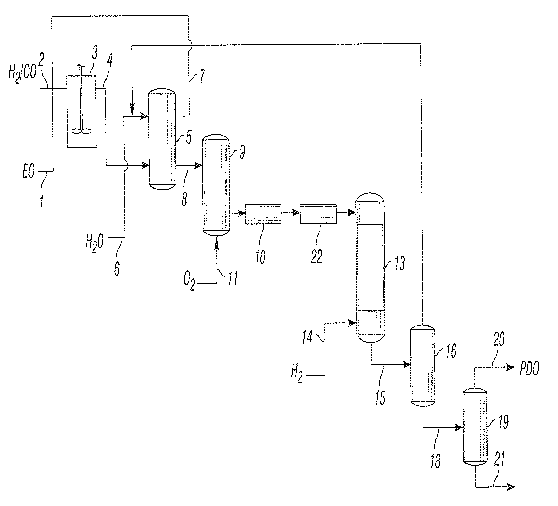Note: Descriptions are shown in the official language in which they were submitted.
CA 02641031 2008-07-30
WO 2007/090158 PCT/US2007/061384
A METHOD OF TREATING AN ALDEHYDE MIXTURE, USE OF THE
TREATED ALDEHYDE, AND AN ALCOHOL
Field of Invention
This invention relates to a method of treating an aldehyde mixture, use of the
treated aldehyde to prepare an alcohol and the alcohol.
B ackground of the Invention
Aldehydes are commonly prepared and hydrogenated into a corresponding alcohol.
A difficulty associated with the process is the oxidation of the aldehyde to
form a
carboxylic acid by-product. The presence of carboxylic acid, especially if
left
unneutralized, may have a negative effect on the performance of most
heterogeneous
hydrogenation catalysts. Additionally, the carboxylic acid may react with the
alcohol
formed during hydrogenation resulting in additional yield losses and
additional separation
costs. The carboxylic acid may cause corrosion of processing equipment,
especially when
present in process streams heated above ambient temperature. Typically, the
carboxylic
acid is partially neutralized prior to hydrogenation. For example, U.S.
2004/0087819
discloses neutralization of an aqueous 3-hydroxypropionaldehyde solution prior
to
hydrogenation. However, partial neutralization through the addition of a base,
typically an
alkali base, is problematic due to the potential degradation of the aldehyde
resulting from
inefficient mixing. The aldehyde, in the presence of excess base occurring
from inefficient
mixing, can combine to form byproducts such as acetals and/or aldols which can
undergo
further condensation to yield polymeric heavy ends. Some of the acids formed
are known
to be hydroxyacids, where neutralization alone may not fully eliminate the
negative impact
on the hydrogenation catalyst. Additionally, the resulting alkali metal salt
formed during
partial neutralization imparts an ash component which substantially reduces
the market
value of the heavy ends co-product, and the alkali metal salt fornied can foul
equipment
downstream such as the reboilers of downstream distillation columns and heat
exchangers.
1,3-propanediol is an industrially important chemical. 1,3-propanediol is used
as a
monomer unit to form polymers such as poly (trimethylene terphthalate) that
are used in
the production of carpets and textiles. 1,3-propanediol is also useful as an
engine coolant,
particularly in cooling systems that require coolants having low conductivity
and low
corrosivity.
1,3-propanediol may be prepared in a two-step process in which ethylene oxide
is
first hydroformylated in an organic solution in the presence of a metal
catalyst such as a
CA 02641031 2008-07-30
WO 2007/090158 PCT/US2007/061384
2
cobalt or rhodium carbonyl to form 3-hydroxypropionaldehyde. The 3-
hydroxypropionaldehyde intermediate is water extracted from the organic phase
under
pressure and the metal catalyst is recycled to the hydroformylation reaction
in the organic
phase. In a subsequent step, the aqueous 3-hydroxypropionaldehyde is
hydrogenated to
1,3-propanediol.
Ideally, the aqueous 3-hydroxypropionaldehyde extract could be routed directly
to
the hydrogenation reactor. However, as discussed above, the carboxylic acid
formed as a
byproduct during hydroformylation may have a negative effect on the
performance of most
heterogeneous hydrogenation catalysts. Additionally, the small amount of metal
from the
hydroformylation catalyst that typically leaches into the water phase during
extraction of 3-
hydroxypropionaldehyde also may have a negative effect on the performance of
most
heterogeneous hydrogenation catalysts.
U.S. 2004/0087819 discloses removing a hydroformylation catalyst from an
aqueous 3-hydroxypropionaldehyde solution by utilizing a cation exchange
resin. As
discussed hereinbefore, the reference also discloses neutralization of the
aqueous 3-
hydroxypropionaldehyde solution. The neutralization occurs after contact with
the cation
exchange resin and before hydrogenation.
It goes without saying that it is highly desirable to improve the process for
preparing an alcohol from an aldehyde.
Summary of the Invention
The present invention provides a method of treating an aldehyde mixture
comprising a carboxylic acid and a metal cation, which method comprises:
- contacting the aldehyde mixture with a basic separating medium, and
- subsequently or simultaneously contacting with an acidic separating medium.
In an
embodiment of the invention, the aldehyde comprises 3-hydroxypropionaldehyde,
the
carboxylic acid comprises 3-hydroxypropionic acid, the metal cation coniprises
a Group
VIII metal cation, and the method additionally comprises controlling the pH of
the mixture
at a value of at most 6, as measured at a temperature of operation.
The present invention also provides a process for preparing 1,3-propanediol,
which
process coniprises hydrogenating a treated aldehyde mixture which has been
obtained by
the treatment method in accordance with this invention.
The present invention also provides a 1,3-propanediol product.
CA 02641031 2008-07-30
WO 2007/090158 PCT/US2007/061384
3
Brief Description of the Drawings
Figure 1 is a schematic illustrating a process for preparing 1,3-propanediol
by
hydroformylation of ethylene oxide, carbon monoxide and hydrogen to form 3-
hydroxypropionaldehyde followed by hydrogenation of the 3-
hydroxypropionaldehyde to
1,3-propanediol which process incorporates the treatment method of the
invention.
Detailed Description of the Invention
In accordance with the present invention, processes for producing an alcohol
from
an aldehyde may be improved by contacting an aldehyde mixture with a basic
separating
medium and then further contacting with an acidic separating medium. In
particular,
contacting the aldehyde mixture with a basic separating medium removes at
least part of
the carboxylic acid present in the mixture such that it may be recovered as a
co-product,
eliminates the attendant production of an ash component and fouling associated
with
neutralization of the carboxylic acid, and reduces the subsequent reaction of
the carboxylic
acid with the alcohol product, as described hereinbefore. Also, contacting the
aldehyde
mixture with a basic separating medium prior to contacting with an acidic
separating
medium improves the removal of metal cations present in the mixture.
