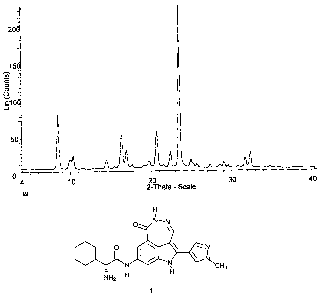
Note: Descriptions are shown in the official language in which they were submitted.
CA 02648369 2008-10-03
WO 2007/113647 PCT/IB2007/000859
-1-
POLYMORPHIC FORMS OF (2R,Z)-2-AMINO-2-CYCLOHEXYL-N-(5-(1-METHYL-1H-
PYRAZOL-4-YL)-1-OXO-2,6-DIHYDRO-1 H-f 1,21DIAZEPINO f4,5,6-CDIINDOL-8-
YL)ACETAMIDE
Field of the Invention
The present invention relates to novel polymorphic forms of (2R,Z)-2-amino-2-
cyclohexyl-
N-(5-(1-methyl-1 H-pyrazol-4-yl)-1-oxo-2,6-dihydro-1 H-[1,2]diazepino[4,5,6-
cd]indol-8-yl)
acetamide and to methods for their preparation. The invention is also directed
to pharmaceutical
compositions containing at least one polymorphic form and to the therapeutic
use of such
polymorphic forms and compositions.
Background of the Invention
The compound (2R,Z)-2-amino-2-cyclohexyl-N-(5-(1-methyl-1 H-pyrazol-4-yl)-1-
oxo-2,6-
dihydro-1 H-[1,2]diazepino[4,5,6-cd]indol-8-yl)acetamide (also referred to as
"Compound 1"),
H
I
O N~N
O ~ \ N
/ N NICH3
NH2 H
1
as well as pharmaceutically acceptable salts thereof, is described in U.S.
Patent No. 6,967,198,
issued November 22, 2005, the disclosure of which is incorporated herein by
reference.
Many anticancer agents, as well as radiation therapy, cause DNA damage to
cells,
especially cancer cells. CHK1 inhibition enhances the anti-cancer effect of
these anti-cancer
agents or radiation therapy by abrogating the S and G2 arrest of those DNA
damaged cells and
thus leading to mitotic catastrophe and cell death of these cells. (2R,Z)-2-
amino-2-cyclohexyl-N-
(5-(1-methyl-1 H-pyrazol-4-yl)-1-oxo-2,6-dihydro-1 H-[1,2]diazepino[4,5,6-
cd]indol-8-yl)acetamide
is a potent CHK1 protein kinase inhibitor. Use of (2R,Z)-2-amino-2-cyclohexyl-
N-(5-(1-methyl-1 H-
pyrazol-4-yl)-1-oxo-2,6-dihydro-1 H[1,2]diazepino [4,5,6-cd]indol-8-
yl)acetamide, a
pharmaceutically acceptable salt or solvates thereof, or a mixture thereof, in
combination with an
anti-cancer agent or radiation therapy will greatly enhance the anti-cancer
effect of the anti-cancer
agent or radiation therapy.
A solid compound may exist in amorphous or crystalline forms. Each of the
different
crystalline forms of the same compound is considered a polymorphic form of the
compound.
Crystalline polymorphs are different crystalline forms of the same compound.
Different
polymorphic forms of the same active pharmaceutical ingredient (API) may have
very different
physical properties, such as thermodynamic stability, solubility,
hygroscopicity as well as
pharmacological properties such as oral bioavailability. These properties
greatly influence the
properties of a drug, in such areas as shelf life, cost of production,
consistent dosage and even
CA 02648369 2008-10-03
WO 2007/113647 PCT/IB2007/000859
-2-
effectiveness of the drug. Thus it is desirable to have polymorphic forms of a
compound having
good physical and pharmacological properties.
Summary of the Invention
In one embodiment, the present invention provides a crystalline form of (2R,Z)-
2-amino-2-
cyclohexyl-N-(5-(1-methyl-1 H-pyrazol-4-yl)-1-oxo-2,6-dihydro-1 H-
[1,2]diazepino[4,5,6-cd]indol-8-
yl)acetamide, represented by Formula 1
H
i
0 N~N
\
0_'AN N
I / N N, CH3
NH2 H H
1
In another embodiment, the present invention provides a crystalline form of
(2R,Z)-2-
amino-2-cyclohexyl-N-(5-(1-methyl-1 H-pyrazol-4-yl)-1-oxo-2,6-dihydro-1 H-
[1,2]diazepino[4,5,6-
cd]indol-8-yi)acetamide. Preferably, the crystalline form is a substantially
pure polymorph of Form
1. In one aspect of the embodiment, the crystalline form has a powder X-ray
diffraction pattern
comprising peaks at diffraction angles (20) of 23.6 0.1 and 8.5 0.1. In
another aspect of the
embodiment, the crystalline form has a powder X-ray diffraction pattern
comprising peaks at
diffraction angles (20) of 23.6 0.1 , 8.5 0.1 and 20.7 0.1. In another
aspect of the
embodiment, the crystalline form has a powder X-ray diffraction pattern
comprising peaks at
diffraction angles (20) of 23.6 0.1, 8.5 0.1, 20.7 0.1 and 16.4 0.1. In
another aspect of the
embodiment, the crystalline form has a powder X-ray diffraction pattern
comprising peaks at
diffraction angles (20) of 23.6 0.1, 8.5 0.1, 20.7 0.1, 16.4 0.1 and
17.0 0.1. In another
aspect of the embodiment, the crystalline form has a powder X-ray diffraction
pattern comprising
peaks at diffraction angles (20) essentially the same as shown in Figure 1. In
another aspect of
this embodiment, the crystalline form has a 13C solid state NMR peak pattern
comprising peaks at
chemical shifts 175.0 0.1, 137.6 0.1, 134.9 0.1, 110.3 0.1, 106.3
0.1, 41.1 0.1 and 32.6
0.1 ppm. In another aspect of this embodiment, the crystalline form has a 13C
solid state NMR
peak pattern comprising at least three of the following seven peaks at
chemical shifts 175.0 0.1,
137.6 0.1, 134.9 0.1, 110.3 0.1, 106.3 0.1, 41.1 0.1 and 32.6 0.1
ppm. In another
aspect of this embodiment, the crystalline form has a 13C solid state NMR peak
pattern
comprising at least four of the following seven peaks at chemical shifts 175.0
0.1, 137.6 0.1,
134.9 0.1, 110.3 0.1, 106.3 0.1, 41.1 0.1 and 32.6 0.1 ppm. In
another aspect of this
embodiment, the crystalline form has a'3C solid state NMR peak pattern
comprising at least five
of the following seven peaks at chemical shifts 175.0 0.1, 137.6 0.1,
134.9 0.1, 110.3 0.1,
106.3 0.1, 41.1 0.1 and 32.6 0.1 ppm. In another aspect of this
embodiment, the crystalline
form has a 13C solid state NMR peak pattern comprising at least six of the
following seven peaks
at chemical shifts 175.0 0.1, 137.6 0.1, 134.9 0.1, 110.3 0.1, 106.3
0.1, 41.1 0.1 and
32.6 0.1 ppm. In another aspect of this embodiment, the crystalline form has
a 13C solid state
CA 02648369 2008-10-03
WO 2007/113647 PCT/IB2007/000859
-3-
NMR peak pattern comprising peaks at chemical shifts 175.0 0.2, 137.6 0.2,
134.9 0.2,
110.3 0.2, 106.3 0.2, 41.1 0.2 and 32.6 0.2 ppm. In another aspect of
this embodiment,
the crystalline form has a'3C solid state NMR peak pattern comprising at least
three of the seven
peaks at chemical shifts 175.0 0.2, 137.6 0.2, 134.9 0.2, 110.3 0.2,
106.3 0.2, 41.1 0.2
and 32.6 0.2 ppm. In another aspect of this embodiment, the crystalline form
has aI 3C solid
state NMR peak pattern comprising at least four of the seven peaks at chemical
shifts 175.0
0.2, 137.6 0.2, 134.9 0.2, 110.3 0.2, 106.3 0.2, 41.1 0.2 and 32.6
0.2 ppm. In another
aspect of this embodiment, the crystalline form has a 13C solid state NMR peak
pattern
comprising at least five of the seven peaks at chemical shifts 175.0 0.2,
137.6 0.2, 134.9
0.2, 110.3 0.2, 106.3 0.2, 41.1 0.2 and 32.6 0.2 ppm. In another aspect
of this
embodiment, the crystalline form has a 13C solid state NMR peak pattern
comprising at least six
of the seven peaks at chemical shifts 175.0 0.2, 137.6 0.2, 134.9 0.2,
110.3 0.2, 106.3
0.2, 41.1 0.2 and 32.6 0.2 ppm. In another aspect of this embodiment, the
crystalline form has
a 13C solid state NMR peak pattern comprising peaks at chemical shifts
position essentially the
same as shown in Figure 4b.
In another embodiment, the present invention provides a crystalline form of
(2R,Z)-2-
amino-2-cyclohexyl-N-(5-(1-methyl-1 H-pyrazol-4-yl)-1-oxo-2,6-dihydro-1 H-
[1,2]diazepino[4,5,6-
cd]indol-8-yl)acetamide. Preferably, the crystalline form is a substantially
pure polymorph of Form
II. In one aspect of this embodiment, the crystalline form has a powder X-ray
diffraction pattern
comprising peaks at diffraction angles (20) of 25.3 0.1 and 16.0 0.1. In
another aspect of the
embodiment, the crystalline form has a powder X-ray diffraction pattern
comprising peaks at
diffraction angles (20) of 25.3 0.1, 16.0 0.1, 13.9 0.1 and 29.2 0.1. In
another aspect of the
embodiment, the crystalline form has a powder X-ray diffraction pattern
comprising peaks at
diffraction angles (20) of 25.3 0.1, 16.0 0.1, 13.9 0.1, 29.2 0.1 and
12.2 0.1. In another
aspect of the embodiment, the crystalline form has a powder X-ray diffraction
pattern comprising
peaks at diffraction angles (26) of 25.3 0.1, 16.0 0.1, 13.9 0.1, 29.2
0.1 12.2 0.1 and 16.8
0.1. In another aspect of the embodiment, the crystalline form has a powder X-
ray diffraction
pattern comprising peaks at diffraction angles (20) of 25.3 0.1, 16.0 0.1,
13.9 0.1, 29.2 0.1
12.2 0.1, 16.8 0.1, 6.9 0.1 and 13.6 0.1. In another aspect of the
embodiment, the crystalline
form has a powder X-ray diffraction pattern comprising peaks at diffraction
angles (20) essentially
the same as shown in Figure 2. In another aspect of the embodiment, the
crystalline form has a
13C solid state NMR peak pattern comprising peaks at chemical shifts 177.7
0.1, 133.2 0.1,
127.8 0.1, 103.8 0.1, and 22.7 0.1ppm. In another aspect of the
embodiment, the
crystalline form has a'3C solid state NMR peak pattern comprising at least
three of the five peaks
at chemical shifts 177.7 0.1, 133.2 0.1, 127.8 0.1, 103.8 0.1, and
22.7 0.1ppm. In
another aspect of the embodiment, the crystalline form has a 13C solid state
NMR peak pattern
comprising at least four of the five peaks at chemical shifts 177.7 0.1,
133.2 0.1, 127.8 0.1,
103.8 0.1, and 22.7 0.1 ppm. In another aspect of the embodiment, the
crystalline form has a
13C solid state NMR peak pattern comprising peaks at chemical shifts 177.7
0.2, 133.2 0.2,
127.8 0.2, 103.8 0.2, and 22.7 0.2ppm. In another aspect of the
embodiment, the
CA 02648369 2008-10-03
WO 2007/113647 PCT/IB2007/000859
-4-
crystalline form has a 13C solid state NMR peak pattern comprising at least
three of the five peaks
at chemical shifts 177.7 0.2, 133.2 0.2, 127.8 0.2, 103.8 0.2, and
22.7 0.2ppm. In
another aspect of the embodiment, the crystalline form has a 13C solid state
NMR peak pattern
comprising at least four of the five peaks at chemical shifts 177.7 0.2,
133.2 0.2, 127.8 0.2,
103.8 0.2, and 22.7 0.2ppm. In another aspect of the embodiment, the
crystalline form has a
13C solid state NMR peak pattern comprising peaks at chemical shifts position
essentially the
same as shown in Figure 5b.
