Note: Descriptions are shown in the official language in which they were submitted.
CA 02687747 2009-11-19
WO 2008/145562
PCT/EP2008/056165
-1-
NON-NUCLEOSIDE REVERSE TRANSCRIPTASE INHIBITORS
The invention relates to the field of antiviral therapy and, in particular, to
non-
nucleoside compounds that inhibit HIV reverse transcriptase and are useful for
treating
Human Immunodeficiency Virus (HIV) mediated diseases. The invention provides
novel
1H-pyrimidine-2,4-dione, dihydro-pyrimidine-2,4-dione and imidazolidine-2,4-
dione
compounds according to formula I, for treatment or prophylaxis of HIV mediated
diseases, AIDS or ARC, employing said compounds in monotherapy or in
combination
therapy.
The invention relates to the field of antiviral therapy and, in particular, to
non-
nucleoside compounds that inhibit HIV reverse transcriptase and are useful for
treating
Human Immunodeficiency Virus (HIV) mediated diseases. The invention provides
novel
heterocyclic compounds according to formula I, for treatment or prophylaxis of
HIV
mediated diseases, AIDS or ARC, employing said compounds in monotherapy or in
combination therapy.
The human immunodeficiency virus HIV is the causative agent of acquired
immunodeficiency syndrome (AIDS), a disease characterized by the destruction
of the
immune system, particularly of the CD4+ T-cell, with attendant susceptibility
to
opportunistic infections. HIV infection is also associated with a precursor
AIDS - related
complex (ARC), a syndrome characterized by symptoms such as persistent
generalized
lymphadenopathy, fever and weight loss.
In common with other retroviruses, the HIV genome encodes protein precursors
known as gag and gag-pol which are processed by the viral protease to afford
the
protease, reverse transcriptase (RT), endonuclease/integrase and mature
structural
proteins of the virus core. Interruption of this processing prevents the
production of
normally infectious virus. Considerable efforts have been directed towards the
control of
HIV by inhibition of virally encoded enzymes.
Two enzyme have been extensively studied for HIV-1 chemotherapy: HIV protease
and HIV reverse transcriptase. (J. S. G. Montaner et at., Antiretroviral
therapy: 'the state
JZ/14.04.2008
CA 02687747 2009-11-19
WO 2008/145562 PCT/EP2008/056165
- 2 -
of the art', Biomed & Pharmacother. 1999 53:63- 72; R. W. Shafer and D. A.
Vuitton,
Highly active retroviral therapy (HAART) for the treatment of infection with
human
immunodeficiency virus type, Biomed. & Pharmacother. 1999 53 :73-86; E. De
Clercq,
New Developments in Anti-HIV Chemotherap. Curr. Med. Chem. 2001 8:1543-1572).
Two general classes of RTI inhibitors have been identified: nucleoside reverse
transcriptase inhibitors (NRTI) and non-nucleoside reverse transcriptase
inhibitors.
Currently the CCR5 co-receptor has emerged as a potential target for anti-HIV
chemotherapy (D. Chantry, Expert Opin. Emerg. Drugs 2004 9(1):1-7; C. G.
Barber,
Curr. Opin. Invest. Drugs 2004 5(8):851-861; D. Schols, Curr. Topics Med.
Chem. 2004
4(9):883-893; N. A. Meanwell and J. F. Kadow, Curr. Opin. Drug Discov. Dev.
2003
6(4):451-461). Drugs targeted at new enzymatic targets have entered the market
including integrase inhibitors typified by Raltegravir (Merck) has been
approved by the
FDA and Elvitegravir (Gilead Sciences and Japan Tobacco) is in phase II
trials. The
CCR5 antagonist maraviroc (SELZENTRYTm, Pfizer) has also been approved by the
FDA for anti-HIV-1 therapy.
NRTIs typically are 2',3'-dideoxynucleoside (ddN) analogs which must be
phosphorylated prior to interacting with viral RT. The corresponding
triphosphates
function as competitive inhibitors or alternative substrates for viral RT.
After
incorporation into nucleic acids the nucleoside analogs terminate the chain
elongation
process. HIV reverse transcriptase has DNA editing capabilities which enable
resistant
strains to overcome the blockade by cleaving the nucleoside analog and
continuing the
elongation. Currently clinically used NRT1s include zidovudine (AZT),
didanosine (ddI),
zalcitabine (ddC), stavudine (d4T), lamivudine (3TC) and tenofovir (PMPA).
NNRTIs were first discovered in 1989. NNRTI are allosteric inhibitors which
bind
reversibly at a nonsubstrate-binding site on the HIV reverse transcriptase
thereby altering
the shape of the active site or blocking polymerase activity (R. W. Buckheit,
Jr., Non-
nucleoside reverse transcriptase inhibitors: perspectives for novel
therapeutic
compounds and strategies for treatment of HIV infection, Expert Opin.
Investig. Drugs
2001 10(8)1423-1442; E. De Clercq, The role of non-nucleoside reverse
transcriptase
inhibitors (NNRTIs) in the therapy of HIV infection, Antiviral Res. 1998
38:153-179; E.
De Clercq, New Developments in Anti-HIV Chemotherapy, Current Med. Chem. 2001
8(13):1543-1572; G. Moyle, The Emerging Roles of Non-Nucleoside Reverse
Transcriptase Inhibitors in Antiviral Therapy, Drugs 2001 61 (1):19-26).
Although over
CA 02687747 2015-02-18
WO 2008/145562 PCT/EP2008/056165
- 3 -
thirty structural classes of NNRTIs have been identified in the laboratory,
only three
compounds have been approved for HIV therapy: efavirenz, nevirapine and
delavirdine.
Initially viewed as a promising class of compounds, in vitro and in vivo
studies
quickly revealed the NNRTIs presented a low barrier to the emergence of drug
resistant
HIV strains and class-specific toxicity. Drug resistance frequently develops
with only a
single point mutation in the RT. While combination therapy with NRTIs, PIs and
NNRTIs has, in many cases, dramatically lowered viral loads and slowed disease
progression, significant therapeutic problems remain. (R. M. Gulick, Eur. Soc.
Clin.
MicrobioL and Inf. Dis. 2003 9(3):186-193) The cocktails are not effective in
all
patients, potentially severe adverse reactions often occur and the rapidly
reproducing
HIV virus has proven adroit at creating mutant drug-resistant variants of wild
type
protease and reverse transcriptase. There remains a need for safer drugs with
activity
against wild type and commonly occurring resistant strains of HIV.
Pyridazinone non-nucleoside reverse transcriptase inhibitors have been
described
by J. P. Dunn etal. in U. S. Patent No. 7,189,718 issued March 13, 2007 and by
J. P.
Dunn et al. in U. S. Publication No. 2005021554 filed March 22, 2005. 5-
Aralky1-2,4-
dihydro-[1,2,4]triazol-3-one, 5-aralky1-3H-[1,3,4]oxadiazol-2-one and 5-
aralky1-3H-
[1,3,4]thiadiazol-2-one non-nucleoside reverse transcriptase inhibitors have
been
disclosed by J. P. Dunn etal. in U. S. Patent No. 7,208,059 issued April 24,
2007, U.S.
Patent Publication 20060225874 published October 5, 2006 and U. S. Publication
No.
20060025462 filed June 27, 2005. Related compounds are disclosed by Y. D.
Saito etal.
in U. S. Publication No. 20070078128 published April 5, 2007. Phenylacetamide
non-
nucleoside reverse transcriptase inhibitors have been disclosed by J. P. Dunn
et al. in
U.S. Patent No. 7,166,738 issued January 23, 2007 and methods for treating
retroviral
infection with phenylacetamide compounds have been disclosed by J. P. Dunn et
al. in U.
S. Publication No. 20050239880 published Oct. 27, 2005; T. Mirzadegan and T.
Silva in
U. S. Publication No. 20070088053 published April 19, 2007; and by Z. K.
Sweeney and
T. Silva in U. S. Publication No. 20070088015 published April 19, 2007.
In W02006/067587 published June 26, 2006, L. H. Jones et al. disclose
phenoxyacetamide derivatives and compositions containing them which bind HI-1
reverse transcriptase and are modulators, especially inhibitors, thereof. K.
