Note: Descriptions are shown in the official language in which they were submitted.
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
SUBSTITUTED PHENYL ACETIC ACIDS AS DP-2 ANTAGONISTS
BACKGROUND OF THE INVENTION
[0001] Prostaglandin D2 (PGD2) is the major proinflammatory mediator
abundantly
secreted by mast cells activated by allergen exposure of a previously
sensitized host. PGD2 is
capable of illiciting a multitude of pathobiological responses relevant to
inflammatory
disorders including constriction of the airways, leukocyte influx, increase in
vascular
permeability, edema, and mucus secretion. The biological actions of PGD2 are
mediated by
at least 3 distinct G-protein coupled receptors: The high affinity receptors
DP-1 (formerly
known as DP) and DP-2 (formerly known as orphan receptor GPR44 and
"chemoattractant
receptor homologue expressed in Th2 cells", CRTH2 (See Hirai, H., et al. J.
Exp. Med. 2001,
193(2): 255-61; Nagata, K., J. Biol. Regul. Homeost. Agents 2003, 17(4):334-7)
and the
thromboxane A2 receptor, TP, to which PGD2 binds with low affinity.
[0002] DP-2 receptor is a major contributor to the pathophysiological actions
of PGD2.
Accordingly, pharmaceuticals that target this receptor are likely to be
therapeutically
beneficial for a host of disorders, specifically inflammatory conditions that
have an allergic
component, such as asthma (See Huang, J., J. Microbiol. Immunol. Infect 2005,
38(3): 158-
63). DP-2 is selectively expressed in Eosinophils, Basophils, and highly
polarized Th2 cells
in humans. These cell types are well known contributors to inflammatory
disorders and other
conditions. Activation of DP-2, a chemoattractant receptor, stimulates
chemotaxis of human
Th2 cells, eosinophils, and basophils both in vitro and in vivo and may
mediate recruitment of
relevant cell types to diseased sites and exacerbate end organ damage.
[0003] DP-2 agonists are capable of directly activating inflammatory cells and
DP-2-mediated activation and mediator release from Eosinophils and Basophils
has been
reported (see Gervais, F. G. et al.., JAllergy Clin Immunol (2004), 108
(6):982-8;
Yoshimura-Uchiyama, C. et al., Clin Exp Allergy 2004, 34(8):1283-90).
Furthermore, Th2
effector T lymphocytes will elaborate inflammatory cytokines IL-4, IL-5 and IL-
13 in
response to DP-2 stimulation (See Xue, L. et al., J. Immunol. 2005, 175(10):
6531-6). These
cytokines in turn act as important regulators of inflammatory responses and
support Th2 cell
differentiation, mast cell growth, differentiation and IgE synthesis, and the
differentiation,
infiltration and survival of eosinophils.
1
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
[0004] This suggests that the PGD2/DP-2 pathway acts as a positive feedback
loop and
augments pathologic responses in disorders associated with excessive or
dysregulated PGD2
production. Therefore, pharmaceutical agents that interfere with this pathway
may have
utility in the treatment of a broad array of allergic and inflammatory
conditions and other
disorders.
[0005] The utility of PGD2 antagonists in the treatment of inflammatory
disorders is
supported by clinical studies with Ramatrobari (Baynas, BAY u3405). Clinical
studies have
demonstrated a beneficial effect of Ramatrobari on rhinitis symptoms as well
as
inflammatory markers in nasal lavages, suggesting anti-inflammatory activity.
Ramatrobari
was initially described as a TP selective antagonist, and its clinical effects
on rhinitis were
believed to be TP mediated. Recent discoveries, however, revealed that
Ramatrobari
possesses dual specificity and antagonizes both TP and DP-2 receptors (See
Sugimoto, H., et
al., J. Pharmacol. Exp. Ther. 2003, 305(1): 347-52). In light of the presence
of DP-2 on
pivotal inflammatory cells involved in allergic rhinitis, and the stimulatory
effects of PGD2
and other DP-2 agonists on theses cells, it is reasonable to postulate that
the clinical benefit of
Ramatrobari in allergic rhinitis is to a large extent due to its activity
against the DP-2
receptor. It can be inferred therefore that DP-2 selective antagonists may be
useful in the
treatment of allergic rhinitis, other inflammatory conditions, other
conditions where the PGD2
pathway is deregulated, as well as other disorders where the utility of
Rainatroban has been
established.
[0006] Minami et al. have demonstrated the effectiveness of Ramatroban on
edema in
experimental allergic conjunctivitis (See Minami, K., et al. Int.
Immtilnopharmacol. 2004,
4(12):1531-5). It has been demonstrated that DP-2 exerts an essential role in
allergic
disorders, specifically, IgE-mediated cutaneous responses that occur in
chronic contact
hypersensitivity (See Mitsumori, S., Curr. Pharm. Des., 2004, 10(28):3533-8);
Moroi, R., et
al. 30th Annu. Meet. Jpn. Soc. Invest. Dermatol. (Apr 20-Apr 22, Yokohama)
2005, Abst.
48).
[0007] Numerous compounds have been reported as modulators of PGD2 receptors
and/or
useful for the treatment of allergic and inflainmatory disorders. WO
2006021418 discloses a
series of sulfamyl-benzoimidazole-l-yl-acetic acid compounds with DP-2 or PGD2
antagonist
activity. WO 2006021759 discloses a series of biphenyloxyacetic acid
derivatives with PGD2
and DP-2 modulating activity said to be useful for the treatment of
respiratory disorders.
2
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
WO 2005019171, WO 2004106302 and WO 2005054232 disclose a series of Acetic
acid-indole, -indazole and -benzimidazole compounds said to be useful for the
treatment of
respiratory disorders. WO 2005105727 discloses phenoxy acetic acid compounds
with DP-2
antagonistic activity. WO 2005018529 discloses phenoxy acetic acid compounds
said to be
useful for the treatment of asthma and rhinitis. WO 2005040114 and WO
2005040112
discloses a series of compounds with DP-2 or PGD2 antagonist activity said to
useful for the
treatment of allergy, asthma and atopic dermatitis. WO 2004058164, U.S. Patent
publication
No. 2005038070 and WO 2005007094 disclose a series of compounds said to be
useful for
the treatment of allergy, asthma, cancer and inflammation. WO 2004096777
discloses a
series of pyrimidine derivatives useful for the treatment of conditions
mediated by DP-2,
including asthma conjunctivitis, dermatitis, atopic rhinitis, allergic
sinusitis.
WO 2004078719 discloses a series of indole compounds said to be useful for the
treatmeant
of asthma and allergic rhinitis. U.S. Patent publication No. 2004132772
discloses a series of
tetrahydroquinoline compounds as DP-2 antagonists said to be useful for the
treatment of
allergic asthma and allergic rhinitis. WO 2003066046, WO 2003066047, WO
2003101961,
WO 2003101981, WO 2004007451 discloses a series of indole-acetic acids said to
be useful
in the treatment of asthma, chronic obstructive pulmonary disease (COPD),
rhinitis and other
conditions. WO 2003097598 discloses a series of compounds said to exhibit PGD2
receptor
antagonism. U.S. Patent No. 4,656,192 discloses a series of Tropolone
compounds said to be
useful a anti-tumor agents. EP 1170594 discloses methods for the
identification of
compounds useful for the treatment of conditions mediated by prostaglandin D2,
a ligand for
orphan receptor DP-2. GB 1356834 discloses a series of compounds said to
possess
anti-inflaininatory, analgesic and antipyretic activity.
[0008] Even so, there is a relative paucity of drugs that selectively modulate
non-aminergic
liganded G-protein -coupled receptors on the market (see Beaumont K et al.,
Bioorg Med
Chem Lett., 2005, 15 (16): 3658-64).
SUMMARY OF THE INVENTION
[0009] It has now surprisingly been found that certain phenyl acetic acids are
potent DP-2
receptor antagonists. In certain embodiments, the phenyl acetic acids are
selective DP-2
receptor antagonists over other PGD2 receptors. The phenyl acetic acid
compounds of the
invention are expected to be potentially useful for the treatment or
prevention of medical
conditions or disorders responsive to DP-2 antagonism, or symptoms associated
with such
3
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
medical conditions or disorders, such as those having an allergic or
inflammatory component.
Examples conditions or disorders treatable or preventable with compounds and
compositions
of the invention are provided below.
[0010] Amongst several aspects of the present invention, the invention
provides
compounds, pharmaceutical compositions and methods useful for treating or
preventing
conditions and disorders associated with inflammation and/or allergic
processes. In particular,
the invention provides compounds, pharmaceutical compositions and methods
useful for
treating or preventing asthma, allergic conditions, inflammatory conditions,
cancer and viral
infection.
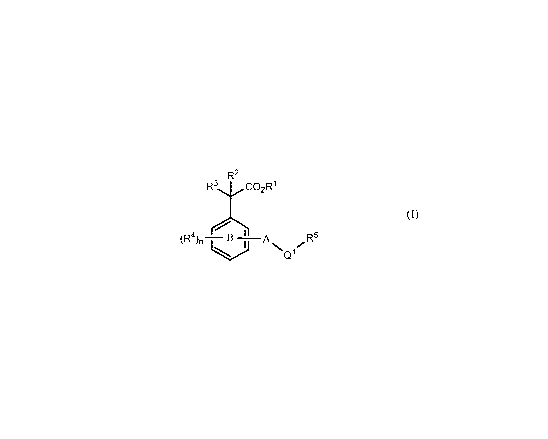
[0011] The compounds of the invention have the general structure (I):
R2
R3 C02R'
5
(R4), \ ~ q\Q1R
(I)
[0012] A is a 5-14-membered heterocyclic ring fused or bonded to phenyl ring B
having
1-4 ring heteroatoms each independently selected from the group consisting of
nitrogen,
oxygen and sulfur, the heterocyclic ring being moncyclic or polycyclic,
optionally substituted
with 1-3 R 8 substituents.
[0013] Ql is selected from the group consisting of: a bond, -Ci-C4alkylene-,
-Ci-C4heteroalkylene-, -CO-, -NH-, -0-, -SOq-, -C(O)O-, -OC(O)-, -CONH-, -NHCO-
,
-NHCONH-, -NHSOq- , -SOqNH- and -COCH2HNSOq.
[0014] Each R', R 2 and R3 is independently selected from the group consisting
of H,
C1_6alkyl, C0_6alkylaryl and C0_6alkylheteroaryl; wherein the aryl or
heteroaryl portions are
optionally substituted with C1_6alkyl, CN, OR, C1_6haloalkyl, C1_6heteroalkyl,
NR2, NO2, halo,
C(O)R, CO2R, CONR2, SOqR, SOqNR2, OC(O)OR, OC(O)R, OC(O)NR2, NRC(O)NR2,
NRC(O)R and NRC(O)OR.
[0015] Each R4 is independently selected from the group consisting of
C1_6alkyl,
C04alkylC3_1ocycloalkyl, C0_4alkylaryl, C0_4alkylheteroaryl, CZ_aalkenylaryl,
C2_4alkynylaryl,
4
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
C0_4alkylheterocyclyl, CN, amino, NHCOR', hydroxy, C1_6alkoxy, OC(O)R1, -OCo-
4alkylaryl,
OC0_4alkylheteroaryl, -OC0_4a1ky1C3_Iocycloalkyl,
OC0_4alky1C3_ioheterocycloalkyl,
OC0_4alky1NR8, nitro, halo and haloC1_6alkyl; or are combined together to form
an aryl or
heterocyclyl ring having 1-2 heteroatoms selected from the group consisting of
nitrogen,
oxygen and sulfur; wherein the alkyl, aryl and heterocyclyl portions are each
optionally
substituted with 1 to 3 substituents each independently selected from the
group consisting of
C1_6alkyl, CN, CONHR', COZR1, amino, C1_6alkoxy, halo, haloC1_6alkyl and
SOgRI.
[0016] R5 is selected from the group consisting of C1_6alkyl, C0_4alkylaryl,
C2_4alkenylaryl,
C2_4alkynylaryl, C0_4alkylheteroaryl, each of which is optionally substituted
with 1-3 R9
substitutents.
[0017] Each R 8 is independently selected from the group consisting of
C1_6alkyl,
C0_6alkylC3_6cycloalkyl, C0_6alkylaryl, C0_6alkylheteroaryl, oxo, C1_6alkyl,
CN, OR,
C1_6haloalkyl, C1_6heteroalkyl, NR2, NOZ, halo, C(O)R, CO2R, CONR2, SOqR,
SOqNR2,
OC(O)OR, OC(O)R, OC(O)NR2, NRC(O)NR2, NRC(O)R and NRC(O)OR.
[0018] Each R9 is independently selected from the group consisting of
C1_6alkyl, CN, OR,
oxo, C1_6haloalkyl, C1_6heteroalkyl, NR2, NO2, halo, C(O)R, CO2R, CONR2, SOqR,
SOqNR2,
OC(O)OR, OC(O)R, OC(O)NR2, NRC(O)NR2, NRC(O)R and NRC(O)OR.
[0019] Each R is independently selected from the group consisting of H,
C1_6alkyl, C0_4
alkylheteroaryl,CO_4heterocyclyl, C3_8cycloalkyl and C0_4alkylaryl or when
attached to the
same nitrogen atom may be combined to fonn a 5-8 membered ring having 1-4 ring
heteroatoms each independently selected from the group consisting of nitrogen,
oxygen and
sulfur.
[0020] The subscript n is independently 0, 1, 2, 3 or 4.
[0021] Each subscript q is independently 0, 1 or 2.
[0022] The invention also provides pharmaceutically acceptable salts,
hydrates, solvates
and prodrugs of compounds of structure I. Examples of prodrugs are compounds
wherein R~
is C1_6alkyl, C0_6alkylaryl or C0_6alkylheteroaryl wherein the aryl or
heteroaryl portions are
optionally substituted as described herein.
[0023] The invention also provides phannaceutical compositions comprising a
compound
of formula I and a pharmaceutically acceptable carrier, excipient or diluent.
5
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
[0024] The invention also provides methods for antagonizing a DP-2 receptor
comprising
contacting a DP-2 receptor with a compound of structure I as well as methods
of selectively
agonizing a DP-2 receptor over one or more PGD2 receptors.
[0025] The invention also provides methods for treating or preventing a
disorder or
condition responsive to the antagonizing a DP-2 receptor as wells as methods
of treating or
preventing a disorder or condition associated with elevated levels of PGD2 or
a metabolite
thereof comprising administering to a subject in need thereof a
therapeutically effective
amount of a compound of structure I.
[0026] The invention further provides methods for treating or preventing an
inflammatory
disorder or condition with an inflammation or allergic component as provided
herein.
[0027] The invention also provides methods for treating or preventing a
condition or
disorder mediated by DP-2 and/or one or more other PGD2 receptors, e.g., DP-1,
comprising
administering to a subject in need thereof a therapeutically effective amount
of a compound
of formula I.
[0028] The invention also provides methods for selectively modulating DP-2 in
the
presence of one or more other PGD2 receptors, e.g., DP-1, comprising
contacting a cell with a
compound of structure I.
[0029] Other objects, features and advantages of the invention will become
apparent to
those skilled in the art from the following description and claims.
DETAILED DESCRIPTION OF THE INVENTION
Abbreviations and Definitions
[0030] The abbreviations used herein are conventional, unless otherwise
defined. The
following abbreviations are used: EtOAc = Ethylacetate, DMF = N,N-Dimethyl
forinainide,
NMP = N-methylpyrrolidine, THF = tetrahydrofuran, RT = room temperature, TFA =
=
trifluoroacetic acid, LDA = lithium diisopropylamine, n-BuLi = n-butyl
lithium, Na2CO3
sodium carbonate, DME = dimethyl ether, K2PO4 = potassiuin phosphate, CHZC12
or DCM =
dichloromethane, Et3N = triethylamine, DIEA = Hunig's base or diisopropyl
ethylamine,
KOH = potassium hydroxide, NaOH = sodiuin hydroxide, TMS = trimethylsilyl, Tf
=
trifluoromethylsulfonyl, Boc = t-butylcarbonyl, Bz - benzyl, IPA = isopropyl
alcohol, NBS =
N-broinosuccinainide, AIBN = azobisisobutyronitrile (also
azobisisobutylonitrile), Pin =
6
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
pinacolato, Cs2CO3 = cesium carbonate, HIV = human immunodeficiency virus, RLV
=
Raucher leukemia virus, IgE = immunoglobulin E.
[0031] It is noted here that as used in this specification and the appended
claims, the
singular forms "a," "an," and "the" include plural reference unless the
context clearly dictates
otherwise.
[0032] The term "alkyl," by itself or as part of another substituent, means,
unless otherwise
stated, a straight or branched chain, or cyclic hydrocarbon radical, or
combination thereof,
which is fully saturated, having the number of carbon atoms designated (i.e.,
C1_8 means one
to eight carbons). Examples of alkyl groups include methyl, ethyl, n-propyl,
isopropyl,
n-butyl, t-butyl, isobutyl, sec-butyl, cyclohexyl, (cyclohexyl)methyl,
cyclopropylmethyl,
homologs and isomers of, for example, n-pentyl, n-hexyl, n-heptyl, n-octyl and
the like.
[0033] The term "alkenyl", by itself or as part of another substituent, means
a straight or
branched chain, or cyclic hydrocarbon radical, or combination thereof, which
may be mono-
or polyunsaturated, having the number of carbon atoms designated (i.e., C2-C8
means two to
eight carbons) and one or more double bonds. Examples of alkenyl groups
include vinyl,
2-propenyl, crotyl, 2-isopentenyl, 2-(butadienyl), 2,4-pentadienyl, 3-(1,4-
pentadienyl) and
higher homologs and isomers thereof.
[0034] The term "alkynyl", by itself or as part of another substituent, means
a straight or
branched chain hydrocarbon radical, or combination thereof, which may be mono-
or
polyunsaturated, having the number of carbon atoms designated (i.e., C2-C8
means two to
eight carbons) and one or more triple bonds. Examples of alkynyl groups
include ethynyl, 1-
and 3-propynyl, 3-butynyl and higher homologs and isomers thereof.
[0035] The tenn "alkylene" by itself or as part of another substituent means a
divalent
radical derived from alkyl, as exemplified by -CH2CH2CH2CH2-. Typically, an
alkyl (or
alkylene) group will have 1 to 24 carbon atoms, with those groups having 10 or
fewer carbon
atoms being preferred in the present invention. A "lower alkyl" or "lower
alkylene" is a
shorter chain alkyl or alkylene group, generally having eight or fewer carbon
atoms.
[0036] The terms "alkoxy," "alkylamino" and "alkylthio" (or thioalkoxy) are
used in their
conventional sense, and refer to those alkyl groups attached to the remainder
of the molecule
via an oxygen atom, an amino group, or a sulfur atom, respectively. Similarly,
the tenn
7
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
dialkylamino refers to an amino group having two attached alkyl groups that
can be the same
or different.
[0037] The term "heteroalkyl," by itself or in combination with another terin,
means, unless
otherwise stated, a stable straight or branched chain, or cyclic hydrocarbon
radical, or
combinations thereof, consisting of the stated number of carbon atoms and from
one to three
heteroatoms selected from 0, N, Si and S, and wherein the nitrogen and sulfur
atoms may
optionally be oxidized and the nitrogen heteroatom may optionally be
quatemized. The
heteroatom(s) 0, N and S may be placed at any interior position of the
heteroalkyl group. The
heteroatom Si may be placed at any position of the heteroalkyl group,
including the position
at which the alkyl group is attached to the remainder of the molecule.
Examples include
-CH2-CH2-0-CH3, -CH2-CH2-NH-CH3, -CH2-CH2-N(CH3)-CH3, -CH2-S-CH2- -CH3,
-CH2-CH2, -S(O)-CH3, -CH2-CH2-S(O) 2-CH3, -CH=CH-O-CH3, -Si(CH3)3,
-CH2-CH=N-OCH3, and -CH=CH-N(CH3)-CH3. Up to two heteroatoms may be
consecutive,
such as, for example, -CH2-NH-OCH3 and -CHZ-O-Si(CH3)3. When a prefix such as
(C2-C8)
is used to refer to a heteroalkyl group, the number of carbons (2-8, in this
example) is meant
to include the heteroatoms as well. For example, a C2-heteroalkyl group is
meant to include,
for example, -CH2OH (one carbon atom and one heteroatom replacing a carbon
atom) and
-CH2SH. The term "heteroalkylene" by itself or as part of another substituent
means a
divalent radical derived from heteroalkyl, as exemplified by -CH2-CH2-S-CH2CH2-
- and
-CH2-S-CH2-CH2-NH-CH2-. For heteroalkylene groups, heteroatoms can also occupy
either
or both of the chain termini (e.g., alkyleneoxy, alkylenedioxy, alkyleneamino,
alkylenediainino, and the like). Still further, for alkylene and
heteroalkylene linking groups,
no orientation of the linking group is implied.
[0038] The tenns "cycloalkyl", "heterocyclyl" and "heterocyclic ring", by
themselves or in
combination with other terms, represent, unless otherwise stated, cyclic
versions of "alkyl"
and "heteroalkyl", respectively. Thus, the terms "cycloalkyl" and
"heterocyclic ring" are
meant to be included in the terms "alkyl" and "heteroalkyl", respectively.
Additionally, for a
heterocyclic ring, a heteroatom can occupy the position at which the
heterocycle is attached
to the remainder of the molecule. Examples of cycloalkyl include cyclopentyl,
cyclohexyl,
1 -cyclohexenyl, 3-cyclohexenyl, cycloheptyl, and the like. Examples of a
heterocyclic ring
include pyrrolidinyl, pyrrolyl, piperadinyl, tetrahydropyridinyl, piperazinyl,
piperazin-l-oxide, morpholinyl, thiomorpholinyl, azepanyl, azepinyl,
oxazepane, thiazepane,
8
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
azocanyl, azocinyl, indolyl, azaindole, tetrahydroquinolinyl,
decahydroquinoliny,
tetrahydrobenzooxazepinyl dihydrodibenzooxepin, and the like.
[0039] The terms "halo" or "halogen," by themselves or as part of another
substituent,
mean, unless otherwise stated, a fluorine, chlorine, bromine, or iodine atom.
Additionally,
terms such as "haloalkyl", are meant to include alkyl substituted with halogen
atoms which
can be the same or different, in a number ranging from one to (2m'+l), where
m' is the total
number of carbon atoms in the alkyl group. For example, the term
"haloC1_6alkyl" is meant to
include trifluoromethyl, 2,2,2-trifluoroethyl, 4-chlorobutyl, 3-bromopropyl,
and the like.
Thus, the term "haloalkyl" includes monohaloalkyl (alkyl substituted with one
halogen atom)
and polyhaloalkyl (alkyl substituted with halogen atoms in a number ranging
from two to
(2m'+1) halogen atoms). The term "perhaloalkyl" means, unless otherwise
stated, alkyl
substituted with (2m'+1) halogen atoms, where m' is the total number of carbon
atoms in the
alkyl group. For example, the term "perhaloC1_6alkyl", is meant to include
trifluoromethyl,
pentachloroethyl, 1,1,1-trifluoro-2-bromo-2-chloroethyl, and the like.
[0040] The term "aryl" means, unless otherwise stated, a polyunsaturated,
typically
aromatic, hydrocarbon substituent which can be a single ring or multiple rings
(up to three
rings) which are fused together or linked covalently. The term "heteroaryl"
refers to aryl
groups (or rings) that contain from one to four heteroatoms selected from the
group
consisting of N, 0 and S, wherein the nitrogen and sulfur atoms are optionally
oxidized, and
the nitrogen atom(s) are optionally quaternized. A heteroaryl group can be
attached to the
remainder of the molecule through a heteroatom. Non-limiting examples of aryl
and
heteroaryl groups include phenyl, 1-naphthyl, 2-naphthyl, 4-biphenyl, 1-
pyrrolyl, 2-pyrrolyl,
3-pyrrolyl, 3-pyrazolyl, 2-imidazolyl, 4-imidazolyl, pyrazinyl, 2-oxazolyl, 4-
oxazolyl,
2-phenyl-4-oxazolyl, 5-oxazolyl, 3-isoxazolyl, 4-isoxazolyl, 5-isoxazolyl, 2-
thiazolyl,
4-thiazolyl, 5-thiazolyl, 2-furyl, 3-furyl, 2-thienyl, 3-thienyl, 2-pyridyl, 3-
pyridyl, 4-pyridyl,
2-pyrimidyl, 4-pyriinidyl, 2-pyrimidinyl, 4-pyrimidinyl, 5- pyrimidinyl, 3-
pyridazinyl, 4-
pyridazinyl, 5-benzothiazolyl, purinyl, 2-benziinidazolyl, 5-indolyl, 1H-
indazole, carbazole,
a-carboline, (3 -carboline, y-carboline, 1-isoquinolyl, 5-isoquinolyl, 2-
quinoxalinyl,
5-quinoxalinyl, 2-quinolyl, 3-quinolyl, 4-quinolyl, 5-quinolyl, 6-quinolyl, 7-
quinolyl and
8-quinolyl.
[0041] In some embodiments, the tenn "aryl" refers to a phenyl or naphthyl
group which is
unsubstituted or substituted. In some embodiments, the tenn "heteroaryl"
refers to a pyrrolyl,
9
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
pyrazolyl, imidazolyl, pyrazinyl, oxazolyl, isoxazolyl, thiazolyl, furyl,
thienyl, pyridyl,
pyrimidyl, benzothiazolyl, purinyl, benzimidazolyl, indolyl, isoquinolyl,
quinoxalinyl or
quinolyl group which is unsubstituted or substituted.
[0042] For brevity, the term "aryl" when used in combination with other terms
(e.g.,
aryloxy, arylthioxy, arylalkyl) includes both aryl and heteroaryl rings as
defined above. Thus,
the term "arylalkyl" is meant to include those radicals in which an aryl group
is attached to an
alkyl group (e.g., benzyl, phenethyl, pyridylmethyl and the like) including
those alkyl groups
in which a carbon atom (e.g., a methylene group) has been replaced by, for
example, an
oxygen atom (e.g., phenoxymethyl, 2-pyridyloxymethyl, 3 -(1 -
naphthyloxy)propyl, and the
like).
[0043] Each of the above terms (e.g., "alkyl," "heteroalkyl," "aryl" and
"heteroaryl") is
meant to include both substituted and unsubstituted forms of the indicated
radical, unless
otherwise indicated. Preferred substituents for each type of radical are
provided below.
[0044] Substituents for the alkyl and heteroalkyl radicals (as well as those
groups referred
to as alkylene, alkenyl, heteroalkylene, heteroalkenyl, alkynyl, cycloalkyl,
heterocyclyl) can
be a variety of groups selected from: -OR', =0, =NR', =N-OR', -NR'R", -SR',
halogen,
-SiR'R" R"', -OC(O)R', -C(O)R', -CO2R', -CONR'R", -OC(O)NR'R", -NR"C(O)R',
-NR'-C(O)NR"R"', -NR'-SOZNR"R"', -NR"CO2R', -NH-C(NH2)=NH, -NR'C(NH2)=NH,
-NH-C(NH2)=NR', -S(O)R', -SOZR', -SO2NR'R", -NR"SO2R, -CN and -NO2, in a
number
ranging from zero to three, with those groups having zero, one or two
substituents being
particularly preferred. R', R" and R"' each independently refer to hydrogen,
unsubstituted Ci_
6alkyl and heteroalkyl, unsubstituted aryl, aryl substituted with one to three
halogens,
unsubstituted alkyl, alkoxy or thioalkoxy groups, or aryl-C1_6alkyl groups.
When R' and R"
are attached to the same nitrogen atom, they can be combined with the nitrogen
atom to forin
a 5-, 6- or 7-membered ring. For example, -NR'R" is meant to include 1-
pyrrolidinyl and
4-morpholinyl. Typically, an alkyl or heteroalkyl group will have from zero to
three
substituents, with those groups having two or fewer substituents being
preferred in the
present invention. More preferably, an alkyl or heteroalkyl radical will be
unsubstituted or
monosubstituted. Most preferably, an alkyl or heteroalkyl radical will be
unsubstituted. From
the above discussion of substituents, one of skill in the art will understand
that the tenn
"alkyl" is meant to include groups such as trihaloalkyl (e.g., -CF3 and -
CH2CF3).
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
[0045] In some embodiments, substituents for the alkyl and heteroalkyl
radicals are
selected from: -OR', =0, -NR'R", -SR', halogen, -SiR'R" R"', -OC(O)R', -
C(O)R', -CO2R',
-CONR'R", -OC(O)NR'R", -NR"C(O)R', -NR"COZR', -NR'-SO2NR"R"', -S(O)R', -SOZR',
-SO2NR'R", -NR"SOZR, -CN and -NOZ, where R' and R" are as defined above. In
some
embodiments, substituents are selected from: -OR', =0, -NR'R", halogen, -
OC(O)R', -COZR',
-CONR'R", -OC(O)NR'R", -NR"C(O)R', -NR"COZR', -NR'-SO2NR"R"', -SO2R', -
SO2NR'R",
-NR"SOZR, -CN and -NOZ.
[0046] Similarly, substituents for the aryl and heteroaryl groups are varied
and are selected
from: -halogen, -OR', -OC(O)R', -NR'R", -SR', -R', -CN, -NOZ, -CO2R', -
CONR'R", -C(O)R',
-OC(O)NR'R", -NR"C(O)R', -NR"C(O) 2R', -NR'-C(O)NR"R"', -NH-C(NH2)=NH,
-NR'C(NH2)=NH, -NH-C(NH2)=NR', -S(O)R', -S(O) ZR', -S(O) 2NR'R", -N3, -
CH(Ph)2,
perfluoroC1_6alkoxy, and perfluoroC1_6alkyl, in a number ranging from zero to
the total
number of open valences on the aromatic ring system; and where R', R" and R"'
are
independently selected from hydrogen, C1_6alkyl and heteroalkyl, unsubstituted
aryl and
heteroaryl, (unsubstituted aryl)- C1_6alkyl, and (unsubstituted aryl)oxy-
C1_6alkyl.
[0047] Two of the substituents on adjacent atoms of the aryl or heteroaryl
ring may
optionally be replaced with a substituent of the formula -T-C(O)-(CH2)q-U-,
wherein T and U
are independently -NH-, -0-, -CH2- or a single bond, and q is 0, 1 or 2.
Alternatively, two of
the substituents on adjacent atoms of the aryl or heteroaryl ring may
optionally be replaced
with a substituent of the formula -A-(CH2),--B-, wherein A and B are
independently -CH2-,
-0-, -NH-, -S-, -S(O)-, -S(O)Z-, -S(O) 2NR'- or a single bond, and r is 1, 2
or 3. One of the
single bonds of the new ring so fonned may optionally be replaced with a
double bond.
Alternatively, two of the substituents on adjacent atoms of the aryl or
heteroaryl ring may
optionally be replaced with a substituent of the fonnula -(CH2)S-X-(CH2)t- -,
where s and t are
independently integers of from 0 to 3, and X is -0-, -NR'-, -S-, -S(O)-, -
S(O)2-, or -S(O)
2NR'-. The substituent R' in -NR'- and -S(O)2NR'- is selected from hydrogen or
unsubstituted
C i_6a1ky1. Otherwise, R' is as defined above.
[0048] As used herein, the term "heteroatom" is meant to include oxygen (0),
nitrogen (N),
sulfur (S) and silicon (Si).
[0049] The term "pharmaceutically acceptable salts" or "phannaceutically
acceptable
carrier" is meant to include salts of the active compounds which are prepared
with relatively
nontoxic acids or bases, depending on the particular substituents found on the
compounds
11
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
described herein. When compounds of the present invention contain relatively
acidic
functionalities, base addition salts can be obtained by contacting the neutral
form of such
compounds with a sufficient amount of the desired base, either neat or in a
suitable inert
solvent. Examples of pharmaceutically acceptable base addition salts include
sodium,
potassium, calcium, ammonium, organic amino, or magnesium salt, or a similar
salt. When
compounds of the present invention contain relatively basic functionalities,
acid addition salts
can be obtained by contacting the neutral form of such compounds with a
sufficient amount
of the desired acid, either neat or in a suitable inert solvent. Examples of
pharmaceutically
acceptable acid addition salts include those derived from inorganic acids like
hydrochloric,
hydrobromic, nitric, carbonic, monohydrogencarbonic, phosphoric,
monohydrogenphosphoric, dihydrogenphosphoric, sulfuric, monohydrogensulfuric,
hydriodic, or phosphorous acids and the like, as well as the salts derived
from relatively
nontoxic organic acids like acetic, propionic, isobutyric, maleic, malonic,
benzoic, succinic,
suberic, fumaric, lactic, mandelic, phthalic, benzenesulfonic, p-
tolylsulfonic, citric, tartaric,
methanesulfonic, and the like. Also included are salts of amino acids such as
arginate and the
like, and salts of organic acids like glucuronic or galactunoric acids and the
like (see, e.g.,
Berge et al., Journal ofPharmaceutical Science 66:1-19 (1977)). Certain
specific
compounds of the present invention contain both basic and acidic
functionalities that allow
the compounds to be converted into either base or acid addition salts. Other
pharmaceutically
acceptable carriers known to those of skill in the art are suitable for the
present invention.
[0050] The neutral forms of the compounds may be regenerated by contacting the
salt with
a base or acid and isolating the parent compound in the conventional manner.
The parent
fonn of the compound differs from the various salt foi-ins in certain physical
properties, such
as solubility in polar solvents, but otherwise the salts are equivalent to the
parent fonn of the
compound for the purposes of the invention.
[0051] In addition to salt forms, the invention provides compounds which are
in a prodrug
fonn. Prodrugs of the compounds described herein are those compounds that
readily undergo
chemical changes under physiological conditions to provide the compounds of
formula I
which are antagonists of the DP-2 receptor. Additionally, prodrugs can be
converted to the
compounds of the invention by chemical or biochemical methods in an ex vivo
environment.
For exainple, prodrugs can be slowly converted to the compounds of the
invention when
placed in a transdermal patch reservoir with a suitable enzyine or chemical
reagent. Prodrugs
are often useful because, in some situations, they may be easier to administer
than the parent
12
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
drug. They may, for instance, be bioavailable by oral administration whereas
the parent drug
is not. The prodrug may also have improved solubility in pharmaceutical
compositions over
the parent drug. A wide variety of prodrug derivatives are known in the art,
such as those that
rely on hydrolytic cleavage or oxidative activation of the prodrug. An
example, without
limitation, of a prodrug would be a compound of the invention which is
administered as an
ester (e.g. wherein R' is substituted or unsubstituted C1_6alkyl,
C0_6alkylaryl or
C0_6alkylheteroaryl, the "prodrug"), but then is metabolically hydrolyzed to
the carboxylic
acid (e.g. wherein R' is H, the "active entity"). Additional examples include
peptidyl
derivatives of a compound of the invention.
[0052] Certain compounds of the invention can exist in unsolvated forms as
well as
solvated forms, including hydrated forms. In general, the solvated forms are
equivalent to
unsolvated forms and are intended to be encompassed within the scope of the
invention.
Certain compounds of the invention may exist in multiple crystalline or
amorphous forms. In
general, all physical forms are equivalent for the uses contemplated by the
invention and are
intended to be within the scope of the invention.
[0053] Certain compounds of the invention possess asymmetric carbon atoms
(optical
centers) or double bonds; the racemates, enantiomers, diastereomers, geometric
isomers and
individual isomers are all intended to be encompassed within the scope of the
invention.
These isomers can be resolved or asymmetrically synthesized using conventional
methods to
render the isomers "optically pure", i.e., substantially free of its other
isomers. If, for instance,
a particular enantioiner of a compound of the present invention is desired, it
may be prepared
by asymmetric synthesis, or by derivation with a chrial auxilliary, where the
resulting
diastereomeric mixture is separated and the auxilliary group cleaved to
provid'2 the pure
desired enantioiners. Alternatively, where the molecule contains a basic
functional group,
such as amino, or an acidic functional group, such as carboxyl,
diastereoineric salts are
formed with an appropriate optically-active acid or base, followed by
resolution of the
diasteromers thus formed by fractional crystallization or chromatagraphic
means well known
in the art, and subsequent recovery of the pure enantiomers.