In another embodiment, the present invention provides an amorphous form of
(2R,Z)-2-
amino-2-cyclohexyl-N-(5-(1-methyl-1 H-pyrazol-4-yl)-1-oxo-2,6-dihydro-1 H-
[1,2]diazepino[4,5,6-
cd]indol-8-yl)acetamide. Preferably, the amorphous form is substantially pure.
In one aspect of
the embodiment, the amorphous form has a 13C solid state NMR peak pattern
comprising peaks
at chemical shifts 163.6 0.2, 138.9 0.2, 131.4 0.2, 129.9 0.2, and
30.8 0.2 ppm. In
another aspect of the embodiment, the amorphous form has a 13C solid state NMR
peak pattern
comprising at least three of the five peaks at chemical shifts 163.6 0.2,
138.9 0.2, 131.4 0.2,
129.9 0.2, and 30.8 0.2 ppm. In another aspect of the embodiment, the
amorphous form has
a 13C solid state NMR peak pattern comprising at least four of the following
peaks at chemical
shifts 163.6 0.2, 138.9 0.2, 131.4 0.2, 129.9 0.2, and 30.8 0.2 ppm.
In another aspect
of the embodiment, the amorphous form has a 13C solid state NMR peak pattern
comprising
peaks at chemical shifts position essentially the same as shown in Figure 6.
In another embodiment, the present invention provides a solid form of (2R,Z)-2-
amino-2-
cyclohexyl-N-(5-(1-methyl-1 H-pyrazol-4-yl)-1-oxo-2,6-dihydro-1 H-
[1,2]diazepino[4,5,6-cd]indol-8-
yI)acetamide (Compound 1), wherein the solid form comprises at least two forms
selected from
polymorphic Form I, polymorphic Form II and an amorphous form of Compound 1.
In one aspect
of this embodiment, the solid form comprises at least 10% of polymorph Form I.
More preferably,
the solid form comprises at least 20% of polymorph Form I. More preferably,
the solid form
comprises at least 30% of polymorph Form I. Even more preferably, the solid
form comprises at
least 30%, at least 40%, or at least 50% of polymorph Form I. Even more
preferably, the solid
form comprises at least 60%, at least 70% or at least 80% of polymorph Form I.
Even more
preferably, the solid form comprises at least 90% of polymorph Form I. Even
more preferably, the
solid form comprises at least 95% of polymorph Form I.
In another embodiment, the present invention provides a pharmaceutical
composition
comprising the polymorphic Form I, the polymorphic Form II, the amorphous form
or the solid
form of (2R,Z)-2-amino-2-cyclohexyl-N-(5-(1-methyl-1 H-pyrazol-4-yl)-1-oxo-2,6-
dihydro-1 H-
[1,2]diazepino[4,5,6-cd]indol-8-yl)acetamide of the invention.
In another embodiment, the present invention provides a pharmaceutical
composition
comprising the solid form of (2R,Z)-2-amino-2-cyclohexyl-N-(5-(1-methyl-1H-
pyrazol-4-yl)-1-oxo-
2,6-dihydro-1H-[1,2]diazepino[4,5,6-cd]indol-8-yl)acetamide of the invention.
In another embodiment, the present invention provides a method of treating
cancer in a
mammal comprising administering to the mammal in need thereof the
pharmaceutical
composition of the invention.
CA 02648369 2008-10-03
WO 2007/113647 PCT/IB2007/000859
-5-
In another embodiment, the present invention provides a method of treating
cancer in a
mammal comprising administering to the mammal in need thereof a
therapeutically effective
amount of the pharmaceutical composition of the invention, in combination with
a therapeutically
effective amount of an anti-cancer treatment selected from an anti-cancer
agent and radiation
therapy. In one aspect of this embodiment, the anti-cancer treatment is an
anti-cancer agent.
Preferably, the anti-cancer agent is selected from the group consisting of Ara-
c, VP-16, cis-platin,
adriamycin, 2-chloro-2-deoxyadenosine, 9- (3-D-arabinosyl-2-fluoroadenine,
carboplatin,
gemcitabine, camptothecin, paclitaxel, BCNU, 5-fluorouracil, irinotecan, and
doxorubicin. In
another aspect of this embodiment, the anti-cancer treatment is radiation
therapy.
In another embodiment, the present invention provides a method of treating a
mammalian
disease condition mediated by CHKI protein kinase activity, comprising
administering to a
mammal in need thereof the polymorphic Form I, the polymorphic Form II, the
amorphous form or
the solid form of (2R,Z)-2-amino-2-cyclohexyl-N-(5-(1-methyl-1H-pyrazol-4-yl)-
1-oxo-2,6-dihydro-
1 H-[1,2]diazepino[4,5,6-cd]indol-8-yl)acetamide of the invention, in
combination with a anti-cancer
treatment selected from anti-cancer agent, radiation therapy and the
combination thereof. In one
aspect of this embodiment, the anti-cancer treatment is an anti-cancer agent.
Preferably, the anti-
cancer agent is selected from the group consisting of Ara-c, VP-16, cis-
platin, adriamycin, 2-
chloro-2-deoxyadenosine, 9- (3-D-arabinosyl-2-fluoroadenine, carboplatin,
gemcitabine,
camptothecin, paclitaxel, BCNU, 5-fluorouracil, irinotecan, and doxorubicin.
In another aspect of
this embodiment, the anti-cancer treatment is radiation therapy.
One of the ordinary skill in the art would appreciate that the chemical shifts
of the 13C
solid state NMR of the polymorphic Form I, II or the amorphous form of
Compound 1 may have
some variance depending on the external reference used. In the claims of the
present invention,
the chemical shifts of the 13C solid state NMR refer to those obtained when
the upfield signal of
adamantine at 29.5ppm is used as external reference.
The term "in combination with" refers to the relative timing of the
administration of a first
therapeutic treatment, such as (2R,Z)-2-amino-2-cyclohexyl-N-(5-(1-methyl-1H-
pyrazol-4-yl)-1-
oxo-2,6-dihydro-lH-[1,2]diazepino[4,5,6-cd]indol-8-yl)acetamide, a
pharmaceutically acceptable
salt or solvate thereof, or a mixture thereof, to the mammal in need, to that
of a second
therapeutic treatment, such as a anti-cancer agent or radiation therapy, the
relative timing being
those normally used in the field of medicine for combination therapy. In
particular, relative timing
can be sequential or simultaneous.
The term "hyperproliferative disorder" refers to abnormal cell growth that is
independent
of normal regulatory mechanisms (e.g., loss of contact inhibition), including
the abnormal growth
of normal cells and the growth of abnormal cells. This includes, but is not
limited to, the abnormal
growth of tumor cells (tumors), both benign and malignant. Examples of such
benign proliferative
diseases are psoriasis, benign prostatic hypertrophy, human papilloma virus
(HPV), and
restinosis.
The term "cancer" includes, but is not limited to, lung cancer, bone cancer,
pancreatic
cancer, skin cancer, cancer of the head or neck, cutaneous or intraocular
melanoma, uterine
CA 02648369 2008-10-03
WO 2007/113647 PCT/IB2007/000859
-6-
cancer, ovarian cancer, rectal cancer, cancer of the anal region, stomach
cancer, colon cancer,
breast cancer, uterine cancer, carcinoma of the fallopian tubes, carcinoma of
the endometrium,
carcinoma of the cervix, carcinoma of the vagina, carcinoma of the vulva,
Hodgkin's Disease,
cancer of the esophagus, cancer of the small intestine, cancer of the
endocrine system, cancer of
the thyroid gland, cancer of the parathyroid gland, cancer of the adrenal
gland, sarcoma of soft
tissue, cancer of the urethra, cancer of the penis, prostate cancer, chronic
or acute leukemia,
lymphocytic lymphomas, cancer of the bladder, cancer of the kidney or ureter,
renal cell
carcinoma, carcinoma of the renal pelvis, neoplasms of the central nervous
system (CNS),
primary CIVS lymphoma, spinal axis tumors, brain stem glioma, pituitary
adenoma, or a
combination of one or more of the foregoing cancers. In another embodiment of
said method,
said abnormal cell growth is a benign proliferative disease, including, but
not limited to, psoriasis,
benign prostatic hypertrophy or restinosis.
The term "mediated by CHK1 protein kinase activity" refers to biological or
molecular
processes that are regulated, modulated, or inhibited by CHK1 protein kinase
activity.
The term "polymorph" refers to different crystalline forms of the same
compound.
"Polymorph" includes, but is not limited to, other solid state molecular forms
including hydrates
(e.g., bound water present in the crystalline structure) and solvates (e.g.,
bound solvents other
than water) of the same compound.
The term "pharmaceutically acceptable, carrier, diluent, or vehicle" refers to
a material (or
materials) that may be included with a particular pharmaceutical agent to form
a pharmaceutical
composition, and may be solid or liquid. Exemplary of solid carriers are
lactose, sucrose, talc,
gelatin, agar, pectin, acacia, magnesium stearate, stearic acid and the like.
Exemplary of liquid
carriers are syrup, peanut oil, olive oil, water and the like. Similarly, the
carrier or diluent may
include time-delay or time-release material known in the art, such as glyceryl
monostearate or
glyceryl distearate alone or with a wax, ethylcellulose,
hydroxypropylmethyicellulose,
methylmethacrylate and the like.
The term "pharmaceutical composition" refers to a mixture of one or more of
the
compounds or polymorphs described herein, or physiologically/pharmaceutically
acceptable salts
or solvates thereof, with other chemical components, such as
physiologically/pharmaceutically
acceptable carriers and excipients. The purpose of a pharmaceutical
composition is to facilitate
administration of a compound to an organism.
The term "radiation therapy" refers to medical use of radiation to control
malignant cells.