The aldehyde mixture may be any aldehyde containing mixture. The aldehyde may
be any aldehyde and may be a alkyl or aryl aldehyde, hydroxyaldehyde,
ketoaldehyde
haloaldehyde, or other substituted aldehyde. Preferably, the aldehyde
comprises at most 12
carbon atoms, more preferably at most 8 carbon atoms, and most preferably at
most 4
carbon atoms. The aldehyde preferably comprises carbon atoms in the range of
from 2 to
10, more preferably in the range of from 2 to 4. Preferably the aldehyde
comprises 3
carbon atoms, in particular the aldehyde may comprise 3-
hydroxypropionaldehyde.
Preferably, the aldehyde mixture may be obtained from the aqueous extraction
of a
hydroformylation product mix. The term "hydroformylation product mix", as used
herein,
is a mixture comprising an aldehyde, a hydroformylation catalyst and a
carboxylic acid.
The hydroformylation product mix may additionally comprise a reaction diluent
or
"solvent", residual reactants coniprising carbon monoxide, hydrogen and an
olefin oxide,
and minor amounts of other by-products.
The aldehyde mixture may contain the aldehyde in a quantity in the range of
from 1
to 99 wt. %, preferably in the range of from 10 to 80 wt. %, more preferably
in the range of
from 15 to 60 wt. %, and most preferably in the range of from 20 to 40 wt. %,
relative to
the total weight of the aldehyde mixture.
CA 02641031 2008-07-30
WO 2007/090158 PCT/US2007/061384
4
The carboxylic acid may be any carboxylic acid, preferably comprising at most
12
carbon atoms, more preferably at most 8 carbon atoms, and most preferably at
most 4
carbon atoms. The carboxylic acid preferably comprises carbon atoms in the
range of from
1 to 10, more preferably in the range of from 1 to 4. The carboxylic acid may
comprise
one or more carboxylic acids. Preferably, the carboxylic acid comprises the
oxidized form
of the aldehyde, more preferably, the carboxylic acid comprises a
hydroxycarboxylic acid,
and most preferably the carboxylic acid comprises 3-hydroxypropionic acid. The
carboxylic acid may comprise acetic and formic acid which may be present with
3-
hydroxypropionic acid. The aldehyde mixture may contain one or more carboxylic
acids in
a total quantity in the range of from 0.1 to 5 wt. %; typically in the range
of from 0.03 to
3.5 wt. %, more typically in the range of from 0.06 to 1.5 wt. %, and most
typically in the
range of from 0.1 to 0.8 wt. %, relative to the total weight of the aldehyde
mixture.
The metal cation may be any metal cation, preferably the metal cation
comprises
one or more of a Group IB through Group VIII metal cation (as defined in the
Periodic
Table of Elements in the "CRC Handbook of Chemistry and Physics", 69ffi ed.
(CRC Press
Inc. 1988)), more preferably one or more of a Group VIII metal cation,
preferably one or
more of cobalt, ruthenium, rhodium, palladium, platinum, osmium, and iridium,
most
preferably rhodium, cobalt, iridium and ruthenium, and in particular cobalt
and/or rhodium
cations. The metal cation may be contained in one or more metal compounds,
complexes
or species. The aldehyde mixture may contain the metal cation in a quantity of
at most
0.03, typically at most 0.02, more typically.at most 0.01, most typically at
most 0.002, in
particular at most 0.001 molar equivalents of metal cation per liter of
aldehyde mixture.
The aldehyde mixture may contain the metal cation in a quantity of at least
0.000001, or at
least 0.00001, or at least 0.0001 molar equivalents of metal cation per liter
of aldehyde
mixture.
In an embodinlent, the aldehyde mixture may be dissolved in one or more liquid
diluents such as water, alcohols, diols, ketones, esters, and glycol ethers.
In particular, the
diluent comprises water which forms an aqueous solution. The aqueous aldehyde
mixture
may be any aqueous solution containing an amount of dissolved aldehyde,
carboxylic acid
and metal cation. Preferably, the aqueous aldehyde mixture may contain from 4
to 60 wt.
% aldehyde, and more typically from 20 to 40 wt. % aldehyde, relative to the
total weight
of the aqueous aldehyde mixture. Preferably, the aqueous aldehyde mixture may
contain
amounts of carboxylic acid and metal cations as discussed hereinbefore.
CA 02641031 2008-07-30
WO 2007/090158 PCT/US2007/061384
The aldehyde mixture may be contacted with a basic separating medium which at
least partially separates the carboxylic acid from the aldehyde mixture. The
aldehyde
mixture resulting from the contacting with the basic separating medium may be
referred to
as a first aldehyde containing effluent. The basic separating medium may be in
any
physical form such as a liquid, or preferably a solid.
In an embodiment, the basic separating medium may comprise a basic, anion
exchange resin. The ion exchange resins which may be used in the present
method may
have any physical structure. Preferably, the ion exchange resins used in the
present method
may have a gel type (microporous) or a macroreticular type (macroporous)
structure. The
major chemical component of the resin may be based on polyphenol, polystyrene,
polyacrylic, or polyvinylpyridine and is typically crosslinked with
divinylbenzene.
Reference may be made, for example, to Kirk-Othmer's Encyclopedia of
Cl2enaical
Technology, 4b Ed., Vol. 14, 1995, pages 737-783.
The basic, anion exchange resin may comprise a weak base anion exchange resin.
In particular, the weakly basic, anion exchange resin may have a pKa of less
than 13, or a
pKb of greater than 1. Weak base anion exchange resins are generally defined
as those
which cannot split a neutral salt such as NaCI (sodium chloride), unlike
strong base anion
exchange resins which can. Preferably, the weak base anion exchange resin may
be an
amine anion exchange resin where the amine is a primary, secondary or tertiary
amine, and
more preferably a tertiary amine anion exchange resin, and most preferably a
dimethylamino styrene divinylbenzene anion exchange resin. Commercially
available
tertiary amine styrene divinylbenzene anion exchange resins useful in the
method of the
present invention include AMBERLYSTTM A21 tertiary amine styrene
divinylbenzene
anion exchange resin, available from Rohm & Haas Company, 5000 Richmond
Street,
Philadelphia, Pennsylvania 19137, USA; and DOWEXTM M-43 tertiary amine styrene
divinylbenzene anion exchange resin, available from the Dow Chemical Conipany,
Liquid
Separations Group, P.O. Box 1206, Midland, Michigan 48641, USA.
The basic, anion exchange resin may comprise a strong base anion exchange
resin.
The strong base anion exchange resin may be buffered, preferably a buffered
quaternary
ammonium anion exchange resin, more preferably a quaternary ammonium anion
exchange resin buffered utilizing a mono or dibasic phosphate or carbonate,
and inost
preferably a quaternary ammonium anion exchange resin buffered with dibasic
phosphate.
CA 02641031 2008-07-30
WO 2007/090158 PCT/US2007/061384
6
The basic, anion exchange resin may also be incorporated into a mixed resin
bed
such as Rohm & Haas's STRATABEDTM (mixture of weak base anion exchange resin
and
strong base anion exchange resin), or in a lesser preferred embodiment, a
MONOBEDTM
(strong base anion exchange resin and a strong acid cation exchange resin).
