Note: Descriptions are shown in the official language in which they were submitted.
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
UNITED STATES PATENT APPLICATION
FOR
HYDROGEN-CATALYST REACTOR
BY
RANDELL L. MILLS
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
2
CROSS-REFERENCES TO RELATED APPLICATIONS
This application claims the benefit of (1) Application No. 60/913,556 filed on
April 24, 2007; (2) Application No. 60/952,305 filed on July 27, 2007; (3)
Application
No. 60/954,426 filed on August 7, 2007; (4) Application No. 60/935,373 filed
on
August 9, 2007; (5) Application No. 60/955,465 filed on August 13, 2007; (6)
Application No. 60/956,821 filed on August 20, 2007; (7) Application No.
60/957,540
filed on August 23, 2007; (8) Application No. 60/972,342 filed on September
14,
2007; (9) Application No. 60/974,191 filed on September 21, 2007; (10)
Application
No. 60/975,330 filed on September 26, 2007; (11) Application No. 60/976,004
filed
on September 28, 2007; (12) Application No. 60/978,435 filed on October 9,
2007;
(13) Application No. 60/987,552 filed on November 13, 2007; (14) Application
No.
60/987,946 filed on November 14, 2007; (15) Application No. 60/989,677 filed
on
November 21, 2007; (16) Application No. 60/991,434 filed on November 30, 2007;
(17) Application No. 60/991,974 filed on December 3, 2007; (18) Application
No.
60/992,601 filed on December 5, 2007; (19) Application No. 61/012,717 filed on
December 10, 2007; (20) Application No. 61/014,860 filed on December 19, 2007;
(21) Application No. 61/016,790 filed on December 26, 2007; (22) Application
No.
61/020,023 filed on January 9, 2008; (23) Application No. 61/021,205 filed on
January 15, 2008; (24) Application No. 61/021,808 filed on January 17, 2008;
(25)
Application No. 61/022,112 filed on January 18, 2008; (26) Application No.
61/022,949 filed on January 23, 2008; (27) Application No. 61/023,297 filed on
January 24, 2008; (28) Application No. 61/023,687 filed on January 25, 2008;
(29)
Application No. 61/024,730 filed on January 30, 2008; (30) Application No.
61/025,520 filed on February 1, 2008; (31) Application No. 61/028,605 filed on
February 14, 2008; (32) Application No. 61/030,468 filed on February 21, 2008;
(33)
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
3
Application No. 61/064,453 filed on March 6, 2008; (34) Application No.
61/xxx,xxx
filed on March 21, 2008, and (35) Application No. 61/xxx,xxx filed on April
17, 2008,
all of which are herein incorporated by reference in their entirety.
DESCRIPTION OF THE INVENTION
1. Field of the Invention:
As disclosed in the paper R. Mills, J. He, Z. Chang, W. Good, Y. Lu, B.
Dhandapani, "Catalysis of Atomic Hydrogen to Novel Hydrogen Species H-(1/4)
and H2(1/4) as a New Power Source", Int. J. Hydrogen Energy, Vol. 32, No. 12,
(2007), pp. 2573-2584 which is herein incorporated by reference, the data from
a
broad spectrum of investigational techniques strongly and consistently
indicates that
hydrogen can exist in lower-energy states then previously thought possible.
The
predicted reaction involves a resonant, nonradiative energy transfer from
otherwise
stable atomic hydrogen to a catalyst capable of accepting the energy. The
product is
H(1 / p) , fractional Rydberg states of atomic hydrogen wherein n=~, 3, ~,...,
1;
p
( p<_ 137 is an integer) replaces the well known parameter n = integer in the
Rydberg
equation for hydrogen excited states. He+, Ar+, and K are predicted to serve
as
catalysts since they meet the catalyst criterion-a chemical or physical
process with
an enthalpy change equal to an integer multiple of the potential energy of
atomic
hydrogen, 27.2 eV. Specific predictions based on closed-form equations for
energy
levels were tested. For example, two H(1 / p) may react to form H2 (1 / p)
that have
vibrational and rotational energies that are p2 times those of H2 comprising
uncatalyzed atomic hydrogen. Rotational lines were observed in the 145-300 nm
region from atmospheric pressure electron-beam excited argon-hydrogen plasmas.
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
4
The unprecedented energy spacing of 42 times that of hydrogen established the
internuclear distance as 1/4 that of H2 and identified Hz (1 / 4).
The predicted products of alkali catalyst K are H- (1 / 4) which form KH * X,
a novel alkali halido (X) hydride compound, and H2 (1 / 4) which may be
trapped in
the crystal. The 'H MAS NMR spectrum of novel compound KH * Cl relative to
external tetramethylsilane (TMS) showed a large distinct upfield resonance at -
4.4
ppm corresponding to an absolute resonance shift of -35.9 ppm that matched the
theoretical prediction of H- (1 / p) with p = 4. The predicted frequencies of
ortho
and para-Hz (1 / 4) were observed at 1943 cm-' and 2012 cm-' in the high
resolution
FTIR spectrum of KH * I having a -4.6 ppm NMR peak assigned to H- (1 / 4). The
1943/2012 cm-'-intensity ratio matched the characteristic ortho-to-para-peak-
intensity ratio of 3:1, and the ortho-para splitting of 69 cm-' matched that
predicted.
KH * Cl having H- (1 / 4) by NMR was incident to the 12.5 keV electron-beam
which
excited similar emission of interstitial HZ (1 / 4) as observed in the argon-
hydrogen
plasma. KNO3 and Raney nickel were used as a source of K catalyst and atomic
hydrogen, respectively, to produce the corresponding exothermic reaction. The
energy balance was OH = -17,925 kcal / mole KNO3 , about 300 times that
expected
for the most energetic known chemistry of KNO3 , and -3585 kcal / mole H2 1
over 60
times the hypothetical maximum enthalpy of -57.8 kcal / mole H2 due to
combustion
of hydrogen with atmospheric oxygen, assuming the maximum possible H2
inventory. The reduction of KNO3 to water, potassium metal, and NH3 calculated
from the heats of formation only releases -14.2 kcal / mole H2 which cannot
account
for the observed heat; nor can hydrogen combustion. But, the results are
consistent
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
with the formation of H- (1 / 4) and H2 (1 / 4) having enthalpies of formation
of over
100 times that of combustion.
In embodiments, the invention comprises a power source and a reactor to
form lower-energy-hydrogen species and compounds. The invention further
5 comprises catalyst reaction mixtures to provide catalyst and atomic
hydrogen.
Preferred atomic catalysts are lithium, potassium, and cesium atoms. A
preferred
molecular catalyst is NaH.
Hydrinos
A hydrogen atom having a binding energy given by
Binding Energy = 13.6 eV (1)
(1/p)z
where p is an integer greater than 1, preferably from 2 to 137, is disclosed
in R. L.
Mills, "The Grand Unified Theory of Classical Quantum Mechanics", October 2007
Edition, (posted at http://www.blacklightpower.com/theory/book.shtml); R.
Mills, The
Grand Unified Theory of Classical Quantum Mechanics, May 2006 Edition,
BlackLight Power, Inc., Cranbury, New Jersey, ("'06 Mills GUT"), provided by
BlackLight Power, Inc., 493 Old Trenton Road, Cranbury, NJ, 08512 (posted at
www.blacklightpower.com); R. Mills, The Grand Unified Theory of Classical
Quantum
Mechanics, January 2004 Edition, BlackLight Power, Inc., Cranbury, New Jersey,
("
'04 Mills GUT"), provided by BlackLight Power, Inc., 493 Old Trenton Road,
Cranbury, NJ, 08512; R. Mills, The Grand Unified Theory of Classical Quantum
Mechanics, September 2003 Edition, BlackLight Power, Inc., Cranbury, New
Jersey,
('"03 Milis GUT"), provided by BlackLight Power, Inc., 493 Old Trenton Road,
Cranbury, NJ, 08512; R. Mills, The Grand Unified Theory of Classical Quantum
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
6
Mechanics, September 2002 Edition, BlackLight Power, Inc., Cranbury, New
Jersey,
(" '02 Mills GUT"), provided by BlackLight Power, Inc., 493 Old Trenton Road,
Cranbury, NJ, 08512; R. Mills, The Grand Unified Theory of Classical Quantum
Mechanics, September 2001 Edition, BlackLight Power, Inc., Cranbury, New
Jersey,
Distributed by Amazon.com (" '01 Mills GUT"), provided by BlackLight Power,
Inc.,
493 Old Trenton Road, Cranbury, NJ, 08512; R. Mills, The Grand Unified Theory
of
Classical Quantum Mechanics, January 2000 Edition, BlackLight Power, Inc.,
Cranbury, New Jersey, Distributed by Amazon.com ("'00 Nlills GUT"), provided
by
BlackLight Power, Inc., 493 Old Trenton Road, Cranbury, NJ, 08512; R.L. Mills,
"Physical Solutions of the Nature of the Atom, Photon, and Their Interactions
to Form
Excited and Predicted Hydrino States," Physics Essay, in press; R. L. Mills,
"Exact
Classical Quantum Mechanical Solution for Atomic Helium which Predicts
Conjugate
Parameters from a Unique Solution for the First Time," Physics Essays, in
press; R.
L. Mills, P. Ray, B. Dhandapani, "Excessive Balmer a Line Broadening of Water-
Vapor Capacitively-Coupled RF Discharge Plasmas," International Journal of
Hydrogen Energy, Vol. 33, (2008), 802-815; R. L. Mills, J. He, M. Nansteel, B.
Dhandapani, "Catalysis of Atomic Hydrogen to New Hydrides as a New Power
Source," International Journal of Global Energy Issues (IJGEI). Special
Edition in
Energy Systems, Vol. 28, issue 2-3, (2007), 304-324; R.L. Mills, H. Zea, J.
He, B.
Dhandapani, "Water Bath Calorimetry on a Catalytic Reaction of Atomic
Hydrogen,"
Int. J. Hydrogen Energy, Vol. 32, (2007), 4258-4266; J. Phillips, C. K. Chen,
R. L.
Mills, "Evidence of Catalytic Production of Hot Hydrogen in RF-Generated
Hydrogen/Argon Plasmas," Int. J. Hydrogen Energy, Vol. 32(14), (2007), 3010-
3025;
R. L. Mills, J. He, Y. Lu, M. Nansteel, Z. Chang, B. Dhandapani,
"Comprehensive
Identification and Potential Applications of New States of Hydrogen," Int. J.
Hydrogen
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
7
Energy, Vol. 32(14), (2007), 2988-3009; R. L. Mills, J. He, Z. Chang, W. Good,
Y.
Lu, B. Dhandapani, "Catalysis of Atomic Hydrogen to Novel Hydrogen Species H"
(1/4) and H2(1/4) as a New Power Source," Int. J. Hydrogen Energy, Vol.
32(13),
(2007), pp. 2573-2584; R. L. Mills, "Maxwell's Equations and QED: Which is
Fact
and Which is Fiction," Physics Essays, Vol. 19, (2006), 225-262; R. L. Nlills,
P. Ray,
B. Dhandapani, Evidence of an energy transfer reaction between atomic hydrogen
and argon II or helium II as the source of excessively hot H atoms in radio-
frequency
plasmas, J. Plasma Physics, Vol. 72, No. 4, (2006), 469-484; R. L. Mills,
"Exact
Classical Quantum Mechanical Solutions for One- through Twenty-Electron
Atoms,"
Physics Essays, Vol. 18, (2005), 321-361; R. L. Mills, P. C. Ray, R. M. Mayo,
M.
Nansteel, B. Dhandapani, J. Phillips, "Spectroscopic Study of Unique Line
Broadening and Inversion in Low Pressure Microwave Generated Water Plasmas,"
J. Plasma Physics, Vol. 71, No 6, (2005), 877-888; R. L. Mills, "The Fallacy
of
Feynman's Argument on the Stability of the Hydrogen Atom According to Quantum
Mechanics," Ann. Fund. Louis de Broglie, Vol. 30, No. 2, (2005), pp. 129-151;
R. L.
Mills, B. Dhandapani, J. He, "Highly Stable Amorphous Silicon Hydride from a
Helium Plasma Reaction," Materials Chemistry and Physics, 94/2-3, (2005),
298-307; R. L. Mills, J. He, Z, Chang, W. Good, Y. Lu, B. Dhandapani,
"Catalysis of
Atomic Hydrogen to Novel Hydrides as a New Power Source," Prepr. Pap.-Am.
Chem. Soc. Conf., Div. Fuel Chem., Vol. 50, No. 2, (2005); R. L. Mills, J.
Sankar, A.
Voigt, J. He, P. Ray, B. Dhandapani, "Role of Atomic Hydrogen Density and
Energy
in Low Power CVD Synthesis of Diamond Films," Thin Solid Films, 478, (2005)
77-90; R. L. Mills, "The Nature of the Chemical Bond Revisited and an
Alternative
Maxwellian Approach," Physics Essays, Vol. 17, (2004), 342-389; R. L. Mills,
P.
Ray, "Stationary Inverted Lyman Population and a Very Stable Novel Hydride
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
8
Formed by a Catalytic Reaction of Atomic Hydrogen and Certain Catalysts," J.
Opt.
Mat., 27, (2004), 181-186; W. Good, P. Jansson, M. Nansteel, J. He, A. Voigt,
"Spectroscopic and NMR Identification of Novel Hydride Ions in Fractional
Quantum
Energy States Formed by an Exothermic Reaction of Atomic Hydrogen with Certain
Catalysts," European Physical Journal: Applied Physics, 28, (2004), 83-104; J.
Phillips, R. L. Mills, X. Chen, "Water Bath Calorimetric Study of Excess Heat
in
`Resonance Transfer' Plasmas," J. Appl. Phys., Vol. 96, No. 6, (2004) 3095-
3102; R.
L. Mills, Y. Lu, M. Nansteel, J. He, A. Voigt, W. Good, B. Dhandapani,
"Energetic
Catalyst-Hydrogen Plasma Reaction as a Potential New Energy Source," Division
of
Fuel Chemistry, Session: Advances in Hydrogen Energy, Prepr. Pap.-Am. Chem.
Soc. Conf., Vol. 49, No. 2, (2004); R. L. Mills, J. Sankar, A. Voigt, J. He,
B.
Dhandapani, "Synthesis of HDLC Films from Solid Carbon," J. Materials Science,
J.
Mater. Sci. 39 (2004) 3309-3318; R. L. Mills, Y. Lu, M. Nansteel, J. He, A.
Voigt, B.
Dhandapani, "Energetic Catalyst-Hydrogen Plasma Reaction as a Potential New
Energy Source," Division of Fuel Chemistry, Session: Chemistry of Solid,
Liquid, and
Gaseous Fuels, Prepr. Pap.-Am. Chem. Soc. Conf., Vol. 49, No. 1, (2004); R. L.
Mills, "Classical Quantum Mechanics," Physics Essays, Vol. 16, (2003), 433-
498; R.
L. Mills, P. Ray, M. Nansteel, J. He, X. Chen, A. Voigt, B. Dhandapani,
"Characterization of an Energetic Catalyst-Hydrogen Plasma Reaction as a
Potential
New Energy Source," Am. Chem. Soc. Div. Fuel Chem. Prepr., Vol. 48, No. 2,
(2003); R. L. Mills, J. Sankar, A. Voigt, J. He, B. Dhandapani, "Spectroscopic
Characterization of the Atomic Hydrogen Energies and Densities and Carbon
Species During Helium-Hydrogen-Methane Plasma CVD Synthesis of Diamond
Films," Chemistry of Materials, Vol. 15, (2003), pp. 1313-1321; R. L. Mills,
P. Ray,
"Extreme Ultraviolet Spectroscopy of Helium-Hydrogen Plasma," J. Phys. D,
Applied
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
9
Physics, Vol. 36, (2003), pp. 1535-1542; R. L. Mills, X. Chen, P. Ray, J. He,
B.
Dhandapani, "Plasma Power Source Based on a Catalytic Reaction of Atomic
Hydrogen Measured by Water Bath Calorimetry," Thermochimica Acta, Vol. 406/1-
2,
(2003), pp. 35-53; R. L. Mills, B. Dhandapani, J. He, "Highly Stable Amorphous
Silicon Hydride," Solar Energy Materials & Solar Cells, Vol. 80, No. 1,
(2003), pp.
1-20; R. L. Mills, P. Ray, R. M. Mayo, "The Potential for a Hydrogen Water-
Plasma
Laser," Applied Physics Letters, Vol. 82, No. 11, (2003), pp. 1679-1681; R. L.
Mills,
P. Ray, "Stationary Inverted Lyman Population Formed from Incandescently
Heated
Hydrogen Gas with Certain Catalysts," J. Phys. D, Applied Physics, Vol. 36,
(2003),
pp. 1504-1509; R. L. Mills, P. Ray, B. Dhandapani, J. He, "Comparison of
Excessive
Balmer a Line Broadening of Inductively and Capacitively Coupled RF,
Microwave,
and Glow Discharge Hydrogen Plasmas with Certain Catalysts," IEEE Transactions
on Plasma Science, Vol. 31, No. (2003), pp. 338-355; R. L. Mills, P. Ray, R.
M.
Mayo, "CW HI Laser Based on a Stationary Inverted Lyman Population Formed from
Incandescently Heated Hydrogen Gas with Certain Group I Catalysts," IEEE
Transactions on Plasma Science, Vol. 31, No. 2, (2003), pp. 236-247; R. L.
Mills, P.
Ray, J. Dong, M. Nansteel, B. Dhandapani, J. He, "Spectral Emission of
Fractional-
Principal-Quantum-Energy-Level Atomic and Molecular Hydrogen," Vibrational
Spectroscopy, Vol. 31, No. 2, (2003), pp. 195-213; H. Conrads, R. L. Mills,
Th.
Wrubel, "Emission in the Deep Vacuum Ultraviolet from a Plasma Formed by
Incandescently Heating Hydrogen Gas with Trace Amounts of Potassium
Carbonate," Plasma Sources Science and Technology, Vol. 12, (2003), pp. 389-
395;
R. L. Mills, J. He, P. Ray, B. Dhandapani, X. Chen, "Synthesis and
Characterization
of a Highly Stable Amorphous Silicon Hydride as the Product of a Catalytic
Helium-
Hydrogen Plasma Reaction," Int. J. Hydrogen Energy, Vol. 28, No. 12, (2003),
pp.
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
1401-1424; R. L. Mills, P. Ray, "A Comprehensive Study of Spectra of the Bound-
Free Hyperfine Levels of Novel Hydride Ion H-(1/2), Hydrogen, Nitrogen, and
Air,"
Int. J. Hydrogen Energy, Vol. 28, No. 8, (2003), pp. 825-871; R. L. Mills, M.
Nansteel, and P. Ray, "Excessively Bright Hydrogen-Strontium Plasma Light
Source
5 Due to Energy Resonance of Strontium with Hydrogen," J. Plasma Physics, Vol.
69,
(2003), pp. 131-158; R. L. Mills, "Highly Stable Novel Inorganic Hydrides," J.
New
Materials for Electrochemical Systems, Vol. 6, (2003), pp. 45-54; R. L. Mills,
P. Ray,
"Substantial Changes in the Characteristics of a Microwave Plasma Due to
Combining Argon and Hydrogen," New Journal of Physics, www.njp.org, Vol. 4,
10 (2002), pp. 22.1-22.17; R. M. Mayo, R. L. Mills, M. Nansteel, "Direct
Plasmadynamic
Conversion of Plasma Thermal Power to Electricity," IEEE Transactions on
Plasma
Science, October, (2002), Vol. 30, No. 5, pp. 2066-2073; R. L. Mills, M.
Nansteel, P.
Ray, "Bright Hydrogen-Light Source due to a Resonant Energy Transfer with
Strontium and Argon Ions," New Journal of Physics, Vol. 4, (2002), pp. 70.1-
70.28;
R. M. Mayo, R. L. Mills, M. Nansteel, "On the Potential of Direct and MHD
Conversion of Power from a Novel Plasma Source to Electricity for
Microdistributed
Power Applications," IEEE Transactions on Plasma Science, August, (2002), Vol.
30,
No. 4, pp. 1568-1578; R. M. Mayo, R. L. Mills, "Direct Plasmadynamic
Conversion of
Plasma Thermal Power to Electricity for Microdistributed Power Applications,"
40th
Annual Power Sources Conference, Cherry Hill, NJ, June 10-13, (2002), pp. 1-4;
R.
L. Mills, E. Dayalan, P. Ray, B. Dhandapani, J. He, "Highly Stable Novel
Inorganic
Hydrides from Aqueous Electrolysis and Plasma Electrolysis," Electrochimica
Acta,
Vol. 47, No. 24, (2002), pp. 3909-3926; R. L. Mills, P. Ray, B. Dhandapani, R.
M.
Mayo, J. He, "Comparison of Excessive Balmer a Line Broadening of Glow
Discharge and Microwave Hydrogen Plasmas with Certain Catalysts," J. of
Applied
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
11
Physics, Vol. 92, No. 12, (2002), pp. 7008-7022; R. L. Mills, P. Ray, B.
Dhandapani,
M. Nansteel, X. Chen, J. He, "New Power Source from Fractional Quantum Energy
Levels of Atomic Hydrogen that Surpasses Internal Combustion," J. Mol.
Struct., Vol.
643, No. 1-3, (2002), pp. 43-54; R. L. Mills, J. Dong, W. Good, P. Ray, J. He,
B.
Dhandapani, "Measurement of Energy Balances of Noble Gas-Hydrogen Discharge
Plasmas Using Calvet Calorimetry," Int. J. Hydrogen Energy, Vol. 27, No. 9,
(2002),
pp. 967-978; R. L. Mills, P. Ray, "Spectroscopic Identification of a Novel
Catalytic
Reaction of Rubidium Ion with Atomic Hydrogen and the Hydride Ion Product,"
Int. J.
Hydrogen Energy, Vol. 27, No. 9, (2002), pp. 927-935; R. L. Nlills, A. Voigt,
P. Ray,
M. Nansteel, B. Dhandapani, "Measurement of Hydrogen Balmer Line Broadening
and Thermal Power Balances of Noble Gas-Hydrogen Discharge Plasmas," Int. J.
Hydrogen Energy, Vol. 27, No. 6, (2002), pp. 671-685; R. L. Mills, N. Greenig,
S.
Hicks, "Optically Measured Power Balances of Glow Discharges of Mixtures of
Argon, Hydrogen, and Potassium, Rubidium, Cesium, or Strontium Vapor," Int. J.
Hydrogen Energy, Vol. 27, No. 6, (2002), pp. 651-670; R. L. Mills, "The Grand
Unified Theory of Classical Quantum Mechanics," Int. J. Hydrogen Energy, Vol.
27,
No. 5, (2002), pp. 565-590; R. L. Mills, P. Ray, "Vibrational Spectral
Emission of
Fractional-Principal-Quantum-Energy-Level Hydrogen Molecular Ion," Int. J.
Hydrogen Energy, Vol. 27, No. 5, (2002), pp. 533-564; R. L. Mills and M.
Nansteel,
P. Ray, "Argon-Hydrogen-Strontium Discharge Light Source," IEEE Transactions
on
Plasma Science, Vol. 30, No. 2, (2002), pp. 639-653; R. L. Mills, P. Ray,
"Spectral
Emission of Fractional Quantum Energy Levels of Atomic Hydrogen from a Helium-
Hydrogen Plasma and the Implications for Dark Matter," Int. J. Hydrogen
Energy,
(2002), Vol. 27, No. 3, pp. 301-322; R. L. Mills, P. Ray, "Spectroscopic
Identification
of a Novel Catalytic Reaction of Potassium and Atomic Hydrogen and the Hydride
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
12
Ion Product," Int. J. Hydrogen Energy, Vol. 27, No. 2, (2002), pp. 183-192; R.
L.
Mills, E. Dayalan, "Novel Alkali and Alkaline Earth Hydrides for High Voltage
and
High Energy Density Batteries," Proceedings of the 17th Annual Battery
Conference
on Applications and Advances, California State University, Long Beach, CA,
(January 15-18, 2002), pp. 1-6; R. L. Mills, W. Good, A. Voigt, Jinquan Dong,
"Minimum Heat of Formation of Potassium lodo Hydride," Int. J. Hydrogen
Energy,
Vol. 26, No. 11, (2001), pp. 1199-1208; R. L. Mills, "The Nature of Free
Electrons in
Superfluid Helium-a Test of Quantum Mechanics and a Basis to Review its
Foundations and Make a Comparison to Classical Theory," Int. J. Hydrogen
Energy,
Vol. 26, No. 10, (2001), pp. 1059-1096; R. L. Mills, "Spectroscopic
Identification of a
Novel Catalytic Reaction of Atomic Hydrogen and the Hydride Ion Product," Int.
J.
Hydrogen Energy, Vol. 26, No. 10, (2001), pp. 1041-1058; R. L. Mills, B.
Dhandapani, M. Nansteel, J. He, A. Voigt, "Identification of Compounds
Containing
Novel Hydride Ions by Nuclear Magnetic Resonance Spectroscopy," Int. J.
Hydrogen
Energy, Vol. 26, No. 9, (2001), pp. 965-979; R. L. Mills, T. Onuma, and Y. Lu,
"Formation of a Hydrogen Plasma from an Incandescently Heated Hydrogen-
Catalyst Gas Mixture with an Anomalous Afterglow Duration," Int. J. Hydrogen
Energy, Vol. 26, No. 7, July, (2001), pp. 749-762; R. L. Mills, "Observation
of
Extreme Ultraviolet Emission from Hydrogen-KI Plasmas Produced by a Hollow
Cathode Discharge," Int. J. Hydrogen Energy, Vol. 26, No. 6, (2001), pp. 579-
592;
R. L. Mills, B. Dhandapani, M. Nansteel, J. He, T. Shannon, A. Echezuria,
"Synthesis
and Characterization of Novel Hydride Compounds," Int. J. of Hydrogen Energy,
Vol.
26, No. 4, (2001), pp. 339-367; R. L. Mills, "Temporal Behavior of Light-
Emission in
the Visible Spectral Range from a Ti-K2CO3-H-Cell," Int. J. Hydrogen Energy,
Vol.
26, No. 4, (2001), pp. 327-332; R. L. Mills, M. Nansteel, and Y. Lu,
"Observation of
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
13
Extreme Ultraviolet Hydrogen Emission from Incandescently Heated Hydrogen Gas
with Strontium that Produced an Anomalous Optically Measured Power Balance,"
Int. J. Hydrogen Energy, Vol. 26, No. 4, (2001), pp. 309-326; R. L. Mills,
"BlackLight
Power Technology-A New Clean Hydrogen Energy Source with the Potential for
Direct Conversion to Electricity," Proceedings of the National Hydrogen
Association,
12th Annual U.S. Hydrogen Meeting and Exposition, Hydrogen: The Common
Thread, The Washington Hilton and Towers, Washington DC, (March 6-8, 2001),
pp.
671-697; R. L. Mills, "The Grand Unified Theory of Classical Quantum
Mechanics,"
Global Foundation, Inc. Orbis Scientiae entitled The Role of Attractive and
Repulsive
Gravitational Forces in Cosmic Acceleration of Particles The Origin of the
Cosmic
Gamma Ray Bursts, (29th Conference on High Energy Physics and Cosmology
Since 1964) Dr. Behram N. Kursunoglu, Chairman, December 14-17, 2000, Lago
Mar Resort, Fort Lauderdale, FL, Kluwer Academic/Plenum Publishers, New York,
pp. 243-258; R. L. Mills, B. Dhandapani, N. Greenig, J. He, "Synthesis and
Characterization of Potassium lodo Hydride," Int. J. of Hydrogen Energy, Vol.
25,
Issue 12, December, (2000), pp. 1185-1203; R. L. Mills, "The Hydrogen Atom
Revisited," Int. J. of Hydrogen Energy, Vol. 25, Issue 12, December, (2000),
pp.
1171-1183; R. L. Mills, "BlackLight Power Technology-A New Clean Energy
Source with the Potential for Direct Conversion to Electricity," Global
Foundation
International Conference on "Global Warming and Energy Policy," Dr. Behram N.
Kursunoglu, Chairman, Fort Lauderdale, FL, November 26-28, 2000, Kluwer
Academic/Plenum Publishers, New York, pp. 187-202; R. L. Mills, J. Dong, Y.
Lu,
"Observation of Extreme Liltraviolet Hydrogen Emission from Incandescently
Heated
Hydrogen Gas with Certain Catalysts," Int. J. Hydrogen Energy, Vol. 25,
(2000), pp.
919-943; R. L. Mills, "Novel Inorganic Hydride," Int. J. of Hydrogen Energy,
Vol. 25,
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
14
(2000), pp. 669-683; R. L. Mills, "Novel Hydrogen Compounds from a Potassium
Carbonate Electrolytic Cell," Fusion Technol., Vol. 37, No. 2, March, (2000),
pp.
157-182; R. L. Mills, W. Good, "Fractional Quantum Energy Levels of Hydrogen,"
Fusion Technology, Vol. 28, No. 4, November, (1995), pp. 1697-1719; R. L.
Mills,
W. Good, R. Shaubach, "Dihydrino Molecule Identification," Fusion Technol.,
Vol. 25,
(1994), 103; R. L. Mills and S. Kneizys, Fusion Technol. Vol. 20, (1991), 65;
and in
prior published PCT application Nos. W090/13126; W092/10838; W094/29873;
W096/42085; W099/05735; W099/26078; W099/34322; W099/35698;
W000/07931; W000/07932; W001 /095944; W001 /18948; W001 /21300;
WO01/22472; WO01/70627; W002/087291; W002/088020; W002/16956;
W003/093173; W003/066516; W004/092058; W005/041368; W005/067678;
W02005/116630; W02007/051078; and W02007/053486; and prior US Patent Nos.
Nos. 6,024,935 and 7,188,033, the entire disclosures of which are all
incorporated
herein by reference (hereinafter "Mills Prior Publications").
The binding energy of an atom, ion, or molecule, also known as the ionization
energy, is the energy required to remove one electron from the atom, ion or
molecule. A hydrogen atom having the binding energy given in Eq. (1) is
hereafter
referred to as a hydrino atom or hydrino. The designation for a hydrino of
radius
a
H,where aH is the radius of an ordinary hydrogen atom and p is an integer, is
p
Hj aH j. A hydrogen atom with a radius aH is hereinafter referred to as
"ordinary
LPJ
hydrogen atom" or "normal hydrogen atom." Ordinary atomic hydrogen is
characterized by its binding energy of 13.6 eV.
Hydrinos are formed by reacting an ordinary hydrogen atom with a catalyst
having a net enthalpy of reaction of about
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
m=27.2eV (2)
where m is an integer. This catalyst has also been referred to as an energy
hole or
source of energy hole in Mills earlier filed Patent Applications. It is
believed that the
rate of catalysis is increased as the net enthalpy of reaction is more closely
matched
5 to m= 27.2 eV. It has been found that catalysts having a net enthalpy of
reaction
within 10%, preferably 5%, of m- 27.2 eV are suitable for most applications.
This catalysis releases energy from the hydrogen atom with a commensurate
decrease in size of the hydrogen atom, rn =na, . For example, the catalysis of
H(n = 1) to H(n = 1/ 2) releases 40.8 eV, and the hydrogen radius decreases
from
10 aõ to 2 aH. A catalytic system is provided by the ionization of t electrons
from an
atom each to a continuum energy level such that the sum of the ionization
energies
of the t electrons is approximately m- 27.2 eV where m is an integer.
One such catalytic system involves lithium metal. The first and second
ionization energies of lithium are 5.39172 eV and 75.64018 eV, respectively
[1]. The
15 double ionization (t = 2) reaction of Li to Liz+, then, has a net enthalpy
of reaction of
81.0319 eV, which is equivalent to m = 3 in Eq. (2).
81.0319eV+Li(m)+Hj Lp jLi2++2e-+Hj (p+3) 1+[(p+3)2-p2]=13.6 eV
(3)
Li2++ 2 e- --) Li(m) + 81.0319 eV (4)
And, the overall reaction is
Hj p j--) HF(p+3)~+[(p+3)2 -p2]=13.6eV (5)
In another embodiment, the catalytic system involves cesium. The first and
second ionization energies of cesium are 3.89390 eV and 23.15745 eV,
respectively.
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
16
The double ionization (t = 2) reaction of Cs to Cs2+, then, has a net enthalpy
of
reaction of 27.05135 eV, which is equivalent to m=1 in Eq. (2).
27.05135eV+Cs(m)+HLraP y J j~Csz++2e +Hj (p+l)1+[(p+1)2-pz].13.6eV
L J
(6)
Cs2++2e -4 Cs(m)+27.05135 eV (7)
And, the overall reaction is
H~a,~~H~(p+l)~l+[(p+1)2_p2].13.6eV (8)
p
An additional catalytic system involves potassium metal. The first, second,
and third ionization energies of potassium are 4.34066 eV, 31.63 eV, 45.806
eV,
respectively [1]. The triple ionization ( t= 3) reaction of K to K3+ , then,
has a net
enthalpy of reaction of 81.7767 eV, which is equivalent to m = 3 in Eq. (2).
81.7767 e V + K (m) + H aH K3, +3e +H a" l+[(p+3)2_p21.13.6 e13.6 eV
p (p+3)
(9)
K3+ +3e -~ K(m)+81.7426 eV (10)
And, the overall reaction is
H aH ~H aH ]+[(P+3)2_P2].13.6 e13.6 eV (11)
p (p+3)
As a power source, the energy given off during catalysis is much greater than
the
energy lost to the catalyst. The energy released is large as compared to
conventional chemical reactions. For example, when hydrogen and oxygen gases
undergo combustion to form water
H2 (S)+ 2 02 (g) -4 H20 (1) (12)
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
17
the known enthalpy of formation of water is 4Hf =-286 kJ / mole or 1.48 eV per
hydrogen atom. By contrast, each ( n=1) ordinary hydrogen atom undergoing
catalysis releases a net of 40.8 eV. Moreover, further catalytic transitions
may
1 1 1 1 1 1
occur: n= 2-4 3' 3~ 4' 4~ 5 and so on. Once catalysis begins, hydrinos
autocatalyze further in a process called disproportionation. This mechanism is
similar to that of an inorganic ion catalysis. But, hydrino catalysis should
have a
higher reaction rate than that of the inorganic ion catalyst due to the better
match of
the enthalpy to m- 27.2 eV.
Further Catalysis Products of the Present Invention
The hydrino hydride ion of the present invention can be formed by the
reaction of an electron source with a hydrino, that is, a hydrogen atom having
a
binding energy of about 13.62eV where n = 1 and p is an integer greater than
1.
n p
The hydrino hydride ion is represented by H- (n =1 / p) or H- (1 / p) :
HLaHJ+e---~H-(n=1/p) (13)
p
HLaHJ+e H-(1/p) (14)
p
The hydrino hydride ion is distinguished from an ordinary hydride ion
comprising an ordinary hydrogen nucleus and two electrons having a binding
energy
of about 0.8 eV. The latter is hereafter referred to as "ordinary hydride ion"
or
"normal hydride ion" The hydrino hydride ion comprises a hydrogen nucleus
including proteum, deuterium, or tritium, and two indistinguishable electrons
at a
binding energy according to Eq. (15).
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
18
The binding energy of a novel hydrino hydride ion can be represented by the
following formula:
3
Binding Energy = h 2 s(s + 1) ~_ 7c oe~ ~i 2 1 22
g a 2 1+ s(s + 1) 'ne aH a~ 1+ s(s + 1)
p p
(15)
where p is an integer greater than one, s=1 / 2,ic is pi, h is Planck's
constant bar,
, is the permeability of vacuum, me is the mass of the electron, e is the
reduced
electron mass given by e = m em where m is the mass of the proton, aN is the
e P
+ mP
441 radius of the hydrogen atom, ao is the Bohr radius, and e is the
elementary charge.
The radii are given by
r2 =r,=ao+ s(s+1)) ;s=2 (16)
The binding energies of the hydrino hydride ion, H- (n =1 / p) as a function
of
p, where p is an integer, are shown in TABLE 1.
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
19
TABLE 1. The representative binding energy of the hydrino hydride ion H- (n =1
/ p)
as a function of p, Eq. (15).
Hydride lon rl Binding Wavelength
( ao )a Energy (eV)b (nm)
H- (n =1) 1.8660 0.7542 1644
H(n = 1 2) 0.9330 3.047 406.9
H-(n=1/3) 0.6220 6.610 187.6
H- (n= 1/4) 0.4665 11.23 110.4
H- (n = 115) 0.3732 16.70 74.23
H-(n=1/6) 0.3110 22.81 54.35
H- (n =1 / 7) 0.2666 29.34 42.25
H- (n =1 / 8) 0.2333 36.09 34.46
H- (n = 1/ 9) 0.2073 42.84 28.94
H- (n =1 / 10) 0.1866 49.38 25.11
H- (n = 1/ 11) 0.1696 55.50 22.34
H(n = 1/ 12) 0.1555 60.98 20.33
H- (n = 1/ 13) 0.1435 65.63 18.89
H-(n=1/14) 0.1333 69.22 17.91
H-(n=1/15) 0.1244 71.55 17.33
H- (n= 1/16) 0.1166 72.40 17.12
H-(n=1/17) 0.1098 71.56 17.33
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
H- (n= 1/18) 0.1037 68.83 18.01
H- (n = 1/ 19) 0.0982 63.98 19.38
H- (n = 1/ 20) 0.0933 56.81 21.82
H(n = 1/ 21) 0.0889 47.11 26.32
5 H- (n = 1/ 22) 0.0848 34.66 35.76
H- (n = 1/ 23) 0.0811 19.26 64.36
H- (n =1 / 24) 0.0778 0.6945 1785
a Eq. (16)
b Eq. (15)
According to the present invention, a hydrino hydride ion (H-) having a
binding
energy according to Eqs. (15-16) that is greater than the binding of ordinary
hydride
ion (about 0.8 eV) for p = 2 up to 23, and less for p = 24 (H ) is provided.
For p = 2
to p = 24 of Eqs. (15-16), the hydride ion binding energies are respectively
3, 6.6,
11.2, 16.7, 22.8, 29.3, 36.1, 42.8, 49.4, 55.5, 61.0, 65.6, 69.2, 71.6, 72.4,
71.6, 68.8,
64.0, 56.8, 47.1, 34.7, 19.3, and 0.69 eV. Compositions comprising the novel
hydride ion are also provided.
The hydrino hydride ion is distinguished from an ordinary hydride ion
comprising an ordinary hydrogen nucleus and two electrons having a binding
energy
of about 0.8 eV. The latter is hereafter referred to as "ordinary hydride ion"
or
"normal hydride ion" The hydrino hydride ion comprises a hydrogen nucleus
including proteum, deuterium, or tritium, and two indistinguishable electrons
at a
binding energy according to Eqs. (15-16).
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
21
Novel compounds are provided comprising one or more hydrino hydride ions
and one or more other elements. Such a compound is referred to as a hydrino
hydride compound.
Ordinary hydrogen species are characterized by the following binding
energies (a) hydride ion, 0.754 eV ("ordinary hydride ion"); (b) hydrogen atom
("ordinary hydrogen atom"), 13.6 eV; (c) diatomic hydrogen molecule, 15.3 eV
("ordinary hydrogen molecule"); (d) hydrogen molecular ion, 16.3 eV ("ordinary
hydrogen molecular ion"); and (e) H; , 22.6 eV ("ordinary trihydrogen
molecular
ion"). Herein, with reference to forms of hydrogen, "normal" and "ordinary"
are
synonymous.
According to a further embodiment of the invention, a compound is provided
comprising at least one increased binding energy hydrogen species such as (a)
a
hydrogen atom having a binding energy of about 13.6 eV preferably within 10%,
P/
more preferably 5%, where p is an integer, preferably an integer from 2 to
137; (b)
a hydride ion ( H- ) having a binding energy of about
Binding Energy = h 2 s(s + 1) 2_ic oe2 t~Z 1 22 3 preferably
g Q~~1+ s(s+1)] me aH a~[ I s(s+1)~
p p
within 10%, more preferably 5%, where p is an integer, preferably an integer
from
2 to 24; (c) H; (1 / p) ;(d) a trihydrino molecular ion, H3 (1 / p), having a
binding
energy of about 22.6 z eV preferably within 10%, more preferably 5%, where p
is
Cp)20 an integer, preferably an integer from 2 to 137; (e) a dihydrino having
a binding
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
22
energy of about 15'3 eV referabl within 10%, more referabl +5% where
2 p Y P Y- , p is
LD
an integer, preferably and integer from 2 to 137; (f) a dihydrino molecular
ion with a
binding energy of about 16.3 eV preferably within 10%, more preferably 5%,
)
P
where p is an integer, preferably an integer from 2 to 137.
According to a further preferred embodiment of the invention, a compound is
provided comprising at least one increased binding energy hydrogen species
such
as (a) a dihydrino molecular ion having a total energy of
F Z 2~i
ET -p2 e (41n3-1-21n3) l+p - 1h k
8i'E aõ mec2
2 (17)
_ -p2 16.13392 eV - p30.118755 eV
preferably within 10%, more preferably 5%, where p is an integer, h is
Planck's
constant bar, me is the mass of the electron, c is the speed of light in
vacuum, is
the reduced nuclear mass, and k is the harmonic force constant solved
previously
[2] and (b) a dihydrino molecule having a total energy of
ez
2h 4~e~a~
z
E, =-p2 e 2~-~+~ ln~+l _~ l+p me - l~i~
8~ ao 2 ~-1 mec 2 p
_ -p231.351 eV - p30.326469 eV
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
23
(18)
preferably within 10%, more preferably 5%, where p is an integer and ao is
the
Bohr radius.
According to one embodiment of the invention wherein the compound
comprises a negatively charged increased binding energy hydrogen species, the
compound further comprises one or more cations, such as a proton, ordinary HZ
, or
ordinary H3 H.
A method is provided for preparing compounds comprising at least one
increased binding energy hydride ion. Such compounds are hereinafter referred
to
as "hydrino hydride compounds". The method comprises reacting atomic hydrogen
with a catalyst having a net enthalpy of reaction of about 2= 27 eV, where m
is an
integer greater than 1, preferably an integer less than 400, to produce an
increased
binding energy h dro en atom having a binding energy of about 13.6 eV where s
Y 9 Z p i
Cpl
an integer, preferably an integer from 2 to 137. A further product of the
catalysis is
energy. The increased binding energy hydrogen atom can be reacted with an
electron source, to produce an increased binding energy hydride ion. The
increased
binding energy hydride ion can be reacted with one or more cations to produce
a
compound comprising at least one increased binding energy hydride ion.
Novel hydrogen species and compositions of matter comprising new forms of
hydrogen formed by the catalysis of atomic hydrogen are disclosed in "Mills
Prior
Publications". The novel hydrogen compositions of matter comprise:
(a) at least one neutral, positive, or negative hydrogen species (hereinafter
"increased binding energy hydrogen species") having a binding energy
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
24
(i) greater than the binding energy of the corresponding ordinary
hydrogen species, or
(ii) greater than the binding energy of any hydrogen species for which
the corresponding ordinary hydrogen species is unstable or is not observed
because
the ordinary hydrogen species' binding energy is less than thermal energies at
ambient conditions (standard temperature and pressure, STP), or is negative;
and
(b) at least one other element. The compounds of the invention are
hereinafter referred to as "increased binding energy hydrogen compounds".
By "other element" in this context is meant an element other than an
increased binding energy hydrogen species. Thus, the other element can be an
ordinary hydrogen species, or any element other than hydrogen. In one group of
compounds, the other element and the increased binding energy hydrogen species
are neutral. In another group of compounds, the other element and increased
binding energy hydrogen species are charged such that the other element
provides
the balancing charge to form a neutral compound. The former group of compounds
is characterized by molecular and coordinate bonding; the latter group is
characterized by ionic bonding.
Also provided are novel compounds and molecular ions comprising
(a) at least one neutral, positive, or negative hydrogen species (hereinafter
"increased binding energy hydrogen species") having a total energy
(i) greater than the total energy of the corresponding ordinary hydrogen
species, or
(ii) greater than the total energy of any hydrogen species for which the
corresponding ordinary hydrogen species is unstable or is not observed because
the
ordinary hydrogen species' total energy is less than thermal energies at
ambient
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
conditions, or is negative; and
(b) at least one other element.
The total energy of the hydrogen species is the sum of the energies to remove
all of
the electrons from the hydrogen species. The hydrogen species according to the
5 present invention has a total energy greater than the total energy of the
corresponding ordinary hydrogen species. The hydrogen species having an
increased total energy according to the present invention is also referred to
as an
"increased binding energy hydrogen species" even though some ernbodiments of
the hydrogen species having an increased total energy may have a first
electron
10 binding energy less that the first electron binding energy of the
corresponding
ordinary hydrogen species. For example, the hydride ion of Eqs. (15-16) for p
= 24
has a first binding energy that is less than the first binding energy of
ordinary hydride
ion, while the total energy of the hydride ion of Eqs. (15-16) for p= 24 is
much
greater than the total energy of the corresponding ordinary hydride ion.
15 Also provided are novel compounds and molecular ions comprising
(a) a plurality of neutral, positive, or negative hydrogen species
(hereinafter
"increased binding energy hydrogen species") having a binding energy
(i) greater than the binding energy of the corresponding ordinary
hydrogen species, or
20 (ii) greater than the binding energy of any hydrogen species for which
the corresponding ordinary hydrogen species is unstable or is not observed
because
the ordinary hydrogen species' binding energy is less than thermal energies at
ambient conditions or is negative; and
(b) optionally one other element. The compounds of the invention are
25 hereinafter referred to as "increased binding energy hydrogen compounds".
