Note: Descriptions are shown in the official language in which they were submitted.
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
FUSED-RING HETEROCYCLE OPIOIDS
FEDERALLY SPONSORED RESEARCH
[0001] The following invention was made with Government support under contract
number RO1 DA12180 awarded by U.S. Dept of Health and Human Services. The
Government has certain rights in this invention.
FIELD OF THE INVENTION
[0002] Invention relates to opioid receptor binding compounds containing a
heterocyclic moiety. The compounds are useful as analgesics, anti-diarrheal
agents,
anticonvulsants, anti-obesity agents, antitussives, anti-cocaine, and anti-
addiction
medications.
BACKGROUND OF THE INVENTION
[0003] Opiates have been the subject of intense research since the isolation
of
morphine in 1805, and thousands of compounds having opiate or opiate-like
activity
L__..,G L,.,.._ '.1,,...+]llF.,.,7 Td..,...lty . opioid nr=;ntar ~ti~1e
Cnm1..r~..nnn.....r.+.,lc inr.l>>dina thnce
11Q.v UGGll luG1A1GU. 1vta opioiu riveptwi uiwaa _a _
used for producing analgesia (e.g., morphine) and those used for treating drug
addiction (e.g., naltrexone and cyclazocine) in humans have limited utility
due to poor
oral bioavailability and a very rapid clearance rate from the body. This has
been
shown in many instances to be due to the presence of the 8-hydroxyl group (OH)
of
2,6-methano-3-benzazocines, also known as benzomorphans [(e.g., cyclazocine
and
EKC (ethylketocyclazocine)] and the corresponding 3-OH group in morphinanes
(e.g.,
morphine).
1
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
N N
/ \ / \
8
HO HO 3 0
benzomorphan morphinan
numbering numbering
[0004] The high polarity of these hydroxyl groups retards oral absorption of
the parent
molecules. Furthermore, the 8-(or 3-)OH group is prone to sulfonation and
glucuronidation (Phase II metabolism), both of which facilitate rapid
excretion of the
active compounds, leading to disadvantageously short half-lives for the active
compounds. Until the publications of Wentland in 2001, the uniform experience
in
the art of the past seventy years had been that removal or replacement of the
8-(or 3-)
OH group had led to pharmacologically inactive compounds.
US patent 6,784,187 (to Wentland) disclosed that the phenolic OH of opioids
could be
replaced by CONH2. In the cyclazocine series of opioids, it was shown that 8-
carboxamidocyclazocine (8-CAC) had high affinity for and x opioid receptors.
In
studies in vivo, 8-CAC showed high antinociception activity and a much longer
duration of action than cyclazocine (15 h vs. 2 h) when both were dosed at 1
mg/kg ip
in mice. Preliminary structure-activity relationship studies for 8-CAC
revealed that
mono-substitution of the carboxamide nitrogen with methyl or phenyl reduced
binding
affinity for guinea pig receptors 75- and 2313-fold, respectively whereas
dimethylation of the carboxamide group reduced binding affinity 9375-fold. The
finding that substitution of the carboxamide nitrogen had such a detrimental
effect
with these groups suggested that the NH2 of the amide was critical to opioid
binding.
SUMMARY OF THE INVENTION
2
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
We have now found that the 8-position can be cyclized back into the aromatic
ring at
either 7 or 9 to provide compounds that exhibit excellent opioid binding and,
presumably, good metabolic stability. The compounds of the invention are
therefore
useful as analgesics, anti-pruritics, anti-diarrheal agents, anticonvulsants,
antitussives,
anorexics and as treatments for hyperalgesia, drug addiction, respiratory
depression,
dyskinesia, pain (including neuropathic pain), irritable bowel syndrome and
gastrointestinal motility disorders. Drug addiction, as used herein, includes
alcohol
and nicotine addiction. There is evidence in the literature that the compounds
may
also be useful as immunosuppressants and antiinflammatories and for reducing
ischemic damage (and cardioprotection), for improving learning and memory, and
for
treating urinary incontinence. Those species that do not cross the blood-brain
barrier
are also useful for treating opioid-induced constipation and urinary
retention.
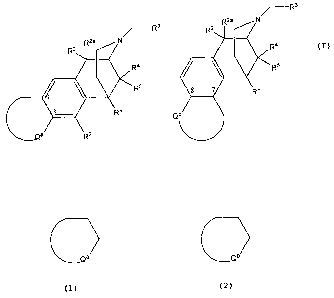
In one aspect, the invention relates to compounds of formula:
/- Rs
R2a N
~-R3 R2
RZ R2a N Ra
R4 Rs
W R5 8 Re
R6 Qb
Ca R7
or
wherein
Qor b
is a heterocyclic ring, which may be substituted or
further fused to form a residue of one to three rings;
3
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
Qa is chosen from
R9
,J=1~ "i.8 N /A "L
HN X-N' \ N N-
O, Re X=N and NHR9
Qb is chosen from
$
$ N
8 rr 8
R9
C ` -- _ N \\
R9HN N -t
~ T z \ /
HN-~ , R9, 0 , xN , and
R9HN
~
X isNorCR9;
R2 and R2a are both hydrogen or taken together R2 and Rza are =0;
R3 is chosen from hydrogen, (Cl-C8)hydrocarbon, heterocyclyl,
heterocyclylalkyl and hydroxyalkyl;
R4 is chosen from hydrogen, hydroxy, amino, (Cl-C6)alkoxy, (CI-CZO)atkyl
and (C 1 -CLV)alkvl suhstih.-ted with hvdroxv or carbonvl;
R5 is (CI-C6)alkyl;
R6 is (C1-C6)alkyl;
R' is chosen from hydrogen, NHR9 and hydroxy; or
together R4, R5, R6 and R' may form from one to three rings, said rings having
optional
additional substitution;
R9 is independently in each of its occurrences H, alkyl or
Rao
~-U Ar
Rit
U is (CH2)r,, wherein one or more CH2 may be replaced by -0-, cycloalkyl or -
CR'aRlb,
,
4
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
R'a and R'b are chosen independently from hydrogen, halogen, (C1-C6)alkyl, (Cl-
C6)alkoxy and (C1-C6)alkylthio;
Ar is an aryl or heteroaryl residue of one to three rings;
R10 is one or two residues chosen independently from hydrogen, hydroxyl,
halogen,
(CI-C6)alkyl, (C1-C6)alkoxy, halo(C1-C6)alkyl and halo(C1-C6)alkoxy and (Cl-
C6)alkylthio;
R15
D u-
R'lis H or
~ D
is an aryl or heteroaryl residue of one to three rings;
U' is (CH2),,,, wherein one or more CH2 may be replaced by -0-, cycloalkyl, -
CR'aR'b, -C(=0)- or -NH-;
R15 is one or two residues chosen independently from hydrogen, hydroxyl,
halogen, (C1-C6)alkyl, (C1-C6)alkoxy, halo(CI-C6)alkyl and halo(C1-C6)alkoxy
and
(C I-C6)alkylthio;
m is zero or an integer from 1 to 6; and
n is an integer from 1 to 6.
Subclasses of the foregoing structure include:
A) 2,6-methano-3-benzazocines of the structure shown above, in which R4, R5,
R6 and R7 do not form additional rings:
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
Ra
R2a N
~- R3 R2
R2a N 4
Ra / \ Rs
P R
R5 R6
Qb
b
or
B) morphinans in which R5 and R6 form one ring:
Rs
R2a N
/-- R3 R2
R2 R2a N
Ra
Ra
Qb
a \ R7 or
C) morphinans in which R5, R6 and R' form two rings:
/- Rs
R2a N
R2
Ra
R1s
R20
Ca 0 R22
R21
6
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
in which
R4 is hydrogen, hydroxy, amino or (C1-C6)alkoxy;
R19 is hydrogen or (C1-C6)alkyl;
R20 is chosen from hydrogen, (CI-C6)alkyl and hydroxy((CI-C6)alkyl); or
together,
R19 and R20 form a spiro-fused carbocycle of 5 to 10 carbons;
R21 is hydrogen;
R22 is chosen from hydroxy, (Cl-C6)alkoxy and -NR13R14; or
together, R21 and R22 form a carbonyl or a vinyl substituent;
D) morphinans in which R5, R6 and R' form two rings and the nitrogen is
quaternized:
H3C\ ~R3
R2a N+
R2 E
R4
R19
R20
R21
a n R22
in which
R4 is hydrogen, hydroxy, amino or (C I -C6)alkoxy;
R19 is hydrogen or (C 1 -C6)alkyl;
R20 is chosen from hydrogen, (Ci-C6)alkyl and hydroxy((C1-C6)alkyl); or
together,
R19 and R20 form a spiro-fused carbocycle of 5 to 10 carbons;
R21 is hydrogen;
R22 is chosen from hydroxy, (Cl-C6)alkoxy and -NR13R14; or
together, R21 and R22 form a carbonyl or a vinyl substituent; and
E- is a pharmaceutically acceptable anion; and
7
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
E) morphinans wherein R4 and Rl' form an additional sixth ring, which may be
saturated or unsaturated:
/- Rs
R2a N
R2
R19
R20
O R22
a
or
Rs
R2a N
R2
R19
R2o
0 R22
a
Tn another aspect; the invention relates to a compound of formula
R3
R2a N
R2
R4
R5
R18 R6
A R17
wherein
A is chosen from -C(=O)NR9R12 and -C(=S)NR9R1z;
R2 and R2' are both hydrogen or taken together RZ and Rza are =O;
8
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
R3 is chosen from hydrogen, (C1-C8)hydrocarbon, heterocyclyl,
heterocyclylalkyl
and hydroxyalkyl;
R4 is chosen from hydrogen, hydroxy, amino, (C]-C6)alkoxy, (Ci-Cz )alkyl and
(C1-C20)alkyl substituted with hydroxy or carbonyl;
R5 is (C I -C6)alkyl;
R6 is (C1-C6)alkyl;
or together R4, R5 and R6 may form from one to three rings, said rings having
optional
additional substitution;
R9 in each of its occurrences is independently chosen from H, alkyl and
R1o
~-U Ar
R11
U is (CHZ),,,wherein one or more CH2 may be replaced by -0-, cycloalkyl or -
CR1aR1b;
Ria and Rlb are chosen independently from hydrogen, halogen, (CI-C6)alkyl, (C1-
C6)alkoxy and (Cl -C6)alkylthio;
Ar is an aryl or heteroaryl residue of one to three rings;
R10 is one or two residues chosen independently from hydrogen, hydroxyl,
halogen,
(CI-C6)alkyl, (CI-C6)alkoxy, halo(Cj-C6)alkyl and halo(C1-C6)alkoxy and (Cl-
C6)alkylthio;
R15
D Ul-
Ri l is H or
D
is an aryl or heteroaryl residue of one to three rings;
U' is (CHZ)wherein one or more CH2 may be replaced by -0-, cycloalkyl, -
CRI aRt b, -C(=0)- or -NH-;
R12 is chosen from hydrogen and (Cl-C6)alkyl;
R15 is one or two residues chosen independently from hydrogen, hydroxyl,
halogen,
(Ci-C6)alkyl, (CI-C6)alkoxy, halo(CI-C6)alkyl and halo(C1-C6)alkoxy and (C1-
C6)alkylthio;
9
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
one of R" or R18 is NIHR9 and the other is hydrogen;
m is zero or an integer from 1 to 6; and
n is an integer from 1 to 6.
In another aspect the invention relates to a pharmaceutical formulation
comprising a
pharmaceutically acceptable carrier and a compound as described above.
In another aspect the invention relates to a method for treating a disease or
condition
by altering a response mediated by an opioid receptor comprising bringing into
contact
with said opioid receptor a compound as described above.
DETAILED DESCRIPTION OF THE INVENTION
From many years of SAR studies, it is known that the hydroxyl of morphinans
and
benzomorphans interacts with a specific site in the opiate receptor. We have
now
surprisingly found that the hydroxyl can be replaced with a heterocycle fused
to the
aromatic ring through a carbon that occupies the position formerly occupied by
the
hydroxyl. A fairly wide range of fused heterocycles exhibit binding to at
least one of
the opioid binding sites ( , 6 or x) in the desired range below 250 nanomolar.
