Note: Descriptions are shown in the official language in which they were submitted.
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
PARATHYROID HORMONE ANALOGUES AND METHODS OF USE
FIELD OF THE INVENTION
This application relates to methods and compositions of treating a subject
with a bone
deficit disorder. The methods generally include administering to a subject in
need thereof a
pharmaceutically acceptable formulation comprising a parathyroid hormone (PTH)
peptide or
analog in a dose sufficient to result in an effective pharmacokinetic profile,
while
simultaneously reducing undesirable side effects. Additionally, the dose can
be optimized
based on the weight, body surface area, body mass index (BMI), lean body mass
or other
body characteristic of the patient to be treated. This dose optimization
approach applies to
PTH peptides and analogs, as well as to other therapeutics as described
herein.
BACKGROUND OF THE INVENTION
Bone remodeling, or turnover, consists of two opposing activities: the
breakdown
(resorption) of old bone by osteoclasts, and the formation of new bone by
osteoblasts. Loss
of bone mass occurs as part of the natural aging process. Calcium is
constantly being added
to and taken away from bone. When calcium is taken away faster than it is
added, the bones
become lighter, less dense, and more porous. This makes the bones weaker and
increases
their risk of fracture.
Bones naturally become thinner (called osteopenia) as people grow older,
because
existing bone is broken down faster than new bone is made. As this occurs, the
bones lose
minerals, heaviness (mass), and structure, making them weaker and more
fragile. With
further bone loss, osteopenia develops into osteoporosis. Accordingly, the
thicker a person's
bones are, the longer it takes to develop osteoporosis. Although osteoporosis
can occur in
men, it is most common in women older than age 65.
Osteoporosis often results in spontaneous fractures of load-bearing bones and
the
physical and mental deterioration characteristic of immobilizing injuries. In
particular,
postmenopausal osteoporosis is caused by the disappearance of estrogens which
triggers an
acceleration of bone turnover with an increased imbalance between resorption
of old bone
and formation of new bone. Instead of bone mass remaining stable, bone loss
results because
osteoclasts, the cells that destroy old bone (resorption of bones), outperform
osteoblasts, the
cells that build new bone (formation of bones). This accelerated bone loss due
to resorption
1
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
without adequate compensation by bone formation results in gradual thinning,
increased
porosity, and depletion of load-bearing bones.
End stage renal disease is invariably associated with bone disease, known as
renal
osteodystrophy (ROD). ROD may exist in a high turnover form characterized by
high
circulating levels of parathyroid hormone (PTH) and overactive bone tissue,
often with
osteitis fibrosa cystica. The low turnover form of the disease, also known as
adynamic bone
disease, is characterized by normal or low circulating levels of PTH.
Histologically, the bone
surfaces are quiescent with little or no cellular activity and osteomalacia
may also be present.
The incidence of the condition is increased with advanced age, presence of
corticosteroid
therapy, presence of calcimimetic therapy, calcium containing phosphate
binders and high
doses of Vitamin D sterols. However, adynamic bone disease is currently
difficult to treat
without leading to an unacceptable increase in serum calcium. Accordingly,
there is a
continuous unmet need for effective therapy.
Among the remedies for osteoporosis (which have historically involved increase
in
dietary calcium, estrogen therapy, and increased doses of vitamin D), human
parathyroid
hormone (hPTH) treatments are used to build bones to compensate for the bone
loss due to
osteoporosis. Parathyroid hormone is produced by the parathyroid gland and is
involved in
the control of calcium levels in blood. It is a hypercalcemic hormone,
elevating blood
calcium levels. PTH is a polypeptide and synthetic polypeptides may be
prepared using the
method disclosed by Erickson and Merrifield, The Proteins, Neurath et al.,
Eds., Academic
Press, New York, 1976, page 257, preferably as modified by the method of
Hodges et al.,
Peptide Research, 1, 19 (1988) or by Atherton, E. and Sheppard, R. C., Solid
Phase Peptide
Synthesis, IRL Press, Oxford, 1989. When serum calcium is reduced to below a
"normal"
level, the parathyroid gland releases PTH and resorption of bone calcium and
increased
absorption of calcium from the intestine, as well as renal reabsorption of
calcium, occur. An
antagonist of PTH is calcitonin, which acts to reduce the level of circulating
calcium.
Although high levels of PTH can remove calcium from the bone, intermittent low
doses can
actually promote bone growth. For example, the native hPTH-(1-84) and its
fragment
hPTH-(1-34) (as sold under the tradename FORTEO by Eli Lilly and Co.) have
been shown
to be useful in the treatment of osteoporosis. The native hPTH-(1-84) and the
hPTH-(1-34)
fragment, however, suffer a drawback that while they promote bone formation,
they
simultaneously activate bone resorption. As a consequence hPTH-(1-34) is
effective in
reducing the fracture frequency of trabecular bone (which make up the bones of
the axial
skeleton, and include the rib cage, the back bones and the skull, and
vertebrate bone), but its
2
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
fracture reduction efficacy on cortical bone (which serves to protect against
torsional loads
and includes, for example, the hip and wrists) is considerably less.
There remains a need for therapeutic approaches employing suitable PTH
analogues
to restore bones and increase bone mineral density in both trabecular and
cortical bones in
patients with osteoporosis or other bone degenerative/deficit disorders. There
further remains
a need for therapeutic approaches employing suitable PTH analogues to restore
bones and
increase bone mineral density and formation without stimulating bone
resorption, and without
significantly increasing the levels of serum calcium in patients with
osteoporosis or other
bone degenerative disorders.
SUMMARY OF THE INVENTION
The present invention provides pharmaceut'ical compositions and formulations
containing suitable PTH peptides or analogs thereof for use in methods
directed to treating
subjects suffering from various bone degenerative or bone deficit disorders.
The PTH
peptides or analogs described herein induce bone formation in both trabecular
and cortical
bones, thereby increasing bone mineral density and restoring bones.
Unexpectedly, the PTH
peptides or analogs described herein induce bone formation while causing less
bone
resorption than previously known PTH analogs, and also demonstrate lower
incidences of
and severity in hypercalcemia.
The PTH peptides or analogs disclosed herein, when administered within the
specified
dosage ranges, are effective in reversing the effects of osteoporosis on
cortical bones in
animals. Righting the imbalance between resorption of old cortical bone and
formation of
new cortical bone, these PTH peptides or analogs have been shown to reverse
the effects of
osteoporosis on bone. Thus, the methods described herein promote cortical bone
growth in
animals without significantly increasing cortical bone porosity.
These PTH peptides or analogs also promote recovery from bone injuries.
Therefore,
administration of the specified dosages of the PTH peptides or analogs of the
present
invention restore osteoporotic cortical bones and promote bone healing in
various
circumstances, such as in the treatment of fractures.
In one aspect, the invention provides a method for the treatment of
osteoporosis, for
treating a bone fracture, for inducing bone formation in trabecular and
cortical bones, for
treating or preventing renal osteodystrophy (ROD) and related disorders,
comprising
administering to a subject in need thereof a pharmaceutically acceptable
formulation
3
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
comprising a parathyroid hormone (PTH) peptide or analog, wherein said PTH
peptide
analogue has a reduced phospholipase-C activity and maintains adenylate
cyclase activity,
wherein the dosage administered results in an effective pharmacokinetic
profile and effective
bioactivity.
Another embodiment provides the use of the PTH peptides or analogs of the
present
invention for treating osteoporosis, for treating or preventing a bone
fracture, for inducing
bone formation in trabecular and cortical bones, for treating or preventing
renal
osteodystrophy (ROD) and related disorders, or for any other therapeutic use
of PTH,
wherein calcium monitoring is not required.
Another embodiment provides the use of the PTH peptides of the present
invention
for treating osteoporosis, for treating or preventing a bone fracture, for
inducing bone
formation in trabecular and cortical bones, for treating or preventing renal
osteodystrophy
(ROD) and related disorders, or for any other therapeutic use of PTH, wherein
a warrming
regarding osteosarcoma formation is not required and wherein administration of
the PTH
peptides of the present invention may lead to lower incidences of osteosarcoma
as compared
to administration of Forteo.
In another embodiment, the invention provides a pharmaceutical formulation for
subcutaneous administration comprising a unit dosage form of a therapeutically
effective
amount of an aqueous formulation of a parathyroid hormone (PTH) peptide or
analog in a
daily dosage range of 2 to 100 g, wherein said PTH peptide or analog has
reduced
phospholipase-C activity and maintains adenylate cyclase activity and has an
effective
pharmacokinetic profile and effective bioactivity; and a pharmaceutically
acceptable
excipient, diluent, or carrier, or combinations thereof. Other therapeutically
effective
amounts within the pharmaceutical formulation include a daily dosage range of
from 0.5 to
50 g of a formulation stabilized with propylene glycol and/or ethanol for sub-
cutaneous
delivery or a daily dosage range of 100 to 3,000 g for inhalation delivery,
and weekly doses
at 3-7 times the daily doses. Other embodiments include any dosage with any
route of
administration which results in an effective pharmacokinetic profile and
effective bioactivity.
Another embodiment of the invention is a kit for treating a bone deficit
disorder
comprising, in one or more containers, a therapeutically effective amount of
the above-
described pharmaceutical composition contained in a device, and a label or
packaging insert
containing instructions for use.
PTH analogues optionally include less than the first 34 amino acids at the N-
terminal
end. The PTH peptide analogues of the present invention, when compared to full-
length PTH
4
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
peptides or other PTH peptide analogues which are 34 amino acid residues or
longer, trigger
less than full activation of phospholipase-C, less bone resorption, and less
incidences or
lower severity of hypercalcemia, while still maintaining increases in bone
mineral density
(BMD) at a variety of sites within the body.
Specific embodiments of PTH peptide analogues of the present invention include
the
following: PTH-(1-31)NH2, Ostabolin; PTH-(1-30)NHZ; PTH-(1-29)NH2; PTH-(1-
28)NH2;
Leu27PTH-(1-31)NH2i Leu27PTH-(1-30)NH2; Leu27PTH-(1-29)NH2; Leu27 cyclo(22-
26)PTH-
(1-31)NH2 Ostabolin-CTM; Leu27cyclo(22-26)PTH-(1-34)NH2i Leu27cyclo(Lys26-
Asp3)PTH-
(1-34)NH2i Cyclo(Lys27-Asp30)PTH-(1-34)NH2i Leu27cyclo(22-26)PTH-(1-31)NH2;
Ala27 or
N1e27 or Tyr27 or I1e27 cyclo(22-26)PTH-(1-31)NH2i Leu27cyclo(22-26)PTH-(1-
32)NH2;
Leu27cyclo(22-26)PTH-(1-31)OH; LeuZ7 cyclo(26-30)PTH-(1-31)NH2i
CysZ2Cys26Leu27cyclo(22-26)PTH-(1-31)NH2, Cys22Cys26Leu27cyclo(26-30)PTH-(1-
31)NH2;
Cyclo(27-30)PTH-(1-31)NH2i Leu27cyclo(22-26)PTH-(1-30)NH2; Cyclo(22-26)PTH-(1-
31)NH2i Cyclo(22-26)PTH-(1-30)NH2; Leu27cyclo(22-26)PTH-(1-29)NH2;
I.eu27cyclo(22-
26)PTH-(1-28)NH2; G1ut7,LeuZ7cyclo(13-17)(22-26)PTH-(1-28)NH2; and
Glu",LeuZ'cyclo(13 -17)(22-26)PTH-(1-31)NH2.
Other embodiments of the invention include compositions and methods for
treating a
bone deficit disorder, while reducing side effects associated with the
administration of a
parathyroid hormone, comprising administering to a subject with a bone deficit
disorder a
pharmaceutically acceptable formulation comprising a parathyroid hormone (PTH)
peptide
analogue in a daily dose sufficient to result in an effective pharmacokinetic
profile and
maintained adenylate cyclase activity, while simultaneously reducing
undesirable side
effects. Optionally, the PTH peptide analogue can result in reduced
phospholipase-C
activity.
In another embodiment, the effective pharmacokinetic profile can be achieved
by a
variety of routes of administration with a variety of different formulations
and comprises a
phanmacokinetic parameter selected from the group consisting of a) a half-life
of said PTH
peptide analogue of between 2 minutes and 60 minutes; b) a duration of
exposure to said PTH
peptide analogue of between 30 minutes and 4 hours; c) a T. of said PTH
peptide analogue
of between 2 minutes and 30 minutes; and d) a C. of said PTH peptide analogue
of between
and 400 pg/ml. In additional embodiments, the half-life is between 5-30
minutes, the
duration of exposure is between one and two hours, the T. is between 15-30
minutes, and
the C. is between 50-200 pg/ml.
5
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
This pharmacokinetic profile can be achieved with any route of administration
known
to those skilled in the art, including oral, topical, transdermal, nasal,
pulmonary,
transpercutaneous (wherein the skin has been broken either by mechanical or
energy means),
rectal, buccal, vaginal, via an implanted reservoir, or parenteral. Parenteral
includes
subcutaneous, intravenous, intramuscular, intraperitoneal, intra-articular,
intra-synovial,
intrasternal, intrathecal, intrahepatic, intralesional, and intracranial
injection or infusion
techniques. More preferably, the route of administration is subcutaneous,
transcutaneous,
intranasal, transdermal, oral, or inhalation administration.
In further embodiments, the undesirable side effects that are reduced are
selected from
the group consisting of bone resorption, feeling cold, fatigue, loose stool,
feeling hot, lower
abdominal pain, injection site reaction, arthralgia, injection site
hemorrhage,
pharyngolaryngeal pain, muscle cramps, and abdominal pain. More specifically,
the
undesirable side effects that are reduced are selected from the group
consisting of
hypercalcemia, increase in mean serum calcium level, headache, nausea, back
pain, dizziness,
and extremity pain.
The PTH peptides of the present invention can also be administered at a
variety of
doses. Effective dosages can vary according to the type of formulation of PTH
peptides or
analogs administered as well as the route of administration. One skilled in
the art can adjust
the dosage by changing the route of administration or formulation, so that the
dosage
administered would result in a similar pharmacokinetic or biological profile
as would result
from the preferred dosage ranges described herein. Exemplary dosages include a
daily dose
of 2 to 100 g for subcutaneous delivery of an aqueous formulation, a daily
dose of 0.5 to 50
g for subcutaneous delivery of a formulation stabilized with propylene glycol
and/or
ethanol, a daily dose of 100 to 3,000 g for inhalation delivery, and weekly
doses at 3-7 times
the daily doses. Other suitable dosages include any dosage with any route of
administration
that results in a bioavailability or pharmacokinetic profile similar to those
yielded by the
above-described dosage ranges.
Preferred dosages for subcutaneous delivery of an aqueous formulation include
dosages between 5-9 g, 10-19 g, 20-30 g, 31-40 g, 42-45 g, 46-50 g, and
more
specifically at 5 g, 7.5 g, 10 g, 15 g, 20 pg, 25 g, 30 g, 35 g, 40 g,
45 g, or 50 g.
Most preferred doses for subcutaneous delivery of an aqueous formulation
include either 7.5,
15, 30, or 45 g . The dosage can also be calculated based on the size of the
patient. The g
dosages can be normalized for patient characteristics such as height, weight,
body surface
area, BMI, lean body mass, etc., by converting the g to g/kg, or g/m2, or
g /ml. The
6
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
PTH peptides of the invention can also be administered at a daily dose of
between 0.20 and
0.90 g/kg, more preferably between 0.30 and 0.70 g/kg, and more preferably
between 0.46
and 0.69 g/kg.
Additional embodiments of the invention include sequential therapy. One
embodiment of such a treatment regimen starts treatment with a high dose of
suitable PTH
peptides or analogs and then after a period of time which could be 1-12 months
but preferably
3-9 months and most preferably 4-8 months converts to a lower dose which
maintains bone
formation at a lower level but does not allow stimulation of bone resorption.
Sequential
therapy could also start treatment with a low dose and then convert to a high
dose. Such
sequential therapy can be administered in all doses disclosed herein.
One suitable dosage regimen includes administering an aqueous formulation of a
PTH
peptide or analog by subcutaneous administration in a first daily dose of from
35 g to 100
g, and then after the termination of the first period of time administering
for a second period
of time a second dose of from 2 g to 35 g of a PTH peptide analog. Another
suitable
dosage regimen includes administering an aqueous formulation of a PTH peptide
or analog
by subcutaneous administration in a first daily dose of from 2 g to 35 jig,
and then after the
termination of the first period of time administering for a second period of
time a second dose
of from 35 jig to 100 g of a PTH peptide analog. Additionally, the PTH
peptides of the
present invention can be administered by inhalation at a first and second
daily dose of
between 100 g -2,000 g, or at a first and second weekly dose of 3-7 times
greater than the
daily dose. In similar ways, dosages for sequential therapy can be calculated
for inhalation
administration or for formulations stabilized with propylene glycol or
ethanol, or for any
other formulations administered by any routes known in the art.
Another embodiment of the present invention is the administration of a dose to
a
patient based on that patient's weight, height, body surface area, BMI, or
other patient
characteristic and/or presentation of symptoms. This weight cut off method
provides a
method for determining a therapeutically effective dosage while maintaining a
low incidence
of side effects for a patient based upon their weight, body surface area, or
BMI. By providing
different doses to patients based on their body weight or mass, the amount of
exposure of
drug to a variety of patients is made more level. The choice of dosage based
on weight, body
surface area, or BMI of the patient improves the benefit to risk profile of
the present peptides
by improving the overall efficacy and proportion of patients who respond to
the dosage while
reducing the side effects of a dose that results in high exposure for an
individual.
7
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
All osteoporosis therapeutics with a predominant action to stimulate bone
fonnation
may be administered in a manner where the dosage is based on the patient's
weight, height,
body surface area, BMI, or other body type characteristic of the patient.
Examples of such
therapeutics within the scope of the present invention include the anti-
sclerostin Mab,
inhibitors of negative regulators of the Wnt signaling pathways, and activin
receptor agonists.
Additionally dosage based on patient weight, body surface area, or BMI is
effective for all
therapeutics whose bone formation effect is mediated by the action of PTH on
its receptor,
including PTH, full-length (1-84) and fragments thereof, PTH analogs, PTHrP,
and PTHrP
analogs. Specific PTH peptides which are effective with dosage based on
patient weight,
body surface area, or BMI include, but not limited to, full length PTH 1-84,
PTH 1-34, PTH-
(1-31)NH2, Ostabolin; PTH-(1-30)NH2; PTH-(1-29)NHZ; PTH-(1-28)NH2i Leu27PTH-(1-
31)NH2i Leu27PTH-(1-30)NH2; Leu27PTH-(1-29)NH2i Leu27cyclo(22-26)PTH-(1-31)NH2
Ostabolin-CTM; Leu27cyclo(22-26)PTH-(1-34)NH2; Leu27cyclo(Lys26-Asp3)PTH-(1-
34)NH2;
Cyclo(Lys27-Asp30)PTH-(1-34)NH2; Leu27cyclo(22-26)PTH-(1-31)NH2; Ala27 or
N1eZ7 or
Tyr27 or I1e27 cyclo(22-26)PTH-(1-31)NH2i Leu27cyclo(22-26)PTH-(1-32)NH2;
Leu27cyclo(22-26)PTH-(1-31)OH; Leu27cyclo(26-30)PTH-(1-31)NH2;
Cys22Cys26Leu27cyclo(22-26)PTH-(1-31)NH2i Cys22Cys26Leu27cyclo(26-30)PTH-(1-
31)NH2;
Cyclo(27-30)PTH-(1-31)NH2i Leu27cyclo(22-26)PTH-(1-30)NH2i Cyclo(22-26)PTH-(1-
31)NH2i Cyclo(22-26)PTH-(1-30)NH2; Leu27cyclo(22-26)PTH-(1-29)NH2i
LeuZ7cyclo(22-
26)PTH-(1-28)NH2i Glu17,LeuZ7cyclo(13-17)(22-26)PTH-(1-28)NH2; and
Glu17,LeuZ7cyclo(13-17)(22-26)PTH-(1-31)NH2.
Additionally, suitable examples of therapeutics which can be administered
based on
the weight, body surface area, or BMI of the patient include any drugs which
have a narrow
therapeutic window, more specifically hormone therapies. Specific therapeutics
also include
calcium receptor antagonists which stimulate endogenous PTH production, such
as those that
act as agonists of the PTH receptor, including PTH, full-length and fragments
thereof, PTH
analogs, PTHrP and analogs thereof. The administration of a dosage based upon
the weight,
body surface area, or BMI of a patient can be used in a variety of
indications, including
osteoporosis, fracture repair, renal bone disease, corticosteroid-induced
osteoporosis,
transplant, and the induction of bone formation in trabecular and cortical
bone.
This invention also pertains to specific formulations of Ostabolin-C,
including an
inhalation powder, and stabilized formulations which have an increased
bioavailability. One
such stabilized formulation includes Ostabolin-C, 40% ethanol and 60% water,
with a pH
between 6.0 and 8.0, wherein the formulation is stable for at least 2 years at
5 C. Another
8
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
formulation includes Ostabolin-C, 40% propylene glycol and 60% water, with a
pH between
6.0 and 8.0, wherein the formulation is stable for at least 2 years at 5 C.
These formulations
may also include, singularly or in combination, methionine, lipoic acid,
sucrose and NaCI.
BRIEF DESCRIPTION OF THE DRAWINGS
For Figures 1-17, if not stated otherwise, the measurements following
administration
of Ostabolin-C were made after a 15 week course of subcutaneous daily
administration of the
stated dose, and the changes were measured as compared to baseline. As used
herein,
baseline is the patient's individual measurement prior to receiving any
treatment.
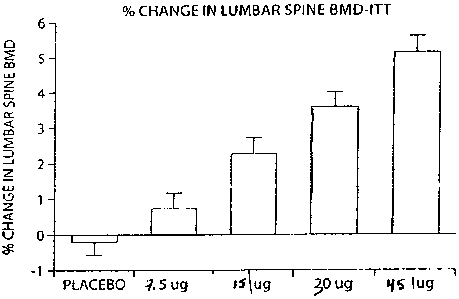
Fig. I is a bar graph showing the percentage change in lumbar spine bone
mineral
density (BMD) in patients with moderate osteoporosis receiving a
pharmaceutical
formulation containing [Leu27]cyclo[G1u22-Lys26]-PTH-(1-31)-NHZ.
Fig. 2 is a graph showing the percentage change in lumbar spine bone mineral
density
(BMD) in patients with moderate osteoporosis receiving the pharmaceutical
formulation
containing hPTH-(1-34) teriparatide, Forteo .
Fig. 3 is a bar graph showing the percentage change in total hip bone mineral
density
(B1VID) in patients with moderate osteoporosis receiving a pharmaceutical
formulation
containing [Leu27]cyclo[GIu22-Lys26]-PTH-(1-31)-NH2.
Fig. 4 is a bar graph showing the percentage change in femoral neck bone
mineral
density (BMD) in patients with moderate osteoporosis receiving a
pharmaceutical
formulation containing [Leu27]cyclo[G1u22-Lys26]-PTH-(1-31)-NHZ.
Fig. 5 is a bar graph showing the percentage change in trochanter bone mineral
density (BMD) in patients with moderate osteoporosis receiving a
phanmaceutical
formulation containing [Leu27]cyclo[G1u22-Lys26]-PTH-(1-31)-NH2.
Fig. 6 is a bar graph showing the percentage change in distal radius bone
mineral
density (BMD) in patients with moderate osteoporosis receiving a
pharmaceutical
formulation containing [Leu27]cyclo[G1u22-Lys26]-PTH-(1-31)-NH2.
9
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
Fig. 7 is a bar graph showing the percentage change in mid-shaft radius bone
mineral
density (BMD) in patients with moderate osteoporosis receiving a
pharmaceutical
formulation containing [Leu27]cyclo[G1u22-Lys26]-PTH-(1-31)-NH2.
Fig. 8 is a bar graph showing the percentage change in the bone formation
marker
amino terminal pro-peptide of type I pro-collagen (P 1NP) in patients with
moderate
osteoporosis receiving a pharmaceutical formulation containing
[Leu27]cyclo[G1u22-Lys26]=
PTH-(1-31)-NH2.
Fig. 9 is a bar graph showing the percentage change in the bone formation
marker
osteocalcin in patients with moderate osteoporosis receiving a pharmaceutical
formulation
containing [Leu27]cyclo[G1u22-Lys26]-PTH-(1-31)-NHZ.
Fig. 10 is a bar graph showing the percentage change in the bone formation
marker
bone-specific alkaline phosphatase (BSAP) in patients with moderate
osteoporosis receiving
a pharmaceutical formulation containing [Leu27]cyclo[G1u22-Lys26]-PTH-(1-31)-
NH2.
Fig. 11 is a bar graph showing the percentage change in the bone resorption
marker
N-telopeptide (NTx) in patients with moderate osteoporosis receiving a
pharmaceutical
formulation containing [Leu27]cyclo[G1u22-Lys26]-PTH-(1-31)-NH2.
Fig. 12 is a bar graph showing the percentage change in the bone resorption
marker C-
terminal telopeptide (CTx) in patients with moderate osteoporosis receiving a
pharmaceutical
formulation containing [Leu27] cyclo [G1u22-Lys26]-PTH-(1-31)-NHZ.
Fig. 13 is a graph showing the percentage change in the bone fonnation and
bone
resorption markers in patients with moderate osteoporosis receiving the
pharmaceutical
formulation containing rhPTH-(1-34), teriparatide, Forteo .
Fig. 14 is a bar graph showing the percentage of abnormal serum calcium levels
in
patients with moderate osteoporosis receiving a pharmaceutical formulation
containing
[Leu27]cyclo[G1u22-Lys26]-PTH-(1-31)-NHZ.
Fig. 15 is a slide showing the Forteo data derived from Deal et al., (2005) J.
Bone
Min. Res. 20, p. 1905-1991.
Fig. 16 is a slide showing the effectiveness of Ostabolin-C and Forteo.
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
Fig. 17 is a slide showing the effectiveness of Ostabolin-C and Forteo.
Fig. 18 is a graph showing the Phase I pharmacokinetics of Ostabolin-C at
different doses.
Fig. 19 is a graph showing the Ostabolin- pharmacokinetics in female rats at
different doses.
Fig. 20 is a graph showing the Ostabolin- pharmacokinetics in monkeys in a 6-
week
subcutaneous study.
Fig. 21 is a graph showing data for the administration of Ostabolin-C at 30
ug, including
effects at the lumbar spine, mid-shaft radius, hypercalcemia, and the anabolic
window.
Fig. 22 is a graph showing data for the administration of Ostabolin-C at 45
ug, including
effects at the lumbar spine, mid-shaft radius, hypercalcemia, and the anabolic
window.