In an alternative embodiment, the basic separating medium may comprise a metal
oxide. Preferably, the metal oxide may be one or more of activated alumina,
titania,
zirconia, chromia and mixtures thereof, and more preferably the metal oxide
comprises
activated alumina. Without wishing to be bound by theoiy, it is believed that
the metal
oxide has basic sites, especially in contact with an aqueous mixture, which
can ion
exchange acidic species when brought into contact with the metal oxide.
Commercially
available metal oxides useful in the method of the present invention include
Alcoa F200 or
LDS aluminas.
The aldehyde mixture may be contacted with the basic separating medium in any
manner sufficient to bring the carboxylic acid in the aldehyde mixture into
contact with the
basic separating medium preferably while minimizing any degradation of the
aldehyde.
The aldehyde mixture and the basic separating medium may be contacted in a
vessel such
as a stirred niixing tank, a HIGGINS LOOPTM, a carousel-type arrangement, an
alternating
dual bed-type arrangement, by flow of the mixture through a fixed bed of basic
separating
medium, or by passing the mixture through a chromatography column containing
the basic
separating medium. Preferably, the aldehyde mixture and the basic separating
medium
may be contacted in a HIGGINS LOOPTM or other moving bed arrangements, a
carousel-
type arrangement, an alternating dual bed-type arrangement, or other fixed-bed
arrangements. Reference may be made, for example, to Perry's Cherfazcal
Eiagineers'
Handbook, 6`i' Ed., 1984, pages 19-40 to 19-45. The HIGGINS LOOPTM is an
example of
a continuous, countercurrent, exchange column loop system and comprises a
closed loop
having an ion exchange/adsorption zone, a rinsing zone, a regenerating zone,
and a pulsing
zone. The carousel-type arrangement may involve the placement of a number of
separating
medium-containing columns on a carousel, or use of valves to switch feeds in a
prescribed
manner between a number of fixed columns. When the basic separating medium
comprises a nletal oxide, the carousel-type arrangement is preferred.
The aldehyde mixture may be contacted with the basic separating medium at a
temperature which minimizes the degradation of the aldehyde when in contact
with the
CA 02641031 2008-07-30
WO 2007/090158 PCT/US2007/061384
7
basic separating medium, preferably at a temperature of from 5 to 45 C, and
more
preferably at a temperature of from 15 to 25 C.
Preferably, the aldehyde mixture may be contacted with the basic separating
medium while controlling the pH of the aldehyde mixture at a value of at most
6, more
preferably at most 5.5, and most preferably at most 5. By controlling the pH
of the
aldehyde mixture, any degradation of the aldehyde may be minimized.
Preferably, only 90
to 98% of the oliginal carboxylic acid is removed by the basic separating
medium, leaving
2 to 10% of the original acid to maintain pH in the range described
hereinbefore.
Depending on the initial acid concentration, this may correspond to a
concentration of
unneutralized acid of between about 1 x 10-6 and 2 x 10-3 molar equivalents of
carboxylic
acid per liter of aldehyde mixture. The desired amount of residual carboxylic
acid may be
readily achieved via a control system based upon direct monitoring of pH.
Unless otherwise stated, the pH values are deemed to be directly measured at
the
temperature of operation, using a conventional standard pH probe immersed in
the
aldehyde mixture.
The amount of the carboxylic acid removed from the aldehyde mixture and the
resulting pH may be dependent on several factors. In particular, the amount of
acid
removal and pH may be dependent on the separating capacity of the basic
separating
medium, the amount of basic separating medium employed in the separation step
relative
to the amount of aldehyde mixture present for contact with the basic
separating medium,
the amount of carboxylic acid present in the aldehyde mixture, the apparatus
used to effect
contact between the aldehyde mixture and the basic separating medium, and the
contact
time for the process. These factors, in particular the time duration of the
contacting step,
may be adjusted to control the acid removal and the pH of the aldehyde
mixture.
For a batch-type process, a sufficient amount of basic separating medium by
weight
of medium to weight of aldehyde mixture may be from 0.1 to 25 weight percent
and
preferably from 1 to 10 weight percent, relative to the weight of the aldehyde
mixture. For
a continuous process, the aldehyde mixture may be passed through the basic
separating
medium at a volume hourly space velocity (volume of aldehyde mixture feed per
volume
of basic separating medium per hour) of from 0.1 h"1 to 40 h-1, preferably
from 0.5 h"1 to 20
h-1, and more preferably from 1 h-1 to 10 h"1.
Preferably, the contact between the aldehyde mixture which has had the
carboxylic
acid removed to pH values as discussed hereinbefore and unneutralized basic
separating
CA 02641031 2008-07-30
WO 2007/090158 PCT/US2007/061384
8
medium is avoided. This may be accomplished by adjusting the factors, as
discussed
hereinbefore, preferably by additionally using continuous ion exchange, in
particular short
beds with high dispersive mixing, or backmixed stages with frequent
regeneration. It is
preferred to apply continuous ion exchange where the total ion exchange bed
volume is
broken up into a series of "N" equivalent stages either by use of discrete
vessels (carousel-
type) or via periodically pulsing the bed through the ion exchange zone
(HIGGINSTM
LOOP). The volume hourly space velocity for a single stage is thus "N"-times
higher than
for a single larger bed. The higher volume hourly space velocity results in
mixing within
stage due to axial dispersion so that high pH regions in the bed, due to
locally complete
acid removal, are avoided. As a result, degradation of the aldehyde is
minimized and the
method operates with a more constant and optimal outlet pH than with a larger
bed.
The first aldehyde containing effluent may contain at least 70 percent of the
aldehyde present in the aldehyde mixture, more preferably at least 80 percent,
and most
preferably at least 90 percent.
The first aldehyde containing effluent may contain a smaller quantity of the
carboxylic acid than the aldehyde mixture, and preferably the first aldehyde
containing
effluent may contain at most 20 percent of the carboxylic acid present in the
aldehyde
mixture, more preferably at most 10 percent, and most preferably at most 5
percent. The
first aldehyde containing effluent may contain at least 1 percent of the
carboxylic acid
present in the aldehyde mixture, more preferably at least 1.5 percent, and
most preferably
at least 2 percent. The pH of the first aldehyde containing effluent may be at
most 6,
preferably at most 5.5, and more preferably at most 5.
The amount of metal cations in the first aldehyde containing effluent may be
the
same as the amount of metal cations in the aldehyde mixture since the metal
cations may
not be removed in any significant quantity by the basic separating medium.
After the separating capacity has diminished, the basic separating medium may
be
subjected to a base treatment to regenerate the basic properties of the
separating medium.
