Note: Descriptions are shown in the official language in which they were submitted.
CA 02635451 2008-06-26
WO 2007/078900 PCT/US2006/048130
TWO-PART SIZING COMPOSITION FOR REINFORCEMENT FIBERS
TECHNICAL FIELD AND INDUSTRIAL
APPLICASILtITY OF THE INVENTION
The present invention relates generally to a sizing composition for a
reinforcing
fiber material, and more particularly, to a two-part sizing formulation that
imparts
improved strength to reinforced composites which includes a size composition
and a
binder composition. A composite article formed from a reinforcing fiber
material sized
with a two-part sizing formulation is also provided.
DACKGROIJND OF THE INVENTION
Glass fibers are useful in a variety of technologies. For example, glass
fibers are
commonly used as reinforcements in polymer matrices to form glass fiber
reinforced
plastics or composites. Glass fibers have been used in the form of continuous
or chopped
filaments, strands, rovings, woven fabrics, nonwoven fabrics, meshes, and
scrims to
reinforce polymers. It is known in th.e art that glass fiber reinforced
polymer composites
possess higher mechanical properties compared to unreinforced polymer
composites,
provided that the reinforcement fiber surface is suitably modified by a sizing
composition.
Thus, better dimensional stability, tensile strength and modulus, flexural
strength and
modulus, impact resistance, and cree:p resistance may be achieved with glass
fiber
reinforced composites.
Chopped glass fibers are conunonly used as reinforcement materials in
reinforced
composites. Conventionally, glass fibers are formed by attenuating streams of
a molten
glass material from a bushing or orifice. The glass fibers may be attenuated
by a winder
that collects gathered filaments into a package or by rollers that pull the
fibers before they
are collected and chopped. An aqueous sizing composition, or chemical
treatment, is
typically applied to the fibers after they are drawn from the bushing. After
the fibers are
treated with the aqueous sizing composition, they may be dried in a package or
chopped
strand form.
Chopped strand segments may be mixed with a polymeric resin and supplied to a
compression- or injection- molding machine to be formed into glass fiber
reinforced
composites. Typically, the chopped strand segments are mixed with pellets of a
thermoplastic polymer resin in an extruder. In one conventional method,
polymer pellets
1
CA 02635451 2008-06-26
WO 2007/078900 PCT/US2006/048130
are fed into a first port of a twin screw extruder and the chopped glass
fibers are fed into a
second port of the extruder with the melted polymer to form a fiber/resin
mixture.
Alternatively, the polymer pellets and chopped strand segments are dry mixed
and fed
together into a single screw extruder where the resin is melted, the integrity
of the glass
fiber strands is destroyed, and the fiber strands are dispersed throughout the
molten resin
to form a fiber/resin mixture. Next, the fiber/resin mixture is degassed and
formed into
pellets. These dry fiber strand/resin dispersion pellets are then fed to a
molding machine
and formed into molded composite articles that have a substantially
homogeneous
dispersion of glass fiber strands throughout the composite article.
Unfortunately, chopped glass l:ibers are often bulky and do not flow well in
automated equipment. As a result, thf~; chopped fiber strands may be compacted
into rod=
shaped bundles or pellets to improve their flowability and to enable the use
of automated
equipment, such as, for example, for transporting the pellets and mixing the
pellets with
the polymer resins. U.S. Patent.No. 5,578,535.to. Hill et al. discloses glass
fiber pellets
.15 that are from about 20% to 30ofo denser than the individual glass strands
from which they
are made, and approximately 5 to 15 times larger in diameter. These pellets
are prepared
by hydrating cut fiber strand segments to a hydration level -sufficient to
prevent separation
of the fiber strand segments into individual filaments but insufficient to
cause the fiber
strand segments to agglomerate into a clump. The hydrated strand segments are
then
mixed for a period of time sufficient for the strand segments to form pellets.
Suitable
mixing methods include processes that keep the fibers inoving over and around
one
another, such as by tumbling, agitating, blending, commingling, stirring
and/or
intermingling the fibers.
Sizing compositions, such as are used in reinforced composites, are well-known
in
the art and conventionally include a polyacid polymeric component, a film
forming
polymeric component, a coupling agent, and a lubricant. A polyacid sizing
composition is
typically added to glass fibers to reduce interfilament abrasion and to make
the glass fibers
compatible with the polymeric matrices they are intended to reinforce. The
sizing
composition also ensures the integrity of the strands of glass fibers,for
example, the
interconnection of the glass filaments that form the strand.
One fundamental problem associated with polyacid conventional sizing
compositions used in reinforced composites is the presence of a di-cationic
species, such
2
CA 02635451 2008-06-26
WO 2007/078900 PCT/US2006/048130
as a metallic salt of a long chain carboxylic acid, in the polymer used to
form a reinforced
composite article. The interaction of the di-cationic lubricant with the
polyacid
conventional sizing compositions causes a decrease in mechanical strength. For
example,
polyamide composites reinforced with conventional sizing compositions often
demonstrate
a reduction in impact strengths (for example, Charpy or Izod, un-notched or
notched) as
well as a reduction in tensile strength and elongation at break when such
tests are run on
hydro-aged composite pieces. Thus, there exists a need in the art for a sizing
composition
that confers improved mechanical strength to reinforced composites that
contain a di-
cationic lubricant.
SUMMARY OF THE INVENTION
It is an object of the present invention to provide a two-part
sizing'formulation that
imparts improved dry-as-molded (DaM) mechanical properties and hydrolysis
resistance to
reinforced polymer composites (for example, reinforced polyamide composites),
even
when such composites contain di-cationic.or higher valency cation additives
such as a. ..... .. ... .
calcium stearate lubricant. The two-part sizing formulation includes a size
composition
and a binder composition. The size composition may be applied to a reinforcing
fiber
material before the binder composition is applied. The reinforcing fiber
material may be
one or more strands of glass (for example, Advantex glass), natural fibers,
carbon fibers,
or one or more synthetic polymers. According to at least one exemplary
embodiment of
the present invention, the size composition includes one or more coupling
agents and one
or more resinous film forming agents. Preferably, the coupling agent is an
aminosilane or
a diaminosilane. In addition, the size composition may optionally include
conventional
additives such as lubricants, wetting agents, pH adjusters, antioxidants,
antifoaming
agents, processing aids, antistatic agents, and/or non-ionic surfactants.
The binder composition includes a high acid number copolymer formed from the
polymerization of maleic anhydride ar maleic acid and at least one other
desired monomer
and/or a high acid number polycarbo:Kylic acid. The maleic anhydride or acid
copolymer
may be a pure copolymer or a derivative in the anhydride, acid, salt, or
partial-ester, -
amide, or -imide form. Suitable copolymers include C2 - C5 a-olefins, such as
butadiene-,
ethylene-, propylene- or (iso)butylene-maleic acid copolymers, and methyl
vinyl ether-
maleic acid copolymers. In a preferred embodiment, the binder composition
includes an
ethylene-maleic acid copolymer forrried by the hydrolysis of an ethylene-
maleic anhydride
3
CA 02635451 2008-06-26
WO 2007/078900 PCT/US2006/048130
copolymer. The binder composition may also include a film forming agent, which
may be
the same as, or different from, the film forming agent in the size
composition.
Conventional additives such as lubricants, surfactants, and anti-static agents
may also be
included in the binder composition.
It is yet another object of the present invention to provide a process for
making a
densified reinforcing fiber product. The process for making a densified
reinforcing fiber
product may be an in-line process that includes applying a two-part sizing
formulation as
described above to a strand of a reinforcing fiber material, chopping the
strand of sized
reinforcing fibers into segments, applying a binder composition as described
above to the
segments, and pelletizing and/or densifying the segments to form the densified
reinforcing
fiber product. Pellet formation and densification may occur in separate
tumbling
apparatuses, such as in a rotary drum ifor example, pelletizer) and rotating
zig-zag tube
(for example, densifier). AlternativelY , pellet formation and densification
may occur in
separate regions within a single apparatus, such as in.a"Zig-Zag"blender
cornm.ercially....
available from Patterson Kelly. The size composition may be applied to the
fibers as they
are being formed and the binder composition may be applied to the sized fibers
in a
pelletizer. By applying the binder cojnposition in the pelletizer, an
application efficiency
of approximately 95% to 100% for th.e binder composition may be obtained. In
addition,
applying the binder composition separately from the size composition outside
the fiber-
forming environment permits the inclusion of materials that are not desirably
applied
during the fiber-forming process because of safety, flammability, irritation,
stability, low
compatibility with aminosilanes, viscosity, toxicity, cleanliness, odor, cost,
and/or shear
sensitivity.
It is an advantage of the present invention that a high loss-on-ignition (LOI)
of the
high acid number maleic anhydride c:opolymer, ethylene maleic acid copolymer,
or
polycarboxylic acid in the sizing composition provides for improved dry-as-
molded
mechanical properties and improved hydrolysis resistance.
It is another advantage of the present invention that polyurethane film
formers
present in the sizing composition demonstrate good compatibility with
polyamide
matrices, which helps to improve the dispersion of the reinforcement fiber
bundles in the
melt (for example, in extrusion process or injection molding process) when
forming a
composite article. This increased fiber dispersion may cause a reduction of
defects such as
4
CA 02635451 2008-06-26
WO 2007/078900 PCT/US2006/048130
visual defects in the final product, a reciuction in processing breaks, and/or
low mechanical
properties in the final article.
lt is a fiirther advantage of the present invention that the polyurethane
dispersion
present in the sizing composition improves the compatibility of the sizing
composition
with the di-cationic species containing polymer to improve the dispersion of
the fibers
during further processing to form a cojnposite article.
It is yet another advantage that the present invention imparts improved
physical
properties such as improved dry-as-molded (DaM) mechanical properties of the
composite
part or after aging the composite part in severe hydrolysis conditions to
composites formed
from industrially processable and easi:ly dispersible pellets.
It is also an advantage of the present irivention that the two-part sizing
formulation has
improved stability over conventional sizing formulations that contain an
aminosilane and a
polyacid in the same mixture.
The foregoing and other.objects,.features, and advantages of the
invention.will appear _ .. __.
more fully hereinafter from a consideration of the detailed description that
follows.
BRIEF DESCR][PTION OF THE DRAWINGS
The advantages of this invention will be apparent upon consideration of the
following detailed disclosure of the irivention, especially when taken in
conjunction with
the accompanying drawings wherein:
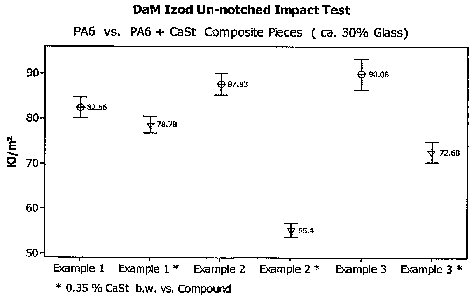
FIG. 1 is a graphical illustration of dry-as-molded (DaM) Izod Un-notched
Impact
tests on polyamide 6 and polyamide Ei/calcium stearate composite pieces;
FIG. 2 is a graphical illustration of dry-as-molded (DaM) Charpy Un-notched
Impact tests on polyamide 6 and polyamide 6/calcium stearate composite pieces;
FIG. 3 is a graphical illustration of Charpy Un-notched Impact tests after
hydro-
aging polyamide 6 and polyamide 6/calcium stearate composite pieces;
FIG. 4 is a graphical illustration of dry-as-molded (DaM) Charpy Un-notched
Impact tests on polyamide 6 and polyamide 6/calcium stearate composite pieces;
and
FIG. 5 is a graphical illustration of dry-as-molded (DaM) Charpy Un-notched
Impact tests on polyamide 6 and pohramide 6/calcium stearate composite pieces.