R. Romines et
al (.1. Med. Chem. 2006 49(2):727-739) and P. Bonneau et al. (U.S. Publication
No.
CA 02687747 2009-11-19
WO 2008/145562 PCT/EP2008/056165
- 4 -
20060069261 published March 30, 2006) describe phenoxyacetamides that inhibit
HIV-1
reverse transcriptase. In U. S. Patent Publication 2007/0021442 published
January 25,
2007, S. A. Saggar et at. disclose diphenyl ether HIV-1 reverse transcriptase
inhibitors.
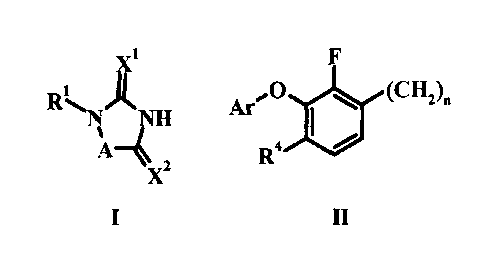
The present invention relates to a compound according to formula I wherein:
x1 F
RiNANH ArA, 0 (CH2).
Al -I
X2 R4
2
I II
A is (a) CHR3=CHR2 ,
CH2R3CH2R2 , or,
CHR2;
either (i) R1 is II and R2 is hydrogen, C1_3 alkyl, C1_3 haloalkyl,
C2_6 alkenyl, or
CH2OW; and, in addition, if A is either (a) or (b) R2 can also be halogen,
NRaltb, CN, or
ORc;
Or, (ii) R1 is C1_6 alkyl and R2 is II;
X1 is 0 or S;
X2 is 0, or, if A is (a), X2 is 0 or NR';
R3 is hydrogen or Ci_3 alkyl;
R4 is halogen, C1_6 alkyl, C3_6 cycloalkyl Ci_6 haloalkyl or Ci_6 alkoxY;
le and Rb are independently hydrogen, Ci_3 alkyl or Ci_6 acyl;
Rc and Rd are independently hydrogen or C1_3 alkyl;
Ar is phenyl substituted with 1 to 3 groups independently selected from
halogen,
cyano, C1_6 haloalkyl, C1_6 alkyl, C1_6 alkoxy or C3_6 cycloalkyl;
n is an integer from 1 to 3; or,
pharmaceutically acceptable salts thereof.
CA 02687747 2009-11-19
WO 2008/145562 PCT/EP2008/056165
- 5 -
Compounds of formula I inhibit HIV-1 reverse transcriptase and afford a method
for prevention and treatment of HIV-1 infections and the treatment of AIDS
and/or ARC.
HIV-1 undergoes facile mutations of its genetic code resulting in strains with
reduced
susceptibility to therapy with current therapeutic options. The present
invention also
relates to compositions containing compounds of formula I useful for the
prevention and
treatment of HIV-1 infections and the treatment of AIDS and/or ARC. The
present
invention further relates to compounds of formula I which are useful in
monotherapy or
combination therapy with other anti-viral agents.
The phrase "a" or "an" entity as used herein refers to one or more of that
entity; for
example, a compound refers to one or more compounds or at least one compound.
As
such, the terms "a" (or "an"), "one or more", and "at least one" can be used
interchangeably herein.
The phrase "as defined herein above" refers to the broadest definition for
each
group as provided in the Summary of the Invention or the broadest claim. In
all other
embodiments provided below, substituents which can be present in each
embodiment and
which are not explicitly defined retain the broadest definition provided in
the Summary
of the Invention.
Technical and scientific terms used herein have the meaning commonly
understood
by one of skill in the art to which the present invention pertains, unless
otherwise
defined. Reference is made herein to various methodologies and materials known
to
those of skill in the art. Standard reference works setting forth the general
principles of
pharmacology include Goodman and Gilman's The Pharmacological Basis of
Therapeutics, 10th Ed., McGraw Hill Companies Inc., New York (2001). Any
suitable
materials and/or methods known to those of skill can be utilized in carrying
out the
present invention. However, preferred materials and methods are described.
Materials,
reagents and the like to which reference are made in the following description
and
examples are obtainable from commercial sources, unless otherwise noted.
As used in this specification, whether in a transitional phrase or in the body
of the
claim, the terms "comprise(s)" and "comprising" are to be interpreted as
having an open-
ended meaning. That is, the terms are to be interpreted synonymously with the
phrases
"having at least" or "including at least". When used in the context of a
process, the term
"comprising" means that the process includes at least the recited steps, but
may include
additional steps. When used in the context of a compound or composition, the
term
CA 02687747 2009-11-19
WO 2008/145562 PCT/EP2008/056165
- 6 -
"comprising" means that the compound or composition includes at least the
recited
features or components, but may also include additional features or
components.
The term "about" is used herein to mean approximately, in the region of,
roughly,
or around. When the term "about" is used in conjunction with a numerical
range, it
modifies that range by extending the boundaries above and below the numerical
values
set forth. In general, the term "about" is used herein to modify a numerical
value above
and below the stated value by a variance of 20%.
The term "optional" or "optionally" as used herein means that a subsequently
described event or circumstance may, but need not, occur, and that the
description
includes instances where the event or circumstance occurs and instances in
which it does
not. For example, "optionally substituted" means that the optionally
substituted moiety
may incorporate a hydrogen or a substituent.
The phrase "optional bond" means that the bond may or may not be present, and
that the description includes single, double, or triple bonds. If a
substituent is designated
to be a "bond" or "absent", the atoms linked to the substituents are then
directly
connected.
When any variable (e.g., R1, R4a, Ar, X1 or Het) occurs more than one time in
any
moiety or formula depicting and describing compounds employed or claimed in
the
present invention, its definition on each occurrence is independent of its
definition at
every other occurrence. Also, combinations of substituents and/or variables
are
permissible only if such compounds result in stable compounds.
A "stable" compound is a compound which can be prepared and isolated and whose
structure and properties remain or can be made to remain essentially unchanged
for a
period of time sufficient to allow the use of the compound for the purposes
described
herein (e.g., therapeutic or prophylactic administration to a subject).
Unless expressly stated to the contrary, all ranges cited herein are
inclusive. For
example, a heterocyclic ring described as containing "1 to 4 heteroatoms"
means the ring
can contain 1, 2, 3 or 4 hetero atoms. It is also to be understood that any
range cited
herein includes within its scope all of the subranges within that range. Thus,
for
example, an aryl or a heteroaryl described as optionally substituted with
"from 1 to 5
substituents" is intended to include as aspects thereof, any aryl optionally
substituted
CA 02687747 2009-11-19
WO 2008/145562 PCT/EP2008/056165
- 7 -
with 1 to 4 substituents, 1 to 3 substituents, 1 to 2 substituents, 2 to 5
substituents, 2 to 4
substituents, 2 to 3 substituents, 3 to 5 substituents, 3 to 4 substituents, 4
to 5
substituents, 1 substituent, 2 substituents, 3 substituents, 4 substituents,
and 5
substituents.
The symbol "*" at the end of a bond or" ----- "drawn through a bond each refer
to the point of attachment of a functional group or other chemical moiety to
the rest of
the molecule of which it is a part. Thus, for example:
MeC(=0)0R4 wherein R4 = or ¨KI MeC(=0)0¨<1
=
It is contemplated that the definitions described herein may be appended to
form
chemically-relevant combinations, such as "heteroalkylaryl,"
"haloalkylheteroaryl,"
"arylalkylheterocyclyl," "alkylcarbonyl," "alkoxyalkyl," and the like. When
the term
"alkyl" is used as a suffix following another term, as in "phenylalkyl," or
"hydroxyalkyl," this is intended to refer to an alkyl group, as defined above,
being
substituted with one to two substituents selected from the other specifically-
named
group. Thus, for example, "phenylalkyl" refers to an alkyl group having one to
two
phenyl substituents, and thus includes benzyl, phenylethyl, and biphenyl. An
"alkylaminoalkyl" is an alkyl group having one to two alkylamino substituents.