Preferably, the base treatment may be a base wash if the basic separating
medium is a
solid. Prior to the base wash, the basic separating medium may be subjected to
a water
wash. The basic separating medium niay be contacted with the base wash for a
sufficient
time and in a sufficient concentration to regenerate the basic properties of
the separating
medium. The base wash may have a pH above that of the pKb of the basic
separating
medium to most fully regenerate the medium. The base wash will preferably have
a pH of
CA 02641031 2008-07-30
WO 2007/090158 PCT/US2007/061384
9
8 or above, more preferably a pH of 10 or above. The base wash is preferably a
potassium
hydroxide solution, more preferably a 4 wt. % potassium hydroxide solution.
Other bases,
however, may be utilized as the base wash, including, but not limited to,
sodiuni
hydroxide, ammonium hydroxide, or other metal hydroxides. The basic separating
medium may be contacted with the base wash at a temperature of from 5 to 45
C.
The first aldehyde containing effluent may be subsequently or simultaneously
contacted with an acidic separating medium to yield a treated aldehyde
mixture. The
treated aldehyde mixture resulting from the contacting with the acidic
separating medium
may be referred to as a second aldehyde containing effluent. Contacting the
first aldehyde
containing effluent with the acidic separating medium at least partially
separates the metal
cations from the first aldehyde containing effluent.
Preferably, the first aldehyde containing effluent may be separated from the
basic
separating medium and subsequently contacted with the acidic separating
medium. The
first aldehyde containing effluent may or may not have undergone further
modification
before contacting the acidic separating medium whereby the concentration of
components
in the effluent is changed. The modification of the effluent may include any
process such
as dilution or concentration. Preferably, the process does not substantially
change the
chemical structure of the aldehyde. A substantial change is understood to mean
typically
there is no more than a 25 percent decrease in the molar quantity of aldehyde
present in the
effluent. The acidic separating medium may be in any physical form such as a
liquid or
preferably a solid.
The acidic separating medium may comprise a carboxylic acid cation exchange
resin, i.e., a weak acid cation exchange resin. Preferably, the carboxylic
acid cation
exchange resin may be an acrylic acid cation exchange resin. The carboxylic
acid cation
exchange resins which may be used in the present method may have any physical
structure,
preferably, a gel type (microporous) or a macroreticular type (macroporous)
structure.
Commercially available acrylic acid cation exchange resins include DOW MAC-3
acrylic
acid cation exchange resin, available from The Dow Chemical Company, Liquid
Separations, P.O. Box 1206, Midland, Michigan 48642, USA; IRC76 acrylic acid
cation
exchange resin, available from Rohm & Haas Company, Ion Exchange Resins, 100
Independence Mall West, Philadelphia, Pennsylvania 19106, USA; and C140E
acrylic acid
cation exchange resin, available fiom The Purolite Company, 150 Monument Road,
Bala
Cynwyd, Pennsylvania 19004, USA.
CA 02641031 2008-07-30
WO 2007/090158 PCT/US2007/061384
The first aldehyde containing effluent may be contacted with the acidic
separating
medium in any manner sufficient to ensure that the metal cations in the first
aldehyde
containing effluent are brought into contact with the acidic separating
medium. For
example, the first aldehyde containing effluent and the acidic separating
medium may be
contacted in a vessel such as a stirred mixing tank, a HIGGINS LOOPTM, a
carousel-type
arrangement, an alternating dual bed-type arrangement, by flow of the mixture
through a
fixed bed of separating medium, or by passing the mixture through a
chromatography
column containing the acidic separating medium.
For a batch-type process, a sufficient amount of acidic separating medium by
weight of medium to weight of first aldeliyde containing effluent may be in
the range of
from 1:5 to 1:25, and preferably in the range of from 1:10 to 1:15. Contact
times may vary
from 30 minutes to several hours, for example from 1 to 50 hours. For a
continuous
process, the first aldehyde containing effluent may be passed through the
acidic separating
medium at a volume hourly space velocity (volume of first aldehyde containing
effluent
feed per volume of acidic separating medium per hour) of from 0.1 h"1 to 100
h"1,
preferably from 2 h-I to 30 h-1.
The first aldehyde containing effluent may be contacted with the acidic
separating
medium at any temperature that minimizes degradation of the separating medium
or the
aldehyde. Preferably, the first aldehyde containing effluent and the acidic
separating
medium may be contacted at a temperature of from 5 to 45 C, and more
preferably of
from 15 to 25 C.
The second aldehyde containing effluent may contain at least 70 percent of the
aldehyde present in the aldehyde mixture, more preferably at least 80 percent,
and most
preferably at least 90 percent.
The second aldehyde containing effluent may contain at least 70 percent of the
aldehyde present in the first aldehyde containing effluent, more preferably at
least 80
percent, and most preferably at least 90 percent. The second aldehyde
containing effluent
may contain a smaller quantity of the metal cations than the aldehyde mixture
and first
aldehyde containing effluent, and preferably the second aldehyde containing
effluent may
contain at most 50 percent, more preferably at most 25 percent, and most
preferably at
most 10 percent of the metal cations present in the aldehyde mixture and first
aldehyde
containing effluent. Preferably, the second aldehyde containing effluent may
contain a
total of at most 0.001, more preferably at most 0.0001, and most preferably at
most
CA 02641031 2008-07-30
WO 2007/090158 PCT/US2007/061384
11
0.00001 molar equivalents of the metal cations per liter of the second
aldehyde containing
effluent.
The second aldehyde containing effluent may or may not have undergone further
modification prior to hydrogenation whereby the concentration of components in
the
effluent is changed. The modification of the effluent may include any process
such as
dilution or concentration.
After the separating capacity has diminished, the acidic separating medium may
be
subjected to an acid treatment to regenerate the acidic properties of the
separating medium.
Preferably, the acid treatment may be an acid wash if the acidic separating
medium is a
solid. The acidic separating medium may be contacted with an acid wash at a
temperature
of at least 45 C, preferably in the range of from 70 to 100 C, most
preferably from 85 to
95 C, for a sufficient time to regenerate the acidic properties of the acidic
separating
medium. The acid wash should have a pH below that of the pKa of the acidic
separating
medium to most fully regenerate the separating medium. Unless otherwise
stated, the pKa
values are deemed to be measured at a temperature of 25 C. The acid wash will
preferably have a pH of 2 or below, more preferably a pH of 1 or below. The
acid wash is
preferably a sulfuric acid solution, more preferably a 10 % sulfuric acid
solution. Other
acids, however, may be utilized as the acid wash, including, but not limited
to,
hydrochloric acid, phosphoric acid, or other mineral acids.
Referring now to Figure 1, Figure 1 is a schematic illustrating a process for
preparing 1,3-propanediol by hydroformylation of ethylene oxide, carbon
monoxide and
hydrogen to form 3-hydroxypropionaldehyde followed by hydrogenation of the 3-
hydroxypropionaldehyde to 1,3-propanediol which process incorporates the
treatment
method of the invention.
Figure 1 depicts an embodiment of the invention within the 1,3-propanediol
process. Separate or combined streams of ethylene oxide (1), carbon monoxide
and
hydrogen (2) are charged to hydroformylation vessel (3) and reacted in the
presence of a
hydroformylation catalyst to produce a hydroformylation product mix.
