Note: Descriptions are shown in the official language in which they were submitted.
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
COLORFAST FABRICS AND GARMENTS OF OLEFIN BLOCK COMPOSITIONS
CROSS-REFERE.NNCE TO RELATED AI'PLICATIONS
[00011 For purposes afC'nited States patent practice, the contents of U.S.
Provisional
Application No. 60;'885,.202, tiled.lanaary 16, 2007, is herein incorporated
by reference in its
entirety.
FIELD OF 'l"HE INVENTION
100021 This invention relates to dyed fabrics that are colorl:ast.
BACKGROLND AND SUMMARY OF TI-IE INVENTION
[00031 Many different materials have been used in making dyed fabrics for use
in, for
example, barments. It is often desirable that such fabrics liave a combination
of properties
including one or more of the following: dimensional stability, heat-set
properties, capability
to be made stretchable in one or both dimensiotis, chemical, heat, and
abrasion resistant,
tenacity, etc. In addition, it is also often important that such dyed fabrics
be able to hold
color, e.-., dve, long;er aiid darker when subje.cted to laundering without
significantly
degrading one or more of tfle aforenientiozied properties. Fut-the.r,
increased throughput with
reduced defects, e.g., fiber breaka~e-, is so~.times desirable if the dyed
i'abric is, for example,
a knitted fabric.
[0004] Improved fabrics have now been discovercd ~khich often have a balanced
combination of desirable properties including beii-ig able to be able to be c-
olored darker and
11oEd color, i.e., colorfast, with laundering. These compositions may also
allow for irnproved
processability in some applications. The fabric of the present invention is
typically a knit or
woven fabric comprisirzt-, elastic fibers. Such knit fabrics include, for
example, polyesters
like microfiber polyesters. "I'be elastic fibers often coinprise the reaction
product of at least
one etliylerie block polymer and at least orre crosslitiking agent. Tbe fibers
are characterized
by an arnount of cross4inkirL~ such that the fabric lias the desired
properties. -1.`bc ethylene
block polyi7ier is usually
(A1 an ethylenc=0.-olefi:j in'.',rpc?l, mer. wherein the ethv lene:'ft-
olct~~al i1iierpolx yi-ie;r
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
(1) an average block index greater than zero and up to about 1.0 and a
molecular Gveiglit distribution, Mw'IV1.n., greater than aboLit 1.3; or
(2) at least one molecular fraction which elutes between 40"C and 1'30"C.
when fractionated using "fREF, cliaracterized in that the fraction has a block
index of at least
0.5 and up to about 1; or
(3) anMwiMn frorn about 1.7 to about 3.5, at least one melting point.. Tm,
in degrees Cefsius, and a density, d, in grams/cubic centimeter, wherein the
numerical values
of Tm and d correspond to the relationship:
T, > -2002.9 + 4538.5(d) - 2422.2(d)2; or
(4) an iv1w!Mn from about 1.7 to about 3.5, and is characterized by a heat
of fusion, AH in J/g, and a delta quantity, AT, in degrees Celsius defined as
the temperature
diflerence between the tallest DSC peak and the tallest CRYSTAF peak, wherein
the
numerical values of AT and Af1 have the following relationships:
AT > -0. 1299(AH) + 62.81 for dFl greater than zero and up to 130 .l/g,
AT > 48 C: for A H greater than 130 J.%g ,
wherein the CRYSTAF peak is determined tising at least 5 percent of the
curnulative
polyiner, and if less than 5 percerlt of the pofymer lias an identifiable
CRYSTAF peak, then
the CRYSTAFtemperature is 5(3'C; or
(5) an elastic rc,covery, Re, in percent at 300 percent strain and I cycle
measured with a conipression-molded film of the ethylene,'a-oletim
interpolyaner, aild has a
clensity. d, in ~,~rams"cubic centimeter, wherein the numerical values of Re
and d satisfy the
following relationship when ethylene,'a-olefin interpolymer is substantially
free of a cross-
linkecl phase:
Re > 1.481-1629(d); or
(6) a inol4cular frac.ticjn v, i-.ich e4utcw betvvt;en 40 C a1id 130 C:
_ = 9
. ... r . I.. _ . . . . . . _ . _ _ . . .. . . . ~ . .. ..
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
interpolymer has the same comonomer(s) and has a melt index, density, and
molar
comonomer content (based on the whole polymer) within 10 percent of that of
the ethylene/u.-
olefin interpolymer; or
(7) a storage modulus at 25 C. G'(25 C), and a storage modulus at 1 00
C. G'(1[f0 C), wherein the ratio of Cr'(?a C) to G'( l00 C) is in the range
of about I:1 to
about 9:1.
(00051 The ethylene/ct-olefin interpolymer characteristics (1) through (7)
above are given
with respect to the ethylene/a-olefin interpolymer before any signiti.cant
crosslinkino, i.e.,
before crosslinking. The etkrylene/a-olefin interpolymers useful in the
present invention are
usually crosslinked to a degree to obtain the desired properties. By using
characteristics (1)
through (7) as measured before crosslinking is not meant to suggest that the
interpolymer is
not required to be crosslinked - only that the characteristic is measured with
respect to the
interpolymer without significant crosstinking. Crosslinking may or may not
change each of
these properties depending upon the specific polymer and degree of
crosslinking. The dyed
fabrics of the present invention may often be characterized by a color change
of greater than
or equal to about 3.0 according to AAT'CC. evalLiation after a first wash by
AATCC61-2003-
2A. The dyed fabrics of the present invention may often be characterized by a
color strength
after dying of greater than or equal to about 600 as rneasured with a spectrum
photometer.
BRIEF DE:SCRIP`I'ION OF Tl.-IE I)RAWING5
100061 Figure I shows the ertelting point/density relationship for the
inventive polyi-ners
(represented by diainonds) as compared to traditional randorn copolymers
(represented by
circles) and Ziegler-Natta copolymers (represeilted by triangles).
100071 Figure 2 shows plots of delta DSC-CRYSTAF as a function of DSC. Melt
Enthalpy for various polymers. The dianronds represent random ethvle,ne/octene
copoiyYilers:
the squares represent poly3ner examples 1-4; the triangles represent polymer
examples 5-9;
and the circles represent polymer examples 10-19. The. "X" svm.bols represent
polyrner
examples A*-F *.
100081 Figure 3 shows the etfect of density on elastic rec;overy for
unoriented films rl1ade
. . , ,
~:(:;r, inventiv.w ira _
Tiel
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
from The Dow Chemical C,ompany)). The squares represent inventive
ethyloueibutene
copolymers; and the circles represent inventive ethvlenc;:'octene copolymers.
100091 Figure 4 is a plot of octene content of TREF fractionated ethylene/ 1-
oetene
copolvmer fractions versus `I`REF elution temperature of the fraction for the
poEvmer of
Example 5 (represented by the circles) and comparative poly-mers E and
F(represented by the
"X" symbols). 'T'he diamonds represent traditional random etlzylene,`octene
copolymers.
100101 Figure 5 is a plot of octene content of TREF fractionated ethyleDe,` l-
octene
copolyrmer fractions versus yI'REF elution temperature of the fraction for the
polymer of
Example 5 (curve 1) and for comparative F(c-urve 2). 'I'he squares represent
Exan-iple F*;
and the triangles represent Example 5.
1001.11 Figure 6 is a graph of the log ofi'storage modulus as a function of
temperature for
comparative ethylene/1-octene copolymer (curve 2) and propylene/ ethylene-
copolymer
(curve 3) and for two ethylerie/ 1-oetene block copolymers of the invention
made, with
differing quantities of chain shuttling agent (curves 1).
100121 Figure 7 shows a plot of TMA (I mm.) versus flex modulus for some
inventive
polymers (repre-sented by the diamonds), as compared to some known polymers.
The
triangles represent various Dow VERSIFY `' polymers(available from The Dow
Chemical
Company); the circles represent various random ethylenei'styrene copolymers;
and the
squares represent various Dow AFFINITYT'll polymers(available from The Dow
Cliemica[
Company).
100131 Figure 8 shows photos of a lab dveing machine.
100141 Figure 9 shows a dyeing and reduction wash process.
DETAILED DESCRII'TION OF 'FHF. INVENTION,
General Definitions
((I0151 "Fiber'" tneans a material in which the length to diameter ratio is
greater titall
about 10. Fiber is typica3ly- classified according to its diameter. Filainent
fiber is generally
defined as having an individual fiber diametcr ~_,reater than about 15 denier,
usuafl;~ oreater
tliai1 about S0 denier per filament. Fine denier tiber generally refers to a
fiber b.avim.): a
diaineter less tb~:n abotit 15 den:. r per fiiament. 'Micrcjdenier fiber is
~:nerally di~;Iined as
y
.~.-: iic,:, ],
a
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
disc:otitinuous strand of material of definite leilgth (i.e., a strand which
has beeri cut or
other",,-ise divided into segments ofapredeterinined length).
[00171 "Elastic" means that a fiber will recover at least about 50 percent of
its stretched
length after the first pull and after the fourth to 100% strain (doubled the
length). Elasticity
can also be described by the "permanent set" of the fiber. Permanent set is
the converse of
elasticity. A liber is strretched to a certain point and subsequently released
to the original
position before stretch, and then stretched again. 'T'he point at which the
fiber begins to ptill a
load is designated as the percent pern-mnent set. "Elastic materials" are also
referred to in the
art as "elastomcrs" and "elastameric". Elastic material (sometimes referred to
as an elastic
article) includes the copolymer itself as well as, but tiot limited to, the
copolymer in the form
of a fiber, filrn, strip, tape, ribbon, sheet, coating, molding and the like.
The preferred elastic
niaterial is fiber. The elastic material can be either cured or uncured,
radiated or un-radiated,
andlor crosslinked or uncrosslinked.
[00181 "Nonelastic material" means a material, e.g., a fiber, that is not
elastic as defined
above.
100191 "Flomofil fiber" means a fiber that has a single polymer region or
damain, and
that does not have any other distinct polyrn.c:r regions (as do bicomponent
fibers).
100201 "Bicomponent fiber" means a iiber that has two or more distinct polymer
regions
or domains. Bicomponent fibers are also know as conjugated or multicompon:erit
fibers. The
polymers are tiisually different f'roin each other altholi(vh two or more
coinponents may
comprise the same polyniir. "l'he polymers are arranged in substaxitially
distinct zones across
the cross-section of the bic<>mponent liber, aiid usually extend continuously
alolag the length
of the bicomponent fiber. '-1'be configuration of a bicomponezit fiber can be,
for example, a
sheath/core arrangement (in which one polymer is surrounded by another), a
side by side
arrangement, a pie arrangement or an `islands-irl-the sea" arrangement.
Biconiponeiit fibers
are further described in U.S. Patents No. 6,225,243, 6,140,442, 5,382,40{),
5,336,552 and
5,1 08.820.
10021.1 -'Meltblor.~,n fibers"- are fibers formed by extruding a molten
therm.op[astic
potyilrer coiiipositic>n through a plurality of fine, usually circalar, die
capillaries as molten
threads or fiianients into converging high velociiy gas strcai-ns (e.g. air)
~.vhich liÃnction to
attetiuafic the tbrG_acis or filarnents to rGducc 1 ".l'he: filaments or
threads are cai-ried
, ._. ,..: ... _ 4,. _I ~_ _. - _.,. . `. .1. i. .. .-.. ....
Sti.1.Ac 4 .. A41 1.~. ,. ~e1s
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
[0022] "Meltspun fibers" are fibers formed by i-nelting at least one polyi-ner
and then
drawing the fiber in the melt to a diameter (or other cross-section shape)
less than the
diameter (or other cross-section shape) of the die.
[0023[ "Spunboncl fibers" are fibers formed by extruding a molten
thermoplastic polymer
composition as filaments through a plurality of fine, usually circLElar. die
capillaries of a
spinrreret. The diameter of the extruded filaments is rapidly reduced, and
then the filaiiients
are deposited onto a collecting surface to form a web of randonily dispersed
fibers with
avera~;e diaaneters generally between about 7 and about 30 microns.
100241 -Nonwoven" means a web or fabric having a structure of individual
fibers or
t.hreads which are randomly interlaid, but not in an identifiable manner as is
the, case of a
knitted fabric. The elastic fiber in accordance with embodiments of the
invention can be
employed to prepare nonwoven structures as well as composite structures of
elastic
nonwoven fabric in. combination with nonelastic materials.
100251 ":Yarn" means a continuous length of twisted or otherwise entangled
filaments
which can be used in the manufacture of woven or knitted fabrics anci other
articles. Yarn
can be covered or uncovered. Covered yarn is yarn at least partially wrapped
within an outer
covering of another fiber or material, typically a natural fiber such as
cotton or wool.
[0026] "Polymer" means a polymeric cotnpound prepared by polymerizing
monomers,
whether of the same or a different type. The generic term "polymer" embraces
the terms
-homop ly mer," '-copolymer,: " .`terpolymer ' as well as "interpolymer."
[0027[ "Interpolyrn.er" means a polymer prepared by the polymerization of at
least two
different types of nionomers. Tl1e generic term '`interpolymer" includes the
term
"copolymer" (wlaich is usually employed to refer to a polymer prepared from
two different
monomers) as well as the term "terpolymer" (which is usually employed to refer
to a poly=ner
prepared from three differerit types of monomers). It also encompasses
polyillers made by
polymerizing four or more types of monomers.
100281 The term "ethvlene%'a-olehn interpolyrner" generally refers to
polyiners
comprising ethylene and an a-oletin having 3 or more carbon atoins.
Preferably, ethylene
comprises the ina.jority z-ae3ie fraction of the whole polymer, i.e.. ethylene
comprises at least
about 50 mole perceiit of the whole polymer. More preferably ethylene
comprises at least
abocjt 60 mole pereent, at least about 70 mole percent. orat least about 80
niole percent. ~~=ith
} .
f >}'5q = ""L- q [[t y(
vi~pt13Yl3Y1J~>-. Lxs. ~ ~L.zWi~Wr+ rr>>A:A~ ..~...:.5. ~.. = e~.i. ._ ._.nl
v~sl _..._i. ` a...:.1 ~ ~__~ i~.. .,.,....ta.
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
n'tole percent of the whole polymer and an octene content of from about 10 to
about 15,
preferably from about 15 to abotit 20 mole percent of the whole polymer. In
some
ernbodiments, the ethylene.`a-oletin interpolymers do not include those
produced in. low
yields or in a rninor amount or as a by-product of a chemical process. While
the ethylene=a-
olcfin interpolymers can be blended with one or more polymcrs, the as-produced
ethylcne,'a-
oletin interpolymers are substantially pure and often comprise a major
component of the
reaction product of a polymerization process.
100291 The ethylene/rx-oletin interpolyniers comprise ethylene and one or more
copolymerizable a-olefin comonomers in polymerized forni, characterized by
multiple blocks
or segnients of two or more polynierizzed monomer units difterinb in chemical
or physical
properties. That is, the ethylene/a-olefin interpolymers are block
interpolymers, preferably
multi-block interpolymers or copolymers. 'I`hc terms "interpolymer" and
"copolyi-ner" are
used interchangeably he-rein. In some embodiments, the multi-block copolymer
can be
represented by the following formula:
(AB)õ
where n is at least 1, preferably an integer greater than 1, such as 2, 3. 4,
5, 10, 15, 20, 30. 40,
50" 60, 70, 80, 90, 100, or higher, :-A" represents a hard block or se-gment
and "B" represents
a soft block or segment. Preferably, As and Bs are linked in a substantially
linear fashion, as
opposed to a substantially branched or substantially star-shaped fashion. In
other
embodiments, A blocks and B blocks are randomly distributed along the polymer
chai-1. In
other words, the block copolymers uscia[Iy do not have a structure as follows.
AAA-----AA-BBB-BB
[0030] In still other embodiments, the block copolyniers do not usually have a
third type
of block, which comprises different comonomer(s). I.n yet other en-
ibodirncnts, each of block
A and block B I-ias monomers or comonomers substantially randomly distributed
~vithin the
block. In other worcls, neither block A nor block B comprises two or more sub-
segments (or
sub-blocks) of distinct composition. such as a tip se~,~ment, which has a
substantialiy different
composition than the rest of the block.
10031.1 -rhe multi-block polymers typically comprise various amounts of `hard
a~1Ãi
soft" se-ments. ` I Iard" segments refer to blocks of polymerized units in
which c.thyletre is
present in an aTtlouiit Lireater than about 95 ~,vei_vht per::erit. ~1nd
prctcrably greater thati abotlt
_'ni,- is s ,". . . ' . :a
'~li.e
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
polymer. In some, enzbodiments, the hard segmen.ts comprises all or
substantially all
ethylene. "Soft'' segments, on the other hand, refer to blocks of polymerized
units in which
the comonomer content (content of monomers other than ethylene) is greater
than about 5
weight percent, preferably greater than about Sweight percent. greater than
about 10 weight
percent, or greater than about 1. 5 weight percent based oii the weil;ht of
the polymer. In some
embodiments, the comonona.er content in the sQft segments can be greater than
about 20
weight percc nt, greater than about 25 weight percent, greater than about 30
weight percelit,
greater than about 35 weight percent, greater thai-i about 40 weight percent,
greater than about
45 weight percent, greater than about 50 weight percent, or greater than about
60 weil;ht
percent.
100321 The soft segments can often be present in a block interpolymer from
about I
weight percent to about 99 weight percent of the total weight of'the block
interpolymer,
preferably from about 5 weight percent to about 95 weight percent, from about
10 weight
percent to about 90 weight percent, from about 15 weight percent to about 85
weight percent,
from about 20 weight percent to about 90 weight percent, from about 25 weight
percent to
about 75 weight percent, from about 30 weight percent to about 70 weight
percent., froin
about 35 weight percent to about 65 weight percent, from about 40 weight
percent to about
60 weight percent, or from about 45 weight percent to about 55 weight percent
of the total
weight of the block interpolymer. C,anversely, the hard segments can be
present in similar
ranges. The soft segment weight percentage and the hard segment weight
percentage can be
calculated based on data obtained from DSC or NMR. Such metliods and
calculations are
disclosed in a concurrently filed U.S. Patent Application Serial No.
11/376,835, Attorney
Docket No. 3850639Ã39558, entitled " Ethyfene!ci-C}letins Block
Iiiterpolymers", tiled on
March 15. 2006, in the iia.me of Co1ii7 L.P. Shan, Lonnie HazlÃtt, et. al. and
assigiled to Dow
Global Technologies Inc., the disclosure of which is incorporated by
referen.cc; herein in its
elitiretv.
100331 The term ``crystallinc." if enaployed. refers to a polymer that
possesses a first order
trailsitiott or crystalline aiielting point (Tm) as detertiiined by
differential scainliilg
calorinietry {DSC} or equivalent tecbniÃ.lue. The term rnay be used
interchangeably witli the,
teri1i "sernicrystalline". Tbe term " amorpbous" refers to a polymer lacking a
crystalline
me.ltino point as determined bv dit-f:erentia.l sÃ:Ginn..rtg calc;rinic,try-
(DSC) or equivalent
~ ~ ~ ,~ ..,_. . ~= . .~
i
I't=c tcr~ii `n- t, -
~
C.,~ ~Jlrxsingf -_kÃ~ or rnor~
_~_
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
preferablyjoit-ieci in a linear manner, that is, a polymer cornprising
chemically differentiated
units which are joined end-to-end with respect to polymerized ethylenic
functionality, rather
than in pendent or grafted fashion. In a preferred embodiment, the blocks
differ in the
a.moun.t or type of comonomer incorporated therein, the de.nsity, the amount
of crystallinity,
the crystallite size attributable to a polymer of such composition, the type
or degree of
tacticity (isotactic or syndiotactic), regio-regularity or regio-irregedarity,
the amount of
branching, including long chain branchiilg or hyper-branching, the
homogene.ity, or anv other
cbei-nical or physical property. The multi-block copolymers are characterized
by uilique
distributions o#'both polydispersity index (PDI or Mw/Mn), block length
distribution, and/or
block number distribution due to the unique, process making of the copolymers.
More
specifically, when produced in a continuous process, the polyniers desirably
possess PDI
f'rom 1.7 to 2.9, preferably Froni 1.8 to 2.5, more preferably from 1.8 to
2.2, and most
preferably from 1.8 to 2.1. Wtien produced in a batch or semi-batch process,
the polymers
possess PDI from 1.0 to 2.9, preferably from 1.3 to 2.5, more preferably from
1.4 to 2.0, and
most pre.ferabEy from 1.4 to 1.8.
10035] In the following description, all numbers disclosed herein are
approximate values,
regardless whether the word "about" or `'approxir~ate' is used in connection
therewith. They
may vary by I percent, 2 percent, 5 percent, or, sornetinies, 10 to 20
percent. Whenever a
numerical range with a lower limit, Rl- and an upper limit, R"", is disclosed,
any number
falling within the range is specilically disclosLd. In particular, the
i'ollowing nunibers within
thc, range are specifically disclosecl: R-R'--k*(R" R1). wherein k is a
variable ranging from 1
percent to 10(1 percent with a 1 percent increment, i.e., k is i percent, 2
percent, 3 percent, 4
percent, 5 perc-ent,..., 50 percent, 51 percent, 52 percent,..., 95 percent,
96 percent, 97 percent,
98 percent, 99 percent, or 100 percent. Moreover, any numerical range defined
by two R
numbers as defined in the above is also specifically disclosed.
