molecules, i.e. the 2 single units (LD014_F1 & LD014_F2) and the 2
concatemers (LD014 C1 & LD014 C2), were digested with Xbal and Xmal and
subcloned in
pBluescriptII SK+ linearised by Xbal and Xmal digests, resulting in
recombinant plasmids
p1, p2, p3, & p4, respectively.
Double-stranded RNA production: dsRNA was synthesized using the commercially
available kit T7 RibomaxTM Express RNAi System (Cat. Nr. P1700, Promega).
First two
separate single 5' T7 RNA polymerase promoter templates were generated in two
separate
PCR reactions, each reaction containing the target sequence in a different
orientation relative
to the T7 promoter. For LD014_F1, the sense T7 template was generated using
the specific
T7 forward primer oGBM159 and the specific reverse primer oGBM164 (represented
herein
as SEQ ID NO:: 204 and SEQ ID NO: 205, respectively) in a PCR reaction with
the
following conditions: 4 minutes at 95 C, followed by 35 cycles of 30 seconds
at 95 C, 30
seconds at 55 C and 1 minute at 72 C, followed by 10 minutes at 72 C. The anti-
sense T7
template was generated using the specific forward primer oGBM163 and the
specific T7
reverse primer oGBM160 (represented herein as SEQ ID NO: 206 and SEQ ID NO::
207,
77
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
respectively) in a PCR reaction with the same conditions as described above.
The resulting
PCR products were analyzed on agarose gel and purified by PCR purification kit
(Qiaquick
PCR Purification Kit, Cat. Nr. 28106, Qiagen) and NaC104 precipitation. The
generated T7
forward and reverse templates were mixed to be transcribed and the resulting
RNA strands
were annealed, Dnase and Rnase treated, and purified by sodium acetate,
following the
manufacturer's instructions. The sense strand of the resulting dsRNA is herein
represented
by SEQ ID NO: 203.
For LD014_F2, the sense T7 template was generated using the specific T7
forward
primer oGBM161 and the specific reverse primer oGBM166 (represented herein as
SEQ ID
NO:: 209 and SEQ ID NO:: 210, respectively) in a PCR reaction with the
following
conditions: 4 minutes at 95 C, followed by 35 cycles of 30 seconds at 95 C, 30
seconds at
55 C and 1 minute at 72 C, followed by 10 minutes at 72 C. The anti-sense T7
template was
generated using the specific forward primer oGBM165 and the specific T7
reverse primer
oGBM162 (represented herein as SEQ ID NO:: 211 and SEQ ID NO:: 212,
respectively) in a
PCR reaction with the same conditions as described above. The resulting PCR
products were
analyzed on agarose gel and purified by PCR purification kit (Qiaquick PCR
Purification Kit,
Cat. Nr. 28106, Qiagen) and NaC104 precipitation. The generated T7 forward and
reverse
templates were mixed to be transcribed and the resulting RNA strands were
annealed, Dnase
and Rnase treated, and purified by sodium acetate, following the
manufacturer's instructions.
The sense strand of the resulting dsRNA is herein represented by SEQ ID NO:
208.
Also for the concatemers, separate single 5' T7 RNA polymerase promoter
templates
were generated in two separate PCR reactions, each reaction containing the
target sequence in
a different orientation relative to the T7 promoter. The recombinant plasmids
p3 and p4
containing LD014_C1 & LD014_C2 were linearised with Xbal or Xmal, the two
linear
fragments for each construct purified and used as template for the in vitro
transcription assay,
using the T7 promoters flanking the cloning sites. Double-stranded RNA was
prepared by in
vitro transcription using the T7 RiboMAXTm Express RNAi System (Promega). The
sense
strands of the resulting dsRNA for LD014_Cl and LD014_C2 are herein
represented by SEQ
ID NO:: 213 and2114, respectively.
Shorter sequences of target LD014 and concatemers were able to induce
lethality in
Leptinotarsa decemlineata, as shown in Figure 4-LD.
G. Screening dsRNAs at different concentrations using
artificial diet
for activity against Leptinotarsa decemlineata
78
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
Fifty .1 of a solution of dsRNA at serial ten-fold concentrations from 1 jig/
1 (for
target LD027 from 0.1 gg,/ 1)down to 0.01 ng/ 1 was applied topically onto the
solid artificial
diet in the wells of a 24-well plate (Nunc). The diet was dried in a laminair
flow cabin. Per
treatment, twenty-four Colorado potato beetle larvae (2nd stage), with two
insects per well,
were tested. The plates were stored in the insect rearing chamber at 25 2
C, 60 % relative
humidity, with a 16:8 hours light:dark photoperiod. The beetles were assessed
as live or dead
at regular intervals up to day 14. After seven days, the diet was replaced
with fresh diet with
topically applied dsRNA at the same concentrations. The dsRNA targets were
compared to
diet only.
Feeding artificial diet containing intact naked dsRNAs of different targets to
L.
decemlineata larvae resulted in high larval mortalities at concentrations as
low as between 0.1
and 10 ng dsRNA/121 as shown in Figure 5-LD.
H. Cloning of a CPS gene fragment in a vector suitable for
bacterial
production of insect-active double-stranded RNA
While any efficient bacterial promoter may be used, a DNA fragment
corresponding
to an MLB gene target was cloned in a vector for the expression of double-
stranded RNA in a
bacterial host (See WO 00/01846).
The sequences of the specific primers used for the amplification of target
genes are
provided in Table 8. The template used is the pCR8/GW/topo vector containing
any of target
sequences. The primers are used in a PCR reaction with the following
conditions: 5 minutes
at 98 C, followed by 30 cycles of 10 seconds at 98 C, 30 seconds at 55 C and 2
minutes at
72 C, followed by 10 minutes at 72 C. The resulting PCR fragment is analyzed
on agarose
gel, purified (QIAquick Gel Extraction kit, Cat. Nr. 28706, Qiagen), blunt-end
cloned into Srf
I-linearized pGNA49A vector (reference to W000188121A1), and sequenced. The
sequence
of the resulting PCR product corresponds to the respective sequence as given
in Table 8. The
recombinant vector harboring this sequence is named pGBNJ003.
The sequences of the specific primers used for the amplification of target
gene
fragment LD010 are provided in Table 8 (forward primer SEQ ID NO:: 191 and
reverse
primer SEQ ID NO:: 190). The template used was the pCR8/GW/topo vector
containing the
LD010 sequence (SEQ ID NO:: 11). The primers were used in a PCR reaction with
the
following conditions: 5 minutes at 98 C, followed by 30 cycles of 10 seconds
at 98 C, 30
seconds at 55 C and 2 minutes at 72 C, followed by 10 minutes at 72 C. The
resulting PCR
79
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
fragment was analyzed on agarose gel, purified (QIAquick Gel Extraction kit,
Cat. Nr. 28706,
Qiagen), blunt-end cloned into Srf I-linearized pGNA49A vector (reference to
WO
00/188121A1), and sequenced. The sequence of the resulting PCR product
corresponds to
SEQ ID NO:: 188 as given in Table 8. The recombinant vector harboring this
sequence was
named pGBNJ003.
I. Expression and production of a double-stranded RNA target
in
two strains of Escherichia coli: (1) AB309-105, and, (2) BL21(DE3)
The procedures described below were followed in order to express suitable
levels of
insect-active double-stranded RNA of target LD010 in bacteria. An RNaseIII-
deficient strain,
AB309-105, was used in comparison to wild-type RNaseIII-containing bacteria,
BL21(DE3).
Transformation of AB309-105 and BL21(DE3)
Three hundred ng of the plasmid was added to and gently mixed in a 50 Ill
aliquot of
ice-chilled chemically competent E. coli strain AB309-105 or BL21(DE3). The
cells were
incubated on ice for 20 minutes before subjecting them to a heat shock
treatment of 37 C for
minutes, after which the cells were placed back on ice for a further 5
minutes. Four
hundred and fifty ill of room temperature SOC medium was added to the cells
and the
suspension incubated on a shaker (250 rpm) at 37 C for 1 hour. One hundred l
of the
bacterial cell suspension was transferred to a 500 ml conical flask containing
150 ml of liquid
Luria-Bertani (LB) broth supplemented with 100 pg/m1 carbenicillin antibiotic.
The culture
was incubated on an Innova 4430 shaker (250 rpm) at 37 C overnight (16 to 18
hours).
Chemical induction of double-stranded RNA expression in AB309-105 and
BL21(DE3)
Expression of double-stranded RNA from the recombinant vector, pGBNJ003, in
the
bacterial strain AB309-105 or BL21(DE3) was made possible since all the
genetic
components for controlled expression are present. In the presence of the
chemical inducer
isopropylthiogalactoside, or IPTG, the T7 polymerase will drive the
transcription of the target
sequence in both antisense and sense directions since these are flanked by
oppositely oriented
T7 promoters.
The optical density at 600 nm of the overnight bacterial culture was measured
using
an appropriate spectrophotometer and adjusted to a value of 1 by the addition
of fresh LB
broth. Fifty ml of this culture was transferred to a 50 ml Falcon tube and the
culture then
centrifuged at 3000 g at 15 C for 10 minutes. The supernatant was removed and
the bacterial
pellet resuspended in 50 ml of fresh S complete medium (SNC medium plus 5
jig/m1
CA 02622660 2008-03-14
PCT/IB2006/004003
WO 2007/074405
cholesterol) supplemented with 100 gg/m1 carbenicillin and 1 mM IPTG. The
bacteria were
induced for 2 to 4 hours at room temperature.
Heat treatment of bacteria
Bacteria were killed by heat treatment in order to minimize the risk of
contamination
of the artificial diet in the test plates. However, heat treatment of bacteria
expressing double-
stranded RNA is not a prerequisite for inducing toxicity towards the insects
due to RNA
interference. The induced bacterial culture was centrifuged at 3000 g at room
temperature for
minutes, the supernatant discarded and the pellet subjected to 80 C for 20
minutes in a
water bath. After heat treatment, the bacterial pellet was resuspended in 1.5
ml MilliQ water
and the suspension transferred to a microfuge tube. Several tubes were
prepared and used in
the bioassays for each refreshment. The tubes were stored at -20 C until
further use.
J.
Laboratory trials to test Escherichia coli expressing dsRNA target
LD010 against Leptinotarsa decendineata
Two bioassay methods were employed to test double-stranded RNA produced in
Escherichia coli against larvae of the Colorado potato beetle: (1) artificial
diet-based
bioassay, and, (2) plant-based bioassay.
Artificial diet-based bioassays
Artificial diet for the Colorado potato beetle was prepared as
described.previously in
Example 4. A half milliliter of diet was dispensed into each of the wells of a
48-well
multiwell test plate (Nunc). For every treatment, fifty Ill of an OD 1
suspension of heat-
treated bacteria (which is equivalent to approximately 5 x 107 bacterial
cells) expressing
dsRNA was applied topically onto the solid diet in the wells and the plates
were allowed to
dry in a laminair flow cabin. Per treatment, forty-eight 2nd stage Colorado
potato beetle
larvae, one in each well containing diet and bacteria, were tested. Each row
of a plate (i.e. 8
wells) was considered as one replicate. The plates were kept in the insect
rearing chamber at
25 2 C, 60 5 % relative humidity, with a 16:8 hours light:dark
photoperiod. After every 4
days, the beetles were transferred to fresh diet containing topically-applied
bacteria. The
beetles were assessed as alive or dead every one or three days post
infestation. For the
survivors, growth and development in terms of larval weight was recorded on
day 7 post
infestation.
For RNaseIII-deficient E. coli strain AB309-105, bacteria containing plasmid
pGBNJ003 and those containing the empty vector pGN29 (reference to WO
00/188121A1)
were tested in bioassays for CPB toxicity. Bacteria harboring the pGBNJ003
plasmid showed
81
CA 02622660 2008-03-14
PCT/IB2006/004003
WO 2007/074405
a clear increase in insect mortality with time, whereas little or no mortality
was observed for
pGN29 and diet only control (Figures 6a-LD & 7a-LD). The growth and
development of
Colorado potato beetle larval survivors, 7 days after feeding on artificial
diet containing
bacteria expressing dsRNA target LD010, was severely impeded (Table 10-LD,
Figure 8a-
LD).
For E. coli strain BL21(DE3), bacteria containing plasmid pGBNJ003 and those
containing the empty vector pGN29 were tested against the Colorado potato
beetle larvae.
Similar detrimental effects were observed on larvae fed diet supplemented with
BL21(DE3)
bacteria as for the RNAseIII-deficient strain, AB309-105 (Figures 6b-LD & 7b-
LD).
However, the number of survivors for the five clones were higher for BL21(DE3)
than for
AB309-105; at day 12, average mortality values were approximately 25 % lower
for this
strain compared to the RNase III deficient strain. Also, the average weights
of survivors fed
on diet containing BL21(DE3) expressing dsRNA corresponding to target LD010
was
severely reduced (Table 10-LD, Figure 8b-LD).
The delay in growth and development of the CPB larvae fed on diet containing
either
of the two bacterial strains harboring plasmid pGBNJ003 was directly
correlated to feeding
inhibition since no frass was visible in the wells of refreshed plates from
day 4 onwards when
compared to bacteria harboring the empty vector pGN29 or the diet only plate.
This
observation was similar to that where CPB was fed on in vitro transcribed
double-stranded
RNA topically applied to artificial diet (see Example 3D); here, cessation of
feeding occurred
from day 2 onwards on treated diet.
Plant-based bioassays
Whole potato plants were sprayed with suspensions of chemically induced
bacteria
expressing dsRNA prior to feeding the plants to CPB larvae. The potato plants
of variety 'line
5' were grown from tubers to the 8-12 unfolded leaf stage in a plant growth
room chamber
with the following conditions: 25 2 C, 60 % relative humidity, 16:8 hour
light:dark
photoperiod. The plants were caged by placing a 500 ml plastic bottle upside
down over the
plant with the neck of the bottle firmly placed in the soil in a pot and the
base cut open and
covered with a fine nylon mesh to permit aeration, reduce condensation inside
and prevent
larval escape. Fifteen Colorado potato beetle larvae at the Li stage were
placed on each
treated plant in the cage. Plants were treated with a suspension of E. coli
AB309-105
harboring the pGBNJ003 plasmids (clone 1; Figure 7a-LD) or pGN29 plasmid
(clone 1; see
Figure 7a-LD). Different quantities of bacteria were applied to the plants:
66, 22, and 7
units, where one unit is defined as 109 bacterial cells in 1 ml of a bacterial
suspension at
82
CA 02622660 2008-03-14
PCT/IB2006/004003
WO 2007/074405
optical density value of 1 at 600 nm wavelength. In each case, a total volume
of 1.6 ml was
sprayed on the plant with the aid of a vaporizer. One plant was used per
treatment in this trial.
The number of survivors were counted and the weight of each survivor recorded.
Spraying plants with a suspension of E. coil bacterial strain AB309-105
expressing
target dsRNA from pGBNJ003 led to a dramatic increase in insect mortality when
compared
to pGN29 control. The mortality count was maintained when the amount of
bacteria cell
suspension was diluted 9-fold (Figure 9-LD). The average weights of the larval
survivors at
day 11 on plants sprayed with bacteria harboring the pGBNJ003 vector were
approximately
10-fold less than that of pGN29 (Figure 10-LD). Feeding damage by CPB larvae
of the
potato plant sprayed with bacteria containing the pGBNJ003 plasmid was much
reduced
when compared to the damage incurred on a potato plant sprayed with bacteria
containing the
empty vector pGN29 (Figure 11-LD).
These experiments showed that double-stranded RNA corresponding to an insect
gene
target sequence produced in either wild-type or RNaseIII-deficient bacterial
expression
systems is toxic towards the insect in terms of substantial increases in
insect mortality and
growth/development delay for larval survivors. It is also clear from these
experiments that an
exemplification was provided for the effective protection of plants/crops from
insect damage
by the use of a spray of a formulation consisting of bacteria expressing
double-stranded RNA
corresponding to an insect gene target.
K. Testing various culture suspension densities of
Eschericizia coil
expressing dsRNA target LD010 against Leptinotarsa decemlineata
Preparation and treatment of bacterial cultures are described in Example 3J.
Three-
fold serial dilutions of cultures (starting from 0.25 unit equivalents) of
Escherichia coil
RNAseIII-deficient strain AB309-105 expressing double-stranded RNA of target
LD010
were applied to foliages of the potato plant of variety 'Bintje' at the 8-12
unfolded leaf stage.
Ten Ll larvae of the L. decemlineata were placed on the treated plants with
one plant per
treatment. Scoring for insect mortality and growth impediment was done on day
7 (i.e., 7
days post infestation).
As shown in Figure 14-LD, high CPB larval mortality (90 to 100 %) was recorded
after 1 week when insects were fed potato plants treated with a topical
application by fine
spray of heat-inactivated cultures of E. coil harboring plasmid pGBNJ003 (for
target 10
dsRNA expression) at densities 0.25, 0.08 and 0.025 bacterial units. At 0.008
units, about a
third of the insects were dead, however, the surviving insects were
significantly smaller than
those in the control groups (E. coil harbouring the empty vector pGN29 and
water only).
83
CA 02622660 2008-03-14
PCT/IB2006/004003
WO 2007/074405
Feeding damage by CPB larvae of the potato plant sprayed with bacteria
containing the
pGBNJ003 plasmid at concentrations 0.025 or 0.008 units was much reduced when
compared
to the damage incurred on a potato plant sprayed with bacteria containing the
empty vector
pGN29 (Figure 15-LD).
L. Adults are extremely susceptible to orally ingested dsRNA
corresponding to target genes.
The example provided below highlights the finding that adult insects (and not
only
insects of the larval stage) are extremely susceptible to orally ingested
dsRNA corresponding
to target genes.
Four targets were chosen for this experiment: targets 2, 10, 14 and 16 (SEQ ID
NO:
168, 188, 198 and 220, respectively). GFP fragment dsRNA (SEQ ID NO: 235) was
used as a
control. Young adults (2 to 3 days old) were picked at random from our
laboratory-reared
culture with no bias towards insect gender. Ten adults were chosen per
treatment. The adults
were prestarved for at least 6 hours before the onset of the treatment. On the
first day of
treatment, each adult was fed four potato leaf discs (diameter 1.5 cm2) which
were pretreated
with a topical application of 25 pl of 0.1 g/ 1 target dsRNA (synthesized as
described in
Example 3A; topical application as described in Example 3E) per disc. Each
adult was
confined to a small petridish (diameter 3 cm) in order to make sure that all
insects have
ingested equal amounts of food and thus received equal doses of dsRNA. The
following day,
each adult was again fed four treated leaf discs as described above. On the
third day, all ten
adults per treatment were collected and placed together in a cage consisting
of a plastic box
(dimensions 30 cm x 20 cm x 15 cm) with a fine nylon mesh built into the lid
to provide good
aeration. Inside the box, some moistened filter paper was placed in the base.
Some
(untreated) potato foliage was placed on top of the paper to maintain the
adults during the
experiment. From day 5, regular assessments were carried out to count the
number of dead,
alive (mobile) and moribund insects. For insect moribundity, adults were laid
on their backs
to check whether they could right themselves within several minutes; an insect
was
considered moribund only if it was not able to turn onto its front.
Clear specific toxic effects of double-stranded RNA correpsonding to different
targets
towards adults of the Colorado potato beetle, Leptinotarsa deceinlineata, were
demonstrated
in this experiment (Figure 12-LD). Double-stranded RNA corresponding to a gfp
fragment
showed no toxicity towards CPB adults on the day of the final assessment (day
19). This
84
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
experiment clearly showed that the survival of CPB adults was severely reduced
only after a
few days of exposure to dsRNA when delivered orally. For example, for target
10, on day 5,
out of 10 adults were moribund (sick and slow moving); on day 6, 4 out of 10
adults were
dead with three of the survivors moribund; on day 9 all adults were observed
dead.
As a consequence of this experiment, the application of target double-stranded
RNAs
against insect pests may be broadened to include the two life stages of an
insect pest (i.e.
larvae and adults) which could cause extensive crop damage, as is the case
with the Colorado
potato beetle.
Example 4: Phaedon cochleariae (mustard leaf beetle)
A. Cloning of a partial sequence of the Phaedon cochleariae (mustard leaf
beetle)
PC001, PC003, PC005, PC010, PC014, PC016 and PCO27 genes via family PCR
High quality, intact RNA was isolated from the third larval stage of Phaedon
cochleariae (mustard leaf beetle; source: Dr. Caroline Muller, Julius-von-
Sachs-Institute for
Biosciences, Chemical Ecology Group, University of Wuerzburg, Julius-von-Sachs-
Platz 3,
D-97082 Wuerzburg, Germany) using TRIzol Reagent (Cat. Nr. 15596-026/15596-
018,
Invitrogen, Rockville, Maryland, USA) following the manufacturer's
instructions. Genomic
DNA present in the RNA preparation was removed by DNase (Cat. Nr. 1700,
Promega)
treatment following the manufacturer's instructions. cDNA was generated using
a
commercially available kit (SuperScript TM III Reverse Transcriptase, Cat. Nr.
18080044,
Invitrogen, Rockville, Maryland, USA) following the manufacturer's
instructions.
To isolate cDNA sequences comprising a portion of the PC001, PC003, PC005,
PC010, PC014, PC016 and PCO27 genes, a series of PCR reactions with degenerate
primers
were performed using Amplitaq Gold (Cat. Nr. N8080240, Applied Biosystems)
following
the manafacturer's instructions.
The sequences of the degenerate primers used for amplification of each of the
genes
are given in Table 2-PC. Table 2-PC displays Phaedon cochleariae target genes
including primer
sequences and cDNA sequences obtained. These primers were used in respective
PCR reactions
with the following conditions: 10 minutes at 95 C, followed by 40 cycles of 30
seconds at
95 C, 1 minute at 55 C and 1 minute at 72 C, followed by 10 minutes at 72 C.
The resulting
PCR fragments were analyzed on agarose gel, purified (QIAquick Gel Extraction
kit, Cat. Nr.