Following the hydroformylation reaction, the hydroformylation product mix
containing 3-hydroxypropionaldehyde may be cooled and passed to extraction
vessel (5)
via line (4), where an aqueous liquid, generally water and optional
miscibilizing diluent,
are added via line (6) for extraction and concentration of the 3-
hydroxypropionaldehyde for
the subsequent hydrogenation step. The organic phase resulting from the liquid-
liquid
CA 02641031 2008-07-30
WO 2007/090158 PCT/US2007/061384
12
extraction may be recycled, with optional purge of heavy ends, from the
extraction vessel
to hydroformylation reaction via line (7). The aqueous 3-
hydroxypropionaldehyde solution
generated from the liquid-liquid extraction may be passed via line (8) to
degasser-stripper-
oxidizer vessel (9) for removal of carbon monoxide and hydrogen and for
oxidation of any
remaining catalyst in carbonyl form. Oxidation may be conveniently carried out
by
introducing an oxygen-containing gas such as air into the aqueous 3-
hydroxypropionaldehyde solution extract. The preferred oxidation technique
involves
sparging air from inlet (11) in an upward direction through degasser-stripper-
oxidizer
vessel (9) as the aqueous 3-hydroxypropionaldehyde solution to be treated
flows in a
downward direction through vessel (9). The stripping gas may be sparged
through the
degasser-stripper-oxidizer vessel (9) through the same inlet (11) as the
oxidizing gas, or
through a separate inlet (not shown) positioned to permit the stripping gas to
flow through
the aqueous 3-hydroxypropionaldehyde solution as the solution flows through
vessel (9).
In the method of the present invention, the aqueous 3-hydroxypropionaldehyde
solution, i.e., the aqueous 3-hydroxypropionaldehyde mixture, may be passed
from vessel
(9) to vessel (10) where carboxylic acid may be separated by contacting the
aqueous 3-
hydroxypropionaldehyde mixture with the basic separating medium. The first 3-
hydroxypropionaldehyde containing effluent resulting from vessel (10) may then
be passed
to vessel (22) where the metal cations may be separated by contacting the
first 3-
hydroxypropionaldehyde containing effluent with an acidic separating medium.
The second 3-hydroxypropionaldehyde containing effluent resulting from vessel
(22) may then be passed to the hydrogenation zone (13) and reacted with
hydrogen (14) in
the presence of a hydrogenation catalyst to produce a hydrogenation product
mixture (15)
containing 1,3-propanediol. In such a process, the illustrated hydrogenation
zone (13)
includes a series of two or more separate reaction vessels.
Residual diluent and extractant water may be recovered by distillation of the
hydrogenation product mixture (15) in column (16) and recycled to a water
extraction
process for further distillation (not shown) and separation and purge of light
ends. 1,3-
propanediol containing product stream (18) may be passed to a distillation
column (19) for
recovery of 1,3 -propanediol (20) from heavy ends (21).
The hydroformlyation vessel may be a pressure reaction vessel such as a bubble
column or agitated tank, operated batchwise or in a continuous manner. The
feed streams
may be contacted in the presence of a hydroformylation catalyst. The
hydroformylation
CA 02641031 2008-07-30
WO 2007/090158 PCT/US2007/061384
13
catalyst may comprise one or more metal cations, as described hereinbefore.
The
hydroformylation catalyst may further comprise a carbonyl, in particular water-
insoluble
cobalt and/or rhodium carbonyls such as Co4(CO)12 (tetracobalt
dodecacarbonyl),
Co2(CO)8 (dicobalt octacarbonyl) and Rh6(CO)16(hexarhodium hexadecacarbonyl).
The
hydroformylation catalyst will typically be present in the reaction mixture in
an amount
within the range of 0.01 to 1 wt. %, preferably from 0.05 to 0.3 wt. %,
relative to the total
weight of the hydroformylation reaction mix. The hydrogen and carbon monoxide
will
generally be introduced into the reaction vessel in a molar ratio within the
range of 1:2 to
8:1, preferably 1:1 to 6:1.
The hydroformylation reaction may be carried out under conditions effective to
produce a hydroformylation product mix containing a major portion of 3-
hydroxypropionaldehyde and a minor portion of acetaldehyde and 1,3-
propanediol. The
level of 3-hydroxypropionaldehyde in the reaction mixture is preferably less
than 15 wt. %,
more preferably within the range of 5 to 10 wt. %, relative to the total
weight of the
hydrofonnylation reaction mixture. To provide for diluents having different
densities, the
desired concentration of 3-hydroxypropionaldehyde in the reaction mixture can
be
expressed in molai.-ity, i.e., less than 1.5M, preferably within the range of
0.5M to 1M.
Generally, the cobalt-catalyzed hydroformylation reaction may be carried out
at a
temperature of less than 100 C, preferably 60 C to 90 C, and most
preferably 75 C to
85 C, with rhodium-catalyzed hydroformylations on the order of about 10 C
higher. The
hydroformylation reaction may generally be carried out at a pressure within
the range of 1
to 35 MPa, preferably (for process economics) 7 to 25 MPa, with higher
pressures
preferred for greater selectivity. The hydroformylation reaction may be
carried out in a
liquid diluent inert to the reactants. By "inert" is meant that the diluent is
not consumed
during the course of the reaction. In general, ideal diluents for the
hydroformylation
process will solubilize carbon monoxide, will be essentially non-water
miscible, and will
dissolve 3-hydroxypropionaldehyde to the desired concentration of at least 5
wt. % under
hydroformylation conditions, while most of the diluent will remain as a
separate phase
upon water extraction. By "essentially non-water miscible" is meant that the
diluent has a
solubility in water at 25 C of less than 25 wt. %, so as to fomi a separate
organic phase
upon water-extraction of 3-hydroxypropionaldehyde from the hydrofornlylation
product
mix. Preferably, the hydroformylation reaction diluents may be alcohols and
ethers. More
preferably, the hydroformylation reaction diluents may be ethers such as
methyl-t-butyl
CA 02641031 2008-07-30
WO 2007/090158 PCT/US2007/061384
14
ether, ethyl-t-butyl ether, diethyl ether, phenylisobutyl ether, ethoxyethyl
ether, diphenyl
ether, and diisopropyl ether. Blends of diluents such as
tetrahydrofuran/toluene,
tetrahydrofuran/heptane, and t-butylalcohol/hexane may also be used. Most
preferably, the
hydroformylation reaction diluent may be methyl-t-butyl ether because of the
high yields
of 3-hydroxypropionaldehyde which can be achieved under moderate reaction
conditions.
To further enhance yields under moderate reaction conditions, the
hydroformylation
reaction mix will preferably include a catalyst promoter to accelerate the
reaction rate.