[0054] The compounds of the invention may also contain unnatural proportions
of atomic
isotopes at one or more of the atoms that constitute such compounds. For
exainple, the
compounds may be radiolabeled with radioactive isotopes, such as for example
tritium (3H),
iodine-125 (1251) or carbon-14 (14C). Radiolabled compounds are useful as
therapeutic or
13
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
prophylactic agents, e.g., cancer therapeutic agents, research reagents, e.g.,
DP-2 assay
reagents, and diagnostic agents, e.g., in vivo imaging agents. All isotopic
variations of the
compounds of the invention, whether radioactive or not, are intended to be
encompassed
within the scope of the invention.
[0055] An "antagonist" or "inhibitor" refers to an agent or molecule that
inhibits or binds
to, partially or totally blocks stimulation or activity, decreases, closes,
prevents, delays
activation or enzymatic activity, inactivates, desensitizes, or down regulates
the activity of a
receptor of the invention. As used herein, "antagonist" also includes a
reverse or inverse
agonist.
[0056] An "agonist" or "activator" refers to an agent or molecule that binds
to a receptor of
the invention, stimulates, increases, opens, activates, facilitates, enhances
activation or
enzymatic activity, sensitizes or up regulates the activity of a receptor of
the invention.
[0057] "Modulators" of activity are used to refer to "ligands", "antagonists"
and "agonists"
identified using in vitro and in vivo assays for activity and their homologs
and mimetics.
Modulators include naturally occurring and synthetic ligands, antagonists,
agonists,
molecules and the like. Assays to identify antagonists and agonists include,
e.g., applying
putative modulator compounds to cells, in the presence or absence of a
receptor of the
invention and then determining the functional effects on a receptor of the
invention activity.
Samples or assays comprising a receptor of the invention that are treated with
a potential
activator, inhibitor, or modulator are compared to control samples without the
inhibitor,
activator, or modulator to examine the extent of effect. Control samples
(untreated with
modulators) are assigned a relative activity value of 100%. Inhibition is
achieved when the
activity value of a receptor of the invention relative to the control is about
80%, optionally
50% or 25-1 %. Activation is achieved when the activity value of a receptor of
the invention
relative to the control is 110%, optionally 150%, optionally 200-500%, or 1000-
3000%
higher.
[0058] The terms "treat", "treating", "treatment" and grammatical variations
thereof as used
herein, includes partially or completely delaying, alleviating, mitigating or
reducing the
intensity of one or more attendant symptoms of a disorder or condition and/or
alleviating,
mitigating or impeding one or more causes of a disorder or condition.
Treatments according
to the invention may be applied preventively, prophylactically, pallatively or
remedially.
14
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
[0059] The terms "prevent", "preventing", "prevention" and grammatical
variations thereof
as used herein, refers to a method of partially or completely delaying or
precluding the onset
or recurrence of a disorder or condition and/or one or more of its attendant
symptoms or
barring a subject from acquiring or reacquiring a disorder or condition or
reducing a subject's
risk of acquiring or reaquiring a disorder or condition or one or more of its
attendant
symptoms.
[0060] The term "therapeutically effective amount" or "therapeutically
effective dose"
refers to the amount of the subject compound that will elicit the biological
or medical
response of a tissue, system, animal or human that is being sought by the
researcher,
veterinarian, medical doctor or other clinician. The term "therapeutically
effective amount"
includes that amount of a compound that, when administered, is sufficient to
prevent
development of, or alleviate to some extent, one or more of the symptoms of
the condition or
disorder being treated. The therapeutically effective amount will vary
depending on the
compound, the disorder or condition and its severity and the age, weight,
etc., of the mammal
to be treated.
[0061] The phrase "selectively" or "specifically" when referring to binding to
a receptor,
refers to a binding reaction that is determinative of the presence of the
receptor, often in a
heterogeneous population of receptors and other biologics. Thus, under
designated
conditions, the compounds bind to a particular receptor at least two times the
background and
more typically more than 10 to 100 times background. Specific binding of a
compound under
such conditions requires a compound that is selected for its specificity for a
particular
receptor. For example, small organic molecules can be screened to obtain only
those
compounds that specifically or selectively bind to a selected receptor and not
with other
receptors or proteins. A variety of assay fonnats may be used to select
compounds that are
selective for a particular receptor. For example, High-throughput screening
assays are
routinely used to select compounds that are selective for a particular a
receptor.
[0062] The "subject" is defined herein to include animals such as mammals,
including, but
not limited to, primates (e.g., humans), cows, sheep, goats, horses, dogs,
cats, rabbits, rats,
mice and the like. In preferred embodiments, the subject is a human.
[0063] As used herein, the term "DP-2" refers to a DP-2 receptor protein
(RefSeq
Accession No. NP-007469) or a variant thereof that is capable of mediating a
cellular
response to PGD2 in vitro or in vivo. DP-2 variants include proteins
substantially homologous
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
to native DP-2, i.e., proteins having one or more naturally or non-naturally
occurring amino
acid deletions, insertions or substitutions (e.g., DP-2 derivatives, homologs
and fragments).
The amino acid sequence of DP-2 variant preferably is at least about 80%
identical to a native
DP-2, more preferably at least about 90% identical, and most preferably at
least about 95%
identical.
[0064] As used herein, the terms "other PGD2 receptor", "another PGD2
receptor" and the
like refer to a prostanoid receptor protein other than DP-2, or variant
thereof, that is capable
of mediating a cellular response to PGD2 in vitro or in vivo. Another PGD2
receptor may be
selective for PGD2, e.g., DP-1 (RefSeq Accession No. NP-000944), or may also
interact with
one or more other prostanoids (e.g., EP1, EP2, EP3 and EP4, FP, IP and TP).
Other PGD2
receptor variants include proteins substantially homologous to a corresponding
native
prostanoid receptor other than DP-2, i.e., proteins having one or more
naturally or
non-naturally occurring amino acid deletions, insertions or substitutions
(e.g., derivatives,
homologs and fragments of another PGD2 receptor). The amino acid sequence of
other PGD2
receptor variants preferably is at least about 80% identical to the
corresponding native other
PGD2 receptors, more preferably at least about 90% identical, and most
preferably at least
about 95% identical. Preferably, another PGD2 receptor is DP-1.
[0065] As used herein, the term "DP-1" refers to a DP-1 receptor protein
(RefSeq
Accession No. NP-000944) or a variant thereof that is capable of mediating a
cellular
response to PGD2 in vitro or in vivo. DP-1 variants include proteins
substantially
homologous to native DP-1, i.e., proteins having one or more naturally or non-
naturally
occurring amino acid deletions, insertions or substitutions (e.g., DP-1
derivatives, homologs
and fraginents). The amino acid sequence of DP-1 variant preferably is at
least about 80%
identical to a native DP-1, more preferably at least about 90% identical, and
most preferably
at least about 95% identical.
[0066] As used herein, the term "TP" refers to a TP protein (RefSeq Accession
No.
NP-963998) or variant thereof that is capable of mediating a cellular response
to PGD2 in
vitro or in vivo. TP variants include proteins substantially homologous to
native TP, i.e.,
proteins having one or more naturally or non-naturally occurring amino acid
deletions,
insertions or substitutions (e.g., TP derivatives, homologs and fraginents).
The amino acid
sequence of TP variant preferably is at least about 80% identical to a native
TP, more
preferably at least about 90% identical, and most preferably at least about
95% identical.
16
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
[0067] The terms "modulate", "modulation" and the like refer to the ability of
a compound
to increase or decrease the function and/or expression of DP-2 and/or one or
more other
PGD2 receptors, e.g., DP-1, where such function may include transcription
regulatory activity
and/or protein-binding. Modulation may occur in vitro or in vivo. Modulation,
as described
herein, includes the inhibition, antagonism, partial antagonism, activation,
agonism or partial
agonism of a function or characteristic associated with DP-2 and/or one or
more other PGD2
receptors, either directly or indirectly, and/or the upregulation or
downregulation of the
expression of DP-2 and/or one or more other PGD2 receptors, either directly or
indirectly. In
a preferred embodiment, the modulation is direct. Inhibitors or antagonists
are compounds
that, e.g., bind to, partially or totally block stimulation, decrease,
prevent, inhibit, delay
activation, inactivate, desensitize, or downregulate signal transduction.
Activators or agonists
are compounds that, e.g., bind to, stimulate, increase, open, activate,
facilitate, enhance
activation, activate, sensitize or upregulate signal transduction. The ability
of a compound to
inhibit the function of DP-2 and/or one or more other PGD2 receptors can be
demonstrated in
a biochemical assay, e.g., binding assay, or a cell-based assay, e.g., a
transient transfection
assay.
[0068] As used herein, the term "condition or disorder responsive to
modulation of PGD2
or a PGD2 receptor" and related terms and phrases refer to a condition or
disorder associated
with inappropriate, e.g., less than or greater than normal, activity of a PGD2
receptor and at
least partially responsive to or affected by modulation of a PGD2 receptor
(e.g., a PGD2
receptor antagonist or agonist results in some improvement in patient well-
being in at least
some patients). Inappropriate functional activity of a PGD2 receptor might
arise as the result
of expression of a PGD2 receptor in cells which noi-mally do not express the
receptor, greater
than nonnal production of PGD2, or slower than nonnal metabolic inactivation
or elimination
of PGD2 or its active metabolites, increased expression of a PGD2 receptor or
degree of
intracellular activation (leading to, e.g., inflaininatory and immune-related
disorders and
conditions) or decreased expression of a PGD2 receptor. A condition or
disorder associated
with a PGD2 receptor may include a"DP-2-mediated condition or disorder".
[0069] As used herein, the phrases "condition or disorder responsive to the
antagonizing a
DP-2 receptor", and related phrases and tenns refer to a condition or disorder
characterized
by inappropriate, e.g., greater than nonnal, DP-2 activity. Inappropriate DP-2
functional
activity might arise as the result of DP-2 expression in cells which normally
do not express
DP-2 or increased DP-2 expression or degree of intracellular activation
(leading to, e.g.,
17
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
inflammatory and immune-related disorders and conditions). A condition or
disorder
responsive to the antagonizing a DP-2 receptor may be completely or partially
mediated by
inappropriate DP-2 functional activity. However, a condition or disorder
responsive to the
antagonizing a DP-2 receptor is one in which modulation of DP-2 results in
some effect on
the underlying condition or disorder (e.g., an DP-2 antagonist results in some
improvement in
patient well-being in at least some patients).
Embodiments of the Invention
[0070] A class of compounds that antagonize DP-2 has been discovered.
Depending on the
biological environment (e.g., cell type, pathological condition of the host,
etc.), these
compounds can antagonize DP-2 and/or one or more other PGD2 receptors (e.g.,
ligand
binding). By antagonizing DP-2 and/or one or more other PGD2 receptors, the
compounds
will find use as therapeutic agents capable of modulating disorders and
conditions responsive
to modulation of DP-2 and/or one or more other PGD2 receptors and/or mediated
by DP-2
and/or one or more other PGD2 receptors. Examples of such conditions and
disorders are
provided below.
[0071] While the compounds of the invention are believed to exert their
effects by
selectively interacting with DP-2, the mechanism of action by which the
compounds act is
not a limiting embodiment of the invention. For example, compounds of the
invention may
interact with PGD2 receptor subtypes other than DP-2. However, as noted
herein, the present
invention specifically contemplates the activity of the disclosed compounds to
selectively
antagonize DP-2 receptor over e.g. DP-1 receptor, and/or other prostanoid
receptors, e.g., TP
receptor.
[0072] Compounds contemplated by the invention include, but are not limited
to, the
exemplary compounds provided herein.
Compounds of the Invention
[0073] In one embodiment, the present invention provides compounds of the
general
structure (I):
18
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
R2
R3 C02R1
(R4),\ i A\Q R5
\1)
[0074] A is a 5-14-membered heterocyclic ring fused or bonded to phenyl ring B
having
1-4 ring heteroatoms each independently selected from the group consisting of
nitrogen,
oxygen and sulfur, the heterocyclic ring being moncyclic or polycyclic,
optionally substituted
with 1-3 R8 substituents.
[0075] Ql is selected from the group consisting of: a bond, -C1-C4alkylene-,
-Cl-C4heteroalkylene-, -CO-, -NH-, -0-, -SOq-, -C(O)O-, -OC(O)-, -CONH-, -NHCO-
,
-NHCONH-, -NHSOq-, -SOqNH- and -COCH2HNSOq.
[0076] Each R', R 2 and R3 is independently selected from the group consisting
of H,
C1_6alkyl, C0_6alkylaryl and C0_6alkylheteroaryl; wherein the aryl or
heteroaryl portions are
optionally substituted with C1_6alkyl, CN, OR, C1_6haloalkyl, C1_6heteroalkyl,
NR2, NO2, halo,
C(O)R, COZR, CONR2, SOqR, SOqNR2, OC(O)OR, OC(O)R, OC(O)NRZ, NRC(O)NR2,
NRC(O)R and NRC(O)OR.
[0077] Each R8 is independently selected from the group consisting of
C1_6alkyl,
C0_6a1ky1C3_6cycloalkyl, C0_6alkylaryl, C0_6alkylheteroaryl, oxo, C1_6alkyl,
CN, OR,
Ci_6haloalkyl, C1_6heteroalkyl, NR2, NOZ, halo, C(O)R, CO2R, CONR2, SOqR,
SOqNR2,
OC(O)OR, OC(O)R, OC(O)NR2, NRC(O)NR2, NRC(O)R and NRC(O)OR.
[0078] Each R4 is independently selected from the group consisting of
C1_6alkyl,
CO_4a1ky1C3_iocycloalkyl, Co_4alkylaryl, C0_4alkylheteroaryl, C2_4alkenylaryl,
C2_4alkynylaryl,
CO_4alkylheterocyclyl, CN, amino, NHCOR', hydroxy, C1_6alkoxy, OC(O)RI, -
OC0_4alkylaryl,
OC0_4alkylheteroaryl, -OC0_4alkylC3_ i ocycloalkyl, OC0_4a1ky1C3_ i
oheterocyclyl,
OC0_4a1ky1NR8, nitro, halo and haloC1_6alkyl; or are combined together to form
an aryl or
heterocyclyl ring having 1-2 heteroatoms selected from the group consisting of
nitrogen,
oxygen and sulfur; wherein the alkyl, aryl and heterocyclyl portions are each
optionally
substituted with 1 to 3 substituents each independently selected from the
group consisting of
C1_6alkyl, CN, CONHRI, C02R', amino, C1_6alkoxy, halo, haloC1_6alkyl and
SOqR'.
19
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
[0079] R5 is selected from the group consisting of C1_6alkyl, C0_4alkylaryl,
C2_4alkenylaryl,
C2_4alkynylaryl and C0_4alkylheteroaryl, each of which is optionally
substituted with 1-3 R9
substitutents.
[0080] Each R9 is independently selected from the group consisting of
C1_6alkyl, CN, OR,
oxo, C1_6haloalkyl, C1_6heteroalkyl, NR2, NO2, halo, C(O)R, CO2R, CONR2, SOqR,
SOqNR2,
OC(O)OR, OC(O)R, OC(O)NR2, NRC(O)NR2, NRC(O)R and NRC(O)OR.
[0081] Each R is independently selected from the group consisting of H,
C1_6alkyl, C0_4
alkylheteroaryl,Co-4heterocyclyl, C3_8cycloalkyl and C0_4alkylaryl or when
attached to the
same nitrogen atom may be combined to form a 5-8 membered ring having 1-4 ring
heteroatoms each independently selected from the group consisting of nitrogen,
oxygen and
sulfur.
[0082] The subscript n is independently 0, 1, 2, 3 or 4;
[0083] The subscript o is independently 0 or 1;
[0084] Each subscript q is independently 0, 1 or 2.
[0085] In another embodiment, the present invention provides pharmaceutically
acceptable
derivatives thereof.
[0086] In another embodiment, A is fused to phenyl ring B. In another
embodiment, A is
bonded to phenyl ring B.
[0087] In another embodiment, R', R 2 and R3 are each independently selected
from the
group consisting of H, C1_6alkyl and C0_6alkylaryl. In one embodiment, R1, R2
and R3 are
each independently selected from the group consisting of H, CH3 and phenyl. In
one
embodiment, R' is H. In another embodiment, R 2 and R3 are H.
[0088] In another embodiment, A has the structure (II):
Ny
R"
(II)
wherein
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
Y is selected from the group consisting of a bond, CH2, N, 0, NO and SOq;
R10 and R' 1 are H or are combined together to form an aryl, heteroaryl or
cycloalkyl ring;
the subscript p is independently 0, 1 or 2;
each dashed ring bond independently indicates the presence of a single, double
or normalized
bond;
the wavy line indicates the point of attachment to QI and the dashed line
indicates the point
of attachment to phenyl ring B.
[0089] In another embodiment, A has the structure (II):
Ny
R"
(II)
wherein
Y is selected from the group consisting of a bond, CH2, N, 0, NO and SOq;
R10 and R' 1 are H or are combined together to fonn an aryl, heteroaryl or
cycloalkyl ring;
the subscript p is independently 0, 1 or 2;
each dashed ring bond independently indicates the presence of a single, double
or normalized
bond;
the dashed line indicates the point of attachment to Ql and the wavy line
indicates the point
of attachment to phenyl ring B.
[0090] In another embodiment, A is selected from the group consisting of
pyrrolidinyl,
pyrrolyl, piperadinyl, tetrahydropyridinyl, piperazinyl, piperazin-l-oxide,
morpholinyl,
thiomorpholinyl, azepanyl, azepinyl, oxazepane, thiazepane, azocanyl,
azocinyl, indolyl,
azaindole, tetrahydroquinolinyl and decahydroquinolinyl.
[0091] In another embodiment, A has a fonnula selected from the group
consisting of:
21
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
(R$)m (R$)m (R$)m (R $)m
\
~-~ N \N3
N
~ i (R$)m (R$)m (R$)m (R$)m
1 ~
r ~s ; r~1o ~
\N)
(R\)m (R\)m (R\)m (R\)m
~N - ~ \N ~N`J-~)
(R)m (R$)m ~ (R$)m ~ (R$)m
~'\~/; -=~/; N,
N N
(R$)m (R$)m (R$)m (R$)m
V N'~ r\--
--~ ~N ~ -~ N --~
and
m is and integer from 0 to 3; and
the dashed line indicates the point of attachment to Ql and the wavy line
indicates the point
of attachment to phenyl ring B.
[0092] In another embodiment, A has a formula selected from the group
consisting of:
22
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
Ho1 (R$)m (R$)m (R$)m (R8)m
-~N F/
(R)m (R)m (R$)m (R)m
rv~'~1
N ~-~N N/
(R$)m (R$)m (R$)m (R8)m
,
~/NN/ ~ N/ (R$)m (R$)m (R$)m (R$)m
N~ N~
N~ N/
i i
(R$)m (R)m (R$)m (R)m
I--~/N
and
/ / .
m is and integer from 0 to 3; and
the wavy line indicates the point of attachment to QI and the dashed line
indicates the point
of attachment to phenyl ring B.
[0093] In another embodiment, Ql is selected from a bond, -Ci-C4alkylene-,
-Ci-C4heteroalkylene-, -CO-, -NH-, -0-, -SOq-, -C(O)O-, -OC(O)-, -CONH-, -NHCO-
,
-NHCONH-, -NHSOq- ,-SOqNH- and -COCH2HNSOq. In another embodiment, Ql is a
bond. In another embodiment, Ql is -Ci-C4alkylene-. In another embodiment, Q,
is
-Ci-C4heteroalkylene-. In another embodiment, Q1 is -CO-. In another
embodiment, Q1 is a
-NH-. In another embodiment, Ql is a -0-. In another embodiment, Q, is -SOq-.
In another
embodiment, Q~ is -C(0)0-. In another embodiment, Q1 is -OC(O)-. In another
embodiment, Q~ is -CONH-. In another embodiment, Ql is -NHCO-. In another
embodiment,
Ql is -NHCONH-. In another embodiment, Ql is -NHSOq-. In another embodiment,
Q, is
-SOqNH-. In another embodiment, Ql is -COCH2HNSOq.
[0094] In another embodiment, the compound has a structure (III):
23
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
O (R)m
HO / ) p
Y ~ R5
R' ~11~1/
(R4)n
(III)
wherein
Y is selected from the group consisting of a bond, CH2, N, 0, NO and SOq;
R10 and Rl l are H or are combined together to form an aryl, heteroaryl or
cycloalkyl ring;
the subscript m is independently 0, 1, 2 or 3;
the subscript p is independently 0, 1 or 2; and
each dashed ring bond independently indicates the presence of a single, double
or normalized
bond.
[0095] In another embodiment, A is fused to phenyl ring B. In another
embodiment, A is
bonded to phenyl ring B.
[0096] In another embodiment, Y is CH2 and p is 0.
[0097] In another embodiment, the compound is 2-(2 -(1 -tosylpiperi din- 3 -
yl)phenyl)aceti c
acid or 2-(2-(1-tosylpiperidin-4-yl)phenyl)acetic acid.
[0098] In another embodiment, the compound has a structure (IV):
0
R'O
(R)m
,\~
p
(R4)~ Y N R5
Q1'
Rlo R"
(IV)
wherein
Y is selected from the group consisting of a bond, CH2, N, 0, NO and SOq;
24
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
R10 and RlI are H or are combined together to form an aryl, heteroaryl or
cycloalkyl ring;
the subscript m is independently 0, 1, 2 or 3;
the subscript p is independently 0, 1 or 2; and
each dashed ring bond independently indicates the presence of a single, double
or normalized
bond.
[0099] In another embodiment, Y is CH2 and p is 0.
[0100] In another embodiment, the compound has the general structure (IVa):
0
OR'
~N Q1_Rs
I /J l ~ IR$)m
~/ Y \R~~
R4)n Rl0
(IVa).
[0101] In another embodiment, Ql is -CO-.
[0102] In another embodiment, the compound is
{3-[1-(4-fluoro-benzoyl)-piperidin-3-yl]-phenyl}-acetic acid.
[0103] In another embodiment, Ql is -SOq-.
[0104] In another embodiment, the compound is selected from the group
consisting of:
{3-[1-(4-fluoro-benzenesulfonyl)-piperidin-2-yl]-phenyl}-acetic acid;
2-(3-(1-(inethylsulfonyl)piperidin-3-yl)phenyl)acetic acid;
2-(4-(4-chlorobenzyloxy)-3-(1-(inethylsulfonyl)piperidin-3-yl)phenyl)acetic
acid;
2-(3-(1-(thiophen-2-ylsulfonyl)piperi din-3-yl)phenyl)acetic acid;
2-(3-(1-(thiophen-3-ylsulfonyl)piperidin-3-yl)phenyl)acetic acid;
2-(3-(1-(5-chlorothiophen-2-ylsulfonyl)piperidin-3-yl)phenyl)acetic acid;
2-(3-(1-(5-bromothiophen-2-ylsulfonyl)piperidin-3-yl)phenyl)acetic acid;
2-(3-(1-(benzofuran-2-ylsulfonyl)piperidin-3-yl)phenyl)acetic acid;
2-(3-(1-(pyridin-3-ylsulfonyl)piperidin-3-yl)phenyl)acetic acid;
2-(3-(1-(benzylsulfonyl)piperidin-3-yl)phenyl)acetic acid;
(E)-2-(3-(1-(styrylsulfonyl)piperidin-3-yl)phenyl)acetic
acid; {3-[ 1-(Toluene-4-sulfonyl)-decahydro-quinolin-3-yl]-phenyl } -acetic
acid;
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
{3-[1-(4-Fluoro-benzenesulfonyl)-1,2,3,4-tetrahydro-quinolin-3-yl]-phenyl}-
acetic acid;
2-(3 -(1 -(phenylsulfonyl)piperidin-3 -yl)phenyl) acetic acid; 2-(3-(1-
tosylpiperidin-3-yl)phenyl)
acetic acid; 2-(4-(4-chlorobenzyloxy)-3-(1-(phenylsulfonyl)piperidin-3-
yl)phenyl)acetic acid;
2-(3-(1-(3,5-dichlorophenylsulfonyl)piperidin-3-yl)phenyl)acetic acid;
(2-(3-(1-(2,3-dichlorophenylsulfonyl)piperidin-3-yl)phenyl)acetic acid;
2-(3-(1-(4-nitrophenylsulfonyl)piperidin-3-yl)phenyl)acetic acid;
2-(3-(1-(naphthalen-1-ylsulfonyl)piperidin-3-yl)phenyl)acetic acid;
{3-[ 1-(4-Fluoro-benzenesulfonyl)-6-methyl-piperidin-3-yl]-phenyl}-acetic acid
methyl
2-(3-(1-(4-fluorophenylsulfonyl)-1,2,5,6-tetrahydropyridin-3-
yl)phenyl)acetate;
2-(3-(1-(4-fluorophenylsulfonyl)-1,2,5,6-tetrahydropyridin-3-yl)phenyl)acetic
acid;
2-(3-(1-(4-fluorophenylsulfonyl)-1,4,5,6-tetrahydropyridin-3-yl)phenyl)acetic
acid;
{3-[1-(4-Fluoro-benzenesulfonyl)-4-methyl-piperidin-3-yl]-phenyl}-acetic acid
methyl ester;
{3-[1-(4-Fluoro-benzenesulfonyl)-4-methyl-piperidin-3-yl]-phenyl}-acetic acid;
{3-[1-(4-Fluoro-benzenesulfonyl)-2-methyl-piperidin-3-yl]-phenyl}-acetic acid;
{3-[1-(4-Fluoro-benzenesulfonyl)-6-methyl-piperidin-3-yl]-phenyl}-acetic acid;
2-(3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)phenyl)acetic acid;
2-(4-(4-chlorobenzyloxy)-3-(1-(4-fluorophenylsulfonyl)piperidin-3-
yl)phenyl)acetic acid;
2-(3 -(1 -(4-chlorophenylsulfonyl)piperi din-3 -yl)phenyl) acetic acid; methyl
2-(4-chloro-3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)phenyl)acetate;
2-(4-chloro-3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)phenyl)acetic acid;
2-(3-chloro-5-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)phenyl)acetic acid;
2-(2-chloro-5-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)phenyl)acetic acid;
2-(3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)-2-inethylphenyl)acetic acid;
{3-[1-(4-Fluoro-benzenesulfonyl)-piperidin-3-yl]-5-hydroxy-phenyl}-acetic
acid;
{3-Benzyloxy-5-[1-(4-fluoro-benzenesulfonyl)-piperidin-3-yl]-phenyl}-acetic
acid;
{3-(4-Chloro-benzyloxy)-5-[ 1-(4-fluoro-benzenesulfonyl)-piperidin-3-yl]-
phenyl } -acetic
acid; {3,4-Dichloro-5-[1-(4-fluoro-benzenesulfonyl)-piperidin-3-yl]-phenyl}-
acetic acid;
{3-Amino-5-[1-(4-fluoro-benzenesulfonyl)-piperidin-3-yl]-phenyl}-acetic acid;
{3-[4-Cyclohexyl-1-(4-fluoro-benzenesulfonyl)-piperi din-3-yl]-phenyl}-acetic
acid;
{3-[ 1-(4-Fluoro-benzenesulfonyl)-4-phenyl-piperidin-3-yl]-phenyl}-acetic
acid;
{3-[ 1-(4-Fluoro-benzenesulfonyl)-4-phenyl-piperidin-3-yl]-phenyl}-acetic
acid;
{3-Acetylamino-5-[1-(4-fluoro-benzenesulfonyl)-piperidin-3-yl]-phenyl}-acetic
acid;
{3-[1-(4-Fluoro-benzenesulfonyl)-piperidin-3-yl]-5-phenoxy-phenyl}-acetic
acid;
2-(3-(1-(4-fluorophenylsulfonyl) piperidin-3-yl)-4-inethylphenyl) acetic acid;
26
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
2-(3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)-5-methoxyphenyl)acetic acid;
2-(3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)-5-hydroxyphenyl)acetic acid;
2-(3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)-5-methylphenyl)acetic acid;
2-(5-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)-2-inethylphenyl)acetic acid;
2-(3-(1-(4-cyanophenylsulfonyl)piperidin-3-yl)phenyl)acetic acid;
2-(3-(1-(4-tert-butylphenylsulfonyl)piperidin-3-yl)phenyl)acetic acid;
2-(3-(1-(2,4-dichlorophenylsulfonyl)piperidin-3-yl)phenyl)acetic acid;
2-(3-(1-(4-methoxyphenylsulfonyl)piperidin-3-yl)phenyl)acetic acid;
2-(3-(1-(o-tolylsulfonyl)piperidin-3-yl)phenyl)acetic acid;
2-(3-(1-(2-chlorophenylsulfonyl)piperidin-3-yl)phenyl)acetic acid;
2-(3-(1-(4-ethylphenylsulfonyl)piperidin-3-yl)phenyl)acetic acid;
2-(3-(1-(phenethylsulfonyl)piperidin-3-yl)phenyl)acetic acid;
2-(3 -(1 -(2-chloro-4-fluorophenylsulfonyl)piperidin-3 -yl)phenyl) acetic
acid;
2-(3-(1-(butylsulfonyl)piperidin-3-yl)phenyl)acetic acid;
2-(3-(1-(4-(methylsulfonyl)phenylsulfonyl)piperidin-3-yl)phenyl)acetic acid;
2-(3-(1-(3,4-dichlorophenylsulfonyl)piperidin-3-yl)phenyl)acetic acid;
2-(3-(1-(4-fluoro-2-methylphenylsulfonyl)piperidin-3-yl)phenyl)acetic acid;
2-(3-(1-(3-chlorophenylsulfonyl)piperi din-3 -yl)phenyl) acetic acid;
2-(3-(1-(m-tolylsulfonyl)piperidin-3-yl)phenyl)acetic acid; Methyl
2-(3-(1-(4-fluorophenylsulfonyl)piperidin-4-yl)phenyl)acetate; and
2-(3-(1-(4-fluorophenylsulfonyl)piperidin-4-yl)phenyl)acetic acid.
[0105] In another embodiment, Y is a bond and p is 0.
[0106] In another embodiment, the compound is selected from the group
consisting of:
2-(3-(1-(4-fluorophenylsulfonyl)pyrrolidin-3-yl)phenyl)acetic acid;
2-(3-(1-(4-fluorophenylsulfonyl)-1 H-pyrrol-3-yl)phenyl)acetic acid;
{3-[1-(4-Fluoro-benzenesulfonyl)-4-phenyl-lH-pyrrol-3-yl]-phenyl}-acetic acid;
[ 3 -(1 -Benzenesulfonyl- I H-indol-3 -yl)-phenyl] -acetic acid;
[3-(1-Methanesulfonyl-lH-indol-3-yl)-phenyl]-acetic acid;
{3-[1-(4-Methoxy-benzenesulfonyl)-1H-indol-3-yl]-phenyl}-acetic acid;
{3-[1-(4-Fluoro-benzenesulfonyl)-1H-indol-3-yl]-phenyl}-acetic acid;
{3-[1-(Toluene-4-sulfonyl)-1H-indol-3-yl]-phenyl}-acetic acid; and
{3-[ 1-(4-Fluoro-benzenesulfonyl)-2-inethyl-1 H-indol-3-yl]-phenyl}-acetic
acid.
27
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
[0107] In another embodiment, Y is selected from the group consisting of N, 0,
NO and
SOq;
[0108] In another embodiment, Ql is -CONH-.
[0109] In another embodiment, the compound is
{3-[1-(4-Fluoro-phenylcarbamoyl)-piperidin-3-yl]-phenyl}-acetic acid.
[0110] In another embodiment, the compound has a structure (V):
O
HO
// ~R$)m
R4
~ )nrp
\ R5
Q1'
R10 R11
(V)
wherein
Y is selected from the group consisting of a bond, CH2, N, 0, NO and SOq;
R10 and RI I are H or are combined together to form an aryl, heteroaryl, or
cycloalkyl ring;
the subscript m is independently 0, 1, 2 or 3;
the subscript p is independently 0, 1 or 2; and
each dashed ring bond independently indicates the presence of a single, double
or nonnalized
bond.
[0111] In another embodiment, Ql is a bond. In another embodiment, Ql is
-C1-C4alkylene-. In another embodiment, Ql is -Ci-C4heteroalkylene-. In
another
embodiment, Q' is -CO-. In another embodiment, Ql is a -NH-. In another
embodiment, Q,
is a -0-. In another embodiment, QI is -SOq-. In another embodiment, Ql is -
C(O)O-. In
another embodiment, Ql is -OC(O)-. In another embodiment, Ql is -CONH-. In
another
embodiment, QI is -NHCO-. In another embodiment, Ql is -NHCONH-. In another
embodiment, Ql is -NHSOq-. In another embodiment, Ql is -SOqNH-. In another
embodiment, QI is -COCH2HNSOq.
28
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
[0112] In another embodiment, the compound is
{4-[1-(Toluene-4-sulfonyl)-piperidin-3-yl]-phenyl}-acetic acid.
[0113] In another embodiment, the compound has the structure (VI):
0
HO
(is)m
(R4)n li \J ~
~Y~ JP
\ R5
Q1'
R10 R11
(VI)
wherein
Y' is selected from the group consisting of a bond, CH2, N, 0, NO and SOq;
R10 and Rl 1 are H or are combined together to form an aryl, heteroaryl, or
cycloalkyl ring;
the subscript m is independently 0, 1, 2 or 3;
the subscript p is independently 0, 1 or 2; and
each dashed ring bond independently indicates the presence of a single, double
or normalized
bond.
[0114] In another embodiment, Ql is a bond. In another embodiment, Ql is
-Ci-C4alkylene-. In another embodiment, Ql is -Ci-C4heteroalkylene-. In
another
embodiment, Ql is -CO-. In another embodiment, Ql is a -NH-. In another
embodiment, Q,
is a -0-. In another embodiment, Ql is -SOq-. In another embodiment, Ql is -
C(O)O-. In
another embodiment, QI is -OC(O)-. In another embodiment, Ql is -CONH-. In
another
embodiment, Ql is -NHCO-. In another embodiment, Ql is -NHCONH-. In another
einbodiinent, Ql is -NHSOq-. In another embodiment, Ql is -SOqNH-. In another
embodiment, Ql is -COCH2HNSOq.
[0115] In another embodiment, the compound has the general structure (VII):
29
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
CO2R'
n(R4)
ON_O1-R
(VII)
wherein R' is H or C1_6alkyl;
each R2 is independently selected from the group consisting of C1_4alkyl,
halo,
5 ary1C1_4alkoxy, optionally substituted with 1-3 R7 substitutents;
R5 is aryl optionally substituted with 1-3 R9 substitutents; and
each R9 is independently selected from the group consisting of halo and
C1_6alkyl.
[0116] In another embodiment, the compound is
2-(4-(2-(4-methylphenylsulfonamido)acetyl)-2,3,4,5-tetrahydrobenzo[f] [
1,4]oxazepin-7-yl)ac
etic acid.
[0117] In another embodiment, the compound has the general structure (VIII):
0
R'O
4
n(R ) ~ ~ Y3
/ Y2
(VIII)
wherein:
each YZ or Y3 is independently CH2 or NQ1 R5;
the subscript n is independently 0, 1, 2, 3 or 4.
[0118] In another embodiment, the compound has the general structure (IX):
0
R'O
n(R4) / N\ R5
Q1"
(IX).
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
[0119] In another embodiment, the compound is selected from the group
consisting
of:methyl 2-(2-(4-fluorophenylsulfonyl)-1,2,3,4-tetrahydroisoquinolin-5-
yl)acetate;
2-(2-(4-fluorophenylsulfonyl)-1,2,3,4-tetrahydroisoquinolin-5-yl)acetic acid;
methyl
2-(2-(2 -(4-fluorophenylsulfonamido) acetyl)- 1,2,3,4-tetrahydroisoquinolin- 5
-yl) acetate; and
2-(2- (2 -(4-fluorophenylsulfonamido)acetyl)- 1,2,3,4-tetrahydroisoquinolin- 5
-yl) acetic acid.
[0120] In another embodiment, the compound has the general structure (X):
O OR'
N.Q1=R5
n(R4)
(X)
wherein R' is H or C1_6alkyl;
each R2 is independently selected from the group consisting of C1_4alkyl,
halo,
ary1C1_4alkoxy, optionally substituted with 1-3 R7 ;
R5 is aryl optionally substituted with 1-3 R9 substitutents;
each R9 is independently selected from the group consisting of halo and
C1_6alkyl; and
the subscript n is independently 0 or 1.
[0121] In another embodiment, the compound is selected from the group
consisting of:
methyl 2-(2-(4-fluorophenylsulfonyl)-1,2,3,4-tetrahydroisoquinolin-7-
yl)acetate;
2-(2-(4-fluorophenylsulfonyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)acetic acid;
and
2-(2-(2-(4-inethylphenylsulfonainido)acetyl)-1,2,3,4-tetrahydroisoquinolin-7-
yl)acetic acid.