28706, Qiagen), cloned into the pCR4/TOPO vector (Cat. Nr. K4530-20,
Invitrogen) and
CA 02622660 2008-03-14
PCT/IB2006/004003
WO 2007/074405
sequenced. The sequences of the resulting PCR products are represented by the
respective
SEQ ID NO:s as given in Table 2-PC and are referred to as the partial
sequences.
The corresponding partial amino acid sequence are represented by the
respective SEQ
ID NO:s as given in Table 3-PC. Table 3-PC provides amino acid sequences of
cDNA
clones, and the start of the reading frame is indicated in brackets.
B. dsRNA production of the Phaedon cochleariae genes
dsRNA was synthesized in milligram amounts using the commercially available
kit
T7 RibomaxTm Express RNAi System (Cat. Nr. P1700, Promega). First two separate
single 5'
T7 RNA polyrnerase promoter templates were generated in two separate PCR
reactions, each
reaction containing the target sequence in a different orientation relative to
the T7 promoter.
For each of the target genes, the sense T7 template was generated using
specific T7
forward and specific reverse primers. The sequences of the respective primers
for amplifying
the sense template for each of the target genes are given in Table 8-PC. Table
8-PC provides
details for preparing ds RNA fragments of Phaedon cochleariae target
sequences, including
primer sequences.
The conditions in the PCR reactions were as follows: 1 minute at 95 C,
followed by
20 cycles of 30 seconds at 95 C, 30 seconds at 60 C and 1 minute at 72 C,
followed by 15
cycles of 30 seconds at 95 C, 30 seconds at 50 C and 1 minute at 72 C followed
by 10
minutes at 72 C. The anti-sense T7 template was generated using specific
forward and
specific T7 reverse primers in a PCR reaction with the same conditions as
described above.
The sequences of the respective primers for amplifying the anti-sense template
for each of the
target genes are given in Table 8-PC. The resulting PCR products were analyzed
on agarose
gel and purified by PCR purification kit (Qiaquick PCR Purification Kit, Cat.
Nr. 28106,
Qiagen) and NaC104 precipitation. The generated T7 forward and reverse
templates were
mixed to be transcribed and the resulting RNA strands were annealed, DNase and
RNase
treated, and purified by sodium acetate, following the manufacturer's
instructions. The sense
strand of the resulting dsRNA for each of the target genes is given in Table 8-
PC.
C. Recombination of the Phaedon cochleariae (mustard leaf beetle)
genes into the
plant vector pIC7GWIWG2D(II)
Since the mechanism of RNA interference operates through dsRNA fragments, the
target nucleotide sequences of the target genes, as selected above, were
cloned in anti-sense
86
1/1
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
and sense orientation, separated by the intron - CmR - intron, whereby CmR is
the
chloramphenicol resistance marker, to form a dsRNA hairpin construct. These
hairpin
constructs were generated using the LR recombination reaction between an attL-
containing
entry clone (see Example 4A) and an attR-containing destination vector (=
pK7GWIWG2D(II)). The plant vector pK7GWIWG2D(II) was obtained from the
VIB/Plant
Systems Biology with a Material Transfer Agreement. LR recombination reaction
was
performed by using LR ClonaseTm II enzyme mix (Cat. Nr. 11791-020, Invitrogen)
following
the manufacturer's instructions. These cloning experiments resulted in a
hairpin construct for
each of the PC001, PC010, PC014, PC016 and PCO27 genes, having the promoter -
sense -
intron - CmR - intron - antisense orientation, and wherein the promoter is the
plant operable
35S promoter. The binary vector pK7GWIWG2D(II) with the 35S promoter is
suitable for
transformation into A. tumefaciens.
Restriction enzyme digests were carried out on pCR8/GW/TOPO plasmids
containing
the different targets (see Example 4B): for PC001, a double digest with BsoBI
& PvuI; for
PC010, a double digest with PINT & PvuII; for PC014, a triple digest with
HincII, PvuI &
XhoI; for PC016, a single digest with ApaLI; for PCO27, a double digest with
Alfa' & DrdI.
The band containing the gene of interest flanked by the attL sites using
Qiaquick Gel
Extraction Kit (Cat. Nr. 28706, Qiagen) was purified. An amount of 150 ng of
purified
fragment and 150 ng pK7GWIWG2D(II) was added together with the LR clonase II
enzyme
and incubated for at least lh at 25 C . After proteinase K solution treatment
(10 min at 37 C),
the whole recombination mix was transformed into Top 10 chemically competent
cells.
Positive clones were selected by restriction digest analyses. The complete
sequence of the
hairpin construct for:
- PC001 (sense - intron - CmR - intron - antisense) is represented in SEQ ID
NO: 508;
- PC010 (sense - intron - CmR - intron - antisense) is represented in SEQ ID
NO: 509;
- PC014 (sense - intron - CmR - intron - antisense) is represented in SEQ ID
NO: 510;
- PC016 (sense - intron - CmR - intron - antisense) is represented in SEQ ID
NO: 511;
- PCO27 (sense - intron - CmR - intron - antisense) is represented in SEQ ID
NO: 512;
Table 9-PC provides sequences for each hairpin construct.
D. Laboratory trials to test dsRNA targets, using oilseed rape leaf discs
for activity
against Phaedon cochleariae larvae
87
CA 02622660 2008-03-14
PCT/IB2006/004003
WO 2007/074405
The example provided below is an exemplification of the finding that the
mustard leaf
beetle (MLB) larvae are susceptible to orally ingested dsRNA corresponding to
own target
genes.
To test the different double-stranded RNA samples against MLB larvae, a leaf
disc
assay was employed using oilseed rape (Brassica napus variety SW Oban; source:
Nick
Balaam, Sw Seed Ltd., 49 North Road, Abington, Cambridge, CB1 6AS, UK) leaf
material as
food source. The insect cultures were maintained on the same variety of
oilseed rape in the
insect chamber at 25 2 C and 60 5 % relative humidity with a photoperiod
of 16h
light/8h dark. Discs of approximately 1.1 cm in diameter (or 0.95 cm2) were
cut out off
leaves of 4- to 6-week old rape plants using a suitably-sized cork borer.
Double-stranded
RNA samples were diluted to 0.1 ps,/ 1 in Milli-Q water containing 0.05%
Triton X-100.
Treated leaf discs were prepared by applying 25 il of the diluted solution of
target PC001,
PC003, PC005, PC010, PC014, PC016, PCO27 dsRNA and control gfp dsRNA or 0.05 %
Triton X-100 on the adaxial leaf surface. The leaf discs were left to dry and
placed
individually in each of the 24 wells of a 24-well multiplate containing 1 ml
of gellified 2%
agar which helps to prevent the leaf disc from drying out. Two neonate MLB
larvae were
placed into each well of the plate, which was then covered with a multiwell
plastic lid. The
plate (one treatment containing 48 insects) was divided into 4 replicates of
12 insects per
replicate (each row). The plate containing the insects and leaf discs were
kept in an insect
chamber at 25 2 C and 60 5 % relative humidity with a photoperiod of 16h
light/8h dark.
The insects were fed leaf discs for 2 days after which they were transferred
to a new plate
containing freshly treated leaf discs. Thereafter, 4 days after the start of
the bioassay, the
insects from each replicate were collected and transferred to a Petri dish
containing untreated
fresh oilseed rape leaves. Larval mortality and average weight were recorded
at days 2, 4 7, 9
and 11.
P. cochleariae larvae fed on intact naked target dsRNA-treated oilseed rape
leaves
resulted in significant increases in larval mortalities for all targets
tested, as indicated in
Figure 1(a). Tested double-stranded RNA for target PC010 led to 100 % larval
mortality at
day 9 and for target PCO27 at day 11. For all other targets, signficantly high
mortality values
were reached at day 11 when compared to control gfp dsRNA, 0.05% Trition X-100
alone or
untreated leaf only: (average value in percentage confidence interval with
alpha 0.05)
PC001 (94.4 8.2); PC003 (86.1 4.1); PC005 (83.3 7.8); PC014 (63.9
20.6); PC016
88
V
CA 02622660 2008-03-14
PCT/IB2006/004003
WO 2007/074405
(75.0 16.8); gfp dsRNA (11.1 8.2); 0.05% Triton X-100 (19.4 1 10.5); leaf
only (8.3
10.5).
Larval survivors were assessed based on their average weight. For all targets
tested,
the mustard leaf beetle larvae had significantly reduced average weights after
day 4 of the
bioassay; insects fed control gfp dsRNA or 0.05% Triton X-100 alone developed
normally, as
for the larvae on leaf only (Figure 1(b)-PC).
E. Laboratory trials to screen dsRNAs at different concentrations
using oilseed
rape leaf discs for activity against Phaedon cochleariae larvae
Twenty-five ul of a solution of dsRNA from target PC010 or PCO27 at serial ten-
fold
concentrations from 0.1 ,g/iil down to 0.1 ng/ 1 was applied topically onto
the oilseed rape
leaf disc, as described in Example 4D above. As a negative control, 0.05%
Triton X-100 only
was administered to the leaf disc. Per treatment, twenty-four mustard leaf
beetle neonate
larvae, with two insects per well, were tested. The plates were stored in the
insect rearing
chamber at 25 2 C, 60 5 % relative humidity, with a 16:8 hours light:dark
photoperiod.
At day 2, the larvae were transferred on to a new plate containing fresh dsRNA-
treated leaf
discs. At day 4 for target PC010 and day 5 for target PCO27, insects from each
replicate were
transferred to a Petri dish containing abundant untreated leaf material. The
beetles were
assessed as live or dead on days 2, 4, 7, 8, 9, and 11 for target PC010, and
2, 5, 8, 9 and 12
for target PCO27.
Feeding oilseed rape leaf discs containing intact naked dsRNAs of the two
different
targets, PC010 and PCO27, to P. cochleariae larvae resulted in high
mortalities at concentrations
down to as low as 1 ng dsRNA/ 1 solution, as shown in Figures 2 (a) and (b).
Average mortality
values in percentage confidence interval with alpha 0.05 for different
concentrations, of dsRNA
for target PC010 at day 11, 0 ,g/ 1: 8.3 9.4; 0.1 ps/ 1: 100; 0.01 Will:
79.2 20.6; 0.001
iug,/ 1: 58.3 9.4; 0.0001 pis/ 1: 12.5 1 15.6; and for target PCO27 at day
12, 0 jig/ 1: 8.3 9.4;
0.1 1.1g4t1: 95.8 1 8.2; 0.01 g/ 1: 95.8 8.2; 0.001 g/ptl: 83.3 13.3;
0.0001 ,g/ 1: 12.5 1 8.2.
F. Cloning of a MLB gene fragment in a vector suitable for bacterial
production of
insect-active double-stranded RNA
What follows is an example of cloning a DNA fragment corresponding to an MLB
gene target in a vector for the expression of double-stranded RNA in a
bacterial host,
89
CA 02622660 2008-03-14
PCT/IB2006/004003
WO 2007/074405
although any vector comprising a T7 promoter or any other promoter for
efficient
=
transcription in bacteria, may be used (reference to W00001846).
The sequences of the specific primers used for the amplification of target
genes are
provided in Table 8. The template used is the pCR8/GW/topo vector containing
any of target
sequences. The primers are used in a PCR reaction with the following
conditions: 5 minutes
at 98 C, followed by 30 cycles of 10 seconds at 98 C, 30 seconds at 55 C and 2
minutes at
72 C, followed by 10 minutes at 72 C. The resulting PCR fragment is analyzed
on agarose
gel, purified (QIAquick Gel Extraction kit, Cat. Nr. 28706, Qiagen), blunt-end
cloned into Srf
I-linearized pGNA49A vector (reference to W000188121A1), and sequenced. The
sequence
of the resulting PCR product corresponds to the respective sequence as given
in Table 8. The
recombinant vector harbouring this sequence is named pGBNJ00(to be completed).
The sequences of the specific primers used for the amplification of target
gene
fragment PC010 are provided in Table 8-PC. The template used was the
pCR8/GW/topo
vector containing the PC010 sequence (SEQ ID NO: 253). The primers were used
in a touch-
down PCR reaction with the following conditions: 1 minute at 95 C, followed by
20 cycles of
30 seconds at 95 C, 30 seconds at 60 C with temperature decrease of -0.5 C
per cycle and 1
minute at 72 C, followed by 15 cycles of 30 seconds at 95 C, 30 seconds at 50
C and 1
minute at 72 C, followed by 10 minutes at 72 C. The resulting PCR fragment was
analyzed
on agarose gel, purified (QIAquick Gel Extraction kit, Cat. Nr. 28706,
Qiagen), blunt-end
, cloned into SrfI-linearized pGNA49A vector (reference to W000188121A1), and
sequenced.
The sequence of the resulting PCR product corresponds to SEQ ID NO: 488 as
given in
Table 8-PC. The recombinant vector harbouring this sequence was named
pGCDJ001.
G. Expression and production of a double-stranded RNA target in two
strains of
Escherichia coli AB309-105
The procedures described below are followed in order to express suitable
levels of
insect-active double-stranded RNA of insect target in bacteria. In this
experiment, an
RNaseIII-deficient strain, AB309-105 is used.
Transformation ofAB309-105
Three hundred ng of the plasmid were added to and gently mixed in a 50 IA
aliquot of
ice-chilled chemically competent E. coli strain AB309-105. The cells were
incubated on ice
for 20 minutes before subjecting them to a heat shock treatment of 37 C for 5
minutes, after
which the cells were placed back on ice for a further 5 minutes. Four hundred
and fifty ul of
room temperature SOC medium was added to the cells and the suspension
incubated on a
CA 02622660 2008-03-14
PCT/IB2006/004003
WO 2007/074405
shaker (250 rpm) at 37 C for 1 hour. One hundred p1 of the bacterial cell
suspension was
transferred to a 500 ml conical flask containing 150 ml of liquid Luria-
Bertani (LB) broth
supplemented with 100 ig/m1 carbenicillin antibiotic. The culture was
incubated on an
Innova 4430 shaker (250 rpm) at 37 C overnight (16 to 18 hours).
Chemical induction of double-stranded RNA expression in AB309-105
Expression of double-stranded RNA from the recombinant vector, pGBNJ003, in
the
bacterial strain AB309-105 was made possible since all the genetic components
for controlled
expression are present. In the presence of the chemical inducer
isopropylthiogalactoside, or
IPTG, the T7 polymerase will drive the transcription of the target sequence in
both antisense
and sense directions since these are flanked by oppositely oriented T7
promoters.
The optical density at 600 nm of the overnight bacterial culture was measured
using
an appropriate spectrophotometer and adjusted to a value of 1 by the addition
of fresh LB
broth. Fifty ml of this culture was transferred to a 50 ml Falcon tube and the
culture then
centrifuged at 3000 g at 15 C for 10 minutes. The supernatant was removed and
the bacterial
pellet resuspended in 50 ml of fresh S complete medium (SNC medium plus 5
pg/ml
cholesterol) supplemented with 100 g/m1 carbenicillin and 1 mM IPTG. The
bacteria were
induced for 2 to 4 hours at room temperature.
Heat treatment of bacteria
Bacteria were killed by heat treatment in order to minimize the risk of
contamination
of the artificial diet in the test plates. However, heat treatment of bacteria
expressing double-
stranded RNA is not a prerequisite for inducing toxicity towards the insects
due to RNA
interference. The induced bacterial culture was centrifuged at 3000 g at room
temperature for
minutes, the supernatant discarded and the pellet subjected to 80 C for 20
minutes in a
water bath. After heat treatment, the bacterial pellet was resuspended in a
total volume of 50
ml of 0.05% Triton X-100 solution. The tube was stored at 4 C until further
use
H. Laboratory trials to test Escherichia call expressing dsRNA targets
against
Phaedon cochleariae
Leaf disc bioassays
The leaf-disc bioassay method was employed to test double-stranded RNA from
target PC010 produced in Escherichia coli (from plasmid pGCDJ001) against
larvae of the
mustard leaf beetle. Leaf discs were prepared from oilseed rape foliage, as
described in
91
CA 02622660 2008-03-14
PCT/IB2006/004003
WO 2007/074405
Example 4. Twenty jtI of a bacterial suspension, with an optical density
measurement of 1 at
600 nm wavelength, was pipetted onto each disc. The leaf disc was placed in a
well of a 24-
multiwell plate containing 1 ml gellified agar. On each leaf disc were added
two neonate
larvae. For each treatment, 3 replicates of 16 neonate larvae per replicate
were prepared. The
plates were kept in the insect rearing chamber at 25 2 C and 60 5 %
relative humidity,
with a 16:8 hours light:dark photoperiod. At day 3 (i.e. 3 days post start of
bioassay), larvae
were transferred to a new plate containing fresh treated (same dosage) leaf
discs. The leaf
material was refreshed every other day from day 5 onwards. The bioassay was
scored on
mortality and average weight. Negative controls were leaf discs treated with
bacteria
harbouring plasmid pGN29 (empty vector) and leaf only.
A clear increase in mortality of P. cochleariae larvae with time was shown
after the insects
were fed on oilseed rape leaves treated with a suspension of RNaseIII-
deficient E. coil strain
AB309-105 containing plasmid pGCDJ001, whereas very little or no insect
mortality was
observed in the case of bacteria with plasmid pGN29 or leaf only control
(Figure 3-PC).
Plant-based bioassays
Whole plants are sprayed with suspensions of chemically induced bacteria
expressing
dsRNA prior to feeding the plants to MLB. The are grown from in a plant growth
room
chamber. The plants are caged by placing a 500 ml plastic bottle upside down
over the plant
with the neck of the bottle firmly placed in the soil in a pot and the base
cut open and covered
with a fine nylon mesh to permit aeration, reduce condensation inside and
prevent insect
escape. MLB are placed on each treated plant in the cage. Plants are treated
with a suspension
of E. coil AB309-105 harbouring the pGBNJ001 plasmids or pGN29 plasmid.
Different
quantities of bacteria are applied to the plants: for instance 66, 22, and 7
units, where one unit
is defined as 109 bacterial cells in 1 ml of a bacterial suspension at optical
density value of 1
at 600 nm wavelength. In each case, a total volume of between 1 and 10 ml s
sprayed on the
plant with the aid of a vaporizer. One plant is used per treatment in this
trial. The number of
survivors are counted and the weight of each survivor recorded.
Spraying plants with a suspension of E. coil bacterial strain AB309-105
expressing
target dsRNA from pGBNJ003 iced to a dramatic increase in insect mortality
when compared
to pGN29 control. These experiments show that double-stranded RNA
corresponding to an
insect gene target sequence produced in either wild-type or RNaseIII-deficient
bacterial
expression systems is toxic towards the insect in terms of substantial
increases in insect
mortality and growth/development delay for larval survivors. It is also clear
from these
92
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
experiments that an exemplification is provided for the effective protection
of plants/crops
from insect damage by the use of a spray of a formulation consisting of
bacteria expressing
double-stranded RNA corresponding to an insect gene target.
Example 5: Epilachna varivetis (Mexican bean beetle)
A. Cloning Epilachna varivetis partial gene sequences
High quality, intact RNA was isolated from 4 different larval stages of
Epilachna
varivetis (Mexican bean beetle; source: Thomas Dorsey, Supervising
Entomologist, New
Jersey Department of Agriculture, Division of Plant Industry, Bureau of
Biological Pest
Control, Phillip Alampi Beneficial Insect Laboratory, PO Box 330, Trenton, New
Jersey
08625-0330, USA) using TRIzol Reagent (Cat. Nr. 15596-026/15596-018,
Invitrogen,
Rockville, Maryland, USA) following the manufacturer's instructions. Genomic
DNA
present in the RNA preparation was removed by DNase treatment following the
manafacturer's instructions (Cat. Nr. 1700, Promega). cDNA was generated using
a
commercially available kit (SuperScript TM III Reverse Transcriptase, Cat. Nr.
18080044,
Invitrogen, Rockville, Maryland, USA) following the manufacturer's
instructions.
To isolate cDNA sequences comprising a portion of the EV005, EV009, EV010,
EV015 and EV016 genes, a series of PCR reactions with degenerate primers were
performed
using Amplitaq Gold (Cat. Nr. N8080240, Applied Biosystems) following the
manufacturer's
instructions.
The sequences of the degenerate primers used for amplification of each of the
genes
are given in Table 2-EV, which displays Epilachna varivetis target genes
including primer
sequences and cDNA sequences obtained. These primers were used in respective
PCR reactions
with the following conditions: for EV005 and EV009, 10 minutes at 95 C,
followed by 40
cycles of 30 seconds at 95 C, 1 minute at 50 C and 1 minute 30 seconds at 72
C, followed by
7 minutes at 72 C; for EV014, 10 minutes at 95 C, followed by 40 cycles of 30
seconds at
95 C, 1 minute at 53 C and 1 minute at 72 C, followed by 7 minutes at 72 C;
for EV010 and
EV016, 10 minutes at 95 C, followed by 40 cycles of 30 seconds at 95 C, 1
minute at 54 C
and 1 minute 40 seconds at 72 C, followed by 7 minutes at 72 C. The resulting
PCR
fragments were analyzed on agarose gel, purified (QIAquick Gel Extraction kit,
Cat. Nr,
28706, Qiagen), cloned into the pCR4/TOPO vector (Cat. Nr. K4530-20,
Invitrogen), and
sequenced. The sequences of the resulting PCR products are represented by the
respective
SEQ ID NO:s as given in Table 2-EV and are referred to as the partial
sequences. The
93
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
corresponding partial amino acid sequences are represented by the respective
SEQ ID NO:s
as given in Table 3-EV, where the start of the reading frame is indicated in
brackets.
B. dsRNA production of the Epilachna varivetis genes
dsRNA was synthesized in milligram amounts using the commercially available
kit
T7 RibomaxTM Express RNAi System (Cat. Nr. P1700, Promega). First two separate
single 5'
T7 RNA polymerase promoter templates were generated in two separate PCR
reactions, each
reaction containing the target sequence in a different orientation relative to
the T7 promoter.