Preferred lipophilic promoters include lipophilic phosphonium salts and
lipophilic amines,
which accelerate the rate of hydroformylation without imparting hydrophilicity
(water
solubility) to the active catalyst. Preferably, the lipophilic promoters may
be
tetrabutylphosphonium and dimethyldodecyl amine. As used herein, "lipophilic"
means
that the promoter tends to remain in the organic phase after extraction of 3-
hydroxypropionaldehyde with water. The promoter will generally be present in
an amount
within the range of 0.01 to 1 mole per mole of metal component of the catalyst
(e.g. cobalt
and/or rhodium).
At low concentrations, water serves as a promoter for the formation of the
desired
carbonyl catalyst species. Optimum water levels for hydroformylation in methyl-
t-butyl
ether diluent are within the range of 1 to 2.5 wt. %, relative to the total
weight of the
hydroformylation reaction mix. An excessive amount of water, however, reduces
3-
hydroxypropionaldehyde selectivity and may induce formation of a second liquid
phase.
Liquid-liquid extraction of the 3-hydroxypropion-aldehyde into the water can
be
effected by any suitable means, such as mixer-settlers, packed or trayed
extraction
columns, or rotating disk contactors. The amount of water added to the
hydroformylation
product mix will generally be within the range of 1:1 to 1:20, preferably 1:5
to 1:15 by
volume. Water extraction may preferably be carried out at a temperature within
the range
of 25 C to 55 C, with a lower temperature preferred. Water extraction under
a partial
pressure for carbon monoxide of 0.5 - 5 MPa at 25 C to 55 C maximizes
catalyst
retention in the organic phase.
Typically, the organic phase resulting from the liquid-liquid extraction
contains a
major portion of the hydroformylation reaction diluent and a major portion of
the catalyst.
The organic phase may be recycled, with optional purge of heavy ends, from the
extraction
vessel to hydrofornrylation reaction.
CA 02641031 2008-07-30
WO 2007/090158 PCT/US2007/061384
Preferably, the aqueous 3-hydroxypropionaldehyde mixture generated from the
liquid-liquid water extraction may contain from 4 to 60 wt. % 3-
hydoxypropionaldehyde,
more preferably from 20 to 40 wt. % 3-hydroxypropionaldehyde, relative to the
total
weight of the aqueous 3-hydroxypropionaldehyde mixture.
The aqueous 3-hydroxypropionaldehyde mixture may have a pH in the range of
from 2 to 4, typically from 2.5 to 3.5, and more typically from 2.9 to 3.3.
The aqueous 3-hydroxypropionaldehyde mixture may contain a quantity of
carboxylic acid in the range of from 0.03 to 3 wt. %; typically in the range
of from 0.06 to
1 wt. %; and more typically in the range of from 0.1 to 0.6 wt. %, relative to
the total
weight of the aqueous 3-hydroxypropionaldehyde mixture.
Typically, the carboxylic acid comprises 3-hydroxypropionic acid in a quantity
of
at least 50 wt. %, more typically at least 60 wt. %, most typically at least
75 wt. %, in
particular at least 90 wt. %, relative to the total weight of the carboxylic
acid present in the
aqueous 3-hydroxypropionaldehyde mixture.
The aqueous 3-hydroxypropionaldehyde mixture may contain a total quantity of
cobalt and/or rhodium cations of at most 0.03, typically at most 0.02, more
typically at
most 0.01, most typically at most 0.002, in particular at most 0.001 molar
equivalents of
cobalt and/or rhodium cations per liter of the aqueous 3-
hydroxypropionaldehyde mixture.
The aqueous 3-hydroxypropionaldehyde mixture may contain a total quantity of
cobalt
and/or rhodium cations of at least 0.00000 1, or at least 0.0000 1, or at
least 0.0001 molar
equivalents of cobalt and/or rhodium cations per liter of the aqueous 3-
hydroxypropionaldehyde mixture. Typically, the aqueous 3-
hydroxypropionaldehyde
mixture may contain a total quantity of cobalt and/or rhodium cations in the
range of from
0.001 to 0.003 molar equivalents of cobalt and/or rhodium cations per liter of
the aqueous
3-hydroxypropionaldehyde mixture. The quantity of cobalt and/or rhodium
cations
includes cobalt and/or rhodium cations from both water soluble and water-
insoluble
complexes, compounds or species.
The aqueous 3-hydroxypropionaldehyde solution generated from the liquid-liquid
water extraction may be oxidized. Preferably, the aqueous 3-
hydroxypropionaldehyde
mixture may be contacted with oxygen under wealdy acidic conditions effective
for
oxidation of insoluble metal compounds, e.g. water insoluble cobalt and/or
rliodium
species, to water soluble metal compounds, e.g. water soluble cobalt and/or
rhodium
CA 02641031 2008-07-30
WO 2007/090158 PCT/US2007/061384
16
cations. The oxidation of insoluble metal compounds facilitates removal of the
metal
compounds in the subsequent ion exchange step.
Typically, the quantity of carboxylic acid produced as a byproduct of ethylene
oxide hydroformlyation under conditions favoring the formation of 3-
hydroxypropionaldehyde generates weakly acidic conditions suitable for
oxidation. If
sufficient acid is not already present as a reaction byproduct, the aqueous 3-
hydroxypropionaldehyde solution may be made acidic by addition of an organic
or
inorganic acid in an amount effective to produce a solution having a pH from 3
to 6,
preferably from 3 to 4. Suitable acids include Cl-4 organic acids.
The oxidation may be carried out at a temperature of from 5 C to 45 C and at
a
pressure in the range of from 50 to 200 kPa, preferably about 101.3 kPa
(atmospheric
pressure). The residence time may typically be in the range of from 1 to 15
minutes.
A stripping gas such as nitrogen or carbon dioxide may also be sparged through
the
aqueous 3-hydroxypropionaldehyde solution in the degasser-stripper-oxidizer to
prevent
formation of flammable mixtures and to assist in removal of carbon monoxide
and
hydrogen from the aqueous 3-hydroxypropionaldehyde solution. It is desirable
to remove
even minor amounts of carbon monoxide remaining in the solution since carbon
monoxide
can interfere with the performance of the hydrogenation catalyst.
After passing through the degasser-stripper-oxidizer, the resulting aqueous 3-
hydroxypropionaldehyde mixture may contain 3-hydroxypropionaldehyde and
byproducts
including, one or more water soluble metal cations and carboxylic acid, the
major
component being 3-hydroxypropionic acid.
The aqueous 3-hydroxypropionaldehyde mixture obtained from the degasser-
stripper-oxidizer may be contacted with a basic separating medium yielding a
first 3-
hydroxypropionaldehyde containing effluent. As discussed hereinbefore, the
basic
separating medium may be contacted with the aqueous 3-hydroxypropionaldehyde
mixture
while controlling the pH of the mixture at a value of at most 6, preferably at
most 5.5, and
more preferably at most 5, in order to minimize the degradation of the 3-
hydroxypropionaldehyde. 3-hydroxypropionaldehyde may be increasingly degraded
above
pH 5 and may be significantly degraded at pH values above 6.