[0122] Other embodiments, are disclosed in U.S. Patent Application., Dudler et
al. and
filed 9 June 2006, entitled "Substituted Phenyl Acetic Acids as DP-2
Antagonists" Attorney
Docket No. 014233-003300US (which is incorporated herein by reference in its
entirety).
[0123] The invention encompasses novel compounds, novel pharmaceutical
compositions
and/or novel methods of use. While some compounds disclosed herein are
available from
commercial sources, the pharmaceutical compositions or methods of using these
compounds
are novel. Unless otherwise indicated, it is to be understood that the
invention includes those
compounds that are novel, as well as pharinaceutical compositions, various
methods (e.g.,
methods of treating or preventing certain conditions and disorders mediated by
DP-2 and/or
31
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
one or more other PGD2 receptors), and the like which include both the novel
compounds of
the invention and compounds that are commercially available.
Preparation of the Compounds
[0124] Synthetic routes to the compounds provided herein are also described in
Schemes
A-D and in the Examples. One of skill in the art will understand that the
synthetic routes can
be modified to use different starting materials and/or alternate reagents to
accomplish the
desired transformations. Additionally, one of skill in the art will recognize
that protecting
groups may be necessary for the preparation of certain compounds and will be
aware of those
conditions compatible with a selected protecting group. Accordingly, the
methods and
reagents described herein are all expressed as non-limiting embodiments.
Scheme A
H3C CH3
9~CH3
HOZC I~ B~0 CH3 COZH CO2Me COZMe
1. TFA, DCM
n)~~ p R" TMSCHNZ ~ 2. EyN, DCM I
Tf0 Y }-__( N, Boc 0 I /~
R,o 1 Pd(PPhg)a, base, p CH3OH, C6H6 Y \~~ p 3. R'Q~CI, DCM 4/ )P
Boc }~ Q R'
DME, 80 C, 1.5 hr R Y~N'Boc }~~N Y
A R,o Rõ R, Rõ R Rõ
B C D
CO2H
R4 Y ry p
QlR`
R1 Rn
D
[0125] In some embodiments, as shown in Scheme A, a triflate A can be obtained
by
treating a oxosubstituted heterocycle with triflic anhydride or N-phenyl
triflimide in an
anhydrous solvent such as THF, in the presence of a base such as LDA or nBuLi,
at
temperatures ranging from -78 C to RT. The triflate can then be cross coupled
with an
available aryl boronate ester in the presence of a palladium (0) source, and a
base such as
Na2CO3, in a solvent system such as DME/water or alternatively under anhydrous
conditions
32
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
such as DME or DMF, and a base such as sodium carbonate Na2CO3, or KZP04,
optionally in
the presence of CsF at temperatures varying from 40-100 C, for 1-6 hours. The
carboxylic
acid B is then esterified using trimethyl silyl diazomethane in a solvent such
as hexanes.
Removal of the protecting group occurs under standard conditions using for
example TFA in
a solvent such as DCM, at room temperature for 1-6 hours. Alkylation or
acylation with a
compound such as halide Q'R 5X under basic conditions such as Et3N or DIEA in
a solvent
such as DCM, or alternatively using pyridine both as a solvent and a base, for
a period of 5-
12 hours, at room temperature leads to substituted heterocycle C.
Saponification with a base
such as KOH or NaOH in a solvent system such as methanol: water, for a period
of 1-6 hours
at temperatures between 35-65 C, followed by a mild hydrogenation with a
catalyst such as
for example Pearlman's catalyst, or 10% palladium on carbon or platinum oxide,
at room
temperature under atmospheric pressure to 50 psi, in a solvent such as
methanol, leads to a
carboxylic acid of formula D.
Scheme B
I. oxalyl chloride 0 ~ AgOBz COzMe
COZH CH,Cl7, cat DMF N~ N_ triethylainine
2. TMS-diazomethane, THF MeOH
R~ I Br R~ Br R~ Br
E F G
COzMe
Heterocyclic boronic acid I. Fl,, PtO2 COk,Yr H
CsF MeOH, cat HCI
Pd(PPh3)a I ~ 2. Q'R'X DME/1PA/FI,O TEA, CI-I,CI, R, y/ NH3 I.iOH THF/H,O P
N,
Q
R10 Rii Ria
R"
H
1
[0126) In some embodiments, as shown in Scheme B, an aryl benzoic acid E can
be
converted to a phenyl acetate G using an Arndt-Eistert reaction. Cross
coupling of the aryl
halide with a heterocyclic boronic acid or stannane in the presence of a
palladium (0) source
such as palladium tetrakis triphenyl phosphine, in a mixed solvent system such
as
DME/water, and a base such as cesium fluoride from 1 to 6 hours, at
temperatures ranging
from 25 to 80 C. The heterocyclic methyl ester H may optionally be reduced
using
hydrogenation conditions such as platinum oxide, in a solvent like methanol,
at room
temperature, under pressure ranging from 10-50 psi for 1 to 9 hours and
converted to
33
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
substituted heterocycle I upon treatment with acylating or alkylating agent
such as halide
Q'R5X, under conditions described in Scheme A, followed by a saponification
described in
Scheme A as well.
Scheme C
CH3 H 0
1. NBS, CCI4, AIBN &--
R4Br 2. Me3NO, CH3CN R4Br
K
MeOzC COZMe
1. Jones oxid. (HO)2B,
Het A , CsF I~
2. (COCI)2
/
3. TMSCHNz Br
4. AgOBz, Et3N R4 Pd(OAc)2, PPh3 R4 Yf P
N
MeOH
L
R10 R"
H
[0127] In some embodiments, as shown in Scheme C, aldehyde J was obtained by
treatment with NBS and AIBN in a solvent such as carbon tetrachloride,
followed by addition
of trimethylainine N-oxide in a solvent such as CH3CN, at temperatures ranging
from room
temperature to 80 C, over 15 hours. Oxidation of K with Jone's reagent in a
solvent such as
acetone, followed by the Arndt-Eistert reaction leads to H. Compound H is then
converted to
other products using chemistry described in Schemes A- B.
Scheme D
34
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
0 ~ 0
1. BoczO / THF O
HO DMAP / tBuOH
~ 2. PdC12 dppf
Br B(Pin)Z B O
a KOAc Ra
R E dioxane M Pd(PPh3)4 80 C DME ~ N.Q'.R
Na2CO3 (2M) R^
Br .~ / 80 C
Q1-NH2 K2CO3/CH3CN ~ Jr
RS Q -N` Br p
RS --~\\
N 2~
O
Br
K2CO3 /CH3CN
1.Grubb's 2nd
generation COZH
(20 mol%) / CHZCIZ
60 C 3h
M, Q1 RS
2. Dioxane /AcOH I
HCI / 80 C R 4
3h
Q
[0128] In some embodiments, as shown in Scheme D, esterification of aryl
bromide E with
di-tert-butyl dicarbonate in a mixed solvent system such as THF/tert-butyl
alcohol (tBuOH),
followed by treatment with bis-pinacolato diboran in the presence of a
palladium (0) source
and a mild base such as potassium acetate for example in dioxane at
temperatures ranging
from room temperature to 80 C leads to boronate ester M. Tri-alkylated amine
0 was
obtained by successive treatment of a amine N with alkyl halides in a solvent
system such as
AcCN at room temperature or acetone under reflux conditions for a period of
time ranging
from 3-12 hours, in the presence of a mild base such as KZCO3 or Cs2CO3. A
standard cross
coupling between boronate ester M and alkenyll bromide 0 leads to ainine P in
the presence
of a palladium (0) source such as palladium tetrakis triphenylphosphine in a
solvent system
such as DME, an aqueous base such as sodium carbonate at temperatures ranging
from 25 to
90 C, for 2-13 hours. A ring cyclization, facilitated by Grubb's second
generation catalyst,
for exainple benzylidene [1,3-bis(2,4,6-trimethylphenyl)-2-
imidazolidinylidene]di(chloro(tricyclohexylphosphine) ruthenium in a solvent
such as DCM
at temperatures varying from 25 to 60 C, for a period of 1 to 6 hours leads
to a seven-
membered. Saponification under acidic conditions, using hydrochloric acid in
dioxane for a
period of 2 to 10 hours at temperatures close to reflux leads to heterocycle
Q.
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
Analysis of the Compounds
[0129] In yet another aspect, the invention includes methods to evaluate
putative specific
agonists or antagonists of DP-2 and/or one or more other PGD2 receptors.
Accordingly, the
invention is directed to the use of these compounds in the preparation and
execution of
screening assays for compounds which modulate the function of DP-2 and/or one
or more
other PGD2 receptors. For example, the compounds of this invention are useful
for DP-2
mutants and/or one or more other PGD2 receptor mutants, which are excellent
screening tools
for potent compounds. Furthermore, the compounds of this invention are useful
in
establishing or determining the binding site of other compounds to DP-2 and/or
one or more
other PGD2 receptors, e.g., by competitive inhibition. The compounds of the
instant invention
are also useful for the evaluation of putative specific modulators of DP-2
over one or more
other PGD2 receptors. One of skill in the art will appreciate that thorough
evaluation of
specific antagonists of PGD2 receptors has been hampered by the lack of
availability of
specific, non-peptidyl (metabolically resistant) compounds with high binding
affinity for
these receptors. The compounds provided herein are particularly useful in this
context.
[0130] The above and other assays described herein are designed to be amenable
to a high
throughput format to detect or quantify the presence, absence, quantification,
or other
properties of particular compounds individually or as library containing a
large number of
potential therapeutic compounds (potential modulator compounds). Any of the
assay steps
may be automated and compounds from any convenient source may be provided to
the assay.
Assays are typically run in parallel (e.g., in microtiter formats on
microtiter plates in robotic
assays). Preferred assays detect enhancement or inhibition of DP-2, DP-2
and/or one or more
other PGD2 receptors function.
[0131] High throughput screening systems are commercially available (see e.g.,
Zymark
Corp., Hopkinton Mass.; Air Technical Industries, Mentor Ohio; Beckman
Instruments, Inc.,
Fullerton Calif.; Precision Systems, Inc., Natick Mass.; etc.). These systems
typically
automate entire procedures, including all sample and reagent pipetting, liquid
dispensing,
timed incubations, and final readings of the microplate in detector(s)
appropriate for the
assay. These configurable systems provide high throughput and rapid start-up
as well as a
high degree of flexibility and customization. The manufacturers of such
systems provide
detailed protocols for various high throughput systems. Thus, for example,
Zymark Corp.
36
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
provides technical bulletins describing screening systems for detecting the
modulation of
gene transcription, ligand binding, and the like.
Methods of Use
[0132] The present invention relates to the identification of phenylacetic
acid derivatives
and their use as functional antagonists of the DP-2 receptor for the treatment
of conditions or
disorders mediated by PGD2, to pharmaceutical compositions containing these
derivatives
and to processes for their preparation.
[0133] In particular, compounds and derivatives of the general formula I have
activity as
modulators of DP-2 receptor activity, and therefore may be used in the
treatment of
conditions or disorders which are caused by the excessive, unbalanced or
deregulated
expression of PGD2 and its metabolites. Non-limiting example of such
conditions and
disorders include:
[0134] 1) Respiratory system conditions or disorders such as Obstructive
airway
diseases such as: asthma, e.g., intermittent and persistent asthma, extrinsic
(allergic) asthma,
intrinsic (non-allergic) asthma, mixed extrinsic-intrinsic asthma, exercise
induced asthma,
nocturnal asthma, bronchial asthma, seasonal asthma, occupational asthma,
cough variant
asthma, chronic severe corticosteroid-dependent asthma, steroid-resistant
asthma, allergic
bronchopulmonary aspergillosis, asthma triad (including asthma nasal polyps,
and aspirin
sensitivity), and allergic airway syndrome; bronchitis, e.g., acute and
chronic bronchitis,
allergic rhinobronchitis, eosinophilic bronchitis, and chronic obstructive
pulmonary disease
(COPD)); rhinitis, including acute and chronic rhinitis, atrophic rhinitis,
allergic and non-
allergic rhinitis, seasonal (e.g., rhinitis nervosa, hay fever, and vasoinotor
rhinitis), perennial,
and vasomotor rhinitis, nasal polyposis, nasal congestion, rhinitis
medicamentosa;
sarcoidosis; fanners lung and related diseases; fibroid lung; cystic fibrosis;
idiopathic
interstitial fibrosis; chronic cough associated with inflammation; and
sinusitis, e.g., allergic,
acute, sub-acute, and chronic sinusitis;
[0135] 2) Skin and Eyes conditions or disorders such as dermatitis, e.g.,
allergic contact
dermatitis, atopic dermatitis (eczema), contact (and irritant contact) det-
inatitis, excematous
dennatitis, neurodermatitis, perioral dermatitis, seborrheic dennatitis,
statsis dennatitis,
diaper dermatitis, dyshidrotic dennatitis (pompholyx), nummular dennatitis,
37
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
autosenstitization dermatitis, lichen simplex chronicus, and urticaria;
conjunctivitis, e.g.,
viral, allergic, bacterial, and chemical/toxic conjunctivitis; psoriasis;
urticaria; erythemas;
cutaneous eosinophilia; and chronic skin ulcers;
[0136] 3) Gastrointestinal System conditions or disorders such as food-induced
allergies
(e.g., those that have effects remote form the gut such as migraine, rhinitis
and eczema);
eosinophilic gastroenteritis; mastocytosis; ulcerative colitis; Crohn's
disease; irritable bowel
syndrome; celiac disease;
[0137] 4) Central nervous system conditions or disorders such asinflammatory
pain,
neuropathic pain;
[0138] 5) Conditions or disorders relating to other systems: e.g.,
eosinophilis fascitis;
hyper IgE syndrome; systemic mast cell disorder; Idopathic thrombocytopenia
purpura;
atherosclerosis; lupus erythematosus; systemic lupus erythematosus; sepsis;
reperfusion
injury; glomeruloephritis; allergic nephritis; nephritic syndrome; eosinophil
related disorders
such as Churg-Strauss syndrome; basophilic leukocytosis and basophilic
leukemia and
acquired immunodeficiency syndrome;
[0139] 6) Conditions or disorders relating to skeletal and joints systems,
e.g., arthritis
and conditions associated therewith, e.g., osteoarthritis (OA), osteonecrosis,
psoriatic
arthritis, Reiter's syndrome (reactive arthritis), tendonitis, bursitis,
inflammation of joint
lining, ankylosing spondylitis, Behcet's disease, childhood arthritis, diffuse
idiopathic
skeletal hyperostosis (DISH), Ehlers-Danlos syndrome, rheumatoid arthritis,
Felty's
syndrome, fibromyalgia, gout, pseudo gout, infectious arthritis, lupus, mixed
connective
tissue disease, osteoarthritis, Paget's disease, polyinyalgia rheuinatica,
polyarteritis nodossa,
Wegener's Granulomatosis, myositis (polymyositis dermatomyositis), psoriatic
arthritis,
Raynoud's phenomenon, and Still's disease;
[0140] 7) Autoimmune conditions or disorders, e.g., systemic lupus
erythematosis, anti-
phospholipid syndrome, rheumatoid arthritis, Sjogren's syndrome, scleroderma,
systemic
vasculitis, e.g., giant cell (temporal) arteritis, takayasu's arteritis,
polyarteritis nodosa,
Kawasaki disease, Wegner's granuloinatosis, Churg Strauss syndrome,
microscopic
polyangiitis, Henoch-Schonlein purpura, essential cryoglobulinemic vasculitis,
cutaneous
lekocytoclastic angiitis, autoimmune hemolytic anemia, idiopathic
thrombocytopenic
purpura, autoimmune neutropenia, Diabetes millitus, Hashimoto's disease,
Grave's disease,
autoimmune polyglandular syndromes, multiple sclerosis, myathenia gravis,
Behcet's
38
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
syndrome, pernicious anemia, primary biliary sclerosis, autoimmune hepatitis,
autoimmune
myocarditis, Goodpasture's syndrome, glomerular nephritis, and
tubulointerstitial nephritis;
and
[0141] 8) Other conditions or disorders associated with raised levels of PGD2
or its
metabolites.
[0142] In yet another aspect, the invention provides methods of treating or
preventing a
disorder or condition associated with DP-2 and/or one or more other PGD2
receptors by
administering to a subject having such a condition or disorder, a
therapeutically effective
amount of a compound or composition of the invention. In one group of
embodiments,
disorders and conditions, including chronic conditions and disorders of humans
or other
species, can be treated with modulators, or antagonists, of DP-2 and/or one or
more other
PGD2 receptors. These disorders and conditions include (1) inflammatory or
allergic diseases
such as systemic anaphylaxis and hypersensitivity disorders, atopic
dermatitis, urticaria, drug
allergies, insect sting allergies, food allergies (including celiac disease
and the like) and
mastocytosis, (2) inflammatory bowel diseases such as Crohn's disease,
ulcerative colitis,
ileitis and enteritis, (3) vasculitis, Behcet's syndrome, (4) psoriasis and
inflammatory
dermatoses such as dermatitis, eczema, atopic dermatitis, allergic contact
dermatitis, urticaria,
viral cutaneous pathologies such as those derived from human papillomavirus,
HIV or RLV
infection, bacterial, fungal and other parasital cutaneous pathologies, and
cutaneous lupus
erythematosus, (5) asthma and respiratory allergic diseases such as allergic
asthma, allergic
rhinitis, otitis media, allergic conjunctivitis, hypersensitivity lung
diseases, chronic
obstructive pulmonary disease and the like, (6) autoimmune diseases, such as
arthritis
(including rheumatoid and psoriatic), systemic lupus erythematosus, type I
diabetes,
myasthenia gravis, multiple sclerosis, Graves' disease, glomerulonephritis,
sclerodenna,
including, e.g., systemic sclerodenna, fasciitis, including, e.g.,
eosinophilia fasciitis
(Schulman's syndrome), Sjogren's syndrome, hyper IgE syndrome, soft tissue
disease, and
inflammatory myopathies and the like, (7) graft rejection (including, e.g.,
allograft rejection
and graft-v-host disease), e.g., skin graft rejection, solid organ transplant
rejection, bone
marrow transplant rejection, (8) fever, (9) cardiovascular disorders such as
acute heart failure,
hypotension, hypertension, angina pectoris, myocardial infaretion,
cardiomyopathy,
congestive heart failure, atherosclerosis, coronary artery disease,
restenosis, thrombosis and
vascular stenosis, (10) cerebrovascular disorders such as traumatic brain
injury, stroke,
ischemic reperfusion injury and aneurysm, (11) cancers of the breast, skin,
prostate, cervix,
39
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
uterus, ovary, testes, bladder, lung, liver, larynx, oral cavity, colon and
gastrointestinal tract
(e.g., esophagus, stomach, pancreas), brain, thyroid, blood and lymphatic
system, (12)
fibrosis, connective tissue disease and sarcoidosis, (13) genital and
reproductive conditions
such as erectile dysfunction, (14) gastrointestinal disorders such as
gastritis, ulcers, nausea,
pancreatitis and vomiting; (15) neurologic disorders, such as Alzheimer's
disease, (16) sleep
disorders such as insomnia, narcolepsy, sleep apnea syndrome and Pickwick
Syndrome, (17)
pain, (18) renal disorders, (19) ocular disorders such as glaucoma, (20)
infectious diseases,
viral infections such as HIV, and bacterial infections such as sepsis, (21)
inflammation, (22)
flushing and (23) nasal congestion.
[0143] In yet another aspect, the invention provides methods of treating or
preventing a
condition or disorder mediated, regulated or influenced by Th2 cells,
eosinophils, basophils,
platelets, Langerhans cells, dendritic cells or mast cells, comprising
administering to a subject
having such as condition or disorder a therapeutically effective amount of one
or more of the
subject compounds or compositions.
[0144] In yet another aspect, the invention provides methods of treating or
preventing a
condition or disorder mediated, regulated or influenced by PGD2 and
metabolites thereof,
such as 13,14-dihydro-15-keto-PGDZ and 15-deoxy-A12 14PGJ2, comprising
administering to a
subject having such as condition or disorder a therapeutically effective
amount of one or
more of the subject compounds or compositions.
[0145] In yet another aspect, the invention provides methods of treating or
preventing a
condition or disorder responsive to modulation of DP-2 and/or one or more
other PGD2
receptors comprising administering to a subject having such a condition or
disorder, a
therapeutically effective amount of one or more of the subject compounds or
compositions.
[0146] In yet another aspect, the invention provides methods of treating or
preventing a
condition or disorder mediated by DP-2 and/or one or more other PGD2 receptors
comprising
administering to a subject having such a condition or disorder, a
therapeutically effective
amount of one or more of the subject compounds or compositions.
[0147] In yet another aspect, the invention provides methods of modulating DP-
2 and/or
one or more other PGD2 receptors comprising contacting a cell with one or more
of the
subject compounds or compositions.
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
[0148] Depending on the disorder to be treated and the subject's condition,
the compounds
of the invention may be administered by oral, parenteral (e.g., intramuscular,
intraperitoneal,
intravenous, ICV, intracisternal injection or infusion, subcutaneous injection
or implant),
inhalation, nasal, vaginal, rectal, sublingual, or topical (e.g., transdermal,
local) routes of
administration and may be formulated, alone or together, in suitable dosage
unit formulations
containing conventional non-toxic pharmaceutically acceptable carriers,
adjuvants and
vehicles appropriate for each route of administration. The invention also
contemplates
administration of the compounds of the invention in a depot formulation, in
which the active
ingredient is released over a defined time period.
[0149] In the treatment or prevention of various conditions and disorders
according to the
invention associated with DP-2 and/or one or more other PGD2 receptors, an
appropriate
dosage level will generally be about 0.001 to 100 mg per kg patient body
weight per day
which can be administered in single or multiple doses. Preferably, the dosage
level will be
about 0.01 to about 25 mg/kg per day; more preferably about 0.05 to about 10
mg/kg per day.
A suitable dosage level may be about 0.01 to 25 mg/kg per day, about 0.05 to
10 mg/kg per
day, or about 0.1 to 5 mg/kg per day. Within this range the dosage may be
0.005 to 0.05, 0.05
to 0.5 or 0.5 to 5.0 mg/kg per day. For oral administration, the compositions
are preferably
provided in the form of tablets containing 1.0 to 1000 milligrams of the
active ingredient,
particularly 1.0, 5.0, 10.0, 15Ø 20.0, 25.0, 50.0, 75.0, 100.0, 150.0,
200.0, 250.0, 300.0,
400.0, 500.0, 600.0, 750.0, 800.0, 900.0, and 1000.0 milligrains of the active
ingredient for
the syinptomatic adjustment of the dosage to the patient to be treated. The
compounds may be
administered on a regimen of 1 to 4 times per day, preferably once or twice
per day.
[0150] It will be understood, however, that the specific dose level and
frequency of dosage
for any particular patient may be varied and will depend upon a variety of
factors including
the activity of the specific compound employed, the metabolic stability and
length of action
of that compound, the age, body weight, general health, sex, diet, mode and
time of
administration, rate of excretion, drug combination, the severity of the
particular condition,
and the host undergoing therapy.
Compositions
[0151] In another aspect, the invention provides phannaceutical compositions
suitable for
pharinaceutical use comprising one or more compounds of the invention and a
41
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
pharmaceutically acceptable carrier, excipient or diluent. The term
"composition" as used
herein is intended to encompass a product comprising the specified ingredients
(and in the
specified amounts, if indicated), as well as any product which results,
directly or indirectly,
from combination of the specified ingredients in the specified amounts. By
"pharmaceutically
acceptable" it is meant that the carrier or excipient is compatible with the
other ingredients of
the formulation and not deleterious to the recipient thereof.
[0152] Formulation may improve one or more pharmacokinetic properties (e.g.,
oral
bioavailabilty, membrane permeability) of a compound of the invention (herein
referred to as
the active ingredient).
[0153] The pharmaceutical compositions for the administration of the compounds
of this
invention may conveniently be presented in unit dosage form and may be
prepared by any of
the methods well known in the art. All methods include the step of bringing
the active
ingredient into association with the can-ier which constitutes one or more
accessory
ingredients. In general, the pharmaceutical compositions are prepared by
uniformly and
intimately bringing the active ingredient into association with a liquid
carrier or a finely
divided solid carrier or both, and then, if necessary, shaping the product
into the desired
formulation. In the pharmaceutical composition the active object compound is
included in an
amount sufficient to produce the desired effect upon the process, condition or
disorder.
[0154] The pharmaceutical compositions containing the active ingredient may be
in a form
suitable for oral use, for example, as tablets, troches, lozenges, aqueous or
oily suspensions,
dispersible powders or granules, emulsions, hard or soft capsules, or syrups
or elixirs.
Compositions intended for oral use may be prepared according to any method
known to the
art for the manufacture of pharmaceutical compositions. Such compositions may
contain one
or more agents selected from sweetening agents, flavoring agents, coloring
agents and
preserving agents in order to provide phannaceutically elegant and palatable
preparations.
Tablets contain the active ingredient in admixture with other non-toxic
pharmaceutically
acceptable excipients which are suitable for the manufacture of tablets. These
excipients may
be, for example, inert diluents, such as calcium carbonate, sodium carbonate,
lactose, calcium
phosphate or sodiuin phosphate; granulating and disintegrating agents, for
exainple, corn
starch, or alginic acid; binding agents, for example starch, gelatin or
acacia, and lubricating
agents, for example magnesium stearate, stearic acid or talc. The tablets may
be uncoated or
they may be coated by known techniques to delay disintegration and absorption
in the
42
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
gastrointestinal tract and thereby provide a sustained action over a longer
period. For
example, a time delay material such as glyceryl monostearate or glyceryl
distearate may be
employed. They may also be coated by the techniques described in U.S. Pat.
Nos. 4,256,108;
4,166,452 and 4,265,874 to form osmotic therapeutic tablets for control
release.
[0155] Formulations for oral use may also be presented as hard gelatin
capsules wherein
the active ingredient is mixed with an inert solid diluent, for example,
calcium carbonate,
calcium phosphate or kaolin, or as soft gelatin capsules wherein the active
ingredient is
mixed with water or an oil-medium, for example peanut oil, liquid paraffin, or
olive oil.
[0156] Aqueous suspensions contain the active materials in admixture with
excipients
suitable for the manufacture of aqueous suspensions. Such excipients are
suspending agents,
for example sodium carboxymethylcellulose, methylcellulose,
hydroxy-propylmethylcellulose, sodium alginate, polyvinyl-pyrrolidone, gum
tragacanth and
gum acacia; dispersing or wetting agents may be a naturally-occurring
phosphatide, for
example lecithin, or condensation products of an alkylene oxide with fatty
acids, for example
polyoxy-ethylene stearate, or condensation products of ethylene oxide with
long chain
aliphatic alcohols, for example heptadecaethyleneoxycetanol, or condensation
products of
ethylene oxide with partial esters derived from fatty acids and a hexitol such
as
polyoxyethylene sorbitol monooleate, or condensation products of ethylene
oxide with partial
esters derived from fatty acids and hexitol anhydrides, for example
polyethylene sorbitan
monooleate. The aqueous suspensions may also contain one or more
preservatives, for
example ethyl, or n-propyl, p-hydroxybenzoate, one or more coloring agents,
one or more
flavoring agents, and one or more sweetening agents, such as sucrose or
saccharin.
[0157] Oily suspensions may be fonnulated by suspending the active ingredient
in a
vegetable oil, for example arachis oil, olive oil, sesaine oil or coconut oil,
or in a mineral oil
such as liquid paraffin. The oily suspensions may contain a thickening agent,
for
example-beeswax, hard paraffin or cetyl alcohol. Sweetening agents such as
those set forth
above, and flavoring agents may be added to provide a palatable oral
preparation. These
compositions may be preserved by the addition of an anti-oxidant such as
ascorbic acid.
[0158] Dispersible powders and granules suitable for preparation of an aqueous
suspension
by the addition of water provide the active ingredient in admixture with a
dispersing or
wetting agent, suspending agent and one or more preservatives. Suitable
dispersing or wetting
43
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
agents and suspending agents are exemplified by those already mentioned above.
Additional
excipients, for example sweetening, flavoring and coloring agents, may also be
present.
[0159] The pharmaceutical compositions of the invention may also be in the
form of
oil-in-water emulsions. The oily phase may be a vegetable oil, for example
olive oil or
arachis oil, or a mineral oil, for example liquid paraffin or mixtures of
these. Suitable
emulsifying agents may be naturally-occurring gums, for example gum acacia or
gum
tragacanth, naturally-occurring phosphatides, for example soy bean, lecithin,
and esters or
partial esters derived from fatty acids and hexitol anhydrides, for example
sorbitan
monooleate, and condensation products of the said partial esters with ethylene
oxide, for
example polyoxyethylene sorbitan monooleate. The emulsions may also contain
sweetening
and flavoring agents.
[0160] Syrups and elixirs may be formulated with sweetening agents, for
example glycerol,
propylene glycol, sorbitol or sucrose. Such formulations may also contain a
demulcent, a
preservative and flavoring and coloring agents.
[0161] The pharmaceutical compositions may be in the form of a sterile
injectable aqueous
or oleagenous suspension. This suspension may be formulated according to the
known art
using those suitable dispersing or wetting agents and suspending agents which
have been
mentioned above. The sterile injectable preparation may also be a sterile
injectable solution
or suspension in a non-toxic parenterally acceptable diluent or solvent, for
example as a
solution in 1,3-butane diol. Among the acceptable vehicles and solvents that
may be
employed are water, Ringer's solution and isotonic sodium chloride solution.
In addition,
sterile, fixed oils are conventionally employed as a solvent or suspending
medium. For this
purpose any bland fixed oil may be employed including synthetic mono- or
diglycerides. In
addition, fatty acids such as oleic acid find use in the preparation of
injectables.
[0162] The phannaceutical compositions may also be administered in the form of
suppositories for rectal administration of the drug. These compositions can be
prepared by
mixing the drug with a suitable non-irritating excipient which is solid at
ordinary
temperatures but liquid at the rectal temperature and will therefore melt in
the rectum to
release the drug. Such materials are cocoa butter and polyethylene glycols.
[0163] For topical use, creams, ointments, jellies, solutions or suspensions,
etc., containing
the compounds of the invention are employed. As used herein, topical
application is also
meant to include the use of mouthwashes and gargles.
44
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
Pulmonary Administration
Inhalable Powder
[0164] In some embodiments, the agents are administered directly to the lung
by inhalation.
Accordingly, the agents for use according to the invention may be formulated
as inhalable
powders in admixture with suitable physiologically acceptable excipients (see,
U.S. Patent
Publication No. 20060034776 which is incorporated herein by reference with
respect to
suitable methods of administering pharmaceutical agents by inhalation).
[0165] For aerosol delivery in humans or other primates and mammals, the
aerosol is
generated by a medical nebulizer system that delivers the aerosol through a
mouthpiece,
facemask, etc. from which the mammalian host can draw the aerosol into the
lungs. Various
nebulizers are known in the art and can be used in the method of the present
invention. The
selection of a nebulizer system depends on whether alveolar or airway delivery
(i.e., trachea,
primary, secondary or tertiary bronchi, etc.), is desired. The composition is
formulated as to
not be too irritating at the required dosage.
[0166] Nebulizers useful for airway delivery include those typically used in
the treatment
of asthma. Such nebulizers are also commercially available. A therapeutic
amount of the
agent is a sufficient amount to prevent, treat, or palliate asthma following
administration of
the composition to the host mammal's lung, particularly the alveoli or
bronchopulmonary and
bronchiolopulmonary smooth muscle and epithelial cells of the trachea,
bronchi, bronchia,
bronchioli, and alveoli. Thus, an effective amount of the aerosolized compound
of the
invention, is a dose sufficient to effect treatment, that is, to cause
alleviation or reduction of
symptoms, to inhibit the worsening of symptoms, to prevent the onset of
symptoms, and the
like. The dosages of the preset compositions that constitute an effective
amount can be
determined in view of this disclosure by one of ordinary skill in the art by
running routine
trials with appropriate controls. Comparison of the appropriate treatment
groups to the
controls will indicate whether a particular dosage is effective in preventing
or reducing
particular symptoms.
[0167] The total amount of compound delivered to a mammalian host will depend
upon
many factors, including the total amount aerosolized, the type of nebulizer,
the particle size,
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
breathing patterns of the mammalian host, severity of lung disease,
concentration of the
compound composition in the aerosolized solution, and length of inhalation
therapy.
[0168] Despite the interacting factors described above, one of ordinary skill
in the art will
be able readily to design effective protocols, particularly if the particle
size of the aerosol is
optimized. Based on estimates of nebulizer efficiency, an effective dose
delivered usually
lies in the range of about 1 mg/treatment to about 500 mg/treatment, although
more or less
may be found to be effective depending on the subject, agent, dosage regimen,
and desired
result. It is generally desirable to administer higher doses when treating
more severe
conditions. If the treatment is repeated, the mammalian host can be monitored
to ensure that
there is no adverse response to the treatment. The frequency of treatments
depends upon a
number of factors, such as the amount of the agent administered per dose, as
well as the
health and history of the subject.
Propellant Gas-Driven Inhalation Aerosols
[0169] Inhalation aerosols containing propellant gas according to the
invention may contain
the agents for use according to the invention dissolved in the propellant gas
or in dispersed
form. The propellant gases which may be used to prepare the inhalation
aerosols according
to the invention are known from the prior art. Suitable propellant gases are
selected from
among hydrocarbons such as n-propane, n-butane or isobutane and
halohydrocarbons such as
fluorinated derivatives of methane, ethane, propane, butane, cyclopropane or
cyclobutane.
The propellant gases mentioned above may be used on their own or in mixtures
thereof.
Particularly preferred propellant gases are halogenated alkane derivatives
selected from
TG134a, TG227, and mixtures thereof. The propellant-driven inhalation aerosols
according
to the invention may also contain other ingredients such as cosolvents,
stabilizers, surfactants,
antioxidants, lubricants, preservatives and pH adjusters. All these
ingredients are known in
the art. When in dispersed fonn, the agents can, for instance, be formulated
to have an
average particle size of up to 10 microns or preferably from 0.1 to 5 microns,
or from 1 to 5
microns.
[0170] The propellant-driven inhalation aerosols according to the invention
mentioned
above may be administered using inhalers known in the art, such as metered
dose inhalers.
Accordingly, in another aspect, the present invention relates to
phannaceutical compositions
46
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
in the form of propellant gas-containing aerosols as hereinbefore described
combined with
one or more inhalers suitable for administering these aerosols.
C. Propellant-Free Inhalable Solutions or Suspensions
[0171] Propellant-free inhalable solutions and suspensions of the agents for
use according
to the invention are contemplated. The solvent used may be an aqueous or
alcoholic,
preferably an ethanolic solution. The solvent may be water on its own or a
mixture of water
and ethanol. The relative proportion of ethanol compared with water is not
limited but the
maximum is up to 70 percent by volume, more particularly up to 60 percent by
volume and
most preferably up to 30 percent by volume. The remainder of the volume is
made up of
water.
Combination Therapy
[0172] The pharmaceutical compositions and methods of the invention may
further
comprise other therapeutically active compounds, as noted herein, useful in
the treatment of
asthma, allergic diseases, inflammatory conditions and cancer and pathologies
associated
therewith (e.g., cardiovascular disease) or other adjuvant. In many instances,
compositions
which include a compounds of the invention and an alternative agent have
additive or
synergistic effects when administered.
[0173] The compounds of the invention can be combined or used in combination
with other
agents useful in the treatment, prevention, suppression or ainelioration of
the disorder or
conditions for which compounds of the invention are useful, including
inflainmatory
conditions, immune disorders, asthma, allergic rhinitis, eczema, psoriasis,
atopic dennatitis,
fever, sepsis, systemic lupus erytheinatosus, diabetes, rheumatoid arthritis,
multiple sclerosis,
atherosclerosis, transplant rejection, inflainmatory bowel disease, cancer,
viral infection,
thrombosis, fibrosis, flushing, Crohn's disease, ulcerative colitis, chronic
obstructive
pulmonary disease, inflammation, pain, conjunctivitis, nasal congestion,
urticaria and those
pathologies noted above.
[0174] Such other agents, or drugs, may be administered, by a route and in an
amount
commonly used therefor, simultaneously or sequentially with a compound of the
invention.