"Hydroxyalkyl" includes 2-hydroxyethyl, 2-hydroxypropyl, 1-(hydroxymethyl)-2-
methylpropyl, 2-hydroxybutyl, 2,3-dihydroxybutyl, 2-(hydroxymethyl), 3-
hydroxypropyl, and so forth. Accordingly, as used herein, the term
"hydroxyalkyl" is
used to define a subset of heteroalkyl groups defined below. The term -
(ar)alkyl refers to
either an unsubstituted alkyl or an aralkyl group. The term (hetero)aryl or
(het)aryl refers
to either an aryl or a heteroaryl group.
The term "alkyl" as used herein denotes an unbranched or branched chain,
saturated, monovalent hydrocarbon residue containing 1 to 10 carbon atoms. The
term
"lower alkyl" denotes a straight or branched chain hydrocarbon residue
containing 1 to 6
carbon atoms. "C1-10 alkyl" as used herein refers to an alkyl composed of 1 to
10
carbons. Examples of alkyl groups include, but are not limited to, lower alkyl
groups
include methyl, ethyl, propyl, i-propyl, n-butyl, i-butyl, t-butyl or pentyl,
isopentyl,
neopentyl, hexyl, heptyl, and octyl.
CA 02687747 2009-11-19
WO 2008/145562 PCT/EP2008/056165
- 8 -
The term "alkylene" as used herein denotes a divalent saturated linear
hydrocarbon
radical of 1 to 10 carbon atoms (e.g., (CH2),i)or a branched saturated
divalent
hydrocarbon radical of 2 to 10 carbon atoms (e.g., -CHMe- or -CH2CH(i-Pr)CH2-
),
unless otherwise indicated. The open valences of an alkylene group are not
attached to
the same atom. Examples of alkylene radicals include, but are not limited to,
methylene,
ethylene, propylene, 2-methyl-propylene, 1,1-dimethyl-ethylene, butylene, 2-
ethylbutylene.
The term "alkenyl" as used herein denotes an unsubstituted hydrocarbon chain
radical having from 2 to 10 carbon atoms having an double bonds. C2-10
alkenyl" as used
herein refers to an alkenyl composed of 2 to 10 carbons. Examples are vinyl, 1-
propenyl,
2-propenyl (ally1) or 2-butenyl (crotyl).
The term "cycloalkyl" as used herein denotes a saturated carbocyclic ring
containing 3 to 8 carbon atoms, i.e. cyclopropyl, cyclobutyl, cyclopentyl,
cyclohexyl,
cycloheptyl or cyclooctyl. "C3_7 cycloalkyl" as used herein refers to an
cycloalkyl
composed of 3 to 7 carbons in the carbocyclic ring.
The term "alkoxy" as used herein means an -0-alkyl group, wherein alkyl is as
defined above such as methoxy, ethoxy, n-propyloxy, i-propyloxy, n-butyloxy, i-
butyloxy, t-butyloxy, pentyloxy, hexyloxy, including their isomers. "Lower
alkoxy" as
used herein denotes an alkoxy group with a "lower alkyl" group as previously
defined.
"C1-10 alkoxy" as used herein refers to an-O-alkyl wherein alkyl is C1_10.
The term "halo alkyl" as used herein denotes a unbranched or branched chain
alkyl
group as defined above wherein 1, 2, 3 or more hydrogen atoms are substituted
by a
halogen. Examples are 1-fluoromethyl, 1-chloromethyl, 1-bromomethyl, 1-
iodomethyl,
difluoromethyl, trifluoromethyl, trichloromethyl, tribromomethyl,
triiodomethyl, 1-
fluoroethyl, 1-chloroethyl, 1-bromoethyl, 1-iodoethyl, 2-fluoroethyl, 2-
chloroethyl, 2-
bromoethyl, 2-iodoethyl, 2,2-dichloroethyl, 3-bromopropyl or 2,2,2-
trifluoroethyl.
The term "halogen" or "halo" as used herein means fluorine, chlorine, bromine,
or
iodine.
The term "acyl" as used herein denotes a group of formula -C(=0)R wherein R is
hydrogen or lower alkyl as defined herein. The term or "alkylcarbonyl" as used
herein
CA 02687747 2009-11-19
WO 2008/145562 PCT/EP2008/056165
- 9 -
denotes a group of formula C(=0)R wherein R is alkyl as defined herein. The
term C1-6
acyl refers to a group -C(=0)R contain 6 carbon atoms.
The terms uracil or 1H-pyrimidine-2,4-dione, dihydro-pyrimidine-2,4-dione and
imidazolidine-2,4-dione or hydantoin refer to a moiety of formula (i), (ii)
and (iii)
respectively.
HANH HN1NHH
0 L/L
0
0
(i) (ii)
Compounds of formula I exhibit tautomerism. Tautomeric compounds can exist as
two or more interconvertable species. Prototropic tautomers result from the
migration of
a covalently bonded hydrogen atom between two atoms. Tautomers generally exist
in
equilibrium and attempts to isolate an individual tautomers usually produce a
mixture
whose chemical and physical properties are consistent with a mixture of
compounds.
The position of the equilibrium is dependent on chemical features within the
molecule.
For example, in many aliphatic aldehydes and ketones, such as acetaldehyde,
the keto
form predominates while; in phenols, the enol form predominates. Common
prototropic
tautomers include keto/enol (-C(=0)-CH- -C(-0H)=CH-), amide/imidic acid (-
C(=0)-
NH- -C(-0H)=N-) and amidine (-C(=NR)-NH- -C(-NHR)=N-) tautomers. The
latter two are particularly common in heteroaryl and heterocyclic rings and
the present
invention encompasses all tautomeric forms of the compounds. Thus a cytosine
derivative can be equivalently depicted as either an enamine (i) or an imine
(ii) tautomer.
NH2
eA
N 0
(i) (ii)
In one embodiment of the present invention there is provided a compound
according to formula I wherein A, X15 x25 R15 R25 R35 R45 Ra, Rib, RC, R",
Ar and n are as
defined herein above. In all embodiments of the present invention when "A" is
ethenylene (a) or ethylene (b), the carbon atom bearing R3 is linked to the
nitrogen and
the carbon atom bearing R2 is linked to a carbonyl or an equivalent thereof.
CA 02687747 2009-11-19
WO 2008/145562 PCT/EP2008/056165
- 10 -
In a second embodiment of the present invention there is provided a compound
according to formula I wherein A is (a); le is (II), Xl and X2 are 0; and n is
1.
In a third embodiment of the present invention there is provided a compound
according to formula I wherein A is (a); le is (II); Xl and X2 are 0; n is 1;
R2 is
hydrogen, halogen, C1_6 alkyl, Ci_6 haloalkyl; R3 is hydrogen; R4 is halogen
or Ci_6 alkyl;
and Ar is phenyl optionally substituted with 1 to 3 groups independently
selected from
cyano, halogen or C1_6 haloalkyl.
In a fourth embodiment of the present invention there is provided a compound
according to formula I wherein A is (a); le is (II); Xl and X2 are 0; n is 1;
R2 is
hydrogen, halogen, C1_6 alkyl, C1_6 haloalkyl; R3 is hydrogen; R4 is Br or Cl;
and Ar is 3-
chloro-5-cyano-phenyl, 3,5-dicyano-phenyl or 3-cyano-5-difluoromethyl phenyl.
In a fifth embodiment of the present invention there is provided a compound
according to formula I wherein A is (a); le is (II); Xl is S; X2 is 0; n is 1;
R2 is
hydrogen, halogen, C1_6 alkyl, Ci_6 haloalkyl; R3 is hydrogen; R4 is halogen
or Ci_6 alkyl;
and, Ar is phenyl optionally substituted with 1 to 3 groups independently
selected from
cyano, halogen or Ci_6 haloalkyl.
In a sixth embodiment of the present invention there is provided a compound
according to formula I wherein A is (a); le is (II), Xl is 0; X2 is NHRd ; Rd
is H; and n
is 1.