DETAILED DESCRIPTION AND
PREFERRED EMBODIMENTS OF THE INVENTION
Unless defined otherwise, all technical and scientific terms used herein have
the
same meaning as commonly understood by one of ordinary skill in the art to
which the
5
CA 02635451 2008-06-26
WO 2007/078900 PCT/US2006/048130
invention belongs. Although any methods and materials similar or equivalent to
those
described herein can be used in the practice or testing of the present
invention, the
preferred methods and materials are described herein. All references cited
herein,
including published or corresponding 'U.S. or foreign patent applications,
issued U.S. or
foreign patents, and any other references, are each incorporated by reference
in their
entireties, including all data, tables, figures, and text presented in the
cited references. The
terms "film forming agent" and "film former" may be used interchangeably
herein. In
addition, the terms "reinforcing fiber material" and "reinforcing fiber" may
be used
interchangeably herein.
The present invention relates to a two-part sizing formulation that improves
the
mechanical performance of reinforced composites. In particular, the two-part
sizing
formulation of the invention imparts improved dry-as-molded mechanical
properties and
hydrolysis resistance to polymer reinforced composites, such as a polyamide
reinforced
composite, even when. such composites. contain di-cationic or higher valency
additives.
such as a calcium stearate lubricant. :[t is to be appreciated that although
any di-cation or
higher valency cation may applicable to the present invention, di-cations are
discussed in
detail herein for ease of discussion. The two-part sizing formulation includes
a size
composition and a binder composition. The two-part sizing formulation may be
applied to
a reinforcing fiber material to form a reinforcing fiber product, which may
then be
densified or compacted to form a densified reinforcing fiber product, such as
pellets. The
densified pellets provide a convenient form for storage and handling of the
chopped fibers
used as reinforcing materials in composite structures.
The size composition may be applied to a reinforcing fiber material before the
binder composition is applied. The reinforcing fiber material may be one or
more strands
of glass formed by conventional techniques such as by drawing molten glass
through a
heated bushing to form substantially continuous glass fibers. These fibers may
subsequently be collected into a glass strand. Any type of glass, such as A-
type glass, C-
type glass, E-type glass, S-type glass., or ECR-type glass such as Owens
Corning's
Advantex(@ glass fibers. Preferably, the reinforcing fiber material is E-type
glass or
Advantex glass.
Alternatively, the reinforcing fiber material may be strands of one or more
synthetic polymers such as polyester-, polyamide, aramid, and mixtures
thereof. The
6
CA 02635451 2008-06-26
WO 2007/078900 PCT/US2006/048130
polymer strands may be used alone as =the reinforcing fiber material, or they
can be used in
combination with glass strands such as those described above. As a further
alternative,
natural fibers may be used as the reinforcing fiber material. The term
"natural fiber" as
used in conjunction with the present ir.ivention refers to plant fibers
extracted from any part
of a plant, including, but not limited tci, the stem, seeds, leaves, roots, or
phloem.
Examples of natural fibers suitable for use as the reinforcing fiber material
include cotton,
jute, bamboo, ramie, bagasse, hemp, coir, linen, kenaf, sisal, flax, henequen,
and
combinations thereof. Carbon or polyaramide fibers may be also used as the
reinforcing
fiber material.
The reinforcing fiber material may include fibers that have a diameter of from
about 6 microns to about 24 microns and may be cut into segments approximately
1 mm to
about 50 mm in length. Preferably, the fibers have a diameter from about 7
microns to
about 14 microns and a length from about 3 mm to about 6 mm. Most preferably,
the
fibers have. a diameter of approximately 10 microns._. Prior to the
densification of the
reinforcing fiber material as described below, each strand may contain from
approximately
500 fibers to approximately 8,000 fibers.
After the reinforcing fibers arE: formed, and prior to their collection into a
strand,
they may be coated with a size composition. A suitable size composition
according to at
least one exemplary embodiment of the present invention includes one or more
coupling
agents and one or more film forming agents. Optionally, conventional additives
such as,
but not limited to, lubricants, wetting agents, pH adjusters, antioxidants,
antifoaming
agents, processing aids, antistatic agents, and non-ionic surfactants may be
present in the
size composition. The size composition may be applied to the reinforcement
fibers in an
amount sufficient to achieve a Loss on Ignition (LOI) of from about 0.05% to
about 1.0%
on the dried fiber, and preferably in an amount of from 0.15% to about 0.40%.
LOI may
be defined as the percentage of organic solid matter deposited on the glass
fiber surfaces
measured by the reduction in weight -experienced by the fibers after heating
them to a
temperature sufficient to bum or pyrolyze the organic size from the fibers.
The size composition includes one or more coupling agents. Preferably, the
coupling agent is a silane coupling agent. Besides their role of coupling the
surface of the
reinforcement fibers and the plastic r.natrix, silanes also function to
enhance the adhesion
of the polycarboxylic acid component to the reinforcement fibers and to reduce
the level of
7
CA 02635451 2008-06-26
WO 2007/078900 PCT/US2006/048130
fuzz, or broken fiber filaments, during subsequent processing. Examples of
silane
coupling agents that may be used in the present size composition may be
characterized by
the functional groups amino, epoxy, vinyl, methacryloxy, ureido, isocyanato,
and azamido.
In preferred embodiments, the silane coupling agents include silanes
containing one or
more nitrogen atoms that have one or rnore functional- groups such as amine
(primary,
secondary, tertiary, and quarternary), amino, imino, amido, imido, ureido,
isocyanato, or
azamido.
Suitable silane coupling agents. include, but are not limited to,
aminosilanes, silane
esters, vinyl silanes, methacryloxy silanes, epoxy silanes, sulfur silanes,
ureido silanes, and
isocyanato silanes. Specific non-limiting examples of silane coupling agents
for use in the
instant invention include y-aminopropyltriethoxysilane (A-1100), n-phenyl-y-
aminopropyltrimethoxysilane (Y-9669), n-trimethoxy-silyl-propyl-ethylene-
diamine (A-
1120), methyl-trichlorosilane (A-154)., y-chl.oropropyl-trimethoxy-silane (A-
143), vinyl-
triacetoxy silane.(A- 1 88); methyltrimethoxysilane (A-1 630), y-
ureidopropyltrimethoxysilane (A-1524). Other examples of suitable silane
coupling agents
are set forth in Table 1. All of the silane coupling agents identified above
and in Table 1
are available commercially from GE Silicones.
TABLE 1
Silanes Label Formula
Silane Esters
oc Itriethox silane A-137 CH3 CHZ , Si OCH2CH3 3
meth ltriethox silane A-162 CH3Si OCHZCH3 3
meth ltrimetho silane A-163 CH3Si OCH3 3
proprietary A-12:i0 proprietary
trrs-[3-(trimethoxysilyl)propyl] y-11597 ---
isoc anurate
Vinyl Silanes
ro rieta RC-1 ro rieta
vin Itriethox silane A-151 CH2=CHSi(OCH2CH3 3
vin ltrimethox silane A-171 CH2=CHSi OCH3 3
vin l-tris- 2-methox etho ) silane A-17'2 CHZ=CHSi OCH2CHZOCH3 3
Methacryloxy Silanes
y-methacryloxypropyl- A-174 CH2=C(CH3)COaCH2CH2CH2Si(OCH3)3
trimethoxysilane
Epoxy Silanes
,6-(3,4-epoxycyclohexyl)- C
ethyltrimethoxysilane A-1 fi6 =:~)_CH 2CH2Si(OCH3)3
O
y-glycidoxypropyltrimethoxysilane A-187 CH CHCH2OCH2CH2CH2Si(OCH3)3
8
CA 02635451 2008-06-26
WO 2007/078900 PCT/US2006/048130
Sulfur Silanes
y-merca to ro ltrimethox silane A-189 HSCH2CH2CH2Si OCH3 3
proprietary ol sulfidesilane RC-2 proprietary
Amino Silanes
y-aminopropyltriethoxysilane A-1101 H2NCHZCHZCHZSi(OCH2CH3)3
A-1102
aminoalkyl silicone A-1106 H2NCH2CH2CHzSiO,,S õ
modified aminoorganosilane A-1108 ---
-amino ro Itrimethox silane A-1110 HzNCH2CH2CH2Si OCH3 3
N -,6-(aminoethyi)-y- A-1120 H2NCH2CH2NHCH2CH2CH2Si(OCH3)3
amino ro ltrimetho silane
modified aminoor anosilane A-1126 ---
inodified aminosilane A-1128 ---
triaminofunctional silane A-1130 H2NCH2CH2NHCH2CH2NHCH2CH2CH2Si OCH3 ,
H~ CH2CH2CH2Si(OCH3)3
bis-(y-trimethoxysilylpropyl)amine A-1170
-CH2GH2CH2Si(OGH3)3
oganomodified CH3SiO[CH3)2SiO],~[CH3SiO]yl[CH3SiO]aSiCH3)3
plydiinethylsiloxane 1'-113~13 I I
NR2 NHR'Si OR' 3
polyazamide silylated silane A-1387 ---
Ureido Silanes
- - - O
y-ureidopropyltrialkoxysilane A-1160 I I
H2AICAIHCH2GH2GH2Si(OCH3)x(OCH3CH2)3 x
0
y-ureidopropyltrimethoxysilane Y- 11542 11
H2HCIIIH C3H6Si(O GH3)3
Isocyanato Silanes
y-isocyanatopropyltriethoxysilane A-1310 O=C=NCH2CHaCHZCH2Si(OCHaCH3)3
Additional examples of suitable silane coupling agents include the products
from
Chisso having the trade designations set forth in Table 1A.
TABLE 1A
S-310 n 2-aminoeth 1-:3-amino ro lrneth Idimethox silane
S-320 n- 2-aminoeth 1-3-amino ro ltrimethox silane
S-350 n-[2-(vinylbenzyl.imino)ethyl]-3-aminopropyl-trimethoxysilane
monoh drochloride (methanol solution)
S-510 3- I cidox ro ltrimethox silane
S-610 3-chloro ro lmothydimethox silane
S-620 3-chloro ro ltrirnethox silane
The silane coupling agents used in the present invention may be replaced by
alternative coupling agents or mixtures. For example, A-1387 may be replaced
by a
version in which the methanol solvent is replaced by ethanol. A-1126, an
aminosilane
coupling agent including a mixture of approximately 24% by weight
diaminosilane
modified by a surfactant in a methanol solution (GE Silicones), may be
replaced with
9
CA 02635451 2008-06-26
WO 2007/078900 PCT/US2006/048130
trimethoxy-silyl-propyl-ethylene-diamine (Z-6020 from Dow Corning). A-1120 or
Z-6020
may be substituted by a pre-hydrolyzeci version. Z-6020 may be replaced by Z-
6137, a
pre-hydrolyzed version lacking the alcohol solvent and including 33%
diaminosilane in
water at a concentration of 24% solids (commercially available from Dow
Corning). In
addition, A-1100 may be replaced by its hydrolyzed form Y-9244, which will
reduce or
eliminate the ethanol emission.
Preferably, the silane coupling agent is an aminosilane or a diaminosilane.
The size composition may include one or more of the above-identified coupling
agents.
The coupling agent may be applied to the fibers in an amount sufficient to
achieve a Loss
on Tgnitiori (LOI) of from about 0.02% to about 0.30% on the dried fiber, and
preferably in
an amount of from about 0.04% to about 0.08%.
In addition, the size composition may include at least one resinous film
forming
agent. Any 'conventional film forming agent known to those of skill in the art
may be
utilized in the size composition. In addition, the film former may be the
same. as or ...
different from the film forming agent present in the binder composition
described in detail
below. In the size composition, the film former acts as a polymeric binding
agent to
provide additional protection to the reinforcing fibers and improves
processability, such as
a reduction in fuzz generated by high speed chopping_ The film forming agent
may be
present on the reinforcement fibers in an amount sufficient to provide an LOI
from 0% to
about 1.0%. Preferably, the film forrning agent is present on the fibers in an
amount
sufficient to provide an LOI from about 0.15 fo to about 0.60%.