Ethy[ene/u-41efin Interpolymers
[00361 `I'he oletin block polymers, e.g. ethylen.eicl-olelin interpol y mers,
used in
ctiibodimetits of the invention (also referred to as "inventive interpolymer"
or'`inve,rrtive
polymer"} comprise ethylene and one or riiore copolymerizable a-olefin
coinonorners in
polymerized l'orr .Ã.',.--~~c1,e-ritcd by multi.ple blocks or sepncnts of Vvo
or z?iore polyrricrired
~~-rabiy a
~.z i: ~r~d se Ã~r
ti .t.xe o.'thi- ~.~slnek_ :., _ . ..s.
-9-
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
[0037] In one aspect, the ethylenc;ra-olefirl interpolymers used in
embodiments of the
invention have aM,:M,, from about 1.7 to about 3.5 and at least one nielting
point, 'I',,, in
degrees Celsius and density, d. in grainsr`ctibic ceatimeter, wherein the
numerical values of
the variables correspond to the relationship:
T,,, > -2002.9 + 45' ) $.5(d) - 2422.2(d)2-, and prelerably
T, ?-6288.1 + 13141(d) - 6720.3(d)2, and more prel:erably
.
"T? 858.91 - 1825.3(d) + 1112.8(d)2
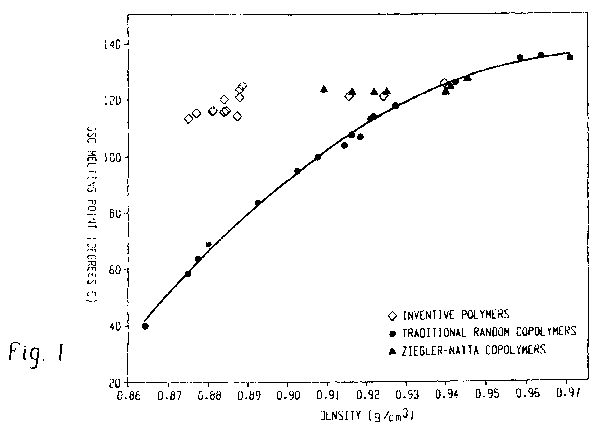
100381 Such meiting point/density relationship is illustrated in Figure 1.
Unlike the
traditional random copolymers of ethylen.e/a-olefins whose rne[ting points
decrease with
decreasing densities, the inventive interpolymers (represented by diamonds)
exhibit melting
points substantially independent of the density, particularly when density is
between about
0.87 g/cc to about 0.95 g'cc. For example, the melting point of such polymers
are in the
range of about 110 C to about 13 W C when density ranges from 0.875 g:~cc to
about 0.945
g1cc. In sozne ernbodirnents, the melting point ol'such polymers are in the
range of about I 15
C to about 125 C wb.en density ranges from 0.875 g.`cc to about 0.945 Z,.`ce.
[00391 In another aspect, the etb.ylene/o.-olefin interpolymers comprise, in
polyrnerized
forrn, ethylene and one or more Ex-oletins and are characterized by a `h, in
degree Celsius,
defined as the temperature for the tallest Differe7itial Scanning Calorimetry
("DSC"') peak
minus the temperature for the tallest Crystalf.i:zation Analysis Fractionation
(`'CRYSJ"AF")
peak and a?aeat of 1'usicin in J;'g, AH, and Al' and AIl satisfy the following
relatioilships:
AT >-0. I 299(Ali) + 62.81, and preferably
AT > -0.1299(AI--I) + 64.38, and more preferably
AT > -0.1299(A,1-I) ~ 65.95,
for Af1 up to 130 Jig. Moreover. A'I' is equal to or greater than 48 C for
A1l greater than 130
J: g. "I"he C'RY STAF peak is deterniined using at least 5 percent of the
cumulative polymer
( . that is, the peak must represent at least 5 percent of the cun-iulative
polymer), and if less than
percent ol`the polymer has an identifiable,~ CRYST,~F peak, then the CRYSTAF
tCt11 i.l? . dAll is the Yl 1 i. JF- ileat .7f i~is ? i;? '. i
t.R`V StA`
~7101_ VL
~.~~.'..
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
examples. Integrated peak areas and peak tei-nperatures are calculated by thc
computerized
drawing pro8rarn supplied by the instrument maker. The diagonal line shown for
the random
ethylene octene comparative polymers corresponds to the equation AT =-0.1299
(AH) T
62.81.
100401 In yet another aspect, the ethylene `u-oletin interpolymers have
ainolecular
fraction which elutes between 40 C and 130 C when fractionated using
Teniperature Rising
Elution Fractionation {"TREF"), characterized in that said fraction has a
molar comonomer
content higher, preferably at least 5 percent higher, more preferably at least
10 percent
higher, than that of a comparable random ethylene interpolymer fraction
eluting bemven tlle
same temperatures, wherein the comparable random ethylene interpolymer
contains the same
cotnonomer(s), and has a melt index, density, and molar comonomer content
(based on the
whole polymer) within 1.0 percent of that of the block interpolymer.
Preferably, the Mw/Mn
of the comparable interpolymer is also within 10 percent of that of the block
interpolymer
andlor the comparable interpolymer has a total comonoiner content within 10
weight percent
of that of the block interpolymer.
[0041] In still another aspect, the ethylen.e,'cr-olefin interpolymers are
characterized by an
elastic recovery, Re, in percent at 340 percent strain and 1 cycle rlleasured
on a compression-
molded filin of an ethylene,'a-olelin interpolymer, and has a density, d. in
grarns/cubic
centimeter, wherein the numerical values of Re and d satisfy the following
relationship when
cthylenela-olefin interpolymer is substantially free of a cross-1i:n.k.ed
phase:
Re >1481-1629(d); and preferably
Re ? 1491-1629(d); and more preferably
Re>15f11-16''9(d) ; arid even rnore: preferably
Re >1 5 11-1629(d).
,ure ; shov,,'s tiie effect of density on elastic recovery ror unoriented
fifin.s niade
100421 Fig
i-rom ce-rtairi inventive interpolymers and traditional randoin copolymers.
For the sainc
density. the invenzize interpolymers have substantially higher elastic
recoveries.
100431 1~1 some em1?odime.nts, the :.'thyiene-'(t-olei-1iFtiterpoly~-ners have
a tensile strength
- _ _ t= t -
, ,, ..~. . 10
s 1
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
percent, highly preferably at least 800 percerit, and most highly preferably
at least 900
percent at a crosshead separation rate of 11 cm:/minute.
(0444] In other embodiments, the e.thyle-neia-oiefin interpolymers have (1) a
storage
modulus ratio, G'(25 C)/G'(1(}0 C), of from I to 5(}, preferably from 1 to 20,
more preferably
from I to 10; and/or (2) a 70 C compression set of less than 80 percent,
preferably less than
70 percent, especially less than 60 percent, less than 50 percent, or less
than 40 percent, down
to a compression set of 0 percent.
[0045] In still other embodimeiits, the ethylenefa-olefin interpolymers have a
70 C
compression set of less than 80 percent, less than 70 percent, less than. 60
percent, or less than
50 percent. Preferably, the 70 C compression set of the interpolymers is less
than 40 percent,
less than 30 percent, less than 20 percent, and may go down to about 0
percent.
[0046] In some embodiments, the ethylene,'ct-oletin interpolymers have a heat
of fusion
of less than 85 Jig and/or a pellet blocking strength of equal to or less than
100 pounds/foot2
(4800 Pa), preferably equal to or Iess than 50 1bs/#i~ (2400 Pa), especially
equal to or less than
lbsi'ft' (240 Pa), and as low as 0 Ibsfft2 (0 Pa).
100471 In other embodiments, the eth.y[ene-/a-olefin interpolymers comprise,
in
polymerized form, at least 50 mole percent ethylene and have a 70 C
compression set of less
than 80 percent, preferably less than. 70 percent or less than 60 percent,
most prelerably less
than 40 to 50 percent and down to close to zero percen.t.
10048] In some embodimejits, the multi-block copolymers possess a PL)I fitting
a
Schtiltz-Flc>ry distribution rather than a Poisson distribution. "hhe
copolymers are i'iErther
characterized as having both a polydisperse b[ock distribution and a
polydisperse distribution
of block sizes and possessing a most probable distribution of block lengths.
Preferred multi-
block copolymers are those containing 4 or more blocks or segments iiieluding
terininal
blocks. More preferably, the copolymers include at least 5, 10 or 20 blocks or
segments
including terminal blocks.
100491 Cornononier contettt may be measured using any suitable tecbniqLie,
with
techniclues based on nuclear magnetic resonaijce ("NMIt") spectroscopy
preferred.
Moreover, ior polymers or bieticls of polymers having retative1v broad 'I"R1'T
curves, the
polviner desirably is first fractionated Using, TREF into fractions eacb ha~-
ing an e-lated
te?"7?t?erxure raT?-C (?f1O"C or 'I hat Is, ctich eautLd tract3(?n has a
collection telnf'. ,i1
. . . . _. . ~ .. . i. -- , ..t_. _ .
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
[00501 In another aspect, the inventive polymer is an olefin interpolymer,
prGferably
comprising ethylene and one or more copolymerizable comonomers in polymerized
form,
characterized by multiple blocks (i.e., at least two blocks) or segments of
two or m.ore
polymerized monomer units differing in chemical or physical properties
(blocked
interpolymer), most preferably a multi-block copolymer. said block
interpolymer hm:fing a
peak (but not.just a molecular fraction) which cIutes between 40 C and 1 3
)0'C: (but without
eollecting andlor isolating individual fractions), characterized in that said
peak, has a
comonomer content estimated by infra-red spectroscopy w-hen expanded using a
full
width'half maximum (:FWIIM area calculation, has an average molar comonomer
content
higher, preferably at least 5 percent higher, more preferably at least 10
percent hitiher_ than
that of a comparable random ethylene interpolymer peak at the same elution
temperature aiid
expanded using a ftill width:'inalf maximum (FWHM) area calculation, wherein
said
comparable random ethylene interpolymer has the same comonomer(s) and has a
melt index,
density, and molar comonomer content (based on the whole polymer) within 10
percent of
that of the blocked interpolymer. Preferably, the Mw/Mn of the comparable
interpolymer is
also within 10 percent of that of the blocked interpolymer and/or the
comparable
interpolymer has a total comonomer content within 10 weight percent of that of
the blocked
interpolymer. The full width/half maximum (F'GUI,IM) calculation is based on
the ratio of
methyl to i-nettrylene response area I'CH;,'CHz] from the ATREb' infra-red
detector, wherein
the tallest (highest) peak is identitied from the base line, and then the
FW1JM area is
determined. For a distribution measured using an A"l`Rl~~~h peak, the FWHM
area is defiiaed
as the area under the curve between T i and '1'2, where T i and T2 are points
determined, to the
left and right of the ATREF peah, by dividing the peak height by two, and then
drawing a line
horizontal to the base line, that intersects the left and right portions of
the A'I'RE-}' curve. A
calibration curve for comonomer content is made using random ethvlene!a-
olefin.
copolyrners, plotting comonomer content from. NMR versus FWIJM area ratio of
the TREF
peak. For this infra-red mthod. the calibration curve is ~:~enc,rated for the
same comonomer
t-vpe of iiiterest. The comonomer content of TtZEF peak of the inventive
polymer can be
determined by referencing this calibration curve using its EWI-1M methyl :
riiettivlene area
ratio [C.l-I;iCH:fl of the TREF peak.
100511 Com.onomer content may be measured tisin~.~ anv suitable technique,
with
t T'.
Ãn
y $:
_Ã~_
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
100521 Preferably, for interpolymers of ethylene ai1d 1-octene, the block
iriterpolymer has
a conionomer content of the TREF fraction eluting between 40 and 13O C greater
than or
equal to the quantity (- (1.2013) T+ 20.07, more preferably greater than or
equal to the
quantity (-0.2013) T-; 21.07, where T is the numerical value of the peak
elution te,niperature
of the TREF fraction being compared. measured in 'C.
[00531 Figure 4 graphically depicts an ernboditnent of the block inter-
polvmers oI'
ethylene and 1-octene where a plot of the comonomer content versus TREF
elution
temperature for several comparable ethylene;`l-octene interpolymers (random
copolyn7ers)
are fit to a Iine representing (-0.2013) T-'- 20.07 (solid line). The line for
the equation (-
0.2013) T + 21.07 is depicted by a dotted line. Also depicted are the
coinonomer contents for
fractions of several block ethylenell-octene interpolymers of the invention
(multi-block
copolymers). All of the block interpolymer fractions have signiliuantly higher
1-octene
content than either line at equivalent e1ution temperatures. "1'his result is
characteristic of the
inventive interpolya.ner and is believed to be due to the presence of
differentiated blocks
within the polymer chains, having both crystalline and amorphous nature.
100541 Figure 5 graphically displays the `1"REF curve and comonoYner contents
of
polymer fractions for Example 5 and Comparative F discussed below. The peak
elutirlg from
40 to 130 C. preferably from 60 C to 95 C for both polymers is fractionated
into three parts,
each part eluting over a temperature range of less than l0 C. Actual data for
Exai7lple 5 is
represented by triangles. The skilled artisan can appreciate that an
appropriate calibration
curve iiaay be- constructed for interpolymers containing different comc>non-
iers aild a line usec3
as a comparison fitted to ihe 'F REF values obtained from conaparative
interpolymers of the
same monomers, preferably random copolymers made usin, a metallocene or other
liomogeneous catalyst composition. Inventive interpolymers are characterized
by ai-no~lar
comonomer content greater than the value determined from the calibration curve
at the same
TREF elution temperature, preferably at least 5 percent cyreater. more
preferably at least 10
percent greater.
[0035] In addition to the above aspects and propez-ties described hc;rein, the
inventive
polymers can be characterized by one or more additional characteristics. ln
one aspect, the
it-iveiitive polviner is an oletin interpolyirier, preferably comprising
et.h3-lene and one or in<>re
c: polyme.ri.f~?ble comc>noiners in lsoly=ner;zed t r:n. cltaracterize.d by
riia?tiple blocks or
.. a . . .~ . . ~ < p y - c
. . . - .., . :. . ...-
. . .
4
ISii:i~.v3FSiiLi4ei
-i`~-
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
fractionated usin~ TRE~ incre~nents, cl~aractc;riz.ed ir~ tbat said iraction
has a molar
como~nomer cont~:~.t bi~er, pre.~l~:rably at least 5 percez~t lri~b.er, more
prc,ferably at least 1 tl,
15, 2{3 or 2~ percent l~igher, than that of a comparable ra.nd.om ethyiene
interpol~.~mer fractio~
eluting bet~vicen tl~e sazne te~mperatures, ~vberein said comparable randoi~
etb.y-lene
ir~terpolymer comprises tl~e s~une comonomer(s}, preferab~y it is the same
comonomer(s), and
a mett ir~dex, dcnsity, a~zd rnolar co~nonorr~er content (based on the ti~-
l~ole poly~ner} ti~.-ithin t Q
percent c~f that of the block~:d interpolytt~er. Preferably, the iviv~~,':'~ln
o~l'~the comparable
interpolymer is alst3 wit.hin 1~ percez~t of that of the b1ocl~ed interpolymer
ar-d~'or tl~e
co~xzparable interpolymer has a total. comonomer content within 10 wei.g[~t
perce~~t o~ tl~at of
the blocked interpolymer.
[00Sb~ Preferably, the above interpolyzners are it~terpolymers o# etbylene and
at feast one
a-olefi~, especially those interpolyi~ners baving a~vhole polyrmer density
frorr~ about (1.855 to
abo~.it 0.935 ~/cm', and more especially for polymers h.avin~ ~nore than about
1 r~ao[e p~reent
comonomer, th~: blocked interpolyrner has a comono~r~er content of the 7'RE~F
1'raction. elutin~;
bet~veer~ 40 and 130 C greater than or equal to the quantity (-0.135fi} T+
13.89, more
preferably ~reater than or equ.al to the quantity (-0.1356) T~!-- 14.93, and
most preferably
~~reater tban or equal to the q~aantity (-0.2013jT ~ 21.O7, v~bere T is the
numerical value of the
peak ~TRIa~ elrltion te~nperature of tl~ze TREI~ fraction bein~ compared,
rneasured in C.
[0057~ Preferably, for tb.e above interpolymers of ethylene and at least one
alpha-oletin
especiall.y those interpolymers ha~~in~ a~;hole polymer de~nsity from abo~at
U.8a~ to abor~t
f1.~35 l;icm~. and ~nore espec,ially for poiymcrs bavin~ more than about 1
rrtofe perce.nt
cornonomer, the blocked interpolymer ~as a coz~nonom.er content of t~e T
IZI~r~ fraction eiutir~~
between 40 and 130 C ~reater than or erlual to tl~e quantity (- 0.2C~13) T
20.07, more
preferably ~;reatc~r tl~an c~r eq~zal to the quantity (-0.2013) T+ 21.07,
~n~he:re T is th~; num.erical
value of the peak elution temperature of tbe 'T'REF f~action being compared,
measured in C.
[~(l~fi~ In sti~l another aspect, tbe iov~:z~tive polyrner is aa~ olelin
i~lterpolyrner, preferably
comprising etl~ylene an.d one or n~ore copoly~r~erizable comonomers in
poly7~~~erized torm,
chara~;teri~ed bv multiple blocks or se~r~~ents c~f t~~~o or tnore
polyrnc~rize.d r~~onomc,r u~lits
differil~g in c~~emical or pl~ysical proper~l:ies (blocke.~d
i~.terpc~lyrt~er~). z~~ost pret~erably a nrulti-
bloc~C cÃ3polyn~er, said blQclZ it~t~e~polyrner ba~.~i~~.~ a n~olec~~lar tl-
action. ~ hi~:b e~rites bet~veen
4~~"C <~~~d 1 3t~"C.'. ~I~~.r~ i~rac;tic~tlat~:d ~asir~i7 "I Rl-:.l='
in~:re~~.n~:nts, ~:Ira~~ac.t~;ri~~:d in ti~at 4very
t ~:.. t~:,at
_. ~.
1<_~~ ~_~1 ~. . _ , ~. _ ~~' :S ~`(~~i i ~ ~,: ~ _ ._ 1 . I t ~ "
iiii~it~', pS:~~:;it l s c. zi s 1.1~Ii)~E q d.: ::'; . s;vt;r's-' ~I :i.:
11;~T? :'~.`.s ~. ,~J~S`_ ~. ~ i:, , i~~~ ~,
- ~ `~
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
or higher. More preferably, said polymer fractions, havino at least I mole
percent
comonomer, has a DSC melting point that corresponds to the equation:
Tm >(-5.Sq26)(mole percent comonomer in the fraction) + 135.90.
[0059] In yet another aspect, the inventive polymer is an olefin interpolymer,
preferably
comprisin(i ethylene and one or more copolymerizable comonomers in polymerized
forrn,
characterized by multiple blocks or segments of two or more polyzneri;red
monornÃ:r units
differing in chemical or physical properties (blocked interpolymer), most
preferabl.y a multi-
block copolymer, said block interpolymer having a molecular fraction which
elutes between
40 C an.d. 130 C, when fractionated using T'RF.F increments, characterized in
that every
fraction that has an ATREF elution temperature greater than or equal to about
76 C, has a
melt enthalpy (heat of fusion) as measttred by DSC, corresponding to the
equation:
Ileat of fusion (J/gm) < (3. i 718}(ATREF elution temperature in Celsius) -
136.58,
100601 The inventive block interpolymers have a molecular fraction which
elutes between
40 C and 130 C, when fractionated using TREF inerernents, characterized in
that every
fraction that has an ATREF elution temperature between 40l C and less than
about 76 C, has
a melt enthalpy (heat of fusion) as measured by T)SC, corresponding to the
equation:
lIeat of fusion (:f.-`~m) <(i _ 13 1 2)(ATREF elution temperature in CelsiLIs)
-~ 22.9'T.
ATREF Peak COnaOnarner Composition Measurement by Infra-Red Detector
100611 "T'he comonomer composition of the 'FRp:F peak can be measured using an
iR4
infra-red detector available from I'olymer Char, Valencia, Spain
(i'=t~~ ~, ~ :, _ ,: 0117;
100621 The "composition mode" of the detector is equipped with a iiieasurement
sensor
(CH2) and composition sensor ('CH;) that are tixed narrow band infra-red
filters in the region.
of 2800-31100 ctri"'. The zn4asurement scnsor detects the methylc.ne (Cf12}
carbons oil tbe
polymer (which directly relates to the polyiner concentration in solution)
ivhile the
composition sensor detects the methyl (CH,) aroaps of the polyme-r. The
mathematical ratio
of the cornposition si~.~~nai (C.1i3) divided by the measurement signal (CI12)
is sensitive to the
comonomer content ofthe measured polymer in solution and its resporise is
calibrated Nvitb.
k~iÃ~vvn Ã;thvlcne z,!i?hn-olefin Ã:opofymÃ:r stanclfm]Q.
-i~-
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
process. A polymer specific calibration can be created by measuring the area
ratio of the CI-I3
to CH2 for polyrners with known comonomer content (preferably measured by
NMR). The
comonorner content of an ATREF peak of a polymer can. be estimated by applying
a the
reference calibration of the ratio of the areas for the individual CH; and CI-
I~ response (i.e.
area ratio CH.;iCI-I, versus camonomer content).