[0005] In one aspect the invention relates to compounds of formula
~-R3
R2 R2a N
R4
R5
C ~ Rs
a R7
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
This subgenus comprises compounds in which the heterocyclic ring is fused at
the 8,9-
a
positions of the benzazocine. In certain embodiments is chosen from
9
< 8 N \ 8 N 9
C R 9 ~ \$
X--~.N N s
HN N-
o R9 XN NHR9 and
/ \ $
N-
NHR9 , and the compounds are of the formulae:
11
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
Rz ~ Ra
Rza N /- R3
Rza N
R4 RZ
N Ra
R5
R / \ R5
'R6
_ Re
HN R7
L
0 N
~R3 ~- Ra
Rza N Rza N
Rz R2
R4 RQ
R5 R5
II - R6 Rs
N N- R7
`9 NHR9
Ra
Rza N
R2
R4
Re
N
\ - 'R6
N R~
and NHR9
[0006] In another aspect the invention relates to compounds of formula
/- R3
R2 R2a N
IR4
R5
8 Rs
Qb
12
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
This subgenus comprises compounds in which the heterocyclic ring is fused at
the 7,8-
positions of the benzazocine. In certain embodiments
r ---~-- ~ \ 7
8 7 ``
Qb
R9HN N X -"-N
is chosen from N--// R9
r-~----~
g 7 R\ g 7
( ~
R9HN
~ N R9HN /
O X=N N
> > >
~ '~ ~ r-----~`----~
8 7 8 7
p p /N
HN and HNJ , and the compounds are of the formulae:
/-R3
R2a N /-Ra
R2~1 R4 Rz-aN
K'
R5 / \ R5
R R6
RsHN N /~N RHN N
!l > >
13
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
R3 R3
R2 R2a N R2 R2a N
R4 R4
R5 / \ Rs
R6 R6
/ \
R9N~XN R9HN 0 /N
> >
R3 3 R3
RZ R2a N/-- O N 2 R2a N/-R ~ R2 R2a Nf
R
R4 Ra R4
R5 / \ R5 R5
R6 Rs R6
NX/NR9 HN and HN
[0007] In another aspect, the invention relates to compounds of formula
/- Rs
R2a N
2
Ra
` R5
R1s
R6
A R17
in which A is -C(=O)NR9R'2 or -C(-S)NR9R12. These compounds are useful both as
intermediates in the synthesis of 7,8-fused pyrimidines and in their own right
as opioid
receptor binding compounds (see example 6 in Table 1 below). Commonly, R12
will
be hydrogen.
[0008] In one major subclass, the groups R9 are biphenyls, diaryl ethers and
the like.
Illustrative formulae are:
14
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
.~- Rs
R2a
R2
R4
Re
R6
R15 R~o
U U-NH
N and
/-Rs
R2a N
R2
R4
Re
R6
R15 R'o
D U, U-NH NH2
O
[0009] Preferred values of R9are hydrogen and those in which
R1: / ~
D A-
(a) 0 is phenyl, R10 is hydrogen and Rl l is , so that R"
represents pyridinyl, phenyl, halophenyl, methylphenyl, methoxyphenyl (in all
of
which A' is a direct bond) and phenoxy (in which A' is -0-).
(b) U is chosen from phenyl, naphthyl, fluorenyl, carbazole, dibenzofuran
and dibenzothiophene, R10 is hydrogen, methoxy, halogen or methyl; and RII is
hydrogen;
(c) is pyridinyl, R10 is hydrogen and Rl' is chosen from phenyl,
halophenyl, methylphenyl, methoxyphenyl and phenoxy.
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
[0010] It is known in the art that compounds that are , 8 and K agonists
exhibit
analgesic activity; compounds that are selective agonists exhibit anti-
diarrheal
activity and are useful in treating dyskinesia; p antagonists and K agonists
are useful in
treating heroin, cocaine, alcohol and nicotine addiction; K agonists are also
anti-
pruritic agents and are useful in treating hyperalgesia. Recently it has been
found
[Peterson et al. Biochem. Pharmacol. 61, 1141-1151 (2001)] that K agonists are
also
useful in treating retroviral infections. In general, the dextrorotatory
isomers of
morphinans of type III above are useful as antitussives and anticonvulsants.
[0011] Opioid receptor ligands having known high affinity are shown in the
following
9 8 7
fl8 Qb
a
charts. Attachment of a fused ring or at the carbon
carrying the phenolic OH (designated 8) and its adjacent carbon (designated 7
or 9) in
these compounds produces compounds that exhibit opioid activity. As will be
apparent to the artisan, the ring containing Qb will not be attached to
morphinanes and
similar compounds in Charts 2 and 3, which already possess substitution at C-
7.
Bmboa+iments oi'tn ie = invention = 1ciuue..1 each o>, ~u F4i..2 compounds set
~~~ L.~ 4~nr4~ in the
ii~~~pounuo ..following charts in which the phenolic hydroxyl is replaced by a
fused ring attached at
the carbon to which the phenolic -OH is attached and the carbon adjacent
thereto.
16
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
Chart 1. Opioid Receptor Ligands
Benzomorphinans (a.k.a. 2,6-Methano-3-benzazocines)
N/ Rs N/ CH2- Nz CH2~
O O
OB2
..,.CH3 CH3
CH3 CH3 CH2CH3
HO HO HO
Cyclazocine, R3 = CH2-c-C3H5 Ketocyclazocine Ethylketocyclazocine (EKC)
Metazocine, R3 = CH3
Phenazocine, R3 = CH2C6H5
SKF 10,047, R3 = CH2CH=CH2
Pentazocine, R3 = CH2CH=C(CH3)2
(all racemic)
HO
CH2- CH2 ~CH2~
N H O N O N
CH3
CH3 ~ ~ ..,.CH2CH3 C~~a
HO CHs HO CH2CH3 HO CH2CH3
MR2034 - "Merz" core MR2266 Bremazocine
structure (opt. active)
/ CH3
N
0
CH3
OC-bCH,
HO
WIN 44,441
17
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
Chart 2. Opioid Receptor Ligands
Morphine and Morphinans
/ R17
~CH3 N
N17
OH
H 14
14
3 6 HO 0 O
HO 0 OH
Naltrexone; R17 = CH2-C-C3H5
Morphine Naloxone; R17 = CH2CH=CH2
Nalmexone; R17 = CH2CH=C(CH3)2
Oxymorphone; R17 = CH3
CHz-< NiCH2-<
OH
CH3
CH3 -
C(CH3)3
CH3
HO 0 OCH3 HO O OCH3
Buprenorphine Diprenorphine
Etorphine (N-Me; n-Pr vs Me)
~CH2-CH=CH2 ~CH2--a CHZ ~
14 H OH AOH
HO 0 OH HO O' N
HO O OH
Nalorphine Naltrindole Nalbuphine
H3C\ i H2~
iCH2~ N/CH2~ N+
N
OH OH OH Br
14 14 14
6 HO 0 O
HO 0 NHy HO 0 CH2
R-Naltrexamine Nalmefene Methylnaltrexone
18
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
Chart 2 (continued). Opioid Receptor Ligands
Morphine and Morphinans
/CHZ-< /CH2-<
N N
AOH OH
/ \ 14
6
HO 0HN~ CO2Me HO O N(CH2CHzCl)2
R-FNA 0 (3-CNA
/CH3
kOH N1~ N
~~ OH HQ
1
~ ~ ~S~ 14
~S~- ~
HO 0 0 3 ~(~
HO 0 N 0 OH
SIOM (S agonist) H
nor-BNI (Norbinaltorphimine)
Reg # = 105618-26-6
/ R17
N /CH3
N
AH
`
~'=.
HO
RO
Levorphanol; R17 = CH3
Cyclorphan; R17 = CH2-c-C3H5 Dextromethorphan; R CH3
MCL 101; R CH --C H7 Dextrorphan; R= H
i~ 2- 4
Butorphanol; R17 = CH2-9-C4H7 (note "opposite" sterochemistry)
and 14-OH
Merz-morphinane hybrid core; R17 _
C H2-(S)-tetrahyd rofu rfu ryI
19
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
Chart 3 - Miscellaneous Opioid Receptor Ligands
N_ N NEt2
HO2C-\-~
N~
OH OH
~N~ ~N
N N
If ~i
Registry Number 216531-48-5 Registry Number 155836-52-5
HO
H
H N
\ OH
CHg H ~ /
Registry number 361444-66-8
OH R = CH3i Registry Number: 69926-34-7
R = CH2CH2CH(OH)C6H11;
cH3 Registry Number: 119193-09-8
CH3 R = CH2CH(CH2Ph)CONHCH2CO2H;
Registry Number: 156130-44-8
N R=(CH2)3CH(CH3)z; Registry Number: 151022-07-0
R R = (CH2)3-2-thienyl; Registry Number: 149710-80-5
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
OH
O
CA N
N
i
CH3 CH3
Meptazinol Ketobemidone
Registry Number 59263-76-2 Registry Number 469-79-4
/CH3
N
Me2N N)
CH3 OH
To
O
CH3OOf-{ N
H
~ N Tramadol active metabolite
I Registry Number 80456-81-1
Registry number 177284-71-8
H3C,
H3CI H N
N H WrN (+)-TAN 67 (-)-TAN 67
OH OH
Registry number 189263-70-5 Registry number 173398-79-3
/CH3
N N
OH OH
HO O N HO O N(/ I
Registry number 189016-07-7 Registry number 189015-08-5
[0012] Other opioid receptor ligands are described in Aldrich, J.V.
"Analgesics" in
Burger's Medicinal Chemistry and Drug Discovery, M.E.Wolff ed., John Wiley &
Sons 1996, pages 321-44, the disclosures of which are incorporated herein by
21
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
reference.
[0013] We have examined the opioid receptor binding of a series of fused-ring
analogs of known compounds that interact at opioid receptors. Binding assays
used to
screen compounds are similar to those previously reported by Neumeyer et al.,
Design
and Synthesis of Novel Dimeric Morphinan Ligands for x and g Opioid Receptors.
J.
Med. Chem. 2003, 46, 5162. Membrane protein from CHO cells that stably
expressed
one type of the human opioid receptor were incubated with 12 different
concentrations
of the compound in the presence of either I nM [3H]U69,59310 (K), 0.25 nM
[3H]DAMGO' 1( ) or 0.2 nM [3H]naltrindole12 (6) in a final volume of 1 mL of
50
mM Tris-HCI, pH 7.5 at 25 C. Incubation times of 60 min were used for
[3H]U69,593
and [3H]DAMGO. Because of a slower association of [3H]naltrindole with the
receptor, a 3 h incubation was used with this radioligand. Samples incubated
with
[3H]naltrindole also contained 10 mM MgClz and 0.5 mM phenylmethylsulfonyl
fluoride. Nonspecific binding was measured by inclusion of 10 M naloxone. The
binding was terminated by filtering the samples through Schleicher & Schuell
No. 32
glass fiber filters using a Brandel 48-well cell harvester. The filters were
subsequently
washed three times with 3 mL of cold 50 mM Tris-HCI, pH 7.5, and were counted
in 2
mL Ecoscint A scintillation fluid. For [3H]naltrindole and [3H]U69,593
binding, the
filters _ were JVarkõacu in vn.tt~ ly..o~.~~hiiy~iit,loõi~.ii=õ~,iiii=õviA for
at. v 1Pau~u~ at 6/1 min hPf/lrA 71CP.. IC5n
oi G pvi u~
values were-calculated by least squares fit to a logarithm-probit analysis. K;
values of
unlabeled compounds were calculated from the equation Ki =(IC50)/1+S where S=
(concentration of radioligand)/(Kd of radioligand).13 Data are the mean SEM
from at
least three experiments performed in triplicate.
[0014] [3SS]GTPyS Binding Assays. In a final volume of 0.5 mL, 12 different
concentrations of each test compound were incubated with 15 gg (x), 10 g (8)
or 7.5
g ( ) of CHO cell membranes that stably expressed either the human x, 8 or
opioid
receptor. The assay buffer consisted of 50 mM Tris-HCI, pH 7.4, 3 mM MgC12,
0.2
mM EGTA, 3 M GDP, and 100 mM NaC1. The final concentration of [35S]GTPyS
was 0.080 nM. Nonspecific binding was measured by inclusion of 10 N.M GTPyS.
22
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
Binding was initiated by the addition of the membranes. After an incubation of
60 min
at 30 C, the samples were filtered through Schleicher & Schuell No. 32 glass
fiber
filters. The filters were washed three times with cold 50 mM Tris-HCI, pH 7.5,
and were
counted in 2 mL of Ecoscint scintillation fluid. Data are the mean E,T,a, and
EC50 values t
S.E.M. from at least three separate experiments, performed in triplicate. For
calculation of the E,naX values, the basal [35 S]GTPyS binding was set at 0%.