In an alternative embodiment of the present invention, the size composition
contains a lubricant to facilitate manufacturing instead of, or in addition
to, a film forming
agent. Examples of suitable lubricants include, but are not limited to,
lubricants such as
water-soluble ethyleneglycol stearates (for example, polyethyleneglycol
monostearate,
butoxyethyl stearate, polyethylene glycol monooleate, and
butoxyethylstearate),
ethyleneglycol oleates, ethoxylated fatty amines, glycerin, emulsified mineral
oils, and
organopolysiloxane emulsions. Other examples of lubricants include alkyl
imidazoline
derivatives (for example, a cationic softener which has a solids content of
approximately
90% and is available commercially fi-om Th. Goldschmidt AG), stearic
ethanolamide (for
example, Lubesize K12 (AOC)), and a polyethyleneimine polyamide salt
commercially
available at 50% active solid from Cognis under the trade name Emery 6760. The
CA 02635451 2008-06-26
WO 2007/078900 PCT/US2006/048130
lubricant may be present on the fibers iin an amount sufficient to provide an
LOI up to
about 0.10%. -
Although the size composition is effective at any pH level, the pH preferably
falls
within the range of from 7 to 11. The pH may be adjusted depending on the
intended
application, or to facilitate the compatibility of the ingredients of the size
composition.
Any suitable pH adjuster (for exaynple; a weak organic acid such as acetic
acid or a base
such as ammonia), may be added to the size composition in an amount sufficient
to adjust
the pH to a desired level.
The size composition may be rnade by dissolving each of the ingredients into a
premix with agitation. The separate premixes may then be combined with
deionized water
to form a main mixture and to achieve: the appropriate concentration and
control the mix of
solids. The premixes may be added ir- any order. If necessary, the pH of the
main mixture
may be adjusted to a desired level. The premixes may be added separately, or
they may be
added at..the. same time to form. the main mixture.
As described above, the two-part sizing formulation also includes a binder
composition. The binder compositior- includes a high acid number copolymer
formed
from the polymerization of maleic anhydride or maleic acid and at least one
other desired
monomer and/or a high acid number polyacid. As used herein, the phrase "high
acid
number" is intended to designate a polyacid with an acid number or acid value
of greater
than about 300. In addition, the bindt.-r composition may include any suitable
additive
identified by one of skill in the art, such as, for example, adhesive film
forming polymers,
lubricants, a surfactant or a mixture of surfactants, antistatic agents, and
crosslinking
agents. The binder composition may be applied to the fiber with an LOI of from
about
0.20% to about 2.0%, depending on the desired application.
The maleic anhydride or maleic acid copolymer may be a pure copolymer or a
derivative in an anhydride, acid, partial-salt, partial-ester, partial-amide,
or partial-imide
form. Suitable copolymers include C2 - C5 a-olefins, such as ethylene-,
propylene-,
(iso)butylene-, or butadiene-maleic acid copolymers, and methyl vinyl ether-
maleic acid
copolymers. The copolymer is poorly soluble when dispersed in water at room
temperature, but when it is heated to temperatures above approximately 90 C,
it dissolves
by virtue of the hydrolysis of the anhydride groups of the polymer to form the
corresponding polyacids. In such a reaction, one mole of anhydride is
hydrolyzed to two
11
CA 02635451 2008-06-26
WO 2007/078900 PCT/US2006/048130
moles of diacid in an exothermic reaction. The aqueous solution formed by the
hydrolysis
may then be used to formulate the binder composition. Similar reactions may be
employed
using ammonia or an amine in water, or an alcohol or an amine in a non-
reactive solvent,
to form, respectively, solutions of the partial-ammonium salt, partial-ester,
partial-amide,
or partial-imide derivatives.
In a preferred embodiment, the: binder composition includes an ethylene-maleic
acid (EMA) copolymer formed by the hydrolysis of an ethylene-maleic anhydride
copolymer. The ethylene-maleic anhydride copolymer may be formed by the
radical
copolymerization between ethylene arid maleic anhydride in the presence of a
peroxide
catalyst. This copolymerization leads to an alternating copolymer that
includes a high
level of maleic units which gives the ethylene-maleic acid copolymer a high
polyacid
functionality.
In at least one exemplary embodiment, the copolymer is an aqueous solution of
the
polyacid, (partial) ammonium salt, partial-ester, partial-amide, or partial-
imide.derivative.
of an alternating block copolymer of maleic anhydride, or mixtures thereof.
Mixtures of
different ethylene maleic acid copolyrners or maleic anhydride copolymers with
other high
acid number polycarboxylic acids such as acrylic or maleic acid homo- or co-
polymers or
derivates thereof such as those descri,bed above may used in the binder
composition to
achieve desired properties, such as iniproved strength in the reinforced fiber
product or
improved fiber processability, and also to reduce the total binder cost.
As discussed above, the binder composition may include a high acid number (for
example, high acid value) polyacid alternatively, or in addition to, the
maleic anhydride or
maleic acid copolymer. Desirably, the polyacid is a polycarboxylic acid. A
suitable
polycarboxylic acid polymer for use in the binder composition is an organic
polymer that
contains numerous pendant carboxylic acid groups and is characterized by a
high acid
number. The acid number or acid vEilue of a substance may be described as a
measure of
the free acid'content, and may be expressed as the number of milligrams of
potassium
hydroxide neutralized by the free acid present in one gram of the substance.
In the binder
composition, the high acid number polyacid may have an acid number of at least
about
300. In at least one exemplary embodiment, the binder composition has an acid
number of
about 300 to about 950.
12
CA 02635451 2008-06-26
WO 2007/078900 PCT/US2006/048130
The polycarboxylic acid polymer may be a homopolymer or copolymer prepared
from unsaturated carboxylic acids including, but not limited to, acrylic acid,
methacrylic
acid, crotonic acid, isocrotonic acid, m,aleic acid, cinnamic acid, 2-
methylmaleic acid,
itaconic acid, 2-methylitaconic acid, and a, (3-methyleneglutaric acid.
Alternatively, the
polycarboxylic acid polymer may be prepared from unsaturated anhydrides such
as maleic
anhydride, itaconic anhydride, acrylic anhydride, methacrylic anhydride, and
mixtures
thereof. Methods for polymerizing these acids and anhydrides are known by
those of
ordinary skill in the art, and will not be discussed in detail herein. In
addition, the
polycarboxylic acid polymer may include a copolymer of one or more of the
unsaturated
carboxylic acids or anhydrides described above and one or more vinyl compounds
including, but not limited to, styrene, cc-ethylstyrene, acrylonitrile,
methacrylonitrile,
methyl acrylate, ethyl acrylate, n-butyl acrylate, isobutyl acrylate, methyl
methacrylate, n-
butyl methacrylate, isobutyl methacrylate, glycidyl methacrylate, vinyl methyl
ether, vinyl
acetate,..1-olefins (for. example, ethylene, isobutene) vinyl and allyl alkyl
ethers (f r.
example, methyl vinyl ether, ethyl vinyl ether), substituted acrylamides, and
sulfonic and
sulfonate monomers (for example, allylsulfonic acid, vinylsulfonic acid). Such
polyacids
are described in detail in U.S. Patent :No. 5,236,982 to Cossement, et al.,
which is hereby
incorporated by reference in its entirety.
The high acid number maleic anhydride copolymer, high acid number ethylene
maleic acid copolymer, or high acid riumber polycarboxylic acid in the binder
composition
may be present on the fibers in an amount sufficient to provide an LOI up to
about 2.0%,
preferably from about 0.10% to about 2.0%, and even more preferably from about
0.40%
to about 1.0%. It is to be appreciated that when the high acid number maleic
anhydride
copolymer, high acid number ethylene maleic acid copolymer, or high acid
number
polycarboxylic acid comes in contacl: with a polymer containing a di-cation,
there is an
interaction between the carboxylic acid moieties and the di-cations in which
the carboxylic
acid moieties are consumed or beconne otherwise unable to interact favorably
with the
polymer matrix. Although not wishing to be bound by theory, it is believed
that a ratio
higher than 2:1 (equivalent carboxylic acid of the polyacid:equivalent di-
cation
respectively) retains the presence of carboxylic acid moieties after the
interaction of the
polyacid moieties with the di-cation. Therefore, it is desirable that an
excess of carboxylic
acid moieties be present in the size/binder composition to compensate for the
polyacid that
13
CA 02635451 2008-06-26
WO 2007/078900 PCT/US2006/048130
is consumed by the di-cations such that improved dry-as-molded mechanical
properties
and improved hydrolysis resistance may be achieved. However, the specific
amount of
carboxylic acid moieties present in the binder composition needed to achieve
an excess
(and improved physical properties) is ctependent upon the amount of di-cations
present in
the polymer. It is believed that it is the difference between the number of
equivalents of
carboxylic acid moieties present on the reinforcing fiber and the number of
equivalents of
di-cationic additive present in the polymer that targets the % LOI of the
maleic anhydride
copolymer, ethylene maleic acid copolymer, or high acid number polycarboxylic
acid. It is
to be noted that the use of a high LOI polyacid does not alter the efficiency
of the calcium
stearate in its primary role as a lubricant/demolding agent.
In addition to a maleic anhydride copolymer, an ethylene maleic acid
copolymer, or
a high acid number polycarboxylic acid, the binder composition may also
include one or
more film forming agents. Film formE:rs are agents which create improved
adhesion
between.the reinforcing fibers, which.results.in improved strand integrity.
Suitable film ...
formers include thermosetting and thermoplastic polymers which promote the
adhesion of
sizing compositions. Polyurethane film formers are a preferred class of film
formers for
use in the binder composition because they demonstrate good compatibility with
polyamide matrices and help to improve the dispersion of the glass fiber
bundles in the
melt (for example, extrusion process or injection molding process) when
forming the
composite article, which causes a reduction or elimination of defects in the
final article
that caused by poor dispersion of the reinforcement fibers (for example,
visual defects,
processing breaks, and/or low mechariical properties). The polyurethane
dispersions
utilized in the two-part sizing forrnule.ition of the present invention may be
part of a
dispersion that either is based or is not based on a blocked isocyanate. In
addition, as
discussed above, the film former present in the binder composition may or may
not be the
same as the film former present in the: size composition.
Examples of suitable urethane film formers that are not based on blocked
isocyanates that may be used in the binder composition include, but are not
limited to,
Baybond XP-2602 (a non-ionic polyurethane dispersion (Bayer Corp.)); Baybond
PU-
401 and Baybond PU-402 (anionic urethane polymer dispersions (Bayer Corp.));
Baybond VP-LS-2277 (an anionic/non-ionic urethane polymer dispersion (Bayer
Corp.));
Aquathane 518 (a non-ionic polyurethane dispersion (Dainippon, Inc.)); and
Witcobond
14
CA 02635451 2008-06-26
WO 2007/078900 PCT/US2006/048130
290H (polyurethane dispersion (Witco Chemical Corp.)). Other examples of
suitable film
forming agents for use in the binder composition include, but are not limited
to,
polyvinylpyrrolidone homo- and co-palymers and other polymers bearing amide-
like
functionality such as polyamide, polyvinylformamide, or polyacrylamide. In at
least one
exemplary embodiment, the film forming agent is an aqueous polyurethane
dispersion that
is not part of a dispersion that includes a polyurethane and a blocked
isocyanate.
Examples of suitable urethane film formers based on blocked isocyanates which
may be used in the binder compositiort include, but are not limited to,
Baybond XW-116;
Baybond XP-055; Baybond PU-330; Baybond PU-400-S; Baybond PU-401
(polyurethane dispersions based on blocked isocyanates (available from Bayer
Corp.));
Baybondo VP-LS-2277 and Baybond'VP-LS-2297 (anionic/non-ionic urethane polymer
dispersions (Bayer Corp.)); Baybond PU-130 (an anionic/non-ionic urethane
polymer
dispersion (Bayer Corp.)); Baybond :PU-403 (a polyurethane dispersion (Bayer
Corp.));
Baybond PU-239 (a crosslinkable anionic/non-ionic urethane. polymer
dispersion (Bayer
Corp.)); Baybond PU-2435 (an anioiiic/non-ionic urethane polymer dispersion
(Bayer
Corp.)); and Witcobond 296B (an aqLieous blocked polyurethane dispersion,
available
from Baxenden Chemicals).