Fig. 23 is a graph showing data for the % change from baseline for lumbar
spine BMD with
increasing Ostabolin-C exposure at 4 months.
Fig. 24 is a linear regression analysis assessing the full range of exposures
of Ostabolin-C and
the % change from baseline for lumbar spine BMD following exposure to
increasing
Ostabolin-C exposure at 4 months and 12 month, as compared to placebo.
Fig. 25 is a graph showing the effects of a 45 ug dose of Ostabolin-C on
lumbar-spine BMD,
s a % change from baseline, at 4 months and 12 months.
Fig. 26 is a linear regression analysis assessing the full range of exposures
of Ostabolin-C and
the % change from baseline for P1NP and CTx following exposure to increasing
Ostabolin-C
exposure at 4 months and 12 months, as compared to placebo.
Fig. 27 is a linear regression analysis assessing the full range of exposures
of Ostabolin-C and
the % change from baseline for total hip BMD following exposure to increasing
Ostabolin-C
exposure at 4 months and 12 months, as compared to placebo.
Fig. 28 is a linear regression analysis assessing the full range of exposures
of Ostabolin-C and
the % change from baseline for senun calcium levels following exposure to
increasing
Ostabolin-C exposure at 4 months and 12 months, as compared to placebos.
11
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
Fig. 29 is a linear regression analysis assessing the full range of exposures
of Ostabolin-C and
the % change from baseline for femoral neck BMD following exposure to
increasing
Ostabolin-C exposure at 4 months, as compared to placebo.
Fig. 30 is a linear regression analysis assessing the full range of exposures
of Ostabolin-C and
the % change from baseline for mid-shaft radius BMD following exposure to
increasing
Ostabolin-C exposure at 12 months, as compared to placebo.
Fig. 31 is a graph of the incidences of hypercalcemia (at least one episode)
following
administration of Ostabolin-C at a variety of doses.
Fig. 32 is a graph of the incidences of hypercalcemia (only I episode compared
to > 1
episode) following administration of Ostabolin-C at a variety of doses.
Fig. 33 is a graph of the exposure range for Ostabolin-C administered at doses
of 7.5, 20, 30,
and 45 ug.
Fig. 34 is a graph of the exposure range for Ostabolin-C administered at doses
of 7.5, 20, 30,
and 45 ug, overlaid with the impact of dose optimization on exposure.
Fig. 35 is a graph of the exposure range for Ostabolin-C administered to women
and men at
30 ug. The women are represented by the solid line and the men are represented
by the
dashed line. The mean weight of the men was 82 kg and the mean weight of the
women was
64 kg.
Fig. 36 is a graph of the exposure range for Ostabolin-C administered to men
and women
using a weight cutoff of 68 kg. The women are represented by the solid line
and the men are
represented by the dashed line. This graph illustrates that with this weight
cutoff the men and
women receive approximately the same exposure.
Fig. 37 is a graph illustrating the incidences of hypercalcemia at 4 months,
applying a weight
cutoff. The graph illustrates that with a weight cutoff of approximately 68
kg, the % of
hypercalcemia seen is less than 15%.
Fig. 38 is a graph illustrating the incidences of hypercalcemia at 12 months,
applying a
weight cutoff.
12
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
Fig. 39 is a graph illustrating more than 1 incidence of hypercalcemia at 4
months, applying a
weight cutoff.
Fig. 40 is a graph illustrating more than 1 incidence of hypercalcemia at 12
months, applying
a weight cutoff.
Fig. 41 is a graph illustrating the effect of a change in weight cutoff on
bone formation.
Fig. 42 is a graph illustrating the effect of a change in weight cutoff on
bone resorption.
Fig. 43 is a graph illustrating the effect of a change in weight cutoff on
bone
formation/resorption ratio.
Fig. 44 is a graph illustrating the incidences of lumbar spine BMD at 4
months, applying a
weight cutoff.
Fig. 45 is a graph illustrating the incidences of lumbar spine BMD at 12
months, applying a
weight cutoff.
Fig. 46 is a graph illustrating the incidences of lumbar spine B1VID > 3%
responders at 4
months, applying a weight cutoff.
Fig. 47 is a graph illustrating the dissociative effect of weight cut-off by
comparing the
hypercalcemia and lurnbar spine B1VID responder rate.
Fig. 48 is a graph illustrating the dissociation of BMD responders by
comparing the site-
specific effect of a weight cutoff change.
Fig. 49 is a graph illustrating the comparative effect of weight cutoff, by
comparing the effect
of a weight cutoff on CTx and hypercalcemia.
Fig. 50 is a graph illustrating the comparative effect of weight cutoff, by
comparing the effect
of a weight cutoff on CTx and serum calcium.
Fig. 51 is a graph illustrating the comparative effect of weight cutoff, by
comparing the effect
of a weight cutoff on serum calcium and lumbar spine BMD.
Fig. 52 is a graph illustrating the incidences of headache and nausea at 4
months, applying a
weight cutoff.
13
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
Fig. 53 is a graph illustrating the incidences of total hip BMD at 4 months,
applying a weight
cutoff.
Fig. 54 is a graph illustrating the incidences of total hip BMD at 12 months,
applying a
weight cutoff.
Fig. 55 is a graph illustrating the biological AUC (0-4) levels achieved with
OCIP, at day 1
following Ostabolin-C inhaled administration.
Fig. 56 is a graph illustrating the biological Cmax levels achieved with OCIP,
at day 1
following Ostabolin-C inhaled administration.
Fig. 57 is a graph illustrating the biological Tmax levels achieved with OCIP,
at day 1
following Ostabolin-C inhaled administration.
Fig. 58 is a graph illustrating the biological Cmax levels achieved with OCIP,
at days 1 and 7
following Ostabolin-C inhaled administration.
Fig. 59 is a graph illustrating the biological AUC (0-4) levels achieved with
OCIP, at days I
and 7 following Ostabolin-C inhaled administration.
Fig. 60 is a graph illustrating the day 1 average blood levels of Ostabolin-C
in pg/ml.
Fig. 61 is a graph illustrating the urinary cAMP levels following Ostabolin-C
inhaled
administration.
Fig. 62 is a graph illustrating the urinary cAMP levels following Ostabolin-C
inhaled
administration.
Fig. 63 is a graph illustrating the relationship between the increase in
cyclic AMP levels and
the increase in AUC (0-2) levels following Ostabolin-C inhaled administration.
Fig. 64 is a graph illustrating the relationship between the increase in
cyclic AMP levels and
the increase in Cmax levels following Ostabolin-C inhaled administration.
Fig. 65 is a graph illustrating the meaci % change in P1NP following Ostabolin-
C inhaled
administration.
14
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
Fig. 66 is a graph illustrating the mean % change in Osteocalcin following
Ostabolin-C
inhaled administration.
Fig. 67 is a graph illustrating the relationship between the increase in P1NP
levels and the
increase in AUC (0-2) levels following Ostabolin-C inhaled administration
Fig. 68 is a graph illustrating the mean % change in CTx following Ostabolin-C
inhaled
administration.
Fig. 69 is a graph illustrating the mean heart rates following Ostabolin-C
inhaled
administration.
Fig. 70 is a graph illustrating the mean heart rates following Ostabolin-C
inhaled
administration
Fig. 71 is a graph illustrating the biological AUC (0-tz) levels achieved with
various
Ostabolin-C formulations, following subcutaneous administration.
Fig. 72 is a graph illustrating the biological Cmax levels achieved with
various Ostabolin-C
formulations, following subcutaneous administration.
Fig. 73 is a graph illustrating the biological AUC (0-tz) levels achieved with
various
Ostabolin-C formulations, following intramuscular and intravenous
administration.
Fig. 74 is a graph illustrating the biological Cmax levels achieved with
various Ostabolin-C
formulations, following intramuscular and intravenous administration.
Fig. 75 is a graph illustrating the plasma concentrations of various Ostabolin-
C formulations
in female rats following intravenous administration.
Fig. 76 is a graph illustrating the plasma concentrations of various Ostabolin-
C formulations
in female rats following subcutaneous administration.
Fig. 77 is a graph illustrating the plasma concentrations of various Ostabolin-
C formulations
in female rats following intramuscular administration
Fig. 78 is a graph illustrating the effects of a dose cutoff with Ostabolin-C
as compared to
Forteo.
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
DETAILED DESCRIPTION OF THE INVENTION
The present invention provides pharmaceutical compositions and formulations
containing suitable PTH peptide analogues for use in methods directed to
treating subjects
suffering from various bone degenerative or bone deficit disorders. The PTH
peptide
analogue compounds described herein induce bone formation in both trabecular
and cortical
bones, thereby increasing bone mineral density and restoring bones.
Unexpectedly, the PTH
peptide analogues described herein induce bone formation while causing less
bone resorption
than previously known PTH analogues, and also demonstrate lower incidences and
severity
of hypercalcemia. Additionally, the PTH peptides of the present invention
provide a shorter
duration PK profile, which allows for maintaining efficacy while reducing side
effects.
The present invention also provides dose optimization as a way to reduce the
side
effects associated with prior art PTHs. The peptides of the present invention
can be
administered in varying doses to patients based on the particular patient's
weight, height,
size, body surface area, or BMI and/or presentation of symptoms. This weight,
body surface
area, or BMI cut off method provides a method for determining a
therapeutically effective
dosage while maintaining a low incidence of side effects for a patient based
upon their
weight, body surface area, or BMI. A dosage that results in high exposure for
a particular
patient will increase the chance of side effects, including hypercalcemia.
Additionally, lack
of efficacy may be observed in certain patients, because the dose for that
particular patient
was too low in a situation for a patient of a certain weight, body surface
area, or BMI who
could have tolerated a higher dosage of the therapeutic agent.
Transient exposure to PTH receptor agonists causes a bone formation response
whereas continuous exposure to some PTH receptor agonists causes a predominant
bone
resorption effect. Even with transient exposure to PTH receptor agonists, as
defined by
conventional subcutaneous injection, some stimulation of bone resorption still
occurs and this
is associated with deleterious clinical effects including hypercalcemia and
increased cortical
porosity. Modifications to drug delivery that decrease the duration of
exposure to PTH
receptor agonists, regardless of the interval between doses and the route of
administration of
the dose, will improve the therapeutic window for PTH receptor agonists by
reducing the
level of stimulation of bone resorption for a given dose while maintaining or
increasing the
level of bone formation.
The present invention relates to PTH analogs, including Ostabolin-C and
related
analogs disclosed herein, which have a shorter duration PK profile than
conventional PTHs.
16
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
Such PTHs generate an increased therapeutic window by reducing the level of
stimulation of
bone resorption for a given dose equivalent while maintaining or enhancing the
stimulation of
bone formation.
The present invention also relates to reducing the undesirable side effects
associated
with the administration of PTH analogues, because of the shorter duration PK
profile.
Undesirable side effects which can be reduced include bone resorption, feeling
cold, fatigue,
loose stool, feeling hot, lower abdominal pain, injection site reaction,
arthralgia, injection site
hemorrhage, pharyngolaryngeal pain, muscle cramps, and abdominal pain. More
specifically, undesirable side effects which can be reduced include
hypercalcemia, increase in
mean serum calcium level, headache, nausea, back pain, dizziness, and
extremity pain.
The invention relates to a method for increasing bone toughness and/or
stiffness,
and/or reducing incidence of fracture in a subject by administering a
parathyroid hormone.
The method can be employed to increase stiffness and/or toughness at a site of
a potential
trauma or at a site of an actual trauma. Trauma generally includes fracture,
surgical trauma,
joint replacement, orthopedic procedures, and the like. Increasing bone
toughness and/or
stiffness generally includes increasing mineral density of cortical bone,
increasing strength of
bone, increasing resistance to loading, and the like. Reducing incidence of
fracture generally
includes reducing the likelihood or actual incidence of fracture for a subject
compared to an
untreated control population.
The present invention includes a method for increasing the toughness and/or
stiffness
of bone, including trabecular and cortical bone, and/or reducing the incidence
and/or severity
of fracture by administering a parathyroid hormone analogue as described
herein. More
particularly, the invention relates to a method for increasing toughness or
stiffriess of bone at
a site of a potential or actual trauma. Increasing toughness and/or stiffness
of bone can be
manifested in numerous ways known to those of skill in the art, such as
increasing bone
mineral density, increasing bone mineral content, increasing work to failure,
and the like. In
one embodiment, the method of the invention reduces the incidence or severity
of vertebral
and/or non-vertebral fractures. The method of the invention can be used to
decrease the risk
of such fractures or for treating such fractures. In particular, the method of
the invention can
reduce the incidence of vertebral and/or non-vertebral fracture, reduce the
severity of
vertebral fracture, reduce the incidence of multiple vertebral fracture,
improve bone quality,
and the like.
The inventors have discovered that PTH peptide analogues that have a reduced
phospholipase-C activity, and which maintain adenylate cyclase activity, are
surprisingly
17
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
useful for inducing bone formation in both trabecular and cortical bones, and
causing less
bone resorption than previous PTH analogues at dosages of about 2 to about 100
g/day,
without significantly increasing levels of serum calcium. The methods provided
by this
invention are generally practiced by administering to an animal in need
thereof a dose of a
PTH compound in the amount of about 2 to about 100 g/day, or weekly at 3 to 7
times
greater than the daily dose to induce bone formation and cause less bone
resorption and lower
incidences of hypercalcemia as compared to the administration of PTH analogues
34 amino
acid residues in length or longer. Additionally, the PTH peptides of the
present invention can
be administered by inhalation at a daily dose of between 100 g -2,000 g.
The PTH peptide analogues, either alone or in combination with other bone
enhancing
agents, of the present invention can be used to treat any mammal, including
humans and
animals, suffering from a disease, symptom, or condition related to bone
deficiency. In an
embodiment of the invention, the subject in need of enhanced bone formation is
a human
patient such as a man or a woman. In a preferred embodiment, the patient is a
post-
menopausal woman.
Definitions
The following definitions are provided to assist the reader. Unless otherwise
defined,
all terms of art, notations and other scientific or medical terms or
terminology used herein are
intended to have the meanings commonly understood by those of skill in the
chemical and
medical arts. In some cases, terms with commonly understood meanings are
defined herein
for clarity and/or for ready reference, and the inclusion of such definitions
herein should not
necessarily be construed to represent a substantial difference over the
definition of the term as
generally understood in the art.
As used herein, the "PTH peptide analogues" of the present invention are
preferably,
but not exclusively, non-naturally occurring and may be obtained either
recombinantly or by
peptide synthesis. The PTH analogues of the present invention include full
length PTH (1-
84), 1-31 and 1-34 fragments, and other fragments or variants of fragments of
human, rat,
porcine, or bovine PTH that have human PTH activity as determined in the
ovarectomized rat
model of osteoporosis (Kimmel et al., Endocrinology, 1993, 32(4):1577). Human
PTH
activity includes the ability of the PTH to increase trabecular and/or
cortical bone growth.
The PTH analogues of the present invention increase AC activity when
administered to a
PTH receptor containing or expressing cell in culture, such as an osteoblast
or an osteoclast.
18
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
The PTH analogues of the present invention have certain additional functional
activities, as
defined below.
As used herein, a PTH peptide analogue that has a "reduced phospholipase-C
activity" refers to a PTH peptide analogue that has been truncated or modified
in some
manner so as to trigger less than full activation of phospholipase-C, as
compared to the full-
length PTH peptide or other PTH peptide analogues which are at least 34 amino
acid residues
in length.
As used herein, a PTH peptide analogue that leads to "reduced bone resorption"
refers
to a PTH peptide analogue that has been truncated or modified in some manner
so as to
trigger less bone resorption, as compared to the full-length PTH peptide or
other PTH peptide
analogues which are at least 34 amino acid residues in length.
As used herein, a PTH peptide analogue that leads to "reduced hypercalcemia
levels"
refers to a PTH peptide analogue that has been truncated or modified in some
manner so as to
trigger less incidences of hypercalcemia, or lower severity of hypercalcemia,
as compared to
the full-length PTH peptide or other PTH peptide analogues which are at least
34 amino acid
residues in length.
As used herein, "treating" or "treatment of' a condition or subject refers to
taking
steps to obtain beneficial or desired results, including clinical results. For
purposes of this
invention, beneficial or desired clinical results include, but are not limited
to, alleviation or
amelioration of one or more disease, symptom, or condition related to bone
deficiency.
Generally, such bone deficit disease, symptoms, and conditions are treated by
inducing bone
formation as measured by an increase in bone mineral density ("BMD"). For
example,
symptoms of osteoporosis include back pain, loss of height and stooped
posture, a curved
backbone (dowager's hump), or fractures that may occur with a minor injury
(especially of
the hip, spine, or wrist). Symptoms of Paget's disease most commonly include
bone pain.
Other symptoms can include: headaches and hearing loss, neck pain, pressure on
nerves,
increased head size or bending of spine, hip pain, damage to cartilage
ofjoints (which may
lead to arthritis), and Barrel-shaped chest. Symptoms of osteoarthritis can
include joint pain
and aching, limited range of motion and instability, radiographic evidence of
the erosion of
the articular cartilage, joint space narrowing, sclerosis of the subchondral
bone, and
osteophytes (spurs). Symptoms for rheumatoid arthritis include painful,
swollen, tender, stiff
joints on both sides of the body (symmetrical), especially the hands, wrists,
elbows, feet,
knees, or neck. Rheumatoid nodules (bumps) ranging in size from a pea to a
mothball
develop in nearly one-third of people who have rheumatoid arthritis. These
nodules usually
19
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
form over pressure points in the body such as the elbows, knuckles, spine, and
lower leg
bones.
As used herein, "reduction" of a symptom or symptoms (and grammatical
equivalents
of this phrase) means decreasing of the severity or frequency of the
symptom(s), or
elimination of the symptom(s).
As used herein, "administering" or "administration of' a drug or
pharmaceutical
composition or formulation described herein to a subject (and grammatical
equivalents of this
phrase) includes both direct administration, including self-administration,
and indirect
administration, including the act of prescribing a drug. For example, as used
herein, a
physician who instructs a patient to self-administer a drug and/or provides a
patient with a
prescription for a drug is administering the drug to the patient.
A variety of administration routes can be used in accordance with the present
invention. An effective amount of the peptide described herein can be
administered
parenterally, orally, by inhalation, topically, rectally, nasally, buccally,
vaginally or via an
implanted reservoir.
In a preferred embodiment of the invention, an effective amount of the peptide
described herein can be administered parenterally. The term "parenteral" as
used herein
includes transdermal, transcutaneous, subcutaneous, intravenous,
intramuscular, intra-
articular, intra-synovial, intrastemal, needle-free injection, intrathecal,
intrahepatic,
intralesional and intracranial injection or infusion techniques. More
preferably, the route of
administration is subcutaneous administration.
As used herein, a "therapeutically effective amount" of a drug or
pharmaceutical
composition or formulation, or agent, described herein is an amount of a drug
or agent that,
when administered to a subject with a disease or condition, will have the
intended therapeutic
effect, e.g., alleviation, amelioration, palliation or elimination of one or
more manifestations
of the disease or condition in the subject. The full therapeutic effect does
not necessarily
occur by administration of one dose and may occur only after administration of
a series of
doses. Thus, a therapeutically effective amount may be administered in one or
more
administrations.
As used herein, a "prophylactically effective amount" of a drug or
pharmaceutical
composition or formulation, or agent, described herein is an amount of a drug
or agent that,
when administered to a subject, will have the intended prophylactic effect,
e.g., preventing or
delaying the onset (or reoccurrence) of disease or symptoms, or reducing the
likelihood of the
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
onset (or reoccurrence) of disease or symptoms. The full prophylactic effect
does not
necessarily occur by administration of one dose and may occur only after
administration of a
series of doses. Thus, a prophylactically effective amount may be administered
in one or
more administrations.
Administration of a bone enhancing agent "in combination with" a drug or
pharmaceutical composition or formulation described herein includes parallel
administration
(i.e., administration of both the drug and the agents to the subject over a
period-of time, co-
administration (in which both the drug and agents are administered at
approximately the same
time, e.g., within about a few minutes to a few hours of one another), and co-
formulation (in
which both the drug and agents are combined or compounded into a single dosage
form
suitable for oral or parenteral administration).
A "subject" is a mammal, preferably a human, but can also be an animal in need
of
veterinary treatment, e.g., companion animals (e.g., dogs, cats, and the
like), farm animals
(e.g., cows, sheep, pigs, horses, and the like) and laboratory animals (e.g.,
rats, mice, guinea
pigs, and the like).
It should be noted that reference to numeric ranges throughout this
specification is
intended to encompass all numbers falling within the disclosed ranges. Thus,
for example,
the recitation of the range of about 1% to about 50% includes 1%, 2%, 3%, 4%,
5%, 6%, 7%,
8%, or 9%, 10%, 12%, 14%, 16%, 20%, 25%, 30%, 35%, 40%, 45%, and 50%, as well
as,
for example, 21.3%, 7.9%, and 44.5%.
Additional active ingredients can be included in the present compositions.
Choices
are not limited, but may be chosen for a desired combined therapeutic effect.
For example,
active ingredients that may be added for a complementary therapeutic effect
include, but are
not limited to, vitamin D and analogs, estrogen, calcitonin, bisphosphonates,
and mixtures
thereof. A particularly desirable choice is calcitonin.
Bone Disorders and Diseases
Bone Deficits
In one aspect, the subject in need has a bone deficit, which means that they
will have
less bone than desirable or that the bone will be less dense or strong than
desired. A bone
deficit may be localized, such as that caused by a bone fracture or systemic,
such as that
caused by osteoporosis. Bone deficits may result from a bone remodelling
disorder whereby
the balance between bone formation and bone resorption is shifted, resulting
in a bone deficit.
Examples of such bone remodelling disorders include, for example,
osteoporosis, Paget's
21
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
disease, renal osteodystrophy, renal rickets, osteoarthritis, rheumatoid
arthritis,
achondroplasia, osteochodrytis, hyperparathyroidism, osteogenesis imperfecta,
congenital
hypophosphatasia, fribromatous lesions, fibrous displasia, multiple myeloma,
abnormal bone
turnover, osteolytic bone disease and periodontal disease. Bone remodelling
disorders
includes metabolic bone diseases which are characterized by disturbances in
the organic
matrix, bone mineralization, bone remodelling, endocrine, nutritional and
other factors which
regulate skeletal and mineral homeostasis. Such disorders may be hereditary or
acquired and
generally are systemic, affecting the entire skeletal system.
Thus, in one aspect the human subject may have a bone remodelling disorder.
Bone
remodelling as used herein refers to the process whereby old bone is being
removed and new
bone is being formed by a continuous turnover of bone matrix and mineral that
involves bone
resorption by osteoclasts and bone formation by osteoblasts.
Osteoporosis is a common bone remodelling disorder characterized by a decrease
in
bone density of normally mineralized bone, resulting in thinning of trabeculae
and increased
porosity of bone cortices. The skeletal fragility caused by osteoporosis
predisposes sufferers
to bone pain and an increased incidence of fractures. Progressive bone loss in
this condition
may result in a loss of up to 50% of the initial skeletal mass. Primary
osteoporosis includes
idiopathic osteoporosis which occurs in children or young adults with normal
gonadal
function, Type I osteoporosis, also described as post-menopausal osteoporosis,
and Type II
osteoporosis, senile osteoporosis, occurs mainly in those persons older than
70 years of age.
Causes of secondary osteoporosis may be endocrine (e.g., glucocorticoid
excess,
hyperparathyroidism, hypoganodism), drug induced (e.g. corticosteroid,
heparin, tobacco)
and miscellaneous (e.g., chronic renal failure, hepatic disease and
malabsorbtion syndrome
osteoporosis).
The phrase "at risk of developing a bone deficit", as used herein, is intended
to
embrace subjects having a higher than average predisposition towards
developing a bone
deficit. As an example, those susceptible towards osteoporosis include post-
menopausal
women, elderly males (e.g., those over the age of 65) and those being treated
with drugs
known to cause osteoporosis as a side-effect (e.g., steroid-induced
osteoporosis). Certain
factors are well known in the art which may be used to identify those at risk
of developing a
bone deficit due to bone remodelling disorders like osteoporosis. Risk factors
for
osteoporosis are known in the art and include hypogonadal conditions in men
and women,
irrespective of age, conditions, diseases or drugs that induce hypogonadism,
nutritional
factors associated with osteoporosis (low calcium or vitamin D being the most
common),
22
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
smoking, alcohol, drugs associated with bone loss (such as glucocorticoids,
thyroxine,
heparin, lithium, anticonvulsants etc.), loss of eyesight that predisposes to
falls, space travel,
immobilization, chronic hospitalization or bed rest, and other systemic
diseases that have
been linked to increased risk of osteoporosis.
Indications of the presence of osteoporosis are known in the art and include
radiological evidence of at least one vertebral compression fracture, low bone
mass (typically
at least 1 standard deviation below mean young normal values), and/or
atraumatic fractures.
Other important factors include family history, life style, estrogen or
androgen deficiency and
negative calcium balance. Postmenopausal women are particularly at risk of
developing
osteoporosis. Hereinafter, references to treatment of bone diseases are
intended to include
management and/or prophylaxis except where the context demands otherwise.
Bone Trauma
The method of the invention is of benefit to a subject that may suffer or have
suffered
trauma to one or more bones. The method can benefit mammalian subjects, such
as humans,
horses, dogs, and cats, in particular, humans. Bone trauma can be a problem
for racing horses
and dogs, and also for household pets. A human can suffer any of a variety of
bone traumas
due, for example, to accident, medical intervention, disease, or disorder. In
the young, bone
trauma is likely due to fracture, medical intervention to repair a fracture,
or the repair of
joints or connective tissue damaged, for example, through athletics. Other
types of bone
trauma, such as those from osteoporosis, degenerative bone disease (such as
arthritis or
osteoarthritis), hip replacement, or secondary conditions associated with
therapy for other
systemic conditions (e.g., glucocorticoid osteoporosis, burns or organ
transplantation) are
found most often in older people.
Osteoporosis can lead, for example, to vertebral and/or non-vertebral
fractures.
Vertebral fractures are those involving the spinal column and non-vertebral
fractures refers to
any fracture not involving the spinal column. Non-vertebral fractures are more
common than
fractures of the vertebrae-an estimated 850,000 non-vertebral compared with
700,000
vertebral fractures occur annually in the United States. Non-vertebral
fractures include more
than 300,000 hip and 250,000 wrist fractures, in addition to 300,000 fractures
at other non-
vertebral sites. Other examples of non-vertebral fractures include a hip
fracture, a fracture of
a distal forearm, a fracture of a proximal humerus, a fracture of a wrist, a
fracture of a radius,
a fracture of an ankle, a fracture of an humerus, a fracture of a rib, a
fracture of a foot, a
fracture of a pelvis, or a combination of these.