To
determine antagonist activity of a compound at the opioid receptors, CHO
membranes expressing the opioid receptor, were incubated with 12 different
concentrations of the compound in the presence of 200 nM of the agonist
DAMGO.
To determine antagonist activity of a compound at the K opioid receptors, CHO
membranes expressing the K opioid receptor, were incubated with the compound
in the
presence of 100 nM of the K agonist U50,488. To determine if a compound was an
antagonist at S receptors, CHO membranes expressing the 6 receptor were
incubated
with 12 different concentrations of the test compound in the presence of 10 nM
of the
S -selective agonist SNC 80.
Examples
CH2 CH2 /CH2-~ CH2
9 / \ --CHg ~ ~ --CHg --CH3 N / \ CH3
HZN
8 7 'CH3 CH3 CH3 CHs
H2N NH2 H2N O N HN
O 0 HN- O
6 7 9
23
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
CH2
N~ NiCH2~ CH2
2
--CH3 --CH3
N 0CH3 / \ CH3
CH3 R9NH N NCH3
/
N NHR9 HN~N,N
10:R9=H 11:R9=H
12; R9 = CH2Ph 13; R9 = CH2Ph 29
14; R9 = BPE 15; R9 = BPE
~CH2~ / CH2~
/CHz--a N N
--CHg --CH3
--CH3 N
" CH3
N J~ CH
// - R9 H H2N ~ N
CH3 3
N - N O
H
34; R9 = H 43
35; R9 = CH3
30 36; R9 = OH
TABLE 1
K; (nM)
Ex. # [3H]DAMGO (p) [3H]Naltrindole (b) [3H]U69,593 (K) GTPyS data
MOR antaaonist. KOR moderate
6 0.55 0.029 35 0.036 0.70 0.036 v agonist
7 88 5.2 2000 12 32 1.7
8 270 33 2000 49 160 7.1
9 170 6.7 780 32 12 0.65
0.55 0.018 120 8.5 1.0 0.071 MOR and KOR antagonist
11 890 39 47% inh. at 10 M 560 23
12 44 0.76 1500 68 240 1.8
13 88 7.2 1000 37 48 2.3
14 6.9 0.33 52 2.6 8.6 1.5
28 1.9 410 61 140 4.4
29 1.4 0.043 46 2.6 0.23 0.009 MOR antagonist; KOR agonist
30 10 1.0 51 7.8 0.81 0.19 MOR antagonist; KOR agonist
MOR weak agonist/antagonist, KOR
34 0.31 0.050 5.1 0.65 0.063 0.0016 and DOR agonist
MOR agonist/weak antagonist; KOR
35 0.77 0.051 18 0.90 0.050 0.002 agonist
36 60 7.9 730 8.3 12 1.3
24
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
K; (nM)
Ex. # [3H]DAMGO (p) [3H]Naltrindole (b) [3H]U69,593 (K) GTPyS data
43 0.75 0.038 99 10 0.65 0.005
[0015] Antinociceptive activity is evaluated by the method described in Jiang
et al. [J.
Pharmacol. Exp. Ther. 264, 1021-1027 (1993), page 1022]. The ED50's of
compounds
of the invention are expected to be under 100 nmol in the mouse acetic acid
writhing
test when administered i.c.v., and an increase in the duration of action is
expected for
compounds of the invention compared to their "parents" when given by i.p.
administration.
Definitions
[0016] Throughout this specification the terms and substituents retain their
definitions.
[0017] Alkyl is intended to include linear, branched, or cyclic hydrocarbon
structures
and combinations thereof. Lower alkyl refers to alkyl groups of from 1 to 6
carbon
atoms. Examples of lower alkyl groups include methyl, ethyl, propyl,
isopropyl,
cyclopropyl, butyl, s-and t-butyl, cyclobutyl and the like. Preferred alkyl
groups are
those of C2o or below. Cycloalkyl is a subset of alkyl and includes cyclic
hydrocarbon
groups of from 3 to 8 carbon atoms. Examples of cycloalkyl groups include c-
propyl,
c-butyl, c-pentyl, norbornyl and the like.
[00181 Alkoxy or alkoxyl refers to groups of from 1 to 8 carbon atoms of a
straight,
branched, cyclic configuration and combinations thereof attached to the parent
structure through an oxygen. Examples include methoxy, ethoxy, propoxy,
isopropoxy, cyclopropyloxy, cyclohexyloxy and the like. Lower-alkoxy refers to
groups containing one to four carbons.
[0019] Aryl and heteroaryl mean a 5- or 6-membered aromatic or heteroaromatic
ring
containing 0-3 heteroatoms selected from 0, N, or S; a bicyclic 9- or 10-
membered
aromatic or heteroaromatic ring system containing 0-3 heteroatoms selected
from 0,
N, or S; or a tricyclic 13- or 14-membered aromatic or heteroaromatic ring
system
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
containing 0-3 heteroatoms selected from 0, N, or S. The aromatic 6- to 14-
membered
carbocyclic rings include, e.g., benzene, naphthalene, indane, tetralin, and
fluorene and
the 5- to 10-membered aromatic heterocyclic rings include, e.g., imidazole,
pyridine,
indole, thiophene, benzopyranone, thiazole, furan, benzimidazole, quinoline,
isoquinoline, quinoxaline, pyrimidine, pyrazine, tetrazole and pyrazole.
[0020] Arylalkyl means an alkyl residue attached to an aryl ring. Examples are
benzyl, phenethyl and the like. Heteroarylalkyl means an alkyl residue
attached to a
heteroaryl ring. Examples include, e.g., pyridinylmethyl, pyrimidinylethyl and
the
like.
[0021] Heterocycle means a cycloalkyl or aryl residue in which one to two of
the
carbons is replaced by a heteroatom such as oxygen, nitrogen or sulfur.
Heteroaryls
form a subset of heterocycles. Examples of heterocycles that fall within the
scope of
the invention include pyrrolidine, pyrazole, pyrrole, indole, quinoline,
isoquinoline,
03tetrahydroisoquinoline, benzofuran, benzodioxan, benzodioxole (commonly
referred
to as methylenedioxyphenyl, when occurring as a substituent), tetrazole,
morpholine,
thiazole, pyridine, pyridazine, pyrimidine, thiophene, furan, oxazole,
oxazoline,
isoxazole, dioxane, tetrahydrofuran and the like.
[0022] Substituted alkyl, aryl, cycloalkyl, or heterocyclyl refer to alkyl,
aryl,
cycloalkyl, or heterocyclyl wherein up to three H atoms in each residue are
replaced
with halogen, alkyl, aryl, cycloalkyl, heterocyclyl, hydroxy, lower-alkoxy,
carboxy,
carboalkoxy, carboxamido, cyano, carbonyl, -NO2, -NR'R2; alkylthio, sulfoxide,
sulfone, acylamino, amidino, phenyl, benzyl, heteroaryl, phenoxy, benzyloxy,
heteroaryloxy, or substituted phenyl, benzyl, heteroaryl, phenoxy, benzyloxy,
or
heteroaryloxy.
[0023] Virtually all of the compounds described herein contain one or more
asymmetric centers and may thus give rise to enantiomers, diastereomers, and
other
stereoisomeric forms that may be defined, in terms of absolute
stereochemistry, as (R)-
or (S)-. The present invention is meant to include all such possible isomers,
as well as
26
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
their racemic and optically pure forms. In general it has been found that the
levo
isomer of morphinans and benzomorphans is the more potent antinociceptive
agent,
while the dextro isomer may be useful as an antitussive or antispasmodic
agent.
Optically active (R)- and (S)- isomers may be prepared using chiral synthons
or chiral
reagents, or resolved using conventional techniques. When the compounds
described
herein contain olefinic double bonds or other centers of geometric asymmetry,
and
unless specified otherwise, it is intended that the compounds include both E
and Z
geometric isomers. Likewise, all tautomeric forms are also intended to be
included.
9
11 \ 8
For example, the structural representation X"-N "R9
9 9
s H; a
is intended to include both tautomers X-NH and X-N
[0024] Some of the compounds oi the inveiition are quaternary salts, i.e.
cati:.ni.^,
species. Therefore they will always be presented as salts, and the term
"pharmaceutically acceptable salt" refers to salts whose counter ion (anion)
derives
from pharmaceutically acceptable non-toxic acids including inorganic acids,
organic
acids and water (which formally furnishes the hydroxide anion). Suitable
pharmaceutically acceptable anions for the compounds of the present invention
include
hydroxide, acetate, benzenesulfonate (besylate), benzoate, bicarbonate,
bisulfate,
carbonate, camphorsulfonate, citrate, ethanesulfonate, fumarate, gluconate,
glutamate,
glycolate, bromide, chloride, isethionate, lactate, maleate, malate,
mandelate,
methanesulfonate, mucate, nitrate, pamoate, pantothenate, phosphate,
succinate,
sulfate, tartrate, trifluoroacetate, p-toluenesulfonate, acetamidobenzoate,
adipate,
alginate, aminosalicylate, anhydromethylenecitrate, ascorbate, aspartate,
calcium
edetate, camphorate, camsylate, caprate, caproate, caprylate, cinnamate,
cyclamate,
27
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
dichloroacetate, edetate (EDTA), edisylate, embonate, estolate, esylate,
fluoride,
formate, gentisate, gluceptate, glucuronate, glycerophosphate, glycolate,
glycollylarsanilate, hexylresorcinate, hippurate, hydroxynaphthoate, iodide,
lactobionate, malonate, mesylate, napadisylate, napsylate, nicotinate, oleate,
orotate,
oxalate, oxoglutarate, palmitate, pectinate, pectinate polymer,
phenylethylbarbiturate,
picrate, pidolate, propionate, rhodanide, salicylate, sebacate, stearate,
tannate,
theoclate, tosylate and the like. The desired salt may be obtained by ion
exchange of
whatever counter ion is obtained in the synthesis of the quat. These methods
are well
known to persons of skill. Although pharmaceutically acceptable counter ions
will be
preferred for preparing pharmaceutical formulations, other anions are quite
acceptable
as synthetic intermediates. Thus X may be pharmaceutically undesirable anions,
such
as iodide, oxalate, trifluoromethanesulfonate and the like, when such salts
are chemical
intermediates. When the compounds of the invention are bisquats, one may
employ as
counter ions either two monoanionic species (e.g. C12) or a single dianionic
species
(e.g. fumarate). Similarly, one could employ oligoanionic species and make
salts
having appropriate ratios of quat to counterion, such as (quat)3 citrates.
These would
be obvious equivalents.
Abbreviations
[0025] The following abbreviations and terms have the indicated meanings
throughout:
Ac = acetyl
BNB = 4-bromomethyl-3-nitrobenzoic acid
Boc = t-butyloxy carbonyl
(CH2)2'
BPE = 2(4-biphenylyl)ethyl=
Bu = butyl
c- = cyclo
DAMGO = Tyr-ala-Gly-NMePhe-NHCH2OH
DBU = diazabicyclo[5.4.0]undec-7-ene
28
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
DCM = dichloromethane = methylene chloride = CH2C12
DEAD = diethyl azodicarboxylate
DIC = diisopropylcarbodiimide
DIEA = N,N-diisopropylethyl amine
DMAP = 4-N,N-dimethylaminopyridine
DMF = N,N-dimethylformamide
DMSO = dimethyl sulfoxide
DOR = delta opioid receptor
DPPF = 1,1'-bis(diphenylphosphino)ferrocene
DVB = 1,4-divinylbenzene
EEDQ = 2-ethoxy-l-ethoxycarbonyl-l,2-dihydroquinoline
Fmoc = 9-fluorenylmethoxycarbonyl
GC = gas chromatography
HATU = O-(7-Azabenzotriazol-1-yl)-1,1,3,3-tetramethyluronium
hexafluorophosphate
HOAc = acetic acid
HOBt = hydroxybenzotriazole
KOR = kappa opioid receptor
Me = methyl
titesyi = :. vthaiesulfonyl
MOR mu opioid receptor
MTBE = methyl t-butyl ether
NMO = N-methylmorpholine oxide
PEG = polyethylene glycol
Ph = phenyl
PhOH = phenol
PfP = pentafluorophenol
PPTS = pyridinium p-toluenesulfonate
PyBroP = bromo-tris-pyrrolidino-phosphonium hexafluorophosphate
rt = room temperature
sat'd = saturated
s- = secondary
29
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
t- = tertiary
TBDMS = t-butyldimethylsilyl
TFA = trifluoroacetic acid
THF = tetrahydrofuran
TMOF = trimethyl orthoformate
TMS = trimethylsilyl
tosyl = p-toluenesulfonyl
Trt = triphenylmethyl
U69,593 = N"'
Ph
C 0
p N~\\
[0026] Terminology related to "protecting", "deprotecting" and "protected"
functionalities occurs throughout this application. Such terminology is well
understood by persons of skill in the art and is used in the context of
processes which
involve sequential treatment with a series of reagents. In that context, a
protecting
group refers to a group which is used to mask a functionality during a process
step in
which it would otherwise react, but in which reaction is undesirable. The
protecting
group prevents reaction at that step, but may be subsequently removed to
expose the
original functionality. The removal or "deprotection" occurs after the
completion of
the reaction or reactions in which the functionality would interfere. Thus,
when a
sequence of reagents is specified, as it is below, the person of ordinary
skill can readily
envision those groups that would be suitable as "protecting groups". Suitable
groups
for that purpose are discussed in standard textbooks in the field of
chemistry, such as
Protective Groups in Organic Synthesis by T.W.Greene [John Wiley & Sons, New
York, 1991], which is incorporated herein by reference.