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
26
The increased binding energy hydrogen species can be formed by reacting
one or more hydrino atoms with one or more of an electron, hydrino atom, a
compound containing at least one of said increased binding energy hydrogen
species, and at least one other atom, molecule, or ion other than an increased
binding energy hydrogen species.
Also provided are novel compounds and molecular ions comprising
(a) a plurality of neutral, positive, or negative hydrogen species
(hereinafter
"increased binding energy hydrogen species") having a total energy
(i) greater than the total energy of ordinary molecular hydrogen, or
(ii) greater than the total energy of any hydrogen species for which the
corresponding ordinary hydrogen species is unstable or is not observed because
the
ordinary hydrogen species' total energy is less than thermal energies at
ambient
conditions or is negative; and
(b) optionally one other element. The compounds of the invention are
hereinafter referred to as "increased binding energy hydrogen compounds".
In an embodiment, a compound is provided, comprising at least one
increased binding energy hydrogen species selected from the group consisting
of (a)
hydride ion having a binding energy according to Eqs. (15-16) that is greater
than
the binding of ordinary hydride ion (about 0.8 eV) for p = 2 up to 23, and
less for
p= 24 ("increased binding energy hydride ion" or "hydrino hydride ion"); (b)
hydrogen atom having a binding energy greater than the binding energy of
ordinary
hydrogen atom (about 13.6 eV) ("increased binding energy hydrogen atom" or
"hydrino"); (c) hydrogen molecule having a first binding energy greater than
about
15.3 eV ("increased binding energy hydrogen molecule" or "dihydrino"); and (d)
molecular hydrogen ion having a binding energy greater than about 16.3 eV
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
27
("increased binding energy molecular hydrogen ion" or "dihydrino molecular
ion").
Characteristics and Identification of Increased Binding Energy Species
A new chemically generated or assisted plasma source based on a resonant
energy transfer mechanism (rt-plasma) between atomic hydrogen and certain
catalysts has been developed that may be a new power source. The products are
more stable hydride and molecular hydrogen species such as H- (1 / 4) and
HZ (1 / 4). One such source operates by incandescently heating a hydrogen
dissociator and a catalyst to provide atomic hydrogen and gaseous catalyst,
respectively, such that the catalyst reacts with the atomic hydrogen to
produce a
plasma. It was extraordinary that intense extreme ultraviolet (EUV) emission
was
observed by Mills et al. [3-10] at low temperatures (e.g. =103 K) and an
extraordinary low field strength of about 1-2 V/cm from atomic hydrogen and
certain
atomized elements or certain gaseous ions which singly or multiply ionize at
integer
multiples of the potential energy of atomic hydrogen, 27.2 eV. A number of
independent experimental observations confirm that the rt-plasma is due to a
novel
reaction of atomic hydrogen which produces as chemical intermediates, hydrogen
in
fractional quantum states that are at lower energies than the traditional
"ground"
( n=1) state. Power is released [3, 9, 11-13], and the final reaction products
are
novel hydride compounds [3, 14-16] or lower-energy molecular hydrogen [17].
The
supporting data include EUV spectroscopy [3-10, 13, 17-22, 25, 27-28],
characteristic emission from catalysts and the hydride ion products [3, 5, 7,
21-22,
27-28], lower-energy hydrogen emission [12-13, 18-20], chemically formed
plasmas
[3-10, 21-22, 27-28], extraordinary (>100 eV) Balmer a line broadening [3-5,
7, 9-
10, 12, 18-19, 21, 23-28], population inversion of H lines [3, 21, 27-29],
elevated
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
28
electron temperature [19, 23-25], anomalous plasma afterglow duration [3, 8],
power
generation [3, 9, 11-13], and analysis of novel chemical compounds [3, 14-16].
The theory given previously [6, 18-20, 30] is based on Maxwell's equations to
solving the structure of the electron. The familiar Rydberg equation (Eq.
(19)) arises
for the hydrogen excited states for n> 1 of Eq. (20).
_ e 2 13.598 eV (19)
E" n287t oaõ nZ n =1, 2,3,... (20)
An additional result is that atomic hydrogen may undergo a catalytic reaction
with
certain atoms, excimers, and ions which provide a reaction with a net enthalpy
of an
integer multiple of the potential energy of atomic hydrogen, m- 27.2 eV
wherein m is
an integer. The reaction involves a nonradiative energy transfer to form a
hydrogen
atom called a hydrino atom that is lower in energy than unreacted atomic
hydrogen
that corresponds to a fractional principal quantum number. That is
n = 2, 3, 4,..., 1; p is an integer (21)
p
replaces the well known parameter n = integer in the Rydberg equation for
hydrogen
excited states. The n=1 state of hydrogen and the n = 1 states of hydrogen
integer
are nonradiative, but a transition between two nonradiative states, say n=1 to
n=112, is possible via a nonradiative energy transfer. Thus, a catalyst
provides a
net positive enthalpy of reaction of m= 27.2 eV (i.e. it resonantly accepts
the
nonradiative energy transfer from hydrogen atoms and releases the energy to
the
surroundings to affect electronic transitions to fractional quantum energy
levels). As
a consequence of the nonradiative energy transfer, the hydrogen atom becomes
unstable and emits further energy until it achieves a lower-energy
nonradiative state
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
29
having a principal energy level given by Eqs. (19) and (21). Processes such as
hydrogen molecular bond formation that occur without photons and that require
collisions are common [31]. Also, some commercial phosphors are based on
resonant nonradiative energy transfer involving multipole coupling [32].
Two H(1 / p) may react to form H2 (1 / p). The hydrogen molecular ion and
molecular charge and current density functions, bond distances, and energies
were
exactly solved previously with remarkable accuracy [30, 33]. Using the
Laplacian in
ellipsoidal coordinates with the constraint of nonradiation, the total energy
of the
hydrogen molecule having a central field of +pe at each focus of the prolate
spheroid molecular orbital is
eZ
4~c8oa~
2~2
ET _-p2 e 2 2~_~+~ ln~+l _~ l+p me - ~i
81ra.ao 2 ~-1 mec 2 j
_ -p2 31.351 eV - p30.326469 eV
(22)
where p is an integer, h is Planck's constant bar, me is the mass of the
electron, c
is the speed of light in vacuum, p is the reduced nuclear mass, k is the
harmonic
force constant solved previously in a closed-form equation with fundamental
constants only [30, 33] and ao is the Bohr radius. The vibrational and
rotational
energies of fractional-Rydberg-state molecular hydrogen H2 (1 / p) are p2
those of
Hz . Thus, the vibrational energies, E;b, for the v = 0 to v=1 transition of
hydrogen-type molecules HZ (1 / p) are [30, 33]
Ey;b = p2 0.515902 eV (23)
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
where the experimental vibrational energy for the v = 0 to v=1 transition of
H2,
EH2(U-ow-,), is given by Beutler [34] and Herzberg [35]. The rotational
energies, E,ot,
for the J to J + 1 transition of hydrogen-type molecules H2 (1 / p) are [30,
33]
z
E,o, = Ej+, - Ej = j[J + 1] = pz (J + 1)0.01509 eV (24)
5 where I is the moment of inertia, and the experimental rotational energy for
the
J = 0 to J=1 transition of H2 is given by Atkins [36]. The p2 dependence of
the
rotational energies results from an inverse p dependence of the internuclear
distance and the corresponding impact on I. The predicted internuclear
distance
2c' for H2 (1/ p) is
10 2c' = a _[2 (25)
p
The rotational energies provide a very precise measure of I and the
internuclear
distance using well established theory [37].
Ar' may serve as a catalyst since its ionization energy is about 27.2 eV. The
catalyst reaction of Ar+ to Ar2+ forms H(1 / 2) which may further serve as
both a
15 catalyst and a reactant to form H(1 / 4) [19-20, 30]. Thus, the observation
of
H(1 / 4) is predicted to be flow dependent since the formation of HZ (1 / 4)
requires
the buildup of intermediates. The mechanism was tested by experiments with
flowing plasma gases. Neutral molecular emission was anticipated for high
pressure
argon-hydrogen plasmas excited by a 12.5 keV electron beam. Rotational lines
for
20 H2 (1 / 4) were anticipated and sought in the 150-250 nm region. The
spectral lines
were compared to those predicted by Eqs. (23-24) corresponding to the
internuclear
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
31
distance of 1/4 that of H2 given by Eq. (25). For p = 4 in Eqs. (23-24), the
predicted
energies for the v=1 -> v= 0 vibration-rotational series of H2 (1 / 4) are
Z 2
Ev,b-rot = p Evib H2 (õ=o~õ=I) p J+ 1 E.o, H,
J = 0,1,2,3... (26)
= 8.254432 eV (J+1)0.24144 eV
He+ also fulfills the catalyst criterion-a chemical or physical process with
an
enthalpy change equal to an integer multiple of 27.2 eV since it ionizes at
54.417 eV
which is 2= 27.2 eV. The product of the catalysis reaction of He', H(1 / 3),
may
further serve as a catalyst to form H(1 / 4) and H(1 / 2) [19-20, 30] which
can lead
to transitions to other states H(1 / p). Novel emission lines with energies of
q- 13.6 eV where q= 1, 2, 3, 4, 6, 7, 8,9, or 11 were previously observed by
extreme
ultraviolet (EUV) spectroscopy recorded on microwave discharges of helium with
2%
hydrogen [18-20]. These lines matched H(1 / p), fractional Rydberg states of
atomic
hydrogen given by Eqs. (19) and (21).
Rotational lines were observed in the 145-300 nm region from atmospheric
pressure electron-beam excited argon-hydrogen plasmas. The unprecedented
energy spacing of 42 times that of hydrogen established the internuclear
distance as
1/4 that of H2 and identified H2 (1 / 4) (Eqs. (23-26)). H2 (1 / p) gas was
isolated by
liquefaction of helium-hydrogen plasma gas using an high-vacuum (10-6 Torr)
capable, liquid nitrogen cryotrap and was characterized by mass spectroscopy
(MS).
The condensable gas had a higher ionization energy than H2 by MS [17]. H2 (1 /
4)
gas from chemical decomposition of hydrides containing the corresponding
hydride
ion H- (1 / 4) as well from liquefaction of the catalysis-plasma gas was also
identified
by `H NMR as an upfield-shifted singlet peak at 2.18 ppm relative to H2 at
4.63 that
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
32
matched theoretical predictions [13, 17]. Hz (1 / 4) was further characterized
by
studies on the vibration-rotational emission from electron-beam maintained
argon-
hydrogen plasmas and from Fourier-transform infrared (FTIR) spectroscopy of
solid
samples containing H- (1 / 4) with interstitial Hz (1 / 4).
Water bath calorimetry was used to determine that measurable power was
developed in rt-plasmas due to the reaction to form states given by Eqs. (19)
and
(21). Specifically, He l HZ (10%) (500 mTorr), Ar l HZ (10%) (500 mTorr), and
H20(g) (500 and 200 mTorr) plasmas generated with an Evenson microwave cavity
consistently yielded on the order of 50% more heat than non rt-plasma
(controls)
such as He, Kr, Kr l H2 (10 /a), under identical conditions of gas flow,
pressure, and
microwave operating conditions. The excess power density of rt-plasmas was of
the
order 10 W- cm-3. In addition to unique vacuum ultraviolet (VUV) lines,
earlier
studies with these same rt-plasmas demonstrated that other unusual features
were
present including dramatic broadening of the hydrogen Balmer series lines [3-
5, 7, 9-
10, 12, 18-19, 21, 23-28], and in the case of water plasmas, population
inversion of
the hydrogen excited states [3, 21, 27-29]. Both the current results and the
earlier
results are completely consistent with the existence of a hitherto unknown
predicted
exothermic chemical reaction occurring in rt-plasmas.
Since the ionization energy of Sr+ to Sr3+ has a net enthalpy of reaction of
2.27.2 eV, Sr+ may serve as catalyst alone or with Ar+ catalyst. It was
reported
previously that an rt-plasma formed with a low field (1 V/cm), at low
temperatures
(e.g. = 103 K), from atomic hydrogen generated at a tungsten filament and
strontium
which was vaporized by heating the metal [4-5, 7, 9-10]. Strong VUV emission
was
observed that increased with the addition of argon, but not when sodium,
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
33
magnesium, or barium replaced strontium or with hydrogen, argon, or strontium
alone. Characteristic emission was observed from a continuum state of Ar2+ at
45.6 nm without the typical Rydberg series of Ar I and Ar II lines which
confirmed the
resonant nonradiative energy transfer of 27.2 eV from atomic hydrogen to Ar+
[5, 7,
22]. Predicted Sr3+ emission lines were also observed from strontium-hydrogen
plasmas [5, 7] that supported the rt-plasma mechanism. Time-dependent line
broadening of the H Balmer a line was observed corresponding to
extraordinarily
fast H (25 eV). An excess power of 20 mW = cm-3 was measured calorimetrically
on
rt-plasmas formed when Ar+ was added to Sr+ as an additional catalyst.
Significant Balmer a line broadening corresponding to an average hydrogen
atom temperature of 14, 24 eV, and 23-45 eV was observed for strontium and
argon-
strontium rt-plasmas and discharges of strontium-hydrogen, helium-hydrogen,
argon-hydrogen, strontium-helium-hydrogen, and strontium-argon-hydrogen,
respectively, compared to = 3 eV for pure hydrogen, xenon-hydrogen, and
magnesium-hydrogen. To achieve that same optically measured light output
power,
hydrogen-sodium, hydrogen-magnesium, and hydrogen-barium mixtures required
4000, 7000, and 6500 times the power of the hydrogen-strontium mixture,
respectively, and the addition of argon increased these ratios by a factor of
about
two. A glow discharge plasma formed for hydrogen-strontium mixtures at an
extremely low voltage of about 2 V compared to 250 V for hydrogen alone and
sodium-hydrogen mixtures, and 140-150 V for hydrogen-magnesium and hydrogen-
barium mixtures [4-5, 7]. These voltages are too low to be explicable by
conventional mechanisms involving accelerated ions with a high applied field.
A low-
voltage EUV and visible light source is feasible [10].
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
34
In general, the energy transfer of m- 27.2 eV from the hydrogen atom to the
catalyst causes the central-field interaction of the H atom to increase by m
and its
electron to drop m levels lower from the radius of the hydrogen atom, af, , to
a
radius of l+m [19-20]. Since K to K3+ provides a reaction with a net enthalpy
equal to three times the potential energy of atomic hydrogen, 3- 27.2 eV, it
may
serve as a catalyst such that each ordinary hydrogen atom undergoing catalysis
releases a net of 204 eV [3]. K may then react with the product H(1 / 4) to
form a
yet lower-state H(1 / 7) or further catalytic transitions may occur:
1--.> 1 1~ 1 1-4 1 and so, involving only hydrinos in a process called
4 5 5 6 6 7
disproportionation. Since the ionization energies and metastable resonant
states of
hydrinos due corresponding to the multipole expansion of the potential energy
are
m- 27.2 eV (Eqs. (19) and (21)) as given previously [19-20, 30] once catalysis
begins, hydrinos autocatalyze further transitions to lower states. This
mechanism is
similar to that of an inorganic ion catalysis. An energy transfer of m- 27.2
eV from
a first hydrino atom to the second hydrino atom causes the central field of
the first
atom to increase by m and its electron to drop m levels lower from a radius of
aH
p
to a radius of aH
p+m
The catalyst product, H(1 / p), may also react with an electron to form a
novel
hydride ion H- (1 / p) with a binding energy EB [3, 14, 16, 21, 30]:
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
E_ ~iZ s(s + 1) _ 7c' oe2~22 1 22
B g ea0 1+ S(S + 1) 2 me aH a~ 1 h S(S + 1) 3 2 p l 1
p
(27)
where p is an integer greater than one, s = 1/ 2, h is Planck's constant bar,
o is
the permeability of vacuum, me is the mass of the electron, e is the reduced
5 electron mass given by P = mem where mp is the mass of the proton, aH is
the
m
e +mP
4
radius of the hydrogen atom, a. is the Bohr radius, and e is the elementary
charge. 50- The ionic radius is r, = p(1+ s(s+1)); s = 2. From Eq. (27), the
calculated
ionization energy of the hydride ion is 0.75418 eV, and the experimental value
given
by Lykke [38] is 6082.99 0.15 cm-' (0.75418 eV).
10 Substantial evidence of an energetic catalytic reaction was previously
reported [3] involving a resonant energy transfer between hydrogen atoms and K
to
form very stable novel hydride ions H- (1 / p) called hydrino hydrides having
a
predicted fractional principal quantum number p = 4. Characteristic emission
was
observed from K3- that confirmed the resonant nonradiative energy transfer of
15 3= 27.2 eV from atomic hydrogen to K. From Eq. (27), the binding energy EB
of
H-(1/4) is
EB =11.232 eV ( AvQc =1103.8 A) (28)
The product hydride ion H- (1 / 4) was observed spectroscopically at 110 nm
corresponding to its predicted binding energy of 11.2 eV [3, 21].
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
36
Upfield-shifted NMR peaks are direct evidence of the existence of lower-
energy state hydrogen with a reduced radius relative to ordinary hydride ion
and
having an increase in diamagnetic shielding of the proton. The total
theoretical shift
MT for H- (1 / p) is given by the sum of the shift of H- (1 / 1) plus the
contribution
B
due to the lower-electronic energy state:
2
ABT =- 0 e (1+a21cp)=-(29.9+1.37p)ppm
B 12meao(1+ s(s+1))
(29)
where p = integer > 1. Corresponding alkali hydrides and alkali hydrino
hydrides
(containing H- (1 / p) ) were characterized by 'H MAS NMR and compared to the
theoretical values. A match of the predicted and observed peaks with no
alternative
represents a definite test.
The 'H MAS NMR spectrum of novel compound KH * Cl relative to external
tetramethylsilane (TMS) showed a large distinct upfield resonance at -4.4 ppm
corresponding to an absolute resonance shift of -35.9 ppm that matched the
theoretical prediction of p = 4 [3, 14-16]. This result confirmed the previous
observations from the rt-plasmas of intense hydrogen Lyman emission, a
stationary
inverted Lyman population, excessive afterglow duration, highly energetic
hydrogen
atoms, characteristic alkali-ion emission due to catalysis, predicted novel
spectral
lines, and the measurement of a power beyond any conventional chemistry [3]
that
matched predictions for a catalytic reaction of atomic hydrogen to form more
stable
hydride ions designated H- (1 / p). Since the comparison of theory and
experimental shifts of KH * Cl is direct evidence of lower-energy hydrogen
with an
implicit large exotherm during its formation, the NMR results were repeated
with the
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
37
further analysis by infrared (FTIR) spectroscopy to eliminate any known
explanation
[39].
Elemental analysis identified [14, 16] these compounds as only containing the
alkaline metal, halogen, and hydrogen, and no known hydride compound of this
composition could be found in the literature which has an upfield-shifted
hydride
NMR peak. Ordinary alkali hydrides alone or mixed with alkali halides show
down-
field shifted peaks [3, 14-16]. From the literature, the list of alternatives
to H-(1 / p)
as a possible source of the upfield NNIR peaks was limited to U centered H.
The
intense and characteristic infrared vibration band at 503 cm-' due to the
substitution
of H- for Cl- in KCl enabled the elimination of U centered H as the source of
the
upfield-shifted NMR peaks [39].
As further characterizations, the X-ray photoelectron spectrum (XPS) of the
hydrino hydride KH * I was performed to determine if the predicted H- (1 / 4)
binding energy given by Eq. (28) was observed, and FTIR analysis of these
crystals
with H- (1 / 4) was performed before and after storage in argon for 90 days to
search
for interstitial H2 (1 / 4) having a predicted rotational energy given by Eq.
(24). The
identification of single rotational peaks at this energy with ortho-para
splitting due to
free rotation of a very small hydrogen molecule would represent definite proof
of its
existence since there is no other possible assignment [39].
Since the rotational emission of H2 (1 / 4) was observed in crystals of KH * I
having a peak assigned to H- (1 / 4) and the vibration-rotational emission of
H2 (1 / 4) was observed from 12.5 keV-electron-beam-maintained plasmas of
argon
with 1% hydrogen due to collisional excitation of HZ (1 / 4), HZ (1 / 4)
trapped in the
lattice of KH * Cl , or H2 (1 / 4) formed from H- (1 / 4) or formed insitu
from K
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
38
catalysis of H via electron bombardment was investigated by windowless EUV
spectroscopy on electron-beam excitation of the crystals using the 12.5 keV
electron
gun at pressures below which any gas could produce detectable emission (<10"5
Torr) [39]. The rotational energy of H2 (1 / 4) was confirmed by this
technique as
well. Consistent results from the broad spectrum of investigational techniques
provide definitive evidence that hydrogen can exist in lower-energy states
then
previously thought possible in the form of H- (1 / 4) and H2 (1 / 4). In an
embodiment, the products of the Li catalyst reaction and NaH catalyst reaction
are
both H- (1 / 4) and H2 (1 / 4) and additionally H- (1 / 3) and H2 (1 / 3) for
NaH. The
present invention provides for their identification and the corresponding
energetic
exothermic reaction by EUV spectroscopy, characteristic emission from
catalysts
and the hydride ion products, lower-energy hydrogen emission, chemically
formed
plasmas, extraordinary Balmer a line broadening, population inversion of H
lines,
elevated electron temperature, anomalous plasma afterglow duration, power
generation, and analysis of novel chemical compounds. Preferred identification
techniques for the species H- (1 / p) and H2 (1 / p) are N MR of H- (1 / p)
and
H2 (1 / p), FTIR of H2 (1 / p) trapped in a crystal, XPS of H- (1 / p), ToF-
SIMs of
H- (1 / p), electron-beam excitation emission spectroscopy of H2 (1 / p),
electron
beam emission spectroscopy of H2 (1 / p) trapped in a crystalline lattice, and
TOF-
SIMS identification of novel compounds comprising H- (1 / p). Preferred
characterization techniques for the energetic catalysis reaction and the power
balance are line broadening, plasma formation, and calorimetry. Preferably,
H- (1 / p) and HZ (1 / p) are H- (1 / 4) and HZ (1 / 4), respectively.
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
39
BRIEF DESCRIPTION OF THE DRAWINGS
FIGLIRE 1A is a schematic drawing of an energy reactor and power plant in
accordance with the present invention.
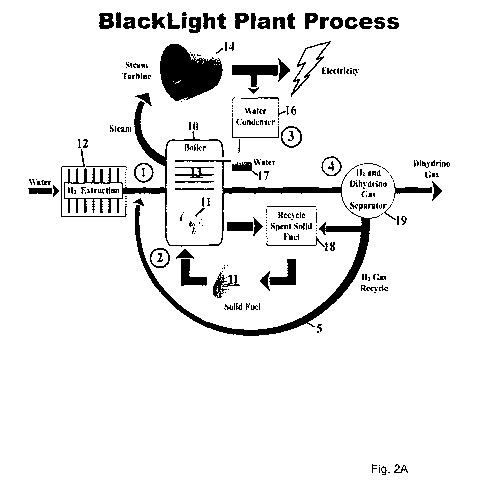
FIGURE 2A is a schematic drawing of an energy reactor and power plant for
recycling or regenerating the fuel in accordance with the present invention.
FIGURE 3A is a schematic drawing of a power reactor in accordance with the
present invention.
FIGURE 4A is a schematic drawing of a discharge power and plasma cell and
reactor in accordance with the present invention.
Figure 1 is the experimental set up comprising a filament gas cell to form
lithium-
argon-hydrogen and lithium-hydrogen rt-plasmas.
Figure 2 is a schematic of the reaction cell and the cross sectional view of
the
water flow calorimeter used to measure the energy balance of the NaH catalyst
reaction to form hydrinos. The components were: 1-inlet and outlet
thermistors;
2-high-temperature valve; 3-ceramic fiber heater; 4-copper water-coolant coil;
5-reactor; 6-insulation; 7-cell thermocouple, and 8-water flow chamber.
Figure 3 is a schematic of the water flow calorimeter used to measure the
energy
balance of the NaH catalyst reaction to form hydrinos.
Figure 4 is a schematic of the stainless steel gas cell to synthesize LiH *
Br,
LiH * I, NaH * Cl and NaH * Br comprising the reaction mixture (i) R-Ni, Li,
LiNH2 ,
and LiBr or Lii or (ii) Pt/Ti dissociator, Na, NaH, and NaCI or NaBr as the
reactants. The components were: 101-stainless steel cell; 117-internal cavity
of
cell; 118-high vacuum conflat flange; 119-mating blank conflat flange;
102-stainless steel tube vacuum line and gas supply line; 103-lid to the kiln
or top
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
insulation, 104-surrounding heaters coverer by high temperature insulation;
108-Pt/Ti dissociator; 109-reactants; 110-high vacuum turbo pump;
112-pressure gauge; 111-vacuum pump valve; 113-valve; 114-valve;
115-regulator, and 116-hydrogen tank.
5 Figure 5 shows the 656.3 nm Balmer a line width recorded with a high-
resolution
visible spectrometer on (A) the initial emission of a lithium-argon-hydrogen
rt-plasma
and (B) the emission at 70 hours of operation. Lithium lines and significant
broadening of only the H lines was observed over time corresponding to an
average
hydrogen atom temperature of >40 eV and fractional population over 90%.
10 Figure 6 shows the 656.3 nm Balmer a line width recorded with a high-
resolution
( 0.006 nm) visible spectrometer on (A) the initial emission of a lithium-
hydrogen rt-
plasma and (B) the emission at 70 hours of operation. Lithium lines and
broadening
of only the H lines was observed over time, but diminished relative to the
case
having the argon-hydrogen gas (95/5%). The Balmer width corresponded to an
15 average hydrogen atom temperature of 6 eV and a 27% fractional population.
Figure 7 shows the results of the DSC (100-750 C) of NaH at a scan rate of
0.1
degree/minute. A broad endothermic peak was observed at 350 C to 420 C which
corresponds to 47 kJ / mole and matches sodium hydride decomposition in this
temperature range with a corresponding enthalpy of 57 kJ / mole. A large
exotherm
20 was observed under conditions that form NaH catalyst in the region 640 C to
825
C which corresponds to at least -354 kJ / moleH2, greater than that of the
most
exothermic reaction possible for H, the -241.8 kJ/ mole H2 enthalpy of
combustion of
hydrogen.
Figure 8 shows the results of the DSC (100-750 C) of MgHZ at a scan rate of
0.1
25 degree/minute. Two sharp endothermic peaks were observed. A first peak
centered
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
41
at 351.75 C corresponding to 68.61 kJ / mole MgH2 matches the 74.4 kJ/ mole
MgH2
decomposition energy. The second peak at 647.66 C corresponding to
6.65 kJ / mole MgH2 matches the known melting point of Mg(m) is 650 C and
enthalpy of fusion of 8.48 kJ/ mole Mg(m). Thus, the expected behavior was
observed for the decomposition of a control, noncatalyst hydride.
Figure 9 shows the temperature versus time for the calibration run with an
evacuated test cell and resistive heating only.
Figure 10 shows the power versus time for the calibration run with an
evacuated
test cell and resistive heating only. The numerical integration of the input
and output
power curves yielded an output energy of 292.2 kJ and an input energy of 303.1
kJ
corresponding to a coupling of flow of 96.4% of the resistive input to the
output
coolant.
Figure 11 shows the cell temperature with time for the hydrino reaction with
the
cell containing the reagents comprising the catalyst material, 1 g Li, 0.5g
LiNH2 1 109
LiBr, and 15g Pd / A1Z03 . The reaction liberated 19.1 kJ of energy in less
than 120 s
to develop a system-response-corrected peak power in excess of 160 W.
Figure 12 shows the coolant power with time for the hydrino reaction with the
cell
containing the reagents comprising the catalyst material, 1 g Li, 0.5g LiNHz ,
lOg
LiBr, and 15g Pd / A1203 . The numerical integration of the input and output
power
curves with the calibration correction applied yielded an output energy of
227.2 kJ
and an input energy of 208.1 kJ corresponding to an excess energy of 19.1 kJ.
Figure 13 shows the cell temperature with time for the R-Ni control power test
with the cell containing the reagents comprising the starting material for R-
Ni, 15g R-
Ni/AI alloy powder, and 3.28g of Na.
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
42
Figure 14 shows the coolant power with time for the control power test with
the
cell containing the reagents comprising the starting material for R-Ni, 15g R-
Ni/Al
alloy powder, and 3.28g of Na. Energy balance was obtained with the
calibration-
corrected numerical integration of the input and output power curves yielding
an
output energy of 384 kJ and an input energy of 385 W.
Figure 15 shows the cell temperature with time for the hydrino reaction with
the
cell containing the reagents comprising the catalyst material, 15g NaOH -doped
R-Ni
2800, and 3.28g of Na. The reaction liberated 36 kJ of energy in less than 90
s to
develop a system-response-corrected peak power in excess of 0.5 M.
Figure 16 shows the coolant power with time for the hydrino reaction with the
cell
containing the reagents comprising the catalyst material, 15g NaOH -doped R-Ni
2800, and 3.28g of Na. The numerical integration of the input and output power
curves with the calibration correction applied yielded an output energy of
185.1 kJ
and an input energy of 149.1 kJ corresponding to an excess energy of 36 U.
Figure 17 shows the cell temperature with time for the hydrino reaction with
the
cell containing the reagents comprising the catalyst material, 15g NaOH -doped
R-Ni
2400. The cell temperature jumped from 60 C to 205 C in 60 s wherein the
reaction
liberated 11.7 kJ of energy in less time to develop a system-response-
corrected
peak power in excess of 0.25 kW.
Figure 18 shows the coolant power with time for the hydrino reaction with the
cell
containing the reagents comprising the catalyst material, 15g NaOH -doped R-Ni
2400. The numerical integration of the input and output power curves with the
calibration correction applied yielded an output energy of 195.7 kJ and an
input
energy of 184.0 kJ corresponding to an excess energy of 11.7 U.
Figure 19 shows the positive ToF-SIMS spectrum (m l e = 0-100 ) of LiBr.
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
43
Figure 20 shows the positive ToF-SIMS spectrum (m l e= 0-100 ) of the LiH * Br
crystals.
Figure 21 shows the negative ToF-SIMS spectrum (m / e = 0-100 ) of LiBr.
Figure 22 shows the negative ToF-SIMS spectrum ( m/ e = 0-100 ) of the
LiH * Br crystals. A dominant hydride, LiHBr-, and Li2H2Br- peaks were
uniquely
observed.
Figure 23 shows the positive ToF-SIMS spectrum (m / e = 0- 200) of Li1.
Figure 24 shows the positive ToF-SIMS spectrum (m / e = 0- 200) of the LiH * I
crystals. LiH1`, Li2Hzi+ , Li4H2i+ , and Li6HZ7` were only observed in the
positive
ion spectrum of the LiH * I crystals.
Figure 25 shows the negative ToF-SIMS spectrum ( m / e = 0-180 ) of Lii.
Figure 26 shows the negative ToF-SIMS spectrum (õ2 / e= 0-180 ) of the LiH * I
crystals. A dominant hydride, LiHi-, LizH21-, and NaHI- peaks were uniquely
observed.
Figure 27 shows the negative ToF-SIMS spectrum (m / e = 20 - 30) of NaH *-
coated Pt lTi following the production of 15 kJ of excess heat. Hydrino
hydride
compounds NaHx were observed.
Figure 28 shows the positive ToF-SIMS spectrum ( m/ e= 0-100 ) of R-Ni reacted
over a 48 hour period at 50 C. The dominant ion on the surface was Na+
consistent
with NaOH doping of the surface. The ions of the other major elements of R-Ni
2400 such as Al', Ni', Cr+, and Fe+ were also observed.
Figure 29 shows the negative ToF-SIMS spectrum ( m/ e = 0-180 ) of R-Ni
reacted over a 48 hour period at 50 C. A dominant hydride, NaH3 and NaH3NaOH-
assigned to sodium hydrino hydride and this ion in combination with NaOH, as
well
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
44
as other unique ions assignable to sodium hydrino hydrides NaHX in
combinations
with NaOH, NaO, OH- and O- were observed.
Figures 30A-B show 'H MAS NMR spectra relative to external TMS. (A)
LiH * Br showing a broad -2.5 ppm upfield-shifted peak and a peak at 1.13 ppm
assigned to H- (1 / 4) and H2 (1 / 4), respectively. (B) LiH * I showing a
broad
-2.09 ppm upfield-shifted peak assigned to H- (1 / 4) and peaks at 1.06 ppm
and
4.38 ppm assigned to HZ (1 / 4) and HZ , respectively.
Figures 31A-B show 'H MAS NMR spectra relative to external TMS. (A) KH* Cl
showing a very sharp -4.46 ppm upfield-shifted peak corresponding to an
environment that is essentially that of a free ion. (B) KH * I showing a broad
-2.31
ppm upfield-shifted peak similar to the case of LiH * Br and LiH * I. Both
spectra
also had a 1.13 ppm peak assigned to H2 (1 / 4).
Figures 32A-B show 'H MAS NMR spectra relative to external TMS showing an
H-content selectivity of LiH * X for molecular species alone based on the
nonpolarizability of the halide and the corresponding nonreactivity towards H-
(1 /4).
(A) LiH * F comprising a nonpolarizable fluorine showing peaks at 4.31 ppm
assigned to H2 and 1.16 ppm assigned to H2 (1 / 4) and the absence of the
H- (1 / 4) ion peak. (B) LiH * Cl comprising a nonpolarizable chlorine showing
peaks at 4.28 ppm assigned to H2 and 1.2 ppm assigned to H2 (1 / 4) and the
absence of the H- (1 / 4) ion peak.
Figure 33 shows the 'H MAS NMR spectra of NaH * Br relative to external TMS
showing a-3.58 ppm upfield-shifted peak, a peak at 1.13 ppm, and a peak at 4.3
ppm assigned to H- (1 / 4), H2 (1 / 4), and H2, respectively.
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
Figures 34A-B show the NaH * Cl 'H MAS NMR spectra relative to external TMS
showing the effect of hydrogen addition on the relative intensities of H2,
H2 (1 / 4),and H- (1 / 4). The addition of hydrogen increased the H- (1 / 4)
peak and
decreased the H2 (1 / 4) while the H2 increased. (A) NaH * Cl synthesized with
5 hydrogen addition showing a -4 ppm upfield-shifted peak assigned to H- (1 /
4), a
1.1 ppm peak assigned to H2 (1 / 4), and a dominant 4 ppm peak assigned to Hz
.
(B) NaH *Cl synthesized without hydrogen addition showing a -4 ppm upfield-
shifted peak assigned to H- (1 / 4), a dominant 1.0 ppm peak assigned to H2 (1
/ 4),
and a small 4.1 ppm assigned to HZ .
10 Figure 35 shows the 'H MAS NMR spectrum relative to external TMS of
NaH * Cl from reaction of NaCI and the solid acid KHSO4 as the only source of
hydrogen showing both the H- (1 / 4) peak at -3.97 ppm and an upfield-shifted
peak
at -3.15 ppm assigned to H- (1 / 3). The corresponding H2 (1 / 4) and H2 (1 /
3)
peaks are shown at 1.15 ppm and 1.7 ppm, respectively. Both fractional
hydrogen
15 states were present and the HZ peak was absent at 4.3 ppm due to the
synthesis of
NaH * Cl using a solid acid as the H source rather that addition of hydrogen
gas
and a dissociator. (SB=side band).
Figures 36A-B show XPS survey spectra (Eb = 0 eVto 1200 eV). (A) LiBr. (B)
LiH * Br.
20 Figure 37 shows the 0-85 eV binding energy region of a high resolution XPS
spectrum of LiH * Br and the control LiBr (dashed). The XPS spectrum of LiH *
Br
differs from that of LiBr by having additional peaks at 9.5 eV and 12.3 eV
that could
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
46
not be assigned to known elements and do not correspond to any other primary
element peak. The peaks match H- (1 / 4) in two different chemical
environments.
Figures 38A-B show the XPS survey spectra ( Eb = 0 eVto 1200 eV). (A) NaBr.
(B) NaH*Br.
Figure 39 shows the 0-40 eV binding energy region of a high resolution XPS
spectrum of NaH * Br and the control NaBr (dashed). The XPS spectrum of
NaH * Br differs from that of NaBr by having additional peaks at 9.5 eV and
12.3 eV
that could not be assigned to known elements and do not correspond to any
other
primary element peak. The peaks match H- (1 / 4) in two different chemical
environments.
Figures 40A-B show XPS survey spectra ( Eb = 0 eV to 1200 eV). (A) Pt l Ti.(B)
NaH * -coated Pt /Ti following the production of 15 kJ of excess heat.
Figures 41A-B show high resolution XPS spectra (Eb = 0 eV to 100 eV). (A)
Pt / Ti.(B) NaH * -coated Pt lTi following the production of 15 kJ of excess
heat.
The Pt 4 f72 , Pt 4 f5,2 , and 0 2s peaks were observed at 70.7 eV, 74 eV, and
23
eV, respectively. The Na 2p and Na 2s peaks were observed at 31 eV and 64 eV
on NaH *-coated Pt l Ti, and a valance band was only observed for Pt l Ti.
Figures 42A-B show high resolution XPS spectra ( Eb = 0 eV to 50 eV). (A)
Pt lTi Ti. (NaH * -coated Pt lTi following the production of 15 kJ of excess
heat.
The XPS spectrum of NaH * -coated Pt lTi differs from that of Pt lTi by having
additional peaks at 6 eV, 10.8 eV, and 12.8 eV that could not be assigned to
known
elements and do not correspond to any other primary element peak. The 10.8 eV,
and 12.8 eV peaks match H- (1 / 4) in two different chemical environments, and
the
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
47
6 eV peak matched and was assigned to H- (1 / 3). Thus, both fractional
hydrogen
states, 1/3 and 1/4, were present as predicted by Eq. (27).
Figure 43 shows XPS survey spectrum (Eb = 0 eV to 120 eV) of NaH *-coated Si
with the primary-element peaks identified.
Figure 44 shows high resolution XPS spectrum (Eb = 0 eV to 120 eV) of NaH *-
coated Si having peaks at 6 eV, 10.8 eV, and 12.8 eV that could not be
assigned to
known elements and do not correspond to any other primary element peak. The
10.8 eV, and 12.8 eV peaks match H- (1 / 4) in two different chemical
environments,
and the 6 eV peak matched and was assigned to H- (1 / 3). Thus, both
fractional
hydrogen states, 1/3 and 1/4, were present as predicted by Eq. (27) matching
the
results of NaH * -coated Pt lTi shown in Figure 42B.
Figures 45A-B show high resolution (0.5 cm-' ) FTIR spectra (490-4000 cm-' ).
(A)
LiBr. .(B) LiH * Br sample having a NMR peak assigned to H-(1 /4) that was
heated to >600 C under dynamic vacuum that retained the -2.5 ppm NMR peak.
The amide peaks at 3314, 3259, 2079(broad), 1567, and 1541 cm-' and the imide
peaks at 3172 (broad), 1953, and 1578 cm-' were eliminated; thus, they were
not
the source of the -2.5 ppm NMR peak that remained. The -2.5 ppm peak in 'H
NMR spectrum was assigned to the H-(1 /4) ion. In addition, the 1989 cm-' FTIR
peak could not be assigned to any know compound, but matched the predicted
frequency of para H2 (1 / 4).
Figure 46 shows the 150-350 nm spectrum of electron-beam excited CsCI
crystals having trapped H2(1/4). A series of evenly spaced lines was observed
in
the 220-300 nm region that matched the spacing and intensity profile of the P
branch
of Hz(1/4) .
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
48
Figure 47 shows the 100-550 nm spectrum of an electron-beam excited silicon
wafer coated with NaH *Cl having trapped HZ(1/4) . A series of evenly spaced
lines
was observed in the 220-300 nm region that matched the spacing and intensity
profile of the P branch of H2(1/4)
DETAILED DESCRIPTION OF THE PREFERRED EMBODIMENTS
Hydrogen Catalyst Reactor
A hydrogen catalyst reactor 50 for producing energy and lower-energy
hydrogen species, in accordance with the invention, is shown in FIGURE 1A and
comprises a vessel 52 which contains an energy reaction mixture 54, a heat
exchanger 60, and a power converter such as a steam generator 62 and turbine
70.
In an embodiment, the catalysis involves reacting atomic hydrogen from the
source
56 with the catalyst 58 to form lower-energy hydrogen "hydrinos" and produce
power. The heat exchanger 60 absorbs heat released by the catalysis reaction,
when the reaction mixture, comprised of hydrogen and a catalyst, reacts to
form
lower-energy hydrogen. The heat exchanger exchanges heat with the steam
generator 62 which absorbs heat from the exchanger 60 and produces steam. The
energy reactor 50 further comprises a turbine 70 which receives steam from the
steam generator 62 and supplies mechanical power to a power generator 80 which
converts the steam energy into electrical energy, which can be received by a
load 90
to produce work or for dissipation.
In an embodiment, the energy reaction mixture 54 comprises an energy
releasing material 56 such as a solid fuel supplied through supply passage 42.
The
reaction mixture may comprise a source of hydrogen isotope atoms or a source
of
molecular hydrogen isotope, and a source of catalyst 58 which resonantly
remove
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
49
approximately m- 27.2 eV to form lower-energy atomic hydrogen where m is an
integer, preferably an integer less than 400 wherein the reaction to lower
energy
states of hydrogen occurs by contact of the hydrogen with the catalyst. The
catalyst
may be in the molten, liquid, gaseous, or solid state. The catalysis releases
energy
in a form such as heat and forms at least one of lower-energy hydrogen isotope
atoms, molecules, hydride ions, and lower-energy hydrogen compounds. Thus, the
power cell also comprises a lower-energy hydrogen chemical reactor.
The source of hydrogen can be hydrogen gas, dissociation of water including
thermal dissociation, electrolysis of water, hydrogen from hydrides, or
hydrogen from
metal-hydrogen solutions. In another embodiment, molecular hydrogen of the
energy releasing material 56 is dissociated into atomic hydrogen by a
molecular
hydrogen dissociating catalyst of the mixture 54. Such dissociating catalysts
may
also absorb hydrogen, deuterium, or tritium atoms and/or molecules and
include, for
example, an element, compound, alloy, or mixture of noble metals such as
palladium
and platinum, refractory metals such as molybdenum and tungsten, transition
metals
such as nickel and titanium, inner transition metals such as niobium and
zirconium,
and other such materials listed in the Prior Mills Publications. Preferably,
the
dissociator has a high surface area such as a noble metal such as Pt, Pd, Ru,
Ir, Re,
or Rh, or Ni on A1203, Si0z , or combinations thereof.
In an embodiment, a catalyst is provided by the ionization of t electrons from
an atom or ion to a continuum energy level such that the sum of the ionization
energies of the t electrons is approximately m- 27.2 eV where t and m are each
an
integer. A catalyst may also be provided by the transfer of t electrons
between
participating ions. The transfer of t electrons from one ion to another ion
provides a
net enthalpy of reaction whereby the sum of the t ionization energies of the
electron-
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
donating ion minus the ionization energies of t electrons of the electron-
accepting
ion equals approximately m- 27.2 eV where t and m are each an integer. In
another
preferred embodiment, the catalyst comprises MH such as NaH having an atom
M bound to hydrogen, and the enthalpy of m- 27.2 eV is provided by the sum of
the
5 M - H bond energy and the ionization energies of the t electrons.
In a preferred embodiment, a source of catalyst comprises a catalytic material
58 supplied through catalyst supply passage 41, that typically provides a net
enthalpy of approximately 2= 27.2 eV plus or minus 1 eV. The catalysts include
those given herein and the atoms, ions, molecules, and hydrinos described in
Mills
10 Prior Publications (e.g. TABLE 4 of PCT/US90/01998 and pages 25-46, 80-108
of
PCT/US94/02219) which are incorporated herein by reference. In embodiments,
the
catalyst may comprise at least one species selected from the group of
molecules of
AIH, BiH, CIH, CoH, GeH, InH, NaH, RuH, SbH, SeH, SiH, SnH, C2, NZ, OZ, COZ,
NO2, and NO3 and atoms or ions of Li, Be, K, Ca, Ti, V, Cr, Mn, Fe, Co, Ni,
Cu, Zn,
15 As, Se, Kr, Rb, Sr, Nb, Mo, Pd, Sn, Te, Cs, Ce, Pr, Sm, Gd, Dy, Pb, Pt, Kr,
2K',
He+, Na+, Rb', Sr+ , Fe3+, MoZ+ , Mo4,, In'+, He+, Ar+, Xe+, Arz'and H+ , and
Ne` and H+.
Hydrogen Catalyst Reactor and Electrical Power System
20 In an embodiment of a power system, the heat is removed by a heat
exchanger having a heat exchange medium. The heat exchanger may be a water
wall and the medium may be water. The heat may be transferred directly for
space
and process heating. Alternatively, the heat exchanger medium such as water
undergoes a phase change such as conversion to steam. This conversion may
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
51
occur in a steam generator. The steam may be used to generate electricity in a
heat engine such as a steam turbine and a generator.
