Note: Descriptions are shown in the official language in which they were submitted.
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-1-
TRIAZOLOPYRIDAZINE DERIVATIVES
This application claims the benefit of U. S. Provisional Application No.
60/803,469 filed on May
30, 2006, the contents of which is hereby incorporated by reference in its
entirety.
Introduction
This invention relates to novel triazolopyridazine derivatives that are useful
in the treatment of
hyperproliferative diseases, such as cancers, in mammals. This invention also
relates to a method of
using such compounds in the treatment of hyperproliferative diseases in
mammals, especially humans,
and to pharmaceutical compositions containing such compounds.
Backciround of the Invention
The hepatocyte growth factor (HGF) receptor (c-MET or HGFR) receptor tyrosine
kinase (RTk)
has been shown in many human cancers to be involved in oncogenesis, tumor
progression with enhanced
cell motility and invasion, as well as metastasis (see, e.g., Ma, P.C.,
Maulik, G., Christensen, J. & Salgia,
R. (2003b). Cancer Metastasis Rev, 22, 309-25; Maulik, G., Shrikhande, A.,
Kijima, T., Ma, P.C.,
Morrison, P.T. & Salgia, R. (2002b). Cytokine Growth Factor Rev, 13, 41-59). c-
MET (HGFR) can be
activated through overexpression or mutations in various human cancers
including small cell lung cancer
(SCLC) (Ma, P.C., Kijima, T., Maulik, G., Fox, E.A., Sattler, M., Griffin,
J.D., Johnson, B.E. & Salgia, R.
(2003a). Cancer Res, 63, 6272-6281).
c-MET is a receptor. tyrosine kinase that is encoded, by the Met proto-
oncogene and transduces
the biological effects of hepatocyte growth factor (HGF), which is also
referred to as scatter factor (SF).
Jiang et aL, Crit. Rev. Onco% Hematol. 29: 209-248 (1999). c-MET and HGF are
expressed in numerous
tissues, although their expression is normally confined predominantly to cells
of epithelial and
mesenchymal origin, respectively. c-MET and HGF are required for normal
mammalian development and
have been shown to be important in cell migration, cell proliferation and
survival, morphogenic
differentiation, and organization of 3-dimensional tubular structures (e.g.,
renal tubular cells, gland
formation, etc.). In addition to its effects on epithelial cells, HGF/SF has
been reported to be an
angiogenic factor, and c-MET signaling in endothelial cells can induce many of
the cellular responses
necessary for angiogenesis (proliferation, motility, invasion).
The c-MET receptor has been shown to be expressed in a number of human
cancers. c-Met and
its ligand, HGF, have also been shown to be co-expressed at elevated levels in
a variety of human
cancers (particularly sarcomas). However, because the receptor and ligand are
usually expressed by
different cell types, c-MET signaling is most commoniy regulated by tumor-
stroma (tumor-host)
interactions. Furthermore, c-MET gene amplification, mutation, and
rearrangement have been observed
in a subset of human cancers. Families with germline mutations that activate c-
MET kinase are prone to
multiple kidney tumors as well as tumors in other tissues. Numerous studies
have correlated the
expression of c-MET and/or HGF/SF with the state of disease progression of
different types of cancer
(including lung, colon, breast, prostate, liver, pancreas, brain, kidney,
ovaries, stomach, skin, and bone
cancers). Furthermore, the overexpression of c-MET or HGF have been shown to
correlate with poor
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-2-
prognosis and disease outcome in a number of major human cancers including
lung, liver, gastric, and
breast. c-MET has also been directly implicated in cancers without a
successful treatment regimen such
as pancreatic cancer, glioma, and hepatocellular carcinoma.
A family of novel compounds have been discovered which exhibit c-Met
modulating ability and
have an ameliorating effect against disorders related to abnormal c-Met
activity. c-Met is an attractive
target from a clinical perspective because: 1) c-Met has been implicated in
the growth and metastases of
most types of cancer; 2) growth at the secondary site appears to be the rate-
limiting step in metastasis;
and 3) by the time of diagnosis, R is likely that the disease has already
spread.
These observations suggest that c-Met kinase inhibitors would be an effective
treatment for
primary tumors that are driven by c-Met, but more importantly, would prevent
disseminated
micrometastases from growing into life-threatening metastases. Therefore, the
utility of a c-Met inhibitor
extends to preventative and adjuvant therapy settings. In addition, certain
cancers (e.g., papillary renal
cell carcinoma, some gastric and lung cancers) can be treated which are
believed to be driven by c-Met
mutation/genetic alteration and dependent on c-Met for growth and survival.
These cancers are expected
to be sensitive to treatment. Furthermore, various human cancers are the
primary target indication for c-
Met antagonists. These cancers include major cancers such as breast, lung,
colorectal, prostate; as well
as pancreatic cancer, glioma, liver cancer, gastric cancer, head and neck
cancers, melanoma, renal
cancer, leukemias, myeloma, and sarcomas. c-Met has been directly implicated
in cancers such as
pancreatic cancer, glioma, and hepatocellular carcinoma.
Accordingly, c-Met (HGFR) inhibitors and methods of using such inhibitors for
the treatment of
abnormal cell growth, such as cancer represent a substantial unmet medical
need in the treatment of
these and possibly other cancers.
Summary of the Invention
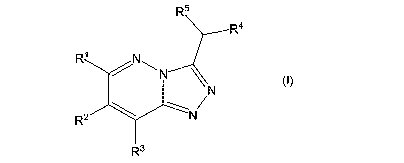
In one embodiment, the present invention relates to a compound of the formula
I:
R5
R4
R NN
N
R2 N
R3
wherein:
R1, R2 and R3 are independently selected from hydrogen, Br, Cl, F, -
O(CH2)nCH3, -O(CH2)õOR6,
-(CH2)nOR6, -C(O)Rs, -C(O)OR6, -C(O)NR6W, -NR6R7, -S(O)2R6, -S(O)R6, -
S(O)2NR6R7, -CF3, -CF2H, -
NR6C(O)NR6R7, -NR6C(O)R7, -NR6S(O)2W, -N(CH2),1(C3-C8 cycloalkyl), -CN, -NO2,
Cl-C6 alkyl, C3-C8
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-3-
cycloalkyl, 3-8 membered heteroalicyclic, 3-8 membered heteroalicyclic-(3-8
membered heteroalicyclic),
8-10 membered heterobicyclic, 5-7 membered heteroaryl, C6-C10 aryl, C2-C6
alkenyl, and C2-C6 alkynyl
wherein C1-C6 alkyl, C3-C8 cycloalkyl, 3-8 membered heteroalicyclic, 8-10
membered heterobicyclic, 5-7
membered heteroaryl, C6-C10 aryl, C2-C6 alkenyl, and C2-C6 alkynyl are
optionally substituted by one or
more moieties selected from the group consisting of Br, Cl, F, -
(CH2)nCH(OR6)CH3, -(CH2)nOR6, -
(CH2)nC(CH3)2OR6, -(CH2)n(3-8 membered heteroalicyclic), -C(O)R6, -C(O)OR6, -
(CR6R')nC(O)OR6,
-C(O)NR6R', -(CR6R')nC(O)NR6W, -(CH2)nNR6R7, -S(O)2R6, -S(O)R6, -S(O)2NR6R7, -
CF3, -CF2H,
-(CH2)nNR6C(O)NR6R7, -(CH2)nNR6C(O)OR7, -NR6C(O)R', -NR6C(O)OR', -NR6S(O)2R', -
CN, -NO2, oXO,
C1-C6 alkyl, C3-C8 cycloalkyl, -(CH2)n(3-8 membered heteroalicyclic), -
(CH2)n(5-7 membered heteroaryl),
-(CH2)n(C6-C10 aryl), C2-C6 alkenyl, and C2-C6 alkynyl;
R4 is a 8-10 membered heterobicyclic optionally substituted by one or more
moieties selected
from the group consisting of Br, Cl, F, -(CH2)nCH(OR6)CH3, -(CH2)nOR6, -
(CH2)nC(CH3)2OR6, -(CH2)n(3-8
membered heteroalicyclic), -C(O)R6, -C(O)OR6, -(CR6R')nC(O)OR6, -C(O)NR6R', -
(CR6R')nC(O)NR6R7, -
(CH2)nNR6R7, -S(O)ZR6, -S(O)R6, -S(O)2NR6R7, -CF3, -CF2H, -(CH2)nNR6C(O)NR6R7,
-(CH2)nNR6C(O)OR7,
-NR6C(O)R', -NR6C(O)OR', -NR6S(O)2R', -CN, -NO2, oxo, C1-C6 alkyl, C3-C8
cycloalkyl, -(CH2)n(3-8
membered heteroalicyclic), -(CH2)n(5-7 membered heteroaryl), -(CH2)n(C6-C10
aryl), C2-C6 alkenyl, and C2-
C6 alkynyl;
R5 is selected from the group consisting of hydrogen, F, -CF3, C1-C6 alkyl and
aryl;
R6 and R' are independently selected from H, -(CH2)nOR8, -(CH2)nC(CH3)2OR8, -
CHR$(CH2)nOR9,
-(CH2)nCHR8OR9, -C(CH3)2(CH2)nOR8, -CH2CF2H, -(CH2)nC(CH3)2NR$R9, -
(CH2)nNR8R9,
-(CH2)nCHOR8(CH2)nOR9, -(CH2)n(NR8R9)C(O)NR8R9, -(CH2)nS(O)2R8, -
(CH2)nC(O)NRaR9, -(CH2)nC(O)R8,
-NR$(CH2)n(5-7 membered heteroaryl), -NR8(CH2)n(3-8 membered heterocycle), -
(CH2)n(8-10 membered
heterobicyclic), -(CH2)n(3-8 membered heteroalicyclic), Cj-C6 alkyl, C3-C8
cycloalkyl, C6-Clo aryl, CZ-C6
alkenyl, 3-8 membered heteroalicyclic and C2-C6 alkynyl, wherein said 5-7
membered heteroaryl, 3-8
membered heterocycle and 8-10 membered heterobicyclic are optionally
substituted by one or more
moieties selected from the group consisting of -(CH2)nOR8, C1-C6 alkyl, C3-C8
cycloalkyl, C6-C10 aryl, C2-
C6 alkenyl, 3-8 membered heteroalicyclic and C2-C6 alkynyi; or when R6 and R7
are attached to the same
atom, R6 and R' optionally combine to form a 3-8 membered heteroalicyclic
ring;
R8 and R9 are independently selected from H, C1-C6 alkyl, -C(O)CH3, C3-C8
cycloalkyl, C6-C10 aryl,
C2-C6 alkenyl, 5-7 membered heteroaryl and C2-C6 alkynyl, wherein said 5-7
membered heteroaryl is
optionally substituted by one or more moieties selected from the group
consisting of C1-C6 alkyl, C3-C8
cycloalkyl, C6-C10 aryl, C2-C6 alkenyl, and C2-C6 alkynyl; or when R12 and R13
are attached to the same
atom, R12 and R13 optionally combine to form a 3-8 membered heteroalicyclic
ring; and
n is 0, 1, 2, 3 or 4;
or a pharmaceutically acceptable salt thereof.
In another embodiment, R1, R2 and R3 are independently selected from hydrogen,
Cl, -OR10,
-O(CH2)nCH3, -OCH2(CH2)nOR10, -C(O)NR10R11, -NR'0R", C1-C6 alkyl, 3-8 membered
heteroalicyclic, 3-8
membered heteroalicyclic-(3-8 membered heteroalicyclic), 8-10 membered
heterobicyclic, 5-7 membered
heteroaryl, C6-C10 aryl and C2-C6 alkenyl, wherein C1-C6 alkyl, 3-8 membered
heteroalicyclic, 3-8
membered heteroalicyclic-(3-8 membered heteroalicyclic), 8-10 membered
heterobicyclic, 5-7 membered
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-4-
heteroaryl, C6-C10 aryl and C2-C6 alkenyl are optionally substituted by one or
more moieties selected from
the group consisting of Br, Cl, F, -(CH2),CH(OR10)CH3, -(CH2)nOR10, -
(CH2),,C(CH3)2OR10, -(CH2)n(3-8
membered heteroalicyclic), -C(O)R10, -C(O)OR10, -(CR10R11)nC(O)OR10, -
C(O)NR1oR11,
- CR10R11 ~,C O NR10R11, - 10R11 10 10 10R11
( ) ( ) (CH2)õNR , -S(O)2R, -S(O)R , -S(O)ZNR , -CF3, -CF2H,
-(CH2)nNR10C(O)NR1oR11, -(CH2)nNR10C(O)OR11, -NR10C(O)R11, -NR10C(O)OR11, -
NR10S(O)2R11, -CN, -
NO2, oxo, C1-C6 alkyl, C3-C8 cycloalkyl, -(CH2)n(3-8 membered
heteroalicyclic), -(CH2)n(5-7 membered
heteroaryl), -(CH2)õ(C6-C10 aryl), C2-C6 alkenyl, and C2-C6 alkynyl.
In a further embodiment, R1 is selected from Cl, 3-8 membered heteroalicyclic-
(3-8 membered
heteroalicyclic), 5-7 membered heteroaryl, and C6-C10 aryl, wherein 3-8
membered heteroalicyclic-(3-8
membered heteroalicyclic), 5-7 membered heteroaryl and C6-C10 aryl are
optionally substituted by one or
more moieties selected from the group consisting of -(CH2)rOR1o, -C(O)OR10, -
(CR10R11)nC(O)NR10R11
, -
(CH2)nNR10R11, -CF3, and -CN.
In another embodiment, R2 and R3 are H. In another embodiment, R5 is H. In
another
embodiment, R5 is C1-C6 alkyl. In another embodiment, R5 is methyl. In another
embodiment, R4 is
selected from
I , I and
F N/ N N H N
In still a further embodiment, the present invention provides for a compound
of the formula (I)
selected from 6-(1-methyl-1 H-pyrazol-4-yl)-3-[(S)-1-(1 H-pyrrolo[2,3-
b]pyridin-3-yl)-ethyl]-
[1,2,4]triazolo[4,3-b]pyridazine, 7-methyl-6-{[6-(1-methyl-1 H-pyrazol-4-
yl)[1,2,4]triazolo[4,3-b]pyridazin-3-
yl]methyl}quinoline, 6-{(S)-1-[6-(1-methyl-1 H-pyrazol-4-yl)-
[1,2,4]triazolo[4,3-b]pyridazin-3-yl]-ethyl}-
quinoline, 6-((6-(1 H-pyrazol-4-yl)-[1,2,4]triazolo[4,3-b]pyridazin-3-
yl)methyl)quinoline, 4-(3-(quinolin-6-
ylmethyl)-[1,2,4]triazolo[4,3-b]pyridazin-6-yl)benzonitrile, 3-[(7-
methylquinolin-6-yl)methyl]-N-
(tetrahydrofuran-3-yl)[1,2,4]triazolo[4,3-b]pyridazin-6-amine, N-cyclopentyl-3-
[(7-methylquinolin-6-
yl)methyl][1,2,4]triazolo[4,3-b]pyridazin-6-amine, 4-{3-[(S)-1-(1 H-
pyrrolo[2,3-b]pyridin-3-yl)-ethyl]-
[1,2,4]triazolo[4,3-b]pyridazin-6-yl}-benzonitrile, isopropyl-[3-((S)-1-
quinolin-6-yl-ethyl)-[1,2,4]triazolo[4,3-
b]pyridazin-6-yl]-amine, [3=(1-quinolin-6-yl-ethyl)-[1,2,4]triazolo[4,3-
b]pyridazin-6-yl]-(tetrahydro-furan-3-
yl)-amine, 2-[3-(1-quinolin-6-yl-ethyl)-[1,2,4]triazolo[4,3-b]pyridazin-6-
ylamino]-ethanol, and 4-[3-((S)-1-
quinolin-6-yl-ethyl)-[1,2,4]triazolo[4,3-b]pyridazin-6-yl]-benzonitrile; or a
pharmaceutically acceptable salt
thereof.
In a further embodiment, the present invention relates to a coumpound selected
from any 10
compounds exemplified in Table 1.
In a further embodiment, the present invention relates to any coumpound
exemplified in Table 1.
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-5-
In another embodiment, the present invention provides a pharmaceutical
composition comprising
a compound according to the formula (I) or a pharmaceutically acceptable salt
thereof, and a
pharmaceutically acceptable excipient.
In another embodiment, the present invention provides for the use of a
compound of the formula
(I) or a pharmaceutically acceptable salt thereof, for the manufacture of a
medicament to treat a c-Met
related disorder in a mammal. In another embodiment, the present invention
provides for the use of a
compound of the formula (I) or a pharmaceutically acceptable salt thereof, for
the manufacture of
medicament for the treatment of cancer in a mammal. In another embodiment, the
cancer is selected from
breast cancer, lung cancer, colorectal cancer, prostate cancer, pancreatic
cancer, glioma, liver cancer,
gastric cancer, head cancer, neck cancer, melanoma, renal cancer, leukemia,
myeloma, and sarcoma.
In another embodiment, the present invention provides a method of treating a
mammal having a
c-Met related disorder, comprising administering to the mammal an effective
amount of a compound of the
formula (I) or with a pharmaceutically acceptable salt thereof.
In another embodiment, the present invention provides a method of treating a
mammal having
.15 cancer, comprising administering to the mammal an effective amount of a
compound of the formula (I) as
defined in any one of claims 1-9 or with a pharmaceutically acceptable salt
thereof.
In another embodiment, the cancer is selected from breast cancer, lung cancer,
colorectal cancer,
prostate cancer, pancreatic cancer, glioma, liver cancer, gastric cancer, head
cancer, neck cancer,
melanoma, renal cancer, leukemia, myeloma, and sarcoma. In another embodiment,
the mammal is a
human. In another embodiment, the mammal is a canine.
It will be understood that each of the embodiments described herein may be
combined with one
or more other embodiments described herein, where reasonable, to form
additional embodiments within
the scope of the invention.
Definitions
"Pharmaceutically acceptable salt" refers to those salts, which retain the
biological effectiveness
and properties of the parent compound. Such salts include:
acid addition salt which is obtained by reaction of the free base of the
parent compound with
inorganic acids such as hydrochloric acid, hydrobromic acid, hydroiodic acid,
nitric acid, phosphoric acid,
sulfuric acid, and perchloric acid and the like, or with organic acids such as
acetic acid, oxalic acid, (D) or
(L) malic acid, maleic acid, methanesulfonic acid, ethanesulfonic acid, p-
toluenesulfonic acid, salicylic
acid, tartaric acid, benzenesulfonic acid (besylate), benzoic acid,
camphorsulfonic add, citric acid, fumaric
acid, gluconic acid, glutamic acid, isethionic acid, lactic acid, maleic acid,
malic acid, mandelic acid, mucic
acid, pamoic acid, pantothenic acid, succinic acid, tartaric acid, or malonic
acid and the like, preferably
hydrochloric acid or (L)-malic acid; or
salts formed when an acidic proton present in the parent compound either is
replaced by a metal
ion, e.g., an alkali metal ion, an alkaline earth ion, or an aluminum ion; or
coordinates with an organic
base such as ethanolamine, diethanolamine, triethanolamine, tromethamine, N-
methylglucamine, and the
like.
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-6-
"Pharmaceutically acceptable excipient" or "excipient" refers to an inert
substance added to a
pharmaceutical composition to further facilitate administration of a compound.
Examples, without
limitation, of excipients include calcium carbonate, calcium phosphate,
various sugars and types of starch,
cellulose derivatives, gelatin, vegetable oils, polyethylene glycols,
diluents, granulating agents, lubricants,
binders, disintegrating agents, and the like.
A "pharmaceutical composition" refers to a mixture of one or more of the
compounds described
herein, or physiologically acceptable salts thereof, with other chemical
components, such as
physiologically acceptable carriers and excipients. The purpose of a
pharmaceutical composition is to
facilitate administration of a compound to an organism.
As used herein, a "physiologically acceptable carrier" refers to a carrier or
diluent that does not
cause significant irritation to an organism and does not abrogate the
biological activity and properties of
the administered compound.
The term "method" refers to manners, means, techniques and procedures for
accomplishing a
given task including, but not limited to, those manners, means, techniques and
procedures either known
to, or readily developed from known manners, means, techniques and procedures
by, practitioners of the
chemical, pharmaceutical, biological, biochemical and medical arts.
As used herein, the term "modulation" or "modulating" refers to the ateration
of the catalytic
activity of c-Met. In particular, modulating refers to the activation of the
catalytic activity of c-Met,
preferably the activation or inhibition of the catalytic activity of c-Met,
depending on the concentration of
the compound or salt to which c-Met is exposed or, more preferably, the
inhibition of the catalytic activity
of c-Met.
The term "contacting" as used herein refers to bringing a compound of this
invention and c-Met
together in such a manner that the compound can affect the catalytic activity
of c-Met, either directly, i.e.,
by interacting with c-Met itself, or indirectly, i.e., by interacting with
another molecule on which the
catalytic activity of c-Met is dependent. Such "contacting" can be
accomplished in vitro, i.e., in a test tube,
a petri dish or the like. In a test tube, contacting may involve only a
compound and c-Met or it may involve
whole cells. Cells may also be maintained or grown in cell culture dishes and
contacted with a compound
in that environment. In this context, the ability of a particular compound to
affect a c-Met related disorder,
i.e., the IC50 of the compound, defined below, can be determined before use of
the compounds in vivo
with more complex living organisms is attempted. For cells outside the
organism, multiple methods exist,
and are well-known to those skilled in the art, to get c-Met in contact with
the compounds including, but
not limited to, direct cell microinjection and numerous transmemtrane carrier
techniques.