In a seventh embodiment of the present invention there is provided a compound
according to formula I wherein A is (a); le is (II); Xl id 0; X2 is NHRd; Rd
is H; n is 1;
R2 is hydrogen, halogen, C1_6 alkyl, C1_6 haloalkyl; R3 is hydrogen; R4 is
halogen or C1-6
alkyl; and Ar is phenyl optionally substituted with 1 to 3 groups
independently selected
from cyano, halogen or C1_6 haloalkyl.
In a eighth embodiment of the present invention there is provided a compound
according to formula I wherein A is (a); le is (II); Xl is 0; X2 is NHRd; Rd
is H; n is 1;
R2 is hydrogen, halogen, C1_6 alkyl, C1_6 haloalkyl; R3 is hydrogen; R4 is Br
or Cl; and
Ar is 3-chloro-5-cyano-phenyl, 3,5-dicyano-phenyl or 3-cyano-5-difluoromethyl-
phenyl.
In a ninth embodiment of the present invention there is provided a compound
according to formula I wherein A is (b); le is (II), Xl and X2 are 0; and n is
1.
CA 02687747 2009-11-19
WO 2008/145562 PCT/EP2008/056165
- 11 -
In a tenth embodiment of the present invention there is provided a compound
according to formula I wherein A is (b); le is (II); Xl and X2 are 0; n is 1;
R2 is
hydrogen, halogen, C1_6 alkyl, Ci_6 haloalkyl; R3 is hydrogen; R4 is halogen
or Ci_6 alkyl;
and Ar is phenyl optionally substituted with 1 to 3 groups independently
selected from
cyano, halogen or C1_6 haloalkyl.
In a eleventh embodiment of the present invention there is provided a compound
according to formula I wherein A is (b); le is (II); Xl and X2 are 0; n is 1;
R2 is
hydrogen, halogen, C1_6 alkyl, C1_6 haloalkyl; R3 is hydrogen; R4 is Br or Cl;
and Ar is 3-
chloro-5-cyano-phenyl, 3,5-dicyano-phenyl or 3-cyano-5-difluoromethyl-phenyl.
In a twelfth embodiment of the present invention there is provided a compound
according to formula I wherein A is (c); le is (II), Xl and X2 are 0; and n is
1.
In a thirteenth embodiment of the present invention there is provided a
compound
according to formula I wherein A is (c); le is (II); Xl and X2 are 0; n is 1;
R4 is halogen
or Ci_6 alkyl; and Ar is phenyl optionally substituted with 1 to 3 groups
independently
selected from cyano, halogen or C1_6 haloalkyl.
In a fourteenth embodiment of the present invention there is provided a
compound
according to formula I wherein A is (c); le is (II); Xl and X2 are 0; n is 1;
R4 is Br or
Cl; and Ar is 3-chloro-5-cyano-phenyl, 3,5-dicyano-phenyl or 3-cyano-5-
difluoromethyl-
phenyl.
In a fifteenth embodiment of the present invention there is provided a
compound
according to formula I wherein A is (a); R2 is (II), Xl and X2 are 0; R3 is
hydrogen.
In a sixteenth embodiment of the present invention there is provided a
compound
according to formula I wherein A is (a); le is hydrogen or methyl; R2 is (II),
Xl and X2
are 0; R3 is hydrogen, R4 is halogen or C1_6 alkyl; and Ar is phenyl
optionally substituted
with 2 groups independently selected from cyano, halogen or C1_6 haloalkyl.
In a seventeenth embodiment of the present invention there is provided a
compound according to formula I wherein A is (a); le is hydrogen or methyl; R2
is (II),
Xl and X2 are 0; R3 is hydrogen, R4 is Br or Cl; and Ar is 3-chloro-5-cyano-
phenyl, 3,5-
dicyano-phenyl or 3-cyano-5-difluoromethyl-phenyl.
CA 02687747 2009-11-19
WO 2008/145562 PCT/EP2008/056165
- 12 -
In a eighteenth embodiment of the present invention there is provided a
compound
according to formula I wherein A is (c); and R2 is (II), Xl and X2 are 0.
In a nineteenth embodiment of the present invention there is provided a
compound
according to formula I wherein A is (c); le is hydrogen or methyl; R2 is (II),
Xl and X2
are 0; R4 is halogen or Ci_6 alkyl; and Ar is phenyl optionally substituted
with 2 groups
independently selected from cyano, halogen or Ci_6 haloalkyl 1.
In a twentieth embodiment of the present invention there is provided a
compound
according to formula I wherein A is (c); le is hydrogen or methyl; R2 is (II),
Xl and X2
are 0; R4 is Br or Cl; and Ar is 3-chloro-5-cyano-phenyl, 3,5-dicyano-phenyl
or 3-cyano-
5-difluoromethyl-phenyl.
In a twenty-first embodiment of the present invention there is provided a
compound selected from compounds I-1 to 1-32 on TABLE 1.
In a twenty-second embodiment of the present invention there is provided a
method
for treating an HIV-1 infection, or preventing an HIV-1 infection, or treating
AIDS or
ARC, comprising administering to a host in need thereof a therapeutically
effective
amount of a compound according to formula I wherein A, X15 x25 R15 R25 R35 R45
Ra, Rib,
R', Rd, Ar and n are as defined herein above.
In a twenty-third embodiment of the present invention there is provided a
method
for treating an HIV-1 infection, or preventing an HIV-1 infection, or treating
AIDS or
ARC, comprising co-administering to a host in need thereof a therapeutically
effective
amount of a compound according to formula I wherein A, X15 x25 R15 R25 R35 R45
Ra, Rb,
R', Rd, Ar and n are as defined herein above and at least one compound
selected from
the group consisting of HIV protease inhibitors, nucleoside reverse
transcriptase
inhibitors, non-nucleoside reverse transcriptase inhibitors, integrase
inhibitors, CCR5
antagonists and viral fusion inhibitors.
In a twenty-fourth embodiment of the present invention there is provided a
method
for treating an HIV-1 infection, or preventing an HIV-1 infection, or treating
AIDS or
ARC, comprising co-administering to a host in need thereof a therapeutically
effective
amount of a compound according to formula I wherein A, X15 x25 R15 R25 R35 R45
Ra, Rb,
le, Rd, Ar and n are as defined herein and at least one compound selected from
the
group zidovudine, lamivudine, didanosine, zalcitabine, stavudine, rescriptor,
sustiva
CA 02687747 2009-11-19
WO 2008/145562 PCT/EP2008/056165
- 13 -
viramune, efavirenz, nevirapine or delavirdine, saquinavir, ritonavir,
nelfinavir,
indinavir, amprenavir, lopinavir, raltegravir potassium, enfuvirtide and
maraviroc.
In a twenty-fifth embodiment of the present invention there is provided a
method
for inhibiting HIV-1 reverse transcriptase in a host infected with HIV-1
comprising
administering a therapeutically effective amount of a compound according to
comprising
administering to a host in need thereof a therapeutically effective amount of
a compound
according to formula I wherein A, Xl, X2, le, R2, R3, R4, Ra, le, le, Rd, Ar
and n are as
defined herein above.
In a twenty-sixth embodiment of the present invention there is provided a
method
for inhibiting HIV-1 reverse transcriptase in a host infected with HIV-1
expressing a
reverse transcriptase with at least one mutation compared to wild type HIV-1
comprising
administering a therapeutically effective amount of a compound according to
comprising
administering to a host in need thereof a therapeutically effective amount of
a compound
according to formula I wherein A, Xl, X2, le, R2, R3, R4, Ra, Rb, le, Rd, Ar
and n are as
defined herein above.
In a twenty-seventh embodiment of the present invention there is provided a
method for inhibiting HIV-1 reverse transcriptase in a host infected with HIV-
1
expressing a reverse transcriptase exhibiting reduced susceptibility to
efavirenz,
nevirapine or delavirdine comprising administering a therapeutically effective
amount of
a compound according to comprising administering to a host in need thereof a
therapeutically effective amount of a compound according to formula I wherein
A, Xl,
X2, le, R2, R3, R4, Ra, Rb, le, Rd, Ar and n are as defined herein above.