23
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
The method of the invention can be used to decrease the risk of such fractures
or for
treating such fractures. The risk of fracture is diminished and the healing of
a fracture is aided
by increasing the strength and/or stiffness of bone, for example, in the hip,
the spine or both.
A typical woman at risk for osteoporosis is a postmenopausal woman or a
premenopausal,
hypogonadal woman. A preferred subject is a postmenopausal woman, and is.
independent of
concurrent hormone replacement therapy (HRT), estrogen or equivalent therapy,
or
antiresorptive therapy. The method of invention can benefit a subject at any
stage of
osteoporosis, but especially in the early and advanced stages.
The present invention provides a method, in particular, effective to prevent
or reduce
the incidence of fractures in a subject with or at risk of progressing to
osteoporosis. For
example, the present invention can reduce the incidence of vertebral and/or
non-vertebral
fracture, reduce the severity of vertebral fracture, reduce the incidence of
multiple vertebral
fracture, improve bone quality, and the like. In another embodiment, the
method of the
present invention can benefit patients with low bone mass or prior fracture
who are at risk for
future multiple skeletal fractures, such as patients in which spinal
osteoporosis may be
progressing rapidly.
Other subjects can also be at risk of or suffer bone trauma and can benefit
from the
method of the invention. For example, a wide variety of subjects at risk of
one or more of the
fractures identified above, can anticipate surgery resulting in bone trauma,
or may undergo an
orthopedic procedure that manipulates a bone at a skeletal site of abnormally
low bone mass
or poor bone structure, or deficient in mineral. For example, recovery of
function after a
surgery such as a joint replacement (e.g. knee or hip) or spine bracing, or
other procedures
that immobilize a bone or skeleton can improve due to the method of the
invention. The
method of the invention can also aid recovery from orthopedic procedures that
manipulate a
bone at a site of abnormally low bone mass or poor bone structure, which
procedures include
surgical division of bone, including osteotomies, joint replacement where loss
of bone
structure requires restructuring with acetabulum shelf creation and prevention
of prosthesis
drift, for example. Other suitable subjects for practice of the present
invention include those
suffering from hypoparathyroidism or kyphosis, who can undergo trauma related
to, or
caused by, hypoparathyroidism or progression of kyphosis.
Bone Toughness and Stiffness
The method of the invention reduces the risk of trauma or aids recovery from
trauma
by increasing bone toughness, stiffness or both. Generally toughness or
stiffness of bone
results from mass and strength of cortical and trabecular (cancellous) bone.
The method of
24
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
the invention can provide levels of bone toughness, stiffness, mass, and/or
strength within or
above the range of the normal population. Preferably the invention provides
increased levels
relative to the levels resulting from trauma or giving rise to risk of trauma.
Increasing
toughness, stiffness, or both decreases risk or probability of fracture
compared to an untreated
control population.
Certain characteristics of bone when increased provide increased bone
toughness
and/or stiffness. Such characteristics include bone mineral density (BMD),
bone mineral
content (BMC), activation frequency or bone formation rate, trabecular number,
trabecular
thickness, trabecular and other connectivity, periosteal and endocortical bone
formation,
cortical porosity, cross sectional bone area and bone mass, resistance to
loading, and/or work
to failure. An increase in one or more of these characteristics is a preferred
outcome of the
method of the invention.
Certain characteristics of bone, such as marrow space and elastic modulus when
decreased provide increased toughness and/or stiffness of bone. Younger
(tougher and stiffer)
bone has crystallites that are generally smaller than crystallites of older
bone. Thus, generally
reducing the size of bone crystallites increases toughness and stiffness of
bone, and can
reduce incidence of fracture. In addition, maturing the crystallites of a bone
can provide
additional desirable characteristics to the bone, including increased
toughness and stiffness of
bone and/or can reduced incidence of fracture. A decrease in one or more of
these
characteristics can be a preferred outcome of the method of the invention:
The method of the invention is effective for increasing the toughness and/or
stiffness
of any of several bones. For example, the present method can increase the
toughness and/or
stiffness of bones including a hip bone, such as an ilium, a leg bone, such as
a femur, a bone
from the spine, such as a vertebra, or a bone from an arm, such as a distal
forearm bone or a
proximal humerus. This increase in toughness and/or stiffness can be found
throughout the
bone, or localized to certain portions of the bone. For example, toughness
and/or stiffness of a
femur can be increased by increasing the toughness and/or stiffness of a femur
neck or a
femur trochanter. Toughness and/or stiffness of a hip can be increased by
increasing the
toughness and/or stiffness of an iliac crest or iliac spine. Toughness and/or
stiffness of a
vertebra can be increased by increasing the toughness and/or stiffness of a
pedicle, lamina, or
body. Advantageously, the effect is on vertebra in certain portions of the
spine, such as
cervical, thoracic, lumbar, sacral, and/or coccygeal vertebrae. Preferably the
effect is on one
or more mid-thoracic and/or upper lumbar vertebrae.
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
The increase in toughness and/or stiffness can be found in each of the types
of bone,
or predominantly in one type of the bone. Types of bone include spongy
(cancellous,
trabecular, or lamellar) bone and compact (cortical or dense) bone and the
fracture callus. The
method of the invention preferably increases toughness and/or stiffness
through its effects on
cancellous and cortical bone, or on cortical bone alone. Trabecular bone, bone
to which
connective tissue is attached can also be toughened and/or stiffened by the
present method.
For example, it is advantageous to provide additional toughness at a site of
attachment for a
ligament, a tendon, and/or a muscle_
In another aspect of the invention, increasing toughness or stiffness can
reduce
incidence of fracture. In this aspect, increasing toughness or stiffiness can
include reducing
incidence of vertebral fracture, reducing incidence of severe fracture,
reducing incidence of
moderate fracture, reducing incidence of non-vertebral fracture, reducing
incidence of
multiple fracture, or a combination thereof.
The methods of the invention may also be used to enhance bone formation in
conditions where a bone deficit is caused by factors other than bone
remodelling disorders.
Such bone deficits include fractures, bone trauma, conditions associated with
post-traumatic
bone surgery (e.g., bone grafts or bone fusions), post-prosthetic joint
surgery, post plastic
bone surgery, post dental surgery, bone chemotherapy, and bone radiotherapy.
Fractures
include all types of microscopic and macroscopic fractures. Examples of
fractures and/or
injuries include avulsion fracture, comminuted fracture, rion-union fracture,
transverse
fracture, oblique fracture, spiral fracture, segmental fracture, a segmental
gap, displaced
fracture, impacted fracture, greenstick fracture, torus fracture, fatigue
fracture, intra-articular
fracture (epiphyseal fracture), closed fracture (simple fracture), open
fracture (compound
fracture), a bone void, and occult fracture in any bones of the subject.
As previously mentioned, a wide variety of bone diseases may be treated in
accordance with the present invention, for example all those bone diseases
connected with the
bone-remodelling cycle. Examples of such diseases include all forms of
osteoporosis,
osteomalacia and rickets. Osteoporosis, especially of the post-menopausal,
male, post-
transplant, and steroid-induced types, is of particular note. In addition, PTH
peptide
analogues find use as bone promotion agents, and as anabolic bone agents. Such
uses form
another aspect of the present invention.
Parathyroid Hormone Analogues
26
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
As active ingredient, the pharmaceutically acceptable composition or solution
described herein may incorporate full-length PTH (1-84), 1-31 and 1-34
fragments, and other
fragments, or variants of fragments, including substitutions, deletions, or
insertions, of human
PTH, or of rat, porcine or bovine PTH that have human PTH activity as
determined in the
ovarectomized rat model of osteoporosis reported by Kimmel et al.,
Endocrinology, 1993,
32(4):1577. Human PTH activity includes the ability of the PTH to increase
trabecular and/or
cortical bone growth. The PTH analogues of the present invention increase AC
activity when
administered to a PTH receptor containing or expressing cell in culture, such
as an osteoblast
or osteoclast. The PTH analogues used in the present invention are naturally
or non-
naturally occurring and desirably incorporate less than the first 34 N-
terminal residues of
PTH.
PTH operates through activation of two second messenger systems, GS-protein
activated adenylyl cyclase (AC) and Gq-protein activated phospholipase C. The
latter results
in a stimulation of membrane-bound protein kinase Cs (PKC) activity. The PKC
activity has
been shown to require PTH residues 29 to 32 (Jouishomme et al (1994) J. Bone
Mineral Res.
9, (1179-1189). It has been established that the increase in bone growth, i.e.
that effect which
is useful in the treatment of osteoporosis, is coupled to the ability of the
peptide sequence to
increase AC activity.
The native PTH sequence, and its truncated 1-34 form, has been shown to have
all of
these activities. The hPTH-(1-34) sequence is:
Ser Val Ser Glu Ile Gln Leu Met His Asn Leu Gly Lys His Leu Asn Ser Met Glu
Arg Val
Glu Trp Leu Arg Lys Lys Leu Gln Asp Val His Asn Phe-OH (SEQ ID NO:1)
AC activity has been shown to require the first few N-terminal residues of the
molecule. Thus, in accordance with this embodiment of the invention, it is
possible to remove
those biological activities associated with the PKC activity by deleting a
selected terminal
portion of the hPTH-(1-34) molecule. In one embodiment, these shortened
analogues are
desirably in the form of carboxyl terminal amides. One feature of the
invention therefore
comprises variants of the human parathyroid analogues PTH(I-25)-NHZ, PTH(1-26)-
NH2,
PTH(1-27)-NHZ, PTH(1-28)-NH2, PTH(1-29)-NH2, PTH(1-30)-NH2, and PTH(1-31)-NH2.
According to another feature of the PTH analogues to be used in the present
invention, it has surprisingly been found that replacing Lys27 with a Leu in
the native hPTH
sequence results in a higher activity for AC stimulation. This analogue also
exhibits its
27
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
maximum activity when in the form of the carboxyl terminal amide. Thus,
another feature of
the invention comprises the use of PTH analogues including all sequences from
[Leu27]-PTH-
(1-25)-NH2 to [Leu27 ]-PTH-(1-31)-NH2.
According to another feature of the present invention, lactams of the PTH
analogues
are formed, for example, by cyclisation involving the coupling of the side-
chains of G1u22
and Lys26, or of the side-chains Lys26 and Asp30, in which Lys27 may be
replaced by a Leu
or by various other hydrophobic residues, and which has either a C-terminal
free amide
ending, or has a C-terminal free carboxyl ending. Such substitutions include
ornithine,
citrulline, alpha-aminobutyric acid, or any linear or branched alpha-amino
aliphatic acid,
having 2-10 carbons in the side chain, any such analogue having a polar or
charged group at
the terminus of the aliphatic chain. Example of polar or charged groups
include amino,
carboxyl, acetamido, guanido and ureido. Ile, norleucine, Met, and ornithine
are expected to
be the most active.
The PTH analogues of the present invention may thus feature the formation of a
lactam, for example, between either residues G1u22 and Lys26, Ly26 and Asp30,
or GIu22
and Lys27. The substitution of Leu for the Lys27 results in a more hydrophobic
residue on
the hydrophobic face of the amphiphilic helix. This resulted in increased
adenylyl cyclase
stimulating activity in the PTH receptor containing rat osteosarcoma (ROS)
cell line. It will
be appreciated by those skilled in the art that other such substitutions would
likely result in
analogues with the same or increased activities. These hydrophobic
substitutions include
residues such as Met or norleucine. The combined effect of substitution and
either lactam
formation is expected to stabilize the alpha-helix and increase bioactivity,
and to protect this
region of the molecule from proteolytic degradation. The presence of the amide
at the C-
terminus is further expected to protect the peptide against exoproteolytic
degradation (Leslie,
F. M. and Goldstein, A. (1982) Neuropeptides 2, 185-196).
In one preferred embodiment of the invention, the peptide used in the
disclosed
method is PTH(1-31)-NH2 with the following sequence:
Ser-Val-Ser-Glu-Ile-Gln-Leu-Met-His-Asn-Leu-Gly-Lys-His-Leu-Asn-Ser-Met-Glu-
Arg-
Val-Glu-Trp-Leu-Arg-Lys-Xaa-Leu-Gln-Asp-Val-NHZ (SEQ ID NO: 2).
Xaa is selected from the group consisting of Lys, Leu, Ile, Nie and Met. In a
preferred
embodiment, Xaa is Lys (SEQ ID NO: 3). This embodiment is also referred to as
OSTABOLIN.
28
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
In another preferred embodiment of the invention, the peptide used in the
disclosed
method is cyclo(22-26)PTH-(1-31)-NH2, cyclized in the form of a lactam between
G1u22 and
Lys26 with the following sequence:
Ser-Val-Ser-Glu-Ile-Gln-Leu-Met-His-Asn-Leu-Gly-Lys-His-Leu-Asn-Ser-Met-Glu-
Arg-
Val-Glu-Trp-Leu-Arg-Lys-Xaa-Leu-Gln-Asp-Val-Y (SEQ ID NO: 4),
Xaa is selected from the group consisting of Leu, Ile, Nle and Met and Y is
NH2 or
OH. When Xaa is Leu and Y is NH2 (SEQ ID NO: 5), the PTH is also referred to
as
OSTABOLIN-CTh'.
The PTH analogues to be used in the present invention can thus be cyclized or
linear,
and can be optionally amidated at the C-terminus. Alternatives in the form of
PTH variants
incorporate from 1 to 5 amino acid substitutions that improve PTH stability
and half-life,
such as the replacement of methionine residues at positions 8 and/or 18 with
leucine or other
hydrophobic amino acid that improves PTH stability against oxidation and the
replacement of
amino acids in the 25-27 region with trypsin-insensitive amino acids such as
histidine or
other amino acid that improves PTH stability against protease. Other suitable
forms of PTH
include PTHrP, PTHrP(1-34), PTHrP(1-36) and analogs of PTH or PTHrP that
activate the
PTH1 receptor. These forms of PTH are embraced by the term "parathyroid
hormone
analogues" as used generically herein. The hormones may be obtained by known
recombinant
or synthetic methods, such as described in U.S. Pat. Nos. 4,086,196;
5,556,940; 5,955,425;
6,541,450; 6,316, 410; and 6,110,892, incorporated herein by reference.
Specific embodiments of PTH peptide analogues of the present invention include
the
following: PTH-(1-31)NH2, Ostabolin; PTH-(1-30)NH2; PTH-(1-29)NHZ; PTH-(1-
28)NHZ;
Leu27PTH-(1-31)NH2; Leu27 PTH-(1-30)NHZ; Leu27PTH-(1-29)NH2; Leu27cyclo(22-
26)PTH-
(1-31)NH2 Ostabolin-CT"t; Leu27cyclo(22-26)PTH-(1-34)NH2; Leu27cyclo(Lys26-
Asp30)PTH-(1-34)NH2; Cyclo(Lys27-Asp30)PTH-(1-34)NHZ; Leu27cyclo(22-26)PTH-(1-
31)NH2; Ala27 or N1e27 or Tyr27 or I1e27 cyclo(22-26)PTH-(1-31)NH2;
Leu27cyclo(22-
26)PTH-(1-32)NH2; Leu27cyclo(22-26)PTH-(1-31)OH; Leu27cyclo(26-30)PTH-(1-
31)NH2;
Cys22Cys26Leu27cyclo(22-26)PTH-(1-31)NHZ; Cys22Cys26LeuZ7cyclo(26-30)PTH-(1-
31)NH2;
Cyclo(27-3 0)PTH-(1-31)NH2i Leu27cyclo (22-26)PTH-(1-3 0)NH2i Cyclo(22-26)PTH-
(1-
31)NH2i Cyclo(22-26)PTH-(1-30)NH2; Leu27cyclo(22-26)PTH-(1-29)NH2i
Leu27cyclo(22-
26)PTH-(1-28)NH2; Glu",LeuZ'cyclo(13-17)(22-26)PTH-(1-28)NH2; and
G1u17,LeuZ7cyclo(13-17)(22-26)PTH-(1-31)NH2.
Generally, preferred embodiments of PTH peptide analogues include those that
when
administered result in reduced phospholipase-C activity, reduced bone
resorption, and
29
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
reduced hypercalcemia levels. As defined in the Definitions section herein,
"reduced
phospholipase-C activity" refers to a PTH peptide analogue that has been
truncated or
modified in some manner so as to trigger less than full activation of
phospholipase-C, as
compared to the full-length PTH peptide or other PTH peptide analogues which
are at least
34 amino acid residues in length; "reduced bone resorption" refers to a PTH
peptide analogue
that has been truncated or modified in some manner so as to trigger less bone
resorption, as
compared to the full-length PTH peptide or other PTH peptide analogues which
are at least
34 amino acid residues in length, and "reduced hypercalcemia levels" refers to
a PTH peptide
analogue that has been truncated or modified in some manner so as to trigger
less incidences
of hypercalcemia, or lower severity of hypercalcemia, as compared to the full-
length PTH
peptide or other PTH peptide analogues which are at least 34 amino acid
residues in length.
The preferred PTH analogues administered in the methods described herein
include
[Leu27]cyclo[G1u22-Lys26]-PTH-(1-31)-NH2, such as advanced by Zelos
Therapeutics, Inc.
under the tradename OSTABOLIN-CTM and [Leu"] PTH-(1-31)-NH2. In another
embodiment of the invention, [Leu27]cyclo[G1u22-Lys26]-PTH-(1-30)-NH2 is used
in the
methods described herein. In another embodiment, the hormone can be the linear
analogue
PTH(1-31), which can have a free carboxyl ending, or be amidated, at the C-
terminus. In yet
another embodiment, the horrnone can be PTH(1-30), which can have a free
carboxyl
ending, or be amidated, at the C-terminus; or [Leu27]-PTH(1-30)- NHZ. Suitable
stabilized
solutions of these and other PTH analogues that can be employed in the present
methods are
described in U.S. Patent Nos. 5,556,940; 5,955,425; 6,541,450; 6,316, 410; and
6;110,892
incorporated herein by reference.
Methods of the Invention and Agents Useful Therein
The methods provided by this invention are generally practiced by
administering to an
animal in need thereof a daily or weekly dose of a PTH compound in an amount
effective to
induce bone formation and inhibit or reduce bone loss or resorption.
One aspect of the present invention provides a method for treating
osteoporosis by
administering to a subject in need thereof a pharmaceutically acceptable
formulation
comprising a PTH peptide analogue in a daily subcutaneous dose of an aqueous
formulation
of 2 g to 100 g or a weekly dose of from 14 g to 700 g , wherein the PTH
peptide
analogue has a reduced phospholipase-C activity but maintains adenylate
cyclase activity.
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
Exemplary dosages include a daily dose of 0.5 to 50 g for subcutaneous
delivery of a
formulation stabilized with propylene glycol and/or ethanol, a daily dose of
100 to 3,000 g
for inhalation delivery, and weekly doses at 3-7 times the daily doses. Other
suitable dosages
include ariy dosage with any route of administration that results in a
bioavailability or
pharmacokinetic profile similar to those yielded by the above-described dosage
ranges.
In one embodiment, the subject is a human man or woman. In a preferred
embodiment the
woman is post-menopausal.
In another embodiment, the osteoporosis can be selected from the group
consisting of
advanced-stage osteoporosis, hypogonadal osteoporosis, spinal osteoporosis,
transplant-
induced osteoporosis, and steroid-induced osteoporosis.
Bone enhancing agents known in the art to increase bone formation, bone
density or
bone mineralisation, or to prevent bone resorption may be used in the methods
and
pharmaceutical compositions of the invention. Those of ordinary skill in the
bone formation
art also recognize that suitable bone enhancing agents include, for example,
natural or
synthetic hormones, such as selective estrogen receptor modulators (SERMs),
estrogens,
androgens, calcitonin, prostaglandins and parathormone; growth factors, such
as platelet-
derived growth factor, insulin-like growth factor, transforming growth factor,
epidermal
growth factor, connective tissue growth factor and fibroblast growth factor;
vitamins,
particularly vitamin D; minerals, such as calcium, aluminum, strontium,
lanthanides (such as
lanthanum (III) compounds as described and used in U.S. Patent No. 7,078,059,
incorporated
herein by reference) and fluoride; isoflavones, such as ipriflavone; statin
drugs, including
pravastatin, fluvastatin, simvastatin, lovastatin and atorvastatin; agonsists
or antagonist of
receptors on the surface of osteoblasts and osteoclasts, including
parathormone receptors,
estrogen receptors and prostaglandin receptors; bisphosphonate and anabolic
bone agents. In
one embodiment, vitamin D, calcium, or both are concurrently administered with
the
pharmaceutical formulations of the present invention.
Generally, preferred embodiments of PTH peptide analogues include those that
when
administered result in reduced phospholipase-C activity, reduced ability to
stimulate bone
resorption, and reduced hypercalcemia levels. As defined in the Definitions
section herein,
"reduced phospholipase-C activity" refers to a PTH peptide analogue that has
been truncated
or modified in some manner so as to trigger less than full activation of
phospholipase-C, as
compared to the full-length PTH peptide or other PTH peptide analogues;
"reduced bone
resorption" refers to a PTH peptide analogue that has been truncated or
modified in some
manner so as to trigger less bone resorption, as compared to the full-length
PTH peptide or
31
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
other PTH peptide analogues, and "reduced hypercalcemia levels" refers to a
PTH peptide
analogue that has been truncated or modified in some manner so as to trigger
less incidences
of hypercalcemia, or lower severity of hypercalcemia, as compared to the full-
length PTH
peptide or other PTH peptide analogues.
The preferred PTH analogues administered in the methods described herein
include
[Leu27]cyclo[Glu22 -Lys26]-PTH-(1-31)-NH2i such as advanced by Zelos
Therapeutics, Inc.
under the tradename OSTABOLIN-CTM and PTH-(1-31)-NH2, such as advanced by
Zelos
Therapeutics, Inc. under the tradename OSTABOLINTM. In another embodiment of
the
invention, [Leu27]cyclo[GIu22-Lys26]-PTH-(1-30)-NH2 is used in the methods
described
herein. In another embodiment, the hormone can be the linear analogue PTH(1-
31), which
can have a free carboxyl ending, or be amidated, at the C-terminus. In yet
another
embodiment, the hormone can be PTH(1-30), which can have a free carboxyl
ending, or be
amidated, at the C-terminus; or [Leu27]-PTH(1-30)- NH2. Suitable stabilized
solutions of
these and other PTH analogues that can be employed in the present methods are
described in
U.S. Patent Nos. 5,556,940; 5,955,425; 6,541,450; 6,316, 410; and 6,110,892
incorporated
herein by reference.
The pharmaceutical compositions and formulations described herein, and in the
doses
and routes of administration described in detail below, further operate to
induce bone
formation by stimulating osteoblast differentiation in trabecular and cortical
bone while
simultaneously reducing the incidence of hypercalcemia (i.e., higher than
normal levels of
calcium in the blood).
In another aspect of the invention, methods for treating a bone fracture in a
subject are
provided. A preferred method can include administering to a subject in need
thereof a
pharmaceutically acceptable formulation of a PTH peptide analogue in a daily
dose of 30 g
for three months, wherein the peptide analogue has reduced phospholipase-C
activity and
maintains adenylate cyclase activity, and wherein the PTH peptide analogue
induces bone
formation. In another embodiment, the method can include administering to a
subject in need
thereof a pharmaceutically acceptable formulation of a PTH peptide analogue in
a daily sub-
cutaneous dose of 30 g - 45 g for 1, 2, or 3 months, wherein the peptide
analogue has
reduced phospholipase-C activity and maintains adenylate cyclase activity, and
wherein the
PTH peptide analogue induces bone formation.
The pharmaceutical formulations described herein can be used to accelerate the
healing of a fracture in any bone of the subject's skeleton. In preferred
embodiments, the
pharmaceutical formulations of the present invention are used to heal
fractures of the hip,
32
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
forearm, humerus, wrist, radius, ankle, rib, femur, tibia, and foot. The
fractures can be of
multiple types as discussed above, and healing can simultaneously occur in a
plurality of
bones that may be fractured.
In another aspect, the invention provides methods for inducing bone formation
in
trabecular and cortical bones, as measured by an increase in BMD by
administering to a
subject in need thereof a daily dose of a pharmaceutically acceptable
formulation of a PTH
peptide analogue, wherein the peptide analogue has reduced phospholipase-C
activity and
maintains adenylate cyclase activity.
In preferred embodiments, the pharmaceutical formulations can be used to
induce
bone formation at the spine, skull, ribs, hips, ankle, and wrists, although
any bone of the
subject's skeleton can be induced to form bone. In another embodiment,
following
administration of the PTH pharmaceutical formulations of the present
invention, the
incidences in the patient population in which the level of serum calcium is
above normal is
less than the those seen with administration of prior art PTH peptides.
In yet another aspect, the present invention provides methods of treating or
preventing'.
renal osteodystrophy (ROD) and related disorders by administering to a subject
in need
thereof a daily dose of a pharmaceutically acceptable formulation of a PTH
peptide analogue,
wherein the peptide analogue has reduced phospholipase-C activity and
maintains adenylate
cyclase activity.
In an embodiment, ROD related disorders are osteitis fibrosa cystica and
adynamic
bone disease.
Unexpected Results
The pharmaceutical compositions and formulations described herein, and in the
doses
and routes of administration described in detail below, operate to induce bone
formation by
stimulating osteoblast differentiation in trabecular and cortical bone while
simultaneously
reducing or inhibiting osteoclast differentiation, and thus, bone resorption.
PTH analogues
less than 34 amino acids in length are preferred, because these truncated
forms maintain the
positive effects of increased bone formation, while minimizing the negative
effects of
increased bone resorption. Minimizing the bone resorption also leads to less
cortical
porosity.
Administration of the PTH analogues of the present invention at a variety of
doses has
led to unexpected and superior results when compared to administration of
prior PTH
analogues. When administered over a course of four months, the PTH analogues
of the
33
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
present invention have been shown to have a similar or greater effect on the
increase in BMD
of lumbar spine, hip, femoral neck, and trochanter as compared to prior art
PTH analogues
which are at least 34 amino acid residues in length given over at least a
course of a year. For
results of prior art PTH analogues, see Neer, N. Eng. J. Med, Vo1344, No. 19,
May 2001, p.
1434-1441. These unexpected results are described in detail in the Examples
and the Figures.
Administration of the peptides of the present invention also has a positive
effect on
cortical bone, specifically the wrist (the distal and mid-shaft radius, Figs 6
and 7).
Historically, PTH has been known to increase bone resorption, which increases
cortical
porosity, thus making it difficult for PTH to increase BMD in cortical bone.