"In vitro" refers to procedures performed in an artificial environment such
as, e.g., without
limitation, in a test tube or culture medium. The skilled artisan will
understand that, for example, isolated c-
Met may be contacted with a modulator in an in vitro environment.
Alternatively, an isolated cell may be
contacted with a modulator in an in vitro environment.
As used herein, "in vivo" refers to procedures performed within a living
organism such as, without
limitation, a mouse, rat, rabbit, ungulate, bovine, equine, porcine, canine,
feline, primate, or human.
As used herein, "c-Met related disorder," refers to a condition characterized
by inappropriate, i.e.,
under-activity or, more commonly, over-activity of the c-Met catalytic
activity. A "c-Met related disorder"
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-7-
also refers to a condition where there may be a mutation in the gene that
produces c-Met, which, in turn,
produces a c-Met that has an increased or decreased c-Met catalytic activity.
Inappropriate catalytic activity can arise as the result of either: (1) c-Met
expression in cells which
normally do not express c-Met, (2) increased c-Met expression leading to
unwanted cell proliferation,
differentiation and/or growth, or, (3) decreased c-Met expression leading to
unwanted reductions in cell
proliferation, differentiation and/or growth. Over-activity of a c-Met refers
to either amplification of the gene
encoding a c-Met or production of a level of c-Met activity which can
correlate with a cell proliferation,
differentiation and/or growth disorder (that is, as the level of the c-Met
increases, the severity of one or
more of the symptoms of the cellular disorder increases). Under-activity is,
of course, the converse,
wherein the severity of one or more symptoms of a cellular disorder increase
as the level of the c-Met
activity decreases.
As used herein, the terms "treat", "treating" and "treatment" refer to a
method of alleviating or
abrogating a c-Met mediated cellular disorder and/or its attendant symptoms.
With regard particularly to
cancer, these terms simply mean that the life expectancy of an individual
affected with a cancer will be
increased or that one or more of the symptoms of the disease will be reduced.
The term "organism" refers to any living entity comprised of at least one
cell. A living organism
can be as simple as, for example, a single eukaryotic cell or as complex as a
mammal. In a preferred
aspect, the organism is a mammal. In a particularly preferred aspect, the
mammal is a human being.
The term "therapeutically effective amount" as used herein refers to that
amount of the compound
being administered which will relieve to some extent one or more of the
symptoms of the disorder being
treated. In reference to the treatment of cancer, a therapeutically effective
amount refers to that amount
which has the effect of (1) reducing the size of the tumor, (2) inhibiting
(that is, slowing to some extent,
preferably stopping) tumor metastasis, (3) inhibiting to some extent (that is,
slowing to some extent,
preferably stopping) tumor growth, and/or, (4) relieving to some extent (or,
preferably, eliminating) one or
more symptoms associated with the cancer.
By "monitoring" is meant observing or detecting the effect of contacting a
compound with a cell
expressing a c-Met. The observed or detected effect can be a change in cell
phenotype, in the catalytic
activity of c-Met or a change in the interaction of c-Met with a natural
binding partner. Techniques for
observing or detecting such effects are well-known in the art. For example,
the catalytic activity of c-Met
may be observed by determining the rate or amount of phosphorylation of a
target molecule.
"Cell phenotype" refers to the outward appearance of a cell or tissue or the
biological function of
the cell or tissue. Examples, without limitation, of a cell phenotype are cell
size, cell growth, cell
proliferation, cell differentiation, cell survival, apoptosis, and nutrient
uptake and use. Such phenotypic
characteristics are measurable by techniques well-known in the art.
A "natural binding partner" refers to a polypeptide that binds to a c-Met in a
cell. Natural binding
partners can play a role in propagating a signal in a c-Met-mediated signal
transduction process. A
change in the interaction of the natural binding partner with c-Met can
manifest itself as an increased or
decreased concentration of the c-Met/natural binding partner complex and, as a
result, in an observable
change in the ability of c-Met to mediate signal transduction.
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-8-
As used herein, "administer" or "administration" refers to the delivery of a
compound or salt of the
present invention or of a pharmaceutical composition containing a compound or
salt of this invention to an
organism for the purpose of prevention or treatment of a c-Met-related
disorder.
The terms "abnormal cell growth" and "hyperproliferative disorder" are used
interchangeably in
this application.
"Abnormal cell growth", as used herein, refers to cell growth that is
independent of normal
regulatory mechanisms (e.g., loss of contact inhibition), including the
abnormal growth of normal cells and
the growth of abnormal cells. This includes, but is not limited to, the
abnormal growth of: (1) tumor cells
(tumors), both benign and malignant, expressing an activated Ras oncogene; (2)
tumor cells, both benign
and malignant, in which the Ras protein is activated as a result of oncogenic
mutation in another gene; (3)
benign and malignant cells of other proliferative diseases in which aberrant
Ras activation occurs.
Examples of such benign proliferative diseases are psoriasis, benign prostatic
hypertrophy, human
papilloma virus (HPV), and restinosis. "Abnormal cell growth" also refers to
and includes the abnormal
growth of cells, both benign and malignant, resulting from activity of the
enzyme farnesyl protein
transferase.
"Alkyl" refers to a saturated aliphatic hydrocarbon including straight chain
or branched chain.
Preferably, the alkyl group has 1 to 20 carbon atoms (whenever a numerical
range; e.g., "1-20", is stated
herein, it means that the group, in this case the alkyl group, may contain 1
carbon atom, 2 carbon atoms,
3 carbon atoms, etc. up to and including 20 carbon atoms). More preferably, it
is a medium size alkyl
having 1 to 10 carbon atoms. Most preferably, it is a lower alkyl having 1 to
6 carbon atoms. The alkyl
group may be substituted or unsubstituted. When substituted, each substituent
group is preferably one or
more individually selected from halogen, -hydroxy, -COR', -COOR', -OCOR', -
CONRR', -RNCOR', -NRR',
-CN, -NOZ, -CF3 -SR', -SOR', -SO2R', -SO2OR', -SO2NRR', thiocarbonyl, -
RNSO2R', perfluoroalkyl, 0-
carbamyl, N-carbamyl, 0-thiocarbamyl, N-thiocarbamyl, silyl, ammonium, lower
alkyl, lower alkenyl, lower
alkynyl, cycloalkyl, heteroalicycle, heteroaryl and aryl. R and R' can be
independently H, alkyl, or aryl,
wherein alkyl or aryl may be further substituted with halogen, (CHAN(R")Z,
(CHACO2R", (CHAOR",
(CHz)õOC(O)R", alkoxycarbonyl, aryloxycarbonyl, aminocarbonyl, a
heteroalicyclic ring, aryl, alkoxy,
-OCF3, aryloxy, C(O)NH2 or heteroaryl. R" can be H, alkyl or aryl. n is 0-3.
"Alkenyl" refers to an aliphatic hydrocarbon having at least one carbon-carbon
double bond,
including straight chain, branched chain or cyclic groups having at least one
carbon-carbon double bond.
Preferably, the alkenyl group has 2 to 20 carbon atoms (whenever a numerical
range; e.g., "2-20", is
stated herein, it means that the group, in this case the alkenyl group, may
contain 2 carbon atoms, 3
carbon atoms, etc. up to and including 20 carbon atoms). More preferably, it
is a medium size alkenyl
having 2 to 10 carbon atoms. Most preferably, it is a lower alkenyl having 2
to 6 carbon atoms. Examples,
without limitation, of alkenyl groups include 1-propenyl, 1- and 2-butenyl,
etc. The alkenyl group may be
substituted or unsubstituted. When substituted, each substituent group is
preferably one or more
individually selected from halogen, -hydroxy, -COR', -COOR', -OCOR', -CONRR', -
RNCOR', -NRR', -CN, -
NO2i -CF3, -SR', -SOR', -SO2R', -SOzOR', -SO2NRR', thiocarbonyl, -RNSO2R',
perfluoroalkyl, O-carbamyl,
N-carbamyl, 0-thiocarbamyl, N-thiocarbamyl, silyl, ammonium, lower alkyl,
lower alkenyl, lower alkynyl,
cycloalkyl, heteroalicycle, heteroaryl and aryl. Wherein R and R' are defined
herein.
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-9-
"Alkynyl" refers to an aliphatic hydrocarbon having at least one carbon-carbon
triple bond,
including straight chain, branched chain or cyclic groups having at least one
carbon-carbon triple bond.
Preferably, the.alkenyl group has 2 to 20 carbon atoms (whenever a numerical
range; e.g., "2-20", is
stated herein, it means that the group, in this case the alkynyl group, may
contain 2 carbon atoms, 3
carbon atoms, etc. up to and including 20 carbon atoms). More preferably, it
is a medium size alkynyl
having 2 to 10 carbon atoms. Most preferably, it is a lower alkynyl having 2
to 6 carbon atoms. Examples,
without limitation, of alkynyl groups include 1-propynyl, 1- and 2-butynyl,
etc. The alkynyl group may be
substituted or unsubstituted. When substituted, each substituent group is
preferably one or more
individually selected from halogen, -hydroxy, -COR', -COOR', -OCOR', -CONRR', -
RNCOR', -NRR', -CN, -
NO2, -CF3, -SR', -SOR', -SO2R', -SO2OR', -SOZNRR', thiocarbonyl, -RNSO2R',
perfluoroalkyl, 0-carbamyl,
N-carbamyl, O-thiocarbamyl, N-thiocarbamyl, silyl, ammonium, lower alkyl,
lower alkenyl, lower alkynyl,
cycloalkyl, heteroalicycle, heteroaryl and aryl. Wherein R and R' are defined
herein.
A "cycloalkyl" or an "alicyclic" group refers to an all-carbon monocyclic or
fused ring (i.e., rings
which share an adjacent pair of carbon atoms) group wherein one of more of the
rings does not have a
completely conjugated pi-electron system. Preferably, the cycloalkyl group has
from 3-8 carbon atoms in
the ring(s). Examples, without limitation, of cycloalkyl groups are
cyclopropane, cyclobutane,
cyclopentane, cyclopentene, cyclohexane, adamantane, cyclohexadiene,
cycloheptane and,
cycloheptatriene. A cycloalkyl group may be substituted or unsubstituted. When
substituted, each
substituent group is preferably one or more individually selected from
halogen, -hydroxy, -COR', -COOR',
-OCOR', -CONRR', -RNCOR', -NRR', -CN, -NO2, -CF3, -SR', -SOR', -SO2R', -
SO2OR', -SO2NRR',
thiocarbonyl, -RNSO2R', perfluoroalkyl, 0-carbamyl, N-carbamyl, 0-
thiocarbamyl, N-thiocarbamyl, silyl,
ammonium, lower alkyl, lower alkenyl, lower alkynyl, cycloalkyl,
heteroalicycle, heteroaryl and aryl.
Wherein R and R' are defined herein.
An "aryl" group refers to an all-carbon monocyclic or fused-ring polycyclic
(i.e., rings which share
adjacent pairs of carbon atoms) groups having a completely conjugated pi-
electron system. Preferably,
the aryl group has from 6 to 12 carbon atoms in the riong(s). Examples,
without limitation, of aryl groups
are phenyl, naphthalenyl and anthracenyl. The aryl group may be substituted or
unsubstituted. When
substituted, each substituted group is preferably one or more selected
halogen, hydroxy, alkoxy, aryloxy, -
COR', -COOR', -OCOR', -CONRR', -RNCOR', -NRR', -CN, -NO2, -CF3, -SR', -SOR', -
SO2R', -SO2OR', -
SO2NRR', thiocarbonyl, -RNSO2R', perfluoroalkyl, 0-carbamyl, N-carbamyl, 0-
thiocarbamyl, N-
thiocarbamyl, silyl, ammonium, lower alkyl, lower alkenyl, lower alkynyl,
cycloalkyl, heteroalicycle,
heteroaryl and aryl. Wherein R and R' are defined herein.
As used herein, a "heteroaryl" group refers to a monocyclic group having in
the ring one or more
atoms selected from the group consisting of nitrogen, oxygen and sulfur with
the proviso that heteroaryl
groups containing highly unstable heteroatom arrangements, such as 0-0, 0-0-0
and the like, are not
contemplated by the present invention. One of ordinary skill in the art will
recognize unstable groups that
are not contemplated by the invention. In addition, the heteroaryl group has a
completely conjugated pi-
electron system. Preferably, the heteroaryl group has from 5 to 7 ring atoms.
Examples of typical monocyclic
heteroaryl groups include, but are not limited to:
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-10-
N O O ~/ ~/N N
pyrrole furan thiophene pyrazole imidazole
(pyrrolyl) (furanyl) (thiophenyl) (pyrazolyl) (imidazolyl)
H
O ON Q
\ /N isoxazole oxazole isothiazole thiazolyl 1,2,3-triazole
(isoxazolyl) (oxazolyl) (isothiazolyl) (thiazolyl) (1,2,3-triazolyl)
H
(N) ON, 011~ "Ioll,
c " N NN
N-N N N
1,3,4-triazole 1-oxa-2,3-diazole 1-oxa-2,4-diazole 1-oxa-2,5-diazole
(1,3,4-triazolyl) (1 -oxa-2,3-diazolyl) (1 -oxa-2,4-diazolyl) (1-oxa-2,5-
diazolyl)
<~~ CC " <~ N~
N-N N N
1-oxa-3,4-diazole 1-thia-2,3-diazole 1 -thia-2,4-diazole 1-thia-2,5-diazole
(1 -oxa-3,4-diazolyl) (1 -thia-2,3-diazolyl) (1 -thia-2,4-diazolyl) (1 -thia-
2,5-diazolyl)
H
S// \\N/N I ~ (JN N
N-N N-N
1-thia-3,4-diazole tetrazole pyridine pyridazine pyrimidine
(1-thia-3,4-diazolyl) (tetrazolyl) (pyridinyl) (pyridazinyl) (pyrimidinyl)
When substituted, each substituted group is preferably one or more selected
from halogen,
hydroxy, -COR', -COOR', -OCOR', -CONRR', -RNCOR', -NRR', -CN, -NO2, -CF3, -
SR', -SOR', -SO2R', -
SO2OR', -SO2NRR', thiocarbonyl, -RNSO2R', perfluoroalkyl, 0-carbamyl, N-
carbamyl, 0-thiocarbamyl, N-
thiocarbamyl, silyl, ammonium, lower alkyl, lower alkenyl, lower alkynyl,
cycloalkyl, heteroalicycle,
heteroaryl and aryl. Wherein R and R' are defined herein.
A "heteroalicyclic ring" or "heteroalicycle" or "heterocyclic" or
"heterocycle" group refers to a
monocyclic group having in the ring one or more atoms selected from the group
consisting of nitrogen,
oxygen and sulfur. The rings may be saturated and also have one or more double
bonds (i.e. partially
unsaturated). However, the rings may not have a completely conjugated pi-
electron system. Preferably,
the heteroalicyclic ring contains from 3 to 8 ring atoms. Examples of suitable
saturated heteroalicyclic
groups include, but are not limited to:
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
- 1 1 -
0 s ~ N H a
oxirane thiarane aziridine oxetane thiatane azetidine tetrahydrofuran
(oxiranyl) (thiaranyl) (aziridinyl) (oxetanyl) (thiatanyl) (azetidinyl)
(tetrahydrofuranyl)
O 000
tetrahydrothiophene pyrrolidine tetrahydropyran tetrahydrothiopyran
(tetrahydrothiophenyl) (pyrrolidinyl) (tetrahydropyranyl)
(tetrahydrothiopyranyl)
H H
N O O C~ N S
C~ C
o S o sC)
piperidine 1,4-dioxane 1,4-oxathiane morpholine 1,4-dithiane
(piperidinyl) (1,4-dioxanyl) (1,4-oxathianyl) (morpholinyl) (1,4-dithianyl)
H H H
N O S N
C;)
SC
)
H
piperazine 1,4-azathiane oxepane thiepane azepane
(piperazinyl) (1,4-azathianyl) (oxepanyl) (thiepanyl) (azepanyl)
O O c c
O S NH S
1,4-dioxepane 1,4-oxathiepane 1,4-oxaazepane 1,4-dithiepane
(1,4-dioxepanyl) (1,4-oxathiepanyl) (1,4-oxaazepanyl) (1,4-dithiepanyl)
H
C c
NH NH
1,4-thieazepane 1,4-diazepane
(1,4-thieazepanyl) (1,4-diazepanyl)
Examples of suitable partially unsaturated heteroalicyclic groups include, but
are not limited to:
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-12-
O O
3,4-dihydro-2H-pyran 5,6-dihydro-2H-pyran 2H-pyran
(3,4-dihydro-2H-pyranyl) (5,6-dihydro-2H-pyranyl) (2H-pyranyl)
H H
ON
1,2,3,4-tetrahydropyridine 1,2,5,6-tetrahydropyridine
(1,2,3,4-tetrahydropyridinyi) (1,2,5,6-tetrahydropyridinyl)
The foregoing groups, as derived from the compounds listed above, may be C-
attached or N-
attached where such is possible. For instance, a group derived from pyrrole
may be pyrrol-1-yl (N-attached)
or pyrrol-3-yl (C-attached). The heteroalicyclic ring may be substituted or
unsubstituted. The heteroalicydic
ring may contain one or more oxo groups. When substituted, the substituted
group(s) is preferably one or
more selected halogen, hydroxy, -COR', -COOR', OCOR', -CONRR', -RNCOR', -NRR',
-CN, -NO2, -CZ3, -
SR', -SOR', -SO2R', -SO2OR', -SO2NRR', thiocarbonyl, -RNSOZR', perfluoroalkyl,
0-carbamyl, N-
carbamyl, 0-thiocarbamyl, N-thiocarbamyl, silyl, ammonium, lower alkyl, lower
alkenyl, lower alkynyl,
cycloalkyl, heteroalicycle, heteroaryl and aryl. Wherein R and R' are defined
herein.
A "3-8 Membered heteroalicyclic-(3-8 membered heteroalicyclic)" group refers
to a group having
two 3-8 membered heteroalicyclic groups covalently bonded to each other
through a single ring atom of
each. The 3-8 membered heteroalicyclic rings may be any heteroalicyclic ring
as defined above.
Furthermore, the heteroalicyclic rings may be substituted or unsubstituted as
defined above.
"Heterobicyclic" or "heterobicycle" refers to a fused ring (i.e., rings which
share an adjacent pair of
atoms) group having in the ring(s) one or more atoms selected from the group
consisting of nitrogen, oxygen
and sulfur and, in addition, having a completely conjugated pi-electron system
(i.e.- aromatic heterobicyclic)
or one or more double bonds that does not create a completely conjugated pi-
electron system, with the
proviso that heterobicyclic groups containing highly unstable heteroatom
arrangements, such as 0-0, 0-0-
0 and the like, are not contemplated by the present invention. One of ordinary
skill in the art will recognize
unstable groups that are not contemplated by the invention. Preferably, the
heterobicyclic group contains
from 8-10 ring atoms. The heterobicyclic ring may be substituted or
unsubstituted. The heterobicyclic ring
may contain one or more oxo groups. Examples of suitable fused ring aromatic
heterobicyclic groups
include, but are not limited to:
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-13-
~ CC\ (:::CN
N
O g N N N
benzofuran benzothiophene indole benzimidazole indazole
(benzofuranyl) (benzothiophenyl) (indolyl) (benzimidazolyl) (indazolyl)
" N
~ ~ ~
(::~:N ( ~ N
~ N N / N
H N H H H
benzotriazole pyrrolo[2,3-b]pyridine pyrrolo[2,3-c]pyridine pyrrolo[3,2-
c]pyridine
(benzotriazolyl) (pyrrolo[2,3-b]pyridinyl) (pyrrolo[2,3-c]pyridinyl)
(pyrrolo[3,2-c]pyridinyl)
N~ \ \ \ \ H
N\
N CXN> N/ N> (~~ N
H H H
pyrrolo[3,2-b]pyridine imidazo[4,5-b]pyridine imidazo[4,5-c]pyridine
pyrazolo[4,3-d]pyridine
(pyrrolo[3,2-b]pyridinyl) (imidazo[4,5-b]pyridinyl) (imidazo[4,5-c]pyridinyl)
(pyrazolo[4,3-d]pyidinyl)
~ N~ N\ N~ N N
Cj::~NH
N / N N N pyrazolo[4,3-c]pyridine pyrazolo[3,4-c]pyridine pyrazolo[3,4-
b]pyridine isoindole
(pyrazolo[4,3-c]pyidinyl) (pyrazolo[3,4-c]pyidinyl) (pyrazolo[3,4-b]pyidinyl)
(isoindolyl)
I\ % N N
N
CTDN
/ NI~, \ N ~ \ N~ N N
H H
indazole purine indolizine imidazo[1,2-a]pyridine imidazo[1,5-a]pyridine
(indazolyl) (purinyl) (indolininyl) (imidazo[1,2-a]pyridinyl) (imidazo[1,5-
a]pyridinyl)
i N
N~ N ~ IN~ N
N N ~
pyrazolo[1,5-a]pyridine pyrrolo[1,2-b]pyridazine imidazo[1,2-c]pyrimidine
(pyrazolo[1,5-a]pyridinyl) (pyrrolo[1-2,b]pyridazinyl) (imidazo[1,2-
c]pyrimidinyl)
Examples of suitable fused ring aromatic heterobicyclic groups include, but
are not limited to:
I / I I NH
N H
3H-Indole Indoline Isoindoline
(3H-indolyl) (indolyl) (isoindolinyl)
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-14-
~
I I O DON
\ O ~
2,3-Dihydrobenzofuran 1, 3-Di hydroisobenzofu ran 1 H-Isoindole
(2,3-dihydrobenzofuranyl) (1,3-dihydroisobenzofuranyl) (1H-isoindolyl)
H
N N\ N
I I I
\ \ \ J
H H H
1,2,3,4-Tetrahydroquinoxaline 1,2-Dihydroquinoxaline 1,2-Dihydroquinazoline
(1,2,3,4-tetrahydroquinoxalinyl) (1,2-dihydroquinoxalinyl) (1,2-
dihydroquinazolinyl)
NH 5:~
O
I
\ J \ \
O O
3,4-Dihydroquinazoline 2,3-Dihydrobenzo[b][1,4]dioxine 4H-Benzo[d][1,3]dioxine
(3,4-dihydroquinazolinyl) (2,3-dihydrobenzo[b][1,4]dioxinyl) (4H-
benzo[d][1,3]dioxinyl)
I I I I
O O O
3,4-Dihydro-2H-chromene 2H-Chromene 4H-Chromene
(3,4-dihydro-2H-chromenyl) (2H-chromenyl) (4H-chromenyl)
When substituted, the substituted group(s) is preferably one or more selected
halogen, hydroxy, -
COR', -COOR', OCOR', -CONRR', -RNCOR', -NRR', -CN, -NO2, -CZ3, -SR', -SOR', -
SOZR', -SO2OR', -
SOZNRR', thiocarbonyl, -RNSO2R', perfluoroalkyl, 0-carbamyl, N-carbamyl, 0-
thiocarbamyl, N-
thiocarbamyl, silyl, ammonium, lower alkyl, lower alkenyl, lower alkynyl,
cycloalkyl, heteroalicycle,
heteroaryl and aryl. Wherein R and R' are defined herein.