In a twenty-eighth embodiment of the present invention there is provided a
pharmaceutical composition comprising a therapeutically effective amount of a
compound according to comprising administering to a host in need thereof a
therapeutically effective amount of a compound according to formula I wherein
A, Xl,
X2, le, R2, R3, R4, Ra, le, le, Rd, Ar and n are as defined herein above and
at least on
carrier, excipient or diluent.
A-M. Vandamme et at. (Antiviral Chemistry & Chemotherapy, 1998 9:187-203)
disclose current HAART clinical treatments of HIV-1 infections in man
including at least
triple drug combinations. Highly active anti-retroviral therapy (HAART) has
traditionally consisted of combination therapy with nucleoside reverse
transcriptase
CA 02687747 2009-11-19
WO 2008/145562 PCT/EP2008/056165
- 14 -
inhibitors (NRTI), non-nucleoside reverse transcriptase inhibitors (NNRTI) and
protease
inhibitors (PI). These compounds inhibit biochemical processes required for
viral
replication. While HAART has dramatically altered the prognosis for HIV
infected
persons, there remain many drawbacks to the current therapy including highly
complex
dosing regimes and side effects which can be very severe (A. Carr and D. A.
Cooper,
Lancet 2000 356(9239):1423-1430). Moreover, these multidrug therapies do not
eliminate HIV-1 and long-term treatment usually results in multidrug
resistance, thus
limiting their utility in long term therapy. Development of new therapeutics
which can
be used in combination with NRTIs, NNRTIs, PIs and viral fusion inhibitors to
provide
better HIV-1 treatment remains a priority.
Typical suitable NRTIs include zidovudine (AZT; RETROVIR ); didanosine (ddl;
VIDEX ); zalcitabine (ddC; HIVID ); stavudine (d4T; ZERIT ); lamivudine (3TC;
EPIVIR ); abacavir (ZIAGEN ); adefovir dipivoxil [bis(P0M)-PMEA; PRE VON ];
lobucavir (BMS-180194), a nucleoside reverse transcriptase inhibitor disclosed
in EP-
0358154 and EP-0736533; BCH-10652, a reverse transcriptase inhibitor (in the
form of a
racemic mixture of BCH-10618 and BCH-10619) under development by Biochem
Pharma; emitricitabine [(-)-FTC] in development by Triangle Pharmaceuticals;
13-L-FD4
(also called 13-L-D4C and named 13 -L-2', 3'-dicleoxy-5-fluoro-cytidene)
licensed Vion
Pharmaceuticals; DAPD, the purine nucleoside, (-)-13-D-2,6-diamino-purine
dioxolane
disclosed in EP-0656778 and licensed to Triangle Pharmaceuticals; and
lodenosine
(FddA), 9-(2,3-dideoxy-2-fluoro-13-D-threo-pentofuranosyl)adenine, an acid
stable
purine-based reverse transcriptase inhibitor under development by U.S.
Bioscience Inc.
Four NNRTIs have been approved in the USA: nevirapine (BI-RG-587;
VIRAMUNE ) available from Boehringer Ingelheim (BI); delaviradine (BHAP, U-
90152; RESCRIPTOR ) available from Pfizer; efavirenz (DMP-266, SUSTIVA ) a
benzoxazin-2-one from BMS and etravirine (TMC-125, INTELENCE ) from Tibotec.
Other NNRTIs currently under investigation include UK-453,061 (L. H. Jones et
at.,
W02005085860), AR806 (J.-L. Girardet et at., W02006026356), IDX899 (R. Storer
et
at. (U52006074054), TMC-278 ( J. E. G. Guillemont et at., W003/16306),PNU-
142721,
a furopyridine-thio-pyrimide under development by Pfizer; capravirine (S-1153
or AG-
1549; 5-(3,5-dichloropheny1)-thio-4-isopropy1-1-(4-pyridyl)methyl-1H-imidazo1-
2-
ylmethyl carbonate) by Shionogi and Pfizer; emivirine [MKC-442; (1-(ethoxy-
methyl)-5-
(1-methylethyl)-6-(phenylmethyl)-(2,4(1H,3H)-pyrimidinedione)] by Mitsubishi
CA 02687747 2009-11-19
WO 2008/145562 PCT/EP2008/056165
- 15 -
Chemical Co. and Triangle Pharmaceuticals; (+)-calanolide A (NSC-675451) and
B,
coumarin derivatives disclosed in NIH U.S. Pat. No. 5,489,697, licensed to
Sarawak/Advanced Life Sciences; DAPY (TMC120; 4- {4-[44(E)-2-cyano-viny1)-2,6-
dimethyl-phenylamino]-pyrimidin-2-ylamino}-benzonitrile) by Tibotec-Virco and
Johnson & Johnson; BILR-355 BS (12-ethy1-8-[2-(1-hydroxy-quinolin-4-yloxy)-
ethy1]-
5-methyl-11,12-dihydro-5H-1,5,10,12-tetraaza-dibenzo[a,e]cycloocten-6-one by
Boehringer-Ingleheim and PHI-236 (7-bromo-3-[2-(2,5-dimethoxy-phenyl) -ethy1]-
3,4-
dihydro-1H-pyrido[1,2-a][1,3,5]triazine-2-thione).
Typical suitable PIs include saquinavir (Ro 31-8959; INVIRASEO;
FORTOVASE ); ritonavir (ABT-538; NORVIR ); indinavir (MK-639; CRIXIVAN );
nelthavir (AG-1343; VIRACEPT ); amprenavir (141W94; AGENERASE ); TMC114
(darunavir, PREZISTA ); lasinavir (BMS-234475); DMP-450, a cyclic urea under
development by Triangle Pharmaceuticals; BMS-2322623, an azapeptide under
development by Bristol-Myers Squibb as a 2nd-generation HIV-1 PI; ABT-378
under
development by Abbott; and AG-1549 an imidazole carbamate under development by
Agouron Pharmaceuticals, Inc.
Pentafuside (FUZEON ) a 36-amino acid synthetic peptide that inhibits fusion
of
HIV-1 to target membranes. Pentafuside (3-100 mg/day) is given as a continuous
sc
infusion or injection together with efavirenz and 2 PI's to HIV-1 positive
patients
refractory to a triple combination therapy; use of 100 mg/day is preferred.
FUZEON
binds to GP41 on the viral coating and prevents the creation of an entry pore
for the
capsid of the virus keeping it out of the cell.
HIV-1 infects cells of the monocyte-macrophage lineage and helper T-cell
lymphocytes by exploiting a high affinity interaction of the viral enveloped
glycoprotein
(Env) with the CD-4 antigen. The CD-4 antigen was found to be a necessary, but
not
sufficient requirement for cell entry and at least one other surface protein
was required to
infect the cells (E. A. Berger et at., Ann. Rev. Immunol. 1999 17:657-700).
Two
chemokine receptors, either the CCR5 or the CXCR4 receptor were subsequently
found
to be co-receptors along with CD4 which are required for infection of cells by
the human
immunodeficiency virus (HIV). Antagonists of CCR5 binding have been sought to
prevent viral fusion. Maraviroc (Pfizer) is a CCR5 antagonists has recently
been
approved by the FDA. Vicriviroc (Schering) by Pfizer is in late development
stage.
Numerous other companies have research programs in various discovery and
CA 02687747 2009-11-19
WO 2008/145562 PCT/EP2008/056165
- 16 -
development stages (see, e.g. A. Palani and J. R. Tagat, J. Med. Chem. 2006
49(10):2851-2857, P. Biswas et at. Expert. Opin. Investig. Drugs 2006
15(5):451-464;
W. Kazmierski et at. Biorg Med. Chem. 2003 11:2663-76). CCR5 antagonists which
reaching the marketplace will likely be useful in combination with NNRTIs,
NRTIs and
PIs.
Other antiviral agents include hydroxyurea, ribavirin, IL-2, IL-12,
pentafuside.