47
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
When a compound of the invention is used contemporaneously with one or more
other drugs,
a pharmaceutical composition containing such other drugs in addition to the
compound of the
invention is preferred. Accordingly, the pharmaceutical compositions of the
invention include
those that also contain one or more other active ingredients or therapeutic
agents, in addition
to a compound of the invention.
[0175] Examples of other therapeutic agents that may be combined with a
compound of the
invention, either administered separately or in the same pharmaceutical
compositions,
include, but are not limited to: (a) VLA-4 antagonists, (b) corticosteroids,
such as
beclomethasone, methylprednisolone, betamethasone, prednisone, prenisolone,
triamcinolone, dexamethasone, fluticasone, flunisolide and hydrocortisone, and
corticosteroid
analogs such as budesonide; (c) immunosuppressants such as cyclosporine
(cyclosporine A,
Sandimmune , Neoral ), tacrolimus (FK-506, Prografo), rapamycin (sirolimus,
Rapamune )
and other FK-506 type immunosuppressants, and mycophenolate, e.g.,
mycophenolate
mofetil (CellCept ); (d) antihistamines (H1-histamine antagonists) such as
bromopheniramine, chlorpheniramine, dexchlorpheniramine, triprolidine,
clemastine,
diphenhydramine, diphenylpyraline, tripelennamine, hydroxyzine, methdilazine,
promethazine, trimeprazine, azatadine, cyproheptadine, antazoline,
pheniramine, pyrilamine,
astemizole, terfenadine, loratadine, cetirizine, fexofenadine,
descarboethoxyloratadine, and
the like; (e) non-steroidal anti-asthmatics such as (32-agonists (e.g.,
terbutaline,
metaproterenol, fenoterol, isoetharine, albuterol, salmeterol, bitolterol and
pirbuterol) and
(32-agonist-corticosteroid combinations (e.g., salmeterol-fluticasone (Advair
),
fonnoterol-budesonid (Symbicort )), theophylline, cromolyn, cromolyn sodium,
nedocromil,
atropine, ipratropium, ipratropiuin bromide, leukotriene antagonists (e.g.,
zafirlukast,
montelukast, montelukast sodium (Singulair ), pranlukast, iralukast,
pobilukast and
SKB-106,203), leukotriene biosynthesis inhibitors (zileuton, BAY-1005); (f)
non-steroidal
antiinflammatory agents (NSAIDs) such as propionic acid derivatives (e.g.,
alminoprofen,
benoxaprofen, bucloxic acid, carprofen, fenbufen, fenoprofen, fluprofen,
flurbiprofen,
ibuprofen, indoprofen, ketoprofen, miroprofen, naproxen, oxaprozin, pirprofen,
pranoprofen,
suprofen, tiaprofenic acid and tioxaprofen), acetic acid derivatives (e.g.,
indomethacin,
acemetacin, alclofenac, clidanac, diclofenac, fenclofenac, fenclozic acid,
fentiazac, furofenac,
ibufenac, isoxepac, oxpinac, sulindac, tiopinac, tolmetin, zidometacin and
zomepirac),
fenamic acid derivatives (e.g., flufenanic acid, meclofenamic acid, mefenamic
acid, niflumic
acid and tolfenainic acid), biphenylcarboxylic acid derivatives (e.g.,
diflunisal and flufenisal),
48
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
oxicams (e.g., isoxicam, piroxicam, sudoxicam and tenoxican), salicylates
(e.g., acetyl
salicylic acid and sulfasalazine) and the pyrazolones (e.g., apazone,
bezpiperylon, feprazone,
mofebutazone, oxyphenbutazone and phenylbutazone); (g) cyclooxygenase-2 (COX-
2)
inhibitors such as celecoxib (Celebrex ) and rofecoxib (Vioxx ); (h)
inhibitors of
phosphodiesterase type IV (PDE-IV); (i) other PGD2 receptor antagonists,
especially DP-1
antagonists; (j) opioid analgesics such as codeine, fentanyl, hydromorphone,
levorphanol,
meperidine, methadone, morphine, oxycodone, oxymorphone, propoxyphene,
buprenorphine,
butorphanol, dezocine, nalbuphine and pentazocine; (k) cholesterol lowering
agents such as
HMG-CoA reductase inhibitors (e.g., lovastatin, simvastatin, pravastatin,
fluvastatin,
atorvastatin and other statins), bile acid sequestrants (e.g., cholestyramine
and colestipol),
vitamin B3 (also known as nicotinic acid, or niacin), vitamin B6 (pyridoxine),
vitamin B12
(cyanocobalamin), fibric acid derivatives (e.g., gemfibrozil, clofibrate,
fenofibrate and
benzafibrate), probucol, nitroglycerin, and inhibitors of cholesterol
absorption (e.g.,
beta-sitosterol and acylCoA-cholesterol acyltransferase (ACAT) inhibitors such
as
melinamide), HMG-CoA synthase inhibitors, squalene epoxidase inhibitors and
squalene
synthetase inhibitors; (1) antithrombotic agents, such as thrombolytic agents
(e.g.,
streptokinase, alteplase, anistreplase and reteplase), heparin, hirudin and
warfarin derivatives,
0-blockers (e.g., atenolol), 0-adrenergic agonists (e.g., isoproterenol), ACE
inhibitors and
vasodilators (e.g., sodium nitroprusside, nicardipine hydrochloride,
nitroglycerin and
enaloprilat); (m) anti-diabetic agents such as insulin and insulin mimetics,
sulfonylureas (e.g.,
glyburide, meglinatide), biguanides, e.g., metformin (Glucophage ), a-
glucosidase inhibitors
(acarbose), thiazolidinone compounds, e.g., rosiglitazone (AvandiaR),
troglitazone
(Rezulino), ciglitazone, pioglitazone (Actos") and englitazone; (n)
preparations of interferon
beta (interferon (3 -1 a, interferon (3 -1 (3); (0) gold compounds such as
auranofin and
aurothioglucose, (p) TNF inhibitors, e.g., etanercept (Enbrel~), antibody
therapies such as
orthoclone (OKT3), daclizumab (Zenapax ), basiliximab (Simulect ), infliximab
(Remicade ) and D2E6 TNF antibody, (q) lubricants or emollients such as
petrolatum and
lanolin, keratolytic agents, vitamin D3 derivatives (e.g., calcipotriene and
calcipotriol
(Dovonex )), PUVA, anthralin (Drithrocreme ), etretinate (Tegison ) and
isotretinoin; (r)
multiple sclerosis therapeutic agents such as interferon (3 -1 (3 (Betaseron
), interferon (3 -1 a
(Avonex ), azathioprine (Imurek , Imuran ), glatiramer acetate (Capoxone ), a
glucocorticoid (e.g., prednisolone) and cyclophosphainide; (s) other compounds
such as
5-aminosalicylic acid and prodrugs thereof; (t) DNA-alkylating agents (e.g.,
49
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
cyclophosphamide, ifosfamide), antimetabolites (e.g., azathioprine, 6-
mercaptopurine,
methotrexate, a folate antagonist, and 5-fluorouracil, a pyrimidine
antagonist), microtubule
disruptors (e.g., vincristine, vinblastine, paclitaxel, colchicine, nocodazole
and vinorelbine),
DNA intercalators (e.g., doxorubicin, daunomycin and cisplatin), DNA synthesis
inhibitors
such as hydroxyurea, DNA cross-linking agents, e.g., mitomycin C, hormone
therapy (e.g.,
tamoxifen, and flutamide), cytostatic agents, e.g., imatinib (ST1571, Gleevec
) and rituximab
(Rituxan ), 5-lipoxygenase activating protein (FLAP) inhibitors, and PLA2
inhibitors. The
weight ratio of the compound of the invention to the second active ingredient
may be varied
and will depend upon the effective dose of each ingredient. Generally, an
effective dose of
each will be used. Thus, for example, when a compound of the invention is
combined with an
NSAID, the weight ratio of the compound of the invention to the NSAID will
generally range
from about 1000:1 to about 1:1000, preferably about 200:1 to about 1:200.
Combinations of a
compound of the invention and other active ingredients will generally also be
within the
aforementioned range, but in each case, an effective dose of each active
ingredient should be
used.
Diagnosis of Asthma
[0176] Methods of diagnosing asthma and other respiratory inflammatory and
obstructive
disorders or conditions are well known to persons of ordinary skill in the
art. For exainple,
spirometry can be used to assess lung function. The diagnosis of asthma, in
particular, may
be made in part based upon fainily history or personal history of a severe and
sudden episode
or recurrent episodes of wheezing, coughing or shortness of breath which may
be associated
with exposure to an allergen or exacerbated or precipitated by moderate
exercise. Typically a
physical exam is involved to detect the disorder or condition.
[0177] Using a nasal speculum, the nose may be examined for signs of allergic
disorder or
condition such as increased nasal secretions, swelling or polyps which may be
triggering
asthma. A stethoscope may be used to listen to the sounds the lungs make
during breathing.
Wheezing sounds are one of the main indicators of the obstructed airways
associated with
asthma. In addition, allergic conditions such as eczema or hives, are often
associated with
asthma.
[0178] Pulmonary function tests are particularly useful in confirming the
diagnosis of
respiratory disorders or conditions. These tests include spirometry to
determine vital
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
capacity, the maximum amount of air that you can inhale and exhale; the peak
expiratory
flow rate, also known as the peak flow rate, which is the maximum flow rate
you can
generate during a forced exhalation; and forced expiratory volume, which is
the maximum
amount of air you can exhale in one second.
[0179] If the measurements are below normal for a person your age, a
bronchodilator drug
used in asthma treatment can be administered to open obstructed air passages
and the
spirometry repeated. If the measurements improve significantly, asthma is
likely.
[0180] In addition, asthma may be diagnosed by challenging the individual with
exercise,
or by inhaling an airway-constricting chemical or taking several breaths of
cold air. After the
challenge with a symptom-producing substance or activity, the spirometry test
is
readministered. If the spirometry measurements fall significantly, asthma is
indicated.
[0181] The following examples are offered by way of illustration and are not
intended to
limit the scope of the invention. Those of skill in the art will readily
recognize a variety of
noncritical parameters that could be modified to yield essentially similar
results.
EXAMPLES
General methods:
[0182] The invention will now be illustrated by the following non-limiting
examples. The
title and sub-titled compounds of the examples and methods were named using
the
ChemDraw Ultra (version 7.0) from CambridgeSoft Inc. Flash column
chromatography
refers to noi-mal phase silica chromatography. Reagents and solvents used can
be obtained
from commercial sources such as Aldrich Chemical Co. (Milwaukee, Wis., USA).
Solvents
were dried with MgSO4 or Na2SO4. Evaporations were carried out by rotary
evaporation in
vacuo and work-up procedures were carried out after removal of residual solids
such as
drying agents by filtration. Unless otherwise stated, operations were carried
out at ambient
teinperature that is in the range 18-25 C and under an atmosphere of an inert
gas such as
argon or nitrogen. Yields are given for illustration only and are not
necessarily the maximum
attainable. The structures of the end-products of the structure (1) were
confirmed by nuclear
(generally proton) magnetic resonance PMR) and mass spectral techniques. I H-
NMR spectra
were recorded on a VarianTM 400 MHz NMR spectrometer. Proton magnetic
resonance
chemical shift values were measured on the delta scale, 8, in parts per
million (ppm).
51
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
Significant peaks are tabulated in the order: multiplicity (s, singlet; d,
doublet; t, triplet; q,
quartet; m, multiplet; br s, broad singlet), coupling constant(s) in Hertz
(Hz) and number of
protons. Intermediates were not generally fully characterised and purity was
assessed by thin
layer chromatography (TLC), high-performance liquid chromatography (BPLC),
mass
spectrometry (MS), infra-red (IR) or NMR analysis. Mass spectra were recorded
by one of
the three Liquid Chromatographic/Mass Spectrometry (LC/MS) methods:
Method A:
[0183] Run on an Agilent 1100 HPLC over a phenomenex Luna C18 3micron 30x2.Omm
id column at a flow rate of 0.300mL/min. The column, at 35 C, was eluted with
a gradient
comprised of increasing AcCN (modified with 0.05% formic acid) and water
(modified with
0.05% formic acid) as described in the table below. The analytes were
monitored at 214nm
and 254nm. The analytes were vaporized in an Agilent electrospray source
charged to 80V
and detected after passing through a single quadrupole.
Gradient
Time % organic Organic Solvent
0.0 10 AcCN
0.2 10 AcCN
3.8 95 AcCN
4.1 95 AcCN
4.4 10 AcCN
6.0 10 AcCN
Method B:
[0184] Run on an Agilent 1100 HPLC over a phenomenex luna C18 3micron 30x2.0mm
id
column at a flow rate of 0.300mL/min. The column, at 35 C, was eluted with a
gradient
comprised of increasing AcCN (modified with 0.05% fonnic acid) and water
(modified with
0.05% formic acid) as described in the table below. The analytes were
monitored at 214mn
and 254nm. The analytes were vaporized in an Agilent multi-mode source in
electrospray
mode charged to 80V and detected after passing through a single quadrupole.
Gradient
Time % organic Organic Solvent
0.0 10 AcCN
0.2 10 AcCN
3.8 95 AcCN
4.1 95 AcCN
4.4 10 AcCN
6.0 10 AcCN
52
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
Method C:
[0185] Run on an Agilent 1100 HPLC over a phenomenex Luna C18 3micron 30x2.Omm
id column at a flow rate of 0.300mL/min. The column, at 35 C, was eluted with
a gradient
comprised of increasing methanol (modified with 0.05% formic acid) and water
(modified
with 0.05% formic acid) as described in the table below. The analytes were
monitored at
214nm and 254nm. The analytes were vaporized in an Agilent multi-mode source
in
atmospheric chemical ionization mode charged to 80V and detected after passing
through a
single quadrupole.
Gradient
Time % organic Organic Solvent
0.0 35 methanol
0.2 35 methanol
3.6 98 methanol
4.1 98 methanol
4.4 35 methanol
6.0 35 methanol
Examples 1-3
[0186] A general procedure for the synthesis of phenyl piperidine series are
shown below
(Scheme 1).
Scheme 1
O Pd(PPh3)4 O Pt02 H2 MeO 1. SOZR5CI HO
MeO MeO 5
OH OSMeOH LiOH Br HOB ~ YS Y4 Y
I Y4
1A DME/2.0 M Na2CO3 1B, Y4 = N, Y5 = CH 1D, Y4 = NH, Y5 = CH2 1F, Y4 = NSOZR5,
1C, Y4 = CH, Y5 = N 1 E, Y4 = CH2, Y5 = NH Y5= CH2
1 G, Y4 = CH2, Y5
= NSO2R5
Example 1
2-(3-(1-(4-fluorophenylsulfonyl)piperidin-4-yl)phenyl)acetic acid (Compound
1G)
53
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
0
OH
F
N %`O
O
Methyl 2-(3-(pyridin-4-yl)phenyl)acetate (Compound 1B)
0
OMe
/N
[0187] 4 -Pyridine boronic acid (325 mg, 2.64 mmol) and palladium tetrakis
(140 mg,
0.12 mmol) were added to a stirring solution of inethyl2-(3-
bromophenyl)acetate (550 mg,
2.4 mmol) in dimethoxy ethane and 2 M Na2CO3 ( 2:1 mixture, 12 mL). The
resulting
suspension was refluxed for 3 h, cooled, and then diluted with ethyl acetate
(EtOAc) (10 mL).
The mixture was washed with H20 (20 mL) and the organic layers were dried over
Na2SO4
(s) and concentrated to give a yellow oil (630 mg). Flash chromatography (3:1
hexanes/EtOAc) afforded pure acetate 2 (221 mg, 41 %) as a clear oil: ES/MS,
calcd for
C14H14N02 228.1, found 228.1 (M+H).
Methyl 2-(3-(piperidin-4-yl)phenyl)acetate (Compound 1D)
O
OMe
NH
[0188] Pt02 (12 mg, 0.053 mmol) and conc. HCl (2 drops) were added to a
solution of 2
(120 mg, 0.53 mmol) in MeOH (5 mL). The resulting mixture was attached to Parr
shaker
and pressurized to 40 psi (H2) and shook for 1 h. Once the reaction is
completed the
54
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
suspension was filtered through CELITE and filter cake was washed with MeOH.
The
combined organic layer was concentrated to give crude piperidine 1D (76 mg) as
a clear oil.
The crude mixture was carried to the next step with no further purification:
ES/MS, calcd for
C14H2ONO2 234.1, found 234.1 (M+H).
Methyl 2-(3 -(1- (4-fluorophenylsulfonyl)piperidin-4-yl)phenyl) acetate
(Compound 1E).
0
0
F
os~'o
[0189] Hunig's base (0.515 mL, 2.96 mmol) and 4-fluorobenzenesulfonyl chloride
(210
mg, 1.08 mmol) were added to a stirring solution of 4 (230 mg, 0.986 mmol) in
CHZCIZ (5
mL) at RT. The resulting suspension was quenched with satd. NaHCO3 (20 mL)
after 15 h
and the aqueous layer was extracted with EtOAc (3 X 20 mL). The combined
organic layers
were washed with brine, dried over Na2SO4, and concentrated to give brown oil
(337 mg).
The chromatography (1:1 hexanes/EtOAc) afforded 6a as a clear oil (155 mg):
ES/MS, calcd
for C20H22FN04S 391.1, found 391.1 (M+H).
2-(3-(1-(4-fluorophenylsulfonyl)piperidin-4-yl)phenyl)acetic acid (Compound
1G)
0
OH
F
OS1O
[0190] Lithium hydroxide (160 mg, 3.83 mmol) was added to a stirring solution
of IE (150
mg, 0.383 mmol) in THF/MeOH/H20 (3:1:1, 5 mL) at RT. The resulting suspension
was
concentrated after 16 h then diluted back with H20 (10 mL). The aqueous was
washed with
ether then acidified with conc. HCI (pH >1). The white precipitate was
filtered (143 mg) and
purified with HPLC to give IG (29 mg); 'H NMR (400 MHz, DMSO-d6) S 12.28 (1H,
bs)
7.86 (2H, m) 7.52 (2H, t, J= 8.4 Hz) 7.22 (1 H, t, J= 8.4 Hz) 7.08 (3H, m)
3.77 (2H, d, J=
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
9.9 Hz) 3.15 (2H, s)2.5 2.33 (2H, t, J= 9.9 Hz) 1.81 (2H, d, J= 11.0 Hz) 1.65
(2H, m);
ES/MS, calcd for C19HZ1FN04S 378.1, found 378.1 (M+H).
Example 2
Methyl2-(2-(1-tosylpiperidin-3-yl)phenyl)acetic acid (Compound 2A) and
2-(2-(1-tosylpiperidin-3-yl)phenyl)acetic acid (Compound 2B):
O
HO
, O
[0191] The compound(s) were prepared by the same procedure as
2-(3-(1-(4-fluorophenylsulfonyl)piperidin-4-yl)phenyl)acetic acid, Compound
IG, using
p-methyl phenylsufonyl chloride; Compound 2B IH NMR (400 MHz, CD3CN) 8 7.63
(2H,
d, J= 8.2 Hz) 7.40 (2H, d, J= 9.3 Hz) 7.18 (4H, m) 3.80-5.65 (4H, m) 2.94 (1
H, m) 2.43
(3H, s) 2.30 (3H, m) 1.78 (2H, m) 1.65 (1H, m) 1.42 (1H, m); ES/MS, m/z 374.1
(M+H).
Example 3
Methyl 2-(2-(1-tosylpiperidin-4-yl)phenyl)acetate (Compound 3A) and
2-(2-(1-tosylpiperidin-4-yl)phenyl)acetic acid (Compound 3B)
o
,o
HO 0O
[0192] The compound(s) were by the same procedure as
2-(2-(1-tosylpiperidin-3-yl)phenyl)acetic acid, Compound I, using methyl
2-(2-bromophenyl)acetate; Compound 3B 1 H NMR (400 MHz, CD3CN) S 7.68 (2H, d,
J
8.2 Hz) 7.44 (2H, dd, J= 0.7, 8.6 Hz) 7.25 (2H, m) 7.15 (2H, m) 3.83 (2H, m)
3.61 (2H, s)
2.61 (IH, m) 2.45 (3H, s) 2.30 (2H, m) 2.16 (1H, brs) 1.74 (4H, m); ES/MS,
calcd for
C2oH23N04S 374.1, found 374.0 (M+H).
56
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
Example 4
Methyl 2-(3-(1-(4-fluorobenzoyl)piperidin-3-yl)phenyl)acetate (Compound 4A)
and
2-(3-(1-(4-fluorobenzoyl)piperidin-3-yl)phenyl)acetic acid (Compound 4B):
Scheme 2
ci
O
0 0
~
O OH
I j
NH Step A Step B O
I~
N I~ F N F
A2 B2 C2
Step A:
Methyl2-(3-(1-(4-fluorobenzoyl)piperidin-3-yl)phenyl)acetate (Compound 4A)
[0193] To a solution of 100 mg (0.429 mmol, 1.0 equivalent) of methyl
2-(3-(piperidin-3-yl)phenyl)acetate (A2) in AcCN (5 ml) was added 1.1
equivalents of
4-fluorobenzoyl chloride (0.47 mmol, .0565 ml) and 3.0 equivalents (177.7 mg)
K2C03.
Reaction was heated in the microwave to 150 C at 300 W power for 5 inin.
Reaction mixture
was washed with water 3 times. Combined aqueous phase was extracted with
EtOAc.
Combined organic phase was washed with brine, dried over sodium sulfate, and
concentrated
to dryness on the RotorVap to yield crude methyl
2-(3-(1-(4-fluorobenzoyl)piperidin-3-yl)phenyl)acetate. MS (m/z) 356 (M+H)
Step B:
2-(3-(1-(4-fluorobenzoyl)piperidin-3-yl)phenyl)acetic acid (Compound 4B)
[0194] Crude methyl 2-(3-(1-(4-fluorobenzoyl)piperidin-3-yl)phenyl)acetate
from step A
was dissolved in THF (3 ml) and aqueous KOH (1.0 N, 3 ml) was added. Reaction
was
stirred for 4 hours. Reaction was acidified to pH 2-4 with 1.0 N aqueous HCl
and extracted
with EtOAc. Organic extracts were washed with brine, dried over sodium
sulfate, and
concentrated to dryness. Crude yield = 150 mg (0.439 mmol, >100%). Final
product was
57
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
purified by HPLC using 0.05% formic acid modifier. Final yield = 70.37 mg
(0.206 mmol).
LC/MS (Method A) Rt = 3.204 min. MS (m/z) 342 (M+H)
Example 5
2-(3-(1-(4-fluorophenylsulfonyl)piperidin-2-yl)phenyl)acetic acid (Compound
5G)
F
O
HO
I ~ 0=5=0
Step A:
tert-butyl 5-oxo-5-m-tolylpentyl carbamate (Compound 5A)
Ny0
O
[01951 To N-Boc valerolactam (0.250g, 1.25mmol) in 3mL THF at -78 C is added
3-tolyl
magnesium bromide (1.OM, 1.5minol). The reaction stirs about 2 hours slowly
warming to
room temperature. The reaction is quenched with sat. NH4C1 (5mL) and extracted
into DCM
3X. The combined extracts are dried over Na2SO4. The reaction is filtered,
dried and passed
over silica eluting with 20% EtOAc in hexanes yielding the title compound.
LC/MS (Method
A) Rt = 4.70 min. MS: 292 m/z (M+H).
Step B:
6-m-tolyl-2,3,4,5-tetrahydropyridine (Compound 5B)
N
58
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
[0196] To tert-butyl 5-oxo-5-m-tolylpentyl carbamate, Compound 5A, (0.30g,
1.03mmo1)
in DCM (2mL) is added TFA (0.5mL). The reaction is judged complete by LC/MS
after 4
hours. The mixture is dried and used without further purification. LC/MS
(Method A) Rt =
0.99 min. MS: 174 m/z (M+H).
Step C:
2-m-tolylpiperidine (Compound 5C)
H
[0197] To compound 8 (0.050g, 0.29mmol) in methanol, Compound 5B, (1mL) is
added
sodium borohydride (0.0055g, 0.145mmol). The reaction is judged complete after
90 min. by
LC/MS and quenched with water. The mixture is extracted into DCM 3X, the
organic layers
combined and dried. The material is used without further purification. LC/MS
(Method A)
Rt = 1.37 min. MS: 176 m/z (M+H).
Step D:
1-(4-fluorophenylsulfonyl)-2-m-tolylpiperidine (Compound 5D)
F
\
O
[0198] To 2-m-tolylpiperidine, Compound 5C, (0.017g, 0.097mmol) in 0.5mL DCM
with
DIEA (0.014g, 0.l0ininol) is added 4-fluoro phenyl sulfonyl chloride (0.019g,
0.l0mmol).
The reaction is judged complete after 1 hour and dried. The resulting oil is
purified over
silica eluting with 25% EtOAc in hexanes. LC/MS (Method A) Rt = 4.30 min. MS:
334
m/z (M+H).
59
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
Step E:
2-(3-(bromomethyl)phenyl)-1-(4-fluorophenylsulfonyl)piperidine (Compound 5E)
F
Br \
i
[0199] To 1-(4-fluorophenylsulfonyl)-2-m-tolylpiperidine, Compound 5C,
(0.090g,
0.27mmol) in 2mL CC14 is added AIBN (0.004g, 0.027mmo1) and NBS (0.058g,
0.32mmol).
The mixture is stirred in a sealed tube at 80 C. Two more equal portions of
NBS are added
over the next 48 hours. At the end of this time the reaction is worked up by
passing over
silica eluting with 50% EtOAc in hexanes. The material is used as a mixture of
the
alpha-bromo and tolyl phenyl piperidines. LC/MS (Method A) Rt = 4.28 min. MS:
410 m/z
(M+H).
Step F:
2-(3-(1-(4-fluorophenylsulfonyl)piperidin-2-yl)phenyl)AcCN (Compound 5F)
F
N~ ~ ~
/
'~
[0200] To 2-(3-(bromomethyl)phenyl)-1-(4-fluorophenylsulfonyl)piperidine
(0.035g,
0.084mmol) in 2mL AcCN is added KZC03 (0.023g, 0.168mmo1) and NaCN (0.005g,
0. 1 Ominol). The mixture is stirred 16 hours at 80 C, then cooled to RT The
title compound
is purified by HPLC eluting with AcCN and water both modified with 0.05%
formic acid.
LC/MS (Method A) Rt = 3.86 min. MS: 359 m/z (M+H).
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
Step G:
2-(3-(1-(4-fluorophenylsulfonyl)piperidin-2-yl)phenyl)acetic acid (Compound
5G)
F
O
HO I
0=5=0
I
N
[0201] To 2-(3-(1-(4-fluorophenylsulfonyl)piperidin-2-yl)phenyl)AcCN, Compound
5F,
(0.005g, 0.014mmol) in 0.25mL methanol is added 0.5mL 3N NaOH. The reaction is
stirred
at 40 C for 48hours, then concentrated in vacuo. The basic solution is
acidified to pH 1 with
1N HCI. The aqueous is extracted into DCM 3X. The dried material is used
without further
purification. LC/MS (Method A) Rt = 3.56 min. MS: 378 m/z (376 m/z M-H).
Example 6
2-(3 -(1 -(4-fluorophenylsulfonyl)- 1,2,5,6-tetrahydropyridin- 3 -yl)phenyl)
acetic acid
(Compound 6)
COZH
O O
I / I N,S I ~
/ F
Scheme 3
9
OTf HOzC B' COzH COkl Me (1) (2) TFAFsCI, , CH,Et3N,CI, CO2H
CH,CI,
~ TMSCHN, (3) INNaOH, CH3OH ~ Neoc Pd(PPhj)q, 2 M Na,CO3, / I NBoc CH3 OH,
C6H6 Boc N S
DME, 80 C, 1.5 hr / F
A3B B3B C3B D3B
61
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
Step A: tert-butyl 5-(trifluoromethylsulfonyloxy)-3,4-dihydropyridine-l-(2H)-
carboxylate
(Compound A3A) and tert-butyl 3-(trifluoromethylsulfonyloxy)-5,6-
dihydropyridine-l-
(2H)-carboxylate (Compound A3B)
OTf OTf
INBoc NBoc
A3A A3B
[0202] The synthesis of the triflates A3A and A3B was based on the procedure
reported
(Vicart, N. et al. Tetrahedron 1996, 52(27): 9101-10) with some modifications:
To a solution
of LDA (2M solution from Aldrich, 14.3 ml) in THF (50m1) at -78 C was added a
solution
of N-Boc-3-piperidone (4 g, 20 mmol) in THF (10 ml) dropwise. After 15 min,
N-phenyltriflimide (8.6 g, 24 mmol) in THF (20 ml) was added. The reaction
mixture was
slowly warmed up to RT, and stirred at RT overnight. After addition of
saturated NH4C1 (15
ml) at 0 C, the mixture was diluted with water (100 ml), and extracted with
CH2C12 (3 x 100
ml). The extracts were dried (Na2SO4), evaporated and flash chromatographied
on silica-gel
with 20% EtOAc/Hexane. Futher purification on silica-gel (5%, 10% and 20%
EtOAc/Hexane) gave the triflate A3A (2.6 g, 44 %) and A3B (2.1 g, 35.6 %) as
yellow oils.
The triflate A3A: 'H NMR (300 MHz, DMSO-d6) S: 7.15 (1H, bs), 3.45 (2H, m),
2.40 (2H, t,
J=5 Hz), 1.86 (2H, m), 1.44 (9H, s). The triflate A3B: 'H NMR (300 MHz, DMSO-
d6)
S:, 6.12 (1H, m), 4.0 (2H, b), 3.39 (2H, t, J=5.5 Hz), 2.26 (2H, m), 1.41 (9H,
s).
Step A:
2-(3-(1-(tert-butoxycarbonyl)-1,2,5,6-tetrahydropyridin-3-yl)phenyl)acetic
acid (Compound
B3)
cOZH
NBoc
[02031 To a degassed mixture of the triflate A3B (100 mg, 0.34 mmol),
2-(3-(4,4,5,5-tetrainethyl-1,3,2-dioxaborolan-2-yl)phenyl)acetic acid (135 mg,
0.51 mmol) in
2M Na2CO3 (aq. 1.1 ml) and DME (1.7 ml) was added Pd(PPh3)4 (20 mg). The
reaction
mixture was stirred at 80 C under N2 for 1.5 h, and diluted with H20 (10 ml)
and acidified
with 10% KHSO4 at 0 C, followed by extraction with EtOAc (3 x 10 ml). The
combined
62
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
organic layers were dried (Na2SO4), evaporated and chromatographed on silica-
gel with 5%
MeOH/CH2C12 to yield the desired product (60 mg, 56%) as a black residue. 'H
NMR (300
MHz, DMSO-d6) 8: 12.3 (1H, bs), 7.28 (3H, m), 7.17 (1H, m), 6.25 (1H, m), 4.19
(2H, b),
3.57 (2H, s), 3.46 (2H, t, J=5.5 Hz), 2.25 (2H, m), 1.42 (9H, s).
Step B:
tert-butyl 3-(3-(2-methoxy-2-oxoethyl)phenyl)-5,6-dihydropyridine-1(2H)-
carboxylate
(Compound C3B)
COkl Me
Boc
[0204] To a solution of
2-(3-(1-(tert-butoxycarbonyl)-1,2,5,6-tetrahydropyridin-3-yl)phenyl)acetic
acid (60 mg, 0.19
mmol) in MeOH (0.42 ml) and C6H6 (1.5 ml) was added TMSCHN2 (2 M solution in
hexane,
0.12 ml, 0.24 mmol). The reaction mixture was stirred at rt for 0.5 hr. The
volatiles were
removed in vacuo, and the resulting residues were co-evaporated with MeOH to
yield the
desired product (60 mg, 100%), which was used in the next step without further
purification.
'H NMR (300 MHz, DMSO-d6) b: 7.30 (3H, m), 7.18 (1H, m), 6.26 (1H, m), 4.19
(2H, b),
3.70 (2H, s), 3.62 (3H, s), 3.46 (2H, t, J=6 Hz), 2.25 (2H, m), 1.43 (9H, s).
Step C: Methyl
2-(3-(1-(4-fluorophenylsulfonyl)-1,2,5,6-tetrahydropyridin-3-yl)phenyl)acetate
(Compound
D3A)
Me
OO
COkN'
S I \
/ F
F
[0205] To a solution of tert-butyl 3-(3-(2-methoxy-2-oxoethyl)phenyl)-5,6-
dihydropyridine
-1(2H)-carboxylate (60 mg, 0.19 mmol) in CH2C12 (1.5 ml) was added TFA (1.5
ml). The
reaction mixture was stirred at rt for 0.5 hr. The volatiles were removed in
vacuo, and the
resulting residues were co-evaporated with CHC13 twice. To a solution of this
ainine as a
TFA salt in CHZCIZ (2.5 ml) was added Et3N (0.066 ml, 0.47 mmol), followed by
63
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
p-fluorobenzenesulfonyl chloride (0.044 g, 0.23 mmol). After stirred at rt
overnight, the
reaction mixture was quenched by addition of saturated NaHCO3 (15 ml). After
stirring at rt
for 1 h with cat. amount of DMAP, the mixture was extracted with EtOAc (3 x 15
ml). The
combined organic layers were washed with 1N HCl (15 ml), saturated NaHCO3 (15
ml) and
saturated NaCl (15 ml), then dried (MgSO4) and evaporated to dryness to yield
the desired
product (63 mg, 85%) as an orange residue. MS (m/z) 390.1 (M+H)/Rt = 3.91 min.
Step D: Methyl
2-(3-(1-(4-fluorophenylsulfonyl)-1,2,5,6-tetrahydropyridin-3-yl)phenyl)acetate
(Compound
D3B)
CoZH
o0
N'S I \
/ F
[0206] To a solution of methyl
2-(3 -(1 -(4-fluorophenylsulfonyl)- 1,2,5,6-tetrahydropyridin-3 -yl)phenyl)
acetate (0.063 g, 0.16
mmol) in MeOH (2 ml) was added 1N NaOH (lml), The reaction mixture was stirred
at rt
for 4 hr, acidified with 1N HCl at 0 C and evaporated to dryness. The
resulting residues
were purified by HPLC (Column: Phenomenex, 250 X 10 mm, 10 micro, Luna 10 ;
Gradient: 90%:10%:0.05% H20/CH3CN/TFA to 5%/95%/0.05% H20/CH3CN/TFA over 16
min) to afford the title product, 36.7 mg (61 %), as a white solid: MS (in/z)
376.1 (M+H)/Rt =
3.43 min.
Example 7
2-(3-(1-(4-fluorophenylsulfonyl)-1,4,5,6-tetrahydropyridin-3-yl)phenyl)acetic
acid
(Compound 7)
CoZH
o0
N2S I \
~F
64
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
Scheme 4
B-~ CozH (1) TFA, CH,CI,
OTf HOZC CO H
I~ p COZMe (2) FsCI, Et;N, CH,CI, Z
~oc ~ I~ TMSCHN, (3) 1N NaOH, CH;OH I~
O, O
NB
Pd(PPhg)q, 2 M NaZCO3, NBoc CH3OH, C6H6 NBoc ~ / N S I
DME, 80 'C, 1.5 hr F
A3A 7A 7B 7C
Steps A and B: tert-butyl 5-(3-(2-methoxy-2-oxoethyl)phenyl)-3,4-
dihydropyridine-l-
(2H)-carboxylate (Compound 7B)
COZMe
~ NBoc
( \
[0207] To a degassed mixture of the triflate A3A (100 mg, 0.34 mmol), the
boronic acid
(135 mg, 0.51 mmol) in 2 M Na2CO3 (1.1 ml) and DME (1.7 ml) was added
Pd(PPh3)4 (20
mg). The reaction mixture was stirred at 80 C under N2 for 1.5 h, and diluted
with H20 (10
ml) and acidified with 10% KHSO4 at 0 C, followed by extraction with EtOAc (3
x 10 ml).
The combined organic layers were dried (Na2SO4), evaporated to yield a black
residue (100
mg). To a solution of the resulting residues in MeOH (0.75 ml) and C6H6 (2.6
ml) was added
TMSCHN2 (2 M solution in hexane, 0.88 ml). The reaction mixture was stirred at
rt for 0.5
hr. The volatiles were removed in vacuo. The resulting residues were co-
evaporated with
MeOH, and chromatographed on silica-gel with 15% EtOAc/hexane to yield the
desired
product (44 mg, 39%) as a colorless residue. MS (m/z) 232.1 (M+-Boc+H)/Rt =
4.25 min.