[00641 The area of the peaks can be calculated using a full r,vidth/half
maximum
(FWI-1M) calculation after applying the appropriate baselines to integrate the
individual
signal responses from the TREF chromatogram. The full width/half maximum
calculation is
based on the ratio of methyl to methylene response area [C.I-Iij'CI-12] from
the ATREF infra-
red detector, wherein the tallest (highest) peak is identified from the base
line, and then the
FVG'HM area is determined. For a distribution measured using an ATREF peak,
the FWHM
area is defined as the area under the curve, between "I'I and T2. wb.ere Tl
and T2 are points
determined, to the left and right of the ATREF peak, by dividing the peak
height by two, and
then drawing a line horizontal to the base line, that intersects the left and
right portions of the
ATREF curve.
100651 The application of infra-red spectroscopy to measure the coiiionorner
content of
polymers in this ATREF-infra-red method is, in principle, similar to that of
GPC /FT1R
systems as described in the following references: Markovich, Ronald P.;
Ilazlitt. Lonnie G.;
Smith, Linley; `Developme nt of gel-permeation chromatography-Eourier
transforn3 inlirared
spectroscopy for characterization of ethylene-based polyc>lef~in copolymers".
Polymeric
Materials Science and Engineering (1991), 65. 98-1 00.; and De.slauricrs,
P.J.;, Rohlfing,
D.C.; Shieh, E.T.; `'Qu~antifyin~ short chain branching microstructures in
etlly[ene- I -oleftl
copolymers iising size exclusion chromatography and Fourier transform infrared
spectroscopy (SFC-wFTIR)". Polymer (2002), 43, 59-170., both of which are
incorporated by
reference herein in their entirety.
100661 tn other embodiments, the inventive ethylene/a,-olef.in interpolymer is
characterized by an average block index. ABI. which is V-reater than zero and
up to about I.f)
axld a molecular weight distribution, M,,,-M,, greater than about 13, The
average block
inde;x, ABI, is the iveight average of the block index (`13I") for each
ot'the. polymer fractions
obtained in preparative. TREF from 2()"C and I I(?"C, with an incremeiit o#~ 5
C:
AI3I 131, 7-
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
where BI, is the block inde-x for the ith 1Taction of the inventive
ethylene."a-oletzn
interpolymer obtained in preparative "I-REF, and w, is the weight pereentage
ofthe ith
fraction.
[00671 For each polymer fraction, BI is defined bv one of the two follorAdng
equations
(both of which give the same BI value):
13I = II'T, - I' T1,r' or BI =_ LnP - Ln.P~-õ
1fT. - IIT,,R LrtP,, -LnP",,,,
where `I`x is the preparative ATREF elution temperature for the itli fraction
(preferably
expressed in Kelvin), Px is the ethylene tnole fraction for tize ith fraction,
which can be
measured byN?VIR or IR as described above. I'Aa is the ethylene mole fraction
of the whole
ethylene..`a-olefin intetpoiymer (before frac-tionation). which also cail be
measured by NMR
or IR. TA and PA are the ATREF elution temperature and the ethylene mole
fraction for pure
"hard segments" (which refer to the crystallitie segments of'the
interpolyrner). As a first
order approximation, the TA and PA values are set to those for high density
polyethylene
homopolymer, if the actual values for the ha.rd segments" are not available.
For calculations
performed herein, "I'A is 372 K, PA is 1.
[00681 'TAa is the ATRET temperature for a randoi:n copolymer of the same
composition
atid having an ethylene mole fraction of I';\jj. T.ka can be calculated from
the following
equation:
Lii PA13 OE/ T ~g
where a and P are two constants which can be determined by calibration using
anurnber of
known random etbylene copolynlers. It should be noted that a and P may vary
from
instrument to instrutnent. Moreover, one twotild need to create their own
calibration curve
with the polviner coinposition of interest and also in a sirnilar molecular
weight range as the
fractions. There is a slight molecular weight effect. If the calibration curve
is obtained 1rom
sin~ilar molecular weight ranges. such effect would be e:ssentially
negligible. In some
embodiments, raridom etily-len.e copolyiiiers satisfy the following
relationship:
Ln P 4~7.8 ~:" l:t" n~:3 0.639
.: . ._. , . _,.
vzm.'. ar
_i~-
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
ConverseCy, Pxo is the ethylene mole fraction for a random copolymer of the
same
composition and having an ATREF temperature of T.X. wliich can be caleulated
from Ln I'xfl
czr I`x + P.
100701 Once the block index (BI ) for each preparative "1 REF fraction is
obtained, the
weight averaoe block index, ABI, for the whole polymer can be calculated. In
some
embodiments, ABI is greater than zero but less than about 0.3 or from about
0.1 to about 0.3.
In other embodiments, ABI is greater than about 0.3 and up to about 1Ø
Preferably, ABI
should be in the range of from about 0.4 to about 0.7, from about 0.5 to about
0.7, or from
about 0.6 to about 0.9. In some errmbodiments.. ABI is in the range of from.
aboLit 0.3 to about
0.9, from abotit 0.3 to about 0.8, or from about 0.3 to about 0.7, from about
0.3 to about 0.6,
from about 0.3 to about 0.5, or from about 0.3 to about 0.4. In other
em.bodiments. ABI is in
the range of from about 0.4 to about 1.0, from about 0.5 to about 1.0, or from
about 0.6 to
about 1.0, from about 0.7 to about 1.0, from about 0.8 to about 1.0, or lroin
about 0.9 to about
1Ø
[0071] Another characteristic of the inventive ethylene/a-oletin interpolymer
is that the
inventive ethylene!a-ole-tin interpolyiner comprises at least one polymer
fraction which can
be obtained by preparative TREF, wherein the fraction has a block index
greater than about
0.1 and up to about 1.0 and a molecular weiaht distribution, M";'MR, greater
than abotit 1.3.
In some embodiments, the polymer fraction has a block index greater than about
0.6 and up
to about 1.0, greater than about 0.7 and up to about 1.0, greater than about
0.8 and Lip to about
1.0, or greater than about 0.9 and up to about 1Ø In other embodiments,
t.kte polymer
fraction has a block index greater than about 0.1 and up to about 1.0, greater
than about 0.2
and up to about 1.0, greater than about 0.3 and up to abottt 1.0, greater than
about 0.4 and up
to about 1.0, or greater than about 0.4 and up to about 1Ø In still other
embodiments, the
polymer fraction has a block index greater than about 0.1 and up to about 0.5,
greater than
greater than about 0.3 and u.p to about 0.5 , or greater than
about 0.2 and up to about 0.5.
about 0.4 and up to about 0.5. In yet other einbodinients, the pr3lyine-r
fraction has a block
index greater than about 02) aiid lip to about {).9, greater than about 0.3 )
and up to about 0.8.
(Treater than about 0.4 and up to about 0.7, or greater thali abolit 0-5 and
tiip to about 0.6.
100721 For copolymers ofethylene and an a-oletin., the inve,ntive polymers
preferably
Pc,,;se;s5 (1) a 1'IN of at least more preferably at least 1.5, sit least
1..7. or at least 2.0, and
, ast L.6m U
~t ~. ~ - Ilyc~pt :. J
s~~~
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
ethylene content of at least 50 weight percent, (~} a(3ass transition
tempcrature. T, of less
than -25 C, more preferably less than -30 C; arzdior (5) one and only one
[0073] Further, the inventive polymers can have, alone or in combination with
anv other
properties disclosed herein, a storage modtilus, G', such that log (G') is
greater than or eclilal
to 4001cPa, preferably greater than or equal to 1.0 MPa, at a#etnperature of
100 C.
Moreover, the inventive polymers possess a relatively flat storage modulus as
a function of
ternperature in the raaigc: from 0 to 100 C (illustrated in Figure 6) that is
characteristic of
block copQlymers, and heretofore unknown for an olefin copolymer, especially a
copolymer
ofethyl.ene and one or more C3-g aliphatic a-olefins. (By the term "relatively
flat" in this
context is meant that log G' (in Pascals) decreases by less than one order of
ma~nitud.e
between 50 and 100 C, preferably between 0 and 100 C).
[11074] The inventive inteFpolymers may be further characterized by a
thermoznechanical
analysis penetration depth of 1 mm at a temperature of at least 90 C as well
as a flexural
modulus of from 3 kpsi (20 MPa) to 13 kpsi (90 MPa). Alternatively, the
inventive
interpolymers can have a thermomechanical analysis penetration depth of 1. mm
at a
temperature of at least 104 C as well as a flexural modulus of at least 3 kpsi
(20 Wa). They
may be characterized as having an abrasion resistance (or volume loss) of less
than 90 mm3.
Figure 7 shows the TMA (1 mm) versus flex modulus for the inventive polymers,
as
compared to other known polymers. The inventive polyniers have significantly
better
flexibility-heat resistance balance than the other polyrn.ers.
[00751 Additionally, the ethyleiieia-olefin interpolymers can have a melt
index, 1,, from
0.(11 to 2000 gI10 niinutes, preferably from 0.01 to 1000 g/ 10 minutes, more
preterab[v from
0.01 to 500 gI10 minutes, and especially from 0.01 to 100 g'10 minutes. In
certain
embodiments, the etbvEene/a-olefin inte.rpolym.ers have a rnelt index. 12,
l:xom 0_01 to 10 gli10
minrites, from 0.5 to 50 gl/40 ininutes, from 1 to 30 g/10 minutes, from 1 to
5g;ll0 miiiutes or
frc~i-n 0.3 to 10 9,=10 minutes. In certain e-mbodianents, the rn.elt
index.for the ethvlene/a-olefin
polvzners is 1g.'10 minutes. 3~ll.0 ininutes or 5g:i10 minutes.
[0076[ The polyniers can have molecular weights, M, 9rotil 1,000 g/mole to
D,000,000
g/mole. preferably frotii 1000 gi:"mole to 1,0(}0,000, more preferably from
1000 g;'inole to
300,000 girnole, and especially from 10,000 g,~mole to 300,000 ~~mole. The
density of the
inventi;-e polvnaers can be from 0.80 to 0.99 fr:'crn 3 and nre#~:er.tblv t:oA
etl}xvlenc: containin<4
thc
~ ~ `~r.
raii gc:- _ ~_ .. _. , . 7 1
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
[00771 The process of rnaking the polymers has been disclosed in the following
patent
applications: U.S. Provisional Application No. 60.`553,906, filed Marcli 17,
20Ã14; U.S.
Provisional Application No. 60r'662,937, filed March 17, 2{}05; U.S.
Provisiotral Application
No. 60=`662,939, filed March 17, 20(}5; UT.S. Provisional Appl.icationNo.
60/662,938, tiled
March 17, 26{}5;1'C`I' Application No. PCT;'US20U51fO[?8916, filed March
17,2005, PCT
Application No. PCTIliS2005`008915, filed March 17,2005; and PCT
Appiication'vo.
PCTtL52005:008917, filed March 17, 2Ã105, all of which are incorporated by
reference
herein in thi~ir entirety. For example, one such method comprises contactin(y
ethylene and
optionally one or more addition polytnerizable monomers other than ethylene,
under addition
polymerization conditions with a catalyst composition comprising:
the admixture or reaction product resulting from combining:
(A) a first olefin polymerization catalyst having a high comonom.er
incoiporation
index,
(B) a second olefin polymerization catalyst Izaving a comonomer incorporation
index less than 90 percent, preferably less than 50 pereent, most preferably
less thaii 5
percent of the comonomer incorporation index of catalyst (A), and
(C) a chain shuttling agent.
100781 Representative catalysts and chain shuttlin.g agent are as follocvs.
100791 Catalyst (A l) is [N-(2,6-di(1-niethylethyl.)phenyl)amido)(2-
isopropy[plienyl)(Ct-
naphthal~.~n-2-diyl(6-pyridin-2-diyl)mcthane)]hafnium dimethyh prepared
according to the
teachings of WO 0340195, 2003liS0204017, USSN 1011429,024, filed May 2, 2003,
and WO
04/24740.
p CH(rr-i3),
(~~~ sc:)-r1c
%t~
Hf o
(H3C)-E[C
C113
A2 !N ¾ ~ ~ I J
m~ ~ ~
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
ofW[) 03i'40195, '2003US02040I7, USSN 10<<`429,024, filed May 2. 2003, and WO
04/24740.
CH3
CH
/
N N
H~
(H,C)21iC CH3 E'H3
[00811 Catalyst (A')) is bis[:,Ni,N""-(2,4,6-
tri(rnethylphcnyl)amida)c.thylenediaminelhaf'nium dibenzvl.
/ C~-13
H3C v---
n
CH~j
I-iN--- WX~ X,, 0.17C6I-.1~
\ CH,
~
H3C'
:
C 1't3
100821 Catalvst (A4) is bis((2-oxcwl-3-(di.bcnzo-If,1-pyzrolc -l-y-l)-5-
(methyl)phcnyl)-2-
pbenox~~mcthyl)cyclz3hexane-l,2-diyl zirconium (IV) dibcnzvl, prepared
substalitially
according to the teachings of L S-A-2{}f)4/04l 01.03),
-~ ~
HXfC11~ eE42ChF15
o~ ~re-~~ ~"~~-~
(~'3~
100831 Catalyst (BI) is l,2-bis-(3,5-cli-t-but.ylplienvie:ne)(1-(N-(1-
~nctI-:N,1c.thvl)iniminc);)niet'nvf )(2..oXovl') r:rcc>nitiam dibenzyi
-`~`~_
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
C(CH;);
f 1AC.H.S)j
C \ ! C(CtF3)3
ZCX"
o / 'k
N-
H(CFF;)2 X..:.-CH,Ct;H;
(CH;};
100841 Catalyst (B2) is 1,2-bis-(3,5-di-t-butylphen.ylene)(1-(iN-(2-
methvlcycloh.exy1)-
immino)rnethyl}(2-oxoy 1) zirconium dibenzyl
C(CH;);
1-i;C
_~v ~ / o C(:CH3)3
zrX2
(H3C)~C~ \ ~ ~
CH~
-
"({-.E~~3)3
100851 Catalyst (C1) is (t-butylamido)dimethyl(3-N-pyrrolyl-1,2.3,3a,7a-rl-
inden-l-
yl)silanctitanium dimethyl prepared substantially according to the techniques
of USP
6,268,444:
(Ff~C.')2~i~ Ti(CH3)2
N
[0086] Catalyst (C?) is (t-hutylamido)di(4-i1iethylplien\~ 1~)(? i~lethvl-1233
)a,7a-rll-inden-
1-vljsilanetitaniuni diine.thyl prcpared substantiallt- according to the
tcachinITs of't;S-;Ik-
?00z/t}04?86:
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
-.,..
HiC
C H,
Si~ R.CH;.32
N
H3C
[00871 Catalyst {C3} is (t-butylamid )di(4-methylpheny[)(2-methyl-1,2;3, 3a,8a-
q-s-
indac-en-I-yl)silanetitanium di.methyl prepared substantially according to the
teachings of US-
A-2D03~'00428fi:
tt;C
Si~ Ti(E'H3)~
1{3C:
[00881 Catalyst (D l) is bis(di.m.cthyldisiloxane)(indenc-l-yl)zirc-onium
diehlQt ide
available from Sigma-Aldrich:
(ti3c)2si~ Zi-c1,
0
[0089[ Shuttling Agents "hhe shuttling a<Z;ents e.mployed include diethylzinc.
di(i-
b tyI)zinc, cli(n-hexyl)zinc-, triethvfaltiminum. trioctylaEuniinum, triethyl-
allium, i-
butyialuminum bis(dimtthylft-butyi)sil xanci. i-butylaluminum
bis(di(triinetb.ylsilyl)an)ide),
n-oÃ:tvlaiuminu1n diÃp}'ridine-2-methoxide?. b'<(n .)ctadÃ.cvl~i-btitN
ls~luminuin, i-
~ ~-
._- 9.
i , _ .. .. _ . _ .
.. .. ,, . ..: , . ... _
L-~~-e .. . .Ii I..;. ., . - . . i.,. ... - . _ i ... . ^ ? , ...- -~-
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
azacycloheptaneamide), n-octylaluminum bis('-.3,6,7-dibenzo-l-
azacycloheptarieamide), n-
octylaluminum bis(dimetb.yl(t-butyl)siloxide. ethylzinc (2,6-
diphenylpbenox.ide), mid
ethylzinc (t-butoxide).
100901 Preferably, the foregoing process takes the form of a continuous
solution process
for forTning block copolymers, especially multi-block copolym.ers, pref:erably
linear multi-
block copolymers of tvv~o or more monomers, more especially ethylene and a C3-
20 olelin or
cycloolefin, and most especially ethylene and a C4_20 a-olefin, using multiple
catalysts that
are incapable of interconversion. That is, the catalysts are cb.eznically
distinct. Under
continuous solution polymerization conditions, the process is ideally suited
for
polymerization of mixtures of monomers at high monomer conversions. Under
these
polymerization conditions, shuttling from the chain shuttling agent to the
catalyst becomes
advantaged compared to chain grouth, ai1d multi-block copolymers. especially -
inear inulti-
block copolymers are formed in high efficiency.
[00911 The inventive interpolymers may be differentiated frotn conve.ntional,
random
copolymers, physical blends of polymers, and block copolymers prepared via
sequential
inonomer addition, fluxional catalysts, anionic or cationic living
polyznerization techniques.
In particular, corrlpared to a random copolymer of the sax-ne monomers and
monomer content
at equivalent crystallinity or modulus, the inventive interpolymers have
better (higher) heat
resistance as measured by melting point, bigh.er TMA penetration ternperature,
higher high-
temperature tensile strength, and/or higher high-temperature torsion storage
modulus as
dLtennined by dynamic mechanical anaiysis. Compared to a random copolymer
containing
the same rnonoi-ners and monomer content, the inventive interpolymers have
lower
compression set, particularly at elevated temperatures, lower stress
relaxation, higher creep
resistance, higher tear strength, higIier blocking resistance, faster setup
due to higher
crystallization (soliditic-atioiz) temperatLire, higher recovery (particularly
at elevated
temperatures), better abrasion resistance, higher retractive force, and better
oil and filler
acceptance.
t00921 The inventive inteipoiytners also exhibit a uniqak~ crystallization and
braiachincv
distribution relationship. 'I"hat is, the inventive interpolymers have a
reiatively large
difference bem,,een the tallest peak tenlperature nieasure~:d tÃsing C.ItYS'1
AF and DSC as a
f`Unction of heat (>l' fiusion, especially as ct. ;:re:i to i=aridoni
cc)polvtrze.rs i:ontaiiaiilg the sarrc
a - -
i~r 2tal. :;" Pa,
:tv. it is b itt ~A.
zx_ this uaiqu~ of the _?~_
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
comonomer in blocks vvithin the polyrner backbone. In particular. the
inventive
interpolymers may c-ornprise alternating blocks of differin.a comonoMer
content (including
homopolymer blocks). The inventive interpolymers may also comprise a
distribution in
number and:`or block size of polymer blocks of differing density or comonomer
content,
which is a Schultz-Floi-v type of distribution. In addition, the inventive
interpolymers also
have a unique peak melting point and crystallization temperature profile that
is substantially
independent of polymer density, modulus, and tnorphology. ln a preferred
embodiment, the
microcrystalline order of the polymers deinon.strates characteristic
spherulites and lamellae
that are distinguishable from random or block copolymers, even at PDI values
that are less
than 1.7. or even less than 1.5, down to less than 1.3.
100931 Moreover, the inventive interpolymers may be prepared using techniques
to
iiifluence the degree or level of blockiness. That is the amount of comonomer
and len~,~th of
each polynier block or segment can be altered by controlling the ratio and
type of catalysts
and shuttling agent as well as the temperature ol~` the polymerization, and
other
polymerization variables. A surprising benefit of this phenomenon is the
discovery that as
tl-te degree of blockiness is increased, the optical properties, tear
strength, and hi"h
temperature recovery properties of the resulting polynler are improved. In
particular, haze
decreases while clarity, tear strength, and high temperature recovery
properties increase as
the average number of blocks in the polymer increases. By selecting shuttling
agents an.d
catalyst combinations having the desired chairi transferring ability (high.
rates of shuttiing,
with low levels of chain te,rmiiiation) other 1"orms of polymer termination
are effectively
suppressed. Accordingly, little if any 13-hy(irid.e eiiinination is observed
in the polymerization
of ethylenei'cc-o[etin comonomer mixtures accordin- to embodiments of the
invention, and the
possessing little or
resulting crystalline blocks are highly, or substantially completely, linear,
no long chain branching.
100941 Polymers with highlv crystalline chain ends can be selectively prepared
in
accordaiiee with embodiments of the invention. In elastomer applications.
reducing the
relative yLiantity of polyrner that terminates with aii ai7iorphous block
redtices the,
intermoleeular dilutive effect on crvstalliiie rc(~ians. "1'his result can be
obtained by cboosing,
chain shuttling agents and c-ataiysts having an appropriate response to
hvdrouen or other
chain terminating agents. SpeciTicall~ , if the c.atalv,t whic;h produces
hiphiy cry4talline
i :. 4;.ISY }? USC ItblC; ~Or .._17.
ic i f.?1cC'i-mxat s.ioI7. Iyf7I<.,r ,'.;-mation). th1',iI th:;
~~a-
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
crystalline polymer segments will preferentially populate the terminal
portions of the
polymer. Not only are the resulting tet-minated groups crystalline, but upon
termination, the
highly crystalline polymer forming catalyst site is once again available for
reinitiation of
polymer forsnation. The initially formed pol~-rner is therefore another highly
crystalline
polymer scanaent. Accordingly, both ends of the resulting multi-block
copolymer are,
preferentially highly crystalline.
[0095] The etliylene a-olefin interpolymers used in the embodiments of the
invention are
preferably interpolymers of ethylene with at least one C3-Q,0 a-olefin.