[0027] The compounds of the invention are synthesized by one of the routes
described
below.
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
Scheme 1.
OCN , CHz-a N CH2 V NICHZ V NICH2~
a b C d
-
-_CH3CH3 --CH3 CH3
X
H3 CH3 CH3 CH3
HO HO Z CF3SOZ0 Z NC Z
1(Cyclazocine) 2: X = H; Z NOZ 16: X = H; Z NOz 18: X= H; Z NOZ
3:X=NO2;Z=H 17:X=NO2;Z=H 19:X=N02;Z=H
NICHp-Q N/CHZ--a N/CHz--< CH2-<
e f
X --CH3 X --CH3 ~ \ --CH3 +/or N ~ \ --CH3
,CH3 , CH3 " CH3 "CH3
H2N Z H2N Z O ~ N HN
O C HN--/ O
4:X=H;Z=N02 6:X=H;Z=NH2 8 9
5:X=N02;Z=H 7:X=NH2;Z=H
Reagents and conditions: (a) 69% HNO3, CH3CO2H, 25 C; (b) PhN(Tf)2, Et3N,
CHZCI2, 25 C; (c) Zn(CN)2, Pd(PPh3)4,
microwaves, 150 C; (d) t-BUGH, KOH, 82 C; (e) MeOH, 10% Pd/C, H2, 25 C; (f)
88% HCO2H; microwaves, 120 C.
Scheme 2.
/CHz-a ~CHZ--a ICH2--<
N N NCHZ -a
/ \ CH3 a --CH3 b ON. CH3 --CH3
_ For21->10:c CH3 CH3 3 For 21 + 12: d
O NHZ
H2N NC NH2 NC N=CHOCH3 For 21 -> 14: e CH3
ReHN ~ N
6 20 21 10:R9=H
12: R9 = CH2CBH5
14: R9 = CH2CH2-4-(C6H4)C6H5
Reagents and conditions: (a) POCI3, pyridine, microwaves, 100 C; (b)
CH(OCH3)3, 4A molecular sieves, 140 C; (c) CH3OH, NH3,
microwaves, 100 C; (d) CH3OH, PhCH2NH2, microwaves, 160 C; (e) CH3OH,
H2NCH2CH2-4-(C6H4)C6H5, microwaves, 160 C.
Scheme 3.
N/CHZ-< N CH2-a N~CH2~ NiCH2~
a b c
__CH3 --CH3 -_~ - CH3 / \ --CH3
02N / ~ HZN ~ ~ H3COCH=N / ~ N
`CH3 `CH3 CH3 CH3
NC NC NC N-
NH2
19 22 23 11
Reagents and conditions: (a) 10% Pd/C, CH3OH, H2, 25 C; (b) CH(OCH3)3, 4A
molecular sieves, 140 C; (c) CH3OH, NH3, microwaves, 120`
31
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
Scheme 4.
N~CH2 N/ CH2-1 iCH2
N
a
--C~.i3 _-
_-CH3 --CH3
H2N (H3C)2NCH=N ~~ For 24 -~ 13: b N
CH3 CH3 For 24 -> 15: c C~ -
CH3
H2N NC N
O NHR9
7 24 13: R9 = CH2C6H5
15: R9 = CH2CH2-4-(C6H4)C6H5
Reagents and conditions: (a) POCI3, DMF, microwaves radiation, 100 C; (b)
CH3CO2H, PhCH2NH2, CH3CN,
microwaves, 160 C; (c) CH3CO2H, H2NCH2CH2-4-(C6H4)C6H5, CH3CN, microwaves, 160
C.
Scheme 5.
N"CH2 N/CH2 ~ /CH2~
b
X --CH3 a X / , --CHs ---- X / \ --CH3
H3 - CH3 - CH3
CF3SO2O Z Ph2C=N Z H2N Z
16:X=H;Z=NO2 25:X=H;Z=NO2 27:X=H;Z=NO2
17:X=NO2;ZH 26:X=NO2;Z=H 28:X=NO2;Z=H
CH2~ CH2~
3 / \ --CH3
--CH
For27->29:c,d OCH,
For 28 -~ 30: c,d N _
N ~ CH3
HN~ N H
N
29 30
Reagents:(a) Pd(OAc)2, BINAP, CsCO3, H2N=C(Ph)2, tol (b) 3N HCI, THF; (c) 10%
Pd/C, MeOH,
H2; (d) NaNO2, CH3CO2H.
32
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
Scheme 6.
N , CH2-< N ICHZ-a ~CH2-a
N
a b
_-CH3 -_CH3 _-CH3
02N ~ ~ OZN J ~ HZN ~ ~
~CH3 ~CH3 ~CH3
CF3SO20 PhCH2NH H2N
17 32 33
iCH2-a
For33->34:c N 34:R9=H
For 33 -> 35: d 35: R9 = CH3
For 33 --> 36: e --CH3 36: R9 = OH
N ~ ~ 37: R9 = CH2CH3
For 33 ~ 37: f 1 - CH 38: R9 = CH(CH3)2
For 33 -4 38: g R9 J1\ N 3 39: R9 = c-C H
For33->39:h H 3 5
Reagents: (a) PhCH2NH2, CH3CN; (b) 10% PdlC, MeOH, HCO2NH4; (c) HCO2H; (d)
CH3CO2H,
microwaves; (e) COCI2 in tol, THF; (f) CH3CH2CO2H, microwaves; (g)
(CH3)2CHCO2H, microwaves;
(h) c-C3H5CO2H, microwaves.
Scheme 7.
N/CH2 N/CH2 /CH2-<
N
For40->41:b
O"CCH3 --CH3 a ~ ~ --CHs For 40 -> 42: c --CH3
CH3 3 , CH3
H2N NO2 H2N NH2 HN~N
28 40 R9 41: R9 = H
42: R9 = CH3
Reagents:(a) 10% Pd/C, MeOH, H2 (b) HCOzH, microwaves; (c) CH3CO2H,
microwaves.
Scheme B.
N /CH2 -< N "CH2--<
CH3 a_~_ --CH3
"CH3 CH3
NC NO2 H2N N
OI
18 43
Reagents and conditions: (a) SnC12=2H20, HCI, 14h, 25 C.
33
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
Experimental Section
[0028] Proton NMR spectra and in certain cases 13C NMR were obtained on a
Varian
Unity-300 or 500 NMR spectrometer with tetramethylsilane as an internal
reference
for samples dissolved in CDC13. Samples dissolved in CD3OD and DMSO-d6 were
referenced to the solvent. Proton NMR multiplicity data are denoted by s
(singlet), d
(doublet), t (triplet), q (quartet), m (multiplet), dd (doublet of doublets),
and br (broad).
Coupling constants are in hertz. Direct insertion probe chemical ionization
mass
spectral data were obtained on a Shimadzu GC-17A GC-MS mass spectrometer.
Direct infusion electrospray ionization (in positively charged ion mode) mass
spectral
data were obtained on an Agilent 1100 series LC/MSD system (Germany). Melting
points were determined on a Meltemp capillary melting point apparatus and were
uncorrected. Infrared spectral data were obtained on a Perkin-Elmer Paragon
1000
FT-IR spectrophotometer. Optical rotation data was obtained from a Perkin-
Elmer
241 polarimeter. The assigned structure of all test compounds and
intermediates were
consistent with the data. Carbon, hydrogen, and nitrogen elemental analyses
for all
novel targets were performed by Quantitative Technologies Inc., Whitehouse,
NJ, and
were within 0.4% of theoretical values except as noted; the presence of
water or
other solvents was confirmed by proton NMR. Reactions were generally performed
in
an argon or iiitrogen atmo. iph vre. C..mmerC:ally p rchaCP.lj (_11eTT11raIs
were used
without purification unless otherwise noted. The following reagents were
purchased
from Aldrich Chemical Company: N-hydroxysuccinimide, phenethylamine, 3-phenyl-
1-propylamine, 4-aminobiphenyl, palladium acetate, 4-phenylbenzylamine and
benzyl
amine. The following reagent was purchased from Trans World Chemicals: 2-(4-
biphenyl ethylamine). The following reagents were purchased from Strem
Chemicals,
Incorporated: 1,1'-bis(diphenyl-phosphino)ferrocene (dppf) and dichloro[1,1'-
bis(diphenylphosphino)-ferrocene]palladium (II) dichloromethane adduct
[PdC12(dppf)]. Pyridine was distilled from KOH. DMF and DMSO were distilled
over CaH2 under reduced pressure. Amines were purchased from Aldrich Chemical
Company and used as received unless otherwise indicated. Toluene and Et20 were
distilled from sodium metal. THF was distilled from sodium/benzophenone ketyl.
Pyridine was distilled from KOH. Methylene chloride was distilled from CaH2.
DMF
34
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
and DMSO were distilled from CaH2 under reduced pressure. Methanol was dried
over 3zL molecular sieves prior to use. Silica gel (Bodman Industries, ICN
SiliTech 2-
63 D 60A, 230-400 Mesh) was used for flash column chromatography.
[0029] Cis-( )-3-(cyclopropylmethyl)-1,2,3,4,5,6-hexahydro-6,11-dimethyl-7-
nitro-2,6-methano-3-benzazocine-8-ol (2) and cis-(:+3-(cyclopropylmethyl)-
1,2,3,4,5,6-hexahydro-6,11-dimethyl-9-nitro-2,6-methano-3-benzazocin-8-ol (3).
A solution of 69% nitric acid (0.20 g) in 2.0 mL glacial acetic acid was added
to a
solution of cylazocine3 (1; 0.542 g, 2.0 mmol) in 3.0 mL glacial acetic acid
at 25 C.
After stirring at 25 C for 3 h, tlc indicated the presence of starting
material and an
additiona10.10 gm of 69% nitric acid was added. After stirring 2 h at 25 C,
tic
indicated all starting material was consumed and the reaction mixture was
poured into
a mixture of ice and excess concentrated ammonium hydroxide. The mixture was
treated with ethyl acetate and the organic phase was washed with brine, dried
over
Na2SO4, filtered, and concentrated to give a crude solid produce which was
purified by
gradient silica gel flash chromatography (CH2C12:CH3OH; 20:1 --> 10:1) to give
2
(0.26 g, 40%) as an off-white solid and 3 (0.35 g, 54%) as an off-white foam:
Recrystallization from MeOH/CH2C12 gave off-white crystals having mp 145 C
and
mp 175 C, respectively.
[0030] For 2: 'H NMR (500 MHz, CDC13) 8 6.98 (d, 1H, J= 8.3 Hz), 6.83 (d, IH,
J=
8.5 Hz), 3.10 (m, IH), 2.84 (d, 1 H, J= 18.8 Hz), 2.81-2.57 (m, 2H), 2.46 (m,
1 H), 2.32
(m, 1H), 2.03 (m, 3H), 1.86-1.66 (m, 1H), 1.31 (s, 3H), 1.25 (m, IH), 0.87 (m,
4H),
0.51 (m, 2H), 0.11 (m, 2H); MS (ESI) mlz 317 (M+H)+; Anal. Caled. for
C18H24N203=0.75 H20: C 65.53, H 7.79, N 8.49. Found: C 65.27, H 7.41, N 8.23.
[0031] For 3: 'H NMR (500 MHz, CDC13) 8 10.36 (s, 1H), 7.80 (s, 1H), 7.03 (s,
1H),
3.16 (m, 1H), 2.95 (d, 1H, J= 18.8 Hz), 2.79-2.56 (m, 2H), 2.48 (m, 1H), 2.32
(m,
1H), 1.96 (m, 3H), 1.39 (s, 3H), 1.36 (m, 1H), 0.85 (m, 4H), 0.52 (m, 2H),
0.11 (m,
2H); MS (ESI) m/z 317 (M+H)+; Anal. Calcd. for C18H24N203-0.5 H20: C 66.44, H
7.74, N 8.61. Found: C 66.03, H 7.33, N 8.48.