Hydroxyurea (Droxia), a ribonucleoside triphosphate reductase inhibitor shown
to have a
synergistic effect on the activity of didanosine and has been studied with
stavudine. IL-2
(aldesleukin; PROLEUKINO) is disclosed in Ajinomoto EP-0142268, Takeda EP-
0176299, and Chiron U.S. Pat. Nos. RE 33,653, 4,530,787, 4,569,790, 4,604,377,
4,748,234, 4,752,585, and 4,949,314. Ribavirin, 1-13-D-ribofuranosy1-1H-1,2,4-
triazole-
3-carboxamide.
Commonly used abbreviations include: acetyl (Ac), atmospheres (Atm), tert-
butoxycarbonyl (Boc), di-tert-butyl pyrocarbonate or boc anhydride (B0C20),
benzyl
(Bn), butyl (Bu), Chemical Abstracts Registration Number (CASRN),
benzyloxycarbonyl (CBZ or Z), 1,5-diazabicyclo[4.3.0]non-5-ene (DBN), 1,8-
diazabicyclo[5.4.0]undec-7-ene (DBU), N,N'-dicyclohexylcarbodiimide (DCC), 1,2-
dichloroethane (DCE), dichloromethane (DCM), diethyl azodicarboxylate (DEAD),
di-
iso-propylazodicarboxylate (DIAD), di-iso-butylaluminumhydride (DIBAL or DIBAL-
H), di-iso-propylethylamine (DIPEA), N,N-dimethyl acetamide (DMA), 4-N,N-
dimethylaminopyridine (DMAP), N,N-dimethylformamide (DMF), dimethyl sulfoxide
(DMSO), 1-(3-dimethylaminopropy1)-3-ethylcarbodiimide hydrochloride (EDCI),
ethyl
(Et), ethyl acetate (Et0Ac), ethanol (Et0H), 2-ethoxy-2H-quinoline-l-
carboxylic acid
ethyl ester (EEDQ), diethyl ether (Et20), 0-(7-azabenzotriazole-1-y1)-N,
N,N'N'-
tetramethyluronium hexafluorophosphate acetic acid (HATU), acetic acid (HOAc),
1-N-
hydroxybenzotriazole (HOBt), high pressure liquid chromatography (HPLC), iso-
propanol (IPA), methanol (Me0H), melting point (mp), MeS02- (mesyl or Ms)õ
methyl
(Me), acetonitrile (MeCN), m-chloroperbenzoic acid (MCPBA), mass spectrum
(ms),
methyl t-butyl ether (MTBE), N-methylmorpholine (NMM), N-methylpyrrolidone
(NMP), phenyl (Ph), propyl (Pr), iso-propyl (i-Pr), pounds per square inch
(psi), pyridine
(pyr), room temperature (rt or RT), tert-butyldimethylsilyl or t-BuMe2Si
(TBDMS),
triethylamine (TEA or Et3N), triflate or CF3S02- (TO, trifluoroacetic acid
(TFA)õ 0-
benzotriazo1-1-yl-N,N,N',N'-tetramethyluronium tetrafluoroborate (TBTU), thin
layer
chromatography (TLC), tetrahydrofuran (THF), trimethylsilyl or Me3Si (TMS), p-
CA 02687747 2009-11-19
WO 2008/145562
PCT/EP2008/056165
- 17 -
toluenesulfonic acid monohydrate (Ts0H or pTs0H), 4-Me-C6H4S02- or tosyl (Ts),
N-
urethane-N-carboxyanhydride (UNCA),. Conventional nomenclature including the
prefixes normal (n-), iso (i-), secondary (sec-), tertiary (tert-) and neo
have their
customary meaning when used with an alkyl moiety. (J. Rigaudy and D. P.
Klesney,
Nomenclature in Organic Chemistry, IUPAC 1979 Pergamon Press, Oxford.).
Examples of representative compounds encompassed by the present invention and
within the scope of the invention are provided in the following Table. These
examples
and preparations which follow are provided to enable those skilled in the art
to more
clearly understand and to practice the present invention. They should not be
considered
as limiting the scope of the invention, but merely as being illustrative and
representative
thereof.
In general, the nomenclature used in this Application is based on AUTONOMTm
v.4.0, a Beilstein Institute computerized system for the generation of IUPAC
systematic
nomenclature. If there is a discrepancy between a depicted structure and a
name given
that structure, the depicted structure is to be accorded more weight. In
addition, if the
stereochemistry of a structure or a portion of a structure is not indicated
with, for
example, bold or dashed lines, the structure or portion of the structure is to
be interpreted
as encompassing all stereoisomers of it.
TABLE I
HIV-1
CPD RT
NAME MS MP
NO. ICso
(1-11\4)
3-[6-Bromo-3-(2,4-dioxo-tetrahydro-pyrimidin-1- 161'5- 0I-1
0287
ylmethyl)-2-fluoro-phenoxy]-5-chloro-benzonitrile 163.0 =
3-[6-Bromo-2-fluoro-3-(5-methy1-2,4-dioxo-3,4-dihydro-
I-2 2H-pyrimidin-1-ylmethyl)-phenoxy]-5-chloro-
223.1- 0 0039
224.2 '
benzonitrile
3-[6-Bromo-3-(2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-
1-3
229.0- 00036
ylmethyl)-2-fluoro-phenoxy]-5-chloro-benzonitrile 230.5 =
3-[6-Bromo-3-(5-chloro-2,4-dioxo-3,4-dihydro-2H-
2608-
1-4 pyrimidin-l-ylmethyl)-2-fluoro-phenoxy]-5-chloro-
261..7 0'0041
benzonitrile
3-[6-Bromo-3-(5-ethy1-2,4-dioxo-3,4-dihydro-2H-
I-5 pyrimidin-l-ylmethyl)-2-fluoro-phenoxy]-5-chloro-
224.6- 0 0016
225.6 '
benzonitrile
3-[6-Bromo-2-fluoro-3-(5-fluoro-2,4-dioxo-3,4-dihydro-
2340-
1-6 2H-pyrimidin-1-ylmethyl)-phenoxy]-5-chloro-
236..0 0'0022
benzonitrile
3-[6-Bromo-3-(2,4-dioxo-5-trifluoromethy1-3,4-dihydro-
110.0-
1-7 2H-pyrimidin-l-ylmethyl)-2-fluoro-phenoxy]-5-chloro-11 06
2.0 0'0
benzonitrile
CA 02687747 2009-11-19
WO 2008/145562
PCT/EP2008/056165
- 18 -
5-[6-Bromo-2-fluoro-3-(5-methy1-2,4-dioxo-3,4-dihydro-
I-8 235.6- 0.0043
2H-pyrimidin-1-ylmethyl)-phenoxy]-isophthalonitrile 236.0
5-[6-Bromo-3-(5-chloro-2,4-dioxo-3,4-dihydro-2H-
I-9 pyrimidin-l-ylmethyl)-2-fluoro-phenoxy]- 255.0- 0.0052
255.6
isophthalonitrile
5-[6-Bromo-3-(5-ethy1-2,4-dioxo-3,4-dihydro-2H-
469 &
1-10 pyrimidin-l-ylmethyl)-2-fluoro-phenoxy]- 471 0.0135
isophthalonitrile
5-[6-Bromo-3-(2,4-dioxo-3,4-dihydro-2H-pyrimidin-1- 132.0- 0.0229
I-11
ylmethyl)-2-fluoro-phenoxy]-isophthalonitrile 133.0
3-[3-(4-Amino-2-oxo-2H-pyrimidin-1-ylmethyl)-6- 240.0- 0.1483
1-12
bromo-2-fluoro-phenoxy]-5-chloro-benzonitrile 245.0
3-[3-(4-Amino-5-methy1-2-oxo-2H-pyrimidin-1-
1-13 ylmethyl)-6-bromo-2-fluoro-phenoxy]-5-chloro- 262.7- 0.0196
263.7
benzonitrile
3- {6-Bromo-2-fluoro-3-[2-(5-methy1-2,4-dioxo-3,4-
1-14 dihydro-2H-pyrimidin-1-y1)-ethyl]-phenoxy{ -5-chloro- 198.0- 0 007
200.0 '
benzonitrile
1-[4-Bromo-3-(3-chloro-5-cyano-phenoxy)-2-fluoro-
I-15 246.9-
benzy1]-2,4-dioxo-1,2,3,4-tetrahydro-pyrimidine-5- 474
247.8 0'0612
carbonitrile
3-[6-Bromo-2-fluoro-3-(6-methy1-2,4-dioxo-3,4-dihydro-
I-16 2H-pyrimidin-1-ylmethyl)-phenoxy]-5-chloro- 227.0- 0.0261
228.0
benzonitrile
3-Chloro-5-[6-chloro-2-fluoro-3-(5-methyl-2,4-dioxo-3,4-
1-17 226.0- 0.0031
dthydro-2H-pyrimidin-l-ylmethyl)-phenoxy]-benzonitrile 227.5
3-Chloro-5-[6-chloro-3-(5-chloro-2,4-dioxo-3,4-dihydro-