When used herein, the R groups on substitutents having two or more R groups on
different
atoms, such as -(CH2)n(NR8R9)C(O)NR8R9 or-NR6C(O)NR6R7, may be the same or
different. Specifically,
in the exemplary substituent -NR6C(O)NR6R', the two R6 groups may be the same
or different with
respect to each other, likewise, the two R6 groups may be the same or
different with respect to the R'
group. In, for example, -(CH2)n(NR8R9)C(O)NR8R9, the two R8 groups may be the
same or different with
respect to each other, and the two R9 groups may be the same or different with
respect to each other.
Likewise, the two R8 groups may be the same or different with respect to the
two R9 groups. In addition,
where a single atom is substituted by more than one group, the groups on that
atom may be the same or
different. So, in -NR6C(O)NR6R', the R6 and R7 on the same nitrogen may be the
same or different from
one another.
An "oxo" group refers to a carbonyl moiety such that alkyl substituted by oxo
refers ro a ketone
group.
A "hydroxy" group refers to an -OH group.
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-15-
An "alkoxy" group refers to both an -Oalkyl and an -Ocycloalkyl group, as
defined herein.
An "alkoxycarbonyl" refers to a -C(O)OR.
An "aminocarbonyl" refers to a -C(O)NRR'.
An "aryloxycarbonyl" refers to -C(O)Oaryl.
An "aryloxy" group refers to both an -Oaryl and an -Oheteroaryl group, as
defined herein.
An "arylalkyl" group refers to -alkylaryl, where alkyl and aryl are defined
herein.
An "aryisulfonyi" group refers to a-SO2aryL
An "alkylsulfonyl" group refer to a-SOZalkyl.
A "heteroaryloxyl" group refers to a heteroaryl group with heteroaryl as
defined herein.
A "heteroalicycloxy" group refers to a heteroalicyclic-O group with
heteroalicyclic as defined
herein.
A "carbonyl" group refers to a -C(=O)R.
An "aldehyde" group refers to a carbonyl group where R is hydrogen.
A "thiocarbonyl" group refers to a -C(=S)-R group.
A "trihalomethanecarbonyl" group refers to a Z3CC(O) group, where Z is
halogen.
A "C-carboxyl" group refers to a -C(O)OR groups.
An "O-carboxyl" group refers to a RC(O)O group.
A "carboxylic acid" group refers to a C-carboxyl group in which R is hydrogen.
A "halo" or "halogen" group refers to fluorine, chlorine, bromine or iodine.
A "trihalom ethyl" group refers to a -CZ3 group.
A"trihalomethanesulfonyl" group refers to a Z 3CS(O) 2 group.
A "trihalomethanesulfonamido" group refers to a Z3CS(O)2NR-group.
A "sulfinyl" group refers to a -S(O)R group.
A "sulfonyl" group refers to a -S(O) 2R group.
An "S-sulfonamido" group refers to a-S(O)2NR-group.
An "N-Sulfonamido" group refers to a-NR-S(O)2R group.
An "O-carbamyl" group refers to a -OC(O)NRR' group.
An "N-carbamyl" group refers to a ROC(O)NR-group.
An "O-thiocarbamyl" group refers to a -OC(S)NRR' group.
An "N-thiocarbamyl" group refers to a ROC(S)NR' group.
An "amino" group refers to an -NH2 or an -NRR'group.
A "C-amido" group refers to a -C(O)NRR'group.
An "N-amido" group refers to a R'C(O)NR group.
A "nitro" group refers to a-NO2 group.
A "cyano" group refers to a -CN group.
A"silyl" group refers to a -Si(R) 3 group.
A "phosphonyl" group refers to a -P(=O)(OR)2 group.
An "aminoalkyl" group refers to an -alkylNRR' group.
An "alkylaminoalkyl" group refers to an -alkyl-NR-alkyl group.
A "dialkylamionalkyl" group refers to an -alkylN-(alkyl)2 group.
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-16-
A "perfluoroalkyl group" refers to an alkyl group where all of the hydrogen
atoms have been
replaced with fluorine atoms.
Compounds that have the same molecular formula but differ in the nature or
sequence of bonding
of their atoms or arrangements of their atoms in space are termed "isomers."
Isomers that differ in the
arrangement of their atoms in space are termed "stereoisomers". Stereoisomers
that are not mirror
images of one another are termed "diastereomers" and those that are non-
superimposable mirror images
of each other are termed "enantiomers". When a compound has an asymmetric
center, for example, it is
bonded to four different groups, a pair of enantiomers is possible. An
enantiomer can be characterized by
the absolute configuration of its asymmetric center and is described by the R-
and S-sequencing rules of
Cahn and Prelog, or by the manner in which the molecule rotates the plane of
polarized light and
designated as dextrorotatory or levorotatory (i.e., as (+) or (-)-isomers
respectively). A chiral compound
can exist as either individual enantiomer or as a mixture thereof. A mixture
containing equal proportions of
the enantiomers is called a "racemic mixture". The chemical formulae referred
to herein may exhibit the
phenomena of tautomerism and structural isomerism. This invention encompasses
any tautomeric or
structural isomeric form and mixtures thereof which possess the ability to
modulate c-Met activity and is
not limited to any one tautomeric or structural isomeric form. This invention
encompasses any tautomeric
or structural isomeric form and mixtures thereof which possess the ability to
modulate c-Met activity and is
not limited to any one tautomeric or structural isomeric form.
The compounds of this invention may possess one or more asymmetric centers;
such compounds
can therefore be produced as individual (R)-or (S)-stereoisomers or as
mixtures thereof. Unless indicated
otherwise, the description or naming of a particular compound in the
specification and claims is intended
to include both individual enantiomers and mixtures, racemic or otherwise,
thereof. The methods for the
determination of stereochemistry and the separation of stereoisomers are well-
known in the art (see
discussion in Chapter 4 of "Advanced Organic Chemistry", 4th edition J. March,
John Wiley and Sons,
New York, 1992). Thus, this invention also encompasses any stereoisomeric
form, their corresponding
enantiomers (d- and I- or (+) and (-) isomers) and diastereomers thereof, and
mixtures thereof, which
possess the ability to modulate c-Met activity and is not limited to any one
stereoisomeric form.
The compounds of the formula (I) may exhibit the phenomena of tautomerism and
structural
isomerism. For example, the compounds described herein may adopt an E or a Z
configuration about a
double bond or they may be a mixture of E and Z. This invention encompasses
any tautomeric or
structural isomeric form and mixtures thereof which possess the ability to
modulate c-Met activity and is
not limited to any one tautomeric or structural isomeric form.
It is contemplated that compounds of the formula (I) would be metabolized by
enzymes in the
body of the organism such as human being to generate a metabolite that can
modulate the activity of c-
Met. Such metabofites are within the scope of the present invention.
Those compounds of the formula (I) that are acidic in nature are capable of
forming base salts with
various pharmacologically acceptable cations. Examples of such salts include
the alkali metal or alkaline
earth metal salts and particularly, the sodium and potassium salts.
The compounds of the present invention have asymmetric centers and therefore
exist in different
enantiomeric and diastereomeric forms. This invention relates to the use of
all optical isomers and
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-17-
stereoisomers of the compounds of the present invention, and mixtures thereof,
and to all pharmaceutical
compositions and methods of treatment that may employ or contain them. The
compounds of formula (I)
may also exist as tautomers. This invention relates to the use of all such
tautomers and mixtures thereof.
This invention also encompasses pharmaceutical compositions containing and
methods of
treating proliferative disorders or abnormal cell growth through administering
prodrugs of compounds of
the formula (I). Compounds of formula (I) having free amino, amido, hydroxy or
carboxylic groups can be
converted into prodrugs. Prodrugs include compounds wherein an amino acid
residue, or a polypeptide
chain of two or more (e.g., two, three or four) amino acid residues is
covalently joined through an amide or
ester bond to a free amino, hydroxy or carboxylic acid group of compounds of
formula (I). The amino acid
residues include but are not limited to the 20 naturally occurring amino acids
commonly designated by
three letter symbols and also includes 4-hydroxyproline, hydroxylysine,
demosine, isodemosine, 3-
methylhistidine, norvalin, beta-alanine, gamma-aminobutyric acid, citrulline
homocysteine, homoserine,
ornithine and methionine sulfone. Additional types of prodrugs are also
encompassed. For instance, free
carboxyl groups can be derivatized as amides or alkyl esters. Free hydroxy
groups may be derivatized
using groups including but not limited to hemisuccinates, phosphate esters,
dimethylaminoacetates, and
phosphoryloxymethyloxycarbonyls, as outlined in Advanced Drug Delivery
Reviews, 1996, 19, 115.
Carbamate prodrugs of hydroxy and amino groups are also included, as are
carbonate prodrugs,
sulfonate esters and sulfate esters of hydroxy groups. Derivatization of
hydroxy groups as
(acyloxy)methyl and (acyloxy)ethyl ethers wherein the acyl group may be an
alkyl ester, optionally
substituted with groups including but not limited to ether, amine and
carboxylic acid functionalities, or
where the acyl group is an amino acid ester as described above, are also
encompassed. Prodrugs of this
type are described in J. Med. Chem. 1996, 39, 10. Free amines can also be
derivatized as amides,
sulfonamides or phosphonamides. All of these prodrug moieties may incorporate
groups including but not
limited to ether, amine and carboxylic acid functionalities.
Detailed Description of the Invention
The compounds presented herein are exemplary and are not to be construed as
limiting the
scope of the invention.
A general synthetic route to the compounds of the present invention is shown
in Scheme 1. One
of skill in the art will recognize that this general scheme may be modified
and yet still produce the
compounds of the present invention. The groups Ra, Rb, Rc and Rd shown in
Scheme 1 include but are not
limited to those R' substituents described herein in connection with the
present invention. Further
exemplary methods for making the compounds of the invention are outlined in
the non-limiting examples
below.
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-18-
Scheme 1
CIIN~ R5 CI N
Amide Coupling I ~ N H R5 Heat
2 / ,NHZ + HO Ra N 1
R N R2 N~ u\Ra
R3 H O R3 H IOI
Ra
Palladium
coupling reactions Ra y N"N x N R. Aryl, Alkynyl,
R2 ~N/ Alkenyl, Alkyl, & Carbonyl
R5
~Ra Various Rb Rc R3 R5
Cl N~N \ diversification N Rb ~Ra
~N N,
Rz N N Rc / N \N
R3 R2 _N
Rd-OH R3 R Ra
base Rd/O N\N X
N
R2 N
R3
In one aspect, this invention is directed to a pharmaceutical composition
comprising one or more
compounds of formula (I) or a pharmaceutically acceptable salt thereof and a
pharmaceutically acceptable
excipient.
5 It is also an aspect of this invention that a compound described herein, or
its salt, might be
combined with other chemotherapeutic agents for the treatment of the diseases
and disorders discussed
above. For instance, a compound or salt of this invention might be combined
with alkylating agents such
as fluorouracil (5-FU) alone or in further combination with leukovorin; or
other alkylating agents such as,
without limitation, other pyrimidine analogs such as UFT, capecitabine,
gemcitabine and cytarabine, the
alkyl sulfonates, e.g., busulfan (used in the treatment of chronic
granulocytic leukemia), improsulfan and
piposulfan; aziridines, e.g., benzodepa, carboquone, meturedepa and uredepa;
ethyleneimines and
methylmelamines, e.g., altretamine, triethylenemelamine,
triethylenephosphoramide,
triethylenethiophosphorami- de and trimethylolmelamine; and the nitrogen
mustards, e.g., chlorambucil
(used in the treatment of chronic lymphocytic leukemia, primary
macroglobulinemia and non-Hodgkin's
lymphoma), cyciophosphamide (used in the treatment of Hodgkin's disease,
multiple myeloma,
neuroblastoma, breast cancer, ovarian cancer, lung cancer, Wilm's tumor and
rhabdomyosarcoma),
estramustine, ifosfamide, novembrichin, prednimustine and uracil mustard (used
in the treatment of
primary thrombocytosis, non-Hodgkin's lymphoma, Hodgkin's disease and ovarian
cancer); and triazines,
e.g., dacarbazine (used in the treatment of soft tissue sarcoma).
Likewise a compound or salt of this invention might be expected to have a
beneficial effect in
combination with other antimetabolite chemotherapeutic agents such as, without
limitation, folic acid
analogs, e.g. methotrexate (used in the treatment of acute lymphocytic
leukemia, choriocarcinoma,
mycosis fungiodes breast cancer, head and neck cancer and osteogenic sarcoma)
and pteropterin; and
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-19-
the purine analogs such as mercaptopurine and thioguanine which find use in
the treatment of acute
granulocytic, acute lymphocytic and chronic granulocytic leukemias.
A compound or salt of this invention might also be expected to prove
efficacious in combination
with natural product based chemotherapeutic agents such as, without
limitation, the vinca alkaloids, e.g.,
vinblastin (used in the treatment of breast and testicular cancer),
vincristine and vindesine; the
epipodophylotoxins, e.g., etoposide and teniposide, both of which are useful
in the treatment of testicular
cancer and Kaposi's sarcoma; the antibiotic chemotherapeutic agents, e.g.,
daunorubicin, doxorubicin,
epirubicin, mitomycin (used to treat stomach, cervix, colon, breast, bladder
and pancreatic cancer),
dactinomycin, temozolomide, plicamycin, bleomycin (used in the treatment of
skin, esophagus and
genitourinary tract cancer); and the enzymatic chemotherapeutic agents such as
L-asparaginase.
In addition to the above, a compound or salt of this- invention might be
expected to have a
beneficial effect used in combination with the platinum coordination complexes
(cisplatin, etc.); substituted
ureas such as hydroxyurea; methylhydrazine derivatives, e.g., procarbazine;
adrenocortical suppressants,
e.g., mitotane, aminoglutethimide; and hormone and hormone antagonists such as
the
adrenocorticosteriods (e.g., prednisone), progestins (e.g.,
hydroxyprogesterone caproate); estrogens
(e.g., diethylstilbesterol); antiestrogens such as tamoxifen; androgens, e.g.,
testosterone propionate; and
aromatase inhibitors (such as anastrozole).
Finally, the combination of a compound of this invention might be expected to
be particularly
effective in combination with mitoxantrone or paclitaxel for the treatment of
solid tumor cancers or
leukemias such as, without limitation, acute myelogenous (non-lymphocytic)
leukemia.
The above method can be carried out in combination with a chemotherapeutic
agent selected
from the group consisting of mitotic inhibitors, alkylating agents,
antimetabolites, cell cycle inhibitors,
enzymes, topoisomerase inhibitors, biological response modifiers, anti-
hormones, antiangiogenic agents
such as MMP-2, MMP-9 and COX-2 inhibitors, and anti-androgens.
Examples of useful COX-II inhibitors include Vioxx T"',, CELEBREX.T"'.
(alecoxib), vaidecoxib,
paracoxib, rofecoxib, and Cox 189. Examples of useful matrix metalloproteinase
inhibitors are described
in WO 96/33172 (published Oct. 24, 1996), WO 96/27583 (published Mar. 7,
1996), European Patent
Application No. 97304971.1 (filed Jul. 8, 1997), European Patent Application
No. 99308617.2 (filed Oct.
29, 1999), WO 98/07697 (published Feb. 26, 1998), WO 98/03516 (published Jan.
29, 1998), WO
98/34918 (published Aug. 13, 1998), WO 98/34915 (published Aug. 13, 1998), WO
98/33768 (published
Aug. 6, 1998), WO 98/30566 (published Jul. 16, 1998), European Patent
Publication 606,046 (published
Jul. 13, 1994), European Patent Publication 931,788 (published Jul. 28, 1999),
WO 90/05719 (published
May 31, 1990), WO 99/52910 (published Oct. 21, 1999), WO 99/52889 (published
Oct. 21, 1999), WO
99/29667 (published Jun. 17, 1999), PCT International Application No.
PCT/IB98/01113 (filed Jul. 21,
1998), European Patent Application No. 99302232.1 (filed Mar. 25, 1999), Great
Britain patent application
number 9912961.1 (filed Jun. 3, 1999), U.S. Provisional Application No.
60/148,464 (filed Aug. 12, 1999),
U.S. Pat. No. 5,863,949 (issued Jan. 26, 1999), U.S. Pat. No. 5,861,510
(issued Jan. 19, 1999), and
European Patent Publication 780,386 (published Jun. 25, 1997), all of which
are incorporated herein in
their entireties by reference.
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-20-
Preferred MMP-2 and MMP-9 inhibitors are those that have little or no activity
inhibiting MMP-1.
More preferred, are those that selectively inhibit MMP-2 and/or MMP-9 relative
to the other matrix-
metalloproteinase- s (i.e. MMP-1, MMP-3, MMP-4, MMP-5, MMP-6, MMP-7, MMP-8,
MMP-10, MMP-11,
MMP-12, and MMP-13). Some specific examples of MMP inhibitors useful in the
present invention are
AG-3340, RO 32-3555, RS 13-0830, and the compounds recited in the following
list:
3-[[4-(4-fluoro-phenoxy)-benzenesulfonyl]-(1-hydroxycarbamoyl-cyclo-pentyl)-am
ino]propionic
acid; 3-exo-3-[4-(4-fluoro-phenoxy)-benzenesulfon-ylamino]-8-oxa-
bicyclo[3.2.1]-octane-3-carboxylic acid
hydroxyamide; (2R, 3R) 1 -[4-(2-chloro-4-fl uoro-benzyloxy)-benzenesulfonyl]-3-
hydroxy-3-m ethyl-
piperidine-2-carboxylic acid hydroxyamide; 4-[4-(4-fluoro-phenoxy)-
benzenesulfonylamino]tetrahydro-
pyran-4-carboxylic acid hydroxyamide; 3-[[4-(4-fluoro-phenoxy)-
benzenesulfonyl]-(1-hydroxy-
carbamoylcyclobutyl)-amino]-propionic acid; 4-[4-(4-chloro-phenoxy)-
benzenesulfonylamino]-tetrahydro-
pyran-4-carboxylic acid hydroxyamide; (R) 3-[4-(4-chloro-phenoxy)-
benzenesulfonyl-amino]-tetrahydro-
pyran-3-carboxylic acid hydroxyamide; (2R, 3R) 1-[4-(4-fluoro-2-
methylbenzyloxy)-benzenesulfonyl]-3-
hydroxy-3-methyl-piperidine-2-carboxylic acid hydroxyamide; 3-[[(4-(4-fluoro-
phenoxy)-benzenesulfonyl]-
(1-hydroxycarbamoyl-l-methylethyl)-amino]propionic acid; 3-[[4-(4-fluoro-
phenoxy)-benzenesulfonyl]-(4-
hydroxycarbamoyl-tetrahydropyran-4-yl)-amino]-propionic acid; 3-exo-3-[4-(4-
chloro-phenoxy)-
benzenesulfonylamino]-8-oxa-bicyclo[3.2.1]octane-3-carboxylic acid hydroxy-
amide; 3-endo-3-[4-(4-
fluoro-phenoxy)-benzenesulfonylamino]-8-oxa-bicylo[3.2.1]octane-3-carboxylic
acid hydroxyamide; and
(R) 3-[4-(4-fluoro-phenoxy)-benzenesulfonylamino]-tetrahydro-furan-3-
carboxylic acid hydroxyamide; and
.20 pharmaceutically acceptable salts and solvates of said compounds.
Other anti-angiogenesis agents, including other COX-II inhibitors and other
MMP inhibitors, can
also be used in the present invention.
Compounds of the formula (I) can also be used with signal transduction
inhibitors, such as agents
that can inhibit EGFR (epidermal growth factor receptor) responses, such as
EGFR antibodies, EGF
antibodies, and molecules that are EGFR inhibitors; VEGF (vascular endothelial
growth factor) inhibitors;
and erbB2 receptor inhibitors, such as organic molecules or antibodies that
bind to the erbB2 receptor, for
example, HERCEPTINTM. (Genentech, Inc. of South San Francisco, Calif., USA).