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
[0032] Trifluoromethanesulfonic acid, cis-(f)-3-(cyclopropylmethyl)-
1,2,3,4,5,6-
hexahydro-6,11-dimethyl-7-nitro-2,6-methano-3-benzazocine-8-yl ester (16).
Triethylamine (0.22 g, 1.48 mmol) was added to a solution of 2 (0.47 g, 1.48
mmol)
dissolved in 20 mL of CHC13. PhN(SO2CF3)2 (0.58 g, 1.63 mmol) was then added
and
the resulting mixture stirred at 25 C for 4 h. The solvent was removed on a
rotary
evaporator and the resulting mixture was purified by gradient silica gel flash
chromatography (CH2C12:CH3OH; 80:1 --> 40:1) to give 16 (0.59 g, 88%) as an
off-
white foam. 'H NMR (500 MHz, CDC13) S 7.30 (d, 1H, J= 8.5 Hz), 7.24 (d, 1H, J=
8.6 Hz), 3.56 (m, 1H), 3.17 (m, 1H), 3.05 (m, 2H), 2.81 (m, 1H), 2.66 (m, 1H),
2.29-
2.04 (m, 2H), 1.90 (m, 1H), 1.34 (m, 4H), 0.87 (m, 4H), 0.69 (m, 2H), 0.28 (m,
2H).
[0033] Trifluoromethanesulfonic acid, cis-(f)-3-(cyclopropylmethyl)-
1,2,3,4,5,6-
hexahydro-6,11-dimethyl-9-nitro-2,6-methano-3-benzazocine-8-yl ester (17).
Using a procedure similar to that used to prepare 16, compound 3 was converted
to 17
(93%) as off-white foam. 'H NMR (500 MHz, CDC13) S 7.94 (s, 1H), 7.27 (s, 1H),
3.60 (m, 1H), 3.22-2.94 (m, 3H), 2.84 (m, 1H), 2.68 (m, IH), 2.30 (m, 1H),
2.11 (m,
2H), 1.41 (s, 3H), 1.38 (m, 1H), 0.84 (m, 414), 0.69 (m, 2H), 0.29 (m, 2H).
[0034] Cis-(+)-3-(cyclopropyhnethyl)-1,2,3,4,5,6-hexahydro-6,11-dimethyl-7-
IIIIrV-nG,Oi-Il '- lelll'd--llO-3-~IJC- -'--~llL'ALVI=Il1G-OO-larIv"o
u;i4u..'ili~, tio., /1 Ql). T. n au t'.:b.P. c.^.ntainina 16
v ..Zj -(0.27 g, .061 mmol) was added under an N2 blanket, Zn(CN)2 (0.14 g,
1.22 mmol) and
Pd(PPh3)4 (0.07 g, 0.061 mmol). DMF (degassed with N2), 3.0 mL) was then added
via a cannula under N2. The resulting mixture was irradiated with microwaves
at 150
C for 15 min. The resulting mixture was partitioned between water and EtOAc.
The
organic phase was washed with water (X2) and brine, and then dried over
Na2SO4,
filtered, and concentrated to give a crude product which was purified by
silica gel flash
chromatography (CH2CI2:CH3OH:NH4OH;80:1:0.1) to give 18 (0.14 g, 70%) as an
off-white foam. 'H NMR (500 MHz, CDC13) S 7.52 (d, 1H, J= 8.1 Hz), 7.35 (d,
1H, J
= 8.1 Hz), 3.20 (m, 1 H), 3.04 (d, 1 H, J= 19.1 Hz), 2.86 (m, 1 H), 2.68 (m,
2H), 2.47
(m, 1H), 2.34 (m, 1H), 2.10-1.74 (m, 3H), 1.34 (m, 4H), 0.89 (m, 1H), 0.84 (d,
3H, J=
7.1 Hz), 0.54 (m, 2H), 0.12 (m, 2H). MS (ESI) m/z 326 (M+H)+.
36
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
[0035] Cis-(f)-3-(cyclopropylmethyl)-1,2,3,4,5,6-hexahydro-6,11-dimethyl-9-
nitro-2,6-methano-3-benzazocine-8-carbonitrile (19). Using a procedure similar
to
that used to prepare 18, compound 17 was converted to 19 (quantitative yield)
as an
off-white foam. 'H NMR (500 MHz, CDC13) S 8.05 (s, 1H), 7.75 (s, 1H), 3.22 (m,
1H), 3.08 (d, 1H, J= 19.8 Hz), 2.79 (m, 2H), 2.47 (m, 1H), 2.32 (m, 1H), 2.10-
1.78
(m, 3H), 1.46 (s, 3H), 1.33 (m, IH), 0.87 (m, 1H), 0.83 (d, 3H, J= 7.1 Hz),
0.54 (m,
2H), 0.12 (m, 2H).
[0036] Cis-(t)-3-(cyclopropylmethyl)-1,2,3,4,5,6-hexahydro-6,11-dimethyl-7-
nitro-2,6-methano-3-benzazocine-8-carboxamide (4). A solution of 18 (0.11 g,
0.33 mmol) dissolved in t-BuOH (2.0 mL) was heated at 82 C and KOH (0.056 g,
1.0
mmol) was added. After stirring at 82 C for lh, brine and EtOAc were added.
The
organic phase was dried over Na2SO4, filtered, and concentrated to give a
crude
product which was purified by silica gel flash chromatography
(CH2C12:CH3OH:NH4OH; 20:1:0.1) to give 4 as an off-white solid (0.098 g, 85%).
Crystallization of this solid from acetone followed by a recrystallization
from i-
PrOH/t-BuOH gave crystals having mp 190 C. IH NMR (500 MHz, CDC13) S 8.03
(s, 1H), 7.54 (s, 1H), 7.38 (s, 2H), 3.04 (m, 1H), 2.96 (d, 1H, J= 19.5 Hz),
2.76 (m,
2H), 2.38 (m, 1H), 2.24 (m, 1H), 2.06-1.56 (m, 3H), 1.19 (m, 4H), 0.79 (m,
1H), 0.73
(d 3H J- v`.e Hz\ v.44'~i, 2H\ v.v7 ~.. 2H~ h1S (ESI) m/z 344 rivr+u~+= Ana1,
> > - J, l , h l l1-__l ,
Calcd. for C19H25N303=0.5 H20: C 64.75, H 7.44, N 11.92. Found: C 64.47, H
7.21, N
11.56.
[0037] Cis-(#)-3-(cyclopropylmethyl)-1,2,3,4,5,6-hexahydro-6,11-dimethyl-9-
nitro-2,6-methano-3-benzazocine-8-carboxamide (5). Using a procedure similar
to
that used to prepare 4, compound 19 was converted to 5 (45%) as an off-white
foam.
1H NMR (500 MHz, CDC13) S 7.80 (s, 1H), 7.42 (s, 1H), 5.89 (m, 2H), 3.20 (m,
11-1),
3.03 (d, 1 H, J= 19.0 Hz), 2.75 (m, 2H), 2.47 (m, 1H), 2.33 (m, 1 H), 2.06-
1.82 (m,
3H), 1.43 (s, 314), 1.34 (m, 114), 0.87 (m, 1H), 0.83 (d, 3H, J= 7.1 Hz), 0.53
(m, 2H),
0.12 (m, 2H); MS (ESI) m/z 344 (M+H)+; Anal. Calcd. for C19H25N303-0.25 H20: C
65.59, H 7.39, N 12.08. Found: C 65.39, H 7.38, N 11.93.
37
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
[0038] Cis-(t)-3-(cyclopropylmethyl)-1,2,3,4,5,6-hexahydro-6,11-dimethyl-7-
amino-2,6-methano-3-benzazocine-8-carboxamide (6). To a solution of 4 (0.15 g,
0.44 mmol) dissolved in MeOH (20 mL) was added 10% Pd/C (0.093 g). The
resulting mixture was subjected to 55 psi H2 in a Parr shaker for 3d at 25 C.
The
mixture was filtered and concentrated to give a crude product that was
purified by
silica gel flash chromatography (CH2C12:CH3OH:NH4OH; 30:1:0.1) giving 6(0A60
g,
44%) as a white foam. 1H NMR (500 MHz, CDC13) S 7.14 (d, 1H, J= 8.1 Hz), 6.42
(d, 1 H, J= 8.1 Hz), 6.12 (s, 1 H), 5.5 8(br s, 2H), 3.09 (m, 1 H), 2.76 (m, 3
H), 2.24 (m,
IH), 2.28 (m, IH), 2.06-1.70 (m, 3H), 1.59 (s, 3H), 1.58 (m, 1H), 0.91 (d, 3H,
J= 7.1
Hz), 0.86 (m, 1H), 0.51 (m, 2H), 0.10 (m, 2H); MS (ESI) m/z 314 (M+H)+; Anal.
Calcd. for C19H27N30=0.25 H20: C 71.78, H 8.72, N 13.22. Found: C 72.00, H
8.84,
N 12.98.
[0039] Cis-(t)-3-(cyclopropylmethyl)-1,2,3,4,5,6-hexahydro-6,11-dimethyl-9-
amino-2,6-methano-3-benzazocine-8-carboxamide (7). Using a procedure similar
to
that used to prepare 6, compound 5 was converted to 7 (63%) as an off-white
foam.
1H NMR (500 MHz, CDC13) 6 7.20 (s, IH), 6.42 (s, 1H), 5.61 (br s, 2H), 5.42
(s, 2H),
3.10 (m, 1H), 2.84 (d, 1H, J= 18.8 Hz), 2.75-2.53 (m, 2H), 2.46 (m, 1H), 2.30
(m,
1H), 2.06-1.80 (m, 3H), 1.35 (s, 3H), 1.27 (m, IH), 0.87 (m, 1H), 0.84 (d, 3H,
J= 7.1
rr_. n c, i.__ ~rr~ n ~ n ul. }~ Rc rTteT m/-, 21d (1~A-1-T4){- An l(~:alrrl.
for
I1G), V.J1 `lll, LJ-1) , V.1V (m, 211), i"viv ~LVi) aaaic. ki.i -/ , a.. .
C 19H27N30 -0.25 1120: C 71.78, H 8.72, N 13.22. Found: C 72.00, H 8.73, N
13.27.
[0040] Cis-(f)-7-Amino-3-(cyclopropylmethyl)-1,2,3,4,5,6-hexahydro-6,11-
dimethyl-2,6-methano-3-benzazocine-8-carbonitrile (20). A mixture of 6 (0.22
g,
0.70 mmol), POC13 (0.11 gm, 0.70 mmol), and pyridine (2.0 mL) was heated at
100 C
for 20 min under microwave radiation and concentrated. The residue was
dissolved in
1.0 N HCI and stirred for 1 h at 25 C. The reaction mixture was made basic
with
saturated Na2CO3 and the organic material was extracted into ethyl acetate.
The
organic layer were washed with brine, dried over Na2SO4, filtered and
concentrated to
give a crude product that was purified by silica gel chromatography
(Combiflash -
CH2CIZ:CH3OH:NH4OH) to give 20 as an off-white solid (0.11 g) in 54% yield: 'H
NMR (500 MHz, CDC13) 6 7.07 (d, 1H, J= 8.5 Hz), 6.41 (d, 1H, J= 8.8 Hz), 3.16
(m,
38
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
1H), 2.70 (m, 4H), 2.46 (m, 1H), 2.30 (m, 1H), 2.07-1.70 (m, 3H), 1.64 (m,
4H), 0.94
(d, 3H, J= 6.8 Hz), 0.88 (m, 1H), 0.53 (m, 2H), 0.12 (m, 2H).
[00411 Cis-(:L)-9-Amino-3-(cyclopropylmethyl)-1,2,3,4,5,6-hexahydro-6,11-
dimethyl-2,6-methano-3-benzazocine-8-carbonitrile (22). A mixture of 19 (0.180
g,
0.55 mmol), 10% Pd/C and CH3OH (20 mL) was subjected to 40 psi H2 in a Parr
shaker at 25 C for 15 h. The mixture was filtered and concentrated to give 22
as a
crude product that was purified by silica gel chromatography (Combiflash -
CH2C12:CH3OH:NH4OH) to give an off-white foam (0.070 g, 47%): 1H NMR (500
MHz, CDC13) 8 7.25 (s, 1H), 6.47 (s, 1H), 4.18 (s, 2H), 3.15 (m, 1H), 2.86 (d,
1H, J=
19.0 Hz), 2.80-2.58 (m, 2H), 2.48 (m, 1H), 2.33 (m, 1H), 1.94 (m, 3H), 1.32
(s, 3H),
1.25 (m, 1H), 0.90 (m, 1H), 0.81 (d, 3H, J- 7.1 Hz), 0.53 (m, 2H), 0.12 (m,
2H); MS
(ESI) m/z 296 (M+H)+.