For each of the target genes, the sense T7 template was generated using
specific T7
forward and specific reverse primers. The sequences of the respective primers
for amplifying
the sense template for each of the target genes are given in Table 8-EV.
The conditions in the PCR reactions were as follows: 1 minute at 95 C,
followed by
20 cycles of 30 seconds at 95 C, 30 seconds at 60 C and 1 minute at 72 C,
followed by 15
cycles of 30 seconds at 95 C, 30 seconds at 50 C and 1 minute at 72 C followed
by 10
minutes at 72 C. The anti-sense T7 template was generated using specific
forward and
specific T7 reverse primers in a PCR reaction with the same conditions as
described above.
The sequences of the respective primers for amplifying the anti-sense template
for each of the
target genes are given in Table 8-EV. The resulting PCR products were analyzed
on agarose
gel and purified by PCR purification kit (Qiaquick PCR Purification Kit, Cat.
Nr. 28106,
Qiagen) and NaC104 precipitation. The generated T7 forward and reverse
templates were
mixed to be transcribed and the resulting RNA strands were annealed, DNase and
RNase
treated, and purified by sodium acetate, following the manufacturer's
instructions. The sense
strand of the resulting dsRNA for each of the target genes is given in Table 8-
EV.
C. Recombination of the Epilachna varivetis genes into the plant
vector
pK7GWIWG2D(II)
Since the mechanism of RNA interference operates through dsRNA fragments, the
target nucleotide sequences of the target genes, as selected above, are cloned
in anti-sense
and sense orientation, separated by the intron - CmR - intron, whereby CmR is
the
chloramphenicol resistance marker, to form a dsRNA hairpin construct. These
hairpin
constructs are generated using the LR recombination reaction between an attL-
containing
entry clone (see Example 1) and an attR-containing destination vector (=
pK7GWIWG2D(II)). The plant vector pK7GWIWG2D(II) is obtained from the
VIB/Plant
Systems Biology with a Material Transfer Agreement. LR recombination reaction
is
94
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
performed by using LR ClonaseTM II enzyme mix (Cat. Nr. 11791-020, Invitrogen)
following
the manufacturer's instructions. These cloning experiments result in a hairpin
construct for
each of the target genes, having the promoter - sense - intron - CmR - intron -
antisense
orientation, and wherein the promoter is the plant operable 35S promoter. The
binary vector
pK7GWIWG2D(II) with the 35S promoter is suitable for transformation into A.
tumefaciens.
Restriction enzyme digests were carried out on pCR8/GW/TOPO plasmids
containing
the different targets (see Example B). The band containing the gene of
interest flanked by the
attL sites using Qiaquick Gel Extraction Kit (Cat. Nr. 28706, Qiagen) is
purified. An amount
of 150 ng of purified fragment and 150 ng pK7GWIWG2D(II) is added together
with the LR
clonase II enzyme and incubated for at least 1h at 25 C . After proteinase K
solution
treatment (10 min at 37 C), the whole recombination mix is transformed into
Top 10
chemically competent cells. Positive clones are selected by restriction digest
analyses.
D. Laboratory trials to test dsRNA targets using bean leaf discs for
activity against
Epilachna varivetis larvae
The example provided below is an exemplification of the finding that the
Mexican
bean beetle (MBB) larvae are susceptible to orally ingested dsRNA
corresponding to own
target genes.
To test the different double-stranded RNA samples against MBB larvae, a leaf
disc
assay was employed using snap bean (Phaseolus vulgaris variety Montano;
source: Aveve
NV, Belgium) leaf material as food source. The same variety of beans was used
to maintain
insect cultures in the insect chamber at 25 2 C and 60 5 % relative
humidity with a
photoperiod of 16h light/8h dark. Discs of approximately 1.1 cm in diameter
(or 0.95 cm2)
were cut out off leaves of 1- to 2-week old bean plants using a suitably-sized
cork borer.
Double-stranded RNA samples were diluted to 1 g/ 1 in Milli-Q water
containing 0.05%
Triton X-100. Treated leaf discs were prepared by applying 25 p.1 of the
diluted solution of
target Ev005, Ev010, Ev015, Ev016 dsRNA and control gfp dsRNA or 0.05 % Triton
X-100
on the adaxial leaf surface. The leaf discs were left to dry and placed
individually in each of
the 24 wells of a 24-well multiplate containing 1 ml of gellified 2 % agar
which helps to
prevent the leaf disc from drying out. A single neonate MBB larva was placed
into each well
of a plate, which was then covered with a multiwell plastic lid. The plate was
divided into 3
replicates of 8 insects per replicate (row). The plate containing the insects
and leaf discs were
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
kept in an insect chamber at 25 2 C and 60 5 % relative humidity with a
photoperiod of
16h light/8h dark. The insects were fed on the leaf discs for 2 days after
which the insects
were transferred to a new plate containing freshly treated leaf discs.
Thereafter, 4 days after
the start of the bioassay, the insects were transferred to a petriplate
containing untreated fresh
bean leaves every day until day 10. Insect mortality was recorded at day 2 and
every other
day thereafter.
Feeding snap bean leaves containing surface-applied intact naked target dsRNAs
to E.
vari-vestis larvae resulted in significant increases in larval mortalities, as
indicated in Figure 1.
Tested double-stranded RNAs of targets Ev010, Ev015, & Ev016 led to 100 %
mortality after
8 days, whereas dsRNA of target Ev005 took 10 days to kill all larvae. The
majority of the
insects fed on treated leaf discs containing control gfp dsRNA or only the
surfactant Triton
X-100 were sustained throughout the bioassay (Figure 1-EV).
E. Laboratory trials to test dsRNA targets using bean leaf discs for
activity against
Epilachna varivestis adults
The example provided below is an exemplification of the finding that the
Mexican
bean beetle adults are susceptible to orally ingested dsRNA corresponding to
own target
genes.
In a similar bioassay set-up as for Mexican bean beetle larvae, adult MBBs
were
tested against double-stranded RNAs topically-applied to bean leaf discs. Test
dsRNA from
each target Ev010, Ev015 and Ev016 was diluted in 0.05 % Triton X-100 to a
final
concentration of 0.1 [tg/fil. Bean leaf discs were treated by topical
application of 30 IA of the
test solution onto each disc. The discs were allowed to dry completely before-
placing each on
a slice of gellified 2 % agar in each well of a 24-well multiwell plate. Three-
day-old adults
were collected from the culture cages and fed nothing for 7-8 hours prior to
placing one adult
to each well of the bioassay plate (thus 24 adults per treatment). The plates
were kept in the
insect rearing chamber (under the same conditions as for MBB larvae for 24
hours) after
which the adults were transferred to a new plate containing fresh dsRNA-
treated leaf discs.
After a further 24 hours, the adults from each treatment were collected and
placed in a plastic
box with dimensions 30 cm x 15 cm x 10 cm containing two potted and untreated
3-week-old
bean plants. Insect mortality was assessed from day 4 until day 11.
96
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
All three target dsRNAs (Ev010, Ev015 and Ev016) ingested by adults of
Epilachna
varivestis resulted in significant increases in mortality from day 4 (4 days
post bioassay start),
as shown in Figure 2(a)-EV. From day 5, dramatic changes in feeding patterns
were
observed between insects fed initially with target-dsRNA-treated bean leaf
discs and those
that were fed discs containing control gfp dsRNA or surfactant Triton X-100.
Reductions in
foliar damage by MBB adults of untreated bean plants were clearly visible for
all three
targets when compared to gfp dsRNA and surfactant only controls, albeit at
varying levels;
insects fed target 15 caused the least damage to bean foliage (Figure 2(b)-
EV).
F. Cloning of a MBB gene fragment in a vector suitable for bacterial
production of
insect-active double-stranded RNA
What follows is an example of cloning a DNA fragment corresponding to an MLB
gene target in a vector for the expression of double-stranded RNA in a
bacterial host,
although any vector comprising a T7 promoter or any other promoter for
efficient
transcription in bacteria, may be used (reference to W00001846).
The sequences of the specific primers used for the amplification of target
genes are
provided in Table 8-EV. The template used is the pCR8/GW/topo vector
containing any of
target sequences. The primers are used in a PCR reaction with the following
conditions: 5
minutes at 98 C, followed by 30 cycles of 10 seconds at 98 C, 30 seconds at 55
C and 2
minutes at 72 C, followed by 10 minutes at 72 C. The resulting PCR fragment is
analyzed
on agarose gel, purified (QIAquick Gel Extraction kit, Cat. Nr. 28706,
Qiagen), blunt-end
cloned into Srf I-linearized pGNA49A vector (reference to W000188121A1), and
sequenced.
The sequence of the resulting PCR product corresponds to the respective
sequence as given in
Table 8-EV. The recombinant vector harbouring this sequence is named
pGBNJOOXX.
G. Expression and production of a double-stranded RNA target in two strains
of
Escherichia coli: (1) AB309-105, and, (2) BL21(DE3)
The procedures described below are followed in order to express suitable
levels of
insect-active double-stranded RNA of insect target in bacteria. An RNaseIII-
deficient strain,
AB309-105, is used in comparison to wild-type RNaseIII-containing bacteria,
BL21(DE3).
Transformation of AB309-105 and BL21(DE3)
Three hundred ng of the plasmid are added to and gently mixed in a 50 1.1.1
aliquot of
ice-chilled chemically competent E. coli strain AB309-105 or BL21(DE3). The
cells are
97
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
incubated on ice for 20 minutes before subjecting them to a heat shock
treatment of 37 C for
minutes, after which the cells are placed back on ice for a further 5 minutes.
Four hundred
and fifty trl of room temperature SOC medium is added to the cells and the
suspension
incubated on a shaker (250 rpm) at 37 C for 1 hour. One hundred 1 of the
bacterial cell
suspension is transferred to a 500 ml conical flask containing 150 ml of
liquid Luria-Bertani
(LB) broth supplemented with 100 p,g/m1 carbenicillin antibiotic. The culture
is incubated on
an Innova 4430 shaker (250 rpm) at 37 C overnight (16 to 18 hours).
Chemical induction of double-stranded RNA expression in AB309-105 and
BL21(DE3)
Expression of double-stranded RNA from the recombinant vector, pGBNJ003, in
the
bacterial strain AB309-105 or BL21(DE3) is made possible since all the genetic
components
for controlled expression are present. In the presence of the chemical inducer
isopropylthiogalactoside, or IPTG, the T7 polymerase will drive the
transcription of the target
sequence in both antisense and sense directions since these are flanked by
oppositely oriented
T7 promoters.
The optical density at 600 nm of the overnight bacterial culture is measured
using an
appropriate spectrophotometer and adjusted to a value of 1 by the addition of
fresh LB broth.
Fifty ml of this culture is transferred to a 50 ml Falcon tube and the culture
then centrifuged
at 3000 g at 15 C for 10 minutes. The supernatant is removed and the
bacterial pellet
resuspended in 50 ml of fresh S complete medium (SNC medium plus 5 g/ml
cholesterol)
supplemented with 100 1.1g/m1 carbenicillin and 1 mM IPTG. The bacteria are
induced for 2
to 4 hours at room temperature.
Heat treatment of bacteria
Bacteria are killed by heat treatment in order to minimize the risk of
contamination of
the artificial diet in the test plates. However, heat treatment of bacteria
expressing double-
stranded RNA is not a prerequisite for inducing toxicity towards the insects
due to RNA
interference. The induced bacterial culture is centrifuged at 3000 g at room
temperature for
minutes, the supernatant discarded and the pellet subjected to 80 C for 20
minutes in a
water bath. After heat treatment, the bacterial pellet is resuspended in 1.5
ml MilliQ water
and the suspension transferred to a microfuge tube. Several tubes are prepared
and used in the
bioassays for each refreshment. The tubes are stored at -20 C until further
use.
H. Laboratory trials to test Escherichia coli expressing dsRNA targets
against
Epilachtza varivetis
98
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
Plant-based bioassays
Whole plants are sprayed with suspensions of chemically induced bacteria
expressing
dsRNA prior to feeding the plants to MBB. The are grown from in a plant growth
room
chamber. The plants are caged by placing a 500 ml plastic bottle upside down
over the plant
with the neck of the bottle firmly placed in the soil in a pot and the base
cut open and covered
with a fine nylon mesh to permit aeration, reduce condensation inside and
prevent insect
escape. MMB are placed on each treated plant in the cage. Plants are treated
with a
suspension of E. coil AB309-105 harbouring the pGBNJ001 plasmids or pGN29
plasmid.
Different quantities of bacteria are applied to the plants: for instance 66,
22, and 7 units,
where one unit is defined as 109 bacterial cells in 1 ml of a bacterial
suspension at optical
density value of 1 at 600 nm wavelength. In each case, a total volume of
between 1 and 10 ml
s sprayed on the plant with the aid of a vaporizer. One plant is used per
treatment in this trial.
The number of survivors are counted and the weight of each survivor recorded.
Spraying plants with a suspension of E. coil bacterial strain AB309-105
expressing
target dsRNA from pGBNJ003 lead to a dramatic increase in insect mortality
when compared
to pGN29 control. These experiments show that double-stranded RNA
corresponding to an
insect gene target sequence produced in either wild-type or RNaseIII-deficient
bacterial
expression systems is toxic towards the insect in terms of substantial
increases in insect
mortality and growth/development delay for larval survivors. It is also clear
from these
experiments that an exemplification is provided for the effective protection
of plants/crops
from insect damage by the use of a spray of a formulation consisting of
bacteria expressing
double-stranded RNA corresponding to an insect gene target.
Example 6: Anthonomus grandis (cotton boll weevil)
A. Cloning Anthonomus grandis partial sequences
High quality, intact RNA was isolated from the 3 instars of Anthonomus grandis
(cotton boll weevil; source: Dr. Gary Benzon, Benzon Research Inc., 7 Kuhn
Drive, Carlisle,
Pennsylvania 17013, USA) using TRIzol Reagent (Cat. Nr. 15596-026/15596-018,
Invitrogen, Rockville, Maryland, USA) following the manufacturer's
instructions. Genomic
DNA present in the RNA preparation was removed by DNase treatment following
the
manafacturer's instructions (Cat. Nr. 1700, Promega). cDNA was generated using
a
commercially available kit (SuperScript TM III Reverse Transcriptase, Cat. Nr.
18080044,
Invitrogen, Rockville, Maryland, USA) following the manufacturer's
instructions.
99
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
To isolate cDNA sequences comprising a portion of the AG001, AG005, AG010,
AG014 and AG016 genes, a series of PCR reactions with degenerate primers were
performed
using Amplitaq Gold (Cat. Nr. N8080240, Applied Biosystems) following the
manafacturer's
instructions.
The sequences of the degenerate primers used for amplification of each of the
genes
are given in Table 2-AG. These primers were used in respective PCR reactions
with the
following conditions: for AG001, AG005 and AG016, 10 minutes at 95 C, followed
by 40
cycles of 30 seconds at 95 C, 1 minute at 50 C and 1 minute and 30 seconds at
72 C,
followed by 7 minutes at 72 C; for AG010, 10 minutes at 95 C, followed by 40
cycles of 30
seconds at 95 C, 1 minute at 54 C and 2 minutes and 30 seconds at 72 C,
followed by 7
minutes at 72 C; for AG014, 10 minutes at 95 C, followed by 40 cycles of 30
seconds at
95 C, 1 minute at 55 C and 1 minute at 72 C, followed by 7 minutes at 72 C.
The resulting
PCR fragments were analyzed on agarose gel, purified (QIAquick Gel Extraction
kit, Cat. Nr.
28706, Qiagen), cloned into the pCR8/GW/TOPO vector (Cat. Nr. 1(2500-20,
Invitrogen) and
sequenced. The sequences of the resulting PCR products are represented by the
respective
SEQ ID NO:s as given in Table 2-AG and are referred to as the partial
sequences. The
corresponding partial amino acid sequence are represented by the respective
SEQ ID NO:s as
given in Table 3-AG.
B. dsRNA production of the Anthonomus grandis (cotton boll weevil) genes
dsRNA was synthesized in milligram amounts using the commercially available
kit
T7 RibomaxTM Express RNAi System (Cat. Nr. P1700, Promega). First two separate
single 5'
T7 RNA polymerase promoter templates were generated in two separate PCR
reactions, each
reaction containing the target sequence in a different orientation relative to
the T7 promoter.
For each of the target genes, the sense T7 template was generated using
specific T7
forward and specific reverse primers. The sequences of the respective primers
for amplifying
the sense template for each of the target genes are given in Table 8-AG. A
touchdown PCR
was performed as follows: 1 minute at 95 C, followed by 20 cycles of 30
seconds at 95 C, 30
seconds at 60 C with a decrease in temperature of 0.5 C per cycle and 1 minute
at 72 C,
followed by 15 cycles of 30 seconds at 95 C, 30 seconds at 50 C and 1 minute
at 72 C,
followed by 10 minutes at 72 C. The anti-sense T7 template was generated using
specific
forward and specific T7 reverse primers in a PCR reaction with the same
conditions as
described above. The sequences of the respective primers for amplifying the
anti-sense
template for each of the target genes are given in Table 8-AG. The resulting
PCR products
100
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
were analyzed on agarose gel and purified by PCR purification kit (Qiaquick
PCR
Purification Kit, Cat. Nr. 28106, Qiagen) and NaC104 precipitation. The
generated T7
forward and reverse templates were mixed to be transcribed and the resulting
RNA strands
were annealed, DNase and RNase treated, and purified by sodium acetate,
following the
manufacturer's instructions. The sense strand of the resulting dsRNA for each
of the target
genes is given in Table 8-AG.
C. Recombination of Anthonomus grandis genes into the plant vector
pK7GWIWG2D(II)
Since the mechanism of RNA interference operates through dsRNA fragments, the
target nucleotide sequences of the target genes, as selected above, are cloned
in anti-sense
and sense orientation, separated by the intron - CmR - intron, whereby CmR is
the
chloramphenicol resistance marker, to form a dsRNA hairpin construct. These
hairpin
constructs are generated using the LR recombination reaction between an attL-
containing
entry clone (see Example 1) and an attR-containing destination vector (=
pK7GWIWG2D(II)). The plant vector pK7GWIWG2D(II) is obtained from the
VIB/Plant
Systems Biology with a Material Transfer Agreement. LR recombination reaction
is
performed by using LR ClonaseTM II enzyme mix (Cat. Nr. 11791-020, Invitrogen)
following
the manufacturer's instructions. These cloning experiments result in a hairpin
construct for
each of the target genes, having the promoter - sense - intron - CmR - intron -
antisense
orientation, and wherein the promoter is the plant operable 35S promoter. The
binary vector
pK7GWIWG2D(II) with the 35S promoter is suitable for transformation into A.
tumefaciens.
Restriction enzyme digests were carried out on pCR8/GW/TOPO plasmids
containing
the different targets (see Example 2). The band containing the gene of
interest flanked by the
attL sites using Qiaquick Gel Extraction Kit (Cat. Nr. 28706, Qiagen) is
purified. An amount
of 150 ng of purified fragment and 150 ng pK7GWIWG2D(II) is added together
with the LR
clonase II enzyme and incubated for at least lh at 25 C . After proteinase K
solution
treatment (10 min at 37 C), the whole recombination mix is transformed into
Top 10
chemically competent cells. Positive clones are selected by restriction digest
analyses.
D. Laboratory trials to test dsRNA targets, using artificial diet for
activity against
the larvae of the house cricket, Acheta domesticus
House crickets, Acheta domesticus, were maintained at Insect Investigations
Ltd.
(origin: Blades Biological Ltd., Kent, UK). The insects were reared on bran
pellets and
101
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
cabbage leaves. Mixed sex nymphs of equal size and no more than 5 days old
were selected
for use in the trial. Double-stranded RNA was mixed with a wheat-based
pelleted rodent diet
(rat and mouse standard diet, B & K Universal Ltd., Grimston, Aldbrough, Hull,
UK). The
diet, BK001P, contains the following ingredients in descending order by
weight: wheat, soya,
wheatfeed, barley, pellet binder, rodent 5 vit min, fat blend, dicalcium
phosphate, mould carb.
The pelleted rodent diet was finely ground and heat-treated in a microwave
oven prior to
mixing, in order to inactivate any enzyme components. All rodent diet was
taken from the
same batch in order to ensure consistency. The ground diet and dsRNA were
mixed
thoroughly and formed into small pellets of equal weight, which were allowed
to dry
overnight at room temperature.
Double-stranded RNA samples from targets and gfp control at concentrations 10
lig/u1 are applied in the ratio 1 g ground diet plus 1 ml dsRNA solution,
thereby resulting in
an application rate of 10 mg dsRNA per g pellet. Pellets are replaced weekly.
The insects are
provided with treated pellets for the first three weeks of the trial.
Thereafter untreated pellets
are provided. Insects are maintained within lidded plastic containers (9 cm
diameter, 4.5 cm
deep), ten per container. Each arena contains one treated bait pellet and one
water source
(damp cotton wool ball), each placed in a separate small weigh boat. The water
is replenished
ad lib throughout the experiment.
Assessments are made at twice weekly intervals, with no more than four days
between
assessments, until all the control insects had either died or moulted to the
adult stage (84
days). At each assessment the insects are assessed as live or dead, and
examined for
abnormalities. From day 46 onwards, once moulting to adult commences, all
insects (live and
dead) are assessed as nyumph or adult. Surviving insects are weighed on day 55
of the trial.
Four replicates are performed for each of the treatments. During the trial the
test conditions
are 25 to 33 C and 20 to 25 % relative humidity, with a 12:12 hour light:dark
photoperiod.
E. Cloning of a MLB gene fragment in a vector suitable for bacterial
production of
insect-active double-stranded RNA
What follows is an example of cloning a DNA fragment corresponding to an MLB
gene target in a vector for the expression of double-stranded RNA in a
bacterial host,
although any vector comprising a T7 promoter or any other promoter for
efficient
transcription in bacteria, may be used (reference to W00001846).