EGFR inhibitors are
described in, for example in WO 95/19970 (published Jul. 27, 1995), WO
98/14451 (published Apr. 9,
1998), WO 98/02434 (published Jan. 22, 1998), and U.S. Pat. No. 5,747,498
(issued May 5, 1998), and
such substances can be used in the present invention as described herein.
EGFR-inhibiting agents include, but are not limited to, the monoclonal
antibodies C225 and anti-
EGFR 22Mab (ImClone Systems Incorporated of New York, N.Y., USA), the
compounds ZD-1839
(AstraZeneca), BIBX-1382 (Boehringer Ingelheim), MDX-447 (Medarex Inc. of
Annandale, N.J., USA),
and OLX-103 (Merck & Co. of Whitehouse Station, N.J., USA), VRCTC-310 (Ventech
Research) and EGF
fusion toxin (Seragen Inc. of Hopkinton, Mass.).
These and other EGFR-inhibiting agents can be used in the present invention.
VEGF inhibitors can also be combined with a compounds of the Formulae (I).
VEGF inhibitors are
described in, for example in WO 99/24440 (published May 20, 1999), PCT
International Application
PCT/IB99/00797 (filed May 3, 1999), in WO 95/21613 (published Aug. 17,1995),
WO 99/61422 (published
Dec. 2,1999), U.S. Pat. No. 5,834,504 (issued Nov. 10, 1998), WO 01/60814,WO
98/50356 (published
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-21 -
Nov. 12,1998), U.S. Pat. No. 5,883,113 (issued Mar. 16, 1999), U.S. Pat. No.
5,886,020 (issued Mar. 23,
1999), U.S. Pat. No. 5,792,783 (issued Aug. 11, 1998), WO 99/10349 (published
Mar. 4, 1999), WO
97/32856 (published Sep. 12, 1997), WO 97/22596 (published Jun.26, 1997), WO
98/54093 (published
Dec. 3, 1998), WO 98/02438 (published Jan. 22, 1998), WO 99/16755 (published
Apr. 8, 1999), and WO
98/02437 (published Jan. 22, 1998), all of which are incorporated herein in
their entireties by reference.
Other examples of some specific VEGF inhibitors useful in the present
invention are IM862 (Cytran Inc. of
Kirkland, Wash., USA); anti-VEGF monoclonal antibody of Genentech, Inc. of
South San Francisco, Calif.;
and angiozyme, a synthetic ribozyme from Ribozyme (Boulder, Colo.) and Chiron
(Emeryville, Calif.).
These and other VEGF inhibitors can be used in the present invention as
described herein.
ErbB2 receptor inhibitors, such as GW-282974 (Glaxo Wellcome pic), and the
monoclonal
antibodies AR-209 (Aronex Pharmaceuticals Inc. of TheWoodlands, Tex., USA) and
2B-1 (Chiron), can
furthermore be combined with a compound of the formula (I) for example those
indicated in WO 98/02434
(published Jan. 22, 1998), WO 99/35146 (published Jul. 15, 1999), WO 99/35132
(published Jul. 15,
1999), WO 98/02437 (published Jan. 22, 1998), WO 97/13760 (published Apr. 17,
1997), WO 95/19970
(published Jul. 27, 1995), U.S. Pat. No. 5,587,458 (issued Dec. 24, 1996), and
U.S. Pat. No. 5,877,305
(issued Mar. 2, 1999), which are all hereby incorporated herein in their
entireties by reference. ErbB2
receptor inhibitors useful in the present invention are also described in U.S.
Provisional Application No.
60/117,341, filed Jan. 27,1999, and in U.S. Provisional Application No.
60/117,346, filed Jan. 27,1999,
both of which are incorporated in their entireties herein by reference. The
erbB2 receptor inhibitor
compounds and substance described in the aforementioned PCT applications, U.S.
patents, and U.S.
provisional applications, as well as other compounds and substances that
inhibit the erbB2 receptor, can
be used with compounds of the formula (I), in accordance with the present
invention.
Compounds of the formula (I) can also be used with other agents useful in
treating cancer,
including, but not limited to, agents capable of enhancing antitumor immune
responses, such as CTLA4
(cytotoxic lymphocite antigen 4) antibodies, and other agents capable of
blocking CTLA4; and anti-
proliferative agents such as other famesyl protein transferase inhibitors, for
example the farnesyl protein
transferase inhibitors described in the references cited in the "Background"
section, of U.S. Pat. No,
6,258,824 BI. Specific CTLA4 antibodies that can be used in the present
invention include those
described in U.S. Provisional Application No. 60/113,647 (filed Dec. 23, 1998)
which is incorporated by
reference in its entirety, however other CTLA4 antibodies can be used in the
present invention.
The above method can be also be carried out in combination with radiation
therapy, wherein the
amount of a compound of the formula (I) in combination with the radiation
therapy, is effective in treating
the above diseases. The level of radiation therapy administered may be reduced
to a sub-efficacy dose
when administered in combination with the compounds of the preferred
embodiments of the present
invention.
Techniques for administering radiation therapy are known in the art, and these
techniques can be
used in the combination therapy described herein. The administration of the
compound of the invention in
this combination therapy can be determined as described herein.
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-22-
Another aspect of the invention is directed ot the use of compounds of the
Formulae (I) in the
preparation of a medicament, which is useful in the treatment of a disease
mediated by abnormal Met
kinase activity.
Indications
A precise understanding of the mechanism by which the compounds of the
invention, in particular,
the compounds generated in vivo from the compounds of the invention, inhibit c-
Met is not required in
order to practice the present invention. However, while not hereby being bound
to any particular
mechanism or theory, it is believed that the compounds interact with the amino
acids in the catalytic
region of c-Met. The compounds disclosed herein may thus have utility as in
vitro assays for c-Met as well
as exhibiting in vivo therapeutic effects through interaction with c-Met.
In another aspect, this invention relates to a method for treating or
preventing a c-Met related
disorder by administering a therapeutically effective amount of a compound of
this invention, or a salt
thereof, to an organism.
It is also an aspect of this invention that a pharmaceutical composition
containing a compound of
this invention, or a salt thereof, is administered to an organism for the
purpose of preventing or treating a
c-Met related disorder.
This invention is therefore directed to compounds that modulate PK signal
transduction by
affecting the enzymatic activity of c-Met, thereby interfering with the signal
transduced by c-Met. More
particularly, the present invention is directed to compounds which modulate c-
Met mediated signal
transduction pathways as a therapeutic approach to treat the many cancers
described herein.
A method for identifying a chemical compound that modulates the catalytic
activity of c-Met is
another aspect of this invention. The method involves contacting cells
expressing c-Met with a compound
of this invention (or its salt) and monitoring the cells for any effect that
the compound has on them.
Alternatively, the method can involve contacting the c-Met protein itself
(i.e., not in a cell) with a chemical
compound of the preferred embodiments of the present invention and monitoring
the protein for any effect
that the compound has on it. The effect may be observable, either to the naked
eye or through the use of
instrumentation. The effect may be, for example, a change or absence in a cell
phenotype. The change or
absence of change in the cell phenotype monitored, for example, may be,
without limitation, a change or
absence of change in the catalytic activity of c-Met in the cells or a change
or absence of change in the
interaction of c-Met with a natural binding partner.
Pharmaceutical Compositions and Use
A compound of the present invention or a physiologically acceptable salt
thereof, can be
administered as such to a human patient or can be administered in
pharmaceutical compositions in which
the foregoing materials are mixed with suitable carriers or excipient(s).
Techniques for formulation and
administration of drugs may be found in "Remington's Pharmacological
Sciences," Mack Publishing Co.,
Easton, Pa., latest edition.
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-23-
Routes of Administration
Suitable routes of administration may include, without limitation, oral,
intraoral, rectal,
transmucosal or intestinal administration or intramuscular, epicutaneous,
parenteral, subcutaneous,
transdermal, intramedullary, intrathecal, direct intraventricular,
intravenous, intravitreal, intraperitoneal,
intranasal, intramuscular, intradural, intrarespiratory, nasal inhalation or
intraocular injections. The
preferred routes of administration are oral and parenteral.
Altematively, one may administer the compound in a local rather than systemic
manner, for
example, via injection of the compound directly into a solid tumor, often in a
depot or sustained release
formulation.
Furthermore, one may administer the drug in a targeted drug delivery system,
for example, in a
liposome coated with tumor-specific antibody. The liposomes will be targeted
to and taken up selectively
by the tumor.
Composition/Formulation
Pharmaceutical compositions of the present invention may be manufactured by
processes well
known in the art, e.g., by means of conventional mixing, dissolving,
granulating, dragee-making,
levigating, emulsifying, encapsulating, entrapping, lyophilizing processes or
spray drying.
Pharmaceutical compositions for use in the methods of the present invention
may be prepared by
any methods of pharmacy, but all methods include the step of bringing in
association the active ingredient
with the carrier which constitutes one or more necessary ingredients. In
particular, pharmaceutical
compositions for use in accordance with the present invention may be
formulated in conventional manner
using one or more physiologically acceptable carriers comprising excipients
and auxiliaries which facilitate
processing of the active compounds into preparations which can be used
pharmaceutically. Proper
formulation is dependent upon the route of administration chosen.
Dosage forms include tablets, troches, dispersions, suspensions, solutions,
capsules, patches,
syrups, elixirs, gels, powders, magmas, lozenges, ointments, creams, pastes,
plasters, lotions, discs,
suppositories, nasal or oral sprays, aerosols and the like.
For injection, the compounds of the invention may be formulated in aqueous
solutions, preferably
in physiologically compatible buffers such buffers with or without a low
concentration of surfactant or
cosolvent, or physiological saline buffer. For transmucosal administration,
penetrants appropriate to the
barrier to be permeated are used in the formulation. Such penetrants are
generally known in the art.
For oral administration, the compounds can be formulated by combining the
active compounds
with pharmaceutically acceptable carriers well known in the art. Such carriers
enable the compounds of
the invention to be formulated as tablets, pills, lozenges, dragees, capsules,
liquids, gels, syrups, slurries,
suspensions and the like, for oral ingestion by a patient. Pharmaceutical
preparations for oral use can be
made using a solid excipient, optionally grinding the resulting mixture, and
processing the mixture of
granules, after adding other suitable auxiliaries if desired, to obtain
tablets or dragee cores. Useful
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-24-
excipients are, in particular, fillers such as sugars, including lactose,
sucrose, mannitol, or sorbitol,
cellulose preparations such as, for example, maize starch, wheat starch, rice
starch and potato starch and
other materiais such as gelatin, gum tragacanth, methyl cellulose,
hydroxypropylmethyl-cellulose, sodium
carboxymethylcellulose, and/or polyvinyl-pyrrolidone (PVP). If desired,
disintegrating agents may be
added, such as cross-linked polyvinyl pyrrolidone, agar, or alginic acid. A
salt such as sodium alginate
may also be used.
Dragee cores are provided with suitable coatings. For this purpose,
concentrated sugar solutions
may be used which may optionally contain gum arabic, talc, polyvinyl
pyrrolidone, carbopol gel,
polyethylene glycol, and/or titanium dioxide, lacquer solutions, and suitable
organic solvents or solvent
mixtures. Dyestuffs or pigments may be added to the tablets or dragee coatings
for identification or to
characterize different combinations of active compound doses.
Pharmaceutical compositions which can be used orally include push-fit capsules
made of gelatin,
as well as soft, sealed capsules made of gelatin and a plasticizer, such as
glycerol or sorbitol. The push-fit
capsules can contain the active ingredients in admixture with a filler such as
lactose, a binder such as
starch, and/or a lubricant such as talc or magnesium stearate and, optionally,
stabilizers. In soft capsules,
the active compounds may be dissolved or suspended in suitable liquids, such
as fatty oils, liquid paraffin,
liquid polyethylene glycols, cremophor, capmui, medium or long chain mono-, di-
or triglycerides.
Stabilizers may be added in these formulations, also.
For administration by inhalation, the compounds for use according to the
present invention are
conveniently delivered in the form of an aerosol spray using a pressurized
pack or a nebulizer and a
suitable propellant, e.g., without limitation, dichlorodifluoromethane,
trichlorofluoromethane, dichlorotetra-
fluoroethane or carbon dioxide. In the case of a pressurized aerosol, the
dosage unit may be controlled by
providing a valve to deliver a metered amount. Capsules and cartridges of, for
example, gelatin for use in
an inhaler or insufflator may be formulated containing a powder mix of the
compound and a suitable
powder base such as lactose or starch.
The compounds may also be formulated for parenteral administration, e.g., by
bolus injection or
continuous infusion. Formulations for injection may be presented in unit
dosage form, e.g., in ampoules or
in multi-dose containers, with an added preservative. The compositions may
take such forms as
suspensions, solutions or emulsions in oily or aqueous vehicles, and may
contain formulating materials
such as suspending, stabilizing and/or dispersing agents.
Pharmaceutical compositions for parenteral administration include aqueous
solutions of a water
solubie form, such as, without limitation, a salt, of the active compound.
Additionally, suspensions of the
active compounds may be prepared in a lipophilic vehicle. Suitable lipophilic
vehicles include fatty oils
such as sesame oil, synthetic fatty acid esters such as ethyl oleate and
triglycerides, or materials such as
liposomes. Aqueous injection suspensions may contain substances which increase
the viscosity of the
suspension, such as sodium carboxymethyl cellulose, sorbitol, or dextran.
Optionally, the suspension may
also contain suitable stabilizers and/or agents that increase the solubility
of the compounds to allow for
the preparation of highly concentrated solutions.
Alternatively, the active ingredient may be in powder form for constitution
with a suitable vehicle,
e.g., sterile, pyrogen-free water, before use.
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-25-
The compounds may also be formulated in rectal compositions such as
suppositories or retention
enemas, using, e.g., conventional suppository bases such as cocoa butter or
other glycerides.
In addition to the formulations described previously, the compounds may also
be formulated as
depot preparations. Such long acting formulations may be administered by
implantation (for example,
subcutaneously or intramuscularly) or by intramuscular injection. A compound
of this invention may be
formulated for this route of administration with suitable polymeric or
hydrophobic materials (for instance, in
an emulsion with a pharmacologically acceptable oil), with ion exchange
resins, or as a sparingly soluble
derivative such as, without limitation, a sparingly soluble salt.
A non-limiting example of a pharmaceutical carrier for the hydrophobic
compounds of the
invention is a cosolvent system comprising benzyl alcohol, a nonpolar
surfactant, a water-miscible organic
polymer and an aqueous phase such as the VPD co-solvent system. VPD is a
solution of 3% w/v benzyl
alcohol, 8% w/v of the nonpolar surfactant Polysorbate 80, and 65% w/v
polyethylene glycol 300, made up
to volume in absolute ethanol. The VPD co-solvent system (VPD:D5W) consists of
VPD diluted 1:1 with a
5% dextrose in water solution. This co-solvent system dissolves hydrophobic
compounds well, and itself
produces low toxicity upon systemic administration. Naturally, the proportions
of such a co-solvent system
may be varied considerably without destroying its solubility and toxicity
characteristics. Furthermore, the
identity of the co-solvent components may be varied: for example, other low-
toxicity nonpolar surfactants
may be used instead of Polysorbate 80, the fraction size of polyethylene
glycol may be varied, other
biocompatible polymers may replace polyethylene glycol, e.g., polyvinyl
pyrrolidone, and other sugars or
polysaccharides may substitute for dextrose.
Alternatively, other delivery systems for hydrophobic pharmaceutical compounds
may be
employed. Liposomes and emulsions are well known examples of delivery vehicles
or carriers for
hydrophobic drugs. In addition, certain organic solvents such as
dimethylsulfoxide also may be employed,
although often at the cost of greater toxicity.
Additionally, the compounds may be delivered using a sustained-release system,
such as
semipermeable matrices of solid hydrophobic polymers containing the
therapeutic agent. Various
sustained-release materials have been established and are well known by those
skilled in the art.
Sustained-release capsules may, depending on their chemical nature, release
the compounds for a few
weeks up to over 100 days. Depending on the chemical nature and the biological
stability of the
therapeutic reagent, additional strategies for protein stabilization may be
employed.
The pharmaceutical compositions herein also may comprise suitable solid or gel
phase carriers or
excipients. Examples of such carriers or excipients include, but are not
limited to, calcium carbonate,
calcium phosphate, various sugars, starches, cellulose derivatives, gelatin,
and polymers such as
polyethylene glycols.
Many of the PK modulating compounds of the invention may be provided as
physiologically
acceptable salts wherein the claimed compound may form the negatively or the
positively charged
species. Examples of salts in which the compound forms the positively charged
moiety include, without
limitation, quaternary ammonium (defined elsewhere herein), salts such as the
hydrochloride, sulfate,
carbonate, lactate, tartrate, maleate, sucinate, malate, acetate and
methylsulfonate (CH3SO3), wherein
the nitrogen atom of the quaternary ammonium group is a nitrogen of the
selected compound of this
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-26-
invention which has reacted with the appropriate acid. Salts in which a
compound of this invention forms
the negatively charged species include, without limitation, the sodium,
potassium, calcium and
magnesium salts formed by the reaction of a carboxylic acid group in the
compound with an appropriate
base (e.g. sodium hydroxide (NaOH), potassium hydroxide (KOH), Calcium
hydroxide (Ca(OH)2), etc.).
Dosage
Pharmaceutical compositions suitable for use in the present invention include
compositions
wherein the active ingredients are contained in an amount sufficient to
achieve the intended purpose, i.e.,
the modulation of PK activity or the treatment or prevention of a PK-related
disorder.
More specifically, a therapeutically effective amount means an amount of
compound effective to
prevent, alleviate or ameliorate symptoms of disease or prolong the survival
of the subject being treated.
- Determination of a therapeutically effective amount is well within the
capability of those skilled in
the art, especially in light of the detailed disclosure provided herein.
For any compound used in the methods of the invention, the therapeutically
effective amount or
dose can be estimated initially from cell culture assays. Then, the dosage can
be formulated for use in
animal models so as to achieve a circulating concentration range that includes
the IC50 as determined in
cell culture (i.e., the concentration of the test compound which achieves a
half-maximal inhibition of c-Met
activity). Such information can then be used to more accurately determine
useful doses in humans.
Toxicity and therapeutic efficacy of the compounds described herein can be
determined by
standard pharmaceutical procedures in cell cultures or experimental animals,
e.g., by determining the IC50
and the LD50 (both of which are discussed elsewhere herein) for a subject
compound. The data obtained
from these cell culture assays and animal studies can be used in formulating a
range of dosage for use in
humans. The dosage may vary depending upon the dosage form employed and the
route of
administration utilized. The exact formulation, route of administration and
dosage can be chosen by the
individual physician in view of the patient's condition. (See e.g., Fingl, et
al., 1975, in "The
Pharmacological Basis of Therapeutics", Ch. 1 p.1).
Dosage amount and interval may be adjusted individually to provide plasma
levels of the active
species which are sufficient to maintain the kinase modulating effects. These
plasma levels are referred to
as minimal effective concentrations (MECs). The MEC will vary for each
compound but can be estimated
from in vitro data, e.g., the concentration necessary to achieve 50-90%
inhibition of a kinase may be
ascertained using the assays described herein. Dosages necessary to achieve
the MEC will depend on
individual characteristics and route of administration. HPLC assays or
bioassays can be used to
determine plasma concentrations.
Dosage intervals can also be determined using MEC value. Compounds should be
administered
using a regimen that maintains plasma levels above the MEC for 10-90% of the
time, preferably between
30-90% and most preferably between 50-90%. At present, the therapeutically
effective amounts of
compounds of the Formulae (I)-(IV) may range from approximately 10 mg/m2 to
1000 mg/mZ perday. Even
more preferably 25 mg/m2 to 500 mg/m2.
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-27-
In cases of local administration or selective uptake, the effective local
concentration of the drug
may not be related to plasma concentration and other procedures known in the
art may be employed to
determine the correct dosage amount and interval.
The amount of a composition administered will, of course, be dependent on the
subject being
treated, the severity of the affliction, the manner of administration, the
judgment of the prescribing
physician, etc.
Packaging
The compositions may, if desired, be presented in a pack or dispenser device,
such as an FDA
approved kit, which may contain one or more unit dosage forms containing the
active ingredient. The pack
may for example comprise metal or plastic foil, such as a blister pack. The
pack or dispenser device may
be accompanied by instructions for administration. The pack or dispenser may
also be accompanied by a
notice associated with the container in a form prescribed by a governmental
agency regulating the
manufacture, use or sale of pharmaceuticals, which notice is reflective of
approval by the agency of the
form of the compositions or of human or veterinary administration. Such
notice, for example, may be of
the labeling approved by the U.S. Food and Drug Administration for
prescription drugs or of an approved
product insert. Compositions comprising a compound of the invention formulated
in a compatible
pharmaceutical carrier may also be prepared, placed in an appropriate
container, and labeled for
treatment of an indicated condition. Suitable conditions indicated on the
label may include treatment of a
tumor, inhibition of angiogenesis, treatment of fibrosis, diabetes, and the
like.
Examples
Compounds of the present invention can be made according to general Methods 1-
13 described
below. It will be understood by those skilled in the art that the following
general methods are not limiting
to the invention. It may be possible to alter exact solvents, conditions and
reagents and quantities without
deleterious effects. Specific embodiments of the present invention are
summarized in Table 1 below.