C.opolyrners of
ethylene and a C;-CZp a-olefin are especially preferred. The interpolymers may
further
comprise C4-C18 diolefin and/or alkenylbenzen.e. Suitable unsaturated
comonomers useful
for polyrnerizing with ethylene include, for example. ethylenically
unsaturated monomers,
conjugated or nonconjugated dienes. polyenes, alkenylbenzenes, etc. Examples
ofsuch
cam.onorners include C3-C20 a-olefins such as propylene, isobutylene, 1-
butene, I -hexene,
1-pentene, 4-methyi-I -pentene, 1-heptene, I -octene, 1-nonene, I-decene, and
the like. 1-
butene and I -octene are especially preferred. Other suitable rnonomers
include styrene, halo-
or alkyl-substitute.d styrenes, vinylbenzocyclobutane, ,4-hexadiene, I.?-
octadiene, and
naph.thenics (e.g., cyclopentene, cyclohexene and cyclooctene).
100961 While ethylene'o,-oletin interpolymers are preferred polymers, other
ethylene/olefin polymers may also be used. Otefins as used herein refer to a
family of
nnsattiirated hydrocarbon-based compounds with at least one carbon-carbozl
double bolid.
Depending on the selection of c-atalysts, at-iy o[etin may be used in
embodiinents of the
invention. Preferably, suitable oEefins are C3-C7o aliphatic and aromatic
compounds
containijig vinylic unsaturation, as well as cycEic comporrnds, such as cvc-
lobutene,
cyclopentene, dicyclope-n.tadiene, and norbornene, including bnt not lirnited
to, norbornene
substituted in the 5 and 6 position with. CI-C2(} hydrocarbyl or
cyclohydrocarbyl groups.
Also included are mixtures of such olefins as well as mixtures of such olefins
with C4-C40
diolefin compounds.
100971 Examples of olefin rnonomers include, but are not limited to
propyleiie,
isobutylene, 1-butene, I-pentene, I-bexene, 1-beptene, 1-octene, 1-noiiene, I -
decene, and I-
dt>dL:;enc. i t~ t~ f w~ e: e, I h~ ~ t~e~ 1~e. 1(~ctaclc c, ne. I-clc(j
=enc.. 3-n ~uthyl- I-but,_: rie. 3-
<
v11~?'
.
11.cI1[ . C i(iiii tt T. CA ~I3 2 d tt3 1
-~,_
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
I.3-pentadiene, 1,4-bexadicne, 1.5-hexadiene, 1,7-oct.adie'ne, 1,9-de.cadiene,
other C4-C40 a-
olefins. and the like. In certain enibodiments, the a-olefin is propylene,1-
buteue, I-
pentene,l -hexene, 1 -octene or a combination thereof. Although any
hydrocarbon containing
a vinyl group potentially may be used in embodiments of the invention,
practical issues such
as monomer availability, cost, and the ability to conveniently remove
unreacted monomer
from the resulting polymer may become more problematic as the molecular weight
of the
inonozner becomes too high.
100981 'The polymerization processes described herein are well suited for the
production
of olefin polymers comprising monovinylidene aromatic monomers including
styrene, o-
methyl styrene., p-methyl styrezie, t-butylstyrene, and the like. In
particular, interpolymers
cornprisin, ethylene and styrene can be prepared by tollowing thc, teachings
herein.
Optionally, copolymers comprising ethylene, styreile and a C3-C20 alpha
olefin, optionally
comprising a C4-C20 diene, having improved properties can be prepared.
[0099] Suitable non-conjugated diene monomers can be a straight chain,
branched chain
or cyclic hydrocarbon diene having from 6 to 15 carbon atoms. Examples of
suitable non-
conjugated dienes include, but are not Iii:nited to, straight chain acyclic
dienes, such as 1,4-
hexadiene, 1.6-octadiene, 1,7-octadiene. 1,9-decadiene, branched chain acyclic
dienes, such
as 5-rnethyl-1,4-hexadiene; 3.7-diniethyl-l,6-octadiene; 3,7-dimethyl-1,7-
octadiene and
mixed isomers of dillydrotnyricene and dihydroocinene, single ring alicyclic
dienes, such as
1,3-c-yclopentadiene; 1,4-eyclohexadienc; 1,5-cycEooctadiene and 1,5-
eyclododecadiene, atid
inulti-rin.g alicyclic f'Lised and bridged ring dienes, such as
tetcahyd:roindene, methyl
tetrahydroindene, dicyclopentacliene, bicyclo-(2,2,1)-bepta-2,5-diene;
alkenyl, alkylidene.
cycioalkenyl and cycloalkylidene norbornenes, such as 5-methylene-2-norbornene
(::41NB); 5-
propenyl-2-norborntne, 5-isopxopyliderle-2-norbornene, 5-(4-cyclopentenyl)=2-
norborrrel3e,
5-cycloh.eYylidene-2-norbornerze, 5-vinyl-2-norbornene. and norbornadiene. Of
the dienes
typically used to prepare EPDMs, the particulariy preferred dienes are 1,4-
bexadiene (HD),
5-ethylidene-2-norbornene (EIvB). 5-vinylidezie-2-norboniene (VNB), 5-
metbvlen.e-2-
norbornene ( MNB), arld dicyclopentadiene (DCPD). '1'he especially preferred
dietie-s are 5-
cthylide.ne-2-norbomene (ENB) and 1.4-hexadieue (1-1D).
[01041 One class of desirable polymers that cati be niade in accordance wit.b
lbod:n ela5tot~-icric 3r:- r-õ mc _ ~'A`
one o
. . , a., e . p
rl~'~~r~1.3
~.Slti:-~SL.~~ ~c
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
where R* is a linear or branched alkyl group of from I to 12 carbon atoms.
Examples of
suitable a-olefins include, but are not limited to, propylene, isobutylene, 1-
butene, 1-pentene,
1-hexene, 4-methyl-l-pentene, and 1-octene. A particularly preferred a-olefin
is propylene.
The propylene based polymers are generally referred to in the art as EP or
EPDM polymers.
Suitable dienes for use in preparing such polymers, especially multi-block
EPDM type
polymers include conjugated or non-conjugated. straight or branched chain-,
cyclic- or
polycyeiic- dienes comprising from 4 to 20 carbons. Preferre-d dienes include
1,4-pentadiene,
1,4-bexadiene, 5-ethylidene-2-norbornene, dicyclopentadiene, cyclohexadi4ne,
and 5-
butylidene-2-norbornen.e. A particularly preferred diene is 5-ethylidenc-2-
norbornene.
[0101] Because the diene containing polymers comprise alternating segments or
Hocks
containing greater or lesser quantities of the diene (including none) and a-
olel-in (including
none), the total quantity of diene and a-olc.fin may be reduced without loss
of subsequent
polymer properties. That is, because the diene and a-olefin monomers are
preferentially
incorporated into one type of block of the polymer rather than uniformly or
randomlv
throughout the polymer, they are more efficiently utilized and subsequently
the crosslink
density of the polymer can be better controlled. Such crosslinkable elastomers
and the cured
products have advantaged properties, including higher tensile stren~th and
better elastic
recoverv.
101021 In some embodiments, the inventive interpolymers made with two
catalysts
incorporating differing quantities of cotnonomer have a weiglit ratio of
blocks f'ortned tflereby
from 95:5 to 5:95. `I`he elastomeric polymers desirably have an ethylene
content of li=om 20
to 90 percent, a diene content of from 0.1 to 10 percent, ancl an a-oiefin
content of from 10 to
80 perce:nt, based on the total weight of the polynaer. Further preferably,
the multi-block
elastomeric polymers have an ethylene content of l:rom 60 to 90 percent, a
diene content of
from 0.1 to 10 percent, and an a-olefin content of from 10 to 40 percent,
based on the total
weight of the polymer. Pref'erred polymers are high molecular weight polymers,
having a
weight average z=nolecular weight (Mw) from 10.000 to about 2,500.000,
preferably from
to 500.000, more preferably froin 20.000 to 350,000, and a polydispersit.y
less than
more prelerably less than 3.0, and aMc>oncy viscosity (N,1L (1.i-4) 125"C..)
from i to 250.
More preferabiy. such poiyme:rs have an ethylenc contetit from 65 to 75
percelit, aÃl.iene
c _ontent froni 0 to 6 perc.ent, and an ft-ole-fin o.-a_ nt frotn 20 to '3 5
percent.
. ,~, ,..
a1.` ..1 ..1 ,i=a:iCl)I'
P
4_1C~L.i~t . st;. '.-'.ip.)u. et7n feI"I1~~ai, a. , 3l~irsaL
~Y#
. , l"
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
ethylenically unsaturated mono- and di-Cunctional carboxylic acid anhydrides.
salts thereof
and esters thereof. Such functional groups may be grafted to an ethylene,`Ot -
olefin
itlterpolymer, or it may be copolymerized with ethylene and an optional
additional
comonomer to form an interpolymer of ethylene. the functional comonomer and
optionally
other comonomer(s). Means for grafting functional groups onto polyetb.ylene
are described
for example in L.S. Patents Nos. 4,762,890, 4,927,888, and 4,950,541, the
disclosures of
these patents are incorporated herein by reference in their entirety. One
particularly useful
functioz-tal group is malic anhydride.
101041 The amount of the functional. group present in the functional
interpolymer can
vary. The functional ;roup can typically be present in a c-opolymer-lype
functionalized
interpolymer in an aznount of at least about 1.0 weight percent, preferably at
least about 5
weight percent, and more preferably at least about 7 weight percent. The
functional group
will typically be present in a copolymer-type l~unctionalized interpolymer in
an amount less
than about 40 weight percent, preferably less than about 30 weight percent,
and more
preferably less than about 25 weight percent.
Testing Methods
(01051 ln the examples that follow, the following analytical techniques are
employed:
GPC Method for Samples 1-4 and A-C
101.(36) An autoinated liquid-handling robot equipped with a heated needle set
to 160 C is
cised to add enough 1,2,4-trichlort>benzene stabilized with 300 pptn lonol to
each dried
polymer sample to give a final. concentration of 30 mg/mL. A small glass stir
rod is placed
into each tube and the samples are heated to 160 C for 2 hours on a heated,
orbital-shaker
rotating at 250 rpm. The concentrated po[viiier solution is then diluted to I
mg%~~nl using the
autornate-d liquid-handling robot and the heated needle set to 160 C.
101071 A Symyx Rapid Cil'C system is used to determine the molecular weight
data for
each saniple. A Gilson 350 pump set at ?.0 rnl:`min flow rate is used to puinp
helium-purged
1.2-dichlorobenzenc; stabilized with 300 ppm lonol as the mobile phase
throu,.h three 1'lg~:l
micrometer {pin} Mixed B 300mrn x 7.5mm columns placed in series and heated to
1 60 C. A Polymer Labs ELS 1000 I3etector is uscd %&-ith the Evaporatoi' <'.~t
to 250"C. the
.
Nebt[l,zt r`" to 16 c'i`. ir,, :l: I .c} St 80 ~. "~"l
d400-66T. "FbC . t.: 1 Ãlito a
2 kj LL~ IC7O`
. ... `~ -
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
polymer samples using two switched loops and overlapping injections are used.
The sample
data is collected and analyzed using Sy~~}x EpochTM sof-tvvare. Peaks are
inanually
integrated and the molecular iveight information reported uncorrected. against
a polystyrene
standard calibration curve.
Standard CRYSTAF Method
(0108) Branching distributions are dettrinined by crystallization. analysis
fractionation
(CRYSTAF) using a CRYSTAF 200 unit commercially available from PolymerChar,
Valencia, Spain. The samples are dissolved in 1,2,=1 trichlorobenzene at 160 C
(0.66 mg/mL)
for I hour and stabilized at 95 C for 45 minutes. The sai-nplinl; temperatures
range t:rom 95
to 30 C at a cooling rate of 0.2 C!min. An infrared detector is used to
measure the polymer
solution concentrations. 'rhe cumtilative soluble concentration is measured as
the polymer
crystallizes while the temperature is decreased. The analytical derivative of
the cumulative.-
proti.le reflects the short chain branching distribtrtion of the polymer.
[01091 The CRYSTAF peak temperature and area are identified by the peak
analysis
module included in the CRYS'I'AF Software (Version 2001.b, Polyir-erChar,
Valencia,
Spain). T'he CRYSTAl' peak finding routine identifies a peak temperature as a
maximum in
the dWIdT curve and the area between the largest positive inflections on
e:ither side of the
identified peak in the derivative curve. To calculate the CRYSTAF curve, the
preferred
processing parameters are with a temperature limit of 70 C and with smoothing,
paraz~.eters
above the temperaturc, Iii-nit of 0.1. and below the temperature limit of 0.3.
DSC Standard Method (Excluding Samples 1T=1 and A-C)
101.101 Differential Scanning Calorimetry results are determined using a TAI
model
Q1000 DSC equipped with an RCS cooling accessory and an autosampier. A
nitrogen pur"e
gas flow of 50 rnI/'min is used. The sample is pressed into a thin film and
melted in tfle press
at about 175'C and then air-cooled to room temperature (25 C). 3-10 mg of
material is then
cut into a 6 nim diameter disk, accurately vveighed. p[aced in a light
aluminum pan (ca 50
rng), and then crimped shut. 7"he thermal behavior of the sample is
investigated with the
tolln,~ving te.niperattiire profile. The sample is rapidly heated to I80 C and
held isotherrnal for
3minute.s in order to reniove any previc>tls thermal historv, -1`he sample is
theii cooled to -
, - . .. 3 ' i v"5t.'sa .y~ ~ i.. , '' ~
a. : ~i ~ : ? ~~I~ ~d I' t ~t~` C t ~"te s~iIY1P,~t is ~a~.t ,'~~~ <~t~ I t
~., ,. .
i 3t.;- ~C at :(~,~...1~~~in. l.~:ati~~~~ rate, 1_1_ . ..r~~ s d re r
.,:~i_
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
[01111 The DSC melting peak is measured as the maximum in heat now rate {W,`g}
with
respect to the linear basc:linedrawn betkNreen --'?O C and end of melting. The
heat of fusion is
measured as the area under the melting curve between -30 C and the end of
melting using a
linear baseline.
GPC Method (Excluding Samples 1-4 and A-C)
101121 The gel permeation ellrom.a.tographic system consists of either a
Polymer
Laboratories Model PL-2 10 or a Polymer Laboratories Model PL-220 instrument.
'l'lie
column and carousel compartments are operated at 140 C. Three Polymer
Laboratories 10-
micron Mixed-B columns are used. The solvent is 1,2,4 trichlorobenzene. The
samples are
prepared at a concentration of 0.1 grams o#'polymer in 50 milliliters of
solvent containing
200 ppm of butylated hydroxytoluene (BHT). Samples are prepared by agitating
lightly for 2
hours at 160 C. The injection volume used is 100 microliters and the flow rate
is 1.0
rnl /rninute.
[01131 Calibration of the GPC column set is pertormed with 21 narrow molecular
weight
distribution polystyrene standards with molecular weights ranging from 580 to
8,400,000,
arranged in 6"cocktail" mixtures with at least a decade of separation between
individual
molecular weights. The standards are purchased from Polymer Laboratories
(Shropshire,
UK). The polystyrene standards are prepared at 0.025 grains in 50 milliliters
of solvent for
molecular weights equal to or greater than 1,000,004, and 0.05 grar-ns in 50
milliliters of
solvent For molec.ul.ar weights less than 1,000,[100. The polystyrene
standards are dissolved at
80 C with gentle agitation for 30 mirtutes. The narrow staridards mixtures are
rur, f'irst and in
order of decreasing highest molecular weight component to minimize
degradation. The
polystyrene standard peak inolecular weights are, converted to polyethylene
molecular
weights using the following equation (as described in Williams and Ward, J.
Polym. Sci.,
Polvm. Let., 6, 621 (1968)): Mpj:jsetj,,.-c õc =- 0.431(Mp,,i}-,tyr nd=
101141 Polyethylene equivalent molecular w-ei~;~.t calculations are performed
using
Viscotek TriSEC- software Version 3Ø
Compression Set
101151 Ctj;rt-i 'on 5 measured according to AS"1'M D-395. The, sample is
prepared
, _. 2.0 .
l _~
~IlId s~ e; [~l .2 Iiii7la ~.'.~~ I~2~"13_ and ~~'.,5 ti:~.I'~i
t:~I~.i~Y~~;1d~it:
, ... l IIN ni:ys of 12. .` . - ,G .~~ . .. , .~ ~cs are ._i'i__ ..~.f ,.. .
...,.~ Ã~M i- }24i3idQ
( 7_ ., . I':, x t'. f
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
minutes at 190 C, followred by 86 MPa for 2 minutes at 190 C. followed by
cooling inside
the press with cold running water at 86 MPa.
Density
[01161 Samples for density measurement are prepared according to ASTM l) 1928.
Measurements are made within one hour of sample pressing using ASTM I3792,
Method B.
F[exural/Secant MOClulus/ Storage Modulus
[0117] Samples are compression molded using ASTM D 1928. Flexural and 2
percent
secant moduli are measured according to ASTM D-790. Storabe modulus is
measured
according to ASTM D 5026-01 or equivalent technique.
Optical properties
[01181 Films of 0.4 mm thickness are compression molded usitag a hot press
(Carver
Model #4095-4PR1001 R). `T`he pellets are placed between
polytetrafluoroethylene sheets,
heated at 190 C at 55 psi {380 kPa) for 3 minutes, followed by 1.3 MPa for 3
minutes, and
then 2.611iPa for 3 minutes. The film is then cooled in the press with running
cold watcr at
1.3 MPa for 1mintite. 'I'lie compression molded films are used for optical
measiire.rrm.ea:ts,
tensile behavior, recovery, and stress relaxation.
[01.191 Clarity is measured using BYK Gardner Haze-gard as specified in AS"1"M
D 1746.
101201 45 gloss is measured using BYK Gardner Cilossinete.rMicrogloss 45 as
specified in ASTM D-2457.
[0121) Internal haze is measured using BYK Gardner Haz.e-gard based on ASTM D
1003
Procedure A. Mineral oil is applied to the filrn surface to remove surface
scratches.
Mechanical Properties - Tensile, Hysteresis, and Tear
[01221 Stress-strain behavior in uniaxial tension is measLired using ASTM D
1708
microter-sitL specimens. Samples are stretched with an. Instron at 500% inir-
I at ? 1 C;.
Tensile strength and elongation at break are reported froni an average of 5
specimens.
[0123] 100% and 300% o I-lyste.resis is determined from cyclic loading to 100%
and 300%
stnains usin _ ASTM D 1-08 microtensilkz sp~ %.in'ie:ns with an ln,tron-1-m
instrument. Th~
3 Cvclic exl-_,
~
aji.t'''ct a?1L trI the 'cFWC expci>~_tii~, ~~1c
sailiple 1a '. ijtl j'.: 1ir the ici cTT2nerattirE be foi ~ ie52Sm, . In the
.~.~-
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
2FC, 300% strain cyclic experiment, the retractive stress at 150% strain irom
the #irst
unloading cycle is recorded. Percent recovery for all experinae=nts are
calculated from the
first unloading cycle using the strain at which the load rettLrned to the base
line. 'The percent
recovery is defined as:
E. ~
%Recoti-ery= x100
where cf is the strain taken for cyclic loading and Fti is the strain where
the load returns to the
baseline during the 1" unloading cycle.
101241 Stress relaxation is measured at 50 percent strain and 37 C for 12
hours using an
InstronTM instrument equipped with an environmental chaniber. The gauge
geometry was 76
m.m x 25 mm x 0.4 mm. After equilibrating at 37 C for 45 rnin in the
environmental
chamber, the sample was stretched to 50% strain at 333% min.-~. Stress was
recorded as a
function of time for 12 hours. 'T'he percent stress relaxation after 12 hours
was calculated
using the formula:
% xS-tress Relaxation ~ l G~~ lv x 100
LO
where Lo is the load at 50% strain at 0 time and L12 is the load at 50 percent
straizl after 12
hours.
101251 Tensile notched tear experimeiits are carried oiit on samples having a
density of
0.88 glcc or less using an In,stronT~ instrument. 'Tlze geomc try consists of
a gauge se ctioii of
76 mm x 13 mm x 0.4 m.rn witli a 2 mm notch c-Lit into the sample at half the
specirnen length.
'I'he saznple, is stretched at 508 rnm min-' at 21 C until it breaks. The
tear energy is
calculated as the area under the stress-elongation curve up to strain at
maximum load. An
average of at least 3 specimens are reported.
TMA
101261 Thermal X-1eclzanical Analysis (Penetration TernperatLrre) is conducted
on 30tnm
diameter x 3.3 mi-n. thick, coilipres5ion molded discs, formed at 18Ã1 C and
10 MPa molding
i tr 111
~ 1i t 4aT'd then air q3It r1Clled. i i i'i2St1'i Tn=: `n a a ImCr. 1.5
)416')is
api)iiCl,i iii talc' ~tI ldi ti of the `iYS.21:1pa.: disc with . ;'~ . I'h.'
is I'aI:~," at _5 CP11I1
34
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
from 25gC. The probe penetration distance is measured as afunction of
temperature. The
experiment ends when the probe has penetrated 1 mm into the sample.
DMA
101271 Dynamic Mechanical Analysis (DMA) is measured on compression molded
disks
formed in a hot press at 180 C at 10 MPa pressure for 5 minutes and then water
cooled in the
press at 90 C f min. Testing is conducted usiiig an ARFS controlled strain
rheometer (TA
instruments) equipped with dual cantilever fixtures for torsion testing.