Step C: 2-(3-(1-(4-fluorophenylsulfonyl)-1,4,5,6-tetrahydropyridin-3-
yl)phenyl)acetic acid
(Compound 7C)
COZH
O 0
N'S aF
[0208] To a solution of tert-butyl 3-(3-(2-methoxy-2-oxoethyl)phenyl)-5,6-
dihydropyridine-l-(2H)-carboxylate (44 mg, 0.133 mmol) in CH2Cl2 (1.5 ml) was
added TFA
(0.75 ml). The reaction mixture was stirred at RT for 0.5 hr. The volatiles
were removed in
vacuo, and the resulting residues were co-evaporated with CHC13 twice. To a
solution of this
amine as a TFA salt in CH2Cl2 (2.0 ml) was added Et3N (0.047 ml, 0.34 mmol),
followed by
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
p-fluorobenzenesulfonyl chloride (0.031 g, 0.16 mmol). After stirred at RT
overnight, the
reaction mixture was quenched by addition of saturated NaHCO3 (10 ml). After
stirring at
RT for 1 h with cat. amount of DMAP, the mixture was extracted with EtOAc (3 x
10 ml).
The combined organic layers were washed with 1N HCl (10 ml), saturated NaHCO3
(10 ml)
and saturated NaCl (10 ml), then dried (MgSO4) and evaporated to dryness to
yield a residue
(44 mg). To a solution of the resulting residue in MeOH (1 ml) was added 1N
NaOH (0.25
ml), The reaction mixture was stirred at RT overnight, acidified with 1N HCl
at 0 C and
evaporated to dryness. The resulting residues were purified by HPLC (Column:
Phenomenex, 250 X 10 mm, 10 micro, Luna 10 ; Gradient: 90%:10%:0.05%
H20/CH3CN/TFA to 5%/95%/0.05% H20/CH3CN/TFA over 16 min) to afford the title
product, 12.9 mg (25.9%), as a white solid: MS (m/z) 376.1 (M++H)/Rt = 3.53
min.
Example 8
2-(3-(1-(phenylsulfonyl)piperidin-3-yl)phenyl)acetic acid (Compound 8)
0
OH
O
/ ~ -
n
N-S
11
O
Scheme 5
0 O 0
OH OMe OMe
Step A Step B
\ I / N N NH
\ I \ I
8A 8B
O O
Step C OMe Step D OH
~ I 0~ R5 I 0 R5
CI-S-R
n N SO N SO
O
8C 8D
66
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
Step A: Methyl 2-(3-(pyridine -3-yl)phenyl)acetate (Compound 8A)
[0209] To 2-(3-(pyridine-3-yl)phenyl) acetic acid (400mg, 1.878mmo1) in MeOH
(5m1)
was added thionyl chloride (0.205m1, 2.817mmol), the solution was brought to
reflux for 5h.
The product (0.426mg, 100%) was afforded after concentration. LC/MS Rt = 2.126
min.
(Method A); MS (m/z) 228 (M +H).
Step B: Methyl 2-(3-(piperidin-3-yl)phenyl)acetate (Compound 8B)
[0210] To the methyl ester (280mg, 1.234mmo1) in MeOH (5m1) was added the
catalytic
amount of Pt02. The suspension was purged 3 times, and was stirred at 1 atm
under H2
overnight. The catalyst was filtered off through CELITE. The mixture was
concentrated to
remove solvent, the product methyl2-(3-(piperidin-3-yl)phenyl) acetate
(Intermediate 1,
287mg, 100%) was obtained. Rt = 1.624 min. (Method A); MS (m/z) 234 (M +H).
Steps C and D: 2-(3-(1-(phenylsulfonyl)piperidin-3-yl)phenyl)acetic acid
(Compound 8D)
0
OH
O
/ ~ -
N-S
=
[0211] To intennediate 1(84.8mg, 0.364mmo1) in DCM (2ml) was added DIEA
(0.095m1,
0.546mmo1), followed by the addition of phenylsulfonyl chloride (0.056m1,
0.437mmo1). The
mixture was stirred at RT overnight. The methyl ester (63.6mg, 47%) was
afforded after flash
column chromatography with EtOAC/hexanes. The product was then dissolved in
THF
(1 ml), 1 ml of aqueous 1 N NaOH was added. The mixture was stirred overnight.
Diluted with
EtAc (15m1), washed with 1N HCI (3 X 2m1), dried over Na2SO4, the desired
material
(55.4mg, 93%) was obtained. LC/MS Rt = 3.486 min. (Method A); MS (m/z) 360 (M
+ H).
Example 9
Methyl 2-(3-(1-tosylpiperidin-3-yl)phenyl) acetate (Compound 9A) and
2-(3-(1-tosylpiperidin-3-yl)phenyl) acetic acid (Compound 9B)
67
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
0
OH
O
/ ~ -
N-S
O
[0212] The title compounds were obtained using the same experimental procedure
described for 2-(3-(1-(phenylsulfonyl)piperidin-3-yl)phenyl)acetic acid using
4-methyl-phenyl sulfonyl chloride. Compound 9B Rt = 3.639 min. (Method A); MS
(m/z)
374 (M + H)
Example 10
Methyl2-(3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)phenyl)acetic acid
(Compound l0A)
and
2-(3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)phenyl)acetic acid (Compound
lOB)
0
OH
O
-
11
~
N-S ~ ~ F
/5 11
O
[0213] The title compounds were obtained using the same experimental procedure
described for 2-(3-(1-(phenylsulfonyl)piperidin-3-yl)phenyl)acetic acid using
4-fluorophenyl
sulfonyl chloride. Compound lOB Rt = 3.562 min. (Method A); MS (m/z) 378 (M +
H).
Exainple 11
Methyl 2-(3-(1-(methylsulfonyl)piperidin-3-yl)phenyl)acetate (Compound 11A)
and
2 -(3 -(1 -(methyl sulfonyl)piperidin-3 -yl)phenyl) acetic acid (Compound 11B)
68
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
0
OH
0
N-S-
[0214] The title compounds were obtained using the same experimental procedure
described for 2-(3-(1-(phenylsulfonyl)piperidin-3-yl)phenyl)acetic acid using
methyl sulfonyl
chloride. Rt = 2.895 min. (Method A); MS (m/z) 298 (M + H).
Example 12
Methyl 2-(4-(4-chlorobenzyloxy)-3 -(1-(4-fluorophenylsulfonyl)piperidin-3 -
yl)phenyl)acetate
(Compound 12A) and
2-(4-(4-chlorobenzyloxy)-3-(1-(4-fluorophenylsulfonyl)piperidin-3-
yl)phenyl)acetic acid
(Compound 12B)
0
OH
O
-
N-S ~ ~ F
O O
CI
69
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
Scheme 6
O O 0 O
OMe OMe OMe HOMe
Step A Step B Step C
-= /
~ NBoc
HBr I N
OH OH OH \ OH
A6 O B6 O C6 D6
OMe HOMe
Step D\ NBoc Step E -- N H
O O
\ I \ I
CI CI
E6 F6
Methyl2-(4-(4-chlorobenzyloxy)-3-(piperidin-3-yl)phenyl)acetate (F6)
Step A:
[0215] To 4-hydroxyphenyl) methyl acetate(6.0g, 36mmol) in DCM (30m1) was
added Br2
(2.22ml, 43.2mmo1) at 0 C. After 30min, the mixture was allowed to warm up to
RT, and
stirred at the temperature for 2h. The mixture was diluted with 80m1 of DCM,
and was
washed with H20 (3 X 20inl), dried over Na2SO4, 3-bromo-4-hydroxyphenyl methyl
acetate
(8.86g, 100%) was yielded.
Step B:
[0216] To the mixture of 3-bromo-4-hydroxyphenyl methyl acetate (1.56g,
6.341mmo1)
and pyridin-3-ylboronic acid in DME (15m1) was added palladium tetrakis
(366mg,
0.317mmol), followed by the addition of CsF (2.89g, 19.Ommo1) in water (5m1).
The mixture
was heated at 85 C overnight. The reaction mixture was diluted with EtAc
(100m1), washed
with sat Na2CO3 (3 X 20m1), dried over Na2SO4. The product (0.818g, 55%) was
afforded
after column chromatography on silica gel.
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
Step C:
[0217] To 4-hydroxy-3-(pyridin-3-yl)phenyl methyl acetate (300mg, 1.234mmo1)
in MeOH
(2m1) was added lml of 1N HCl in Et20, after stirred for 5min, the solvents
were pumped off.
The residue was dissolved in MeOH (5m1), and was added the catalytic amount of
Pt02. The
suspension was purged 3 times, and was stirred at 1 atm under H2 for 3h. The
catalyst was
filtered off through CELITE. Concentrated to remove solvent, the product amine
was
obtained and dissolved in DCM (4m1). DIEA (0.644m1, 3.70mmo1) and Boc
anhydride
(404mg, 1.852mmo1) were added into the solution. The mixture was stirred at RT
overnight.
The product (223mg, 50%) was obtained after flash column chromatography on
silica gel.
Step D:
[0218] To the previous product (223mg, 0.640mmol) in CH3CN (3m1) were added
K2CO3
and chlorobenzyl chloride (124mg, 0.768mmo1). The reaction mixture was heated
at 80 C
overnight. The desired product (217mg, 72%) was yielded after flash column
chromatography, and was treated with TFA (0.35m1) in 4ml DCM. After lh
stirring at RT,
the SM had disappeared, intermediate 2 was used without further purification.
Rt = 2.553
min. (Method A); MS (m/z) 374 (M+ + H).
Step E:
[0219] To intennediate F6 (56.9mg, 0.152mmo1) in DCM (2ml) was added DIEA
(0.133m1, 0.763ininol), followed by the addition of the 4-fluorophenylsulfonyl
chloride
(35.6mg, 0.183mmo1). The mixture was stirred at RT for 3h. The product (80mg,
99%) was
afforded after flash column chromatography on silica gel. The product was then
dissolved in
THF (1 ml), 1 ml of aqueous 1 N NaOH was added. The mixture was stirred
overnight. Diluted
with EtOAc (15m1), washed with 1N HCl (3 X 2ml), dried over Na2SO4, the final
product
(73.5 mg, 95%) was obtained. Rt = 4.209 min. (Method A); MS (m/z) 518 (M + H).
Exainple 13
Methyl 2-(4-(4-chlorobenzyloxy)-3-(1-(phenylsulfonyl)piperidin-3-
yl)phenyl)acetate
(Compound 13A) and
71
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
2-(4-(4-chlorobenzyloxy)-3-(1-(phenylsulfonyl)piperidin-3-yl)phenyl)acetic
acid
(Compound 13B)
0
OH
O
/ ~ -
N-S
O O
CI
[02201 The desired material using the procedure described for
2-(4-(4-chlorobenzyloxy)-3-(1-(4-fluorophenylsulfonyl)piperidin-3-
yl)phenyl)acetic acid
using phenyl sulfonyl chloride. Compound 13B LC/MS Rt = 4.156 min. (Method A);
MS
(m/z) 501 (M + H)
Example 14
Methyl 2-(4-(4-chlorobenzyloxy)-3-(1-(methylsulfonyl)piperidin-3-
yl)phenyl)acetate
(Compound 14A) and
2-(4-(4-chlorobenzyloxy)-3-(1-(methylsulfonyl)piperidin-3-yl)phenyl)acetic
acid
(Compound 14B)
0
OH
0
N-S-
O O
CI
[02211 The desired material using the procedure described for
2-(4-(4-chlorobenzyloxy)-3-(1-(4-fluorophenylsulfonyl)piperidin-3-
yl)phenyl)acetic acid
using methyl sulfonyl chloride. Compound 14B LC/MS Rt = 3.699 min. (Method A);
MS
(m/z) 438 (M+ H)
72
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
Examples 15-20
2-(4-chloro-3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)phenyl)acetic acid
(Compound 15)
and analogs
[0222] Substituted analogues of were prepared according to Scheme 7. The
benzoic acids
were homologated using an Arndt-Eistert protocol. The esters were then coupled
to a boronic
acid, which was reduced and sulfonylated. Finally, the ester was saponified to
produce the
free acid.
Scheme 7
1. oxalyl chloride O AgOBz CO2Me
CO2H CH2CI2, cat DMF +NIIN_ triethylamine
1 2. TMS-diazomethane, THF MeOH
R Br RN Br R Br
3-pyridine boronic acid CO2Me I. Hz, Pt02 COZH
CsF MeOH, cat HCI
Pd(PPh3)4 2. p-fluorobenzene sulfonyl chloride / DME/IPA/H,O .TEA, CH,CI, R 3.
LiOH THF/H,O R
N N
I
0=S=0
F
Example 15
2-(4-chloro-3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)phenyl)acetic acid
(Compound 15F)
0
OH N-S ~ / F
O
O
CI
Step A: 1-(3-bromo-4-chlorophenyl)-2-diazoethanone (Compound 15A)
1. oxalyl cliloride O
COzH CH,CIZ, cat DMF +N~ N_
2. TMS-diazometliane, THF
Br Br
CI CI
73
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
[0223] 1.5 g of 3-bromo-4-chlorobenzoic acid (6.4 mmol, 1.0 Eq) was dissolved
in 25 mL
anhydrous CH2C12 and cooled to -5 C (ice/brine). 831 L oxalyl chloride (9.6
mmol, 1.5 eq)
was then added dropwise, along with 4 drops anhydrous DMF. The reaction was
warmed to
25 C overnight and concentrated to dryness. The resulting oil was then
dissolved in 50 mL
anhydrous THF, and cooled to -5 C (ice/brine), and 7.2 mL TMS-diazomethane
(2.0 M in
hexanes, 2.25 eq) was added via syringe. The reaction was allowed to warm to
25 overnight,
and concentrated to dryness. The resulting yellow oil was then subjected to
silica flash
chromatography (90:10 hexanes:EtOAc) to provide 930 mg of bright yellow solid.
(58%
yield over two steps). 'H NMR (400 MHz, CDC13) 6 5.95 (s, 1H), 7.35 (m, 1H),
7.60 (m,
1 H), 8.05 (m, 1 H).
Step B: Methyl 2-(3-bromo-4-chlorophenyl)acetate (Compound 15B)
O ~ AgOBz O
+NI _ triethylamine = O/
N MeOH
Br
Br
CI CI
[0224] 930 mg of the diazoketone (3.58 mmol, 1.0 eq) was dissolved in 35 mL
dry
methanol; in a separate round bottom, 492 mg of silver benzoate (2.15 minol,
0.6 eq) was
dissolved in 8 mL anhydrous triethylamine. This solution was then added
dropwise at room
temperature, to the diazoketone solution via syringe. The resulting black
solution was stirred
at room temperature overnight. The solvent was evaporated, and the residue
dissolved in
EtOAc, washed with saturated aqueous NH4C1(x3) and brine (x2), dried over
MgSO4, and
concentrated to give 790 rng of yellow oil. (83% yield). IH NMR (400 MHz,
CDC13) 6 3.45
(s, 2H), 3.70 (s, 3H), 7.17 (app d, 1H), 7.4 (m, 1H), 7.55 (m, 1H).
Step C: Methyl 2-(4-chloro-3-(pyridin-3-yl)phenyl)acetate (Compound 15C)
O Pd(PPh3)4 0
O CsF
+ B(OH)2 DME/IPA/H,O
Br N
CI CI I i
N
74
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
[0225] Into a 100 mL sealed reaction flask was placed 390 mg of methyl
2-(3-bromo-4-chlorophenyl)acetate (1.48 mmol, 1.0 eq), 279 mg of 3-pyridine
boronic acid
(1.9 mmol, 1.3 eq), 790 mg of CsF (5.2 mmol, 3.5 eq), 119 mg Pd(PPh3)4 (0.12
mmol, 0.07
eq), and 4 mL dimethoxyethane, 2 mL isopropyl alcohol, and 2 mL distilled
water. The
reaction was sealed and heated to 115 C (oil bath) overnight. The reaction was
cooled,
partitioned between EtOAc and brine, and washed brine (x2). After drying with
MgSO4 and
concentrating, the resulting oil was then subjected to silica flash
chromatography (1:1
hexanes:EtOAc) to give 278 mg of yellow oil. (72% yield). 'H NMR (400 MHz,
CDC13) 6
3.65 (s, 2H), 3.75 (s, 3H), 7.27 (m, 2 H), 7.4-7.55 (m, 2H), 7.85 (m, 1H),
8.65 (m, 1H), 8.78
(m, 1 H).
Step D:
Methyl 2-(4-chloro-3-(piperidin-3-yl)phenyl)acetate (Compound 15D)
0
Pt02
O Hz O
MeOH, cat. HCI
\ \ \
CI I i CI
N N
H
[0226] 278 mg ofinethyl 2-(4-chloro-3-(pyridin-3-yl)phenyl)acetate (1.06 mmol,
1.0 eq)
was dissolved in 7 mL anhydrous methanol. About 10 mg of Pt02 was added, along
with 4
drops concentrated HCI. A hydrogen balloon was attached, the round bottom was
evacuated
and backflushed with H2 (x3), and stirred at room temperature for 7 hrs. The
methanol was
evaporated, the residue diluted with EtOAc, washed with sat. aqueous NaHCO3
(x2) and
brine (x2), dried over MgSO4, and concentrated to give 265 mg of clear oil.
(93% yield).
LC/MS (Standard Method B). Rt = 1.42 min. MS 269.1 (M +H).
Step E:
Methyl 2-(4-chloro-3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)phenyl)acetate
(Compound
15E)
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
0 p-fluorobenzenesulfonyl
O/ chloride _
triethylamine ~
/ THF O N-S ~ ~ F
I O
O
\
CI
CI
N
H
[0227] 100 mg of 2-(4-chloro-3-(piperidin-3-yl)phenyl)acetate (0.37 mmol, 1.0
eq) was
dissolved in 7 mL anhydrous CH2C12. 77 L triethylamine (0.56 mmol, 1.5 eq)
was added,
and the reaction cooled to 0 C, and 92 mgp-fluorobenzenesulfonyl chloride
(0.47 mmol, 1.25
eq) was added, and the reaction allowed to warm to 25 C overnight. The
reaction was
concentrated and purified via silica flash chromatography (90:10 hexanes:
EtOAc) to give
112 mg clear oil. (71 % yield). LC/MS (Method A). Rt = 3.99 min. MS= 426.1 (M
+H).
Step F:
2-(4-chloro-3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)phenyl)acetic acid
(Compound 15F)
0 0 11 _
11 ~ N-S \/ F LiOH OH N O F
p ~ THF / H2O
--~ O
O -
CI CI
~ ~
[0228] 100 ing of the methyl ester (0.24 inmol, 1.0 eq) was suspended in 5 inL
1:1
THF/H20, and 17 ing LiOH (0.72 mmol, 3.0 eq) was added, and the reaction
stirred at 25 C
overnight. The THF was then evaporated, the reaction acidified (0 C, conc.
HCl), and
extracted with EtOAc (x3). The organic were combined, washed with brine, dried
over
MgSO4, and purified via reverse phase HPLC, and lyophilized to give 17 mg of
white
amorphous powder. LC/MS (Method B). Rt = 3.59 min. MS= 412.1 (M+H). 'H NMR
(400
MHz, d6-DMSO) 6 1.56-1.85 (m, 4H), 2.07 (s, 1H), 2.23-2.40 (m, 2H), 3.54 (s,
2H), 3.73 (m,
2H), 7.16 (d of d, 1 H), 7.23 (d, 1 H), 7.39 (d, 1 H), 7.44-7.48 (m, 2H), 7.81-
7.85 (m, 2H).
Example 16
Methyl 2-(3 -Chloro- 5 -(1 -(4-fluorophenylsul fonyl)piperidin-3 -yl)phenyl)
acetate (Compound
16A) and 2-(3-chloro-5-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)phenyl)acetic
acid
(Compound 16B):
76
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
o _
N-S ~ / F
O
O OH
cl
[0229] The title materials were obtained using the same procedure described
for
2-(4-chloro-3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)phenyl)acetic acid,
starting with
5-chloro-3-bromo benzoic acid. Compound 16B LC/MS (Method B). Rt = 3.52 min.
MS
412.1 m/z (M +H). 'H NMR (400 MHz, d6-DMSO) S 1.55 (m, 2.H), 1.79 (m, 2H),
2.82 (m,
1 H), 3.56 (s, 2H), 3.65 (m, 2H), 7.13 (s, 1 H), 7.21 (s, 1 H), 7.25 (s, 1 H),
7.47 (app t, 2H), 7.83
(app d of d, 2H).
Example 17
Methyl 2-(2-chloro-5-(1-(4-fluorophenylsulfonyl)piperidin-3 -yl)phenyl)acetate
(Compound
17A) and 2-(2-chloro-5-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)phenyl)acetic
acid
(Compound 17B)
o _
OH N-S ~ / F
O
O
cl
[0230] The title material was obtained using the same procedure described for
2-(4-chloro-3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)phenyl)acetic acid,
starting with
6-chloro-3-bromo benzoic acid. LC/MS (Method B). Compound 17B Rt = 3.47 min.
MS
412.1 m/z (M+H). 'H NMR (400 MHz, d6-DMSO) 8 1.57 (in, 2H), 1.79 (m, 2H), 2.28
(m,
2H), 2.80 (m, 1 H), 3.62 (m, 2H), 3.66 (s, 2H), 7.20 (app d of d, 1 H), 7.31
(m, 1 H), 7.36 (d,
1 H), 7.48 (app t, 2H), 7.82 (m, 2H).
Exainple 18
Methyl 2-(3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)-2-methylphenyl)acetate
(Compound
18A) and 2-(3-(1-(4-fluorophenylsulfonyl)piperi din- 3 -yl) -2-methylphenyl)
acetic acid
(Compound 18B)
77
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
o
_
11
N F
OH I~ /
0
O
~ ~
[0231] The title material was obtained using the same procedure described for
2-(4-chloro-3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)phenyl)acetic acid
using
2-methyl-3-bromo benzoic acid. LC/MS (Method B). Compound 18B Rt = 3.54 min.
MS
392.3 m/z (M +H). 'H NMR (400 MHz, d6-DMSO) 6 1.57 (m, 1H), 1.72 (m, 2H), 1.85
(1H),
2.22 (s, 3H), 2.30 (m, 1H), 2.35 (t, 1H), 3.05 (t, 1H), 3.62 (m, 1H), 3.64 (s,
2H), 3.75 (m,
1H), 7.06 (app s, 3H), 7.46 (m, 2H), 7.83 (m, 2H).
Example 19
Methyl2-(3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)-4-methylphenyl)acetate
(Compound
19A) and 2-(3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)-4-methylphenyl)acetic
acid
(Compound 19B)
HO2C
a F
~
\
O / NSO
Me
[0232] The title material was obtained using the same procedure described for
2-(4-chloro-3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)phenyl)acetic acid,
using
4-methyl-3-bromo benzoic acid. LC/MS (Method B). Compound 19B Rt = 33.40 min.
MS
392.3 rn/z (M +H). 'H NMR (400 MHz, d6-DMSO) 6 1.48-1.1.88 (m, 4H), 2.25 (s,
3H), 2.35
(m, 1 H), 2.97 (m, 1 H), 3.48 (s, 2H), 3.62 (m, 1 H), 3.66 (m, 1 H), 7.02 (m,
2H), 7.15 (m, 1 H),
7.47 (app t, 1 H), 7.83 (in, 1 H).
Example 20
Methyl 2-(3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)-5-
inethoxyphenyl)acetate
(Compound 20A) and
2-(3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)-5-methoxyphenyl)acetic acid
(Compound
20B)
78
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
o _
N-S ~ / F
O
HOZC -
0
~
[0233] The title material was obtained using the same procedure described for
2-(4-chloro-3 -(1-(4- fluorophenylsulfonyl)piperidin-3 -yl)phenyl) acetic
acid, using
5-methoxy-3-bromo benzoic acid. LC/MS (Method B). Compound 20B Rt = 3.51 min.
MS
408.1 m/z (M +H). 'H NMR (400 MHz, CDC13) S 1.55 (br m, 2H), 1.76 (m, 2H),
2.30 (app t,
2H), 2.75 (m, 1 H), 3.49 (s, 2H), 3.68 (br m, 2 H), 3.73 (s, 3H), 6.70 (m, 3
H), 7.45 (m, 2H),
7.82 (m, 2H).
Example 21
Methyl 2-(3-(1-(4-fluorophenylsulfonyl)piperidin-3 -yl)-5-
hydroxyphenyl)acetate
(Compound 21A) and
2-(3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)-5-hydroxyphenyl)acetic acid
(Compound
21B)
o _
11
N-II ~ ~ F
O
HOzC
OH
[0234] The methyl ester of
2-(3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)-5-inethoxyphenyl)acetic acid
was treated
with 3.0 eq of BBr3 at 0 C in CHZCIZ. Following an aqueous workup, the free
phenol was
obtained as a white foain in 92% yield. This was then saponified as described
in Step F of
2-(4-chloro-3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)phenyl)acetic acid to
give the title
compound. Compound 21B LC/MS (Method B). Rt = 3.20 min. MS 394.1 m/z (M+H). 'H
NMR (400 MHz, d6-DMSO) S 1.57 (br m, 2H), 1.79 (m, 2H), 2.22 (m, 2 H), 2.65
(m, 1 H),
3.30 (v broad s, Ar-OH), 3.40 (s, 2H), 3.62 (m, 1 H), 6.50 (m, 3H), 7.46 (m
2H), 7.82 (m,
2H).
79
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
Example 22
Methyl 2-(3-(benzyloxy)-5-(1-(4-fluorophenylsulfonyl)piperidin-3-
yl)phenyl)acetate
(Compound 22A) and
2-(3-(benzyloxy)-5-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)phenyl)acetic
acid
(Compound 22B):
CO2H
O
N
0=S=0
F
[0235] Methyl- 2-(3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)-5-
hydroxyphenyl)acetate
was treated with benzyl bromide in presence of K2CO3 (2.0 eq) in DMF at RT,
overnight.
EtOAC was added, and the organic layer washed with brine (x3), dried over
MgSO4. Upon
filtration and evaporation of the solvent under reduced pressure, the crude
material was
submitted to hydrolysis with NaOH (1.0 eq) in THF:water. The reaction was
stirred at RT for
four hours, and submitted to an acid/base work-up. Upon extraction with EtOAc,
the desired
material was obtained as a foam. Compound 22B LC/MS (Standard Method B). Rt=
3.91
min. MS 484.2 m/z (M+H). 'H NMR (400 MHz, CDC13) 8 1.42 (m, 1H), 1.85 (in,
2H), 1.88
(m, 2 H), 2.26 (m, 2H), 2.88 (in, 1 H), 3.61 (s, 2H), 3.78 (m, 1 H), 5.15 (s,
2H), 6.41 (m, 1 H),
6.82 (m, 1 H), 7.20 (in, 1 H), 7.31-7.85 (in, 8H), 7.76 (in, 1 H).
Example 23
Methyl 2-(3-(4-chlorobenzyloxy)-5-(1-(4-fluorophenylsulfonyl)piperidin-3 -
yl)phenyl)acetate
(Compound 23A) and
2-(3-(4-chlorobenzyloxy)-5-(1-(4-fluorophenylsulfonyl)piperidin-3-
yl)phenyl)acetic acid
(Compound 23B)
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
COZH
I
~O \
N
CI I
0=S=0
F
[0236] The desired material was obtained using the procedure described for the
synthesis of
2-(3-(benzyloxy)-5-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)phenyl)acetic
acid using
4-chloro benzyl bromide. Compound 23B LC/MS (Standard Method B). Rt = 4.2 min.
MS
516.0 m/z (M -H). 'H NMR (400 MHz, CDC13) b 1.51 (m, 1H), 1.82 (m, 2H), 1.88
(m, 2 H),
2.26 (m, 2H), 2.78 (m, 1 H), 3.61 (s, 2H), 3.78 (m, 1 H), 5.05 (s, 2H), 6.77
(m, 3H), 7.43 (m, 6
H), 7.81 (m, 2H).
Example 24
Methyl2-(3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)phenyl)acetate (Compound
24A) and
2-(3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)phenyl)acetic acid (Compound
24B)
CO2H
H2N
N
I
0=S=0
F
[0237] These compound(s) were synthesized starting with 3-bromo-5-nitrobenzoic
acid,
using the procedure described for
2-(4-chloro-3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)phenyl)acetic acid,
followed by a
hydrogenation in MeOH using a catalytic ainount of Pt02, under reduced
pressure. Upon
filtration over CELITE, the material was isolated. LC/MS (Standard Method B).
Compound
24B Rt = 3.43 min. MS 393.10 m/z (M -H). 'H NMR (400 MHz, d6-DMSO) b 1.55 (m,
2H),
81
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
1.79 (m, 2H), 2.22 (m, 2 H), 2.65 (m, 1 H), 3.52 (s, 2H), 3.62 (m, 2H), 6.92
(app t, 3H), 7.43
(app t, 2H), 7.77 (m, 2H).
Example 25
2-(3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)-5-methylphenyl)acetic acid
(Compound
25B)
Br I. aq. NaNO2 / H2SO4 Br standard COZH O
procedure N i~
/~NHZ 2. toluene, reflux \
\ I I~ O ~
Me \ C02H Me COZH ~ F
Me
Step A:
3-bromo-5-methylbenzoic acid (Compound 25A)
[0238] 2.0 g of 2-amino-3-bromo-5-methylbenzoic acid (8.7 mmol, 1.0 eq) was
dissolved
in 45 mL toluene and 15 mL reagent grade ethanol. The reaction was cooled to 0
C, and 1.5
mL concentrated H2SO4 was added dropwise. 1.32 g NaNO2 was added portionwise,
and then
stirred at the same temperature for 35 min., and finally refluxed for 1.5
hours. The mixture
was cooled to room temperature, diluted with EtOAc and brine, and extracted
(x3) with
EtOAc. After combining the organics, these were washed with 1 M HCI (x3),
brine (x3) and
dried over MgSOa. After filtering and concentrating, 910 mg of yellow solid
was obtained.
(49% yield). LC/MS (Method B). Rt = 3.24 min. MS 215.2 m/z (M +H).
Step B:
2-(3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)-5-inethylphenyl)acetic acid
(Compound
25B)
HOzC
F
\
I / 0
S
Me N' %
[0239] The title material was obtained from 3-bromo-5-methylbenzoic acid using
the
procedure described for -(4-chloro-3-(1-(4-fluorophenylsulfonyl)piperidin-3-
yl)phenyl)acetic
acid. LC/MS (Method B). Rt = 3.82 min. MS 392.1 m/z (M +H). 'H NMR (400 MHz,
82
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
d6-DMSO) S 1.42-1.68 (m, 2H), 1.8 (m, 2H), 2.24 (s, 3H), 2.32 (m, 2H), 2.73
(m, 1H), 3.49
(s, 2H), 3.66 (m, 2H), 6.85 (m, 3H), 7.47 (app t, 2H), 7.85 (m, 2H).
Example 26
2-(5-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)-2-methylphenyl)acetic acid
(Compound
26B)
Br Br Br std conditions
NaOH H2SO4
I/ --= I/ NH2 OH ~
CN
Me Me 0 Me 0
COzH
F
Me
O~
NO
Step A: 5-bromo-2-methylbenzoic acid
[0240] 3.75 g of with 5-bromo-2-methylbenzonitrile (19,0 mmol, 1.0 eq) was
dissolved in
175 mL ethanol, and heated to 40 C. 70 mL of 1 N aqueous NaOH was then added,
followed
by 70 mL 10% H202, and heating continued for 35 min. After cooling, half the
ethanol was
evaporated, and the reaction was partitioned between EtOAc and brine. After
standard
workup and drying, 3.4 g yellow orange solid was obtained. This material was
then heated to
110 C in 30 mL conc. H2SO4 and 60 mL H20 overnight. After cooling, this was
diluted with
water and extracted with EtOAc. After washing with brine and drying over
MgSOa, 2.02 g of
beige crystals were obtained. (49% over two steps). 'H NMR (400 MHz, CDC13) 6
2.53 (s,
3H), 7.33 (m, 1H), 7.51 (m, 1H), 7.8 (m, 1H). 13C NMR ((100 MHz, CDC13) b
20.57,
129.54, 130.25, 131.41, 132.25, 133.46, 138.00, 167.41.
Step B: 2-(5-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)-2-methylphenyl)acetic
acid
(Compound 26B)
[0241] The title material was obtained from 5-bromo-2-methylbenzoic acid using
the
procedure described for
2-(4-chloro-3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)phenyl)acetic acid.
LC/MS (Method
B) Rt = 3.46 min. MS 392.2 m/z (M +H). 'H NMR (400 MHz, d6-DMSO) 8 1.52 (m, 1
H),
83
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
1.61 (m, 1H), 1.80 (m, 2H), 2.17 (s, 3H), 2.34 (m, 2 H), 3.55 (s, 2H), 3.66
(m, 2H), 7.05 (m,
3H), 7.46 (m, 2H), 7.81 (m, 2H).
Example 27
2-(2-chloro-3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)phenyl)acetic acid
(Compound
27B)
NHZ standard conditions
Br ~
~(C1 12.. NCuBr,aN0, HCI, HNO~ HZO ~cC1
COZH COZH HO2C
F
CI
01,
/ N~S O
Step A: 3-amino-2-chlorobenzoic acid (Compound 27A)
[0242] 2.0 g of 3-amino-2-chlorobenzoic acid (11.7 mmol, 1.0 eq) was dissolved
in 35 mL
H20 and 9 mL conc HCI at 0 C. To this was added a solution of 849 mg NaNO2
(12.3 mmol,
1.05 eq) dissolved in 3 mL H20, dropwise; this solution was stirred at 0 C for
20 min. 1.8 g
CuBr (12.9 mmol, 1.1 eq) was slurried in 10 mL H20, and heated to 95 C in a
separate round
bottom. The solution of the diazonium salt was then added via pipette to the
CuBr slurry
dropwise. After the addition was complete, heating continued for 30 inin.,
when the reaction
was cooled to RT Extraction into EtOAc, washing with brine, drying a
concentration give
2.25 g of a thick brown gum. LC/MS (Method B, negative mode). Rt = 2.96 inin.
MS 234.3
m/z (M - H).
Step B: 2-(2-chloro-3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)phenyl)acetic
acid
(Compound 27B)
[0243] The title material was obtained from 3-amino-2-chlorobenzoic acid using
the
procedure described for
2-(4-chloro-3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)phenyl)acetic acid.
LC/MS (Method
B). Rt = 3.56 min. MS 412.0 m/z (M +H). 'H NMR (400 MHz, d,-DMSO) S 1.56-1.85
(m,
84
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
4H), 2.07 (s, 1H), 2.23-2.40 (m, 2H), 3.54 (s, 2H), 3.73 (m, 2H), 7.24 (m,
3H), 7.42 (app t,
2H), 7.83 (m, 2H).
Example 28
2-(3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)-5-phenoxyphenyl)acetic acid
(Compound
28A) and 2-(3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)-5-
phenoxyphenyl)acetic acid
(Compound 28B)
HOZC
F
/ I I \ O\
\ O N/SO
[0244] Methyl2-(3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)-5-
hydroxyphenyl)acetate
(400 mg, 0.97 mmol, 1.0 eq) was dissolved in 10 mL anhydrous CH2C12. To this
was added
236 mg PhB(OH)2 (1.94 mmol, 2.0 eq), 176 mg anhydrous Cu(OAc)2 (0.97 mmol, 1.0
eq),
338 L diisopropylamine (1.94 mmol, 2.0 eq), and a spatula tip of 4A molecular
sieves. The
reaction was then stirred under Ar at RT for 48 hrs. After filtering, washing
with brine,
drying and concentrating, the resulting dark residue was subjected to the
general
saponification conditions to give the title compound 28B. LC/MS (Method B). Rt
= 3.99
min. MS 470.10 m/z (M +H). 'H NMR (400 MHz, d6-DMSO) S 1.52 (br s, 2H), 1.79
(m,
2H), 2.31 (m, 2H), 2.77 (s, 1 H), 3.52 (s, 2H), 3.65 (m, 2H), 6.75 (m, 1 H),
6.83 (m, 1 H), 6.93
(m, 1 H), 6.99 (m, 1 H), 7.13 (m, 1 H), 7.3 8 (m, 2H), 7.47 (m, 2H), 7.82 (m,
2H).