Abbreviations:
TLC: thin layer chromatography
aq.: aqueous
DMF: N,N-dimethylformamide
HPLC: High-performance liquid chromatography (also known as high-pressure
liquid
chromatography)
AcOH: Acetic acid
HATU: 2-(7-Aza-1 H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium
hexafluorophosphate
DME: Dimethyl ether
EtOAc: Ethyl acetate
ACN: Acetonitrile
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-28-
MeOH : Methanol
DMSO: Dimethylsulfoxide
THF: Tetrahydrofuran
LDA: Lithium diisopropylamide
EDCI: 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride
Method 1:
HO--'--rOH
OH OH OH SOCI2
I/ O FeSO4/H2SO4 N O MeOH
HzN Step I Step 2
2
/ I\ O~ 1. LDA/THF \ O-, LiOH
2. Mel / 0 Me0 /HH O
N Step 3 Step 4 ~
3 4
OH
/ CI N~ N 1. EDC/DMF
~ + I NH2 > CI N~N
N / O /N 2. AcOH ~ N
H Step 5 N
6 7
Step 1.
A mixture of compound 1 (276 g, 1.8 mol), ferrous sulfate (63.6 g, 0.22 mol),
glycerol (696 g, 7.56
mol), nitrobenzene (138 g, 1.12 mol) and conc. sulfuric acid (324 mL) was
heated gently. After the first
vigorous reaction, the mixture was refluxed for five hours and then was
treated with aq. sodium hydroxide
solution (2 N, 1320 mL), stirred with kieselguhr, and filtered. The filtrate
was basified with aq. sodium
hydroxide solution to pH 5-6, and a dark brown precipitate formed. The
precipitate was filtered, washed
with water, taken up with aq. sodium hydroxide solution (0.82 N, 3000 mL),
then boiled with carbon (150
g). The mixture was filtered and the filtrate was treated with glacial acetic
acid (240 mL), and the mixture
was left standing overnight during which time dark-brown crystalline
precipitate formed. The precipitate
was collected and dried in vacuo to give crude compound 2 (60 g, 17.8%).
Step 2.
To a suspension of compound 2 (60 g, 0.32 mol) in MeOH (600 mL) cooled to 0-5
C, SOCI2 (30
mL, 0.35 mol) was added dropwise. After the mixture was heated to reflux for 2
h, the mixture was
evaporated under reduced pressure, and the residue was taken up with EtOAc
(600 mL). The mixture was
washed with aq. NaHCO3 and brine, dried over Na2SO4 and concentrated to give
crude product, which
was purified via a silica column chromatography (EtOAc: Petroleum ether = 1:5)
to give pure compound 3
(50 g, 72.6%) as a yellow oil. 1 H NMR (400 MHz, CDCI3): 6 8.898-8.878 (dd,
1H), 8.130-8.055 (m, 2H),
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-29-
7.718 (s, 1 H), 7.670-7.634 (dd, 1 H), 7.407-7.365 (q, 1 H), 4.207-4.135(q, 2
H), 3.799(s, 2H), 1.279-1.232
(t, 3H).
Step 3.
To a solution of quinolin-6-yl-acetic acid methyl ester 3 (20.00 g, 99.54
mmol) in anhydrous
tetrahydrofuran (200 mL) was added LDA (1.8 M THF solution, 61 mL, 109.5 mmol)
drop wise at -78 C
under nitrogen. The reaction mixture was stirred at at -78 C under nitrogen
for half an hour. To the
reaction mixture was added methyl iodide (6.20 mL, 99.54 mmol), and the
mixture was stirred under
nitrogen from -78 C to ambient temperature overnight. The reaction was
quenched with the careful
addition of water. The product was extracted with ethyl acetate. The combined
extracts were washed
with water and brine, dried over Na2SO4, and concentrated to provide 2-
quinolin-6-yl-propionic acid
methyl ester, 4 (21.49 g, -100% yield). MS mle 216 [M+1]+; 'H NMR (400 MHz,
DMSO-D6) 6 1.49 (d,
J=7.07 Hz, 3 H), 3.60 (s, 3 H), 4.03 (q, J=7.07 Hz, 1 H), 7.51(dd, J=8.34,
4.04 Hz, 1 H), 7.68 (dd, J=8.59,
2.02 Hz, 1 H), 7.86 (d, J=1.77 Hz, 1 H), 7.98 (d, J=8.59 Hz, 1 H), 8.33 (d,
J=7.58 Hz, 1 H), 8.87 (dd,
J=4.17, 1.64 Hz, 1 H).
Step 4.
To a solution of 4 (21.17 g, 98.35 mmol) in methanol (200 mL) and water (50
mL) was added
lithium hydroxide (12.02 g, 491.75 mmol). The reaction mixture was stirred at
65 C oil bath for 4 hours,
cooled to ambient temperature, and adjusted the acidity to pH -7 with 6N HCI
(65 mL). A lot of precipitate
was formed. After filtration, the solid was washed with water, and the
filtrate was concentrated to remove
methanol. The solid was filtrated and washed with water. The combined solid
product was dried under
high vacuum to provide 2-quinolin-6-yl-propionic acid, 5 (19.09 g, 90% yield).
MS m/e 202 [M+1]+;'H NMR
(400 MHz, DMSO-D6) 5 1.37 (d, J=7.07 Hz, 3 H), 3.54 (q, J=7.07 Hz, 1 H), 7.43
(dd, J=8.34, 4.04 Hz, 1
H), 7.72 - 7.80 (m, 2 H), 7.82 - 7.89 (m, 1 H), 8.20 - 8.26 (m, 1 H), 8.78
(dd, J=4.04, 1.77 Hz, I H).
Step 5.
To a solution of 5 (3.00 g, 14.9 mmol) in DMF (75 mL) was added 1-(3-
dimethylaminopropyl)-3-
ethylcarbodiimide hydrochloride, 6 (3.14 g, 16.4 mmol). The reaction mixture
was stirred under nitrogen
for half an hour and then (6-chloro-pyridazin-3-yl)-hydrazine (2.22 g, 14.9
mmol) was added. The reaction
mixture was stirred under nitrogen for overnight, diluted with ethyl acetate,
washed with water, dried over
Na2SO4, and concentrated to get the crude intermediate, which was dissolved in
acetic acid (20 mL). The
acetic acid solution was refluxed for 2 hours, and concentrated. The residue
was purified on a silica gel
column eluting with 5% methanol in ethyl acetate to provide 6-[1-(6-chloro-
[1,2,4]triazolo[4,3-b]pyridazin-
3-yl)-ethyl]-quinoline, 7 (1.16 g, 25% yield).
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-30-
Method 2:
\ ~ \
N
\ N N
CI N~ Chiral resolution CI N~ CI N
i N N' N
N Step 6 I` L N '~N
2 3
The racemic 6-[1-(6-chloro-[1,2,4]triazolo[4,3-b]pyridazin-3-yl)-ethyl]-
quinoline was resolved with a
chiral column (Chiralcel AD-H) eluting with 45% methanol in liquid carbon
dioxide (100 bar, 2.5 mL/min).
6-[(S)-1-(6-Chloro-[1,2,4]triazolo[4,3-b]pyridazin-3-yl)-ethyl]-quinoline had
an optical rotation of -0.125 in
methanol (5.22 mg/mL), and 6-[(R)-1-(6-chloro-[1,2,4]triazolo[4,3-b]pyridazin-
3-yl)-ethyl]-quinoline had an
optical rotation of +0.157 in methanol (5.53 mg/mL).
Method 3:
CN
N Pd(dppf)aCla NC N
CI N N +
~ Cs2CO3 N" N \ N
N ~B\ DME/H20 ~ ~N
HO OH
3
2
To a solution of 6-[1-(6-chloro-[1,2,4]triazolo[4,3-b]pyridazin-3-yl)-ethyl]-
quinoline (1) (50 mg, 0.16
mmol) and the boronic acid (2) (26.4 mg, 0.18 mmol) in 1,2-dimethoxyethane
(1.5 mL) was added a
freshly prepared solution of Cs2CO3 (186.3 mg, 0.528 mmol) in water (0.5 mL),
and the catalyst
Pd(dppf)2CI2.CH2CI2 (6.5 mg, 0.008 mmol). The reaction mixture was degassed
and charged with
nitrogen for three times, and heated at 80 C oil bath for overnight. The
reaction solution was diluted with
methanol, and filtered through a celite pad. The filtrate was concentrated and
purified on a reverse-phase
C-18 preparative HPLC eluting with acetonitrile-water containing 0.1% acetic
acid to provide 4-[3-(1-
quinolin-6-yl-ethyl)-[1,2,4]triazolo[4,3-b]pyridazin-6-yl]-benzonitrile (27
mg, 45% yield).
Method 4:
/ HN
N N Z
0 1. n-BuOH / N
CI N~ + ~ ~ ~ N N~
N~ N H O 2. HCI/Methanol
~N \ N
N
A solution of 6-[1-(6-chloro-[1,2,4]triazolo[4,3-b]pyridazin-3-yl)-ethyl]-
quinoline (50.0 mg, 0.16
mmol) and (R)-pyrrolidin-3-yl-carbamic acid tert-butyl ester (60.0 mg, 0.32
mmol) in n-butanol (1.5 mL)
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-31-
was microwaved at 125 C for 50 minutes. After evaporation of solvents, the
residue was suspended in
methanol (2 mL) and HCI dioxane solution (4.0 N, 4 mL) until the de-protection
was complete. The
solvents were evaporated, and the residue was purified on a reverse-phase C-18
column eluting with
acetonitrile-water system containing 0.1% acetic acid to provide (R)-1-[3-(1-
quinolin-6-yl-ethyl)-
[1,2,4]triazolo[4,3-b]pyridazin-6-yl]-pyrrolidin-3-ylamine as an acetic acid
salt (47.2 mg, 70% yield).
Method 5.
N O P\N/
ACI H
H2N N,
- 'YN NN
N\ N Pyridine 0 N N,
To a solution of 3-(1-quinolin-6-yl-ethyl)-[1,2,4]triazolo[4,3-b]pyridazin-6-
ylamine (30.0 mg, 0.103
mmol) in anhydrous pyridine (1 mL) was added acetyl chloride (0.015 mL, 0.207
mmol). The reaction
mixture was stirred under nitrogen at ambient temperature for overnight. After
evaporation of solvent, the
residue was purified on a reverse-phase C-18 preparative HPLC eluting with
acetonitrile-water containing
0.1 % acetic acid to provide N-[3-(1-quinolin-6-yl-ethyl)-[1,2,4]triazolo[4,3-
b]pyridazin-6-yl]-acetamide (27.1
mg, 79% yield).
Method 6:
o
N C C ~ N
0 NaH/DMF ~
N ~
CI N~N N + 850 N~N N~
H N N
To a solution of I H-imidazole-4-carboxylic acid methyl ester (83.1 mg, 0.646
mmol) in anhydrous
DMF (3 mL) was added sodium hydride (60% oil suspension, 28.4 mg, 0.71 mmol).
The reaction mixture
was stirred at ambient temperature for one hour, and then 6-[1-(6-chloro-
[1,2,4]triazolo[4,3-b]pyridazin-3-
yl)-ethyl]-quinoline (200.0 mg, 0.646 mmol) was added. The reaction was heated
in an 85 C oil bath
under nitrogen for overnight. LC-MS showed the reaction was not complete, and
additional portions of
sodium hydride (14.2 mg, 0.35 mmol) and 1 H-imidazole-4-carboxylic acid methyl
ester (41.5 mg, 0.323
mmol) were added. The reaction was continued for another 2 hours at 85 C under
nitrogen. After
cooling, the reaction was quenched with an addition of saturated aqueous
ammonium chloride solution,
and a lot of precipitate was observed. The solid was filtered, washed with
water, methanol and ether to
provide 1-[3-(1-quinolin-6-yl-ethyl)-[1,2,4]triazolo[4,3-b]pyridazin-6-yl]-1 H-
imidazole-4-carboxylic acid
methyl ester (164.9 mg, 64% yield).
Method 7:
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-32-
N N Pd(PPh3)2CI2 J
N
` // ~ - ~ I NN
CI NN \ +N Sn(nBu)3 CuUACN N ~ N
N reflux
2 3
To a solution of 6-[1-(6-chloro-[1,2,4]triazolo[4,3-b]pyridazin-3-yl)-ethyl]-
quinoline (50.0 mg, 0.16
mmol) in ACN (2 mL) was added Pd(PPh3)zCI2 (5.6 mg, 0.008 mmol), Cul (4.6 mg,
0.024 mmol), and 1-
methyl-5-tributylstannyl-1 H-imidazole (74 mg, 0.194 mmol). The reaction
solution was degassed and
charged with nitrogen for three times, and heated at 85 C oil bath under
nitrogen for overnight. The
reaction was evaporated and purified on a reverse-phase C-18 preparative HPLC
eluting with acetonitrile-
water containing 0.1% acetic acid to provide 6-{1-[6-(3-methyl-3H-imidazol-4-
yl)-[1,2,4]triazolo[4,3-
b]pyridazin-3-yl]-ethyl}-quinoline (22 mg, 38% yield).
Method 8:
" o-
N
/ / _'A 0 Cs2C03 % ~ O N
/B\ + Br O DMF + CI N~N
O O OB,O N
O ~
OH
N
Pd(dPPf)2C'2 N
N
Cs2C03 N + '-O N~N \
DME/H20 N,N N
N N
To a solution of 4-(4,4,5,5-Tetramethyl-[1,3,2]dioxaborolan-2-yl)-1 H-pyrazole
(5.0 g, 25.8 mmol) in
DMF (52 mL) was added Cs2CO3 (8.396 g, 25.8 mmol) and bromo-acetic acid methyl
ester (2.52 mL, 25.8
mmol). The reaction mixture was heated at 90 C under nitrogen for overnight.
After cooling, the reaction
mixture was diluted with water, and extracted with ethyl acetate. The combined
extracts were washed
with water for three times and brine, dried over Na2SO4., and concentrated to
provide 4-(4,4,5,5-
tetramethyl-[1,3,2]dioxaborolan-2-yl)-pyrazol-1-yi]-acetic acid methyl ester
(4.27 g, 62% yield).
To a solution of 4-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-pyrazol-1-
yl]-acetic acid methyl
ester (644.2 mg, 2.42 mmol) and 6-[1-(6-chloro-[1,2,4]triazolo[4,3-b]pyridazin-
3-yl)-ethyl]-quinoline (500.0
mg, 1.61 mmol) in 1,2-dimethoxyethane were added a freshly prepared solution
of CS2CO3 (1.574 g, 4.83
mmol) in water (2.3 mL) and Pd(dppf)2CIz.CH2CI2 (40 mg, 0.048 mmol). The
reaction mixture was
degassed and charged with nitrogen for three times and then heated at 85 C oil
bath for overnight. After
cooling, the residue was dissolved in methanol and filtered through a celite
pad. The filtrate was
concentrated and purified on a reverse-phase preparative HPLC eluting with
acetonitrile-water containing
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-33-
0.1% acetic acid to provide {4-[3-(1-quinolin-6-yl-ethyl)-[1,2,4]triazolo[4,3-
b]pyridazin-6-yl]-pyrazol-l-yl}-
acetic acid (193 mg, 30% yield).
Method 9:
, NJ
/
0 N
OH O
N N HATU N N
N~ N, + HZN__~ N DMF N\ N,N
N
N N NN
To a solution of {4-[3-(1-quinolin-6-yi-ethyl)-[1,2,4]triazolo[4,3-b]pyridazin-
6-yl]-pyrazol-l-yl}-acetic
acid (50.0 mg, 0.13 mmol) in DMF (2 mL) was added HATU (52.4 mg, 0.138 mmol).
The reaction mixture
was stirred at ambient temperature under nitrogen for half an hour, and then 2-
pyrrolidin-1-yl-ethylamine
(0.03 mL, 0.25 mmol) was added. The reaction was continued for overnight and
purified on a reverse-
phase C-18 preparative HPLC eluting with acetonitrile-water containing 0.1%
acetic acid to provide N-(2-
Pyrrolidin-1-yl-ethyl)-2-{4-[3-(1-quinolin-6-yl-ethyl)-[1,2,4]triazolo[4,3-
b]pyridazin-6-yl]-pyrazol-1-yl}-
acetamide (12 mg, 19% yield).
Method 10.
N
O
OH CI N~N 1. HATU/DMF_ CI N NH
CD + N, N
N N N
NH2 2. Heat
H H N
1 2 3
(1 H-Pyrrolo[2,3-b]pyridin-3-yl)-acetic acid (1) was prepared according to M.
M. Robinson's
method (J. Am. Chem. Soc. 78 (1956) 1247-1249). To a solution of (1 H-
pyrrolo[2,3-b]pyridin-3-yl)-acetic
acid (1) (283 mg, 1.61 mmol) and (6-chloro-pyridazin-3-yl)-hydrazine (233 mg,
1.61 mmol) in DMF (8 mL)
was added HATU (612 mg, 1.61 mmol). The reaction mixture was stirred at
ambient temperature for one
hour and then heated at 120 C for two hours. After cooling, the reaction was
concentrated and purified on
a reverse-phase C-18 preparative HPLC eluting with acetonitrile-water
containing 0.1% acetic acid to
provide 6-chloro-3-[1-(1H-pyrrolo[2,3-b]pyridin-3-yl)-ethyl]-
[1,2,4]triazolo[4,3-b]pyridazine (99 mg, 20%
yield).
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-34-
Method 11.
O 0 O
O (Boc)20/THF O 1. LDA/THF \ O
( \ ~ I \ ~ --> I
N DMAP N ~ 2. Mel N~ ~
N H
O O O O O ~
O CI N'~N
HCI/Dioxane LiOH OH
--~ \
CH2CI2 N N MeOH/H20 I ~ ~ + \ I N~NH2
H N H H
eN
1. SOCIz
= ~ \ NH
2. Heat/DMF CI NN
N
N
Step 1.
To a solution of (1 H-pyrrolo[2,3-b]pyridin-3-yl)-acetic acid methyl ester
(5.10 g, 28.78 mmol) and
4-dimethylaminopyridine (175.8 mg, 1.44 mmol) in anhydrous THF (100 mL) was
added di-tert-butyl
dicarbonate (34.6 g, 34.54 mmol). The reaction mixture was stirred for
overnight, diluted with ethyl
acetate, washed with water and brine, dried over Na2SO4r and concentrated. The
residue was suspended
in hexane, and the solid was filtered and dried to provide a white solid of 3-
methoxycarbonylmethyl-
pyrrolo[2,3-b]pyridine-l-carboxylic acid tert-butyl ester (6.07 g). The
filtrate was concentrated and purified
on a silica gel column eluting with hexane-ethyl acetate to provide additional
product (1.71 g, total 7.78 g,
97% yield). 1 H NMR (400 MHz, CHLOROFORM-D) 51.60 (s, 9 H) 3.62 (s, 3 H) 3.83
(s, 2 H) 7.28 (dd,
J=7.83, 4.80 Hz, 1 H) 7.73 (s, 1 H) 7.99 (dd, J=7.83, 1.52 Hz, I H) 8.38 (dd,
J=4.55, 1.52 Hz, 1 H).
Step 2.
To a solution of 3-methoxycarbonylmethyl-pyrrolo[2,3-b]pyridine-l-carboxylic
acid tert-butyl ester
(6.07 g, 20.9 mmol) in anhydrous THF (100 mL) was added LDA (1.8 M THF
solution, 12.7 mL, 22.99
mmol) at-78 C under nitrogen. The reaction mixture was stirred at -78 C under
nitrogen for half an hour
and then methyl iodide was added. The reaction mixture was stirred from -78 C
to ambient temperature
overnight under nitrogen, quenched with an addition of saturated ammonium
chloride, and diluted with
ethyl acetate. The ethyl acetate layer was washed with brine, dried over
Na2SO4, concentrated, and dried
under high vacuum to provide 3-(1-methoxycarbonyl-ethyl)-pyrrolo[2,3-
b]pyridine-l-carboxylic acid tert-
butyl ester (6.35 g, -100% yield).
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-35-
Step 3.
To a solution of 3-(1-methoxycarbonyl-ethyl)-pyrrolo[2,3-b]pyridine-1-
carboxylic acid tert-butyl
ester (6.35 g, 20.9 mmol) in dichloromethane (50 mL) was added HCI dioxane
solution (4N, 20 mL). The
reaction mixture was stirred for overnight. After evaporation and high vacuum
dry, the product was used
directly for the next step.
Step 4.
To a solution of 2-(1 H-Pyrrolo[2,3-b]pyridin-3-yl)-propionic acid methyl
ester (4.26 g, 20.9 mmol)
in methanol (45 mL) and water (15 mL) was added LiOH (2.503 g, 104.5 mmol).
The reaction mixture
was stirred at 60oC oil bath for 2 hours. After cooling, the reaction mixture
was neutralized to pH -6 with
6N HCI solution. The solid was filtered and washed with water to provide 2-(1
H-pyrrolo[2,3-b]pyridin-3-yl)-
propionic acid (2.45 g). The filtrate was concentrated and purified on a
reverse-phase C-18 pad to
provide addition portion of the product (1.05 g, total 3.50 g).
Step 5.