The dosages and
formulations of the present invention have a positive effect on cortical bone
growth as
compared to both placebo and to teriparatide, Forteo . This is an
unprecedented finding,
demonstrating a statistically significant difference from placebo for 3 active
doses.
Administration of the PTHs of the present invention also have unexpected
results on
bone formation and bone resorption markers. The bone formation markers include
P1NP,
osteocalcin, and BSAP and the bone resorption markers include NTx and CTx. As
compared
to placebo, the bone formation markers have a greater % change when Ostabolin-
CTM is
administered at 15, 30, and 45 g. Figs 8-10. There is a robust effect in the
increase in the
bone formation markers when Ostabolin-CTM is administered at 30 and 45 g. The
bone
resorption markers in Figures 11-13 demonstrate that although there is some
increase in bone
resorption following the administration of Ostabolin-CTM, this increase is
less than that which
follows administration of the prior art teriparatide, Forteo PTH. Neer et
al., 2001.
Administration of the PTH peptides of the present invention has also been
shown to
unexpectedly result in a much lower incidence and severity of hypercalcemia as
compared to
PTHs known in the art. Hypercalcemia for a patient being administered the PTH
peptides
means the occurrence of at least one serum calcium value for the patient above
the upper
limit of normal (2.64 mmol/L; 10.6 mg/dL). Neer et al., 2001.
Administration of Forteo resulted in an increased level of incidences of
hypercalcemia as compared to placebo. FDA approval of Forteo was based on the
results of
treatment of 1637 postmenopausal women (with prior vertebral fractures) with
20 or 40
g/day of Forteo for an average of 19 months. See Forteo package insert,
incorporated by
reference in its entirety, and Neer. While the medication was generally well-
tolerated,
hypercalcemia was seen at least once in 11% of the 20 g group subjects and in
28% of the
40 g group subjects as compared with 2% in the placebo group. In contrast,
the
34
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
administration of low doses of the PTH peptides of the present invention (7.5,
15, and 30 g)
resulted in only a negligible increase in the incidences of hypercalcemia as
compared to
placebo. As an example, hypercalcemia was seen at least once in 5% of the
placebo group
and in 5% of the group being administered 15 g doses, resulting in no net
increase of
hypercalcemia.
Accordingly, administration of the PTH analogues of the present invention at a
variety of doses leads to following unexpected results: 1) similar or greater
effect on the
increase in B1VID of lumbar spine, hip, femoral neck, and trochanter when
given over a course
of only four months as compared to prior art PTH analogues given over a course
of at least a
year; 2) increase in B1VID on cortical bone, specifically the wrist (the
distal and mid-shaft
radius), whereas prior art PTH peptides have resulted in decease in BMD of
cortical bone;
and 3) lower amount of incidences and severity of hypercalcemia as compared to
prior art
PTH peptides.
The PTH peptides of the present invention offer substantial improvements over
currently available therapy, as they are an anabolic agent that lead to much
lower incidences
and severity of hypercalcemia. Based on preclinical and clinical experience to
date, the
present PTH peptides are a safe and highly effective anabolic agent for
treating osteoporosis,
without inducing hypercalcemia. Due to its reduced impact on bone resorption,
the present
PTH peptides also have an improved clinical profile with respect to its
effects on bone
quality.
The decrease in bone resorption can be measured by a reduction in the level of
bone
resorptive markers. Although biochemical markers of bone tumover cannot reveal
how much
bone is present in the skeleton at any given time, and thus, cannot be used to
diagnosis
osteoporosis or to tell how severe the disease may be, biochemical markers can
be used in
conjunction with the pharmaceutical compositions and formulations of the
present invention
to (1) predict bone loss in peri- and post-menopausal women and to (2) monitor
the skeletal
response to treatment. Unlike bone mineral density (B1VID) measurements,
biochemical
markers are able to detect acute changes in bone turnover. While BMD tests
typically detect
bone density changes in years, markers are able to detect changes in bone
metabolism in
weeks or months. Bone tumover can be assessed via the measurement of various
biochemical markers. There are two basic types of markers: markers of bone
formation and
markers of bone resorption. Additionally, these markers can be categorized
into two groups:
markers that measure substances released by osteoblasts and osteoclasts and
markers that
measure substances produced during the formation or breakdown of collagen, a
primary
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
protein found in bone. As bone remodeling occurs, these substances are
released into the
blood and, eventually, excreted in the urine. Many biochemical markers can be
detected and
measured in both the blood (serum) and urine.
The most commonly used assays for bone formation are serum tests of bone-
specific
alkaline phosphatase (BSAP), osteocalcin and procollagen peptides, proteins
produced by
osteoblasts and released into the bloodstream during bone formation. Bone
resorption
markers typically measure the breakdown of products of collagen, the major
protein of bone.
These include pyridinoline, deoxypyridinoline, urinary deoxypyridinoline
(urinary DPD), N-
telopeptides (NTX) and C-telopeptides (CTX) of Type I collagen crosslinks.
Earlier assays, such as total alkaline phosphatase and hydroxyproline, are
still used in
monitoring such metabolic bone diseases as Paget's disease. However, these
tests are not
sensitive enough to be used in monitoring the more subtle bone remodeling
changes that tend
to occur in osteoporosis, as levels tend to be within normal limits in
individuals with the
disease.
An additional unexpected result is the lack of occurrence of osteosarcoma
formation
with long term administration of the PTH peptides of the present invention. In
its packaging,
the prior art Forteo includes a warning label that Forteo caused an increase
in incidence of
osteosarcoma in rats. The label warns that Forteo should not be prescribed
for patients who
are at increased baseline risk for osteosarcoma. In contrast, the risk of
osteosarcoma
occurrence with the long term use of the PTH peptides of the present invention
is. minimal.
The present PTH peptides may have no, or less, incidence of osteosarcoma based
on a
different sequence and different signaling as compared to PTH (1-34). The
phospholipase-C
and downstream protein kinase C activity, which are minimized with
administration of the
PTH peptides of the present invention, may be involved in ostoeoblast growth.
Another unexpected result with the PTH peptides of the present invention is
the lack
of need to monitor serum calcium levels in patients taking these peptides for
possible
occurrences of hypercalcemia. Serum calcium levels in patients taking the
prior art Forteo
is monitored through samples of blood and/or urine during the course of
treatment. The
Forteo package insert warns that administration of Forteo may "exacerbate
hypercalcemia." Use of Forteo is not recommended for patients with high
amounts of
calcium in their blood (hypercalcemia), bone cancer or other bone disorders.
In contrast,
administration of the PTH peptides of the present invention leads to lower
incidences of
hypercalcemia, as compared to administration of Forteo . Accordingly, calcium
monitoring
may not be required with administration of the PTH peptides of the present
invention.
36
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
Another unexpected result with the peptides of the present invention is the
ability to
tailor the dose administered to a patient based on that patient's weight, body
surface area, or
BMI and presentation of symptoms. The weight, body surface area, or BMI cut
off method
provides a method for determining a therapeutically effective dosage while
maintaining a low
incidence of side effects for a patient based upon their weight, body surface
area, or BMI. A
dosage that results in high exposure for a particular patient will increase
the chance of side
effects, including hypercalcemia. Additionally, lack of efficacy may be
observed in certain
patients, especially men, because the dose for that particular patient was too
low in a situation
for a patient of a certain weight, body surface area, or BMI who could have
tolerated a higher
dosage of the therapeutic agent.=
All osteoporosis therapeutics with a predominant action to stimulate bone
formation
may be administered in a manner where the dosage is based on the weight, body
surface area,
or BMI of the patient. Examples of such therapeutics within the scope of the
present
invention include the anti-sclerostin Mab, inhibitors of negative regulators
of the Wnt
signaling pathways, and activin receptor agonists. Additionally dosage based
on patient
weight, body surface area, or BMI is effective for all therapeutics whose bone
formation
effect is mediated by the action of PTH on its receptor, including PTH, full-
length and
fragments thereof, PTH analogs, PTHrP, and PTHrP analogs. Specific PTH
peptides which
are effective with dosage based on patient weight, body surface area, or BMI
include, but not
limited to, full length PTH 1-84, PTH 1-34, PTH-(1-31)NH2, Ostabolin; PTH-(1-
30)NH2;
PTH-(1-29)NHZ; PTH-(1-28)NHZ; Leu27PTH-(1-31)NH2; Leu27PTH-(1-30)NH2i Leu27
PTH-
(1-29)NH2, Leu27cyclo(22-26)PTH-(1-31)NH2 Ostabolin-CTM; LeuZ7cyclo(22-26)PTH-
(1-
34)NH2i Leu27cyclo(Lys26-Asp3)PTH-(1-34)NH2; Cyclo(Lys27-Asp3)PTH-(1-34)NH2i
Leu27cyclo(22-26)PTH-(1-31)NH2; Ala27 or Nle27 or Tyr-z7 or I1e27 cyclo(22-
26)PTH-(1-
31)NH2; Leu27cyclo(22-26)PTH-(1-32)NH2i Leu27cyclo(22-26)PTH-(1-31)OH;
Leu27cyclo(26-30)PTH-(1-31)NH2; Cys22Cys26Leu27cyclo(22-26)PTH-(1-31)NH2i
Cys22Cys26Leu27cyclo(26-30)PTH-(1-31)NH2i Cyclo(27-30)PTH-(1-31)NH2;
Leu27cyclo(22-
26)PTH-(1-30)NHz; Cyc1o(22-26)PTH-(1-31)NH2; Cyclo(22-26)PTH-(1-30)NH2;
Leu27cyclo(22-26)PTH-(1-29)NHZ; Leu27cyclo(22-26)PTH-(1-28)NH2;
G1u17,Leu27cyclo(13-
17)(22-26)PTH-(1-28)NH2; and Glu17,Leu27cyclo(13-17)(22-26)PTH-(1-31)NH2.
Additionally, suitable examples of therapeutics which can be administered
based on
the weight, body surface area, or BMI of the patient include calcium receptor
antagonists
which stimulate endogenous PTH production, such as those that act as agonists
of the PTH
receptor, including PTH, full-length and fragments thereof, PTH analogs, PTHrP
and analogs
37
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
thereof. The administration of a dosage based upon the weight, body surface
area, or BMI of
a patient can be used in a variety of indications, including osteoporosis,
fracture repair, renal
bone disease, corticosteroid-induced osteoporosis, transplant, and the
induction of bone
formation in trabecular and cortical bone.
By evaluating the efficacy of a range of doses of the peptides of the present
invention,
the low doses can be eliminated to reduce the number of non-responders and the
high doses,
that nonnally raise safety concerns, can also be eliminated. The choice of
multiple effective
dosages improves the benefit to risk profile of the present peptides by
improving the overall
efficacy and proportion of patients who respond to the dosage while reducing
the side effects
of a dose that results in high exposure for an individual.
Improved PK Profile
Administration of the PTH analogs of the present invention has led to a PK
profile of
shorter duration (sharper peak) as compared to previously known PK profiles.
This shorter
PK profile results in maintaining the positive effects of PTH treatment, while
simultaneously
reducing side effects. Suitable PK parameters within the scope of the present
invention
include a half life of the PTH peptide analogue of between 2 minutes and 60
minutes; a
duration of exposure to the PTH peptide analogue of between 30 minutes and 4
hours; a
Tmax of the PTH peptide analogue of between 2 minutes and 30 minutes; and a
Cmax of the
PTH peptide analogue of between 10 and 400 pg/ml. More preferred PK ranges
include a
half-life of between 15-30 minutes, a duration of exposure between one and 2
hours, a Tmax
of between 15-30 minutes, and a Cmax of between 50-200 pg/ml.
Details of the pharmacokinetics within the scope of the present invention are
shown in
the figures, both for human and animal administration. For the PTH
administered to humans,
Ostabolin-C was administered in a liquid formulation with a buffer, a polyol,
and a stabilizer,
with a pH of between 3 and 5. Alternative embodiments of PTHs can also be
administered
which result in a similar pharmacokinetic profile. For the PTH administered to
animals,
Ostabolin-C was formulated in acidified saline and adjusted with phosphates to
a pH of 7.2
+/- 0.4. The pharmacokinetics using the above-described Ostabolin-C PTH
formulation is
shown in figures 18-20.
Pharmaceutical Compositions/Formulations, Dosing, and Administration
A range of PTH peptide analogue compounds can be used in the methods and
compositions of the present invention. Generally, preferred embodiments of PTH
peptide
38
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
analogues include those that when administered result in reduced phospholipase-
C activity,
reduced bone resorption, and reduced hypercalcemia levels. As defined in the
Definitions
section herein, "reduced phospholipase-C activity" refers to a PTH peptide
analogue that has
been truncated or modified in some manner so as to trigger less than full
activation of
phospholipase-C, as compared to the full-length PTH peptide or other PTH
peptide analogues
which are at least 34 amino acid residues; "reduced bone resorption" refers to
a PTH peptide
analogue that has been truncated or modified in some manner so as to trigger
less bone
resorption, as compared to the full-length PTH peptide or other PTH peptide
analogues which
are at least 34 amino acid residues, and "reduced hypercalcemia levels" refers
to a PTH
peptide analogue that has been truncated or modified in some manner so as to
trigger less
incidences of hypercalcemia, or lower severity of hypercalcemia, as compared
to the full-
length PTH peptide or other PTH peptide analogues which are at least 34 amino
acid
residues.
The preferred PTH analogues administered in the methods described herein
include
[Leu27]cyclo[G1u22-Lys26]-PTH-(1-31)-NH2, such as advanced by Zelos
Therapeutics, Inc.
under the tradename OSTABOLIN-CTM and PTH-(1-31)-NH2, such as advanced by
Zelos
Therapeutics, Inc. under the tradename OSTABOLINTM. In another embodiment of
the
invention, [Leu27]cyclo[G1u22-Lys26]-PTH-(1-30)-NH2 is used in the methods
described
herein. In another embodiment, the hormone can be the linear analogue PTH(1-
31), which
can have a free carboxyl ending, or be amidated, at the C-terminus. In yet
another
embodiment, the hormone can be PTH(1-30), which can have a free carboxyl
ending, or be
amidated, at the C-terminus; or [Leu27]-PTH(1-30)- NH2. Suitable stabilized
solutions of the
PTH peptide analogues that can be employed in the present methods are
described in U.S.
Patent Nos. 5,556,940; 5,955,425; 6,541,450; 6,316, 410; and 6,110,892
incorporated herein
by reference.
Dosages
An effective amount of a PTH peptide analogue for use in the present invention
is an
amount that will provide the desired benefit or therapeutic effect upon
administration
according to the prescribed regimen. Effective dosages can vary according to
the type of
formulation of PTH peptides or analogs administered as well as the route of
administration.
One skilled in the art can adjust the dosage by changing the route of
administration or
formulation, so that the dosage administered would result in a similar
pharmacokinetic or
biological profile as result from the preferred dosage ranges described
herein. Exemplary
dosages include a daily dose of 2 to 100 g for subcutaneous delivery of an
aqueous
39
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
formulation, a daily dose of 0.5 to 50 jig for subcutaneous delivery of a
formulation stabilized
with propylene glycol and/or ethanol, a daily dose of 100 to 3,000 gg for
inhalation delivery,
and weekly doses at 3-7 times the daily doses. Other suitable dosages include
any dosage
with any route of administration that results in a bioavailability or
pharmacokinetic profile
similar to those yielded by the above-described dosage ranges.
Nonlimiting examples of an effective amount of PTH analog administered
subcutaneously in an aqueous formulation may range from about 2 g/day to
about 100
g/day, preferably from about 5 jig/day to about 45 g/day, more preferably
from about 7.5
g/day to about 20 g/day, more preferably from about 20 jig/day to about 30
g/day, more
preferably from about 30 g/day to about 45 jig/day, and more preferably 5,
7.5, 10, 15, 20,
25, 30, 35, 40, 45, or 50 g/day. Additional preferred dosages include dosages
of 5, 6, 7,
7.5, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26,
27, 28, 29, 30, 31,
32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, or 45 g/day. Additional
examples of an
effective amount of PTH analogue administered subcutaneously in an aqueous
formulation
may range from about 14 g/week to about 420 g/week, preferably from about 35
g/week
to about 280 g/week, more preferably from about 70 jig/week to about 140
jig/week, more
preferably from about 140 g/week to about 210 jig/week, and more preferably
35, 70, 105,
140, 175, 205, or 245 jig/week. The dosages can be administered every day,
every two days,
every three days, every four days, every five days, every six days, or every
seven days
(once/week). These dosages can also be adjusted to correct for
bioavailability. The doses
can also be measured in mmol, taking into account the molecular weight of the
PTH peptides
used.
The dosage can also be calculated based on the size of the patient. The g
dosages
can be normalized for patient characteristics such as height, weight, body
surface area, BMI,
lean body mass, etc., by converting the g to g/kg, jig/m2, or g /ml, or
other suitable
conversions known in the art. In suitable embodiments, dosages can also be
administered
subcutaneously on a g/kg basis. This calculation is performed as follows
using 30 g and
45 g as exemplary doses. Based on the assumption that the average human
subject weighs
about 65 kg, the 30 g and 45 g doses are converted to 0.46 g/kg and 0.69
g/kg. To
convert jig to g/kg, the g dose is divided by 65 kg, to give a g/kg dose
(dose/weight =
g/kg). For a dose of 30 g, 30 g per 65 kg average human weight gives a g/kg
dose of
about 0.46 gg/kg. For 45 g, 45 g per 65 kg average human weight gives a
g/kg dose of
about 0.69 gg/kg. Dosages within the scope of the present invention for
subcutaneous
delivery of an aqueous formulation include from 0.20-0.90 g/kg, more
preferably 0.30-0.70
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
g/kg, and still more preferably 0.46-0.69 g/kg. Dosages within the preferred
ranges
maximize the effectiveness of PTH therapy while simultaneously reducing side
effects.
For inhalation therapy, the PTH peptide or analogue can be administered at
doses
between 100 g and 3,000 g per day. More specifically, the PTH peptide or
analogue can
be administered at doses of 100 g, 200 g, 300 g, 400 g, 500 g, 600 g,
700 g, 800 g,
900 g, 1000 g, 1100 g, 1200 g, 1300 g, 1400 g, 1500 g, 1600 g, 1700
g, 1800 g,
1900 g, 2000 g, 2100 g, 2200 g, 2300 g, 2400 pg, 2500 g, 2600 g, 2700
pg, 2800
g, 2900 g, or 3000 g per day. Inhalation therapy can also be administered
weekly at 3 to
7 times greater than the daily dose.
A Phase I clinical study was undertaken using Ostabolin-CTm Inhalation Powder
(OCIP) to establish an MTD in post-menopausal women, to compare its PK profile
with
Phase II sub-cutaneous doses, and to evaluate biological activity with cAMP
and biomarkers
of bone turnover. Preferred doses of 300 g and 800 g resulted in a PK
profile similar to
subcutaneous administration. This pk profile included a half life of the PTH
peptide
analogue of between 2 minutes and 60 minutes; a duration of exposure to the
PTH peptide
analogue of between 30 minutes and 4 hours; a Tmax of the PTH peptide analogue
of
between 2 minutes and 30 minutes; and a Cmax of the PTH peptide analogue of
between 10
and 400 pg/ml. More preferred PK ranges include a half-life of between 15-30
minutes, a
duration of exposure between one and 2 hours, a Tmax of between 15-30 minutes,
and a
Cmax of between 50-200 pg/ml. Details of inhalation dosing is shown in Example
14.
Dosages can also be administered in varying amounts based on the presentation
of
symptoms and the weight, body surface area, or BMI of the subject. Such dose
optimization
is discussed in detail below.
Dose Optimization
Dose optimization is important for all drugs, especially for those with a
narrow
therapeutic window. Hormones in general, including PTH and its analogs, are
such drugs
which have a narrow therapeutic window. Because of this narrow therapeutic
window, a
standardized single dose for all patients presenting with a variety of
symptoms may not
always be effective. Our dose optimization approach of two separate doses, 30
g and 45 pg,
for patients with different symptoms and different weights, body surface area,
or BMI,
addresses this problem.
Optimizing the dose for the administration of PTH analogs to human patients
with an
appropriate risk/benefit ratio has proven difficult with prior art PTHs. The
administration and
41
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
potential commercialization of PTH analogs has been analyzed by many
companies, with the
most effective and least toxic dose being difficult to obtain. Two such
companies who have
studied PTH dosing are Lilly (with Forteo 1-34 hPTH) and NPS (Preos -
recombinant full-
length human parathyroid hormone, rhPTH 1-84).
Dose optimization of the PTH peptides of the present invention provides a
benefit
over the single dose currently available with Forteo. Lilly's Forteo has only
been approved
for a 20 g dose. Although Lilly studied the possibility of administering
doses of both 20
and 40 g, only the 20 g dose was approved. The risk/benefit ratio of the 40
g dose was
too great, as hypercalcemia was seen at least once in 28% of the subjects
administered 40 g
of Forteo. Accordingly, the 20 gg dose of Forteo (hPTH 1-34) is the only dose
available for
all symptoms of all patients. There are some patients for whom a 20 g dose
will not give
enough benefit. Additionally, in a Summary Basis of Approval for Forteo, the
FDA
indicated that the correct dose for men should be 30 g. Our dose optimization
approach of
two distinct dosages, 30 g and 45 jig, will overcome both the risks of
Forteo's 40 gg dose
and the lack of efficacy in certain patients with Forteo's 20 g dose.
Additionally, NPS has studied a variety of doses for its recombinant full-
length
human parathyroid hormone, rhPTH 1-84, Preos. In Phase 2 clinical trials, NPS
analyzed
does of 50, 75, and 100 g/day. In Phase 3 clinical trials, the dose
administered was limited
to a single dose of 100 gg. This dose has not yet been approved due to high
incidence levels
of hypercalcemia.
The presently described dose/weight cutoff will help avoid the prior art
problems of
the risks of PTH administration of a single dose outweighing the benefits
achieved.
The dosages can thus be administered based on the weight, body surface area,
or BMI of a
subject. As shown in the data below, the greatest benefits, with the least
side effects, are
obtained when Ostabolin-C is administered at a dose of about 30 g for a
patient below the
weight cutoff and at a dose of about 45 g for a patient above the weight
cutoff. These same
benefits of dose/weight cutoff occur with PTH analogs described herein in
addition to
Ostabolin-C, and other therapeutics, including the anti-sclerostin Mab,
inhibitors of negative
regulators of the Wnt signaling pathways, activin receptor agonists,
therapeutics whose bone
formation effect is mediated by the action of PTH on its receptor, including
PTH, full-length
and fragments thereof, PTH analogs, PTHrP, and PTHrP analogs, and calcium
receptor
antagonists which stimulate endogenous PTH production, such as those that act
as agonists of
the PTH receptor, including PTH, full-length and fragments thereof, PTH
analogs, PTHrP
and analogs thereof.
42
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
The 4 month and 12 month Phase 11 data from the Ostabolin-C sub-cutaneous
program, detailed in the Examples herein, indicate that a daily dose of 30-45
pg has an
appropriate risk to benefit profile for the treatment of osteoporosis. The 30
g dose
demonstrates clinically beneficial increases in lumbar spine BMD with a low
incidence of
hypercalcemia and reduced incidence of side effects. The 45 g dose causes
larger increases
in lumbar spine BMD and also produces an increase in BMD of the hip. It is
therefore of
interest to explore whether dose optimization could capture some or all of the
upside efficacy
benefits of the high dose without incurring the increased side effects that
are also observed at
that dose. This analysis is described below.
At the 30 g dose, lumbar spine BMD increases, mid-radius BMD increases, the
percent incidences of hypercalcemia is relatively low, and the increase in
bone formation
versus bone resorption is great, as demonstrated by the anabolic window. The
advantages of
this 30 g dose include early and large increase in bone formation with
evidence of reduced
bone resorption relative to Forteo. The PTHs within the scope of the present
invention which
have a shorter duration PK profile, provide a benefit similar to that seen
with Forteo in the
positive BMD change at lumbar spine and hip, but the present invention reduces
the side
effects with a reduced potential for cortical bone loss. The present invention
also exhibits a
decreased propensity to cause hypercalcemia. Data for the administration of 30
and 45 g
doses of Ostabolin-C is shown in figures 21 and 22.
Administration of PTH analogs at 45 g dosages also provides additional
benefits.
These same benefits occur with PTH analogs described herein in addition to
Ostabolin-C.
This 45 g dose has superior efficacy, but with the emergence of side effects.
Administration
of a 45 g dose of Ostabolin-C results in strong early BMD change at hip or
lumbar spine
allied with powerful bone formation effects. This positive effect is also
combined with.some
evidence of bone resorption stimulation and hypercalcemia. These results are
shown in
figure 22.
Based on the effects demonstrated above with dosages of 30 and 45 g, it is
evident
that dose optimization is important in order to obtain the benefits while
simultaneously
minimizing adverse effects. One method of optimizing dose is to administer a
dose which
results in a shorter PK profile. Suitable PK parameters within the scope of
the present
invention include a half life of the PTH peptide analogue of between 2 minutes
and 60
minutes; a duration of exposure to the PTH peptide analogue of between 30
minutes and 4
hours; a Tmax of the PTH peptide analogue of between 2 niinutes and 30
minutes; and a
Cmax of the PTH peptide analogue of between 10 and 400 pg/ml. More preferred
PK ranges
43
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
include a half-life of between 15-30 minutes, a duration of exposure between
one and two
hours, a Tmax of between 15-30 minutes, and a Cmax of between 50-200 pg/ml.
Another method of optimizing dose is to administer different dosages,
depending on
the weight, height, body surface area, or BMI of the subject. As shown by the
data herein,
the high dose of 45 g has efficacy greater than previously seen with Forteo,
particularly at
the hip. But this high dose also presents a risk factor of hypercalcemia for
certain patients.
The low dose of 30 g has efficacy at least as good as has been seen with
Forteo, and does
not present a risk factor for hypercalcemia. Neither of the 30 or 45 g doses
are optimal for
all patients. The dose optimization presented herein provides a dosing regimen
that combines
the positive features of both the 30 and 45 g doses, namely superior efficacy
with low risks
of adverse effects. This dosing regimen provides for different doses based on
the patient's
body characteristics, including weight, height, BMI, lean body mass, or body
surface area.
In this way, the potential for non response in a subject is mitigated by
avoidance of
low dose in large individuals and the potential of adverse effects in a
subject is mitigated by
avoidance of high doses in small individuals. Dosages within the scope of the
present
invention include administering 45 g to a subject weighing 50 kg or more,
more specifically
65 kg or more, more specifically 68 kg or more, more specifically 70 kg or
more and
administering 30 g to a subject weighing less than 90 kg, more specifically
less than 75 kg,
more specifically less than 68 kg, more specifically less than 65 kg, more
specifically less
than 59 kg, and more specifically less than 50 kg.