The term "substantially pure" with reference to particular polymorphic forms
or an
amorphous form of (2R,Z)-2-amino-2-cyclohexyl-N-(5-(1-methyl-1H-pyrazol-4-yl)-
1-oxo-2,6-
dihydro-1H-[1,2]diazepino[4,5,6-cd]indol-8-yl)acetamide means the polymorphic
form or the
amorphous form includes less than 10%, prefereably less than 5%, prefereably
less than 3%,
preferably less than 1% by weight of impurities, including other polymorphic
forms of (2R,Z)-2-
amino-2-cyclohexyl-N-(5-(1-methyl-1 H-pyrazol-4-yl)-1-oxo-2,6-dihydro-1 H-
[1,2]diazepino[4,5,6-
cd]indol-8-yl)acetamide. Such purity may be determined, for example, by X-ray
powder
diffraction.
CA 02648369 2008-10-03
WO 2007/113647 PCT/IB2007/000859
-7-
The term "therapeutically effective amount" generally refers to an amount of a
compound,
a pharmaceutically acceptable salt or solvate thereof, or a mixture thereof,
being administered
which will relieve to some extent one or more of the symptoms of the disorder
being treated. In
particular, when the term is used in describing a combination therapy,
"therapeutically effective
amount" refers to the amount of a particular therapeutic which will 1) enhance
the therapeutic
effect of another therapeutic such as an anti-cancer agent or radiation
therapy, or 2) in
combination with the other therapeutic, relieve to some extent one or more of
the symptoms of the
disorder being treated. In reference to the treatment of cancer, symptoms of
the disease being
treated includes a) reducing the size of the tumor; b) inhibiting (that is,
slowing to some extent,
preferably stopping) tumor metastasis; c) inhibiting to some extent (that is,
slowing to some
extent, preferably stopping) tumor growth, and d) relieving to some extent
(or, preferably,
eliminating) one or more symptoms associated with the cancer.
The term "2 theta value" or "20" refers to the peak position based on the
experimental
setup of the.X-ray diffraction experiment described in the present invention,
including but not
limited to the radiation source used and the wavelength of the radiation
source, and is a common
abscissa unit in diffraction patterns. The experimental setup requires that if
a reflection is
diffracted when the incoming beam forms an angle theta (0) with a certain
lattice plane, the
reflected beam is recorded at an angle 2 theta (20).
The terms "treat", "treating" and "treatment" refer to a method of alleviating
or abrogating
a cancer and/or its attendant symptoms. With regard particularly to cancer,
these terms simply
mean that the life expectancy of an individual affected with a cancer will be
increased or that one
or more of the symptoms of the disease will be reduced.
As used herein, the term "essentially the same" with reference to X-ray
diffraction peak
positions means that typical peak position and intensity variability are taken
into account. For
example, one skilled in the art will appreciate that the peak positions (26)
will show some inter-
apparatus variability, typically as much as 0.10. Further, one skilled in the
art will appreciate that
relative peak intensities will show inter-apparatus variability as well as
variability due to degree of
crystallinity, preferred orientation, prepared sample surface, and other
factors known to those skilled
in the art, and should be taken as qualitative measures only. Similarly, as
used herein, "essentially
the same" with reference to solid state NMR spectra and Raman spectra is
intended to also
encompass the variabilities associated with these analytical techniques, which
are known to those
of skill in the art. For example, 13C chemical shifts measured in solid state
NMR will typically have a
variability of 0.1 ppm, while Raman shifts will typically have a variability
of 1 cm"'.
Brief Description of the Drawings
Figure 1 is a X-Ray Powder Diffraction Pattern of polymorphic Form I of (2R,Z)-
2-amino-
2-cyclohexyl-N-(5-(1-methyl-1 H-pyrazol-4-yl)-1-oxo-2,6-dihydro-1 H-
[1,2]diazepino[4, 5,6-cd]indol-
8-yl)acetamide.
CA 02648369 2008-10-03
WO 2007/113647 PCT/IB2007/000859
-8-
Figure 2 is a X-Ray Powder Diffraction Pattern of polymorphic Form II of
(2R,Z)-2-amino-
2-cyclohexyl-N-(5-(1-methyl-1 H-pyrazol-4-yl)-1-oxo-2,6-dihydro-1 H-
[1,2]diazepino[4,5,6-cd]indol-
8-yl)acetamide.
Figure 3a is a X-Ray Powder Diffraction Pattern of an amorphous form (the
first batch) of
(2R,Z)-2-amino-2-cyclohexyl-N-(5-(1-methyl-1 H-pyrazol-4-yl)-1-oxo-2,6-dihydro-
1 H-
[1,2]diazepino[4,5,6-cd]indol-8-yl)acetamide.
Figure 3b is a X-Ray Powder Diffraction Pattern of an amorphous form (the
second batch)
of (2R,Z)-2-amino-2-cyclohexyl-N-(5-(1-methyl-1H-pyrazol-4-yl)-1-oxo-2,6-
dihydro-1H-
[1,2]diazepino[4,5,6-cd]indol-8-yl)acetamide.
Figure 4a is a Solid State 13C Cross Polarization and Magic Angle Spinning
(CP/MAS)
NMR of polymorphic Form I (the first spectrum) of (2R,Z)-2-amino-2-cyclohexyl-
N-(5-(1-methyl-
1 H-pyrazol-4-yl)-1-oxo-2,6-dihydro-1 H-[1,2]diazepino[4,5,6-cd]indol-8-
yl)acetamide.
Figure 4b is a Solid State 13C Cross Polarization and Magic Angle Spinning
(CP/MAS)
NMR of polymorphic Form I (the second spectrum) of (2R,Z)-2-amino-2-cyclohexyl-
N-(5-(1-
methyl-1 H-pyrazol-4-yl)-1-oxo-2,6-dihydro-1 H-[1,2]diazepino[4,5,6-cd]indol-8-
yl)acetamide.
Figure 5a is a Solid State 13C CP/MAS NMR of polymorphic Form II (the first
spectrum) of
(2R,Z)-2-am ino-2-cyclohexyl-N-(5-(1-methyl-1 H-pyrazol-4-yl)-1-oxo-2,6-
dihydro-1 H-
[1,2]diazepino[4,5,6-cd]indol-8-yl)acetamide.
Figure 5b is a Solid State 13C CP/MAS NMR of polymorphic Form II (the second
spectrum) of (2R,Z)-2-amino-2-cyclohexyl-N-(5-(1-methyl-1 H-pyrazol-4-yl)-1-
oxo-2,6-dihydro-1 H-
[1,2]diazepino[4, 5,6-cd]indol-8-yl)acetamide.
Figure 6 is a solid State 13C CP/MAS NMR of the amorphous form (the second
batch) of
(2R,Z)-2-amino-2-cyclohexyl-N-(5-(1-methyl-1 H-pyrazol-4-yl)-1-oxo-2,6-dihydro-
1 H-
[1,2]diazepino[4,5,6-cd]indol-8-yl)acetamide.
Detailed Description of the Invention
In this section, "BOC", "Boc" or "boc" refers to N-tert-butoxycarbonyl, "CBZ"
refers to
carbobenzyloxy, "DCE" refers to dichloroethane, "DCM" refers to
dichloromethane, "DCC" refers
to 1,3-dicyclohexylcarbodiimide, DIC" refers to diisopropylcarbodiimide,
"DIPEA" or "DIEA" refers
to diisopropyl ethyl amine, DMA refers to N,N-dimethylacetamide, "DMAP" refers
to 4-dimethyl
amino pyridine, "DME" refers to 1,2-dimethoxyethane, "DMF" refers to dimethyl
formamide,
"DMSO" refers to dimethylsulfoxide, "DPPP" refers to 1,3-
bis(diphenylphosphino)propane, "EDC"
refers to 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride, "HATU"
refers to O-(7-
azabenzotriazol-l-yl)-N,N,N;N'-tetramethyluronium hexafluorophosphate, "HBTU"
refers to O-
benzotriazol-l-yl-N,N,N;N'-tetramethyluronium hexafluorophosphate, "HOAc"
refers to acetic
acid, "HOBt" refers to 1-hydroxybenzotriazole hydrate, "IPA" refers to
isopropyl alcohol, "LAH"
refers to lithium aluminum hydride, "LiHMDS" refers to lithium
bis(trimethylsilyl)amide, "MSA"
means methanesulfonic acid; "MTBE" refers to methyl t-butyl ether, "NMP"
refers to 1-methyl 2-
pyrrolidinone, "TEA" refers to triethyl amine, "TFA" refers to trifluoro
acetic acid, "TIPS" referst to
triisopropylsilyl-. TMSCI refers to trimethyl silyl chloride, and "Trt" refers
to triphenylmethyl-.
CA 02648369 2008-10-03
WO 2007/113647 PCT/IB2007/000859
-9-
It has been found that the Compound 1 can exist in more than one polymorphic
crystalline form as well as amorphous form. Processes for producing these
polymorphic forms in
high purity and characterization of these different polymorphic forms are
described herein.
Pharmaceutical formulations comprising Compound 1 are also provided.
1. Synthesis of Compound 1:
Synthetic route to make compound 12, the HCI salt of (2R,Z)-2-amino-2-
cyclohexyl-N-(5-
(1-methyl-1 H-pyrazol-4-yl)-1-oxo-2,6-dihydro-1 H-[1,2]diazepino[4,5,6-
cd]indol-8-yl) acetamide,
was described in United Patent No. 6,967,198, issued November 22, 2005. In the
present
invention, the said synthetic route was modified. In particular, the reaction
conditions were
improved, to fit the scale of the reactions. As shown in Scheme 1, Compound I
can be made
directly from compound 11, the N-Boc precursor of compound 12. Compound 1 can
also be
made from compound 12 by neutralizing the HCI salt. In either approach,
depending on the work
up and purification condition, different polymorphic forms of Compound 1 can
be obtained.
Scheme 1
OMe
CO2Me
C02H Me0 N- CH3 NMe2
CH3 /
H3C MeOH, Me3SiC1
THF, 50 C O2N N02 60 C, 20 h
(
OaN NOZ 8 h
2
CO2Me 1. NH4OCHO, Pd/C C02Me
\ OMe MeOH/THF, 45 oC Boc20,
I ~ 4 M NaOH,THF
/ OMe 2. MeOH, Conc HCI
02N N02 HCIH2N H
3 4
OMe OMe
O O CHO
35 wt% aq NH2NH2
POCIa.DMF, TH F I\ ~ AcOH, MeOH
BocHN H N BocHN N
5 6
H3C
H3C 7 0
0 N-N HN-N H3C g CN
Py.HBr3 0 0 N, CH
\ DMF, -14 C Br H3C 2 3
I ~ NH Pd(PPh3)4, K3PO4
BocHN / H 80% o
7 BocHN DMA, 90 C
8
CA 02648369 2008-10-03
WO 2007/113647 PCT/IB2007/000859
-10-
0 H
N_ N 0 N-N.HCI
4N HCI in dioxane 01"C00H
GH2CI2
N NHBoc
BocHN N N
H N HCI H N N ,CH3 EDC, DMAP, DMF
2 H 1h,50oC
\ CH3
9 10
H
H
0 N-N 0 N-N
N MSA, THF, reflux N
NI N N\ CH OiN N N,
H 3 H k CH3
= H H
NHBoc NHZ 1
11
H HCI
0 N-N
I N
,
W
H N N CH3
H
NHZ HCI
12
As illustrated in Scheme 1, 2-methyl-3,5-dinitrobenzoic acid reacts with N,N-
dimethylformamide dimethyl acetal under heat to give compound 2. Solvents
suitable for this
reaction are DMF, DMA, MTBE, toluene and THF. Preferably, THF is used.