101281 A 1.5mm plaque is pressed and cut in a bar of dimensions 32x12mm. The
sample
is clamped at both ends between fixtures separated by lflmm (grip separation
AL) and
subjected to successive temperature steps from -lOQ C to 200` C(S"C per step).
At each
temperature the torsion modulus G' is measured at an angular frequency of 10
radfs, the
strain amplitude being maintained between 0.1 percent and 4 percent to ensure
dlat the torque
is sufficient and that the measurement remains in the linear regime.
101291 An initial static force of 10 g is maintained (auto-tension mode) to
prevent slack in
the sample when thernial expansion occurs. As a consequence, the grip
separation. AL
increases with the temperature, particularly above the melting or softening
point of the
polymer sample. "rhe test stops at the maximum temperature or when the gap
betvvc:en the
fixtures reaches 65 mm.
Melt Index
[01301 ti=leit index, or I2, is measured in accordance with ASEM D 1238,
Condition
190"C!2.16 kg. Melt index, or I70 is also measured in accordance with ASTM D
1238,
Condition 1)0 C110 kg.
ATREF
101311 Analytical temperature risintz elution fractionation (ATREF) analysis
is conducted
accordinc, to the method described in U.S. I'a.teiit No. 4.798,081 and Wilde,
L.; Ryie, 1".R.;
Knobeloc-h, D.C-.; 1'eat. l.R.. Determination qf'Brcrnchinl,; Dislt-ihittion.S
in Pol-teth.ylere and
Ethy,Iene C."opolylm~.~r-s. J. Polym. Sci., 20, =141-455 (1 982), which are
incorporated bv
reference hereira i i t1-1: r:~ ~tir::ty. The cc~i-npositi.oii to be analyzed
is dissolved in
trichl~?r l~h~~~ed c: ='l=~~e i11 ,~ ~,ti = 4teÃ,1 sli".) i: ltiilng E l~; i
;:vZ~3 ~~e =_. `LK ~. _
. . . . ,
c5~ ~1.$ili~1 .e'... ~ ~.
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
generated by eluting the crystallizeti pÃ~lyiner sample from the column by
slowly increasing
the temperature of the eluting solvent (trichlorobenrene) from 20 to 120 C at
a rate of
13C NMR A.nalysis
101321 The samples are prepared by addin~,7approximatelv 3a of a 50.1150
mixture of
tetrachloroethane-d`: orthodichlorobenzcrre to 0_4 g sample in a 10 mm NM.R
tube. The
samples are dissolved and homogenized by heating the tube and its contents to
150 C. The
data are collected using a JEOI_ F'clipseT'" 400MHz spectrometer or a Varian
Unity I'lusTM
400MHz spectrometer, corresponding to a$3C resonance frequency of 100.5 MHz.
The, data
are acquired usin, 4000 transients per data file with a 6 second pulse
repetition delav. To
achieve minimum signal-to-noise for qLiantitative analysis, multiple data
files are added
together. The spectral width is 25,000 I-Iz with a minimum tile size of 32K
data points. The
samples are analyzed at 130 C in a 10 mm broad band probe. The comonomer
incorporation
is determined using Randall's triad method (Randall, J.C.; JMS-Rev. Macromol_
Chem.
Phys,, C29, 201-317 (I989), which is incorporated by reference herein in its
entirety.
POtymer Fractionation by TREF
[01331 Large-scale TREF fractionation is carried by dissolving 15-20 g of
polymer in 2
liters of l,2,4-trichlorobenzene (TCB)by stirring for 4 hours at 1 60 C. The
polymer soIutioll
is forced by 15 psig (100 kPa) nitro(len onto a 3 inch by 4 foot (7.6 c-m x 12
cnr) steel column
packed with a 60:40 (v:v) mix of 30-40 mesh (600-425 lzm) spherical, technical
quality glass
beads (available from Potters Industries, H.C 30 Box 20, Brownwood, TX, 76801)
and
stainless steel, 0.028" (0.7mm) diameter cut wire shot (available from
Pellets, Inc. 63
Industrial Drive, North Tonawanda, NY, 14120). The column is immersed in a
thermally
controlled oil jackEt, set initially to 160 C:. The column is first cooled
ballistically to 125 C,
then slow cooled to 20 C at 0.04 C per ii7iuute and held for one hour. Fresh
"I`C.B is
introduced at about 65 ml;r$nin while the teinperature is increased at 0.167 C
per minute.
101341 Approximately 2000 inI portions of eltza.nt from the preparative TREF
column are
collected in a 16 station, heated fractiori collector. 'rhe poiN"mer is
concentrated in each
fraction using, a rotary evaporator until about 50 to 100 nil of the polynier
solution renlains.
-,cbor %~ sitl~.r$'-L~ ~.,~i e, s 6t<,';.;$
` $S ! !';: ~. ' LC$$1
:i.. _4 1
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
polytetrafluoroethylene coated i:ilter paper (available from Osmonics Inc.,
Cat4
I50WP0475Q). Tbe; filtrated fractions are dried overnight in a vacuum oven at
60 C and
weighed on an analytical balance before further testing.
Melt Strength
(01.351 Melt Strength (MS) is measured by using a capillary rlieometer fitted
with a 2.1
mm diameter, 20:1 die with an entrance angle of approximately 45 degrees.
After
equilibrating the samples at 190 C for 10 minutes. the piston is rtzn at a
speed of 1
inch/minute (2.54 cmiminute). The standard test temperature is 190 C. 'T-he
sample is drawn
uniaxially to a set of accelerating nips located 100 mm below the die with an
acceleration of
2.4 mm/sec2. "[`he required tensile force is recorded as a function of the
take-up speed of the
iiip rolls. 't'he maximum tensile force attained during the test is defined as
the melt strength.
In the case of polymer melt exhibiting draw resonance, the tensile force
before the onset of
draw resonance was taken as melt strength. The melt strength is recorded in
centiNetutons
("cN"").
Catalysts
[0136) The term "overniglZt", ifused, refers to a time of approximately 1.6-18
hours, the
term "room temperature", refers to a temperature of 20-25 C, and the term
`n3ixed alkanes"
refers to a commercially obtained mixture of C6-9 aliphatic hydrocarbons
available ujider the
trade desionation Isopar E'( , .from. Exxon..Mobil Chemical Company. In the
event the name of
a compoLind herein does not conforrn to the structural representation
thereot'. the structural
representation shall control. The synthesis of all metal cornplexcs and the
preparation of all
screening experiments were carried out in a dry nitrogen atmosphere using dty
box
techniques. All solvents used were H1'LC grade and were dried before their
use.
[0137] MMAO refers to modified methylalumoxane, a triisobutylaluminum modified
met17vlalumoxane available commercially from Akzo-Noble Corporation.
101381 'l'he preparation ol' catalyst (B 1) is conducted as fol lows.
a) Yreparation ot (1-methvlethvl)('-bydroxv-3,5-di(t-
but~~l)phenvl)methti~linrine
S-Di-t-but% lsalie,,rlaldeh.yde (3.00 g) is added to 10 mL of isopropylam.ine.
'I'he
i' ~,r s ~t3tiirs.
Ã:;:u,r vaiC:uum C3 i ~.'.`b. St,lld ~Ã}t percent
-.~:
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
b) Preparation of 1,2-bis-"3,5-di-t-butvl henvlene) l- N- I-
methvlethvl immino Ãnethvl "2-oxovl zirconium dibenzvl
A solution of (I-m ethviethyl)(2-hydroxy-3,5-di(t-butyt)phen yl)iÃnirÃe (605
mg. 2.2
mmol) in w mL toluene is slowly added to a solution of Zr(CH2Ph)4 (500 mg, 1.1
rnmol) in 50
mL toluene. The resulting dark yellow solution is stirred for 30 minutes.
Solvent is removed
under reduced pressure to yield the desired product as a reddish-brown solid.
[0I391 The preparation. of catalyst (132) is conducted as follows.
a) Pre ara.tion of 1- 2-methvlcvclohex 1 eth l(2-oxovl-3.5-di t-butvl hen yl
imine
2-Methylcycl.ohexylamine (8.44 mL, 64.0 mmol) is dissolved in methanol (90
mL),
and di-t-butylsalicaldehyde (10.00 ~, 42.67 mmol) is added. The reaction
mixture is stirred
for three hours and then cooled to -25 C for 12 hours. The resulting yellow
solid precipitate
is collected by filtration and washe.-d with cold methanol (2 x 15 mL), and
then dried under
reduced pressure. The yield is 11. 17 g of a yellow solid. 'T-I NMR is
consistent with the
desired product as a mixture of isomers.
b) Preparation of bis- l- 2-metilvlcvclohex rl e.th -1 2-oxo yl-3.5-di t-but 1
hen l
immino zircorÃium dibenzyl
A solution of (1-(2-Ãnethti-lcyclohexyl)ethyl)(2-ox,oyl-3.5-di(t-
butyl)phÃ:nyl)im.ine
(7.6:3 g, 23.2 nimol) in 200 mL toluene is slowly added to a solution of
Zr(CH2Ph)4 (5.28 ~.
I 1.6 mmoi) in 600 niL toluene. 'The resultinl; dark yellow solution is
stirred for 3 liour at
25 C. The solution is diluted further with 680 mL toluene to give a solution
having a
concentration of 0.00783 M.
[01401 Cocatalyst I A mixture of rnethyldi(Ci4_1 y alkyl)aÃnmonium salts of
tetrakis(pentaflLÃorophenyl)borate (here-in-after armeenium boratc). prepared
by reaction of a
I.on, chain trialkylamine (Armeen"'4 M2H"T'. available froÃn. Akro-Niabel,
Inc.). FiCI and
Li[T3(Cf;P,)41, substantially as disclosed in USP 5,919,9883, Ex. 2.
101411 Cocatalyst 2 Mixed C14-,s alhyldimethvlanmonium salt of
bis(.tris{Pentli:luorophenyl)-alÃtmane)-2-undecvli.midarolide, prepared
according to USP
6e' )9 ~ f_... .5.
10I421 DF/ 1
SA~A2;, di{o lt~~:, iC ~;~.~,. =~)a
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
trioetylaluminum (SA5), trietbylgallium (SA6), i-butylalurninum bis(dimethyl(t-
butyl)siloxane) (SA7), i-butylaluminum bzs(di(trimetbylsilyl)arriide) (SA8), n-
octylaluminum
di(pyridine-2-r-nethoxide) (SA9), bis(n-octadecyl)i-butylalun.-iarzum (SA10),
i-butylaluminum
bis(di(n-pentvl)arnide) (SAl I), n-octylaluminurn bis(2,6-di-t-butylphenoxide)
(SA1~.?), n-
octylaluminunr di(ethyl(1-naphthyl)amide) (SA13), ethylaluminum bis(t-
butyldimethylsiloxide) (SA 14), ethylaluminum di(bis(trimethylsilyl)amide) (SA
15),
ethylaluminum bis(2.3,6,7-dibenzo-l-azacycloheptarzeamide) (SAl6), n-
octylaluminum
bis(2,3,6,7-dibenzo- l-azacycloheptaneamide) (SA 17), n-octylaltiminum
bis(.dimethyl{t-
butyl}siloxide(SA 18), ethylzinc (2,6-diphenyiplrc:noxide) (SA 19), and
ethylzizrc (t-butoxide)
{SA20}.
Exaanples T.-4. Comparative A-C
General High Throughput Parallel Polymerization Conditions
[01431 Polymerizations are conducted using a high throughput, parallel
polymerization
reactor (PPR) available from Syrnyx "T-eclrnologies, Inc. and operated
substantially according
to US Patents No. 6,248,540, 6,030,917, 6,362,309, 6,306,658, and 6,316,663.
Ethylene
copolymerizations are conducted at 130 C and 200 psi (1.4 MPa) with ethylene
on demand
using 1.2 equivalents of cocatalyst 1 based on total catalyst used (1.1
equivalents when
MMAO is preseiit). A series ot'polymerizations are conducted in a parallel
pressure reactor
(PPR) contained of 48 individual reactor cells in a 6 x 8 array that are
fitted with a pre-
weighed glass tLibe. The working volume in each reactor cell is 6000 ~iL. Each
cell is
temperature and pressure controlled with stirring provided by individual
stirring paddles.
'T"he monomer gas and quench gas are plumbed directly into the PPR unit and
controlled by
automatic valves. Liquid reagents are robotically added to each reactor cell
by syringes and
the reservoir solvent is niixed alkanes. l`he order of addition is mixed
alkanes solvent (4 ml),
ethylene, T-octe-ne comojiomer (t ml), cocatalyst I or cocatalyst 1/E9?1!IAO
mixture, sliuttling
agent, and catalyst or catalyst niixture. When a mixture of cocatalyst I and
MMAO or a
mixture of t,~vo catalysts is Ãised, the reagents are premixed in a sniall
vial immediately prior
to addition to the reactor. When a reagent is omitted in an expe.riment, the
above order of
addition is otherwise rnaintaizied. Polymerizations are conducted for
approxirnate-ly 1-2
nii: 1 nii l prt ri-: ~n~:!:,ie cc>ns_il t1 ls a_ reac.bi J.
.L`,:._ . ~ . .. ..... , , . .i.. .... ~ _..., ..,
. . . . Y 3 ,. ... , r. . .._,~s
_..,
zi ut"twC~ ~t~i i_ =_ts~it~ c~t ["3 ~ ~.. ~ e dried
.~~)~
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
polymer are weighed and the difference between this weight and the tare wei-ht
gives the net
yield of polymer. Results are contained in`T'able 1. In Table 1. and elsetivl-
ierc in the
application, comparative compounds are indicated by an asterisk (*).
[0144] Examples 1-4 demonstrate the synthesis of linear block copolymers by
the present
invention as evidenced by the t'orinat.iou of a very narrow MWD, essentially
monomodal
copo[ynier when DEZ is present and a bimodal, broad molecular weight
distribution product
(a mixture of separately produced polymers) in tbe absence of DEZ. Due to the
fact that
Catalyst (Al ) is knotkPn. to incorporate more octene than Catalyst (B 1), the
different blocks or
segments of the resultinM copolymers of the invention are distinguishable
based on branching
or density.
Table I
Cat. (A 1) Cat (B1) Cocat M.V1AO shuttling
Ex. mo[ unol ( znol) Lmo[ agYÃ.n.t (vmoL) Yield ~, Mn Mw,M17 heylsl
A* 0.06 - 0.066 0.3 - 0.1363 300502 3.32 -
B* - 0.1 0.110 0.5 - 0.1581 36957 1.22 2.5
C* 0.06 0.1 0,176 0.8 - 0.2038 45526 5.30' 5.5
1 0.06 0.1 0.192 - DEZ (8.0) 0.1974 28715 1.19 4.8
2 0.06 0.1 0.192 - DBZ (80.0) 0.1468 2161 1.12 14.4
3 0.06 0.1 0.192 - '1"C:A (8.0) 0.208 22675 1.71 4.6
4 0.06 0.1 0.192 - TEA (80.0) 0.1879 3338 1.54 9.4
1 C6 or higher chain content per 1000 carbons
2 Bimodal molecular weight distribution
[0145] It may be seen the polymers produced according to the invention have a
relatively
narrow polvdispersi.ty (Mw/Mn) and larger block-copolymer content (trimer,
tetramer, or
larger) than polymers prepared in the abseiace of the shuttling agent.
101461 F'ur-ther characterizing data for the polvmers of Table 1 are
determined by
reference to the figures. More specifically DSC and A`C'REF results show the
following:
101471 The DSC curve for the polvrner of example 1 shows a l 15.7 C melting
point (Tzn)
with a heat of fusion of 158.1 .l;'g. The corresponding CRYSTAF curve shows
the tallest
peak at 34.5 C with a peak area of 52.9 percent. -hbe difFerence between the
DSC Tm and
tbe Tcrystaf is 81.2 C.
101481 The DSC curve for the polymer of example 2 shows a peak with a 11)9.7 C-
me-lting point (Trn) with a heat of fusion of 214.0 .i.'g- Tbe corresponding
CRYSTAF curve
the tallest peak x '0.2'C with a pcak areao;~57.() percent. The di.fic.reiiwe
betv"een the
~ C, 0
11 alnd ~.-
40-
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
(0149] The DSC curve for the polymer of example 3 shows a peak with a 120.7 C
melting point (Tm) with a heat of fusion of 160.1 Jtg. The corresponding
C.RYSTAF curve
shows the tallest peak at 66.1 C with a peak area of 71.8 percent. Tbe
difference between the
DSC Tm and the Tcrystaf is 54.6 C.
(0150] The DSC curve for the polymer of example 4 shows a peak with a 104.5 C
melting point (Tm) with a heat of fusion of 170.7 JIg. The corresponding
CRYSTAF curve
shows the tallest peak at 30 C with a peak area of 18.2 percent. The
difference between the
DSC 'I'm and the Tcrystaf is 74.5 C.
101511 The DSC curve for comparative A shows a 90.0 C melting point (Tm) with
a heat
of fusion of 86.7 Jib. "I"he corresponding CRYSTAF curve shows the tallest
peak at 48.5 C
with a peak area of 29.4 percent. Both of these values are consistent with a
resin that is low
in density. The difference between the DSC Tm and the Tcrystaf is 41.8 C.
101521 The DSC curve for comparative B shows a 129.8 C melting point (Tm) with
a
heat of fusion of 237.0 J/g. The corresponding CRYSTAF curve shows the tallest
peak at
82.4 C witlr a peak area of 83.7 percent. Both of these values are consistent
with a resin that
is high in density. The difference between the DSC Tm and the T ervstaf is
47.4 C.
101531 The DSC curve for comparative C shows a 125.3 C meltint, point ("1'ni)
with a
heat of fusion of 143.0 Jj`g. '~t'he correspondin- CRYSTAF curve shows the
tallest peak at
81.8 C with a peak area of 34.7 percent as well as a lower crystalline peak
at 52.4 C-. The
separation between the two peaks is consistent with the presc ce of a high
crystalline and a
Eow crystalline polymer. The difference between the DSC 'htn and the 4'crystaf
is 43.3 C.
Examples 5-19 Comparatives D-F Continuous Solution P Ivmerization Catalyst
A1/B2 + DEZ
[0154] Continuous solution palyn-ierizations are carried out in a computer
controlled
autoclave reactor equipped with an internal stirrer. Purified mixed alkanes
solvent (lsapar~'m
E available from ExxonMobil Chemical C:nmpany), ethylene at 2.70 lbs/hour
(1.22 kg./hour),
1-octene, and by-drooen (where used) are snppfied to a 3.8 L reactor equipped
with a jacket
for telnperature control and an internal thermocouple. The solvent feed to the
reactor is
measured by ama.ss-f1ow controller. A variable speed Ãliaphragzn pump controls
the solv~:nt
Ae a I _ re to t~,~ At th_
;ide flush 1:1 t
Ov L,ont$roA
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
valves or by the rnanual adjustment of needle valves. "1'he remaining solvent
is combined
with l-octene., etb.ylene, and hydrogen (where used) and fed to the reactor. A
mass flow
controller is used to deliver hydrogen to the reactor as needed. The
temperature of the
solvent,'monomer solution is controlled by use of a heat exchanger before
entering the
reactor. This stream enters the bottom of the reactor. The catalNrst component
solutions are
metered using pumps and mass flow meters and are combined ~~ith the catalyst
flush solvent
and introduced into the bottom of the reactor. The reactor is run liquid-full
at 500 psig (3.45
MPa) with vigorous stirring. Product is removed through exit lines at the top
of the reactoz--
All exit lines from the reactor are steam traced and insulated. Polymerization
is stopped by
the addition of a small amount of water into the exit line along with any
stabilizers or other
additives and passing the mixture through a static rnixer. "1'he product
stream is then heated
by passing through a heat exchanger before devolatilization. The polymer
product is
recovered by extrusion using a devolatilizinl; extruder and water cooled
pelletizer. Process
details and results are contained in Table 2. Selected polymer properties are
provided in
'T'able 3.
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
= .r, rv L-,
..... ,.... - -. r^-3 v-~; - G3
, _.. N ..... ...E hr r, c~.i ., -- 11
[ C/? p ~ .....~ r.. ..... .... ,., " . ..... ..... .... .... =,-~ %'~ . ~ .-
~ ~ `
[( 7
~ .....
v ~ ^ ~ C'~.7 e+7 , .. ^Jp M co c~~ f^> h~ n rv r , -t m -7 C~ r9
~ aocrr~ ~, o"~ oa~ c; (7~.~.nc c o3_
~
r-- kn -rEn-^Nr-,r-ooea~.-o:~~n~r;:c,
- - - - _.. - - - - - - - .,., -
(.= ,J ^ t~7 CR ^ t`~ ~ C^~ V; d' G't ~F 6`GC7 CC C('rs 'L^
nt rn e - - rn N't V-)
ry
~~' .~ (~ C~ ^-^= Vr OG C3 f V ~4 ^~ L`= v t C C~+ .['~ Q GO ,r",'^~õ
p =.-~- "~ ^^ C~I -^^ Q ^-~ O C =- C? C ^ ~ ^^^
C.J i.zõ
M f^~ f+l rr7 m f^. fn rrl M 111 M CV
t-
~
rn N 41.
'i' "CS J
Lr 5 S C~ ~O e+~ 00 v~"
N O 7 J .T.. ^^ '"J N ^ ^ N =~
"~ ^"'W... X i C> C~..= O O , .. ^J C C v^ ... C7 ~.. C.=. . ~ """"
-Z crr fn r*; r+; . , rr? .. . , , , . .. _ . ~'.