This dose/weight cutoff optimization provides the ability to tailor the dose
administered to a patient based on that patient's weight, body surface area,
or BMI and
presentation of symptoms. The weight cut off method provides a method for
determining a
therapeutically effective dosage while maintaining a low incidence of side
effects for a
patient based upon their weight, body surface area, or BMI. A dosage that
results in high
exposure for a particular patient will increase the chance of side effects,
including
hypercalcemia. Additionally, lack of efficacy may be observed in certain
patients, because
the dose for that particular patient was too low in a situation for a patient
of a certain weight,
body surface area, or BMI who could have tolerated a higher dosage of the
therapeutic agent.
By evaluating the efficacy of a ranges of doses, the low doses can be
eliminated to
reduce the number of non-responders and the high doses, that normally raise
safety concerns,
can also be eliminated. The weight, body surface area, or BMI cutoff limits
the dosage to be
administered to a patient to either the high or low dose. The choice of
multiple effective
dosages improves the benefit to risk profile of the present peptides by
improving the overall
44
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
efficacy and proportion of patients who respond to the dosage while reducing
the side effects
of a dose that results in high exposure for an individual.
To explore the dosing range between 30 and 45 g/day, response to Ostabolin-C
was
analyzed based on individual exposure to drug, with the doses converted to
g/kg (daily dose
divided by weight in kg x 100 (for avoidance of decimals)). All patients who
received active
treatment were converted to an individual dose exposure and then regrouped
into ascending
exposure dose cohorts of g/kg (0-15, 16-25, 26-35, 36-45, 46-55, >56 g/kg).
The mean
changes for primary and secondary endpoints after 4 months of treatment in
these new dose
exposure cohorts were calculated. A linear regression analysis enabled the
effect of
exposure to Ostabolin-C to be assessed across the full range of dose
exposures. The group
means are presented as a linear regression analysis with 95% CI for continuous
variables.
The data are shown in the figures for effects on the lumbar spine BMD (figures
23-24),
femoral neck BMD (figure 29), forearm midshaft radius (figure 30), total hip
(figure 27, 53-
54), serum calcium (figure 28), hypercalcemia (figures 31-32), and bone
formation and bone
resorption markers (figure 26). The results indicate that all of the biomarker
and BMD
endpoints and serum calcium (all continuous variables) change in a linear
pattem across the
entire dose range tested in this study.
Based on the assumption that the average human subject weighs about 65 kg, the
30
g and 45 g doses were converted to 0.46 g/kg and 0.69 g/kg. To convert g
to g/kg,
the g dose is divided by 65 kg, to give a g/kg dose (dose/weight = g/kg).
For a dose of 30
g, 30 g per 65 kg average human weight gives a g/kg dose of about 0.46
g/kg. For 45
g, 45 g per 65 kg average human weight gives a g/kg dose of about 0.69
g/kg. These
doses are demonstrated in the figures. Dosages within the scope of the present
invention
include from 0.20-0.90 pg/kg, more preferably 0.30-0.70 g/kg, and still more
preferably
0.46-0.69 pg/kg. Dosages within the preferred ranges maximize the
effectiveness of PTH
therapy while simultaneously reducing side effects.
One method of reducing the range of dose exposures in a study population is to
utilize
two dose strengths with a single weight cutoff (i.e. all patients who weigh
less than the cutoff
receive the low dose (30 pg), whereas all those above the weight cutoff
receive the high dose
(45 pg). The impact of this dose optimization strategy on Ostabolin-C exposure
is illustrated
in figures 33-36.
The effect of a weight cutoff at 68 kg reduces the spread of exposures in the
30 and 45
g dose groups. This is shown in detail in the examples. Comparison of modeled
data for
three different BMD parameters illustrates that progressively higher dose
exposures are
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
required to affect total hip and femoral neck BMD compared to lumbar spine
BMD. This will
enable therapy to be tailored to the individual needs of the patient. For
lumbar spine BMD,
the proposed weight cutoff is 68kg, so that patients with lumbar spine BMD who
weigh less
than 68 kg would be administered the 30 g dose and patients with lumbar spine
BMD
weighing 68 kg or more would be administered the 45 g dose. For hip and
femoral neck,
the proposed cutoff weight is lower than 68 kg, since higher doses are
required to have a
similar positive effect.
Plasma Concentration - AUC
The dose may also be selected to provide an effective plasma concentration of
PTH
analogue or other osteoporosis therapeutic. Examples of an effective maximum
plasma
concentration of peptide concentration may range from about 10 pg/mL to about
400 pg/mL,
preferably from about 20 pg/mL to about 300 pg/mL; from about 50 pg/mL to
about 280
pg/mL; from about 80 pg/mL to about 250 pg/mL; from about 100 pg/mL to about
150
pg/mL. Other suitable dosage ranges for maximum plasma concentration of PTH
peptide
analogues include 20-40 pg/mL, 40-60 pg/mL, 60-80 pg/mL, 80-100 pg/mL, 100-120
pg/mL,
120-140 pg/mL, 140-160 pg/mL, 160-180 pg/mL, 180-200 pg/mL, 200-230 pg/mL, 230-
260
pg/mL, 260-300 pg/mL, 300-350 pg/mL, and 350-400 pg/mL.
In another specific embodiment of the invention, the peptide is administered
in an
effective amount that results in the value for area under the curve (herein
referred to as
"AUC") in the plasma peptide concentration versus time curve in the range of 5
pg=h/mL -
400 pg=h/mL. More preferably, the range of AUC is between 10 pg=h/mL - 350
pg=h/mL.
More preferably, AUC is in the range of 20 pg=h/mL - 300 pg=h/mL. More
preferably, AUC
is in the range of 50 pg=h/mL - 250 pg-h/mL. More preferably, AUC is in the
range of 70
pg-h/mL - 200 pg=h/mL. More preferably, AUC is in the range of 90 pg=h/mL -
150 pg=h/mL.
Even more preferably, AUC is in the range of 95 pg-h/mL - 125 pg=h/mL. Other
suitable
range for AUC is 5 pg=h/mL - 20 pg=h/mL, 20 pg=h/mL - 50 pg=h/mL, 50 pg=h/mL -
70
pg=h/mL, 70 pg.h/mL - 90 pg-h/mL, 90 pg=h/mL - 100 pg=h/mL, 100 pg-h/mL -110
pg=h/mL, 110 pg=h/mL - 120 pg-h/mL, 120 pg=h/mL - 130 pg=h/mL, 130 pg=h/mL -
150
pg=h/mL, 150 pg=h/mL - 175 pg=h/mL, 175 pg=h/mL - 200 pg-h/mL, 200 pg=h/mL -
225
pg.h/mL, 225 pg=h/mL - 250 pg=h/mL, 250 pg-h/mL -275 pg=h/mL, 275 pg-h/mL -
300
pg=h/mL, 300 -350 pg-h/mL, or 350 pg-h/mL - 400 pg-h/mL.
46
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
Accordingly, in one aspect, the invention provides a pharmaceutical
formulation
comprising a therapeutically effective amount of a PTH peptide analogue as the
active
ingredient in a daily dosage range of 2 g to 60 g or a weekly dosage range
of 14 g to 420
g, wherein the PTH peptide analogue has reduced phospholipase-C activity and
maintains
adenylate cyclase activity, in admixture with a pharmaceutically acceptable
excipient,
diluent, or carrier, or combinations thereof. Effective dosages can vary
according to the type
of formulation of PTH peptides or analogs administered as well as the route of
administration. One skilled in the art can adjust the dosage by changing the
route of
administration or formulation, so that the dosage administered would result in
a similar
pharmacokinetic or biological profile as result from the preferred dosage
ranges described
herein. Exemplary dosages include a daily dose of 2 to 100 g for subcutaneous
delivery of
an aqueous formulation, a daily dose of 0.5 to 50 jig for subcutaneous
delivery of a
formulation stabilized with propylene glycol and/or ethanol, a daily dose of
100 to 3,000 g
for inhalation delivery, and weekly doses at 3-7 times the daily doses. Other
suitable dosages
include any dosage with any route of administration that results in a
bioavailability or
pharmacokinetic profile similar to those yielded by the above-described dosage
ranges.
Routes of Administration
Administration of the PTH peptide analogues of the present invention includes
both
direct administration, including self-administration, and indirect
administration, including the
act of prescribing a drug. For example, as used herein, a physician who
instructs a patient to
self-administer a drug and/or provides a patient with a prescription for a
drug is administering
the drug to the patient.
A variety of administration routes can be used in accordance with the present
invention, including oral, topical, transdermal, nasal, pulmonary,
transpercutaneous (wherein
the skin has been broken either by mechanical or energy means), rectal,
buccal, vaginal, via
an implanted reservoir, or parenteral. Parenteral includes subcutaneous,
intravenous,
intramuscular, intraperitoneal, intra-articular, intra-synovial, intrasternal,
intrathecal,
intrahepatic, intralesional and intracranial injection or infusion techniques.
More preferably,
the route of administration is subcutaneous, transcutaneous, intranasal,
transdermal, oral, or
inhalation administration.
Formulations
47
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
A stabilized solution of a parathyroid hormone can include a stabilizing
agent, a
buffering agent, a preservative, an antibacterial agent and the like. The
stabilizing agent
incorporated into the solution or composition includes alcohols, ethanol or a
polyol which
includes a saccharide, preferably a monosaccharide or disaccharide, e.g.,
glucose, trehalose,
raffinose, or sucrose; a sugar alcohol such as, for example, mannitol,
sorbitol or inositol, and
a polyhydric alcohol such as glycerine or propylene glycol or mixtures
thereof. A preferred
polyol is mannitol or propylene glycol. The concentration of polyol may range
from about 1
to about 20 wt-%, preferably about 3 to 10 wt-% of the total solution.
The buffering agent employed in the solution or composition of the present
invention
may be any acid or salt combination which is pharmaceutically acceptable.
Useful buffering
systems are, for example, acetate, tartrate or citrate sources. Preferred
buffer systems are
acetate or tartrate sources, most preferred is an acetate source. The
concentration of buffer
may be in the range of about 2 mM to about 500 mM, preferably about 2 mM to
100 mM.
The stabilized solution or composition of the present invention may also
include a
parenterally acceptable preservative. Such preservatives include, for example,
cresols, benzyl
alcohol, phenol, benzalkonium chloride, benzethonium chloride, chlorobutanol,
phenylethyl
alcohol, methyl paraben, propyl paraben, thimerosal and phenylmercuric nitrate
and acetate.
A preferred preservative is m-cresol or benzyl alcohol; most preferred is m-
cresol. The
amount of preservative employed may range from about 0.1 to about 2 wt-%,
preferably
about 0.3 to about 1.0 wt-% of the total solution.
The parathyroid hormone compositions can, if desired, be provided in a
powder form containing not more than 2% water by weight, that results from the
freeze-
drying of a sterile, aqueous hormone solution prepared by mixing the selected
parathyroid
hormone, a buffering agent and a stabilizing agent as above described.
Especially useful as a
buffering agent when preparing lyophilized powders is a tartrate source.
Particularly useful
stabilizing agents include glycine, sucrose, trehalose and raffinose.
Ready to use formulations containing hPTH, or more specifically, Ostabolin-C,
are
not stable at room temperature and must be stored under refrigerated
conditions (2-8 C).
Since hPTH undergoes hydrolysis, oxidation and deamidation in aqueous media,
it is difficult
to develop a solution formulation for room temperature storage. Although, the
formulation is
stable at 5 C, it is preferred that the formulation is stable at about pH 7.5,
as the pH is closer
to physiological pH. Studies have indicated that an Ostabolin-C solution is
less stable at pH
7.5 compared to the ready-to-use formulation. Oxidation and deamidation both
occur and
takes place above pH 7Ø As such, a 100% aqueous formulation above pH 7 under
48
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
refrigerated conditions may not be feasible. Hence mixtures of ethanol/water
or
propylene/water systems were used with the antioxidants methionine or lipoic
acid to
evaluate the stability of the formulations of this invention.
Another additive to help maintain the stability of an hPTH formulation is
Methionine.
Methionine has been shown to be a potential antioxidant and improve hPTH
stability.
Additionally, polyols have the potential to stabilize peptide and protein
formulations and
sucrose concentrations up to 1 M at pH 5.5 have been found to reduce the rate
of both
deamidation and oxidation of hPTH.
In addition, parathyroid hormones formulated with typical buffers and
excipients
employed in the art to stabilize and solubilize proteins for parenteral
administration. Buffers
also have an effect on stability. Previous models showed that for pHs above 7,
TRIS buffer
had a much lower deamidation rate constant than a corresponding phosphate
buffer. Adding
NaC1 also has a positive effect of the formulation because of its
physiological ionic strength.
Art recognized pharmaceutical carriers and their formulations are described in
Martin,
"Remington's Pharmaceutical Sciences," 15th Ed.; Mack Publishing Co., Easton
(1975).
More details of stabilizing additives to a hPTH formulation are shown in
Examples 15 and
16.
The PTH peptide analogue may also be formulated into a composition suitable
for
administration by any convenient route, e.g., orally (including sublingually),
topically,
transdermally (including percutaneous absorption of the composition through
the skin, such
as by patches, ointments, creams, gels, salves and the like), intranasally,
rectally or inhaled as
a dry powder, aerosol, or mist, for pulmonary delivery.
Such forms of the compounds of the invention may be administered by
conventional
means for creating aerosols or a.dministering dry powder medications using
devices such as
for example, metered dose inhalers, nasal sprayers, dry powder inhaler, jet
nebulizers, or
ultrasonic nebulizers. Such devices optionally may include a mouthpiece fitted
around an
orifice. It should be understood, however, that the invention embraces all
forms of
administration which make the PTH peptide analogues systemically or locally
available.
In addition to the usual meaning of administering the formulations described
herein to
any part, tissue or organ whose primary function is gas exchange with the
extemal
environment, for purposes of the present invention, "pulmonary" is also meant
to include a
tissue or cavity that is contingent to the respiratory tract, in particular,
the sinuses. For
pulmonary administration, an aerosol formulation containing the active agent,
a manual pump
spray, nebulizer or pressurized metered-dose inhaler as well as dry powder
formulations are
49
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
contemplated. Suitable formulations of this type can also include other
agents, such as
antistatic agents, to maintain the disclosed compounds as effective aerosols.
A drug delivery device for delivering aerosols comprises a suitable aerosol
canister
with a metering valve containing a pharmaceutical aerosol formulation as
described and an
actuator housing adapted to hold the canister and allow for drug delivery. The
canister in the
drug delivery device has a head space representing greater than about 15% of
the total
volume of the canister. Often, the polymer intended for pulmonary
administration is
dissolved, suspended or emulsified in a mixture of a solvent, surfactant and
propellant. The
mixture is maintained under pressure in a canister that has been sealed with a
metering valve.
Orally administrable compositions may, if desired, contain one or more
physiologically compatible carriers and/or excipients and may be solid or
liquid. Intranasal
administration to the subject includes administering a therapeutically
effective amount of the
PTH peptide analogue to the mucous membranes of the nasal passage or nasal
cavity of the
subject. Pharmaceutical compositions for nasal administration can include, for
example,
nasal spray, nasal drops, suspensions, gels, ointments, creams, or powders.
Pharmaceutically acceptable compositions of the peptide described herein can
be used
according to the method of the present invention. The pharmaceutical
compositions described
herein can optionally include one or more pharmaceutically acceptable
excipients. Such
pharmaceutically acceptable excipients are well known in the art and include,
for example,
salts (such as protamine sulfate, disodium hydrogen phosphate, potassium
hydrogen
phosphate, sodium chloride, zinc salts, colloidal silica and magnesium
trisilicate),
surfactant(s), water-soluble polymers (such as polyvinyl pyrrolidone,
cellulose based
substances, polyethylene glycol, polyethylene glyco1400, polyacrylates, sodium
carboxymethylcellulose, waxes and polyethylene-polyoxypropylene-block
polymers),
preservatives, antimicrobials, antioxidants, cryo-protectants, wetting agents,
viscosity agents,
tonicity modifying agents, levigating agents, absorption enhancers,
penetration enhancers, pH
modifying agents, muco-adhesive agents, coloring agents, flavoring agents,
diluting agents,
emulsifying agents, suspending agents, solvents, co-solvents, buffers (such as
phosphates,
glycine, sorbic acid, potassium sorbate and partial glyceride mixtures of
saturated vegetable
fatty acids), serum proteins (such as human serum albumin), ion exchangers,
tacopherol
polyethylene glycol 1000 succinate (TPGS) mygly oil, labrosol, labrofac,
ethanol, fillers such
as sugars, including lactose sucrose, mannitol, or sorbitol; cellulose
preparations such as, for
example, maize starch, wheat starch, rice starch, potato starch, gelatin, gum
tragacanth,
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
methyl cellulose, hydroxypropylmethyl-cellulose, sodium
carboxymethylcellulose, and/or
polyvinylpyrrolidone (PVP), and combinations of these excipients. If desired,
disintegrating
agents may be added, such as the cross-linked polyvinylpyrrolidone, agar, or
alginic acid or a
salt thereof such as sodium alginate. Further examples of such carriers or
excipients include
but are not limited to, calcium carbonate, calcium phosphate, various sugars,
starches,
cellulose derivatives, gelatin, and polymers such as polyethylene glycols.
Examples of surfactants suitable for use in the formulations of the present
invention
include, but are not limited to, cholic acid and salts of cholic acid,
deoxycholic acid and salts
of deoxycholic acid, taurocholic acid and salts of taurocholic acid,
polyvinylpyrrolidone,
PEG compounds such as cocamines, glyceryl stearates, glyceryl oleates,
hydrogenated
lanolins, lanolins, laurates and oleates, sorbitan laurates, sorbitan
palmitates, sorbitan
stearates, quaternium surfactants, sodium sulfates, glyceryl compounds,
palmitic acid and its
derivatives and oleic acid and its derivatives.
The excipient included within the pharmaceutical compositions of the invention
is
chosen based on the expected route of administration of the composition in
therapeutic
applications. Accordingly, compositions designed for oral, lingual,
sublingual, buccal and
intrabuccal administration can be made without undue experimentation by means
well known
in the art, for example, with an inert diluent or with an edible carrier. The
compositions may
be enclosed in gelatin capsules or compressed into tablets. For the purpose of
oral
therapeutic administration, the pharmaceutical compositions of the present
invention may be
incorporated with excipients and used in the form of tablets, troches,
capsules, elixirs,
suspensions, syrups, wafers, chewing gums and the like.
Solid dosage forms, such as tablets, pills and capsules, may also contain one
or more
binding agents, filling agents, suspending agents, disintegrating agents,
lubricants,
sweetening agents, flavoring agents, preservatives, buffers, wetting agents,
disintegrants,
effervescent agents, and other excipients. Such excipients are known in the
art. Examples of
filling agents are lactose monohydrate, lactose anhydrous, and various
starches. Examples of
binding agents are various celluloses and cross-linked polyvinylpyrrolidone,
microcrystalline
cellulose, and silicifized microcrystalline cellulose (SMCC). Suitable
lubricants, including
agents that act on the flowability of the powder to be compressed, are
colloidal silicon
dioxide, talc, stearic acid, magnesium stearate, calcium stearate, and silica
gel. Examples of
sweeteners are any natural or artificial sweetener, such as sucrose, xylitol,
sodium saccharin,
cyclamate, aspartame, and accsulfame K. Examples of flavoring agents are
bubble gum
flavor, fruit flavors, and the like. Examples of preservatives are potassium
sorbate,
51
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
methylparaben, propylparaben, benzoic acid and its salts, other esters of
parahydroxybenzoic
acid such as butylparaben, alcohols such as ethyl or benzyl alcohol, phenolic
compounds
such as phenol, or quarternary compounds such as benzalkonium chloride.
Suitable diluents
include pharmaceutically acceptable inert fillers, such as microcrystalline
cellulose, lactose,
dibasic calcium phosphate, saccharides, and/or mixtures of any of the
foregoing. Examples of
diluents include microcrystalline cellulose, lactose such as lactose
monohydrate, lactose
anhydrous, dibasic calcium phosphate, mannitol, starch, sorbitol, sucrose and
glucose.
Suitable disintegrants include corn starch, potato starch, and modified
starches,
crosspovidone, sodium starch glycolate, and mixtures thereof. Examples of
effervescent
agents are effervescent couples such as an organic acid and a carbonate or
bicarbonate.
Suitable organic acids include, for example, citric, tartaric, malic, fumaric,
adipic, succinic,
and alginic acids and anhydrides and acid salts. Suitable carbonates and
bicarbonates include,
for example, sodium carbonate, sodium bicarbonate, potassium carbonate,
potassium
bicarbonate, magnesium carbonate, sodium glycine carbonate, L-lysine
carbonate, and
arginine carbonate. Alternatively, only the acid component of the effervescent
couple may be
present.
Various other materials may be present as coatings or to modify the physical
form of
the dosage unit. For instance, tablets may be coated with shellac, sugar or
both. A syrup or
elixir may contain, in addition to the active ingredient, sucrose as a
sweetening agent, methyl
and propyl parabens as preservatives, a dye and a flavoring such as cherry or
orange flavor,
and the like.
The compositions may take any convenient form including, for example, tablets,
coated tablets, capsules, lozenges, aqueous or oily suspensions, solutions,
emulsions, syrups,
elixirs and dry products suitable for reconstitution with water or another
suitable liquid
vehicle before use. The compositions may advantageously be prepared in dosage
unit form.
Tablets and capsules according to the invention may, if desired, contain
conventional
ingredients such as binding agents, for example syrup, acacia, gelatin,
sorbitol, tragacanth or
polyvinyl-pyrollidone; fillers, for example lactose, sugar, maize-starch,
calcium phosphate,
sorbitol or glycine; lubricants, for example magnesium stearate, talc,
polyethylene glycol or
silica; disintegrants, for example potato starch; or acceptable wetting agents
such as sodium
lauryl sulphate. Tablets may be coated according to methods well known in the
art.
Liquid compositions may contain conventional additives such as suspending
agents,
for example sorbitol syrup, methyl cellulose, glucose/sugar syrup, gelatin,
hydroxymethylcellulose, carboxymethylcellulose, aluminium stearate gel or
hydrogenated
52
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
edible fats; emulsifying agents, for example lecithin, sorbitan monooleate or
acacia; non-
aqueous vehicles, which may include edible oils, for example vegetable oils
sucl2 as arachis
oil, almond oil, fractionated coconut oil, fish-liver oils, oily esters such
as polysorbate 80,
propylene glycol, or ethyl alcohol; and preservatives, for example methyl or
propyl p-
hydroxybenzoates or sorbic acid. Liquid compositions may conveniently be
encapsulated in,
for example, gelatin to give a product in dosage unit form.
Formulations for oral delivery may be formulated in a delayed release
formulation
such that the PTH peptide analogue is delivered to the large intestine.
Delayed release
formulations are well known in the art and include for example, delayed
release capsules or
time pills, osmotic delivery capsules etc.
Compositions for parenteral administration may be formulated using an
injectable
liquid carrier such as sterile pyrogen-free water, sterile peroxide-free ethyl
oleate, dehydrated
alcohol or propylene glycol or a dehydrated alcohol/propylene glycol mixture,
and may be
injected intravenously, intraperitoneally, subcutaneously or intramuscularly.
Sterile
injectable solutions are prepared by incorporating the active compounds in the
required
amount in the appropriate solvent with various of the other ingredients
enumerated above, as
required, followed by filtered sterilization. Generally, dispersions are
prepared by
incorporating the various sterilized active ingredients into a sterile vehicle
which contains the
basic dispersion medium and the required other ingredients from those
enumerated above. In
the case of sterile powders for the preparation of sterile injectable
solutions, the preferred
methods of preparation are vacuum-drying and freeze-drying techniques which
yield a
powder of the active ingredient plus any additional desired ingredient from a
previously
sterile-filtered solution thereof.
Compositions for rectal administration may be formulated using a conventional
suppository base such as cocoa butter or another glyceride.
Compositions for topical administration include ointments, creams, gels,
lotions,
shampoos, paints, powders (including spray powders), pessaries, tampons,
sprays, dips,
aerosols, pour-ons and drops. The active ingredient may, for example, be
formulated in a
hydrophilic or hydrophobic base as appropriate.
It may be advantageous to incorporate an antioxidant, for example ascorbic
acid,
butylated hydroxyanisole or hydroquinone in the compositions of the invention
to enhance
their storage life. The pharmacokinetic profiles of various formulations
containing
Ostabolin-C are detailed in Example 17.
53
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
Dosing Regimen
Administration in this invention may consist of one or more cycles; during
these
cycles one or more periods of osteoclastic and osteoblastic activity will
occur, as well as one
or more periods when there is neither osteoclastic nor osteoblastic activity.
Alternatively,
administration may be conducted in an uninterrupted regimen; such a regimen
may be a long
term regimen, e.g., a permanent regimen.
It will be understood that the dosages of compositions and the duration of
administration according to the invention will vary depending on the
requirements of the
particular subject. The precise dosage regime will be determined by the
attending physician
or veterinary surgeon who will, inter alia, consider factors such as body
weight, age and
symptoms (if any). The compositions may if desired incorporate one or more
further active
ingredients.
During the dosing regimen, the hormone can be administered regularly (e.g.,
once or
more each day or week), intermittently (e.g., irregularly during a day or
week), or cyclically
(e.g., regularly for a period of days or weeks followed by a period without
administration).
Regular administration can include once daily, once every two days, once every
three days,
once every four days, once every five days, once every six days, or once every
seven days
(once/week). Preferably PTH is administered once daily for 1-7 days for a
period ranging
from 3 months for up to 3 years in osteoporotic patients. In additional
embodiments, PTH is
administered for no less than 8 days. The present invention also encompasses
embodiments
wherein PTH is administered on a weekly basis.
Preferably, cyclic administration includes administering a parathyroid hormone
for at
least 2 bone remodeling cycles and withdrawing parathyroid hormone for at
least 1 bone
remodeling cycle. Another preferred regime of cyclic administration includes
administering
the parathyroid hormone for at least about 12 to about 24 months and
withdrawing
parathyroid hormone for at least 6 months. Typically, the benefits of
administration of a
parathyroid hormone persist after a period of administration. The benefits of
several months
of administration can persist for as much as a year or two, or more, without
additional
administration.
If desired, the PTH peptide analogue compound may be administered
simultaneously
or sequentially with other active ingredients, e.g., bone enhancing agents.