Preferably, the
reaction is carried out at around 50 C. The reaction mixture is concentrated
under reduced
pressure followed by addition of methanol. Compound 2 will then precipitate
out as the product.
Compound 2 is then treated with an acid in methanol under heat to give
compound 3.
Typical acids that can be used here are TMSCI, HCI, MSA, H2SO4 and TFA.
Preferably, TMSCI is
used. During the course of the reaction, compound 3 crystallizes out from the
reaction solution,
thus significantly simplifies the isolation process.
In US 6,967,198, compound 3 was converted to compound 4 by a conventional
hydrogenation reaction under Pd/C and H2. However, this conventional
hydrogenation reaction
condition is difficult to control on large scale due to the fact that
reduction of the two nitro groups
is highly exothermic. In the present invention, compound 3 is converted to
compound 4 by a
transfer hydrogenation reaction followed by an acid-promoted cyclization
reaction. Transfer
hydrogenation reaction of compound 3 provides a dose-controlled procedure and
therefore
reduces thermal hazard. Different transfer hydrogenations reagents can be
used, such as
HCO2NH4 with Pd/C, BH3.NMe3 with Pd(OH)2, hydrazine with FeCl3/C and ammonium
formate
with Pd/C. Preferably, ammonium formate with Pd/C is used. Compound 3 can be
reduced using
ammonium formate with Pd/C in THF. The crude product is then cyclized to give
compound 4
under a strong acid. A typical condition for this acid catalyzed cyclization
is to use concentrated
HCI in MeOH.
CA 02648369 2008-10-03
WO 2007/113647 PCT/IB2007/000859
-11-
The primary amine group of compound 4 is then BOC-protected to give compound
5. In
US 6, 967,198, this reaction was carried out using triethylamine as base and
the reaction was
carried out in acetonitrile. In the present invention, this reaction is
carried out using more benign
conditions of an inorganic base such as NaOH, and a solvent such as THF.
Compound 5 is converted to compound 6 by a Vilsmeier formylation reaction. In
US
6,967,198, this reaction was carried out in methylene chloride, using 3
equivalents of the
Vilsmeier reagent. Since neither compound 5 nor the reaction intermediate was
very soluble in
methylene chloride, this transformation in methylene chloride was a solid-to-
solid reaction, which
was quite often problematic in large scale. Furthermore, di-formylation
occurred when compound
5 was treated with 3 equivalents of Vilsmeier reagent and additional
hydrolysis step was required
to convert di-formylated indole to compound 6. In the present invention, the
reaction is carried out
in a solvent in which the solubility of compound 5 is better. A typical
solvent here is THF.
Compound 5 is soluble in THF and therefore a solution-to-solid reaction is
achieved. Mono-C-
formylation takes place in preference to N-formylation to generate compound 6
directly when the
amount of Vilsmeier reagent is reduced to 1.1 equivalents, thus simplified the
reaction procedure
significantly.
Compound 6 is converted to compound 7 by treating compound 6 with hydrazine
under
acidic condition. Hydrazine monohydrate as well 30% hydrazine aqueous solution
can be used for
this reaction. More preferably, the reaction is carried out in a dilute MeOH
solution with excess
hydrazine, preferably more than 5 equivalents.
Compound 7 is then brominated to give compound 8. In US 6,967,198, NBS was
used
as the bromination reagent. In the present invention, pyridinium tribromide is
used instead as a
cheaper alternative.
Compound 8 is then converted to compound 9 through Suzuki coupling. In US
6,967,198, Pd(dppf)CI2 was used. In the present invention, cheaper alternative
Pd(PPh3)4 is
used. Compound 8 is treated with 3 mol % Pd(PPh3)4 and K3PO4 in DMA. The
residue palladium
content in the crude product is typically very high (6000-8000 ppm). Due to
the poor solubility of
compound 9 in regular organic solvent, Florisol treatment for Pd removal
proved to be not
effective and required a large amount of solvent. A simple and effective water
precipitation Pd
removing procedure is developed in the present invention: 1) dissolve the
crude product in a
water immiscible solvent such as DMF, DMA, THF, DMSO DMA, preferably DMA, then
add N-
acetylcysteine to the resulting solution and stir for 1 hour; 2) add water to
precipitate out product
while the N-acetylcysteine-palladium complex remained in the aqueous solution.
After the N-
acetylcysteine treatment, the residual Pd level dropped below 400 ppm, which
leads to the API
Compound 1 with less than 20 ppm residual Pd.
Compound 9 is then treated with 4M HCI in dioxane and methylene chloride to
give
compound 10 as hydrogen chloride salt. It was found by Ion Chromatograph
analysis that the
hydrogen chloride content varied from batch to batch ranging from 2.1 to 2.8
ratio of hydrogen
chloride to the corresponding free base. Other acids may also be used for this
reaction, for
example, sulfuric acid, sulfonic acids and TFA.
CA 02648369 2008-10-03
WO 2007/113647 PCT/IB2007/000859
-12-
Compound 10 is then coupled with the boc protected hexyl amino acid to give
compound
11. EDC is chosen as coupling agent. Many coupling conditions can be used such
as EDC,
HATU and DCC. In the present invention, EDC is used as the coupling agent,
together with
DMAP as catalyst. The required amount of DMAP should preferably be the same
molar amount
of hydrogen chloride presented in the starting amine salt. Large excess of
DMAP in reaction
mixture could cause significant racemization. Compound 11 is isolated by
adding water into
reaction mixture. Chiral HPLC analysis indicated the existence of -1%
racemized byproduct,
which is removed by crystallization in the final step.
Compound 11 is then treated with a strong acid to remove the boc group.
Conditions
such as HCI/MeOH, HCI/EtOH, TFA/CH2CI2 or MSA/THF can all be used. When
compound 11 is
treated with HCI/MeOH, or HCI/EtOH, the reaction mixture can be evaporated
under reduced
pressure to give compound 12, the HCI salt of (2R,Z)-2-amino-2-cyclohexyl-N-(5-
(1-methyl-1 H-
pyrazol-4-yl)-1-oxo-2,6-dihydro-1 H-[1,2]diazepino[4,5,6-cd]indol-8-yl)
acetamide.
Compound I can be obtained directly from compound 11 by a basic aqueous work
up of
the de-boc reaction. For example, compound 11 is mixed with MSA and refluxed
in THF to
remove the Boc group. Upon completion of the reaction and cooling of the
reaction mixture,
aqueous solution of Na2CO3, K2CO3 or NaHCO3 can be added to the reaction
mixture.
Subsequent standard work up will give Compound 1 in good purity and yield.
Compound I can also be obtained from compound 12 by mixing compound 12 with a
basic aqueous solution, for example aqueous NaHCO3, vigorously stirring the
mixture followed by
extracting the mixture with an organic solvent.
II. Preparation of Polymorphic forms I, II and the amorphous form of Compound
1.
A. Polymorphic Form I
Polymorphic Form I of Compound 1 can be prepared directly from compound 11 by
removing the boc group under strong acidic condition followed by a basic
aqueous work up.
Typical strong acidic conditions to be used here are HCI/MeOH, HCI/EtOH,
MSA/THF or TFA.
Preferably, compound 11 is treated with methanesulfonic acid in THF to remove
the boc group.
After the reaction is complete, the mixture is basified with an aqueous
inorganic base solution,
such as aqueous solution of NaOH, Na2CO3, NaHCO3, K2CO3 or KHCO3. Preferably,
2 M
aqueous sodium hydroxide or saturated NaHCO3 solution is used. The organic
phase is
separated from aqueous phase and dried with magnesium sulfate. The organic
solution is then
reduced to a smaller volume and ethanol is added. The resulting solution is
reduced to a smaller
volume followed by further addition of ethanol. This volume reduction and
addition of ethanol
process is repeated until Compound I precipitates out as polymorphic Form I.
Polymorphic Form I of Compound 1 can also be prepared from compound 12.
Compound 12 is dissolved in an aqueous inorganic base solution portion wise
with vigorous
stirring. Typical aqueous inorganic base solution used here are aqueous
solution of NaOH,
Na2CO3, NaHCO3, K2CO3 or KHCO3, Preferably, NaHCO3 aqueous solution is used
here. The
mixture is extracted with large amount of an organic solvent such as EtOAc.
The resulting
organic solution is washed with brine and dried over Na2SO4 and filtered. The
filtrate is
CA 02648369 2008-10-03
WO 2007/113647 PCT/IB2007/000859
-13-
concentrated on vacuo to afford a yellow solid which is the amorphous form of
Compound 1. This
yellow solid is then dissolved in EtOH under heat, preferably around 75-80 C
with vigorous
stirring. The solution is cooled, preferably to room temperature of around 22
C and kept at 22 C
for 12 hours. Polymorphic Form I will precipitate out.
B. Polymorphic Form II.
Compound 1 in polymorphic Form II may be obtained from compound 12 or compound
11
following similar procedure as described in previous paragraphs except that
during the final
working up procedure, polymorphic Form II forms in methanol at around 4 C.
C. The amorphous form.
Amorphous form of Compound 1 can be prepared directly from compound 12
following
similar procedure of preparation of polymorphic Form I. After compound 12 is
dissolved in an
aqueous base solution portion wise with vigorous stirring, the mixture is
extracted with large
amount of EtOAc. The resulting EtOAc solution is washed with brine and dried
and filtered. The
filtrate is concentrated on vacuo to afford a yellow solid which is the
amorphous form of
Compound 1.
Amorphous form of Compound 1 can also be prepared from compound 11 following
similar procedure of preparation of polymorphic Form I. Use EtOAc as the
organic phase during
the work up. After drying the organic phase, evaporating the solvent at around
40-70 C affords
Compound 1 in amorphous from.
The amorphous form of Compound 1 can also be made from polymorphic Form I of
Compound 1 by dissolving the polymorphic Form I in THF until saturation
followed by removing
the solvent at 50 C.
Ill. Characterization and properties of different polymorphic forms of
Compound 1:
Each solid form of Compound 1 can be characterized by one or more of the
following: X-
ray powder diffraction pattern (i.e., X-ray diffraction peaks at various
diffraction angles (20)),
melting point onset (and onset of dehydration for hydrated forms) as
illustrated by endotherms of
a Differential Scanning Calorimetry (DSC) thermogram, Raman spectral diagram
pattern,
aqueous solubility, light stability under International Conference on
Harmonization (ICH) high
intensity light conditions, and physical and chemical storage stability. For
example, samples of
polymorphic forms I, II and the amorphous form, of Compound 1 were each
characterized by the
positions and relative intensities of peaks in their X-ray powder diffraction
patterns.