102
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
The sequences of the specific primers used for the amplification of target
genes are
provided in Table 8. The template used is the pCR8/GW/topo vector containing
any of target
sequences. The primers are used in a PCR reaction with the following
conditions: 5 minutes
at 98 C, followed by 30 cycles of 10 seconds at 98 C, 30 seconds at 55 C and 2
minutes at
72 C, followed by 10 minutes at 72 C. The resulting PCR fragment is analyzed
on agarose
gel, purified (QIAquick Gel Extraction kit, Cat. Nr. 28706, Qiagen), blunt-end
cloned into Srf
I-linearized pGNA49A vector (reference to W000188121A1), and sequenced. The
sequence
of the resulting PCR product corresponds to the respective sequence as given
in Table 8. The
recombinant vector harbouring this sequence is named pGBNJOOXX.
F. Expression and production of a double-stranded RNA target in two
strains of
Escherichia coli: (1) AB309-105, and, (2) BL21(DE3)
The procedures described below are followed in order to express suitable
levels of
insect-active double-stranded RNA of insect target in bacteria. An RNaseIII-
deficient strain,
AB309-105, is used in comparison to wild-type RNaseIII-containing bacteria,
BL21(DE3).
Transformation of AB309-105 and BL21(DE3)
Three hundred ng of the plasmid are added to and gently mixed in a 50 IA
aliquot of
ice-chilled chemically competent E. coli strain AB309-105 or BL21(DE3). The
cells are
incubated on ice for 20 minutes before subjecting them to a heat shock
treatment of 37 C for
minutes, after which the cells are placed back on ice for a further 5 minutes.
Four hundred
and fifty Ill of room temperature SOC medium is added to the cells and the
suspension
incubated on a shaker (250 rpm) at 37 C for 1 hour. One hundred 1.11 of the
bacterial cell
suspension is transferred to a 500 ml conical flask containing 150 ml of
liquid Luria-Bertani
(LB) broth supplemented with 100 jig/ml carbenicillin antibiotic. The culture
is incubated on
an Innova 4430 shaker (250 rpm) at 37 C overnight (16 to 18 hours).
Chemical induction of double-stranded RNA expression in AB309-105 and
BL21(DE3)
Expression of double-stranded RNA from the recombinant vector, pGBNJ003, in
the
bacterial strain AB309-105 or BL21(DE3) is made possible since all the genetic
components
for controlled expression are present. In the presence of the chemical inducer
isopropylthiogalactoside, or IPTG, the T7 polymerase will drive the
transcription of the target
sequence in both antisense and sense directions since these are flanked by
oppositely oriented
T7 promoters.
103
CA 02622660 2008-03-14
WO 2007/074405
PCT/1B2006/004003
The optical density at 600 nm of the overnight bacterial culture is measured
using an
appropriate spectrophotometer and adjusted to a value of 1 by the addition of
fresh LB broth.
Fifty ml of this culture is transferred to a 50 ml Falcon tube and the culture
then centrifuged
at 3000 g at 15 C for 10 minutes. The supernatant is removed and the
bacterial pellet
resuspended in 50 ml of fresh S complete medium (SNC medium plus 5 ,g/m1
cholesterol)
supplemented with 100 jig/m1 carbenicillin and 1 mM IPTG. The bacteria are
induced for 2
to 4 hours at room temperature.
Heat treatment of bacteria
Bacteria are killed by heat treatment in order to minimise the risk of
contamination of
the artificial diet in the test plates. However, heat treatment of bacteria
expressing double-
stranded RNA is not a prerequisite for inducing toxicity towards the insects
due to RNA
interference. The induced bacterial culture is centrifuged at 3000 g at room
temperature for
minutes, the supernatant discarded and the pellet subjected to 80 C for 20
minutes in a
water bath. After heat treatment, the bacterial pellet is resuspended in 1.5
ml MilliQ water
and the suspension transferred to a microfuge tube. Several tubes are prepared
and used in the
bioassays for each refreshment. The tubes are stored at -20 C until further
use.
G.
Laboratory trials to test Escherichia colt expressing dsRNA targets against
Anthonomus grandis
Plant-based bioassays
Whole plants are sprayed with suspensions of chemically induced bacteria
expressing
dsRNA prior to feeding the plants to CBW. The are grown from in a plant growth
room
chamber. The plants are caged by placing a 500 ml plastic bottle upside down
over the plant
with the neck of the bottle firmly placed in the soil in a pot and the base
cut open and covered
with a fine nylon mesh to permit aeration, reduce condensation inside and
prevent insect
escape. CBW are placed on each treated plant in the cage. Plants are treated
with a
suspension of E. coli AB309-105 harbouring the pGBNJ001 plasmids or pGN29
plasmid.
Different quantities of bacteria are applied to the plants: for instance 66,
22, and 7 units,
where one unit is defined as 109 bacterial cells in 1 ml of a bacterial
suspension at optical
density value of 1 at 600 nm wavelength. In each case, a total volume of
between 1 and 10 ml
s sprayed on the plant with the aid of a vaporizer. One plant is used per
treatment in this trial.
The number of survivors are counted and the weight of each survivor recorded.
104
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
Spraying plants with a suspension of E. coil bacterial strain AB309-105
expressing
target dsRNA from pGBNJ003 lead to a dramatic increase in insect mortality
when compared
to pGN29 control. These experiments show that double-stranded RNA
corresponding to an
insect gene target sequence produced in either wild-type or RNaseIII-deficient
bacterial
expression systems is toxic towards the insect in terms of substantial
increases in insect
mortality and growth/development delay for larval survivors. It is also clear
from these
experiments that an exemplification is provided for the effective protection
of plants/crops
from insect damage by the use of a spray of a formulation consisting of
bacteria expressing
double-stranded RNA corresponding to an insect gene target.
Example 7: Tribolium castaneum (red flour beetle)
A. Cloning Tribolium castaneum partial sequences
High quality, intact RNA was isolated from all the different insect stages of
Tribolium
castaneum (red flour beetle; source: Dr. Lara Senior, Insect Investigations
Ltd., Capital
Business Park, Wentloog, Cardiff, CF3 2PX, Wales, UK) using TRIzol Reagent
(Cat. Nr.
15596-026/15596-018, Invitrogen, Rockville, Maryland, USA) following the
manufacturer's
instructions. Genomic DNA present in the RNA preparation was removed by DNase
treatment following the manafacturer's instructions (Cat. Nr. 1700, Promega).
cDNA was
generated using a commercially available kit (SuperScript TM III Reverse
Transcriptase, Cat.
Nr, 18080044, Invitrogen, Rockville, Maryland, USA) following the
manufacturer's
instructions.
To isolate cDNA sequences comprising a portion of the TC001, TC002, TC010,
TC014 and TC015 genes, a series of PCR reactions with degenerate primers were
performed
using Amplitaq Gold (Cat. Nr. N8080240, Applied Biosystems) following the
manafacturer's
instructions.
The sequences of the degenerate primers used for amplification of each of the
genes
are given in Table 2-TC. These primers were used in respective PCR reactions
with the
following conditions: 10 minutes at 95 C, followed by 40 cycles of 30 seconds
at 95 C, 1
minute at 50 C and 1 minute and 30 seconds at 72 C, followed by 7 minutes at
72 C (TC001,
TC014, TC015); 10 minutes at 95 C, followed by 40 cycles of 30 seconds at 95
C, 1 minute
at 54 C and 2 minutes and 30 seconds at 72 C, followed by 7 minutes at 72 C
(TC010); 10
minutes at 95 C, followed by 40 cycles of 30 seconds at 95 C, 1 minute at 53 C
and 1 minute
105
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
at 72 C, followed by 7 minutes at 72 C (TC002) . The resulting PCR fragments
were
analyzed on agarose gel, purified (QIAquick Gel Extraction kit, Cat. Nr.
28706, Qiagen),
cloned into the pCR8/GW/TOPO vector (Cat. Nr. K2500-20, Invitrogen), and
sequenced. The
sequences of the resulting PCR products are represented by the respective SEQ
ID NO:s as
given in Table 2-TC and are referred to as the partial sequences. The
corresponding partial
amino acid sequences are represented by the respective SEQ ID NO:s as given in
Table 3-
TC.
B. dsRNA production of the Tribolium castaneum genes
dsRNA was synthesized in milligram amounts using the commercially available
kit
T7 RibomaxTM Express RNAi System (Cat. Nr. P1700, Promega). First two separate
single 5'
T7 RNA polyrnerase promoter templates were generated in two separate PCR
reactions, each
reaction containing the target sequence in a different orientation relative to
the T7 promoter.
For each of the target genes, the sense T7 template was generated using
specific T7
forward and specific reverse primers. The sequences of the respective primers
for amplifying
the sense template for each of the target genes are given in Table 8-TC. The
conditions in the
PCR reactions were as follows: 1 minute at 95 C, followed by 20 cycles of 30
seconds at
95 C, 30 seconds at 60 C (-0.5 C/cycle) and 1 minute at 72 C, followed by 15
cycles of 30
seconds at 95 C, 30 seconds at 50 C and 1 minute at 72 C, followed by 10
minutes at 72 C.
The anti-sense T7 template was generated using specific forward and specific
T7 reverse
primers in a PCR reaction with the same conditions as described above. The
sequences of the
respective primers for amplifying the anti-sense template for each of the
target genes are
given in Table 8-TC. The resulting PCR products were analyzed on agarose gel
and purified
by PCR purification kit (Qiaquick PCR Purification Kit, Cat. Nr. 28106,
Qiagen) and NaC104
precipitation. The generated T7 forward and reverse templates were mixed to be
transcribed
and the resulting RNA strands were annealed, DNase and RNase treated, and
purified by
sodium acetate, following the manufacturer's instructions. The sense strand of
the resulting
dsRNA for each of the target genes is given in Table 8-TC.
C. Recombination of Tribolium castaneum genes into the plant vector
pK7GWIWG2D(II)
Since the mechanism of RNA interference operates through dsRNA fragments, the
target nucleotide sequences of the target genes, as selected above, are cloned
in anti-sense
and sense orientation, separated by the intron - CmR - intron, whereby CmR is
the
106
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
chloramphenicol resistance marker, to form a dsRNA hairpin construct. These
hairpin
constructs are generated using the LR recombination reaction between an attL-
containing
entry clone (see Example 1) and an attR-containing destination vector (=
pK7GWIWG2D(II)). The plant vector pK7GWIWG2D(II) is obtained from the
VIB/Plant
Systems Biology with a Material Transfer Agreement. LR recombination reaction
is
performed by using LR ClonaseTM II enzyme mix (Cat. Nr. 11791-020, Invitrogen)
following
the manufacturer's instructions. These cloning experiments result in a hairpin
construct for
each of the target genes, having the promoter - sense - intron - CmR - intron -
antisense
orientation, and wherein the promoter is the plant operable 35S promoter. The
binary vector
pK7GWIWG2D(II) with the 35S promoter is suitable for transformation into A.
tumefaciens.
Restriction enzyme digests were carried out on pCR8/GW/TOPO plasmids
containing
the different targets (see Example 2). The band containing the gene of
interest flanked by the
attL sites using Qiaquick Gel Extraction Kit (Cat. Nr. 28706, Qiagen) is
purified. An amount
of 150 ng of purified fragment and 150 ng pK7GWIWG2D(II) is added together
with the LR
clonase II enzyme and incubated for at least lh at 25 C . After proteinase K
solution
treatment (10 min at 37 C), the whole recombination mix is transformed into
Top 10
chemically competent cells. Positive clones are selected by restriction digest
analyses.
D. Laboratory trials to test dsRNA targets, using artificial diet
for activity
against Tribolium castaneum larvae
The example provided below is an exemplification of the finding that the red
flour
beetle (RFB) larvae are susceptible to orally ingested dsRNA corresponding to
own target
genes.
Red flour beetles, Tribolium castaneum, were maintained at Insect
Investigations Ltd.
(origin: Imperial College of Science, Technology and Medicine, Silwood Park,
Berkshire,
UK). Insects were cultured according to company SOP/251/01. Briefly, the
beetles were
housed in plastic jars or tanks. These have an open top to allow ventilation.
A piece of netting
was fitted over the top and secured with an elastic band to prevent escape.
The larval rearing
medium (flour) was placed in the container where the beetles can breed. The
stored product
beetle colonies were maintained in a controlled temperature room at 25 3 C
with a 16:8
hour light:dark cycle.
107
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
Double-stranded RNA from target TC014 (with sequence corresponding to SEQ ID
NO: -799) was incorporated into a mixture of flour and milk powder (wholemeal
flour:
powdered milk in the ratio 4:1) and left to dry overnight. Each replicate was
prepared
separately: 100 IA of a 10 pg/Ial dsRNA solution (1 mg dsRNA) was added to 0.1
g flour/milk
mixture. The dried mixture was ground to a fine powder. Insects were
maintained within Petri
dishes (55 mm diameter), lined with a double layer of filter paper. The
treated diet was placed
between the two filter paper layers. Ten first instar, mixed sex larvae were
placed in each dish
(replicate). Four replicates were performed for each treatment. Control was
Milli-Q water.
Assessments (number of survivors) were made on a regular basis. During the
trial, the test
conditions were 25 ¨33 C and 20 ¨ 25 % relative humidity, with a 12:12 hour
light:dark
photoperiod.
Survival of larvae of T. castaneum over time on artificial diet treated with
target
TC014 dsRNA was significantly reduced when compared to diet only control, as
shown in
Figure 1.
E. Cloning of a RFB gene fragment in a vector suitable for bacterial
production of
insect-active double-stranded RNA
What follows is an example of cloning a DNA fragment corresponding to an RFB
gene target in a vector for the expression of double-stranded RNA in a
bacterial host,
although any vector comprising a T7 promoter or any other promoter for
efficient
transcription in bacteria, may be used (reference to W00001846).
The sequences of the specific primers used for the amplification of target
genes are
provided in Table 8-TC. The template used is the pCR8/GW/topo vector
containing any of
target sequences. The primers are used in a PCR reaction with the following
conditions: 5
minutes at 98 C, followed by 30 cycles of 10 seconds at 98 C, 30 seconds at 55
C and 2
minutes at 72 C, followed by 10 minutes at 72 C. The resulting PCR fragment is
analyzed
on agarose gel, purified (QIAquick Gel Extraction kit, Cat. Nr. 28706,
Qiagen), blunt-end
cloned into Srf I-linearized pGNA49A vector (reference to W000188121A1), and
sequenced.
The sequence of the resulting PCR product corresponds to the respective
sequence as given in
Table 8-TC. The recombinant vector harbouring this sequence is named pGBNJO0
X_X.
F. Expression and production of a double-stranded RNA target in two
strains of
Escherichia coil: (1) AB309-105, and, (2) BL21(DE3)
108
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
The procedures described below are followed in order to express suitable
levels of
insect-active double-stranded RNA of insect target in bacteria. An RNaseIII-
deficient strain,
AB309-105, is used in comparison to wild-type RNaseIII-containing bacteria,
BL21(DE3).
Transformation of AB309-105 and BL21(DE3)
Three hundred ng of the plasmid are added to and gently mixed in a 50 ill
aliquot of
ice-chilled chemically competent E. coli strain AB309-105 or BL21(DE3). The
cells are
incubated on ice for 20 minutes before subjecting them to a heat shock
treatment of 37 C for
minutes, after which the cells are placed back on ice for a further 5 minutes.
Four hundred
and fifty 1.1.1 of room temperature SOC medium is added to the cells and the
suspension
incubated on a shaker (250 rpm) at 37 C for 1 hour. One hundred 1 of the
bacterial cell
suspension is transferred to a 500 ml conical flask containing 150 ml of
liquid Luria-Bertani
(LB) broth supplemented with 100 1.tg/m1 carbenicillin antibiotic. The culture
is incubated on
an Innova 4430 shaker (250 rpm) at 37 C overnight (16 to 18 hours).
Chemical induction of double-stranded RNA expression in AB309-105 and
BL21(DE3)
Expression of double-stranded RNA from the recombinant vector, pGBNJ003, in
the
bacterial strain AB309-105 or BL21(DE3) is made possible since all the genetic
components
for controlled expression are present. In the presence of the chemical inducer
isopropylthiogalactoside, or IPTG, the T7 polymerase will drive the
transcription of the target
sequence in both antisense and sense directions since these are flanked by
oppositely oriented
T7 promoters.
The optical density at 600 nm of the overnight bacterial culture is measured
using an
appropriate spectrophotometer and adjusted to a value of 1 by the addition of
fresh LB broth.
Fifty ml of this culture is transferred to a 50 ml Falcon tube and the culture
then centrifuged
at 3000 g at 15 C for 10 minutes. The supernatant is removed and the
bacterial pellet
resuspended in 50 ml of fresh S complete medium (SNC medium plus 5 prg/m1
cholesterol)
supplemented with 100 1.1g/m1 carbenicillin and 1 mM IPTG. The bacteria are
induced for 2
to 4 hours at room temperature.
Heat treatment of bacteria
Bacteria are killed by heat treatment in order to minimise the risk of
contamination of
the artificial diet in the test plates. However, heat treatment of bacteria
expressing double-
stranded RNA is not a prerequisite for inducing toxicity towards the insects
due to RNA
interference. The induced bacterial culture is centrifuged at 3000 g at room
temperature for
minutes, the supernatant discarded and the pellet subjected to 80 C for 20
minutes in a
109
CA 02622660 2008-03-14
WO 2007/074405
PCT/1B2006/004003
water bath. After heat treatment, the bacterial pellet is resuspended in 1.5
ml MilliQ water
and the suspension transferred to a microfiige tube. Several tubes are
prepared and used in the
bioassays for each refreshment. The tubes are stored at -20 C until further
use.
G.
Laboratory trials to test Escherichia coli expressing dsRNA targets against
Tribolium castaneum
Plant-based bioassays
Whole plants are sprayed with suspensions of chemically induced bacteria
expressing
dsRNA prior to feeding the plants to RFB. The are grown from in a plant growth
room
chamber. The plants are caged by placing a 500 ml plastic bottle upside down
over the plant
with the neck of the bottle firmly placed in the soil in a pot and the base
cut open and covered
with a fine nylon mesh to permit aeration, reduce condensation inside and
prevent insect
escape. RFB are placed on each treated plant in the cage. Plants are treated
with a suspension
of E. coil AB309-105 harbouring the pGBNJ001 plasmids or pGN29 plasmid.
Different
quantities of bacteria are applied to the plants: for instance 66, 22, and 7
units, where one unit
is defined as 109 bacterial cells in 1 ml of a bacterial suspension at optical
density value of 1
at 600 nm wavelength. In each case, a total volume of between 1 and 10 ml s
sprayed on the
plant with the aid of a vaporizer. One plant is used per treatment in this
trial. The number of
survivors are counted and the weight of each survivor recorded.
Spraying plants with a suspension of E. coil bacterial strain AB309-105
expressing
target dsRNA from pGBNJ003 leed to a dramatic increase in insect mortality
when compared
to pGN29 control. These experiments show that double-stranded RNA
corresponding to an
insect gene target sequence produced in either wild-type or RNaseIII-deficient
bacterial
expression systems is toxic towards the insect in terms of substantial
increases in insect
mortality and growth/development delay for larval survivors. It is also clear
from these
experiments that an exemplification is provided for the effective protection
of plants/crops
from insect damage by the use of a spray of a formulation consisting of
bacteria expressing
double-stranded RNA corresponding to an insect gene target.
Example 10: Mvzus persicae (green peach aphid)
A. Cloning Myzus persicae partial sequences
High quality, intact RNA was isolated from nymphs of Myzus persicae (green
peach
aphid; source: Dr. Rachel Down, Insect & Pathogen Interactions, Central
Science Laboratory,
110
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
Sand Hutton, York, Y041 1LZ, UK) using TRIzol Reagent (Cat. Nr. 15596-
026/15596-018,
Invitrogen, Rockville, Maryland, USA) following the manufacturer's
instructions. Genomic
DNA present in the RNA preparation was removed by DNase treatment following
the
manafacturer's instructions (Cat. Nr. 1700, Promega). cDNA was generated using
a
commercially available kit (SuperScript TM III Reverse Transcriptase, Cat. Nr.
18080044,
Invitrogen, Rockville, Maryland, USA) following the manufacturer's
instructions.
To isolate cDNA sequences comprising a portion of the MP001, MP002, MP010,
MP016 and MP027 genes, a series of PCR reactions with degenerate primers were
performed
using Amplitaq Gold (Cat. Nr. N8080240, Applied Biosystems) following the
manafacturer's
instructions.
The sequences of the degenerate primers used for amplification of each of the
genes
are given in Table 2-MP. These primers were used in respective PCR reactions
with the
following conditions: for MP001, MP002 and MP016, 10 minutes at 95 C, followed
by 40
cycles of 30 seconds at 95 C, 1 minute at 50 C and 1 minute 30 seconds at 72
C, followed by
7 minutes at 72 C; for MP027, a touchdown program was used: 10 minutes at 95
C, followed
by 10 cycles of 30 seconds at 95 C, 40 seconds at 60 C with a decrease in
temperature of 1 C
per cycle and 1 minute 10 seconds at 72 C, followed by 30 cycles of 30 seconds
at 95 C, 40
seconds at 50 C and 1 minute 10 seconds at 72 C, followed by 7 minutes at 72
C; for
MP010, 10 minutes at 95 C, followed by 40 cycles of 30 seconds at 95 C, 1
minute at 54 C
and 3 minutes at 72 C, followed by 7 minutes at 72 C. The resulting PCR
fragments were
analyzed on agarose gel, purified (QIAquick Gel Extraction kit, Cat. Nr.
28706, Qiagen),
cloned into the pCR8/GW/TOPO vector (Cat. Nr. K2500-20, Invitrogen), and
sequenced. The
sequences of the resulting PCR products are represented by the respective SEQ
ID NO:s as
given in Table 2-MP and are referred to as the partial sequences. The
corresponding partial
amino acid sequences are represented by the respective SEQ ID NO:s as given in
Table 3-
MP.