[0042] 7,8-Fused pyrimidinone derivative 8 and 8,9-Fused pyrimidinone
derivative 9. A mixture of 6 (0.035 g, 0.11 mmol) and 2.0 mL of 88% formic
acid
was heated at 120 C under microwave radiation for 30 min. The reaction
mixture was
basified using excess NH4OH and the organic material was extracted into ethyl
acetate.
The organic phase was washed with brine, dried over Na2SO4 and concentrated
giving
a
a trri.iue prou a==~ul.+l + Lt-L +_~ was ~ puril f+ar1lli4 1v,yo <. gv
..b...i,..~ al Ohrnmatnoranhv (( nm.. hiflach -
ial. u..ava _____.__
CH2C12:CH3OH:NH4OH) giving 8 (0.020 gm, 54%). In similar fashion, 7 (0.021 g,
0.067 mmol) was converted to 9(0.016 gm, 50%). Alternatively, the product from
the
nitration of cyclazocine containing the 7- and 9-nitro-cyclazocine derivatives
2/3 was
converted to a mixture of the 7- and 9-nitro-carbonitrile derivatives 18/19
using the
same method as described above for the individual regioisomers. The mixture of
18/19 (1.00 gm, 3.08 mmol) was dissolved in CH3OH (50 mL) and 10% Pd/C (0.065
g) was added. The resulting mixture was subjected to 20 psi hydrogen in a Parr
shaker
for 20 h, filtered and concentrated giving a crude product consisting of 6/7
contaminated with 20/22. This crude reaction product (1.01 g) was treated with
10 mL
88% formic acid at 100 C for 37 h and made basic with excess NaOI-I/H2O. The
organic materials were extracted into ethyl acetate, washed with brine, dried
over
NaZSO4 and concentrated giving a mixture that was separated by silica gel
flash
39
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
chromatography (hexane:acetone:NH4OH) to provide 8 (0.298 g) and 9 (0.348 gm)
as
off-white solids in overall yields (two steps) of 30% and 35%, respectively.
[0043] For 8: 'H NMR (500 MHz, CDC13) S 10.90 (br s, 1H), 8.08 (d, 1H, J= 8.1
Hz), 7.99 (s, 1H), 7.25 (d, 1H, J= 8.1 Hz), 3.19 (m, 1H), 2.93 (m, 2H), 2.77
(m, 1H),
2.48 (m, IH), 2.29 (m, 1H), 2.06 (m, IH), 1.90 (m, 2H), 1.81 (s, 3H), 1.64 (m,
1H),
0.90 (d, 3H, J= 7.1 Hz), 0.88 (m, IH), 0.51 (m, 2H), 0.10 (m, 2H); MS (ESI)
m/z 324
(M+H)+; Anal. Calcd. for C20H25N30: C 74.27, H 7.79, N 12.99. Found: C 73.95,
H
7.86, N 12.78.
[0044] For 9: 'H NMR (500 MHz, CDC13) 8 11.10 (br s, 1H), 8.19 (s, IH), 8.05
(s,
1 H), 7.48 (s, 1 H), 3.23 (m, 1H), 3.14 (d, 1 H, J= 19.3 Hz), 2.91-2.72 (m,
2H), 2.51 (m,
IH), 2.35 (m, 1H), 2.08-1.86 (m, 3H), 1.52 (s, 3H), 1.38 (m, 1H), 0.90 (m,
1H), 0.87
(d, 3H, J= 7.1 Hz), 0.88 (m, 1H), 0.53 (m, 2H), 0.13 (m, 2H); MS (ESI) m/z 324
(M+H)+; Anal. Calcd. for C20H25N30-0.25 HZO: C 73.25, H 7.84, N 12.81. Found:
C
73.14, H 7.90, N 12.38.
[0045] 7,8-Fused aminopyrimidine derivative 10. A mixture of 20 (0.11 g, 0.38
mmol), CH(OCH3)3 (2 mL) and 4A molecular sieves was heated at 140 C for 48 h.
mt F1~õ .,,]tcu and 4 A ==n Rt~IP mi~la e:ne
Pd1atP 21
i Ile rCa(:t1Ui! fi111illlre waS till.c aiu ivnienii atiu w~i v v 1 t
(0.120 g) which, without further purification, was combined with methanol
saturated
with ammonia gas. The resulting mixture was heated for 1 h at 100 C under
microwave radiation and then made basic with concentrated ammonia. After
dilution
with H20, the organic material was extracted into CHZC12 and the organic layer
was
washed with brine, dried over Na2SO4 and concentrated to give mixture that was
purified by silica gel chromatography (Combiflash - CH2C12:CH3OH:NH4OH) and
crystallization. The desired product 10 (0.074 gm) was obtained in 56% yield
(2 steps)
as an off-white solid: mp 190 C: NMR (500 MHz, CDC13) 6 8.56 (s, 1H), 7.49
(d,
1H, J= 8.3 Hz), 7.20 (d, 1H, J= 8.3 Hz), 5.54 (s, 2H), 3.20 (m, 1H), 2.92 (m,
2H),
2.76 (m, 1H), 2.48 (m, 1H), 2.29 (m, 1H), 2.19 (m, IH), 1.94 (m, 4H), 1.89 (s,
3H),
0.91 (d, 3H, J= 7.1 Hz), 0.89 (m, 1H), 0.51 (m, 2H), 0.10 (m, 2H); MS (ESI)
m/z 323
(M+H){; Anal. Caled. for C2oH26N4=0.25 H20: C 73.47, H 8.17, N 17.14. Found: C
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
73.59, H 8.04, N 16.92.
[0046] 7,8-Fused benzylaminopyrimidine derivative 12. Using a procedure
similar
to that used to prepare 10, compound 21 was treated with benzylamine to
provide 12
(69%) as an off-white foam: NMR (500 MHz, CDC13) 8 8.64 (s, 1H), 7.44 (d, 1H,
J=
8.5 Hz), 7.40-7.30 (m, 5H); 7.15 (d, 1H, J= 8.3 Hz), 5.81 (m, 1H), 4.83 (d,
2H, J= 5.4
Hz), 3.20 (m, 1H), 2.91 (m, 2H), 2.77 (m, 1H), 2.48 (m, 1H), 2.29 (m, 1H),
2.22 (m,
1H), 1.93 (m, 2H), 1.90 (s, 3H), 1.88 (m, 1H); 0.90 (d, 3H, J= 7.1 Hz), 0.88
(m, IH),
0.51 (m, 2H), 0.10 (m, 2H); MS (ESI) m/z 413 (M+H)+; Anal. Calcd. for
C27H32N4=0.5 H20: C 76.92, H 7.89, N 13.29. Found: C 76.77, H 7.99, N 12.90.
[0047] 7,8-Fused biphenylethylaminopyrimidine derivative 14. Using a procedure
similar to that used to prepare 10, compound 21 was treated with 4-
biphenylethylamine to provide to 14 (71%) as an off-white foam: NMR (500 MHz,
CDC13) S 8.64 (s, 1H), 7.58 (m, 4H), 7.45 (m, 21-1), 7.34 (m, 3H), 7.29 (d,
IH, J= 8.5
Hz), 7.13 (d, 1 H, J= 8.5 Hz), 5.5 6 (m, 1 H), 3.93 (m, 2H), 3.19 (m, 1 H),
3.06 (t, 2H, J
= 6.6 Hz), 2.89 (m, 2H), 2.77 (m, 1H), 2.47 (m, 1H), 2.28 (m, 1H), 2.21 (m,
1H), 1.90
(s, 3H), 1.87 (m, 1H); 1.63 (m, 2H), 0.90 (d, 3H, J= 7.1 Hz), 0.88 (m, 1H),
0.51 (m,
2H), 0.10 (m, 2H); MS (ESI) m/z 503 (M+H)+; Anal. Caled. for C34H38N4=0.5 H20:
C
n + rr n ~~ wr In nc r. .. a. ^7[l 00 LT 7 ti~ N 111 Q2
7J.81, t1 7.68, tv tv.75. rvuttu. V i7.oo, ti i.v~, i. iv.u..,.
[0048] 8,9-Fused aminopyrimidine derivative 11. Using a procedure similar to
that
used to prepare 10, compound 22 was converted to imidate interemdiate 23 which
was
then converted to 11 (86%) as an off-white foam: 1H NMR (500 MHz, CDC13) 6
8.56
(s, 1H), 7.62 (s, 1H), 7.58 (s, 1H), 6.00 (s, 2H), 3.23 (m, IH), 3.18 (d, IH,
J= 19.0
Hz), 2.89 (m, 1H), 2.73 (m, 1H), 2.51 (m, 1H), 2.35 (m, 1H), 2.01 (m, 3H),
1.48 (s,
3H), 1.35 (m, IH), 0.89 (m, IH), 0.87 (d, 3H, J= 7.3 Hz), 0.53 (m, 2H), 0.13
(m, 2H);
MS (ESI) m/z 323 (M+H)+; C20H26N4=0.25 H20: C 73.47, H 8.17, N 17.14. Found: C
73.33, H 8.03, N 16.85.
[0049] 8,9-Fused benzylaminopyrimidine derivative 13. A mixture of 7 (0.084 g,
0.27 mmol), POC13 (0.41 g, 2.7 mmol), and DMF (3.0 mL) was heated at 100 C
under
41
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
microwave radiation for 10 min and concentrated. The resulting dark oil was
dissolved in H20, made basic with Na2CO3 and extracted (X3) with CH2Cl2. The
combined organic extracts were dried over Na2SO4 and concentrated to give
mixture
that was purified by silica gel chromatography (CH2Cl2:CH3OH:NH4OH) giving the
desired amidine intermediate 24 in 89% yield. Treatment of 24 (0.12 g, 0.34
mmol)
with benzylamine (0.044 g, 0.41 mmol) and excess 30% HOAc in CH3CN at 160 C
under microwave radiation for 20 min provided, after concentration, an oil
that was
partitioned between saturated Na2CO3 and CH2C12. The organic phase was washed
with brine, dried over Na2SO4 and concentrated to give a crude product that
was
purified by silica gel chromatography (Combiflash - CH2C12:CH3OH:NH4OH) giving
13 (0.16 g) 92% yield as an off-white solid: NMR (500 MHz, CDC13) 6 8.64 (s,
IH),
7.56 (m, IH), 7.50 (s, 1H), 7.44 (d, 2H, J= 7.3 Hz), 7.39 (t, 2H, J= 7.3 Hz),
7.34 (d,
1H, J= 7.3 Hz), 5.94 (br s, IH), 4.90 (m, 2H), 3.22 (m, I H), 3.17 (d, IH, J=
19.0
Hz), 2.88 (m, 1H), 2.72 (m, 1H), 2.51 (m, IH), 2.34 (m, 1H), 1.99 (m, 3H),
1.47 (s,
3H), 1.33 (m, 1H), 0.88 (m, 1H), 0.86 (d, 3H, J= 7.1 Hz), 0.52 (m, 2H), 0.12
(m, 2H);
MS (ESI) m/z 413 (M+H)+; C2H32N4= H20: C 75.31, H 7.96, N 13.01. Found: C
75.64, H 7.73, N 13.02.