A. X-ray power diffraction pattern of the polymorphic forms.
The X-ray powder diffraction pattern for polymorphic Form I, From II and the
first batch of
the amorphous form of Compound 1(Figure 3a) was measured on a Bruker AXS D8-
Discover
diffractometer with a Cu Ka"1ea" radiation source of 1.5418Q wavelength
operated at 40 kV and 40
mA. The samples were analyzed from angles 4-40 degrees (20) using a general
area diffraction
detector. The detector was set 30 cm from the sample. One of the ordinary
skill in the art will
appreciate that the peak positions (20) will show some inter-apparatus
variability, typically as
much as 0.1 degrees. Accordingly, where peak positions (20) are reported, such
numbers are
intended to encompass such inter-apparatus variability. Furthermore, where the
crystalline forms
CA 02648369 2008-10-03
WO 2007/113647 PCT/IB2007/000859
-14-
of the present invention are described as having a powder X-ray diffraction
pattern essentially the
same as that shown in a given figure, the term "essentially the same" is also
intended to
encompass such inter-apparatus variability in diffraction peak positions.
Table 1 shows the 20
values of polymorphic Form I and Form II. The amorphous form shows a
continuous X-Ray
power diffraction spectrum, where no 20 value is collected.
The X-Ray powder diffraction patterns of the second batch of the amorphous
form of
Compound 1 is shown in Figure 3b. The preparation of the second batch of the
amorphous form
is described in Example 4b. Here, the powder X-ray diffraction pattern was
generated with a
Bruker D5000 diffractometer using Cu Ka radiation (wavelength = 1.5406 A). The
instrument was
equipped with a line focus X-ray tube. The tube voltage and amperage were set
to 38 kV and 38
mA, respectively. The divergence and scattering slits were set at 1 mm, and
the receiving slit was
set at 0.6 mm. Diffracted radiation was detected by a Sol-X energy dispersive
X-ray detector. A
theta two theta continuous scan at 2.4 20 /min (1 sec/0.04 2A step) from 3.0
to 40 28 was used.
Experiments were carried out at ambient temperature. An alumina standard (NIST
standard
reference material 1976) was analyzed to check the instrument alignment. Data
were collected
and analyzed using BRUKER AXS DIFFRAC PLUS software Version 2Ø The PXRD peak
was
selected using the peak maximum peak height.
Table 1: X-Ray Powder Diffraction of Polymorphic Forms I and II.
Polymorphic Form I Polymorphic Form II
29 Relative intensity 26 Relative intensity
8.5 33.5 6.9 21.2
10.4 10.1 10.8 9.8
14.4 7.2 12.2 26.3
16.4 21.5 13.6 21.2
17.0 12.6 13.9 28.7
20.7 23.1 16.0 40.0
22.4 11.5 16.8 23.8
23.6 100.0 18.2 11.8
25.0 6.8 20.1 12.0
31.8 8.3 20.8 11.9
32.4 10.9 21.9 10.2 '
25.3 100.0
27.9 13.1
29.2 28.5
One skilled in the art will appreciate that relative peak intensities will
show inter-apparatus
variability as well as variability due to degree of crystallinity, preferred
orientation, prepared
sample surface, and other factors known to those skilled in the art, and
should be taken as
qualitative measures only.
Where a solid form comprises two or more polymorphs of the present invention,
the X-ray
diffraction pattern will have peaks characteristic of each of the individual
polymorphs of the
present invention. For example, a solid form that comprises two polymorphs
will have a powder
CA 02648369 2008-10-03
WO 2007/113647 PCT/IB2007/000859
-15-
X-ray diffraction pattern that is a convolution of the two X-ray diffraction
patterns that correspond
to the substantially pure polymorphic forms.
B. Solid State NMR (SSNMR) of the polymorphic forms.
Solid state NMR is a powerful tool to analyze solids. Different polymorphs
often show
significant chemical shifts differences in solid state 13C cross polarization
and magic angle
spinning (CP/MAS) NMR. Solid state 13C CP/MAS NMR was performed on polymorphic
Form I
and Form II, as well as the amorphous form.
For the polymorphic Form I, a first 13C SSNMR spectrum was collected on a 600
MHz
Bruker spectrometer and that chemical shifts were externally referenced to the
methyl resonance
of hexamethyl bezene at 17.36 ppm. A 500 MHz Bruker spectrometer was used for
a second 13C
SSNMR spectrum of polymorphic Form I, a first and a second spectrum of
polymorphic Form II
and the spectrum of the second batch of the amorphous form of Compound 1 and
the chemical
shifts were externally referenced to the upfield signal of adamantine at
29.5ppm. Table 2a shows
the chemical shifts of the first 13C SSNMR spectrum of polymorphic Form I and
the first 13C
SSNMR spectrum of polymorphic Form II. Table 2b shows the chemical shift of
the second
spectrum of polymorphic Form I, the second spectrum of polymorphic Form II and
the spectrum of
the second batch of the amorphous form of Compound 1. In Table 2b, peak
intensity is defined
as peak heights and can vary depending on the actual setup of the CPMAS
experimental
parameters.
Table 2a: Solid state 13C CP/MAS NMR Chemical Shifts for polymorphic Form I
(first
spectrum) and polymorphic Form II (first spectrum) of Compound 1.
Peak Chemical Shift Chemical Shift
number Form I (ppm) Form II (ppm)
1 177.1 177.7
2 168.0 165.7
3 142.8 140.7
4 139.6 136.6
5 137.0 135.3
6 130.7 133.2
7 128.3 127.8
8 127.3 125.2
9 116.0 114.4
10 112.4 112.5
11 108.5 108.9
12 64.6 103.8
13 43.2 58.7
14 40.9 39.7
15 34.8 37.9
16 28.9 30.3
17 22.7
CA 02648369 2008-10-03
WO 2007/113647 PCT/IB2007/000859
-16-
Table 2b: Solid state 13C CP/MAS NMR Chemical Shifts for polymorphic Form I
(second
spectrum), polymorphic Form II (second spectrum) and the amorphous form
(second batch) of
Compound 1.
13C 13C C Chemical
Peak Chemical Chemical Shifts
Number Shifts Form I Intensity Shifts Form II Intensity amorphous Intensity
m [ppml m
1 175.0 4.65 177.7 6.59 175.8 2.53
2 166.0 4.17 165.7 7.78 166.2 1.3
3 140.8 3.46 140.7 10.95 163.6 5.6
4 137.6 3.59 136.6 9.45 138.9 4.45
134.9 6.91 135.3 8.67 135.5 7.42
6 128.7 3.83 133.2 8.21 131.4 6.89
7 126.3 4.89 127.8 12 129.9 7.19
8 125.2 7.25 125.2 8.34 125.9 5.58
9 113.7 6 114.4 10.58 112.2 6.9
110.3 4.64 112.5 7.72 108.6 6.9
11 106.3 3.53 108.9 10.43 61.3 2.37
12 62.4 3.62 103.8 6.17 40.1 3.42
13 41.1 4.76 58.7 5.68 37.7 5.75
14 38.8 5.4 39.7 5.57 30.8 4.42
32.6 4.35 37.9 8.77 27.1 12
16 26.8 12 30.3 5.62
17 22.7 3.66
5 C. Differential Scanning Calorimetry test of the polymorphic forms.
Different polymorphic forms of Compound 1 were also distinguished using
differential
scanning calorimetry (DSC). DSC is a thermoanalytical technique in which the
difference in the
amount of heat required to increase the temperature of a sample and reference
are measured as
a function of temperature. DSC test is also frequently used to test the
melting point of a solid.
10 In the present invention, DSC was performed using a TA Instruments Q-1000
DSC. The
scanning rate for polymorphic Form I was 10 C/min from 25 C, 'to 300 C and
under such
condition, Polymorphic Form I has an onset melting point of 272 C. The
scanning rate for
polymorphic Form II was 40 C/min from 25 C to 300 C, and under such
condition, polymorphic
Form II started melting at 242 C, rapidly formed a new crystal form. We
believe that the new
15 crystal formed was polymorphic Form I. This was proved by the following
test: Polymorphic Form
II was heated to 260 C and the resulting solid was examined by PXRD. The PXRD
result was
identical to the PXRD result of polymorphic Form I. When the scanning rate for
polymorphic Form
II was set at 10 C/min from 25 C to 300 C, polymorphic form II started
melting at 243 "C, rapidly
formed a new crystal form and then melted at 274 C and we believed the new
crystal form was
also polymorphic Form I.
D. Thermal Gravimetric Analysis of the polymorphic forms.
Thermal gravimetric analysis (TGA) is a testing procedure in which changes in
weight of a
specimen are recorded as the specimen is heated in air or in a controlled
atmosphere such as
nitrogen. Thermogravimetric curves (thermograms or TGA scan graphs) provide
information
regarding solvent and water content and the thermal stability of materials.
CA 02648369 2008-10-03
WO 2007/113647 PCT/IB2007/000859
-17-
In the present invention, TGA was performed on Compound 1 polymorphic Form I
and II
using a TA Instruments TGA Q 500. The temperature was increased at 10 C/min
from 25 C to
310 C and 350 C for polymorphic Form I and Form II, respectively. TGA analysis
of polymorphic
Form I showed that the crystalline Form I lost approximately 0.26% of total
weight by the time the
temperature reached 265 C. The degradation of polymorphic Form I occurred
rapidly just after
melt. TGA analysis of polymorphic Form II showed a loss of approximately 0.85%
of the total
weight by the time the temperature reached 275 C. The degradation of
polymorphic Form II
occurred rapidly after 275 C. These TGA results are consistent with the
conclusion that both
polymorphic Form I and polymorphic Form II are anhydrous/non-solvated forms.
E. Solubility of the different polymorphic forms.
Equilibrium solubility was tested on both polymorphic Form I and polymorphic
Form II.
Samples were prepared by dissolving each corresponding polymorph into or
ethonal. Samples
were stirred overnight at room temperature and centrifuged to get rid of any
solid which was not
dissolved. The solution was then analyzed by HPLC. The results are listed in
Table 3.
Table 3: Solubility of Polymorphic Form I and Form II
Solvent Polymorphic Form I solubility Polymorphic Form II solubility
m/mL m/mL
Water (pH= 8.35) 0.0015 0.0014
Ethanol 3.81 3.84
F. Hygroscopicity of the different polymorphic forms.
Different polymorphic forms of a compound may have different hygroscopic
properties.
Isothermal water sorption and desorption experiments of both polymorphic Form
I and II of
Compound 1, were performed on a Surface Measurements Systems Dynamic Vapor
Sorption-
1000 (DVS). Each polymorphic Form I and Form II was loaded on to the DVS
instrument starting
at 25 C of 0% RH. The humidity was increased step-by-step from 0% to 90%RH by
stepping at
10% RH increments. After the sample weight stabilized at 90% RH, the humidity
was decreased
from 90%RH to 0% RH, stepwise, to complete a full cycle. The temperature
remained constant of
25 C during the whole procedure. Weight gain during this experiment was used
to determine
hygroscopicity. A sample with weight gain of less than 2.0% from 0-90% RH is
considered non-
hygroscopic.