These active
ingredients may, for example include other medicaments or compositions capable
of
interacting with the bone remodelling cycle and/or which are of use in
fracture repair. Such
54
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
medicaments or compositions may, for example, be those of use in the treatment
of
osteoarthritis or osteoporosis as discussed above.
In yet a further aspect, the invention provides a method of treatment or
prevention of
bone-related diseases, in particular osteoporosis, which comprises
administering to a
mammal, including humans, in need of such treatment (a) an effective amount of
PTH
peptide analogues during a period of approximately 6 to 24 months; and (b)
after the
administration of PTH has been tenninated, an effective amount of a bone
resorption
inhibitor during a period of approximately 12 to 36 months. The bone
resorption inhibitor
can be a bisphosphonate, e.g. alendronate; or a substance with estrogen-like
effect, e.g.
estrogen; or a selective estrogen receptor modulator, e.g. raloxifene,
tamoxifene, droloxifene,
toremifene, idoxifene, or levormeloxifene; or a calcitonin-like substance,
e.g. calcitonin; or a
vitamin D analog; or a calcium salt.
As discussed above, high dose Ostabolin-C and other PTH analogs of the present
invention give a marked early bone formation and BNID response but are
associated with
stimulated bone resorption that have the potential to decrease the rate of
improvement in bone
strength by reducing the level of bone mineralization and by increasing
cortical porosity.
Lower doses of Ostabolin-C and other PTH analogs also cause increases in bone
formation at
a lower level but are free of bone resorption stimulating activity. In order
to obtain the early
benefits of the higher dose therapy while simultaneously minimizing side
effects, a suitable
treatment regimen within the present invention is sequential therapy. One
embodiment of
such a treatment regimen starts treatment with a high dose of Ostabolin-C or
suitable PTH
analogs and then after a period of time which could be 1-12 months but
preferably 3-9
months and most preferably 4-8 months converts to a lower dose which maintains
bone
formation at a lower level but does not allow stimulation of bone resorption.
Sequential
therapy could also start treatment with a low dose and then convert to a high
dose_ Such a
dosing regimen should be superior to high dose and low dose therapy because it
will allow
continued bone formation and the full maturation of the early bone fonnation
caused by the
high dose treatment without degradation by stimulated bone resorption. It will
also be
superior to high dose therapy in that the incidence of safety and tolerability
adverse events
will be reduced. Such sequential therapy will thus be an effective therapy
while
simultaneously minimizing side effects.
Such sequential therapy can be administered in all doses disclosed herein. One
suitable dosage regimen includes administering a first daily dose
subcutaneously of an
aqueous formulation in a dosage range of from 35 g to 100 g of a PTH peptide
analogue to
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
said human, and then after the termination of the first period of time
administering for a
second period of time a second dose of from 2 g to 35 g of a PTH peptide
analogue to said
human.
For human administration, preparations should meet sterility, pyrogenicity,
general
safety and purity standards as required by the FDA.
Kits
The present invention also encompasses a kit including the present
pharmaceutical
compositions and to be used with the methods of the present invention. The kit
can contain a
vial, for example, which contains a formulation of the present invention and
suitable carriers,
either dried or in liquid form. The kit further includes instructions in the
form of a label on
the vial and/or in the form of an insert included in a box in which the vial
is packaged, for the
use and administration of the compounds. The instructions can also be printed
on the box in
which the vial is packaged. The instructions contain information such as
sufficient dosage
and administration information so as to allow a worker in the field to
administer the drug. It
is anticipated that a worker in the field encompasses any doctor, nurse, or
technician who
might administer the drug, or a patient who might self-administer the
pharmaceutical
composition.
In one embodiment the kit contains a medication delivery pen that houses a
cartridge
assembly containing a vial or cartridge that has the capability of holding
about a 60 day
supply of daily doses of the pharmaceutical compositions described herein. In
additional
embodiments, the pen has the capability of holding a 1, 2, 3, 4, 5, 6, 7, or 8
week supply of
daily doses of the phannaceutical compositions described herein. In preferred
embodiments,
the pen has the capability of holding a 2 or 4 week supply of daily doses of
the
pharmaceutical compositions described herein. Such a device provides ease of
use for self-
administration of the pharmaceutical compositions described herein.
In a further embodiment, the cartridge can contain a liquid dosage of the
pharmaceutical composition, or a lyophilized dosage, which is reconstituted by
the user prior
to injection. Those of skill in the pharmaceutical arts will recognize that
medication delivery
pens, cartridge assemblies for holding a liquid or lyophilized pharmaceutical
dosage
formulation for same, and methods of lyophilizing and sealing an injectable
composition are
known in the art, as evidenced by U.S. Patent Nos. 5,334,162; 6,053,893; and
6,648,859 the
teachings of which are incorporated herein by reference.
56
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
The examples which follow are illustrative of the invention and are not
intended to be
limiting.
EXAMPLE 1 Synthesis and Purification of [LeuZ7]cyclo[G1u22 -Lys26]-hPTH-(1-31)-
NH2 I
This peptide was synthesized and purified as described in U.S. Patent No.
5,955,425,
the teachings of which are incorporated herein by reference, with Lys-Alloc
and Glu-OA11
substituted at position 26 and 22, respectively. After the addition of Fmoc-
Ser17, the peptide-
resin was removed from the column to a reaction vial (Minivial, Applied
Science), suspended
in 1.7 ml of a solution of tetrakis(triphenylphosphine)palladium(0) (0.24
mmol), 5% acetic
acid and 2.5% N-methylmorpholine (NMM) in dichloromethane (DCM) under argon,
then
shaken at 20 C for 6 hr to remove the allyl and alloc protecting groups
(Sole, N. A. et al
(1993) In Peptides: Chemistry, Structure, and Biology, Smith, J. And Hodges,
R. (Eds),
ESCOM pp. 93-94, incorporated herein by reference). The peptide resin was then
washed
with 0_5% diethyldithiocarbamate (DEDT), 0.5% NMM in DMF (50 ml), followed by
DMF
(50 ml) and DCM (50 ml). The peptide (0.06 mmot) was cyclized by shaking with
0.06 mmol
of 1-hydroxy-7-azabenzotriazole (HOAt)/0.12 mmol NMM in 2 ml DMF for 14 h at
20 C
(Carpino, L. A_ (1993) J. Am. Chem. Soc. 115, 4397-4398). The peptide-resin
was filtered,
then washed once with DMF, repacked into the column, and washed with DMF until
bubbles
were removed from the suspension. The remaining synthesis was carried out as
the linear
counterpart above except that the N-terminal Fmoc group was not removed. The
Fmoc-
peptide was cleaved from the resin with reagent K as described above. The HPLC
was
carried out as the linear counterpart above, with the Fmoc group removed prior
to the final
HPLC.
Other suitable stabilized solutions of the PTH peptide analogues that can be
employed
in the present methods can be synthesized and purified as described in U.S.
Patent Nos.
5,556,940; 5,955,425; 6,541,450; 6,316, 410; and 6,110,892 the teachings of
which are
incorporated herein by reference.
EXAMPLE 2[Leu271cyclo[G1u22 -Lys261-hPTH-(1-31)-NH2 Promotes Growth in Both
Trabecular and Cortical Bones in a Monkey Model
The peptide [Leu27]cyclo[G1u22 -Lys26]-hPTH-(1-31)-NH2 Ostabolin-C TMwas
administered daily by subcutaneous injection to gonad-intact cynomolgus
monkeys
57
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
(4/sex/group) at dose levels of 0, 2, 10 and 25 g/kg for 52 weeks. Monkeys
were 30 to 40
months of age (2.3-3.5 kg) at treatment start. Tibiae were retained for
histomorphometry
following labeling with calcein green 15 and 5 days prior to euthanasia. Bone
mass, as
measured by DXA (dual-energy x-ray absorptiometry) and QCT (quantitative
computed
tomography), was increased at the lumbar spine, femur and tibia. Changes in
vertebral BMD
(bone mineral density) translated into significant increases in bone strength.
The peptide
[Leu27]cyclo[Glu2Z -Lys26]-hPTH-(1-31)-NH2 substantially increased osseous
accretion in the
cancellous and endocortical bone compartments of the proximal tibia at all
doses. Tibial
cancellous bone volume increased by more than 50% in all the peptide
[Leu27]cyclo[G1u2Z -
Lys26]-hPTH-(1-31)-NHZ treated groups compared to controls and in the tibial
mid-diaphysis,
increases in cortical width and relative cortical area with concurrent
decreases in medullary
area were observed. Only minor increases in cortical porosity were observed at
the two
highest dose levels. The increase in bone mass appeared to be related to
increases in bone
formation and decreases in bone resorption as measured by a significant
reduction in
osteoclast surface. Increases in indices of bone formation were associated
with decreases in
indices of bone resorption (decreased bone resorption markers, decreased
osteoclast surface
area, minimal cortical porosity), consistent with the uncoupling of these
events. This
combination of anabolic and anti-catabolic actions may have significant
therapeutic value in
the treatment of osteoporosis.
Example 3 - Pre-Clinical Cortical Porosity Data
Comparative data regarding increase in cortical bone porosity in monkey
subjects
using Ostabolin C at a variety of doses and using the prior art PTHs 1-34 is
shown below.
Molecule Model Site M/F Dose % Cortical Study
Porosi Reference
Ostabolin-C Gonad Tibial M Control 3.4t0.89 Zelos
intact young Mid- 2 pg/kg/day 4.2 0.29
Cynomolgus Diaphysis 10 5.1 1.08
monkeys pg/kg/day 8.0 5.54
treated daily 25
for 12 pg/kg/day
months
Gonad Tibial F Control 2.0 0.32 Zelos
intact young Mid- 2 pg/kg/day 2.5 0.41
Cynomolgus Diaphysis 10 2.6t0.85
monkeys pg/kg/day 3.2t0.87
treated daily 25
for 12 /k /da
58
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
months
Ostabolin-C Gonad Tibial M Control .3.5t1.18 Zelos
intact young Mid- 10 3.7t0.70
Cynomolgus Diaphysis pg/kg/day 5.8 1.82
monkeys 25 16.4 7.14*
treated dilay pg/kg/day
for 6 weeks 80
pg/kg/day
Gonad Tibial F Control 3.3t0.90 Zelos
intact young Mid- 10 3.2 0.97
Cynomoigus Diaphysis pg/kg/day 4.0 1.25
monkeys 25 10.6 0.35
treated dilay pg/kg/day
for 6 weeks 80
29/kg/day
PTH 1-34 OVX adult Humerus F Control -5.0 Burr et al.,
Cynomolgus Mid- 1 pg/kg/day -15.0* JBMR
monkeys Diaphysis 5 pg/kg/day -25'' 16:157-165,
treated daily 2001
for 18
months
OVX adult Femoral F Control 6.7 0.7 Sato et al.,
Cynomolgus Neck 1 pg/kg/day 8.5 0.8* JBMR
monkeys 5 pg/kg/day 8.9 0.6* 19:623-629,
treated daily 2004
for 18
months
Example 4- Pre-Clinical Toxicity Data
The below table demonstrates that the prior art PTH, 1-34, teriparatide,
Forteo , is
more nephrotoxic than Ostabolin-CTM, the difference possibly being linked to
the different
hypercalcemic states. As shown below, PTH-(1-34) induces a mineralizing
nephropathy in
monkeys and possibly rats. A NoAEL was not established for the monkey.
Ostabolin-CTM
was nephrotoxic only in monkeys and a NoAEL was established. Ostabolin-CTM is
at least 4-
fold safer than PTH-(1-34).
TERIPARATIDE, FORTEO OSTABOLIN C DIFFERENCES
Study Doses g/kg Results Doses Results
pg/kg
Toxicity, 12 0, 0.5, 2, 10 Free Ca increased 0, 2, Variable free Ca: Ostabolin-
C not
mth monkey all doses; tubulo- 10, 25 increased week 31, bypercalaemic and
interstitial decreased week >4-fold less
nephritis all doses; 52. tubulo- nephrotozic than
serum neutralising interstitial PTH-(1-34)
antibodies nephritis mid and
detected all doses high dose. Bone
most frequently hypertrophy all
high dose at wk doses. NoAEL 2
50 pg/kg
NoAEL
59
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
<0.5pg/kg
EXAMPLE 5 Clinical Study of OSTABOLIN-CTM
A four month Phase II clinical study was undertaken to investigate the safety,
tolerability and efficacy of Ostabolin-CTM in post-menopausal women with low
bone mineral
density (BMD). Comparative data from this study demonstrates that the use of
Ostabolin-
CTM has many advantages over the current therapy, use of 1-34 PTH,
teriparatide, Forteo .
The clinical protocol is a 16-week phase II randomized, double-blind, placebo-
controlled,
parallel group, dose finding study to investigate the safety, tolerability and
efficacy of
Ostabolin-CTM in post-menopausal women with low bone mineral density (BMD). In
this
study, 261 patients underwent four months of daily dosing of placebo and four
active groups.
The active groups included daily administration of Ostabolin-CTM in doses of
7.5, 15, 30, and
45 g. Ostabolin-CTM is formulated as a clear, colorless liquid provided in
pre-filled syringes
and injected subcutaneously (SC). Subjects self-administer SC 0.1 mL
injections of their
assigned dose of Ostabolin-CTM 7.5, 15, 30, and 45 g or placebo daily for 16
weeks in
rotating quadrants of the abdomen. The subjects were post-menopausal women
(for at least 5
years) with moderate osteoporosis.
The key endpoints for the study include change in mean BMD at the lumbar
spine, as
assessed by dual energy x-ray absorptiometry (DEXA), and measured by change
from the
Baseline visit. The Baseline visit is the first visit of the patient, before
undergoing any
treatment. Secondary efficacy endpoints include the following, as measured by
change from
Baseline visit:
DEXA: Bone formation and resorption markers:
= Mean femoral neck BMD = Serum osteocalcin
= Mean trochanter BMD = Serum amino terminal pro-peptide of
= Mean total hip BMD type 1 pro-collagen (P1NP)
= Mean radial BMD (distal and = Bone specific alkaline phosphatase
midshaft) (BSAP)
= Bone mineral content (BMC) = Serum C-telopeptide (CTx)
= Bone area = Serum N-telopeptide (NTx)
Other measurements:
= Lateral thoracic, lumbar spine and left antero-posterior hip radiographs
= Height
EXAMPLE 6 Clinical Results - Effects of Low Dose (7.5, 15, and 30 pg)
Ostabolin-CTM
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
Administration of a daily dosage of 7.5, 15, and 30 g of Ostabolin-CTM as
described
above in Example 5 demonstrates robust bone anabolic effects at multiple sites
in the body,
including the spine, the hip, and the wrist without the concomitant negative
effects previously
seen with the use of prior art PTHs. The unprecedented BNID increases at the
mid-radius and
the lower incidence and severity of hypercalcemia make these highly attractive
doses.
As shown in Figure 1, administration of 7.5, 15, and 30 g daily dosages of
Ostabolin-CTM over a course of 15 weeks results in an increase in lumbar spine
BMD.
Figures 3, 4, and 5 demonstrate mild B1VID increase in hip, femoral neck, and
trochanter
BMD following administration of Ostabolin-CTM for 15 weeks.
Figures 6 and 7 demonstrate that daily administration of 7.5, 15, and 30 g of
Ostabolin-CTM has an unexpectedly positive effect on cortical bone,
specifically the wrist (the
distal and mid-shaft radius). There were statistically significant effects at
the mid-radius at
daily dosages of 7.5, 25, and 30 g with no negative effect of bone
resorption. Historically,
PTH has been known to increase bone resorption, which leads to increased
cortical porosity,
and decreased BMD in radius cortical bone. Neer et al., 2001. As described in
Neer, the
administration of prior art Forteo PTH 1-34 led to a decrease in BMD
(increased cortical
porosity) in the distal and mid-shaft radius as compared to placebo. In
contrast, the dosages
and formulation of the present invention, namely administration of 7.5, 15,
and 30 g
Ostabolin-CTM, actually increases cortical BMD in the distal and mid-shaft
radius as
compared to both placebo and to teriparatide, Forteo . This is an
unprecedented finding,
demonstrating a statistically significant difference from placebo for 3 active
doses (7.5, 15,
and 30 g).. Figures 8-13 demonstrate the effect which the PTHs of the present
invention
have on bone formation and bone resorption markers. The bone formation markers
include
P1NP, osteocalcin, and BSAP and the bone resorption markers include NTx and
CTx. As
compared to placebo, the bone formation markers have a greater % change when
Ostabolin-
CTM is administered at 15 and 30 g.
The bone resorption markers in Figures 11-13 demonstrate that although there
is some
increase in bone resorption following the administration of Ostabolin-CTM,
this increase is
less than that which follows administration of the prior art teriparatide,
Forteo PTH.
Daily dosages of 7.5, 15, and 30 g Ostabolin-CTM has also been shown to have
a
much lower incidence of hypercalcemia as compared to PTHs known in the art.
Figure 14
demonstrates that there was no notable difference from placebo on the per cent
of abnormal
serum calcium for doses of Ostabolin-CTM up to and including 30 g. In
comparison,
teriparatide, Forteo is shown to have a much higher effect at similar doses.
For patients
61
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
receiving Forteo , hypercalcemia was seen at least once in 11 % of the 20 g
group subjects
and in 28% of the 40 g group subjects, as compared with 2% in the placebo
group. Neer et
al., 2001. The administration of low doses of the PTH peptides of the present
invention (7.5,
15, and 30 g) resulted in no significant increase in the incidences of
hypercalcemia as
compared to placebo. Hypercalcemia was seen at least once in 5% of the placebo
group and
in the group being administered 30 g doses, resulting in no net increase.
This is in
comparison to the 11% seen with Forteo administered at 20 g.
Accordingly, the above results demonstrate that administration of Ostabolin-
CTM at
7.5, 15, and 30 g daily dosages provides many advantages over the
administration of
Forteo at 20 g. The unexpected results include increased cortical B1VID in
the distal and
mid-shaft radius as compared to placebo, less bone resorption than prior art
PTH, and lower
incidence and severity of hypercalcemia, while maintaining anabolic bone
growth as
measured by increased BMD at a variety of sites, including spine and hip.
EXAMPLE 7 Pre-Clinical Results - Effects of High Dose (45 pg) Ostabolin-CTM
Administration of a daily dosage of 45 g of Ostabolin-CTM has demonstrated an
unprecedented ability to build bone at different sites, including the spine
and hip, with early
onset of effect in combination with only a mild hypercalcemia signal. This is
an
improvement over the prior art teriparatide, Forteo 1-34 PTH.
Figures 1 and 2 demonstrate that administration of 45 g Ostabolin-CTM leads
to an
increase in BMD in the lumbar spine. Figure 2 shows the increase in lumbar
spine BMD
with administration of 20 and 40 g Forteo .
Figures 3, 4, and 5 and the table below demonstrate that a daily dosage of 45
g
Ostabolin-CTM has a positive effect on bone formation at the hip, femoral
neck, and
trochanter. This is an unprecedented finding, demonstrating a statistically
significant and
clinically meaningful benefit at 45 g at 15 weeks. The table below
demonstrates the change
in hip, femoral neck, and trochanter BMD, comparing the administration of
teriparatide,
Forteo (20 g) over a course of at least 12 months versus Ostabolin-CTM (45
g) at 15
weeks. As shown below, for hip and trochanter, administration of 45 g
Ostabolin-CTM
achieved results in 15 weeks similar to the results obtained with
administration of Forteo over
a course of at least 12 months. Regarding femoral neck, Ostabolin-CTM shows a
much gteater
increase in BMD in a shorter period of time.
62
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
Ostabolin-C ERIPARATIDE,
45 gg For 15 Weeks ORTEO
0 g for at least 12
onths
ean % Change In 1.44 1.70
otal Hip
ean % Change In 2.75 1.54
emoral Neck
4ean % Change In 2.24 .68
rochanter
Figures 8-13 demonstrate the effect which the PTHs of the present invention
have on
bone formation and bone resorption markers. The bone formation markers include
P1NP,
osteocalcin, and BSAP and the bone resorption markers include NTx and CTx. As
compared
to placebo, the bone formation markers have a greater % change when Ostabolin-
CTM is
administered at 45 g. There is a robust effect in the increase in the bone
formation markers
when Ostabolin-CTM is administered at 45 g. The bone resorption markers in
Figures 11-13
demonstrate that although there is some increase in bone resorption following
the
administration of Ostabolin-CTM, this increase is less than that which follows
administration
of the prior art teriparatide, Forteo PTH.
Accordingly, the above results demonstrate that administration of Ostabolin-
CTM at 45
g daily dosages provides many advantages over the administration of rhPTH 1-34
teriparatide, Forteo at 20 and 40 g . The unexpected results include
increased BMD in the
spine and hip, with less bone resorption and lower incidences of hypercalcemia
than prior art
PTH.
EXAMPLE 8 Pharmacokinetic (PK) Evaluation of Ostabolin-C
The objective of this portion of the study was to evaluate the
pharmacokinetics of
Ostabolin-C under steady state conditions when given subcutaneously (sc) once
a day to post-
menopausal female subjects with low bone mineral density.
This study was a Phase H, multicenter, randomized, double-blind, placebo-
controlled,
parallel group dose-finding study in post-menopausal female subjects. After
Screening
procedures and a 2-week placebo run-in phase, subjects were to be dosed once a
day for 16
weeks with either Placebo or Ostabolin-C (7.5, 15, 30 or 45 g). A subset of
subjects from
63
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
all treatment groups had blood collected for measurements of Ostabolin-C in
order to
determine PK parameters and compare them to prior studies.
The full study duration of the study was 22 weeks, which included a 6-week
screening
period involving a 2-week placebo run-in and then 16 weeks of treatment. The
subset of
subjects for this component of the study was treated the same as all other
subjects with the
exception of the additional blood collections at baseline and Week 12.
DATA HANDLING AND PK Analyses
All of the values from the Placebo subject except one (Baseline 2 hour time-
point)
were below the assay level of detection (i.e., 10 pg/ml). With very limited
exceptions, all
values from Placebo subjects in prior trials have also been below the levels
of detection.
Thus, this one value was considered to be a laboratory error and PK parameters
for this
placebo subject were not calculated.
No samples were obtained for Pre-dose at either Baseline or Week 12. In all
prior
studies, pre-treatment values have been below the levels of detection and 24-
hour time points
at doses of 40 ug and below have been below the levels of detection. Thus, no
observable
values were anticipated and for calculating PK the pre-treatment values for
Baseline visit
were set to zero.
For pre-dose at Week 12, it was also anticipated that the values would be
below the
levels of detection based on prior studies and that the 24-hour post dose
value would verify
this. Only two 24-hour time-point values were above the level of detection;
i.e. 24 hours post
dosing at Baseline for Subject 030-003 and 24 hours post dosing at Week 12 for
Subject 030-
0004. The values of both of these 24-hour time-points were marginally above
the assay level
of detection. Also, both of these subjects had values below the levels of
detection for both
the 4 hour and 6 hour time-points after dosing on the prior day. Thus, these
values are most
likely artifacts and not real values. No valid 24 hour sample for subject 032-
0001 at Week
12 was obtained. However, the 6 hour time point after dosing at Week 12 was
below the
levels of detection, and thus the value for 24-hours was assumed to be also
below the level of
detection to estimate the AUC(0-24) value. Thus, for calculating PK
parameters, the Pre-
dose values for Week 12 were also set to zero.
The pharmacokinetic parameters that were estimated at Baseline and Week 12 are
as
follows:
64
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
- The area under the drug concentration-time curve from time zero to time 4
hours
(AUC(o-4))
- The area under the drug concentration-time curve from time zero to time 24
hours
(AUC(o-24))
- The maximum observed drug concentration (C.)
- The time of the maximum drug concentration (T
Since so few subjects were included in this subset of subjects and the time
points used
for collections were limited, additional PK parameters were not calculated.
AUC values were estimated by a simple summation of trapezoidal areas from each
time period. Data from each dose group were summarized using simple statistics
on an
Excel spreadsheet; i.e., average (AVG) and Standard Deviation (STD). It
should be noted
that particularly with the lower doses and associated low blood levels and at
late time points,
those values just above versus just below the assay limits of detection can
have a
disproportionate impact to AUC calculations. This adds to that variability of
the calculated
numbers.
PK VALUES
The following table summarizes the estimated PK parameters.
BASELINE PK PARAMETERS
Site/sub Date Cmax Tmax AUC 0-24 AUC 0-4
Dose Group = 7.5 u
006-0063 18-Oct-05 20.7 0.25 11.71 11.71
030-0005 19-Oct-05 27.75 0.25 16.25 16.25
038-0010 11-Jan-06 33.15 1.00 99.67 88.35
AVG 27.20 0.50 42.54 38.77
STD 6.24 0.43 49.52 43.00
Dose Group = 15 u
030-0003 10-Aug-05 30.6 0.50 65.98 65.98
032-0001 13-Jun-05 57.54 0.25 82.31 82.31
038-0001 8-Nov-05 46.59 0.25 74.38 74.38
AVG 44.91 0.33 74.22 74.22
STD 13.55 0.14 8.17 8.17
Dose Group = 30 ug
006-0144 16-Dec-05 63.93 0.50 104.11 104.11
Dose Grou = 45 u
006-0141 13-Dec-05 114.54 0.50 469.78 337.16
030-0004 16-Nov-05 233.33 0.50 370.47 344.39
AVG 173.94 0.50 420.12 340.77
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
Since there are so few subjects that actually participated in this part of the
study and
since the values for Cmax, Tmax, and AUC appeared to be very similar for both
Baseline and
Week 12, the values for all times were averaged to obtain another estimate of
these
parameters; see table below. Similar PK values for Baseline and Day 7 when
steady state
kinetics should have been have reached equilibrium have been seen in two
previous Phase 1
studies involving this dose range.
WEEK 12 PK PARAMETERS
Sitelsub Date Cmax Tmax AUC 0-24 AUC 0-4
Dose Group = 7.5 ug
006-0063 17-Jan-06 21.04 0.25 9.10 9.10
030-0005 11-Jan-06 37.84 0.25= 28.83 28.83
038-0010 5-Apr-06 40.38 0.25 64.83 64.83
AVG 33.09 0.25 34.25 34.25
STD 10_51 0.00 28.26 28.26
Dose Group = 15 ug
030-0003 2-Nov-05 41.98 0.25 164.36 68.69
032-0001 14-Sep-05 33.33 0.25 54.29" 54.29
038-0001 24-Jan-06 69.33 0.50 110.80 110.80
AVG 48.21 0.33 109.82 77.93
STD 18_79 0.14 55.04 29.37
Dose Group = 30 ug
006-0144 13-Mar-06 76.25 0.25 153.89 142.45
Dose Group = 45 ug
006-0141 15-Mar-06 46.75 0.25 214.03 70.73
030-0004 8-Feb-06 87.1 0.25 128.37 128.37
AVG 66.93 0.25 171.20 99.55
*Note -no valid 24 hour sample jor subject 032-0001 was obtained but since the
6 hour time point was below the levels of
detection, the value jor 24-hours was assumed to be also below the level
ofdetection to estimate the AUC(0-24) value.