I-18 267.2- 0.0037
2H-pyrimidin-1-ylmethyl)-2-fluoro-phenoxy]-benzonitrile 268.0
3-[6-Bromo-2-fluoro-3-(5-methoxy-2,4-dioxo-3,4-
190.0- 029
1-19 dihydro-2H-pyrimidin-1-ylmethyl)-phenoxy]-5-chloro-
191.1 0.0029
benzonitrile
3-[6-Bromo-3-(2,4-dioxo-imidazolidin-l-ylmethyl)-2-
I-20 131'5- 0.009
fluoro-phenoxy]-5-chloro-benzonitrile 132.8
3-[6-Bromo-2-fluoro-34(R)-5-methy1-2,4-dioxo- 119.0- 0.0169
1-21
imidazolidin-l-ylmethyl)-phenoxy]-5-chloro-benzonitrile 120.0
3-[3-(5-Ally1-2,4-dioxo-imidazolidin-l-ylmethyl)-6- 200.0-
1-22 1
bromo-2-fluoro-phenoxy]-5-chloro-benzonitrile 201.0
3-[6-Bromo-2-fluoro-3-(5-methyl-4-oxo-2-thioxo-3,4-
I-23 dihydro-2H-pyrimidin-1-ylmethyl)-phenoxy]-5-chloro- 225.0- 0 0055
226.0 '
benzonitrile
3-[6-Bromo-2-fluoro-3-(5-hydroxymethy1-2,4-dioxo-3,4-
I-24 dihydro-2H-pyrimidin-1-ylmethyl)-phenoxy]-5-chloro- 226.0- 0.0111
227.0
benzonitrile
3-Chloro-5-[6-ethyl-2-fluoro-3-(5-methyl-2,4-dioxo-3,4-
I-25 180.5- 0.0018
dthydro-2H-pyrimidin-l-ylmethyl)-phenoxy]-benzonitrile 181.5
3-Chloro-5-[3-(5-chloro-2,4-dioxo-3,4-dihydro-2H-
434 &
1-26 pyrimidin-l-ylmethyl)-6-ethyl-2-fluoro-phenoxy]- 0.0014
436
benzonitrile
3-[6-Bromo-2-fluoro-3-(5-methy1-2,4-dioxo-3,4-dihydro-
I-27 2H-pyrimidin-1-ylmethyl)-phenoxy]-5-difluoromethyl- 232.3- 0 0033
234.8 '
benzonitrile
3-[6-Bromo-2-fluoro-3-(4-oxo-2-thioxo-imidazolidin-1- 172.0- 86
1-28
ylmethyl)-phenoxy]-5-chloro-benzonitrile 173.0 0.0
3-[6-Bromo-2-fluoro-3-(5-methy1-2,4-dioxo-tetrahydro-
I-29 227.8- 0.0858
pyrimidin-l-ylmethyl)-phenoxy]-5-chloro-benzonitrile 228.6
CA 02687747 2009-11-19
WO 2008/145562 PCT/EP2008/056165
- 19 -
3-[6-Bromo-2-fluoro-3-(5-hydroxy-2,4-dioxo-3,4-
238.0-
1-30 dihydro-2H-pyrimidin-1-ylmethyl)-phenoxy]-5-chloro-
240.0 0'0345
benzonitrile
3-[6-Bromo-2-fluoro-3-(1-methy1-2,4-dioxo-1,2,3,4-
226.0-
1-31 tetrahydro-pyrimidin-5-ylmethyl)-phenoxy]-5-chloro-
227.0 0'0059
benzonitrile
3-[6-Bromo-2-fluoro-3-(3-methyl-2,5-dioxo- 452 &
1-32 . .
0.0063
nnklazolidin-4-ylmethyl)-phenoxy]-5-chloro-benzonitrile 454
Compounds of the present invention can be made by a variety of methods
depicted
in the illustrative synthetic reaction schemes shown and described below. The
starting
materials and reagents used in preparing these compounds generally are either
available
from commercial suppliers, such as Aldrich Chemical Co., or are prepared by
methods
known to those skilled in the art following procedures set forth in references
such as
Fieser and Fieser's Reagents for Organic Synthesis; Wiley & Sons: New York,
Volumes
1-21; R. C. LaRock, Comprehensive Organic Transformations, 2nd edition Wiley-
VCH,
New York 1999; Comprehensive Organic Synthesis, B. Trost and I. Fleming (Eds.)
vol.
1-9 Pergamon, Oxford, 1991; Comprehensive Heterocyclic Chemistry, A. R.
Katritzky
and C. W. Rees (Eds) Pergamon, Oxford 1984, vol. 1-9; Comprehensive
Heterocyclic
Chemistry II, A. R. Katritzky and C. W. Rees (Eds) Pergamon, Oxford 1996, vol.
1-11;
and Organic Reactions, Wiley & Sons: New York, 1991, Volumes 1-40. The
following
synthetic reaction schemes are merely illustrative of some methods by which
the
compounds of the present invention can be synthesized, and various
modifications to
these synthetic reaction schemes can be made and will be suggested to one
skilled in the
art having referred to the disclosure contained in this Application.
The starting materials and the intermediates of the synthetic reaction schemes
can
be isolated and purified if desired using conventional techniques, including
but not
limited to, filtration, distillation, crystallization, chromatography, and the
like. Such
materials can be characterized using conventional means, including physical
constants
and spectral data.
Unless specified to the contrary, the reactions described herein preferably
are
conducted under an inert atmosphere at atmospheric pressure at a reaction
temperature
range of from about -78 C to about 150 C, more preferably from about 0 C to
about
125 C, and most preferably and conveniently at about room (or ambient)
temperature,
e.g., about 20 C.
CA 02687747 2009-11-19
WO 2008/145562
PCT/EP2008/056165
- 20 -
Some compounds in following schemes are depicted with generalized
substituents;
however, one skilled in the art will immediately appreciate that the nature
and number of
the R groups can be varied to afford the various compounds contemplated in
this
invention. The general formulae in the schemes are intended to be illustrative
and are not
intended to imply a limitation to the scope of the invention which is defined
by the
appended claims. Moreover, the reaction conditions are exemplary and
alternative
conditions are well known. The reaction sequences in the following examples
are not
meant to limit the scope of the invention as set forth in the claims.
SCHEME A
F F F 0
F 0 Br step 1 Cl 0 R step 5 Cl 0
NANH
Br Br Br 0
CN CN
A-1 step 2 A-2a: R = Br 1-3
1¨ A-2b: R = CHO
step 3
1-0. A-2c: R = CH2OH
step 4 E. A-2d: R = CH2Br
Compounds of the present invention are prepared by alkylation of a suitably
substituted benzyl bromide A-2d with either uracil or cytosine or a
substituted derivative
thereof. (SCHEME A) The requisite benzyl bromide is prepared by formylation of
the
Grignard salt derived from A-2a which, in turn, was prepared by nucleophilic
displacement of one of the fluorine substituents of A-1 (CASRN 1566882-52-9).
The
preparation of diaryl ethers has been reviewed (J. S. Sawyer, Recent Advances
in Diary'
Ether Synthesis, Tetrahedron 2000 56:5045-5065). Introduction of the aryloxy
ether can
often be accomplished by direct SNAr displacement reaction on an aromatic ring
bearing
a leaving group and electronegative substituents. Fluoroaromatic compounds
with
electronegative substituents are known to be sensitive to nucleophilic attack
by soft
nucleophiles. Fluorine substituents are generally significantly more labile
than other
halogen substituents. While hard nucleophiles like water and hydroxide fail to
displace
fluoride, soft nucleophiles like phenols, imidazoles, amines, thiols and some
amides
undergo facile displacement reactions even at room temperature (D. Boger et
at., Biorg.