The film former(s) may be present in the binder composition in an amount
sufficient to provide an LOI up to about 1.0%, and preferably from about 0.10%
to about
0.40%. It is desirable that the amount of film former present in the two-part
sizing
formulation is such that the two-part sizing formulation provides the desired
level of
compatibility for the reinforcement fibers and the polymer matrix to help
fiber dispersion
during processing to form a composite part without affecting the positive
effect of the
polyacid moieties to improve dry-as-molded and hydro-aged mechanical
properties and
without developing static and/or an undesirable color in the reinforcing fiber
product.
The binder composition may be formed by mixing a solution of the desired high
acid number polyacid(s) (for example, a polycarboxylic acid obtained by
hydrolysis of the
parent polyanhydride in hot water) with a solution of the film forming agent.
Optionally,
the solution may be diluted with water. It may be necessary to adjust the pH
of the
polyacid solution with a base before mixing it with the film former solution
to reduce or
eliminate the occurrence of destabilization phenomena. Any suitable pH
adjuster may be
added to the binder composition to adjust the pH to a desired level; however,
it is preferred
CA 02635451 2008-06-26
WO 2007/078900 PCT/US2006/048130
that ammonia (NH4) be added to adjust the pH. The pH of the binder composition
may fall
in the range of from 2.5 - 10.
The total LOI for the two-partsizing formulation on the reinforcing fiber
material
may be from about 0.25% to about 2.05%, preferably from about 0.60% to about
1.10%.
One advantage of the two-part sizing formulation of the present invention is
that
the product resulting from the application of the two-part sizing formulation,
which may
contain up to about 2.0% LOI of the high acid number maleic anhydride
copolymer, high
acid number ethylene maleic acid copolymer, or high acid number polycarboxylic
acid,
provides improved mechanical properties, even when the polyamide matrix
contains a di-
cation additive such as calcium stearate.
In addition, the two-part sizing formulation of the present invention imparts
improved physical properties, such as improved dry-as-molded (DaM) mechanical
properties of the composite part after aging the composite part in severe
hydrolysis
conditions, to.composites formed.frorn industrially processable and easily
dispersible
pellets.
The present invention is also advantageous in that the polyurethane film
formers
present in the two-part sizing formulation demonstrate good compatibility with
polymer
matrices (such as polyamide matrices) that contain di-cationic lubricants,
which helps to
improve the dispersion of the reinforcement fiber bundles in the melt (for
example, in
extrusion process or injection molding process) when forming a composite
article. This
increased dispersion of the reinforcement fibers causes a reduction of defects
such as
visual defects, processing breaks, and/or low mechanical properties in the
final product.
Further, the two-part sizing formulation of the present invention has improved
stability over conventional sizing fortnulations containing an aminosilane and
a polyacid
in the same mixture. Mixtures containing both aminosilanes and polyacids may
not be
stable due to chemical interactions between the two compounds. By placing
substantially
all (if not all) of the aminosilane in the size composition and the polyacid
in the binder
composition, both the size composition and the binder composition have
increased shelf
lives.
The process for making a densified reinforcing fiber product may be an in-line
process that permits the application of the size composition, the chopping of
the glass
fibers, the application of the binder composition, and pelletizing the
reinforcing fiber
16
CA 02635451 2008-06-26
WO 2007/078900 PCT/US2006/048130
material. Such an in-line process forrns a pellet product that exhibits
superior physical
properties, such as improved strength, when integra#ed into a composite (for
example,
when compared to pellets produced by processes previously known in the art).
Although
not wishing to be bound by theory, such superior properties are believed to be
due to the
improved compatibility of the size composition and binder composition, which
permits a
better coating of the reinforcing fiber inaterial.
The process for making a densified reinforcing fiber product according to the
invention may employ an apparatus that includes: (a) an apparatus for applying
a size
composition to a continuous fiber material; (b) an apparatus for cutting the
glass fiber
strands to form chopped strand segments; (c) an apparatus for conveying the
chopped
strand segments to a first tumbling apparatus; (d) an apparatus for applying a
binder
composition to the chopped strand segments; (e) a first tumbling apparatus for
imparting a
tumbling action to the chopped strand segments to disperse the binder
composition and
cause.the. chopped strand.segments to align.and coalesce into pellets; (f)
optionally, an
apparatus for conveying the pellets to a second tumbling apparatus; (g)
optionally, a
second tumbling apparatus for tumbling the pellets to compact them and
increase their
density; (h) an apparatus for conveying the densified pellets to a drying
apparatus; and (i) a
drying apparatus adapted to receive aiid dry the pellets.
Initially, the size composition may be applied to the reinforcing fiber
material by
any conventional means, including kiss roll, dip-draw, and slide or spray
applicators.
Preferably, the precursor size is applied by passing the reinforcing fiber
material,for
example, strands of glass or polymer, over a kiss roll applicator. The size
composition is
preferably applied to the strands in ar.i amount sufficient to provide the
strands with a
moisture content of from about 8% by weight to about 13% by weight, more
preferably
about 10% to about 11% by weight.
The sized strands may then be: chopped into strand segments. The strand
segments
may have a length of from approximately 2 mm to approximately 50 mm.
Preferably, the
strands have a length of from about 3 to about 4 mm. Any suitable method or
apparatus
known to those of ordinary skill for chopping glass fiber strands into
segments may be
used.
Next, the binder composition may be applied to the chopped strand segments.
The
coated chopped strand segments are -then pelletized by any suitable method
known to those
17
CA 02635451 2008-06-26
WO 2007/078900 PCT/US2006/048130
of ordinary skill in the art, such as, for example, tumbling or otherwise
agitating the
chopped strand segments in a pelletizer. Processes suitable for pelletizing
the chopped
strand segments are disclosed in U.S.1'atent Nos. 5,868,982, 5,945,134,
6,365,090, and
6,659,756 to Strait et al., and U.S. Patent No. 5,693,378 to Hill et al., all
of which are
incorporated by reference in their entireties. The amount of moisture in the
binder
composition serves to adjust the moisture content of the strand segments to a
level suitable
for the formation of pellets when the strand segments are tumbled in the
pelletizer.
Although the moisture content of the strand segments can be adjusted prior to
their
introduction into the pelletizer, it is preferred that the segments are
hydrated to a moisture
content suitable for pellet formation in the pelletizer itself.
Preferably, the moisture conteiit of the chopped strand segments in the
pelletizer is
from about 12% by weight to about 16% by weight, and more preferably from
about 13%
by weight to about 14% by weight, based on the total weight of the binder-
sized, chopped
strand segments. If.the.moisture content is too low, the strand.segments tend
not to ._.._.
combine into pellets and will remain in a strand formation. On the other hand,
if the
moisture content is too high, the strands tend to agglomerate or clump or form
pellets
having a large diameter and an irregul.ar, non-cylindrical shape.
The binder composition may lie applied to the chopped strand segments as they
enter the pelletizer, or after the chopped segments are placed in the
pelletizer but prior to
tumbling. In an alternative embodiment, the binder composition may be sprayed
onto the
strands before they are chopped. In this alternative embodiment, it is
preferable to use a
pelletizer that is specially equipped with tumbling means such as baffles to
ensure
adequate tumbling and formation of the pellets.
To ensure good coverage of the chopped segments, the binder composition is
preferably applied to the chopped strand segments as they enter the pelletizer
but before
they begin to coalesce into pellets. If the binder composition is applied at
other locations
within the pelletizer, pellets may forr.n before the strand segments are
completely coated
with the binder composition, which results in pellets containing fibers that
are not coated
with the binder composition. When such pellets are used in the manufacture of
fiber
reinforced composite articles, the uncoated fibers lack the interfacial
coating required to
provide good reinforcing characteristics, and the resulting composite article
will have less
than optimal properties. Preferably, the pelletizer is equipped with a spray
nozzle, located
ls
CA 02635451 2008-06-26
WO 2007/078900 PCT/US2006/048130
adjacent to the strand segment inlet, for spraying the binder size onto the
strand segments
as they enter the pelletizer.
The pelletizer may be any apparatus capable of tumbling the strand segments in
such a way that: (1) they become substantially uniformly coated with the
binder
composition, and (2) multiple chopped strand segments align and coalesce into
pellets
having a desired dimension. Such a tumbling apparatus should have an average
residence
time sufficient to insure that the strand. segments become substantially
coated with the
binder size and form pellets, but insufi:icient for the pellets to be damaged
or degraded
through abrasion (for exarnple, by rubbing against one another). Preferably,
the residence
time in the tumbling apparatus is from about 1 minute to about 10 minutes.
More
preferably, the residence time in the tumbling apparatus is from about 1
minute to about 3
minutes.
A preferred pelletizer is a rotating drum, such as that disclosed in U.S.
Patent No.
5,868,9.82,.as_referenced.herein above.. U.S._Patent No. 5,868,982 discloses
an-apparatus
for making reinforcing fiber pellets, which is preferably provided with a
system for
monitoring and/or adjusting various process parameters. The moisture content
of the
strand segment input may be monitored and controlled using suitable method. In
one
embodiment in which the binder composition is applied to the strand segments
before they
are placed in the pelletizer, the rotating drum is adapted to accommodate a
spray head for
applying the binder composition to the strand segments as they enter the drum.
The binder
composition and a solvent, such as water, may be combined into one fluid
stream and
dispersed through the nozzle orifice. This stream may be combined with two
jets of air
positioned approximately 180 degrees apart and at an angle of 60 degrees to
the direction
of the stream flow. Mixing the binder composition with the forced air streams
effectively
creates a mist that is propelled onto tLte surface of the tumbling strand
segments in the
drum.
Rotation of the drum causes the wet strand segments to tumble around one
another
while the surface tension created by the wet sizing or coating causes strand
segments
contacting one another over a substantial portion of their length to align
with one another
and coalesce into a cylindrically shaped pellet. By such action, any fines or
single fibers
created during the chopping operatiori are recombined with and incorporated
into the
forming pellets to essentially eliminate individual fine fibers from the
resulting pellets.
19
CA 02635451 2008-06-26
WO 2007/078900 PCT/US2006/048130
Preferably, the drum is tilted slightly s+) that the end of the drum from
which the pellets
exit is lower than the end in which they enter to ensure that the pellets
formed in the drum
do not remain in the drum for an excessive period of time.
The size of the pellets formed in the drum is controlled primarily by the
moisture
content of the strand segments. If the moisture content is maintained at a
high level, a
greater number of strand segments will coalesce into a pellet and the pellet
will have a
larger diameter. On the other hand, if the moisture is maintained at a lower
level, fewer
strand segments will coalesce into a pellet and the pellet will have a smaller
diameter. The
amount of binder composition that is clischarged onto the strands may be
controlled by a
computer which monitors the weight of wet glass entering the pelletizer and
adjusts the
amount of the binder composition to obtain a final chopped strand having a
strand solids
content of from about 0.25% to about 2.05% by weight.
Preferably the pellets formed have a diameter of from about 20% to about 65%
of
their length. Such.pellets are typically formed by. .combining from
about.70.strand . . . . . .. . ..
segments to about 175 strand segments, each containing from about 500
individual
filaments per strand to about 8000 individual filaments per strand.
The size of the pellets may also be affected by drum throughput. For example,
the
higher the drum throughput, the shorter the residence time of the strand
segments in the
drum. As a result, smaller pellets may be formed because the binder
composition is not
adequately dispersed on the strands which may cause the strands not to
coalesce into a
pellet. In addition, pellets that are forrned in the drum for a shorter period
of time are less
compacted than those pellets that formed in the drum for a longer period of
time.