An embodiment of an hydrogen catalyst energy and lower-energy-hydrogen
species-producing reactor 5, for recycling or regenerating the fuel in
accordance with
the Invention, is shown in FIGURE 2A and comprises a boiler 10 which contains
a
solid fuel reaction mixture 11, a hydrogen source 12, steam pipes and steam
generator 13, a power converter such as a turbine 14, a water condenser 16, a
water-make-up source 17, a solid-fuel recycler 18, and a hydrogen-dihydrino
gas
separator 19. At Step 1, the solid fuel comprising a source of catalyst and a
source
of hydrogen reacts to form hydrinos and lower-energy hydrogen products. At
Step 2,
the spent fuel is reprocessed to re-supply the boiler 10 to maintain thermal
power
generation. The heat generated in the boiler 10 forms steam in the pipes and
steam
generator 13 that is delivered to the turbine 14 that in turn generates
electricity by
powering a generator. At Step 3, the water is condensed by the water condensor
16. Any water loss may be made up by the water source 17 to complete the cycle
to
maintain thermal to electric power conversion. At Step 4, lower-energy
hydrogen
products such as hydrino hydride compounds and dihydrino gas may be removed,
and unreacted hydrogen may be returned to the fuel recycler 18 or hydrogen
source
12 to be added back to spent fuel to make-up recycled fuel. The gas products
and
unreacted hydrogen may be separated by hydrogen-dihydrino gas separator 19.
Any product hydrino hydride compounds may be separated and removed using
solid-fuel recycler 18. The processing may be performed in the boiler or
externally to
the boiler with the solid fuel returned. Thus, the system may further comprise
at
least one of gas and mass transporters to move the reactants and products to
achieve the spent fuel removal, regeneration, and re-supply. Hydrogen make-up
for
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
52
that spent in the formation of hydrinos is added from the source 12 during
fuel
reprocessing and may involve recycled, unconsumed hydrogen. The recycled fuel
maintains the production of thermal power to drive the power plant to generate
electricity.
In a preferred embodiment, the reaction mixture comprises species that can
generate the reactants of atomic or molecular catalyst and atomic hydrogen
that
further react to form hydrinos, and the product species formed by the
generation of
catalyst and atomic hydrogen can be regenerated by at least the step of
reacting the
products with hydrogen. In an embodiment, the reactor comprises a moving bed
reactor that may further comprise a fluidized-reactor section wherein the
reactants
are continuously supplied and side products are removed and regenerated and
returned to the reactor. In an embodiment, the lower-energy hydrogen products
such as hydrino hydride compounds or dihydrino molecules are collected as the
reactants are regenerated. Furthermore, the hydrino hydride ions may be formed
into other compounds or converted into dihydrino molecules during the
regeneration
of the reactants.
The power system may further comprise a catalyst condensor means to
maintain the catalyst vapor pressure by a temperature control means which
controls
the temperature of a surface at a lower value than that of the reaction cell.
The
surface temperature is maintained at a desired value which provides the
desired
vapor pressure of the catalyst. In an embodiment, the catalyst condensor means
is
a tube grid in the cell. In an embodiment with a heat exchanger, the flow rate
of the
heat transfer medium may be controlled at a rate that maintains the condensor
at the
desired lower temperature than the main heat exchanger. In an embodiment, the
working medium is water, and the flow rate is higher at the condensor than the
water
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
53
wall such that the condensor is the lower, desired temperature. The separate
streams of working media may be recombined to be transferred for space and
process heating or for conversion to steam.
The present energy invention is further described in Mills Prior Publications
which are incorporated herein by reference. The cells of the present invention
include those described previously and further comprise the catalysts,
reaction
mixtures, methods, and systems disclosed herein. The electrolytic cell energy
reactor, plasma electrolysis reactor, barrier electrode reactor, RF plasma
reactor,
pressurized gas energy reactor, gas discharge energy reactor, microwave cell
energy reactor, and a combination of a glow discharge cell and a microwave and
or
RF plasma reactor of the present invention comprises: a source of hydrogen;
one of
a solid, molten, liquid, and gaseous source of catalyst; a vessel containing
hydrogen
and the catalyst wherein the reaction to form lower-energy hydrogen occurs by
contact of the hydrogen with the catalyst or by reaction of MH catalyst; and a
means for removing the lower-energy hydrogen product. For power conversion,
each cell type may be interfaced with any of the converters of thermal energy
or
plasma to mechanical or electrical power described in Mills Prior Publications
as well
as converters known to those skilled in the Art such as a heat engine, steam
or gas
turbine system, Sterling engine, or thermionic or thermoelectric converter.
Further
plasma converters comprise the magnetic mirror magnetohydrodynamic power
converter, plasmadynamic power converter, gyrotron, photon bunching microwave
power converter, charge drift power, or photoelectric converter disclosed in
Mills
Prior Publications. In an embodiment, the cell comprises at least one cylinder
of an
internal combustion engine as given in Mills Prior Publications.
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
54
Hydrogen Gas Cell and Solid Fuel Reactor
According to an embodiment of the invention, a reactor for producing hydrinos
and power may take the form of a hydrogen gas cell. A gas cell hydrogen
reactor of
the present invention is shown in FIGLIRE 3A. Reactant hydrinos are provided
by a
catalytic reaction with catalyst. Catalysis may occur in the gas phase or in
solid or
liquid state.
The reactor of FIGURE 3A comprises a reaction vessel 207 having a
chamber 200 capable of containing a vacuum or pressures greater than
atmospheric. A source of hydrogen 221 communicating with chamber 200 delivers
hydrogen to the chamber through hydrogen supply passage 242. A controller 222
is
positioned to control the pressure and flow of hydrogen into the vessel
through
hydrogen supply passage 242. A pressure sensor 223 monitors pressure in the
vessel. A vacuum pump 256 is used to evacuate the chamber through a vacuum
line 257.
In an embodiment, the catalysis occurs in the gas phase. The catalyst may
be made gaseous by maintaining the cell temperature at an elevated temperature
that, in turn, determines the vapor pressure of the catalyst. The atomic
and/or
molecular hydrogen reactant is also maintained at a desired pressure that may
be in
any pressure range. In an embodiment, the pressure is less than atmospheric,
preferably in the range about 10 millitorr to about 100 Torr. In another
embodiment,
the pressure is determined by maintaining a mixture of source of catalyst such
as a
metal source and the corresponding hydride such as a metal hydride in the cell
maintained at the desired operating temperature.
A source of catalyst 250 for generating hydrino atoms can be placed in a
catalyst reservoir 295, and gaseous catalyst can be formed by heating. The
reaction
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
vessel 207 has a catalyst supply passage 241 for the passage of gaseous
catalyst
from the catalyst reservoir 295 to the reaction chamber 200. Alternatively,
the
catalyst may be placed in a chemically resistant open container, such as a
boat,
inside the reaction vessel.
5 The source of hydrogen can be hydrogen gas and the molecular hydrogen.
Hydrogen may be dissociated into atomic hydrogen by a molecular hydrogen
dissociating catalyst. Such dissociating catalysts or dissociators include,
for
example, Raney nickel (R-Ni), precious or noble metals, and a precious or
noble
metal on a support. The precious or noble metal may be Pt, Pd, Ru, Ir, and Rh,
and
10 the support may be at least one of Ti, Nb, A1203, Si02 and combinations
thereof.
Further dissociators are Pt or Pd on carbon that may comprise a hydrogen
spillover
catalyst, nickel fiber mat, Pd sheet, Ti sponge, Pt or Pd electroplated on Ti
or Ni
sponge or mat, TiH, Pt black, and Pd black, refractory metals such as
molybdenum
and tungsten, transition metals such as nickel and titanium, inner transition
metals
15 such as niobium and zirconium, and other such materials listed in the Prior
Mills
Publications. In a preferred embodiment, hydrogen is dissociated on Pt or Pd.
The
Pt or Pd may be coated on a support material such as titanium or A1203. In
another
embodiment, the dissociator is a refractory metal such as tungsten or
molybdenum,
and the dissociating material may be maintained at elevated temperature by
20 temperature control means 230, which may take the form of a heating coil as
shown
in cross section in FIGURE 3A. The heating coil is powered by a power supply
225.
Preferably, the dissociating material is maintained at the operating
temperature of
the cell. The dissociator may further be operated at a temperature above the
cell
temperature to more effectively dissociate, and the elevated temperature may
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
56
prevent the catalyst from condensing on the dissociator. Hydrogen dissociator
can
also be provided by a hot filament such as 280 powered by supply 285.
In an embodiment, the hydrogen dissociation occurs such that the dissociated
hydrogen atoms contact gaseous catalyst to produce hydrino atoms. The catalyst
vapor pressure is maintained at the desired pressure by controlling the
temperature
of the catalyst reservoir 295 with a catalyst reservoir heater 298 powered by
a power
supply 272. When the catalyst is contained in a boat inside the reactor, the
catalyst
vapor pressure is maintained at the desired value by controlling the
temperature of
the catalyst boat, by adjusting the boat's power supply. The cell temperature
can be
controlled at the desired operating temperature by the heating coil 230 that
is
powered by power supply 225. The cell (called a permeation cell) may further
comprise an inner reaction chamber 200 and an outer hydrogen reservoir 290
such
that hydrogen may be supplied to the cell by diffusion of hydrogen through the
wall
291 separating the two chambers. The temperature of the wall may be controlled
with a heater to control the rate of diffusion. The rate of diffusion may be
further
controlled by controlling the hydrogen pressure in the hydrogen reservoir.
To maintain the catalyst pressure at the desire level, the cell having
permeation as the hydrogen source may be sealed. Alternatively, the cell
further
comprises high temperature valves at each inlet or outlet such that the valve
contacting the reaction gas mixture is maintained at the desired temperature.
The
cell may further comprise a getter or trap 255 to selectively collect the
lower-energy-
hydrogen species and/or the increased-binding-energy hydrogen compounds and
may further comprise a selective valve 206 for releasing dihydrino gas
product.
The catalyst may be at least one of the group of atomic lithium, potassium, or
cesium, NaH molecule and hydrino atoms wherein catalysis comprises a
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
57
disproportionation reaction. Lithium catalyst may be made gaseous by
maintaining
the cell temperature in the 500-1000 C range. Preferably, the cell is
maintained in
the 500-750 C range. The cell pressure may be maintained at less than
atmospheric, preferably in the range about 10 millitorr to about 100 Torr.
Most
preferably, at least one of the catalyst and hydrogen pressure is determined
by
maintaining a mixture of catalyst metal and the corresponding hydride such as
lithium and lithium hydride, potassium and potassium hydride, sodium and
sodium
hydride, and cesium and cesium hydride in the cell maintained at the desired
operating terriperature. The catalyst in the gas phase may comprise lithium
atoms
from the metal or a source of lithium metal. Preferably, the lithium catalyst
is
maintained at the pressure determined by a mixture of lithium metal and
lithium
hydride at the operating temperature range of 500-1000 C and most preferably,
the
pressure with the cell at the operating temperature range of 500-750 C. In
other
embodiments, K, Cs, and Na replace Li wherein the catalyst is atomic K, atomic
Cs,
and molecular NaH.
In an embodiment of the gas cell reactor comprising a catalyst reservoir or
boat, gaseous Na, NaH catalyst, or the gaseous catalyst such as Li, K, and Cs
vapor
is maintained in a super-heated condition in the cell relative to the vapor in
the
reservoir or boat which is the source of the cell vapor. In one embodiment,
the
superheated vapor reduces the condensation of catalyst on the hydrogen
dissociator
or the dissociator of at least one of metal and metal hydride molecules
disclosed
infra. In an embodiment comprising Li as the catalyst from a reservoir or
boat, the
reservoir or boat is maintained at a temperature at which Li vaporizes. H2 may
be
maintained at a pressure that is lower than that which forms a significant
mole
fraction of LiH at the reservoir temperature. The pressures and temperatures
that
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
58
achieve this condition can be determined from the data plots of Mueller et al.
such as
Figure 6.1 [40] of H2 pressure versus LiH mole fraction at given isotherms. In
an
embodiment, the cell reaction chamber containing a dissociator is operated at
a
higher temperature such that the Li does not condense on the walls or the
dissociator. The H2 may flow from the reservoir to the cell to increase the
catalyst
transport rate. Flow such as from the catalyst reservoir to the cell and then
out of
the cell is a means to remove hydrino product to prevent hydrino product
inhibition of
the reaction. In other embodiments, K, Cs, and Na replace Li wherein the
catalyst is
atomic K, atomic Cs, and molecular NaH.
Hydrogen is supplied to the reaction from a source of hydrogen. Preferably
the hydrogen is supplied by permeation from a hydrogen reservoir. The pressure
of
the hydrogen reservoir may be in the range of 10 Torr to 10,000 Torr,
preferably 100
Torr to 1000 Torr, and most preferably about atmospheric pressure. The cell
may be
operated in the temperature of about 100 C to 3000 C, preferably in the
temperature of about 100 C to 1500 C, and most preferably in the temperature
of
about 500 C to 800 C.
The source of hydrogen may be from decomposition of an added hydride. A
cell design that supplies H2 by permeation is one comprising an internal metal
hydride placed in a sealed vessel wherein atomic H permeates out at high
temperature. The vessel may comprise Pd, Ni, Ti, or Nb. In an embodiment, the
hydride is placed in a sealed tube such as a Nb tube containing a hydride and
sealed at both ends with seals such as Swagelocks. In the sealed case, the
hydride
could be an alkaline or alkaline earth hydride. Or, in this as well as the
internal-
hydride-reagent case, the hydride could be at least one of the group of saline
hydrides, titanium hydride, vanadium, niobium, and tantalum hydrides,
zirconium and
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
59
hafnium hydrides, rare earth hydrides, yttrium and scandium hydrides,
transition
element hydrides, intermetalic hydrides, and their alloys given by W. M.
Mueller et al.
[40].
In an embodiment the hydride and operating temperature 200 C, based on
each hydride decomposition temperature is at least one of the list of:
a rare earth hydride with an operating temperature of about 800 C;
lanthanum hydride with an operating temperature of about 700 C; gadolinium
hydride with an operating temperature of about 750 C; neodymium hydride with
an
operating temperature of about 750 C; yttrium hydride with an operating
temperature of about 800 C; scandium hydride with an operating temperature of
about 800 C; ytterbium hydride with an operating temperature of about 850-900
C;
titanium hydride with an operating temperature of about 450 C; cerium hydride
with
an operating temperature of about 950 C; praseodymium hydride with an
operating
temperature of about 700 C; zirconium-titanium (50%/50 /o) hydride with an
operating temperature of about 600 C; an alkali metal/alkali metal hydride
mixture
such as Rb/RbH or K/KH with an operating temperature of about 450 C, and an
alkaline earth metal/alkaline earth hydride mixture such as Ba/BaH2 with an
operating temperature of about 900-1000 C.
Metals in the gas state comprise diatomic covalent molecules. An objective of
the present Invention is to provide atomic catalyst such as Li as well as K
and Cs.
Thus, the reactor may further comprise a dissociator of at least one of metal
molecules ("NIM") and metal hydride molecules ("IVIH"). Preferably, the source
of
catalyst, the source of H2, and the dissociator of NIM, NIH, and HH, wherein M
is the
atomic catalyst are matched to operate at the desired cell conditions of
temperature
and reactant concentrations for example. In the case that a hydride source of
H2 is
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
used, in an embodiment, its decomposition temperature is in the range of the
temperature that produces the desired vapor pressure of the catalyst. In the
case of
that the source of hydrogen is permeation from a hydrogen reservoir to the
reaction
chamber, preferable sources of catalysts for continuous operation are Sr and
Li
5 metals since each of their vapor pressures may be in the desired range of
0.01 to
100 Torr at the temperatures for which permeation occurs. In other embodiments
of
the permeation cell, the cell is operated at a high temperature permissive of
permeation, then the cell temperature is lowered to a temperature which
maintains
the vapor pressure of the volatile catalyst at the desired pressure.
10 In an embodiment of a gas cell, a dissociator comprises a means to generate
catalyst and H from sources. Surface catalysts such as Pt on Ti or Pd,
iridium, or
rhodium alone or on a substrate such as Ti may also serve the role as a
dissociator
of molecules of combinations of catalyst and hydrogen atoms. Preferably, the
dissociator has a high surface area such as Pt/AI2O3 or Pd/AI203.
15 The H2 source can also be H2 gas. In this case, the pressure can be
monitored and controlled. This is possible with catalyst and catalyst sources
such as
K or Cs metal and LiNH2, respectively, since they are volatile at low
temperature
which is permissive of using a high-temperature valve. LiNH2 also lowers the
necessary operating temperature of the Li cell and is less corrosive which is
20 permissive of long-duration operation using a feed through in the case of
plasma and
filament cells wherein a filament serves as a hydrogen dissociator.
Further embodiments of the gas cell hydrogen reactor having NaH as the
catalyst comprise a filament with a dissociator in the reactor cell and Na in
the
reservoir. H2 may be flowed through the reservoir to main chamber. The power
may
25 be controlled by controlling the gas flow rate, H2 pressure, and Na vapor
pressure.
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
61
The latter may be controlled by controlling the reservoir temperature. In
another
embodiment, the hydrino reaction is initiated by heating with the external
heater and
an atomic H is provided by a dissociator.
The invention is also directed to other reactors for producing increased
binding energy hydrogen compounds of the invention, such as dihydrino
molecules
and hydrino hydride compounds. A further products of the catalysis is plasma,
light,
and power. Such a reactor is hereinafter referred to as a"hydrogen reactor" or
"hydrogen cell". The hydrogen reactor comprises a cell for making hydrinos.
The
cell for making hydrinos may take the form of a gas cell, a gas discharge
cell, a
plasma torch cell, or microwave power cell, for example. These exemplary cells
which are not meant to be exhaustive are disclosed in Mills Prior Publications
and
are incorporated by reference. Each of these cells comprises: a source of
atomic
hydrogen; at least one of a solid, molten, liquid, or gaseous catalyst for
making
hydrinos; and a vessel for reacting hydrogen and the catalyst for making
hydrinos.
As used herein and as contemplated by the subject invention, the term
"hydrogen",
unless specified otherwise, includes not only proteum (`H ), but also
deuterium (zH )
and tritium (3H).
Hydrogen Gas Discharge Power and Plasma Cell and Reactor
A hydrogen gas discharge power and plasma cell and reactor of the present
invention is shown in FIGURE 4A. The hydrogen gas discharge power and plasma
cell and reactor of FIGURE 4A, includes a gas discharge cell 307 comprising a
hydrogen gas-filled glow discharge vacuum vessel 315 having a chamber 300. A
hydrogen source 322 supplies hydrogen to the chamber 300 through control valve
325 via a hydrogen supply passage 342. A catalyst is contained in the cell
chamber
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
62
300. A voltage and current source 330 causes current to pass between a cathode
305 and an anode 320. The current may be reversible.
In an embodiment, the material of cathode 305 may be a source of catalyst
such as Fe, Dy, Be, or Pd. In another embodiment of the hydrogen gas discharge
power and plasma cell and reactor, the wall of vessel 313 is conducting and
serves
as the cathode which replaces electrode 305, and the anode 320 may be hollow
such as a stainless steel hollow anode. The discharge may vaporize the
catalyst
source to catalyst. Molecular hydrogen may be dissociated by the discharge to
form
hydrogen atoms for generation of hydrinos and energy. Additional dissociation
may
be provided by a hydrogen dissociator in the chamber.
Another embodiment of the hydrogen gas discharge power and plasma cell
and reactor where catalysis occurs in the gas phase utilizes a controllable
gaseous
catalyst. The gaseous hydrogen atoms for conversion to hydrinos are provided
by a
discharge of molecular hydrogen gas. The gas discharge cell 307 has a catalyst
supply passage 341 for the passage of the gaseous catalyst 350 from catalyst
reservoir 395 to the reaction chamber 300. The catalyst reservoir 395 is
heated by a
catalyst reservoir heater 392 having a power supply 372 to provide the gaseous
catalyst to the reaction chamber 300. The catalyst vapor pressure is
controlled by
controlling the temperature of the catalyst reservoir 395, by adjusting the
heater 392
by means of its power supply 372. The reactor further comprises a selective
venting
valve 301. A chemically resistant open container, such as a stainless steel,
tungsten
or ceramic boat, positioned inside the gas discharge cell may contain the
catalyst.
The catalyst in the catalyst boat may be heated with a boat heater using an
associated power supply to provide the gaseous catalyst to the reaction
chamber.
Alternatively, the glow gas discharge cell is operated at an elevated
temperature
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
63
such that the catalyst in the boat is sublimed, boiled, or volatilized into
the gas
phase. The catalyst vapor pressure is controlled by controlling the
temperature of
the boat or the discharge cell by adjusting the heater with its power supply.
To
prevent the catalyst from condensing in the cell, the temperature is
maintained
above the temperature of the catalyst source, catalyst reservoir 395 or
catalyst boat.
In a preferred embodiment, the catalysis occurs in the gas phase, lithium is
the catalyst, and a source of atomic lithium such as lithium metal or a
lithium
compound such as LiNH2 is made gaseous by maintaining the cell temperature in
the range of about 300-1000 C. Most preferably, the cell is maintained in the
range
of about 500-750 C. The atomic and/or molecular hydrogen reactant may be
maintained at a pressure less than atmospheric, preferably in the range of
about 10
millitorr to about 100 Torr. Most preferably, the pressure is determined by
maintaining a mixture of lithium metal and lithium hydride in the cell
maintained at
the desired operating temperature. The operating temperature range is
preferably in
the range of about 300-1000 C and most preferably, the pressure is that
achieved
with the cell at the operating temperature range of about 300-750 C. The cell
can
be controlled at the desired operating temperature by the heating coil such as
380 of
FIGURE 4A that is powered by power supply 385. The cell may further comprise
an
inner reaction chamber 300 and an outer hydrogen reservoir 390 such that
hydrogen
may be supplied to the cell by diffusion of hydrogen through the wall 313
separating
the two chambers. The temperature of the wall may be controlled with a heater
to
control the rate of diffusion. The rate of diffusion may be further controlled
by
controlling the hydrogen pressure in the hydrogen reservoir.
An embodiment of the plasma cell of the present invention regenerates the
reactants such as Li and LiNH2. In an embodiment, the reaction given by Eqs.
(32)
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
64
and (37) occurs to generate the hydrino reactants Li and H with a large excess
of
energy released due to hydrino production. The products are then hydrogenated
by
a hydrogen source. In the case that LiH is formed, one reaction to regenerate
the
lower-energy-hydrogen-catalysis reactants is given by Eq. (66). This may be
achieved with the reactants placed in a reactive region in the plasma cell
such as at
the cathode region in a hydrogen plasma cell. The reaction may be
LiH + e- to Li and H- (30)
and then the reaction
Li2NH + H- to Li + LiNH2 (31)
may occur to some extent to maintain a steady-state level of Li + LiNH2. The
H2
pressure, electron density, and energy may be controlled to achieve the
maximum or
desired extent of the reaction to regenerate hydrino reactants Li + LiNHz.
In an embodiment, the mixture is stirred or mixed during the plasma reaction.
In a further embodiment of the plasma regeneration system and method of the
present invention, the cell comprises a heated flat-bottom stainless steel
plasma
chamber. LiH and Li2NH comprise a mixture in molten Li. Since stainless steel
is
not magnetic, the liquid mixture may be stirred with a stainiess-steel-coated
stirring
bar driven by a stirring motor upon which the flat-bottom plasma reactor sits.
The Li-
metal mixture may serve as a cathode. The reduction of LiH to Li and H" and
the
further reaction of H" + Li2NH to Li and LiNH2 can be monitored by XRD and
FTIR of
the product.
In another embodiment of a system having a reaction mixture comprising
species of the group of Li, LiNH2, Li2NH, Li3N, LiNO3, LiX, NH4X (X is a
halide), NH3,
and H2, at least one of the reactants is regenerated by adding one or more of
the
reagents and by a plasma regeneration. The plasma may be one of the gases such
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
as NH3 and H2. The plasma may be maintained in situ (in the reaction cell) or
in an
external cell in communication with the reaction cell. In other embodiments,
K, Cs,
and Na replace Li wherein the catalyst is atomic K, atomic Cs, and molecular
NaH.
To maintain the catalyst pressure at the desire level, the cell having
5 permeation as the hydrogen source may be sealed. Alternatively, the cell
further
comprises high temperature valves at each inlet or outlet such that the valve
contacting the reaction gas mixture is maintained at the desired temperature.
The plasma cell temperature can be controlled independently over a broad
range by insulating the cell and by applying supplemental heater power with
heater
10 380. Thus, the catalyst vapor pressure can be controlled independently of
the
plasma power.
The discharge voltage may be in the range of about 100 to 10,000 volts. The
current may be in any desired range at the desired voltage. Furthermore, the
plasma may be pulsed as disclosed in Mills Prior Publications such as
15 PCT/USO4/10608 entitled "Pulsed Plasma Power Cell and Novel Spectral Lines"
which is herein incorporated by reference in its entirety.
Boron nitride may comprise the feed-throughs of the plasma cell since this
material is stable to Li vapor. Crystalline or transparent alumina are other
stable
feed-through materials of the present invention.
Solid Fuels and Hydrogen Catalyst Reactor
Metals in the gas state comprise diatomic covalent molecules. An objective of
the present Invention is to provide atomic catalyst such as Li as well as K
and Cs
and molecular catalyst NaH. Thus, in a solid-fuels embodiment, the reactants
comprise alloys, complexes, or sources of complexes that reversibly form with
a
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
66
metal catalyst M and decompose or react to provide gaseous catalyst such as
Li. In
another embodiment, at least one of the catalyst source and atomic hydrogen
source
further comprises at least one reactant which reacts to form at least one of
the
catalyst and atomic hydrogen. In an embodiment, the source or sources comprise
at
least one of amides such as LiNH2, imides such as Li2NH, nitrides such as
Li3N, and
catalyst metal with NH3. Reactions of these species provide both Li atoms and
atomic hydrogen. These and other embodiments are given infra., wherein,
additionally, K, Cs, and Na may replace Li and the catalyst is atomic K,
atomic Cs,
and molecular NaH.
The present invention comprises an energy reactor comprising a reaction
vessel constructed and arranged to contain pressures lower, equal to, and
higher
than atmospheric pressure, a source of atomic hydrogen for chemically
producing
atomic hydrogen in communication with the vessel, a source of catalyst
comprising
at least one of atomic lithium, atomic cesium, atomic potassium, and molecular
NaH
in communication with the vessel, and may further comprise a getter such as
source
of an ionic compound for binding or reacting with a lower-energy hydride. The
source of catalyst and reactant atomic hydrogen may comprise a solid fuel that
may
be continuously or batch-wise regenerated inside or outside of the cell
wherein a
physical process or chemical reaction generates the catalyst and H from a
source
such that H catalysis occurs and hydrinos are formed. Thus, embodiments of the
present invention of hydrino reactants comprise solid fuels, and preferable
embodiments comprise those solid fuels that can be regenerated. Solid fuels
can
used in many applications ranging from space and process heating, electricity
generation, motive applications, propellants, and others applicants well known
to
those skilled in the Art.
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
67
A gas cell or plasma cell of the present invention such as those shown in
FIGURES 3A and 4A comprises a means for the formation of catalyst and H atoms
from sources. In solid-fuels embodiments, the cell further comprises reactants
to
provide catalyst and H upon initiation of a chemical or physical process. The
initiation may be by means such as heating or plasma reaction. Preferably the
external power requirement to maintain the production of hydrinos is low or
zero
based on the large power of the H catalysis reaction to form hydrinos. With a
large
energy gain, the reactants can be regenerated with a net release of energy for
each
cycle of reaction and regeneration.
In other embodiments, the reactor shown in FIGURE 3A comprises a solid-
fuels reactor wherein a reaction mixture comprises a source of catalyst and a
source
of hydrogen. The reaction mixture can be regenerated by supplying a flow of
reactants and by removing products from the corresponding product mixture. In
an
embodiment, the reaction vessel 207 has a chamber 200 capable of containing a
vacuum or pressures equal to or greater than atmospheric. At least one source
of
reagent such a gaseous reagent 221 is in corrimunication with chamber 200 and
delivers reagent to the chamber through at least one reagent supply passage
242. A
controller 222 is positioned to control the pressure and flow of reagent into
the
vessel through reagent supply passage 242. A pressure sensor 223 monitors
pressure in the vessel. A vacuum pump 256 is used to evacuate the chamber
through a vacuum line 257. Alternatively, line 257 represents at least one
output
path such as a product passage line to remove material from the reactor. The
reactor further comprises a source of heat such as a heater 230 to bring the
reactants up to a desired temperature that initiates the solids fuel chemistry
and the
hydrino-forming catalysis reaction. In an embodiment, the temperature is in
the
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
68
range of about 50 to 1000 C; preferably it is in the range of about 100-600
C, and
for reactants comprising at least the Li/N-alloy system, the desired
temperature is in
the range of about 100-500 C.
The cell may further comprise a source of hydrogen gas and dissociator to
form atomic hydrogen. The vessel may further comprise a source of hydrogen 221
in communication with the vessel for regenerating at least one of the source
of
atomic catalyst such as atomic lithium and the source of atomic hydrogen. The
hydrogen source may be hydrogen gas. The H2 gas may be supplied by a hydrogen
line 242 or by permeation from a hydrogen reservoir 290. In exemplary
regeneration
reactions, the source of atomic lithium and atomic hydrogen may be generated
by
hydrogen addition according to Eqs. (66-71). The first step of an alternative
regeneration reaction may given by Eq. (69).
In an embodiment, the cell size and materials are such that a high operating
temperature is archived. The cell may be appropriately sized to the power
output to
achieve the desired operating temperature. High-temperature materials for the
cell
construction are niobium and a high-temperature stainless steel such as
Hastalloy.
The source of H2 may be an internal metal hydride that does not react with
LiNH2,
but releases H only at very high temperature. Also, even in the cases that the
hydride does react with LiNH2, it can be separated from the reagents such as
Li and
LiNH2 by placing it in an open or closed vessel in the cell. A cell design
that supplies
H2 by permeation is one comprising an internal metal hydride placed in a
sealed
vessel wherein atomic H permeates out at high temperature.
The reactor may further comprise means to separate components of a
product mixture such as sieves for mechanically separating by differences in
physical properties such as size. The reactor may further comprise means to
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
69
separate one or more components based on a differential phase change or
reaction.
In an embodiment, the phase change comprises melting using a heater, and the
liquid is separated from the solid by means known in the Art such as gravity
filtration,
filtration using a pressurized gas assist, and centrifugation. The reaction
may
comprise decomposition such as hydride decomposition or reaction to from a
hydride, and the separations may be achieved by melting the corresponding
metal
followed by its separation and by mechanically separating the hydride,
respectively.
The latter may be achieved by sieving. In an embodiment, the phase change or
reaction may produce a desired reactant or intermediate. In embodiments, the
regeneration including any desired separation steps may occur inside or
outside of
the reactor.
Chemical Reactor
A chemical reactor of the present invention further comprises a source of
inorganic compound such as MX wherein M is an alkali metal and X is a halide.
Additionally to halides, the inorganic compound may be an alkali or alkaline
earth
salt such a hydroxide, oxide, carbonate, sulfate, phosphate, borate, and
silicate
(other suitable inorganic compounds are given in D. R. Lide, CRC Handbook of
Chemistry and Physics, 86th Edition, CRC Press, Taylor & Francis, Boca Raton,
(2005-6), pp. 4-45 to 4-97 which is herein incorporated by reference). The
inorganic
compound may further serve as a getter in the generation of power by
preventing
product accumulation and a consequent back reaction or other product
inhibition. A
preferred Li chemical-type power cell comprises Li, LiNH2, LiBr or Lil, and R-
Ni in a
hydrogen cell run at about 760 Torr H2 and about 700+ C. A preferred NaH
chemical-type power cell comprises Na, NaX (X is a halide, preferably Br or I)
and R-
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
Ni in a hydrogen cell run at about 760 Torr H2 and about 700+ C. The cell may
further comprise at least one of NaH and NaNH2. A preferred K chemical-type
power cell comprises K, KI, and Ni screen or R-Ni dissociator in a hydrogen
cell run
at about 760 Torr H2 and about 700+ C. In an embodiment, the H2 pressure
range
5 is about 1 Torr to 105 Torr. Preferably, the H pressure is maintained in the
range of
about 760-1000 Torr. LiHX such as LiHBr and LiHI is typically synthesized in
the
temperature range of about 450-550 C, but can be run at lower temp (-350 C)
with
LiH present. NaHX such as NaHBr and NaHI is typically synthesized in the
temperature range of about 450-550 C. KHX such as KHI is preferably
synthesized
10 in the temperature range of about 450-550 C. In embodiments of the NaHX
and
KHX reactors, NaH and K are supplied from a source such as catalyst reservoir
wherein the cell temperature is maintained at a higher level than that of the
catalyst
reservoir. Preferably, the cell is maintained at the temperature range of
about 300-
550 C and the reservoir is maintained in a temperature range of about 50 to
200 C
15 lower.
Another embodiment of the hydrogen reactor having NaH as the catalyst
comprises a plasma torch for the production of power and increased-binding-
energy
hydrogen compounds such as NaHX wherein H is increased-binding-energy
hydrogen and X is a halide. At least one of NaF, NaCI, NaBr, Nal may be
20 aerosolized in the plasma gas such as H2 or a noble gas/hydrogen mixture
such as
He/H2 or Ar/H2.
General Solid Fuels Chemistry
A reaction mixture of the present invention comprises a catalyst or a source
of
25 catalyst and atomic hydrogen or a source of atomic hydrogen (H) wherein at
least
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
71
one of the catalyst and atomic hydrogen is released by a chemical reaction of
at
least one species of the reaction mixture or between two or more reaction-
mixture
species. Preferably, the reaction is reversible. Preferably, the energy
released is
greater than the enthalpy of reaction of the formation of catalyst and
reactant
hydrogen, and in the case that the reactants of the reaction mixture are
regenerated
and recycled, preferably, net energy is given off over the cycle of reaction
and
regeneration due to the large energy of formation of product H states given by
Eq.
(1). The species may be at least one of an element, alloy, or a compound such
as a
molecular or inorganic compound wherein each may be at least one of a reagent
or
product in the reactor. In an embodiment, the species may form an alloy or
compound such as a molecular or inorganic compound with at least one of
hydrogen
and the catalyst. One or more of the reaction-mixture species may form one or
more
reaction product species such that the energy to release H or free catalyst is
lowered
relative to the case in the absence of the formation of the reaction product
species.
In embodiments of the reactants to provide a catalyst and atomic hydrogen to
form
states with energy levels given by Eq. (1), the reactants comprise at least
one of
solid, liquid (including molten), and gaseous reactants. The reactions to form
the
catalyst and atomic hydrogen to form states with energy levels given by Eq.
(1)
occurs in one or more of the solid, liquid (including molten), and gaseous
phase.
Exemplary solid-fuels reactions are given herein that are certainly not meant
to be
limiting in that other reactions comprising additional reagents are within the
scope of
the Invention.
In an embodiment, the reaction product species is an alloy or compound of at
least one of the catalyst and hydrogen or sources thereof. In an embodiment,
the
reaction-mixture species is a catalyst hydride and the reaction product
species is a
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
72
catalyst alloy or compound that has a lower hydrogen content. The energy to
release H from a hydride of the catalyst may be lowered by the formation of an
alloy
or second compound with the at least one another species such as an element or
first compound. In an embodiment, the catalyst is one of Li, K, Cs, and NaH
molecule and the hydride is one of LiH, KH, CsH, NaH(s) and the at least one
other
element is selected from the group of M (catalyst), Al, B, Si, C, N, Sn, Te,
P, S, Ni,
Ta, Pt, and Pd. The first and the second compound may be one of the group of
H2,
H20, NH3, NHaX, (X is a couterion such as halide (other anions are given in D.
R.
Lide, CRC Handbook of Chemistry and Physics, 86th Edition, CRC Press, Taylor &
Francis, Boca Raton, (2005-6), pp. 4-45 to 4-97 which is herein incorporated
by
reference) IVIX, NINO3, MAIH4, M3AIH6, MBH4, M3N, M2NH, and MNH2 wherein M is
an alkali metal that may be the catalyst. In another embodiment, a hydride
comprising at least one other element than the catalyst element releases H by
reversible decomposition.
One or more of the reaction-mixture species may form one or more reaction
product species such that the energy to release free catalyst is lowered
relative to
the case in the absence of the formation of the reaction product species. A
reaction
species such as an alloy or compound may release free catalyst by a reversible
reaction or decomposition. Also, the free catalyst may be formed by a
reversible
reaction of a source of catalyst with at least one other species such as an
element or
first compound to form a species such as an alloy or second compound. The
element or alloy may comprise at least one of M (catalyst atom), H, Al, B, Si,
C, N,
Sn, Te, P, S, Ni, Ta, Pt, and Pd. The first and the second compound may be one
of
the group of H2, NH3, NH4X where X is a couterion such as halide, MMX, MNO3,
MAIH4, M3AIH6, MBH4, M3N, M2NH, and MNH2, wherein M is an alkali metal that
may
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
73
be the catalyst. The catalyst may be one of Li, K, and Cs, and NaH molecule.
The
source of catalyst may be M-M such as LiLi, KK, CsCs, and NaNa. The source of
H
may be MH such as LiH, KH, CsH, or NaH(s).
Li catalyst may be alloyed or react to form a compound with at least one other
element or compound such that the energy barrier for the release of H from LiH
or Li
from LiH and LiLi molecules is lowered. The alloy or compound may also release
H
or Li by decomposition or reaction with further reaction species. The alloy or
compound may be one or more of LiAIH4, Li3AIH6, LiBH4, Li3N, Li2NH, LiNH2,
LiX,
and LiNO3. The alloy or a compound may be one or more of Li/Ni, Li/Ta, Li/Pd,
Li/Te, Li/C, Li/Si, and Li/Sn wherein the stoichiometry of Li and any other
element of
the alloy or compound is varied to achieve the optimal release of Li and H
which
subsequently react during the catalysis reaction to form lower energy states
of
hydrogen. In other embodiments, K, Cs, and Na replace Li wherein the catalyst
is
atomic K, atomic Cs, and molecular NaH.
In an embodiment, the alloy or compound has the formula IVIXEy wherein M is
the catalyst such as Li, K, or Cs, or it is Na, E is the other element, and x
and y
designate the stoichiometry. M and Ey may be in ant desired molar ratio. In an
embodiment x is in the range of 1 to 50 and y is in the range of 1 to 50, and
preferably x is in the range of 1 to 10 and y is in the range of 1 to 10.
In another embodiment, the alloy or compound has the formula MXEyEZ
wherein M is the catalyst such as Li, K, or Cs, or it is Na, Ey is a first
other element,
EZ is a second other element, and x, y, and z designate the stoichiometry. M,
Ey,
and Ey may be in any desired molar ratio. In an embodiment, x is in the range
of 1 to
50, y is in the range of 1 to 50, and z is in the range of 1 to 50, and
preferably x is in
the range of 1 to 10, y is in the range of 1 to 10, and z is in the range of 1
to 10. In
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
74
preferred embodiments, EY and EZ are selected from the group of H, N, C, Si,
and
Sn. The alloy or compound may be at least one of Li,CySiZ, Li,SnYSiZ,
LiXNySiZ,
LiXSnyCZ, LiXNYSnZ, Li,CyNZ , LixCyHZ , LiXSnyHZ, Li,,NyHZ, and LiXSiYHZ. In
other
embodiments, K, Cs, and Na replace Li wherein the catalyst is atomic K, atomic
Cs,
and molecular NaH.
In another embodiment, the alloy or compound has the formula M,EWEyEZ
wherein M is the catalyst such as Li, K, or Cs, or it is Na, EW is a first
other element,
EY is a second other element, EZ is a third other element, and x, w, y, and z
designate the stoichiometry. M, Ey, Ey, and EZ may be in any desired molar
ratio. In
an embodiment, x is in the range of 1 to 50, w is in the range of 1 to 50, y
is in the
range of 1 to 50, and z is in the range of 1 to 50, and preferably x is in the
range of 1
to 10, w is in the range of 1 to 10, y is in the range of 1 to 10, and z is in
the range of
1 to 10. In preferred embodiments, EW, EY, and EZ are selected from the group
of H,
N, C, Si, and Sn. The alloy or compound may be at least one of LixHWCYSiZ ,
LiXHWSnySiZ, LiXHWNySiZ, LiXHWSnYCZ, LiXHWNYSnZ, and Li,HH,CyNZ. In other
embodiments, K, Cs, and Na replace Li wherein the catalyst is atomic K, atomic
Cs,
and molecular NaH. Species such as MXEWEyEz are exemplary and are certainly
not
meant to be limiting in that other species comprising additional elements are
within
the scope of the Invention.
In an embodiment, the reaction contains a source of atomic hydrogen and a
source of Li catalyst. The reaction contains one or more species from the
group of a
hydrogen dissociator, H2, a source of atomic hydrogen, Li, LiH, LiNO3, LiNH2,
Li2NH,
Li3N, LiX, NH3, LiBH4, LiAIH4, Li3AIH6, NH3, and NH4X wherein X is a
counterion
such halide and those given in the CRC [41]. The weight % of the reactants may
be
in any desired molar range. The reagents may be well mixed using a ball mill.
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
In an embodiment, the reaction mixture comprises a source of catalyst and a
source of H. In an embodiment, the reaction mixture further comprises
reactants
which undergo reaction to form Li catalyst and atomic hydrogen. The reactants
may
comprise one or more of the group of H2, hydrino catalyst, MNH2, M2NH, M3N,
NH3,
5 LiX, NH4X (X is a couterion such as a halide), MNO3, MAIH4, M3AIH6, and
MBH4,
wherein M is an alkali metal that may be the catalyst. The reaction mixture
may
comprise reagents selected from the group of Li, LiH, LiNO3, LiNO, LiNO2,
Li3N,
Li2NH, LiNH2, LiX, NH3, LiBH4, LiAIH4, Li3AIH6, LiOH, Li2S, LiHS, LiFeSi,
Li2CO3,
LiHCO3, Li2SO4, LiHSO4, Li3PO4, LizHPO4, LiH2PO4, Li2MoO4, LiNbO3, LizB4O7
10 (lithium tetraborate), LiBO2, Li2WO4, LiAICl4, LiGaC14, Li2CrO4, Li2Cr2O7,
LiZTiO3,
LiZrO3, LiAIO2, LiCoOz, LiGaO2, Li2GeO3, LiMn2O4, Li4SiO4, Li2SiO3, LiTaO3,
LiCuCl4, LIPdCl4, LIVO3, L1103, LiFeO2, Li104,LICIO4, LiScO, LiTIO, LiVO,
LiCrO,,,
LiCr2On, LiMn2On, LiFeO, LiCoO, LiNiOn, LiNi2O,, LiCuOr, and LiZnOn, where
n=1,
2,3, or 4, an oxyanion, an oxyanion of a strong acid, an oxidant, a molecular
oxidant
15 such as V203i 1205, Mn02, Re207, Cr03, Ru02, AgO, PdO, Pd02, PtO, Pt02, and
NH4X wherein X is a nitrate or other suitable anion given in the CRC [41], and
a
reductant. In each case, the mixture further comprises hydrogen or a source of
hydrogen. In other embodiments, other dissociators are used or one may not be
used wherein atomic hydrogen, and, optionally, atomic catalyst, are generated
20 chemically by reaction of the species of the mixture. In a further
embodiment, the
reactant catalyst may be added to the reaction mixture.
The reaction mixture may further comprise an acid such as H2SO3, H2SO4,
H2CO3, HNO2, HNO3, HCIO4, H3PO3, and H3PO4 or a source of an acid such as an
anhydrous acid. The latter may comprise at least one of the list of SO2, SO3,
CO2,
25 NO2, N2O3, N2O5, C1207, P02, P203, and P2O5.
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
76
In an embodiment, the reaction mixture further comprises a reactant catalyst
to generate the reactants that serve as a lower-energy-hydrogen catalyst or a
source
of lower-energy-hydrogen catalyst and atomic hydrogen or a source of atomic
hydrogen. Suitable reactant catalysts comprise at one of the group of acids,
bases,
halide ions, metal ions and free radical sources. The reactant catalyst may be
at
least one of the group of a weak-base-catalysts such as Li2SO4, a weak-acid
catalyst
such as a solid acid such as LiHSO4, a metal ion source such as TiCl3 or AICI3
which
provide Ti3+ and AI3+ ions, respectively, a free radical source such a CoX2
wherein X
is a halide such as Cl wherein Co2+ may react with 02 to form the OZ radical,
metals
such as Ni, Fe, Co preferably at a concentration of about 1 mol%, a source of
X" ion
(X is halide) such as CI" or F from LiX, a source of free radical
initiators/propagators
such as peroxides, azo-group compounds, and UV light.
In an embodiment, the reactant mixture to form lower-energy hydrogen
comprises a source of hydrogen, a source of catalyst, and at least one of a
getter for
hydrino and a getter for electrons from the catalyst as it is ionized to
resonantly
accept energy from atomic hydrogen to form hydrinos having energies given by
Eq.