7 7 7 7 -
y-
I,,,E ~n
r
=~ .:~ ~ ^
r= e^a "= ;~
rn
~v . . U (`i . . . . = . . hd F^7 C 1 t"~ C~7 N C'l C`f f`.E Ã
à . . . . . . . .~ ~. .....i J ~~ .~ ~
~.w .... :r h;
J
171 j ^' C3
~'-
i
7-
.: ~ ~ . .. = , , , , . , ~ . , t^Y . . - - _ _.. _
~ :~ ,- - -
. - .__. .- ,_, ._, - - -
43
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
I i ~ E E ~][ 3
n~~!ÃI~~O~O~C^, CS~!"'': ",J"N~Ãf;' ~N'^IÃ1'~~{~-~j'~= ~[3p'
à E 7 E
~- E
f
~~~) n a-~t3 ~~N ch ~ n et r~ ~^ oo N~ m;~;vy
N ^
]~.G~:aO`ao i~^1.-~ ~ o o^ cr c~t~x r~ ^
^ rn -- M L CJ ~O L` N
~ V r ~ rt CJ .^~ O N c' G~~ ^^
~l' Ãn O Ãr ~-. ~f' ÃYt ~G E ~t M ~' ^ ,~~./ r N'N~N ~-';N~N N -=~~ --~lCk ~
~fN N
. rh = .~ I ~" ^` '_'
,^~ ...
i. L ~ M ~ -~ ' v , U / . ~ `.C [~ n ~D ~I' 7 3 .~ g c'1 y" W
E~ T ri e~ ~~ r, r r= C7 0~ s~ w~ e^ e~ ^~1 ~t r~ .~
_ _
S. = O[O[Q O~_
,C+ -==..ltr, M ~J' - =N CyE~ S"'ijO-C^, '.~`i "~' .^~.lN `.:
G ~../,....:~ ~-f . v.^e: 'JQ [~Ã~ -2``~~1~-~
n !
;e! r{ II = .
s~""
~
~~,,,, '~. E r^ r G't I`^ ~ [ - ~~.,~ ~ ; t`-~ r. < - ~=~= . 73 ~ ~ . N
r~ I Ã ; E ! ~ ~ ~
L Ev':C ~ .~~, ~ ~C^d !=- ^~,~)v~F(--~ ~~
: ; `[-. -;=--k- --i~-~- r"~'r~, ...=~.~ ~r.
.~-__...._......._........._._ ..~
..,.___,..._.____,..~_~....~..,......_......._....,..s.._.......s.,~..rr....._.
_z._~.~ -
44
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
[01551 "I`he resultinL, polymers are tested by DSC and A.'I-'REE as with
previous exai-nples.
Resuits are as follows:
101561 The DSC curve for the polymer of example 5 shows a peak with a 119.6 C
melting point (Tm) with a heat of fusion of 60.0 J/g. The corresponding
CRYS`I"Ap' c-urve
shows the tallest peak at 47.6 C with a peak area of 59.5 percent. "I'he delta
between the DSC.
Tm and the Tcrvstaf is 72.0 C.
101571 The DSC curve for the polymer of example 6 shows a peak with a 11.5.2
C
melting point (Tm) with a heat of fusion of 60.4 J/g. "T'he corresponding
CRYSTAF curve
shows the tallest peak at 44.2 C with a peak area of 62.7 percent. The delta
between the DSC
Tm and the Tcrystaf is 71.0 C.
101581 The DSC curve for the polymer of exarnple 7 shows a peak with a 121.3
C
melting point with a heat of ftision of 69.1 J'g. The corresponding CRYSTAk'
curve shows
the tallest peak at 49.2 C with a peak area of 29.4 percent. The delta between
the DSC Tm
and the Tcrystaf is 72.1 C.
101591 The DSC curve for the polymer of'exarnple 8 shows a peak with a 123.5
C
melting point (Tm) with a heat of fusion of 67.9 11g. The corresponding
CRYSTAF curve
shows the tallest peak at 801.1 C with a peak area of 12.7 percent. The delta
between the DSC
Ti:n and the "Tcrvstaf is 43.4 C.
101601 The DSC curvc for the polymer of example 9 shows a peak with a 124.6 C
melting point (Taii) with a heat of tiusion of73.5 Jlg. The correspondilig
CRYSTAF curve
shows the tallest peak at 80.8 C with a peak area of 16.0 percent. The
deltabetwer;ii the DSC
"T'ir3 and the Tcrtistaf is 43.8 C.
101611 `I'he DSC curve for the polymer of example 10 shows a peak with a 115.6
C
rneltin- point (Tm) with a heat of ftision of 60.7 JAY. The corresponding
CRYSTAF curve
shows the tallest peak at 40t.9 C with a peak area of 521.4 percent. The delta
between the DSC
"I'm and the Tcrystaf is 74.7 C.
101671 Tbe DSC curve for the polymer of e.xa.mple. 11 shows a peak with a 1 B
.fi `C
l-nclting point (Tm) with a heat of ftision of 70.4 if-. The corresponding
CRYSTAF curve
sho-~vs the tallest peak at 39.6 C with a peak area of 25.2 percent. 'I`he
delta bew;=e:en the DSC
land the lcr~ i * s 74.1r.'.
ri ) with a heat t.i ! L: I i i}il.`% , i ti[. .: IRAf;tdrVe
_~. _
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
shows no peak equal to or above 30 C. (Tcrystaf for purposes of further
calculation is
therefore set at 34 C). The delta between the DSC Tm and the Tcrystaf is 83.2
C.
t01641 The DSC curve for the polymer of example 13 shows a peak with a 114.4
C
melting point (Tm) with a heat of fusion of 49.4 i'g. The corresponding
CRYSTAF curvYe
shows the tallest peak at 33.8 C with a peak area of 7.7 percent. The delta
between the DSC
Tm and the Terystaf is 84.4 C.
101651 The DSC for the polvmer of example 1.4 shows a peak with a 120.8 C
melting
point (Tm) with a heat of fusion of 127.9 J/(,,. The corresponding CRYSTAF
curve shows the
tallest peak at 72.9 C with a peak area of 92.2 percent. The delta between.
the DSC Tm and
the, Terystaf is 47.9 C.
[0166] The DSC curve for the polymer of example 15 shows a peak with a 1] 4.3
C
melting point (Tm) with a heat of fusion of 36.2 Pa. The corresponding CRYSTAF
curve
shows the tallest peak at 32.3 C with a peak area of 9.8 percent. The delta
between the DSC
Tm and the Tcrystaf is 82.0 C.
101671 The DSC curve for the polymer of example 16 shows a peak with a 116.6
C
melting point (; I'm) with a heat of fusion of 44.9 ,lIg. The corresponding
CRYSTAF curve
shows the tallest peak at 48.0 C with a peak area of 65.0 percent. The delta
between the DSC
Tm and the Tcrystaf is 68.6 C.
101681 The DSC curvc for the polymer of example 17 shows a peak with a 116.0
GC
meltin~ point (Tm) E~-ith a heat of fusion of 47.0 T/~,t. 'I`hc corresponding
CRYSTAF curve
shows the tallest peak at 43.1 C with a peak area of 56.8 percent. The: deita
between the
DSC Tm and the I'crystaf is 72.9 C.
101691 The DSC curve for the polymer of example 18 shows a peak witli a 120.5
C
melting point ("l`m) with a heat of fusion of 141.8 Jig. The corresponding
CRYSTAF curve
shows the tallest peak at 70.0 C with a peak area of 94.0 percent. The delta
between the
DSC Tm and the Tcrystaf is 50.5 C.
101701 'T'he DSC curve for the polymer of example 19 shows a peak with a 124.8
C
melting point (Trz) wit1i a heat of fusion of 174.8 J/g. l'be corresponding l;
CRYS"i,Af' curve
shows the tall;.st peak at '19.9 C with a peak area of 87.9 percent. The
delta between the
DSC ..i...I-1? . %,staf:=_s 45.tJ C
9
1~~1 74 ~ 4"h, l~~`t; ~- ~ ~t~. c.omp-arative D ~ neak
iiltCl= 'j.;= " ~' ~ ~ ; ~.~~
:~ ~ ~vi~
-416-
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
shows no peak equal to and above 30 C. Both of these values are consistent
with a resin that
is low in density. The delta betvveen the DSC Tm and the Tcrystaf is 7.3 C.
[01721 The DSCcurve for the polymer of comparative E shows a peak with a 1~4.0
C
melting point (Tm) with a heat of fusion of 179.3 Jig. The corresponding
CR.YSTAF curve
shows the tallest peak at 79.3 C with a peak area of 94.6 percent. Both of
these values are
consistent with a resin that is high in density. The delta between the DSC Tm
and the
Tcrystaf is 44.6 C.
101731 "I`he DSC curve for the polymer of comparative F shows a peak with a
11.4.8 C
meltin- point (Tm) with a heat of t'usion of 90.4 J/g. The corresponding
CRYSTAF curve
shows the tallest peak at 77.6 C with a peak area of 19.5 percent. The
separation. between the
two peaks is consistent with the presence of both a high crystalline and a low
crystalline
polymer. The delta between the DSC Tm and the Tcrystaf is 47.2 C.
Physical Property Testing
101741 Polymer samples are evaluated for physical properties such as high
temperature
resistance properties, as evidenced by TMA temperature tzstinf-), pellet
blocking strength,
high temperature recovery, high temperature compression set and storage
modulus ratio,
G'(25"C )'G'( l QO C)- Several coinmercialiy available polymers are included
in the tests:
Comparative G* is a substantially linear ethyle:ne/ 1-octerie copolymer
(A.pp'IN'I"I`Y?J,
available 1'rom"I'he Dow Chemical Company), Comparative H* is an elastomeric,
substantially linear ethylene/ 1-octene copolymer (AhTINITY'3~EG8if)0,
available frorn `I"he
Dow Chemica[ Company), Comparative I is a substantially linear ethylene%1-
octene
copolymer {AFFTtiITY*PLI840, available from The Dow Chemical Company),
Comparative J is a hydrogenated styrene/butadienelstyrene triblock copolymer
(KRA"I'ONTM
G1652, available from KRA'I'ON Polymers). Comparative K is a thermoplastic
vulcanizate
('I'PV, a polyolefin blend containing dispersed therein a crosslinked
elastomer). Restilts are
presentecl in Table 4.
-47-
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
`1`able 4 l-ligb. Temperature Mechanical Properties
~ T1vIA-imrn Pellet Blocking 300 % Strain Coinpression
penetration Strengtkt G'(25 C); Recoverv (80 C) Set (70 C) Ex. { C) lb/Ft
(kPa) G'(1Ã)0 C) ( erce:nt) recr~t)
D* 51 9 Failed
E* 130 18
F~ 1
141(6,8) 9 Failed 1.00
5 104 (}(0) 6 81 49
6 110 - 5 i 152
7 li3 - 4 84 43
18 1.11 4 Failed j41
9 97 4 66
10 108 - 5 81 55
11 100 - 8 - 68
12 88 - 8 - 79
13 95 - 6 84 71
14 125 - 7 - -
96 5 58
16 113 4 - 42
17 108 0(4) 4 82 47
18 125 - 10 - -
19 133 - 9 - -
G* 75 463 (22.2) 89 Failed 100
H* 70 213 (14.2) 29 Failed 100
1* H1 tl - -
J* 107 5 Failed 100
K* 152 - 3 40
101751 In Table 4, Comparative F (which is a physical blend of the two
polymers
resultino froni simultaneous polymerizations using catalyst A1 and 131) has a
1 m.ni
penetration temperature of about 70 C, while Examples 5-9 have a lmin
penetration
temperature: of 100 C or greater. Further. examples 10-19 all have a 1 mm
penetration
temperature of greater than 85 C-, with most having 1 mrn "i`MA temperature of
greater than
90"C or even greater than 100 C. This shows that the novel polymers have
better
dimensional stability at higher temperatures compared to a physical blenc[.
Comparative J (a
commercial SEBS) has agooci 1 mm TMA temperature of about 107'C, but it has
very poor
(high temperature 70"C) compression set of about 100 percent and it also
failed to recover
(sample broke) during a high temperature (80'C. ) 300 percent strain recovery.
Tlius the
exemplified polymers have aunique, combination of properties unavailable even
in some
cominerciallv available, high performance thermoplastic elastomers.
'01761 Sinnila=1~. i _ a'. (ot)od: tz . { ._ r.._. -..
?)`"C"}, f-t
(Comparative F) ba:s a st(iri:tE?;"s.', I.i.l{7(.it,IeL1` eiii'k) =. 'a
~cxtttiC)z.i, ~ iiz` ~,A~'YI"t:r
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
(Comparative G) of similar density has a storage modulus ratio an order of
tnagnitude greater
(89). It is desirable that the storage modulus ratio of a polymer be as close
to 1 as possible.
Such polymers will be relatively unatTected by temperature, and fabricated
articles made
from such polymers can be usefully employed over a broad temperature range.
This feature
of low storaue anodulus ratio and temperature independence is particularly
useful in elastomer
applications such as in pressure sensitive adhesive formulations.
101771 The data in Table 4 also demonstrate that the polymers of the invention
possess
improved pellet blocking strengtb. In particular, Example 5 has a pellet
blocking strength of
0 MPa, meaning it is free t7.owing under the conditions tested, compared to
Comparatives F
and G which show considerable blocking. Blocking strength is important since
bulk
shipment of polymers having large blocking strengths can result in product
clumping or
sticking together upon storage or shipping, resulting in poor handling
properties.
[01781 High temperature (70 C) compression set for the inventive polymers is
generall.y,
good, meaning generally less than about 80 percent, preferably less than about
70 percent and
especially less than about 60 percent. In contrast, Comparatives F, G, 11 and
J all have a 70 C
compression set of 100 percent (the max:imum possible value, indicatino no
recovery). Good
high temperature compression set (low numerical values) is especiallv needed
for
applications such as gaskets, window profiles, o-rings, and the like.
-49-
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
à _ 3 t 4 ? E 9 u _ [[ ~-~ à à 3 ~ "" [
/- ~.^~'~ .- t{ I 3~M[ 1 1 F I. s~M~ s 1Ã -~
E (h E I! Ã ~ Ã
, ra <r~r~; ^ c~~, n ri r=n -~ ~t F r~ ~ ~ ~c~
~J V:~ --.<<3' ~'-' C'd C`3 { C`3 r-. =====' C`~ i'~"f' '- i 1~ hI f! f~ N M
L c ^ ^c -c'c ^c~c ^ ~- ~ca c
~ ca ~c~~ o0
IC
~'~ CV /~ xi 1`i n n n r~ n.t~.. n x Pa Co ~] :c J~
Y. {
tN ~w^ c~
~/.= I.'L C~F ~_. ~ n~ 00 ~O -Y~. ~9 ~O Ci`-. Cf . 1 ~0 GG ^ I x CO 3
cl~
~ L ^~ ~ aC w
! > F (C ~' ~
on[~n 00 t-- "-.
C
co ~a . x o~ ..... ..~~. [ - oc x
~ Ã [ Ã~
ci ~r ~.~-^Ã h ( [
o~=ri `[~[ t' r~r~E r~^ n lr~:j~. :N~r;3^ v(n r;~^ N
~.. . a c,;
:ct
>: ~ 1 ~`` IIE E
{ f = j f ~ - :~J
. . . - - - . , - . - .. - . .. ~.y.i r . C1 50
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
101791 Table: 5 shows results for mechanical properties for the new pollvmers
as well as
for various comparison polymers at ambient temperatures. It may be seen that
the inventive
polyrners have very good abrasion resistance when tested aecordint-, to ISO
4649, generally
showing a volunie loss of less than about 90 mzn', preferably less than about
80 mm', and
especially less thazz about 50 mm'. In this test, higher numbers indicate
higher volume loss
and consequently lower abrasion resistance,
t0I801 Tear strength as measured by tensile notched tear strength of the
inventive
polymers is generally 1000 mJ or higher, as shown in Table 5. Tear strength
for the
inventive polymers can be as high as 3000 mJ, or even as high as 5000 mJ.
Comparative
polymers generally have tear strengths no higher than 750 W.
101811 Table 5 also shows tl-iat the polyniers of the invention have better
retractive
stress at 150 percent strain (demonstrated by higher retractive stress values)
than some of'
the comparative samples. Comparative Examples F, G and li have retractive
stress value at
150 percent strain of 400 kPa or less, while the inventive polymers have
retractive stress
values at 150 percent strain of 500 kPa (Ex. 1.1) to as high as about l 100
1CI'a. (Ex. 17)-
I'olymers havinc) higher than 150 percei-zt retractive stress values would be
quite usefiil for
elastic applications, such as elastic fibers and fabrics, especially nonwoven
fabrics. Other
applications include diaper, hygiene, aiid medical garment waistband
applications, such as
tabs and elastic bands.
101:821 Table 5 also shows that stress relaxation (at 50 percent strain) is
also improved
(less) for the inventive polyr-ne.rs as coinpared to, for Lxample, Comparative
G. Lowe-r
stress relaxation nieans that the polymer retains its force better in
applications such as
diapers and other garments where retention of elastic properties over long
time periods at
body temperatures is desired.
_~~-
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
O ti~esting
Table 6 f'oEymer Optical Properties
Ex. Internal Haze (percent) Clarity ( ercetit) 45 Gloss (percent)
p* 84 22 49
G* 5 73 56
13 72 1 60
' 6 33 69 53
7 28 57 59
8 20 65 62
9 61 38 49
15 73 67
11 13 69 67
12 8 75 72
1 a 7 74 69
14 59 15 62
IS 11 74 66
16 39 70 65
17 29 73 66
18 61 22 60
19 74 11 52
G* 5 73 56
il* 12 76 59
1* 20 75 59
101831 The optical properties rcporteci in Table 6 are, based on compression
molded
films substantially laekin!"; in orientation. Optical properties of the
polyiners may be varied
over wide ranges, due to variation in crystallite size, resulting from
variaÃiori in the cltianTity
ofc:hain shuttling agent employed in the polymerization.
Extractions of Multi-Block Co iFiners
[0184] Extraction studies of the polymers of.'examples 5, 7 and Comparative E
are
conducted. In the experiments, the polymer sample is weighed into a glass
fritted e_xtraction
thimble and fitted into a Kumagakva type extractor. The extractor with sample
is purged
with nitrogen, and a 50OmL round bottom flask is charged with 350 rnL of
diethyl ether.
The flask is then rit.teÃI to the extractor. "1`he ether is heated while being
stirred. Tiineis
noted ~N-b.en the ether begins to condense into the tbftnble, and the
extraction is allowed to
proceed under n.ifrogen for 24 hours. At this time, heating is stopped and the
soluti.on is
allowed to cool. Any etlier reniaining in the extractor is returned to the
flask. 'I'he ether in
tl n a. 4N..i-" 7~ _ rc3t121 t~, ~~ _ s a
nse C 5 ',
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
pur,(-,e, and the residue dried under vacuum ove,rrriaht at 40 C. Anv
remaining ether in the
extractor is purged dry with nitrogen.
101851 A second clean round bottom flask charged with 350 ml, of'hexane is
then
connected to the extractor. The hexane is heated to reflux with stirring and
maintained at
reflux for 24 hours after hexane is first noticed condensing into the thimble.
Heating is then
stopped and the flask is allowed to cool. Any hexane remaining in the
extractor is
transferred back to the flask. The hexane is removed by evaporation under
vacuum at
ambient temperature, and any residue remaining in the flask is transferred to
a weighed
bottle using successive hexane washes. The hexane in the flask is evaporated
bv a nitrogen
purge, and the residue is vacuum dried overnight at 40 C.
[01861 The polymer sanlple remaining in the thimble after the extractions is
transferred
from the thimble to a weighed bottle and vacuum dried overnight at 40 C.
Results are
contained in "Fable 7.
Table 7
etlaer ether C8 hex aai hexane C~ residue
~~t. soluble solLible mole soluE~le soluble i~aolc. C~ mole
Sani Ee (g) (g) (ercent) percent' f(percent) ercerlt perccnt
C.omp. 1.097 0.063 5.69 12.2 0.245 22.35 13.6 6.5
F*
k~x. 5 1.006 0.041 4.08 - 0.040 3.98 14.2
1 1.6
F>x.7 1.092 0.017 1.59 13.3 0.012 1.10 11.7 9.9
Determined by "C: NMR
Additional POlvmer Exam le"9 A-J, Continuous Solution Potvmerizati n Catalvst
A1./B2 + DEZ
For Exam les 19A-I
101871 Continuous solution polvmerizations are carried out in a camputer
controlled
well-mixed reactor. Purified mixed alkanes solvent (Isopar''m E available
froin Exxon
Mobil. Inc.). ethylene, 1-octeire. and lrvdrogen (dvhere used) are carn.bined
ancl fed to a 27
~_Ya11on reactor. The feeds to the reactor are measured by mass-flow
controllers. Tl-le
temperatr.u=e of the feed stream is controlled bv use of aolvcol cooled heat
exchanger before
etate.rin~,r the reactor. The catalyst component solutions are metered using
pumps and mass
flow meters. 'f he reactor is run liquid-full at approxitnatelv 550 psi,
pressure. Upol1
11-t '"F I('tr'r, i'.'_?t r a1?d ?ddi ` ,: ar e ?3o1
; ; . , ; - = _:. _
._ _ .,.
: . ` _ ._. , . _ ., x :. ... _
.~:_
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
unreacted monomers are removed during the devolatization process. 'I'he
polymer melt is
pumped to a die for underwater pellet cutting.