Although some compaction of the formed pellets invariably occurs in the
pelletizer,
it is typically insufficient to increase the pellet density to a level
providing optimum
flowability. For this reason, after thei.r formation in the pelletizer, the
pellets may
optionally be fed into a second tumbl:ing apparatus or densifier, wherein the
pellets are
further compacted and densified. An;y low-impact tumbling apparatus that will
compact
the pellets without degrading them through abrasion or otherwise damaging the
pellets
may be used. Preferably, the densifier is a zig-zag tube adapted to be rotated
about its
longitudinal axis, such as is describecl in U.S. Patent Nos. 5,868,982,
5,945,134,
6,365,090, and 6,659,756 to Strait et al.
CA 02635451 2008-06-26
WO 2007/078900 PCT/US2006/048130
Preferably, the densifier has a gentler, less vigorous tumbling action than
that of the
pelletizer to minimize degradation of the pellets. As the zig-zag tube is
rotated, pellets
placed therein are gently tumbled abou.t by the tube's rotation as they are
pulled through the
tube by gravity. As with the rotating drum.described above, the zig-zag tube
densifier is
preferably tilted at a slight angle to ensure that the pellets flow through
the apparatus
without excessive residence times. Furthermore, the densifier preferably has
an average
residence time of less than approximalely 5 minutes to reduce any abrasion
that may occur.
Preferably, the average residence time: in the densifier is from about 1
minute to about 2
minutes.
Although pellet formation and densification may occur in separate apparatuses,
such as a separate rotary drum and a rotating zig-zag tube linked by a
conveyor, the
pelletizing process may be accomplished using any suitable apparatus. For
example, pellet
formation and densification may occur in separate tumbling regions or zones
within a
single apparatus.. A.preferred example of such an apparatus is a"Zig-
Zag".blender_
commercially available from Patterson Kelly. In a preferred embodiment of this
device, a
drum is equipped with an interior baffle to reduce the free-fall distance of
the glass pellets
and strand segments during rotation o:f the drum. By reducing this distance,
less
deterioration of the glass fibers and pe:llets through impact and abrasion
occurs, resulting
in improved physical properties in the glass fiber reinforced molded articles
manufactured
therefrom.
After densification, the pellets may be delivered onto a conveyor belt and
dried, for
example, using a hooded oven supplied with hot air and cooling air or any
other suitable
drying apparatus easily identified by one of skill in the art. To reduce
drying time to a
level acceptable for commercial mass production, it is preferred that the
fibers are dried at
elevated temperatures of up to approximately 260 C in a fluidized-bed oven.
After
drying, the densified pellets may be classified by size using a screen or
other suitable
device.
By varying the throughput and moisture content of the strand segments, glass
fiber
pellets can be made that are from about 13% to about 60% denser than the
corresponding
unpelleted strand segments, and from about 10 times to about 65 times larger
in dianleter.
For example, chopped 4 mm (length) segments of a 2000 filament strand composed
of 14
micron (diameter) fibers typically have a bulk density of from about 33 lb/ft3
(528.66
21
CA 02635451 2008-06-26
WO 2007/078900 PCT/US2006/048130
kg/m3) to 36 lb/ft3 (576.72 kg/m3). After being hydrated to a moisture content
of from
about 13% to about 14% and formed into densified pellets such as is described
above;
according to the process of the invention, the resulting dried pellets
typically have a bulk
density of from about 40 lb/ft3 (640.8 l.cg/m) to about 55 lb/ft3 (881.1
kg/m3). As a result
of their increased diameter-to-length ratio and increased density, the
resulting pellets
exhibit significantly improved flowability in comparison to the unpelleted
chopped strand
product.
The size composition and the binder composition facilitate treating
reinforcing
fiber materials,for example, glass, during a continuous process that includes
forming the
fibers as well as subsequent processing or handling. By applying the binder
composition
in the pelletizer, an application efficie-ncy of about 95% to about 100% for
the binder
composition may be obtained. This h:igh application efficiency reduces waste
water
contamination in the plant. Further, because the binder composition can be
applied
efficientl.y, the binder. composition may.be applied. with a reduction in
cost.
In addition, by applying the binder composition separately from the sizing
composition outside the fiber-forming environment permits, materials that are
not
desirably applied during the fiber-forrning process because of toxicity,
safety,
flammability, irritation, stability, low compatibility with aminosilanes,
viscosity, toxicity,
cleanliness, odor, cost, or shear sensitivity may be applied to the glass
fibers. Also,
because the polyacid in the binder coinposition can be applied to the glass
fibers in a more
concentrated form in the pelletizer than if it were applied directly to the
glass strands as
they are being formed, there is reduced fiber logging and waste as compared to
conventional in-line processes.
Although the invention is highly suitable for in-line manufacturing processes,
such
as described above, it may also be used in an off-line process in which the
size
composition and the binder composition are applied to previously formed and
packaged
reinforcing fiber materials, or in which the size composition and the binder
composition
are applied to the reinforcing fiber material at different times. For example,
the size
composition may be applied to a forn:ied fiber strand, after which the strand.
may be wound
and stored before subsequent unwinding, chopping into segments and application
of the
binder composition.
22
CA 02635451 2008-06-26
WO 2007/078900 PCT/US2006/048130
Having generally described this invention, a, further understanding can be
obtained
by reference to certain specific examples illustrated below which are provided
for purposes
of illustration only and are not intended to be all inclusive or limiting
unless otherwise =.
specified.
EXAMPLES
Two-part sizing formulations
TABLE 2
Ex. Ex. Ex. Ex. Ex. Ex. Ex. Ex. Ex. Ex. Ex.
1 2 3 4 5 6 7 8 9 10 11
Size Composition
A3C-I 100 Silane 0.07 0.07 0.07 0.04 0.04 0.04 0.04 0.04 0.07 0.06 0.06
Lubesize 2tb) 0.02
Zonyl FS-300 ' 0.005
Baybond PU403 0.31 0.31 0.31 0.31 0.18
Witcobond 296B ' 0.53 0.53 0.53
Ba bond XP-2602( 0.16 0.16
Binder Composition
Glascol E5(8) 0.28 0.40 0.65 0.95 0.14
Glascol C95 0.04 = .= . .. . = 0.12
ZeMac E4000.30 0.24 0.47 0.38
ZeMac E60 0.40
Ammonia
Baybond PU-403 0.40
Baybond XP-2602 ' 0.21
Aguathane 518 0.19
AX-1100 Silane 0.02
Jeffamine ED2003 0.09
Pluronic F-77 "' 0.0008
Pluronic PE-103 " 0.0022
Pluronic L-101 0.0009
Triton X-100 P 0.0022
Polyacid LOI 0.00 0.28 0.30 0.00 0.40 0.65 0.95 0.28 0.61 0.40 0.50
Total LOI (%) 0.60 0.88 0.90 0.75 0.75 0.95 1.30 0.65 0.86 0.83 0.72
(') A3{-1 100 Silane y-aminopropyltriethoxysilane (GE Silicones)
(b) K 12 Lubesize tetraethylenepentamine reacted with stearic acid (AOC)
(C) Zonyl FS-300 fluoroalkyl alcohol substituted polyethylene glycol (DuPont)
(d) Baybond PU-403 blocked isocyanate polyurettiane dispersion (Bayer Corp.)
W Witcobond 296B blocked polyurethane dispersion (Baxenden Chemicals)
Baybond XP-2602 non ionic polyurethane dispersion (Bayer Corp.)
t8~ Glascol E5 solution of low Mw acrylic acid homopolymer (Ciba)
(h) Glascol C95 solution of low Mw acrylic acid homopolymer partially
neutralized by ammonia (Ciba)
t', ZeMac E400 alternating copolymer of ethylene, and maleic anhydride with a
Mw of approx 400,000 (Zeeland
Chemicals)
6, ZeMac E60 alternating copolymer of ethylene and maleic anhydride with a Mw
of approximately 60,000 (Zeeland
Chemicals)
'k) Aquathane 518 polyurethane dispersion (D.I.C..)
(1) Jeffamine ED2003 water-soluble aliphatic dianiine derived from a propylene
oxide-capped poly(ethyleneoxide)
(Huntsman Corp.)
Pluronic F-77 oxirane (EO-PO copolymer) (BASF)
Pluronic PE-103 oxirane (EO-PO copolymer) (BASF)
tO~ Pluronic L-101 oxirane (EO-PO copolymer) (13ASF)
(P) Triton X-100 octylphenoxypolyethoxyethanol (Union Carbide Corp.)
The two part sizing compositions set forth in Table 2 were prepared as
described
23
CA 02635451 2008-06-26
WO 2007/078900 PCT/US2006/048130
below.
Preparation of Examples 1-- 3 of Ta ble 2
In Examples 1- 3, the components of Table 3 were mixed to prepare the size
composition. The size composition was applied to 10 m Advantex glass fibers
to
achieve a strand Loss-On-Ignition (LOI) of 0.60% on the glass fibers. A
conventional loss
on ignition (LOI) method, ASTM 2854, was used to determine how much of the
applied
size composition was on the glass fibers. The glass fibers were then collected
into a strand
and chopped wet in-line by a chopper i'tnto segments of approximately 4 mm in
length.
TABLE 3
Com onent Amount of Component
Witcobond 296B 78.4 k: as received in 400 L of deionized water
AX-1100 Silane" 19.91 kg as received in 500 L of deionized water
Deionized Water to 1000 L
a blocked polyurethane dispersion (Baxenden Chemicals)
(b) y-aminopropyltriethoxysi].ane (GE Silicones)
The chopped segments were th.en.collected and approximately 1- 2 days later
treated under rotation in a lab pelletizer where the cor=responding.binder set
forth in Table
2 was sprayed onto the chopped segments. In Example 1, no binder was applied
during
the pelletization. In Example 2, Glascol E5, a 25% solids solution of a low
molecular
weight acrylic acid homopolymer available from Ciba, was diluted to 15% solids
by
deionized water and used as the binder. In Example 3, the binder composition
was
prepared by dispersing 132 g of ZeMa.c 400, an alternating copolymer of
ethylene and
maleic anhydride available from Zeeland Chemicals, in 870 g of deionized water
and
dissolved by heating to approximately 95 C under agitation. The various
binder
compositions were applied to achieve the corresponding LOI % set forth in
Table 2. The
densified glass pellets were collected and dried in a moving belt lab oven for
approximately 16 minutes at a maximum temperature of 220 C.
Preparation of Examples 4- 7 of Table 2
In Examples 4- 7, the comporients of Table 4 were mixed to prepare the size
composition. The size composition was applied to 10 m Advantex glass fibers
to
achieve a strand Loss-On-Ignition (LOI) of 0.35% on the glass fibers. A
conventional loss
on ignition (LOI) method, ASTM 2854, was used to determine how much of the
applied
size composition was on the glass fibers. The glass fibers were then collected
into a strand
and chopped wet in-line by a chopper into segments of approximately 4 mm in
length.
24
CA 02635451 2008-06-26
WO 2007/078900 PCT/US2006/048130
TABLE 4
Component Amount of Component
Baybond PU-403 a 160.20 kg as received in 450 L of deionized water
AX-1100 Silane( 14.5 kg as received h drol zed in 65 L of deionized water
Deionized Water to 1400 L
a blocked isocyanate polyuretharie dispersion (Bayer Corp.)
(b) y-aminopropyltriethoxysilane (GE Silicones)
The chopped segments were then collected and approximately 1- 2 days later
treated under rotation in a lab pelletizer where the corresponding binder set
forth in Table
2 was sprayed onto the chopped segments. In Example 4, Baybond PU-403, a 39%
solids
blocked isocyanate polyurethane dispersion available from Bayer Corp., was
diluted to
15% solids by deionized water and used as the binder. In Examples 5 - 7, the
binder was
prepared by diluting Glascol E5, a 25 0o solids solution of a low molecular
weight acrylic
acid homopolymer, to 15% solids by deionized water. The various binder
compositions
were applied to achieve the corresponding LOI % set forth in Table 2. The
densified glass
pellets were collected and dried in a rnoving belt lab oven for approximately
16 minutes at .
a maximum temperature of 220 C.