(1). The hydrino getter may bind to lower-energy hydrogen to prevent the
reverse
reaction to ordinary hydrogen. In an embodiment, the reaction mixture
comprises a
getter for hydrino such as LiX or Li2X (X is haiide or other anion such anions
from the
CRC [41]). The electron getter may perform at least one of accepting electrons
from
the catalyst and stabilizing the catalyst-ion intermediate such as a Li2+
intermediate
to allow the catalysis reaction to occur with fast kinetics. The getter may be
an
inorganic compound comprising at least one cation and one anion. The cation
may
be Li+. The anion may be a halide or other anion given in the CRC [41] such as
one
of the group comprising F, CI", Br, I-, N03 , N02 , S042-, HS04 , CoOz , 103 ,
104 ,
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
77
Ti03 , Cr04 , Fe02", P043-, HP042-, H2PO4 , V03 , C104 and Cr207 2- and other
anions
of the reactants. The hydride binder and/or stabalizer may be at least one of
the
group of LiX (X = halide) and the other corripounds comprising the reactants.
In an embodiment of the reaction mixture such as Li, LiNH2, and X wherein X
is the hydride binding compound, X is at least one of LiHBr, LiHI, a hydrino
hydride
compound, and a lower-energy hydrogen compound. In an embodiment, the
catalyst reaction mixture is regenerated by addition of hydrogen from a source
of
hydrogen.
In an embodiment, the hydrino product may bind to form a stable hydrino
hydride compound. The hydride binder may be LiX wherein X is a halide or other
anion. The hydride binder may react with a hydride that has an NMR upfield
shift
greater than that of TMS. The binder may be an alkali halide, and the product
of
hydride binding may be an alkali hydride halide having an NMR upfield shift
greater
than that of TMS. The hydride may have a binding energy determined by XPS of
11
to 12 eV. In an embodiment, the product of the catalysis reaction is the
hydrogen
molecule H2(1/4) having an solid NMR peak at about 1 ppm relative to TMS and a
binding energy of about 250 eV that is trapped in a crystalline ionic lattice.
In an
embodiment, the product H2(1/4) is trapped in the crystalline lattice of an
ionic
compound of the reactor such that the selection rules for infrared absorption
are
such that the molecule becomes IR active and a FTIR peak is observed at about
1990 cm"'.
Additional sources of atomic Li of the present invention comprise additional
alloys of Li such as these comprising Li and at least one of alkali, alkaline
earth
metals, transitions, metals, rare earth metals, noble metals, tin, aluminum,
other
Group III and Group IV metal, actinides, and lanthanides. Some representative
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
78
alloys comprise one or more members of the group of LiBi, LiAg, Liln, LiMg,
LiAI,
LiMgSi, LiFeSi, LiZr, LiAICu, LiAlZr, LiAIMg, LiB, LiCa, LiZn, LiBSi, LiNa,
LiCu, LiPt,
LiCaNa, LiAICuMgZr, LiPb, LiCaK, LiV, LiSn, and LiNi. In other embodiments, K,
Cs, and Na replace Li wherein the catalyst is atomic K, atomic Cs, and
molecular
NaH.
In another embodiment, an anion can form a hydrogen-type bond with a Li
atom of a covalently bound Li-Li molecule. This hydrogen-type bond can weaken
the Li-Li bond to the point that a Li atom is at vacuum energy (equivalent to
free a
atom) such that it can serve as a catalyst atom to form hydrinos. In other
embodiments, K, Cs, and Na replace Li wherein the catalyst is atomic K, atomic
Cs,
and molecular NaH.
In an embodiment, the function of the hydrogen dissociator is provided by a
chemical reaction. Atomic H is generated by the reaction of at least two
species of
the reaction mixture or by the decomposition of at least one species. In an
embodiment, Li-Li reacts with LiNH2 to form atomic Li, atomic H, and LizNH.
Atomic
Li may also form by the decomposition or reaction of LiNO3. In other
embodiments,
K, Cs, and Na replace Li wherein the catalyst is atomic K, atomic Cs, and
molecular
NaH.
In further embodiments, in addition to a catalyst or source of catalyst to
form
lower-energy hydrogen, the reaction mixture comprises heterogeneous catalysts
to
dissociate MM and MH such as LiLi and LiH as to provide M and H atoms. The
heterogeneous catalyst may comprise at least one element from the group of
transition elements, precious metals, rare earth and other metals and elements
such as Mo, W, Ta, Ni, Pt, Pd, Ti, Al, Fe, Ag, Cr, Cu, Zn, Co, and Sn.
In an embodiment of the Li carbon alloy, the reaction mixture comprises an
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
79
excess of Li over the Li-carbon intercalation limit. The excess may be in the
range
of 1% and 1000% and preferably in the range of 1% to 10%. The carbon may
further comprise a hydrogen spillover catalyst having a hydrogen dissociator
such
as Pd or Pt on activated carbon. In a further embodiment, the cell temperature
exceeds that at which Li is completely intercalated into the carbon. The cell
temperature may be in the range of about 100 to 2000 C, preferably in the
range of
about 200 to 800 C, and most preferably in the range of about 300 to 700 C.
In
other embodiments, K, Cs, and Na replace Li wherein the catalyst is atomic K,
atomic Cs, and molecular NaH.
In an embodiment of the Li silicon alloy, the cell temperature is in the range
over which the silicon alloy further comprising H releases atomic hydrogen.
The
range may be about 50-1500 C, preferably about 100 to 800 C, and most
preferably in the range of about 100 to 500 C. The hydrogen pressure may be
in
range of about 0.01 to 105 Torr, preferably in the range of about 10 to 5000
Torr, and
most preferably in the range of about 0.1 to 760 Torr. In other embodiments,
K, Cs,
and Na replace Li wherein the catalyst is atomic K, atomic Cs, and molecular
NaH.
The reaction mixture, alloys, and compounds may be formed by mixing the
catalyst such as Li or a source of catalyst such as catalyst hydride with the
other
element(s) or compound(s) or a source of the other element(s) or compound(s)
such
as a hydride of the other element(s). The catalyst hydride may be LiH, KH,
CsH, or
NaH. The reagents may be mixed by ball milling. An alloy of the catalyst may
also
be formed from a source of alloy comprising the catalyst and at least one
other
element or compound.
In an embodiment, the reaction mechanism for the Li/N system to form
hydrino reactants of atomic Li and H is
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
LiNH2 + Li-Li to Li + H + Li2NH (32)
In embodiments of the other Li-alloy systems, the reaction mechanism is
analogous
to that of the Li/N system with the other alloy element(s) replacing N.
Exemplary
reaction mechariisms to carryout the reaction to form hydrino reactants,
atomic Li
5 and H, involving the reaction mixtures comprising Li with at least one of S,
Sn, Si,
and C are
SH + Li-Li to Li + H + LiS (33)
SnH + Li-Li to Li + H + LiSn (34)
SiH + Li-Li to Li + H + LiSi, and (35)
10 CH + Li-Li to Li + H + LiC, (36)
Preferred embodiments of the Li/S alloy-catalyst system comprises Li with
Li2S and Li with LiHS. In other embodiments, K, Cs, and Na replace Li wherein
the
catalyst is atomic K, atorriic Cs, and molecular NaH.
15 Primary Li/Nitrogen Alloy Reactions
Lithium in the solid and liquid states is a metal, and the gas comprises
covalent Li2 molecules. In order to generate atomic lithium, the reaction
mixture of
the solid fuel comprises Li/N alloy reactants. The reaction mixture may
comprise at
least one of the group of Li, LiH, LiNH2, Li2NH, Li3N, NH3, a dissociator, a
hydrogen
20 source such as H2 gas or a hydride, a support, and a getter such as LiX (X
is a
halide). The dissociator is preferably Pt or Pd on a high surface area support
inert to
Li. It may comprise Pt or Pd on carbon or Pd/A1203. The latter support may
comprise a protective surface coating of a material such as LiAIO2. Preferred
dissociators for a reagent mixture comprising a Li/N alloy or Na/N alloy are
Pt or Pd
25 on A1203, Raney nickel (R-Ni), and Pt or Pd on carbon. In the case that the
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
81
dissociator support is AI203, the reactor temperature may be maintained below
that
which results in its substantial reaction with Li. The temperature may be
below the
range of about 250 C to 600 C. In another embodiment, Li is in the form of LiH
and
the reaction mixture comprises one or more of LiNH2i Li2NH, Li3N, NH3, a
dissociator, a hydrogen source such as H2 gas or a hydride, a support, and a
getter
such as LiX (X is a halide) wherein the reaction of LiH with AI203 is
substantially
endothermic. In other embodiments, the dissociator may be separate from the
balance of the reaction mixture wherein the separator passes H atoms.
Two preferred embodiments comprise the first reaction mixture of LiH, LiNH2,
and Pd on A1203 powder and a second reaction mixture of Li, Li3N, and hydrided
Pd
on A1203 powder that may further comprise H2 gas. The first reaction mixture
can be
regenerated by addition of H2, and the second mixture can be regenerated by
removing H2 and hydriding the dissociator or by reintroducing H2. The
reactions to
generate catalyst and H as well as the regeneration reactions are given infra.
In an embodiment, LiNH2 is added to the reaction mixture. LiNH2 generates
atomic hydrogen as well as atomic Li according to the reversible reactions
Liz + LiNH2 ~ Li + LizNH + H (37)
and
Liz + Li2NH ~ Li + Li3N + H (38)
In an embodiment, the reaction mixture comprises about 2:1 Li and LiNH2. In
the hydrino reaction cycle, Li-Li and LiNH2 react to form atomic Li, atomic H,
and
Li2NH, and the cycle continues according to Eq. (38). The reactants may be
present
in any wt%.
The mechanism of the formation of Li2NH from LiNH2 involves a first step that
forms ammonia [42]:
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
82
2LiNH2 to Li2NH + NH3 (39)
With LiH present, the ammonia reacts to release H2
LiH + NH3 to LiNH2 + H2 (40)
and the net reaction is the consumption of LiNH2 with the formation of H2:
LiNH2 + LiH to Li2NH + H2 (41)
With Li present, the amide is not consumed due to the energetically much more
favorable back reaction of Li with ammonia:
Li-Li + NH3 to LiNH2 + H + Li (42)
Thus, in an embodiment, the reactants comprise a mixture of Li and LiNHz to
form
atomic Li and atomic H according to Eqs. (37-38).
The reaction mixture of Li and LiNH2 that serves as a source of Li catalyst
and
atomic hydrogen may be regenerated. During the regeneration cycle, the
reaction
product mixture comprising species such as Li, LizNH, and Li3N can be reacted
with
H to form LiH and LiNH2. LiH has a melting point of 688 C; whereas, LiNHZ
melts at
380 C, and Li melts at 180 C. LiNH2 liquid and any Li liquid that forms can
be
physically removed from the LiH solid at about 380 C, and then LiH solid can
be
heated separately to form Li and H2. The Li and LiNH2 can be recombined to
regenerate the reaction mixture. And, the excess H2 from LiH thermal
decomposition can be reused in the next regeneration cycle with some make-up
H2
to replace any H2 consumed in hydrino formation.
In a preferred embodiment, the competing kinetics of the hydriding or
dehydridirtg of one reactant over another is exploited to achieve a desired
reaction
mixture comprising hydrided and non-hydrided compounds. For example, hydrogen
can be added under appropriate temperature and pressure conditions such that
the
reverse of reactions of Eqs. (37) and (38) occur over the competing reaction
of the
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
83
formation of LiH such that the hydrogenated products are predominantly Li and
LiNH2. Alternatively, a reaction mixture comprising compounds of the group of
Li,
Li2NH, and Li3N may be hydrogenated to form the hydrides and the LiH can be
selectively dehydrided by purriping at the temperature and pressure ranges and
duration which achieves the selectivity based on differential kinetics.
In an embodiment, Li is deposited as a thin film over a large area and a
mixture of LiH and LiNH2 is formed by addition of ammonia. The reaction
mixture
may further comprise excess Li. Atomic Li and H are formed according to Eqs.
(37-
38) with the subsequent reaction to form states with energies given by Eq.
(1).
Then, the mixture can be regenerated by H2 addition followed by heating and
pumping with selective pumping and removal of H2.
A reversible system of the present invention to generate atomic lithium
catalyst is the Li3N + H system which can be regenerated by pumping. The
reaction
mixture comprises at least one of Li3N and a source of Li3N such as Li and N2,
and
a source of H such as at least one of H2 and a hydrogen dissociator, LiNHZ 1
LiZNH , LiH, Li, NH3 1 and a metal hydride. The reaction of H2 with Li3N gives
LiH
and Li2NH; whereas, the reaction of Li3N and H from an atomic hydrogen source
such a H2 and a dissociator or form a hydride undergoing decomposition gives
Li3N + H to Li2NH + Li (43)
The atomic Li catalyst can then react with additional atomic H to form
hydrinos. The
side products such as LiH, Li2NH, and LiNH2 can be converted to Li3N by
evacuating
the reaction vessel of H2. Representative Li/N alloy reactions are as follows:
Li3N + H -4 LiZNH + Li (44)
Li3N + LiH -> Li2NH + 2Li (45)
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
84
LizNH+LiH-4 Li3N+Hz (46)
LiZNH + H -4 LiNH2 + Li (47)
Li2NH + LiH ---> LiNH2 + 2Li (48)
Li3N, a source of H, and a hydrogen dissociator are in any desired molar
ratio.
Each are in molar ratios of greater than 0 and less than 100%. Preferably the
molar
ratios are similar. In an embodiment, the ratios of Li3N, at least one of
LiNHr,
Li2NH, LiH, Li, and NH3 1 and a H source such as a metal hydride are similar.
In
other embodiments, K, Cs, and Na replace Li wherein the catalyst is atomic K,
atomic Cs, and molecular NaH.
In an embodiment, lithium amide and hydrogen is reacted to form ammonia
and lithium:
1/ 2H2 + LiNHz -4 NH3 + Li (49)
The reaction can be driven to form Li by increasing the H2 concentration.
Alternatively, the forward reaction can be driven via the formation of atomic
H using
a dissociator. The reaction with atomic H is given by
H + LiNHZ -> NH3 + Li (50)
In an embodiment of the reaction mixture that comprises one or more compounds
that react with a source of Li to form Li catalyst, the reaction mix comprises
at least
one species from the group of LiNH2, Li2NH, Li3N, Li, LiH, NH3, H2 and a
dissociator.
In an embodiment, Li catalyst is generated from a reaction of LiNH2 and
hydrogen,
preferably atomic hydrogen as given in reaction Eq. (50). The ratios of
reactants
may be any desired amount. Preferably the ratios are about stoichiometric to
those
of Eqs. (49-50). The reactions to form catalyst are reversible with the
addition of a
source of H such as H2 gas to replace that reacted to form hydrinos wherein
the
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
catalyst reactions are given by Eqs. (3-5), and lithium amide forms by the
reaction of
ammonia with Li:
NH3+Li-4 LiNHZ+H (51)
In other embodiments, K, Cs, and Na replace Li wherein the catalyst is atomic
5 K, atomic Cs, and molecular NaH. In a preferred embodiment, the reaction
mixture
comprises a hydrogen dissociator, a source of atomic hydrogen, and Na or K and
NH3. In an embodiment, ammonia reacts with Na or K to form NaNH2 or KNH2 that
serves as a source of catalyst. Another embodiment comprises a source of K
catalyst such as K metal, a hydrogen source such as at least one of NH3, H2,
and a
10 hydride such as a metal hydride, and a dissociator. A preferred hydride is
one
comprising R-Ni that also may serve as a dissociator. Additionally, a hydrino
getter
such as KX may be present wherein X is preferably a halide such as Cl, Br, or
I. The
cell may be run continuously with the replacement of the hydrogen source. The
NH3
may act as a source of atomic K by the reversible formation of KN alloy
compounds
15 from K-K such as at least one of amide, imide, or nitride or by formation
of KH with
the release of atomic K.
In a further embodiment, the reactants comprise the catalyst such as Li and
an atomic hydrogen source such H2 and a dissociator or a hydride such as
hydrided
R-Ni. H can react with Li-Li to form LiH and Li which can further serve as the
20 catalyst to react with additional H to form hydrinos. Then, Li can be
regenerated by
evacuating H2 released from LiH. 'rhe plateau temperature at 1 Torr for LiH
decomposition is about 560 C. LiH can be decomposed at about 0.5 Torr and
about 500 C, below the alloy-formation and sintering temperatures of R-Ni.
The
molted Li can be separated from R-Ni, the R-Ni may be rehydrided, and Li and
25 hydrided R-Ni can be returned to another reaction cycle.
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
86
In an embodiment, Li atoms are vapor deposited on a surface. The surface
may support or be a source of H atoms. The surface may comprise at least one
of a
hydride and hydrogen dissociator. The surface may be R-Ni which may be
hydrided.
The vapor deposition may be from a reservoir containing a source of Li atoms.
The
Li source may be controlled by heating. One source that provides Li atoms when
heated is Li metal. The surface may be maintained at a low temperature such as
room temperature during the vapor deposition. The Li-coated surface may be
heated to cause the reaction of Li and H to form H states given by Eq. (1).
Other
thin-film deposition techniques that are well known in the ART comprise
further
embodiments of the Invention. Such embodiments comprise physical spray,
electro-
spray, aerosol, electro-arching, Knudsen cell controlled release, dispenser-
cathode
injection, plasma-deposition, sputtering, and further coating methods and
systems
such as melting a fine dispersion of Li, electroplating Li, and chemical
deposition of
Li. In other erribodiments, K, Cs, and Na replace Li wherein the catalyst is
atomic K,
atomic Cs, and molecular NaH.
In the case of vapor-deposited Li on a hydride surface, regeneration can be
achieved by heating with pumping to remove LiH and Li, the hydride can be
rehydrided by introducing H2, and Li atoms can be redeposited onto the
regenerated
hydride after the cell is evacuated in an embodiment. In other embodiments, K,
Cs,
and Na replace Li wherein the catalyst is atomic K, atomic Cs, and molecular
NaH.
Li and R-Ni are in any desired molar ratio. Each of Li and R-Ni are in molar
ratios of greater than 0 and less than 100%. Preferably the molar ratio of Li
and R-
Ni are similar.
In a preferred embodiment, the competing kinetics of the hydriding or
dehydriding of one reactant over another is exploited to achieve a reaction
mixture
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
87
comprising hydrided and non-hydrided compounds. For example, the formation of
LiH is thermodynamically favored over the formation of R-Ni hydride. However,
the
rate of LiH formation at low temperature such as the range of about 25 C-100
C is
very low; whereas, the formation of R-Ni hydride proceeds at a high rate in
this
temperature range at modest pressures such as the range of about 100 Torr to
3000
Torr. Thus, the reaction mixture of Li and hydrided R-Ni can be regenerated
from
LiH R-Ni by pumping at about 400-500 C to dehydride LiH, cooling the vessel
to
about 25-100 C, adding hydrogen to preferentially hydride R-Ni for a duration
that
achieves the desired selectivity, and then removing the excess hydrogen by
evacuating the cell. While excess Li is present or is added to be in excess,
the R-Ni
can be used in repeated cycles by selectively hydriding alone. This can be
achieved
by adding hydrogen in the temperature and pressure ranges that achieve the
selective hydriding of R-Ni and then by removing the excess hydrogen before
the
vessel is heated to initiate the reactions that form atomic H and atomic Li
and the
subsequent hydrino reaction. Further hydrides and sources of catalysts can be
used
in place of Li and R-Ni in this procedure. In a further embodiment, the R-N is
hydrided to a great extent in a separate preparation step using elevated
temperature
and high-pressure hydrogen or by using electrolysis. The electrolysis may be
in
basic aqueous solution. The base may be a hydroxide. The counter electrode may
be nickel. In this case, R-Ni can provide atomic H for a long duration with
the
appropriate temperature, pressure, and temperature ramp rate.
LiH has a high melting point of 688 C which may be above that which sinters
the dissociator or causes the dissociator metal to form an alloy with the
catalyst
metal. For example, an alloy of LiNi may form at temperatures in excess of
about
550 C in the case that the dissociator is R-Ni and the catalyst is Li. Thus,
in another
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
88
embodiment, LiH is converted to LiNH2 that can be removed at its lower melting
point such that the reaction mixture can be regenerated. The reaction to form
lithium
amide from lithium hydride and ammonia is given by
LiH + NH3 -4 LiNH2 + H2 (52)
Then, molten LiNH2 can be recovered at the melting point of 380 C. LiNH2 may
be
converted to Li by decomposition.
In an erribodiment comprising the recovery of molten LiNH2, gas pressure is
applied to the mixture comprising LiNH2 to increase the rate of its separation
from
solid components. A screen separator or semi-permeable membrane may retain the
solid components. The gas may be an inert gas such as a noble gas or a
decomposition product such as nitrogen to limit the decomposition of LiNH2.
Molten
Li can be separated using gas pressure as well. To clean any residue from a
dissociator, gas flow can be used. An inert gas such as a noble gas is
preferable. In
the case that residual Li adheres to the dissociator such as R-Ni, the residue
can be
removed by washing with a basic solution such as a basic aqueous solution
which
may also regenerate the R-Ni. Alternatively, the Li may be hydrided and the
solids
of LiH and R-Ni and any additional solid compounds present may be separated
mechanically by methods such as sieving. In another embodiment, the
dissociator
such as R-Ni and the other reactants may be physically separated but
maintained in
close proximity to permit diffusion of atomic hydrogen to the balance of
reactant
mixture. The balance of reaction mixture and dissociator may be placed in open
juxtaposed boats, for example. In other embodiments, the reactor further
comprises
multiple compartments independently containing the dissociator and balance of
the
reaction mixture. The separator of each compartment allows for atomic hydrogen
formed in a dissociator compartment to flow to the balance-of-reaction-mixture
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
89
compartment whiie maintaining the chemical separation. The separator may be a
metallic screen or semipermeable, inert membrane which may be metallic. The
contents may be mechanically mixed during the operation of the reactor. The
separated balance of the reaction mixture and its products can be removed and
reprocessed outside of the reaction vessel and returned independently of the
dissociator, or either may be independentiy reprocessed within the reactor.
Other embodiments of systems to generate atomic catalyst Li and atomic H
involve Li, ammonia, and LiH. Atomic Li catalyst and atomic H can be generated
by
reaction of LiZ and NH3:
Li2 + NH3 to LiNH2 + Li + H (53)
LiNH2 is a source of NH3 by the reaction:
2LiNHZ to Li2NH + NH3 (54)
In a preferred embodiment, the Li is dispersed on a support having a large
surface
area to react with ammonia. Ammonia can also react with LiH to generate LiNH2:
LiH + NH3 to LiNHz + H2 (55)
And, H2 can react with Li2NH to regenerate LiNH2:
H2+ Li2NH to LiNH2 + LiH (56)
In another embodiment, the reactants comprise a mixture of LiNH2 and a
dissociator. The reaction to form atomic lithium is:
LiNH2 + H to Li + NH3 (57)
The Li can then react with additional H to form hydrino.
Other embodiments of systems to generate atomic catalyst Li and atomic H
involve Li and LiBHa or NH4X (X is an anion such as halide). Atomic Li
catalyst and
atomic H can be generated by reaction of LiZ and LiBH4:
Liz + LiBH4 to LiBH3 + Li + LiH (58)
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
NH4X can generate LiNH2 and H2
Li2 + NH4X to LiX + LiNH2 + H2 (59)
Then, atomic Li can be generated according to the reaction of Eqs. (32) and
(37). In another embodiment, the reaction mechanism for the Li/N system to
form
5 hydrino reactants of atomic Li and H is
NH4X + Li-Li to Li + H + NH3 + LiX (60)
where X is a counterion, preferably a halide.
Atomic Li catalyst can be generated by reaction of Li2NH or Li3N with atomic
H formed by the dissociation of H2:
10 Li2NH + H to LiNH2 + Li (61)
Li3N + H to Li2NH + Li (62)
In a further embodiment, the reaction mixture corriprises nitrides of metals
in
addition to Li such as those of Mg Ca Sr Ba Zn and Th. The reaction mixture
may
comprise metals that exchange with Li or form mixed-metal compounds with Li.
The
15 metals may be from the group of alkali, alkaline earth, and transition
metals. The
compounds may further comprise N such as amides, imides, and nitrides.
In an embodiment, the catalyst Li is generated chemically by an anion
exchange reaction such as a halide (X) exchange reaction. For example, at
least
one of Li metal and Li-Li molecules are reacted with a halide compound to form
20 atomic Li and LiX. Alternatively, LiX is reacted with a metal M to form
atomic Li and
MX. In an embodiment, lithium metal is reacted with a lanthanide halide to
form Li
and the LiX where X is halide. An example is the reaction of CeBr3 with Li2 to
form Li
and LiBr. In other embodiments, K, Cs, and Na replace Li wherein the catalyst
is
atomic K, atomic Cs, and molecular NaH.
25 In another embodiment, the reaction mixture further comprises the reactants
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
91
and products of the Haber process [43]. The products may be NH, x=0,1,2,3,4.
These products may react with Li or compounds comprising Li to form atomic Li
and
atomic H. For example, Li-Li may react with NHX to form Li and possibly H:
Li-Li + NH3 to Li + LiNH2 + H (63)
Li-Li + NH2 to LiNH2 + Li (64)
Li-Li + NH2 to Li2NH + H (65)
In other embodiments, K, Cs, and Na replace Li wherein the catalyst is atomic
K,
atomic Cs, and molecular NaH.
A mixture of compounds may be used which melts at a lower temperature
than that of one or more of compounds individually. Preferably, a eutectic
mixture
may form that is a molten salt that mixes the reactants such as Li and LiNH2.
The chemistry of the reaction mixture can change very substantially based on
the physical state of the reactants and the presence or absence of a solvent
or
added solute or alloy species. Objectives of the present invention for
changing the
physical state are to control the rate of reaction and to alter the
thermodynamics to
achieve a sustainable lower-energy hydrogen reaction with the addition of H
from a
source of H. For the Li/N alloy system comprising reactants such as Li and
LiNH2,
alkali metals, alkaline earth metals, and their mixtures may serve as the
solvent. For
example, excess Li can serve as a molten solvent for LiNH2 to comprise
solvated Li
and LiNH2 reactants that will have different kinetics and thermodynamics of
reaction
relative to those of the solid-state mixture. The former effect, control of
the kinetics
of the lower-energy hydrogen reaction, can be adjusted by controlling the
properties
of the solute and solvent such as temperature, concentration, and molar
ratios.
Following the reaction to generate atomic catalyst and atomic hydrogen, the
latter
effect can be used to regenerate the initial reactants. This is a route when
the
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
92
products cannot be directly regenerated by hydrogenation.
One embodiment where the regeneration of the reactants is faciiitated by a
solvent or added solute or alloy species involves lithium metal wherein the
hydriding
of Li is not to completion so that Li remains a solvent and a reactant. In Li
solvent,
the following regeneration reaction may occur with the addition of H from a
source to
form LiH:
LiH + Li2NH to 2Li + LiNH2 (66)
For the Li/N alloy system comprising reactants such as Li and LiNH2, alkali
metals, alkaline earth metals, and their mixtures may serve as the solvent. In
an
embodiment, the solvent is selected such that it can reduce LiH to Li and form
an
unstable solvent hydride with the release of H. Preferably, the solvent may be
one
or more of the group of Li (excess), Na, K, Rb, Cs, and Ba that have the
ability to
reduce Li+ and a corresponding hydride having a low thermal stability. In a
case that
the melting point of the solvent is higher than desired such as in the case of
Ba with
a high melting point of 727 C, the solvent can be mixed with other solvents
such as
metals to from a solvent with a lower melting point such as one comprising a
eutectic
mixture. In an embodiment, one or more alkaline earth metals can be mixed with
one or more alkali metals to lower the melting point, add the capability to
reduce Li+,
and decrease the stability of the corresponding solvent hydride.
Another embodiment where the regeneration of the reactants is facilitated by
a solvent or added solute or alloy species involves potassium metal. Potassium
metal in a mixture of LiH and LiNHz may reduce LiH to Li and form KH. Since KH
is
thermally unstable at intermediate temperatures such as 300 C, it may
facilitate the
further hydrogenation of Li2NH to Li and LiNH2.
Thus, K may catalyze the reaction given by Eq. (66). The reaction steps are
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
93
LiH + K to Li + KH (67)
KH + Li2NH to K + Li + LiNHZ (68)
wherein H is added at the rate at which it is consumed by lower-energy
hydrogen
production. Alternatively, K catalytically generates Li and H from LiH wherein
LiNH2
is formed directly from hydrogenation of Li2NH. The reactions steps are
Li2NH + 2H to LiH + LiNH2 (69)
LiH + K to KH + Li (70)
KH to K + H(g) (71)
In addition to the favorable condition of the instability of the hydride (KH),
the amide
(KNH2) is also unstable so that the exchange of lithium amide with potassium
amide
is not thermodynamically favorable. In addition to K, Na is a preferred metal
solvent
since it can reduce LiH and has a lower vapor pressure. Other examples of
suitable
metal solvents are Rb, Cs, Mg. Ca, Sr, Ba, and Sn. The solvent may comprise a
mixture of metals such as a mixture of two or more alkaline or alkaline earth
metals.
Preferable solvents are Li (excess) and Na above 380 C since Li is miscible
in Na
above this temperature.
In another erribodiment, an alkali or alkaline earth metal serves as a
regeneration catalyst according to Eqs. (70-71). In an embodiment, LiNH2 is
first
removed from the LiH/LiNH2 mixture by melting the LiNH2= Then, the metal M may
be added to catalyze the LiH to Li conversion. M can be selectively removed by
distillation. Na, K, Rb, and Cs form hydrides that decompose at relatively low
temperatures and form amides that thermally decompose; thus, in another
embodiment, at least one can serve as a reactant for the catalytic conversion
of LiH
to Li and H according to the corresponding reaction for K given by Eqs. (67-
71). In
addition, some alkaline earths such as Sr can form very stable hydrides which
can
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
94
serve to convert LiH to Li by reaction of LiH and an alkaline earth metal to
form the
stable alkaline earth hydride. By operating at an elevated temperature,
hydrogen
may be supplied from the alkaline earth hydride via decomposition with the
lithium
inventory being primarily as Li. The reaction mixture may comprise Li, LiNH2,
X, and
a dissociator wherein X may be a lithium compound such as LiH, Li2NH, Li3N
with a
small amount of an alkaiine earth metal that forms a stable hydride to
generate Li
from LiH. The source of hydrogen may be H2 gas. The operating temperature may
be sufficient such that H is available.
In an embodiment, LiNO3 can serve to generate the LiNH2 source of Li and
H in a set of coupled reactions. Consider an embodiment of the catalysis
reaction
mixture comprising Li, LiNHZ , and LiNO3 . The reaction of Li and LiNH2 to
Li3N
and release H2 is
LiNHz + 2Li -4 H2 + Li3N (72)
The balanced Hz reduction reaction of the released H2 (Eq. (72)) with LiNO3 to
form water and lithium amide is
4H 2 + LiNO3 -~ LiNHz + 3Hz0 (73)
Then, reaction Eq. (72) can proceed with the generated LiNH 2 and the balance
of
Li, and the coupled reactions given by Eqs. (72) and (73) can occur until the
Li is
completely consumed. The overall reaction is given by
LiNO3 + 8Li + 3LiNHz -> +3H20 + 4Li3N (74)
The water may be dynamically removed by methods such as condensation or
reacted with a getter to prevent its reaction with species such as Li, LiNH2,
Li2NH,
and Li3N.
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
Exemplarv Regeneration of Li Catalyst Reactants
The present invention further comprises methods and systems to generate, or
regenerate the reaction mixture to form states given by Eq. (1) from any side
5 products that form during said reaction. For example, in an embodiment of
the
energy reactor, the catalysis reaction mixture such as Li, LiNHZ 1 and LiNO3
is
regenerated from any side products such as LiOH and Li20 by methods known to
those skilled in the Art such as given in Cotton and Wilkinson [43].
Components of
the reaction mixture including side products may be liquid or solids. The
mixture is
10 heated or cooled to a desired temperature, and the products are separated
physically by means known by those skilled in the Art. In an embodiment, LiOH
and Liz0 are solid, Li, LiNH2, and LiNO3 are liquid, and the solid components
are
separated from the liquid ones. The LiOH and Li20 may be converted to lithium
metal by reduction with H2 at high temperature or by electrolysis of the
molten
15 compounds or a mixture containing them. The electrolysis cell may comprise
a
eutectic molten salt comprising at least one of LiOH, Li20, LiCI, KCl, CaC12
and
NaCI. The electrolysis cell is comprised of a material resistant to attack by
Li such
as a BeO or BN vessel. The Li product may be purified by distillation. LiNH2
is
formed by means known in the Art such as reaction of Li with nitrogen followed
by
20 hydrogen reduction. Alternatively, LiNH2 can be formed directly by reaction
of Li
with NH3.
In the case that the initial reaction mixture comprises at least one of Li,
LiNH2,
and LiNO3, Li metal may be regenerated by methods such as electrolysis, LiNO3
can
be generated from Li metal. One key step that eliminates the difficult
nitrogen
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
96
fixation step is the reaction of Li metal with N2 to form Li3N even at room
temperature. Li3N can be reacted with H2 to form Li2NH and LiNH2. Li3N can be
reacted with an oxygen source to form LiNO3. In an embodiment, Li3N is used in
the
synthesis of lithium nitrate (LiNO3) involving reactants or intermediates of
at least
one or more of lithium (Li), lithium nitride (Li3N), oxygen (02), an oxygen
source,
lithium imide (Li2NH), and lithium amide (LiNH2).
In an embodiments, the oxidation reactions are
LiNHz + 20Z -4 LiNO3 + H2O (75)
Li2NH + 20Z -4 LiNO3 + LiOH (76)
Li3N + 202 -> LiNO3 + Li2O (77)
Lithium nitrate can be regenerated from LiZ0 and LiOH using at least one of
NOZ , NO, and 02 by the following reactions
3LizO + 6N02 + 3 / 202 --~ 6LiNO3 (78)
Liz0 + 3NO2 -~ 2LiNO3 + NO (79)
NO + 1/ 2O2 ~ NOz (80)
LiOH + NOz + NO -4 2LiNOz + H20 (industrial process) (81)
2LiOH + 2N0z -4 LiNO3 + LiNOz + HzO (82)
Lithium oxide can be converted to lithium hydroxide by reaction with steam:
Liz0 + H20 -> 2LiOH (83)
In an embodiment, Liz0 is converted to LiOH followed by reaction with NOz and
NO according to Eq. (81).
Both lithium oxide and lithium hydroxide can be converted to lithium nitrate
by
treatment with nitric acid followed by drying:
Li2O + 2HNO3 -) 2LiNO3 + HZO (84)
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
97
LiOH + HNO3 -> LiNO3 + H20 (85)
LiNO3 can be made by treatment of lithium oxide or lithium hydroxide with
nitric acid. Nitric acid, in tern, can be generated by known industrial
methods such
as by the Haber process followed by the Ostwald process and then by hydration
and
oxidation of NO as given in Cotton and Wilkinson [43]. In one embodiment, the
exemplary sequence of steps are:
N "j )NH - ' )
NO 'HNO (86)
Z Hnber 3 Osnvald 3
process process
LiOH + HNO3 -> LiNO3 + H20 (87)
Specifically, the Haber process may be used to produce NH3 from N2 and H2 at
elevated temperature and pressure using a catalyst such as a -iron containing
some
oxide. The ammonia may be used to form LiNH2 from Li. The Ostwald process
may be used to oxidize the ammonia to NO at a catalyst such as a hot platinum
or
platinum-rhodium catalyst. The NO may be further reacted with oxygen and water
to
form nitric acid which can be reacted with lithium oxide or lithium hydroxide
to form
lithium nitrate. The crystalline lithium nitrate reactant is then obtained by
drying. In
another embodiment, NO and NO2 are reacted directly with the one or more of
lithium oxide and lithium hydroxide to form lithium nitrate. The regenerated
Li,
LiNH2 , and LiNO3 are then returned to the reactor in desired molar ratios.
In further exemplary regeneration reactions, an embodiment of the reactor
comprises the reactants of Li, LINH2, and LiCo02. LiOH, Li20, and Co and its
lower
oxides are the side products. The reactants can be regenerated by electrolysis
of
LiOH and Li20 to Li. LiNH2 can be regenerated by reaction of Li with NH3 or N2
and
then H2. The Co02 and its lower oxides can be regenerated by reaction with
oxygen. The LiCoO2 can be formed by reaction of Li with Co02. Li, LiNH2, and
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
98
LiCoO2 are then returned to the cell in a batch or continuous regeneration
process.
In the case that Li103 or Li104 is a reagent of the mixture, 103 and or 104
may be
regenerated by reaction of iodine or iodide ion with base and may further
undergo
electrolysis to the desired anion which may be precipitated out as Li103 or
Li104,
dried, and dehydrated.
NaH Molecular Catalyst
In a further embodiment, a compound comprising hydrogen such as MH
where H is hydrogen and M is another element serves as a source of hydrogen
and
a source of catalyst. In an embodiment, a catalytic system is provided by the
breakage of the M-H bond plus the ionization of t electrons from an atom M
each to
a continuum energy level such that the sum of the bond energy and ionization
energies of the t electrons is approximately one of m= 27.2 eV and m- 222 eV
where m is an integer.
One such catalytic system involves sodium. The bond energy of NaH is
1.9245 eV [44]. The first and second ionization energies of Na are 5.13908 eV
and
47.2864 eV, respectively ['']. Based on these energies NaH molecule can serve
as
a catalyst and H source since the bond energy of NaH plus the double
ionization
(t = 2) of Na to Naz+, is 54.35 eV (2X27.2 eV) which is equivalent to m = 2 in
Eq.
(2). The catalyst reactions are given by
54.35 eV + N a H -4 Na2+ + 2e + H ~ (3) ~ + [(3)2 -121 -13.6 eV (88)
Naz+ + 2e + H---) NaH + 54.35 eV (89)
And, the overall reaction is
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
99
H-> HL(3) I+[(3)2 -1Z] = 13.6 eV (90)
As given in Chp. 5 of Ref [30], and Ref. [20], hydrogen atoms
H(1 / p) p=1,2, 3,...137 can undergo further transitions to lower-energy
states given
by Eq. (1) wherein the transition of one atom is catalyzed by a second that
resonantly and nonradiatively accepts m- 27.2 eV with a concomitant opposite
change in its potential energy. The overall general equation for the
transition of
H(1 / p) to H(1 /(p + m)) induced by a resonance transfer of m= 27.2 eV to H(1
! p')
is represented by
H(1/p')+H(1/p)-4 H++e +H(1/(p+m))+[ 2pm+m2 -pi2 1=13.6eV
(91)
In the case of high hydrogen concentrations, the transition of H(1 / 3) ( p=
3) to
H(114) ( p+ m= 4) with H as the catalyst ( p=1; m=1) can be fast:
H(1/3)--L-4H(1/4)+81.6 eV (92)
Due to the stable binding of H- (1 / 4) in halides and its stability to
ionization relative
to other reaction species, it and the corresponding molecule formed by the
reactions
2H(1/4)---) H2 (1/4) and H-(1/4)+H+->Hz(1/4) are favored products of the
catalysis of hydrogen.
The NaH catalyst reaction may be concerted since the sum of the bond
energy of NaH, the double ionization (t = 2) of Na to Na2, , and the potential
energy
of H is 81.56 eV ( 3= 27.2 eV) which is equivalent to m = 3 in Eq. (2). The
catalyst
reactions are given by
81.56 eV+NaH+H-4 Na2++2e-+Hfns,+e-+HL~4)J+[(4)z -1Z]=13.6 eV
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
100
(93)
Na2l +2e-+H+H~s +e--~NaH+H+81.56 eV (94)
And, the overall reaction is
H -> H [~4) ] + [(4)2 -12] - 13.6 eV (95)
where H fas, is a fast hydrogen atom having at least 13.6 eV of kinetic energy
In an embodiment, the reaction mixture comprises at least one of a source of
NaH molecules and hydrogen. The NaH molecules may serve as the catalyst to
form H states given by Eq. (1). A source of NaH molecules may comprise at
least
one of Na metal, a source of hydrogen, preferably atomic hydrogen, and NaH(s).
The source of hydrogen may be at least one of H2 gas and a dissociator and a
hydride. Preferably, the dissociator and hydride may be R-Ni. Preferably, the
dissociator may also be Pt/Ti, Pt/Al2O3, and Pd/AI203 powder. Solid NaH may be
a
source of at least one of NaH molecules, H atoms, and Na atoms.
In a preferred embodiment, one of atomic sodium and molecular NaH is
provided by a reaction between a metallic, ionic, or molecular form of Na and
at least
one other compound or element. The source of Na or NaH may be at least one of
metallic Na, an inorganic compound comprising Na such as NaOH, and other
suitable Na compounds such as NaNH2, Na2CO3, and Na20 which are given in the
CRC [41], NaX (X is a halide), and NaH(s). The other element may be H, a
displacing agent, or a reducing agent. The reaction mixture may comprise at
least
one of (1) a source of sodium such as at least one of Na(m), NaH, NaNH2,
Na2CO3,
Na20, NaOH, NaOH doped-R-Ni, NaX (X is a halide), and NaX doped R-Ni, (2) a
source of hydrogen such as H2 gas and a dissociator and a hydride, (3) a
displacing
agent such as an alkali or alkaline earth metal, preferably Li, and (4) a
reducing
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
101
agent such as at least one of a metal such as an alkaline metal, alkaline
earth metal,
a lanthanide, a transition metal such as Ti, aluminum, B, a metal alloy such
as AIHg,
NaPb, NaAI, LiAI, and a source of a metal alone or in combination with
reducing
agent such as an alkaline earth halide, a transition metal halide, a
lanthanide halide,
and aluminum halide. Preferably, the alkali metal reductant is Na. Other
suitable
reductants comprise metal hydrides such as LiBH4, NaBH4, LiAIH4, or NaAIH4.
Preferably, the reducing agent reacts with NaOH to form a NaH molecules and a
Na
product such as Na, NaH(s), and Na20. The source of NaH may be R-Ni comprising
NaOH and a reactant such as a reductant to form NaH catalyst such as an alkali
or
alkaline earth metal or the Al intermetallic of R-Ni. Further exemplary
reagents are
an alkaline or alkaline earth metal and an oxidant such as AIX3, MgX2, LaX3,
CeX3,
and TiX, where X is a halide, preferably Br or I. Additionally, the reaction
mixture
may comprise another compound comprising a getter or a dispersant such as at
least one of Na2CO3, Na3SO4, and Na3PO4 that may be doped into the dissociator
such as R-Ni. The reaction mixture may further comprise a support wherein the
support may be doped with at least one reactant of the mixture. The support
may
have preferably a large surface area that favors the production of NaH
catalyst from
the reaction mixture. The support may comprise at least one of the group of R-
Ni,
Al, Sn, A1203 such as gamma, beta, or alpha alumina, sodium aluminate
(according
to Cotton [45] beta-aluminas have other ions present such as Na+ and possess
the
idealized composition Na2O = 11A1203 ), lanthanide oxides such as M203
(preferably
M= La, Sm, Dy, Pr, Tb, Gd, and Er), Si, silica, silicates, zeolites,
lanthanides,
transition metals, metal alloys such as alkali and alkali earth alloys with
Na, rare
earth metals, Si02-AI2O3 or Si02 supported Ni, and other supported metals such
as
at least one of alumina supported platinum, palladium, or ruthenium. The
support
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
102
may have a high surface area and comprise a high-surface-area (HSA) materials
such as R-Ni, zeolites, silicates, aluminates, aluminas, alumina
nanoparticies,
porous A1203, Pt, Ru, or Pd/A1203, carbon, Pt or Pd/C, inorganic compounds
such as
Na2CO3, silica and zeolite materials, preferably Y zeolite powder. In an
embodiment,
the support such as AI203 (and the A1203 support of the dissociator if
present) reacts
with the reductant such as a lanthanide to form a surface-modified support. In
an
embodiment, the surface Al exchanges with the lanthanide to form a lanthanide-
substituted support. This support may be doped with a source of NaH moiecuies
such as NaOH and reacted with a reductant such as a lanthanide. The subsequent
reaction of the lanthanide-substituted support with the lanthanide will not
significantly
change it, and the doped NaOH on the surface can be reduced to NaH catalyst by
reaction with the reductant lanthanide.
In an embodiment, wherein the reaction mixture comprises a source of NaH
catalyst, the source of NaH may be an alloy of Na and a source of hydrogen.
The
alloy may comprise at least one of those known in the Art such as an alloy of
sodium
metal and one or more other alkaline or alkaline earth metals, transition
metals, Al,
Sn, Bi, Ag, In, Pb, Hg, Si, Zr, B, Pt, Pd, or other metals and the H source
may be H2
or a hydride.
The reagents such as the source of NaH molecules, the source of sodium, the
source of NaH, the source of hydrogen, the displacing agent, and the reducing
agent
are in any desired molar ratio. Each is in a molar ratio of greater than 0 and
less
than 100%. Preferably, the molar ratios are similar.
A preferred embodiment comprises the reaction mixture of NaH and Pd on
A1203 powder wherein the reaction mixture may be regenerated by addition of
H2.