Example 29
(S)-2-(3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)phenyl)acetic acid
(Compound 29C) and
(R)-2-(3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)phenyl)acetic acid
(Compound 29D)
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
Scheme 8
O Pd(PPh3)4 0 1. MeOH / HCI (g) 0 0 F
OH Toluene/
CO BAB/ OH 2. PtOZ , H2 / MeOH Oi 1. RSO2C1 OH -
~ CH2CI2/
N~ 3. L-Tartaric acid DIPEA S O
O
I N 4. Recrystallize I NH 2. LiOH N~
Br ~
MeOH/H20
rBj 2
3 4
Step A: 2-(3-(pyridin-4-yl)phenyl)acetic acid (Compound 29A)
[0245] To a dry 3-neck reaction flask was added 3-bromophenyl acetic acid (1,
lOg,
46.6mmol), tetrabutyl ammonium bromide (TBAB, 1.3g, 4.Olmmol),
diethyl-(3-pyridyl)borane (6.83g, 46.4mmol) and toluene (60mL). The resulting
suspension
was stirred for 15 min., then a solution of K2CO3 (19.12g, 138.4mmol) in H20
(60mL) was
added. Finally a slurry of tetrakis(triphenylphosphine) palladium (0) (0.36g,
0.31mmol) in
toluene (5mL) was added and the suspension heated on oil bath to 84 C and let
reflux 16h.
The reaction mixture was cooled and transferred to a separatory funnel. The
organic layer
(20mL) was separated, the aqueous layer was washed with CH2C12 (2 x100mL),
acidified and
washed with CH2ClZ (2 x100mL) and concentrated in vacuo to give a paste of
salts. This was
completely dried and extracted with methanol to give 9.83 g of product upon
concentration.
MS 214.1m/z (M+H).
Step B: (S)-methyl 2-(3-(piperidin-3-yl)phenyl)acetate (Compound 29B)
0
o1~
NH
[0246] The crude product 2-(3-(pyridin-4-yl)phenyl)acetic acid was taken up in
MeOH
(80inL) and hydrogen chloride gas bubbled through it until gas absorption
ceased - light
yellow solution turns brown. MS 228.09 m/z(M+H). The ester product was
filtered to
remove residual salts. The solution was transferred to a PARR hydrogenation
bottle, Platinum
(IV) oxide catalyst (300mg) added and Parr shaken at 10 psi (H2) for 12 h. The
suspension
was filtered through a pad of CELITE and concentrated to give crude oily
product (8.04g).
86
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
This was taken up in CH2C12 (300mL) and 1.0M NaOH (150mL) added to achieve a
basic
pH. Separation of organic layer, drying with anhydrous Na2SO4 followed by
concentration in
vacuo afforded 5.7g of an amber oily product.
[0247] A solution of L-tartaric acid (1.2g, 8.02mmo1) in MeOH (7mL) was added
to a
solution of product above ( 1.7g, 7.29mmol) in MeOH (3 mL) and heated to 70
C. Let stir
for 20 min. and forced into clear solution by addition of H20 (4.5mL). Let
cool slowly with
stirring. After 14 h, the crystals were filtered into a white powder (1.2g).
This was
recrystallized from 18mL of MeOH-H20 to give pure product (411mg). 500mg of
this
product was free-basified to give 222mg of product 3 (analyzed on chiral LC
column to give
a purity of 98.6%ee). The mother liquor was free-based and treated analogously
with
D-tartaric acid to give the corresponding enantiomer of 99.6% ee purity.
Step C: (S)-2-(3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)phenyl)acetic acid
(Compound
29C)
O
F
HO
N SO
[0248] (S)-methyl 2-(3-(piperi din-3-yl)phenyl)acetate (3, 222mg, 0.95mmo1)
and
N,N-diisopropylethylamine (0.5mL, 2.85mmo1) were taken up in CHZC12 (3mL) and
cooled
to 0 C on ice-bath. To this was added a solution of 4-
fluorophenylsulfonylchloride
(203.7mg, 1.05mmo1) in CHzCIZ (1 mL). Let wann to room temperature. After 2 h,
the
reaction was diluted with EtOAc (60mL) and washed with brine (30mL), dried
(Na2SO4) and
concentrated into oily product (366mg). Recrystallized from hot MeOH into
needles (Melting
point 100.57 C). LC/MS (Method A) MS 393.2 in/z (M+H), (Rt = 4.023 inin).
[0249] The crystalline ester (160mg, 0.409mmol) and lithium hydroxide (50mg,
excess)
were suspended in THF-H20 mixture (1:2) and stirred for 4 hours. The mixture
was then
diluted with EtOAc (60inL), neutralized with 1.OM HCI to pH <7 and washed with
brine. The
solution was then dried and concentrated to give crystalline product
(109.7mg). LC/MS
(Method A) MS m/z 378.1 (M+H), Rt = 3.47 min)..
87
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
(R)-2-(3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)phenyl)acetic acid
(Compound 29D)
O
F
HO
11-z~
.~O
S
GN O
[0250] (R)-2-(3-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)phenyl)acetic acid
(5) was
prepared following the procedure described for the S-enantiomer (4) but
substituting
D-tartaric acid for the S-tartaric acid in the chiral salt formation step).
LC/MS (Method A)
MS m/z 378.1 (M+H), (Rt = 3.426 min).
Example 30
Methyl 2-(3-(1-(4-cyanophenylsulfonyl)piperidin-3-yl)phenyl)acetic acid
(Compound 30A)
and 2-(3-(1-(4-cyanophenylsulfonyl)piperidin-3-yl)phenyl)acetic acid (Compound
30B)
O
OH N
N1O
[0251] The title compound(s) were obtained using the same experimental
procedure
described for 2-(3-(1-(phenylsulfonyl)piperidin-3-yl)phenyl)acetic acid using
4-cyano
phenylsulfonyl chloride. Compound 30B LC/MS (Method A) Rt = 3.467 min. MS
(m/z)
385 (M + H).
Example 31
Methyl 2-(3-(1-(4-tert-butylphenylsulfonyl)piperidin-3-yl)phenyl)acetate
(Compound 31A)
and 2-(3-(1-(4-tert-butylphenylsulfonyl)piperidin-3-yl)phenyl)acetic acid
(Compound 31B)
88
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
O
OH
N.O
[0252] The title compound(s) were obtained using the same experimental
procedure
described for 2-(3 -(1 -(phenylsulfonyl)piperidin-3 -yl)phenyl)acetic acid
using 4-tert-butyl
phenyl sulfonyl chloride. LC/MS (Method A) Rt = 4.110 min. MS (m/z) 416 (M+H).
Example 32
Methyl 2-(3-(1-(2,4-dichlorophenylsulfonyl)piperidin-3-yl)phenyl)acetate
(Compound 32A)
and 2-(3-(1-(2,4-dichlorophenylsulfonyl)piperidin-3-yl)phenyl)acetic acid
(Compound 32B)
O
OH CI
O~
N0 CI
[0253] The title compound(s) were obtained using the same experimental
procedure
described for 2-(3-(1-(phenylsulfonyl)piperidin-3-yl)phenyl)acetic acid using
2,4-dichloro
phenyl sulfonyl chloride. Compound 32B LC/MS (Method A) Rt = 3.921 inin. MS
(m/z)
428 (M+H).
Example 33
Methyl 2-(3-(1-(4-methoxyphenylsulfonyl)piperidin-3-yl)phenyl)acetate
(Compound 33A)
and 2-(3-(1-(4-methoxyphenylsulfonyl)piperidin-3-yl)phenyl)acetic acid
(Compound 33B)
O
OH
O
O\ ~ ~
NO
89
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
[0254] The title compound(s) were obtained using the same experimental
procedure
described for 2-(3 -(1 -(phenylsulfonyl)piperidin-3 -yl)phenyl)acetic acid
using 4-methoxy
phenylsulfonyl chloride. Compound 33B LC/MS (Method A) Rt = 3.518 min. MS
(m/z)
390 (M+H).
Example 34
Methyl2-(3-(1-(o-tolylsulfonyl)piperidin-3-yl)phenyl)acetate (Compound 34A)
and
2-(3-(1-(o-tolylsulfonyl)piperidin-3-yl)phenyl)acetic acid (Compound 34B)
0
OH
N1SO
[0255] The title compound(s) were obtained using the same experimental
procedure
described for 2-(3-(1-(phenylsulfonyl)piperidin-3-yl)phenyl)acetic acid using
2-methyl
phenylsulfonyl chloride. LC/MS (Method A) Compound 34B Rt = 3.614 min. MS
(m/z)
374 (M+H)
Example 35
Methyl 2-(3-(1-(2-chlorophenylsulfonyl)piperidin-3-yl)phenyl)acetate (Compound
35A) and
2-(3-(1-(2-chlorophenylsulfonyl)piperidin-3-yl)phenyl)acetic acid (Compound
35B)
0
OH
/ N,SO CI
[0256] The title compound(s) were obtained using the same experimental
procedure
described for 2-(3-(1-(phenylsulfonyl)piperidin-3-yl)phenyl)acetic acid using
2-chloro
phenylsulfonyl chloride. Compound 35B LC/MS (Method A) Rt = 3.659 min. MS
(m/z)
394 (M+H)
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
Example 36
Methyl2-(3-(1-(4-ethylphenylsulfonyl)piperidin-3-yl)phenyl)acetate (Compound
36A) and
2-(3 -(1 -(4- ethylphenylsul fonyl)piperidin-3 -yl)phenyl) acetic acid
(Compound 36B)
0
OH
O~
No
[0257] The title compound(s) were obtained using the same experimental
procedure
described for 2-(3-(1-(phenylsulfonyl)piperidin-3-yl)phenyl)acetic acid using
4-ethyl phenyl
sulfonyl chloride. Compound 36B LC/MS (Method A) Rt = 3.849 min. MS (m/z) 388
(M+H).
Example 37
Methyl 2-(3-(1-(phenethylsulfonyl)piperidin-3-yl)phenyl)acetate (Compound 37A)
and
2-(3-(1-(phenethylsulfonyl)piperidin-3-yl)phenyl)acetic acid (Compound 37B)
0
OH
O~
N.O
[0258] The title compound(s) were obtained using the same experimental
procedure
described for 2-(3 -(1 -(phenylsulfonyl)piperidin-3-yl)phenyl)acetic acid
using phenethyl
sulfonyl chloride. Compound 37B LC/MS (Method A) Rt = 3.628 min. MS (m/z) 388
(M+H).
Exainple 38
91
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
Methyl2-(3-(1-(2-chloro-4-fluorophenylsulfonyl)piperidin-3-yl)phenyl)acetate
(Compound
38A) and 2-(3-(1-(2-chloro-4-fluorophenylsulfonyl)piperidin-3-yl)phenyl)acetic
acid
(Compound 38B)
O
OH F
O~ \
N/0 CI
[0259] The title compound(s) were obtained using the same experimental
procedure
described for 2-(3 -(1 -(phenylsulfonyl)piperidin-3-yl)phenyl)acetic acid
using
2-chloro-4-fluoro phenylsulfonyl chloride. Compound 38B LC/MS (Method A) Rt =
3.696
min. MS (m/z) 412 (M+H).
Example 39
Methyl2-(3-(1-(butylsulfonyl)piperidin-3-yl)phenyl)acetate (Compound 39A) and
2-(3-(1-(butylsulfonyl)piperidin-3-yl)phenyl)acetic acid (Compound 39B)
0
OH
~ 0 N0
O
[0260] The title compound(s) were obtained using the same experimental
procedure
described for 2-(3-(1-(phenylsulfonyl)piperidin-3-yl)phenyl)acetic acid using
4-butylsulfonyl
chloride. Compound 39B LC/MS (Method A) Rt = 3.454 min. MS (m/z) 340 (M+H).
Example 40
Methyl 2-(3-(1-(4-(inethylsulfonyl)phenylsulfonyl)piperidin-3-
yl)phenyl)acetate (Compound
40A) and 2-(3-(1-(4-(inethylsulfonyl)phenylsulfonyl)piperidin-3-
yl)phenyl)acetic acid
(Compound 40B)
92
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
0
OH 0S
ia ~0
N'O
[0261] The title compound(s) were obtained using the same experimental
procedure
described for 2-(3-(1-(phenylsulfonyl)piperidin-3-yl)phenyl)acetic acid using
(methylsulfonyl)phenylsulfonyl chloride. Compound 40B LC/MS (Method A) Rt =
3.293
min. MS (m/z) 438 (M+H).
Example 41
Methyl2-(3-(1-(3,4-dichlorophenylsulfonyl)piperidin-3-yl)phenyl)acetate
(Compound 41A)
and 2-(3-(1-(3,4-dichlorophenylsulfonyl)piperidin-3-yl)phenyl)acetic acid
(Compound 41B)
0
CI
OH
CI
O~ \
N/\O
[0262] The title compound(s) were obtained using the same experimental
procedure
described for 2-(3-(1-(phenylsulfonyl)piperidin-3-yl)phenyl)acetic acid using
3,4-dichlorophenyl sulfonyl chloride. Compound 41B LC/MS (Method A) Rt = 3.928
inin.
MS (m/z) 428 (M+H).
Example 42
Methyl 2-(3-(1-(4-fluoro-2-methylphenylsulfonyl)piperidin-3-yl)phenyl)acetate
(Compound
42A) and 2-(3-(1-(4-fluoro-2-methylphenylsulfonyl)piperidin-3-yl)phenyl)acetic
acid
(Compound 42B)
93
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
O
OH F
N.O
[0263] The title compound(s) were obtained using the same experimental
procedure
described for 2-(3-(1-(phenylsulfonyl)piperidin-3-yl)phenyl)acetic acid using
2-methyl-4-fluoro phenylsulfonyl chloride. Compound 42B LC/MS (Method A) Rt =
3.686
min. MS (m/z) 392 (M+H).
Example 43
Methyl 2-(3-(1-(3-chlorophenylsulfonyl)piperidin-3-yl)phenyl)acetate (Compound
43A) and
2-(3-(1-(3-chlorophenylsulfonyl)piperidin-3-yl)phenyl)acetic acid (Compound
43B)
0
OH
/
NS\ \ CI
[0264] The title compound(s) were obtained using the same experimental
procedure
described for 2-(3-(1-(phenylsulfonyl)piperidin-3-yl)phenyl)acetic acid using
3-chloro
phenylsulfonyl chloride. Compound 43B LC/MS (Method A) Rt = 3.706 inin. MS
(in/z)
394 (M+H).
Example 44
Methyl 2-(3-(1-(in-tolylsulfonyl)piperidin-3-yl)phenyl)acetate (Compound 44A)
and
2-(3-(1-(in-tolylsulfonyl)piperidin-3-yl)phenyl)acetic acid (Compound 44B)
0
OH
O /
\ ~
N.SO
94
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
[0265] The title compound(s) were obtained using the same experimental
procedure
described for 2-(3-(1-(phenylsulfonyl)piperidin-3-yl)phenyl)acetic acid using
3-methyl
phenylsulfonyl chloride. Compound 44B LC/MS (Method A) Rt = 3.654 min. MS
(m/z)
374 (M+H).
Example 45
2-(3-(1-(4-fluorophenylcarbamoyl)piperidin-3-yl)phenyl)acetic acid (Compound
45B)
0
OH
O
N N/ F
~~
H
Scheme 9
O OCN
O O
o / \
O OH
NH Step A Step B \ ~
N N N N
H H
A9 B9 C9
Step A: Methyl 2-(3-(1-(4-fluorophenylcarbamoyl)piperi din- 3 -yl)phenyl)
acetate
(Compound 45A)
[0266] To a solution of 100 mg (0.429 mmol, 1.0 equivalent) of methyl
2-(3-(piperidin-3-yl)phenyl)acetate (A9) in EtOAc (5 ml) was added 1.2
equivalents of
1-fluoro-4-isocyanatobenzene (0.514 mmol, 70.5 mg) and 2.0 equivalents (0.120
ml) of
triethylamine. Reaction was heated in the microwave to 150 C at 300 W power
for 10 min.
Reaction mixture was washed with water 3 times. Combined aqueous phase was
extracted
with EtOAc. Combined organic phase was washed with brine, dried over sodium
sulfate, and
concentrated to dryness on the RotorVap. Yield = 160 mg of crude methyl
2 -(3 -(1 -(4-fluorophenyl carbamoyl)piperidin- 3 -yl)phenyl) acetate as an
orange oil. MS (m/z)
371 (M+H)
Step B: Methyl 2-(3-(1-(4-fluorophenylcarbainoyl)piperidin-3-yl)phenyl)acetate
(Compound 45B)
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
[0267] Crude methyl 2 -(3 -(1 -(4-fluorophenylcarbamoyl)piperidin-3 -
yl)phenyl) acetate from
step A was dissolved in THF (3 ml) and aqueous KOH (1.0 N, 3 ml) was added.
Reaction
was stirred for 4 hours. Reaction was acidified to pH 2-4 with 1.0 N aqueous
HCl and
extracted with EtOAc. Organic extracts were washed with brine, dried over
sodium sulfate,
and concentrated to dryness. Crude yield = 185 mg (0.52 mmol, >100%). Final
product was
purified by HPLC using 0.05% formic acid modifier. Final yield = 29.35 mg
(0.08 mmol).
LC/MS (Method A) Rt = 3.274 min. MS (m/z) 357 (M+H)
Example 46
2-(3-(1-(4fluorophenylsulfonyl)-4-methylpiperidin-3-yl)phenyl)acetic acid
(Compound
46D)
0
HO
F
O
SII \
N~
Step A: Methyl 2-(3-(4-methylpyridin-3-yl)phenyl)acetate (Compound 46A)
0
N
[0268] To 3 phenyl acetic acid boronic ester (0.15g, 0.57 mmol) in 0.5mL DME
and
0.25mL water is added 4-methyl 3-bromo pyridine (0.119g, 0.69ininol) sodium
carbonate
(0.121 g, 1.14mrnol) and palladium tetrakis (0.032g, 0.028mmo1) and stirred at
90 C for 3
hours. The base is filtered away, and the solvents removed in vacuo. The
resulting material
is resuspended in MeOH (5mL) and HCl (g) is bubbled through. The solvent is
evaporated
away, and the material taken up in water and extracted 2X with DCM. The pH is
increased to
14 and the aqueous is extracted 3X with DCM. The combined basic extracts are
dried and
96
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
the resulting material used without further purification. LC/MS (Method A) Rt
= 1.97 min.
MS: 242 m/z. (M+H)
Step B: Methyl 2-(3-(4-methylpiperidin-3-yl)phenyl)acetate (Compound 46B)
0
NH
[0269] To methyl 2-(3-(4-methylpyridin-3-yl)phenyl)acetate (0.060g, 0.25mmo1)
in MeOH
(3mL) is added catalytic Pt02. The mixture is evacuated 3X and flushed with
H2. The
mixture is stirred under balloon pressure for 16 hours at which time the
reaction is judged
complete by LC/MS (small amount of over-reduction observed as well). The
catalyst is
filtered away and the solvents removed in vacuo. The title compound is
achieved without
further purification. LC/MS (Method A) Rt 2.01 min. MS: 248 m/z. (M+H).
Step C: Methyl 2-(3-(1-(4fluorophenylsulfonyl)-4-methylpiperidin-3-
yl)phenyl)acetate
(Compound 46C)
0
F
O
SI I \
N_1 O
[0270] To methyl 2-(3-(4-methylpiperidin-3-yl)phenyl)acetate (0.036g,
0.14mmol) in linL
DCM is added DIEA (0.036g, 0.28mmol) and 4-fluoro phenyl sulfonyl chloride
(0.029g,
0.15mmo1). The reaction stirs 16 hours and is then worked up by drying and
purifying by
HPLC eluting with AcCN/water both modified with 0.05% formic acid. LC/MS
(Method A)
Rt 4.11 inin. MS: 406 m/z (M+H).
97
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
Step D:
2-(3-(1-(4fluorophenylsulfonyl)-4-inethylpiperidin-3-yl)phenyl)acetic acid
(Compound 46D)
0
HO
F
g
I I N[0271] To methyl2-(3-(1-(4fluorophenylsulfonyl)-4-methylpiperidin-3-
yl)phenyl)acetate
(0.008g, 0.02mmol) in 2mL MeOH is added 1mL 3N NaOH. The reaction stirs 16
hours.
Acidify with 1N HCl to pH 1, and then dry. The title compound is extracted
into DCM and
used without further purification. LC/MS (Method A) Rt 3.69 min. MS: 391 m/z
(390 m/z
negative ion). 'H NMR (300MHz, CDC13) 7.8-7.85 (2H, m); 7.1-7.3 (6H, m); 3.65
(3H, s);
3.2-3.3 5(2H, m); 3.1-3.18 (2H, m); 2.95-3.05 (1 H, m); 1.95-2.05 (1 H, m);
2.8-2.9 (1 H, m);
2.6-2.7 (1H, m); 0.7 (0.3H, d J=15Hz); 0.6 (2.7H, d J=15Hz).
Example 47
Methyl 2-(3-(1-(4fluorophenylsulfonyl)-2-methylpiperi din-3 -yl)phenyl)
acetate (Compound
47A) and 2-(3-(1-(4fluorophenylsulfonyl)-2-methylpiperidin-3-yl)phenyl)acetic
acid
(Compound 47B)
0
HO
F
O
II
N~0
[0272] 2-(3-(1-(4fluorophenylsulfonyl)-2-inethylpiperidin-3-yl)phenyl)acetic
acid is
prepared using the same methodology as was used to prepare using 2-methyl-3-
bromo
pyridine (steps A-D). LC/MS (Method A) Rt 3.64min; MS: 391 m/z (390 m/z
negative ion).
Example 48
98
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
Methyl2-(3-(1-(4fluorophenylsulfonyl)-6-methylpiperidin-3-yl)phenyl)acetate
(Compound
48A) and 2-(3-(1-(4fluorophenylsulfonyl)-6-methylpiperidin-3-yl)phenyl)acetic
acid
(Compound 48B)
0
HO
F
II ~
N/
[0273] 2-(3 -(1 -(4fluorophenylsulfonyl)-6-methylpiperidin-3 -yl)phenyl)
acetic acid is
prepared using the same methodology as was used to prepare 46D using 6-methyl-
3-bromo
pyridine (steps A-D). LC/MS (Method A) Rt 3.64 min; MS: 391 m/z (390 m/z
negative ion).
Example 49
Scheme 5
0 0 0
CH3 CH3
0' 0 Step A 0~ Step B OH
n
+ CI-S-Rs \
~ I ~ Rs ~N Rs
NH ~ N-SO N.SO
A5 B5 C5
General procedure for Step A:
[0274] To a solution of 100 mg (0.429 mmol, 1.0 equivalent) of inethyl
2-(3-(piperidin-3-yl)phenyl)acetate (A5) in DCM (4 ml) was added 1.2
equivalents of the
sulfonyl chloride and 2.5-10 equivalents (150- of triethyl amine. Reaction was
stirred at
room temperature for 12-18 hours. Reaction mixture was concentrated to dryness
on
RotorVap. The residue was brought up in EtOAc, washed with water and brine,
dried over
sodium sulfate and concentrated to dryness. The crude product was taken to
next step as is.
99
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
General procedure for Step B:
[0275] The intermediate from step A was dissolved in THF (2-3 ml) and aqueous
KOH (1.0
N, 3 ml) was added. Reaction was stirred for 30 min. to 12 hours until
hydrolysis was
complete. Reaction was acidified to pH 2-4 with 1.0 N aqueous HCl and
extracted with
EtOAc. Organic extracts were washed with brine, dried over sodium sulfate, and
concentrated to dryness. Final products were purified by HPLC.
Example 49
Methyl 2-(3-(1-(4-chlorophenylsulfonyl)piperidin-3-yl)phenyl)acetate (Compound
49A) and
2-(3-(1-(4-chlorophenylsulfonyl)piperidin-3-yl)phenyl)acetic acid (Compound
49B):
O
OH CI
O /
\
NIO
[0276] The title compound(s) were obtained using the same general experimental
procedure described for step A and step B for making
2-(3-(1-(phenylsulfonyl)piperi din-3 -yl)phenyl) acetic acid, except 4-chloro
phenyl sulfonyl
chloride was used. Crude yield = 81 mg (0.206 mmol, 48% over 2 steps). Crude
product
Compound 49B was purified by HPLC using 0.05% TFA modifier. Final yield = 26.1
mg
(0.066 mmol). LC/MS (Method A) Rt = 3.792 min. MS (m/z) 394 (M+H).
Example 50
Methyl 2-(3-(1-(3,5-dichlorophenylsulfonyl)piperidin-3-yl)phenyl)acetate
(Compound 50A)
and 2-(3-(1-(3,5-dichlorophenylsulfonyl)piperidin-3-yl)phenyl)acetic acid
(Compound 50B):
O
OH CI
CI
100
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
[0277] The title compound(s) were obtained using the same general experimental
procedure described for step A and step B for making
2-(3-(1-(phenylsulfonyl)piperidin-3-yl)phenyl)acetic acid, except 3,5-
dichlorophenylsulfonyl
chloride was used. Compound 50B: Crude yield = 150 mg (0.35 mmol, 81.6% over 2
steps). Crude product was purified by HPLC using 0.05% TFA modifier. Final
yield = 15
mg (0.035 mmol). LC/MS (Method A) Rt = 3.993 min. MS (m/z) 428 (M + H).
Example 51
Methyl 2-(3-(1-(2,3-dichlorophenylsulfonyl)piperidin-3-yl)phenyl)acetate
(Compound 51A)
and 2-(3 -(1-(2,3 -dichlorophenylsulfonyl)piperidin-3 -yl)phenyl) acetic acid
(Compound 51B)
0
OH
CI
O CI
[0278] The title compound(s) were obtained using the same general experimental
procedure described for step A and step B for making
2-(3-(1-(phenylsulfonyl)piperidin-3-yl)phenyl)acetic acid, except 3,5-
dichlorophenylsulfonyl
chloride was used. Compound 51B: Crude yield = 180 mg (0.42 mmol, 97% over 2
steps).
Crude product was purified by HPLC using 0.05% TFA modifier. Final yield = 75
mg (0.175
mmol). LC/MS (Method A) Rt = 3.870 min. MS (m/z) 429 (M+H)
Example 52
Methyl 2-(3-(1-(thiophen-2-ylsulfonyl)piperidin-3-yl)phenyl)acetate (Compound
52A) and
2-(3-(1-(thiophen-2-ylsulfonyl)piperidin-3-yl)phenyl)acetic acid (Compound
52B)
0
OH
O DN
N.SO
101
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
[0279] This compound(s) were using General procedures for step A and step B
except
thiophen-2-ylsulfonyl chloride was used. Compound 52B: Crude yield = 101 mg
(0.276
mmol, 64.4% over 2 steps). Crude product was purified by HPLC using 0.05%
Formic Acid
modifier. Final yield = 49 mg (0.134 inmol). LC/MS (Method A) Rt = 3.426 min.
MS (m/z)
366 (M + H).
Example 53
Methyl2-(3-(1-(thiophen-3-ylsulfonyl)piperidin-3-yl)phenyl)acetate (Compound
53A) and
2-(3-(1-(thiophen-3-ylsulfonyl)piperidin-3-yl)phenyl)acetic acid (Compound
53B)
0
OH
S
N.S
[0280] The title compound(s) were obtained using the same general experimental
procedure described for step A and step B for making
2-(3-(1-(phenylsulfonyl)piperidin-3-yl)phenyl)acetic acid, except starting
with 133 mg of
intermediate A5 (0.57 mmol) and thiophen-3-ylsulfonyl chloride. All other
reagents were
scaled up accordingly. Compounds 53B: Crude yield = 185.6 mg (0.35 mmol, 61.4%
over
2 steps). Crude product was purified by HPLC using 0.05% TFA modifier. Final
yield =
80.5 mg (0.22 mmol). LC/MS (Method A) Rt = 3.374 min. MS (m/z) 366 (M +H)
Example 54
Methyl2-(3-(1-(5-chlorothiophen-2-ylsulfonyl)piperidin-3-
yl)phenyl)acetate(Compound
54A) and 2-(3-(1-(5-chlorothiophen-2-ylsulfonyl)piperidin-3-yl)phenyl)acetic
acid
(Compound 54B)
O
OH CI
O S
~
N.SO
102
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
[0281] The title compound(s) were obtained using the same general experimental
procedure described for step A and step B for making
2-(3-(1-(phenylsulfonyl)piperidin-3-yl)phenyl)acetic acid, except
5-chlorothiophen-2-ylsulfonyl chloride was used. Compound 54B: Crude yield =
111 mg
(0.277 mmol, 65% over 2 steps). Crude product was purified by HPLC using 0.05%
TFA
modifier. Final yield = 32 mg (0.08 mmol). LC/MS (Method A) Rt = 3.798 min. MS
(m/z)
400 (M+H).
Example 55
Methyl2-(3-(1-(5-bromothiophen-2-ylsulfonyl)piperidin-3-yl)phenyl)acetate
(Compound
55A) and 2-(3-(1-(5-bromothiophen-2-ylsulfonyl)piperidin-3-yl)phenyl)acetic
acid
(Compound 55B)
0
OH Br
O S \
,. ~
N.So
[0282] The title compound(s) were obtained using the same general experimental
procedure described for step A and step B for making
2-(3-(1-(phenylsulfonyl)piperidin-3-yl)phenyl)acetic acid, except
5-bromothiophen-2-ylsulfonyl chloride was used. Compound 55B: Crude yield =
124 mg
(0.279 mmol, 65% over 2 steps). Crude product was purified by HPLC using 0.05%
TFA
modifier. Final yield = 50 mg (0.113 mmol). MS (m/z) 446 (M + 2).
103
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
Example 56
Methyl 2-(3-(1-(4-nitrophenylsulfonyl)piperidin-3-yl)phenyl)acetate (Compound
56A) and
2-(3-(1-(4-nitrophenylsulfonyl)piperidin-3-yl)phenyl)acetic acid (Compound
56B):
O
2
OH NO
O\
~ \
/ N
O
O
[0283] The title compound(s) were obtained using the same general experimental
procedure described for step A and step B for making
2-(3-(1-(phenylsulfonyl)piperidin-3-yl)phenyl)acetic acid, except 4-
nitrophenylsulfonyl
chloride was used. Compound 56B: Crude yield = 160 mg (0.396 mmol, 92% over 2
steps).
Crude product was purified by HPLC using 0.05% TFA modifier. Final yield = 7
mg (0.017
mmol). LC/MS (Method A) Rt = 3.591 min. MS (m/z) 405 (M + H)
Example 57
Methyl 2-(3-(1-(benzofuran-2-ylsulfonyl)piperidin-3-yl)phenyl)acetate
(Compound 57A)
and 2-(3 -(1 -(benzofuran-2-ylsulfonyl)piperidin-3 -yl)phenyl)acetic acid
(Compound 57B):
O
OH
O
O~
N SO
[0284] The title compound(s) were obtained using the same general
experiinental
procedure described for step A and step B for making
2-(3-(1-(phenylsulfonyl)piperidin-3-yl)phenyl)acetic acid, except benzofuran-2-
ylsulfonyl
chloride was used. Compound 57B: Crude yield = 66 mg (0.165 mmol, 38.5% over 2
steps). Crude product was purified by HPLC using 0.05% TFA modifier. Final
yield = 25
mg (0.0625 mmol). LC/MS (Method A) Rt = 3.759 min. MS (m/z) 400 (M +H)
104
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
Example 58
Methyl 2-(3-(1-(pyri din-3-ylsulfonyl)piperidin-3-yl)phenyl)acetate (Compound
58A) and
2-(3-(1-(pyridin-3-ylsulfonyl)piperidin-3-yl)phenyl)acetic acid (Compound 58B)
0
OH N
O~
N5 O
[0285] The title compound(s) were obtained using the same general experimental
procedure described for step A and step B for making
2-(3 -(1 -(phenylsulfonyl)piperidin-3 -yl)phenyl) acetic acid, except pyridin-
3-ylsulfonyl
chloride was used. Compound 58B: Crude yield = 108.5 mg (0.30 mmol, 70.2% over
2
steps). Crude product was purified by HPLC using 0.05% TFA modifier. Final
yield = 23
mg (0.064 mmol). LC/MS (Method A) Rt = 3.080 min. MS (m/z) 361 (M +H).
Example 59
Methyl 2-(3-(1-(naphthalen-1-ylsulfonyl)piperidin-3-yl)phenyl)acetate
(Compound 59A)
and 2-(3-(1-(naphthalen-1-ylsulfonyl)piperidin-3-yl)phenyl)acetic acid
(Compound 59B)
0
OH
O\
NO
[0286] The title compound(s) were obtained using the same general experimental
procedure described for step A and step B for making
2-(3 -(1 -(phenylsulfonyl)piperidin-3 -yl)phenyl)acetic acid, except
naphthalen-l-ylsulfonyl
chloride was used. Crude yield = 105 mg (0.257 mmol, 59.8% over 2 steps).
Crude product
was purified by HPLC using 0.05% Formic Acid modifier. Final yield = 60 mg
(0.147
mmol). MS data is not available. 'H NMR (300 MHz, CDC13) b; 8.76 (d, 1H,
aromatic);
8.22 (dd, 1 H, aromatic); 8.07 (d, 1 H, aromatic); 7.95 (d, 1 H, aromatic);
7.7-7.51 (m, 3H,
aromatic); 7.32-7.23 (in, 1H); 7.16 (d, 1H, aromatic); 7.07 (d, 2H, aromatic);
3.97 (m, 2H);
105
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
3.62 (s, 2H); 2.87-2.72 (tt, 1H); 2.68-2.53 (m, 2H); 1.95 (d, 1H); 1.88-1.59
(m, 2H):
1.56-1.38 (qd, 1H).
Example 60
Methyl {3-[1-(Naphthalene-2-sulfonyl)-piperidin-3-yl]-phenyl}-acetate
(Compound 60A)
and {3-[1-(Naphthalene-2-sulfonyl)-piperidin-3-yl]-phenyl}-acetic acid
(Compound 60B)
HO2C
0~O
N.S
[0287] The title compound(s) were obtained using the same general experimental
procedure described for step A and step B for making
2-(3-(1-(phenylsulfonyl)piperidin-3-yl)phenyl)acetic acid, except naphthalen-1-
ylsulfonyl
chloride was used. MS m/z 410 (M+H).
Example 61
Methyl 2-(3-(1-(benzylsulfonyl)piperidin-3-yl)phenyl)acetate (Compound 61A)
and
2-(3-(1-(benzylsulfonyl)piperidin-3-yl)phenyl)acetic acid (Compound 61 B):
0
OH
O
N.SO
[0288] The title compound(s) were obtained using the same general experimental
procedure described for step A and step B for making
2-(3-(1-(phenylsulfonyl)piperidin-3-yl)phenyl)acetic acid. Crude yield = 200
mg (0.42
mmol, >100% over 2 steps). Crude product was purified by HPLC using 0.05% TFA
modifier. Final yield = mg (0.175 mmol). LC/MS (Method A) Rt = 3.423 min. MS
(m/z)
374 (M +H)
106
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
Example 62
Methyl (E)-2-(3-(1-(styrylsulfonyl)piperidin-3-yl)phenyl)acetate (Compound
62A) and
(E)-2-(3-(1-(styrylsulfonyl)piperidin-3-yl)phenyl)acetic acid (Compound 62B):
0
OH
O~
NO
[0289] The title compound(s) were obtained using the same general experimental
procedure described for step A and step B for making
2-(3-(1-(phenylsulfonyl)piperidin-3-yl)phenyl)acetic acid, except styryl
sulfonyl chloride was
used. The product after step A was purified by HPLC using 0.05% TFA modifier.
Final
product was also purified by HPLC using 0.05% TFA modifier. Final yield = 42
mg (0.109
mmol). LC/MS (Method A) Rt = 3.663 min. MS (m/z) 386 (M +H).