Preparation of Examale 8 of Table :L
The size composition of Example 8 was prepared according to the description of
Table 7 set forth in U.S. Patent Publication No. 2004/0209991 Al to Piret et
aL, which=is
hereby incorporated by reference in its entirety. In particular, the size
composition as set
forth in Table 5 was prepared and applied to 10 m Advantex glass fibers as
they were
produced in a continuous in-line process. The glass fibers were then formed
into strands
and chopped into strand segments having a length of approximately 4.0 mm. The
molten
glass was fed through the bushing at approximately 215 lbs/hour. The size
composition
was applied with a conventional kiss roll type applicator turning in the
direction of the
strand at 15 meters per minute. The size composition was applied at a
concentration of
0.69% to achieve a strand LOI of 0.06% solid on the glass.
TABLE 5
Component % by Weight of Active Solids
AX-1100 Silane .'O 51
Lubesize K12 40
Zonyl FS-300 ' 9
(a) y-aminopropyltriethoxysilane (GE Silicones)
(b) tetraethylenepe:ntamine reacted with stearic acid (AOC)
fluoroalkyl alcohol substituted polyethylene glycol (DuPont)
CA 02635451 2008-06-26
WO 2007/078900 PCT/US2006/048130
The chopped segments were then conveyed to a pelletizer where the binder
composition according to Table 6 was sprayed onto the chopped segments. The
binder
composition was applied to achieve a strand LOI of 0.59% solid on the glass
fibers. The
total Loss-On-Ignition (LOI) of the glass was 0.65%. The densified glass
pellets were
conveyed to a conveyor-type or fluidized bed oven for drying.
TABLE 6
Component % by Weight of Active Solids
ZeMac E400 a 40
Jeffarnine ED-2003( 14
Glascol C95 ' 7
AX-1100 Silane 7
A uathane 518 e 31
Pluronic F-77 0.13
Pluronic PE-103 g 0.36
Pluronic L-101 0.15
Triton X-100' 0.36
a alternating copolymer of ethylene and maleic anhydride with a Mw of approx
400,000 (Zeeland Chem_icals)
(b) water-soluble aliphatic diamine derived from a propylene oxide-capped
poly(ethyleneoxide) (Huntsman Corp.)
( ) solution of low Mw acrylic acid homopolymer partially neutralized by
ammonia
(d) Y-aminopropyltrietho.xysilane (GE Silicones)
(e) polyurethane dispersion (D.I.C.)
(n oxirane (EO-PO copolymer) (BASF)
(g) oxirane (EO-PO copolymer) (BASF)
("' oxirane (EO-PO copolymer) (BASF)
0) octylphenoxypolyethaxyethanol (Union Carbide Corp.)
Preparation of Example 9 of Table 2
In Example 9, components of 'Cable 7 were mixed to prepare the size
composition.
The size composition was applied to 10 m Advantex glass fibers to achieve a
strand
Loss-On-Ignition (LOI) of 0.25% on the glass fibers. A conventional loss on
ignition
(LOI) method, ASTM 2854, was used to determine how much of the applied size
composition was on the glass fibers. The glass fibers were then collected into
a strand and
chopped wet in-line by a chopper into segments of approximately 4 mm in
length.
TABLE 7
Component Amount of Component
Baybond PU-403 131.10 k a:a received in 1000 L of deionized water
AX-1100 Silane( 33.80 kg as received h drol zed in 1000 L of deionized water
Deionized Water to 2440 L
~a blocked isocyanate polyurethane dispersion (Bayer Corp.)
(b) y-aminopropyltriethoxysilane (GE Silicones)
26
CA 02635451 2008-06-26
WO 2007/078900 PCT/US2006/048130
The chopped segments were then collected and approximately 1- 2 days later
treated under rotation in a lab pelletizer where the binder was sprayed onto
the chopped
segments. The binder composition was prepared by dispersing 13.43 kg of ZeMac
400
EMA powder in 47.85 kg of deionized water and dissolving the powder by heating
the
water to approximately 95 C under agitation. After the ZeMac 400 EMA powder
was
dissolved, the solution was further dilutted by adding 19 kg of deionized
water. 18.61 kg
of Glascol C95, a solution of a low molecular weight acrylic acid homopolymer
partially
neutralized by ammonia which is available from Ciba, was then added with
agitation at a
temperature lower than 60 C. The binder composition was applied to achieve a
strand LOI
of 0.61 % solid on the glass fibers. The total Loss-On-Ignition (LOI) of the
glass fibers
was 0.86%. The densified glass pellets were collected and dried in a moving
belt lab oven
for approximately 16 minutes at a maximum temperature of 220 C.
Preparation of Example 10 of Table 2
In Example 10, components of Table 8 were mixed to prepare the size
composition. The size composition was applied to 10 m Advantex glass fibers
to
achieve a strand Loss-On-Ignition (LO-I) of 0.22% on the glass fibers. A
conventional loss
on ignition (LOI) method, ASTM 2854, was used to determine how much of the
applied
size composition was on the glass fibers. The glass fibers were then collected
into a strand
and chopped wet in-line by a chopper :into segments of approximately 4 mm in
length.
TABLE 8
Component Amount of Component
Baybond XP-2602 a 143.23 kg as received in 800 L of deionized water
AX-1100 Silane 34.81 kg as received h drol zed in 1000 L of deionized water
Deionized Water to 2200 L
(a) non-ionic polyurethane dispersion (Bayer Corp.)
(') y-aminopropyltriethoxysilane (GE Silicones)
The chopped segments were then conveyed to a pelletizer where the binder was
sprayed onto the chopped segments as they passed through the entrance chamber
of the
pelletizer. The binder composition was prepared by dispersing 32.00 kg of
ZeMac 60
EMA powder in 80.80 kg of deionized water and dissolving the powder by heating
the
water to approximately 95 C with agitation. After the ZeMac 60 EMA powder was
dissolved, the solution was cooled to 25 C and the pH was adjusted to about
3.5 by the
addition of approximately 9.10 kg of a. 25% ammonia solution. 52.37 kg of
Baybond XP-
2602, a non-ionic polyurethane dispersion available from Bayer Corp., was then
added
27
CA 02635451 2008-06-26
WO 2007/078900 PCT/US2006/048130
slowly with agitation. The densified glass pellets were conveyed to a conveyor
or
conveyor-type or fluidized bed oven for drying. The binder composition was
applied to
achieve a strand LOI of 0.61% solid on the glass fibers. The total LOI of the
glass fibers
was 0.83%.
Preparation of Examule 11 of Table 2
In Example 11, components of Table 9 were mixed to prepare the size
composition. The size composition was applied to 10 m Advantex glass fibers
to
achieve a strand Loss-On-Ignition (LOI) of 0.22% on the glass fibers. A
conventional loss
on ignition (LOI) method, ASTM 2854, was used to determine how much of the
applied
size composition was on the glass fibers. The glass fibers were then collected
into a strand
and chopped wet in-line by a chopper into segments of approximately 4 mm in
length.
TABLE 9
Component Amount of Component
Baybond XP-2602 143.23 kg as received in 800 L of deionized water
AX-I 100 Silane 34.81 kg as received h drol zed in 1000 L of deionized water
Deionized Water to 2200 L
(a) non-ionic polyurethane dispersion (Bayer Corp.)
(b) y-aminopropyltriethoxys i lane (GE Silicones)
The chopped segments were then conveyed to a pelletizer where the binder was
sprayed onto the chopped segments as they passed through the entrance chamber
of the
pelletizer. The binder composition was prepared by dispersing 13.43 kg of
ZeMac 400
EMA powder in 47.85 kg of deionized water and dissolving the powder by heating
the
water to approximately 95 C under agitation. 'After the ZeMac 400 EMA powder
was
dissolved, the solution was further diluted by adding 19 kg of deionized
water. 18.61 kg
of Glascol C95, a solution of a low molecular weight acrylic acid homopolymer
partially
neutralized by ammonia which is available from Ciba, was then added with
agitation at a
temperature lower than 60 C. The densified glass pellets were then conveyed to
a
conveyor-type or fluidized bed oven far drying. The binder composition was
applied to
achieve a strand LOI of 0.50% solid on the glass fibers. The total LOI of the
glass fibers
was 0.72%.
Comparative Testin~
The glass fiber pellets were compounded with molding pellets of polyamide 6
(Ultramid B3 from BASF with or witliout added Calcium Stearate) using a twin
screw co-
rotating intermeshing extruder (ZSK30 Werner-Pfleiderer (Coperion)) while
feeding the
28
CA 02635451 2008-06-26
WO 2007/078900 PCT/US2006/048130
pellets into the melt of the second port of the extruder. The fiber/resin
mixture was then
degassed and formed into compounded glass/resin pellets.
Next, the compounded glass/resin pellets were dried for 12 hours at 95 C by a
molecular sieve circulating hot air dryer. After drying, the pellets were
injection-molded
by an injection-molding machine (Den:iag DC80 or Arburg 420C) into Axxicon ISO
molds
to form standardized composite specin:iens. The molded specimens were placed
in
metallic vessels containing silicagel to keep them dry.
A portion of the composite specimens were cross-mixed between different
autoclaves to simulate the same aging for each composite specimen. During
hydrolysis
(hydro-aging) testing, the composites were fully immersed in an ethylene
glycol/water
(50/50) mixture and placed under pressure for 500 hours at a temperature of
120 C.
When each of the hydrolysis tests were completed, the vessel was cooled to
room
temperature before the tensile strengths and the Charpy un-notched impact
strengths were
determined. Dry-as-Molded (DaM) properties such as Tensile Strength, Charpy un-
notched impact strength, and Izod un-inotched and notched impact strength were
measured
on the dry, molded specimens. The results are set forth below.
The tests to determine tensile strength were conducted according to the
procedures
set forth in ISO 527-4/1B/10. The Charpy un-notched impact strength was
measured
according to the procedures set forth in ISO 179-1/1 Eu. The Izod un-notched
and notched
(2 mm notch) were measured according to ISO 180/A. The lab conditions were set
as
described in ISO 291.
Eacamnle 1: Loss of Properties of Polyacid-Based Glass Fibers in the Presence
of
Calcium Stearate
Table 10
Dry-as-Molded Properties of (Polyamide 6) vs. (Polyamide 6+ 0.35 fo Calcium
Steara.te Com osite Pieces
Glass Tensile Charpy Un- Izod Un- Izod
Content Strength notched notched. Notched
(% (MPa) (KJ/m2) KJ/mz) KJ/m2
Example ~ ~ f polyamide 6 30.1 178.4 95.1 82.6 13.6
Table 2
Polyamide 6
+ Calcium 30.0 175.8 90.5 78.8 13.6
Stearate
Delta( ~) -1.4% -4.8% -4.6% 0%
Example 2v of polyamide 6 30.1 176.8 103.5 87.8 14.2
Table 2
29
CA 02635451 2008-06-26
WO 2007/078900 PCT/US2006/048130
Polyamide 6
+ Calciuin 29.9 150.2 64.3 55.4 9.3
Stearate
Delta -15% -37.8 l0 -36.9% -34.7%
Example 3~ f Polyamide 6 29.6 176.2 99.7 90.1 14.5
Table 2
Polyamide 6
+ Calcium 29.2 171.0 82.8 72.7 12.8
Stearate
Delta -3.0% -17% -19.3% -11.6%
blocked polyurethane dispersion (]3axenden Chemicals)/ y-
aminopropyltriethoxysilane (GE
Silicones)
(6) blocked polyurethane dispersion (1Baxenden Chemicals)/y-
aminopropyltriethoxysilane (GE
Silicones) + polyacrylic acid (low polyacid-content)
( ) blocked polyurethane dispersion (]Baxenden Chemicals)/ y-
aminopropyltriethoxysi lane (GE
Silicones) + EMA (low polyacid content)
(a) value of property in (Polyamide 6 + calcium stearate) minus the value of
the property in
Polyamide 6/value of property in Polyamide 6
In Table 10, the general negative effects of the presence of calcium stearate
on
physical properties such as Tensile strength, Charpy un-notched and Izod
notched and un-t
notched impact strengths can be seen.. As shown in Table 10, when calcium
stearate is
present in the Polyamide 6, such as iri Examples 2 and 3, the Tensile, Charpy
and Izod
properties are significantly lower conipared to when there is no calcium
stearate present in
the polyamide. For example, in Exar.nples 2 and 3, the Izod un-notched impact
strengths
decreased from 87.8 KJ/m2 to 55:4 KJ/m2 (Example 2) and from 90.1 KJ/m2 to
72.7
KJ/mz* (Example 3) when calcium stearate was present. Similar decreases in the
Charpy
un-notched and Izod notched impact strengths were obtained when calcium
stearate was
present. As indicated by the strongly negative values of the delta for
Examples 2 and 3,
the losses were most severe in Example 2. Although the negative delta values
for
Example 3 were less severe, they are still significant when compared to the
smaller
negative delta values obtained for the polyacid-free composite of Example 1.