For Exam le 19J
[01881 Continuous solution polymerizations are carried out in a computer
controlled
autoclave reactor equipped with an internal stirrer. Purified mixed alk:anes
solvent
(IsoparTM E available from ExxonMobil Chemical Company). ethylene at 2.70
ibsihour
(1.22 kg,Ihour), 1-octene, and hydrogen (where used) are supplied to a 3.8 L
reactor
equipped with a jacket for temperature control and an internal thermocouple.
The solvent
feed to the reactor is measured by a mass-flow controller. A variable speed
diaphragm
pump controls the solvent flow rate and pressure to the reactor. At the
discharge of the
pump, a side stream is taken to provide flush flows for the catalyst and
cocatalyst injection
lines and the reactor agitator. "I`llese flows are measured b_v Micro-Motion
mass flow meters
and controlled by control valves or by the manual adjustment of needle valves.
The
remaining solvent is combined with 1 -octene, ethylene, and liydrogen (where
used) and fed
to the reactor. A mass flow controller is used to deliver hydrogen to the
reactor as needed.
The temperature of the solvent/monomer solution is controlled by use of a heat
exchanger
before entering the reactor. This stream enters the bottom of the reactor. The
catalyst
component solutions are metered using pumps and mass flow meters and are
combined with.
the catalyst flush solvent atid introduced into the bottom of the reactor. The
reactor is ri-n
liquid-full at 500 psig (3.45 MPa) with vigorous stirring. Product is reinoved
through exit
lines at the top o#'the reactor. All exit lines from the reactor are steam
traced and insuiated..
Polymerization is stopped by the addition of a small amount of water into the
exit line along
with any stabilizers or other additives and passing the Ãnix.ture through a
static mixer, The
product stream is then heated by passin~ thro~~~;h a heat exchanger before
devolatilization.
The polymer product is recovered by extrusion using a devolatili:zing
extrtiidcr and water
cooled pelletizer.
101891 Process details and results are contained inTable 8. Selected polymer
properties
are provided in Tables 9A-C.
10I901 In Table 9B, inventive examples 19F and 19C`~ show low iiiiznccliate
set of
around 65 70 % strain aftcr 500% d-:iongation.
-54-
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
a c.
Z~
_ r n r n
~~~@ rf c x x x x c x c
G. )
'c 't i^ r.^, ur En
V J
G _ 'Ct - ,O L`7 ^ 1V '^ u
~+ w w r ... .... ~. -.. w
-y w ~S- . <r ~r er -t =7 .'t -3.
~.
.~ _
_ -
ra ra ,~~ et rv ca ra
L .~ 1
fJ h rF ~^1 r1' iV 114 C'3 n7 - ~
,, r a
-
~
r ~ 3 ^.)
r, - .-. . t , . . . . . . . - . ~ .~
. - _ ~r - - - -
_
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
TI
/` M:'Cy~(~lÃhl3d-~w ,C'",C ~.
] ~iif = 3 E ! -
E
9
s~ 0 7 r~, e^. 't rn n cn ~( <1
{'"= ~~ r r^'..G C`] GC s^. C
crx,
u a c~r-ao',.-~~71 oxcrr~
^^ C~I tV ~ i`3 -- t`~1 =- h3
kn ^#' v; `7' N N N -r ~
~; r~ ~=i ~-.~ ~s r-; ri ri r-i ri
C~ o
cs ~~^Vc~^~~~~
~'V ' `v.J` 7. rn (`1
~
[-- [``xi;..~. v; _= ~'r,
I~,r E"d cr7 oc apc oo c~a r~~ r~ }~ ~'
!(~ ` C ~ xy-~ r-xiy - ^
I^ nj I'Ã W ,
1~. -- /~r~~ E ...~,.~~.-.j(^
. . .
.... . ~a..._._~_.~
,..~ -
-` -.,~ _- ~ - - - -
;..~ _....-... ._,._ ,
56
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
~
~n r3- _ Ã- =
- ` .. I- +~C oO OG
ti ~'-
'
= ~~:...., Ã~ 70 00 ` D^ ~a = -
,^~i N
> 21
>
ewõ , ; ? ?
44 /~
'2
.." '7:; '4-
- r
G
7; ,
~.t
>w
>
:3 kx x x :A x
õ~-
57
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
Examples 20 and 21
10:1911 'I'he ethvleneia-olelin. interpolyrrier ot'Examples'0 and 21 Niv ere
made in a
substantially similar manner as Examples 19A-1 above with the polymerization
conditions shown in Table 11 below. The polymers exhibited the properties
showii in
Table 10. Table 10 also shows any additives to the polymer.
Tahle 10 - Properties and Additives of Examples 20-21
ExatnpEe 20 Example 21
Density (g/cc) 0.8800 0.8800
mI: 1.3 1.3
DI Water 100 DI Water 75
Irgafos 168 1000 Irgafos 168 1000
Additives Irganox 1076 250 Irganox 1076 250
Irganox 1010 200 Irganox 1010 400
Chimmasorb Chimmasorb
2020 100 2020 80
I-iard segment split
(FVI /tr) 35% 35%
101921 Irganox 1.010 is Tetrakisinethylene(3,5-di-t-butyl-4-
hydroxyhydrocinnamaie)rnethane. Irganox 1076 is Octadeeyl-3-(3`,5-di-t-butyl-
4`-
hydroxyphenyl)propionate. Irgafos 168 is Tris(2,4-di-t-butylphenyl)phosphite.
Cb.itnasorb 2020 is 1,6-Ilexanedia.mine, N,N'-bis('-',2,6,6-tetra7nethvl-4-
piperidiiiyl)-
polyiner with 2,3.6-trichloro-1.3.5-triazine, reaction products with, N-butyl-
l-
butanaz-nine and N-butyl-2,2,6,6-tetramethyl-4-piperidinamine.
<;g
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
~.
à C C t;
=,"` wa
x ti
flw ,y - =-'
( G
[
$ L .....
~Yx
~ .r '...=J "~
r~-
:J
-Jy J
a
~ J ft
555
~ r .mm
=~ ~ j l+
"iYYll ~ tt m -
~v !~ ~ _. - ..... .. . _
59
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
Fibers Suitable for the D'veci Fabrics and Textile Articles of the Present
Invention
101331 The present invention relates to dyed fabrics sLaitable for textile
articles
such as shirts, pants, socks, swimsuits_ etc. The fabrics may be made in any
manner
but typically are either woven or knit. Woven fabrics of the present invention
are
typically characterized by a stretch of at least about about 10 percent
measured
according to ASTM D3107 whereas knit fabrics of the present invention are
typically
characterized by a stretch of at least about 30 percent measared according to
ASTM
D2594.
[0194) The dyed fabrics are usually comprised of one or more elastic fibers
wherein the elastic fibers comprise the reaction product of at least one
ethylene olefin
block polymer and at least one suitable crosslinking agent. As used herein,
<`crosslinkinb agent" is any means which cross-links one or more, preferably a
majority, of the fibers. Thus, crosslinking agents may be chemical compounds
but are
not necessarily so. Crosslinking agents as used herein also include electron-
beam
irradiation, beta irradiation, garnma irradiation, corona irradiation,
silanes, peroxides,
allyl compounds and UV radiation with or without crosslinking catalyst. U.S.
Patents
No, 6,803,014 and 6,667,351 disclose electron-beam irradiation tnethods that
can be
tised in einboclii-nents of the invention. Typicallv, enough fibers are
crosslinked in an
anrount sLich that the fabric is capable of being dyed. This amount varies
depending
Lipon the specific polymer employed and the desired properties. However, in
some
embodiments, the percent of cross-linked polymer is at least aboLit 5 percent,
preferably at least abotit 10, more preferably at least about 15 weight
percent to about
at most 75, preferably at most 65, preferably at most about 50 percent, more
preferably at most about 40 percent as measured by the wei(,?ht percent of
gels formed
according to the method described in Example 25.
[01951 The fibers typica]ly have a filament elongation to brea.k of greater
than
aboLIt 200%, preterably greate;r thari about 2I0%, preferably -reater than
about 220%,
preterab[y greater than aborit 230 fl, preferablv greater than about 240 o,
pre#erablygreater than about 250%. preferably- greater than about 760 o,
preferably greater than
about 2~1"'. r e1erab3. _re:ater tha.n,',out 280" n , .1_
itlli- _a.
-60-
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
elongation t' load at 100% elongation of greater than or equal to about 1.5,
preferably
greater than or equal to about 1.6, preferably 4,reater than or equal to about
1.7.
preferably greater than or equal to about 1.8.
preferabiv greater than or equal to about
1.9, preferably greater than or equal to about 2.0_ preterabl.y greater than
or equal to
about 2.1. preferably ~reater than or equal to about 2.2, preferably greater
than or
equal to about 2.3, preferably greater than or equal to about 2.4, and nlay be
as high as
4 according to ASTM D2731-01. (under force at specified elongation in the
finished
fiber form).
[01961 The poEyolefin may be selected from any suitable ethylene olefin block
polymer. A particularly preferable olefin block polymer is an ethylene/a-
olefin
interpolymer, wherein the ethyleneia-olefin interpolymer has one or more of
the
following characteristics beforc crosslin.king:
(1) an average block index greater than zero and up to about 1.0
and a molecular weight distribution, ;VIw: VIn, greater than about 1.3; or
(2} at least one molecular fraction cMhich elutes between 40 C- and
130 C when fractionated using TREF, characterized in that the fraction has a
block
index of at least 0.5 and up to about l; or
(3) an.Mw/Mn from abotrt 1.: to about 3.5, at least one melting
pc>int, "I'm, in degrees C:elsius, and a density, d, in grams/cubic
eentimeter, wherei.n thenuniericai values of:'Trn and d correspond to the
relation.ship:
T,ry, > -2002.9 - 4538.5(cl) - 2422.2(d) 2 ; or
(4) an Mw/Mn from about 1.7 to about 3.5, and is characterized by
a heat of fusion, ~l-I in Jfg, and a delta qtiiantity, AT, in dearees Celsius
defined as the
temperature dif#erence between the tallest DSC peak and the tallest CRYSTAF
peak.
tiN-herein the niirnerical values of AT and AH have the f'ollowina
relationships:
AT >-D.1299(A.1l) 62.81 for Af-f greater than zero and up to 130 J/
A'I' > 48 C for AH greater than 130 .l1'g ,
-61-
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
wherein the C.RYSTAF peak is determined using at least 5 percent of the
cuznulative polymer, and if less than 5 percent of the polymer has an
identifiable
CRYS`1:AF peak, then the CRYSTAF temperature is 30"C; or
(5) an elastic recovery, Re, in percent at 300 percent strain and I
cycle measured with a cornpression-molded film of the ethylene/n,-olefin
interpolymer, and has a density, d, in granas;`cubic. centimeter, wherein the
numerical
valties of Re and d satisfy the l'ollowing relationship when etb.vlene/a-
olefin
interpolymer is substantially free of a cross-Iiiiked phase:
Re > 1 481-162g(d); or
(6) a molecular fraction which elutes betweert 40 C and 130"C
vuhen ftaetionated usini! TREF, characterized in that the fraction has a molar
comonQmer content of at least 5 percent higher than that of a comparable
random
ethylene interpolymer fraction eluting between the same temperatures, wherein
said
comparable random ethylene interpolymer has the same comonomer(s) and has a
melt
index, density, and molar comonomer content (based on the vhole polymer)
within 10
percent of that of the etbylene/a-olctin interpolynner; or
(7) a storage modulus at 25 C, G'(25 C), and a storage modulLrs
at 100 C. G'(10t) C), wherein the ratio of G'(25 C) to G'(100 "C:) is in
the range of
about 1:1 to about 9: i.
[01971 The fibers may be made into any desirable size and cross-sectional
shape
depending upon the desired application. For nranv applications approxiti-
iately round
cross-section is desirable due to its reduced friction. 11owever, other shapes
such as a
trilobal shape, or a flat (i.e., "ribban" like) sliape can also be employed.
Denier is a
textile term i~~hiclr is defined as the ,rams of the fiber per 9000 nreters of
that tiber's
pen(yth. Preferred denier sizes depend upon. the type of fabric and desired
applications.
Tvpically, knit fabric-s comprise a majority of the fibers having a denier fi-
om at least
about I. preferably at Icast about 20. pre,ferablv at least aboLit 50, to at
most about
180. pref:c:rabt~, ~i -nost 150, r~referab1N- ,ifi raiost aborat 100 .?. T1
1?ret r5I.;' at
-62-
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
101981 Dependin(i upon the application the fiber may take ar,y suitable form
including a staple fiber or binder fiber. Typical exaniples may include a
homofil
fiber, a bicomponent fiber, a meltblow-n fiber, a nieltspun tiber. or a
spunbond fiber.
In the case of a bicomponent fiber it inav have a sheath-core structure; a sea-
island
structcre; a side-by-side structure; a matrix-fibril structure; or a segmented
pie
struetu.re. Adirantaneously, conventional fiber form.in") processes may be
employed to
make the aforementioned fibers. Such processes include those described in, for
example, U.S. Patents No. 4,340,563; 4,663,220; 4,668,566; 4,322,027. and
4,413,110}.
[01991 Depending upon. their composition, the fibers may be made to facilitate
processing and unwind the same as or better t`roin a spool than other tibers.
Ordinarv
fibers when in round cross section often fail to provide satisfactory
unwinding
perforrnance due to their base polymer excessive stress relaxation. This
stress
relaxation is proportional to the age of the spool and causes filaments
located, at the
very surface of the spool to lose grip on the surface, becoming loose filament
strands.
I_ater, when such a spool containing conventional fibers is placed over the
rolls of
positive feeders, i.e. Memminger-IRO, and starts to rotate to industrial
speeds, i.e. 100
to 300 rotations/minute, the loose fibers are thrown to the sides of the spool
surface
and ultimately fall off the edge of the spool. This failure is ku.own as
derails which
denotes the tendency of conventional fibers to slip off the shoulder or edge
of the
package which disrtiipts the unwinding process and ultimately causes machine
stops.
`I'he above fibers may exhibit dera.ilinc, to the same or a much less
signiticant degree
which possibly allows greater throughput.
[02001 Another advantage of the fibers is that defects such as fabric faults
and
elastic filament or fiber breakage may be equivalent or reduced as compared to
conventional fibers. That is, use of the above ti.bers may reduce, buildup of
fiber
fragments on a needle bed -- a problem that often occurs in circular knit
inachines
when polymer residue adheres to the needle surface. `1'lrus, the fibers may
redti.ce the
corresponding f:abric breaks caused by the residue when the #ibers are being
made
into, e.u. fabrics oi1 a circular knitting tnachiile.
,,.: , ... . _
11)2t3I1 'latti;~
t, .
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
elastic olctin fibers require that tlaese guides be i-nade of rotating
eletne.nts such as
pulleys as to m.inimize friction as machine pdrts, such as evelets, are heated
up so that
machine stops or filament breaks could be avoided ciurin4 the circular
knittinfz
process. That is, the frictioti against the guiding elements of the machine is
reduced
by using the inventive fibers. Further information concerrzi g circular
knitting is
found in. for example, Bamberg Meisenbach. "Cir-cular Knitting: TechrzoIvk,
Process, Structures, Yarns, Qiialrty ", 1995, incorporated herein by reference
in its
entiretv.
Additives
[0202] Antioxidants, e.g., 1RGAFOS'-X 168, 1RGANOXfg) 101.0, IRGANGX*1,
3790, and C.1:TIMASSORB~~~ 944 iiiade by Ciba Geigy C,orp., may be added to
the
ethylene polymer to protect against undo degradation during shaping or
fabrication
operation andlor to better control the extent of grafting or crosslinking
(i.e., inhibit
excessive gelation). In-process additives, e,g. calcium stearate, water,
lluoropolymers, etc., may also be used for purposes such as for the
deactivation of
residual catalyst and/or improved processability. TIXliVIN~,R~ 770 (from Ciba-
Geigy)
can be used as a light stabilizer.
102031 The copolymer can be #i[led or unfilled. lf 17illed, then the amount of
filler
present should not exceed an amount that would adversely affect either lieat-
resistance or elasticity at an elevated teniperature. If present, typically
the amount of
filler is between 0.01 atrd 80 wt % a based on the total weight of the copoi3-
mer (or if a
blend of a copolymer and one or more other polymers, then the total weight of
the
blend). Representative fillers include kaolin clay, magnesium hydroxide, zinc
oxide,
silica and calcium carbonate. In a preferred embodimeiit, in which a filler is
present,
the filler is coated with a material that will prevent or retard any tendency
that the
filler mi-ht otlierwise have to interfere with the crosslinlcitig reactions.
Stearic acid is
illustrative of sLÃch a filler coating.
[02041 To reduce the 1'riction coefficient of the f bers. various spin finish
formulations can be used, such as naetallic soaps dispersed in textile oils
(see l~or
exaniple. t.:.S. I'atentNo_ 3,039,40~5 or U.S. 1'~, .jt _X:-a. 6,65,2e599",
suri-actants in a
base <~ ~~~~ssc t;S
fo
. ..- ~. _'. ._ . . '~~. ~ _. . ~
-~~=t-
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
Application No. 10/933,721 (published as LS200501423360) discloses spin finish
compositions that can also be used.
Fabrics
[0205] '1"he present invention is directed to improved, dyed textile articles
comprising an olefin block copolNFmer. For purposes of the present invention,
"textile
articles" includes fabric as well as articles, i.e., garments, made from the
fabric
ineluding, for e.xarnple, clothing and other items in need of coloring. By
knitting it is
meant intertwininc, yarn or thread in a series of connected loops either by
hand, with
knitting needles, or on a machine. The present invention may be applicable to
any
type ol'knitting including, for example, warp or weft knitting, flat knitting,
and
circular knitting. Particularly preferred warp knits inclLid.e tricot and
raschel while
preferred weft knits include circular, flat, and seamless. However, the
invention is
particularly advantageous wben. employed in circular knitting, i.e., knitting
in the
round, in. which a circular needle is employed. The present invention may also
be
applicable to any type of woven fabric.
102061 'f he dyed fabrics oi'the present invention preferably comprise one or
more
elastic fibers wherein the elastic fibers comprise the reaction product ot'at
least one
ethylene olefin block polymer and at least one crosslinking agent wherein the
ethylene
olefin block polymer is an ethylene!a-olefn interpolymer, wherein the
ethylene/c1-
olefin interpolymer has one or anore of the following characteristics prior to
crosslinking:
(1) an average block index greater than zero and up to abotLt 1.0
and a moleci.rlar weight distribution, Mw/Mn, grreater thaii about 1.3; or
(2) at least one molecular tiaction which elute5 between 40 C, and
1' WC when fractionated using TREF, characterized in that the fraction has a
block
i7idex of at least 0.5 and up to about 1; or
(3) an Mw/Mn from about 1.7 to aboLit 3.5, at least one ine,lting
point, T'm. in degrees Celsius, and a dcnsitv, d. in gams,-cubic centimeter,
w1lerein the
rit alues of ":: m and d correspond to th _ .-,ship:
_~~~;_
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
(4) an iv1w:/Mn from about 1.7 to about 3.5, and is characterized by
a heat of fusion, AH in Ji`g, and a delta quantity, AT, in degrees Celsius
defined as the
temperature difference between the tallest DSC peak and the tallest CRYSTAF
peak,
wherein the numerical values of AT and AH have the following relationships:
AT --0.1Z99(AH) = 62.81 for AH greater than zero and np to 130 Jrs_=,
AT ? 48 C for AH greater than 130 J/g,
wherein the CRYSTAF peak is defier-mined using at least 5 percent of the
cuniulative polymer, and if less than 5 percent of the polymer has an
identifiable
C:RYSTAF pcak, then the CRYSTAF temperature is i0 C; or
(5) an elastic recovery, Re, in percent at 300 percent strain and 1
cycle measured with a compressiorr-molded film of the ethylenela-olelin
interpolymer, and has a density, d, in grams/cubic centimeter, wherein the
numerical
values of Re and d sati5fy the following relationship when ethylene/a-olefin
inte.rpolymer is substantially free of a cross-linked phase:
Re >1481-1629(d); or
(6) a molecular fraction which elutes between 40'C. and 130j C
when fractionated using TREF, characterized in that the fraction has a--izolar
comonomer content of at lea~st 5 percent laig.ber than that of a comparable
randoni
ethylene interpolymer fraction elating between the same temperatures, wherein
said
comparable random ethylene interpolymer has the same comonomer(s) and has a
melt
index, density, and m.olar comonomer content (based on the whole polymer)
within 10
percent of that of the ethylenefa-oleiin interpolymer; or
(7) a storage inodulus at 25 C, G'(25 C.), and a storaue modulus
at 100 C., G'(100 C:), wherein the ratio ofG'(?5 C) to Ci '(100 "C.} is in
the range of
about 1:1 to about 9:1.
102071 "fhe amount of polynier in the d_ct;il-,ric :ries depcn.din<~,~ upon
the
1)(~;t ~ r r~: ;aõ ttitl fl8e d Cs fat11'
;_)C:"IlI 5.
;t .. = } e t:
-66-
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
typically comprise less than about 50, preferably less than about 40,
preferably less
than about 30, prefera.bly less than about 20, more preferably less than about
1Ã1
weight percent e:thylene/o-olelin interpolymer. The ethylene;'u-olefin
interpolymer
may be in the form of a fiber and may be blended with another suitable
pcslyrner, e.g.
polyolefins such as random ethylene copolymers, HDPE, LLDPE. LDPE. liI.DPE-
polypropylene homopolymers, copolymers, plastomers and elastomers, lastol, a
polyamide, etc.