Example 63
Methyl 2-(3 -(1 -tosyldecahydroquinolin-3 -yl)phenyl) acetate (Compound 63C)
and
2-(3 -(1 -tosyldecahydroquinolin-3 -yl)phenyl)acetic acid (Compound 63D):
Scheme 10
0 0
0
N OI O
O (HO)2B \ \ I \ \
Step B
Step A N NH
Br
0
O o
CIO2S OH
O O /
Step D O /J:
Step C ~S \ I ~ N SO
NH N ~O
107
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
Step A: Methyl2-(3-(quinolin-3-yl)phenyl)acetate (Compound 63A)
[0290] Dissolved in 2 ml of DME in a microwave reactor were 200 mg (1.0 eq,
0.87 mmol)
of methyl 2-(3-bromophenyl)acetate, 180 mg (1.2 eq, 1.04 mmol) of quinolin-3-
ylboronic
acid, 2 ml (4.5 eq) of 2M aq. sodium carbonate, and 48 mg (5 mol%, 0.043 mmol)
of
palladium tetrakis, with the catalyst added last. Reaction was heated in the
microwave to
180 C at 300 W power for 7 min. Reaction was quenched with water, extracted
with EtOAc,
and concentrated to dryness on RotorVap. Yield = 413 mg (1.49 mmol, >100%) of
crude
methyl 2-(3-(quinolin-3-yl)phenyl)acetate as a thin yellow oil. MS (m/z) 278
(M+H).
Step B: Methyl 2-(3-(decahydroquinolin-3-yl)phenyl)acetate (Compound 63B)
[0291] Crude methyl 2-(3-(quinolin-3-yl)phenyl)acetate from step A was
dissolved in 10
ml methanol. To this solution were added catalytic amounts of concentrated HC1
and
platinum (IV) oxide hydrate. The vessel was charged to 10 psi on the Parr
hydrogenator and
agitated for 5 hours. Reaction was filtered through a pad of CELITE and the
filtrate was
concentrated to dryness. Yield = 145 mg (0.5 mmol) crude methyl
2-(3-(decahydroquinolin-3-yl)phenyl)acetate as a yellow oil. MS (m/z) 288
(M+H).
Step C: Methyl 2-(3-(1-tosyldecahydroquinolin-3-yl)phenyl)acetate (Compound
63C)
[0292] Crude methyl 2-(3-(decahydroquinolin-3-yl)phenyl)acetate (145 mg, 1.0
eq, 0.5
mmol) from step B was dissolved in 10 ml of DCM. To this solution was added
105.8 mg
(0.55 mmol, 1.1 eq) of 4-methylbenzene-l-sulfonyl chloride, and 0.176 ml (1.26
mmol, 2.5
eq) of triethylainine. Reaction was stirred for 18 hours at room temperature.
Reaction was
quenched with water and extracted with EtOAc 3 times. Combined organic phase
was dried
over sodium sulfate and concentrated to dryness. Yield = 171 mg (0.38 mmol) of
crude
methyl 2-(3-(1-tosyldecahydroquinolin-3-yl)phenyl)acetate as a yellow oil. MS
(m/z) 442
(M+H).
Step D: 2-(3-(1-tosyldecahydroquinolin-3-yl)phenyl)acetic acid (Compound 63D)
108
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
[0293] Crude methyl2-(3-(1-tosyldecahydroquinolin-3-yl)phenyl)acetate from
step 3 (171
mg) was dissolved in 3 ml of THF and 3 ml of 1 N aq. KOH was added. Reaction
was stirred
for 18 hours at room temperature. Reaction was acidified to pH 2-4 with 1.0 N
aqueous HCl
and extracted with EtOAc. Organic extracts were washed with brine, dried over
sodium
sulfate, and concentrated to dryness. Final product was purified by HPLC using
0.05%
formic Acid modifier. Final yield = 9.27 mg (0.021 mmol). LC/MS (Method A) RT
= 4.048
min. MS (m/z) 428 (M+H).
Example 64
Methyl2-(3,4-dichloro-5-(1-(4-fluorophenylsulfonyl)piperidin-3-
yl)phenyl)acetate
(Compound 6C) and
2-(3,4-dichloro-5-(1-(4-fluorophenylsulfonyl)piperidin-3-yl)phenyl)acetic acid
(Compound
64D)
Scheme 11:
O
1. Br2, MeOH, HOAc 1. NBS, CC14, AIBN
CI 2. tBuONO, CuCl2 CI Br 2. Me3NO, CH3CN CI Br
NH2 CI ci
Me02C (HO)2g ~ Me02C
1. Jones oxid ~ N
2. (COCI)2 I CsF I
3. TMSCHN2
4. AgOBz, Et3N CI Br Pd(OAc)2, PPh3 CI I N
CI CI
MeOH
1. H2, Pt20 HO2C
2. O, O,Et3N O, 10
CI'S/ CI N'
S/
F F
3. LiOH
Step A: 1-bromo-2,3-dichloro-5-methylbenzene (Compound 64A)
109
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
CI Br
CI
[0294] To a solution of commercially available 2-chloro-4-methylaniline (25.2
g, 177.8
mmol) in MeOH (80 mL)-HOAc (26 mL) at 0 C was added dropwise bromine (9.1 mL,
177.8 mmol) in HOAc (80 mL). The mixture was stirred for 3 hours before NaOH
(10%, 100
mL) and water were added. The mixture (pH 5) was extracted with EtOAc, and
dried over
Na2SO4. Solvent was removed to give a dark brown solid. A solution of the
above solid
(7.81 g, 35.4 mmol) in CH3CN (50 mL) was added dropwise in 25 min. to a
solution of CuC12
(5.71 g, 42.5 mmol) and t-butyl nitrite (t-BuONO) (6.32 mL, 53.1 mmol) at 65
C. Gas
evolution was observed. Upon completion of addition, gas evolution ceased and
the mixture
was cooled to room temperature and stirred for 15 h. Solvent was removed and
the residue
purified on silica gel to give a white needle (6.35 g, 75%). MS (Method B)
Rt=4.47 min.
Step B: 2-(3 -bromo-4,5 -dichlorophenyl) acetic acid (Compound 64B)
HO2C
I
CI Br
CI
[0295] To 1-bromo-2,3-dichloro-5-inethylbenzene (2.81 g, 11.7 inmol) was added
NBS
(2.29 g, 12.8 mmol), AIBN (192 mg, 1.17 mmol), and CC14 (50 mL). The mixture
was
stirred at rt for 30 min. and then at 80 C for another 17 h. Solvent was
removed and the
residue purified on silica gel to give 3.32 g (89%) of a white solid. To a
solution of the above
solid (674 mg, 2.1 mmol) in CH3CN (5 mL) at 0 C was added trimethyl amine N-
oxide (317
mg, 4.2 mmol). The mixture was wanned up to rt and stirred for 30 min. and
then purified
using silica gel chromatography to give 3-bromo-4,5-dichlorobenzaldehyde as a
white solid
(219 mg). To thus obtained aldehyde (219 mg, 0.86 mmol) was added acetone (6
mL) and
Jone's reagent (1.35 mL, -0.7 M) and stirred for 40 min. before MeOH (6 mL)
was added
and the mixture stirred for another 5 min. CHZCl2 and water were added and the
aqueous
layer was extracted with CH2C12. The combined organic layer was dried over
Na2SO4.
110
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
Removal of solvent gave a white solid (226 mg, 0.837 mmol) in 97% yield. MS
(Method B)
Rt=3.86 min, (m/z) 268.9 (M-H).
Step C: Methyl2-(3,4-dichloro-5-(pyridin-3-yl)phenyl)acetate (Compound 64C)
MeO2C
CI ~ I ~ N
CI
[0296] The above acid was dissolved in CH2C12 (8 mL) and oxalyl chloride (95
L, 1.09
mmol) was added, followed by a drop of DMF. After 16 h, solvent was removed
and the
residue was subjected to vacuum for 20 min. and dissolved in THF (8 mL) and
cooled to 0 C.
DIEA (291 gL, 1.67 mmol) was then added, followed by TMSCHN2 (1 mL, 2.09
mmol).
After 2 h, solvent was removed and residue purified on silica gel to give 97
mg (39% for 2
steps) of an off-white solid. To a solution of this solid (97 mg, 0.33 mmol)
in MeOH (4.6
mLO was added dropwise a solution of AgOBz (45 mg, 0.198 mmol) in Et3N (0.9
mL). After
3 d, solvent was removed and the residue purified on silica gel to give methyl
2-(3-bromo-4,5-dichlorophenyl)acetate as a colorless oil (34.5 mg). To thus
obtained methyl
ester methyl (34.5 mg, 0.13 mmol) was added pyridin-3-ylboronic acid (32 mg,
0.26 mmol),
Pd(OAc)Z (2 mg, 0.0091 mmol), PPh3 (7 mg, 0.027 mmol), CsF (69 mg, 0.455
mmol), DME
(1 inL), isopropyl alcohol (0.5 mL), and water (0.5 inL). The reaction vial
was heated at
95 C for 20 h. The mixture was purified directly on silica gel to give methyl
2-(3,4-dichloro-5-(pyridin-3-yl)phenyl)acetate as a colorless oil (11.5 mg,
30% for 2 steps).
MS (Method B) Rt=3.27 min, (m/z) 296 (M).
Step D: 2-(3,4-dichloro-5-(1-(4-fluorophenylsulfonyl)piperidin-3-
yl)phenyl)acetic acid
(Compound 64D)
HO2C
,1S
O
CI .
CI F
C
111
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
[0297] To a solution of inethyl2-(3,4-dichloro-5-(pyridin-3-yl)phenyl)acetate
(11.5 mg,
0.0388 mmol) in MeOH (2 mL) was added concentrated HC1 (200 L) and Pt20
(cat.). The
mixture was stirred under H2 (1 atm) for 1 h before filtered through a plug of
CELITE with
EtOAc-MeOH. Solvent was removed to give a colorless oil (18.5 mg). The crude
product
was dissolved in CH2C12 (3 mL), and Et3N (43 L, 0.31 mmol) was added,
followed by
4-fluorobenzene-l-sulfonyl chloride (15 mg, 0.0776 mmol). The mixture was
stirred for 17 h
before it was diluted with CHZC12 and water and extracted with CH2C12. The
combined
organic layer was dried over Na2SO4. Solvent was removed and the crude product
was
disoolved in THF-water (2 mL-0.5 mL). LiOH=H2O (13 mg, 0.31 mmol) was added.
After
16 h, the mixture was acidified with 1N HCI and extracted with EtOAc.
Purification on
reverse phase HPLC yielded the title compound as a white solid. MS (Method B),
Rt=3.87
min, (m/z) 445 (M-H). 'H NMR (DMSO-d6): ppm 12.4 (s, 1H), 7.83 (m, 2H), 7.5
(m, 3H),
7.2 (s, IH), 3.7 (m, 2H), 3.56 (s, 2H), 3.2 (m, 1 H), 2.3 (m, 2H), 1.8 (m,
2H), 1.6 (m, 2H).
Example 65
{5-[1-(4-Fluoro-benzenesulfonyl)-piperidin-3-yl]-biphenyl-3-yl}-acetic acid
(Compound 65)
H02C
O
N.S
0
F
[0298] Purification on reverse phase HPLC yielded the title compound as a
white solid.
MS in/z 454 (M+H).
Exainple 66
2-(3-(1-(4-fluorophenylsulfonyl)-4-phenylpiperidin-3-yl)phenyl)acetic acid
(Compound 66):
112
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
Scheme 12:
CN CN
Br Step A\ Br Step B I Step C~
\ -- \ \ -N N
N N I/ I I/
A12 B12 C12 D12
0 0
1 1
Step D OMe Step E OH
-' , O
õ - \ ~ O
H
N-S F F
O O
I I \
E12 F12
Step A: 3-bromo-4-phenylpyridine (B12)
[0299] To 3-bromopyridine (2m1, 20mmo1) in THF (25m1) was slowly injected LDA
in
THF (12m1, 24mmol) at -95 C. The resulting solution was stirred at -95 C for
30 min. At
this time, anhydrous ZnC12 (24m1, 24mmol) in Et20 was added dropwise at the
temperature,
and the solution was allowed to warm to RT to provide 3-bromo-4-pyridyl zinc
chloride. To
this solution, was added iodobenzene (2.2m1, 20minol) followed by a solution
of Pd(PPh3)4
(500mg, 0.43mmol) in dry THF (5ml) solution was then heated to reflux for 4h.
After
aqueous work up, the product was afforded after flash chroinotagraphy on
silica gel. LC/MS
Rt = 3.578 min. LC/MS (Method A); MS (m/z) 234.00 (M+ +H).
Step B: 2-(3-(4-phenylpyridin-3-yl)phenyl)acetonitrile (C12)
[0300] To a mixture of 3-bromo-4-phenyl pyridine (610mg, 2.62mmol) and
3-(cyanomethyl)phenylboronic acid (533mg, 3.31mmol) in DME (lOml) was added
Pd(PPh3)4 (150mg, 0.131mmo1), followed by addition of Na2CO3 (555mg, 5.24mmol)
in
water (3ml). The mixture was heated at 85 C overnight. The reaction mixture
was diluted
with EtOAc (100m1), washed with sat Na2CO3 (3 X 20in1), dried over Na2SO4. The
product
113
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
was afforded after flash chromatography on silica gel. RT = 2.969 min. LC/MS
(Method A);
MS (m/z) 271.1(M+ + H).
Step C:
2-(3-(1-benzyl-4-phenyl-1,2,5,6-tetrahydropyridin-3-yl)phenyl)acetonitrile
(D12)
[0301] To intermediate C12 (245.8mg, 0.91mmo1) in CH3CN (5m1) was added benzyl
bromide (0.13m1, 1.09mmo1), and the solution was refluxed for 2h. The solvent
was removed
under the reduced pressure.
[0302] The salt (150mg, 0.418mmo1) was then dissolved in THF (2m1), NaBH4
(32mg,
0.836mmo1) was added at 0 C. After lh, the reaction was quenched with H20
(0.5m1).
Diluted with EtOAc (15m1), washed with H20 (3 X 3m1), dried over Na2SO4, the
desired
product was obtained after the solvent was removed. Rt = 2.564 min. LC/MS
(Method A);
MS (m/z) 365.2(M+ +H).
Step D: Methyl2-(3-(1-benzyl-4-phenyl-1,2,5,6-tetrahydropyridin-3-
yl)phenyl)acetate (E12
[0303] To intermediate D12 (152mg, 0.418mmo1) in MeOH (5m1) was bubbled HCI
gas.
The solution was refluxed overnight. The desired material was obtained after
the solvent was
removed. Rt = 2.564 min. LC/MS (Method A); MS (m/z) 398.2(M+ +H).
[0304] To this product in MeOH (5m1) was added a catalytic amount of 10%
Pd(OH)2/C.
After purging 3 times with H2, the reaction was run under a H2 balloon for
12h. The solution
was concentrated under reduced pressure, the residue was then dissolved in DCM
(5in1).
DIEA (0.29m1, 1.67mmo1) was added, followed by the addition of
4-fluorobenzene-1-sulfonyl chloride (122mg, 0.627mmol). The mixture was
stirred at RT
overnight. The product (73mg) was afforded after flash column chromatography
on silica gel.
Rt = 4.297 min. LC/MS (Method A); MS (m/z) 468.1(M+ +H).
[0305] Step E: 2-(3-(1-(4-fluorophenylsulfonyl)-4-phenylpiperidin-3-
yl)phenyl)acetic acid
(Compound 66)
114
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
[0306] To intermediate 4 (73mg, 0.156mmol) dissolved in THF (lml), lml of
aqueous 1N
NaOH was added. The mixture was stirred overnight. Diluted with EtOAc (15m1),
washed
with 1N HCl (3 X 2ml), dried over NaZSO4, the final product (72.2mg) was
obtained. Rt =
3.886 min. LC/MS (Method A); MS (m/z) 454.1(M+ + H).
Example 67
2-(3-(4-cyclohexyl-l-(4-fluorophenylsulfonyl)-1,2,5,6-tetrahydropyridin-3-
yl)phenyl)acetic
acid (Compound 67D)
O
I
OMe
O
1 -
11
I N-S ~ ~ F
11
O
Scheme 13
CN OI O
HOMe
OMe I Step A~ I Step B/ Step C
CNCJN I
C12 A13
B13
O O
I OMe
OMe I + I NH NH
C13 D13
Step A: Methyl 2-(3-(4-cyclohexylpyridin-3-yl)phenyl)acetate (A13)
[0307] To interlnediate 2 (135mg, 0.50mmol) in MeOH (5m1) was bubbled HCI gas.
The
solution was refluxed overnight. LC/MS was used to monitor the reaction. A
catalytic
115
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
amount of Pt02 was added to the solution. After purging 3 times with H2, the
reaction was
run under H2 balloon for 12h. The catalyst was filtered through CELITE, the
product was
obtained after removal of the solvent. Rt = 2.898 min. LC/MS (Method A); MS
(m/z)
310.2(M+ + H).
Step B: Methyl2-(3-(1-benzyl-4-cyclohexyl-1,2,5,6-tetrahydropyridin-3-
yl)phenyl)acetate
(B13)
[0308] Same experimental procedure as for
2-(3-(1-benzyl-4-phenyl-1,2,5,6-tetrahydropyridin-3-yl)phenyl)acetonitrile.
LC/MS Rt =
2.849 min. (Method A); MS (m/z) 404.2(M+ + H).
[0309] Step C: Methyl 2 -(3 -(4- cyclohexyl- 1,2,5,6-tetrahydropyridin-3 -
yl)phenyl) acetate
(C13) & Methyl 2-(3-(4-cyclohexylpiperidin-3-yl)phenyl)acetate (D13)
[0310] To the intermediate B13 (157mg, 0.388mmo1)in MeOH (5m1) was added a
catalytic
amount of 10% Pd(OH)2/C. After purging 3 times with H2, the reaction was run
under a H2
balloon for 12h. The solution was concentrated under reduced pressure, giving
both
intermediate C13. LC/MS (Method A) Rt = 2.522 min.; MS (m/z) 314.2(M+ +H) and
intennediate D13. LC/MS (Method A) Rt = 2.688 min.; MS (m/z) 316.2(M+ + H).
[0311] Step D:
2-(3-(4-cyclohexyl-l-(4-fluorophenylsulfonyl)-1,2,5,6-tetrahydropyridin-3-
yl)phenyl)acetic
acid (Compound 67D)
[0312] Intennediate C13 was dissolved in DCM (5ml). Hunig base (0.20m1,
1.165mmo1)
was added, followed by the addition of the 4-fluorobenzene-l-sulfonyl chloride
(91mg,
0.466mmo1). The mixture was stirred at RT overnight. The product was afforded
after flash
column chromatography. To the product in THF (linl), was added aqueous 1N NaOH
(lml).
The mixture was stirred overnight. Diluted with EtAc (15m1), washed with 1N
HCI (3 X
2m1), dried over Na2SO4, the final product (40mg) was obtained. Rt = 4.283
min. LC/MS
(Method A); MS (m/z) 458.2(M+ + H). 'H NMR (300 MHz, CDC13) S 7.80 (m, 2H),
7.31
116
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
(m, 1H), 7.22 (m, 3H), 7.02 (m, 2H), 3.67 (s, 4H), 3.27 (t, 2H), 2.30(t, 2H),
2.11 (m, 1H),
1.68-1.52 (m, 3H), 1.38 (m, 2H), 1.25 (m, 2H), 1.06 (in, 3H).
2-(3-(4-cyclohexyl-l-(4-fluorophenylsulfonyl)piperidin-3-yl)phenyl)acetic acid
(Compound
67E)
O
OH
O
ft-F
[0313] The same experimental procedure described for
2-(3 -(4-cyclohexyl-l-(4-fluorophenylsulfonyl)-1,2, 5,6-tetrahydropyridin-3 -
yl)phenyl)acetic
acid was followed starting with methyl2-(3-(4-cyclohexylpiperidin-3-
yl)phenyl)acetate. Rt =
4.378 min. LC/MS (Method A); MS (m/z) 460.2 (M+ +H). 'H NMR (300 MHz, CDC13) 8
7.80 (m, 2H), 7.58 (d, 1H), 7.40 (s, 1H), 7.31 (m, 2H), 7.22 (m, 2H), 4.02(d,
1H), 3.92 (d,
1 H), 3.67 (s, 2H), 3.09 (s, 1 H), 2.58 (dd, 1 H), 2.25 (m, 1 H), 1.90 (m, 1
H), 1.68-0.20 (m,
13H).
Exainple 68
2-(3-(1-(tosyl)-1H-indol-3-yl)phenyl)acetic acid (Compound 68)
o
HO
[0314] To 3-bromo phenyl acetic acid (0.215g, 1.Ommo1) in 2mL dimethoxyethane
and
1mL water was added N-tosyl indole 3-boronic acid (0.315g, 1.Ominol),
palladium tetrakis
(0.058g, 0.05mmo1) and sodium carbonate (0.211g, 2.Ommol). The mixture was
heated to 65
C and stirred for 18 hours at which time the reaction was deemed complete by
LC/MS. The
reaction mixture was diluted with water and extracted into EtOAc 2X. The
aqueous acidified
117
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
to pH 1 and was extracted 3X with EtOAc. The dried material was purified over
HPLC to
yield the title compound. MS m/z 406 (M+H); LC/MS (Method A) Rt= 4.01 min.
Example 69
2-(3-hydroxy-5(1-(phenylsulfonyl)-1H-indol-3-yl)phenyl acetic acid (Compound
69C)
O
HO
I \ O ~
HO / / ry III \ /
Step A: Methyl 3-hydroxy-5-trifluoromethane sulfonyloxy-phenyl acetate
(Compound
69A)
0
I \ \\ / \ F
HO / / ~\ F
O
[0315] To 3,5 dihydroxy phenyl acetic acid methyl ester (5.0g, 27.Ommol) in
lOOmL DCM
at 0 C was added DIEA (4.71mL, 27.0mmo1) and triflic anhydride (11.4mL,
67.5mmol)
dropwise. The reaction was allowed to slowly wann to room temperature and stir
3 days at
RT at which time it was deemed complete by LC/MS. It was used without further
purification. MS m/z 315.0 (M+H); LC/MS(Method A) Rt= 3.50min
Step B: Methyl 2-(3-hydroxy-5(1-(phenylsulfonyl)-1 H-indol-3-yl)phenyl)acetate
(Compound 69B)
O
\O
I \
HO /
/ N III \ /
118
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
[0316] The title compound(s) were synthesized using the procedure described
for
2-(3-(1-(phenylsulfonyl)-1H-indol-3-yl)phenyl)acetic acid, starting with
methyl
3-hydroxy-5-trifluoroinethane sulfonyloxy-phenyl acetate. MS m/z 422.0 (M+H);
LC/MS
(Method A) Rt= 3.94min
Step C: 2-(3-hydroxy-5(1-(phenylsulfonyl)-1H-indol-3-yl)phenyl acetic acid
(Compound
69C)
0
HO
I \ H O
HO / III
[0317] To the previous ester (0.016g, 0.038mmo1) in 1mL methanol was added
0.5mL 3N
NaOH. The reaction stirred at room temperature for 18 hours. The completed
reaction was
acidified to pH 1 and extracted into DCM 3X. The combined organic layers were
dried to
yield the title compound requiring no further purification. MS m/z 408.0
(M+H),
LC/MS(Method A) Rt= 3.54 min.
Examples 70-74
Scheme 14
0 0 0
OMe Pd(PPh3)4 OMe LiOH OH
DME/Na2CO3
R4 Br HO, R4 N-S02Rs R4 N-S02R5
B-OH
\
WN R 8 R8
R8 S0zR5
Example 70
119
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
Methyl2-(3-(1-(phenylsulfonyl)-1H-indol-3-yl)phenyl)acetate (Compound 70A) and
2-(3-(1-(phenylsulfonyl)-1H-indol-3-yl)phenyl)acetic acid (Compound 70B).
Step A: Methyl 2-(3-(1-(phenylsulfonyl)-1H-indol-3-yl)phenyl)acetate (Compound
70A).
O
O
N-S=0
O
[0318] 1-(phenylsulfonyl)-1H-indol-3-ylboronic acid (620 mg, 2.07 mmol) and
Pd(PPh3)4
(109 mg, 0.0939 mmol) were added to a stirring solution ofinethyl 2-(3-
bromophenyl)acetate
(430 mg, 1.88 mmol) in dimethoxy ethane/ 2 M Na2CO3 (2:1, 12 mL). The
resulting solution
was refluxed for 3 h, cooled to RT, then diluted EtOAc (10 mL). The organic
layer was
washed with H20 (10 mL), dried over Na2SO4, and concentrated to give the crude
material(1.13 g) as a green oil. A column chromatography on silica gel (3:1,
hexanes/EtOAc)
afforded pure material (760 mg, 99 %) as a light turquoise oil.
Step B: 2-(3-(1-(phenylsulfonyl)-1H-indol-3-yl)phenyl)acetic acid (Compound
70B).
O
OH
I I /
N-S=0
O
[0319] Solid LiOH (227 ing, 5.43 ininol) was added to a stirring solution of
methyl
2-(3-(1-(phenylsulfonyl)-1H-indol-3-yl)phenyl)acetate (220 mg, 0.543 mmol) in
THF/MeOH/H20 (5 mL, 3:1:1) at RT. After stirring over night, the resulting
mixture was
quenched with 1 N HCl (<pH 1). The aqueous layer was extracted with EtOAc (3 X
20 mL),
dried over Na2SO4, and concentrated to give crude acid (260 mg) as a light
brown oil. HPLC
purification afforded pure compound: ES/MS 392.1 (M+H); LC/MS (Method B) Rt =
3.849
min.
120
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
Example 71
Methyl 2-(3-(1-(methylsulfonyl)-1H-indol-3-yl)phenyl)acetate, (Compound 71A)
and
2-(3-(1-(methylsulfonyl)-1H-indol-3-yl)phenyl)acetic acid, (Compound 71B)
0
OH
N-S=0
O
[0320] The title material was obtained using the procedure described for
2-(3-(1-(phenylsulfonyl)-1H-indol-3-yl)phenyl)acetic acid, starting from 1-
(methyl
sulfonyl)-1H-indol-3-ylboronic acid. Compound 71B: ES/MS, m/z found 330.1
(M+H);
LC/MS (Method B) Rt = 3.285 min.
Example 72
Methyl 2-(3-(1-(4-fluorophenylsulfonyl)-1H-indol-3-yl)phenyl)acetate (Compound
72A)
and
2-(3-(1-(4-fluorophenylsulfonyl)-1H-indol-3-yl)phenyl)acetic acid (Compound
72B)
O
F
OH
N-S=0
O
X /
[0321] The title material was obtained using the procedure described for
2-(3-(1-(phenylsulfonyl)-1 H-indol-3 -yl)phenyl) acetic acid, starting from 1-
(4-fluorophenyl
sulfonyl)-1H-indol-3-ylboronic acid, which was synthesized using the procedure
described in
Garg., N.K., et al., J. Am. Chem. Soc., 2002, 124:1317984. 'H NMR (400 MHz,
DMSO-db)
8 12.36 (1 H, brs) 8.18 (2H, m) 8.10 (1 H, s) 8.04 (1 H, m) 7.86 (1 H, m) 7.65-
7.60 (2H, m)
7.48-7.42 (4H, m) 7.36 (1H, m) 7.29 (1H, m) 3.68 (2H, s); ES/MS, m/z 419.1
(M+H);
LC/MS (Method A) Rt = 3.909 min.
121
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
Example 73
Methyl 2-(3-(1-(4-methoxyphenylsulfonyl)-1H-indol-3-yl)phenyl)acetate
(Compound 73A)
and 2-(3-(1-(4-methoxyphenylsulfonyl)-1H-indol-3-yl)phenyl)acetic acid
(Compound 73B)
0
-_o
OH
~
I ~
N-S=0
O
X /'
[0322] The title material was obtained using the procedure described for
2-(3-(1-(phenylsulfonyl)-1H-indol-3-yl)phenyl)acetic acid, starting from 1-(4-
methoxyphenyl
sulfonyl)-1H-indol-3-ylboronic acid, which was synthesized using the procedure
described in
Garg., N.K., et al., J. Am. Chem. Soc, 2002, 124:1317984. Compound 73B: ES/MS,
m/z
422.1 (M+H); LC/MS (Method A) Rt = 3.878 min.
Example 74
Methyl 2-(3-chloro-5-(1-(phenylsulfonyl)-1H-indol-3-yl)phenyl)acetate
(Compound 74A)
and 2-(3-chloro-5-(1-(phenylsulfonyl)-1H-indol-3-yl)phenyl)acetic acid
(Compound 74B)
0
OH
CI N S=0
0
[0323] The title material was obtained using the procedure described for
2-(3-(1-(phenylsulfonyl)-1H-indol-3-yl)phenyl)acetic acid, starting from 1-(4-
phenyl
sulfonyl)-1H-indol-3-ylboronic acid and methyl 2-(3-broino-5-
chlorophenyl)acetate.
Compound 74B: ES/MS, m/z 426.1 (M+H); LC/MS (Method A) Rt = 3.97 min.
Example 75
122
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
(Z)-2-(3-(1-(4-fluorophenylsulfonyl)-2,5,6,7-tetrahydro-lH-azepin-3-
yl)phenyl)acetic acid
(Compound 75G)
Scheme 15:
O O
1. Boc20 / THF "k
O
HO DMAP / tBuOH
2. PdCl2 dppf ,O
Br B(Pin)2 B
KOAc 0
dioxane Pd(PPh3)4 O"O
80 C DME N'S
Br Na2CO3 (2M) I i
O`3-NH O 80 oC F
,~1i
2 K2CO3/CH3CN 0
-N Br
2. Br ~ I \\
F
--I)
Br F
K2CO3 /CH3CN
1.Grubb's 2nd
generation COZH
(20mo1%) / CH2CI2
60 C 3h
S
0 0
2. Dioxane /AcOH
HCI / 80 C F
3h
Step A: tert-butyl 2-(3-bromophenyl)acetate (Compound 75A)
,kO 0
Br
[0324] To a mixture of 2-(3-bromophenyl)acetic acid (10.0g, 0.046mo1), `BuOH (
34.0 g,
0.46mo1), di-tert-butyl dicarbonate (20.4g 0.094mo1) in THF (50mL) was added
DMAP
(1.7g, 0.014ino1) portion wise, slowly due to effervescence. The reaction was
stirred for 24h
then concentrated in vacuo. The residue was passed through a silica plug (9:1
hexane/EtOAc) and the fractions concentrated and the residue subjected to
vacuum
distillation to give tert-butyl 2-(3-bromophenyl)acetate as a colorless oil
(9.9g, 0.036, 78%):
123
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
b.p 122 C at 0.05mmHg; 'H NMR (300MHz, CDC13) 7.45-7.37 (m, 2H), 7.22-7.17
(m., 2H),
3.49 (s, 2H), 1.4 (s, 9H).
Step B: tert-Buty12-(3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-
yl)phenyl)acetate
(Compound 75B)
)<1O O
B,
6
[0325] A mixture of tert-butyl 2-(3-bromophenyl)acetate (9.5g, 0.035 mol),
bis(pinacolato)diboroane (10.6g, 0.042mo1) and potassium acetate (10.6g, 0.11
mol) in
dioxane (270mL) was purged with argon for 30 min. To the mixture was added
dichloro[1,1'-bis(diphenylphosphino)ferrocene] palladium (II) DCM adduct
(1.2g, 1.7mmo1)
and 1,1'-bis(diphenylphosphino)ferrocene (0.94g, 1.7mmol) and then vigorously
stirred at 80
C for 4h. The solution was cooled, concentrated in vacuo to which was added 5g
of activated
charcoal and 150mL of hexanes. The mixture was passed through a plug of CELITE
and the
filtrate concentrated in vacuo. The volatile impurities were removed with
kugelrohr ditillation
(135 C at 0.02mmHg) and the resulting residue was passed through a silica
plug (9:1
EtOAc/hexane) to give tert-butyl
2-(3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)acetate as a
colorless solid (9.1g,
0.028mol, 82%); 'H NMR (300MHz, CDC13) 7.75-7.65 (m, 2H), 7.41-7.30 (m., 2H),
3.51 (s,
2H), 1.42 (s, 9H), 1.35 (s, 12H).
Step C: 4-fluoro-N-(pent-4-enyl)benzenesulfonainide (Compound 75C)
O
O,n
~S-NH
F
124
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
[0326] A mixture of 4-fluorobenzenesulfonamide (5g, 0.03mo1), 5-bromopentene
(3.5mL,
0.03mo1) and K2CO3 (4.27g, 0.031mol) in acetone (75mL) was heated to reflux
for 14h. The
resulting suspension was cooled and passed through a plug of CELITE and the
concentrated
in vacuo. Column chromatography (silica gel, 0-60% hexane / EtOAc), furnished
4-fluoro-N-(pent-4-enyl)benzenesulfonamide as a colorless oil (1.7g, 6.95mmo1,
23%); 1 H
NMR (300MHz, CDC13) 7.9-7.8 (m, 2H), 7.25-7.1 (m, 2H), 5.8-5.6 (m, 1H), 5.0-
4.9 (m, 2H),
4.6-4.5 (m, IH), 2.95 (q, 2H), 2.05 (m, 2H), 1.6-1.5 (m, 2H).
Step D: N-(2-bromoallyl)-4-fluoro-N-(pent-4-enyl)benzenesulfonamide (Compound
75D)
~
O.~
`S-N Br
F
[0327] A mixture of 4-fluoro-N-(pent-4-enyl)benzenesulfonamide (1.7g, 0.0069
mol),
2,3-dibromoprop-l-ene (1.99g, O.Olmo1) and Cs2CO3 (4.55g, 0.014 mol) in CH3CN
(20mL)
was stirred at RT for 12h. The mixture was filtered through a plug of CELITE
and
concentrated in vacuo. Column chromatography (silica gel, 0-30% hexane/EtOAc)
gave
N-(2-bromoallyl)-4-fluoro-N-(pent-4-enyl)benzenesulfonainide (2.1 g,
0.0058ino1, 84%) as a
colorless oil; 'H NMR (300MHz, CDC13) 7.9-7.8 (m, 2H), 7.20-7.1 (m., 2H), 5.9
(s, 1H),
5.8-5.6 (m, IH), 5.6 (d, 1 H), 5.0 (m, 2H), 4.05 (s, 2H), 3.2-3.1 (m, 2H),
2.05 (m, 2H), 1.7-1.5
(m, 2H).
Step E: tert-Butyl
2-(3-(3-(4-fluoro-N-(pent-4-enyl)phenylsulfonamido)prop-l-en-2-
yl)phenyl)acetate
(Compound 75E)
125
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
0
0
.,0
N~
F
[0328] To a mixture of N-(2-bromoallyl)-4-fluoro-N-(pent-4-
enyl)benzenesulfonamide
1.6g, 4.4mmol) and tert-butyl
2-(3 -(4,4,5,5 -tetramethyl- 1,3,2-dioxaborolan-2-yl)phenyl) acetate (2.1g,
6.6mmo1) was added
degassed Na2CO3 (15mL, 2M), degassed DME (30 mL) and
tetrakis(triphenylphosphine)palladium (0) (254mg, 0.22mmol). The solution was
vigorously
stirred at 90 C for 4h. The mixture was cooled, diluted with EtOAc (30mL) and
the organic
layers separated, washed with brine (l OmL), dried (Na2SO4) and concentrated
in vacuo.
Column chromatography (silica gel, 0-60% hexane / EtOAc), furnished tert-butyl
2-(3-(3-(4-fluoro-N-(pent-4-enyl)phenylsulfonamido)prop-l-en-2-
yl)phenyl)acetate as a
colorless oil (1.63g, 3.4mmol, 78%); 'H NMR (300MHz, CDC13) 7.8-7.7 (m, 2H),
7.30-7.1
(m., 6H), 5.7-5.6 (m, 1 H), 5.49 (s, 111), 5.2 (s, 1H), 5.0-4.9 (m, 2H), 4.2
(s, 2H), 3.5 (s, 2H),
3.05 (m, 2H), 2.0-1.85 (m, 2H), 1.6-1.4 (m, 11H), 1.4-1.3 (in, 2H).