It is to be
noted that although Examples 2 and 3 contain a polyacid, the amount of
polyacid moieties
present in the two-part sizing formulation is not sufficient to overcome the
negative effects
of the di-cations present in the Polyamide 6.
It can also be seen from Table 10 that when no calcium stearate is present in
the
Polyamide 6, Examples 2 and 3, which contain a polyacid, perform as well as,
and
sometimes better than, the polyacid=free composite of Example 1. For example,
the results
of the Charpy impact strength in Examples 2 and 3 and the Izod un-notched and
notched
CA 02635451 2008-06-26
WO 2007/078900 PCT/US2006/048130
impact tests were superior to the corresponding results obtained for Example
1. The
improved performance of Examples 2 and 3 in Polyamide 6 without calcium
stearate with
respect to the Izod un-notched impact strength can be seen graphically in FIG:
1, as well as
the strong losses in the same properties when calcium stearate is present in
the Polyamide
6.
Example 2: Influence of Polyacid Level on Performance of Calcium Stearate
Containing Polyamide 6 Composites
TABLE 11
Dry-as-Molded Properties of (Polyamide 6) vs. (Polyamide 6+ 0.35 0o Calcium
Stearate Com osite Pieces
Glass Tensile Charpy Un- Izod Un- Izod
Content Strength notched notched. Notched
(%') MPa (KJ/m2) (KJ/m2) (KJ/mz
Example 4 pol amide 6 29.7 176.6 98.4 77.3 14.4
of Table 2( )
y
Polyamide 6
+ Calcium 30.0 180.6 90.1 78.1 14.6
Stearate
Delta' +2.3% -8.4% 1.0% +1.2%
Example 5 pol amide 6 29.8 181.4 98.4 87.0 14.8
ofTable 2(b) y
Polyamide 6
+ Calcium 30.0 179.4 82 72.1 14
Stearate
Delta -1.1 fo -16.7% -17.1% -5.3%
Example 6 pol amide 6 29.9 183.4 101.3 90.4 15.8
ofTable 2()
y
Polyainide 6
+ Calcium 29.9 183.1 94.8 82.2 15.1
Stearate
Delta -0.2% -6.4% -9.0% -4.3%
Example a
of Table 2 ) Polyamide 6 29.7 183.9 104.0 92.9 15.2
Polyamide 6
+ Calcium 29.4 183.9 94.7 84.1 14.7
Stearate
Delta 0% - 8.9 % - 9.5 % =3.7%
(a) y-aminopropyltriethoxysilane (GE Silicones) + blocked isocyanate
polyurethane dispersion
(Bayer Corp.)
(b) y-aminopropyltriethoxysilane (GE's Silicones) + blocked isocyanate
polyurethane dispersion
(Bayer Corp.) + solution of low Mw acrylic acid homopolymer (Ciba) (low
polyacid content)
tc1 y-aminopropyltriethoxysilane (GE Silicones) + blocked isocyanate
polyurethane dispersion
(Bayer Corp.) + solution of low Mw acrylic acid homopolymer (Ciba) (medium
polyacid
content)
(d) y-aminopropyltriethoxysilane (GE Silicones) + blocked isocyanate
polyurethane dispersion
(Bayer Corp.) + solution of low Mw acrylic acid homopolymer (Ciba) (high
polyacid content)
(') value of property in (Polyamide 6 + calcium stearate) minus the value of
the property in
Polyamide 6/value of property in Po:lyamide 6
31
CA 02635451 2008-06-26
WO 2007/078900 PCT/US2006/048130
- TABLE 12
Properties of (Polyamide 6) vs. (Polyamide 6+ 0.35% Calcium Stearate)
Composite
Pieces After A in in Water/Glycol Mixture for 500 Hours at 120 C
Glass Tensile Charpy Un-
Content Strength notched
(%) (MPa) KJ/m2
Example 4 of Table 2~ ) Pol amide 6 29.7 69.4 76.6
Polyamide 6+ Calcium 30.0 68.4 75.8
Stearate
DeltEi' -1.3% -1%
Exam le 5 of Table 2(b! Polyamide 6 29.8 81.0 80.1
Polyamide 6 + Calcium 30.0 66.5 65.4
Stearate
Del1;a - 17.8 % - 18.3 %
Example 6 of Table 2~ ) Pol amide 6 29.9 81.7 83.4
Polyamide 6+ Calcium 29 9 78 5 82.5
Stearate
Delta - 3.9 % - 1.1 %
Example 7 of Table 2(d) Pol am- ide 6 29.7 79.5 87.4
Polyamide 6 + Calcium 29.4 82.3 85.9
Stearate
Delta + 3.5 % - 1.8 Jo
(e) y-aminopropyltriethoxysilane (GE Silicones)/blocked isocyanate
polyurethane dispersion - -
(Bayer Corp.)
(b) y-aminopropyltriethoxysilane (GE Silicones) + blocked isocyanate
polyurethane dispersion
(Bayer Corp.) + solution of low Mw acrylic acid homopolymer (Ciba) (low
polyacid content)
( ) y-aminopropyltriethoxysilane (GE Silicones) + blocked isocyanate
polyurethane dispersion
(Bayer Corp.) + solution of low Mw acrylic acid homopolymer (Ciba) (medium
polyacid
content)
(d) y-aminopropyltriethoxysilane (GE Silicones) + blocked isocyanate
polyurethane dispersion
(Bayer Corp.) + solution of low Mw acrylic acid homopolymer (Ciba) (high
polyacid content)
(e) value of property in (Polyamide 6 + calcium stearate) minus the value of
the property in
Polyamide 6/value of property in Polyamide 6
It can be seen from the data obtained and illustrated in Tables 11 and 12 that
the
negative effects caused by the presence of calcium stearate in the Polyamide 6
on the
Tensile strength, the Charpy un-notched, and Izod notched and un-notched
impact
strengths are reduced by the presence of a suitable amount of polyacid in the
two part
inventive sizing composition. It is to be noted that Examples 5, 6, and 7
differ only in the
amount of polyacid content, and increElse in polyacid content from Example 5
(low acid
content) to Example 6 (medium acid content) to Example 7 (high acid content)
respectively. Referring to Table 11, oiie example of the reduction of the
negative effects=
caused by the presence of calcium stearate in the polyamide is shown by
Example 6.
Although the Charpy un-notched impact strength decreased from 101.3 KJ/m2 to
94.8
KJ/m2 when calcium stearate was present, this data demonstrates an improvement
over the
Charpy un-notched impact strength of the polyacid-free composite of Example 4
in which
32
CA 02635451 2008-06-26
WO 2007/078900 PCT/US2006/048130
the Charpy un-notched impact strength, decreased from 98.4 KJ/m2 to 90.1 KJ/m2
when
calcium stearate was present. By including a suitable amount of polyacid in
the inventive
two-part sizing formulation, the Charpy un-notched impact strength was higher
(94.8
KJ/m2) than the Charpy impact strength of the polyacid-free composite of
Example 4 (90.1
KJ/m2) in the presence of calcium stearate. The reduction in the negative
effects of the
presence calcium stearate is also demonstrated by Example 7, in which the
Charpy un-
notched impact strength decreased frorn 104.0 KJ/ma to 94.7 KJ/m2 in the
presence of
calcium stearate. The data obtained from Examples 6 and 7 show a small
difference in the
Charpy un-notched impact strength (94.8 KJ/ma for Example 6 and 94.7 KJ/m2 for
Example 7), indicating that there may be a plateau for the number of polyacid
moieties
needed to achieve improved dry-as-molded mechanical properties in the
composite
product. Similar results demonstratinl; a decrease in the negative effects of
calcium
stearate in the polyacid were obtained with respect to the Tensile strength
and Izod .
notched and.un-notched impact strengths.
With respect to Example 5, it can be seen that the relatively low level
polyacid-
containing composite resulted in better mechanical performance than the
polyacid-free,
polyurethane-based composite formed by the two-part sizing formulation of
Example 4.
This is particularly noticeable for the dry-as-molded (DaM) Izod un-notched
properties
(Table 11) and for the Tensile strengtli properties after hydro-aging when no
calcium
stearate is present (Table 12). However, when calcium stearate was present in
the
Polyamide 6, the polyacid-containing composite of Example 5 showed a loss of
mechanical properties. For example, the polyacid-containing composite of
Example 5
demonstrated a loss of dry-as-molded Charpy and Izod (notched and un-notched)
impact
strengths (Table 11) and Tensile strength and Charpy un-notched impact
strength after
hydro-aging (Table 12) compared to these same mechanical properties in the
absence of
calcium stearate. These losses of mechanical properties are clearly
highlighted by the
strongly negative delta values associated with the separate properties.
Although not
wishing to be bound by theory, it is bi-Ilieved that the amount of polyacid
present in the
binder of the two-part sizing formulation forming the composite product of
Example 5 is
not high enough to compensate for the carboxylic acid moieties consumed in the
negative
interaction with the calcium stearate, and as a result, there is a decrease in
the mechanical
properties tested. On the other hand, as discussed above, Examples 6 and 7
contain a
33
CA 02635451 2008-06-26
WO 2007/078900 PCT/US2006/048130
higher polyacid content than Example 5 and are less sensitive to the presence
of calcium
stearate, as indicated by the smaller negative delta values for the dry-as-
molded or
properties after hydro-aging. In Examples 6 and 7, the amount of polyacid
present in the
two-part sizing formulatiozi is high enough to compensate for the negative
interaction with
the calcium stearate. This excess in polyacid moieties result in improved and
superior
mechanical properties, as can be seen in FIGS. 2 and 3.