The suspension of 2-(1 H-pyrrolo[2,3-b]pyridin-3-yl)-propionic acid (542 mg,
2.85 mmol) in thionyl
chloride (6 mL) was stirred at ambient temperature for two hours, and then
thionyl chloride was removed
in vacuum. To the residue was added a solution of (6-chloro-pyridazin-3-yl)-
hydrazine (412 mg, 2.85
mmol) in anhydrous DMF (5 mL). The reaction solution was stirred for 5 minutes
at ambient temperature
and then heated at.100 C oil bath for 30 minutes. After cooling, DMF was
removed with vacuum. The
residue was dissolved in water, washed with ethyl acetate, and the water layer
was lyophilized to provide
6-chloro-3-[1-(1H-pyrrolo[2,3-b]pyridin-3-yl)-ethyl]-[1,2,4]triazolo[4,3-
b]pyridazine (610 mg, 71.5% yield).
Method 12:
Synthesis of 7-methyl-6-{[6-(1-methyl-1 H-pyrazol-4-yl)[1,2,4]triazolo[4,3-
b]pyridazin-3-
yl]methyl}quinoline (11)
CH3
CH3 &NHAc O CH3 HN~; s ::1 H3C 1 NHAc
NHZ 1 AICI3 NHAc Sulfur J
Step 2 Step 3 0
A B C D
CH3 CH3
HCI glycerol ~ \ \
(.II.COOH + \ \ COOH
Step 4 H N I/ Step 5 N CH3 I N /
2
E F F1
CH3 CH3
SOCIz I\ \ COOEt + (r5'COOEt NH3 -' I N CH
Step 7 3 + (5C0OH
N Step 6 G G1 H H1
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-36-
CI N,\N CIINN H CI N~N H CH3
I NHNHa / NN + I N.N
H O N H O I/ N
Step 8 3C
~ I1
H3C H3C
~ N Pd(PPh3)2CI2, Na2CO3, H3~ N
AcOH DME, H2O, 80 C N
reflux CI N.N \ N o N\ I N, ~
~ N \
NN NBo -NN
Step 9 K
Step 10
Step I
To a solution of compound A (321 g, 3 mol) in CH2CI2 (3.5 L) cooled to 0-10 C
was added
dropwise AcCI (259 g, 3.3 moI) while keeping the temperature at 0-10 C. During
the addition, white solid
deposited. After the addition was complete, the resulting mixture was stirred
at room temperature
overnight. TLC (EtOAc: Petroleum ether = 1:2) indicated the reaction was
complete, the solvent and
excessive AcCI were removed under reduced pressure to give crude compound B
(500 g, 100%), which
was used directly in next step.
Step 2
To a suspension of compound B(500 g, 3 mol in theory) in CH2CI2 (6 L) cooled
to 0-10 C was
added AICI3 (1400 g, 10.5 mol) portionwise. The mixture turned clear and a few
minutes later turned
cloudy. AcCI (282.6 g, 3.6 mol) was added dropwise while keeping the
temperature below 20 C. Then the
resulting mixture was stirred at room temperature overnight. TLC (EtOAc:
Petroleum ether = 1:2) indicated
the reaction was complete. The mixture was poured carefully into a mixture of
conc. HCI (1 L) and ice (2
Kg) with strong stirring. The solid was collected via filtration to give
compound C(100 g). The organic
layer was separated from the filtrate, washed with water (1.5 L) and brine,
dried over Na2SO4 and
concentrated until about 150 mL of CHZCIZ was left. The mixture was filtered
to give compound C(180 g).
The total yield is 48.8%.
Step 3
A mixture of compound C (96 g, 0.5 mol), morpholine (48.5 g, 0.558 mol) and
sulfur (17.9 g, 0.558
mol) was heated to 110-130 C and stirred overnight. TLC (EtOAc: Petroleum
ether = 1:2) indicated the
reaction was complete, the mixture was powered into hot water (70-80 C, 1 L)
with strong stirring. A few
minutes later, some solid deposited. The solid was colleted via filtration to
give crude compound D, which
was re-crystallized from EtOH (150 ml) at 70-80 C to give compound D(66 g,
45.2%).
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-37-
'H NMR (400 MHz, CDC13): b 7.443 (s, 1H), 7.254-7.131 (m, 2H), 7.131-7.080 (d,
1H), 4.440-
4.340 (t, 2H), 4.179 (s, 2H), 3.818-3.786 (t, 2H), 3.497 (s, 4H), 2.231 (s,
3H), 2.187 (s, 3H).
Step 4
A suspension of compound D (151 g, 0.517 mol) in conc. HCI (500 mL) and water
(1 L) was
heated to 100-110 C and stirred for 2 days. The solvent was removed under
reduce pressure. 900 mL of
hot water (50-60 C) was added and filtered. The filtrate was adjusted pH 9-10
with 20% aq. NaOH, and
there was solid deposited. EtOAc (250 mL) was added and the mixture was
filtered. The aqueous layer
was separated from the filtrate, acidified with 10% aq. HCI to pH 5. The solid
was filtered and dried to give
compound E (60 g, 70.3%).
'H NMR (400 MHz, DMSO): b 6.945-6.868 (d, 1 H), 6.519 (s, 1 H), 6.490-6.464
(dd, 1 H), 3.391 (s,
2H), 2.098 (s, 3H).
Step 5
A mixture of compound E (60 g, 0.364 mol), ferrous sulfate (13 g, 0.047 mol),
glycerol (140 g,
1.52 mol), nitrobenzene (27.5 g, 0.22 mol) and concentrated sulfuric acid (65
mL) was heated gently. After
the first vigorous reaction, the mixture was refluxed for five hours and then
treated with aq. sodium
hydroxide solution (2 N, 250 mL) and 4 N aq. sodium hydroxide solution until
pH = 5-6. A dark brown
precipitate formed. The precipitate was filtered, washed with water and taken
up with aq. sodium
hydroxide solution (4 N, 250 mL). The mixture was adjusted to pH 5-6 with
glacial acetic acid. A dark-
brown precipitate formed. The precipitate was collected and dried in vacuo to
give crude compound F&
El (100 g, 100%).
Step 6
To a suspension of compound F& F1 (100 g, 0.364 mol in theory) in EtOH (600
mL) cooled to
0-5 C was added dropwise SOCI2 (37 mL, 0.437 mol). After the mixture was
heated to reflux for 2 h, the
mixture was evaporated under reduced pressure, and the residue was taken up
with EtOAc (300 mL) and
aq. NaHCO3 (300 mL). (If there was solid appeared, filtered). The organic
layer was separated, washed
with brine, dried over Na2SO4 and concentrated to give crude product, which
was purified via a silica
column chromatography (EtOAc: Petroleum ether = 1:10 -1:5) to give compound G&
G1 (50 g, 60%) as
a red oil.
'H NMR (400 MHz, CDCI3): S 8.896-8.886 (d, 0.45H), 8.858-8.848 (d, 1H), 8.416-
8.394 (d,
0.45H), 8.098-8.078 (d, 1 H), 7.958-7.923 (m, 1.4H), 7.658 (s, 1 H), 7.585-
7.563 (d, 0.45H), 7.448-7.414 (q,
0.45H), 7.351-7.320 (q, 1 H), 4.206-4.106 (m, 3.1 H), 3.860 (s, 0.9H), 3.812
(s, 2H), 2.641 (s, 1.3H), 2.520
(s, 3H), 1.276-1.231 (m, 4.8H).
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-38-
Step 7
A mixture of compound G & GI (14 g, 0.06 mol) and sodium hydroxide solution
(20%, 160 mL)
was heated at 90 C for 3 hours. The mixture was diluted with water (160 mL),
and adjusted to pH = 6 with
15% aq. HCI. The white precipitate was collected, washed with water and dried
in vacuo to give
compound H & H1 (11.7 g, 97%) as a white solid.
'H NMR (400 MHz, DMSO): 8 12.510- (s, 1.2H), 8.863-8.844 (dd, 0.3H), 8.820-
8.800 (dd, IH),
8.560-8.505 (d, 0.3H), 8.258-8.233 (d, 1 H), 7.812 (s, IH), 7.793-7.762 (d,
0.3H), 7.697 (s, IH), 7.608-
7.579 (d, 0.3H), 7.550-7.508 (q, 0.3H), 7.452-7.411 (q, 1 H), 3.844 (s, 0.6H),
3.799 (s, I H), 2.550 (s, 0.9H),
2.433 (s, 3H).
Step 8
To a suspension of compound H & HI (11.7 g, 0.058 mol) and (6-chloro-pyridazin-
3-yi)-hydrazine
(8.4 g, 0.058 mol) in DMF (150 mL) was added EDCI (16.7 g, 0.087 mol) at room
temperature. The
mixture turned clear and stirred for 2 days. TLC (CH2CI2: MeOH = 10:1) showed
the reaction was
complete. DMF was removed under reduced pressure, and water (100 mL) and EtOAc
(20 mL) were
added to the mixture and filtered. (If there was no solid separated, the
mixture was evaporated under
reduced pressure for a while and solid would appear). The cake was washed with
water and dried in
vacuum to give compound I& 11 (17 g, 89.5%) as a yellow solid.
'H NMR (400 MHz, DMSO): b 10.289 (s, 1.2H), 9.179 (s, 1.2H), 8.844-8.816 (m,
0.36H), 8.803-
8.797 (m, 1 H), 8.570-8.490 (d, 0.36H), 8.254-8.228 (d, IH), 7.852-7.806 (m,
2.2H), 7.679 (m, 0.38H),
7.558-7.515 (m, 1.7H), 7.458-7.417 (q, IH), 7.035-6.975 (m, 1.3H), 3.836 (s,
0.7H), 3.780 (s, 2H),
2.638(s, 0.96H), 2.509 (s, 3H).
Step 9
A suspension of compound I& 11 (17 g, 0.052 mol) in AcOH (150 mL) was heated
to reflux and
stirred overnight. After most of the solvent was removed in vacuum, water (300
mL) was added to the
residue and stirred strongly. The mixture was filtered, and the cake was
washed with water and dried in
vacuum to give J(6 g, 37.4%) as a grey solid.
'H NMR (400 MHz, DMSO): b 8.816-8.797 (q, 1 H), 8.485-8.453 (d, 1 H), 8.210-
8.182 (d, 1 H),
7.859 (s, 1 H), 7.645 (s, 1 H), 7.510-7.478 (d, 1 H), 7.430-7.388(q, 1 H),
4.638 (s, 2H), 2.544 (s, 3H).
Step 10
Compound J (200mg, 0.646mmo!), pyrazole boronic ester (162mg, 0.779mmol), and
sodium
carbonate (205mg, 1.94 mmol) were mixed in DME (8mL) and H20 (2mL), after
degassed three times, Pd
catalyst (22.7mg, 0.0323mmol) was added, and the reaction mixture was stirred
at 80 C for 3h. After
cooled to room temperature, the reaction mixture was diluted with EtOAc (50m1)
and water (20ml). The
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-39-
aqueous layer was extracted with EtOAc (25m1). The combined organic layers
were dried over MgSO4,
and evaporated in vacuo to afford the crude product. The crude product was
purified by Flash column
chromatography eluting with 70:25:5 CH2CI2:EtOAc:MeOH first and then 70:20:10
CH2CI2:EtOAc:MeOH to
afford 83mg of compound K.
'H NMR (400 MHz, DMSO-d6) 8 ppm 2.60 (s, 3 H) 3.93 (s, 3 H) 4.70 (s, 2 H) 7.42
(dd, J=8.21,
4.17 Hz, 1 H) 7.68 (d, J=9.60 Hz, 1 H) 7.85 (s, I H) 7.88 (s, I H) 8.16 (s, I
H) 8.27 (d, J=8.08 Hz, I H)
8.34 (d, J=9.60 Hz, I H) 8.53 (s, I H) 8.80 (dd, J=4.17, 1.64 Hz, 1 H).
Method 13
_N N ~ ~ B(OH)2
H
z H2N
Cl j N /N~N
NN N
To a flask containing 6-[(6-chloro[1,2,4]triazolo[4,3-b]pyridazin-3-
yl)methyl]quinoline (75 mg, 0.25
mmol), (4-aminomethylphenyl) boronic acid hydrochloride (52 mg, 0.28 mmol),
and cesium carbonate
(284 mg, 0.761 mmol) was added 1:3 water:dimethoxyethane (2.0 mL, degassed by
bubbling with
nitrogen gas for 15 minutes) followed by 1,1'-
bis(diphenylphosphino)ferrocene]dichloropalladium(II) (1:1
with dichloromethane, 8 mg, 0.01 mmol). The resulting mixture was then heated
to 70 C overnight. The
reaction was cooled to room temperature, filtered and the filtrate was
concentrated.
A mixture of 6-[(6-chloro[1,2,4]triazolo[4,3-b]pyridazin-3-yl)methyl]quinoline
(100 mg, 0.338
mmol), (4-aminomethylphenyl) boronic acid hydrochloride (70 mg, 0.37 mmol),
and cesium fluoride (154
mg, 1.01 mmol) in water (1.0 mL) and dimethoxyethane (3.4 mL) was degassed by
alternating between
vacuum and nitrogen (5x), then bis(triphenylphosphine)palladium(ll)chloride (7
mg, 0.01 mmol) was
added and the mixture was heated to 80 C. After 1 hour, sodium carbonate (1 M
in water, I mL) was
added and the reaction mixture continued to heat overnight. The mixture was
cooled to room temperature
diluted with dichloromethane and concentrated.
To a microwave vial containing 6-[(6-chloro[1,2,4]triazolo[4,3-b]pyridazin-3-
yl)methyl]quinoline (75
mg, 0.25 mmol), (4-aminomethylphenyl) boronic acid hydrochloride (52 mg, 0.28
mmol), and sodium
carbonate (1 M in water, 761 pL) was added dimethoxyethane (2.0 mL, degassed
by bubbling with
nitrogen gas for 15 minutes) followed by 1,1'-
bis(diphenylphosphino)ferrocene]dichloropalladium(II) (1:1
with dichloromethane 8 mg, 0.01 mmol). The resulting mixture was heated in the
microwave for 20
minutes at 80 C, 20 minutes at 120 C, then 30 minutes at 160 C. The reaction
mixture was diluted with
dichloromethane, filtered and the filtrate was concentrated.
The concentrated reaction mixtures were combined and purified by flash
chromatography using a
Horizon purification system on a 40S column eluting with chloroform/7 N
ammonia in methanol (0.5-10%),
followed by a second column on a 25S column eluting with chloroform/methanol
(0.1-10%), then
chloroform/7 N methanolic ammonia (0-8%), followed by preparative TLC eluting
(2x) with chloroform/7 N
ammonia in methanol (7%). The peak of interest was scraped and the silica gel
was slurried in
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-40-
chloroform/7 N ammonia in methanol (10%), filtered and concentrated to afford
the title compound (29
mg, 9%).
Method 14:
CI NC _
N NC &B(OH)a_
~~~(((% PdCI2(PPhs)2 ,N, N
N*N N %
~ N ~ ,N
N
A mixture of 3 mL of DMF, Cs2CO3 (0.98 g, 0.003 mol) and 1 mL of water was
degassed for 5
minutes. 6-((6-chloro-[1,2,4]triazolo[4,3-b]pyridazin-3-yl)methyl)quinoline
(0.30 g, 0.001 mol) and
cyanophenyl boronic acid (0.164 g, 0.001 mol) were added. A catalytic amount
of PdCl2(PPh3)2 was
added. The mixture was heated to 80 C and stirred overnight. The mixture was
diluted with CH2CI2 (10
mL), and then filtered. The organic phase was separated, dried over Na2SO4 and
evaporated in vacuum.
The residue was washed with DMF and EtOAc to give 4-(3-(quinolin-6-ylmethyl)-
[1,2,4]triazolo[4,3-
b]pyridazin-6-yl)benzonitrile (90.0 mg, 25.0%) as a white solid.
Method 15:
-.N=0
~N \ O N
CI ~
NS~NN_-, CF3COOH HN `
N, N
-~ O N, N ~
\` N
N~N N PdClz(PPhs)2 "NN ~ ~N N
6-((6-chloro-[1,2,4]triazolo[4,3-b]pyridazin-3-yl)methyl)quinoline (0.50 g,
1.7 mmol) and Cs2CO3
(1.64 g, 5.0 mmol) were dissolved in 5 mL of DMF and 2.5 mL of water. The
resulting solution was
degassed three times. Then catalytic amount of Pd(PPh3)ZCI2 and compound N,N-
dimethyl-4-(4,4,5,5-
tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole-l-sulfonamide (0.60 g, 2.0
mmol) were added. The
reaction mixture was heated to 80 C and stirred overnight. The mixture was
diluted with CH2CI2, and then
filtered. The organic phase was separated and concentrated to dryness in-
vacuum. The residue was
washed with DMF and EtOAc to give N,N-dimethyl-4-(3-(quinolin-6-ylmethyl)-
[1,2,4]triazolo[4,3-
b]pyridazin-6-yl)-1 H-pyrazole-1 -sulfonamide (190 mg, 25.0%) as a white
solid.
N,N-dimethyl-4-(3-(quinolin-6-ylmethyl)-[1,2,4]triazolo[4,3-b]pyridazin-6-yl)-
1 H-pyrazole-l-
sulfonamide (0.19 g, 0.437 mmol) was added to 1 mL of ice-cold CF3COOH. The
resulting mixture was
stirred at room temperature for 3 h. Then CF3COOH was removed in vacuum. 10 mL
of saturated
aqueous NaHCO3 was added carefully. A lot of white solid was formed, which was
filtered and dried to
give 6-((6-(1 H-pyrazol-4-yl)-[1,2,4]triazolo[4,3-b]pyridazin-3-
yl)methyl)quinoline (80 mg, 60.0%) as a white
solid.
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-41 -
Table 1
Example Structure Name NMR/LC-MS Method
1H NMR (400 MHz, DMSO-D6) S
ppm 1.89 (d, J=7.33 Hz, 3 H) 5.00
6-[(S)-1-(6- (q, J=7.07 Hz, 1 H) 7.46 (d, J=9.60
H3C N Chloro- Hz,1 H) 7.51 (dd, J=8.34, 4.29 Hz, 1
[1,2,41triazolo[4 H) 7.78 (dd, J=8.84, 2.02 Hz, 1 H)
1 ,3-b]pyridazin- 7.87 (d, J=1.77 Hz, 1 H) 7.98 (d, 2
Cl N~N 3-yi)-ethyl]- J=8.84 Hz, 1 H) 8.33 (d, J=8.34 Hz,
N quinoline I H) 8.45 (d, J=9.60 Hz, I H) 8.86
N (dd, J=4.17, 1.64 Hz, 1 H). LC-MS
310, 312.
1H NMR (400 MHz, DMSO-D6) S
4-[3-((S)-1- ppm 1.96 (d, J=7.33 Hz, 3 H) 5.17
N~ Quinolin-6-yi- (dd, J=7.07 Hz, 1 H) 7.51 (dd,
H3
C N ethyl)- J=8.21, 4.17 Hz, 1 H) 7.84 (dd,
N\ [1,2,4]triazolo[4 J=8.84, 2.02 Hz, 1 H) 7.94 - 8.02 (m, 2
O
N N ,3-b]pyridazin- 3 H) 8.05 (d, J=8.59 Hz, 2 H) 8.23
- N 6-yl]- (d, J=8.59 Hz, 2 H) 8.32 - 8.42 (m, I
benzonitrile H) 8.50 (d, J=9.60 Hz, 1 H) 8.85 (dd,
J=4.17, 1.64 Hz, 1 H. LC-MS 377.
1 H NMR (400 MHz, DMSO-D6) 6
~ ppm 1.61 - 1.75 (m, I H) 1.86 (d,
H2N (R)-1-[3-(1- J=7.33 Hz, 3 H) 1.95 - 2.09 (m, I H)
H3C ~ N Quinolin-6-yl- 3.26 - 3.50 (m, 3 H) 3.51 - 3.61 (m,
ethyl)- J=5.81, 5.81 Hz, 2 H) 4.75 - 4.90 (m,
3 ~)N N [1,2,4]triazolo[4 I H) 6.92 (d, J=10.11 Hz, 1 H) 7.48 4
N 3-b]pyridazin- (dd, J=8.34, 4.29 Hz, 1 H) 7.73 -
N 6-yl]-pyrrolidin- 7.81 (m, 1 H) 7.89 (s, I H) 7.94 (dd,
3-ylamine J=9.35, 4.55 Hz, 2 H) 8.31 (d,
J=8.34 Hz, 1 H) 8.83 (dd, J=4.17,
1.64 Hz, 1 H. LC-MS 360.
1H NMR (400 MHz, DMSO-D6)
~ S ppm 1.85 (d, J=7.33 Hz, 3 H) 3.18
OH / 2-[3-(1- - 3.27 (m, 2 H) 3.40 - 3.56 (m, 2 H)
~ ~ 4.68 - 4.77 (m, 1 H) 4.81 (q, J=7.24
H3 N Quinolin-6-yl- Hz, I H) 6.76 (d, J=9.85 Hz, 1 H)
~~ ethyl)- 7.36 (t, J=5.31 Hz, I H) 7.48 (dd,
4 [1,2,4]triazolo[4 4
HN N" J=8.34, 4.29 Hz, 1 H) 7.74 (dd,
,3-b]pyridazin
N 6-ylamino]- J=8.59, 2.02 Hz, I H) 7.82 (d,
ethanol J=9.85 Hz, 1 H) 7.89 (d, J=1.77 Hz,
N I H) 7.93 (d, J=8.84 Hz, I H) 8.31
(d, J=7.58 Hz, 1 H) 8.83 (dd, J=4.17,
1.64 Hz, 1 H. LC-MS 335.
1 H NMR (400 MHz, DMSO-D6) S
~ 6-{(S)-1-[6-(1- ppm 1.92 (d, J=7.33 Hz, 3 H) 3.90
(s, 3 H) 5.04 (q, J=6.91 Hz, I H)
H3R N Methyl-1 H-
N HaC 7.49 (dd, J=8.34, 4.29 Hz, 1 H) 7.62
/ pyrazol-4-yl) (d, J=9.85 Hz, I H) 7.83 (d, J=8.59
N~ N [1,2,4]triazolo[4 3
N~ ,3-b]pyridazin- Hz, 1 H) 7.96 (d, J=8.84 Hz, I H)
N 3 yl]-ethyl}- 8.00 (s, 1 H) 8.08 (s, I H) 8.30 (d,
N J=9.60 Hz, 1 H) 8.35 (d, J=8.08 Hz,
quinoline 1 H) 8.46 (s, 1 H) 8.83 (d, J=2.78
Hz, 1 H. LC-MS 356.