In an embodiment, Na atoms are vapor deposited on a surface. The surface
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
103
may support or be a source of H atoms to form NaH molecules. The surface may
comprise at least one of a hydride and hydrogen dissociator such as Pt, Ru, or
Pd/AI203 which may be hydrided. Preferably, the surface area is large. The
vapor
deposition may be from a reservoir containing a source of Na atoms. The Na
source
may be controlled via heating. One source that provides Na atoms when heated
is
Na metal. The surface may be maintained at a low temperature such as room
temperature during the vapor deposition. The Na-coated surface may be heated
to
cause the reaction of Na and H to form NaH and may further cause the NaH
molecules to react to form H states given by Eq. (1). Other thin-film
deposition
techniques that are well known in the ART comprise further embodiments of the
Invention. Such embodiments comprise physical spray, electro-spray, aerosol,
electro-arching, Knudsen cell controlled release, dispenser-cathode injection,
plasma-deposition, sputtering, and further coating methods and systems such as
melting a fine dispersion of Na, electroplating Na, and chemical deposition of
Na.
Na metal may be dispersed on a high-surface area material, preferably Na2CO3,
carbon, silica, alumina, R-Ni, and Pt, Ru, or Pd/AIz03, to increase the
activity to form
NaH when reacted with another reagent such as H or a source of H. Other
dispersion materials are known in the Art such as those given in Cotton et al.
[46].
In an embodiment, at least one reactant comprising the reductant or source of
NaH such as Na and NaOH undergoes aerosolization to create a corresponding
reactant vapor to react to form NaH catalyst. Na and NaOH may react in the
cell to
form NaH catalyst wherein at least one species undergoes aerosolization. The
aerosolized species may be transported into the cell to react to form NaH
catalyst.
The means to carry the aerosolized species may be a carrier gas. The
aerosolization of the reactant may be achieved using a mechanical agitator and
a
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
104
carrier gas such as a noble gas to carry the reactant into the cell to form
NaH
catalyst. In an embodiment, Na which may serve as a source of NaH and a
reductant is aerosolized by becoming charged and electrically dispensed. The
reactants such as at least one of Na and NaOH may be aerosolized mechanically
in
a carrier gas or they may undergo ultrasonic aerosolization. The reactant may
be
forced through an orifice to form a vapor. Alternatively, the reactant may be
heated
locally to very high temperature to be vaporized or sublimed to form a vapor.
The
reactants may further comprise a source of hydrogen. The hydrogen may react
with
Na to form NaH catalyst. The Na may be in the form of a vapor. The cell may
comprise a dissociator to from atomic hydrogen from H2. Other means of
achieving
aerosolization that are known to those skilled in the Art are part of the
Invention.
In an embodiment, the reaction mixture comprises at least one species of the
group comprising Na or a source of Na, NaH or a source of NaH, a metal hydride
or
source of a metal hydride, a reactant or source of a reactant to form a metal
hydride,
a hydrogen dissociator, and a source of hydrogen. The reaction mixture may
further
comprise a support. A reactant to form a metal hydride may comprise a
lanthanide,
preferably La or Gd. In an embodiment, La may reversibly react with NaH to
form
LaHn (n=1,2,3). In an embodiment, the hydride exchange reaction forms NaH
catalyst. The reversible general reaction may be given by
NaH+Mk-=~ Na+MH (96)
The reaction given by Eq. (96) applies to other MH -type catalysts given in
TABLE 2.
The reaction may proceed with the formation of hydrogen that may be
dissociated to
form atomic hydrogen that reacts with Na to form NaH catalyst. The dissociator
is
preferably at least one of Pt, Pd, or Ru/AI2O3 powder, Pt/Ti, and R-Ni.
Preferentially,
the dissociator support such as A1203 comprises at least surface La
substitution for
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
105
Al or comprises Pt, Pd, or Ru/M203 powder wherein M is a lanthanide. The
dissociator may be separated from the rest of the reaction mixture wherein the
separator passes atomic H.
A preferred embodiment comprises the reaction mixture of NaH, La, and Pd
on AI203 powder wherein the reaction mixture may be regenerated in an
embodiment, by adding H2, separating NaH and lanthanum hydride by sieving,
heating lanthanum hydride to form La, and mixing La and NaH. Alternatively,
the
regeneration involves the steps of separating Na and lanthanum hydride by
melting
Na and removing the liquid, heating lanthanum hydride to form La, hydriding Na
to
NaH, and mixing La and NaH. The mixing may be by ball milling.
In an embodiment, a high-surface-area material such as R-Ni is doped with
NaX (X=F, Cl, Br, l). The doped R-Ni is reacted with a reagent that will
displace the
halide to form at least one of Na and NaH. In an embodiment, the reactant is
at
least an alkali or alkaline earth metal, preferably at least one of K, Rb, Cs.
In
another embodiment, the reactant is an alkaline or alkaline earth hydride,
preferably
at least one of KH, RbH, CsH, MgH2 and CaH2. The reactant may be both an
alkali
metal and an alkaline earth hydride. The reversible general reaction may be
given
by
NaX+MH~NaH+MX (97)
NaOH Catalyst Reactions to Form NaH Catalyst
The reaction of NaOH and Na to Na20 and NaH is
NaOH + 2Na ---~ NaZ0 + NaH (98)
The exothermic reaction can drive the formation of NaH(g). Thus, Na metal can
serve as a reductant to form catalyst NaH(g). Other examples of suitable
reductants
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
106
that have a similar highly exothermic reduction reaction with the NaH source
are
alkali metals, alkaline earth metals such as at least one of Mg and Ca, metal
hydrides such as LiBH4, NaBH4, LiAIH4, or NaAIH4, B, Al, transition metals
such as
Ti, lanthanides such as at least one of La, Sm, Dy, Pr, Tb, Gd, and Er,
preferably La,
Tb, and Sm. Preferably, the reaction mixture comprises a high-surface-area
material
(HSA material) having a dopant such as NaOH comprising a source of NaH
catalyst.
Preferably, conversion of the dopant on the material with a high surface area
to the
catalyst is achieved. The conversion may occur by a reduction reaction. The
reductant may be provided as a gas stream. Preferably, Na is flowed into the
reactor as a gas stream. In addition to the preferred reductant, Na, other
preferred
reductants are other alkali metals, Ti, a lanthanide, or Al. Preferably, the
reaction
mixture comprises NaOH doped into a HSA material preferably R-Ni wherein the
reductant is Na or the intermetallic Al. The reaction mixture may further
comprise a
source of H such as a hydride or H2 gas and a dissociator. Preferably the H
source
is hydrided R-Ni.
In an embodiment, the reaction temperature is maintained below that at which
the reductant such as a lanthanide forms an alloy with the source of catalyst
such as
R-Ni. In the case of lanthanum, preferabiy the reaction temperature does not
exceed 532 C which is the alloy temperature of Ni and La as shown by Gasser
and
Kefif [47]. Additionally, the reaction temperature is maintained below that at
which
the reaction with the A1203 of R-Ni occurs to a significant extent such as in
the range
of 100 C to 450 C.
In an embodiment, Na20 formed as a product of a reaction to generate NaH
catalyst such as that given by Eq. (98), is reacted with a source of hydrogen
to form
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
107
NaOH that can further serve as a source of NaH catalyst. In an embodiment, a
regenerative reaction of NaOH from Eq. (98) in the presence of atomic hydrogen
is
NaZO + H-~ NaOH + Na AH =-11.6 kJ / mole NaOH (99)
NaH -4Na + H(1 / 3) AH =-10,500 kJ / mole H (100)
and
NaH-~Na+H(1/4) AH=-19,700kJ/moleH (101)
Thus, a small amount of NaOH and Na with a source of atomic hydrogen or atomic
hydrogen serves as a catalytic source of the NaH catalyst, that in turn forms
a large
yield of hydrinos via multiple cycles of regenerative reactions such as those
given by
Eqs. (98-101). In an embodiment, from the reaction given by Eq. (102), AI(OH)3
can serve as a source of NaOH and NaH wherein with Na and H, the reactions
given by Eqs. (98-101) proceed to form hydrinos.
3Na+ Al (OH)3 -+ NaOH+ NaAIOZ + NaH + 1/ 2HZ (102)
In an embodiment, the Al of the intermetallic serves as the reductant to form
NaH
catalyst The balanced reaction is given by
3NaOH+2Al -~ A1203 +3NaH (103)
This exothermic reaction can drive the formation of NaH(g) to drive the very
exothermic reaction given by Eqs. (88-92) wherein the regeneration of NaH
occurs
from Na in the presence of atomic hydrogen.
Two preferred embodiments comprise the first reaction mixture of Na and R-
Ni comprising about 0.5 wt% NaOH wherein Na serves as the reductant and a
second reaction mixture of R-Ni comprising about 0.5 wt% NaOH wherein
intermetallic AI serves as the reductant. The reaction mixture may be
regenerated
by adding NaOH and NaH that may serve as an H source and a reductant.
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
108
In an embodiment, of the energy reactor, the source of NaH such as NaOH is
regenerated by addition of a source of hydrogen such as at least one of a
hydride
and hydrogen gas and a dissociator. The hydride and dissociator may be
hydrided
R-Ni. In another embodiment, the source of NaH such as NaOH-doped R-Ni is
regenerated by at least one of rehydriding, addition of NaH, and addition of
NaOH
wherein the addition may be by physical mixing. The mixing may be performed
mechanically by means such as by ball milling.
In an embodiment, the reaction mixture further comprises oxide-forming
reactants that react with NaOH or Na20 to form a very stable oxide and NaH.
Such
reactants comprises a cerium, magnesium, lanthanide, titanium, or aluminum or
their
compounds such as AIX3, MgX2, LaX3, CeX3, and TiXn where X is a halide,
preferably Br or I and a reducing compound such as an alkaii or alkaline earth
metal.
In an embodiment, the source of NaH catalyst comprises R-Ni comprising a
sodium
compound such as NaOH on its surface. Then, the reaction of NaOH with the
oxide-
forming reactants such as AIX3, MgX2, LaX3, CeX3, and TiXn, and alkali metal M
forms NaH, MX, and A1203, MgO, La203, Ce203, and Ti203, respectively.
In an embodiment, the reaction mixture comprises NaOH doped R-Ni and an
alkaline or alkaline earth metal added to form at least one of Na and NaH
molecules.
The Na may further react with H from a source such as H2 gas or a hydride such
as
R-Ni to form NaH catalyst. The subsequent catalysis reaction of NaH forms H
states
given by Eq. (1). The addition of an alkali or alkaline earth metal M may
reduce Na+
to Na by the reactions:
NaOH + M to MOH + Na (104)
2NaOH + M to M(OH)2 + 2Na (105)
M may also react with NaOH to form H as well as Na
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
109
2NaOH + M to Na20 + H2 + MO (106)
Na20 + M to M20 + 2Na (107)
Then, the catalyst NaH may be formed by the reaction
Na + H to NaH (108)
by reacting with H from reactions such as that given by Eq. (106) as well as
from R-
Ni and any added source of H. Na is a preferred reductant since it is a
further
source of NaH.
Hydrogen may be added to reduce NaOH and form NaH catalyst:
NaOH + H2 to NaH + H20 (109)
The H in R-Ni may reduce NaOH to Na metal, and water that may be removed by
pumping.
In an embodiment, the reaction mixture comprises one or more compounds that
react with a source of NaH to form NaH catalyst. The source may be NaOH. The
compounds may comprise at least one of a LiNH2, Li2NH, and Li3N. The reaction
mixture may further comprise a source of hydrogen such as H2. In embodiments,
the reaction of sodium hydroxide and lithium amide to form NaH and lithium
hydroxide is
NaOH + LiNHZ -4 LiOH + NaH + 1/ 2N2 + LiH (110)
The reaction of sodium hydroxide and lithium imide to form NaH and lithium
hydroxide is
NaOH+Li2NH--> Li20 +NaH+1/2N2+1/2H2 (111)
And, the reaction of sodium hydroxide and lithium nitride to form NaH and
lithium
oxide is
NaOH+Li3N--> Li20+NaH+1/2N2+Li (112)
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
110
Alkaline Earth Hydroxide Catalyst Reactions to Form NaH Catalyst
In an embodiment, a source of H is provided to a source of Na to form the
catalyst
NaH. The Na source may be the metal. The source of H may be a hydroxide. The
hydroxide may be at least one of alkali, alkaline earth hydroxide, a
transition metal
hydroxide, and AI(OH)3. In an embodiment, Na reacts with a hydroxide to form
the
corresponding oxide and NaH catalyst. In an embodiment wherein the hydroxide
is
Mg(OH)2, the product is MgO. In an embodiment wherein the hydroxide is
Ca(OH)2,
the product is CaO. Alkaline earth oxides may be reacted with water to
regenerate
the hydroxide as given in Cotton [48]. The hydroxide can be collected as a
precipitate by means such as filtration and centrifugation.
For example, in an embodiment, the reaction to form NaH catalyst and
regeneration cycle for Mg(OH)2, are given by the reactions:
3Na+ Mg(OH)z -4 2NaH+ MgO+ NazO (113)
MgO+H2O-~ Mg(OH)z (114)
In an embodiment, the reaction to form NaH catalyst and regeneration cycle
for Ca(OH)2 , are given by the reactions:
4Na+Ca(OH)2 -> 2NaH+CaO+Na2O (115)
CaO+Hz0-4 Ca(OH)z (116)
Na/N Alloy Reactions to Form NaH Catalyst
Sodium in the solid and liquid states is a metal, and the gas comprises
covalent Na2 molecules. In order to generate NaH catalyst, the reaction
mixture of
the solid fuel comprises Na/N alloy reactants. In an embodiment, the reaction
mixture, solid-fuel reactions, and regeneration reactions comprise those of
the Li/N
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
111
system wherein Na replaces Li and the catalyst is molecular NaH except that
the
solid fuel reaction generates molecular NaH rather than atomic Li and H. In an
embodiment, the reaction mixture comprises one or more compounds that react
with
a source of NaH to form NaH catalyst. The reaction mixture may comprise at
least
one of the group of Na, NaH, NaNH2, Na2NH, Na3N, NH3, a dissociator, a
hydrogen
source such as H2 gas or a hydride, a support, and a getter such as NaX (X is
a
halide). The dissociator is preferably Pt, Ru, or PdIA1203 powder. For high-
temperature operation, the dissociator may comprise Pt or Pd on a high surface
area
support suitably inert to Na. The dissociator may be Pt or Pd on carbon or
Pd/AI203.
The latter support may comprise a protective surface coating of a material
such as
NaAIO2. The reactants may be present in any wt%.
A preferred embodiment comprises the reaction mixture of Na or NaH,
NaNH2, and Pd on AI203 powder wherein the reaction mixture may be regenerated
by addition of H2.
In an embodiment, NaNH2 is added to the reaction mixture. NaNH2
generates NaH according to the reversible reactions
Na2 + NaNH2 --4 NaH + Na2NH (117)
and
2NaH + NaNH2 -4 NaH (g) + Na2NH + H2 (118)
In the hydrino reaction cycle, Na-Na and NaNH2 react to form NaH molecule
and Na2NH, and the NaH forms hydrino and Na. Thus, the reaction is reversible
according to the reactions:
Na2 NH + H2 -> NaNH2 + NaH (119)
and
NazNH + Na + H-4 NaNHz + Naz (120)
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
112
In an embodiment, NaH of Eq. (119) is molecular such that this reaction is
another to generate the catalyst.
The reaction of sodium amide and hydrogen to form ammonia and sodium hydride
is
H2 + NaNH 2 ---~ NH3 + NaH (121)
In an embodiment, this reaction is reversible. The reaction can be driven to
form
NaH by increasing the H2 concentration. Alternatively, the forward reaction
can be
driven via the formation of atomic H using a dissociator. The reaction is
given by
2H + NaNH2 -> NH3 + NaH (122)
The exothermic reaction can drive the formation of NaH(g).
In an embodiment, NaH catalyst is generated from a reaction of NaNH2 and
hydrogen, preferably atomic hydrogen as given in reaction Eqs. (121-122). The
ratios of reactants may be any desired amount. Preferably the ratios are about
stoichiometric to those of Eqs. (121-122). The reactions to form catalyst are
reversible with the addition of a source of H such as H2 gas or a hydride to
replace
that reacted to form hydrinos wherein the catalyst reactions are given by Eqs.
(88-
95), and sodium amide forms with additional NaH catalyst by the reaction of
ammonia with Na:
NH3 + Naz -~ NaNH2 + NaH (123)
In an embodiment, a HSA material is doped with NaNH2. The doped HSA
material is reacted with a reagent that will displace the amide group to form
at least
one of Na and NaH. In an embodiment, the reactant is an alkali or alkaline
earth
metal, preferably Li. In another embodiment, the reactant is an alkaline or
alkaline
earth hydride, preferably LiH. The reactant may be both an alkali metal and an
alkaline earth hydride. A source of H such as H2 gas may be further provided
in
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
113
addition to that provided by any other reagent of the reaction mixture such as
a
hydride, HSA material, and dispiacing reagent.
In an embodiment, sodium amide undergoes reaction with lithium to form lithium
amide, imide, or nitride and Na or NaH catalyst. The reaction of sodium amide
and
lithium to form lithium imide and NaH is
2Li + NaNH2 -+ LiZNH + NaH (124)
The reaction of sodium amide and lithium hydride to form lithium amide and NaH
is
LiH + NaNHz -+ LiNH2 + NaH (125)
The reaction of sodium amide, lithium, and hydrogen to form lithium amide and
NaH
is
Li+1/2HZ+NaNH2 -4 LiNHz+NaH (126)
In an embodiment, the reaction of the mixture forms Na, and the reactants
further
comprise a source of H that reacts with Na to form catalyst NaH by a reaction
such
as the following:
Li + NaNH2 to LiNH2 + Na (127)
and
Na + H to NaH (128)
LiH + NaNH2 to LiNH2 + NaH (129)
In an embodiment, the reactants comprise NaNH2, a reactant to displace the
amide
group of NaNH2 such as an alkali or alkaline earth metal, preferably Li, and
may
additionally comprise a source of H such as at least one of MH (M=Li, Na, K,
Rb, Cs,
Mg, Ca, Sr, and Ba), H2 and a hydrogen dissociator, and a hydride.
The reagents of the reaction mixture such as M, MH, NaH, NaNH2, HSA
material, hydride, and the dissociator are in any desired molar ratio. Each of
M, MH,
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
114
NaNH2,and the dissociator are in molar ratios of greater than 0 and less than
100%,
preferably the molar ratios are similar.
Other embodiments of systems to generate molecular catalyst NaH involve
Na and NaBH4 or NH4X (X is an anion such as halide). Molecular NaH catalyst
can
be generated by reaction of Na2 and NaBH4:
Na2 + NaBH4 to NaBH3 + Na + NaH (130)
NH4X can generate NaNH2 and H2
Na2 + NH4X to NaX + NaNH2 + H2 (131)
Then, NaH catalyst can be generated according to the reaction of Eqs. (117-
129). In another embodiment, the reaction mechanism for the Na/N system to
form
hydrino catalyst NaH is
NH4X + Na-Na to NaH + NH3 + NaX (132)
Preparation and Regeneration of NH Catalyst Reactants
In an embodiment NaH molecules or Na and hydrided R-Ni can be
regenerated by systems and methods after those disclosed for the Li-based
reactant
systems. In an embodiment, Na can be regenerated from solid NaH by evacuating
H2 released from NaH. The plateau temperature at about 1 Torr for NaH
decomposition is about 500 C. NaH can be decomposed at about I Torr and 500
C, below the alloy-formation and sintering temperatures of R-Ni. The molten Na
can be separated from R-Ni, the R-Ni may be rehydrided, and Na and hydrided R-
Ni
can be returned to another reaction cycle. In the case of vapor-deposited Na
on a
hydride surface, regeneration can be achieved by heating with pumping to
remove
Na, the hydride can be rehydrided by introducing H2, and Na atoms can be
redeposited onto the regenerated hydride after the cell is evacuated in an
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
115
embodiment.
In a preferred embodiment, the competing kinetics of the hydriding or
dehydriding of one reactant over another is exploited to achieve a reaction
mixture
comprising hydrided and non-hydrided compounds. For example, the formation of
NaH solid is thermodynamically favored over the formation of R-Ni hydride.
However, the rate of NaH formation at low temperature such as the range of
about
25 C-100 C is low; whereas, the formation of R-Ni hydride proceeds at a high
rate
in this temperature range at modest pressures such as the range of about 100
Torr
to 3000 Torr. Thus, the reaction mixture of Na and hydrided R-Ni can be
regenerated from NaH solid and R-Ni by pumping at about 400-500 C to
dehydride
NaH, cooling the vessel to about 25-100 C, adding hydrogen to preferentially
hydride R-Ni for a duration that achieves the desired selectivity, and then
removing
the excess hydrogen by evacuating the cell. While excess Na is present or is
added
to be in excess, the R-Ni can be used in repeated cycles by selectively
hydriding
alone. This can be achieved by adding hydrogen in the temperature and pressure
ranges that achieve the selective hydriding of R-Ni and then by removing the
excess
hydrogen before the vessel is heated to initiate the reactions that form
atomic H and
molecular NaH and the subsequent reaction to yield H states given by Eq. (1).
Alternatively, a reaction mixture comprising Na and a hydrogen source such as
R-IVi
may be hydrogenated to form the hydrides, and the NaH solid can be selectively
dehydrided by pumping at the temperature and pressure ranges and durations
which
achieve the selectivity based on differential kinetics.
In an embodiment having powder reactants such as a powder source of
catalyst and a reductant, the reductant powder is mixed with the catalyst-
source
powder. For example, NaOH-doped R-Ni that provides NaH catalyst is mixed with
a
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
116
metal or metal hydride powder such as a lanthanide or NaH, respectively. In an
embodiment of the reaction mixture having a solid material such as a
dissociator,
support, or HSA material that is doped or coated with at least one other
species of
the reaction mixture, the mixing may be achieved by ball milling or the method
of
incipient wetness. In an embodiment, the surface may be coated by immersing
the
surface into a solution of the species such as NaOH or NaX (X is a counter
anion
such as halide) followed by drying. Alternatively, NaOH may be incorporated
into
Ni/Al alloy or R-Ni by etching with concentrated NaOH (deoxygenated) using the
same procedure as used to etch R-Ni as is well known in the Art [49]. In an
embodiment, the HSA material such as R-Ni doped with a species such as NaOH is
reacted with a reductant such as Na to form NaH catalyst that reacts to form
hydrinos. Then, the excess reductant such as Na may be removed from the
products by evaporation, preferably, under vacuum at elevated temperature. The
reductant may be condensed to be recycled. In another embodiment, at least one
of
the reductant and a product species is removed by using a transporting medium
such as a gas or liquid such as a soivent, and the removed species is isolated
from
the transporting medium. The species can be isolated by means well known in
the
Art such as precipitation, filtration, or centrifugation. The species may be
recycled
directly or further reacted to a chemical form suitable for recycling. In
addition, the
NaOH may be regenerated by H reduction or by reaction with a water-vapor gas
stream. In the former case, excess Na may be removed by evaporation,
preferably,
under vacuum at elevated temperature. Altenatively, the reaction products can
be
removed by rinsing with a suitable solvent such as water, the HSA material may
be
dried, and the initial reactants may be added. Separately, the products may be
regenerated to the original reactants by methods known to those skilled in the
Art.
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
117
Or, a reaction product such as NaOH separated by rinsing R-Ni can be used in
the
process of etching R-Ni to regenerate it. In an embodiment comprising a
reactant
that reacts with the HSA material, the product such as an oxide may be treated
with
a solvent such as dilute acid to remove the product. The HSA material may then
be
re-doped and reused while the removed product may be regenerated by known
methods.
The reductant such as an alkali metal can be regenerated from the product
comprising a corresponding compound, preferably NaOH or Na20, using methods
and systems known to those skilled in the Art as given in Cotton [48]. One
method
comprises electrolysis in a mixture such as a eutectic mixture. In a further
embodiment, the reductant product may comprise at least some oxide such as a
lanthanide metal oxide (e.g. La203). The hydroxide or oxide may be dissolved
in a
weak acid such as hydrochloric acid to form the corresponding salt such as
NaCI or
LaCI3. The treatment with acid may be a gas phase reaction. The gases may be
streaming at low pressure. The salt may be treated with a product reductant
such as
an alkali or alkaline earth metal to form the original reductant. In an
embodiment,
the second reductant is an alkaline earth metal, preferably Ca wherein NaCI or
LaCl3
is reduced to Na or La metal. Methods known to those skilled in the Art are
given in
Cottorl [48] which is herein incorporated by reference in its entirety. The
additional
product of CaCi3 is recovered and recycled as well. In alternative embodiment,
the
oxide is reduced with H2 at high temperature.
In an embodiment wherein NaAIH4 is the reductant, the product comprises Na
and Al that need not be separated from the R-Ni product. The R-Ni is
regenerated
as a source of catalyst without separation. Regeneration may be by the
addition of
NaOH. The NaOH may partially etch Al of R-Ni [49] which is dried [50] for
reuse.
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
118
Alternatively, Na and Al are reacted insitu or separated from the reaction
product
mixture and reacted with H2 to form NaAIH4 directly as given by Cotton [51] or
by
reaction of the recovered NaH with Al to form NaAIH4.
R-Ni is a preferred HSA material having NaOH as a source of NaH catalyst.
In an embodiment, the Na content from the manufacturer is in the range of
about
0.01 mg to 100 mg per gram of R-Ni, preferably in the range of about 0.1 mg to
10
mg per gram of R-Ni, and most preferably in the range of about 1 mg to 10 rog
Na
per gram of R-Ni. The R-Ni or an alloy of Ni may further comprise promoters
such
as at least one of Zn, Mo, Fe, and Cr. The R-Ni or alloy may be at least one
of W. R.
Grace Davidson Raney 2400, Raney 2800, Raney 2813, Raney 3201, and Raney
4200, preferably 2400, or etched or Na-doped erribodiments of these materials.
The
NaOH content of the R-Ni may be increased by a factor in the range of about
1.01 to
1000 times. Solid NaOH may added by mixing by means such as ball milling, or
it
may be dissolved in a solution to achieve a desired concentration or pH. The
solution may be added to R-Ni and the water evaporated to achieve the doping.
The
doping may be in the range of about 0.1 g to 100 mg per gram of R-Ni,
preferably
in the range of about I g to 100 g per gram of R-Ni, and most preferably in
the
range of about 5 g to 50 g per gram of R-Ni. In an embodiment, 0.1 g of NaOH
is dissolved in 100 ml of distilled water and 10 ml of the NaOH solution is
added to
500g of non-decanted R-Ni from W. R. Grace Chemical Company such lot
#2800/05310 having an initial total content of Na of about 0.1 wt%. The
mixture is
then dried. The drying may be achieved by heating at 50 C under vacuum for 65
hours. In another embodiment, the doping may be achieved by ball milling NaOH
with the R-Ni such as about 1 to 10 mg of NaOH per gram of R-Ni.
The R-Ni may be dried dry according to the standard R-Ni drying procedure
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
119
[50]. The R-Ni may be decanted and dried in the temperature range of about 10-
500
C under vacuum, preferably, it is dried at 50 C. The duration may be in the
range
of about 1 hr to 200 hours, preferably, the duration is about 65 hours. In an
embodiment, the H content of the dried R-Ni is in the range of about 1 m1-100
ml H/g
R-Ni, preferably the H content of the dried R-Ni is in the range of about 10-
50 ml H/g
R-Ni (where ml gas are at STP). The drying temperature, time, vacuum pressure
and flow of gases, if any, such as He, Ar, or H2 during and after drying is
controlled
to achieve dryness and the desired H content.
In an embodiment of the R-Ni doped with a source of NaH catalyst such as
NaOH, the preparation of R-Ni from Ni/Al alloy comprises the step of etching
the
alloy with aqueous NaOH solution. The concentration of NaOH, etching times,
and
rinsing exchanges, may be varied to achieve the desired level of incorporation
of
NaOH. In an embodiment, the NaOH solution is oxygen free. The molarity is in
the
range of about 1 to 10 M, preferably in the range of about 5 to 8 M, and most
preferably about 7 M. In an embodiment, the alloy is reacted with the NaOH for
about 2 hours at about 50 C. The solution is then diluted with water such as
deionized water until Al(OH)3 precipitate forms. In that case, the amphoteric
reaction
of NaOH with AI(OH)3 to form water-soluble Na[AI(OH)4] is at least partially
prevented such that NaOH is incorporated into the R-Ni. The incorporation may
be
achieved by drying the R-Ni without decanting. The pH of the diluted solution
may
be in the range of 8 to 14, preferably in the range of 9 to 12, and most
preferably
about 10-11. Argon may be bubbled through the solution for about 12 hours, and
then the solution may be dried.
Following the reaction of the reductant and source of catalyst to form hydrino
(H with states given by Eq. (1)), the reductant and catalyst source are
regenerated.
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
120
In an embodiment, the reaction products are separated. The reductant product
may
be separated from the product of the source of catalyst. In an embodiment
wherein
at least one of the reductant and source of catalyst are powders, the products
are
separated mechanically based on at least one of particle size, shape, weight,
density, magnetism, or dielectric constant. Particles having a significant
difference in
size and shape can be mechanically separated using sieves. Particles with
large
differences in density can be separated by buoyancy differences. Particles
having
large differences in magnetic susceptibility can be separated magnetically.
Particles
with large differences in dielectric constant can be separated
electrostatically. In an
embodiment, the products are ground to reverse any sintering. The grinding may
be
with a ball mill.
Methods known by those skilled in the Art that can be applied to the
separations of the present Invention by application of routine
experimentation. In
general, mechanical separations can be divided into four groups:
sedimentation,
centrifugal separation, filtration, and sieving as described in Earle [52]
which is
incorporated herein in its entirety by reference. In a preferred embodiment,
the
separation of the particles is achieved by at least one of sieving and use of
classifiers. The size and shape of the particle may be selected in the
starting
materials to achieve the desired separation of the products.
In a further embodiment, the reductant is a powder or is converted to a
powder and mechanically separated from the other components of the product
reaction mixture such as a HSA material. In embodiments, Na, NaH, and a
lanthanide comprise at least one of the reductant and a source of the
reductant, and
a HSA material component is R-Ni. The reductant product may be separated from
the product mixture by converting any unreacted non-powder reductant metal to
the
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
121
hydride. The hydride may be formed by the addition of hydrogen. The metal
hydride
may be ground to form a powder. The powder may then be separated from the
other products such as that of the source of the catalyst based on a
difference in the
size of the particles. The separation may be by agitating the mixture over a
series of
sieves that are selective for certain size ranges to cause the separation.
Altematively, or in combination with sieving, the R-Ni particles are separated
from
the metal hydride or metal particles based on the large magnetic
susceptibility
difference between the particles. The reduced R-Ni product may be magnetic.
The
unreacted lanthanide metal and hydrided metal and any oxide such as La203 are
weakly paramagnetic and diamagnetic, respectively. The product mixture may be
agitated over a series of strong magnets alone or in combination with one or
more
sieves to cause the separation based on at least one of the stronger adherence
or
attraction of the R-Ni product particles to the magnet and a size difference
of the two
classes of particles. In an embodiment of the use of sieves and an applied
magnetic
field, the latter adds an additional force to that of gravity to draw the
smaller R-Ni
product particles through the sieve while the weakly paramagnetic or
diamagnetic
particles of the reductant product are retained on the sieve due to their
larger size.
The alkali metal may be recovered from the corresponding hydride by heating
and
optionally by appiying vacuum. The evolved hydrogen can be reacted with alkali
metal in another batch of a repetitive reaction-regeneration cycle. There may
be
more than one batch in the cycle at various stages. The hydride and any other
compound(s) may be separated, and then reacted to form the metal separately
from
the formation of the metal from the hydride.
In an embodiment, the reaction mixture is regenerated by vapor deposition
techniques, preferably in the case that the reactants are on the surface of a
HSA
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
122
material such as R-Ni. In further embodiments, having other coated desired
reactants comprising at least one of a source of NaH catalyst on a surface and
a
material that supports the formation of NaH catalyst such as a HSA material,
the
reactants are provided by reacting gas streams with the HSA material such as R-
Ni.
The deposited reactants may comprise at least one of the group of Na, NaH,
Na20,
NaOH, Al, Ni, NiO, NaAI(OH)4, fi -alumina, NaZO=nAl2O3 (n= integer from 1 to
1000,
preferably 11), Al(OH)3, and AI203 in alpha, beta, and gamma forms. Vapor-
deposited elements, compounds, intermediates, and species that are the desired
reactants or are converted into the desired reactants as well as the sequence
and
composition of the gas streams and the chemistry to form the reactants from
the gas
streams are well by those skilled in the Art of vapor deposition. For example,
alkali
metals can be directly vapor deposited and any metals with low vapor pressure
such
as Al can be vapor deposited from the gaseous halide or hydride. Furthermore,
oxide products such as Na20 may be reacted with a source of hydrogen to form
the
hydroxide such as NaOH. The source of hydrogen may comprise a water-vapor gas
stream to regenerate NaOH. Alternatively, the NaOH can be formed using H2 or a
source of H2. In addition, the hydriding of the HSA material such as R-Ni can
be
achieved by supplying hydrogen gas, and removing excess hydrogen by means
such as pumping. The NaOH may be regenerated stoichiometrically by precisely
controlling the total moles of reacted H from a source such as water vapor or
hydrogen gas. Any additional Na or NaH formed at this stage may be removed by
evaporation, and decomposition and evaporation, respectively. Alternative, an
oxide
or hydroxide product such as Na20 or excess NaOH can be removed. This can be
achieved by conversion to a halide such as Nal which may be removed by
distillation
or vaporization. The vaporization can be achieved with heating and by
maintaining a
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
123
vacuum at elevated temperature. The conversion to a halide may be achieved by
reaction with an acid such as HI. The treatment may be by a gas stream
comprising
the acid gas. In another embodiment, any excess NaOH is removed by
sublimation.
This occurs under vacuum in the temperature range of 350-400 C as given by
Cotton [53]. Any evaporation, distillation, transport, gas-stream process, or
related
processes of the reactants may further comprise a carrier gas. The carrier gas
may
be an inert gas such as a noble gas. Further steps may comprise mechanical
mixing
or separation. For example, NaOH and NaH can be also be deposited or removed
mechanically by methods such as ball milling and sieving, respectively.
In the case that the redundant is an element other than a desired first
element
such as Na, the other element may be replaced by a second such as Na using
methods known in the Art. A step may comprise evaporation of excess reductant.
The large surface-area material such as R-Ni may be etched. The etching may be
with a base, preferably NaOH. The etched product may be decanted with
substantially all of any solvent such as water removed mechanically such as by
decanting and possibly centrifugation. The etched R-Ni may be dried under
vacuum
and recycled.
Additional MH-Type Catalysts and Reactions
Another catalytic system of the type MH involves aluminum. The bond energy
of AIH is 2.98 eV [44]. The first and second ionization energies of Al are
5.985768
eV and 18.82855 eV, respectively [1]. Based on these energies AIH molecule can
serve as a catalyst and H source since the bond energy of AIH plus the double
ionization ( t= 2) of Al to Alz+ , is 27.79 eV (27.2 eV) which is equivalent
to m=1 in
Eq. (2). The catalyst reactions are given by
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
124
27.79 eV+AIH--> Al2++2e-+HL~2)J+[(2)Z-12]=13.6 eV (133)
Al2+ + 2e- + H---> AlH + 27.79 eV (134)
And, the overall reaction is
H -4 HL ) J + [(2)2 -12 ] - 13.6 eV (135)
In an embodiment, the reaction mixture comprises at least one of AIH
molecules and a source of AIH molecules. A source of AIH molecules may
comprise
Al metal and a source of hydrogen, preferably atomic hydrogen. The source of
hydrogen may be a hydride, preferably R-Ni. In another embodiment, the
catalyst
AIH is generated by the reaction of an oxide or hydroxide of Al with a
reductant. The
reductant comprises at least one of the NaOH reductants given previously. In
an
embodiment, a source of H is provided to a source of Al to form the catalyst
AIH.
The Al source may be the metal. The source of H may be a hydroxide. The
hydroxide may be at least one of alkali, alkaline earth hydroxide, a
transition metal
hydroxide, and AI(OH)3.
Raney nickel can be prepared by the following two reaction steps:
Ni + 3Al ---> NiA13 (or NizAl3) (136)
~
NiAl3 + 2NaOH + 6H20 CNiAlx (skeleton, porous Ni) (137)
~ +2Na[Al(OH),]+3Hz
Na[AI(OH)4] is readily dissolved in concentrated NaOH. It can be washed in de-
oxygenated water. The prepared Ni contains Al (-10 wt%, that may vary), is
porous,
and has a large surface area. It contains large amounts of H, both in the Ni
lattice
and in the form of Ni-AIHX (x=1,2,3).
R-Ni may be reacted with another element to cause the chemical release of
AIH molecules which then undergo catalysis according to reactions given by
Eqs.
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
125
(133-135). In an embodiment, the AIH release is caused by a reduction
reaction,
etching, or alloy formation. One such other element M is an alkali or alkaline
earth
metal which reacts with the Ni portion of R-Ni to cause the AIH, component to
release AIH molecules that subsequently under go catalysis. In an embodiment,
M
may react with Al hydroxides or oxides to form Al metal that may further react
with H
to form AIH. The reaction can be initiated by heating, and the rate may be
controlled
by controlling the temperature. M (alkali or alkaline earth metal) and R-Ni
are in any
desired molar ratio. Each of M and R-Ni are in molar ratios of greater than 0
and
less than 100%. Preferably the molar ratio of M and R-Ni are similar.
In an embodiment, Al atoms are vapor deposited on a surface. The surface
may support or be a source of H atoms to form AIH molecules. The surface may
comprise at least one of a hydride and hydrogen dissociator. The surface may
be R-
Ni which may be hydrided. The vapor deposition may be from a reservoir
containing
a source of Al atoms. The Al source may be controlled by heating. One source
that
provides Al atoms when heated is AI metal. The surface may be maintained at a
low
temperature such as room temperature during the vapor deposition. The Al-
coated
surface may be heated to cause the reaction of Al and H to form AIH and may
further
cause the AIH molecules to react to form H states given by Eq. (1). Other thin-
film
deposition techniques that are well known in the ART to form layers of at
least one of
Al and other elements such as metals comprise further embodiments of the
Invention. Such embodiments comprise physical spray, electro-spray, aerosol,
electro-arching, Knudsen cell controlled release, dispenser-cathode injection,
plasma-deposition, sputtering, and further coating methods and systems such as
melting a fine dispersion of Al, electroplating Al, and chemical deposition of
Al.
In an embodiment, the source of AIH comprises R-Ni and other Raney metals
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
126
or alloys of Al known in the Art such as R-Ni or an alloy comprising at least
one of Ni,
Cu, Si, Fe, Ru, Co, Pd, Pt, and other elements and compounds. The R-Ni or
alloy
may further comprise promoters such as at least one of Zn, Mo, Fe, and Cr. The
R-
Ni may be at least one of W. R. Grace Raney 2400, Raney 2800, Raney 2813,
Raney 3201, Raney 4200, or an etched or Na doped embodiment of these
materials.
In another embodiment of the AIH catalyst system, the source of catalyst
comprises
a Ni/Al alloy wherein the Al to Ni ratio is in the range of about 10-90%,
preferably
about 10-50%, and more preferably about 10-30%. The source of catalyst may
comprise palladium or platinum and further comprise Al as a Raney metal.
The source of AIH may further comprise AfH3. The AIH3 may be deposited on
or with Ni to form a NiAIH, alloy. The alloy may be activated by the addition
of a
metal such as an alkali or alkaline earth metal. In an embodiment the reaction
mixture comprises AIH3, R-Ni, and a metal such as an alkali metal. The metal
may
be supplied by vaporization from a reservoir or by gravity feed from a source
that
flows down on the R-Ni at an elevated temperature. In an embodiments, AIH
molecules or Al and hydrided R-Ni can be regenerated by systems and methods
after those disclosed for the other reactant systems.
Another catalytic system of the type MH involves chlorine. The bond energy
of HCI is 4.4703 eV [44]. The first, second, and third ionization energies of
CI are
12.96764 eV, 23.814 eV, and 39.61 eV, respectively [1]. Based on these
energies
HCI can serve as a catalyst and H source since the bond energy of HCI plus the
triple ionization ( t= 3) of Cl to Cl3+ , is 80.86 eV ( 3= 27.2 eV) which is
equivalent to
m = 3 in Eq. (2). The catalyst reactions are given by
80.86eV+HCl-~ Cl3++3e +HL~4)I+[(4)2-lZ] 13.6eV (138)
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
127
Cl3+ + 3e- + H--> HCl + 80.86 eV (139)
And, the overall reaction is
H -~ H[)] +[(4)2 -1Z]= 13.6 eV (140)
In an embodiment, the reaction mixture comprises HCI or a source of HCI. A
source may be NH4CI or a solid acid and a chloride such as an alkali or
alkaline
earth chloride. The solid acid may be at least one of MHSO4, MHCO3, NIH2PO4,
and
MHPO4 wherein M is a cation such as an alkali or alkaline earth cation. Other
such
solid acids are known to those skilled in the Art. In an embodiment, the
reactants
comprise HCI catalyst in an ionic lattice such as HCI in an alkali or alkaline
earth
halide, preferably a chloride. In an embodiment, the reaction mixture
comprises a
strong acid such as H2SO4 and an ionic compound such as NaCI. The reaction of
the acid with the ionic compound such as NaCI generates HCI in the crystalline
lattice to serve as a hydrino catalyst and H source.
In general, MH type hydrogen catalysts to produce hydrinos provided by the
breakage of the NI-H bond plus the ionization of t electrons from the atom M
each to
a continuum energy level such that the sum of the bond energy and ionization
energies of the t electrons is approximately m- 27.2 eV where m is an integer
are
given in TABLE 2. Each MH catalyst is given in the first column and the
corresponding M-H bond energy is given in column two. The atom M of the MH
species given in the first column is ionized to provide the net enthalpy of
reaction of
m- 27.2 eV with the addition of the bond energy in column two. The enthalpy of
the
catalyst is given in the eighth column where m is given in the ninth column.
The
eiectrons, that participate in ionization are given with the ionization
potential (also
called ionization energy or binding energy). For example, the bond energy of
NaH,
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
128
1.9245 eV [44], is given in column two. The ionization potential of the nth
electron of
the atom or ion is designated by IP and is given by the CRC [1]. That is for
example, Na + 5.13908 eV --~ Na' + e and Na+ + 47.2864 eV --4 Na2+ + e. The
first
ionization potential, IP = 5.13908 eV, and the second ionization potential,
IPz = 47.2864 eV , are given in the second and third columns, respectively.
The net
enthalpy of reaction for the breakage of the NaH bond and the double
ionization of
Na is 54.35 eV as given in the eighth column, and m = 2 in Eq. (2) as given in
the
ninth column. Additionally, H can react with each of the MH molecules given in
TABLE 2 to form a hydrino having a quantum number p increased by one (Eq. (1))
relative to the catalyst reaction product of MH alone as given by exemplary
Eq. (92).
TABLE 2. MH type hydrogen catalysts capable of providing a net enthalpy of
reaction of approximately m- 27.2 eV .
Catalyst M-H IP1 IPz IP3 IP4 IPS Enthalpy m
Bond
Energy
AIH 2.98 5.985768 18.82855 27.79 1
BiH 2.936 7.2855 16.703 26.92 1
CIH 4.4703 12.96763 23.8136 39.61 80.86 3
CoH 2.538 7.88101 17.084 27.50 1
GeH 2.728 7.89943 15.93461 26.56 1
InH 2.520 5.78636 18.8703 27.18 1
NaH 1.925 5.139076 47.2864 54.35 2
RuH 2.311 7.36050 16.76 26.43 1
SbH 2.484 8.60839 16.63 27.72 1
SeH 3.239 9.75239 21.19 30.8204 42.9450 107.95 4
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
129
SiH 3.040 8.15168 16.34584 27.54 1
SnH 2.736 7.34392 14.6322 30.50260 55.21 2
In other embodiments of the MH type catalyst, the reactants comprise sources
of SbH, SiH, SnH, and InH. In embodiments providing the catalyst MH, the
sources
comprise at least one of M and a source of H2 and MHX such as at least one of
Sb,
Si, Sn, and In and a source of H2, and SbH3,, SiH4, SnH4,and InH3.
The reaction mixture may further cornprise a source of H and a source of
catalyst wherein the source of at least one of H and catalyst may be a solid
acid or
NH4X where X is a halide, preferably Cl to form HCI catalyst. Preferably, the
reaction mixture may comprise at least one of NH4X, a solid acid, NaX, LiX,
KX,
NaH, LiH, KH, Na, Li, K, a support, a hydrogen dissociator and H2 where X is a
halide, preferably Cl. The solid acid may be NaHSO4, KHSO4, LiHSO4, NaHCO3,
KHCO3, LiHCO3, Na2HPO4, K2HPO4, Li2HPO4, NaH2PO4, KH2PO4, and LiH2PO4.
The catalyst may be at least one of NaH, Li, K, and HCI. The reaction mixture
may
further comprise at least one of a dissociator and a support.
Other thin-film deposition techniques that are well known in the ART comprise
further embodiments of the Invention. Such embodiments comprise physical
spray,
electro-spray, aerosol, electro-arching, Knudsen cell controlled release,
dispenser-
cathode injection, plasma-deposition, sputtering, and further coating methods
and
systems such as melting a fine dispersion of M, electroplating M, and chemical
deposition of M where MH comprises a catalyst.