B. dsRNA production of Myzus persicae genes
dsRNA was synthesized in milligram amounts using the commercially available
kit
T7 RibomaxTM Express RNAi System (Cat. Nr. P1700, Promega). First two separate
single 5'
T7 RNA polymerase promoter templates were generated in two separate PCR
reactions, each
reaction containing the target sequence in a different orientation relative to
the T7 promoter.
For each of the target genes, the sense T7 template was generated using
specific T7
forward and specific reverse primers. The sequences of the respective primers
for amplifying
111
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
the sense template for each of the target genes are given in Table 8-MP. A
touchdown PCR
was performed as follows: 1 minute at 95 C, followed by 20 cycles of 30
seconds at 95 C, 30
seconds at 55 C (for MP001, MP002, MP016, MP027 and gfp) or 30 seconds at 50 C
(for
MP010) with a decrease in temperature of 0.5 C per cycle and 1 minute at 72 C,
followed by
15 cycles of 30 seconds at 95 C, 30 seconds at 45 C and 1 minute at 72 C
followed by 10
minutes at 72 C. The anti-sense T7 template was generated using specific
forward and
specific T7 reverse primers in a PCR reaction with the same conditions as
described above.
The sequences of the respective primers for amplifying the anti-sense template
for each of the
target genes are given in Table 8-MP. The resulting PCR products were analyzed
on agarose
gel and purified by PCR purification kit (Qiaquick PCR Purification Kit, Cat.
Nr. 28106,
Qiagen) and NaC104 precipitation. The generated T7 forward and reverse
templates were
mixed to be transcribed and the resulting RNA strands were annealed, DNase and
RNase
treated, and purified by sodium acetate, following the manufacturer's
instructions. The sense
strand of the resulting dsRNA for each of the target genes is given in Table 8-
MP.
C. Recombination of Myzus persicae genes into the plant vector
pK7GWIWG2D(II)
Since the mechanism of RNA interference operates through dsRNA fragments, the
target nucleotide sequences of the target genes, as selected above, were
cloned in anti-sense
and sense orientation, separated by the intron - CmR - intron, whereby CmR is
the
chloramphenicol resistance marker, to form a dsRNA hairpin construct. These
hairpin
constructs were generated using the LR recombination reaction between an attL-
containing
entry clone (see Example A) and an attR-containing destination vector (=
pK7GWIWG2D(II)). The plant vector pK7GWIWG2D(II) was obtained from the
VIB/Plant
Systems Biology with a Material Transfer Agreement. LR recombination reaction
was
performed by using LR ClonaseTM II enzyme mix (Cat. Nr. 11791-020, Invitrogen)
following
the manufacturer's instructions. These cloning experiments resulted in a
hairpin construct for
each of the MP001, MP002, MP010, MP016 and MP026 genes, having the promoter -
sense
- intron - CmR - intron - antisense orientation and wherein the promoter is
the plant operable
35S promoter. The binary vector pK7GWIWG2D(II) with the 35S promoter is
suitable for
transformation into A. tumefaciens.
A digest with restriction enzyme Alw44I was done for all the targets cloned
into
pCR8/GW/topo (see Example B). The band containing the gene of interest flanked
by the
attL sites using Qiaquick Gel Extraction Kit (Cat. Nr. 28706, Qiagen) was
purified. An
amount of 150 ng of purified fragment and 150 ng pK7GWIWG2D(II) was added
together
112
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
with the LR clonase II enzyme and incubated for at least lh at 25 C . After
proteinase K
solution treatment (10 min at 37 C), the whole recombination mix was
transformed into Top
chemically competent cells. Positive clones were selected by restriction
digest analysis.
The complete sequence of the hairpin construct for:
- MP001 (sense - intron - CmR - intron - antisense) is represented in SEQ
ID NO:
1066;
- MP002 (sense - intron - CmR - intron - antisense) is represented in SEQ
ID NO:
1067;
- MP010 (sense - intron - CmR - intron - antisense) is represented in SEQ ID
NO:
1068;
- MP016 (sense - intron - CmR - intron - antisense) is represented in SEQ
ID NO:
1069;
- MP027 (sense - intron - CmR - intron - antisense) is represented in SEQ ID
NO:
1070.
Table 9-MP provides complete sequences for each hairpin construct.
D. Laboratory trials to test dsRNA targets using liquid artificial
diet for
activity against Myzus persicae
Liquid artificial diet for the green peach aphid, Myzus persicae, was prepared
based
on the diet suitable for pea aphids (Acyrthosiphon pisum), as described by
Febvay et al.
(1988) [Influence of the amino acid balance on the improvement of an
artificial diet for a
biotype of Acyrthosiphon pisum (Homoptera: Aphididae). Can. J. Zool. 66: 2449-
2453], but
with some modifications. The amino acids component of the diet was prepared as
follows: in
mg/100m1, alanine 178.71, beta-alanine 6.22, arginine 244.9, asparagine
298.55, aspartic acid
88.25, cysteine 29.59, glutamic acid 149.36, glutamine 445.61, glycine 166.56,
histidine
136.02, isoleucine 164.75, leucine 231.56, lysine hydrochloride 351.09,
methionine 72.35,
omithine (HC1) 9.41, phenylalanine 293, proline 129.33, serine 124.28,
threonine 127.16,
tryptophane 42.75, tyrosine 38.63, L-valine 190.85. The amino acids were
dissolved in 30 ml
Milli-Q H2O except for tyrosine which was first dissolved in a few drops of 1
M HC1 before
adding to the amino acid mix. The vitamin mix component of the diet was
prepared as a 5 x
concentrate stock as follows: in mg/L, amino benzoic acid 100, ascorbic acid
1000, biotin 1,
calcium panthothenate 50, choline chloride 500, folic acid 10, myoinositol
420, nicotinic acid
100, pyridoxine hydrochloride 25, riboflavin 5, thiamine hydrochloride 25. The
riboflavin
113
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
was dissolved in 1 ml H20 at 50 C and then added to the vitamin mix stock. The
vitamin
mix was aliquoted in 20 ml per aliquot and stored at -20 C. One aliquot of
vitamin mix was
added to the amino acid solution. Sucrose and MgSO4.7H20 was added with the
following
amounts to the mix: 20 g and 242 mg, respectively. Trace metal stock solution
was prepared
as follows: in mg/100m1, CuSO4.5H20 4.7, FeC13.6H20 44.5, MnC12.4H20 6.5, NaC1
25.4,
ZnC12 8.3. Ten ml of the trace metal solution and 250 mg KH2PO4 was added to
the diet and
Milli-Q water was added to a final liquid diet volume of 100 ml. The pH of the
diet was
adjusted to 7 with 1 M KOH solution. The liquid diet was filter-sterilised
through an 0.22 gm
filter disc (Millipore).
Green peach aphids (Myzus persicae; source: Dr. Rachel Down, Insect & Pathogen
Interactions, Central Science Laboratory, Sand Hutton, York, Y041 1LZ, UK)
were reared
on 4- to 6-week-old oilseed rape (Brassica napus variety SW Oban; source: Nick
Balaam, Sw
Seed Ltd., 49 North Road, Abington, Cambridge, CB1 6AS, UK) in aluminium-
framed cages
containing 70 gm mesh in a controlled environment chamber with the following
conditions:
23 2 C and 60 5 % relative humidity, with a 16:8 hours light:dark
photoperiod.
One day prior to the start of the bioassay, adults were collected from the
rearing cages
and placed on fresh detached oilseed rape leaves in a Petri dish and left
overnight in the
insect chamber. The following day, first-instar nymphs were picked and
transferred to
feeding chambers. A feeding chamber comprised of 10 first instar nymphs placed
in a small
Petri dish (with diameter 3 cm) covered with a single layer of thinly
stretched parafilm M
onto which 50 pi of diet was added. The chamber was sealed with a second layer
of parafilm
and incubated under the same conditions as the adult cultures. Diet with dsRNA
was
refreshed every other day and the insects' survival assessed on day 8 i.e. 8th
day post bioassay
start. Per treatment, 5 bioassay feeding chambers (replicates) were set up
simultaneously.
Test and control (gfp) dsRNA solutions were incorporated into the diet to a
final
concentration of 2 ps/11. The feeding chambers were kept at 23 2 C and 60 5
% relative
humidity, with a 16:8 hours light:dark photoperiod. A Mann-Whitney test was
determined by
GraphPad Prism version 4 to establish whether the medians do differ
significantly between
target 27 (MP027) and gfp dsRNA.
In the bioassay, feeding liquid artificial diet supplemented with intact naked
dsRNA
from target 27 (SEQ ID NO: 1061) to nymphs of Myzus persicae using a feeding
chamber,
resulted in a significant increase in mortality, as shown in Figure 1. Average
percentage
114
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
survivors for target 27, gfp dsRNA and diet only treatment were 2, 34 and 82,
respectively.
Comparison of target 027 with gfp dsRNA groups using the Mann-Whitney test
resulted in an
one-tailed P-value of 0.004 which indicates that the median of target 027 is
significantly
different (P < 0.05) from the expected larger median of gfp dsRNA. The green
peach aphids
on the liquid diet with incorporated target 27 dsRNA were noticeably smaller
than those that
were fed on diet only or with gfp dsRNA control (data not presented).
E. Cloning of a GPA gene fragment in a vector suitable for bacterial
production of
insect-active double-stranded RNA
What follows is an example of cloning a DNA fragment corresponding to a GPA
gene
target in a vector for the expression of double-stranded RNA in a bacterial
host, although any
vector comprising a T7 promoter or any other promoter for efficient
transcription in bacteria,
may be used (reference to W00001846).
The sequences of the specific primers used for the amplification of target
genes are
provided in Table 8-MP. The template used is the pCR8/GW/topo vector
containing any of
target sequences. The primers are used in a PCR reaction with the following
conditions: 5
minutes at 98 C, followed by 30 cycles of 10 seconds at 98 C, 30 seconds at 55
C and 2
minutes at 72 C, followed by 10 minutes at 72 C. The resulting PCR fragment is
analyzed
on agarose gel, purified (QIAquick Gel Extraction kit, Cat. Nr. 28706,
Qiagen), blunt-end
cloned into Srf I-linearized pGNA49A vector (reference to W000188121A1), and
sequenced.
The sequence of the resulting PCR product corresponds to the respective
sequence as given in
Table 8-MP. The recombinant vector harbouring this sequence is named
pGBNJOOXX.
F. Expression and production of a double-stranded RNA target in two
strains of
Escherichia coli: (1) AB309-105, and, (2) BL21(DE3)
The procedures described below are followed in order to express suitable
levels of
insect-active double-stranded RNA of insect target in bacteria. An RNaseIII-
deficient strain,
AB309-105, is used in comparison to wild-type RNaseIII-containing bacteria,
BL21(DE3).
Transformation of AB309-105 and BL21(DE3)
Three hundred ng of the plasmid are added to and gently mixed in a 50 1
aliquot of
ice-chilled chemically competent E. coli strain AB309-105 or BL21(DE3). The
cells are
incubated on ice for 20 minutes before subjecting them to a heat shock
treatment of 37 C for
minutes, after which the cells are placed back on ice for a further 5 minutes.
Four hundred
and fifty 1 of room temperature SOC medium is added to the cells and the
suspension
115
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
incubated on a shaker (250 rpm) at 37 C for 1 hour. One hundred ill of the
bacterial cell
suspension is transferred to a 500 ml conical flask containing 150 ml of
liquid Luria-Bertani
(LB) broth supplemented with 100 g/ml carbenicillin antibiotic. The culture
is incubated on
an Innova 4430 shaker (250 rpm) at 37 C overnight (16 to 18 hours).
Chemical induction of double-stranded RNA expression in AB309-105 and
BL21(DE3)
Expression of double-stranded RNA from the recombinant vector, pGBNJ003, in
the
bacterial strain AB309-105 or BL21(DE3) is made possible since all the genetic
components
for controlled expression are present. In the presence of the chemical inducer
isopropylthiogalactoside, or IPTG, the T7 polymerase will drive the
transcription of the target
sequence in both antisense and sense directions since these are flanked by
oppositely oriented
T7 promoters.
The optical density at 600 nm of the overnight bacterial culture is measured
using an
appropriate spectrophotometer and adjusted to a value of 1 by the addition of
fresh LB broth.
Fifty ml of this culture is transferred to a 50 ml Falcon tube and the culture
then centrifuged
at 3000 g at 15 C for 10 minutes. The supernatant is removed and the
bacterial pellet
resuspended in 50 ml of fresh S complete medium (SNC medium plus 5 [Tim'
cholesterol)
supplemented with 100 jig/m1 carbenicillin and 1 mM IPTG. The bacteria are
induced for 2
to 4 hours at room temperature.
Heat treatment of bacteria
Bacteria are killed by heat treatment in order to minimise the risk of
contamination of
the artificial diet in the test plates. However, heat treatment of bacteria
expressing double-
stranded RNA is not a prerequisite for inducing toxicity towards the insects
due to RNA
interference. The induced bacterial culture is centrifuged at 3000 g at room
temperature for
minutes, the supernatant discarded and the pellet subjected to 80 C for 20
minutes in a
water bath. After heat treatment, the bacterial pellet is resuspended in 1.5
ml MilliQ water
and the suspension transferred to a microfuge tube. Several tubes are prepared
and used in the
bioassays for each refreshment. The tubes are stored at -20 C until further
use.
G. Laboratory trials to test Escherichia coli expressing dsRNA targets
against
Myzus persicae
Plant-based bioassays
Whole plants are sprayed with suspensions of chemically induced bacteria
expressing
dsRNA prior to feeding the plants to GPA. The are grown from in a plant growth
room
116
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
chamber. The plants are caged by placing a 500 ml plastic bottle upside down
over the plant
with the neck of the bottle firmly placed in the soil in a pot and the base
cut open and covered
with a fine nylon mesh to permit aeration, reduce condensation inside and
prevent insect
escape. GPA are placed on each treated plant in the cage. Plants are treated
with a suspension
of E. coil AB309-105 harbouring the pGBNJ001 plasmids or pGN29 plasmid.
Different
quantities of bacteria are applied to the plants: for instance 66, 22, and 7
units, where one unit
is defined as 109 bacterial cells in 1 ml of a bacterial suspension at optical
density value of 1
at 600 nm wavelength. In each case, a total volume of between 1 and 10 ml s
sprayed on the
plant with the aid of a vaporizer. One plant is used per treatment in this
trial. The number of
survivors are counted and the weight of each survivor recorded.
Spraying plants with a suspension of E. coil bacterial strain AB309-105
expressing
target dsRNA from pGBNJ003 lead to a dramatic increase in insect mortality
when compared
to pGN29 control. These experiments show that double-stranded RNA
corresponding to an
insect gene target sequence produced in either wild-type or RNaseIll-deficient
bacterial
expression systems is toxic towards the insect in terms of substantial
increases in insect
mortality and growth/development delay for larval survivors. It is also clear
from these
experiments that an exemplification is provided for the effective protection
of plants/crops
from insect damage by the use of a spray of a formulation consisting of
bacteria expressing
double-stranded RNA corresponding to an insect gene target.
Example 11: Nilaparvata Wens (Brown Plant Hopper)
A. Cloning Nilaparvata lugens partial sequences
From high quality total RNA of Nilaparvata lugens (source: Dr. J. A.
Gatehouse,
Dept. Biological Sciences, Durham University, UK) cDNA was generated using a
commercially available kit (SuperScriptTM III Reverse Transcriptase, Cat N .
18080044,
Invitrogen, Rockville, Maryland, USA) following the manufacturer's protocol.
To isolate cDNA sequences comprising a portion of the Nilaparvata lugens
NL001,
NL002, NL003, NL004, NL005, NL006, NL007, NL008, NL009, NL010, NL011, NL012,
NL013, NL014, NL015, NL016, NL018, NL019, NL021, NL022, and NL027 genes, a
series
of PCR reactions with degenerate primers were performed using Amplitaq Gold
(Cat N .
N8080240; Applied Biosystems) following the manufacturer's protocol.
The sequences of the degenerate primers used for amplification of each of the
genes
are given in Table 2-NL. These primers were used in respective PCR reactions
with the
following conditions: for NL001: 5 minutes at 95 C, followed by 40 cycles of
30 seconds at
117
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
95 C, 1 minute at 55 C and 1 minute at 72 C, followed by 10 minutes at 72 C:
for NL002: 3
minutes at 95 C, followed by 40 cycles of 30 seconds at 95 C, 1 minute at 55 C
and 1 minute
at 72 C, followed by 10 minutes at 72 C; for NL003: 3 minutes at 95 C,
followed by 40
cycles of 30 seconds at 95 C, 1 minute at 61 C and 1 minute at 72 C,
followed by 10
minutes at 72 C; for NL004: 10 minutes at 95 C, followed by 40 cycles of 30
seconds at 95
C, 1 minute at 51 C and 1 minute at 72 C; for NL005: 10 minutes at 95 C,
followed by 40
cycles of 30 seconds at 95 C, 1 minute at 54 C and 1 minute at 72 C,
followed by 10
minutes at 72 C; for NL006: 10 minutes at 95 C, followed by 40 cycles of 30
seconds at 95
C, 1 minute at 55 C and 3 minute 30 seconds at 72 C, followed by 10 minutes
at 72 C; for
NL007: 10 minutes at 95 C, followed by 40 cycles of 30 seconds at 95 C, 1
minute at 54 C
and 1 minute 15 seconds at 72 C, followed by 10 minutes at 72 C; for NL008:
10 minutes at
95 C, followed by 40 cycles of 30 seconds at 95 C, 1 minute at 53 C and 1
minute at 72
C, followed by 10 minutes at 72 C; for NL009: 10 minutes at 95 C, followed by
40 cycles
of 30 seconds at 95 C, 1 minute at 55 C and 1 minute at 72 C, followed by
10 minutes at
72 C; for NL010: 10 minutes at 95 C, followed by 40 cycles of 30 seconds at
95 C, 1
minute at 54 C and 2 minute 30 seconds at 72 C, followed by 10 minutes at 72
C; for
NL011: 10 minutes at 95 C, followed by 40 cycles of 30 seconds at 95 C, 1
minute at 55 C
and 1 minute at 72 C; for NL012: 10 minutes at 95 C, followed by 40 cycles
of 30 seconds
at 95 C, 1 minute at 55 C and 1 minute at 72 C; for NL013: 10 minutes at 95
C, followed
by 40 cycles of 30 seconds at 95 C, 1 minute at 54 C and 1 minute 10 seconds
at 72 C,
followed by 10 minutes at 72 C; for NL014: 10 minutes at 95 C, followed by 40
cycles of
30 seconds at 95 C, 1 minute at 53 C and 1 minute at 72 C, followed by 10
minutes at
72 C; for NL015: 10 minutes at 95 C, followed by 40 cycles of 30 seconds at
95 C, 1
minute at 54 C and 1 minute 40 seconds at 72 C, followed by 10 minutes at 72
C; for
NL016: 10 minutes at 95 C, followed by 40 cycles of 30 seconds at 95 C, 1
minute at 54 C
and 1 minute 40 seconds at 72 C, followed by 10 minutes at 72 C; for NL018:
10 minutes at
95 C, followed by 40 cycles of 30 seconds at 95 C, 1 minute at 54 C and 1
minute 35
seconds at 72 C, followed by 10 minutes at 72 C; for NL019: 10 minutes at 95
C, followed
by 40 cycles of 30 seconds at 95 C, 1 minute at 55 C and 1 minute at 72 C,
followed by 10
minutes at 72 C; for NL021: 10 minutes at 95 C, followed by 40 cycles of 30
seconds at 95
C, 1 minute at 54 C and 1 minute 45 seconds at 72 C, followed by 10 minutes
at 72 C: for
NL022: 10 minutes at 95 C, followed by 40 cycles of 30 seconds at 95 C, 1
minute at 54 C
and 1 minute 45 seconds at 72 C, followed by 10 minutes at 72 C; and for
NL027: 10
minutes at 95 C, followed by 40 cycles of 30 seconds at 95 C, 1 minute at 54
C and 1
118
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
minute 45 seconds at 72 C, followed by 10 minutes at 72 C. The resulting PCR
fragments
were analyzed on agarose gel, purified (QIAquick Gel Extraction kit, Cat. Nr.
28706,
Qiagen), cloned into the pCR8/GW/topo vector (Cat. Nr. I(2500 20, Invitrogen),
and
sequenced. The sequences of the resulting PCR products are represented by the
respective
SEQ ID NO:s as given in Table 2-NL and are referred to as the partial
sequences. The
corresponding partial amino acid sequences are represented by the respective
SEQ ID NO:s
as given in Table 3-NL.
B. Cloning of a partial sequence of the Nilaparvata lugens NL023 gene via
EST
sequence
From high quality total RNA of Nilaparvata lugens (source: Dr. J. A.
Gatehouse,
Dept. Biological Sciences, Durham University, UK) cDNA was generated using a
commercially available kit (SuperScriptTM III Reverse Transcriptase, Cat N .
18080044,
Invitrogen, Rockville, Maryland, USA) following the manufacturer's protocol.
A partial cDNA sequence, NL023, was amplified from Nilaparvata lugens cDNA
which corresponded to a Nilaparvata lugens EST sequence in the public database
Genbank
with accession number CAH65679.2. To isolate cDNA sequences comprising a
portion of the
NL023 gene, a series of PCR reactions with EST based specific primers were
performed
using PerfectShotTM ExTaq (Cat N . RROO5A, Takara Bio Inc.) following the
manafacturer's
protocol.