ESTIMATED PK PARAMETERS - AVERAGE OF BASELINE AND WEEK 12
DATA
Cmax Tmax AUC 0-24 AUC 0-4
Dose [NJ= pg/ml Hours pg'hr/ml 'hr/mi
7.5 3 Mean 30.14 0.38 38.40 36.51
Std 8.38 0.31 36.35 32.63
15 3 Mean 46.56 0.33 92.02 76.07
Std 14.76 0.13 40.23 19.38
30 1 Mean 70.09 0.38 129.00 123.28
Std 8.71 0.18 35.20 27.11
45 2 Mean 120.43 0.38 295.66 220.16
Std 80.25 0.14 153.37 141.28
'Note: N= number ofsubjects; data from both Baseline and Week 12 combined,
each subject had two values for each
parameter.
Since T. seems to be dose independent in this study as well as in previous
studies,
the Tma,, from all doses in this study determined at Baseline and Week 12 were
averaged to
obtain an overall estimated value of 0.34 hours with a STD of 0.21 hrs.
66
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
Discussion
The very limited numbers of subjects involved in this study limit the
statistical
confidence in the conclusions drawn from the data in this study. However, the
data are
basically consistent with prior studies.
As seen in prior studies, there was no evidence of accumulation. The PK
parameters
after 12 weeks of dosing were very similar to those on Day 1 at Baseline.
The Tmax was independent of dose and the overall average form all doses and
times
was 0.34 hours (srd = 0.21 hrs).
The Cmax and the AUC values increased with dose. There is a rough dose
relationship with Cmax and AUVC values in the averaged data.
EXAMPLE 9 Treatment Of Renal Osteodystrophy
End stage renal disease is invariably associated with bone disease, known as
renal
osteodystrophy (ROD) (for account of pathogenesis see Primer on Metabolic Bone
Diseases
and Disorders of Mineral Metabolism Chapter 74). ROD may exist in a high
turnover form
characterized by high circulating levels of PTH (secondary
hyperparathyroidism) and
overactive bone tissue. This condition is frequently associated with bone
pain, muscle
weakness, extraskeletal calcification and deformities and growth retardation
in children.
Reduction in PTH levels is considered necessary to treat these problems. The
low tumover
form of the disease, also known as adynamic bone disease, is characterized by
normal or low
circulating levels of PTH and is increasing in incidence due to the increasing
use of therapies
to effectively control secondary hyperparathyroidism such as Vitamin D
sterols, calcium
based phosphate binding agents and calcimimetic dr gs. Histologically the bone
surfaces are
quiescent with little or no osteoblast cellular activity. Clinical
consequences of this
histological state include increased risk of fractures and growth retardation
in prepubertal
children.
Adynamic bone disease is currently difficult to treat. The use of parathyroid
hormone is
contraindicated since reducing parathyroid hormone levels is one of the
important goals of
the therapies that lead to adynamic disease. Hypercalcemia is a frequent
complication of
current therapeutic strategies and this would be exacerbated by the use of
exogenous PTH.
Restoration of normal levels of bone formation activity is therefore difficult
to achieve in this
67
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
setting and there is an unmet need for effective therapy. Agonists of the PTH
receptor,
exemplified by cyclized or linear PTH (1-31) analogs but also including other
cyclic and
linear analogs of smaller size and analogs of PTHrP have been shown to
increase bone
formation but do not have the propensity to stimulate bone resorption that is
seen with other
PTH fragments and with the naturally occurring hormone. PTH receptor agonists
of this type
may be able to stimulate osteoblastic function and bone formation and thus
effectively treat
adynamic bone disease without exacerbation of the risk of hypercalcemia. The
use of low
doses of these agents may be particularly effective in prevention and
treatment of adynamic
bone disease to provide restoration of normal osteoblast activity with
ininimal bone
resorption stimulating activity. Specific treatment scenarios in which PTH
receptor agonists
of this type are used in combination with calcimimetic dr gs, Vitamin D
sterols or other
agents known to increase the occurrence and/or severity of adynamic bone
disease to prevent
this occurrence or exacerbation could be created.
PTH receptor agonists could be used in dialysis patients at increased risk of
developing
adynamic bone disease to prevent the occurrence of adynamic bone disease.
PTH receptor agonists of the type described above could also be used to treat
patients with
osteoporosis and renal disease who have a particularly high risk of fracture
due to adynamic
bone disease.
Example 10 Rat Oncogenicity Study
Prior art PTHs cause osteosarcomas in animals if administered over a course of
two
years. The PTH peptides of the present invention, including Ostabolin-CTM and
PTH 1-30,
are administered subcutaneously to rats for 104 weeks at doses of 0.5, 5, 30,
and 50
g/kg/day. The test article is administered subcutaneously. Analysis of the
incidence and
morphology of tumours following administration may demonstrate that
administration of the
PTH peptides of the present invention over the course of two years may lead to
lower
incidences of osteosarcomas as compared to administration of a similar
duration of prior art
PTH peptides. This difference could be due to the different amino acids
sequences and/or to
the different signalling pathways activated by the PTH molecules.
EXAMPLE 11 COMPARISON OF OSTABOLIN-C AND FORTEO
The below table illustrates a comparison of Ostabolin-C with Forteo data
derived
from Deal et al., (2005) J. Bone Min. Res. 20, p. 1905-1991. As shown below,
bone
68
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
resorption stimulation with 30 g Ostabolin-C is approximately 50% of the
expected effect of
20 g Forteo despite similar effects on LS-BMD. The effect of 30 jig Ostabolin-
C on serum
calcium and incidence of hypercalcemia are both diminished. The effect of 45
jig Ostabolin-
C on bone formation and BMD is greater than the effect of 20 jig Forteo
despite similar
effects on bone resorption and calcium. Both 30 g and 45 g Ostabolin-C doses
have an
improved therapeutic window compared to Forteo. These results are also
represented in
Figures 15, 16, and 17.
Ostabolin-C 30 jig Ostabolin-C 45 g Forteo 20 g
(4 months) (4 months) (6 months)
LS-BMD (%) 3.6 (4.51) 5.2 (5.9 ) 5.2
FN-BMD (%) -0.05 2.75 1.02
TH-BMD (%) 0.06 1.45 0.6
P1NP ( g/L) 50.0 79.5 71
CTx (pM/L) 1400 2900 3300
Mean [Ca](mmol/L) 0.040 0.070 0.075
Pts>2.75 mmol/L 1(0) 6(0) 5(2)
(sustained)
EXAMPLE 12- ANALYSIS OF SIDE EFFECTS WITH
DOSE ADMINISTERED BASED ON PATIENT WEIGHT
To confirm that g/kg exposure is a valid way of analyzing the effect of
Ostabolin-C,
the cumulative response and side effects profiles were assessed. The
cumulative response
was analyzed by looking at the proportion of patients who achieved a>3% change
in lumbar
spine BMD with increasing Ostabolin-C exposure. This analysis illustrates that
assessing the
effect of Ostabolin-C exposure on an individual patient basis follows a
similar pattern to the
linear regression analysis from dose groups created on the basis of increasing
g/kg exposure
(see figure 23-25 on lumbar spine). Similar trends were also demonstrated for
side effect
profiles as is illustrated in a cumulative incidence analysis, shown in
figures 31, 32, for
headache, nausea and hypercalcemia. Increasing exposure was generally
associated with a
higher incidence of side effects and hypercalcemia.
1 Data from subset with LS-BMD T score <-2.5.
2 Inferred from graphed data in Deal et at. (2005) J. Bone Min. Res. 20, p.
1905-1991.
69
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
The linear progression of efficacy endpoints coupled with the association of
higher
side effect rates with increased exposure raised the possibility that if the
exposure to
Ostabolin-C could be maintained between narrower limits than would be produced
by
administration of a single daily dose to the entire cohort, the observed
clinical profile might
be superior. Such a strategy could reduce low efficacy responses by
eliminatirig low
exposure and also reduce unwanted or excessive responses by eliminating high
exposure.
EXAMPLE 13 DOSE OPTIMIZATION -
TWO DOSE STRENGTHS WITH WEIGHT CUT OFF
An analysis of the four month data from the Phase II Trial of the sub-
cutaneous
Formulation of Ostabolin-C as described in Example 5 demonstrates that a high
dose of
Ostabolin-C has efficacy greater than Forteo, particularly at the hip, but
also presents with
some hypercalcemia. The data from Example 5 also demonstrate that the low dose
of
Ostabolin-C has efficacy similar to Forteo, and without hypercalcemia. Based
on these
results, we conducted an analysis to identify a dose that combines the
favorable clinical
features of both Phase H high and low doses, resulting in a dose that both has
superior
efficacy, and is well tolerated.
One method of reducing the range of dose exposures in a study population is to
utilize
two dose strengths with a single weight cutoff (i.e. all patients who weigh
less than the cutoff
receive the low dose (30 pg), whereas all those above the weight cutoff
receive the high dose
(45 pg). The impact of this dose optimization strategy on Ostabolin-C exposure
is illustrated
in figures 33-36.
In contrast to the actual single dose exposure distribution observed in the
table shown
below, the effect of a weight cutoff at 68 kg reduces the spread of exposures
in the 30 and 45
g dose groups.
Exposure Ranges for Different Doses
Treatment
Group N Mean Minimum Maximum
Control 59 0.00 0.000 0.00
7.5 g 49 0.12 0.07 0.16
15 g 48 0.25 0.17 0.33
30 g 51 0.48 0.30 0.69
45 pg 54 0.71 0.48 0.99
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
The effect of the dose optimization strategy was evaluated by performing
subgroup
analyses using actual data from the 30 and 45 pg cohorts of the study (i.e.
creation of a new
subgroup by combining all patients below the weight cutoff from the 30 g dose
group with
all patients above the weight cutoff from the 45 g dose group, shown in the
table below).
A variety of new subgroups were created out of the patients who participated
in the
Phase II clinical trial. The groups from Phase II are shown above, where each
group received
a single dose, either 7.5 g, 15 g, 30 g, 45 g, or placebo. The 30 g and
45 g dose
groups were re-grouped, according to the weight of the patient, and over 100
new sub-groups
were created. This approach of creating new sub-groups out of dosages which
are relatively
close to each other, is a novel way of conducting Phase II studies.
For the 105 patients who were originally given 30 or 45 g, new groups were
created
by utilizing multiple weight cut-offs in increments of 0.5 kg. For example,
one new group
had the weight cut-off at 59 kg, meaning that every patient from the original
low dose (30 g)
group who weighed less than 59 kg and every patient who weighed more than 59
kg from the
original high dose (45 g) group formed a new cohort. This new cohort
represents a
dose/weight cut-off of 59 kg, a group in which patients weighing less than 59
kg receive the
low dose (30 g) and patients weighing more than 59 kg receive the high dose
(45 g).
Another example can be illustrated using a weight cutoff of 68 kg. Such a
group would
include all those patients who weigh less than 68 kg and who originally
received the low dose
(30 g) as well as all those patients who weigh more than 68 kg and who
originally received
the high dose (45 g). By using this method, over 100 separate cohorts were
created by
altering the dose/weight cut-off in increments of 0.5 kg. A summary of the
data generated by
these new dose/weight cut-off cohorts, using 68 kg and 59 kg as the
dose/weight cut-off, is
shown in the table below.
68 59
30 ug cut cut 45 ug
Mean LS-BMD 3.72 4.65 4.95 5.59
LS-BMD 3% 59.6 78.0 80.8 79.5
LS-BMD 6% 19.1 29.3 28.8 36.4
FN-BMD -0.06 0.29 1.5 3.0
TH-BMD 0.07 0.40 0.69 1.6
P 1 NP 117.2 132.1 143.5 160.5
CTx 30.6 32.1 57.5 60.3
71
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
HyperCa >2.65 4.5 12.8 22.0 27.3
HyperCa >2.75 2.3 7.7 6.0 9.1
Sitting HR 2.4 3.1 4.1 4.0
Nausea 25.5 31.9 31.7 29.1
Headache 19.6 12.8 20.6 21.8
N+H 7.8 10.6 12.7 12.7
The table above illustrates the actual clinical profile obtained for two
dose/weight
cutoffs (59 kg which produces a higher exposure profile since more patients
receive the
higher dose and 68 kg in which more patients receive the lower dose). The
profile described
in the table indicates that these two cutoffs produce an intermediate clinical
profile between
30 and 45 g. One problem with simple subgroup analysis is that endpoints with
a small
number of events can skew the observed profile because of large changes in
incidence when
one of these events moves from one side of the weight cutoff to the other.
Therefore, the
dose/weight cutoff methodology was applied systematically to the data from the
30 and 45 g
dose groups starting at the lowest weight patient in the study (corresponds to
the 45 g
profile because everyone receives the high dose) and increasing the cutoff by
0.5 kg
increments through 100 kg (corresponding to the 30 g profile because everyone
receives the
low dose). The multiple cutoff profiles thus obtained when graphed can then
smooth out the
effect of individual events as illustrated for the effect on hypercalcemia.
The effect of weight
cutoff on clinical profile can then be modeled to eliminate the skewing
effects of individual
data points as illustrated in figure 37-40. This approach has been applied to
primary and
secondary endpoints in the study.
Several interesting and unexpected conclusions can be drawn from this
analysis. The
first is that change in biomarkers (both of formation and resorption) is
highly sensitive to
change in weight cutoff between 60 and 70 kg, as shown in figures 41-43, 49-
50.
Although these changes appear coincident, the ratio of P1NP to CTx also
displays a
marked shift in this cutoff range (see figures 41-43, 49-50), reflecting the
already established
bone formation (P1NP) effect at lower doses. The change in incidence of
hypercalcemia and
in serum calcium also show large increases in this cutoff range, further
strengthening the link
between the stimulation of bone resorption and the emergence of hypercalcemia
in the
clinical profile.
By comparing the modeled curves for different clinical variables it is
possible to
establish whether or not Ostabolin-C will differentially affect clinical
parameters if a specific
72
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
weight cutoff is applied to the dosing regimen. A differential effect can be
inferred if the
sigmoidal plots of two different clinical variables are not overlapping, as
noted with change
in incidence of hypercalcemia and lumbar spine BMD responder rate and between
change in
serum calcium and change in lumbar spine BMD, as shown in figures 44-47, 51.
These
comparisons illustrate that a cutoff at approximately 68 kg will provide
maximal BMD
benefits with minimal impact on serum calcium metabolism.
The selection of a definition for BMD responder is different for different BMD
sites
because the magnitude of change at each site is also different. Different
definitions of BMD
`response' have been evaluated based on a separate analysis that demonstrated
that actively
treated patients had greater BMD changes than placebo at every levels of BMD
change
(figures 46-48).
Therefore response definitions for the lumbar spine, femoral neck and total
hip BMD
were selected that produced approximately equal numbers of responders for each
BMD rate
(?3% for lumbar spine and 2!:0% for total hip and femoral neck BMD).
Comparison of
modeled data for three different BMD parameters illustrates that progressively
higher dose
exposures are required to affect total hip and femoral neck BMD compared to
lumbar spine
BMD. This will enable therapy to be tailored to the individual needs of the
patient (see figure
48).
An important benefit of this modeling is that it is possible to project a
clinical profile
for a specific weight cutoff/dose exposure window with a high degree of
accuracy since the
effect of individual events is eliminated. The anticipated hypercalcemia and
lumbar spine
BMD profile for a 68 kg cutoff is illustrated in the table below, which
compares the modeled
hypercalcemia and lumbar spine BMD changes and the actual Phase II data for
the 68 kg
weight cutoff. It will also be possible to model the effects of even narrower
exposure
windows by using multiple dose cutoffs and different doses.
68 68
cut modeled
Mean LS-BMD 4.65 4.57
LS-BMD 3% 78.0 76.5
HyperCa >2.65 12.8 14.8
HyperCa >2.75 7.7 7.6
The analysis of the effects of g/kg exposure to Ostabolin-C has yielded a
simple but
powerful approach to dose optimization which will provide superior efficacy
responses while
73
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
minimizing the emergence of adverse effects. It is anticipated that this
dosing approach will
provide additional flexibility to the use of PTH analogs and a superior
clinical profile for the
treatment of osteoporosis in males and females.
These dose optimization benefits illustrated above for Ostabolin-C will also
apply to
other therapeutics, including the anti-sclerostin Mab, inhibitors of negative
regulators of the
Wnt signaling pathways, activin receptor agonists, therapeutics whose bone
formation effect
is mediated by the action of PTH on its receptor, including PTH, full-length
and fragments
thereof, PTH analogs, PTHrP, and PTHrP analogs, and calcium receptor
antagonists which
stimulate endogenous PTH production, such as those that act as agonists of the
PTH receptor,
including PTH, full-length and fragments thereof, PTH analogs, PTHrP and
analogs thereof.
EXAMPLE 14 OSTABOLIN-CTM INHALATION POWDER PHASE I
CLINICAL TRIAL
A Phase I clinical study was undertaken using Ostabolin-CTm Inhalation Powder
(OCIP) to establish an MTD in post-menopausal women, to compare its PK profile
with
Phase II sub-cutaneous doses, and to evaluate biological activity with cAMP
and biomarkers
of bone turnover. The OCIP was administered using the Nektar T-326 dry powder
inhaler
(DPI), which is well-accepted by osteoporosis patients in focus groups. An
ascending dose
tolerance was used. Each cohort was randomized and included 6 active patients
and 2
placebo patients. The patients were post menopausal healthy females that were
older than 40
years of age and had no known history of osteoporosis or other bone disease.
The primary measurements were taken at clinical laboratories where patient
surveys
were taken and the patient could describe any adverse events. Additionally the
patients' vital
signs were taken with Holter monitoring of EKGs and Spirometry.
Pharmacokinetics, such
as Ostabolin-C blood levels, PK parameters after a single dose and after 7
days of daily
dosing were also measured. Pharmacodynamics were also monitored for changes in
bone
markers including P1NP, Osteocalcin, NTx, CTx, and urine cyclic AMP.
The overall design of the trial is as follows:
SD SA SA SA
' aiiy'd ,sing 28 d ~ i (
Cohort I
SD
Cohort 2 R ily ing 28 days
Week 1 2 3 4 5 6
74
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
The table below shows the dosing summary.
Dosing Summary
Daily Dosing Formula Fill Wt Number of
Dose Schedule Strength (mg) Capsules
~x~rx ; ax~
0 1 m~ SD ~c~;.xÃ~at .~~ . , ss~'a: yE . : '~ sv'" p
~Coh~~1 28 da,y~ 4%:~~ 2 5= Ã 1 t;~ :.
Cohort 2 0.2 mg SD + 28 days 4% 5.0 1
3
Cohort 4 0.4 mg SD + 28 days 4% 5.0 2
.
Coho ;5 p~6 ~ ~ SD:~+ 28~ria:ys ~ , ~F `~~ : .-; ~6t0~
.. = .~..r. _. .-... ~s.__ ~ ~ - ,..~: =
Cohort 6 0.8 mg SD + 28 days 16% 5.0 1
'4L A2Yy~ =I
+~28 as 1 s% ~' ~SKp ~~AF ~ .. 2
Cohort 8 0.8 mg SD only 4% 5.0 4
~ro;
~Cohort 9 12-rn SL1 o ;nly
SC 3 0 pg SD comparator - - -
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
The chart below demonstrates that administration of OCIP results in PK
parameters
similar to those achieved when Ostabolin-C is administered sub-cutaneously.
Day I Normalized Doses*
OCIP ,
~~PK armeter~; ~300"u9 . 600 N9
~._. ~~:_ ~.... Q~. ~, _~ .~
..
AUC(O-4) 30.4 (+/-20.0) 59.6 (+/-41.0) 41.2 (+/-26.7)
~ ~"..`.
7~,.x t~ .. k '.i 7
GmaX' q ~'':' ~7x 50 RM+/ 25*93 ) F' - 1 OrC).6 (~+f=5a1 *)~ i .74 7+/ 348;).
X
Tmax 0.24 (+/-0.08) 0.22 (+/-0.11)
0~69 (+/-0 94)~ MW W 0 62J (+~ ~0 g2) ;~
*AUC and Cmax values normalized to indicated doses
OCIP used 0.3 mg to 1.0 mg (4%) values; SC Used 30 & 45 ug values
**Tmax and T! /2 dose independent - averaged for all subjects
The chart below shows that PK parameters at Day 1 and Day 7 administration of
OCIP are
very similar.
OCIP Normalized to 0.6 mg
~~ ~ ~ ~ ?~=~~ ~~~ ~ A .~ s `7 8~#m~' zv,:r a 1' i- ~ ia4
ir<'
~~
~ ~Day~1 ~^'~ ay"7
AUC(0-4) 49.6 (+/-42.2) 42.5 (+/-27.0)
MMCma895 - 8 5) 629Tmax** 0.23 (+/-0.08) 0.31. (+/-0.20)
A
*AUC and Cmax values normalized to 0.6 mg dose
OCIP used 0.3 mg to 0.6 mg (4%) values
Tmax and T1/2 dose independent - averaged for all subjects
Figures 55-60 illustrate that the pharmacokinetic parameters achieved with
OCIP
administration are similar to those achieved with sub-cutaneous
administration. They also
demonstrate that the OCIP PK profile is dose proportional. These figures
include the
76
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
following measurements: therapeutic AUC levels, therapeutic Cmax levels,
overall cmax and
tTõ. , the ctõ., AUC (0-4) values for the OCIP 4% formulation, and that the pk
profiles of the
4% ocip formulation.
Generally, the PK trial identified that the Phase II trial would utilize the
4%
fonnulation and the therapeutic range would be dose proportional and there
would be single
and repeat dosing with no accumulation.
The data in Figures 55-60 demonstrate that lung delivery of Ostabolin-C is
biologically active, since cAMP stimulation can be maintained with repeat
dosing and bone
formation biomarkers increase with delivery to the tung. Additionally, day 1
PK parameters
are predictive of cAMP change. The figures also show the urinary cAMP
increases within
therapeutic ranges on day 1, that there is a consistent cAMP response with
repeat dosing at
days 1, 7 and 28 respectively, that the cAMP generation correlates with AUC
and C,r. on
day 1.
The figures also demonstrate that the levels of P1NP increased from 25 up to
100%
by day 28 as compared to baseline and that the levels of osteocalcin increased
from 25 up to
100% by day 28 as compared to baseline. The figures also show that the
increase in P1NP
correlates with AUC. The figures also show that the OCIP administration had no
effect on
bone resorption markers by showing the percent change in CTx. Accordingly, the
data
demonstrates that there is a robust urinary cAMP and bone biomarker response
with the
administration of OCIP. There is therefore a high likelihood of Phase H
efficacy comparable
with subcutaneous administration, especially since OCIP cAMP dose response
exceeds
subcutaneous response, the biomarker response correlates with cAMP responses,
and the
biomarker responses are consistent and clinically relevant.
Adverse Events Profile
The preliminary results of the subjects to whom OCIP was administered as
described
above showed that the AE profile was consistent with PTH class effects, in
that greater than
95% were mild and there were no pulmonary or cardiovascular AEs. Moreover,
there were
no serious adverse effects, no effects on spirometry parameters or vital
signs. The table
below shows the distribution of adverse events.
77
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
No. No.. No. No.
Subjects Events Mild Mod
Placebo~' 30
K16,11 /'t~4 I -~ ~.
0.1 mg (4%) 5/6 31 31 0
N~
~(4%) 1 ~~E
0.3 mg. (4%) 4/6 33 28 5
~~
0.6 mg. (4%) 5/6 13 13 0
J
s.....'' ~.i.. . ~_k.:.
~6
1.6. mg (16%) 6/6 36 - 35 1
%) ~ ~0,
]KI ~~ '.~i, #
1.2 mg. (4%) (SD) , 3/6 11 10 1
The summary of adverse events in the table below shows that the vast majority
(>95%) of
adverse events were mild. No adverse events were rated as severe.
No. of Sub. With No. of No. No.
AE / Group Size AEs Mild Moderate
Pla ebo~~ . L~83; , 6 E~ =,5 1~(sBack~pany)~
0.1 mg (4%) 2/6 8 8 0
_ 4.
0V2 rn9 (~,4% ) L ';~
0.3 mg (4%) 1/6 1 1 0
LQ 4 i 9~~4%3~ ~~ g y6 A u~~.
'e~
0.6 mg (4%) 2/6 3 3 0
V +xt a fiõ~~ ~` r.. ... : ... ~kw,'.k~.. " ~a. 1 am .... ..
~ 0~8~ (~16 ~) ~
1.6mg(16%) 2/6 4 4 0
-~r md'FF.-) x
8 rn9 (~4 ~~ ry~~'~ ~`~6 0
1.2 mg (4%) 3/6 11 10 1(Vomiting)
78
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
The table below shows a sununary of adverse events experienced, including
headaches,
nausea, and vomiting.
Headaches Nrqausea Vonmiting
No. No. No. No. No. No.
Subjects Events Subjects Events Subjects Events
~
~.~.7,-, ~: ~..
~~~ .
Placebo ~r 1~~j ~2 3 ~
~
0.1 mg (4%) 1 4 1 1 0 0
U2 g~/o} m 2 2 Oa ~, ~ 0' 0 ~Q`
~' . ~ ~s~,E~.:
0.3mg(4%) 4 8 0 0 2 2
Y` V,~-"
{04m 4/0 2 5 0 0 0 ~ 0
u
_
0.6 mg (4%) 1 3 1 1 0 0
g (. 6~/0) ~ , 0~.=,
U.8 m
~1`'~ ~ 0 1.6mg(16%) I I 2 4 4 7 1 1
. .,,.
~ _ -...;.~,,... .. ,
.--
1.2 mg (4%) (SD) 1 1 1 1 1 1
Overall the OCIP is Phase II ready and with an appropriate, transient PK
profile with
acceptable variability and with a biological activity predictive of
therapeutic benefit that
established a therapeutic window. Moreover, there is a comparability with the
SC
formulation with a high probability of a late phase success.
Example 15: Formulations
Formulation screening studies on Ostabolin-C solution to develop a stable
formulation
are detailed. Previous formulations experience oxidation and deamidation at a
pH above 7Ø
Mixtures of ethanol/water or propylene/water systems with the antioxidants
methionine or
lipoic acid were evaluated and their stability was accessed.