In each case of a source of MH comprising an M alloy such as AIH and Al,
respectively, the alloy may be hydrided with a source of H2 such as H2 gas. H2
can
be supplied to the alloy during the reaction, or H2 may be supplied to form
the alloy
of a desired H content with the H pressure changed during the reaction. In
this case,
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
130
the initial H2 pressure may be about zero. The alloy may be activated by the
addition
of a metal such as an alkali or alkaline earth metal. For MH catalysts and
sources of
MH, the hydrogen gas may be maintained in the range of about 1 Torr to 100
atm,
preferably about 100 Torrto 10 atm, more preferably about 500 Torrto 2 atm. In
other embodiments, the source of hydrogen is from a hydride such as an alkali
or
alkaline earth metal hydride or a transition metal hydride.
Atomic hydrogen in high density can undergo three-body-collision reactions to
form hydrinos wherein one H atom undergoes the transition to form states given
by
Eq. (1) when two additional H atoms ionize. The reaction are given by
27.21eV+2H[aH]+H[at,] -~ 2H++2e + H 2 ) +[(2)2-12]=13.6eV (141)
2H'+2e -~ 2H[ay]+27.21 eV (142)
And, the overall reaction is
) + [(2)2 -12] 13.6 eV (143)
H[aH H
[ -~H-- I
In another embodiment, the reaction are given by
13.6eV (144)
54.4eV+2H[aH]+H[aH]-a2Hfas,+2e +H 3)
2Hfpsr+2e -~2H[aH]+54.4eV (145)
And, the overall reaction is
H[aH~ H~(3)I+[(3)2 -12] = 13.6 eV (146)
In an embodiment, the material that provides H atoms in high density is R-Ni.
The atomic H may be from at least one of the decomposition of H within R-Ni
and the
dissociation of H2 from an H2 source such as H2 gas supplied to the cell. R-Ni
may
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
131
be reacted with an alkali or alkaline earth metal M to enhance the production
of
layers of atomic H to cause the catalysis. R-Ni can be regenerated by
evaporating
the metal M followed by addition of hydrogen to rehydride the R-Ni.
References
1. D. R. Lide, CRC Handbook of Chemistry and Physics, 78 th Edition, CRC
Press,
Boca Raton, Florida, (1997), p. 10-214 to 10-216; hereafter referred to as
"CRC".
2. R. L. Mills, "The Nature of the Chemical Bond Revisited and an Alternative
Maxwellian Approach", Physics Essays, Vol. 17, No. 3, (2004), pp. 342-389.
Posted at
http://www.blacklightpower.com/pdf/technical/H2PaperTableFiguresCaptions111
303.pdf which is incorporated by reference.
3. R. Mills, P. Ray, B. Dhandapani, W. Good, P. Jansson, M. Nansteel, J. He,
A.
Voigt, "Spectroscopic and NMR Identification of Novel Hydride Ions in
Fractional
Quantum Energy States Formed by an Exothermic Reaction of Atomic Hydrogen
with Certain Catalysts", European Physical Journal-Applied Physics, Vol. 28,
(2004), pp. 83-104.
4. R. Mills and M. Nansteel, P. Ray, "Argon-Hydrogen-Strontium Discharge Light
Source", IEEE Transactions on Plasma Science, Vol. 30, No. 2, (2002), pp. 639-
653.
5. R. Mills and M. Nansteel, P. Ray, "Bright Hydrogen-Light Source due to a
Resonant Energy Transfer with Strontium and Argon Ions", New Journal of
Physics, Vol. 4, (2002), pp. 70.1-70.28.
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
132
6. R. Mills, J. Dong, Y. Lu, "Observation of Extreme Ultraviolet Hydrogen
Emission
from Incandescently Heated Hydrogen Gas with Certain Catalysts", Int. J.
Hydrogen Energy, Vol. 25, (2000), pp. 919-943.
7. R. Mills, M. Nansteel, and P. Ray, "Excessively Bright Hydrogen-Strontium
Plasma Light Source Due to Energy Resonance of Strontium with Hydrogen", J.
of Plasma Physics, Vol. 69, (2003), pp. 131-158.
8. H. Conrads, R. Mills, Th. Wrubel, "Emission in the Deep Vacuum Ultraviolet
from
a Plasma Formed by Incandescently Heating Hydrogen Gas with Trace Amounts
of Potassium Carbonate", Plasma Sources Science and Technology, Vol. 12,
(3003), pp. 389-395.
9. R. L. Mills, J. He, M. Nansteel, B. Dhandapani, "Catalysis of Atomic
Hydrogen to
New Hydrides as a New Power Source", submitted.
10. R. L. Mills, M. Nansteel, J. He, B. Dhandapani, "Low-Voltage EUV and
Visible
Light Source Due to Catalysis of Atomic Hydrogen", submitted.
11. J. Phillips, R. L. Nlills, X. Chen, "Water Bath Calorimetric Study of
Excess Heat in
'Resonance Transfer' Plasmas", Journal of Applied Physics, Vol. 96, No. 6, pp.
3095-3102.
12. R. L. Mills, X. Chen, P. Ray, J. He, B. Dhandapani, "Plasma Power Source
Based on a Catalytic Reaction of Atomic Hydrogen Measured by Water Bath
Calorimetry", Thermochimica Acta, Vol. 406/1-2, (2003), pp. 35-53.
13. R. L. Mills, Y. Lu, M. Nansteel, J. He, A. Voigt, B. Dhandapani,
"Energetic
Catalyst-Hydrogen Plasma Reaction as a Potential New Energy Source", Division
of Fuel Chemistry, Session: Chemistry of Solid, Liquid, and Gaseous Fuels,
227th American Chemical Society National Meeting, March 28-April 1, 2004,
Anaheim, CA.
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
133
14. R. Mills, B. Dhandapani, M. Nansteel, J. He, T. Shannon, A. Echezuria,
"Synthesis and Characterization of Novel Hydride Compounds", Int. J. of
Hydrogen Energy, Vol. 26, No. 4, (2001), pp. 339-367.
15. R. Mills, B. Dhandapani, M. Nansteel, J. He, A. Voigt, "Identification of
Compounds Containing Novel Hydride Ions by Nuclear Magnetic Resonance
Spectroscopy", Int. J. Hydrogen Energy, Vol. 26, No. 9, (2001), pp. 965-979.
16. R. Mills, B. Dhandapani, N. Greenig, J. He, "Synthesis and
Characterization of
Potassium lodo Hydride", Int. J. of Hydrogen Energy, Vol. 25, Issue 12,
December, (2000), pp. 1185-1203.
17. R. L. Mills, Y. Lu, J. He, M. Nansteel, P. Ray, X. Chen, A. Voigt, B.
Dhandapani,
"Spectral Identification of New States of Hydrogen", submitted.
18. R. L. Mills, P. Ray, "Extreme Ultraviolet Spectroscopy of Helium-Hydrogen
Plasma", J. Phys. D, Applied Physics, Vol. 36, (2003), pp. 1535-1542.
19. R. L. Mills, P. Ray, B. Dhandapani, M. Nansteel, X. Chen, J. He, "New
Power
Source from Fractional Quantum Energy Levels of Atomic Hydrogen that
Surpasses Internal Combustion", J Mol. Struct., Vol. 643, No. 1-3, (2002), pp.
43-
54.
20. R. Mills, P. Ray, "Spectral Emission of Fractional Quantum Energy Levels
of
Atomic Hydrogen from a Helium-Hydrogen Plasma and the Implications for Dark
Matter", Int. J. Hydrogen Energy, Vol. 27, No. 3, (2002), pp. 301-322.
21. R. L. Mills, P. Ray, "A Comprehensive Study of Spectra of the Bound-Free
Hyperfine Levels of Novel Hydride lon H-(1/2), Hydrogen, Nitrogen, and Air",
Int.
J. Hydrogen Energy, Vol. 28, No. 8, (2003), pp. 825-871.
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
134
22. R. Mills, "Spectroscopic Identification of a Novel Catalytic Reaction of
Atomic
Hydrogen and the Hydride Ion Product", Int. J. Hydrogen Energy, Vol. 26, No.
10,
(2001), pp. 1041-1058.
23. R. L. Nlills, P. Ray, B. Dhandapani, R. M. Mayo, J. He, "Comparison of
Excessive
Balmer a Line Broadening of Glow Discharge and Microwave Hydrogen
Plasmas with Certain Catalysts", J. of Applied Physics, Vol. 92, No. 12,
(2002),
pp. 7008-7022.
24. R. L. Mills, P. Ray, B. Dhandapani, J. He, "Comparison of Excessive Balmer
a
Line Broadening of Inductively and Capacitively Coupled RF, Microwave, and
Glow Discharge Hydrogen Plasmas with Certain Catalysts", IEEE Transactions
on Plasma Science, Vol. 31, No. (2003), pp. 338-355.
25. R. L. Mills, P. Ray, "Substantial Changes in the Characteristics of a
Microwave
Plasma Due to Combining Argon and Hydrogen", New Journal of Physics,
www.njp.org, Vol. 4, (2002), pp. 22.1-22.17.
26. J. Phillips, C. Chen, "Evidence of Energetic Reaction Between Helium and
Hydrogen Species in RF Generated Plasmas", submitted.
27. R. Nlills, P. Ray, R. M. Mayo, "CW HI Laser Based on a Stationary Inverted
Lyman Population Formed from Incandescently Heated Hydrogen Gas with
Certain Group I Catalysts", IEEE Transactions on Plasma Science, Vol. 31, No.
2, (2003), pp. 236-247.
28. R. L. Mills, P. Ray, "Stationary Inverted Lyman Population Formed from
Incandescently Heated Hydrogen Gas with Certain Catalysts", J. Phys. D,
Applied Physics, Vol. 36, (2003), pp. 1504-1509.
29. R. Mills, P. Ray, R. M. Mayo, "The Potential for a Hydrogen Water-Plasma
Laser", Applied Physics Letters, Vol. 82, No. 11, (2003), pp. 1679-1681.
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
135
30. R. Mills, The Grand Unified Theory of Classical Quantum Mechanics; October
2007 Edition, posted at
http://www.blacklightpower.com/theory/bookdownload.shtml.
31. N. V. Sidgwick, The Chemical Elements and Their Compounds, Volume I,
Oxford, Clarendon Press, (1950), p.17.
32. M. D. Lamb, Luminescence Spectroscopy, Academic Press, London, (1978), p.
68.
33. R. L. Mills, "The Nature of the Chemical Bond Revisited and an Alternative
Maxwellian Approach", submitted; posted at
http://www.blacklightpower.com/pdf/technical/H2PaperTableFiguresCaptions111
303.pdf.
34. H. Beutler, Z. Physical Chem., "Die dissoziationswarme des
wasserstoffmolekuls
H2, aus einem neuen ultravioletten resonanzbandenzug bestimmt", Vol. 27B,
(1934), pp. 287-302.
35. G. Herzberg, L. L. Howe, "The Lyman bands of molecular hydrogen", Can. J.
Phys., Vol. 37, (1959), pp. 636-659.
36. P. W. Atkins, Physical Chemistrys Second Edition, W. H. Freeman, San
Francisco, (1982), p. 589.
37. M. Karplus, R. N. Porter, Atoms and Molecules an Introduction for Students
of
Physical Chemistry, The Benjamin/Cummings Publishing Company, Menlo Park,
California, (1970), pp. 447-484.
38. K. R. Lykke, K. K. Murray, W. C. Lineberger, "Threshold photodetachment of
H-", Phys. Rev. A, Vol. 43, No. 11, (1991), pp. 6104-6107.
39. R. Mills, J. He, Z. Chang, W. Good, Y. Lu, B. Dhandapani, "Catalysis of
Atomic
Hydrogen to Novel Hydrogen Species H(1/4) and H2(1/4) as a New Power
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
136
Source", Int. J. Hydrogen Energy, Vol. 32, No. 12, (2007), pp. 2573-2584.
40. W. M. Mueller, J. P. Blackledge, and G. G. Libowitz, Metal Hydrides,
Academic
Press, New York, (1968), Hydroaen in Intermetalic Compounds I, Edited by L.
Schlapbach, Springer-Verlag, Berlin, and HydroQen in Intermetalic Compounds
II,
Edited by L. Schlapbach, Springer-Verlag, Berlin which is incorporate herein
by
reference.
41. D. R. Lide, CRC Handbook of Chemistry and Physics, 86th Edition, CRC
Press,
Taylor & Francis, Boca Raton, (2005-6), pp. 4-45 to 4-97 which is herein
incorporated by reference.
42. W. I. F. David, M. O. Jones, D. H. Gregory, C. M. Jewell, S. R. Johnson,
A.
Walton, P. Edwards, "A Mechanism for Non-stoichiometry in the Lithium
Amide/Lithium Imide Hydrogen Storage Reaction," J. Am. Chem. Soc., 129,
(2007), 1594-1601.
43. F. A. Cotton, G. Wilkinson, Advanced Inorganic Chemistry, lnterscience
Publishers, New York, (1972).
44. D. R. Lide, CRC Handbook of Chemistry and Physics, 86th Edition, CRC
Press,
Taylor & Francis, Boca Raton, (2005-6), pp. 9-54 to 9-59.
45. F. A. Cotton, G. Wilkinson, C. A. Murillo, M. Bochmann, Advanced Inorganic
Chemistry, Sixth Edition, John Wiley & Sons, Inc., New York, (1999), Chp 6.
46. F. A. Cotton, G. Wilkinson, C. A. Murillo, M. Bochmann, Advanced Inorganic
Chemistry, Sixth Edition, John Wiley & Sons, Inc., New York, (1999), p. 95.
47. J-G. Gasser, B. Kefif, "Electrical resistivity of liquid nickel-lanthanum
and nickel-
cerium alloys", Physical Review B, Vol. 41, No. 5, (1990), pp. 2776-2783.
48. F. A. Cotton, G. Wilkinson, C. A. Murillo, M. Bochmann, Advanced Inorganic
Chemistry, Sixth Edition, John Wiley & Sons, Inc., New York, (1999).
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
137
49. V. R. Choudhary, S. K. Chaudhari, "Leaching of Raney Ni-Al alloy with
alkali;
kinetics of hydrogen evolution", J. Chem. Tech. Biotech, Vol. 33a, (1983), pp.
339-349.
50. R. R. Cavanagh, R. D. Kelley, J. J. Rush, "Neutron vibrational
spectroscopy of
hydrogen and deuterium on Raney nickel", J. Chem. Phys., Vol. 77(3), (1982),
pp. 1540-1547.
51. F. A. Cotton, G. Wilkinson, C. A. Murillo, M. Bochmann, Advanced Inorganic
Chemistry, Sixth Edition, John Wiley & Sons, Inc., New York, (1999), pp. 190-
191.
52. R.L. Earle, M.D. Earle, Unit Operations in Food Processing, The New
Zealand
Institute of Food Science & Technology (Inc.), Web Edition 2004, available at
http://www.nzifst.orq.nz/unitoperations/.
53. F. A. Cotton, G. Wilkinson, C. A. Murillo, M. Bochmann, Advanced Inorganic
Chemistry, Sixth Edition, John Wiley & Sons, Inc., New York, (1999), p. 98.
EXPERIMENTAL
Equation numbers, section numbers, and reference numbers given hereafter in
this
Experimental section refer to those given in this Experimental section of the
Disclosure.
Abstract
The data from a broad spectrum of investigational techniques strongly and
consistently indicates that hydrogen can exist in lower-energy states than
previously
thought possible. The predicted reaction involves a resonant, nonradiative
energy
transfer from otherwise stable atomic hydrogen to a catalyst capable of
accepting
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
138
the energy. The product is H(1/p), fractional Rydberg states of atomic
hydrogen
called "hydrino 1 1 1 1 ~ 137 is an inte er re laces the
atoms" wherein n = 2' 3' 4'~~~' (p - g) p
p
well-known parameter n = integer in the Rydberg equation for hydrogen excited
states. Atomic lithium and molecular NaH served as catalysts since they meet
the
catalyst criterion-a chemical or physical process with an enthalpy change
equal to
an integer multiple m of the potential energy of atomic hydrogen, 27.2 eV
(e.g.
m = 3 for Li and m = 2 for NaH). Specific predictions based on closed-form
equations for energy levels of the corresponding hydrino hydride ions H- (1 /
4) of
novel alkali halido hydrino hydride compounds (MH * X; M = Li or Na, X =
halide)
and dihydrino molecules H2 (1 / 4) were tested using chemically generated
catalysis
reactants.
First, Li catalyst was tested. Li and LiNHZ were used as a source of atomic
lithium and hydrogen atoms. Using water-flow, batch calorimetry, the measured
power from 1 g Li, 0.5g LiNHZ 1 10g LiBr, and 15g Pd / A1Z03 was about 160W
with
an energy balance of AH = -19.1 kJ . The observed energy balance was 4.4 times
the maximum theoretical based on known chemistry. Next, Raney nickel (R-Ni)
served as a dissociator when the power reaction mixture was used in chemical
synthesis wherein LiBr acted as a getter of the catalysis product H(1 / 4) to
form
LiH * X as well as to trap H2 (1J 4) in the crystal. The ToF-SIMs showed LiH *
X
peaks. The 'H MAS NMR LiH * Br and LiH * I showed a large distinct upfield
resonance at about -2.5 ppm that matched H- (1 / 4) in a LiX matrix. An NMR
peak
at 1.13 pprri matched interstitial H2 (1 / 4), and the rotation frequency of
HZ (1 / 4) of
42 times that of ordinary H2 was observed at 1989 cm-` in the FTIR spectrum.
The
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
139
XPS spectrum recorded on the LiH * Br crystals showed peaks at about 9.5 eV
and
12.3 eV that could not be assigned to any known elements based on the absence
of
any other primary element peaks, but matched the binding energy of H- (1 / 4)
in two
chemical environments. A further signature of the energetic process was the
observation of the formation of a plasma called a resonant transfer- or rt-
plasma at
low temperatures (e.g. =103 K) and very low field strengths of about 1-2 V/cm
when
atomic Li was present with atomic hydrogen. Time-dependent line broadening of
the H Balmer a line was observed corresponding to extraordinarily fast H (>40
eV).
NaH uniquely achieves high kinetics since the catalyst reaction relies on the
release of the intrinsic H, which concomitantly undergoes the transition to
form
H(1/3) that further reacts to form H(1/4). High-temperature differential
scanning
calorimetry (DSC) was performed on ionic NaH under a helium atmosphere at an
extremely slow temperature ramp rate (0.1 C/min) to increase the amount of
molecular NaH formation. A novel exothermic effect of -177 kJ / moleNaH was
observed in the temperature range of 640 C to 825 C. To achieve high power, R-
Ni
having a surface area of about 100 mz I g was surface-coated with NaOH and
reacted with Na metal to form NaH. Using water-flow, batch calorimetry, the
measured power from 15g of R-Ni was about 0.5 kW with an energy balance of
AH = -36 kJ compared to AH = 0 kJ from the R-Ni starting material, R-NiAI
alloy,
when reacted with Na metal. The observed energy balance of the NaH reaction
was -1.6X104 kJ / mole H2 1 over 66 times the -241.8 kJ / mole H2 enthalpy of
combustion.
The ToF-SIMs showed sodium hydrino hydride, NaHx , peaks. The 'H MAS
NMR spectra of NaH * Br and NaH * Cl showed large distinct upfield resonance
at
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
140
-3.6 ppm and -4 ppm, respectively, that matched H- (1 / 4), and an NMR peak at
1.1 ppm matched H2 (1 / 4). NaH * Cl from reaction of NaCI and the solid acid
KHSO4 as the only source of hydrogen comprised two fractional hydrogen states.
The H- (1 / 4) NMR peak was observed at -3.97 ppm, and the H- (1 / 3) peak was
also present at -3.15 ppm. The corresponding H2 (1 / 4) and HZ (1 / 3) peaks
were
observed at 1.15 ppm and 1.7 ppm, respectively. The XPS spectrum recorded on
NaH * Br showed the H- (1 / 4) peaks at about 9.5 eV and 12.3 eV that matched
the
results from LiH * Br and KH * I; whereas, sodium hydrino hydride showed two
fractional hydrogen states additionally having the H- (1 / 3) XPS peak at 6 eV
in the
absence of a halide peak. The predicted rotational transitions having energies
of 42
times those of ordinary HZ were also observed from H2 (1 / 4) which was
excited
using a 12.5 keV electron beam.
1. Introduction
Mills [1-12] solved the structure of the bound electron using classical laws
and
subsequently developed a unification theory based on those laws called the
Grand
Unified Theory of Classical Physics (GUTCP) with results that match
observations
for the basic phenomena of physics and chemistry from the scale of the quarks
to
cosmos. This paper is the first in a series of two that covers two specific
predictions
of GUTCP involving the existence of lower-energy states of the hydrogen atom,
which represents a powerful new energy source and the transitions of atomic
hydrogen to lower-energy states [2].
GUTCP predicts a reaction involving a resonant, nonradiative energy transfer
from otherwise stable atomic hydrogen to a catalyst capable of accepting the
energy
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
141
to form hydrogen in lower-energy states than previously thought possible.
Specifically, the product is H(1/ p), fractional Rydberg states of atomic
hydrogen
wherein n 1 1 1 1( p ~ 137 is an integer) replaces the well known parameter
= 2' 3' 4
p
n = integer in the Rydberg equation for hydrogen excited states. He+, Ar+,
Sr+, Li,
K, and NaH are predicted to serve as catalysts since they meet the catalyst
criterion-a chemical or physical process with an enthalpy change equal to an
integer multiple of the potential energy of atomic hydrogen, 27.2 eV. The data
from
a broad spectrum of investigational techniques strongly and consistently
support the
existence of these states called hydrino, for "small hydrogen", and the
corresponding
diatomic molecules called dihydrino molecules. Some of these prior related
studies
supporting the possibility of a novel reaction of atomic hydrogen, which
produces
hydrogen in fractional quantum states that are at lower energies than the
traditional
"ground" ( n=1) state, include extreme ultraviolet (EUV) spectroscopy,
characteristic
emission from catalysts and the hydride ion products, lower-energy hydrogen
emission, chemically-formed plasmas, Balmer a line broadening, population
inversion of H lines, elevated electron temperature, anomalous plasma
afterglow
duration, power generation, and analysis of novel chemical compounds [13-40].
Recently, there has been the announcement of some unexpected
astrophysical results that support the existence of hydrinos. In 1995, Mills
published the GLITCP prediction [41] that the expansion of the universe was
accelerating from the same equations that correctly predicted the mass of the
top
quark before it was measured. To the astonishment of cosmologists, this was
confirmed by 2000. Mills made another prediction about the nature of dark
matter
based on GUTCP that may be close to being confirmed. Based on recent evidence,
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
142
Bournaud et al. [42-43] suggest that dark matter is hydrogen in dense
molecular
form that somehow behaves differently in terms of being unobservable except by
its
gravitational effects. Theoretical models predict that dwarfs formed from
collisional
debris of massive galaxies should be free of nonbaryonic dark matter. So,
their
gravity should tally with the stars and gas within them. By analyzing the
observed
gas kinematics of such recycled galaxies, Bournaud et al. [42-43] have
measured
the gravitational masses of a series of dwarf galaxies lying in a ring around
a
massive galaxy that has recently experienced a collision. Contrary to the
predictions of Cold-Dark-Matter (CDM) theories, their results demonstrate that
they
contain a massive dark component amounting to about twice the visible matter.
This baryonic dark matter is argued to be cold molecular hydrogen, but it is
distinguished from ordinary molecular hydrogen in that it is not traced at all
by
traditional methods, such as emission of CO lines. These results match the
predictions of the dark matter being dihydrino molecules.
Erriission lines recorded on cold interstellar regions containing dark matter
matched H(1 / p), fractional Rydberg states of atomic hydrogen given by Eqs.
(2a)
and (2c) [29]. Such emission lines with energies of q- 13.6 eV, where
q=1,2,3,4,6,7,8,9, or 11 were also observed by extreme ultraviolet (EUV)
spectroscopy recorded on microwave discharges of helium with 2% hydrogen [27-
29]. These He+ fulfills the catalyst criterion-a chemical or physical process
with an
enthalpy change equal to an integer multiple of 27.2 eV since it ionizes at
54.417 eV,
which is 2- 27.2 eV. The product of the catalysis reaction of He+, H(1 / 3),
may
further serve as a catalyst to lead to transitions to other states H(1 / p) .
J. R. Rydberg showed that all of the spectral lines of atomic hydrogen were
given by a completely empirical relationship:
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
143
1 1
v = R nf - z (1)
where R=109,677 cm-', nf =1,2,3,..., n, = 2,3,4,... and n; > n f. Bohr,
Schrodinger,
and Heisenberg, each developed a theory for atomic hydrogen that gave the
energy
levels in agreement with Rydberg's equation.
13.598 eV (2a)
E_- e 2
n n2p~oaH nZ
n=1,2,3,... (2b)
where e is the elementary charge, E. is the permittivity of vacuum, and aH is
the
radius of the hydrogen atom. The excited energy states of atomic hydrogen are
given by Eq. (2a) for n > 1 in Eq. (2b). The n=1 state is the "ground" state
for "pure"
photon transitions (i.e. the n=1 state can absorb a photon and go to an
excited
electronic state, but it cannot release a photon and go to a lower-energy
electronic
state). However, an electron transition from the ground state to a lower-
energy state
may be possible by a resonant nonradiative energy transfer such as multipole
coupling or a resonant collision mechanism. Processes such as hydrogen
molecular
bond formation that occur without photons and that require collisions are
common
[44]. Also, some commercial phosphors are based on resonant nonradiative
energy
transfer involving multipole coupling [45].
The theory reported previously [1, 13-40] predicts that atomic hydrogen may
undergo a catalytic reaction with certain atoms, excimers, ions, and diatomic
hydrides which provide a reaction with a net enthalpy of an integer multiple
of the
potential energy of atomic hydrogen, Eh = 27.2 eV where E,, is one Hartree.
Specific
species (e.g. He+, Ar`, Sr`, K, Li, HCl, and NaH) identifiable on the basis of
their known electron energy levels are required to be present with atomic
hydrogen
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
144
to catalyze the process. The reaction involves a nonradiative energy transfer
followed by q- 13.6 eV emission or q= 13.6 eV transfer to H to form
extraordinarily hot,
excited-state H [13-17, 19-20, 32-39] and a hydrogen atom that is lower in
energy
than unreacted atomic hydrogen that corresponds to a fractional principal
quantum
number. That is
n 1 1 1 1 , . p < 137 is an integer (2c)
=1,2,3,4,...,-
p
replaces the well known parameter n = integer in the Rydberg equation for
hydrogen
excited states. The n=1 state of hydrogen and the n = 1 states of hydrogen
integer
are nonradiative, but a transition between two nonradiative states, say n=1 to
n = 1/ 2, is possible via a nonradiative energy transfer. Thus, a catalyst
provides a
net positive enthalpy of reaction of m- 27.2 eV (i.e. it resonantly accepts
the
nonradiative energy transfer from hydrogen atoms and releases the energy to
the
surroundings to affect electronic transitions to fractional quantum energy
levels). As
a consequence of the nonradiative energy transfer, the hydrogen atom becomes
unstable and emits further energy until it achieves a lower-energy
nonradiative state
having a principal energy level given by Eqs. (2a) and (2c).
The catalyst product, H(1 / p), may also react with an electron to form a
novel
hydride ion H-(1 / p) with a binding energy E, [1, 13-14, 18, 30]:
E _ ~iZ s(s + 1) ~ oeZh Z 1 2 2
(3)
2 3 aH 3[1 + S(S + 1) 3
B 2 1+ s(S + 1) 2 'ne
8 ea0 1 p a0 p
where p = integer > 1, s = 1/ 2, h is Planck's constant bar, , is the
permeability of
vacuum, me is the mass of the electron, e is the reduced electron mass given
by
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
145
e = memp where mP is the mass of the proton, ao is the Bohr radius, and the
m
+mp
ionic radius is r, = p(] + s(s+1)). From Eq. (3), the calculated ionization
energy of
the hydride ion is 0.75418 eV, and the experimental value given by Lykke [46]
is
6082.99 0.15 cm-` (0.75418 eV).
Upfield-shifted NMR peaks are a direct evidence of the existence of lower-
energy state hydrogen with a reduced radius relative to ordinary hydride ion
and
having an increase in diamagnetic shielding of the proton. The shift is given
by the
sum of that of ordinary hydride ion H and a component due to the lower -energy
state [1, 15]:
2
A` =- 0 e (1+a2np)=-(29.9+1.37p)ppm (4)
B 12meao(1+ s(s +1))
where for H p = 0 and p = integer > 1 for H- (1 / p) and a is the fine
structure
constant.
H(1 / p) may react with a proton and two H(1 / p) may react to form
H2 (1 / p)+ and H2 (1 / p), respectively. The hydrogen molecular ion and
molecular
charge and current density functions, bond distances, and energies were solved
previously [1, 6] from the Laplacian in ellipsoidal coordinates with the
constraint of
nonradiation.
~)R4 a~(Rga~)+~~-~)~~(R,,~)+~~-t1)R~a~(R~~)=0 (5)
The total energy E, of the hydrogen molecular ion having a central field of
+pe at
each focus of the prolate spheroid molecular orbital is
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
146
2ez
z 47Ce.~2aH~3
z 6
~ -pe (41n3-1-21n3) l+p 2~ me _ 1 h k O
81cEoaõ mec 2 ~
_ -pz 16.13392 eV - p30.118755 eV
where p is an integer, c is the speed of light in vacuum, p is the reduced
nuclear
mass, and k is the harmonic force constant solved previously in a closed-form
equation with fundamental constants only [1, 6]. The total energy of the
hydrogen
molecule having a central field of +pe at each focus of the prolate spheroid
molecular orbital is
F~'=rElal
E_ z T ez
-p 2~-~+~ ~+1-~ l+p 2~ -1 ~ k
81rEao 2 [2 -1 mec 2
_ -p2 31.351 eV - p30.326469 eV
(7)
The bond dissociation energy, ED , of hydrogen molecule H2 (1 / p) is the
difference between the total energy of the corresponding hydrogen atoms and ET
Eo = E(2H(1/ p)) - ET (8)
where [47]
E(2H (1 / p)) _ -pz 27.20 eV (9)
ED is given by Eqs. (8-9) and (7):
Eo = -pz 27.20 eV - ET
=-pz 27.20 eV -(-pz 31.351 eV - p30.326469 eV) (10)
= pz4.151 eV+p30.326469 eV
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
147
The calculated and experimental parameters of H2, D2, HZ , and DZ from Ref.
[1,
6] are given in TABLE 3.
TABLE 3. The Maxwellian closed-form calculated and experimental parameters of
Hz,Dz,Hz andD2+.
Parameter Calculated Experimental
H2 Bond Energy 4.478 eV 4.478 eV
D2 Bond Energy 4.556 eV 4.556 eV
HZ Bond Energy 2.654 eV 2.651 eV
DZ Bond Energy 2.696 eV 2.691 eV
HZ Total Energy 31.677 eV 31.675 eV
D2 Total Energy 31.760 eV 31.760 eV
H2 Ionization Energy 15.425 eV 15.426 eV
D2 Ionization Energy 15.463 eV 15.466 eV
Hz Ionization Energy 16.253 eV 16.250 eV
DZ Ionization Energy 16.299 eV 16.294 eV
Hz Magnetic Moment 9.274 X 10-2' JT ' 9.274 X 10-24 JT-`
( s) ( 8)
Absolute H2 Gas-Phase -28.0 ppm -28.0 ppm
NMR Shift
H2 Internuclear Distancea 0.748 A 0.741 A
fao
D2 Internuclear Distancea 0.748 A 0.741 A
,r2 aa
Hz Internuclear Distance 1.058 A 1.06 A
2 ao
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
148
DZ Internuclear Distancea 1.058 A 1.0559 A
2 ao
H2 Vibrational Energy 0.517 eV 0.516 eV
D2 Vibrational Energy 0.371 eV 0.371 eV
H2 wexe 120.4 cm-' 121.33 cm-'
D2 CUexe 60.93 cm-1 61.82 cm-'
HZ Vibrational Energy 0.270 eV 0.271 eV
DZ Vibrational Energy 0.193 eV 0.196 eV
Hz J=1 to J=0 Rotational Energya 0.0148 eV 0.01509 eV
D2 J=1 to J=0 Rotational Energya 0.00741 eV 0.00755 eV
HZ J=1 to J=0 Rotational Energy 0.00740 eV 0.00739 eV
Dz J=1 to J=0 Rotational Energya 0.00370 eV 0.003723 eV
a Not corrected for the slight reduction in internuclear distance due to Eos,.
The 'H NMR resonance of HZ(1/ p) is predicted to be upfield from that of H,
due to the fractional radius in elliptic coordinates [1, 6] wherein the
electrons are
significantly closer to the nuclei. The predicted shift, B for H2(1/ p)
derived
previously [1, 6] is given by the sum of that of H2 and a term that depends on
p = integer > 1 for H, (1 / p) :
2
A = -,uo 4- F2 ln~+l e (1+7cop) (11)
B f -1 36aome
ABT
B = 428.01 +0.64p)ppm (12)
where for Hz p= 0 .
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
149
The vibrational energies, Ev;b , for the v = 0 to v=1 transition of hydrogen-
type molecules HZ (I l p) are [1, 6]
E,;b = p20.515902 eV (13)
where p is an integer and the experimental vibrational energy for the v = 0 to
v = 1
transition of Hz , EH, (U-o~u-1), is given by Beutler [48] and Herzberg [49].
The rotational energies, E,o,, for the J to J + 1 transition of hydrogen-type
molecules H2 (1 / p) are [1, 6]
E~, =E,,,-E,= j [J+1]=p2(J+1)0.01509eV (14)
where p is an integer, I is the moment of inertia, and the experimental
rotational
energy for the J = 0 to J=1 transition of H2 is given by Atkins [50].
The p2 dependence of the rotational energies results from an inverse p
dependence of the internuclear distance and the corresponding impact on the
moment of inertia I. The predicted internuclear distance 2c' for HZ (1 / p) is
2c'= a _[2 (15)
P
The formation of new states of hydrogen is very energetic. A new chemically
generated or assisted plasma source based on the resonant energy transfer
mechanism (rt-plasma) has been developed that may be a new power source. One
such source operates by incandescently heating a hydrogen dissociator and a
catalyst to provide atomic hydrogen and gaseous catalyst, respectively, such
that the
catalyst reacts with the atomic hydrogen to produce a plasma. It was
extraordinary
that intense EUV emission was observed by Mills et al. [13-21, 38-39] at low
temperatures (e.g. =103 K), as well as an extraordinary low field strength of
about 1-
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
150
2 V/cm from atomic hydrogen and certain atomized elements or certain gaseous
ions, which singly or multiply ionize at integer multiples of the potential
energy of
atomic hydrogen, 27.2 eV.
K to K3+ provides a reaction with a net enthalpy equal to three times the
potential energy of atomic hydrogen. It was reported previously [13-21, 38-39]
that
the presence of these gaseous atoms with thermally dissociated hydrogen formed
an rt-plasma having strong EUV emission with a stationary inverted Lyman
population. Other noncatalyst metals such as Mg produced no plasma.
Significant
line broadening of the Balmer a, /3, and y lines of 18 eV was observed.
Emission
from rt-plasmas occurred even when the electric field applied to the plasma
was
zero. Since a conventional discharge power source was not present, the
formation
of a plasma would require an energetic reaction. The origin of Doppler
broadening is
the relative thermal motion of the emitter with respect to the observer. Line
broadening is a measure of the atom temperature, and a significant increase
was
expected and observed for catalysts, K as well as Sr+ or Ar' [13-21, 38-39],
with
hydrogen. The observation of a high hydrogen temperature with no conventional
explanation would indicate that an rt-plasma must have a source of free
energy. An
energetic chemical reaction was further implicated since it was found that the
broadening is time dependent [13-14, 20]. Therefore, the thermal power balance
was measured calorimetrically. The reaction was exothermic since excess power
of
20 mW =cm-3 was measured by Calvet calorimetry [20]. In further experiments,
KNO3 and Raney nickel were used as a source of K catalyst and atomic hydrogen,
respectively, to produce the corresponding exothermic reaction. The energy
balance
was AH =-17, 925 kcal / mole KNO3, about 300 times that expected for the most
energetic known chemistry of KNO3, and -3585 kcal / mole H2, over 60 times the
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
151
hypothetical maximum enthalpy of -57.8 kcal Imole Hz due to combustion of
hydrogen with atmospheric oxygen, assuming the maximum possible H2 inventory
[14]. Additional substantial evidence of an energetic catalytic reaction was
previously reported [13-15, 24-26, 30-31] involving a resonant energy transfer
between hydrogen atoms and K to form very stable novel hydride ions and
molecules H-(1/4) and H2(1/4), respectively. Characteristic emission was
observed from K3+ that confirmed the resonant nonradiative energy transfer of
3.27.2 eV from atomic hydrogen to K that served as a predicted catalyst. From
Eq.
(3), the binding energy EB of H-(1/4) is
E, = 11.232 eV (Ava, =110.38 nm ) (16)
The product hydride ion H(1/4) was observed by EUV spectroscopy at
110 nm corresponding to its predicted binding energy of 11.2 eV [13-15, 24-26,
30-
31]. The identification of H(1/4) was confirmed previously by the XPS
measurement of its binding energy. The XPS spectrum of KH* I differed from
that
of KI by having additional features at 8.9 eV and 10.8 eV that did not
correspond to
any other primary element peaks but did match the H- (1 / 4) Eb =11.2 eV
hydride
ion (Eq. (3)) in two different chemical environments. The 'H MAS NMR spectrum
of
novel compound KH* Cl relative to external tetramethylsilane (TMS) showed a
large
distinct upfield resonance at -4.4 ppm corresponding to an absolute resonance
shift
of -35.9 ppm that matched the theoretical prediction of p = 4 [13-15, 25-26,
30-31].
Elemental analysis identified [13-15, 25-26, 30-31] these compounds as only
containing the alkaline metal, halogen, and hydrogen, and no known hydride
compound of this composition could be found in the literature that had an
upfield-
shifted hydride NMR peak. Ordinary alkali hydrides alone or mixed with alkali
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
152
halides show down-field shifted peaks [13-15, 25-26, 30-31]. From the
literature, the
list of alternatives to H-(1 / p) as a possible source of the upfield NMR
peaks was
limited to U centered H. This was eliminated by the absence of the intense and
characteristic infrared vibration band at 503 cm-' due to the substitution of
H- for
Cl- in KCl [5'1 ].
As a further characterization, FTIR analysis of KH*1 crystals with H"(1/4)
was performed and interstitial H2(1/4) having a predicted rotational energy
given by
Eq. (14) was observed. Rotational lines were observed previously [13-14] in
the
145-300 nm region from atmospheric pressure electron beam-excited
argon-hydrogen plasmas. The unprecedented energy spacing of 42 times that of
hydrogen established the internuclear distance as 1/4 that of Hz and
identified
Hz(1/4) (Eqs. (13-15)). The spectrum was asymmetric with the P branch dominant
corresponding to the absence of populated rotational states in the exited v=1
vibrational state. This was due to the high rotational energy (10 times the
thermal
energy), the short lifetime of the rotational excited states, and the low
cross section
for electron-beam rotational excitation; whereas, the vibrational dipole
excitation was
allowed. Thus, only the v = 1, J = 0 state was populated significantly from e-
beam
excitation, and transitions occurred with dI > 0 during the v=1 to v = 0
transition.
KH*Cl having H(1/4) by NMR was incident to the 12.5 keV electron beam, which
excited similar emission of interstitial HZ(1/4) as observed in the argon-
hydrogen
plasma [13-14]. Specifically, H2(1/4) trapped in the lattice of KH*Cl was
investigated by windowless EUV spectroscopy on electron-beam excitation of the
crystals using the 12.5 keV electron gun at pressures below which any gas
could
produce detectable emission (<10-5 Torr). The rotational energy of HZ(1/4) was
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
153
confirmed by this technique as well. These results confirmed the previous
observations from the plasmas formed by the energetic hydrino-forming reaction
having intense hydrogen Lyman emission, a stationary inverted Lyman
population,
excessive afterglow duration, highly energetic hydrogen atoms, characteristic
alkali-
ion emission due to catalysis, predicted novel spectral lines, and the
measurement
of a power beyond any conventional chemistry [13-40] that matched predictions
for a
catalytic reaction of atomic hydrogen to form more stable hydride ions
designated
H-(1/p). Since the comparison of theory and experimental energies is direct
evidence of lower-energy hydrogen with an implicit large exotherm during its
formation, we report in this paper the results when these experiments were
repeated
with additionally predicted catalysts Li and NaH.
A catalytic system used to make and analyzed predicted hydride compounds
involves lithium atoms. The first and second ionization energies of lithium
are
5.39172 eV and 75.64018 eV, respectively [52]. The double ionization ( t= 2)
reaction
of Li to Li2+ then, has a net enthalpy of reaction of 81.0319 eV, which is
equivalent
to 3- 27.2eV.
81.0319eV+Li(m)+H~aH~Li2++2e-+Hj (p~3)j+[(p+3)z -pz ]- 13.6 eV
p L J
(17)
Li2++2e- -4 Li(m) + 81.0319 eV (18)
And, the overall reaction is
H~a P H ] ' HL(p+ 3)]+[(p+ 3)z - pz ] -13.6 eV (19)
Lithium is a metal in the solid and liquid states, and the gas comprises
covalent Li2 molecules [53], each having a bond energy of 110.4 kJ/mole [54].
In
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
154
order to generate atomic lithium, LiNH2was added to the reaction mixture.
LiNH2
generates atomic hydrogen as well, according to the reversible reactions [55-
64]:
Liz + LiNHz ~ Li + Li2NH + H (20)
and
LiZ + LizNH ~ Li + Li3N + H (21)
The energy for the reaction of lithium amide to lithium nitride and lithium
hydride is
exothermic [65-66]:
4Li + LiNH2 ---~ Li3N + 2LiH OH =-198.5 kJ / mole LiNH2 (22)
Thus, it should occur to a significant extent. The specific predictions of the
energetic
reaction given by Eqs. (17-19) were tested by rt-plasma formation and H line
broadening. The power developed was measured using water-flow, batch
calorimetry. Then, the predicted products of H-(1/4) and H2 (1/4) having the
energies given by Eqs. (3) and (5-15), respectively, were tested by magic
angle solid
proton nuclear magnetic resonance spectroscopy (MAS 'H NMR), X-ray
photoelectron spectroscopy (XPS), time of flight secondary ion mass
spectroscopy
(ToF-SIMs), and Fourier transform infrared spectroscopy (FTIR).
A compound comprising hydrogen such as MH, where M is element other
than hydrogen, serves as a source of hydrogen and a source of catalyst. A
catalytic
reaction is provided by the breakage of the M - H bond plus the ionization of
t
electrons from the atom M each to a continuum energy level such that the sum
of
the bond energy and ionization energies of the t electrons is approximately
m- 27.2 eV , where m is an integer. One such catalytic system involves sodium.
The bond energy of NaH is 1.9245 eV [54], and the first and second ionization
energies of Na are 5.13908 eV and 47.2864 eV, respectively [52]. Based on
these
energies NaH molecule can serve as a catalyst and H source, since the bond
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
155
energy of NaH plus the double ionization (t = 2) of Na to Na2+ is 54.35 eV
(2. 27.2 eV). The catalyst reactions are given by
54.35 eV + NaH -) Na2+ + 2e- + H P 3 ~ + [32 -1'] 13.6 eV (23)
Na2+ + 2e + H-> NaH + 54.35 eV (24)
And, the overall reaction is
H-4 H[ 3 ] +[32 -12]=13.6eV (25)
As given in Chp. 5 of Ref [1], and Ref. [29], hydrogen atoms
H(1 / p) p=1, 2, 3,...137 can undergo further transitions to lower-energy
states given
by Eqs. (2a) and (2c) wherein the transition of one atom is catalyzed by a
second
that resonantly and nonradiatively accepts m- 27.2 eV with a concomitant
opposite
change in its potential energy. The overall general equation for the
transition of
H(1 / p) to H(1 /(p+ m)) induced by a resonance transfer of m- 27.2 eV to H(1
/ p')
is represented by
H(1/p')+H(1/p) -4 H++e +H(1/(p+m)) +12pm+m2-pi2].13.6eV (26)
In the case of a high hydrogen atom concentration, the transition of H(1 / 3)
( p= 3)
to H(1 / 4) ( p+ m = 4) with H as the catalyst ( p=1; m = 1) can be fast:
H(1 / 3)--L-4 H(1 / 4)+81.6 eV (27)
The NaH catalyst reactions may be concerted since the sum of the bond energy
of
NaH, the double ionization (t = 2) of Na to Na2+ , and the potential energy of
H is
81.56 eV ( 3= 27.2 eV). The catalyst reactions are given by
81.56 eV + NaH + H -4 Na2++2e-+Hfa, +e- +HL 4 J+[42-1z]=13.6eV
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
156
(28)
Na2++2e +H+Hfas, +e -4 NaH + H + 81.56 eV (29)
And, the overall reaction is
H-> HL 4 J+[42-12]=13.6eV (30)
where H f~t is a fast hydrogen atom having at least 13.6 eV of kinetic energy.