~ 6-[6-(1-Methyl- 1 H NMR (300 MHz, DMSO-d6) S
H3R N 1H pyrazol-4 ppm 8.84 (dd, J=4.24, 1.60 Hz, 1 H)
N 8.52 (s, 1 H) 8.26 - 8.39 (m, 2 H)
6 N\ ~ N [1,2,4]tr azolo[4 8.16 (s, 1 H) 7.93 - 8.03 (m, 2 H) 3
, N ~ 3-b]pyridazin Hz?1 H) 750 (dd, J> 8.29,(4.33 Hz,11
N N 3-ylmethyl]- H) 4.73 (s, 2 H) 3.93 (s, 3 H). LC-MS
quinoline 341.
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-42-
Example Structure Name NMRILC-MS Method
1H NMR (400 MHz, DMSO-D6) S
6-{1-[6-(4- ppm 1.92 (d, J=7.33 Hz, 3 H) 2.16
Methyl- (s, 3 H) 5.05 (q, J=6.99 Hz, 1 H)
H3 N 7.49 (dd, J=8.34, 4.29 Hz, I H) 7.60
N_ imidazol-1-yl)-H 7 H3C _Il N\ [1,2,4]triazolo[4 Js=9. 5)Hz,1 H)7 94 6 (d1
J=8 84 Hz, 6
\ ,3-b]pyridazin-
N 3-yl]-ethyl)- I H) 8.02 (s, I H) 8.35 (d, J=8.08
N Hz, 1 H) 8.48 (s, 1 H) 8.53 (d,
quinoline J=g 85 Hz, 1 H) 8.78 - 8.87 (m, 1 H).
LC-MS 356.
O [3-(1-Quinolin- 1H NMR (400 MHz, DMSO-D6) S
6-yl-ethyl)- ppm 1.51 -1.82 (m, 2 H) 1.82 - 1.89
HaC N [1,2,4]triazolo[4 (m, 3 H) 3.50 - 3.93 (m, 4 H) 4.00 -
3-b]pyridazin- 4.18 (m, 1 H) 4.81 (dd, J=7.20, 3.92
8 HN N~ ' 6-yl]" 7.44 17.56 (m,(2 H) 7.66 H 4
7.75 (m,
~ (tetrahydro-
1 H) 7.82 - 7.89 (m, J=11.37 Hz, 2 H)
N furan-3-yl)-
N amine 7.93 (d, J=8.59 Hz, I H) 8.30 (dd,
J=7.71, 3.92 Hz, I H). LC-MS 361.
F F 6-{1-[6-(2- 1H NMR (400 MHz, DMSO-D6) S
Methyl-5- ppm 1.92 (d, J=6.82 Hz, 3 H) 3.98
F H3C N trifluoromethyl- (s, 3 H) 5.10 (q, J=7.07 Hz, I H)
2H-pyrazol-3- 7.48 (dd, J=8.08, 3.79 Hz, I H) 7.65
9 N` ~ N\ yl)- (s, 1 H) 7.74 (d, J=8.59 Hz, 1 H) 7
N
N N [1,2,4]triazolo[4 7.79 (d, J=9.60 Hz, 1 H) 7.87 (s, 1
,3-b]pyridazin- H) 7.96 (d, J=8.59 Hz, I H) 8.31 (d,
H3C N 3-yl]-ethyl)- J=8.08 Hz, 1 H) 8.51 (d, J=9.85 Hz,
quinoline 1 H) 8.83 (s, 1 H. LC-MS 424.
1 H NMR (400 MHz, DMSO-D6) S
ppm 0.87 (d, J=6.32 Hz, 3 H) 1.08
' (d, J=6.57 Hz, 3 H) 1.79 (d, J=7.33
/ Isopropyl-[3- Hz, 3 H) 3.60 - 3.80 (m, 1 H) 4.74 (q,
HsC ~ N ((S)-1-quinolin- J=7.24 Hz, 1 H) 6.59 (d, J=9.85 Hz,
H N~ 6-yl-ethyl)- I H) 7.06 (d, J=6.82 Hz, I H) 7.42 4
H3C [1,2,4]triazolo[4 (dd, J=8.34, 4.29 Hz, I H) 7.64 (dd,
N\N ,3-b]pyridazin- J=8.72, 1.89 Hz, I H) 7.76 (d,
CH3 ~ ~N 6-yl]-amine J=9.85 Hz, I H) 7.81 (s, 1 H) 7.86
(d, J=8.84 Hz, 1 H) 8.24 (d, J=8.34
Hz, 1 H) 8.73 - 8.81 (m, I H). LC-MS
333.
HO
1H NMR (400 MHz, DMSO-D6) S
O {4-[3-(1- ppm 1.92 (d, J=7.07 Hz, 3 H) 5.00 -
Quinolin-6-yl- 5.15 (m, 3 H) 7.45 - 7.51 (m, 1 H)
HsC N ethyl)- 7.65 (d, J=9.85 Hz, 1 H) 7.83 (d,
11 NN [1,2,4]triazolo[4 J=8.34 Hz, 1 H) 7.96 (d, J=8.84 Hz, 8
N,N \ 3-b]pyridazin- 1 H) 8.01 (s, I H) 8.13 (s, 1 H) 8.31
N 6-yl]-pyrazol-1- (d, J=9.60 Hz, 1 H) 8.35 (d, J=8.34
N yl)-acetic acid Hz, I H) 8.51 (s, 1 H) 8.83 (s, I H)
13.39 (s, I H). LC-MS 400.
1H NMR (400 MHz, DMSO-D6) S
N-(2-Pyrrolidin- ppm 1.74 (s, 4 H) 1.90 (s, 2 H) 1.93
HN 1-yl-ethyl)-2-{4- (d, J=7.07 Hz, 3 H) 2.67 - 2.81 (m, 4
[3-(I-quinolin- H) 4.89 (s, I H) 5.05 (q, J=7.33 Hz,
O 6-yl-ethyl)- 1H) 7.48 (dd, J=8.21, 4.17 Hz, 1 H)
12 7.66 (d, J=9.85 Hz, 1 H) 7.83 (dd, 9
H C N [1,2,4]triazolo[4
3 3-b]pyridazin- J=8=72, 1.89 Hz, I H) 7.96 (d,
J=8.59 Hz, I H) 7.99 (s, I H) 8.11
N ~ 6-yl]-pyrazol-1 - (s, 1 H) 8.26 (s, 1 H) 8.29 - 8.37 (m,
~ NN \ yl} acetamide 2 H) 8.49 (s, I H) 8.83 (dd, J=4.17,
N N 1.64 Hz, I H). LC-MS 496.
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-43-
Example Structure Name NMR/LC-MS Method
1H NMR (400 MHz, MeOD) 6 ppm
6-{1-[6-(1H- 2.03 (d, J=7.07 Hz, 3 H) 5.17 (q,
H N J=7.33 Hz, 1 H) 7.54 (dd, J=8.34,
HN 3 Pyrazol-4-yl)- 4.55 Hz, 1 H) 7.68 (d, J=9.85 Hz, 1
[1,2,4]triazolo[4
13 H) 7.86 - 7.94 (m, 1 H) 7.98 - 8.05 3
N N\N ,3-b]pyridazin- (m, 2 H) 8.13 (d, J=9.60 Hz, I H)
N 3-yl]-ethyl)- 8.20 - 8.29 (m, 2 H) 8.40 (d, J=8.34
N quinoline Hz, I H) 8.78 - 8.85 (m, 1 H). LC-MS
342.
/ N 6-(1-Methyl- 1H NMR (400 MHz, DMSO-d6) S
H3C I H-pyrazol-4- ppm 11.50 (d, J=1.01 Hz, 1 H) 8.56
N yI)-3-(1H- (s, 1 H) 8.27 - 8.33 (m, 1 H) 8.15 -
14 / NH pyrrolo[2,3- 8.22 (m, 2 H) 8.04 (d, J=6.57 Hz, 1 3
b]pyridin-3- H) 7.66 (d, J=9.85 Hz, 1 H) 7.53 (d,
N N~N \ ylmethyl)- J=2.53 Hz, 1 H) 7.03 (dd, J=7.83,
N [1,2,4]triazolo[4 4.80 Hz, 1 H) 4.60 (s, 2 H) 3.94 (s, 3
3-b]pyridazine H). LC-MS 330.
6-(1-Methyl- 1 H NMR (300 MHz, DMSO-d6) S
N 1H-pyrazol-4- ppm 11.47 (s, I H) 8.50 (s, I H) 8.27
H3C H3C -' yl)-3-[(S)-1- (d, J=9.61 Hz, I H) 8.08 - 8.15 (m, 2
N (1 H- H) 7.92 - 8.00 (m, 1 H) 7.61 (d,
15 N I NH pyrrolo[2,3- J=9.80 Hz, 1 H) 7.54 (s, 1 H) 6.97 2
~ N, N b]pyridin-3-yl)- (dd, J=7.72, 4.90 Hz, 1 H) 5.00 -
N ethyl]- 5.14 (m, J=7.35 Hz, I H) 3.92 (s, 3
N [1,2,4]triazolo[4 H) 1.91 (d, J=7.35 Hz, 3 H). LC-MS
,3-b]pyridazine 345.
n\//-N 4-{3-[(S)-1-(1H- 1H NMR (300 MHz, DMSO-d6) S
Pyrrolo[2,3- ppm 11.54 (s, I H) 8.47 (d, J=9.61
N H3C b]pyridin 3 yl) Hz, I H) 8.25 - 8.32 (m, 2 H) 8.16
(dd, J=4.71, 1.51 Hz, 1 H) 8.03 -
16 NH ethyl]- 8.09 (m, 2 H) 7.95 - 8.01 (m, 2 H) 2
N~N ~ [1,2,4]triazolo[4 7.51 (d, J=2.26 Hz, I H) 7.01 (dd,
3-b]pyridazin- N J=7.91, 4.71 Hz, 1 H) 5.14 - 5.24 (m,
N 6-yl)_ J=7.16 Hz, 1 H) 1.91 - 1.99 (m,
benzonitrile J=7,35, 7.35 Hz, 3 H. LC-MS 366.
1H NMR (400 MHz, DMSO-d6)
N Dimethyl-(3- S ppm 8.85 (dd, J=4.28, 1.76 Hz, I
quinolin-6- H) 8.28 - 8.37 (m, I H) 8.00 (d,
17 CH3 ylmethyl- J=10.07 Hz, 1 H) 7.87 - 7.97 (m, 2 4
~N N, [1,2,4]triazolo[4 H) 7.76 (dd, J=8.81, 2.01 Hz, I H)
H3C N\N ,3-b]pyridazin- 7.50 (dd, J=8.31, 4.28 Hz, 1 H) 7.21
N 6-yl)-amine (d, J=10.32 Hz, I H) 4.57 (s, 2 H)
3.07 (s, 6 H). LC-MS 304.
N 1H NMR (400 MHz, DMSO-d6) S
3-Quinolin-6- ppm 8.85 (dd, J=4.29, 1.77 Hz, I H)
ylmethyl- 8.29 (d, J=7.33 Hz, I H) 7.94 (t,
18 [1,2,4]triazolo[4 J=8.84 Hz, 2 H) 7.78 (s, 1 H) 7.71 4
H2N N~N 3-b]pyridazin- (dd, J=8.84, 2.02 Hz, 1 H) 7.50 (dd,
6-ylamine J=8.21, 4.17 Hz, I H) 6.71 - 6.84 (m,
N 3 H) 4.52 (s, 2 H). LC-MS 276.
N
H 1 H NMR (400 MHz, DMSO-D6) S
N ppm 1.84 (d, J=7.33 Hz, 3 H) 3.24 -
Azetidin-3-yl- 3.33 (m, I H) 3.43 - 3.52 (m, I H)
~ H3C N [3 (1-quinolin- 3.56 - 3.65 (m, I H) 3.76 - 3.84 (m, 1
H) 4.35 - 4.46 (m, J=6.32 Hz, I H)
19 6-yl-ethyl)- 4.80 (q, J-6.99 Hz, 1 H) 6.69 (d, 4
HN N" [1,2,4]triazolo[4 J=9.85 Hz, I H) 7.48 (dd, J=8.21,
N 3-b]pyridazin 4.17 Hz, I H) 7.74 (dd, J=8.72, 1.64
N 6 yl] amine Hz, I H) 7.82 - 7.99 (m, 4 H) 8.33 (d,
J=8.08 Hz, I H) 8.73 - 8.87 (m, 1 H).
LC-MS 346.
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-44-
Example Structure Name NMRILC-MS Method
~ 1H NMR (400 MHz, DMSO-D6) 5
~ h ppm 1.81 (d, J=7.33 Hz, 3 H) 4.81
H3C \ N 3-(1-Quinolin- (q, J=7.07 Hz, 1 H) 6.62 - 6.80 (m,
~ 6-yl-ethyl)- 3H) 7.48 (dd, J=8.21, 4.17 Hz, 1 H)
20 [1,2,4]triazolo[4 7.74 (d, J=8.59 Hz, I H) 7.77 (s, 1 4
H2N N~N \ 3-b]pyridazin- H) 7.91 (d, J=9.85 Hz, 1 H) 7.95 (d,
N 6-ylamine J=8.59 Hz, 1 H) 8.29 (d, J=8.08 Hz,
N 1 H) 8.84 (d, J=2.78 Hz, 1 H). LC-
MS 291.
1 H NMR (400 MHz, DMSO-D6)
N 3-(1- S ppm 1.88 (d, J=7.33 Hz, 3 H) 2.11
H3 N Quinolin-6-yl- (s, 3 H) 4.92 (q, J=6.95 Hz, 1 H)
21 H ethyl)- 7.49 (dd, J=8.34, 4.29 Hz, 1 H) 7.77
H3C~N YN~ [1,2,4]triazolo[4 (dd, J=8.84, 2.02 Hz, I H) 7.81 (d, 5
NN 3blpYridazin - J=1=77 Hz, 1 H) 7.90 (d, J=9.85 Hz,
O \~~N 6 yl] acetamide I H) 7.96 (d, J=8.59 Hz, 1 H) 8.25 -
8.33 (m, 2 H) 8.84 (dd, J=4.29, 1.77
Hz, 1 H 10.95 s, 1 H. LC-MS 333.
H3C 0 1-[3-(1- 1 H NMR (400 MHz, DMSO-D6) S
Quinolin-6-yl- ppm 1.92 (d, J=7.07 Hz, 3 H) 3.82
ethyl) (s, 3 H) 5.14 (q, J=7.16 Hz, 1 H)
H3 N [1,2,4]triazolo[4 7.49 (dd, J=8.34, 4.29 Hz, I H) 7.80
22 N 3-b]pyridazin- - 7.87 (m, 1 H) 7.92 - 8.01 (m, 2 H) 6
~N N- N 6-yl]-1H- 8.04 (s, 1 H) 8.33 (d, J=8.08 Hz, 1
NN imidazole-4- H) 8.59 - 8.64 (m, 2 H) 8.67 (d,
carboxylic acid J=1.26 Hz, 1 H) 8.83 (dd, J=4.17,
methyl ester 1.64 Hz, 1 H. LC-MS 400.
1H NMR (400 MHz, DMSO-D6) S
6-[1-(6- ppm 1.89 (d, J=7.33 Hz, 3 H) 3.87
H3 N Methoxy- (s, 3 H) 4.92 (q, J=7.07 Hz, I
[1,2,4]triazolo[4 H) 6.97 (d, J=9.85 Hz, 1 H) 7.49 (dd,
23 O N~ ,3-b]pyridazin J=8.34, 4.29 Hz, 1 H) 7.74 - 7.82 (m, 4
H3C~ ~ N 3-yl)-ethyl]- 1 H) 7.89 - 8.01 (m, 2 H) 8.19 (d,
N quinoline J=9=85 Hz, I H) 8.33 (d, J=8.34 Hz,
N 1 H) 8.84 (dd, J=4.17, 1.64 Hz, I H).
LC-MS 306.
1H NMR (400 MHz, DMSO-D6) 6
C-) ppm1.66-1.81(m,1H)1.81-1.96
6-{1-[6-((R)-3- (m, 4 H) 2.04 - 2.21 (m, 1 H) 2.25 -
Morpholin 4 yl 2.46 (m, 4 H) 2.71 - 2.91 (m, J=8.34
Ha N pyrrolidin-1-yl)- Hz, 1 H) 2.98 - 3.19 (m, I H) 3.45 -
24 [1,2,4]triazolo[4 3.74 (m, 6 H) 4.75 - 4.92 (m, J=7.07, 4
3-b]pyridazin- 2.02 Hz, 1 H) 6.94 (d, J=10.11 Hz, 1
N NN ~N 3-yl]-eth I H) 7.48 (dd, J=8.21, 4.17 Hz, 1 H)
N quinoline 7.71 (d, J=8.59 Hz, I H) 7.87 - 7.94
(m, 2 H) 7.97 (d, J=10.11 Hz, 1 H)
8.29 (d, J=8.34 Hz, 1 H) 8.79 - 8.87
m, 1 H. LC-MS 430.
~ 1H NMR (400 MHz, DMSO-D6) S
6-{1-[6-(3- ppm 1.89 (d, J=7.33 Hz, 3 H) 3.66
N H3C N Methyl-3H- (s, 3 H) 5.04 (q, J=7.24 Hz, 1 H)
imidazol-4-yl)- 7.48 (dd, J=8.34, 4.29 Hz, 1 H) 7.66
25 [1,2,4]triazolo[4 - 7.77 (m, 2 H) 7.80 - 7.92 (m, 3 H) 7
N N \ ,3-b]pyridazin- 7.95 (d, J=8.59 Hz, 1 H) 8.30 (d,
3 y]I eth I
H3C N - y}- J=8.34 Hz, 1 H) 8.33 (d, J=9.60 Hz,
N quinoline I H) 8.80 - 8.87 (m, 1 H). LC-MS
356.
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-45-
Example Structure Name NMRILC-MS Method
/ CH3 1H NMR (400 MHz, DMSO-D6) S
H3C-N N,N-Dimethyl- ppm 1.93 (d, J=7.07 Hz, 3 H) 2,81 -
p 2-{4-[3-(1- 2.91 (m, 3 H) 3.04 (s, 3 H) 5.06 (q,
/ quinolin-6-yl- J=7.07 Hz, 1 H) 5.20 (s, 2 H) 7.43 -
26 N H3C ~ N ethyl)- 7.52 (m, J=8.21, 4.17 Hz, I H) 7.66 9
\~ [1,2,4]triazolo[4 (d, J=9.60 Hz, I H) 7.83 (d, J=8.84
N\ N, 3-b]pyridazin- Hz, 1 H) 7.96 (d, J=8.59 Hz, 1 H)
N~N 6-yl]-pyrazol-l- 8.00 (s, 1 H) 8.10 (s, I H) 8.32 (dd, 2
\ ~N yl)-acetamide H) 8.42 (s, I H) 8.83 (d, J=4.04 Hz,
1 H). LC-MS 427.
/CH3 1H NMR (400 MHz, DMSO-D6) S
HN ppm 1.93 (d, J=7.07 Hz, 3 H) 2.62
O ~ N-Methyl-2-{4- (d, J=4.29 Hz, 3 H) 4.85 (s, 2 H)
[3-(1-quinolin- 5.06 (q, J=7.07 Hz, 1 H) 7.48 (dd,
6-yl-ethyl)- J=8.21, 4.17 Hz, I H) 7.66 (d,
27 H3 N [1,2,4]triazolo[4 J=9.85 Hz, I H) 7.83 (d, J=8.84 Hz, 9
N ,3b]pyridazin-6- 1 H) 7.96 (d, J=8.59 Hz, I H) 8.00
N~N y!]-pyrazol-l- (s, I H) 8.07 - 8.14 (m, 2 H) 8.33
N ylj-acetamide (dd, J=17.31, 8.97Hz, 2 H) 8.50 (s, 1
~N H) 8.83 (d, J=3.03 Hz, 1 H). LC-MS
413.
HO 1 H NMR (400 MHz, DMSO-D6) S
ppm 1.89 (d, J=7.33 Hz, 3 H) 4.38 -
2-{4-[3-(1- 4.73 (m, 4 H) 4.92 (dd, J=7.33 Hz, 1
Quinolin-6-yl- H) 6.18 (t, J=1.89 Hz, 1 H) 6.91 (d,
H3C ethyl)- J=9.85 Hz, 1 H) 7.38 - 7.44 (m, 1 H)
28 N I \~ [1,2,4]triazolo[4 7.48 (dd, J=8.21, 4.17 Hz, 1 H) 7.68 4
N, 3-b]pyridazin- (d, J=2.27 Hz, 1 H) 7.78 (dd, J=8.72,
N
N 6-yi]-pyrazol-1- 1.89 Hz, 1 H) 7.91 - 8.01 (m, 2 H)
N yl)-ethanol 8.15 - 8.20 (m, I H) 8.31 (d, J=8.08
Hz, 1 H) 8.83 (dd, J=4.17, 1.64 Hz, 1
H . LC-MS 386.