The conditions and vessels for contacting the aqueous 3-hydroxypropionaldehyde
mixture with the basic separating medium may be as discussed hereinbefore for
the
aldehyde mixture.
CA 02641031 2008-07-30
WO 2007/090158 PCT/US2007/061384
17
The first 3-hydroxypropionaldehyde containing effluent may contain at least 70
percent of the aldehyde present in the aqueous 3-hydroxypropionaldehyde
mixture, more
preferably at least 80 percent, and most preferably at least 90 percent.
The first 3-hydroxypropionaldehyde containing effluent may contain a smaller
quantity of the carboxylic acid than the aqueous 3-hydroxypropionaldehyde
mixture, and
preferably the first 3-hydroxypropionaldehyde containing effluent may contain
at most 20
percent of the carboxylic acid present in the aqueous 3-hydroxypropionaldehyde
mixture,
more preferably at most 10 percent, and most preferably at most 5 percent. The
first 3-
hydroxypropionaldehyde containing effluent may contain at least 1 percent of
the
carboxylic acid present in the aqueous 3-hydroxypropionaldehyde mixture, more
preferably at least 1.5 percent, and most preferably at least 2 percent. The
pH of the first 3-
hydroxypropionaldehyde containing effluent may be at most 6, preferably at
most 5.5, and
more preferably at most 5.
The amount of metal cations in the first 3-hydroxypropionaldehyde containing
effluent may be the same as the amount of metal cations in the aqueous 3-
hydroxypropionaldehyde mixture since the metal cations are not removed in any
significant quantity by the basic separating medium.
The first 3-hydroxypropionaldehyde containing effluent may be contacted with
an
acidic separating medium yielding a second 3-hydroxypropionaldehyde containing
effluent. The conditions and vessels for contacting the first 3-
hydroxypropionaldehyde
containing effluent with the acidic separating medium may be as discussed
hereinbefore for
the first aldehyde containing effluent.
The second 3-hydroxypropionaldehyde containing effluent may contain at least
70
percent of the aldehyde present in the aqueous 3-hydroxypropionaldehyde
mixture, more
preferably at least 80 percent, and most preferably at least 90 percent.
The second 3-hydroxypropionaldehyde containing effluent may contain at least
70
percent of the aldehyde present in the first 3-hydroxypropionaldehyde
containing effluent,
more preferably at least 80 percent, and most preferably at least 90 percent.
The second 3-hydroxypropionaldehyde containing effluent may contain a smaller
quantity of the metal cations than the aqueous 3-hydroxypropionaldehyde
mixture or first
3-hydroxypropionaldehyde containing effluent, and preferably the second 3-
hydroxypropionaldehyde containing effluent may contain at most 50 percent,
more
preferably at most 25 percent, and most preferably at most 10 percent of the
metal cations
CA 02641031 2008-07-30
WO 2007/090158 PCT/US2007/061384
18
present in the aqueous 3-hydroxypropionaldehyde mixture or first 3-
hydroxypropionaldehyde containing effluent. Preferably, the second 3-
hydroxypropionaldehyde containing effluent may contain a total of at most
0.001, more
preferably at most 0.0001, and most preferably at most 0.0001 molar
equivalents of the
metal cations per liter of the second 3-hydroxypropionaldehyde containing
effluent.
The pH of the second 3-hydroxypropionaldehyde containing effluent may be in
the
range of from 3 to 6, preferably in the range of from 3.5 to 5.5.
The hydrogenation catalyst may preferably be a fixed-bed supported nickel
catalyst,
such as is available commercially as Calsicat E-475SR and R-3142 from W.R.
Grace.
The hydrogenation process may be carried out in one stage or two or more
sequential temperature stages. In a preferred embodiment, hydrogenation may be
carried
out as described above at a temperature within the range of 50 C to 130 C,
followed by a
second stage carried out at a temperature higher than that of the first stage
and within the
range of 70 C to 155 C, and then optionally a third stage at a temperature
greater than
120 C for reversion of heavy ends to 1,3-propanediol.
Having generally described the invention, a further understanding may be
obtained
by reference to the following examples, which are provided for purposes of
illustration
only and are not to be construed as limiting the scope of the invention
described herein.
EXAMPLE 1
Preparation of an aqueous 3-hydroxypropionaldehyde mixture for use in Examples
2-4:
An ethylene oxide hydroformylation product mix was water extracted under 1350
psi (9300 kPa) of 4:1 hydrogen:carbon monoxide at 35 C, the aqueous extract
layer
forming an aqueous solution. After depressuring to atmospheric pressure, the
aqueous
extract layer was separated and sparged with a dilute air/nitrogen mixture to
convert the
cobalt carbonyl catalyst to a water-soluble cationic cobalt metal species. The
aqueous 3-
hydroxypropionaldehyde mixture was analyzed by gas chromatography to determine
a 3-
hydroxypropionaldehyde concentration of 12.25 wt.% for the aqueous 3-
hydroxypropionaldehyde mixture. A colorimetric technique based on
derivitization of
thiocyanate was used to determine a cobalt concentration of 77 ppmw. Titration
of acidity
witli a 0.1N KOH titrant resulted in a concentration of 0.051 meq of
carboxylic acid per
gram of 3-hydroxypropionaldehyde mixture at an equivalence point between pH =
8 and 9
CA 02641031 2008-07-30
WO 2007/090158 PCT/US2007/061384
19
when measured at a temperature of 25 C. The pH of the aqueous 3-
hydroxypropionaldehyde mixture was 3.3, when measured at a temperature of 25
C. The
aqueous 3-hydroxypropionaldehyde mixture was divided into a number of aliquots
used in
examples 2-4.
EXAMPLE 2
In accordance with the method of the invention, the effectiveness of a basic
separating medium, in particular a tertiary amine ion exchange resin, to
remove carboxylic
acid from an aqueous 3-hydroxypropionaldehyde mixture and the effectiveness of
an acidic
separating medium, in particular a carboxylic acid cation exchange resin, to
remove cobalt
cations from a first 3-hydroxypropionaldehyde containing effluent was
determined. 10
grams of the aqueous 3-hydroxypropionaldehyde mixture from example 1 were
contacted
with 1 wet grams of AMBERLYSTTM A-21 (a dimethylamino macroreticular styrene
divinylbenzene available from Rohm and Haas Company) ion exchange resin, via
tumbling
for 18 hours at 24 C in a glass vial to insure liquid-solid equilibration. A
separate sample
of the resin was dried via vacuum oven overnight at 65 C to establish a dry
solids content
of 52 wt. % of the wet resin. Analysis of the liquid phase after contact with
the resin (i.e.,
the first 3-hydroxypropionaldehye containing effluent) revealed a pH of 5, as
measured at
25 C, a carboxylic acid concentration of 0.002 meq/g, a cobalt concentration
of 76 ppmw,
and a 3-hydroxypropionaldehyde concentration of 11.9 wt. %. Any difference of
less than
3 wt. % between the initial and final concentration of 3-
hydroxypropionaldehyde is
negligible given normal experimental error in gas chromatography analysis of
this reactive
intermediate.