Polymorphic Form I and Form II showed an approximate 1.7% and 1.3% moisture
gain
from 0-90% RH, respectively.
F. Stability of the different polymorphic forms.
Solid state stability of polymorphic Form I of Compound I was investigated at
40 C, at
75% relative humidity (RH), in both open and closed vials for six weeks. The
samples were
analyzed by X-ray powder diffraction to check physical stability and HPLC for
chemical stability.
The results are presented in Table 4.
CA 02648369 2008-10-03
WO 2007/113647 PCT/IB2007/000859
-18-
Table 4: Solid State Stability of Polymorphic Form I
Close vial sample Open vial sample
Physical stability determined No change No change
by PXRD and DSC
Chemical stability determined 99.3% unchanged 99.3% unchanged
by HPLC
Polymorphs are considered enantiotropic when one form is more
thermodynamically
stable at one temperature and the other form is more stable at another
temperature. The
temperature at which both polymorphs are equally stable is known as the
transition temperature
(T). Enantiotropy study was carried out for polymorphic Form I and Form II of
Compound 1. A
one-to-one mixture of Form I and Form II of 10-20 mg was added 1-2 mL of
ethanol to form a
slurry. Samples of such slurry were prepared and stirred at 70 C, 60 C, 50 C,
40 C, 30 C, room
temperature (about 23"C) and 3.5 C respectively. The samples were then
centrifuged. The
supernatant was decanted and the remaining material was left to dry under
vacuum at room
temperature. The resulting materials were analyzed by PXRD. The results are
summarized in
the Table 5.
Table 5: Conversion between Form I and Form II
Temperature Conversion direction Time Completion of conversion
period
70 C Form II --> Form I 1 day Complete
60 C Form 114 Form I 1 day Complete
50 C Form 114 Form I 6 days Complete
40 C Form 114 Form I 5 days complete
30 C Form 114 Form I 11 days Small amount of Form II
remaining
23 C (RT) Form 14 Form II 12 days Incomplete
3.5 C Form 14 Form II 7 days Small amount of Form I
remaining.
IV. Methods of Using the Polymorphs of the Invention
The inventive polymorphic forms of Compound 1 may be useful in all aspects
that
Compound I may be useful. The method of using Compound 1 was described in US
6,967,198
as method to use genus of compounds which contain Compound 1. It was described
in US
6,967,198 that compounds of the invention therein may be used in combination
with a
therapeutically effective amount of an anti-neoplastic agent or radiation
therapy to treat neoplasm
in a mammal. It is within contemplation of the present invention, that the
polymorphic forms and
the pharmaceutical compositions of Compound 1 of the current invention may be
used in
combination with a therapeutically effective amount of an anti-neoplastic
agent or radiation
CA 02648369 2008-10-03
WO 2007/113647 PCT/IB2007/000859
-19-
therapy, as described in US 6,967,198 that could be used in combination with
the compounds of
the invention therein.
Examples
Example 1: Preparation of (2R,Z)-2-amino-2-cyclohexyl-N-(5-(1-methyl-1H-
pyrazol-4-yl)-1-
oxo-2,6-dihydro-1H-[1,2]diazepino[4,5,6-cd]indol-8-yl)acetamide HCI salt (HCI
salt of
Compound 1)
Preparation of 2-(2-Dimethylamino-vinyl)-3,5-dinitro-benzoic acid methyl
ester, compound
2: 2-Methyl-3,5-dinitro-benzoic acid (102.62 g, 0.45 mol) was dissolved in
anhydrous THF (1200
mL). N,N-dimethylformamide dimethyl acetal (Aldrich, 94% purity, 3.0 eq, 172.4
g, 1.35 mol) was
added under nitrogen atmosphere over 10 minutes at room temperature with
stirring.
Temperature rose to 29 C from 22 C. The solution was immediately heated to
50 C and was
stirred at this temperature for 8 hours behind a shield. The reaction solution
was then cooled to
room temperature and concentrated in a rotorvap to remove most of the THF and
the unreacted
N,N-dimethylformamide dimethyl acetal until -250 g of crude material remained
in the flask (water
bath temperature was not allowed to exceed 30 C). Methanol (400 ml) was added
and the slurry
was stirred at room temperature for 1 hour. The solid was collected by
filtration, washed with cold
methanol (60 ml) and dried in airflow to afford 109.08 g of purple solid (84%
yield). 'H NMR (300
MHz, CDCI3) 8 3.01(s, 6H), 3.93(s, 3H), 5.99(d, 1H, J= 13.5 Hz), 6.75(d, 1 H,
J= 13.2 Hz), 8.49(d,
1 H, J= 2.4 Hz), 8.57(d, 1 H, J= 2.4 Hz).
Preparation of 2-(2,2-Dimethoxy-ethyl)-3,5-dinitro-benzoic acid methyl ester
compound 3:
The purple solid 2 (92.25 g, 0.313 mol) was suspended in anhydrous MeOH (1000
mL). TMSCI
(70.25 g, 0.65 mol, 2.1 eq) was added over 10 min. A clear solution was
formed. The solution was
heated at 60 C (slow reflux) for 20 hours. HPLC analysis indicated the
disappearance of starting
material. Reaction mixture was cooled to room temperature and white solid
precipitated out (often
times, the product crystallized from the reaction mixture when reaction was
near completion). The
reaction mixture was stirred at room temperature for 1 h. The precipitated
solid was collected by
filtration, washed with cold MeOH (50 mL) and dried in airflow to afford 82.71
g of white solid. The
mother liquor was concentrated to remove -800 ml of solvent. Additional
product precipitated out.
The mixture was cooled to 0 C and stirred for 1 h. The orange solid was then
collected, washed
with cold methanol (25 ml) and dried in airflow to giving another 5.58 g of
the product. The
combined yield for compound 3 was 88.29 g (90%). 'H NMR (300 MHz, CDCI3) 8
3.30(s, 6H),
3.73(d, 2H, J= 5.1 Hz), 4.00(s, 3H), 4.51(t, 1 H, J= 5.1 Hz), 8.65(d, 1 H, J=
2.4 Hz), 8.76(d, 1 H, J=
2.4 Hz).
Preparation of 6-amino-1H-indole-4-carboxylic acid methyl ester hydrochloride
compound
4: A 2-L Erlenmeyer flask was charged with 10% Pd/C wet catalyst (8.6 g),
methanol (800 ml) and
THF (160 ml). Ammonium formate (139.1 g, 2.21 mol) was then added with
stirring and the
mixture was heated to 35 C. Water (100 ml) was added. Compound 3 was then
added in small
portions over 10 minutes. The reaction temperature was maintained between 40
C and 45 C by
controlling the addition rate of 3 and proper cooling. The reaction mixture
was stirred for 30
minutes after addition is complete. HPLC analysis indicated that the reaction
was complete. The
CA 02648369 2008-10-03
WO 2007/113647 PCT/IB2007/000859
-20-
reaction mixture was cooled to room temperature. The catalyst was filtered off
through a Celite
pad and the Celite pad was washed with MeOH (50 ml). The filtrate was
concentrated to remove
volatile components under reduced pressure. EtOAc (500 ml) and water (75 ml)
were then added
and the mixture was stirred for 5 minutes. Organic phase was separated and the
aqueous phase
was extracted with EtOAc (200 ml). The combined organic solution was
concentrated to dryness
to afford light-yellow oil (79.17 g). MeOH (100 ml) was added to the oil
intermediate. The
resulting solution was then added to the solution of concentrated aqueous HCI
(37 wt%, 82.8 g) in
MeOH (600 ml) while the temperature was maintained at 32 C. The reaction
mixture was stirred
for 3 hours at 35 C. Solid product precipitated out during this period. HPLC
analysis indicated
that the reaction was complete. The reaction mixture was cooled to room
temperature. EtOAc
(500 ml) was added and the resulting mixture was stirred for 30 minutes. The
solid was collect by
filtration to afford 45.07 g of product. The filtrate was concentrated to
afford a paste (331 g).
EtOAc (400 ml) was added to the paste and the mixture was stirred for 30
minutes. The solid was
then collect by filtration to afford the 2"d crop of product 4 (16.28 g). ).
'H NMR (300 MHz,
DMSO-d6) is 3.93(s, 3H), 6.97(d, 1 H, J= 1.8 Hz), 7.65(m, 1 H), 7.72(d, 1 H,
J= 1.8 Hz), 7.80(s, 1 H),
11.81(s, br, I H).
Preparation of 6-amino-1H-indole-4-carboxylic acid methyl ester hydrochloride
4 via direct
hydrogenation of 2-(2-Dimethylamino-vinyl)-3,5-dinitro-benzoic acid methyl
ester compound 2:
Methanol (27 kg) and 2-(2,2-dimethoxy-vinyl)-3,5-dinitrobenzoic acid methyl
ester 2 (10 kg, 33.87
mol) were charged into a reactor through the charge port. 10 % Palladium on
Carbon Catalyst
(0.4 kg) was then charged to the reactor as a slurry in water (2.5 0.5 Kg).
Ethyl Acetate (123
3 kg) was added followed by addition of hydrogen portion wise. The internal
temperature was
maintained at approximately 0 C through cooling and controlling the rate of
hydrogen addition.
After pressure was constant for more than 30 min, hydrogen pressure was kept
at 50-60 psi and
the temperature was maintained at 0 C for 2 hours. The catalyst was filtered
off through an in-
line filter (Note: Catalyst is pyrrophoric. Do not allow the catalyst to pull
dry.) The filtrate was
concentrated to remove approximately 95 % of the solvent volume by vacuum
distillation at an
internal temperature of less than 35 C. Ethyl acetate (130L ) was added with
stirring. 37% HCI
(10kg) was then added slowly below 10 C. The resulting mixture was stirred at
10'C for 1 hour.
The solids was filtered onto a plate filter and washed with methyl t-butyl
ether (15L). The product
(wet cake) was transferred to a vacuum dryer and dried at 23 3 C to afford
4.6 kg of product 4
(60% yield).
Preparation of 6-tert-Butoxycarbonylamino-1H-indole-4-carboxylic acid methyl
ester 5:
THF (600 ml) and 4 M aqueous NaOH solution (138 ml, 0.55 mol) were charged
into a reaction
flask. Indole HCI salt 4 (61.0 g, 0.269 mol) was then added and the mixture
was stirred until solid
was dissolved completely. (BOC)20 (70.43 g, 0.32 mol) was added slowly as
solution in THF (50
ml) while temperature was maintained between 25 C and 32 C during addition.
The resulting
reaction mixture was stirred for 3 hours at room temperature. HPLC analysis
indicated that the
reaction was complete. Aqueous phase was separated. Organic phase was washed
with
saturated ammonium chloride aqueous solution (15 ml). The organic solution was
then
CA 02648369 2008-10-03
WO 2007/113647 PCT/IB2007/000859
-21 -
concentrated to afford a paste (176 g). THF (40 ml) and heptane (800 ml) were
added to the
paste and the resulting mixture was stirred for 30 minutes. The solid was
collected by filtration to
afford 69.86 g of product 5(90% yield). 'H NMR (300 MHz, DMSO-d6) b 1.50(s,
9H), 3.89(s, 3H),
6.83(s, 1 H), 7.41(m, 1 H), 7.89(s, 1 H), 7.91(s, 1 H), 9.38(s, 1 H), 11.24(s,
br, 1 H).