For NL023, the specific primers oGBKW002 and oGBKW003 (represented herein as
SEQ ID NO: 1157 and SEQ ID NO: 1158, respectively) were used in two
independent PCR
reactions with the following conditions: 3 minutes at 95 C, followed by 30
cycles of 30
seconds at 95 C, 30 seconds at 56 C and 2 minutes at 72 C, followed by 10
minutes at
72 C. The resulting PCR products were analyzed on agarose gel, purified
(QIAquick Gel
Extraction Kit; Cat. N . 28706, Qiagen), cloned into the pCR4-TOPO vector (Cat
N . K4575-
40, Invitrogen) and sequenced. The consensus sequence resulting from the
sequencing of
both PCR products is herein represented by SEQ ID NO: 1111 and is referred to
as the
partial sequence of the NL023 gene. The corresponding partial amino acid
sequence is herein
reperesented as SEQ ID NO: 1112.
C. dsRNA production of Nilaparvata lugens genes
dsRNA was synthesized in milligram amounts using the commercially available
kit
T7 RibomaxTM Express RNAi System (Cat. Nr. P1700, Promega). First two separate
single 5'
119
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
T7 RNA polymerase promoter templates were generated in two separate PCR
reactions, each
reaction containing the target sequence in a different orientation relative to
the T7 promoter.
For each of the target genes, the sense T7 template was generated using
specific T7
forward and specific reverse primers. The sequences of the respective primers
for amplifying
the sense template for each of the target genes are given in Table 4. The
conditions in the
PCR reactions were as follows: for NL001: 4 minutes at 94 C, followed by 35
cycles of 30
seconds at 94 C, 30 seconds at 60 C and 1 minute at 72 C, followed by 10
minutes at
72 C; for NL002: 4 minutes at 94 C, followed by 35 cycles of 30 seconds at 94
C, 30
seconds at 60 C and 1 minute at 72 C, followed by 10 minutes at 72 C; for
NL003: 4
minutes at 94 C, followed by 35 cycles of 30 seconds at 94 C, 30 seconds at
66 C and 1
minute at 72 C, followed by 10 minutes at 72 C; for NL004: 4 minutes at 95
C, followed
by 35 cycles of 30 seconds at 95 C, 30 seconds at 54 C and 1 minute at 72
C, followed by
minutes at 72 C; for NL005: 4 minutes at 95 C, followed by 35 cycles of 30
seconds at
95 C, 30 seconds at 57 C and 1 minute at 72 C, followed by 10 minutes at 72
C; for
NL006: 4 minutes at 95 C, followed by 35 cycles of 30 seconds at 95 C, 30
seconds at 54
C and 1 minute at 72 C, followed by 10 minutes at 72 C; for NL007: 4 minutes
at 95 C,
followed by 35 cycles of 30 seconds at 95 C, 30 seconds at 51 C and 1 minute
at 72 C,
followed by 10 minutes at 72 C; for NL008: 4 minutes at 95 C, followed by 35
cycles of 30
seconds at 95 C, 30 seconds at 54 C and 1 minute at 72 C, followed by 10
minutes at
72 C; for NL009: 4 minutes at 95 C, followed by 35 cycles of 30 seconds at 95
C, 30
seconds at 54 C and 1 minute at 72 C, followed by 10 minutes at 72 C; for
NL010: 4
minutes at 95 C, followed by 35 cycles of 30 seconds at 95 C, 30 seconds at
54 C and 1
minute at 72 C, followed by 10 minutes at 72 C; for NL011: 4 minutes at 95
C, followed
by 35 cycles of 30 seconds at 95 C, 30 seconds at 53 C and 1 minute at 72
C, followed by
10 minutes at 72 C; for NL012: 4 minutes at 95 C, followed by 35 cycles of
30secondes at
95 C, 30 seconds at 53 C and 1 minute at 72 C, followed by 10 minutes at 72
C; for
NL013: 4 minutes at 95 C, followed by 35 cycles of 30 seconds at 95 C, 30
seconds at 55
C and 1 minute at 72 C, followed by 10 minutes at 72 C; for NL014: 4 minutes
at 95 C,
followed by 35 cycles of 30 seconds at 95 C, 30 seconds at 51 "DC and 1
minute at 72 C,
followed by 10 minutes at 72 C; for NL015: 4 minutes at 95 C, followed by 35
cycles of 30
seconds at 95 C, 30 seconds at 55 C and 1 minute at 72 C, followed by 10
minutes at
72 C; for NL016: 4 minutes at 95 C, followed by 35 cycles of 30 seconds at 95
C, 30
seconds at 57 C and 1 minute at 72 C, followed by 10 minutes at 72 C; for
NL018: 4
minutes at 95 C, followed by 35 cycles of 30 seconds at 95 C, 30 seconds at
55 C and 1
120
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
minute at 72 C, followed by 10 minutes at 72 C; for NL019: 4 minutes at 95
C, followed
by 35 cycles of 30 seconds at 95 C, 30 seconds at 54 C and 1 minute at 72
C, followed by
minutes at 72 C; for NL021: 4 minutes at 95 C, followed by 35 cycles of 30
seconds at
95 C, 30 seconds at 55 C and 1 minute at 72 C, followed by 10 minutes at 72
C; for
NL022: 4 minutes at 95 C, followed by 35 cycles of 30 seconds at 95 C, 30
seconds at 53
C and 1 minute at 72 C, followed by 10 minutes at 72 C; for NL023: 4 minutes
at 95 C,
followed by 35 cycles of 30 seconds at 95 C, 30 seconds at 52 C and 1 minute
at 72 C,
followed by 10 minutes at 72 C; and for NL027: 4 minutes at 95 C, followed by
35 cycles of
30secondes at 95 C, 30 seconds at 52 C and 1 minute at 72 C, followed by 10
minutes at
_ 72 C. The anti-sense T7 template was generated using specific forward and
specific T7
reverse primers in a PCR reaction with the same conditions as described above.
The
sequences of the respective primers for amplifying the anti-sense template for
each of the
target genes are given in Table 4-NL. The resulting PCR products were analyzed
on agarose
gel and purified by PCR purification kit (Qiaquick PCR Purification Kit, Cat.
Nr. 28106,
Qiagen). The generated T7 forward and reverse templates were mixed to be
transcribed and
the resulting RNA strands were annealed, DNase and RNase treated, and purified
by sodium
acetate, following the manufacturer's instructions, but with the following
modification: RNA
peppet is washed twice in 70% ethanol. The sense strand of the resulting dsRNA
for each of
the target genes is given in Table 8-NL.
The template DNA used for the PCR reactions with T7 primers on the green
fluorescent protein (gfp) control was the plasmid pPD96.12 (the Fire Lab,
http://genome-
www.stanford.edu/group/fire/), which contains the wild-type gfp coding
sequence
interspersed by 3 synthetic introns. Double-stranded RNA was synthesized using
the
commercially available kit T7 RiboMAXTm Express RNAi System (Cat.N . P1700,
Promega). First two separate single 5' T7 RNA polymerase promoter templates
were
generated in two separate PCR reactions, each reaction containing the target
sequence in a
different orientation relative to the T7 promoter. For gfp, the sense T7
template was
generated using the specific T7 FW primer oGAU183 and the specific RV primer
oGAU182
(represented herein as SEQ ID NO: 236 and SEQ ID NO: 237 , respectively) in a
PCR
reaction with the following conditions: 4 minutes at 95 C, followed by 35
cycles of 30
seconds at 95 C, 30 seconds at 55 C and 1 minute at 72 C, followed by 10
minutes at
72 C. The anti-sense T7 template was generated using the specific FW primer
oGAU181 and
the specific T7 RV primer oGAU184 (represented herein as SEQ ID NO: 238 and
SEQ ID
NO: 239 , respectively) in a PCR reaction with the same conditions as
described above. The
121
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
resulting PCR products were analyzed on agarose gel and purified (QIAquick
PCR
Purification Kit; Cat. N . 28106, Qiagen). The generated T7 FW and RV
templates were
mixed to be transcribed and the resulting RNA strands were annealed, DNase and
RNase
treated, and purified by precipitation with sodium acetate and isopropanol,
following the
manufacturer's protocol, but with the following modification: RNA peppet is
washed twice in
70% ethanol. The sense strands of the resulting dsRNA is herein represented by
SEQ ID NO:
235.
D. Laboratory trials to screen dsRNA targets using liquid artificial diet
for activity
against Nilaparvata lugens
Liquid artificial diet (MMD-1) for the rice brown planthopper, Nilaparvata
lugens,
was prepared as described by Koyama (1988) [Artificial rearing and nutritional
physiology of
the planthoppers and leafhoppers (Homoptera: Delphacidae and Deltocephalidae)
on a holidic
diet. JARQ 22: 20-27], but with a modification in final concentration of diet
component
sucrose: 14.4 % (weight over volume) was used. Diet components were prepared
as separate
concentrates: 10 x mineral stock (stored at 4 C), 2 x amino acid stock
(stored at -20 C) and
x vitamin stock (stored at -20 C). The stock components were mixed
immediately prior to
the start of a bioassay to 4/3 x concentration to allow dilution with the test
dsRNA solution (4
x concentration), pH adjusted to 6.5, and filter-sterilised into approximately
500 1 aliquots.
Rice brown planthopper (Nilaparvata lugens) was reared on two-to-three month
old
rice (Oryza sativa cv Taichung Native 1) plants in a controlled environment
chamber: 27 2
C, 80 % relative humidity, with a 16:8 hours light:dark photoperiod. A feeding
chamber
comprised 10 first or second instar nymphs placed in a small petri dish (with
diameter 3 cm)
covered with a single layer of thinly stretched parafilm M onto which 50 1.11
of diet was
added. The chamber was sealed with a second layer of parafilm and incubated
under the same
conditions as the adult cultures but with no direct light exposure. Diet with
dsRNA was
refreshed every other day and the insects' survival assessed daily. Per
treatment, 5 bioassay
feeding chambers (replicates) were set up simultaneously. Test and control
(gfp) dsRNA
solutions were incorporated into the diet to a final concentration of 2 mg/ml.
The feeding
chambers were kept at 27 2 C, 80 % relative humidity, with a 16:8 hours
light:dark
photoperiod. Insect survival data were analysed using the Kaplan-Meier
survival curve model
and the survival between groups were compared using the logrank test (Prism
version 4.0).
Feeding liquid artificial diet supplemented with intact naked dsRNAs to
Nilaparvata
lugens in vitro using a feeding chamber resulted in significant increases in
nymphal
122
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
mortalities as shown in four separate bioassays (Figures 1(a)-(d)-NL; Tables
la-d-NL).
These results demonstrate that dsRNAs corresponding to different essential BPH
genes
showed significant toxicity towards the rice brown planthopper.
Effect of gfp dsRNA on BPH survival in these bioassays is not significantly
different
to survival on diet only
Tables 10a-d-NL show a summary of the survival of Nilaparvata lugens on
artificial
diet supplemented with 2 mg/m1 (final concentration) of the following targets;
in Table
10(a)-NL: NL002, NL003, NL005, NL010; in Table 10(b)-NL NL009, NL016; in Table
10(c)-NL NL014, NL018; and in Table 10(d)-NL NL013, NL015, NL021. In the
survival
analysis column, the effect of RNAi is indicated as follows: + = significantly
decreased
survival compared to gfp dsRNA control (alpha < 0.05); - = no significant
difference in
survival compared to gfp dsRNA control. Survival curves were compared (between
diet only
and diet supplemented with test dsRNA, gfp dsRNA and test dsRNA, and diet only
and gfp
dsRNA) using the logrank test.
E. Laboratory trials to screen dsRNAs at different concentrations using
artificial
diet for activity against Nilaparvata lugens
Fifty ill of liquid artificial diet supplemented with different concentrations
of target
NL002 dsRNA, namely 1, 0.2, 0.08, and 0.04 mg/ml (final concentration), was
applied to the
brown planthopper feeding chambers. Diet with dsRNA was refreshed every other
day and
the insects' survival assessed daily. Per treatment, 5 bioassay feeding
chambers (replicates)
were set up simultaneously. The feeding chambers were kept at 27 2 C, 80 %
relative
humidity, with a 16:8 hours light:dark photoperiod. Insect survival data were
analysed using
the Kaplan-Meier survival curve model and the survival between groups were
compared
using the logrank test (Prism version 4.0).
Feeding liquid artificial diet supplemented with intact naked dsRNAs of target
NL002 at
different concentrations resulted in significantly higher BPH mortalities at
final
concentrations of as low as 0.04 mg dsRNA per ml diet when compared with
survival on diet
only, as shown in Figure 2-NL and Table 9-NL. Table 9-NL summarizes the
survival of
Nilaparvata lugens artificial diet feeding trial supplemented with 1, 0.2,
0.08, & 0.04 mg/ml
(final concentration) of target NL002. In the survival analysis column the
effect of RNAi is
indicated as follows: + = significantly decreases survival compared to diet
only control (alpha
< 0.05); - = no significant differences in survival compared to diet only
control. Survival
curves were compared using the logrank test.
123
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
F. Cloning of a BPH gene fragment in a vector suitable for bacterial
production of
insect-active double-stranded RNA
What follows is an example of cloning a DNA fragment corresponding to a BPH
gene
target in a vector for the expression of double-stranded RNA in a bacterial
host, although any
vector comprising a T7 promoter or any other promoter for efficient
transcription in bacteria,
may be used (reference to W00001846).
The sequences of the specific primers used for the amplification of target
genes are
provided in Table 8. The template used is the pCR8/GW/topo vector containing
any of target
sequences. The primers are used in a PCR reaction with the following
conditions: 5 minutes
at 98 C, followed by 30 cycles of 10 seconds at 98 C, 30 seconds at 55 C and 2
minutes at
72 C, followed by 10 minutes at 72 C. The resulting PCR fragment is analyzed
on agarose
gel, purified (QIAquick Gel Extraction kit, Cat. Nr. 28706, Qiagen), blunt-end
cloned into Srf
I-linearized pGNA49A vector (reference to W000188121A1), and sequenced. The
sequence
of the resulting PCR product corresponds to the respective sequence as given
in Table 8-NL.
The recombinant vector harbouring this sequence is named pGBNJ00.
G. Expression and production of a double-stranded RNA target in two strains of
Escherichia coli: (1) AB309-105, and, (2) BL21(DE3)
The procedures described below are followed in order to express suitable
levels of
insect-active double-stranded RNA of insect target in bacteria. An RNaseIII-
deficient strain,
AB309-105, is used in comparison to wild-type RNaseIII-containing bacteria,
BL21(DE3).
Transformation of AB309-105 and BL21(DE3)
Three hundred ng of the plasmid are added to and gently mixed in a 50 I.
aliquot of
ice-chilled chemically competent E. coli strain AB309-105 or BL21(DE3). The
cells are
incubated on ice for 20 minutes before subjecting them to a heat shock
treatment of 37 C for
minutes, after which the cells are placed back on ice for a further 5 minutes.
Four hundred
and fifty p.1 of room temperature SOC medium is added to the cells and the
suspension
incubated on a shaker (250 rpm) at 37 C for 1 hour. One hundred 1 of the
bacterial cell
suspension is transferred to a 500 ml conical flask containing 150 ml of
liquid Luria-Bertani
(LB) broth supplemented with 100 g/ml carbenicillin antibiotic. The culture
is incubated on
an Innova 4430 shaker (250 rpm) at 37 C overnight (16 to 18 hours).
Chemical induction of double-stranded RNA expression in AB309-105 and
BL21(DE3)
124
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
Expression of double-stranded RNA from the recombinant vector, pGBNJ003, in
the
bacterial strain AB309-105 or BL21(DE3) is made possible since all the genetic
components
for controlled expression are present. In the presence of the chemical inducer
isopropylthiogalactoside, or IPTG, the T7 polymerase will drive the
transcription of the target
sequence in both antisense and sense directions since these are flanked by
oppositely oriented
T7 promoters.
The optical density at 600 nm of the overnight bacterial culture is measured
using an
appropriate spectrophotometer and adjusted to a value of 1 by the addition of
fresh LB broth.
Fifty ml of this culture is transferred to a 50 ml Falcon tube and the culture
then centrifuged
at 3000 g at 15 C for 10 minutes. The supernatant is removed and the
bacterial pellet
resuspended in 50 ml of fresh S complete medium (SNC medium plus 5 ug/m1
cholesterol)
supplemented with 100 g/ml carbenicillin and 1 mM IPTG. The bacteria are
induced for 2
to 4 hours at room temperature.
Heat treatment of bacteria
Bacteria are killed by heat treatment in order to minimise the risk of
contamination of
the artificial diet in the test plates. However, heat treatment of bacteria
expressing double-
stranded RNA is not a prerequisite for inducing toxicity towards the insects
due to RNA
interference. The induced bacterial culture is centrifuged at 3000 g at room
temperature for
minutes, the supernatant discarded and the pellet subjected to 80 C for 20
minutes in a
water bath. After heat treatment, the bacterial pellet is resuspended in 1.5
ml MilliQ water
and the suspension transferred to a microfuge tube. Several tubes are prepared
and used in the
bioassays for each refreshment. The tubes are stored at -20 C until further
use.
H. Laboratory trials to test Escherichia coli expressing dsRNA targets against
Nilaparvata lugens
Plant-based bioassays
Whole plants are sprayed with suspensions of chemically induced bacteria
expressing
dsRNA prior to feeding the plants to BPH. The are grown from in a plant growth
room
chamber. The plants are caged by placing a 500 ml plastic bottle upside down
over the plant
with the neck of the bottle firmly placed in the soil in a pot and the base
cut open and covered
with a fine nylon mesh to permit aeration, reduce condensation inside and
prevent insect
escape. BPH are placed on each treated plant in the cage. Plants are treated
with a suspension
of E. colt AB309-105 harbouring the pGBNJ001 plasmids or pGN29 plasmid.
Different
125
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
quantities of bacteria are applied to the plants: for instance 66, 22, and 7
units, where one unit
is defined as 109 bacterial cells in 1 ml of a bacterial suspension at optical
density value of 1
at 600 nm wavelength. In each case, a total volume of between 1 and 10 ml s
sprayed on the
plant with the aid of a vaporizer. One plant is used per treatment in this
trial. The number of
survivors are counted and the weight of each survivor recorded.
Spraying plants with a suspension of E. coli bacterial strain AB309-105
expressing
target dsRNA from pGBNJ003 leed to a dramatic increase in insect mortality
when compared
to pGN29 control. These experiments show that double-stranded RNA
corresponding to an
insect gene target sequence produced in either wild-type or RNaseIII-deficient
bacterial
expression systems is toxic towards the insect in terms of substantial
increases in insect
mortality and growth/development delay for larval survivors. It is also clear
from these
experiments that an exemplification is provided for the effective protection
of plants/crops
from insect damage by the use of a spray of a formulation consisting of
bacteria expressing
double-stranded RNA corresponding to an insect gene target.
Example 10: Chilo suppressalis (rice striped stem borer)
A. Cloning of partial sequence of the Chilo suppressalis genes via family
PCR
High quality, intact RNA was isolated from the 4 different larval stages of
Chilo
suppressalis (rice striped stem borer) using TRIzol Reagent (Cat. Nr. 15596-
026/15596-018,
Invitrogen, Rockville, Maryland, USA) following the manufacturer's
instructions. Genomic
DNA present in the RNA preparation was removed by DNase treatment following
the
manafacturer's instructions (Cat. Nr. 1700, Promega). cDNA was generated using
a
commercially available kit (SuperScript TM III Reverse Transcriptase, Cat. Nr.
18080044,
Invitrogen, Rockville, Maryland, USA) following the manufacturer's
instructions.
To isolate cDNA sequences comprising a portion of the CS001, CS002, CS003,
CS006, CS007, CS009, CS011, CS013, CS014, CS015, CS016 and CS018 genes, a
series of
PCR reactions with degenerate primers were performed using Amplitaq Gold (Cat.
Nr.
N8080240, Applied Biosystems) following the manafacturer's instructions.
The sequences of the degenerate primers used for amplification of each of the
genes
are given in Table 2-CS. These primers were used in respective PCR reactions
with the
following conditions: 10 minutes at 95 C, followed by 40 cycles of 30 seconds
at 95 C, 1
minute at 55 C and 1 minute at 72 C, followed by 10 minutes at 72 C. The
resulting PCR
fragments were analyzed on agarose gel, purified (QIAquick Gel Extraction kit,
Cat. Nr.
126
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
28706, Qiagen), cloned into the pCR4/TOPO vector (Cat. Nr. K2500-20,
Invitrogen), and
sequenced. The sequences of the resulting PCR products are represented by the
respective
SEQ ID NO:s as given in Table 2-CS and are referred to as the partial
sequences. The
corresponding partial amino acid sequences are represented by the respective
SEQ ID NO:s
as given in Table 3-CS.
B. dsRNA production of the Club suppressalis suppressalis
genes
dsRNA was synthesized in milligram amounts using the commercially available
kit
T7 RibomaxTm Express RNAi System (Cat. Nr. P1700, Promega). First two separate
single 5'
T7 RNA polymerase promoter templates were generated in two separate PCR
reactions, each
reaction containing the target sequence in a different orientation relative to
the T7 promoter.
For each of the target genes, the sense T7 template was generated using
specific T7
forward and specific reverse primers. The sequences of the respective primers
for amplifying
the sense template for each of the target genes are given in Table 8-CS. The
conditions in the
PCR reactions were as follows: 4 minutes at 95 C, followed by 35 cycles of 30
seconds at
95 C, 30 seconds at 55 C and 1 minute at 72 C, followed by 10 minutes at 72 C.
The anti-
sense T7 template was generated using specific forward and specific T7 reverse
primers in a
PCR reaction with the same conditions as described above. The sequences of the
respective
primers for amplifying the anti-sense template for each of the target genes
are given in Table
8-CS. The resulting PCR products were analyzed on agarose gel and purified by
PCR
purification kit (Qiaquick PCR Purification Kit, Cat. Nr. 28106, Qiagen) and
NaC104
precipitation. The generated T7 forward and reverse templates were mixed to be
transcribed
and the resulting RNA strands were annealed, DNase and RNase treated, and
purified by
sodium acetate, following the manufacturer's instructions. The sense strand of
the resulting
dsRNA for each of the target genes is given in Table 8-CS.