Examnle 3: Performance of High vs. Low Polyacid Content Glass Fiber in Calcium
Stearate Containing Polyamide 6 Composites
TABLE 13
Dry-as-Molded Properties of (Pcilyamide 6) vs. (Polyamide 6+ 0.35 lo Calcium
Stearate Composite Pieces
Glass Tensile Charpy Un- Izod Un- Izod
Content Strength notched notched. Notched
(% (MPa) (KJ/mz) KJ/mz) (KJ/m2
Example I Pol ainide 6 29.5 180.4 95.7 83.1 13.0
of Table 2(a)
y
Polyamide 6
+ Calcium 28.9 179.4 88.4 75.4 11.9
Stearate
Delta(d) -0.6% -7.6% -9.3% -8.6%
Example 8~ Polyamide 6 29.3 180.5 104.9 92.6 14.4
of Table 2<
Polyamide 6
+ Calcium 29.9 180.8 91.2 80.6 13.2
Stearate
Delta +0.2% -13.1% -12.9% -8.4%
Example 9 Pol amide 6 29.4 186.8 109.0 95.0 15.3
of Table 2( )
y
Polyamide 6
+ Calcium 29.6 190.4 105.4 91.2 14.6
Stearate
Delta +1.9% -3.4% -4.0% -5.1%
y-aminopropyltriethoxysilane (GE Silicones)/ blocked polyurethane dispersion
(Baxenden
Chemicals)
(b) y-aminopropyltriethoxysiiane (GE Si.licones) + tetraethylenepentarnine
reacted with stearic
acid (AOC) + fluoroalkyl alcohol substituted polyethylene glycol (DuPont) +
solution of low Mw
acrylic acid homopolymer partially neutralized by ammonia (Ciba) + alternating
copolymer of
ethylene and maleic anhydride (Zeeland Chemicals) + polyurethane dispersion
(D.I.C.) + water-
soluble aliphatic diamine derived from a propylene oxide-capped
poly(ethyleneoxide) (Huntsman
Corp.) + mixture of oxiranes (EO-PO copolymers) (BASF) (low polyacid content)
(') y-aminopropyltriethoxysilane (GE Silicones) + blocked isocyanate
polyurethane dispersion
(Bayer Corp.) + solution of low Mw aciylic acid homopolymer (Ciba) +
alternating copolymer of
ethylene and maleic anhydride (Zeeland Chemicals) (high polyacid content)
(d) value of property in (Polyamide 6 + calcium stearate) minus the value of
the property in
Polyamide 6/value of property in Polyainide 6
34
CA 02635451 2008-06-26
WO 2007/078900 PCT/US2006/048130
Table 14
Properties of (Polyamide 6) vs. (Polyamide 6+ 0.35% Calcium Stearate)
Composite
Pieces After A in in Water/Gl col Mixture for 500 hours at 120 C
Glass Tensile Charpy Un-
Content Strength notched
(%) (MPa) (KJ/m:I)
Exam le I of Table 2c , Polyamide 6 29.5 56.2 63.7
Polyamide 6 + Calcium 28.9 55.6 57.6
Stearate
Delta( - 1.1 % - 9.5 %
Example 8 of Table 2( ) Pol ai'nide 6 29.3 82.2 88.4
Polyamide 6 + Calcium 29.9 68.9 60:7
Stearate
Delta -16.5% - 3 l .3 %
Example 9 of Table 2( ) Pol arnide 6 29.4 83.7 88.1
Polyamide 6 + Calcium 29.6 77.3 72.4
Stea.rate
Delta - 7.6 % - 17.8 oo
(a) y-am inopropyltriethoxysi lane (GE Silicones)/ blocked polyurethane
dispersion (Baxenden
Chemicals)
(b) y-aminopropyltriethoxysilane (GE Silicones) + tetraethylenepentamine
reacted with stearic
acid (AOC) + fluoroalkyl alcohol substituted polyethylene glycol (DuPont) +
solution of low Mw
acrylic acid homopolymer partially neui~ralized by amrnonia (Ciba) +
alternating copolymer of
ethylene and maleic anhydride (Zeeland Chemicals) + polyurethane dispersion
(D.I.C.) + water-
soluble aliphatic diamine derived from ~i propylene oxide-capped
poly(ethyleneoxide) (Huntsman
Corp.) + mixture of oxiranes (EO-PO copolymers) (BASF) (low polyacid content)
(c) y-aminopropyltriethoxysilane (GE Silicones) + blocked isocyanate
polyurethane dispersion
(Bayer Corp.) + solution of low Mw aciylic acid homopolymer (Ciba) +
alternating copolymer of
ethylene and maleic anhydride (Zeelancl Chemicals) (high polyacid content)
(d) value of property in (Polyamide 6+ calcium stearate) minus the value of
the property in
Polyamide 6/value of property in Polyainide 6
It can be seen from the data obtained and illustrated in Tables 13 and 14 that
the
negative effects caused by the presence of calcium stearate in the Polyamide 6
on the
Tensile strength, the Charpy un-notched, and Izod notched and un-notched
impact
strengths are reduced by the presence of a suitable amount of polyacid in the
two-part
inventive sizing composition. In the absence of calcium stearate, the polyacid-
based glass
fiber composite of Example 8 demonstrated an improvement in the dry-as-molded
Charpy
un-notched and Izod notched and un-notched impact properties (Table 13) and
Tensile
strength and Charpy un-notched impact strength after hydro-aging (Table 14)
versus the
polyacid-free, polyurethane-based coinmercial glass fiber of Example 1. Thus,
the
polyacid performed its expected function of improving the mechanical
properties when no
calcium stearate was present.
In the presence of calcium stearate, however, the level of polyacid present in
the
two-part sizing formulation of Example 8 was not enough to compensate for the
level of
CA 02635451 2008-06-26
WO 2007/078900 PCT/US2006/048130
calcium stearate present in the polyamide, and, as a result, the polyacid
moieties were
consumed by the negative interaction tvith the calcium stearate di-cation. The
low level of
polyacid present in Example 8 results i.n lower-than-expected performance (for
exarnple,
results that are well below the results that are observed with Polyamide 6
without calcium
stearate) of the dry-as-molded Charpy un-notched and Izod (notched and un-
notched)
impact strengths (Table 13) and the Tensile strength and Charpy un-notched
impact
strength after hydro-aging (Table 14). The high sensitivity to calcium
stearate of the low
polyacid level of Example 8 is also clearly highlighted by the high negative
delta value for
these properties. On the other hand, the composite product of Example 9
contained a
higher level of polyacid in the two-part sizing formulation. It can be seen
from Tables 13
and 14 that when calcium stearate was present in the Polyamide 6, the losses
induced by
the calcium stearate in the dry-as-molded Charpy un-notched and Izod (notched
and un-
notched) impact strengths (Table 13) and the Tensile strength and Charpy un-
notched
impact strength after hydro-aging (Table 14) of Example 9.were less severe
compared to
the composite product of Example 8. The improvement of the dry-as-molded
Charpy un-
notched impact strength of Example 9 is illustrated graphically in FIG. 4. The
excess of
polyacid present in the two-part sizing formulation resulted in a high
mechanical
performance of the composite pieces tested both before and after hydro-aging
both with
and without the presence of calcium stearate in the Polyamide 6. Such improved
mechanical properties demonstrated by the composite product of Example 9
confirms the
superior and unexpected properties of the two-part sizing formulation of the
present
invention.
Examnle 4: Influence of Calcium Sitearate Level Present in Polyamide 6 on the
Performance of Polyamide 6 Reinfo:rced by Polyacid-Based Fibers
Table 15
Dry-as-Molded Properties of (Polyamide 6) vs. Polyamide 6+ 0.14% Calcium
Stearate 1 and Polyamide 6+0.35% Calcium Stearate 2 Com osite Pieces
Glass Tensile Charpy Un- Izod
C:ontent Strength notched Notched
(%) (MPa) (KJ/m2) (KJ/m2)
Example I
of Table 2( ) Polyamide 6 30.3 185.9 96.8 13.3
Polyamide 6+ 30.4 187.5 97.0 13.8
Calcium Stearate (1)
Polyamide 6-+- 30.4 186.5 93.5 13.9
Calcium Stearate (2)
Delta(') +0.3 fo -3.4% +4.0%
36
CA 02635451 2008-06-26
WO 2007/078900 PCT/US2006/048130
Bxample 8
of Table 2(b) Polyamide 6 30.2 185.0 106.8 15.8
Polyamide 6+ 305 187.6 97.2 14.6
Calcium Stearate 1
Polyamide 6+ 30.3 186.0 87.6 13.8
Calciuin Stearate (2)
Delta + 0.5 % - 17.9 % - 12.8 %
Exainple 10 polyamide 6 30.6 184.7 103.2 15.8
of Table 2
Polyamide 6+ 30.4 186.5 103.1 15.4
Calcium Stearate (1
Polyamide 6 + 30.4 185.5 93.4 14.1
Calcium Stearate 2)
Delta +0.4% -9.4 fo -10.8%
Example ~~1, Polyamide 6 30.3 183.1 106.5 16.3
of Table 2
Polyamide 6 +
30.2 185.1 104.4 15.4
Calcium Stearate (1)
Polyamide 6 }
Calcium Stearate (2) 30.2 184.6 98.8 14.8
Delta + 0.8 % = 7.3 % - 9.4 %
(a) y-aminopropyltriethoxysilane (GE Silicones)/ blocked polyurethane
dispersion (Baxenden
Chemicals)
(b) y-aminopropyltriethoxysilane (GE Silicones) + tetraethylenepentamine
reacted with stearic
acid (AOC) + fluoroalkyl alcohol substituted polyethylene glycol (DuPont) +
solution of low Mw
acrylic acid homopolymer partially neutralized by ammonia (Ciba) + alternating
copolymer of
ethylene and maleic anhydride (Zeeland. Chemicals) + polyurethane dispersion
(D.I.C.) + water-
soluble aliphatic diamine derived from a propylene oxide-capped
poly(ethyleneoxide) (Huntsman
Corp.) + mixture of oxiranes (EO-PO copolymers) (BASF) (low polyacid content)
(c) y-aminopropyltriethoxysilane (GE Silicones) + non ionic polyurethane
dispersion (Bayer
Corp.) + alternating copolymer of ethylene and maleic anhydride (Zeeland
Chemicals)
(d) y-aminopropyltriethoxysilane (GE Si.licones) + non ionic polyurethane
dispersion (Bayer
Corp.) + solution of low Mw acrylic acid homopolymer partially neutralized by
ammonia +
alternating copolymer of ethylene and nialeic anhydride (Zeeland Chemicals)
(e) value of property in (Polyamide 6+calcium stearate(2) minus the value of
the property in
Polyamide 6/value of property in Polyainide 6
Another way to exemplify the results of the high polyacid content of glass
fibers
versus traditional polyacid-free or lomT polyacid-based *glass fibers is to
compare their
performance in polyamide formulations that contain increasing levels of
calcium stearate.
Table 15 shows the mechanical properties from Polyamide 6 composite pieces
based on
5, four different glass fibers that were produced on an industrial scale. For
each example,
data on the mechanical properties of the composites were collected for
Polyamide 6 (with
no calcium stearate), for Polyamide 6 containing 0.14% by weight vs. compound
of
calcium stearate (1), and for Polyamicie 6 containing 0.35% by weight vs.
compound of
calcium stearate (2). The level of polyacid (as LOI) increases from Example 1
to Example
37
CA 02635451 2008-06-26
WO 2007/078900 PCT/US2006/048130
8 to Example 10 to Example 11 respectively. The specific LOI for each example
is set
forth in Table 2.
It can be seen in Table 15, that when calcium stearate is not present, all of
the
polyacid-based products perform as well as (for example, tensile strength) or
better than
(for example, Charpy and Izod notchecl impact strength) the polyacid-free,
polyurethane-
based glass fiber of Example 1. At intermediate levels of calcium stearate,
the impact
properties of the low polyacid content candidate of Example 8 are markedly
affected. On
the other hand, the higher polyacid coritent composites of Examples 10 and 11
present
acceptable impact properties under intermediate levels of calcium stearate, as
well as in -
presence of even higher amount of calcium stearate. These results are
illustrated in FIG. 5,
which shows the relative performance of the glass fibers in the related Charpy
un-notched
property of DaM-tested polyamide 6 composites containing differing levels of
calcium
stearate. It is to be noted that the tensile strength properties are less
dependent on the level
of calcium stearate present in tha. compound..
The foregoing description of the specific embodiments will so fully reveal the
general nature of the invention that others can, by applying knowledge within
the skill of
the art (including the contents of the re:ferences cited herein), readily
modify and/or adapt
for various applications such specific E;mbodiments, without undue
experimentation,
without departing from the general concept of the present invention.
Therefore, such
adaptations and modifications are intended to be within the meaning and range
of
equivalents of the disclosed embodimp-nts, based on the teaching and guidance
presented
herein. It is to be understood that the phraseology or terminology herein is
for the purpose
of description and not of limitation, such that the terminology or phraseology
of the
present specification is to be interprete;d by the skilled artisan in light of
the teachings and
guidance presented herein, in combination with the knowledge of one of
ordinary skill in
the art.
The invention of this application has been described above both generically
and
with regard to specific embodiments. Although the invention has been set forth
in what is
believed to be the preferred embodiments, a wide variety of alternatives known
to those of
skill in the art can be selected within the generic disclosure. The invention
is not
otherwise limited, except for the recitation of the claims set forth below.
38