Two ethanol/water formulations with methionine were examined. The first set of
experiments examined the stability of Ostabolin-C solution in the mixture of
40%
ethanol/60% water (hPTH #1) above pH 7Ø One mg/ml methionine wasincluded in
the
formulation to control oxidation. The drug and methionine were dissolved in
water and pH
was adjusted to 7 with 0.1N NaOH and then ethanol was added to obtain the
target ratio of
ethanol and water. Ostabolin-C showed excellent stability in 40% ethanol/60%
water system.
The stability data of hPTH#1 are presented in the Table below.
Solution Stability of Ostabolin C in 40% Ethanol/Water with 1 mg/mL of
methionine at pH 7 (Batch
hPTH#1)
Storage Time Concentration % of Degradation Degradation De adation
79
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
Te erature Da s m cnl initial Peaks RRT Peak Peak Area%
Initial 0.26 1o0 0.59 Oxidation 1 1.08
0.65 Oxidation 2 2.03
9 0.25 94 0.59 Oxidation 1 1.16
0.65 Oxidation 2 1.58
12 0.24 91 0.59 Oxidation 1 .1.46
0.65 Oxidation 2 1.24
0.58 Oxidation 1 0.87
40 C 15 0.23 87 0.65 Oxidation 2 1.13
0.90 Hydrolysis 2.31
0.56 Oxidation 1 1.27
45 0.21 78 0.62 Oxidation 2 1.37
0.90 Hydrolysis 5.21
0.56 Oxidation 1 3.65
106 0.16 62 0.62 Oxidation 2 6.22
0.90 H drol sis 12.2
0.59 Oxidation 1 1.27
14 0.26 97 0.65 Oxidation 2 1.37
21 0.26 97 0.59 Oxidation 1 0.77
25C 0.65 Oxidation 2 1.70
0.56 Oxidation 1 1.13
45 0.25 96 0.63 Oxidation 2 1.58
106 0.26 100 0.56 Oxidation 1 1.32
0.63 Oxidation 2 1.95
0.56 Oxidation 1 0.87
45 0.25 96 0.63 Oxidation 2 1.85
5C
106 0.26 100 0.56 Oxidation 1 0.88
0.63 Oxidation 2 1.71
Ostabolin-C showed excellent stability in the 40% ethanol /60% water system.
The
degradation peaks eluting at RRT of 0.56 to 0.59 and 0.62 to 0.65 are two
oxidative
degradation peaks. They are present in the initial sample and did not change
significantly
during the stability study under all conditions with the exception of the 106-
day sample
stored at 40 C. These data suggest that ethanol is stabilizing the
formulation. The only other
degradant observed (RRT 0.90) is a hydrolysis product which is increasing at
15, 45, and 106
days storage at 40 C. The hydrolysis product was not observed at 45 days/25 C
suggesting
that the formulation is robust and one could project a shelf life of 2 year
under refrigeration
conditions.
The other approach was to exaniine the use of a diol (propylene glycol) to
further
stabilize the structure of the peptide and thus enhance stability.
Additionally, the antioxidant
lipoic acid was included. Sample solutions were prepared by dispersing lipoic
acid in water,
adjusting the pH to 8.0 and then adding drug. The solution pH was adjusted to
pH 7.5 and
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
propylene glycol added to obtain a final solution of 60% propylene glycoU40%
water. The
stability data is presented in the Table, below.
Stability of Ostabolin-C in 60% propylene glycol/40% water containing 5 mg/ml
of lipoic
acid at pH 7.5 (hPTH#2)
Storage Time Concentration % of Degradation Peak Degradation
Temperature (Days) (mg/ml) initial RRT Peak
Area%
Initial 0.167 100 -- --
3 0.172 103 -- --
9 0.169 101 H drol sis 0.91
12 0.162 97 Hydrolysis 1.15
40 C 15 0.162 97 -- --
45 0.109 65 H drol sis 5.23
106 0.067 40 Oxidation 1 2.8
Oxidation 2 1.4
H drol sis 15.9
7 0.151 93 -- --
14 0.146 87 -- --
25 C 21 0.150 90 -- --
45 0.150 90 -- --
106 0.141 84 -- --
C 45 0.173 104 -- --
106 0.148 89 -- --
The stability of Ostabolin-C in the above solvent system is outstanding. No
significant
degradant peaks were found at 25 C and 5 C storage samples after 106 days.
The only
degradation we observed is the hydrolysis product at 40 C. Some potency loss
was observed
in 25 C storage sample as well as the 5 C sample after 106 days. However no
degradation
was observed and this loss of potency may be attributable to adherence of drug
to the vials.
Both hPTH#1 and hPTH#2 showed excellent stability and both formulations will
be stable
for two years under refrigeration storage.
Example 16: Stability Enhancement
Stability studies conducted to data indicate that it is possible to obtain a
refrigerator-
stable formulation of Ostabolin-C either with a propylene glycol/water or
ethanol/water
mixture. The addition of other potential stability modifiers is determined. It
is well know
that hPTHs are susceptible to oxidation during storage. Methionine has been
shown to be a
potential antioxidant and improve hPTH stability. Additionally, it is well
known that polyols
have the potential to stabilize peptide and protein formulations. Previously
the effect of
81
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
sucrose on the stability of hPTH (1-34) was examined. Sucrose concentrations
up to IM at
pH 5.5 were found to reduce the rate of both deamidation and oxidation of hPTH
(1-34).
Buffer type could also have an effect on stability. The intent was to examine
these
solutions at a more physiological relevant pH (pH 7.5). Previously for model
compounds it
was shown that for pHs above 7, TRIS buffer had a much lower deamidation rate
constant
than a corresponding phosphate buffer. Lastly, the effect of the addition of 9
mg/mi NaCI
was examined as this more represents the physiological ionic strength. The
stability data on
Ostabolin-C solution formulations in the presence of inethionine, Tris buffer,
sucrose and
NaCI in the solvent system consisting of 60% propylene glycol and 40% water at
pH 7.5 is
examined
Description of Formulations:
Form #1: 250 g/ml of Ostabolin-C in 0.05M Tris buffer in 60% propylene glycol
and 40%
water with pH adjusted to 7.5 with HCl
Form #2: 250 g/ml of Ostabolin-C in 0.05M Tris buffer in 60% propylene glycol
and 40%
water and 5 mg/ml of inethionine with pH adjusted to 7.5 with HCl
Form #3: 250 g/ml of Ostabolin-C in 0.05M Tris buffer in 60% propylene glycol
and 40%
water, 5 mg/ml methionine and 200 mg/mi sucrose with pH adjusted to 7.5 with
HCl
Form #4: 250 g/ml of Ostabolin-C in 0_05M Tris buffer in 60% propylene glycol
and 40%
water, 5 mg/ml methionine, 200 mg/ml sucrose and 9 rng/ml of NaCI with pH
adjusted to 7.5
with HCI
In a one liter volumetric flask, 600 ml of propylene glycol was added. The
contents
of the flask were made up to one liter by adding Milli Q water. The resulting
solution is 60%
propylene glycol and 40% water. Using this as a stock solution, each batch of
formulation
was prepared by adding and dissolving the inactive ingredients and adjusting
the pH of the
resultant solution to about 8.0 prior to adding drug. The drug was added and
the pH was
readjusted to 7.5 with 0. iN HCI.
All test batches were placed at 40 C, 25 C, and 5 C for stability testing.
From earlier
stability studies, it was observed that the principal degradation peaks of
Ostabolin-C are three
oxidation peaks OXl (RRT -0.59), OX2 (RRT -0.65), OX3 (RRT -0.7), two
hydrolysis
peak (RRT - 0.90 (HYD1) and 0.98 (HYD2)), three late eluting peaks (RRT
between 1.01 to
1.25; DEG1, DEG2 and DEG3) and additional peaks (RRT > 1.4, eluting after the
45 minutes
during the gradient wash out period; identified as gradient eluting peaks,
GEP(#). The
number in parenthesis denotes the number of peaks that are eluting with the
gradient change.
82
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
The degradation peaks observed during the stability analyses will be presented
using the
above defined nomenclature.
The drug appears to also adhere to the glass and hence 100% of drug is not
recovered.
Loss in potency is observed with time without any increase in the degradation
peak area
percent. Four weeks stability of the aforementioned formulations at 40 C and
25 C has been
complete and the data summarized in the Tables below.
Stabilit of Ostabolin-C Formulations #1-4 at 400C/4 Weeks Storage
Batch Number Form # 1 Form #2 Form #3 Form #4
Nature of Test TEST RESULTS
POTENCY 65 64 61 72
HYD 1 7.0 8.3 6.9 5.3
DEG1 1.0 2.1 1.6 1.2
OX 1 2.1 0.4 0.5 0.4
OX2 2.8 1.0 1.0 0.9
OX3 1.7 1.4 1.3 1.7
OX (total) 5.6 2.8 2.8 3.0
# GEPs 8 4 5 4
Area % 1.0 2.1 1.6 1.2
Stability of Ostabolin-C Formulations #1-4 at 25'C/4 Weeks Storage
Batch.Number Form #1 Form #2 Form #3 Form #4 ND: Not
Nature of Test TEST RESULTS detected
POTENCY 90 88 81 91
HYD 1 1.4 1.2 0.9 0.3
DEG1 ND ND ND ND
OX1 0.5 0.2 ND ND
OX2 0.7 0.3 ND ND
OX3 ND ND ND ND
# GEPs 4 2 2 3
Area % 0.6 0.4 0.5 0.5
All four tested formulations showed HYD1 ranging 5.3 to 8.3 area percent upon
storage 40 C for 4 weeks. Hydrolysis degradation is lowest in the formulation
containing
sucrose and sodium chloride (Form #4) suggesting that the presence of sodium
chloride
retards or slows down hydrolytic degradation. However, potency of Ostabolin
ranges from
65 to 72 for the same storage period, suggesting variable recovery of drug
from formulation
to formulation. As shown in the Tables above, all formulations showed very
little
degradation on storage at 25 C for 4 weeks. Hydrolytic degradation is still
lowest in the
formulation that contains both sucrose and NaCl (Form#4). Other formulations
had much
higher levels of hydrolytic degradation (about 1 area %) suggesting that the
hydrolytic
83
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
degradation affected by the presence of sodium chloride or the ionic strength
of the reaction
medium.
With regard to the oxidative degradation, formulations #1-4 showed a small
percent
of oxidative degradation despite the presence of high level (5 mg/ml) of the
antioxidizing
agent methionine. However, the addition of antioxidizing agent, methionine in
the
formulation slowed down the oxidative degradation by 50% (Form#1 vs Form #2-
4).
Interestingly, it was not found that oxidative degradation for the first three
weeks in the
formulations that contained antioxidizing agent. However, the stability
analyses of 4 weeks
data of 40 C samples indicated that all formulation showed the presence of all
three oxidative
degradants.
Stability analyses of 25 C/4 weeks sample showed oxidative degradation in
formulations #1-2. Additionally, only two oxidative degradation peaks were
observed. In
addition to hydrolytic and oxidative degradations, it was also observed late
eluting peak
presented as DEG1 in the tables. The area percent of this degradation peak
varies from 1 to
2% at 40 C over a storage period of four weeks, and wasn't observed at 25 C
under the same
condition.
Stability Data of Ostabolin-C Formulations #1-4
Storage Storage % Ostabolin-C Identification of Total area%
Formulation (C) Time retained degradant of
degradant
Initial m ml 100 GEP 1 0.6
1 week 88 HYD1 1.3
GEP 2 0.3
OX1 1.5
2 weeks 80 OX2 1.3
HYD 1 4.1
40 GEP 4 1.0
OX 1 2.7
3 weeks 72 OX2 2.2
Formulation #1 HYDI 6.0
Base Case GEP 4 1.0
OX 1 2.1
OX2 2.8
4 weeks 65 OX3 1.7
HYD 1 7.0
DEG1 1.0
GEP 8 1.7
1 week 98 GEP 0.1
25 OXl 0.5
4 weeks 90 OX2 0.7
HYD1 1.4
GEP 4 0.6
Initial 100 GEP 2 0.2
1 week 86 HYD1 1.7
GEP 3 0.5
84
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
40 2 weeks 78 GEPD3 0.6
3 weeks 71 HYD 1 6.2
GEP 4 1.8
OX 1 0.4
OX2 1.0
4 weeks 64 OX3 1.4
HYDI 8.3
Formulation #2 DEG 1 2.1
Base Case plus GEP 4 1.5
Methionine I week 93 GEP 2 0.3
OX 1 0.2
4 weeks 88 OX2 0.3
25 HYD1 1.2
GEP 2 0.4
Initial 100 GEP 2 0.3
1 week 81 HYD 1 1.8
GEP 2 0.4
2 weeks 73 HYDI 5.1
GEP 3 0.6
3 weeks 67 HYD 1 5.0
GEP 3 1.4
40 OX 1 0.5
Formulation #3 OX2 1.0
Base Case plus OX3 1.3
Methionine and Sucrose 4 weeks 61 HYD 1 6.9
DEG 1 1.6
GEP 5 1.8
25 1 week 88 GEP 2 0.3
4 weeks 81 HYDI 0.9
GEP 2 0.5
Initial 100 GEP 2 0.2
l week 91 HYD 1 2.1
GEP 2 0.4
Formulation #4 40 2 weeks HYD 1 3.7
Base Case plus 85 GEP 3 0.6
Methionine and Sucrose HYD 1 3.9
and NaCI 3 weeks 77 GEP 3 1.1
OX 1 0.4
OX2 0.9
4 weeks OX3 1.7
72 HYD 1 5.3
DEG 1 1.2
GEP 4 1.5
25 1 week 99 GEP 2 0.3
4 weeks 91 HYD1 0.3
GEP 3 0.5
In addition to the above experiments, the stability of Ostabolin in water
without pH
adjustment either without or with the addition of sucrose (200 mg/ml) and
sodium chloride (9
mg/ml), formulations #A and #B, respectively was conducted. The measured pH of
these
formulations is about 5.6.
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
One week stability data at either 40 C or 25 C indicated severe degradation
for the
water control sample (Form #A). Major degradation products are oxidative
degradants and
other degradation products that are eluting before the oxidative degradants.
Unlike the
stability at pH 7.5 system, there were no peaks observed at RRT >1Ø The
stability of the
same water formulation in the presence of sucrose and sodium chloride (Form
#B) has
improved significantly. Oxidative degradants levels are significantly lowered
compared to
Form #A. However, it was observed several late eluting peaks. The stability
data are
summarized in the Table below.
Stability Data of Control Formulations
Formulation Storage Storage % hPTH RRT Total area% of
C Time retained of degradant degradant
Initial mg0.22 /mL GEP 0.2
0.1 -0.3 17.2
0.33 6.54
0.35 3.38
0.45 1.34
0.47 1.01
40 1 week 0.53 1.99
0.55 0.56
Form #A 0.57(oxl) 8.89
Ostabolin-C in water 0.63 (ox2) 36.0
0.70 (ox3) 23.2
0.1 - 0.3 6.0
0.33 2.85
0.34 0.44
0.35 0.40
25 1 week 14 0.41 1.85
0.42 1.11
0.51 19.3
0.53 18.0
0.56 (oxl) 25.6
0.63 (ox2) 7.9
Initial m0'212L GEP 0.2
Form #B 0.33 1.06
Ostabolin-C in water plus 0.35 0.93
sucrose plus NaCI 40 1 week 39 0.51 1.91
0.53 5.35
0.63 (ox2) 1.95
GEP 9Ø
0.52 2.41
0.53 0.44
0.54 1.78
25 1 week 73 0.56 1.30
0.57(oxl) 0.87
0.65 (ox2) 5.35
0.70 (ox3) 0.61
86
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
These data as a whole indicate that propylene glycol/water mixtures
significantly
enhance the solution stability of Ostabolin-C (Form#A vs. Form #1). Additions
of both
sucrose and NaCI confer additional stability (Form #1 vs. Form #4 and Form #A
vs. Form
#B).
Example 17: Pharmacokinetic Profile of Above Formulations
The following plasma pharmacokinetics profile of various formulations of
Ostabolin-
C, were studied and compared; subcutaneous, intramuscular and intravenous
administrations
(clinical formulation) and also subcutaneous administrations (new
formulations) with
subcutaneous administrations (pre-clinical formulation) to rats. The following
shows the
details of the formulations used:
Formulations were prepared once only as follows: The bulk freeze-dried peptide
was
dissolved in 0.01 M acetic acid (supplied by Fisher Scientific, Loughborough,
UK) in a 1:1
ratio, re-aliquotted into dosing vials and deep frozen (approximately -70 C).
The vials were
freeze-dried at <-20 C and stored frozen until required. The freeze-dried
aliquots for
injection were re-constituted with water for injection (supplied by Animalcare
Ltd, York,
UK). The peptide was dissolved in an appropriate volume of purified water to
an
approximate concentration of 2 to 3 mg/mL. Phosphate buffered saline (pH 7.4)
was added
to give the final required concentration (at approximately pH 7.2). The capped
vial(s) were
mixed thoroughly to ensure the peptide was fully dissolved. Aseptic techniques
and glass
vials were used throughout dose preparation. The dose was not sterile
filtered. Clinical
formulation (50 mg/mL mannitol, 0.166 mg/mL sodium acetate trihydrate, 0.4
mg/mL glacial
acetic acid, pH 4.5). The calculated amounts of mannitol, sodium acetate
trihydrate and
glacial acetic acid were weighed and then dissolved in the correct amount of
water for
injection. The test article was weighed, added to the solution and stirred to
dissolve. The pH
was adjusted to 4.5 with 0.1N NaOH or HCl (supplied by Covance, Harrogate)
depending on
initial pH. Water was added to obtain the required volume.
New formulation 1 (40% propylene glycol)
The calculated amount of lipoic acid was weighed and transferred to a suitable
container and then dispersed with water for injection. The pH was adjusted to
7.7 with 1N
NaOH (supplied by BDH Laboratory Supplies, a division of Merck Ltd, Poole,
UK). The test
article was weighed, added and stirred to dissolve. The pH was adjusted to 7.5
with 1N
NaOH. Propylene glycol was added to obtain the correct volume. The recorded
final pH was
6.6.
87
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
New formulation 2 (40% ethanol in water pH 7.5)
The calculated amount of DL-Methionine was weighed and transferred to a
suitable container
and dispersed in water for injection. The test article was weighed and added
to the solution.
The pH was adjusted to 7.5 with 0.1N NaOH. Ethanol was added to make the
required
volume.
The following dose levels were selected:
Main Study
Group Dose level Number of animals in group
Number Description Dose Route ( gtkg) Main study
Female
1 Treated-] SC (interscapular) 200 12
2 Treated-2 SC (interscapular) 200 12
3 Treated-3 SC (interscapular) 200 12
4 Treated-4 SC (interscapular) 200 12
Control IM (calf muscle) 0 12
6 Treated-5 IM (calf muscle) 200 12
7 Treated-6 IV 200 12
Treated 1= Standard formulation (acidified saline)
Treated 2 = Clinical formulation (50 mg/mL mannitol, 0.166 mg/mL sodium
acetate trihydrate, 0.4 mg/mL glacial acetic
acid, pH 4.5)
Treated 3 = New formulation 1 (40% propylene glycol)
Treated 4 = New formulation 2 (40% ethanol in water pH 7.5)
Control = Clinical formulation (vehicle)
Treated 5 = Clinical formulation
Treated 6 = Clinical formulation
Repeat Study
Group Dose level Number of animals in group
Number Description Dose Route ( g/kg) Repeat study
Female
8 Control-2 IM (calf muscle) 0 12
9 Treated-7 IM (calf muscle) 200 12
Treated-8 IV 200 12
Control 2 = Clinical formulation (vehicle)
Treated 7 = Clinical formulation
Treated 8 = Clinical formulation
The relative bioavailability of Ostabolin-C in the test formulations compared
to the
standard formulation was markedly higher, being 1.5-, 27.1- and 37.7 fold
higher for the
clinical, new 2 and new 1 formulations, respectively. The relative
bioavailability of
Ostabolin-C administered intramuscularly as the clinical formulation was
markedly higher
than all the SC administered formulations being approximately 200-fold higher
than the
standard formulation and 5-fold higher than new formulation 1.
Ostabolin-C was not quantifiable in the plasma samples from the intramuscular
(IM) control
animals (Group 8).
88
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
Following a single approximate four minute IV infusion of Ostabolin-C at 200
g/kg
to female rats, maximum quantifiable plasma concentrations of Ostabolin-C
occurred at a
tmax of 5 minutes post the end of the infusion. However, the plasma
concentrations at 2
minutes post the end of the infusion were not quantified, being >610000 pg/mL,
and suggest
that the true Cmax occurred at this earlier time point.
Following single SC administration of Ostabolin-C as four different
formulations (standard,
clinical, new formulations 1 and 2), plasma drug concentrations increased
rapidly to reach
maximum levels at the initial blood sampling time of 5 minutes post dose for
all
formulations. Single IM administration of the clinical formulation resulted in
a slightly later
tmax of 10 minutes post dose.
The total plasma clearance (CL) of Ostabolin-C was 45.4 mUmin/kg and is
similar to
hepatic blood flow. The volume of distributions (Vz and Vss) were similar
being 0.471 and
0.479 L/kg, respectively, and suggested extensive distribution of Ostabolin-C.
The toxicokinetic parameters for Ostabolin-C following IV administration are
presented
below:
Treatment AUCo.e. AUCo._ C~ (p9.lUmL) (p9-h/mL) AUCx.,nv (p9/mL) (m n) (m n)
(mLJmin/k V vp
9) (uk9) (L&9)
6 4390000 4410000 0.308 423000 5.0 7.19 45.4 0.471 0.479
Treatment 6= IV Clinical formulation
The pharmacokinetic parameters for Ostabolin-C following SC and IM
administration are
resented below:
rreatment Rout ( g.hlmL) (gh/mL) AUC,c.~o (
Crrox mL) (m n) (min) ~ /k91 in (L/k9) M M
1 SC 1890 NC NC 285 5.00 NC NC NC 0.0431 NA
2 SC 2850 5470 47.9 329 5.00 42.4 36600 2240 0.0648 151
3 SC 71400 NC NC 4650 5.00 NC NC NC 1.63 3770
4 SC 51300 51900 1.26 3700 5.00 9.10 3850 50.6 1.17 2710
IM 380000 NC NC 33900 10.0 NC NC NC 8.66 20100
Treatment 1= SC Standard formulation (acidified saline)
Treatment 2 SC Clinical formulation (50 mg/mL mannitol, 0.166 mg/mL sodium
acetate
tihydrate, 0.4 mg/mL glacial acetic acid, pH 4.5)
Treatment 3 = SC New formulation 1 (40% propylene glycol)
Treatment 4 = SC New formulation 2 (40% ethanol in water pH 7.5)
Treatment 5 = IM Clinical formulation
At necropsy, redness or red area was recorded in the subcutaneous injection
site of
some animals dosed with new formulation 2 (Group 4), which generally
correlated with
89
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
findings seen microscopically. Microscopic findings in animals treated by the
subcutaneous
dose route were generally infrequent and of a minor nature. The microscopic
finding of
congestion/haemorrhage was consistent with minor mechanical damage at the time
of
injection. A minor level of myositis/myopathy was recorded in most animals
treated by the
intramuscular dose route (control & clinical formulation, Groups 5 & 6); this
was consistent
with low-grade mechanical damage due to injection and is not considered to be
test article
related.
Following single SC administration of Ostabolin-C as four different
formulations
(standard, clinical, new formulations 1 and 2), plasma drug concentrations
increased rapidly
to reach maximum levels at the initial blood sampling time of 5 minutes post
dose for all
formulations. Single IM administration of the clinical formulation resulted in
a slightly later
tmax of 10 minutes post dose.
After attainment of Cmax, plasma concet}trations of Ostabolin-C appeared to
decline
in a generally bi phasic manner after both SC and IM administration. The
terminal
elimination half-life was only able to be tentatively defined for the clinical
formulation and
new formulation 2 after SC administration and was 42.4 and 9.1 minutes,
respectively. The
determination of tl /2 for the clinical formulation is not considered to be
robust as it was
determined over less than one half-life in duration.
The apparent clearance (CL/F) and volume of distribution (Vz/F), calculated
where
possible for the formulations administered subcutaneously, were formulation
dependent being
3850 and 36600 mUmin/kg and 50.6 and 2240 L/kg, respectively, for the new
formulation 2
and clinical formulation.
Absolute bioavailability of each SC formulation was very low (<2%) being
lowest for
the standard formulation (Fabs = 0.04%) and highest for the new formulation
1(Fabs =
1.6%). The remaining SC formulations (new formulation 2 and clinical
formulation) both had
a Fabs of approximately 1%. In contrast, the absolute bioavailability of the
IM formulation
was much greater being 8.7%.
The relative bioavailability of Ostabolin-C in the test formulations compared
to the
standard formulation was markedly higher, being 1.5-, 27.1- and 37.7 fold
higher for the
clinical, new 2 and new I formulations, respectively. The relative
bioavailability of
Ostabolin-C administered intramuscularly as the clinical formulation was
markedly higher
than all the SC administered formulations being approximately 200-fold higher
than the
standard formulation and 5-fold higher than new formulation 1. The plasma
concentrations
for Ostabolin-C following IV administration are shown in Figure 75 (treatment
6), and for
CA 02659846 2009-02-02
WO 2008/016404 PCT/US2007/010720
Ostabolin-C sub-cutaneously and intramuscularly are shown in Figures 76 and 77
(treatments
1-5). This figures show the increased bioavailability with the new
formulations.
The AUCO-tz and Cmax of Ostabolin-C after SC administration of the standard,
clinical, new 1 and new 2 formulations, as well as the AUCO-tz and Cmax of
Ostabolin-C
after IM and IV administration of the clinical formulation are shown in
figures 70-74.
Overall, following single dose administration of 200 g/kg Ostabolin-C to
female rats
by SC, IM and 1V dose routes, maximum plasma levels of Ostabolin-C were
rapidly reached
by 5 minutes after SC administration of all formulations. Tmax occurred at 10
minutes after
IM dosing of the clinical formulation. The terminal elimination half life,
where calculable,
ranged from 7.2 to 42.4 minutes.
Absolute bioavailability of Ostabolin-C was very low (<2%) after SC
administration
being lowest for the standard formulation (Fabs = 0.04%) and highest for the
new
formulation 1(Fabs = 1.6%). Absolute bioavailability following IM dosing was
markedly
higher being 8.7%.
While this invention has been particularly shown and described with references
to
preferred embodiment thereof, it will be understood by those skilled in the
art that various
changes in form and details may be made therein without departing from the
scope of the
invention encompassed by the appended claims.
91