C. Recombination of the Chilo suppressalis genes into the plant
vector
pK7GWIWG2D(H)
Since the mechanism of RNA interference operates through dsRNA fragments, the
target nucleotide sequences of the target genes, as selected above, are cloned
in anti-sense
and sense orientation, separated by the intron - CmR - intron, whereby CmR is
the
thloramphenicol resistance marker, to form a dsRNA hairpin construct. These
hairpin
constructs are generated using the LR recombination reaction between an attL-
containing
entry clone (see Example 1) and an attR-containing destination vector (=
127
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
pK7GWIWG2D(II)). The plant vector pK7GWIWG2D(II) is obtained from the
VIB/Plant
Systems Biology with a Material Transfer Agreement. LR recombination reaction
is
performed by using LR ClonaseTm II enzyme mix (Cat. Nr. 11791-020, Invitrogen)
following
the manufacturer's instructions. These cloning experiments result in a hairpin
construct for
each of the target genes, having the promoter - sense - intron - CmR - intron -
antisense
orientation, and wherein the promoter is the plant operable 35S promoter. The
binary vector
pK7GWIWG2D(II) with the 35S promoter is suitable for transformation into A.
tumefaciens.
Restriction enzyme digests were carried out on pCR8/GW/TOPO plasmids
containing
the different targets (see Example B). The band containing the gene of
interest flanked by the
attL sites using Qiaquick Gel Extraction Kit (Cat. Nr. 28706, Qiagen) is
purified. An amount
of 150 ng of purified fragment and 150 ng pK7GWIWG2D(II) is added together
with the LR
clonase II enzyme and incubated for at least lh at 25 C . After proteinase K
solution
treatment (10 min at 37 C), the whole recombination mix is transformed into
Top 10
chemically competent cells. Positive clones are selected by restriction digest
analyses.
D. Laboratory trials to test dsRNA targets, using artificial diet for
activity against Chilo
suppressalis larvae
Rice striped stem borers, Chilo suppressalis, (origin: Syngenta, Stein,
Switzerland)
were maintained on a modified artificial diet based on that described by Kaman
and Sato,
1985 (in: Handbook of Insect Rearing. Volumes I & II. P Singh and RF Moore,
eds., Elsevier
Science Publishers, Amsterdam and New York, 1985, pp 448). Briefly, a litre
diet was made
up as follows: 20 g of agar added to 980 ml of Milli-Q water and autoclaved;
the agar
solution was cooled down to approximately 55 C and the remaining ingredients
were added
and mixed thoroughly: 40 g corn flour (Polenta), 20 g cellulose, 30 g sucrose,
30 g casein, 20
g wheat germ (toasted), 8 g Wesson salt mixture, 12 g Vanderzant vitamin mix,
1.8 g sorbic
acid, 1.6 g nipagin (methylparaben), 0.3 g aureomycin, 0.4 g cholesterol and
0.6 g L-cysteine.
The diet was cooled down to approx. 45 C and poured into rearing trays or
cups. The diet
was left to set in a horizontal laminair flow cabin. Rice leaf sections with
oviposited eggs
were removed from a cage housing adult moths and pinned to the solid diet in
the rearing cup
or tray. Eggs were left to hatch and neonate larvae were available for
bioassays and the
maintenance of the insect cultures. During the trials and rearings, the
conditions were 28 2
C and 80 5 % relative humidity, with a 16:8 hour light:dark photoperiod.
128
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
The same artificial diet is used for the bioassays but in this case the diet
is poured
equally in 24 multiwell plates, with each well containing 1 ml diet. Once the
diet is set, the
test formulations are applied to the diet's surface (2 cm2), at the rate of 50
ill of 1 ii,g/p,1
dsRNA of target. The dsRNA solutions are left to dry and two first instar moth
larvae are
placed in each well. After 7 days, the larvae are transferred to fresh treated
diet in multiwell
plates. At day 14 (i.e. 14 days post bioassay start) the number of live and
dead insects is
recorded and examined for abnormalities. Twenty-four larvae in total are
tested per treatment.
An alternative bioassay is performed in which treated rice leaves are fed to
neonate
larvae of the rice striped stem borer. Small leaf sections of Indica rice
variety Taichung
native 1 are dipped in 0.05 % Triton X-100 solution containing 1 ilg/i.11 of
target dsRNA, left
to dry and each section placed in a well of a 24 multiwell plate containing
gellified 2 % agar.
Two neonates are transferred from the rearing tray to each dsRNA treated leaf
section (24
larvae per treatment). After 4 and 8 days, the larvae are transferred to fresh
treated rice leaf
sections. The number of live and dead larvae are assessed on days 4, 8 and 12;
any
abnormalities are also recorded.
E. Cloning of a SSB gene fragment in a vector suitable for bacterial
production of
insect-active double-stranded RNA
What follows is an example of cloning a DNA fragment corresponding to an SSB
gene target in a vector for the expression of double-stranded RNA in a
bacterial host,
although any vector comprising a T7 promoter or any other promoter for
efficient
transcription in bacteria, may be used (reference to W00001846).
The sequences of the specific primers used for the amplification of target
genes are
provided in Table 8. The template used is the pCR8/GW/topo vector containing
any of target
sequences. The primers are used in a PCR reaction with the following
conditions: 5 minutes
at 98 C, followed by 30 cycles of 10 seconds at 98 C, 30 seconds at 55 C and 2
minutes at
72 C, followed by 10 minutes at 72 C. The resulting PCR fragment is analyzed
on agarose
gel, purified (QIAquick Gel Extraction kit, Cat. Nr. 28706, Qiagen), blunt-end
cloned into Srf
I-linearized pGNA49A vector (reference to W000188121A1), and sequenced. The
sequence
of the resulting PCR product corresponds to the respective sequence as given
in Table 8-CS.
The recombinant vector harbouring this sequence is named pGBNJOOXX.
F. Expression and production of a double-stranded RNA target in two strains
of
Escherichia coil: (1) AB309-105, and, (2) BL21(DE3)
129
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
The procedures described below are followed in order to express suitable
levels of
insect-active double-stranded RNA of insect target in bacteria. An RNaseIII-
deficient strain,
AB309-105, is used in comparison to wild-type RNaseIII-containing bacteria,
BL21(DE3).
Transformation of AB309-105 and BL21(DE3)
Three hundred ng of the plasmid are added to and gently mixed in a 50 pl
aliquot of
ice-chilled chemically competent E. coli strain AB309-105 or BL21(DE3). The
cells are
incubated on ice for 20 minutes before subjecting them to a heat shock
treatment of 37 C for
minutes, after which the cells are placed back on ice for a further 5 minutes.
Four hundred
and fifty IA of room temperature SOC medium is added to the cells and the
suspension
incubated on a shaker (250 rpm) at 37 C for 1 hour. One hundred 1 of the
bacterial cell
suspension is transferred to a 500 ml conical flask containing 150 ml of
liquid Luria-Bertani
(LB) broth supplemented with 100 lig/m1 carbenicillin antibiotic. The culture
is incubated on
an Innova 4430 shaker (250 rpm) at 37 C overnight (16 to 18 hours).
Chemical induction of double-stranded RNA expression in AB309-105 and
BL21(DE3)
Expression of double-stranded RNA from the recombinant vector, pGBNJ003, in
the
bacterial strain AB309-105 or BL21(DE3) is made possible since all the genetic
components
for controlled expression are present. In the presence of the chemical inducer
isopropylthiogalactoside, or IPTG, the T7 polymerase will drive the
transcription of the target
sequence in both antisense and sense directions since these are flanked by
oppositely oriented
T7 promoters.
The optical density at 600 nm of the overnight bacterial culture is measured
using an
appropriate spectrophotometer and adjusted to a value of 1 by the addition of
fresh LB broth.
Fifty ml of this culture is transferred to a 50 ml Falcon tube and the culture
then centrifuged
at 3000 g at 15 C for 10 minutes. The supernatant is removed and the
bacterial pellet
resuspended in 50 ml of fresh S complete medium (SNC medium plus 5 g/m1
cholesterol)
supplemented with 100 g/ml carbenicillin and 1 mM IPTG. The bacteria are
induced for 2
to 4 hours at room temperature.
Heat treatment of bacteria
Bacteria are killed by heat treatment in order to minimise the risk of
contamination of
the artificial diet in the test plates. However, heat treatment of bacteria
expressing double-
stranded RNA is not a prerequisite for inducing toxicity towards the insects
due to RNA
interference. The induced bacterial culture is centrifuged at 3000 g at room
temperature for
minutes, the supernatant discarded and the pellet subjected to 80 C for 20
minutes in a
130
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
water bath. After heat treatment, the bacterial pellet is resuspended in 1.5
ml MilliQ water
and the suspension transferred to a microfuge tube. Several tubes are prepared
and used in the
bioassays for each refreshment. The tubes are stored at -20 C until further
use.
G. Laboratory trials to test Escherichia coli expressing dsRNA targets
against Chilo
suppressalis
Plant-based bioassays
Whole plants are sprayed with suspensions of chemically induced bacteria
expressing
dsRNA prior to feeding the plants to SSB. The are grown from in a plant growth
room
chamber. The plants are caged by placing a 500 ml plastic bottle upside down
over the plant
with the neck of the bottle firmly placed in the soil in a pot and the base
cut open and covered
with a fine nylon mesh to permit aeration, reduce condensation inside and
prevent insect
escape. SSB are placed on each treated plant in the cage. Plants are treated
with a suspension
of E. coli AB309-105 harbouring the pGBNJ001 plasmids or pGN29 plasmid.
Different
quantities of bacteria are applied to the plants: for instance 66, 22, and 7
units, where one unit
is defined as 109 bacterial cells in 1 ml of a bacterial suspension at optical
density value of 1
at 600 nm wavelength. In each case, a total volume of between 1 and 10 ml s
sprayed on the
plant with the aid of a vaporizer. One plant is used per treatment in this
trial. The number of
survivors are counted and the weight of each survivor recorded.
Spraying plants with a suspension of E. coli bacterial strain AB309-105
expressing
target dsRNA from pGBNJ003 leed to a dramatic increase in insect mortality
when compared
to pGN29 control. These experiments show that double-stranded RNA
corresponding to an
insect gene target sequence produced in either wild-type or RNaseIII-deficient
bacterial
expression systems is toxic towards the insect in terms of substantial
increases in insect
mortality and growth/development delay for larval survivors. It is also clear
from these
experiments that an exemplification is provided for the effective protection
of plants/crops
from insect damage by the use of a spray of a formulation consisting of
bacteria expressing
double-stranded RNA corresponding to an insect gene target.
Example 9: Plutella xylostella (Diamondback moth),
A. Cloning of a partial sequence of the Plutella xylostella
High quality, intact RNA was isolated from all the different larval stages of
Plutella
xylostella (Diamondback moth; source: Dr. Lara Senior, Insect Investigations
Ltd., Capital
131
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
Business Park, Wentloog, Cardiff, CF3 2PX, Wales, UK) using TRIzol Reagent
(Cat. Nr.
15596-026/15596-018, Invitrogen, Rockville, Maryland, USA) following the
manufacturer's
instructions. Genomic DNA present in the RNA preparation was removed by DNase
treatment following the manufacturer's instructions (Cat. Nr. 1700, Promega).
cDNA was
generated using a commercially available kit (SuperScript 114 III Reverse
Transcriptase, Cat.
Nr. 18080044, Invitrogen, Rockville, Maryland, USA) following the
manufacturer's
instructions.
To isolate cDNA sequences comprising a portion of the PX001, PX009, PX010,
PX015, PX016 genes, a series of PCR reactions with degenerate primers were
performed
using Amplitaq Gold (Cat. Nr. N8080240, Applied Biosystems) following the
manufacturer's
instructions.
The sequences of the degenerate primers used for amplification of each of the
genes
are given in Table 2-PX. These primers were used in respective PCR reactions
with the
following conditions: 10 minutes at 95 C, followed by 40 cycles of 30 seconds
at 95 C, 1
minute at 50 C and 1 minute and 30 seconds at 72 C, followed by 7 minutes at
72 C (for
PX001, PX009, PX015, PX016); 10 minutes at 95 C, followed by 40 cycles of 30
seconds at
95 C, 1 minute at 54 C and 2 minute and 30 seconds at 72 C, followed by 7
minutes at 72 C
(for PX010). The resulting PCR fragments were analyzed on agarose gel,
purified (QIAquick
Gel Extraction kit, Cat. Nr. 28706, Qiagen), cloned into the pCR8/GW/TOPO
vector (Cat.
Nr. 1(2500-20, Invitrogen) and sequenced. The sequences of the resulting PCR
products are
represented by the respective SEQ ID NO:s as given in Table 2-PX and are
referred to as the
partial sequences. The corresponding partial amino acid sequence are
represented by the
respective SEQ ID NO:s as given in Table 3-PX.
B. dsRNA production of the Plutella xylostella genes
dsRNA was synthesized in milligram amounts using the commercially available
kit
T7 RibomaxTM Express RNAi System (Cat. Nr. P1700, Promega). First two separate
single 5'
T7 RNA polymerase promoter templates were generated in two separate PCR
reactions, each
reaction containing the target sequence in a different orientation relative to
the T7 promoter.
For each of the target genes, the sense T7 template was generated using
specific T7
forward and specific reverse primers. The sequences of the respective primers
for amplifying
the sense template for each of the target genes are given in Table 8-PX. The
conditions in the
PCR reactions were as follows: 1 minute at 95 C, followed by 20 cycles of 30
seconds at
95 C, 30 seconds at 60 C (-0.5 C/cycle) and 1 minute at 72 C, followed by 15
cycles of 30
132
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
seconds at 95 C, 30 seconds at 50 C and 1 minute at 72 C, followed by 10
minutes at 72 C.
The anti-sense T7 template was generated using specific forward and specific
T7 reverse
primers in a PCR reaction with the same conditions as described above. The
sequences of the
respective primers for amplifying the anti-sense template for each of the
target genes are
given in Table 8-PX. The resulting PCR products were analyzed on agarose gel
and purified
by PCR purification kit (Qiaquick PCR Purification Kit, Cat. Nr. 28106,
Qiagen) and NaC104
precipitation. The generated T7 forward and reverse templates were mixed to be
transcribed
and the resulting RNA strands were annealed, DNase and RNase treated, and
purified by
sodium acetate, following the manufacturer's instructions. The sense strand of
the resulting
dsRNA for each of the target genes is given in Table 8-PX.
C. Recombination of the Plutella xylostella genes into the plant
vector
pK7GWIWG2D(II)
Since the mechanism of RNA interference operates through dsRNA fragments, the
target nucleotide sequences of the target genes, as selected above, are cloned
in anti-sense
and sense orientation, separated by the intron - CmR - intron, whereby CmR is
the
chloramphenicol resistance marker, to form a dsRNA hairpin construct. These
hairpin
constructs are generated using the LR recombination reaction between an attL-
containing
entry clone (see Example 1) and an attR-containing destination vector (=
pK7GWIWG2D(II)). The plant vector pK7GWIWG2D(II) is obtained from the
VIB/Plant
Systems Biology with a Material Transfer Agreement. LR recombination reaction
is
performed by using LR ClonaseTM II enzyme mix (Cat. Nr. 11791-020, Invitrogen)
following
the manufacturer's instructions. These cloning experiments result in a hairpin
construct for
each of the target genes, having the promoter - sense - intron - CmR - intron -
antisense
orientation, and wherein the promoter is the plant operable 35S promoter. The
binary vector
pK7GWIWG2D(II) with the 35S promoter is suitable for transformation into A.
tumefaciens.
Restriction enzyme digests were carried out on pCR8/GW/TOPO plasmids
containing
the different targets (see Example 2). The band containing the gene of
interest flanked by the
attL sites using Qiaquick Gel Extraction Kit (Cat. Nr. 28706, Qiagen) is
purified. An amount
of 150 ng of purified fragment and 150 ng pK7GWIWG2D(II) is added together
with the LR
clonase II enzyme and incubated for at least lh at 25 C . After proteinase K
solution
treatment (10 min at 37 C), the whole recombination mix is transformed into
Top 10
chemically competent cells. Positive clones are selected by restriction digest
analyses.
133
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
D. Laboratory trials to test dsRNA targets, using artificial diet for
activity against
Plutella xylostella larvae
Diamond-back moths, Plutella xylostella, were maintained at Insect
Investigations
Ltd. (origin: Newcastle University, Newcastle-upon-Tyne, UK). The insects were
reared on
cabbage leaves. First instar, mixed sex larvae (approximately 1 day old) were
selected for use
in the trial. Insects were maintained in Eppendorf tubes (1.5 ml capacity).
Commercially
available Diamond-back moth diet (Bio-Serv, NJ, USA), prepared following the
manafacturer's instructions, was placed in the lid of each tube (0.25 ml
capacity, 8 mm
diameter). While still liquid, the diet was smoother over to remove excess and
produce an
even surface.
Once the diet has set the test formulations are applied to the diet's surface,
at the rate
of 25 I undiluted formulation (1 g/ 1 dsRNA of targets) per replicate. The
test formulations
are allowed to dry and one first instar moth larva is placed in each tube. The
larva is placed
on the surface of the diet in the lid and the tube carefully closed. The tubes
are stored upside
down, on their lids such that each larva remains on the surface of the diet.
Twice weekly the
larvae are transferred to new Eppendorf tubes with fresh diet. The insects are
provided with
treated diet for the first two weeks of the trial and thereafter with
untreated diet.
Assessments are made twice weekly for a total of 38 days at which point all
larvae are
dead. At each assessment the insects are assessed as live or dead and examined
for
abnormalities. Forty single larva replicates are performed for each of the
treatments. During
the trial the test conditions are 23 to 26 C and 50 to 65 % relative
humidity, with a 16:8 hour
light:dark photoperiod.
E. Cloning of a DBM gene fragment in a vector suitable for bacterial
production of
insect-active double-stranded RNA
What follows is an example of cloning a DNA fragment corresponding to a DBM
gene target in a vector for the expression of double-stranded RNA in a
bacterial host,
although any vector comprising a T7 promoter or any other promoter for
efficient
transcription in bacteria, may be used (reference to W00001846).
The sequences of the specific primers used for the amplification of target
genes are
provided in Table 8-PX. The template used is the pCR8/GW/topo vector
containing any of
target sequences. The primers are used in a PCR reaction with the following
conditions: 5
minutes at 98 C, followed by 30 cycles of 10 seconds at 98 C, 30 seconds at 55
C and 2
134
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
minutes at 72 C, followed by 10 minutes at 72 C. The resulting PCR fragment is
analyzed
on agarose gel, purified (QIAquick Gel Extraction kit, Cat. Nr. 28706,
Qiagen), blunt-end
cloned into SrfI-linearized pGNA49A vector (reference to W000188121A1), and
sequenced.
The sequence of the resulting PCR product corresponds to the respective
sequence as given in
Table 8-PX. The recombinant vector harbouring this sequence is named
pGBNJOOXX.
F. Expression and production of a double-stranded RNA target in two
strains of
Escherichia coli: (1) AB309-105, and, (2) BL21(DE3)
The procedures described below are followed in order to express suitable
levels of
insect-active double-stranded RNA of insect target in bacteria. An RNaseIII-
deficient strain,
AB309-105, is used in comparison to wild-type RNaseIII-containing bacteria,
BL21(DE3).
Transformation of AB309-105 and BL21(DE3)
Three hundred ng of the plasmid are added to and gently mixed in a 50 [il
aliquot of
ice-chilled chemically competent E. coli strain AB309-105 or BL21(DE3). The
cells are
incubated on ice for 20 minutes before subjecting them to a heat shock
treatment of 37 C for
minutes, after which the cells are placed back on ice for a further 5 minutes.
Four hundred
and fifty pi of room temperature SOC medium is added to the cells and the
suspension
incubated on a shaker (250 rpm) at 37 C for 1 hour. One hundred jul of the
bacterial cell
suspension is transferred to a 500 ml conical flask containing 150 ml of
liquid Luria-Bertani
(LB) broth supplemented with 100 [tg/m1 carbenicillin antibiotic. The culture
is incubated on
an Innova 4430 shaker (250 rpm) at 37 C overnight (16 to 18 hours).
Chemical induction of double-stranded RNA expression in AB309-105 and
BL21(DE3)
Expression of double-stranded RNA from the recombinant vector, pGBNJ003, in
the
bacterial strain AB309-105 or BL21(DE3) is made possible since all the genetic
components
for controlled expression are present. In the presence of the chemical inducer
isopropylthiogalactoside, or IPTG, the T7 polymerase will drive the
transcription of the target
sequence in both antisense and sense directions since these are flanked by
oppositely oriented
T7 promoters.
The optical density at 600 nm of the overnight bacterial culture is measured
using an
appropriate spectrophotometer and adjusted to a value of 1 by the addition of
fresh LB broth.
Fifty ml of this culture is transferred to a 50 ml Falcon tube and the culture
then centrifuged
at 3000 g at 15 C for 10 minutes. The supernatant is removed and the
bacterial pellet
resuspended in 50 ml of fresh S complete medium (SNC medium plus 5 ps/m1
cholesterol)
135
CA 02622660 2008-03-14
WO 2007/074405
PCT/1B2006/004003
supplemented with 100 fig/m1 carbenicillin and 1 mM IPTG. The bacteria are
induced for 2
to 4 hours at room temperature.
Heat treatment of bacteria
Bacteria are killed by heat treatment in order to minimise the risk of
contamination of
the artificial diet in the test plates. However, heat treatment of bacteria
expressing double-
stranded RNA is not a prerequisite for inducing toxicity towards the insects
due to RNA
interference. The induced bacterial culture is centrifuged at 3000 g at room
temperature for
minutes, the supernatant discarded and the pellet subjected to 80 C for 20
minutes in a
water bath. After heat treatment, the bacterial pellet is resuspended in 1.5
ml MilliQ water
and the suspension transferred to a microfuge tube. Several tubes are prepared
and used in the
bioassays for each refreshment. The tubes are stored at -20 C until further
use.