[0050] 8,9-Fused biphenylethylaminopyrimidine derivative 15. Using a procedure
stai tU ittat iiscu .,] to ,. prepare 24 v:~a.~; treated with 4-
biphenylethylamine ~.,, compound u
biphenylethylamine to provide to 15 (86%) as an off-white foam: NMR (500 MHz,
CDC13) 8 8.63 (s, 1H), 7.58 (m, 4H), 7.54 (s, IH), 7.45 (m, 2H), 7.36 (m, 4H),
5.72 (m,
I H), 3,96 (m, 2H), 3.20 (m, ,1 H), 3.16 (d, I H, J= 19.1 Hz), 3.09 (t, 2H, J=
7.1 Hz),
2.80 (m, 1H), 2.71 (m, IH), 2.50 (m, IH), 2.34 (m, IH), 2.04-1.94 (m, 3H),
1.43 (s,
3H), 1.30 (m, 1H), 0.88 (m, IH), 0.85 (d, 3H, J= 7.1 Hz), 0.52 (m, 2H), 0.12
(m, 2H);
MS (ESI) m/z 503 (M+H)+; Anal. Calcd. for C34H38N4-0.5 H20: C 79.81, H 7.68, N
10.95. Found: C 79.52, H 7.64, N 10.83.
[0051] Cis-(:E)-3-(cyclopropylmethyl)-N-(diphenylmethylene)-1,2,3,4,5,6-
hexahydro-6,11-dimethyl-7-nitro-2,6-methano-3-benzazocine-8-amine 25. To a
tube containing benzophenoneimine (0.10 g, 0.56 mmol), Pd(OAc)2 (0.010 g,
0.045
mmol), BINAP (0.014 g, 0.022 mmol), and CsZCO3 (0.18 g, 0.56 mmol) was added
16
42
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
(0.20 g, 0.45 mmol) dissolved in 5 mL toluene. The reaction mixture was heated
at
150 C for 15 min under microwave radiation. Upon cooling to 25 C, the
mixture was
diluted with EtOAc, filtered and concentrated in vacuo. The resulting residue
was
purified by silica gel flash chromatography giving 25 (0.12 g, 56%) as an off-
white
solid. 'H NMR (500 MHz, CDC13) 8 7.73-7.28 (m, 10H), 6.78 (d, 1H, J= 8.7 Hz),
6.18 (d, 1 H, J= 8.7 Hz), 3.05 (m, IH), 2.80 (m, 1 H), 2.63 (m, 1 H), 2.57 (m,
1 H), 2.44
(m, 1H), 2.30 (m, 1H), 1.82 (m, 2H), 1.35 (s, 3H), 1.25 (m, 1H), 0.84 (d, 3H,
J- 7.0
Hz), 0.51 (m, 2H), 0.10 (m, 2H). MS (ESI) m/z 480 [(M+H)+].
[0052] Cis-(f)-3-(cyclopropylmethyl)-N-(diphenylmethylene)-1,2,3,4,5,6-
hexahydro-6,11-dimethyl-9-nitro-2,6-methano-3-benzazocine-8-amine 26. Using a
procedure similar to that used to prepare 25, compound 17 was converted to 26
(88%)
as an off-white foam. MS (ESI) m/z 480 [(M+H)+].
[0053] Cis-(f)-3-(cyclopropylmethyl)-1,2,3,4,5,6-hexahydro-6,11-dimethyl-7-
nitro-2,6-methano-3-benzazocine-8-amine 27. Compound 25 (0.13 g, 0.26 mmol)
was dissolved in 2 mL THF and 4 mL of 3N HCl was added. The reaction mixture
was stirred at 25 C for 30 min and was made basic through the addition of
conc.
NH4OH. The mixture was treated with ethyl acetate and the organic phase was
dried
nr111C`.P which was
A7_[LZJlJC~l14, 1:t1111a....,.G1G,7U, ait,U7 concentrated tnw give crude i~~a
a u rr~il~a soliwl p,r.,.. ....
over 1V
purified by silica gel flash chromatography to give 27 (0.080 g, 98%) as an
off-white
solid. 'H NMR (500 MHz, CDCl3) S 6.98 (d, 1H, J= 8.1 Hz), 6.63 (d, 1H, J= 8.1
Hz), 3.93 (s, 2H), 3.10 (m, 1H), 2.80 (m, 2H), 2.66 (m, 1H), 2.60 (m, 1H),
2.48 (m,
1H), 2.43 (m, 1H), 2.36 (s, 1H), 2.32 (m, 1H), 2.00 (m, 1H), 1.82 (m, 2I-I),
1.30 (s,
3H), 0.82 (d, 3H, J= 7.2 Hz), 0.52 (m, 2H), 0.11 (m, 2H). MS (ESI) m/z 316
[(M+H)+].
[0054] Cis-(t)-3-(cyclopropylmethyl)-1,2,3,4,5,6-hexahydro-6,11-dimethyl-9-
nitro-2,6-methano-3-benzazocine-8-amine 28. Using a procedure similar to that
used
to prepare 27, compound 26 was converted to 28 (88%) as an off-white foam. 'H
NMR (500 MHz, CDC13) 8 7.81 (s, 1H), 6.67 (s, 1H), 5.88 (s, 2H), 3.25 (m, 1H),
2.87
(d, 1H, J= 18.0 Hz), 2.70 (m, 2H), 2.48 (dd, 1H, J= 5.0 Hz), 2.33 (dd, 1H, J=
5.0
43
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
Hz), 2.04 (m, 1H), 1.92 (m, 1H), 1.31 (s, 3H), 1.25 (m, 1H), 0.83 (d, 3H, J=
8.1 Hz),
0.50 (m, 2H), 0.09 (m, 2H). MS (ESI) m1z 316 [(M+H)+].
[0055] 7,8-Fused triazole derivative 29. Compound 27 (0.060 g), 10% Pd/C
(0.006
gm) and methanol (2 mL) was subjected to 42 psi H2 in a Parr shaker for 8 h at
25 C.
The mixture was filtered and the filtrate concentrated giving a somewhat
unstable
diamine product (0.056 gm, 100%). A portion (0.025 gm) of this crude product
was
dissolved in 1.0 mL HOAc. To this solution was added NaNO2 (0.006 g) and the
resulting mixture stirred for 1 h at 25 C. The mixture was basified with
excess cone.
NH4OH and the organic material was extracted into ethyl acetate. The extracts
were
dried over Na2SO4, filtered, and concentrated to give a crude solid produce
which was
purified by silica gel flash chromatography to give 29 (0.016 g, 62%) as an
off-white
foam. 'H NMR (500 MHz, CDC13) 5 10.68 (br, 1H), 7.63 (d, 1H, J= 8.4 Hz), 7.17
(d,
1 H, J= 8.4 Hz), 3.3 0(s, 114), 3.10 (d, 1 H, J= 18.6 Hz), 2.92 (m, 1 H), 2.86
(m, IH),
2.60 (m, 1H), 2.43 (m, 1H), 2.11 (m, 2H), 2.03 (m, 2H), 1.91 (s, 311), 1.79
(d, 1H, J=
8.1 Hz), 0.94 (d, 3H, J= 7.2 Hz),0.52 (m, 2H), 0.13 (m, 2H). MS (ESI) m/z 296
[(M+H)+]. Anal. Calcd. for C18H24N4=0.33 H20: C 71.48, H 8.14, N 18.14. Found:
C
71.48, H 8.24, N 18.53.
rr..u
[00506] an,y n- ____ ~~eu =iriazol ~_C ,Sõ uCi~_iv~,a~ ~iv av~õ=cZ(~. voiõ~,
TTn'nn a a procedure nna~.,.,..iira Simila:'t that .
.n. 11cP.({ tn
prepare 29, compound 28 was converted to 30 (68% overall yield) as an off-
white
foam. 'H NMR (500 MHz, CDC13) S 8.40-8.80 (m, 1H), 7.82 (d, 1H, J= 3.0 Hz),
7.58
(s, 1H), 3.23 (d, 1H, J= 18.0 Hz), 2.88 (d, 1H, J= 18.5 Hz), 2.84 (d, 1H, J=
12.0 Hz),
2.62 (m, 1H), 2.49 (m, IH), 2.10 (s, 3H), 2.04 (m, 2H), 1.48 (s, 311), 1.39
(d, 1H, J=
12.5 Hz), 0.93 (m, 1H), 0.89 (d, 3H, J= 7.0 Hz), 0.52 (d, 2H, J= 7.5 Hz), 0.14
(m,
2H). MS (ESI) m/z 296 [(M+H)+]. Anal. Calcd. for C18H24N4=0.70 H20: C 70.00, H
8.30, N 18.13. Found: C 70.44, H 8.10, N 17.78.
[0057] Cis-(=L)-3-(cyclopropylmethyl)-N-(phenylmethyl)-1,2,3,4,5,6-hexahydro-
6,11-dimethyl-9-nitro-2,6-methano-3-benzazocine-8-amine 32. Benzylamine (0.18
mL, 1.65 mmol) was added to a flask containing the solution of 17 (0.248 gm,
0.55
mmol) in 5 mL CH3CN, under argon at room temperature. A reflux condenser was
44
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
attached and the reaction mixture was stirred at reflux for 18 hours. TLC
showed the
completion of reaction. The cooled reaction mixture was diluted with methylene
chloride and washed with 0.1M NaOH solution and brine. The combined organic
layer
was dried over sodium sulfate and concentrated to give orange colored oil,
which was
purified by flash chromatography (CH2ClZ: CH30H: NH40H; I0:1:0.1) to give 32
as
an orange foam (0.171 gm, 77%); mp 215- 216 C. 'H NMR (500MHz, CDC13) 8 8.23
(t, 1H), 7.88 (s, 1H), 7.36 (m, 4H), 7.29 (m, IH), 6.65 (s, 1H), 3.12-3.10 (m,
1H), 2.90
(d, 1 H, J= 18.5 Hz), 2.70-2.67 (m, I H), 2.61 (d, 0.5H, J= 1 Hz), 2.60 (d,
0.5H, J= 1
Hz), 2.48-2.44 (m, 1H), 2.31-2.28 (m, 1H), 2.01-1.96 (m, 1H), 1.90-1.83 (m,
2H),
1.22-1.20 (m, 3H), 1.19-1.I8 (m, IH), 0.83 (m, IH), 0.82 (s, 1.5H), 0.81 (s,
1.5H),
0.51-0.49 (m, 2H), 0.11-0.09 (m, 2H) ppm. MS (ESI) m/z 406 [(MfH)+].
[0058] Cis-(f)-3-(cyclopropylmethyl)-1,2,3,4,5,6-hexahydro-6,11-dimethyl-2,6-
methano-3-benzazocine-8,9-diamine 33. 15 mL Methanol was added to the reaction
flask containing compound 32 (0.17 gm, 0.42 mmol), ammonium formate (0.264 gm,
4.20 mmol) and 10% Pd/C (0.046 gm, 0.042 mmol). The reaction mixture was then
stirred at reflux for 20 hours. After the completion of reaction, the mixture
was filtered
through celite and the residue was washed with excess methanol and the
combined
methanol layer was concentrated. This concentrated matter was then partitioned
between methylene chloride ar~d satui ated sodiunn bicarbonate. The Co:ubined -
r~,^anic
layer was dried over sodium sulfate, filtered, and concentrated to give crude
solid
produce which was purified by silica gel flash chromatography (CH2C12: CH3OH:
NH4OH; 10:1:0.1) to give the somewhat unstable diamino compound 33 (0.080 gm,
67%) as an off-white foam. 'H NMR (500MHz, CDC13) S 6.57 (s, 1H), 6.41 (s,
IH),
3.28 (s, 4H), 3.09-3.07 (m, 1H), 2.78-2.74 (d, 1H, J= 18 Hz), 2.74-268 (m,
0.5H),
2.67-2.66 (m, 0.514), 2.56-2.55 (d, 0.511, J= 6 Hz), 2.52-2.51 (d, 0.514, J=
5.5 Hz),
2.48-2.44 (m, 1H), 2.31-2.28 (m, 1H), 1.86-1.78 (m, 2H), 1.29 (m, 4H), 0.87-
0.83 (m,
4H), 0.50-0.48 (m, 2H), 0.10-0.08 (m, 2H) ppm. HRMS m/z Calcd, 286.2283;
Found,
286.2264 for C18H27N3.
[0059] 8,9-Fused imidazole derivative 34. A solution of compound 33 (0.240 gm,
0.84 mmol) in 10 mL formic acid was stirred at refluxed for 20 hours under
argon.
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
After the completion of the reaction, it was cooled to 0 C and carefully
basified with
conc. NH4OH. The organic matter was then extracted into methylene chloride.
The
extracts were dried over sodium sulfate, filtered, and concentrated to give
crude solid
produce which was purified by silica gel flash chromatography (CH2CI2: CH3OH:
NH4OH; 40:9:1) to give compound 34 (0.170 gm, 69%) as an off-white foam. This
product was converted to its HCI salt by treatment with 1M HCl in Et20. The
crude
salt was crystallized with EtOAC/MeOH to give white crystalline solid having
mp
218 C. 'H NMR (500MHz, CD3OD) 8 9.32 (s, 1H), 7.85 (s, 1H), 4.89 (s, 1H), 4.04
(s,
1H), 3.52-3.49 (m, 1H), 3.42 (m, 1H), 3.35 (m, 1H), 3.12-3.08 (m, 1H), 2.60-
2.58 (m,
1H), 2.37 (m, 111), 2.21 (m, 1H), 1.73 (m, 1H), 1.62 (s, 3H), 1.22 (m, 1H),
1.00 (d, 3H,
J= 7 Hz), 0.94 (m, 1H), 0.80-0.77 (m, 2H), 0.50 (m, 2H) ppm. MS (ESI) m/z 296
[(M+H)+]. Anal. Calcd. for C19H25N3-2HCI-0.5H2O: C 60.48, H 7.48, N 11.14.