[0208] The ethylenet'a-olefin interpolymer of the fabric may have any density
but
is usually at least about 0.85 and preferably at least about 0.865 1;/cm'
(ASTM D
792). Correspondingly, the density is usually less than about 0.93, preferably
less
than about 0.92 g/cm3 (ASTM D 792). The etliylene/cc-olefin interpolymer of
the
fabric is eharacterized by an uncrosslinked melt index of from about 0.1 to
about 10
g!10 minutes. If crosslinking is desired, then the percent of cross-linked
polymer is
often at least 10 percent, preferably at least about 20, more preferably at
least about
25 weight percent to about at most 90, preferably at most about 75, as
measured by
the weiglit percent of -;e1s formed.
[02091 The fabrics often comprise another material selected from the group
consisting of rayon, nylon, viscose. polyester such as microfiber polyester,
polyamide,
polypropylene, cellulose, cotton, flax, rarmie, hern.p, svool, silk, linen,
bamboo, tencel,
mohair, other natural fibers, other sythetic fibers, and mixtures thereof.
Often the
other niaterial comprises the majority of the fabric, ln such case it is
preferred that
the other material comprise from at least about 50, preferably at least about
60.
preferably at least about 70, preferably at least about 80, sometimes as much
as 90-95,
percent by weight o.f the fabric.
102101 'I'hc ethvlene/a-olefi.n interpolymer, the other material or both may
be in
the fornl of a Iiber. Preferred sizes include a denier from at least about L.
preferably
at least about 20, preferably at least about 50. to at most about 180,
preferably at most
about 150, preferably at most about 100, preFerably at most about 80 clenier.
102111 Particularly prefcrred circular knit fabrics comprise ethylene;'et-
olefin
interpolymer in the form of a fiber in an amount of from about 5 to about'fl
percent
(b~- 4 the = .' ' , . l~..t: _ 1_ .? = )retcrrew s. , r, f" or.=s
6f
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
about 30 percent (by weight) of the fabric in the fornt of a fiber. Often such
warp knit
and circular knit fabrics also comprise polyester or aticroliber polyester.
102121 The fabric, particularly knit fabrics, often have less than about 5,
preferably less than 4, preferably less than 3, preferably less than 2,
preferably less
than 1, preferably less than 0.5, preferably less than 0.25. percent shrinkage
after wash
according to AATCC 135 in either the horizontal direction, the veqical
direction, or
both. '1iore specifically, the fabric (after heat setting) often has a
dimensional
stability of from about 7% to about +7%, preferably -5% to about S%,
preferably
from about -3% to about 4-3%, preferably -2% to about --2%, more preferably -I
% to
about -1 /a in the lengthwise direction, the widthwise direction, or both
according to
AATCC135 IVAi. In addition, the fabrics often have less shrinkage after wash
aecording to AATCC 135 IVAi than a comparable fabric of elastic fibers with a
higher amount of crosslinking.
102131 Knit fabrics can be made to stretch in two dimensions if desired by
controlling the type and arriount of eth.ylene/a-ole~~in interpolymer and
other materials.
Knit fabrics may sometimes be characterized by a stretch of at least about 30
percent
measured according to AS"I-M D2594. Similarly, the fabric can be made such
that the
&uowth in the lengthwise and widthwise directions is less than about 7,
preferably less
than about 5, preferably less than about 4, preferably less t.han about 3,
preferably less
than about 2, preferably less thair about 1, to as little as 0.5 percent
according to
ASTM D 2594. Using the same test (AS"1'M D 2594) the lengthwise growth at b0
seconds can be less than about I5, preferably less than about 12, preferably
less than
about 10, preferably less than about 8%. Correspondin-ly, using the same test
(AS"I'M D 2594) the widthwise growth at 60 seconds can be less than about 20,
preferably less than about 18, preferably less than about 16, preferably less
than abotit
13%. In regard to the 60 minute test of ASTM D21594, the widthwise growth can
be
less than about I0, preferably less tlaan about 9, preferably less than about
8,
preferably less than about 6% ~~-hile the lengthwise growth at 60 minutes can
be less
than about 8, preferably less than about 7, preferably less tlian about 6,
preferably less
than about 5%. "fhe- lo .er iyrowth described above allows the fabrics of the
inventiori
to t' .., ..~ ;t l~.ss t': a , ,u I. " I
- - . ,.- _ ` .... . .. _.. i . . .
.,~,8m
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
size. In contrast to knit fabrics, tv oven fabrics ma.y be characterized bv a
stretch of at
least about 10 percent measured according to ASTM D3107.
142141 Advantageously. knit fabrics of the present iaivention caii be made
without
a substantial number of breaks and using a knitting machine comprising an
eyelet
feeder system, a pulley system, or a combination thereof. Thus, the circular
knitted
stretch fabrics having improved tnoldabi.lity while having acceptable
dimensional
stability (lengthwise and widthwise), acceptable growth and sla.rinkage, the
ability to
be heat set at low temperatures while controlling size, .iow moisture regain
can be
made without significant breaks, with high throughput, and without derailing
in a
wide variety of circular knitting machines.
Dyeing
[02151 The dyed fabrics of the present invention may be made by virtually any
dyeing process. For example, many useful techniques are described in
Fundamentals of Dyeing and Printing, by Garry Mock, North Carolina State
Ciniversity 2002, ISBN 9780000033871. One advantage of the fabrics of the
present
invention is that they may often be contacted with the dye at a temperature of
at least
about 130 C to produce a dyed fabric wherein the fabric exhibits a growth to
stretch
ratio of less than 0.5, preferably less than 0.4, prefcrably less than 0.35,
pref'erably
less than 0.3, preferably less than 0.25, preferably less than 0.2, preferably
less than
0.15, preferably less than 0.1, preferably less than 0,05. Advantaf;eously,
the
resulting dyed fabrics of the present invention are often characterized by a
color
change of greater than or equal to about 3 a.0, preferably areater thayr or
equal to about
3.5, more preferably greater than. or equal to about 4.0 according to AATCC
evaluation after a first wash by AA`I'CC6I-2003-2A. Another advantage is that
the
fabrics of the present invention may sometimes exhibit a color change of
greater than
or equal to about 2.5, preferably ~.,.reater than or equal to about 3Ø more
preferably
(yreatir than or equal to about 3.5 according to r1ATCC eva.luation after a
second
wash by AATCC.6I-2003-2rL. In essence this a-neans that the dyed I'abric5 of
the
present invention ina~,,' exhibit less fading when subjjected to launderin-
than
conventional d~:d fabrics.
107
d~'Ã liczg;rlcs t11"s.tS' C=tce?i a coI; atl(:' u'
-69-
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
or equal to about 600, preferably of greater than or equal to about 650,
preferably of
greater th.an or equal to about 700, preferably of greater than or equal to
about 750, as
measured with a spectrum photometer. Advantageousiv, the color is
substantially
retained even after a first and second wash. For examplÃ;. the dyed fabrics
may be
characterized by a color strength after a first wash by AATCC6I-2003-2A that
is at
least about 90, pref.'erably at least about 95, more preferably at least about
97 percent
of the color strength after dying whereiii each color strength is measured
with a
spectrum photometer. The dyed fabrics may sometimes also be characterized by a
color strength afler a second wash by AATCC61-2003-2A that is at least about
90,
preferably at least about 92.5, niore preferably at least about 94 percent of
the color
strength after dying wherein each color strength is measured with a spectrum
photometer.
[0217] While not wishing to be bound by any theory it is believed that the
reason
the dyed fabrics of the present invention dye darker is due to the fibers of
the olefin
block polymer. 'I'hat is the olefin block polynler fzbers dye, to a lesser
extent allowing
the other rnaterial to get darker. Also, a higher dycing temperature can be
employed
with less fiber breakaue when oletin block polymers are used as the fibers. In
a
similar manner it is believed that the reason the dyed fabrics fade less upon
laundering
is that the olefin block polymer fibers are not dyed to as great of an extent
as fibers
made with other polymers. In this manner, the olefin block polymers cannot
fade or
bleed as much.
EXAMPLES
Exampte 22 - Fibers of elastic ethylenela-Olefin interpolymer
j02181 The elastic ethyItne/a-olefan interpolymer (oIe-fin block polymer) of
Example. 20was used to make monofilament fibers of 40 denier having an
approximately round cross-section. 13efore the fiber was made the following
additives
were added to the polymer: 7000 ppm f'DMSO(polydimethvl siloxane), 3000 ppm
CYANOX 1790 (1.3,4-tris-(4-t-butyC-3-hydroxy-2,6-dimethylhenzyl)-1.3,5-
triazine-
2,4 b-(1H,3H,5H)-trione, and 3000 ppm CH1iL9<1SOR13 944 Poly-[[6-(I,1,3.3-
Te I 1.
pi
,~
< Fxt ':3 ',`,~x ~,iiuu ~ca x73a's a ~z9ti =~i wNIila ci t:ttef;,2' ~'P
'TÃ}
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
diarncter, a spin temperature of 299~C, a winder speed of 650zn'mi.nute. a
spin finish
of 2%, a cold draw of 6%, and a spool weight of 150g. "I"he fibers were then
crosslinked using a total of 176A kGv irradiation as the crosslinking agent.
Example 23 - Hard yarns of fibers
[0219] A hard yam was made that comprised the elastic fibers ohl;xample 22 and
150 den.ier, 288 filament polyester. 'I'he filament of microfiber is fine as
0.52 denier
per filament. Two comparative examples were also made. One comparative example
hard yam employed 40 denier Eibers of a random ethylene-octene copolymer made
with a line speed of 450 rnlmin and the same 150 denier, 288 tilarnent
polyester
f1bers. The ran.dom ethylcrre-octene copolyrner had an average nrelt index of
3.0
;Il Omin, a density of 0.875 g/cm3 and was crosslinked with a dosage of of
166.4 kGy
irradiation as the crosslinking agent. The second comparative example hard
yarn was
made with multi-filament fibers of Lycrarm 162 C polymer and. the, same 150
denier,
288 filainent polyester fibers.
Example 24 - Dyeing
102201 An experiment was designed to evaluate the elastic fiber color stainin.-
and
the color darkness of microtiber polyester based fabrics. The experiment
evaluated
the disperse dyestufi'staining on the ribers coniprised of olefiin block
polymers, the
fibers comprised of Lycra" 162C, and the fibers comprised of random ethylene-
octene copolymer. 1grarn of each of the three different types of fibers and 9
(Trams of
micro-fiber polyester fabric (witness fabric) that made the hard yam was
loaded in a
lab rapid dycing machine tliat is shown in Figure 8. 'I:'he dyeing and
reduction wash
process as shown in l~igure 9 was then conducted on each of the three
different types
of libers. Clariant dyestuff Foron Black S-WF was used to dye the fibers and
fabrics
into black. The Lycra based fibers were dyed at 125 C since this elastic
tiber may
uildergo severe damage at higher temperatures. The other two types of (7ibers
~-ere
dyed at 135 'C. The specin-ten.s of alter dyeing, after Is` reduction Aash and
after 2"d
reduction ik-ash were collected for evaltiiation.
(0221] The three dif.'ferent t}-pes of fibers after dycing and reduction wash
were
evaluated visnaIl,. The po]ycster micro Eiber fabric was also tesiud to get
color
f -
Ti b E ~~
-
~ ~ I~ i~b - , ~Tl,yth { N ~' Ii~ '` ~ -:}~-
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
photometer (Datacolor mociel-600PLUS). 1-ligh KiS value represented darker
color.
Color change was measured according to AATCC 61-2003 LA that repoz-ts the
color
difference between ori-inal specimen and the speciinen. after wash. The
quotation
ranges #ron3 1-5 by arey scale according to AATCC evaluation procedure. A Iow
er
grade indicates a bigger color change and therefore less colorfastness. The
specimens
after dyeing, aftcr I5Ã reduction wash. and after 2rt reduction wash were
washed by
AATCC b 1-2003-2 A and the color change before and after was measured.
102221 Color staining is also based on the test of AATCC 61-2003-2A. A multi-
fiber test Eabric that consists of acetate, cotton, polyamide, acrylic and
wool fiber, is
attached to the specimen to wash. The test grades 1-5 and a lower grade means
heavier color staining. Textile industry practice is to use the grade result
of
polyamide as an indication of color staining.
102231 Lab dips of the three different types of elastic fibers after dyeing,
after 1 S`
reduction wash and after 2d reduction wash were done. The results indicated
good
colorfastness and darker color for the fabrics comprising the olefin block
polymer.
Table 12 shows the elastic fiber color staining after dyeing, after
I"reduction wash
and after 2 d reduction wash. More dyestuff uptake makes darker color on
elastic
fiber itself. High dyesttrff uptake is positive to obtain dark colors but can
be
detrimental if it bleeds out during washing (home laundry). "I`he I..ycrarl"
fiber sllows
darkest color after dy'eing, after I" redtaction wash and after 2"" reduction
wash. The
raiidom ethyleine-octene copolymer and olefin block polymer fiber shows
lighter color
staining. The specimens are very siiirilar after dyeing, after I'r reduction
wash and
after 2" d reduction wash. The olefin block polymer fiber shows less dyestuff
uptake
that helps better colorfastness in micro fiber polyester fabric colorfastness.
Table 12 Color staining
Random
Testing item Lycra 162C ethylene Olefin block
octene polymer
copnlr mer
i Elastic Fiber color stain after Darkest Li=Yhter Zi,~hter
4
; dyeing
h1astic liber Color stait-t after Darkest I..iLhter I,iuyhter
1.~'R ci ~
~ .
~.~er C:3t,,,. ;:ffs,f
:C
- i".'. . t . .. . i i -
^7'~
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
[02241 Table 13 shows the color strenath. (K; S) value of rnicro fiber
polyester
tabric. The higher value otKiS represented darker color. Witness micropoly-
ester
fabrics d.ved with random ethylene-octene copolymer and olefin block polymer
tibers
showed darker black compared with L,ycra. While not wishing to be bound to any
theory it is believed that this result is due to the higher dyein~ temperature
employed.
There were no significant differences among the sanipies after dyeing, after 1
reduction wash and after 2"d reduction wash. However, the microfiber polyester
of
olefin block polymer can reach a darker color.
Table 13 Color strength(K/S) value of fabrics
Ranciam
Testing item Lycra 162C ethylene- Olefin block
octene polymer
c ol rner
Micro PES witness fabric 423.38 755.77 774.71
Color strength (K/S) after
dveing
Micro PES witness fabric K/S 414.68 783.83 753.67
after l" RC
Micro PES ~-itness fabric K/S 411.73 75s.00 739.86
affier 2"d RC
102251 "I"able 14 shows the color change value otmicro fiber polyester after
dycing, after lst reduction ikash and after 2" reduction wash. 'I"he higher
value t-nea s
lighter color change. All specimens show good color change results.
Tab[e 14 Result of color change of micro polyester
Random
Olefin Testin~ item Lyera ethylene- block
162C octene
co~salymer ~olyrner
Micro PES witness fabric Color 4 4 4
change after dyeing
%Iicro 1'I_:S witness fabric Color
4 4 4
chanue after I't RC
'vficro PES witness fabric Color [ 4 4 4 chan,~,e after 2"d RC - . ~ le
1ess ~: ir
--,
,,_
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
color staining. There is no obvious difference between the results shown after
1.'t and
2"d reduction wash_ The dyed, witness fabrics of random. ethylene-octene
copolymer
and olefin block polymer are darker than the dyed Lycra fabric and this has an
effect
on the color fastness as given in Table 12 . None of the results involve
fabric heat
setting.
Table 15 Color staining to polvarmide fabric
Randona. Olefin
ethy lene-
Testing item Lycra 162C block
octene polymer
co . ol ymer
Micro PES witness fabric Color ~ 2.5 2.5
fastness to wash after dyeing
Micro PES witness fabric Color 3.5 3.5 3.5
fastness to wash after 1 st RC
Micro PES witness fabric Color 3.5 3.5 3.5
fastness to wash after 2nd RC
[0227] Three single jersey knits are use in this test. They are micro fiber
polyester
hard yarn knitted with 40 denier Lycra, 40 denier random ethylen.e-oetene
copolymer
and 40 denier olefin block polymer fiber. The knitting speed, elastic draft
and the
fabric weight of ereige are given in Table 16.
Table 16 Fabric description of various elastic fiber contented fabric
Sample Speed ~ D.R. Greige
r mLycra 162C 12.4 2.6 203 bl rti
Random ethylene- 12.4 2.6 1701;/mi
ctene co olvmer
lerin Block 20 3 .2 186g,='rt1
Polymer
102281 Random ethylei.it-oc-tr;ne copoltirner and olefin block polymer greige
are
scoured at 85~C for 1-0 minntes, dried at 13 5"C for 45 mintites, tensionless
dryed at
13C}C for 60 minutes, set at 165'C for 120 seconds (15 yards per minute) a120%
overtee.d, and tinished. The and reduction conditions are -i,, cli in Fioure 9
for
ra l 3 . _h ;
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
Table 17 Fabric weight of various elastic fiber contented fabric
Sample ID Finished weight
Lycra 162C 269 g/ t71
andom ethylene- 210 gr m
ctene co olvmer
lefin Block
190gi M2
Polymer
[02291 Table 18 shows the test result of AATCC 61-21003-2A, random ethylene-
octene copolymer and olefin block polymer both have excellent performance in
color
chan~;e compared with I..vcra 1.62 before or after heat setting. The reason is
random
ethylene-octene copolymer and olefin block polymer fiber were dyed at 135 C.
the
disperse dyestuff has better reaction in this temperature. In the dye lot of
micro-fiber
polyesteriLycra, there is un-reacted disperse dyestuff because of low dyein.g
temperature that stained on fabric and bleeds out that znakes specimen color
fading
during testing. Random ethylene-octene copolyiner and OBC both has good color
fastness to polyamide compared with Lyera. Lycra shows poor color fastness
after
heat settina. 'l`he reason is the disperse dyes migrated durin.g 185"C high
temperature
heat setting.
Table 18 Test result of color change and color fastness of fabrics
Random
ethylene- Olefin Block
Testing item Lycra 162C octene
Pa[vmer
co olvmer
Color change after RC 3 4 4
Color change after heat setting 2.5 4 4
Color fastness to wash after 3 3.5 3.5
RC
Color fastness to wash aftc;r ? ~ 3.5 4
heat settini
102301 Three finished fabrics after heat setting v,,-ere tested by spectrum
photometer (Da,tacolor naodel-6(I0hLt S }. Tabi~ 19 s}iows the color strength
(K/S)
ti~a:tt i: 1 =ts C~~ll doni ~ it~c.
dild .<Lc:cdarker coÃ~ -vith the k e_.~~
'-mtaiation.
m~~_
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
Table 19 Eabrie width of crreicre and finished goods
Rat-ldom
ethylene- OBC EXP
Testing item Lvcra 162C octene 6116
co olvmer
Color strength (K;`S) 4 54.23 747.55 774.18
Example 25 - Varying Amounts of Fiber Crosslinking
[0231] The elastic ethylenefo,-olefi n interpolymer of Example 20 was used to
make monofilament fibers of 40 denier having an approximately round cross-
section.
Before the fiber was made the following additives were added to the polymer:
7000
ppm. PDMSO (polydimethyl siloxane). 3000 ppm CYANOX 1790 (1,3,5-tris-(4-t-
butyl-3-hydroxy-2,6-dimethylbenzyl)-1,3,5-triazine-2,4,6-(1H,3.14,5H)-trione.
and
3000 ppm CHI.MASORB 944 Poiy-[[6-(1,1,3,3-tetrarrnethy-lbutyl)amino]-s-
triazine-
2,4-diyll[2,2,6,6-tetramethy1-4-piperidyl)iminojhexamethylene[(2,2,6,6-
tetramethyl-
4-piperidyl)iminoJ] and 0.5% by weiglat "1"i0,. The fibers were produced using
a die
profile with circular 0.8 mm diameter, a spin temperature of 299 C, a winder
speed of
650numinute., a spin finish of 2%, a cold draw of 6%, and a spool weight of
150g.
Fibers were then crosslinked using varying amounts of irradiation from an e-
beam as
the crosslinking acent.
[02321 'I"he gel content versus the amount of irradiation is shown in Figure
11.
The gel content was determined by weighing out an approximately 2-i nrg iiber
sample to 4 significant tigure accuracy. The sample is then combined with 7 ml
xylene in a capped 2-dratn vial. The vial is heated 90 mint.ites at 125 C to
135 C,
with inversion mixing (i.e. turrting vial upside down) every 15 minutes, to
extract
essentially all the non-crosslinked polymer. Once the vial has cooled to
approxiniately 25 C, the xylene is decanted from the gel. The grel is rinsed
in the, vial
with a small portion of fresh xylenes. The rinsed gel is transferred to a
tared
aluminum weighing pan. The tared dish with gel is vacuum dried at 125"C: for
30
minutes to remove the xyleiie by evaporation. The pan with dried gel is
weighed on
an analytical balatice. The gel content is calculated based ojr the extracted
gel '~kreipht
cndori==inal fiib':'rA. i~; :e i' shov".E tK ';,ncl;":,I
-76-
CA 02674597 2009-07-06
WO 2008/089220 PCT/US2008/051142
that the precise relationship between the amount of crosslinking and e-beam
dosage
may be atficted by a~,7ivcn polvmer's properties, e.g.. molecular rveight or
melt index.
ml7