G.
Laboratory trials to test Escherichia coli expressing dsRNA targets against
Plutella xylostella
Plant-based bioassays
Whole plants are sprayed with suspensions of chemically induced bacteria
expressing
dsRNA prior to feeding the plants to DBM. The are grown from in a plant growth
room
chamber. The plants are caged by placing a 500 ml plastic bottle upside down
over the plant
with the neck of the bottle firmly placed in the soil in a pot and the base
cut open and covered
with a fine nylon mesh to permit aeration, reduce condensation inside and
prevent insect
escape. DBM are placed on each treated plant in the cage. Plants are treated
with a
suspension of E. coli AB309-105 harbouring the pGBNJ001 plasmids or pGN29
plasmid.
Different quantities of bacteria are applied to the plants: for instance 66,
22, and 7 units,
where one unit is defined as 109 bacterial cells in 1 ml of a bacterial
suspension at optical
density value of 1 at 600 nm wavelength. In each case, a total volume of
between 1 and 10 ml
s sprayed on the plant with the aid of a vaporizer. One plant is used per
treatment in this trial.
The number of survivors are counted and the weight of each survivor recorded.
Spraying plants with a suspension of E. coli bacterial strain AB309-105
expressing
target dsRNA from pGBNJ003 leed to a dramatic increase in insect mortality
when compared
to pGN29 control. These experiments show that double-stranded RNA
corresponding to an
insect gene target sequence produced in either wild-type or RNaseIII-deficient
bacterial
expression systems is toxic towards the insect in terms of substantial
increases in insect
mortality and growth/development delay for larval survivors. It is also clear
from these
136
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
experiments that an exemplification is provided for the effective protection
of plants/crops
from insect damage by the use of a spray of a formulation consisting of
bacteria expressing
double-stranded RNA corresponding to an insect gene target.
Example 12: Acheta domesticus (house cricket)
A. Cloning Acheta domesticus partial sequences
High quality, intact RNA was isolated from all the different insect stages of
Acheta
domesticus (house cricket; source: Dr. Lara Senior, Insect Investigations
Ltd., Capital
Business Park, Wentloog, Cardiff, CF3 2PX, Wales, UK) using TRIzol Reagent
(Cat. Nr.
15596-026/15596-018, Invitrogen, Rockville, Maryland, USA) following the
manufacturer's
instructions. Genomic DNA present in the RNA preparation was removed by DNase
treatment following the manafacturer's instructions (Cat. Nr. 1700, Promega).
cDNA was
generated using a commercially available kit (SuperScript Tm III Reverse
Transcriptase, Cat.
Nr. 18080044, Invitrogen, Rockville, Maryland, USA) following the
manufacturer's
instructions.
To isolate cDNA sequences comprising a portion of the AD001, AD002, AD009,
AD015 and AD016 genes, a series of PCR reactions with degenerate primers were
performed
using Amplitaq Gold (Cat. Nr. N8080240, Applied Biosystems) following the
manafacturer's
instructions.
The sequences of the degenerate primers used for amplification of each of the
genes
are given in Table 2-AD. These primers were used in respective PCR reactions
with the
following conditions: 10 minutes at 95 C, followed by 40 cycles of 30 seconds
at 95 C, 1
minute at 50 C and 1 minute and 30 seconds at 72 C, followed by 7 minutes at
72 C. The
resulting PCR fragments were analyzed on agarose gel, purified (QIAquick Gel
Extraction
kit, Cat. Nr. 28706, Qiagen), cloned into the pCR8/GW/topo vector (Cat. Nr.
K2500 20,
Invitrogen) and sequenced. The sequences of the resulting PCR products are
represented by
the respective SEQ ID NO:s as given in Table 2-AD and are referred to as the
partial
sequences. The corresponding partial amino acid sequence are represented by
the respective
SEQ ID NO:s as given in Table 3-AD.
B. dsRNA production of the Acheta domesticus genes
dsRNA was synthesized in milligram amounts using the commercially available
kit
T7 RibomaxTM Express RNAi System (Cat. Nr. P1700, Promega). First two separate
single 5'
137
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
T7 RNA polymerase promoter templates were generated in two separate PCR
reactions, each
reaction containing the target sequence in a different orientation relative to
the T7 promoter.
For each of the target genes, the sense T7 template was generated using
specific T7
forward and specific reverse primers. The sequences of the respective primers
for amplifying
the sense template for each of the target genes are given in Table 8-AD. The
conditions in the
PCR reactions were as follows: 1 minute at 95 C, followed by 20 cycles of 30
seconds at
95 C, 30 seconds at 60 C (-0.5 C/cycle) and 1 minute at 72 C, followed by 15
cycles of 30
seconds at 95 C, 30 seconds at 50 C and 1 minute at 72 C, followed by 10
minutes at 72 C.
The anti-sense T7 template was generated using specific forward and specific
T7 reverse
primers in a PCR reaction with the same conditions as described above. The
sequences of the
respective primers for amplifying the anti-sense template for each of the
target genes are
given in Table 8-AD. The resulting PCR products were analyzed on agarose gel
and purified
by PCR purification kit (Qiaquick PCR Purification Kit, Cat. Nr. 28106,
Qiagen) and NaC104
precipitation. The generated T7 forward and reverse templates were mixed to be
transcribed
and the resulting RNA strands were annealed, DNase and RNase treated, and
purified by
sodium acetate, following the manufacturer's instructions. The sense strand of
the resulting
dsRNA for each of the target genes is given in Table 8-AD.
C. Recombination of the Acheta domesticus genes into the plant vector
pK7GWIWG2D(II)
Since the mechanism of RNA interference operates through dsRNA fragments, the
target nucleotide sequences of the target genes, as selected above, are cloned
in anti-sense
and sense orientation, separated by the intron - CmR - intron, whereby CmR is
the
chloramphenicol resistance marker, to form a dsRNA hairpin construct. These
hairpin
constructs are generated using the LR recombination reaction between an attL-
containing
entry clone (see Example 1) and an attR-containing destination vector (=
pK7GWIWG2D(II)). The plant vector pK7GWIWG2D(II) is obtained from the
VIB/Plant
Systems Biology with a Material Transfer Agreement. LR recombination reaction
is
performed by using LR ClonaseTM II enzyme mix (Cat. Nr. 11791-020, Invitrogen)
following
the manufacturer's instructions. These cloning experiments result in a hairpin
construct for
each of the target genes, having the promoter - sense - intron - CmR - intron -
antisense
orientation, and wherein the promoter is the plant operable 35S promoter. The
binary vector
pK7GWIWG2D(II) with the 35S promoter is suitable for transformation into A.
tumefaciens.
138
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
Restriction enzyme digests were carried out on pCR8/GW/TOPO plasmids
containing
the different targets (see Example 2). The band containing the gene of
interest flanked by the
attL sites using Qiaquick Gel Extraction Kit (Cat. Nr. 28706, Qiagen) is
purified. An amount
of 150 ng of purified fragment and 150 ng pK7GWIWG2D(II) is added together
with the LR
clonase II enzyme and incubated for at least lh at 25 C . After proteinase K
solution
treatment (10 min at 37 C), the whole recombination mix is transformed into
Top 10
chemically competent cells. Positive clones are selected by restriction digest
analyses.
D. Laboratory trials to test dsRNA targets, using artificial diet for
activity against
Acheta domesticus larvae
House crickets, Acheta domesticus, were maintained at Insect Investigations
Ltd.
(origin: Blades Biological Ltd., Kent, UK). The insects were reared on bran
pellets and
cabbage leaves. Mixed sex nymphs of equal size and no more than 5 days old
were selected
for use in the trial. Double-stranded RNA is mixed with a wheat-based pelleted
rodent diet
(rat and mouse standard diet, B & K Universal Ltd., Grimston, Aldbrough, Hull,
UK). The
diet, BK001P, contains the following ingredients in descending order by
weight: wheat, soya,
wheatfeed, barley, pellet binder, rodent 5 vit min, fat blend, dicalcium
phosphate, mould carb.
The pelleted rodent diet is finely ground and heat-treated in a microwave oven
prior to
mixing, in order to inactivate any enzyme components. All rodent diet is taken
from the same
batch in order to ensure consistency. The ground diet and dsRNA are mixed
thoroughly and
formed into small pellets of equal weight, which are allowed to dry overnight
at room
temperature.
Double-stranded RNA samples from targets and gfp control at concentrations 10
i.tg/i.t1 were applied in the ratio 1 g ground diet plus 1 ml dsRNA solution,
thereby resulting in
an application rate of 10 mg dsRNA per g pellet. Pellets are replaced weekly.
The insects are
provided with treated pellets for the first three weeks of the trial.
Thereafter untreated pellets
are provided. Insects are maintained within lidded plastic containers (9 cm
diameter, 4.5 cm
deep), ten per container. Each arena contains one treated bait pellet and one
water source
(damp cotton wool ball), each placed in a separate small weigh boat. The water
is replenished
ad lib throughout the experiment.
Assessments are made at twice weekly intervals, with no more than four days
between
assessments, until all the control insects had either died or moulted to the
adult stage (84
days). At each assessment the insects are assessed as live or dead, and
examined for
139
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
abnormalities. From day 46 onwards, once moulting to adult has commenced, all
insects (live
and dead) are assessed as nymph or adult. Surviving insects are weighed on day
55 of the
trial. Four replicates are performed for each of the treatments. During the
trial the test
conditions are 25 to 33 C and 20 to 25 % relative humidity, with a 12:12 hour
light:dark
photoperiod.
E. Cloning of a HC gene fragment in a vector suitable for bacterial
production of
insect-active double-stranded RNA
What follows is an example of cloning a DNA fragment corresponding to a HC
gene
target in a vector for the expression of double-stranded RNA in a bacterial
host, although any
vector comprising a T7 promoter or any other promoter for efficient
transcription in bacteria,
may be used (reference to W00001846).
The sequences of the specific primers used for the amplification of target
genes are
provided in Table 8. The template used is the pCR8/GW/topo vector containing
any of target
sequences. The primers are used in a PCR reaction with the following
conditions: 5 minutes
at 98 C, followed by 30 cycles of 10 seconds at 98 C, 30 seconds at 55 C and 2
minutes at
72 C, followed by 10 minutes at 72 C. The resulting PCR fragment is analyzed
on agarose
gel, purified (QIAquick Gel Extraction kit, Cat. Nr. 28706, Qiagen), blunt-end
cloned into Srf
I-linearized pGNA49A vector (reference to W000188121A1), and sequenced. The
sequence
of the resulting PCR product corresponds to the respective sequence as given
in Table 8-AD.
The recombinant vector harbouring this sequence is named pGBNJOOXX.
F. Expression and production of a double-stranded RNA target in two strains
of
Escherichia coli: (1) AB309-105, and, (2) BL21(DE3)
The procedures described below are followed in order to express suitable
levels of
insect-active double-stranded RNA of insect target in bacteria. An RNaseIII-
deficient strain,
AB309-105, is used in comparison to wild-type RNaseIII-containing bacteria,
BL21(DE3).
Transformation of AB309-105 and BL21(DE3)
Three hundred ng of the plasmid are added to and gently mixed in a 50 1.11
aliquot of
ice-chilled chemically competent E. coli strain AB309-105 or BL21(DE3). The
cells are
incubated on ice for 20 minutes before subjecting them to a heat shock
treatment of 37 C for
minutes, after which the cells are placed back on ice for a further 5 minutes.
Four hundred
and fifty l of room temperature SOC medium is added to the cells and the
suspension
incubated on a shaker (250 rpm) at 37 C for 1 hour. One hundred 1 of the
bacterial cell
suspension is transferred to a 500 ml conical flask containing 150 ml of
liquid Luria-Bertani
140
CA 02622660 2008-03-14
WO 2007/074405
PCT/1B2006/004003
(LB) broth supplemented with 1001.tg/m1 carbenicillin antibiotic. The culture
is incubated on
an Innova 4430 shaker (250 rpm) at 37 C overnight (16 to 18 hours).
Chemical induction of double-stranded RNA expression in AB309-105 and
BL21(DE3)
Expression of double-stranded RNA from the recombinant vector, pGBNJ003, in
the
bacterial strain AB309-105 or BL21(DE3) is made possible since all the genetic
components
for controlled expression are present. In the presence of the chemical inducer
isopropylthiogalactoside, or IPTG, the T7 polymerase will drive the
transcription of the target
sequence in both antisense and sense directions since these are flanked by
oppositely oriented
T7 promoters.
The optical density at 600 nm of the overnight bacterial culture is measured
using an
appropriate spectrophotometer and adjusted to a value of 1 by the addition of
fresh LB broth.
Fifty ml of this culture is transferred to a 50 ml Falcon tube and the culture
then centrifuged
at 3000 g at 15 C for 10 minutes. The supernatant is removed and the
bacterial pellet
resuspended in 50 ml of fresh S complete medium (SNC medium plus 5 ug/m1
cholesterol)
supplemented with 100 1.1s/m1 carbenicillin and 1 mM IPTG. The bacteria are
induced for 2
to 4 hours at room temperature.
Heat treatment of bacteria
Bacteria are killed by heat treatment in order to minimise the risk of
contamination of
the artificial diet in the test plates. However, heat treatment of bacteria
expressing double-
stranded RNA is not a prerequisite for inducing toxicity towards the insects
due to RNA
interference. The induced bacterial culture is centrifuged at 3000 g at room
temperature for
minutes, the supernatant discarded and the pellet subjected to 80 C for 20
minutes in a
water bath. After heat treatment, the bacterial pellet is resuspended in 1.5
ml MilliQ water
and the suspension transferred to a microfuge tube. Several tubes are prepared
and used in the
bioassays for each refreshment. The tubes are stored at -20 C until further
use.
G.
Laboratory trials to test Escherichia coli expressing dsRNA targets against
Acheta domesticus
Plant-based bioassays
Whole plants are sprayed with suspensions of chemically induced bacteria
expressing
dsRNA prior to feeding the plants to HC. The are grown from in a plant growth
room
chamber. The plants are caged by placing a 500 ml plastic bottle upside down
over the plant
with the neck of the bottle firmly placed in the soil in a pot and the base
cut open and covered
with a fine nylon mesh to permit aeration, reduce condensation inside and
prevent insect
141
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
escape. HC are placed on each treated plant in the cage. Plants are treated
with a suspension
of E. coil AB309-105 harbouring the pGBNJ001 plasmids or pGN29 plasmid.
Different
quantities of bacteria are applied to the plants: for instance 66, 22, and 7
units, where one unit
is defined as 109 bacterial cells in 1 ml of a bacterial suspension at optical
density value of 1
at 600 nm wavelength. In each case, a total volume of between 1 and 10 ml s
sprayed on the
plant with the aid of a vaporizer. One plant is used per treatment in this
trial. The number of
survivors are counted and the weight of each survivor recorded.
Spraying plants with a suspension of E. coil bacterial strain AB309-105
expressing
target dsRNA from pGBNJ003 leed to a dramatic increase in insect mortality
when compared
to pGN29 control. These experiments show that double-stranded RNA
corresponding to an
insect gene target sequence produced in either wild-type or RNaseIII-deficient
bacterial
expression systems is toxic towards the insect in terms of substantial
increases in insect
mortality and growth/development delay for larval survivors. It is also clear
from these
experiments that an exemplification is provided for the effective protection
of plants/crops
from insect damage by the use of a spray of a formulation consisting of
bacteria expressing
double-stranded RNA corresponding to an insect gene target.
Example 13: Ppricularia Rrisea (rice blast)
A. Cloning P. grisea partial sequences
High quality, intact RNA is isolated from different growth stages of P. grisea
using
TRIzol Reagent (Cat. Nr. 15596-026/15596-018, Invitrogen, Rockville, Maryland,
USA)
following the manufacturer's instructions. Genomic DNA present in the RNA
preparation is
removed by DNase treatment following the manafacturer's instructions (Cat. Nr.
1700,
Promega). cDNA is generated using a commercially available kit (SuperScript TM
III Reverse
Transcriptase, Cat. Nr. 18080044, Invitrogen, Rockville, Maryland, USA)
following the
manufacturer's instructions.
To isolate cDNA sequences comprising a portion of a target gene, PCR is
performed
with degenerate primers using Amplitaq Gold (Cat. Nr. N8080240, Applied
Biosystems)
following the manafacturer's instructions. The resultant PCR products are
fractionated and
sequenced.
B. dsRNA production of P. grisea genes
142
CA 02622660 2008-03-14
WO 2007/074405
PCT/1B2006/004003
dsRNA is synthesized in milligram amounts using a commercially available kit,
such
as T7 RibomaxTM Express RNAi System (Cat. Nr. P1700, Promega), following the
manufacturer's instructions. The resulting PCR products are analyzed on an
agarose gel and
purified by a PCR purification kit (e.g. Qiaquick PCR Purification Kit, Cat.
Nr. 28106,
Qiagen) and NaC104 precipitation. The producr T7 forward and reverse templates
are mixed
and the resulting RNA strands are annealed, then DNase and RNase treated, and
purified by
sodium acetate, following the manufacturer's instructions.
C.
Recombination of P. grisea target into the plant vector pK7GWIWG2D(II)
Since the mechanism of RNA interference operates through dsRNA fragments, the
target nucleotide sequences of the target genes, as selected above, are cloned
in anti-sense
and sense orientation, separated by the intron - CmR - intron, whereby CmR is
the
chloramphenicol resistance marker, to form a dsRNA hairpin construct. These
hairpin
constructs are generated using the LR recombination reaction between an attL-
containing
entry clone (see Example A) and an attR-containing destination vector (=
pK7GWIWG2D(II)). The plant vector pK7GWIWG2D(II) is obtained from the
VIB/Plant
Systems Biology with a Material Transfer Agreement. LR recombination reaction
is
performed by using LR ClonaseTm II enzyme mix (Cat. Nr. 11791-020, Invitrogen)
following
the manufacturer's instructions. These cloning experiments result in a hairpin
construct for
the target gene, having the promoter - sense - intron - CmR - intron -
antisense orientation,
and wherein the promoter is the plant operable 35S promoter. The binary vector
pK7GWIWG2D(II) with the 35S promoter is suitable for transformation into A.
tumefaciens.
Restriction enzyme digests are carried out on pCR8/GW/TOPO plasmids containing
the target (see Example B). The band containing the gene of interest flanked
by the attL sites
using Qiaquick Gel Extraction Kit (Cat. Nr. 28706, Qiagen) is purified. An
amount of 150 ng
of purified fragment and 150 ng pK7GWIWG2D(II) is added together with the LR
clonase II
enzyme and incubated for at least lh at 25 C. After proteinase K solution
treatment (10 min
at 37 C), the whole recombination mix is transformed into Top 10 chemically
competent
cells. Positive clones are selected by restriction digest analyses.
D. Expression and production of a double-stranded RNA target in two
strains of
Escherichia coli: (1) AB309-105, and, (2) BL21(DE3)
The procedures described below are followed in order to express suitable
levels of
fungal double-stranded RNA of fungal target in bacteria. An RNaseIII-deficient
strain,
AB309-105, is used in comparison to wild-type RNaseIII-containing bacteria,
BL21(DE3).
143
CA 02622660 2008-03-14
WO 2007/074405 PCT/1B2006/004003
Transformation of AB309-105 and BL21(DE3)
Three hundred ng of the plasmid are added to and gently mixed in a 50 pi
aliquot of
ice-chilled chemically competent E. coli strain AB309-105 or BL21(DE3). The
cells are
incubated on ice for 20 minutes before subjecting them to a heat shock
treatment of 37 C for
minutes, after which the cells are placed back on ice for a further 5 minutes.
Four hundred
and fifty pi of room temperature SOC medium is added to the cells and the
suspension
incubated on a shaker (250 rpm) at 37 C for 1 hour. One hundred pi of the
bacterial cell
suspension is transferred to a 500 ml conical flask containing 150 ml of
liquid Luria-Bertani
(LB) broth supplemented with 100 g/ml carbenicillin antibiotic. The culture
is incubated on
an Innova 4430 shaker (250 rpm) at 37 C overnight (16 to 18 hours).
Chemical induction of double-stranded RNA expression in AB309-105 and
BL21(DE3)
Expression of double-stranded RNA from the recombinant vector, pGBNJ003, in
the
bacterial strain AB309-105 or BL21(DE3) is made possible since all the genetic
components
for controlled expression are present. In the presence of the chemical inducer
isopropylthiogalactoside, or IPTG, the T7 polymerase will drive the
transcription of the target
sequence in both antisense and sense directions since these are flanked by
oppositely oriented
T7 promoters.
The optical density at 600 nm of the overnight bacterial culture is measured
using an
appropriate spectrophotometer and adjusted to a value of 1 by the addition of
fresh LB broth.
Fifty ml of this culture is transferred to a 50 ml Falcon tube and the culture
then centrifuged
at 3000 g at 15 C for 10 minutes. The supernatant is removed and the
bacterial pellet
resuspended in 50 ml of fresh S complete medium (SNC medium plus 5 p.g/m1
cholesterol)
supplemented with 100 ug/m1 carbenicillin and 1 mM IPTG. The bacteria are
induced for 2
to 4 hours at room temperature.
Heat treatment of bacteria
Bacteria are killed by heat treatment in order to minimise the risk of
contamination of
the artificial diet in the test plates. However, heat treatment of bacteria
expressing double-
stranded RNA is not a prerequisite for inducing toxicity towards the insects
due to RNA
interference. The induced bacterial culture is centrifuged at 3000 g at room
temperature for
minutes, the supernatant discarded and the pellet subjected to 80 C for 20
minutes in a
water bath. After heat treatment, the bacterial pellet is resuspended in 1.5
ml MilliQ water
and the suspension transferred to a microfuge tube. Several tubes are prepared
and used in the
bioassays for each refreshment. The tubes are stored at -20 C until further
use.
144