Found
C 60.14, H 7.63, N 10.84.
[0060] 8,9-Fused imidazole derivative 35. A solution of compound 33 (0.090 gm,
0.32 mmol) in 1.5 mL acetic acid was heated under microwave radiation at 130
C for
30 min. After the completion of the reaction, it was cooled to 0 C and
carefully
basified with conc. NH4OH. The organic matter was then extracted into
methylene
chloride. The extracts were dried over sodium sulfate, filtered, and
concentrated to
give crude solid produce which was purified by silica gel flash chromatography
(CHZC12: CH3OH: NH4OH; 10:1:0.1) to give the compound 35 (0.061 gm, 62%) as an
off-white foam. 'H NMR (500MHz, CDC13) 6 10.40 (s, br, 1H), 6.56 (s, 1H), 6.41
(s,
1H), 3.28 (s, 3H), 3.11-3.09 (m, 1H), 2.78-2.74 (d, 1H, J= 18 Hz), 2.71-2.68
(m, 1H),
2.58-2.53 (m, 1H), 2.49-2.46 (m, 1H), 2.33-2.29 (m, 1H), 2.08-2.03 (m, 1H),
1.88-1.79
(m, 2H), 1.29 (s, 3H), 1.27-1.23 (m, 1H), 0.87 (m, 114), 0.85 (d, 314, J= 7
Hz), 0.50-
0.48 (m, 214), 0.11-0.09 (m, 2H) ppm. MS (ESI) m/z 310 [(M+H)+]. Anal. Calcd.
for
C20H27N3-0.63H20: C 74.67, H 9.16, N 13.10. Found C 74.90, H 8.90, N 13.10.
[0061] 8,9-Fused imidazole derivative 36. 2 mL of phosgene solution in toluene
was
added to a THF solution of compound 33 (0.075 gm, 0.26 mmol) and the reaction
was
stirred for 16 hours at room temperature. The reaction mixture was diluted
with water
46
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
and basified with conc. NH4OH. The organic matter was then extracted into
methylene
chloride. The extracts were dried over sodium sulfate, filtered, and
concentrated to
give crude solid produce which was purified by silica gel flash chromatography
(CH2C12: CH3OH: NH4OH; 10:1:0.1) to give the compound 36 (0.064 gm, 78%) as an
off-white solid, which was crystallized with EtOAc/MeOH to give a solid having
mp
of 172 C. 'H NMR (500MHz, CDC13) 8 8.63 (s, 1H), 8.52 (s, IH), 6.93 (s, 1H),
6.75
(s, 1H), 3.12 (s, IH), 2.95 (d, 1H, J= 18 Hz), 2.74-2.69 (m, 2H), 2.50-2.46
(m, 1H),
2.33-2.29 (m, 1H), 1.98-187 (m, 3H), 1.37 (s, 3H), 1.29 (m, 1H), 0.86 (d, 3H,
J= 7
Hz), 0.51 (m, 2H), 0.11 (m, 2H) ppm. MS (ESI) m/z 312 [(M+H)+]. Anal. Calcd.
for
C19H25N3O-0.25H2O: C 73.28, H 8.09, N 13.49. Found C 72.23, H 8.14, N 13.30.
[0062] 8,9-Fused imidazole derivative 37. Using a procedure similar to that
used to
prepare 35, compound 33 was treated with CH3CH2CO2H to provide 37 (88% yield)
as
an off-white foam. 1H NMR (500 MHz, CDC13) 8 9.04 (s, br, 1H), 7.62 (s, IH),
7.06
(s, IH), 3.17 (s, br, 1 H), 3.12-3.04 (m, IH), 2.94-2.80 (m, 3H), 2.69 (d,
114, J= 12 Hz),
2.53-2.48 (m, 1H), 2.37-2.30 (m, IH), 2.05-1.78 (m, 3H), 1.73 (s, br, 2H),
1.43 (t, 3H,
J= 9 Hz), 1.37-1.23 (m, 3H), 0.87 (d, 3H, J= 7 Hz), 0.54-0.48 (m, 2H), 0.19-
0.08 (m,
2H) ppm.
[0063 j 8,9-Fused imidazole derivative 38. Usi~ig a procedure similar to that
used to
prepare 35, compound 33 was treated with (CH3)2CHCO2H to provide 38 (71%
yield)
as an off-white foam. 'H NMR (500 MHz, CDC13) 6 9.79 (s, br, 1H), 7.64 (s,
1H),
7.08 (s, 1H), 3.18 (s, br, 2H), 3.07 (d, 1H, J= 18 Hz), 2.87 (dd, IH, J1,2 =
6, 19 Hz),
2.71(dd, 1H, JI,Z = 2, 12 Hz), 2.54-2.48 (m, 1H), 2.39-2.32 (m, 1H), 2.07-1.85
(m, 3H),
1.44 (d, 6H, J= 7 Hz), 1.42-1.15 (m, 5H), 0.87 (d, 3H, J= 7 Hz), 0.56-0.48 (m,
2H),
0.13-0.08 (m, 2H) ppm.
[00641 8,9-Fused imidazole derivative 39. Using a procedure similar to that
used to
prepare 35, compound 33 was treated with c-C3H5CO2H to provide 39 (86% yield)
as a
yellow foam. 'H NMR (500 MHz, CDC13) 8 8.95 (s, br, 1H), 7.57 (s, IH), 7.03
(s,
IH), 3.17 (s, br, 1H), 3.06 (dd, 1H, J1,2 = 8, 18 Hz), 2.90-2.80 (m, IH), 2.73-
2.65(m,
1H), 2.55-2.46 (m, IH), 2.40-2.30 (m, lI-I), 2.10-1.80 (m, 4H), 1.75-1.60 (m,
2H),
47
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
1.47-1.40 (m, 2H), 1.39-1.23 (m, 2H), 1.20-1.13 (m, 2H), 1.10-1.05 (m, 1 H),
0.95-0. 8
(m, 3H), 0.56-0.47 (m, 2H), 0.19-0.07 (m, 2H) ppm.
[0065] Cis-( )-3-(cyclopropylmethyl)-1,2,3,4,5,6-hexahydro-6,11-dimethyl-2,6-
methano-3-benzazocine-7,8-diamine 40. To a solution of compound 28 (0.080 gm,
0.25 mmol) in 10 mL of methanol was added 10% Pd/C (0.008 gm). The suspension
was placed in a Parr hydrogenation apparatus and shaken for 8 hours at 25 C
at a
pressure of 40 psi. The reaction mixture was then filtered over Celite. The
filtrate was
concentrated in vacuo, to give compound 40 as a somewhat unstable off-white
foam in
quantitative yield. 1H NMR (500MHz, CDC13) S 6.60 (d, 1H, J= 8 Hz), 6.46 (d,
1H, J
= 8 Hz), 5.00-4.00 (s, br, 2H), 3.46 (s, 2 H), 3.09 (d, 1 H, J= 11 Hz), 2.99
(dd, 1H, J1,2
= 6, 14 Hz), 2.80 (d, 1H, J= 14 Hz), 2.73-2.68 (m, IH), 2.64-2.59 (m, 1H),
2.27 (t,
1H, J= 11 Hz), 2.14 (d, 1H, J= 5 Hz), 2.02 (s, 2H), 1.80 (d, 1 H, J= 13 Hz),
1.62 (s,
3H), 1.01-1.00 (m, 1H), 0.96 (d, 3H, J= 7 Hz), 0.61 (d, 2H, J= 8 Hz), 0.25
(dd, 2H,
JI,2 = 4, 13 Hz) ppm.
[00661 7,8-Fused imidazole derivative 41. Using a procedure similar to that
used to
prepare 35, compound 40 was treated with HCOZH to provide 41 (80% yield) as an
off-white foam. 'H NMR (500MHz, CDC13) 8 9.18 and 9.02 (s, br, IH), 7.97 and
7.95
i., , 111~u~, -7 lJ co Q.11,a ^7.'77 (,a, 1 u, 7= o u`l, 7=na and 7 Q? (1 FT
d, :. 1= 9 Hzly 3,19-3.17 (m,
/./U U /4/ u 111
1H), 3.03-2.95 (d, 1H, J= 18 Hz), 2.88-2.81 (m, 1H), 2.73-2.69 (m, 1H), 2.52-
2.48
(dd, 1H, J= 7, 13 Hz), 2.35-2.28 (dd, IH, J= 6, 13 Hz), 2.02-1.88 (m, 3H),
1.94 and
1.69 (s, 3H), 1.60-1.59 (m, 1H), 0.96 and 0.93 (d, 3H, J= 7 Hz), 0.89-0.86 (m,
1H),
0.52-0.50 (m, 2H), 0.13-0.10 (m, 2H) ppm.; MS (ESI) m/z 296 [(M+H)+].; Anal.
Calcd. for C19H25N3=0.5H2O=0.4CH2C12: C 68.86, H 7.98, N 12.42. Found C 69.19,
H
7.53, N 11.98.
[00671 7,8-Fused imidazole derivative 42. Using a procedure similar to that
used to
prepare 35, compound 40 was treated with CH3CO2H to provide 42 (97% yield) as
an
off-white foam. 'H NMR (500 MHz, CDCl3) 8 8.80 (s, br, 1H), 7.44 (d, 1H, J= 9
Hz),
6.97 (d, 1H, J= 9 Hz), 3.20 (s, br, 1H), 3.02-2.94 (m, 1H), 2.90-2.70 (m, 3H),
2.55-
2.48(m, 1H), 2.05-1.82 (m, 4H), 1.66 (s, 3H), 1.60-1.55 (m, 1H), 0.93 (d, 3H,
J= 7
48
CA 02686851 2009-11-05
WO 2008/144394 PCT/US2008/063713
Hz), 0.92-0.83 (m, 1H), 0.56-0.47 (m, 2H), 0.18-0.08 (m, 2H) ppm. Anal. Calcd.
for
C201-127IN3 0.5 H20: C 75.43, H 8.86, N 13.19. Found C 74.99, H 8.51, N 12.98.
[0068] 7,8-Fused isoxazole derivative 43. To a solution of compound 18 (0.48
gm,
1.50 mmol) in 10 mL of HC1(37%) was added SnC12-2H2O (1.67 gm, 7.50 mmol).
The reaction mixture was stirred overnight at 25 C and then carefully
basified with 2
N NaOH solution. The organic matter was then extracted into EtOAc. The
extracts
were dried over sodium sulfate, filtered, and concentrated to give crude solid
produce
which was purified by silica gel flash chromatography (CH2CI2: CH3OH: NH4OH;
10:1:0.1) to give compound 43 (0.18 gm, 39%) as an off-white foam. 'H NMR
(500MHz, CDCl3) S 6.98 (d, IH, J= 9 Hz), 6.44 (d, 1H, J= 9 Hz), 5.04 (s, 2H),
3.14-
3.11 (m, I H), 2.78-2.57 (m, 3 H), 2.48-2.43 (m, 1H), 2.30-2.26 (m, 1H), 2.03-
1.96 (m,
IH), 1.94-1.88 (m, 1H), 1.86-1.80 (m, 1H), 1.80-1.72 (m, 1H), 1.67 (s, 3H),
0.94 (d,
3H, J= 7 Hz), 0.91-0.82 (m, 1H), 0.56-0.46 (m, 2H), 0.14-0.06 (m, 2H) ppm. MS
(ESI) m/z 312 [(M+H)+]. Anal. Calcd. for C19H25N30=0.1 H20: C 72.86,14 8.11, N
13.42. Found C 72.62, H 8.20, N 13.19.
[0069] In general, the chemistry described above works in the presence of the
variety
of functional groups found on known core structures. The exceptions would be
morphine ~ and congeners tii~~..irn au~ faee 6-CTi, protected õ,hi~h r-.an be
nrnte~c~bv a TRDPS (t-
i.ia .rJ uvua~ a L
butyldiphenylsilyl) group [see Wentland et al., "Selective Protection and
Functionalization of Morphine. ..", J. Med. Chem. 43, 3558-3565 (2000)], the
entire
contents of which are incorporated herein by reference.
49