Step F: (Z)-tert-butyl
2-(3-(1-(4-fluorophenylsulfonyl)-2,5,6,7-tetrahydro-1 H-azepin-3-
yl)phenyl)acetate
(Compound 75F)
0
OSO
N' \
F
[0329] To a solution of tert-butyl
2-(3-(3-(4-fluoro-N-(pent-4-enyl)phenylsulfonainido)prop-I-en-2-
yl)phenyl)acetate (1.5g,
0.032mo1) in CH2C12 (330mL) was added
benzylidene[ 1,3-bis(2,4,6-triinethylphenyl)-2-iinidazolidinylidene]
126
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
dichloro(tricyclohexylphosphine) ruthenium (537mg, 0.63mmol). The solution was
heated at
reflux 4h, then cooled and concentrated in vacuo. Column chromatography
(silica gel,
0-50% hexane / EtOAc), furnished (Z)-tert-butyl
2-(3-(1-(4-fluorophenylsulfonyl)-2,5,6,7-tetrahydro-lH-azepin-3-
yl)phenyl)acetate as a
colorless oil (1.25g, 0.028 mol, 88%); 'H NMR (300MHz, CDC13) 7.9-7.7 (m, 2H),
7.40-7.1
(m, 6H), 5.9 (t, 1H), 4.4(s, 2H), 3.55-3.34 (m, 4H), 2.3-2.2 (m, 2H), 1.9-1.8
(m, 2H), 1.45 (s,
9H)
Step G:
(Z)-2-(3-(1-(4-fluorophenylsulfonyl)-2,5,6,7-tetrahydro-1 H-azepin-3-
yl)phenyl)acetic acid
(Compound 75G)
0
OH
I ~ O
/S
N
F
[0330] A mixture of (Z)-tert-butyl
2-(3-(1-(4-fluorophenylsulfonyl)-2,5,6,7-tetrahydro-1 H-azepin-3-
yl)phenyl)acetate (1.25g,
0.028mo1), AcOH (4mL), dioxane (40mL) and HC1(15mL, 2M) was heated to 80 C
for 4h,
cooled, then concentrated in vacuo. Column chromatography (silica gel, 0-> 10%
CHZC12 /
metahnol),
(Z)-2-(3-(1-(4-fluorophenylsulfonyl)-2,5,6,7-tetrahydro-1 H-azepin-3-
yl)phenyl)acetic acid as
a colorless solid (0.9g, 0.023mo1, 82%); LC/MS (Method A) Rt = 3.681 min., MS
m/z 390
(M+ H).
Example 76
2-(3-(1-(4-fluorophenylsulfonyl)pyrrolidin-3-yl)phenyl)acetic acid (Compound
76A) and
2-(3-(1-(4-fluorophenylsulfonyl)-1H-pyrrol-3-yl)phenyl)acetic acid (Compound
76B)
127
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
0
OH
~ I N-O F
~, \ /
O
Scheme 16
0 0 O
OMe OMe OMe
Step A Step B
~
~ I /
Br NH ~ NH
A16 B16 C16
Step A: Methyl2-(3-(1H-pyrrol-3-yl)phenyl)acetate (B16)
[0331] To the mixture of methyl 2-(3-bromophenyl)acetate (400mg, 1.747mmo1)
and
1-(triisopropylsilyl)-1H-pyrrol-3-ylboronic acid (467mg, 1.747mmo1) in DME
(4m1) was
added palladium tetrakis (100mg, 0.087mmo1), followed by the addition of CsF
(796mg,
5.24mmol) in water (lml). The mixture was heated at 90 C for 4ht. The
reaction mixture was
diluted with EtAc (30m1), washed with sat H20 (3 X l Oml), dried over Na2SO4.
The product,
intermediate 3 (0.232g, 64%) was afforded after column chromatography on
silica gel.
Step B: Methyl 2-(3-(1H-pyrrolidin-3-yl)phenyl)acetate (C16)
[0332] To intermediate B16 (92.7mg, 0.431mmo1) in MeOH (2m1) was added lml of
IN
HC1 in Et20, after stirred for 5min, the solvents were pumped off. The residue
was dissolved
in MeOH (5m1), and was added the catalytic amount of Pt02. The suspension was
purged 3
times, and was stirred at 1 atm under H2 for 3h. The catalyst was filtered off
through
CELITE . Concentrated to remove solvent, interinediate 4 (94.2mg. 100%) was
obtained. Rt
= 0.545 inin. (Method A); MS (m/z) 220 (M + H).
2-(3-(1-(4-fluorophenylsulfonyl)pyrrolidin-3-yl)phenyl)acetic acid (Compound
76A)
128
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
0
OH
O
N S F
- ~ ~
O
[0333] To intermediate C16 (66.4mg, 0.302mmol) in DCM (2ml) was added Hunig's
base
(0.21m1, 1.207mmo1), followed by the addition of the 4-fluorophenylsulfonyl
chloride
(117mg, 0.604mmo1). The mixture was stirred at RT overnight. The product was
afforded
after flash column chromatography on silica gel. The product was then
dissolved in THF
(1 ml), 1 ml of aqueous 1 N NaOH was added. The mixture was stirred overnight.
Diluted with
EtAc (15m1), washed with 1N HCl (3 X 2m1), dried over Na2SO4, the final
product (18.5mg)
was obtained after HPLC. Rt = 3.358 min. (Method A); MS (m/z) 364 (M + H)
2-(3-(1-(4-fluorophenylsulfonyl)-1H-pyrrol-3-yl)phenyl)acetic acid (Compound
76B)
0
OH
O
n
N-S F
O
[0334] To intennediate B16 (42.5mg, 0.198mmo1) in DCM (2m1) was added NaOH
(40mg,
0.989mmol), followed by the addition of the 4-fluorophenylsulfonyl chloride
(46mg,
0.604mmo1). The mixture was stirred at RT for 2 days. 0.5m1 of H20 was added
to the
mixture, the product was obtained after HPLC (3.2mg). MS (m/z) 360 (M + H); 'H
NMR
(300 MHz, CDC13) S 7.93 (m, 2H), 7.42 (m, 3H), 7.35 (m, 1H), 7.20 (m, 4H),
6.63 (s, 1H),
3.68 (s, 2H).
Example 77
2-(4-(1-(4fluorophenylsulfonyl) piperidin-3-yl)phenyl)acetic acid (Compound
77)
129
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
F
0
OH \
N
O
[0335] 2-(4-(1-(4fluorophenylsulfonyl) piperidin-3-yl)phenyl)acetic acid is
prepared using
the same methodology as was used to prepare 2-(3-(1-(4fluorophenylsulfonyl)
piperidin-3-yl)phenyl)acetic acid using methyl-4-bromo phenyl acetic acid
ester (steps A-D).
LC/MS (Method A) Rt = 3.54 min; MS: 378 m/z (376 m/z negative ion).
Example 78
2-(2-(4-fluorophenylsulfonyl)-1,2,3,4-tetrahydroisoquinolin-5-yl)acetic acid
(Compound 78)
Scheme 17
0 OH CI
NaBH4, EtOH SOCI2, CH2CI2 NaCN
N N pyr. N DMF
CN C02Me COzMe
HC H2, Pt20 Et3N, CH2CI2
N MeOH N MeOH NHHCI C102S
HCI
F
CO2Me CO2H
F LiOH F
N,
S\O 'S"O
130
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
Step A. Preparation of isoquinolin-5-ylmethanol (Compound 78A)
OH
/ ~N
[0336] To a solution of commercially available isoquinoline-5-carbaldehyde
(808 mg, 5.1
mmol) in EtOH (10 mL) at 0 C was added NaBH4 (194 mg, 5.1 mmol). The mixture
was
stirred at 0 C for 2.5 hours before 10% NaOH solution was added. It was
stirred for
additional 18 hours. Solvent was removed under reduced pressure and the
mixture extracted
with CHZCIZ. The crude mixture was purified using silica gel chromatography to
give a pale
yellowish oil (690 mg, 4.3 mmol). MS (m/z) 160.1 (M++H).
Step B. Preparation of 5-(chloromethyl)isoquinoline (Compound 78B)
CI
6CN
[0337] To isoquinolin-5-ylmethanol was added the thus obtained oil was added
CHZCI2 (10
mL), SOC12 (2.52 mL, 34.6 mmol), and pyridine (1.4 mL, 17.3 mmol). The mixture
was
stirred for 20 hours before being cooled to 0 C and quenched with H20. The
mixture was
basified with 10% NaOH and extracted with CH2C12, and dried over Na2SO4. The
crude
chloride was purified using silica gel chromatography to give an off-white
solid (464 mg,
2.61 mmol). MS (m/z) 178.6 (M++H).
Step C: Preparation of 2-(isoquinolin-5-yl)acetonitrile (Compound 78C)
CN
I \ \
/ N
[0338] To 5-(chloromethyl)isoquinoline (174 mg, 0.98 mmol) was added thus
obtained
chloride (174 mg, 0.98 mmol) was added NaCN (98 mg, 2 mmol) and DMF (6 mL).
The
131
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
mixture was heated at 70 C for 1.5 hours and purified directly using silica
gel
chromatography to give an off-white solid (194 mg, 1.15 mmol). MS (m/z) 169.2
(M++H).
Step D. Preparation of methyl 2-(isoquinolin-5-yl)acetate hydrochloride
(Compound 78D)
CO2Me
I \ \
N
HCI
[0339] 2-(isoquinolin-5-yl)acetonitrile was dissolved in MeOH (5 mL) and
HCl(g) was
bubbled for 5 min. (exothermic). The resulting clear solution was stirred for
3 hours.
Solvent was removed and the crude hydrochloride salt was used in the next step
as is. MS
(m/z) 202.2 (M++H).
Step E.. Preparation of inethyl2-(1,2,3,4-tetrahydroisoquinolin-5-yl)acetate
hydrochloride
(Compound 78E)
CO2Me
NHHCI
[0340] To methyl 2-(isoquinolin-5-yl)acetate hydrochloride (125 mg, 0.618
mmol) was
added Pt20 (cat.) and MeOH (4 inL), and it was hydrogenated under H2 balloon
for 19 hours.
The mixture was filtered through a plug of CELITE and flushed with MeOH.
Removal of
solvent gave methyl 2-(1,2,3,4-tetrahydroisoquinolin-5-yl)acetate
hydrochloride as an
off-white solid (125 mg, 0.51 mmol). MS (m/z) 206.2 (M++H).
132
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
Step F..Preparation of methyl
2-(2-(4-fluorophenylsulfonyl)-1,2,3,4-tetrahydroisoquinolin-5-
yl)acetate_(Compound 78F)
CO2Me
F
N,
O~S~-O
[0341] Methyl 2-(1,2,3,4-tetrahydroisoquinolin-5-yl) acetate hydrochloride (59
mg, 0.24
mmol) was dissolved in CH2C12 (4 mL). TEA (134 L, 0.96 mmol) was then added
followed
by 4-fluorobenzene-l-sulfonyl chloride (71 mg, 0.36 mmol). The mixture was
stirred for 19
hours before it was diluted with CH2C12 and H20. The aqueous layer was
extracted with
CH2C12 and the crude mixture was purified using silica gel chromatography to
give a white
solid (48 mg, 0.13 mmol). MS (m/z) 364.4 (M++H).
Step G. Preparation of
2-(2-(4-fluorophenylsulfonyl)-1,2,3,4-tetrahydroisoquinolin-5-yl)acetic acid
(Compound
78G)
CO2H
F
N //S\\
O
[0342] To methyl 2-(2-(4-fluorophenylsulfonyl)-1,2,3,4-tetrahydroisoquinolin-5-
yl) acetate
was added THF-H20 (2 mL-0.5 rnL) and LiOH=HZO (45 mg, 1.06 mmol). It was
stirred for
16 hours before THF was removed and mixture acidified with 1N HC1. It was
extracted with
EtOAc, dried over Na2SO4. Removal of solvent gave 46 mg (100%) of the title
compound
2-(2-(4-fluorophenylsulfonyl)-1,2,3,4-tetrahydroisoquinolin-5-yl)acetic acid
as a white solid.
LC/MS (m/z) 350.00 (M++H); Rt = 3.13 min.
Example 79
133
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
2-(2-(2-(4-fluorophenylsulfonamido)acetyl)-1,2,3,4-tetrahydroisoquinolin-5-
yl)acetic acid
(Compound 79G)
Scheme 18
C02Me C02Me
Boc-Gly, HATU TFA-CH2C12
NHHCI N
-r---NHBoc
O
C02Me C02Me
Et3N, CH2C12
N~NHZ TFA C102S \ I/ N O\S/,
O
O ~
0 F F
CO2H
LiOH
N O'l S~O
0 H
F
Step A. Preparation of methyl
2-(2-(2-(teYt-butoxycarbonylamino)acetyl)-1,2,3,4-tetrahydroisoquinolin-5-
yl)acetate
(Compound 79A)
CO2Me
N-IrNHBoc
O
[0343] To the previously obtained methyl 2-(1,2,3,4-tetrahydroisoquinolin-5-
yl)acetate
hydrochloride (66 mg, 0.272 mmol) was added Boc-glycine (95 mg, 0.544 mmol),
CH3CN (4
mL), DIEA (237 L, 1.36 mmol) and HATU (207 mg, 0.544 mmol). The mixture was
stirred
for 18 hours before the solvent was removed and the mixture diluted with EtOAc
and washed
with NaHCO3 (sat.) and brine. The combined organic layer was dried over
Na2SO4. Solvent
was removed to give an oil. It was used as is in the next step. MS (m/z) 363.4
(M++H).
134
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
Step B. Preparation of methyl
2-(2-(2-aminoacetyl)-1,2,3,4-tetrahydroisoquinolin-5-yl)acetate TFA salt
(Compound 79B)
CO2Me
N-jrNH2 TFA
O
[0344] To the above crude methyl 2-(2-(2-(tert-butoxycarbonylamino)
acetyl)-1,2,3,4-tetrahydroisoquinolin-5-yl) acetate was added CHZC12 (2 mL)
and TFA (1
mL) and stirred for 2.5 hours. Solvent was removed to give the TFA amine salt
as an oil. It
was used as is in the next step. MS (m/z) 263.3 (M++H).
Step C. Preparation of methyl
2-(2-(2-(4-fluorophenylsulfonamido)acetyl)-1,2,3,4-tetrahydroisoquinolin-5-
yl)acetate
(Compound 79C)
CO2Me
N O~~S%O
,lrW
O H
F
[0345] To this crude methyl
2-(2-(2-aininoacetyl)-1,2,3,4-tetrahydroisoquinolin-5-yl)acetate TFA salt was
added CH2C12
(4 inL). TEA (335 L, 2.4 mmol) was then added followed by 4-fluorobenzene-l-
sulfonyl
chloride (84 mg, 0.43 mmol). The mixture was stirred for 16 hours before it
was diluted with
CH2C12 and H20. The organic layer was dried over Na2SO4. Removal of solvent
gave the
sulfonainide as a brown oil. It was used in the next step without further
purification. MS
(m/z) 421.4 (M++H).
135
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
Step D. Preparation of
2-(2-(2-(4-fluorophenylsulfonamido)acetyl)-1,2,3,4-tetrahydroisoquinolin-5-
yl)acetic acid
(Compound 79D)
CO2H
N O.~S%O
.
O I a F
H
[0346] The subsequent hydrolysis was conducted in THF-HZO (2 mL-0.5 mL) and
LiOH=HZO (114 mg, 2.72 mmol). It was stirred for 3 days before being quenched
with 1N
HCl and extracted with EtOAc. Reverse phase HPLC purification yielded the
title compound
2-(2-(2-(4-fluorophenylsulfonamido)acetyl)-1,2,3,4-tetrahydroisoquinolin-5-
yl)acetic acid as
a white solid. MS (m/z) 407.20 (M++H); Rt = 2.73 min.
Example 80
Preparation of 2-(2-(4-fluorophenylsulfonyl)-1,2,3,4-tetrahydroisoquinolin-7-
yl)acetic acid
(Compound 80)
Scheme 19
MeOzC NH HCI Et3N, CHZCIZ MeOZC N O '~S'O
~
I ~~vJ C102S I /
F
F
LiOH HO2C O1,1O
N S' I~
~ ~
F
136
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
OH
H02C ~;'O BH3 ~ 0O ~ SOC12, CH2CI2
THF I I pyr.
F ~
F
CI OO CN O O
NaCNHCI (9)
MeOH
)aF DMF F
CO2Me CO2H
OO LiOH O~O
N/ N' ~
~
F F
Step A. 2-(4-fluorophenylsulfonyl)-1,2,3,4-tetrahydroisoquinoline-7-
carboxylate
(Compound 80A)
MeO2C O~\~O
F
[0347] To 560 mg (2.45 mmol) of commercially available methyl
1,2,3,4-tetrahydroisoquinoline-7-carboxylate hydrochloride was added CH2C12
(20 mL).
TEA (1.36 mL, 9.8 mmol) was then added followed by 4-fluorobenzene-l-sulfonyl
chloride
(718 mg, 3.7 mmol). The mixture was stirred for 17 hours before it was
purified directly with
silica gel chromatography to give 798 mg (93%) of the sulfonamide as a white
solid. MS
(m/z) 350.3 (M++H).
Step B. 2-(4-fluorophenylsulfonyl)-1,2,3,4-tetrahydroisoquinoline-7-carboxylic
acid
(Compound 80B)
HO2C 0;'0
F
[0348] To the methyl
2-(4-fluorophenylsulfonyl)-1,2,3,4-tetrahydroisoquinoline-7-carboxylate was
added
137
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
THF-H20 (8 mL-2 mL) and LiOH=H2O (765 mg, 18.2 mmol). It was stirred for 1 day
at
ambient temperature and then heated at 60 C for 4 h before THF was removed
carefully by
blowing a stream of N2. The mixture was acidified with 1N HC1 and extracted
with EtOAC.
The combined organic layer was dried over Na2SO4. Removal of solvent yielded
760 mg
(99%) of the title compound
2-(4-fluorophenylsulfonyl)-1,2,3,4-tetrahydroisoquinoline-7-carboxylic acid as
a white solid.
MS (m/z) 334.10 (M-H); Rt = 3.07 min.
Step C. (2-(4-fluorophenylsulfonyl)-1,2,3,4-tetrahydroisoquinolin-7-
yl)methanol
(Compound 80C)
OH
0l ~~0
N /
~ XITQF
[0349] To the previously obtained
2-(4-fluorophenylsulfonyl)-1,2,3,4-tetrahydroisoquinoline-7-carboxylic acid
(463 mg, 1.38
mmol) in THF at 0 C was added BH3=THF (4.14 mL, 4.14 mmol). The mixture was
stirred at
0 C for 2.5 hours then at ambient temperature for 4 hours before being
quenched with
MeOH. Solvent was removed and 1N HCI and EtOAc were added and the slurry
stirred for
overnight. The aqueous layer was basified with 10% NaOH to pH=9 and extracted
with
CH2Cl2 and dried over Na2SO4. Removal of solvent gave 320 mg of a white solid
(72%).
MS (m/z) 322.3 (M++H).
Step D. 7-(chloroinethyl)-2-(4-fluorophenylsulfonyl)-1,2,3,4-
tetrahydroisoquinoline
(Compound 80D)
CI
~S%O~
~ N O
I ~ I ~ F
[0350] To (2-(4-fluorophenylsulfonyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)
methanol was
added CH2C12 (4 mL), SOCI2 (1 mL), and pyridine (2 mL). The mixture was
stirred for 18
hours before solvent was removed. The mixture was basified with 10% NaOH and
extracted
138
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
with CH2C12, and purified using silica gel chromatography to give 60 mg of a
white solid
(18%). MS (m/z) 340.8 (M++H).
Step E. 2-(2-(4-fluorophenylsulfonyl)-1,2,3,4-tetrahydroisoquinolin-7-
yl)acetonitrile
(Compound 80E)
CN
O'1S1,O
j\ N/
~ F
[0351] To 7-(chloromethyl)-2-(4-fluorophenylsulfonyl)-1,2,3,4-
tetrahydroisoquinoline was
added NaCN (18 mg, 0.36 mmol) and DMF (2 mL). The mixture was heated at 70 C
for 2
hours and purified directly using silica gel chromatography to give 37 mg of a
white solid
(65%). MS (m/z) 331.3 (M++H).
Step F. 2-(2-(4-fluorophenylsulfonyl)-1,2,3,4-tetrahydroisoquinolin-7-
yl)acetate (Compound
80F)
CO2Me
~ N~ ~
I ~ I ~ F
[0352] 2-(2-(4-fluorophenylsulfonyl)-1,2,3,4-tetrahydroisoquinolin-7-
yl)acetonitrile
dissolved in MeOH (4 mL) and EtOAc (3 mL). HC1(g) was bubbled for 1 min.
(exothennic). The resulting clear solution was stirred for 35 min. Solvent was
removed and
the crude product was used as is. MS (m/z) 364.4 (M++H).
139
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
Step G. 2-(2-(4-fluorophenyl sulfonyl)- 1,2,3,4-tetrahydroisoquinolin-7-yl)
acetic acid
(Compound 80G)
Ol S ,O
HO2C N~
[0353] To methyl 2-(2-(4-fluorophenylsulfonyl)-1,2,3,4-tetrahydroisoquinolin-7-
yl)acetate
was added THF-H20 (2.5 mL-0.5 mL) and LiOH=H2O (149 mg, 3.56 mmol). It was
stirred
for 18 hours before being acidified with 1N HCI. It was extracted with EtOAc.
Reverse
phase HPLC purification yielded the title compound
2-(2-(4-fluorophenylsulfonyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)acetic acid
as a white solid.
MS (m/z) 350.05 (M++H); Rt = 3.08 min.
Example 81
2-(2-(2-(4-methylphenylsulfonamido)acetyl)-1,2,3,4-tetrahydroisoquinolin-7-
yl)acetic acid
(Compound 81)
Scheme 20
Me02C NH HCI Boc20 Me02C GDIBAL-H
NH B o c
NaHCO3
OH I
~ NHBoc PPh3, 12 NaCN
I I NHBoc
/ imidazole / DMF
CN CO2Me
I~ NHBoc HCI (g) NH HCI
/ MeOH
i. Ts Gly, HATU COZH O N
ii. LiOH N
140
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
Step A. 2-tert-butyl 7-methyl 3,4-dihydroisoquinoline-2,7(1 H)-dicarboxylate
(Compound
81A)
HBoc
MeO2C icc N
[0354] To 616 mg (2.7 mmol) of commercially available methyl
1,2,3,4-tetrahydroisoquinoline-7-carboxylate hydrochloride was added THF-H20
(16 mL-4
mL), NaHCO3 (1.36 g) and Boc2O (1.18 g). The mixture was stirred for 17 hours
and
extracted with EtOAc. Silica gel chromatography gave a colorless oil (100%).
MS (m/z)
293.3 (M++H).
Step B. tert-butyl 7-(hydroxymethyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate
(Compound 81B)
OH
I ~ NHBoc
~
[0355] 2-tert-butyl 7-methyl-3,4-dihydroisoquinoline-2, 7-(1H)-dicarboxylate
was
dissolved in THF (30 mL) and cooled to 0 C. DIBAL-H (8.1 mL, 8.1 mmol, 1 M in
THF)
was added. The mixture was stirred at ambient temperature for 16 hours before
a solution of
Na-K tartrate was added and the mixture stirred for 5 hours. The mixture was
extracted with
EtOAc and purification by silica gel chromatography gave 294 mg the desired
alcohol (41 %)
as well as recovered starting ester (320 mg, 41 %). MS (m/z) 265.3 (M++H).
Step C. tert-butyl 7-(iodoinethyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate
(Compound
81C)
I ~ NHBoc
/
[0356] To tert-butyl 7-(hydroxymethyl)-3,4-dihydroisoquinoline-2 (1H)-
carboxylate in
THF (10 mL) at 0 C was added PPh3 (441 mg, 1.68 mmol), imidazole (190 mg, 2.8
mmol)
and IZ (426 mg,1.68 mmol). The mixture was stirred at 0 C for 30 min. and then
at ambient
141
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
temperature for 3 more hours. Solvent was removed and the residue purified by
silica gel
chromatography to give 65 mg of the desired iodide (16%) as well as the
recovered starting
alcohol (116 mg, 40%). MS (m/z) 375.2 (M++H).
Step D. 7-(cyanomethyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (Compound
81D)
CN
I ~ NHBoc
/
[0357] To tert-butyl7-(iodomethyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate
was added
NaCN (17 mg, 0.34 mmol) and DMF (2 mL). The mixture was heated at ambient
temperature for 30 min. and then purified directly using silica gel
chromatography to give 41
mg of a colorless oil (89%). MS (m/z) 274.3 (M++H).
Step E. Methyl 2-(1,2,3,4-tetrahydroisoquinolin-7-yl)acetate hydrochloride
(Compound
80E)
CO2Me
I NH HCI
[0358] tert-butyl 7-(cyanomethyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate
was
dissolved in MeOH (3 mL) and HCl(g) was bubbled for 20 seconds (exothennic).
The
resulting clear solution was stirred for 16 hours. Solvent was removed and the
crude product
was used as is. MS (m/z) 206.2 (M++H).
142
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
Step F.
2-(2-(2-(4-methylphenylsulfonamido)acetyl)-1,2,3,4-tetrahydroisoquinolin-7-
yl)acetic acid
(Compound 81F)
CO2H O ~
H
N
O
[0359] To methyl2-(1,2,3,4-tetrahydroisoquinolin-7-yl) acetate hydrochloride
was added
added Ts-glycine (15 mg, 0.065 mmol), CH3CN (2 mL), DIEA (45 L, 0.26 mmol)
and
HATU (33 mg, 0.0868 mmol). The mixture was stirred for 3 days before THF-H20
(2
mL-0.5 mL) was added, followed by LiOH=HZO (40 mg, 0.95 mmol). It was stirred
for 3
days before being acidified with 1N HC1 and extracted with EtOAc. Reverse
phase HPLC
purification yielded the title compound
2-(2-(2-(4-methylphenylsulfonamido)acetyl)-1,2,3,4-tetrahydroisoquinolin-7-
yl)acetic acid as
a white solid. MS (m/z) 403.10 (M++H); Rt = 2.80 min.
Example 82
2-(4-(2-(4-methylphenylsulfonamido)acetyl)-2,3,4,5-
tetrahydrobenzo[f][1,4]oxazepin-7-yl)ac
etic acid (Compound 82)
Scheme 21
COZMe CO2Me CO2Me
HONH2 Boc2O
\ _ I _ I
(CHO)r,, i-PrOH NaHCO3
HO NH HO NBoc
OH
HO HO
CO2Me CO2H
PPh3, DIAD i. TFA-CH2CI2 _ I\ 0
S'
THF ii. Ts-Gly, HATU
O\.NBOc iii. LiOH
O
143
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
Step A. Methyl 2- (4-hydroxy-3 -((2-hydroxyethyl amino)methyl)phenyl) acetate
(Compound
82A)
CO2Me
HO NH
HO
[0360] To 7.82 g (47 mmol) of commercially available methyl 2-(4-
hydroxyphenyl)acetate
was added ethanol amine (2.8 mL, 47 mmol), paraformaldehyde (1.55 g, 52 mmol)
and
isopropyl alcohol (100 mL). The slurry was refluxed at 95 C for 19 hours.
Solvent was
removed and silica gel chromatography purification gave 1.4 g of a colorless
oil (12%). MS
(m/z) 240.2 (M++H).
Step B. Methyl
2-(3 -( (tert-butoxycarbonyl (2-hydroxyethyl)amino)methyl)-4-
hydroxyphenyl)acetate
(Compound 82B)
CO2Me
HONBoc
HO
[0361] To 252 mg of methyl 2-(4-hydroxy-3-((2-hydroxyethylamino) methyl)
phenyl)
acetate was added added THF-H20 (8 mL-2 mL), NaHCO3 (441 mg) and Boc2O (345
mg).
The mixture was stirred for 18 hours and extracted with EtOAc. Silica gel
chromatography
gave 214 mg of N-Boc derivative as a white solid (60%). MS (m/z) 340.3 (M++H).
144
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
Step C. tert-butyl
7-(2-methoxy-2-oxoethyl)-2,3-dihydrobenzo[f] [ 1,4]oxazepine-4(5H)-carboxylate
(Compound 82C)
CO2Me
ON Boc
[0362] To 115 mg of inethyl2-(3-((tert-butoxycarbonyl(2-hydroxyethyl) amino)
methyl)-4-hydroxyphenyl) acetate in THF (3.5 mL) was added PPh3 (267 mg, 1.02
mmol)
and DIAD (197 L, 1.02 mmol, slow addition in 14 min.). The mixture was
stirred for 30
min. and quenched with NaHCO3 (sat.) and extracted with EtOAc. Silica gel
chromatography gave 44 mg of the cyclized product as a colorless oil (40%). MS
(m/z)
322.3 (M++H).
Step D.
2-(4-(2-(4-methylphenylsulfonamido)acetyl)-2,3,4,5-tetrahydrobenzo[f][
1,4]oxazepin-7-yl)ac
etic acid (Compound 82D)
CO2H
O_S~ ~
O N H ~ ~
~~~
[0363] To the above oil was added added CH2C12 (2 mL) and TFA (1 mL). The
mixture
was stirred for 2 hours and solvent was removed. To this hydrochloride salt
was added added
Ts-glycine (47 mg, 0.2 mmol), CH3CN (4 mL), DIEA (120 L, 0.685 mmol) and HATU
(104
ing, 0.27 mmol). The mixture was stirred for 6 hours before solvent was
removed and
THF-H20 (4 mL-0.8 mL) was added, followed by LiOH=H2O (86 mg, 2.06 mmol). It
was
stirred for 20 hours before THF was carefully blown off by a stream of N2. The
mixture was
acidified with 1N HCl and extracted with EtOAc and dried over Na2SO4. Removal
of solvent
gave 37 mg of the title compound
145
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
2-(4-(2-(4-methylphenylsulfonamido)acetyl)-2,3,4,5-tetrahydrobenzo[f] [ 1,4]
oxazepin-7-yl)ac
etic acid as a white solid (64%). MS (m/z) 419.10 (M++H); Rt = 2.69 min.
Pharmacological Data:
Receptor Interaction Assay
Cell culture:
[0364] Human Jurkat cells transfected with DP-2, DP-1 or TP receptors were
maintained in
culture in a humidified atmosphere at 37 C ( 5% CO2) in RPMI 1640 media
(Gibco ,
Invitrogen, USA) with 10% fetal bovine serum (Hyclone, Logan, UT, USA) plus
penicillin-streptomycin (Gibco), L-Glutamine (Gibco ), sodium pyruvate and
100ug/ml
G418. Cells were grown in T225 flasks (Corning ) and harvested by
centrifugation. Cell
pellets were collected from approximately 200 ml of cell suspension, pelleted
by
centrifugation and stored at -20 C until processed into membranes.
Preparation of cell membranes:
[0365] Frozen Jurkat cell pellets expressing either DP-2 , DP-1 or TP were
thawed on ice.
Each pellet was suspended in membrane buffer (25mM Hepes pH7.2, 6mM MgC12,
1mM
EDTA) plus Complete protease inhibitor cocktail tablets (Roche Mannheim
Gennany). The
pellets were dounce homogenized and centrifuged at 1900 RPM for 10 min. in a
table top
centrifuge (Beckman Coulter Allegra 6R). The supernatants were collected and
pellets
resuspended in l Omis of membrane buffer, dounce homogenized again and
centrifuged as
above. The supernatants were pooled and centrifuged in a Beckman J2-21 M
centrifuge using
a JA20 rotor at 20,000 RPM for 1.5 hours at 4 C. The supernatants were
discarded and the
membrane pellets suspended in membrane buffer and pooled. Protein
concentration was
determined and membranes adjusted to approximately 1.5ings/hnl.
DP-2 Binding Assay:
[0366] Compound interactions with the DP-2 receptors were detennined by means
of
competitive radioligand binding assays using membranes prepared from DP-2
expressing
cells (prepared as above) and 3[H] PGD2 (166Ci/mmol) as a radioactive tracer.
Assays were
perfonned in a final volume of 150 1 of assay buffer (10mM Hepes , 10mM MnC12,
1mM
146
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
EDTA and 1% DMSO). Test article serially diluted in assay buffer was incubated
with 1nM
radioactive tracer and 10ug/well of the membranes prepared from DP-2
expressing cells in a
96 well polypropylene plate for one hour at room temperature. The reaction
mixture was then
transferred to a Millipore(Bedford, MA) MultiScreen , FC MAFCNOB glass fiber
filter
plate. The plate was vacuum aspirated, and washed 2 times with 200u1 of
binding buffer
vacuum aspirating between each wash. The plate was allowed to dry and 50u1 of
Optiphase
`Super Mix' (Wallac Oy Turku, Finland) scintillation cocktail was added to
each well. The
plate was counted on a Wallac TM(Wallac Oy Turku, Finland) 1450 micro beta
liquid
scintillation counter
DP-2 Chemotaxis Assay
[0367] The ability of compounds of the invention to antagonize DP-2 receptor
function was
examined in chemotaxis assays using DP-2 transfected Jurkat cells. Compounds
were serially
diluted into complete media containing I nM PGD2 as a chemoattractant, and
600u1 of this
mixture were transferred into the bottom wells of a Costar Transwell plate (8
m pore size).
DP-2 transfected Jurkat cells were harvested, re-suspended at 7.5x106/ml
complete media,
and 100 L of this cell suspension was added into the pore filter inserts.
After equilibration of
all the components to 37 C in a cell incubator for 15 min, chemotaxis was
initiated by
transfer of the filter inserts onto the bottom wells. Following 2 hr
incubation in a 37 C
incubator the filter inserts were removed, the media with cells were collected
from lower
wells and transferred to FACS tubes. Cells in each sample were then enumerated
on
FACScan using CellQuest software.
Selectivity assay
DP-1 binding assay
[0368] DP-1 binding assays were perfonned in a manner substantially identical
to the DP-2
binding assay, except that DP-1 transfected cell membranes were used.
Human TP binding assay
[0369] TP receptor interaction was assessed in competition binding assays
using
membranes from TP receptor transfected cells (prepared as above) and 3[H]
SQ29,548
147
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
(48.2uCi/mmol) as a TP-selective tracer. Assays were performed in a final
volume of 150u1
of binding buffer (10mM Hepes, 10mM MnC12, 1mM EDTA and 1% DMSO. Duplicate
samples of serially diluted test compound were incubated with l Oug/well of TP
membranes
in the presence of 3nM 3[H] SQ 29,548. Following a one hour incubation at room
temperature the reaction mixture was transferred to a Millipore (Bedford, MA)
MultiScreen , FC MAFCNOB glass fiber filter plate. The mixture was vacuum
aspirated,
and washed 2 times with 200u1 of binding buffer vacuum aspirating between each
wash.
After air drying 50u1 of Optiphase Super MixTM (Wallac Oy Turku, Finland)
scintillation
cocktail was added to each well and radioactivity was quantified on a Wa11acTM
(Wallac Oy
Turku, Finland) 1450 micro beta liquid scintillation counter.
[0370] All of the acid compounds of the Examples that were tested in the assay
exhibited
IC50 values less than 10 M, for example the acid compounds of examples 2, 4,
6, 7, 8, 9, 10,
11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29,
29, 30, 31, 32, 33, 34,
35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 49, 50, 51, 52, 53, 54,
55, 56, 57, 58, 59, 60,
61, 62, 63, 64, 65, 69, 70, 71, 72, 73, 76. In some embodiments, the compounds
of the
invention exhibited IC50 values less than 1 M. In some embodiments, the
compounds of the
invention exhibited IC50 values less than 0.1 M.
[0371] All of the acid compounds of the Examples that were tested in the above-
described
ligand binding assays exhibited an average IC50 value which was least 2-fold
lower for DP-2
over DP-1 or TP, for example the acid compounds of examples 8, 9, 10, 11, 12,
13, 14, 16,
17, 18, 21, 22, 29, 33, 34, 44, 46, 47, 49, 50, 52, 53, 54, 55, 57, 58, 59,
61, 63, 64, 69 and 76.
In some embodiments, the acid compounds of the invention exhibited an average
IC50 value
which was least 10-fold lower for DP-2 over DP-1 or TP, for example the acid
compounds of
examples 8, 9, 10, 11, 12, 13, 14, 16, 17, 18, 21, 22, 29, 33, 34, 44, 46, 47,
49, 52, 53, 54, 55,
57, 59, 63, 64, 69 and 76. In some embodiments, the acid compounds of the
invention
exhibited an average IC50 value which was least 50-fold lower for DP-2 over DP-
1 or TP, for
example the compounds of examples 8, 9, 10, 11, 12, 13, 14, 16, 17, 21, 22,
29, 33, 34, 44,
46, 47, 49, 59, 64, 69 and 76.
[0372] All publications and patent applications cited in this specification
are herein
incorporated by reference as if each individual publication or patent
application were
specifically and individually indicated to be incorporated by reference.
Although the
foregoing invention has been described in some detail by way of illustration
and example for
148
CA 02654927 2008-12-09
WO 2007/146838 PCT/US2007/070805
purposes of clarity of understanding, it will be readily apparent to those of
ordinary skill in
the art in light of the teachings of this invention that certain changes and
modifications may
be made thereto without departing from the spirit or scope of the appended
claims.
149