H- (1 / 4) forms stable halidohydrides and is a favored product together with
the
corresponding molecule formed by the reactions 2H (1 / 4) --~ H2 (1 / 4) and
H-(1/4)+H+ -4 H2(1 /4) [13-15, 24-26, 30-31]. The corresponding hydrino atom
H(1 / 4) is a preferred final product consistent with observation since the p
= 4
quantum state has a multipolarity greater than that of a quadrupole giving it
a long
theoretical lifetime. H(1 / 4) may be formed directly from H (e.g. Eqs. (36-
38)) or
via multiple transitions (e.g. Eqs. (23-27)). In the latter case, the higher-
energy
H(1 / p) states with quantum numbers p= 2; f = 0,1 and p= 3; f = 0,1,2
corresponding to dipole and quadrupole transitions, respectively, have
theoretically
allowed, fast transitions.
Sodium hydride is typically in the form of an ionic crystalline compound
formed by the reaction of gaseous hydrogen with metallic sodium. And, in the
gaseous state, sodium comprises covalent Na2 molecules [53] with a bond energy
of
74.8048 kJ/mole [54]. It was found that when NaH(s) was heated at a very slow
temperature ramp rate (0.1 C/min) under a helium atmosphere to form NaH(g),
the
predicted exothermic reaction given by Eqs. (23-25) was observed at high
temperature by differential scanning calorimetry (DSC). To achieve high power,
a
chemical system was designed to greatly increase the amount and rate of
formation
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
157
of NaH(g). The reaction of NaOH and Na to Na2O and NaH(s) calculated from
the heats of formation [54, 65] releases AH =-44.7 kJ / mole NaOH :
NaOH+2Na-~ Na2O+NaH(s) OH=-44.7 kJ/moleNaOH (31)
This exothermic reaction can drive the formation of NaH(g) and was exploited
to
drive the very exothermic reaction given by Eqs. (23-25). The regenerative
reaction
in the presence of atomic hydrogen is
NazO + H-4NaOH + Na OH =-11.6 kJ / mole NaOH (32)
NaH -4Na+ H(1 / 3) AH =-10,500 kJ / mole H (33)
and
NaH-4Na+H(114) AH=-19,700kJ/moleH (34)
Thus, a small amount of NaOH, Na, and atomic hydrogen serves as a catalytic
source of the NaH catalyst that in turn forms a large yield of hydrinos via
multiple
cycles of regenerative reactions such as those given by Eqs. (31-34). R-Ni
having a
high surface area of about 100 m2 / g and containing H was surface coated with
NaOH and reacted with Na metal to form NaH(g). Since the energy balance in the
formation of NaH(g) was negligible due to the small amounts involved, the
energy
and power due to the hydrino reactions given by Eqs. (23-25) were specifically
measured using water-flow, batch calorimetry. Next, R-Ni 2400 was prepared
such
that it comprised about 0.5 wt% NaOH, and the Al of the intermetallic served
as the
reductant to form NaH catalyst during calorimetry measurement. The reaction of
NaOH + Al to A1203 + NaH calculated from the heats of formation [65] is
exothermic by OH = -189.1 kJ / mole NaOH. The balanced reaction is given by
3NaOH + 2A1 -~ A1203 + 3NaH OH =-189.1 kJ / mole NaOH (35)
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
158
This exothermic reaction can drive the formation of NaH(g) and was exploited
to
drive the very exothermic reaction given by Eqs. (23-25) wherein the
regeneration of
NaH occurred from Na in the presence of atomic hydrogen. For 0.5wt% NaOH, the
exothermic reaction given by Eq. (35) gave a negligible OH = -0.024 kI
background
heat during measurement.
It was reported previously [28-29] that the reaction products H(1 / p) may
undergo further reaction to lower-energy states. For example, the catalyst
reaction
of Ar+ to ArZ, forms H(1 / 2), which may further serve as both a catalyst and
a
reactant to form H(1/4) [1, 13-14, 28-29] and the corresponding favored
molecule
Hz (1 / 4) , observed using different catalysts [13-14]. Thus, predicted
products of
NaH catalyst from Eqs. (23-25) and Table 1 of Ref. [29] are H- (1 / 3) and H2
(1 / 4)
having the energies given by Eqs. (3) and (5-15), respectively. They were
tested by
MAS `H NMR and ToF-SIMs.
Another catalytic system of the type MH involves chlorine. -rhe bond energy
of HCl is 4.4703 eV [54]. The first, second, and third ionization energies of
Cl are
12.96764 eV, 23.814 eV, and 39.61 eV, respectively [52]. Based on these
energies,
HCl can serve as a catalyst and H source, since the bond energy of HCl plus
the
triple ionization ( t= 3) of Cl to Cl3+ is 80.86 eV ( 3= 27.2 eV ). The
catalyst reactions
are given by
80.86 eV+HCI->Cl3++3e-+HL 4 J+[42-12]- 13.6 eV (36)
Cl3+ + 3e- + H---> HCl + 80.86 eV (37)
And, the overall reaction is
H HL 4 J+[42-12]. 13.6eV (38)
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
159
The anticipated product then is H2(1 / 4).
Alkali chlorides contain both Cl and H, typically from H20 contamination.
Thus, some HCl can form interstitially in the crystalline matrix. Since H' can
most
easily substitute for Li', and the substitution is least likely in the case of
Cs+, it was
anticipated that alkali chlorides may form HC1 that undergoes catalysis to
form
HZ (1 / 4) with the trend of the rate of formation increasing in the order of
the Group I
elements. Due to the difference in lattice structure, MgCl2 may not form HCl
catalyst; thus, it serves as a chlorine control. This condition applies to
other alkaline
earth halides and transition metal halides such as those of copper that can
serve as
controls for the formation of HZ (1 / 4). One exception from this set is Mg2+
in a
suitable lattice, since the ionization of Mgz+ to Mg3+ is 80.1437 eV [52]
which is
close to 3- 27.2 eV . These hypotheses were tested by electron beam-excitation
emission spectroscopy on alkali halides, MgX2 ( X= F,C1,Br,I), and CuX2
( X= F,CI,Br ) with the goal of determining whether the predicted emission of
H2014) is selectively observed when a catalyst reaction is possible and not
otherwise. NMR was recorded on these compounds to search for the corresponding
predicted H2 (1 / 4) peak to be compared with the emission results.
II. Experimental Methods
Rt-plasma and Line Broadening Measurements. LiNH2 argon-hydrogen
(95/5%) and LiNH2 hydrogen rt-plasmas was generated in the experimental set up
described previously [15-21] (Figure 1) comprising a thermally insulated
stainless
steel cell with a cap that incorporated ports for gas inlet, and outlet. A
titanium
filament (55 cm long, 0.5 mm diameter) that served as a heater and hydrogen
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
160
dissociator was in the cell. 1 g of LiNHZ (Alfa Aesar 99.95%) was placed in
the
center of the cell under 1 atm of dry argon in a glove box. The cell was
sealed and
removed from the glove box. The cell was maintained at 50 C for 4 hours with
helium flowing at 30 sccm at a pressure of 1 Torr. The filament power was
increased to 200 W in 20 W increments every 20 minutes. At 120 W, the filament
temperature was estimated to be in the range 800 to 1000 C. The external cell
wall
temperature was about 700 C. The cell was then operated with and without an
argon-hydrogen (95/5%) flow rate of 5.5 sccm maintained at 1 Torr.
Additionally, the
cell was operated with hydrogen gas flow replacing argon-hydrogen (95/5%). The
LiNH2 was vaporized by the filament heater as evidence the presence of Li
lines.
The presence of an argon-hydrogen or hydrogen plasma was determined by
recording the visible spectrum over the Balmer region with a Jobin Yvon Horiba
1250
M spectrometer with a CCD detector described previously [15-21 ] using
entrance/exits slits of 80/80 ,um and a 3 second integration time. The width
of the
656.3 nm Balmer a line emitted from the argon-hydrogen (95/5%)-LiNHz or
hydrogen-LiNH2 rt-plasma having a titanium filament was measured initially and
periodically during operation. As further controls, the experiment was run
with each
of the flowing gases in the absence of LiNHz .
Differential Scanning Calorimetry (DSC) Measurements. Differential scanning
calorimeter (DSC) measurements were performed using the DSC mode of a
Setaram HT-1 000 calorimeter (Setaram, France). Two matched alumina glove
fingers were used as the sample compartment and the reference compartment. The
fingers permitted the control of the reaction atmosphere. 0.067 g NaH was
placed
in a flat-base Al-23 crucible (Alfa-Aesar, 15 mm high x 10 mm OD x 8 mm ID).
The
crucible was then placed in the bottom of the sample alumina glove finger
cell. As a
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
161
reference, an aluminum oxide sample (Alfa-Aesar, -400 Mesh powder, 99.9%) with
matching weight of the sample was placed in a matched Al-23 crucible. All
samples
were handled in a glove box. Each alumina glove finger cell was sealed in the
glove
box, removed from the glove box, and then quickly attached to the Setaram
calorimeter. The system was immediately evacuated to pressure of 1 mTorr or
less.
The cell was back filled with 1 atm of helium, evacuated again, and then
refilled with
helium to 760 Torr. The cells were then inserted into the oven, and secured to
their
positions in the DSC instrument. The oven temperature was brought to the
desired
starting temperature of 100 C. The oven temperature was scanned from 100 C
to
750 C at a ramp rate of 0.1 degree/minute. As a control, MgH2 replaced NaH. A
0.050 g MgH2 sample (Alfa-Aesar, 90%, reminder Mg) was added to the sample
cell, while a similar weight of aluminum oxide (Alfa-Aesar) was added to the
reference cell. Both samples were also handled in a glove box.
Water-Flow, Batch Calorimetry. The cylindrical stainless steel reactor of
approximately 60 cm3 volume (1.0" outside diameter (OD), 5.0" length, and
0.065"
wall thickness) is shown in Figure 2. The cell further comprised a welded-in
2.5"
long, cylindrical thermocouple well with a wall thickness of 0.035" along the
centerline that held a Type K thermocouple (Omega) read by a meter (DAS). For
the cell sealed with a high temperature valve, a 3/8" OD, 0.065" thick SS tube
welded at the end of the cell 1/4" off-center served as a port to introduce
combinations of the reagents comprising the group of (i) 1 g Li, 0.5g LiNHz 1
10g
LiBr, and 15g Pd / A1203 ,(ii) 3.28 g Na, 15g Raney (R-) Ni / Al alloy, (iii)
15g R- Ni
doped with NaOH, and (iv) 3 wt%Al(OH)3 doped Ni / Al alloy . In the case that
this
port was spot-weld sealed, the SS tube had a 1/4" OD and a 0.02" wall-
thickness.
The reactants were loaded in a glove box, and a valve was attached to the port
tube
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
162
to seal the cell before it was removed from the glove box and connected to a
vacuum pump. The cell was evacuated to a pressure of 10 mTorr and crimped. The
cell was then sealed with the valve or hermetically sealed by spot-welding
1/2" from
the cell with the remaining tube cut off.
The reactor was installed inside a cylindrical calorimeter chamber shown in
Figure 3. The stainless steel chamber had 15.2 cm ID, 0.305 cm wall thickness,
and
40.4 cm length. The chamber was sealed at both ends by removable stainless
steel
plates and Viton o-rings. The space between the reactor and the inside surface
of
the cylindrical chamber was filled with high temperature insulation. The gas
composition and pressure in the chamber was controlled to modulate the thermal
conductance between the reactor and the chamber. The interior of the chamber
was
first filled with 1000 Torr helium to allow the cell to reach ambient
temperature, the
chamber was then evacuated during the calorimetric run to increase the cell
temperature. Afterwards, 1000 Torr helium was added to increase the heat
transfer
rate from the hot cell to the coolant and balance any heat associated with P-V
work.
The relative dimensions of the reactor and the chamber were such that heat
flow
from the reactor to the chamber was primarily radial. Heat was removed from
the
chamber by cooling water which flowed turbulently through 6.35 mm OD copper
tubing, which was wound tightly (63 turns) onto the outer cylindrical surface
of the
chamber. The reactor and chamber system were designed to safely absorb a
thermal power pulse of 50 kW with one a minute duration. The absorbed energy
was subsequently released to the cooling water stream in a controlled manner
for
calorimetric measurement. The temperature rise of the cooling water was
measured
by precision thermistor probes (Omega, OL-703-PP, 0.01 C) at the cooling coil
inlet
and exit. The inlet water temperature was controlled by a Cole Parmer (digital
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
163
Polystat, model 12101-41) circulating bath with 0.01 C temperature stability
and 900
W cooling capacity at 20 C. A well insulated eight-liter damping tank was
installed
just downstream of the bath in order to reduce temperature fluctuations caused
by
cycling of the bath. Coolant flow through the system was maintained by an FMI
model QD variable flow rate positive displacement lab purrip. Cooling water
flow
rate was set by a variable area flow meter with a high-resolution control
valve. The
flow meter was calibrated directly by water collection in situ. A secondary
flow rate
measurement was performed by a turbine flow meter (McMillan Co., G111
Flometer,
1 %) which continuously output the flow rate to the data acquisition system.
The
calorimeter chamber was installed in a covered HDPE tank which was filled with
melamine foam insulation to minimize heat loss from the system. Careful
measurement of the thermal power release to the coolant and comparison with
the
measured input power indicated that thermal losses were less than 2-3 %.
The calorimeter was calibrated with a precision heater applied for a set time
period to determine the percentage recovery of the total energy applied by the
heater. The energy recovery was determined by integrating the total output
power
PT over time. The power was given by
PT = r'rcCPOT (39)
where th was the mass flow rate, CP was the specific heat of water, and AT was
the absolute change in temperature between the inlet and outlet where the two
thermistors were matched to correct any offset using a constant flow with no
input
power. In first step of the calibration test, an empty reaction cell, that was
identical
to the latter tested power cell containing the reactants, was evacuated to
below 1
Torr and inserted into the calorimeter vacuum chamber. The chamber was
evacuated and then filled with helium to 1000 Torr. The unpowered assembly
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
164
reached equilibrium over an approximately two-hour period at which time the
temperature difference between the thermistors became constant. The system was
run another hour to confirm the value of the difference due to absolute
calibrations of
the two sensors. The magnitude of the correction was 0.036 C, and it was
confirmed to be consistent over all of the tests performed over the reported
data set.
To increase the temperature of the cell per input power, ten minutes before
the end of the ten-hour equilibration period, helium was evacuated from the
chamber
by the vacuum pump, and the chamber was maintained under dynamic pumping at a
pressure below 1 Torr. 100.00 W of power was supplied to the heater (50.23 V
and
1.991 A) for a period of 50 minutes. During this period, the cell temperature
increased to approximately 650 C, and the maximum change in water temperature
(outlet minus inlet) was approximately 1.2 C. After 50 minutes, the program
directed the power to zero. To increase the rate of heat transfer to the
coolant, the
chamber was re-pressurized with 1000 Torr of helium and the assembly was
allowed
to fully reach equilibrium over a 24-hour period as confirmed by the
observation of
full equilibrium in the flow thermistors.
The hydrino-reaction procedure followed that of the calibration run, but the
cell contained the reagents. The equilibration period with 1000 Torr helium in
the
chamber was 90 minutes. 100.00 W of power was applied to the heater, and after
10 minutes, the helium was evacuated from the chamber. The cell heated at a
faster
rate post evacuation, and the reagents reached a hydrino reaction threshold
temperature of 190 C at 57 minutes. The onset of reaction was confirmed by a
rapid rise in cell temperature that reached 378 C at about 58 minutes. After
ten
minutes, the power was terminated, and helium was reintroduced into the cell
slowly
over a period of 1 hour at a rate of 150 sccm.
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
165
The reactants 0.1 wt% NaOH -doped R-Ni 2800 or 0.5 wt% NaOH -doped R-
Ni 2400 (elemental analysis was provided by the manufacturer, W. R. Grace
Davidson, and the wt% NaOH was confirmed by elemental analysis (Galbraith)
performed on samples handled in an inert atmosphere) and the products
following
the reaction of these reactants as well as those of the reaction mixture
comprising
Li (1 g) and LiNH2 (0.5 g) (Alfa Aesar 99%), LiBr (10g) (Alfa Aesar ACS grade
99+%), and Pd / AlzO3 (15g) (1 % Pd, Alfa Aesar) were analyzed by quantitative
X-
ray diffraction (XRD) using hermetically sealed sample holders (Bruker Model
#A100B37) loaded in a glove box under argon and analyzed with a Siemens D5000
diffractometer using Cu radiation at 40kV/30mA over the range 10 - 70 with a
step
size of 0.02 and a counting time of eight hours. In addition, a weighed
sample of R-
Ni in a 16.5 cc stainless steel cell connected to a vacuum system having a
total
volume of 291cc was heated with a temperature ramp from 25 C to 550 C to
decompose any physically absorbed or chemisorbed gasses and to identify and
quantify the released gasses. The hydrogen content was determined by mass
spectroscopy, quantitative gas chromatography (HP 5890 Series II with a
ShinCarbon ST 100/120 micropacked column (2 m long, 1/16" OD), N2 carrier gas
with a flow rate of 14 ml/min, an oven temperature of 80 C, an injector
temperature
of 100 C, and thermal-conductivity detector temperature of 100 C), and by
using
the ideal gas law and the measured pressure, volume, and temperature. Hydrogen
dominated each analysis with trace water only detected by mass spectroscopy,
and
<2% methane was also quantified by gas chromatography. The trace water of the
R-Ni and controls was quantified independently of the hydrogen by liquefying
the
H20 in a liquid nitrogen trap, pumping off the hydrogen, and allowing all the
water to
vaporize by using a sample size of 0.5 g which is less than that which gives
rise to a
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
166
saturated water-vapor pressure at room temperature.
Synthesis and Solid 'H MAS NMR of LiH * Br, LiH * 1, NaH * Cl and
NaH * Br. Lithium bromo and iodohydrinohydride (LiH * Br and LiH * I) were
synthesized by reaction of hydrogen with Li (1 g) and LiNH2 (0.5 g) (Alfa
Aesar
99%) as a source of atomic catalyst and additional atomic H with the
corresponding
alkali halide (10 g), LiBr (Alfa Aesar ACS grade 99+%) or LiI (Alfa Aesar 99.9
%),
as an additional reactant. The compounds were prepared in a stainless steel
gas
cell (Figure 4) further containing Raney Ni (15 g) (W. R. Grace Davidson) as
the
hydrogen dissociator according to the methods described previously [13-14].
The
reactor was run at 500 C in a kiln for 72 hours with make-up hydrogen
addition such
that the pressure ranged cyclically between 1 Torr to 760 Torr. Then, the
reactor
was cooled under helium atmosphere. The sealed reactor was then opened in a
glove box under an argon atmosphere. NMR samples were placed in glass
ampules, sealed with rubber septa, and transferred out of the glove box to be
flame
sealed. `H MAS NMR was performed on solid samples of LiH * X (X is a halide)
at Spectral Data Services, Inc., Champaign, Illinois as described previously
[13-14].
Chemical shifts were referenced to external TMS. XPS was also performed on
crystalline samples that were handled as air-sensitive materials.
Since the synthesis reaction comprised LiNHz , and Li2NH was a reaction
product, both were run as controls alone and in a LiBr or LiI matrix. The
LiNH2
was the commercial starting material, and LiZNH was synthesized by the
reaction of
LiNH2 and LiH [67] and by decomposition of LiNH2 [68] with the LiZNH product
confirmed by X-ray diffraction (XRD). To eliminate the possibility that the
alkali
halide influenced the local environment of the protons or that any given known
species produced an NMR resonance that was shifted upfield relative to the
ordinary
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
167
peak, controls comprising LiH (Aldrich Chemical Company 99%), LiNH2, and
Li2NH with an equimolar mixture of LiX were run. The controls were prepared by
mixing equimolar amounts of compounds in a glove box under argon. To further
eliminate F centers as a possible contributor to the local environment of the
protons
of any given known species to produce an upfield-shifted NNIR resonance,
electron
spin resonance spectroscopy (ESR) was performed on the LiH * Br and LiH * 1
samples. For the ESR studies, the samples were loaded into 4 mm OD Suprasil
quartz tubes and evacuated to a final pressure of 10-4 Torr. ESR spectra were
recorded with a Bruker ESP 300 X-band spectrometer at room temperature and 77
K. The magnetic field was calibrated with a Varian E-500 gauss meter. The
microwave frequency was measured by a HP 5342A frequency counter.
Elemental analysis was performed at Galbraith Laboratories to confirm the
product composition and to eliminate the possibility of NMR-detectable amounts
of
any transition metal hydrides or other exotic hydrides that may give rise to
upfield-
shifted peaks. Specifically, the abundance of all elements present in the
product
(Li,H,X) and the stainless steel reaction vessel and R-Ni (Ni,Fe,Cr,Mo,Mn,AI )
were determined.
NaH * Cl and NaH * Br were synthesized by reaction of hydrogen with Na
(3.28 g) and NaH (1 g) (Aldrich Chemical Company 99%) as a source of NaH
catalyst and intrinsic atomic H with the corresponding alkali halide (15 g),
NaCI or
NaBr (Alfa Aesar ACS grade 99+%), as an additional reactant. The compounds
were prepared in a stainless steel gas cell (Figure 4) further containing Pt /
Ti (Pt
coated Ti (15 g); Titan Metal Fabricators, platinum plated titanium mini-
expanded
anode, 0.089 cm x 0.5 cm x 2.5 cm with 2.54 um of platinum) as the hydrogen
dissociator. Each synthesis was run according to the methods described for Li
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
168
except that the kiln was maintained at 500 C, and, the NaH * Cl synthesis was
repeated without the addition of hydrogen gas to determine the effect of using
NaH(s) as the sole hydrogen source. XPS was performed on NaH *Cl since no
primary element peaks were possible in the region for H- (1 / 4), and NMR
investigations of both products were preformed.
NaH *Cl was also synthesized from NaCI (10 g) and the solid acid KHSO4
(1.6 g) as the only source of hydrogen with the kiln maintained at 580 C. NMR
was
performed to test whether H- (1 / 3) formed by the reactions of Eqs. (23-25)
could be
observed when the rapid reaction to H- (1 / 4) according to Eq. (27) was
partially
inhibited due to the absence of a high concentration of H from a dissociator
with H2
or a hydride.
A silicon wafer (2 g, 0.5 x 0.5 x 0.05 cm, Silicon Quest International,
silicon
(100), boron-doped, cleaned by heating to 700 C under vacuum) was coated by
the
product NaH * Cl and NaH * by placing it in reactants comprising Na (1.7 g),
NaH
(0.5 g), NaCI (10 g), and Pt /Ti (15 g) wherein the NaCI that was initially
heated to
400 C under vacuum to remove any Hz (1 / 4). The reaction was run at 550 C in
the
kiln for 19 hours with an initial hydrogen pressure of 760 Torr. XPS was
performed
on a spot comprising only sodium hydrino hydride coated silicon wafer (NaH *-
coated Si). The NaH * Cl -coated silicon wafer (NaH * Cl -coated Si) was
investigated by electron-beam excitation spectroscopy. An emission spectrum of
a
pressed pellet of the NaH * Cl crystals was also recorded.
ToF-SIMS Spectra. The crystalline samples of LiH * Br, LiH * I, NaH * Cl,
NaH * Br, and the corresponding alkali halide controls were sprinkled onto the
surface of a double-sided adhesive tape and characterized using a Physical
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
169
Electronics TFS-2000 ToF-SIMS instrument. The primary ion gun utilized a 69Ga+
liquid metal source. A region on each sample of (60 m)2 was analyzed. In order
to
remove surface contarninants and expose a fresh surface, the samples were
sputter-
cleaned for 60 seconds using a 180 m X 100 m raster. The aperture setting was
3, and the ion current was 600 pA resulting in a total ion dose of 1015 ionsl
cm2.
During acquisition, the ion gun was operated using a bunched (pulse width 4
ns bunched to 1 ns) 15 kV beam [69-70]. The total ion dose was 1012 ions l cm2
.
Charge neutralization was active, and the post accelerating voltage was 8000
V.
The positive and negative SIMS spectra were acquired. Representative post
sputtering data is reported.
In addition, 0.1g Na, 0.5g NaH, and 15g Pt lTi were loaded into the water
flow calorimetry cell, and water flow calorimetry was performed under the same
conditions as described for Na and R-Ni. The cell generated 15 kJ of excess
energy; whereas, the theoretical energy balance from the decomposition of NaH
is
endothermic by +1.2 W. Thus, to confirm the presence of hydrino hydrides
corresponding to the reactions given by Eqs. (23-25) as the source of the
excess
heat, a sample of the Pt / Ti coated with sodium hydrino hydride (NaH * -
coated
Pt lTi ) was analyzed directly by the same procedure as for the crystalline
samples
except that the sputtering was for 100s. Unreacted Pt lTi coated with the
starting
materials served as a control. XPS was also performed.
ToF-SIMS of R-Ni 2400 reacted over a 48 hour period at 50 C was also
performed by the same procedure as for the crystalline samples. The reactions
to
form hydrinos are given by Eqs. (32-35). Since the surface was coated with
NaOH,
sodium hydrino hydride compounds with NaOH were predicted.
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
170
FTIR Spectroscopy. FTIR analysis was performed on solid-sample-KBr
pellets of LiH * Br using the transmittance mode at the Department of
Chemistry,
Princeton University, New Jersey using a Nicolet 730 FTIR spectrometer with
DTGS
detector at resolution of 4 cm-` as described previously [13-14]. The samples
were
handled under an inert atmosphere. The resolution was 0.5 cm-'. Controls
comprised LiNH2, Li2NH , and Li3Nthat were commercially available except Li2NH
that was synthesized by the reaction of LiNH2 and LiH [67] and by
decomposition
of LiNH2 [68] with the Li2NH product confirmed by X-ray diffraction (XRD).
XPS Spectra. A series of XPS analyses were made on the crystalline
samples using a Scienta 300 XPS Spectrometer. The fixed analyzer transmission
mode and the sweep acquisition mode were used. The step energy in the survey
scan was 0.5 eV, and the step energy in the high-resolution scan was 0.15 eV.
In
the survey scan, the time per step was 0.4 seconds, and the number of sweeps
was
4. In the high-resolution scan, the time per step was 0.3 seconds, and the
number of
sweeps was 30. C ls at 284.5 eV was used as the internal standard.
UV Spectroscopy of Electron-Beam Excited Interstitial H2 (1 / 4). Vibration-
rotational emission of H2(1/4) trapped in the lattice of alkali halides,
MgCl2, and in a
silicon wafer was investigated via electron bombardment of the crystals.
Windowless UV spectroscopy of the emission from electron-beam excitation of
the
crystals was recorded using a 12.5 keV electron gun at a beam current of 10-20
,uA
in the pressure range of <10-5 Torr. The UV spectrum was recorded with a
photomultiplier tube (PMT). The wavelength resolution was about 2 nm (FWHM)
with an entrance and exit slit width of 300 m. The increment was 0.5 nm and
the
dwell time was 1 second.
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
171
III. Results and Discussion
A. RT-plasma Emission and Balmer a Line Widths. An argon-hydrogen (95/5%)-
lithium rt-plasma formed with a low field (1 V/cm), at low temperatures (e.g.
= 103 K),
from atomic hydrogen generated at a titanium filament and LiNH2 that was
vaporized by heating. Lithium and H emission were observed that confirmed
LiNH2
and its decomposition product Li served as a source of atomic Li and H. Argon
of
the argon-hydrogen mixture increased the amount of atomic H as evidenced by
the
significantly decreased H emission in the absence of argon. H Balmer emission
corresponding to population of a level with energy > 12 eV was observed, as
shown
in Figures 5 and 6, which also requires that Lyman emission was present.
No plasma formed with argon/hydrogen alone. No possible chemical reaction
of the titanium filament, the vaporized LiNH2 1 and 0.6 Torr argon-hydrogen
mixture
at a cell temperature of 700 C could be found to account for the Balmer
emission. In
fact, no known chemical reaction releases enough energy to excite Balmer and
Lyman emission from hydrogen. In addition to known chemical reactions,
electron
collisional excitation, resonant photon transfer, and the lowering of the
ionization and
excitation energies by the state of "non ideality" in dense plasmas were also
rejected
as the source of ionization or excitation to form the hydrogen plasma [21].
The
formation of an energetic reaction of atomic hydrogen was consistent with a
source
of free energy from the catalysis of atomic hydrogen by Li.
The energetic hydrogen atom energies were calculated from the width of the
656.3 nm Balmer a line emitted from RF rt-plasmas. The full half-width A.~, of
each
Gaussian results from the Doppler (A2,o ) and instrumental (AA,) half-widths:
0A~ = OAo + AA,; (40)
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
172
0,~, in our experiments was 0.006 nm. The temperature was calculated from the
Doppler half-width using the formula:
1 2
A,~, =7.16X10-'/1o~ T) (41)
where ~,o is the line wavelength, T is the temperature in K(1 eV= 11,605 K),
and
is the molecular weight (=1 for atomic hydrogen). In each case, the average
Doppler
half-width that was not appreciably changed with pressure, varied by 5%
corresponding to an error in the energy of 10%.
The 656.3 nm Balmer a line widths recorded on the argon-hydrogen (95/5%)-
lithium rt-plasma, initially and after 70 hours of operation, are shown in
Figures 5A
and 5B, respectively. The Balmer a line profile of the plasma emission at both
time
points comprised two distinct Gaussian peaks, an inner, narrower peak
corresponding to a slow component of less than 0.5 eV and an outer,
significantly
broadened peak corresponding to a fast component of >40 eV. The fast component
accounted for 90% of the n = 3 excited-state H population initially and
increased to
97% at 70 hours. Only the hydrogen lines were broadened. As shown previously,
the source of energy of the fast H cannot be attributed to any applied
electric field,
but is predicted by the mechanism of the catalysis of hydrogen to lower-states
[32-
37].
A lithium rt-plasma also formed in the case of pure HZ gas at a pressure of 1
Torr, except that the line broadening and populations were less, about 6 eV
with only
a 27% population, at the initial and 70-hour time points as shown in Figures
6A and
6B, respectively. This result was expected, since the excess H2 can react with
Li
to form LiH that catalyzes the destruction of LiNHZ by the reaction:
LiH + LiNH2 -4 Li2NH + H2 (42)
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
173
Thus, the reactions to produce atomic Li and H are diminished. In addition,
argon
of the argon-hydrogen mixture can increase the amount of atomic H by
preventing its
recombination, and Ar+ generated by the plasma can participate as a catalyst
as
well as Li.
We have assumed that Doppler broadening due to thermal motion was the
dominant source to the extent that other sources may be neglected. This
assumption was confirmed when each source was considered. In general, the
experimental profile is a convolution of two Doppler profiles, an instrumental
profile,
the natural (lifetime) profile, Stark profiles, van der Waals profiles, a
resonance
profile, and fine structure. The contribution from each source was determined
to be
below the limit of detection [13-21, 38-39].
The formation of fast H can be explained by a resonant energy transfer from
hydrogen atoms to Li atoms, of three times the potential energy of atomic
hydrogen,
to form a short-lived intermediate H*(1 / 4) having a central field equivalent
to four
times that of a proton and a radius of the hydrogen atom. The intermediate
spontaneously decays by a collisional or through-space energy transfer as the
radius
decreases to ao / 4 yielding fast H(n = 1), as well as the emission of q= 13.6
eV
photons reported previously [27-29]. Collisional energy transfer including
through-
space coupling is common. For example, the exotherrriic chemical reaction of
H + H to form HZ does not occur with the emission of a photon. Rather, the
reaction
requires a collision with a third body, M, to remove the bond energy-
H + H + M --> H2 + M * [44]. The third body distributes the energy from the
exothermic reaction, and the end result is the H2 molecule and an increase in
the
temperature of the system. In the case of the catalytic reaction with the
formation of
states given by Eqs. (2a) and (2c), the temperature of H becomes very high.
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
174
B. Differential Scanning Calorimetry (DSC) Measurements. The DSC (100-750 C)
of NaH is shown in Figure 7. A broad endothermic peak was observed at 350 C to
420 C which corresponded to 47 kJ / mole. Sodium hydride decomposes in this
temperature range with a corresponding enthalpy of 57 kJ / mole [71 ]. A large
exotherm was observed in the region 640 C to 825 C which corresponded to
-177 ki / mole . The DSC (100-750 C) of MgH2 is shown in Fjgure 8. Two sharp
endothermic peaks were observed. A first peak was observed centered at 351.75
C corresponding to 68.61 kJ / mole MgH2 . The decomposition of MgH2 is
observed
at 440 C to 560 C corresponding to 74.4 kJl mole MgH2 [71 ]. In Figure 8, a
second
peak was observed centered at 647.66 C corresponding to 6.65 U / mole MgH2.
The known melting point of Mg(m) is 650 C corresponding to an enthalpy of
fusion
of 8.48 kJ/ mole Mg(m) [72]. Thus, the expected behavior was observed for the
decomposition of a control, noncatalyst hydride. In contrast, a novel
exothermic
effect of -177 kJ / moleNaH or at least -354 kJ / moleH2 was observed under
conditions that form NaH catalyst with some portion of the H undergoing the
catalysis reactions given by Eqs. (23-25). The observed enthalpy was greater
than
that of the most exothermic reaction possible for H, the -241.8 kJ / mole H2
enthalpy
of combustion of hydrogen.
C. Water-Flow Calorimetry Power Measurements. In each test, the energy input
and energy output were calculated by integration of the corresponding power.
For
the input power, the voltage and current measured at the end of each time
interval
were multiplied by the time interval (typically 10 seconds) to obtain the
energy
increment in Joules. All energy increments were summed over the entire
experiment
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
175
after the equilibration period to obtain total energy. For output energy, the
thermistor
offset was calculated after each test assuming that the final readings of
inlet and
outlet temperature were identical. This offset was calculated to be 0.036 C.
The
thermal energy in the coolant flow in each time increment was calculated using
Eq.
(39) by multiplying volume flow rate of water by the water density at 19 C
(0.998
kg/liter), the specific heat of water (4.181 kJ/kg- C), the corrected
temperature
difference, and the time interval. Values were summed over the entire
experiment to
obtain the total energy output. The total energy from cell E, must equal the
energy
input E. and any excess energy Ee,:
E,. = E;,, + EPZ (43)
From the energy balance, any excess heat was determined.
The calibration test results are shown in Figures 9 and 10. In the plot of
Figure 10, there is a time point at which the slope of the coolant power
changes
almost discontinuously. This point at about one hour corresponds to the helium
addition enhancing heat transfer from the cell to the chamber wall. The
numerical
integration of the input and output power curves yielded an output energy of
292.2 kJ
and an input energy of 303.1 kJ corresponding to a coupling of flow of 96.4%
of the
resistive input to the output coolant.
The cell temperature with time and the coolant power with time for the hydrino
reaction with the cell containing the reagents comprising the catalyst
material, 1 g Li,
0.5g LiNHZ , 10g LiBr, and 15g Pd / Alz0, are shown in Figures 11 and 12,
respectively. The numerical integration of the input and output power curves
with
the calibration correction applied yielded an output energy of 227.2 kJ and an
input
energy of 208.1 kJ. Thus, from Eq. (43), the excess energy was 19.1 kJ. In the
plot
of Figure 12, there is a point at which the slope of the temperature changes
almost
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
176
discontinuously. The slope change occurs just slightly after 1 hour, and this
corresponds to the cell temperature rising rapidly with the onset of reaction.
Based
on the system response to a power pulse, the excess energy of 19.1 kJ occurred
in
less than 2 minutes which places the power for the reaction at over 160 W.
The quantitative XRD of the composition of the products following the reaction
showed that the LiBr and Pd / A1Z03 were unchanged. Thus, assuming a 100%
yield, the maximum theoretical energy released by known chemistry is 4.3 kJ
from
the formation lithium nitride and hydride according to Eq. (22); whereas, the
observed energy balance was 4.4 times this maximum. The only exothermic
reaction possible to account for the energy balance is that given by Eqs. (17-
19).
The hydrogen content of the 0.5g LiNH2 was 22 mmoles H2. Thus, the observed
energy balance is -870 kJ / mole Hz , over 3.5 times the -241.8 kJ / mole H2
enthalpy
of combustion, the most energetic reaction of hydrogen assuming the maximum
possible Hz inventory.
The cell temperature with time and the coolant power with time for the R-Ni
control power test with the cell containing the reagents comprising the
starting
material for R-Ni, 15g R-Ni/Al alloy powder, and 3.28g of Na are shown in
Figures 13
and 14, respectively. The temperature and coolant power time profiles curves
were
very similar to the calibration. The numerical integration of the input and
output
power curves with the calibration correction applied yielded an output energy
of 384
kJ and an input energy of 385 kJ. Energy balance was obtained.
The cell temperature with time and the coolant power with time for the hydrino
reaction with the cell containing the reagents comprising the catalyst
material, 15g
NaOH -doped R-Ni, and 3.28g of Na are shown in Figures 15 and 16,
respectively.
The numerical integration of the input and output power curves with the
calibration
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
177
correction applied yielded an output energy of 185.1 kJ and an input energy of
149.1
kJ. Thus, from Eq. (43), the excess energy was 36 kJ. In the plot of Figure
15, there
is a point at which the slope of the temperature changes almost
discontinuously.
The slope change occurs just slightly before 1 hour, and this corresponds to
the cell
temperature rising rapidly with the onset of reaction. Based on the system
response
to a power pulse, the excess energy of 36 kJ occurred in less than 1.5 minutes
which places the power for the reaction at over 0.5 kW.
The composition of the reactant NaOH -doped R-Ni and the product following
the reaction with the alkali metal determined by quantitative XRD was Ni with
trace
Bayerite and Ni with trace alkali hydroxide, respectively. The formation of a
sodium-
Ni alloy or the reaction of sodium with A1203 of R-Ni [73-74] is significantly
endothermic ( OH = +138 kJ / mole Na [75] and OH = +72.18 kJ / mole Na [65],
respectively). Using the heat of formations, the reaction of Bayerite with
sodium to
form NaOH (AH =-15.6 kJ / mole Al (OH)3 [65, 76]) contributes negligibly to
the
energy balance based on the XRD analysis showing trace Bayerite initially and
the
corresponding NaOH product from reaction with Na. Consistent with the
literature
[74], the H20 content from Bayerite decomposition was 47.7 moles H20/g R-Ni
corresponding to a negligible contribution due to the formation of NaOH
( OH =-184.0 kI / mole HZO [65]) from the decomposition of Al(OH)3
( 2Al (OH)3 -4 A1203 + 3H2O OH = +92.45 kI / mole Al). The overall reaction is
the
reaction of Bayerite with sodium to form NaOH (AH = -15.6 kJ / mole Al (OH)3
).
The only exothermic reaction possible to account for the energy balance is
that given by Eqs. (23-25). The hydrogen content of the R-Ni determined using
quantitative GC and by using the ideal gas law on the measured P, V, and T was
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
178
150 moles Hz lg R-Ni. Thus, the observed energy balance is
-1.6X104 kJ / mole H2 , over 66 times the -241.8 kJ / mole HZ enthalpy of
combustion,
the most energetic reaction of hydrogen assuming the maximum possible H2
inventory. The conservative theoretical energy yield for the reaction of Eq.
(44) is
259 eV / H2 or 25 MJ / moleH2 (Eq. (7)).
Hz -> Hz (1 / 3) (44)
Among the most energetic known oxidation reactions involving a solid fuel is
the
reaction Be + 1/ 202 -> Be0, which has a heat of combustion of 24 kJ/g, and
there
are very few known fuel/oxidizer systems producing greater than 10 kJ/g [65].
As a
comparison, even without possibly going to completion, the H content of the
recyclable catalyst NaH produced energy of over 300 times that of the best
known
solid fuel per weight.
With increased NaOH doping and a switch to R-Ni 2400, the catalytic material
generated high power and energy without requiring the addition of Na. The cell
temperature with time and the coolant power with time for the hydrino reaction
with
the cell containing the catalyst material, 15g NaOH -doped R-Ni 2400, are
shown in
Figures 17 and 18, respectively. The numerical integration of the input and
output
power curves with the calibration correction applied yielded an output energy
of
195.7 kJ and an input energy of 184.0 kJ corresponding to an excess energy of
11.7
kJ, and the power was over 0.25 kW.
The composition of the reactant NaOH -doped R-Ni and the product following
the reaction determined by quantitative XRD was R-Ni with 3.7wt% Bayerite and
R-
Ni, respectively. The measured H20 content from Bayerite decomposition of the
initial R-Ni was 32.8 moles Hz0/g R-Ni compared to the measured H20 content
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
179
from Bayerite decomposition of 34.0 moles H2O1g for 3 wt%Al(OH)3 doped
Ni / Al alloy. The most exothermic reaction possible was the reaction of Al
(OH)3 to
A1203. The balanced reaction is given by [65, 75, 77]:
2A1(OH)3 + 2Ni5Al -> 2A1203 + Ni1oH6 AH = -263.9 kJ / mole Al (OK (45)
For 3.7wt% Al(OH)3, the maximum theoretical energy from the reaction given by
Eq. (45) is OH=-1.88 kJ. This was confirmed by the heat measurement of 15g of
3
wt%Al(OH)3 doped Ni / Al alloy that showed and average energy of OH =-1.1 kJ
compared to the theoretical energy of AH = -1.7 k/ (AH = -300 kJ / mole Al
(OH)3
using Eq. (45) with AHf (NiAlcrystal) = -96 kJ/mole [75]). Thus, the observed
energy
from the NaOH -doped R-Ni was 4.4 times the theoretical; thus, it was
predominantly
attributable to the catalysis reaction given by Eqs. (23-25).
D. ToF-SIMS Spectra. The positive ToF-SIMS spectrum obtained from LiBr and
the LiH * Br crystals are shown in Figures 19 and 20, respectively. The
positive ion
spectrum of the LiH * Br crystals and that of the LiBr control were dominated
by the
Li' ion. Li2 , Na+, Ga+, and Li(LiBr)+ were also observed.
The negative ion ToF-SIMS of LiBr and the LiH *Br crystals are shown in
Figures 21 and 22, respectively. The LiH * Br spectrum was dominated by H- and
Br- peaks with the intensity of H- > Br. Bromide alone dominated the negative
ion ToF-SIMS of the LiBr control. For both, O- , OH-, Cl-, and LiBr- were also
observed. In addition to the increased hydride, other unique peaks of the LiH
* Br
sample were LiHBr- and Li2H2Br- consistent with the formation of novel lithium
bromohydride.
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
180
The positive ToF-SIMS spectrum obtained from Li1 and the LiH * I crystals
are shown in Figures 23 and 24, respectively. The positive ion spectrum of the
LiH * I crystals and that of the Lii control were dominated by the Li+ ion.
Liz ,
Na', Ga', and a series of positive ions Li[LiI]n were also observed. Unique
peaks
of the LiH * 1 sample were LiHI+, Li2HZI` , Li4H21' , and Li6H2I+ .
The negative ion ToF-SIMS of Lil and the LiH * 1 crystals are shown in
Figure 25 and 26, respectively. The LiH * I spectrum was dominated by H- and I-
peaks with the intensity of H- > I-. Iodide alone dominated the negative ion
ToF-
SIMS of the Li7 control. For both, O- , OH-, Cl", and a series of negative
ions
I[Li1]n were also observed. In addition to the increased hydride, other unique
peaks of LiH * I sample were LiHr, LiZHzI- , and NaHl- consistent with the
formation of novel lithium iodohydride.
The negative ToF-SIMS spectrum (m / e = 20 - 30) of NaH * -coated Pt / Ti
following the production of 15 kJ of excess heat is shown in Figure 27.
Hydrino-
hydride-compound series NaHX was observed wherein the mass deficit from the
hjgh resolution (10,000) mass determination definitively distinguished this
assignment over the CZHx series observed in controls. The XPS spectrum showed
that NaH * -coated Pt / Ti comprised two fractional hydrogen states, H- (1 /
3) and
H- (1 / 4) (Sec. I I IF).
NaHx having the mass-deficit series was also observed in the spectrum of R-
Ni from the Na/R-Ni water-flow calorimetric run that produced 36 kJ of excess
heat.
The positive ToF-SIMS spectrum obtained from R-Ni reacted over a 48 hour
period
at 50 C is shown in Figure 28. 'rhe dominant ion on the surface was Na`
consistent
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
181
with NaOH doping of the surface. The ions of the other major elements of R-Ni
2400 such as Al` , Ni+, Cr+, and Fe` were also observed.
The negative ion ToF-SIMS of R-Ni reacted over a 48 hour period at 50 C is
shown in Figure 29. The spectrum showed a very large H- peak as well as
hydroxide fragments OH- and O- . Two other dominant peaks matched the high
resolution mass of NaH3 and NaH3NaOH- to 10,000 and were assigned to sodium
hydrino hydride and this ion in combination with NaOH. Other unique ions
assignable to sodium hydrino hydrides NaHX in combinations with NaOH, NaO,
OH- and O- were observed.
E. NMR Identification of H- (1 / 3), H-(1 / 4), H2 (1 / 3) and H2 (1 / 4). The
`H MAS
NMR spectra of LiH * Br and LiH * I relative to external TMS are shown in
Figures
30A and 30B, respectively. LiH * X samples showed a large distinct upfield
resonance at -2.51 ppm and -2.09 ppm for X = Br and X= I, respectively. None
of the controls comprising LiH, equal molar mixtures of LiH and LiBr or Lii,
LiNH2, Li2NH, and equal molar mixtures of LiNH2 or LiZNH and LiBr or LiI
showed an upfield-shifted peak. Since the upfield peak of LiH * X at about -
2.2
ppm was very broad, it is useful to compare these results to those of the
prior
identification of H- (1 / 4) of KH * Cl and KH * I.