This demonstrates that a basic separating medium may be used to remove 96 % by
weight of the carboxylic acid present in the aqueous 3-hydroxypropionaldehyde
mixture
with negligible degradation of the 3-hydroxypropionaldehyde.
4.55 grams of residual liquid supernatant (i.e., the first 3-
hydroxypropionaldehyde
containing effluent) was contacted with 0.038 wet grams of DOWEXTM Mac-3 (a
macroreticular acrylic acid available from The Dow Chemical Company) ion
exchange
resin, via tumbling for 18 hours at 24 C in a glass vial. A separate sample
of the resin was
dried via vacuum oven overnight at 65 C to establish a dry solids content of
53 wt. % of
the wet resin. Analysis of the liquid phase after contact with the resin
(i.e., the second 3-
hydroxypropionaldehye containing effluent) revealed a pH of 4.4, a cobalt
concentration of
18 ppmw, and a 3-hydroxypropionaldehyde concentration of 11.5 wt. %.
CA 02641031 2008-07-30
WO 2007/090158 PCT/US2007/061384
The conlbined contact with a basic separating medium and an acidic separating
medium removed 96 % by weight of the carboxylic acid and 77 % by weight of the
cobalt
with only a 7 % by weight degradation of the 3-hydroxypropionaldehyde,
relative to the
aqueous 3-hydroxypropionaldehyde mixture. The ratio of the concentration of
cobalt on
the resin (based on dry gram) per concentration of cobalt remaining in the
second 3-
hydroxypropionaldehyde containing effluent was 741.
Example 3 (comparative)
4.34 grams of the first 3-hydroxypropionaldehyde containing effluent from
example
2 was contacted with an additiona10.56 wet grams of AMBERLYSTTM A-21 (a
dimethylamino macroreticular styrene divinylbenzene available from Rohm and
Haas
Company) ion exchange resin, via tumbling for 18 hours at 24 C in a glass
vial. Analysis
of the liquid phase after continued contact with the resin revealed complete
removal of the
carboxylic acid, a pH of 7.07, a cobalt concentration of 76 ppmw, and a 3-
hydroxypropionaldehyde concentration of 3.17 wt. %.
This example demonstrates that not controlling the pH of the aqueous 3-
hydroxypropionaldehyde mixture results in degradation of 3-
hydroxypropionaldehyde once
the carboxylic acid has been removed from the aqueous 3-hydroxypropionaldehyde
mixture.
Example 4 (comparative)
A 10 gram aliquot of the aqueous 3-hydroxypropionaldehyde mixture from
example 1 was contacted with 0.063 dry grams of DOWEXTM Mac-3 (a
macroreticular
acrylic acid available from The Dow Chemical Company) ion exchange resin, via
tumbling
for 18 hours at 24 C in a glass vial. Analysis of the liquid phase after
contact with the
resin revealed a pH of 3.3, a cobalt concentration of 48 ppmw, and a 3-
hydroxypropionaldehyde concentration of 12.45 wt. %. The ratio of the
concentration of
cobalt on the resin (based on dry gram) per concentration of cobalt remaining
in the
aqueous mixture of 3-hydroxypropionaldehyde was 96.
Comparison of Example 2 with Example 4 demonstrates that treatment with a
basic
separating medium before contact with an acidic separating medium
substantially improves
the amount of cobalt removed by the acidic separating medium while minimizing
the
degradation of the 3-hydroxypropionaldehyde.
Example 5
CA 02641031 2008-07-30
WO 2007/090158 PCT/US2007/061384
21
The effectiveness of metal oxide to remove carboxylic acid from an aqueous 3-
hydroxypropionaldehyde mixture in accordance with the method of the invention
is
determined.
An aqueous 3-hydroxypropionaldehyde mixture was prepared similar to Example 1
except the aqueous 3-hydroxypropionaldehyde mixture had a concentration of 3-
hydroxypropionaldehyde of 22 wt. %, and a concentration of carboxylic acid of
0.041
meq/g of solution.
2 grams of LDS, an activated alumina available from Coastal Chemical
Corporation, was water washed to remove residual base and air dried. The water
washed
LDS was added to a vial containing 18 grams of the aqueous 3-
hydroxypropionaldehyde
mixture obtained from an ethylene oxide hydroformylation product mix. The vial
was
rotated on a rack for 68 hours at room temperature. The LDS alumina removed 79
% by
weight of the acid which resulted in a pH of 3.3 for the first 3-
hydroxypropionaldehyde
containing effluent and less than 4 wt. % of the 3-hydroxypropionaldehyde was
degraded
after contact with the LDS alumina.
This demonstrates that a metal oxide may be used to remove carboxylic acid
present in the aqueous 3-hydroxypropionaldehyde mixture with minimal
degradation of the
3-hydroxypropionaldehyde.
The first 3-hydroxypropionaldehyde containing effluent is then contacted with
a
quantity of DOWEXTM Mac-3 (a macroreticular acrylic acid available from The
Dow
Chemical Company) ion exchange resin to yield a second 3-
hydroxypropionaldehyde
containing effluent. The second 3-hydroxypropionaldehyde containing effluent
will
contain a lesser quantity of cobalt than present in the first 3-
hydroxypropionaldehyde
containing effluent.
Example 6
The effectiveness of metal oxide in a continuous process to remove carboxylic
acid
from an aqueous 3-hydroxypropionaldehyde mixture in accordance with the method
of the
invention is determined.
1300 grams of LDS alumina were packed into a 2-inch (5 cm) by 18-inch (46 cm)
column. Aqueous 3-hydroxypropionaldehyde mixture was fed to the column at a
weight
hourly space velocity (WHSV hr-1) of between 0.4 and 0.6 hr"1. The aqueous 3-
hydroxypropionaldehyde mixture contained a carboxylic acid concentration in
the range of
from 0.021 to 0.038 meq/g of solution and a quantity of 3-
hydroxypropionaldehyde of 17
CA 02641031 2008-07-30
WO 2007/090158 PCT/US2007/061384
22
to 23 wt. %. The first 3-hydroxypropionaldehyde containing effluent showed
negligible
degradation of 3-hydroxypropionaldehyde after contact with the alumina while
still
removing 92 % by weight of the acid present in the aqueous 3-
hydroxypropionaldehyde
mixture. After breakthrough of the acid, regeneration of the alumina was
performed using
4 wt. % potassium hydroxide solution.
The first 3-hydroxypropionaldehyde containing effluent is then contacted with
a
quantity of DOWEXTM Mac-3 (a macroreticular acrylic acid available from The
Dow
Chemical Company) ion exchange resin to yield a second 3-
hydroxypropionaldehyde
containing effluent. The second 3-hydroxypropionaldehyde containing effluent
will
contain a lesser quantity of cobalt than present in the first 3-
hydroxypropionaldehyde
containing effluent.