Preparation of 6-tert-butoxycarbonylamino-3-formyl-1 H-indole-4-carboxylic
acid methyl
ester compound 6: N, N-dimethylformamide (85 ml) was charged to a 250-mI flask
and then
cooled to -5 C with an ice-acetone bath. Phosphorus oxychloride (35.15 g,
0.23 mol) was
syringed into the flask over 10 min at 4 C. After the addition was complete,
the mixture was
stirred at 0 C for an additional 30 min. In another 2L 3-necked flask equipped
with overhead
stirrer and thermometer, indole 5 (60.57 g, 0.21 mol) and anhydrous THF (600
ml) were added
and a clear solution was formed. The solution was cooled to -7 C with an ice-
acetone bath. To
the stirred THF solution was cannulatede the cold pre-formed Vilsmeier reagent
over 10 min while
the temperature was maintained at -3 C. After the addition was complete, a
heavy precipitate
formed within minutes. The reaction mixture was further stirred at 0 C for 45
minutes. EtOAc
(600 ml) was added to the reaction mixture followed by addition of cold
aqueous NaOAc solution
(3 M, 310 ml, 0.93 mol) with vigorously stirring. Temperature rose to 10 C
after quench. Cooling
bath was removed. The reaction mixture was warmed to 21 C over 20 minutes and
stirred at this
temperature for 2.5 h. Solids were completely dissolved. HPLC analysis
indicated that all the
intermediate was converted to product. Agitation was stopped and aqueous layer
was separated.
Organic layer was concentrated under reduced pressure to remove volatiles. A
paste (226 g) was
obtained. Water (30 ml) and EtOAc (70 ml) were added to the paste. The
resulting mixture was
stirred for 30 minutes. Solid was collected by filtration, washed with EtOAc
(25 ml) and dried to
afford 62.98 g of product (95% yield, 97% purity). 'H NMR (300 MHz, DMSO-d6) b
1.50(s, 9H),
3.86(s, 3H), 7.68(d, 1 H, J= 1.8 Hz), 7.97(s, IH), 8.23(s, 1 H), 9.56(s, 1 H),
10.10(s, 1 H), 12.25(s,
br, I H).
Preparation of tert-butyl 1-oxo-2,6-dihydro-1 H-[1,2]diazepino[4,5,6-cd]indol-
8-ylcarbamate
compound 7: MeOH (1200 ml), acetic acid (100 ml) and aldehyde 6 (62.98 g, 0.20
mol) were
charged into a 2L flask. Aqueous hydrazine (35 wt%, 88.8 g, 0.97 mol) was then
add with stirring.
The mixture was heated to 60 C and stirred for 3 hours. HPLC analysis
indicated that the
reaction was complete. The heating was stopped and the reaction mixture was
cooled to room
temperature. The mixture was stirred slowly at room temperature for 2 hours.
The solid was
collected by filtration to afford 46.0 g of product. The filtrate was
concentrated to afford a paste
(223 g). Water (250 ml) was added slowly with stirring. The mixture was
stirred for additional 50
minutes to granulate solid. The solid was collected and dried to afford the
2"d crop of product
(13.0 g). The combined yield for compound 7 was 99%. 'H NMR (300 MHz, DMSO-d6)
b 1.49(s,
9H), 7.44(s, IH), 7.51(d, 1 H, J= 2.1 Hz), 7.62(s, 1 H), 7.76(s, 1 H), 9.45(s,
1 H), 10.19(s, 1 H),
11.63(s, br, IH).
Preparation of tert-butyl 5-bromo-l-oxo-2,6-dihydro-1 H-[1,2]diazepino[4,5,6-
cd]indol-8-
yicarbamate 8: Compound 7(45.0g, 0.15 mol) and N,N-dimethylformamide (0.45 L)
were charged
into a 5L-flask equipped with overhead stirrer, thermometer and nitrogen gas
inlet. The mixture
CA 02648369 2008-10-03
WO 2007/113647 PCT/IB2007/000859
-22-
was stirred to form a clear solution. The solution was then cooled to -14 C
under protection of
nitrogen. Pyridinium tribromide (57.6g, 0.18 mol) was added portion-wise over
5 minutes under
protection of nitrogen. Reaction temperature was controlled below -5 C during
addition. After
addition was complete, the reaction mixture was stirred at 0 C for 1.5 hours.
HPLC analysis
indicated that the reaction was complete. Water (0.225L) was added slowly to
the reaction
mixture while the temperature was maintained below 20 C during addition.
EtOAc (0.225L) was
added to the reaction mixture at 15 C followed by addition of aqueous
potassium carbonate
(Preparation: dissolve 20.73g K2C03 in 60ml water, cool to 10 C for use) in
one portion. The
mixture was stirred for 15 minutes to dissolve solid. Aqueous sodium sulfite
solution (Preparation:
dissolve 3.78g Na2SO3 in 18m1 water) was then added and stirred for 10
minutes. Water (0.5 L)
was added via addition funnel over 3 minutes while the temperature was
maintained at 25-30 C.
Solid gradually precipitated out. The mixture was stirred for 20 minutes to
granulate the solid.
Additional water (0.625 L) was added to drive the precipitation to completion.
The mixture was
stirred for 60 minutes. The solid was collected by filtration, washed with
water (0.3L) and dried in
airflow for 90 minutes. The solid was dried in a vacuum oven at 60 C with
nitrogen breeze for 12
hours The crude product was suspended in acetone (0.19L) and stirred for 1
hour. Heptane
(0.19L) was added slowly into the mixture. The resulting mixture was stirred
for 1.5 hour. The
solid was collected by filtration, washed with mixture of acetone (30m1) and
heptane (30m1) and
dried in airflow for 2 hours. The product was further dried in a vacuum oven
at 50 C for 12 hours.
The solid weighed 45.5g (80% yield, 98.3% purity). 'H NMR (300 MHz, DMSO-d6) b
1.53(s, 9H),
7.31(s, 1 H), 7.70(d, 1 H, J= 1.7 Hz), 7.77(s, 1 H), 9.57(s, 1 H), 10.47(s, 1
H), 12.54(s, br, 1 H).
Preparation of tert-butyl 5-(1-methyl-1 H-pyrazol-4-yl)-1-oxo-2,6-dihydro-1 H-
[1, 2]diaze pi no[4,5,6-cd]indol-8-ylcarbam ate 9: Compound 8 (308.24g, 0.81
mol) and N,N-
dimethylacetamide (2.5L) were added to a 10L flask equipped with overhead
stirrer, thermometer
and nitrogen gas inlet. A clear solution was formed. Water (0.625L) was added
to the DMA
solution with stirring while the temperature was controlled at 30 C. 1-methyl-
4-(4,4,5,5-
tetramethyl-1,3,2-dioxaborolan-2-yl)-1 H-pyrazole (186g, 0.89 mol) and
potassium phosphate
(431.3g, 2.03 mol) were then added to the flask with stirring. The mixture was
degassed by
vacuuming the flask followed by nitrogen flush. The degassing process was
repeated 3 times
over 20 minutes. Tetrakis(triphenylphosphine) palladium (28.2g, 0.024 mol) was
added under
protection of nitrogen. The reaction mixture was degassed 3 times by
vacuum/nitrogen cycle over
20 minutes. The reaction was heated at 90 C under protection of nitrogen for
5 hours. HPLC
analysis indicates the reaction is complete. The reaction mixture was cooled
to room
temperature. N-acetylcysteine (30g, 0.18 mol) was added into the flask and the
mixture was
stirred for 2 hours. Water (5L) was added over 30 minutes with stirring while
the temperature was
controlled at 30 C. Solid precipitated out. The mixture was stirred for
additional 2 hours to
granulate solid. Solid was collected by filtration and washed with a mixture
of DMA (0.1 L) and
water (0.3L), water (0.7L), and a mixture of acetone (0.3L) and water (0.3L).
The filter cake was
dried in airflow overnight and then dried in vacuum oven at 60 C for 16
hours. The crude product
was dissolved in DMA (1.5L). N-acetylcysteine (30g, 0.18 moI) was added into
the DMA solution
CA 02648369 2008-10-03
WO 2007/113647 PCT/IB2007/000859
-23-
and the solution was stirred for 2 hours. Water (0.8L) was added over 10
minutes with stirring
while the temperature was controlled at 30 C. Solid precipitated out. The
mixture was stirred for
20 minutes to granulate solid. Additional water (2.2L) was added over 5
minutes to drive the
precipitation to completion. The mixture was stirred for 1 hour. Solid was
collected by filtration
and washed sequentially with a mixture of DMA (0.2L) and water (0.4L), water
(0.8L), and a
mixture of acetone (0.4L) and water (0.4L). The compound was dried in vacuum
oven at 60 C
until the water content is reduced to less than 1.5 wt%. The product weighed
286g (93% yield,
98% apparent purity). 'H NMR (300 MHz, DMSO-d6) S 1.49(s, 9H), 3.92(s, 3H),
7.57(s, IH),
7.65(d, 1 H, J= 1.5 Hz), 7.68(d, 1 H, J= 1.5 Hz), 7.90(s, 1 H), 8.27(s, 1 H),
9.40(s, 1 H), 10.14(s, 1 H),
11.75(s, br, 1 H).
Preparation of 8-amino-5-(1-methyl-1 H-pyrazol-4-yl)-2H-[1,2]diazepino[4,5,6-
cd]indol-
1(6H)-one hydrochloride 10: tert-butyl 5-(1-methyl-1H-pyrazol-4-yl)-1-oxo-2,6-
dihydro-lH-
[1,2]diazepino[4,5,6-cd]indol-8-ylcarbamate 9 (236.06g, 0.62 mol) was charged
into a IOL flask
equipped with equipped with overhead stirrer, thermometer, and a bubbler.
Dichloromethane
(3.5L) was added and the mixture was stirred for 10 minutes. The suspension
was then cooled to
10 C followed by addition of 4M HCI in dioxane (2.4L) over 10 minutes. The
temperature was not
allowed to exceed 25 C. The reaction mixture was stirred at room temperature
for 16 hours.