The `H MAS NMR spectra relative to TMS of KH* Cl samples (Figure 31A)
from independent syntheses and controls were given previously [13-15, 24-26].
The
experimental absolute resonance shift of TMS is -31.5 ppm relative to the
proton's
gyromagnetic frequency [78-79]. The KH experimental shift of +1.1 ppm relative
to
TMS corresponding to absolute resonance shift of -30.4 ppm matches very well
the
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
182
predicted shift of H-(1 / 1) of -30 ppm given by Eq. (4) wherein p= 0. The
novel
peak at -4.46 ppm relative to TMS corresponding to an absolute resonance shift
of
-35.96 ppm indicates that p = 4 in Eq. (4). H-(1/4) is the hydride ion
predicted by
using K as the catalyst [1, 15, 30]. Furthermore, the extraordinarily narrow
peak-
width is indicative of a small hydride ion that is a free rotator. In
contrast, KH * I
(Figure 31 B) shows a very broad peak at -2.31 ppm. The predicted product
hydride
ion H-(1/4) of the reaction with K catalyst to form KH * I was observed by XPS
[13-
15, 26, 30] at its predicted binding energy of 11.2 eV. Thus, the diamagnetic
shift
due to the larger halide is +2.15 ppm. The corrected upfield NMR peaks for LiH
* X
are each about -4.46 ppm which matches the predicted shift of the free ion
given by
Eq. (4).
The elemental analysis of LiH * Br by wt% was Li (8%), H(1.1 %), 1(90.9%)
corresponding stoichiometrically to LiHBr with the stainless steel and R-Ni
components at less than detectable levels. The elemental analysis of LiH * I
by
wt% was Li (5.2%), H(0.8%), I(94 /a) corresponding stoichiometrically to LiHI
with
the stainless steel and R-Ni components at less than detectable levels. Thus,
no
hydrides other than those of Li are possible assqgnments. U H does not have an
upfield-shifted NMR peak as determined previously [13-14]. F centers could not
have been the source since no ESR signal was detectable in LiH * Br or LiH * I
at
room temperature or 77 K. `H MAS NMR spectra obtained on LiNH2, LiZNH , and
these compounds in a LiBr or Lil matrix also showed that neither of these
compounds have an upfield-shifted NMR peak. To further eliminate LiNHZ and
Li2NH as the source of the -2.5 ppm peak, LiH * Br samples with the -2.5 ppm
peak were heated to >600 C under dynamic vacuum to decompose LiNH2 and
Li2NH. The heat-treated samples were analyzed by FTIR spectroscopy to confirm
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
183
that the amide and imide were eliminated as indicated by the absence of the
amide
peaks at 3314, 3259, 2079(broad), 1567, and 1541 cm-' and the imide peaks at
3172
(broad), 1953, and 1578 cm-' while the -2.5 ppm peak remained upon reanalysis
by
NMR. The FTIR spectrum shown in Figure 45B shows the elimination of these
species while the corresponding NMR showed the -2.5 ppm peak. Since the past
and present NMR and FTIR analysis leads to the conclusion that the -2.5 ppm
peak
in 'H NMR spectrum is not associated with the U H, LiNH2 , Li2NH, or any other
known species, the -2.5 ppm peak in 'H NMR spectrum is assigned to the H(1/4)
ion which matches theoretical prediction and is direct evidence of a lower-
energy
state hydride ion.
In addition to the -2.5 ppm and -2.09 ppm peaks assigned to H-(1/4) , a 1.3
ppm peak was observed in the `H MAS NMR spectra of LiH * Br and LiH * I
shown in Figures 30A. and 30B, respectively. None of the controls showed this
peak
which eliminated any of the starting compounds or their possible known
products.
However, the peak may be due to the H2 (1 / 4) molecule corresponding to H- (1
/ 4).
HZ has been characterized by gas-phase `H NMR. The experimental
absolute resonance shift of gas-phase TMS relative to the proton's
gyromagnetic
frequency is -28.5 ppm [80]. Hz was observed at 0.48 ppm compared to gas phase
TMS set at 0.00 ppm [81]. Thus, the corresponding absolute H2 gas-phase
resonance shift of -28.0 ppm (-28.5 + 0.48) ppm was in excellent agreement
with
the predicted absolute gas-phase shift of -28.01 ppm given by Eq. (12).
The absolute H2 gas-phase shift can be used to determine the matrix shift for
H2 in a lithium-compound matrix. The correction for the matrix shift can then
be
applied to the 1.3 ppm peak to determine the gas-phase absolute shift to
compare to
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
184
Eq. (12). The shifts of all of the peaks were relative to liquid-phase TMS
which has
an experimental absolute resonance shift of --31.5 ppm relative to the
proton's
gyromagnetic frequency [78-79]. The experimental shift of H2 in a lithium-
compound
matrix of 4.06 ppm relative to liquid-phase TMS is shown in Figure 7 of Lu et
al. [82]
and corresponds to an absolute resonance shift of -27.44 ppm (-31.5 ppm + 4.06
ppm). Using the absolute HZ gas-phase resonance shift of -28.0 ppm
corresponding to 3.5 ppm (-28.0 ppm - 31.5 ppm) relative to liquid TMS, the
lithium-
compound matrix effect is +0.56 ppm (4.06 ppm - 3.5 ppm) requiring a
correction of
the measured shift of -0.56 ppm. Then, the peak upfield of H2 at 1.26 ppm peak
relative to TMS corresponds to a matrix-corrected absolute resonance shift of -
30.8
ppm (-31.5 ppm + 1.26 ppm - 0.56 ppm). Using Eq. (12), the data indicates p =
4
and matches H2 (1 / 4) :
ABT = -(28.01+ 0.64p)ppm
B
_ -(28.01 +0.64(4)) ppm (46)
_ -30.6 ppm
Lu et al. [82] also observed a peak at this position that increased in
intensity relative
to H2 with the duration of in situ heating of LiH+LiNH2 (1.1/1). They were
unable
to assign the peak labeled unknown in their Figures 6 and 7. The assignment of
the
peak that matched the theoretical shift of Hz (1 / 4) extremely well, was
confirmed by
FTIR (Sec. IIIG) and electron beam-excitation emission spectroscopy (Sec.
IIIH).
The presence of the H- (1 / 4) ion in LiH * X was found to depend on the
polarizability of the halide ion. The 'H MAS NMR spectra of LiH * F and LiH *
Cl
are shown in Figures 32A and 32B, respectively. Peaks at 4.3 ppm and 1.2 ppm
matched theoretical predictions of molecular hydrogen in two different quantum
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
185
states [1, 6]. The 4.3 ppm peak matched the assignment of Lu et al. [82] for
H2 1
and the 1.2 ppm peak labeled unknown by Lu et al. [82] matched H2 (1 / 4). The
H2 (1 / 4) assignment was confirmed by the observation of the predicted
rotational
transition in the FTIR spectrum (Sec. IIIG) and the predicted rotational
spacing by
electron beam-excitation emission spectroscopy (Sec. IIIH). The H- (1 / 4) ion
peak
was absent in LiH * F comprising a nonpolarizable fluorine as well as in LiH *
Cl
comprising a nonpolarizable chlorine; whereas, it was the dominant peak in
both
LiH * Br and LiH * I as shown in Figures 30A and 30B, respectively. These
results
indicate that a polarizable halide is required for LiX to react with the H- (1
/ 4) ion to
form the corresponding lithium halidohydride. Since molecular species are
nonspecifically trapped in the crystalline lattice, the H-content selectivity
of LiH * X
for molecular species alone or in combination with H- (1 / 4) ions is based on
the
polarizability of the halide and the corresponding reactivity towards H-(1/4)
.
Potassium catalyst formed HZ (1 / 4) as well, but in KCl and KI matrices with
H- (1 / 4), as shown in Figures 31 A and 31 B.
The 'H MAS NMR spectra of NaH * Br relative to external TMS is shown in
Figure 32. NaH * Br showed a large distinct upfield resonance at -3.58 ppm.
None
of the controls comprising NaH or equal molar mixtures of NaH and NaBr showed
an upfield-shifted peak. The -3.58 ppm upfield peak of NaH * Br was broadened,
but not significantly as in the case of KH * I; thus, the matrix may not have
as large
an effect as in the prior case of the identification of H- (1 / 4) in KH * I.
Thus, the
measured shift is directly compared to theory with the expectation of that it
is the
peak shifted downfield due to the matrix effect. The experimental absolute
resonance shift of TMS is -31.5 ppm relative to the proton's gyromagnetic
frequency
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
186
[78-79]. The novel peak at -3.58 ppm relative to TMS corresponding to an
absolute
resonance shift of -35.08 ppm indicates that p = 4 in Eq. (4). H- (1 / 4) is
the
favored hydride ion predicted by using NaH as the catalyst (Eqs. (3-4) and (23-
27)).
Similar to the case of LiH * X, the 4.3 ppm peak shown in Figure 33 is
assigned to
Hz , and the 1.13 ppm peak is assigned to H2 (1 / 4). The latter is commonly
observed as a favored catalysis molecular product [29].
NaH *Cl 'H MAS IVMR spectra relative to external TMS showing the effect of
hydrogen addition on the relative intensities of HZ , H2 (1 / 4),and H- (1 /
4) is shown
in Figures 34A-B. The addition of hydrogen increased the H- (1 / 4) peak and
decreased the HZ (1 / 4) while the Hz increased. (A) NaH * Cl synthesized with
hydrogen addition showing a -4 ppm upfield-shifted peak assigned to H- (1 /
4), a
1.1 ppm peak assigned to Hz (1 / 4), and a dominant 4 ppm peak assigned to H2
.
(B) NaH * Cl synthesized without hydrogen addition showing a -4 ppm upfield-
shifted peak assigned to H" (1 / 4), a dominant 1.0 ppm peak assigned to Hz (1
/ 4),
and a small 4.1 ppm assigned to HZ .
The effect of hydrogen addition on the relative 'H MAS NMR intensities of
H2 1 H2 (1 / 4), and H- (1 / 4) in NaH * Cl is shown in Figures 34A-B. The
dominant
peak switched from being HZ to H2 (1 / 4) with the addition of external
hydrogen
indicating that H2 may occupy sites in the lattice that are filled by HZ (1 /
4) when
H2 is less abundant. However, the addition of hydrogen increased the relative
intensity of the H-(1 / 4) peak, mostly likely by increasing the hydrino
reactant
concentration.
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
187
NMR was performed on NaH * Cl synthesized from NaCI and the solid acid
KHSO4 as the only source of hydrogen to test whether H- (1 / 3) formed by the
reactions of Eqs. (23-25) could be observed when the rapid reaction to H- (1 /
4)
according to Eq. (27) was partially inhibited due to the absence of a high
concentration of H from a dissociator with H2 or a hydride. The 1H MAS NMR
spectrum of NaH * Cl formed using the solid acid relative to external TMS is
shown
in Figure 35. Peaks at -3.97 ppm and 1.15 ppm matched the -4 ppm and 1.1 ppm
peaks of Figures 34A-B that were assigned to H- (1 / 4) and HZ (1 / 4),
respectively,
of NaH * Cl synthesized using H from a dissociator with H2 or a hydride. The
close match was expected since the KHSO4 was only 6.5 mole% of the mixture
with
NaCI such that the matrix effect was essentially constant between samples.
Uniquely, another set of peaks at -3.15 ppm and 1.7 ppm was observed for the
solid-acid product. Using Eqs. (4) and (12) with the matrix shift given
previously for
NaH * Cl, these peaks matched and were assigned to H- (1 / 3) and H2 (1 / 3),
respectively. Curve fitting of two peaks put the peaks at about -3 ppm and -4
ppm,
the theoretical values with experimental error. Thus, both fractional hydrogen
states
were present, and the H2 peak was absent at 4.3 ppm due to the synthesis of
NaH * Cl using a solid acid as the only H source which confirms the reactions
given
by Eqs. (23-30). The presence of H- (1 / 4) and H2 (1 / 4) in NaH * Cl from
reaction
of NaCI and the solid acid KHSO4 was confirmed by XPS and electron beam-
excitation emission spectroscopy.
Helium is another catalyst that can cause a transition reaction to [-]
because the second ionization energy is 54.4 eV, (2= 27.2 eV). The catalyst
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
188
reactions are given by
54.4eV+He++H kH-1 HeZ++e +H aN +[(p+2)2-p2]13.6eV (47)
(p + 2)
Hez + e-4 He+ + 54.4 eV (48)
And, the overall reaction is
H p -) H (p~2) ]+[(P+2)2 -p2]=13.6eV (49)
As in the case of the NaH catalyst reaction, the subsequently rapid transition
of the
He' catalysis product [-] to [-] may occur via further catalysis by atomic
hydrogen that first accepts 27.2 eV from [-] as given by Eq. (27).
Characteristic
broad emission starting at 46.5 nm and continuing to shorter wavelengths is
predicted for this transition reaction as the energetic H catalyst decays. The
emission has been observed by EUV spectroscopy recorded on microwave
discharges of helium with 2% hydrogen [27-29]. The spectroscopic and NMR data
provide strong support for the catalyst mechanism of the formation of [-] with
the
subsequent transition to L 4 J. Additional evidence is the observation of both
H- (1 / 3) and H- (1 / 4) in NaH * Cl as given in Sec. IIIF.
F. XPS Identification of H-(1 /4) and H- (1 / 3). A survey spectrum was
obtained
on each of LiBr and LiH * Br over the region Eb = 0 eV to 1200 eV (Figures 36A-
B).
The primary element peaks allowed for the determination of all of the elements
present in the LiH * Br crystals and the control LiBr. No elements were
present in
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
189
the survey scan which could be assigned to peaks in the low binding energy
region
(Figure 37) with the exception of the Li is peak at 55 eV (shifted I eV lower
compared to LiBr), the 0 2s at 23 eV, the Br 3ds,Z and Br 3d3,2 peaks at 69 eV
and
70 eV, respectively, the Br 4s at 15 eV, and the Br 4d at 5 eV. Accordingly,
any
other peaks in this region must be due to novel species. As shown in Figure
37, the
XPS spectrum of LiH * Br differs from that of LiBr by having additional peaks
at 9.5
eV and 12.3 eV that do not correspond to any other primary element peaks but
do
match the H- (1 / 4) Eb =11.2 eV hydride ion (Eqs. (4) and (16)). The
literature was
searched for elements having a peak in the valance-band region that could be
assigned to these peaks. Given the primary element peaks present, there was no
known alternative assignment. Thus, the 9.5 eV and 12.3 eV peaks that could
not
be assigned to known elements and do not correspond to any other primary
element
peak were assigned to the H- (1 / 4) in two different chemical environments.
These
features closely matched those for H- (1 / 4) of KH * I reported previously
[13-15,
26, 30].
A survey spectrum was obtained on each of NaBr and NaH * Br over the
region Eb = 0 eVto 1200 eV (Figures 38A-B). The primary element peaks allowed
for
the determination of all of the elements present in the NaH * Br crystals and
the
control NaBr. No elements were present in the survey scan which could be
assigned to peaks in the low binding energy region (Figure 39) with the
exception of
the Na 2p and Na 2s peaks at 30 eV and 63 eV (shifted 1 eV lower compared to
NaBr), the 0 2s at 23 eV, the Br 3d5,2 and Br 3d3,2 peaks at 69 eV and 70 eV,
respectively, the Br 4s at 15.2 eV, and the Br 4d at 5 eV. Accordingly, any
other
peaks in this region must be due to novel species. As shown in Figure 39, the
XPS
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
190
spectrum of NaH * Br differs from that of NaBr by having additional peaks at
9.5 eV
and 12.3 eV that do not correspond to any other primary element peaks but do
match the H-(1 /4) Eb =11.2 eV hydride ion (Eqs. (4) and (16)). The literature
was
searched for elements having a peak in the valance-band region that could be
assigned to these peaks. Given the primary element peaks present, there was no
known alternative assignment. Thus, the 9.5 eV and 12.3 eV peaks that could
not
be assigned to known elements and do not correspond to any other primary
element
peak were assigned to the H- (1 / 4) in two different chemical environments.
Survey spectra over the region Eb = 0 eVto 1200 eV were obtained on each of
Pt lTi and NaH * -coated Pt lTi following the production of 15 kJ of excess
heat
(Figures 40A-B). The primary element peaks allowed for the determination of
all of
the elements present in the NaH * -coated Pt / Ti and the control Pt / Ti. No
elements were present in the survey scan which could be assigned to peaks in
the
low binding energy region (Figures 41A-B) with the exception of the Pt 4 f72
and
Pt 4 f5,z peaks at 70.7 eV and 74 eV, respectively, and the 0 2s at 23 eV. The
Na 2p and Na 2s peaks were observed at 31 eV and 64 eV on NaH * -coated
Pt lTi , and a valance band was only observed for Pt lTi . Accordingly, any
other
peaks in this region must be due to novel species. As shown in Figures 42A-B,
the
XPS spectrum of NaH * -coated Pt / Ti differs -From that of Pt / Ti by having
additional peaks at 6 eV, 10.8 eV, and 12.8 eV that do not correspond to any
other
primary element peaks but do match the H- (1 / 3) Eb = 6.6 eV and
H- (1 / 4) Eb =11.2 eV hydride ions (Eqs. (4) and (16)). The literature was
searched
for elements having a peak in the valance-band region that could be assigned
to
these peaks. Given the primary element peaks present, there was no known
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
191
alternative assignment. Thus, the 10.8 eV, and 12.8 eV peaks that could not be
assigned to known elements and do not correspond to any other primary element
peak were assigned to the H- (1 / 4) in two different chemical environments.
The 6
eV peak matched and was assigned to H- (1 / 3). Thus, in the absence of a
halide
peak in this region, both fractional hydrogen states, 1/3 and 1/4, were
observed as
predicted by Eq. (27). The absence of a valance band due to the high-binding
energies was also consistent with the hydrino hydride assignments of NaH * -
coated
PtlTi.
The results of the NaH *-coated Pt lTi shown in Figure 42B were replicated
with NaH * -coated Si. As shown in Figures 43 and 44, the XPS spectra of NaH *
-
coated Si showed peaks at 6 eV, 10.8 eV, and 12.8 eV that could not be
assigned to
known elements and do not correspond to any other primary element peak, but
matched H- (1 / 3) and H- (1 / 4). Thus, both fractional hydrogen states, 1/3
as
H- (1 / 3) at the 6 eV and 1/4 as H- (1 / 4) at 10.8 eV and 12.8 eV, were
present as
predicted by Eq. (27).
G. FTIR Identification of HZ (1 / 4). Samples of LiH * Br having an upfield-
shifted
'H NMR peak at -2.5 ppm assigned to H- (1 / 4) and an NMR peak at 1.3 ppm
assigned to the corresponding molecule HZ (1 / 4) were analyzed by high
resolution
FTIR spectroscopy. As shown in Figure 45B, a single narrow peak was observed
at
1989 cm-'. The compounds, LiNH21 Li2NH, and Li3N are possible, based on the
staring materials and predicted reactions, but none of these compounds showed
peaks in the region of 1989 cm-'. No additional peaks other than those easily
assignable to LiBr were observed (Figure 45A). An exhaustive list of species
that
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
192
have features in this region were considered, including exotic species such as
azide,
metal carbonyls, and metaborate ion. The former were eliminated based on their
known spectra, which have very broad bands. Metaborate ion was eliminated by
ToF-SIMs analysis, which showed a total boron content that was not detectable
at
the ppb level which is orders of magnitude below its FTIR detection limit and
the
absence of two peaks corresponding to the boron isotopes 10B (20% N.A.) and "B
(80% N.A.).
Considering a possible matrix effect, the peak at 1989 cm-' (0.24 eV )
matched the theoretical prediction of 1947 cm-' for H2(1/4). From Eqs. (14-
15), the
unprecedented rotational energy of 42 times that of ordinary hydrogen
establishes
the internuclear distance of H2 (1/4) as 1/4 that of H2. Interstitial H2 in
silicon and
GaAs is a nearly free rotator showing single rovibrational transitions [83-
87]. H2 is
FTIR active as well as Raman active due to the induced dipole from
interactions with
the crystalline lattice [83]. The crystalline lattice may also influence the
selection
rules to permit an otherwise forbidden transition in H2 (1/4). Considering a
matrix
effect, the match to the predicted 1943 cm-' peak and the relatively narrow
peak
width, indicates that H2 (1 / 4) can rotate essentially freely inside of the
crystal and
confirms its small size corresponding to 1/4 the dimensions of ordinary
hydrogen.
Ordinary hydrogen shows a 3:1 ortho-para ratio at non-cryogenic temperatures;
whereas, a single peak of Hz (1 / 4) formed under the synthesis conditions is
assigned
to the para form only due to the 64 times increase in stability due to the 1/4
relative
internuclear separation. Given the frequency match of the 1989 cm-' peak and
the
absence of any known alternative, wherein hydrogen is the only known species
that
exhibits single rovibrational transitions in a solid matrix, the 1989 cm-'
peak is
assigned to the J = 0 to J=1 rotational transitions of para H2 (1 / 4).
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
193
H. HZ (1 / 4) Rotational UV Spectrum by Electron Beam Excitation. HZ (1 / 4)
trapped in the lattice of alkali halides, MgX2 ( X= F,CI,Br,I ), and CuXz
( X= F,CI,Br ) was investigated by windowless UV spectroscopy on electron beam
excitation of the crystals using the 12.5 keV electron gun at a beam current
of 10-20
,uA in the pressure range of <10-5 Torr. Of the alkali metals, it was found
that only
alkali chlorides showed the peaks predicted by Eq. (14), and the intensity
roughly
matched the order predicted, increasing intensity down the column of the Group
I
elements. In all cases, the peaks could be eliminated by heating with the loss
of the
Lyman a peak, and no other peaks were observed in the UV. The on-line mass
spectrometer recorded hydrogen only. Of the compounds of the series MgX2
( X= F,CI, Br, I) and CuX, ( X= F,CI, Br ), the predicted band was just
detectable
only for MgI2 which, in this case, can be attributed to Mg2+ as the catalyst.
NMR on
these crystals showed the H2 (1 / 4) peak at 1.13 ppm only in MgX2 with
relative
intensities F,CI,Br, I that matched the detection of the band by electron
beam-
excitation emission for Mglz only.
The 100-350 nm spectrum of electron beam-excited CsCI crystals having
trapped H2 (1/4) is shown in Figure 46. A series of evenly spaced lines was
observed in the 220-300 nm region as shown in Figure 46. The series matched
the
spacing and intensity profile of the P branch of H2(1/4) given by Eq. (14).
P(1), P(2),
P(3), P(4), P(5), and P(6) were observed at 226.0 nm, 237.0 nm, 249.5 nm,
262.5
nm, 277.0 nm, and 292.5 nm, respectively. The slope of the linear curve-fit of
the
energies of the peaks shown in Figure 46 is 0.25 eV with an intercept of 5.73
eV and
a sum of residual errors r2 < 0.0000. The slope matches the predicted
rotational
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
194
energy spacing of 0.241 eV (Eq. (14); p= 4) with AJ =+1; J = 1, 2,3,4,5,6
where J is
the rotational quantum number of the final state. H2(114) is a free rotator,
but is not
a free vibrator which is similar to the case of interstitial hydrogen in
silicon discussed
previously [83-87] . The observed intercept of 5.73 eV is shifted from the
predicted
v=1--> v = 0 vibrational energy of H2 (1 / 4) of 8.25 eV (Eq. (13)) by about
twice the
percentage as that of interstitial HZ in silicon [83-87]. In the latter case,
vibrational
energy of free HZ is 4161 cm-', whereas the vibrational peaks in silicon are
observed at 3618 and 3627 cm-' corresponding to ortho and para-HZ ,
respectively
[83]. In the former case the shift is about 30% lower, possibly due to an
increase in
the effective mass from coupling of the molecular vibrational mode with the
crystal
lattice.
Using Eqs. (14) and (15) with the measured rotational energy spacing of 0.25
eV establishes an internuclear distance of 1/4 that of the ordinary H2 for H2
(1 / 4).
A corresponding weak band was observed from NaH * Br, and a more intense band
was observed from NaH * Cl. Regarding the latter case, the intensity of the
emission was significantly increased by trapping H2 (1 / 4) in a silicon
matrix. The
100-550 nm spectrum of an electron beam-excited silicon wafer coated with
NaH * Cl having trapped Hz (1 / 4) is shown in Figure 47. The series matching
the
spacing and intensity profile of the P branch of HZ(1/4) given by Eq. (14) was
observed. P(1), P(2), P(3), P(4), P(5), and P(6) were observed at 222.5 nm,
233.4
nm, 245.2 nm, 258.2 nm, 272.2 nm, and 287.4 nm, respectively. The slope of the
linear curve-fit of the energies of the peaks shown in Figure 47 is 0.25 eV
with an
intercept of 5.82 eV and a sum of residual errors r2 < 0.0000. The linearity
is
characteristic of rotation, and the results again match HZ (1 / 4). This
technique
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
195
confirms the solid NMR and FTIR results given in Secs. IIIE and IIIG,
respectively. It
was reported previously [13-14] that when KH* Cl having H-(1/4) by NMR was
incident to the 12.5 keV electron beam, similar excited emission of
interstitial
H2 (1 / 4) was observed as that from electron-beam excited alkali chlorides,
NaH * Cl -coated Si, and argon-hydrogen plasmas [13-14]. It was further
observed
that the band assigned to H2 (1 / 4) was eliminated from the KC1 stating
material by
heating to high temperature. KH * Cl was then synthesized from the heat-
treated
KC1, and H2 (1 / 4) trapped in the lattice of KH * Cl was then observed in
addition to
H-(114) demonstrating that multiple catalysts, HCl, NaH, K, and Ar+, can give
rise to Hz (1 / 4).
Experimental References
1. R. Mills, The Grand Unified Theory of Classical Quantum Mechanics; October
2007 Edition, posted at
http://www.blacklightpower.com/theory/bookdownload.shtml.
2. R. Mills, K. Akhar, Y. Lu, " Spectroscopic Observation of Helium- and
Hydrogen-
Catalyzed Hydrino Transitions ", to be submitted.
3. R. L. Mills, "Classical Quantum Mechanics", Physics Essays, Vol. 16, No. 4,
December, (2003), pp. 433-498.
4. R. Mills, "Physical Solutions of the Nature of the Atom, Photon, and Their
Interactions to Form Excited and Predicted Hydrino States", in press.
5. R. L. Mills, "Exact Classical Quantum Mechanical Solutions for One- Through
Twenty-Electron Atoms", Physics Essays, Vol. 18, (2005), pp. 321-361.
6. R. L. Mills, "The Nature of the Chemical Bond Revisited and an Alternative
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
196
Maxwellian Approach", Physics Essays, Vol. 17, (2004), pp. 342-389.
7. R. L. Mills, "Maxwell's Equations and QED: Which is Fact and Which is
Fiction", in
press.
8. R. L. Mills, "Exact Classical Quantum Mechanical Solution for Atomic Helium
Which Predicts Conjugate Parameters from a Unique Solution for the First
Time",
submitted.
9. R. L. Mills, "The Fallacy of Feynman's Argument on the Stability of the
Hydrogen
Atom According to Quantum Mechanics," Annales de Ia Fondation Louis de
Broglie, Vol. 30, No. 2, (2005), pp. 129-151.
10. R. Mills, "The Grand Unified Theory of Classical Quantum Mechanics", Int.
J.
Hydrogen Energy, Vol. 27, No. 5, (2002), pp. 565-590.
11. R. Mills, The Nature of Free Electrons in Superfluid Helium-a Test of
Quantum
Mechanics and a Basis to Review its Foundations and Make a Comparison to
Classical Theory, Int. J. Hydrogen Energy, Vol. 26, No. 10, (2001), pp. 1059-
1096.
12. R. Nlills, "The Hydrogen Atom Revisited", Int. J. of Hydrogen Energy, Vol.
25,
Issue 12, December, (2000), pp. 1171-1183.
13. R. L. Mills, J. He, Y. Lu, M. Nansteel, Z. Chang, B. Dhandapani,
"Comprehensive
Identification and Potential Applications of New States of Hydrogen", Int. J.
Hydrogen Energy, Vol. 32(14), (2007), pp. 2988-3009.
14. R. Mills, J. He, Z. Chang, W. Good, Y. Lu, B. Dhandapani, "Catalysis of
Atomic
Hydrogen to Novel Hydrogen Species H-(1/4) and H2(1/4) as a New Power
Source", Int. J. Hydrogen Energy, Vol. 32, No. 12, (2007), pp. 2573-2584.
15. R. Mills, P. Ray, B. Dhandapani, W. Good, P. Jansson, M. Nansteel, J. He,
A.
Voigt, "Spectroscopic and NMR Identification of Novel Hydride Ions in
Fractional
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
197
Quantum Energy States Formed by an Exothermic Reaction of Atomic Hydrogen
with Certain Catalysts", European Physical Journal-Applied Physics, Vol. 28,
(2004), pp. 83-104.
16. R. Mills and M. Nansteel, P. Ray, "Argon-Hydrogen-Strontium Discharge
Light
Source", IEEE Transactions on Plasma Science, Vol. 30, No. 2, (2002), pp. 639-
653.
17. R. Mills and M. Nansteel, P. Ray, "Bright Hydrogen-Light Source due to a
Resonant Energy Transfer with Strontium and Argon Ions", New Journal of
Physics, Vol. 4, (2002), pp. 70.1-70.28.
18. R. Mills, J. Dong, Y. Lu, "Observation of Extreme Ultraviolet Hydrogen
Emission
from Incandescently Heated Hydrogen Gas with Certain Catalysts", Int. J.
Hydrogen Energy, Vol. 25, (2000), pp. 919-943.
19. R. Nlills, M. Nansteel, and P. Ray, "Excessively Bright Hydrogen-Strontium
Plasma Light Source Due to Energy Resonance of Strontium with Hydrogen", J.
of Plasma Physics, Vol. 69, (2003), pp. 131-158.
20. R. L. Mills, J. He, M. Nansteel, B. Dhandapani, "Catalysis of Atomic
Hydrogen to
New Hydrides as a New Power Source", International Journal of Global Energy
Issues (IJGEI), Special Edition in Energy Systems, Vol. 28, Nos. 2/3 (2007),
pp.
304-324.
21. H. Conrads, R. Mills, Th. Wrubel, "Emission in the Deep Vacuum Ultraviolet
from
a Plasma Formed by Incandescently Heating Hydrogen Gas with Trace Amounts
of Potassium Carbonate", Plasma Sources Science and Technology, Vol. 12,
(3003), pp. 389-395.
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
198
22. J. Phillips, R. L. Mills, X. Chen, "Water Bath Calorimetric Study of
Excess Heat in
'Resonance Transfer' Plasmas", Journal of Applied Physics, Vol. 96, No. 6, pp.
3095-3102.
23. R. L. Mills, X. Chen, P. Ray, J. He, B. Dhandapani, "Plasma Power Source
Based on a Catalytic Reaction of Atomic Hydrogen Measured by Water Bath
Calorimetry", Thermochimica Acta, Vol. 406/1-2, (2003), pp. 35-53.
24. R. Mills, B. Dhandapani, M. Nansteel, J. He, T. Shannon, A. Echezuria,
"Synthesis and Characterization of Novel Hydride Compounds", Int. J. of
Hydrogen Energy, Vol. 26, No. 4, (2001), pp. 339-367.
25. R. Mills, B. Dhandapani, M. Nansteel, J. He, A. Voigt, "Identification of
Compounds Containing Novel Hydride Ions by Nuclear Magnetic Resonance
Spectroscopy", tnt. J. Hydrogen Energy, Vol. 26, No. 9, (2001), pp. 965-979.
26. R. Nlills, B. Dhandapani, N. Greenig, J. He, "Synthesis and
Characterization of
Potassium lodo Hydride", Int. J. of Hydrogen Energy, Vol. 25, Issue 12,
December, (2000), pp. 1185-1203.
27. R. L. Mills, P. Ray, "Extreme Ultraviolet Spectroscopy of Helium-Hydrogen
Plasma", J. Phys. D, Applied Physics, Vol. 36, (2003), pp. 1535-1542.
28. R. L. Mills, P. Ray, B. Dhandapani, M. Nansteel, X. Chen, J. He, "New
Power
Source from Fractional Quantum Energy Levels of Atomic Hydrogen that
Surpasses Internal Combustion", J Mol. Struct., Vol. 643, No. 1-3, (2002), pp.
43-
54.
29. R. Mills, P. Ray, "Spectral Emission of Fractional Quantum Energy Levels
of
Atomic Hydrogen from a Helium-Hydrogen Plasma and the Implications for Dark
Matter", Int. J. Hydrogen Energy, Vol. 27, No. 3, (2002), pp. 301-322.
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
199
30. R. L. Mills, P. Ray, "A Comprehensive Study of Spectra of the Bound-Free
Hyperfine Levels of Novel Hydride Ion H-(1/2), Hydrogen, Nitrogen, and Air",
Int.
J. Hydrogen Energy, Vol. 28, No. 8, (2003), pp. 825-871.
31. R. Mills, "Spectroscopic Identification of a Novel Catalytic Reaction of
Atomic
Hydrogen and the Hydride Ion Product", Int. J. Hydrogen Energy, Vol. 26, No.
10,
(2001), pp. 1041-1058.
32. R. L. Mills, P. Ray, B. Dhandapani, R. M. Mayo, J. He, "Comparison of
Excessive
Balmer a Line Broadening of Glow Discharge and Microwave Hydrogen
Plasmas with Certain Catalysts", J. of Applied Physics, Vol. 92, No. 12,
(2002),
pp. 7008-7022.
33. R. L. Mills, P. Ray, B. Dhandapani, J. He, "Comparison of Excessive Balmer
a
Line Broadening of Inductively and Capacitively Coupled RF, Microwave, and
Glow Discharge Hydrogen Plasmas with Certain Catalysts", IEEE Transactions
on Plasma Science, Vol. 31, No. (2003), pp. 338-355.
34. R. L. Mills, P. Ray, "Substantial Changes in the Characteristics of a
Microwave
Plasma Due to Combining Argon and Hydrogen", New Journal of Physics,
www.njp.org, Vol. 4, (2002), pp. 22.1-22.17.
35. R. L. Mills, P. Ray, B. Dhandapani, "Excessive Balmer a Line Broadening of
Water-Vapor Capacitively-Coupled RF Discharge Plasmas" Int. J. Hydrogen
Energy, in press.
36. R. Mills, P. Ray, B. Dhandapani, "Evidence of an Energy Transfer Reaction
Between Atomic Hydrogen and Argon II or Helium II as the Source of Excessively
Hot H Atoms in RF Plasmas", Journal of Plasma Physics, (2006), Vol. 72, Issue
4, pp. 469-484.24.
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
200
37. J. Phillips, C-K Chen, K. Akhtar, B. Dhandapani, R. Mills, "Evidence of
Catalytic
Production of Hot Hydrogen in RF Generated Hydrogen/Argon Plasmas",
International Journal of Hydrogen Energy, Vol. 32(14), (2007), 3010-3025.
38. R. Mills, P. Ray, R. M. Mayo, "CW HI Laser Based on a Stationary Inverted
Lyman Population Formed from Incandescently Heated Hydrogen Gas with
Certain Group I Catalysts", IEEE Transactions on Plasma Science, Vol. 31, No.
2, (2003), pp. 236-247.
39. R. L. Mills, P. Ray, "Stationary Inverted Lyman Population Formed from
Incandescently Heated Hydrogen Gas with Certain Catalysts", J. Phys. D,
Applied Physics, Vol. 36, (2003), pp. 1504-1509.
40. R. Mills, P. Ray, R. M. Mayo, "The Potential for a Hydrogen Water-Plasma
Laser", Applied Physics Letters, Vol. 82, No. 11, (2003), pp. 1679-1681.
41. R. L. Mills, The Grand Unified Theory of Classical Quantum Mechanics,
November 1995 Edition, HydroCatalysis Power Corp., Malvern, PA, Library of
Congress Catalog Number 94-077780, ISBN number ISBN 0-9635171-1-2, Chp.
22.
42. F. Bournaud, P. A. Duc, E. Brinks, M. Boquien, P. Amram, U. Lisenfeld, B.
Koribalski, F. Walter, V. Charmandaris, "Missing mass in collisional debris
from
galaxies", Science, Vol. 316, (2007), pp. 1166-1169.
43. B. G. Elmegreen, "Dark matter in galactic collisional debris", Science,
Vol. 316,
(2007), pp. 32-33.
44. N. V. Sidgwick, The Chemical Elements and Their Compounds, Volume I,
Oxford, Clarendon Press, (1950), p.17.
45. M. D. Lamb, Luminescence Spectroscopy, Academic Press, London, (1978), p.
68.
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
201
46. K. R. Lykke, K. K. Murray, W. C. Lineberger, "Threshold photodetachment of
H", Phys. Rev. A, Vol. 43, No. 11, (1991), pp. 6104-6107.
47. D. R. Lide, CRC Handbook of Chemistry and Physics, 79 th Edition, CRC
Press,
Boca Raton, Florida, (1998-9), p. 10-175.
48. H. Beutler, Z. Physical Chem., "Die dissoziationswarme des
wasserstoffmolekuls
Hz , aus einem neuen ultravioletten resonanzbandenzug bestimmt", Vol. 27B,
(1934), pp. 287-302.
49. G. Herzberg, L. L. Howe, "The Lyman bands of molecular hydrogen", Can. J.
Phys., Vol. 37, (1959), pp. 636-659.
50. P. W. Atkins, Physical Chemistry, Second Edition, W. H. Freeman, San
Francisco, (1982), p. 589.
51. F. Abeles (Ed.), Optical Properties of Solids, (1972), p. 725.
52. D. R. Lide, CRC Handbook of Chemistry and Physics, 86th Edition, CRC
Press,
Taylor & Francis, Boca Raton, (2005-6), pp. 10-202 to 10-204.
53. F. A. Cotton, G. Wilkinson, C. A. Murillo, M. Bochmann, Advanced Inorganic
Chemistry, Sixth Edition, John Wiley & Sons, Inc., New York, (1999), p. 92.
54. D. R. Lide, CRC Handbook of Chemistry and Physics, 86th Edition, CRC
Press,
Taylor & Francis, Boca Raton, (2005-6), pp. 9-54 to 9-59.
55. P. Chen, Z. Xiong, J. Luo, J. Lin, K. L. Tan, "Interaction of Hydrogen
with Metal
Nitrides and Amides," Nature, 420, (2002), 302-304.
56. P. Chen, Z. Xiong, J. Luo, J. Lin, K. L. Tan, "Interaction between Lithium
Amide
and Lithium Hydride," J. Phys. Chem. B, 107, (2003), 10967-10970.
57. W. I. F. David, M. O. Jones, D. H. Gregory, C. M. Jewell, S. R. Johnson,
A.
Walton, P. Edwards, "A Mechanism for Non-stoichiometry in the Lithium
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
202
Amide/Lithium Imide Hydrogen Storage Reaction," J. Am. Chem. Soc., 129,
(2007), 1594-1601.
58. D. B. Grotjahn, P. M. Sheridan, I. Al Jihad, L. M. Ziurys, "First
Synthesis and
Structural Determination of a Monomeric, Unsolvated Lithium Amide, LiNHz," J.
Am. Chem. Soc., 123, (2001), 5489-5494.
59. F. E. Pinkerton, "Decomposition Kinetics of Lithium Amide for Hydrogen
Storage
Materials," J. Alloys Compd., 400, (2005), 76-82.
60. Y. Kojima, Y. Kawai, "IR Characterizations of Lithium Imide and Amide," J.
Alloys
Compd., 395, (2005), 236-239.
61. T. Ichikawa, S. Isobe, N. Hanada, H. Fujii, "Lithium Nitride for
Reversible
Hydrogen Storage," J. Alloys Compd., 365, (2004), 271-276.
62. Y. H. Hu, E. Ruckenstein, "Ultrafast Reaction between Li3N and LiNH2 to
Prepare
the Effective Hydrogen Storage Material Li2NH," Ind. Eng. Chem. Res., 45,
(2006), 4993-4998.
63. Y. H. Hu, E. Ruckenstein, "Hydrogen Storage of LiNH2 Prepared by Reacting
Li
with NH3," Ind. Eng. Chem. Res., 45, (2006), 182-186.
64. Y. H. Hu, E. Ruckenstein, "High Reversible Hydrogen Capacity of LiNH2/Li3N
Mixtures," Ind. Eng. Chem. Res., 44, (2005), 1510-1513.
65. D. R. Lide, CRC Handbook of Chemistry and Physics, 86th Edition, CRC
Press,
Taylor & Francis, Boca Raton, (2005-6), pp. 5-4 to 5-18; 9-63.
66. P. Chen, Z. Xiong, J. Luo, J. Lin, K.L. Tan, "Interaction of hydrogen with
metal
nitrides and imides", Nature, Vol. 420, (2002), pp. 302-304.
67. Yun Hang Hu, Eli Ruckenstein, "Hydrogen Storage of Li2NH Prepared by
Reacting Li with NH3," Ind. Eng. Chem. Res., Vol. 45, (2006), pp. 182-186.
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
203
68. K. Ohoyama, Y. Nakamori, S. Orimo, "Characteristic Hydrogen Structure in
Li-N-
H Complex Hydrides," Proceedings of the International Symposium on Research
Reactor and Neutron Science-In Commemoration of the 10th Anniversary of
HANARO-Daejeon, Korea, April 2005, pp. 655-657.
69. Microsc. Microanal. Microstruct., Vol. 3, 1, (1992).
70. For specifications see PHI Trift II, ToF-SIMS Technical Brochure, (1999),
Eden Prairie, MN 55344.
71. W. M. Muller, J. P. Blackledge, G. G. Libowitz, Metal Hydrides, Academic
Press,
New York, (1968), p 201.
72. David R. Lide, CRC Handbook of Chemistry and Physics, 79 th Edition, CRC
Press, Boca Raton, Florida, (1998-9), p. 12-191.
73. R. R. Cavanagh, R. D. Kelley, J. J. Rush, "Neutron vibrational
spectroscopy of
hydrogen and deuterium on Raney nickel," J. Chem. Phys., 77(3), (1982),
1540-1547.
74. I. Nicolau, R. B. Andersen, "Hydrogen in a commercial Raney nickel," J.
Catalysis, Vol. 68, (1981), 339-348.
75. K. Niessen, A. R. Miedema, F. R. de Boer, R. Boom, "Enthalpies of
formation of
liquid and solid binary alloys based on 3d metals," Physica B, Vol. 152,
(1988),
303-346.
76. B. S. Hemingway, R. A. Robie, "Enthalpies of formation of low albite (
NaAISi,Ox ),
gibbsite (Al(OH)3), and NaAIOz; revised values for AHf298' and OGf29,o of some
aluminosilicate minerals", J. Res. U.S. Geol. Surv., Vol. 5(4), (1977), pp.
413-
429.
77. B. Baranowski, S. M. Filipek, "45 years of nickel hydride-history and
perspectives", Journal of Alloys and Compounds, 404-406, (2005), pp. 2-6.
CA 02684952 2009-10-21
WO 2008/134451 PCT/US2008/061455
204
78. K. K. Baldridge, J. S. Siegel, "Correlation of empirical 8(TMS) and
absolute NMR
chemical shifts predicted by ab initio computations", J. Phys. Chem. A, Vol.
103,
(1999), pp. 4038-4042.
79. J. Mason, Editor, Multinuclear NMR, Plenum Press, New York, (1987), Chp.
3.
80. C. Suarez, E. J. Nicholas, M. R. Bowman, "Gas-phase dynamic NMR study of
the internal rotation in N-trifluoroacetlypyrrolidine", J. Phys. Chem. A, Vol.
107,
(2003), pp. 3024-3029.
81. C. Suarez, "Gas-phase NMR spectroscopy", The Chemical Educator, Vol. 3,
No.
2, (1998).
82. C. Lu, J. Hu, J. H. Kwak, Z. Yang, R. Ren, T. Markmaitree, L. Shaw, "Study
the
Effects of Mechanical Activation on Li-N-H Systems with'H and 6Li Solid-State
NMR," J. Power Sources, Vol. 170, (2007), 419-424.
83. M. Stavola, E. E. Chen, W. B. Fowler, G. A. Shi, "Interstitial H2 in Si:
are All
Problems Solved?" Physica B, 340-342, (2003), pp. 58-66.
84. E. V. Lavrov, J. Weber, "Ortho and Para Interstitial H2 in Silicon," Phys.
Rev.
Letts., 89(21), (2002), pp. 215501 to 1-215501-4.
85. E. E. Chen, M. Stavola, W. B. Fowler, J. A. Zhou, "Rotation of Molecular
Hydrogen in Si: Unambiguous Identification of Ortho-H2 and Para-Dz," Phys.
Rev.
Letts., 88(24), (2002), pp. 245503-1 to 245503-4.
86. E. E. Chen, M. Stavola, W. B. Fowler, P. Walters, "Key to Understanding
Interstitial H2 in Si," Phys. Rev. Letts., 88(10), (2002), pp. 105507-1 to
105507-4.
87. A. W. R. Leitch, V. Alex, J. Weber, "Raman Spectroscopy of Hydrogen
Molecules in Crystalline Silicon," Phys. Rev. Letts., 81(2), (1998), pp. 421-
424.