~ 1 H NMR (400 MHz, DMSO-D6) S
6-{1-[6-(2H- ppm 1.93 (d, J=7.33 Hz, 3 H) 5.08
H3C N Pyrazol-3-yl)- (q, J=7=07 Hz, 1 H) 6.69 - 6.75 (m, 1
[1,2,4jtriazolo[4 H) 7.48 (dd, J=8.21, 4,17 Hz, 1 H)
29 N 7.86 (dd, 1 H) 7.90 - 7.99 (m, 3 H) 3
\ 3-b]pyridazin-
N ~N, N\ 3-yl]-ethyl8=03 (s, 1 H) 8.34 (d, J=8.34 Nz, 1
H N H) 8.47 (d, J=9.85 Hz, 1 H) 8.60 (d,
N quinoline J=2.53 Hz, I H) 8.80 - 8.88 (m, I H).
LC-MS 342.
1H NMR (400 MHz, DMSO-D6)
Dimethyl-[3-(1- S ppm 1.86 (d, J=7.33 Hz, 3 H) 2.99
H3C N quinolin-6-yl- (s, 6 H) 4.85 (q, J=7.20 Hz, I H)
CH3 7.15 (d, J=1 0.11 Hz, 1 H) 7.48 (dd,
1 ethyl)-
30 4]triazolo[4 J=8.34, 4.29 Hz, 1 H) 7.74 (dd, 4
~N N~ [1,2, J=8.72, 1.89 Hz, I H) 7.86 8.03 (m,
H3C N \ 3-b]pyridazin-
N 6-ylj-amine 3 H) 8.30 (d, J=8.08 Hz, 1 H) 8.83
N (dd, J=4.17, 1.64 Hz, 1 H). LC-MS
319.
N 6 Chloro-3- 1 H NMR (400 MHz, DMSO-d6) S
(1H- ppm 11.68 (s, 1 H) 8.36 - 8.53 (m, 1
pyrrolo[2,3- H) 8.23 (d, J=4.80 Hz, I H) 8.03 -
31 NH b]pyridin-3- 8.13 (m, 1 H) 7.40 - 7.51 (m, 2 H) 10
CI N~ ylmethyl)- 7.11 (dd, J=7.83, 4.80 Hz, 1 H) 4.59
N \ [1,2,4]triazolo[4
~ N 3-b]pyridazine (s, I H). LC-MS 285, 287.
~
1 H NMR (400 MHz, DMSO-D6) S
/ 1N 6-Chloro-3-[1- ppm 1.87 (d, J=7.07 Hz, 1 H) 4.86 -
(1H- 5.24 (m, J=7.33 Hz, 1 H) 7.01
H3C pyrrolo[2,3- (dd,J=7.83, 4.80 Hz, 1 H) 7.39 (d,
32 \ NH b]pyridin-3-yl)- J=2.27 Hz, I H) 7.45 (d, J=9.85 Hz, 11
ethylj- I H) 7.94 (d, J=8.08 Hz, 1 H) 8.17
CI N \ [1,2,4]triazolo[4 (d, J=4.80 Hz, 1 H) 8.42 (d, J=9.60
N 3-bjpyridazine Hz, I H)11.53 (s, I H). LC-MS 299,
I~__N 301.
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-46-
Example Structure Name NMR/LC-MS Method
~ N 6-(1-Methyl- 1H NMR (300 MHz, DMSO-d6) S
1H-pyrazol-4- ppm 11.47 (s, 1 H) 8.50 (s, 1 H) 8.27
H3 N yl)-3-[(R)-1- (d, J=9.61 Hz, 1 H) 8.08 - 8.15 (m, 2
H3C~, NH (1 H- H) 7.92 - 8.00 (m, 1 H) 7.61 (d,
33 N / pyrrolo[2,3- J=9.80 Hz, 1 H) 7.54 (s, I H) 6.97 2
N, N blpyridin-3-yl)- (dd, J=7.72, 4.90 Hz, 1 H) 5.00 -
N ethyl]- 5.14 (m, J=7.35 Hz, 1 H) 3.92 (s, 3
N [1,2,4]triazolo[4 H) 1.91 (d, J=7.35 Hz, 3 H). LC-MS
,3-b]pyridazine 345.
4-{3-[(R)-1- 1 H NMR (300 MHz, DMSO-d6) S
N (1H- ppm 11.54 (s, 1 H) 8.47 (d, J=9.61
Pyrrolo[2,3- Hz, I H) 8.25 - 8.32 (m, 2 H) 8.16
H3c>' NH b]pyridin-3-yl)- (dd, J=4.71, 1.51 Hz, 1 H) 8.03 -
34 ethyl]- 8.09 (m, 2 H) 7.95 - 8.01 (m, 2 H) 2
N [1,2,4]triazolo[4 7.51 (d, J=2.26 Hz, 1 H) 7.01 (dd,
N ,3-b]pyridazin- J=7.91, 4.71 Hz, 1 H) 5.14 - 5.24 (m,
6-yl)- J=7.16 Hz; 1 H) 1.91 - 1.99 (m,
benzonitrile J=7.35, 7.35 Hz, 3 H. LC-MS 366.
H3C 7-methyl-6-{[6- 'H NMR (400 MHz, DMSO-d6) S ppm
H3Ck _N (1-methyl-lH- 2.60 (s, 3 H) 3.93 (s, 3 H) 4.70 (s, 2
N pyrazol-4- H) 7.42 (dd, J=8.21, 4.17 Hz, I H)
N I \~ yl)[1,2,4]triazol 7.68 (d, J=9.60 Hz, I H) 7.85 (s, 1 12
35 \ N o[4,3- H) 7.88 (s, I H) 8.16 (s, 1 H) 8.27 (d,
N b]pyridazin-3- J=8.08 Hz, 1 H) 8.34 (d, J=9.60 Hz,
N yl]methyl}quino 1 H) 8.53 (s, I H) 8.80 (dd, J=4.17,
line 1.64 Hz, 1 H). LC-MS 356.
1-{4-[3- (400 MHz, MeOD) S ppm 3.89 (s, 2
HZN N (quinolin-6- H) 4.88 (s, 2 H) 7.50 - 7.57 (m, 3 H)
ylmethyl)[1,2,4] 7.86 7.89 (m, I H) 7.91 (d, J=3.28
36 N`N triazolo[4,3- Hz, 1 H) 7.98 - 8.08 (m, 4 H) 8.24 (d, 13
N N b]pyridazin-6- J=9.60 Hz, 1 H) 8.34 (d, J=8.34 Hz,
yl]phenyl}meth 1 H) 8.81 (dd, J=4.17, 1.39 Hz, 1 H).
anamine LC-MS 367.
1 H NMR (400 MHz, MeOD) S ppm
(3R)-1-[3- 2=04 - 2.09 (m, I H) 2.14 (dt, J=8.91,
HO N (quinolin-6- 4.52 Hz, 1 H) 3.51 (d, J=11.87 Hz, 1
ylmethyl)[1,2,4] H) 3.64 (dt, J=9.09, 4.55 Hz, 3 H)
37 triazolo[4,3- 4.51 - 4.57 (m, I H) 4.61 - 4.66 (m, 2 4
N NN b]pyridazin 6 H) 7.06 (d, J=9.85 Hz, 1 H) 7.51 (dd,
N yl]pyrrolidin-3- J=8=34, 4.29 Hz, 1 H) 7.81 - 7.88 (m,
N ol 2 H) 7.90 - 7.99 (m, 2 H) 8.31 (d,
J=8.08 Hz, I H) 8.76 - 8.81 (m, 1 H).
LC-MS 347.
I H NMR (400 MHz, MeOD) S ppm
DN (3S)-1-[3- 2=04 - 2.09 (m, 1 H) 2.14 (ddd,
HO, (quinolin-6 J=13.33, 8.91, 4.29 Hz, I H) 3.46 -
~ ylmethyl)[1,2,4] 3.55 (m, I H) 3.60 - 3.68 (m, 3 H)
38 cNN)\N triazolo[4,3- 4.51 - 4.57 (m, 1 H) 4.65 (s, 2 H) 7.06 (d, J10.10 Hz,
1 H) 7.51 (dd,
b]pyridazin-6
J8.34, 4.55 Hz, 1 H) 7.81 - 7.87 (m,
yl]pyrrolidinol 3 2 H) 7.90 - 7.99 (m, 2 H) 8.31 (d,
J=7.58 Hz, I H) 8.79 (dd, J=4.29,
1.77 Hz, 1 H. LC-MS 347.
1H NMR (400 MHz, DMSO-d6) S
H3C N-cyclopentyl- ppm 1.38 -1.49 (m, J=1 1.97,11.97,
3-[(7- 6.32, 6.13 Hz, 2 H) 1.53 -1.61 (m, 2
N methylquinolin- H) 1.62 -1.67 (m, 2 H) 1.85 -1.96
39 HN N~ 6- J=6.57, 6.38, (6.38H6.38, 6.38 Hz, 1 4
(
~ yl)methyl][1,2,4 H) 4.51 (s, 2 H) 6.69 - 6.74 (m, 1 H)
N ]triazolo[4,3- 7.33 (d, J=6.57 Hz, I H) 7.42 (dd,~
N b]pyridazin-6- J=8.21, 4.17 Hz, 1 H) 7.80 - 7.88 (m,
amine 3 H) 8.18 - 8.22 (m, 1 H) 8.80 (dd,
J=4.17, 1.64 Hz, I H. LC-MS 359.
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-47-
Example Structure Name NMR/LC-MS Method
1 H NMR (400 MHz, DMSO-d6) S
7-methyl-6-({6- ppm 1.75 - 1.86 (m, 1 H) 2.13 - 2.23
~ H3C [(3R)-3- (m, I H) 2.37 - 2.48 (m, 3 H) 2.57 (s,
morpholin-4- 3 H) 2.84 - 2.95 (m, I H) 3.13 - 3.22
~N (m, I H) 3.37 - 3.43 (m, 2 H) 3.58 -
r~ ylpyrrolidin-l- 3.66 (m, 5 H) 3.69 (d, J=8.84 Hz, 1
yl][1,02,[44,3-]triazol H) 4.54 (s, 2 H) 6.99 (d, J=9.85 Hz, 4
40 \\\sss N~N N
N b]pyridazin-3- 1 H) 7.43 (dd, J=8.34, 4.04 Hz, I H)
yl)methyl)quino 7.84 (d, J=18.19 Hz, 2 H) 8.01 (d,
line J=10.11 Hz, 1 H) 8.21 (d, J=8.08 Hz,
I H) 8.80 (dd, J=4.29, 1.52 Hz, I H).
LC-MS 430.
(400 MHz, DMSO-d6) S ppm 1.74 -
1.82 (m, 1 H) 2.12 - 2.22 (m, 1 H)
7 3-(quinolin-6- 3.51 (dd, J=9.09, 3.28 Hz, I H) 3.70
~ ylmethyl)-N- (td, J=8.15, 5.68 Hz, 1 H) 3.75 - 3.85
N/ (tetrahydrofura (m, 2 H) 4.18 - 4.26 (m, I H) 4.54 (s,
41 H n-3- 2 H) 6.74 (d, J=9.85 Hz, 1 H) 7.50 4
N yl)[1,2,4]triazol (dd, J=8.21, 4.17 Hz, 1 H) 7.60 (d,
Oa NN o[4,3- J=5.81 Hz, 1 H) 7.73 (dd, J=8.59,
N b]pyridazin-6- 1.77 Hz, I H) 7.87 - 7.92 (m, 2 H)
amine 7.95 (d, J=8.59 Hz, I H) 8.30 (d,
J=8.08 Hz, 1 H) 8.85 (dd, J=4.17,
1.39 Hz, 1 H. LC-MS 347.
(400 MHz, DMSO-d6) S ppm 1.75 -
H3C 3-[(7- 1.84 (m, 1 H) 2.11 - 2.23 (m, 1 H)
methylquinolin- 2.58 (s, 3 H) 3.53 (dd, J=8.97, 3.16
~JN\ 6-yl)methyl]-N- Hz, 1 H) 3.66 - 3.75 (m, 1 H) 3.75 -
H (tetrahydrofura 3.86 (m, 2 H) 4.17 - 4.28 (m, 1 H)
42 ~N Nn-3- 4.51 (s, 2 H) 6.74 (d, J=9.85 Hz, 1 4
p NN yl)[1,2,4]triazol H) 7.42 (dd, J=8.08, 4.29 Hz, 1 H)
N o[4,3- 7.60 (d, J=6.06 Hz, I H) 7.78 (s, I
bjpyridazin-6- H) 7.83 (s, I H) 7.90 (d, J=9.85 Hz,
amine 1 H) 8.21 (d, J=7.83 Hz, I H) 8.80
d, J=3.79 Hz, 1 H. LC-MS 361.
N\ _ 4-(3-(quinolin- 1H NMR (300 MHz, CDCI3-D1) S
6-ylmethyl) ppm 8.89 (d, J= 1.8 Hz, 1H), 8.24 (d,
N [1,2,4]triazolo[4 J= 7.5 Hz, 1 H), 8.17 (m, 2H), 8.02
43 N 3-blpyridazin- (d, J= 6.3, 2H), 7.91 (s, 1H), 7.87 14
N 6 (m, 1H), 7.83 (d, J= 6.3 Hz, 2H),
N yl)benzonitrile 7.54 (d, J = 7.5 Hz, 1 H), 7.44 (m,
I H), 4.86 (s, 2H). LC-MS 363.
6-((6-(1H- 1H NMR (300 MHz, DMSO-D6)
N~ pyrazol-4-yl)
' N [1,2,4]triazolo[4 S ppm, 13.41 (bs, 1H), 8.84 (m,
HN 1 H), 8.39 (bs, 2H), 8.32 (d, J= 5.8
44 N.N N 3 b]p3ridazin- Hz, 2H), 7.96 (m, 2H), 7.82 (m, 1H), 15
N yl)methyl)quino 7.72 (d, J= 9.2 Hz, 1 H), 7.49 (m,
line 1H), 4.73 (s, 2H). LC-MS 328.
Biological Assays
General
In vitro assays may be used to determine the level of activity and effect of
the different
compounds of the present invention on one or more of the PKs. Similar assays
can be designed along
the same lines for any PK using techniques well known in the art. See for
example, Technikova-Dobrova
Z, Sardanelli AM, Papa S FEBS Lett. 1991 Nov 4; 292: 69-72.
A general procedure is as follows: compounds and kinase assay reagents are
introduced into test
wells. The assay is initiated by addition of the kinase enzyme. Enzyme
inhibitors reduce the measured
activity of the enzyme.
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-48-
Presently, the continuous-coupled spectrophotometric assay was used to
determine the level of
activity and effect of the different compounds of the present invention on the
tyrosine kinase activity of
HGFR on the Met-2 substrate peptide. In the continuous-coupled
spectrophotometric assay the time-
dependent production of ADP by the kinase is determined by analysis of the
rate of consumption of NADH
by measurement of the decrease in absorbance at 340 nm. As the PK produces ADP
it is re-converted to
ATP by reaction with phosphoenol pyruvate and pyruvate kinase. Pyruvate is
also produced in this
reaction. Pyruvate is subsequently converted to lactate by reaction with
lactate dehydrogenase, which
simultaneously converts NADH to NAD. NADH has a measurable absorbance at 340
nm whereas NAD
does not.
The presently preferred protocol for conducting the continuous-coupled
spectrophotometric
experiments for specific PKs is provided below. However, adaptation of this
protocol for determining the
activity of compounds against other RTKs, as well as for CTKs and STKs, is
well within the scope of
knowledge of those skilled in the art.
HGFR Continuous-coupled Spectrophotometric Assay
This assay was used to analyze the tyrosine kinase activity of HGFR on the Met-
2 substrate
peptide, a peptide derived from the activation loop of the HGFR. Assay results
in the form of Ki values
(pM) are summarized in Table 2.
Materials and Reagents:
1. HGFR enzyme from Upstate (Met, active) Cat. # 14-526
2. Met-2 Peptide (HGFR Activation Loop) Ac-ARDMYDKEYYSVHNK (MW = 1960).
Dissolve up in
200 mM HEPES, pH 7.5 at 10 mM stock.
3. 1 M PEP (phospho-enol-pyruvate) in 200 mM HEPES, pH 7.5
4. 100 mM NADH (B-Nicotinamide Adenine Dinucleotide, Reduced Form) in 200mM
HEPES, pH 7.5
5. 4 M MgC12 (Magnesium Chloride) in ddH2O
6. 1 M DTT (Dithiothreitol) in 200 mM HEPES, pH 7.5
7. 15 Units/mL LDH (Lactic Dehydrogenase)
8. 15 Units/mL PK (Pyruvate Kinase)
9. 5M NaCI dissolved in ddH2O
10. Tween-20 (Protein Grade) 10% Solution
11. 1 M HEPES buffer: (N-[2-Hydroxethyl]piperazine-N-[2-ethanesulfonic acid])
Sodium Salt.
Dissolve in ddH2O, adjust pH to 7.5, bring volume to 1 L. Filter at 0.1 pm.
12. HPLC Grade Water; Burdick and Jackson #365-4, 1 X 4 liters (or equivalent)
13. 100% DMSO (SIGMA)
14. Costar # 3880 - black clear flat bottom half area plates for Ki
determination and % inhibition
15. Costar # 3359 - 96 well polypropylene plates, round bottom for serial
dilutions
16. Costar # 3635 - UV-plate clear flat bottom plates for % inhibition
17. Beckman DU-650 w/ micro cell holders
18. Beckman 4-position micro cell cuvette
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-49-
Procedure:
Prep Dilution Buffer (DB) for Enzyme (For 30 mL prep)
1. DB final concentration is 2 mM DTT, 25 mM NaCI2, 5 mM MgCl2, 0.01% Tween-
20, and 50 mM
HEPES buffer, pH 7.5.
2. Make up 50 mM HEPES by adding 1.5 mL I M HEPES into 28.1 mL of ddH2O. Add
rest of the
reagents. Into 50 mL conical vial, add 60 L of 1 M DTT, 150 L 5M NaCIZ, 150
L 1 M MgCIZ, and
30 L of 10% Tween-20 to give total volume of 30 mL.
3. Vortex for 5-10 seconds.
4. Aliquot out DB at 1 mL/tube and label tubes as "DB HGFR"
5. Note: This can be prepared and stored ahead of time.
6. Freeze un-used aliquots in microcentrifuge tubes at -20 C freezer.
Prep Compounds
1. For compound dilution plate, add 4 L of 10 mM stock into column I of
plate, and bring volume to
100 L with 100% DMSO.
2. Set up the Precision 2000 dilution method. A final concentration of 200 M
compound in 50%
DMSO, 100 mM HEPES (1:2 serial dilution).
Prep Coupled Enzymatic Buffer:
1. Final concentration in assay:
Reagent (Stock Conc.) Final Conc. In Assay
a. PEP (1 M) 1 mM
b. NADH (100 mM) 300 M
c. MgCI2 (4 M) 20 mM
d. DTT (1 M) 2mM
e. ATP (500 mM) 300 M
f. HEPES 200 mM (pH 7.5) 100 mM
g. Pyruvate Kinase (PK) 15 units/mL
h. Lactic Dehydrogenase (LDH) 15 units/mL
i. Met-2 peptide (10 mM) 0.500 mM
j. HGFR 50 nM
2. For a 10 mL reaction buffer add 10 L of 1M PEP, 33 L of 100 mM NADH, 50
L of 4M MgCI2,
20 L of 1 M DTT, 6 L of 500 mM ATP, and 500 L of 10 mM Met-2 peptide into
100 mM HEPES
buffer pH 7.5 and vortex/mix.
3. Add coupling enzymes, LDH and PK, into reaction mix. Mix by gentle
inversion.
Running samples
1. Spectrophotometer settings:
i. Absorbance wavelength (A): 340 nm
ii. Incubation time: 10 min
iii. Run time: 10 min
CA 02651979 2008-11-12
WO 2007/138472 PCT/IB2007/001446
-50-
iv. Temperature: 37 C
2. Add 85 L of CE reaction mix into each well of assay plate.
3. Add 5 pL of diluted compound into a well of the assay plate.
4. Add 5 NL of 50% DMSO for negative control into last column of assay plate.
5. Mix with multi-channel pipettor or orbital shaker.
6. Pre-incubate for 10 minutes at 37 C.
7. Add 10 laL of 500 nM HGFR to each well of assay plate; the final HGFR
concentration is 50 nM in
a total final volume of 100 pL.
8. Measure activity for 10 minutes at A= 340 nm and 37 C.
Table 2
Example Ki (pM) Example Ki M Example Ki M
1 0.052 16 0.015 31 0.326
2 0.008 17 0.25 32 0.130
3 0.058 18 0.379 33 0.187
4 0.046 19 0.214 34 0.332
5 0.01 20 0.163 35 0.02
6 0.074 21 0.487 36 0.46
7 0.08 22 0.171 37 0.112
8 0.033 23 0.162 38 0.7
9 0.09 24 0.151 39 0.007
10 0.007 25 0.142 40 0.154
11 0.054 26 0.105 41 0.105
12 0.089 27 0.157 42 0.022
13 0.064 28 0.66 43 0.024
14 0.09 29 0.306 44 0.038
0.017 30 0.109