HPLC analysis indicates -98% of starting material was consumed. The solid was
collected by
centrifuge under protection of nitrogen and washed with dichloromethane
(2.2L). The product
was then dried in vacuum oven at 40 C to afford product (239.77g). 'H NMR
(300 MHz, DMSO-
d6) 8 3.94(s, 3H), 7.47(d, 1 H, J= 1.8 Hz), 7.58(d, 1 H, J= 1.8 Hz), 7.66(s, 1
H), 8.00(s, 1 H), 8.40(s,
1 H), 10.42(s, 1 H), 12.51(s, br, 1 H).
Preparation of tert-butyl (R)-1-cyclohexyl-2-(5-(1-methyl-1 H-pyrazol-4-yl)-1-
oxo-2,6-
dihydro-1 H-[1,2]diazepino[4,5,6-cd]indol-8-ylamino)-2-oxoethylcarbamate 11: 8-
amino-5-(1-
methyl-1 H-pyrazol-4-yl)-2H-[1,2]diazepino[4,5,6-cd]indol-1(6H)-one
hydrochloride 10 (224.35g,
0.64 mol, containing 25.7% of chloride determined by Ion Chromatography) was
suspended in
anhydrous DMF(2.2 L). 4-dimethylaminopyridine (197.08g, 1.65 mol) was added
and the mixture
was stirred for 30 minutes. Boc-D-cyclohexylglycine (179.80 g, 0.7 mol) was
then added and the
mixture was heated to 35 C. 1-(3-dimethylaminopropyl)-3-ethylcarbondiimide
(158.5 g, 0.83 mol)
was added in one portion and the reaction mixture was heated at 50 C for 1
hour. HPLC
analysis indicates the reaction was complete. The reaction mixture was cooled
to room
temperature. Water (2.2 L) was added over 10 minutes while the temperature was
maintained at
30 C. The mixture was stirred for 20 minutes to granulate solid. Additional
water (4.4 L) was
added over 5 minutes to drive the precipitation to completion and the mixture
was stirred for
another 30 minutes. The solid was collect by filtration and washed with a
mixture of DMF (0.5 L)
and water (1.5 L), then water (1.0 L). The product was dried in vacuum oven at
60 C for at least
48 hours to afford 293.91 g of product (94.2% HPLC purity). 'H NMR (300 MHz,
DMSO-d6) b
1.1(m, 5H), 1.36(s, 9H), 1.60(m, 6H), 3.93(s, 3H), 3.96(m, 1 H), 7.59(s, 1 H),
7.60(s, 1 H), 7.92(s,
1 H), 8.07(s, 1 H), 8.30(s, 1 H), 10.05(s, 1 H), 10.20(s, 1 H), 11.86(s, br, 1
H).
CA 02648369 2008-10-03
WO 2007/113647 PCT/IB2007/000859
-24-
Preparation of (2R)-2-amino-2-cyclohexyl-N-(5-(1-methyl-1 H-pyrazol-4-yi)-1-
oxo-2,6-
dihydro-1 H-[1,2]diazepino[4,5,6-cd]indol-8-yl)acetamide 12: Methansulfonic
acid (44.4 g, 0.46
mol) was added to THF (690 mL) in a 2-L flask. tert-Butyl (R)-1-cyclohexyl-2-
(5-(1-methyl-1H-
pyrazol-4-yl)-1-oxo-2,6-dihydro-1 H-[1, 2]diazepino[4, 5,6-cd]indol-8-ylamino)-
2-oxoethylcarbamate
11 (30.0 g, 57.74 mmol) was added via powder funnel. THF (60 mL) was used to
rinse the funnel
and the sides of the flask. The flask was purged with nitrogen, and the
reaction was heated to 65
C and stirred for 18-24h. HPLC analysis revealed that the reaction was
complete. The reaction
was cooled to room temperature using a water bath. 2M NaOH (255 mL) was added
over 30
minutes while maintaining the temperature at 20 5 C. After stirring for 5
minutes, the mixture
was transferred to a 2-L separatory funnel using THF for the rinse. The layers
were separated.
The aqueous phase was extracted with THF (60 mL). The organic fractions were
combined and
washed twice with saturated aqueous NaCI (2 x 60 mL). MeOH (80 mL) and MgSO4
(42 g) were
added. The mixture was stirred for 75 minutes, and then filtered through
celite. The cake was
washed with 9:1 THF/MeOH (3 x 50 mL), and the solution was transferred to a 2-
L distillation
flask. The solution was concentrated to a volume of 300 mL by distillation at
1 atmosphere.
EtOH (450 mL) was added slowly, and the solution was cooled to 50 C to
crystallize the product.
Once the product had crystallized, the mixture was re-heated and distilled
down to a volume of
450 mL. The reaction solvent ratio was monitored by 'H NMR. The distillation
was stopped once
THF content was reduced to 5 mol%. The resulting yellow suspension was cooled
to 25 C over
60 minutes and vacuum-filtered on paper. The filter cake was washed with EtOH
(2 x 75 ml).
The solids were transferred to a crystallizing dish and dried under vacuum at
55 C for 16 hours.
13.55 g of compound 12 (54%) was obtained as a yellow solid. 'H NMR (300 MHz,
DMSO-d6) 8
1.14(m, 5H), 1.62(m, 6H), 3.10(d, 1 H, J= 5.7 Hz), 3.93(s, 3H), 7.58(s, 1 H),
7.59(s, IH), 7.91(s,
1 H), 8.11(s, 1 H), 8.29(s, 1 H), 10.19(s, 1 H), 11.83(s, br, 1 H).
Example 2: preparation of (2R)-2-amino-2-cyclohexyl-N-(5-(1-methyl-1H-pyrazol-
4-yl)-1-oxo-
2,6-dihydro-1H-[1,2] diazepino[4,5,6-cd]indol-8-yl)acetamide polymorphic Form
I:
From HCI salt of Compound 1: To an aqueous solution of sodium bicarbonate (5%;
5 eq;
-18 mL) was added portion wise and with vigorous stirring, (2R)-2-amino-2-
cyclohexyl-N-(5-(1-
methyl-1 H-pyrazol-4-yl)-1-oxo-2,6-dihydro-1 H-[1,2] diazepino[4,5,6-cd]indol-
8-yl)acetamide
hydrochloride (1 g). To the mixture were added 500 mL of ethyl acetate and 80
mL of water. The
suspension was vigorously stirred for 10-15 minutes and stirring was stopped.
The organic phase
was decanted carefully into another erlenmeyer and the process was repeated 3
more time, each
time with 500 mL of ethyl acetate. The combined organic phase (-2000 mL) was
washed with
brine (100 mL), dried over sodium sulfate and filtered. The volatiles were
removed in vacuo and
the resulting yellow solid dried overnight to afford 670 mg of polymorphic
Form I of Compound 1
(-84% yield).
From the amorphous form of Compound 1: amorphous form of (2R)-2-amino-2-
cyclohexyl-N-(5-(1-methyl-1 H-pyrazol-4-yl)-1-oxo-2, 6-dihydro-1 H-[1,
2]diazepino[4, 5, 6-ct!]indol-8-
yl)acetamide (100 mg) was mixed with ethyl alcohol (1.5 mL) to form a
suspension. The
suspension was heated at 75-80 C for five hours with vigorous stirring and
let sit at 22 C for
CA 02648369 2008-10-03
WO 2007/113647 PCT/IB2007/000859
-25-
twelve hours. The solid was filtered and washed with 0.5 mL of ethyl alcohol.
It was then dried
with house vacuum for 24 hours and later, additionally dried at 25-30 C on a
vacuum pump for 12
hours to afford 66 mg of crystalline material.
Example 3: preparation of (2R)-2-amino-2-cyclohexyl-N-(5-(1-methyl-1H-pyrazol-
4-yl)-1-oxo-
2,6-dihydro-1H-[1,2] diazepino[4,5,6-cd]indol-8-yl)acetamide polymorphic Form
II:
Polymorphic Form I of Compound 1, (2R)-2-amino-2-cyclohexyl-N-(5-(1-methyl-lH-
pyrazol-4-yl)-1-oxo-2,6-dihydro-lH-[1,2] diazepino[4,5,6-cd]indol-8-
yl)acetamide (50mg) was
slurred in 1 mL of methanol at 4 C for 23 days. The slurry was centrifuged at
14,000 RPM to
separate the solid. The solid was dried in a low temperature vacuum oven to
give Form II.
Example 4a: Preparation of the first batch of amorphous form of (2R)-2-amino-2-
cyclohexyl-N-(5-(1-methyl-1 H-pyrazol-4-yi)-1-oxo-2,6-dihydro-1 H-[1,2]
diazepino[4,5,6-
cd]indol-8-yl)acetamide amorphous form:
1.3 g of compound 11 (2 HCI) was dissolved in water (200 mL) at 22 C followed
by the
addition of a saturated aqueous sodium bicarbonate solution (50 mL) and 500 mL
of ethyl
acetate. Two more spatulas of sodium bicarbonate were added and another 50 mL
of water. After
vigorous stirring, the mixture was filtered and the phases separated. The
aqueous phase was re-
extracted with ethyl acetate (two times 200 mL). The combined organic phase
was washed with
brine and dried over sodium sulfate. Filtration followed by evaporation of the
volatiles up to 10 mL
afforded a yellow solid, which was, filtered and dried. Total arriount of
amorphous material
obtained was 690 mg.
Example 4b: Preparation of the second batch of amorphous form of (2R)-2-amino-
2-
cyclohexyl-N-(5-(1-methyl-1 H-pyrazol-4-yl)-1-oxo-2,6-dihydro-1 H-[1,2]
diazepino[4,5,6-
cal]indol-8-yl)acetamide amorphous form:
749.2 mg of polymorphic Form I of Compound 1 was dissolved in 850 mL USP Grade
ethanol. The solution was vacuum filtered using a Buchner funnel and Whatman
Type 2 filter
paper. The filtered solution was transferred to a.190 mm x 100 mm
crystallization dish and
allowed to evaporate under ambient conditions for 63 hours. 589.1 mg of an
orange solid were
recovered as the second batch of amorphous form of Compound 1.
Example 5. 13C Solid State NMR of polymorphic Form I (the second spectrum),
Form II (the
second spectrum) and the amorphous form (the second batch) of Compound 1.
Samples were packed into a Zr02 rotors. Form I and amorphous samples were
packed
into 4mm rotors and the sample of form II was packed into 7mm rotor. The
carbon spectra were
collected at ambient conditions on a Bruker-Biospin 4 and 7mm CPMAS probes
positioned into a
wide-bore Bruker-Biospin Avance DSX 500 MHz NMR spectrometer. The rotors were
placed at
the magic angle and spun at 15.0 kHz (4mm rotors) and 7.0 kHz (7mm rotor). The
fast spinning
speed minimized the intensities of the spinning side bands. The number of
scans was adjusted to
obtain adequate S/N. The one dimensional 13C spectra of form I and amorphous
samples were
collected using'H-13C Cross-Polarization Magic Angle Spinning (CPMAS) which
was followed by
Total Suppression of Spinning side bands (TOSS) in the case of form II sample.
The TOSS was
applied to suppress the spinning side bands. To optimize the signal
sensitivity, the cross-
CA 02648369 2008-10-03
WO 2007/113647 PCT/IB2007/000859
-26-
polarization contact time was adjusted to 2.0 ms, and the decoupling field was
set to
approximately 75 kHz. 512 scans were acquired with recycle delay of 30s for
form I, 256 scans
were acquired with recycle delay of 30s for form II and 2048 scans were
acquired with recycle
delay of 3s for the amorphous sample. All spectra were referenced using an
external sample of
adamantane with its upfield signal set to 29.5 ppm.