Med. Chem. Lett. 2000 10: 1471-75; F. Terrier Nucleophilic Aromatic
Displacement:
The Influence of the Nitro Group VCH Publishers, New York, NY 1991). One
skilled in
the art will appreciate that while the reaction sequence in SCHEME A is
exemplified
with 3-chloro-5-hydroxybenzonitrile, other phenols could be used analogously.
CA 02687747 2009-11-19
WO 2008/145562 PCT/EP2008/056165
- 21 -
Monometallation of A-2a with iso-PrMgCl/LiCl/THF and formylation of the
resulting magnesium salt with DMF afforded A-2b. Reduction of the resulting
aldehyde
to A-2c can be achieved by utilizing well established reagents which allow the
selective
reduction of the aldehyde. Sodium borohydride is known to reduce aldehydes and
ketones selectively in the presence of cyano substituent. Sodium borohydride
reductions
are typically carried out in alcoholic or aqueous media. The conversion of an
alcohol (A-
2c) to a halide (A-2d) is a well-established procedure which can be carried
out with a
variety reagents. Commonly used reagents include SOBr2, PBr3 , POBr3 and
phosphorus
derived halogenating agents such as (R0)3PRX and R3PX2 are examples of
commonly
used reagents. In the present instance, carbon tetrabromide and
triphenylphosphine or
PBr3 produce an effective brominating agent (A. R. Katritzky et at. Chem Scr.
1987
27:477). Treatment of a uracil with hexamethyldisilazane affords the 2,4-bis-
(trimethylsilyloxy)pyrimidine which reacts with the alkyl halide exclusively
at the N-1
position. (H. Singh et at., Synthesis 1990 520)
The corresponding compounds with an ethylene linker (formula II, n = 2) can be
prepared from the corresponding 3-aryloxy-phenylacetic acid or their
corresponding
esters. 3-Aryloxy-2-fluoro-4-substituted-phenylacetic acids can be prepared
conveniently by the sequential condensation of 2,3,4-trifluoro-l-nitro-benzene
with a
suitably substituted phenol and a malonic acid diester. The ester is
hydrolyzed and
decarboxylated to afford the phenyl-acetic acid. The use of tert-butyl ethyl
(or methyl)
malonate allows for selective acid-catalyzed hydrolysis of the tert-butyl
ester and
decarboxylation to afford the ethyl (or methyl) ester directly. The nitro
group can be
reduced and the corresponding amine replaced by halogen utilizing the
Sandmeyer
reaction which is well known in the art to afford the 4-chloro or 4-bromo
derivatives.
Phenols used in the initial condensation are readily available, however,
referential
example 1 provides routes which are provided for example and are not meant to
limit the
scope of the invention. This methodology has been described by J. P. Dunn et
at. in
U.S. Patent No. 7,166,730 published January 23, 2007 and by D. Kertesz et at.
in U.S.
Publication No. 2005/0234236 published September 29, 2005. Reduction of the of
the
aryloxyphenyl acetic acid to the corresponding alcohol, conversion of the
alcohol to the
corresponding halide and displacement of the halide with a cytosine or uracil
can be
carried out as described above.
Compounds of the present invention wherein R4 is C1_6 alkyl can be prepared
from
a 4-bromo intermediate, e.g. A-2b, utilizing the Negishi coupling procedure.
The Negishi
CA 02687747 2009-11-19
WO 2008/145562 PCT/EP2008/056165
- 22 -
coupling of organozinc halides or dialkylzinc with haloarenes and aryl
triflates is an
effective means for attachment of an alkyl group to an arene (E.-I. Negishi,
Acc. Chem.
Res. 1982 15:340-348). The reaction is catalyzed by Pd(0) and palladium is
preferably
ligated to a bidentate ligand including Pd(dppf)C12 and Pd(dppe)C12. (J. M.
Herbert
Tetrahedron Lett. 2004 45:817-819) Typically the reaction is run an inert
aprotic solvent
and common ethereal solvents including dioxane, DME and THF are suitable. The
reaction is commonly run at elevated temperature.
Embodiments of the present invention wherein R4 is cyclopropyl are prepared by
introduction of a vinyl sub stituent utilizing tri-n-butyl-vinyl-tin in the
Stille reaction
followed by Pd(II)-mediated cyclopropanation of he resulting styrene with
diazomethane.
SCHEME B
Cl F HO 0 Me step 1 CI
Cl 0 r R
1W _2N.
step 3
1W +
IW
CN CN
B-1 B-2B-3a: R = Me
step 2 1¨
311.. B-3b: R = CH2Br
Cl r 0
IW I
0 NriNTL (1-17)
CI 0
CN Me
Embodiments of the present invention wherein R4 is chloride and n = 1, were
prepared from 3-chloro-5-(6-chloro-2-fluoro-3-methyl-phenoxy)-benzonitrile (B-
3a)
which was prepared by condensation of 3-chloro-5-fluoro-benzonitrile (B-1,
CASRN
327056-73-05) and 6-chloro-2-fluoro-3-methyl-pheno1 (B-2). Free radical
bromination
of the methyl substituent with NBS and AIBN affords B-3b which is converted to
compounds of the present invention were prepared as described above. (SCHEME
B)
Embodiments of the invention containing a hydantoin ring are prepared by base-
catalyzed cyclization an N-carbamoyl-N-substituted alpha amino acid which are
in turn
available from reductive alkylation of
CA 02687747 2009-11-19
WO 2008/145562
PCT/EP2008/056165
- 23 -
SCHEME C
A-2b Ar0 OR2
Ar0 = H
R 0
R4 R4 0
C-la: R1= H C-2
C-lb: R1= C(=0)NH2
A-2b and an alpha-amino acid ester as depicted in SCHEME C. One skilled in he
art will appreciate that the ready availability of alpha amino acids allows
the introduction
of a variety of substituents at the 5-position of the hydantoin ring.
SCHEME D
Ar 110 step 3 Ar/0
4
NH
R4 R Me
0
step 6
D-2
D-la: Y Br
step 1 ______________ =
D-lb: Y = CH(NMeBoc)CO2Me
step 2
D-lc: Y = CH(NHMe)CO2Me
Ar
NH
1-37. D-la: Y = Br
step 4 _________ D-ld: Y = CH(CO2tBu)CO2Et R4 NX
step 5 D-le: Y = CHCO2Et
D-3a: X = S, R = H
step 7
D-3b: X = 0, R = H
step 8 E D-3e: x = R = me
Compounds of the present invention in which the C-5 carbon of either the
uracil or
the hydantoin ring is linked to a 3-aryloxy-benzyl moiety require the
introduction of a C-
C bond rather than the C-N bond required when the 3-aryloxy-benzyl moiety was
attached to a nitrogen atom (SCHEME D). R4 and Ar in SCHEME D are as described
in
claim 1. The hydantoins were prepared by alkylation of A-2b with anion derived
from
an N-protected amine add ester which introduces two carbon atoms and one
nitrogen
atom of the hydantoin ring. The example in SCHEME D utilizes the N-Boc methyl
ester
of sarcosine. After the urethane protecting group is removed, D-lc is treated
with
trimethylsilyl isocyanate which results in acylation of the amine and
intramolecular
cyclization of the newly introduced nitrogen and the ester. The C-linked
uracils are
prepared by homologation of A-2b via a malonic ester condensation and
decarboxylation
CA 02687747 2009-11-19
WO 2008/145562
PCT/EP2008/056165
- 24 -
to afford the 3-aryl propionate ester D-le. Acylation of the ester with ethyl
formate
affords a 13-formyl-ester which was cyclized with thiourea to afford D-3a
which can be
hydrolyzed to afford the urea D-3b which is alkylated by silylation of the
more acidic
imide nitrogen and alkylation of the second nitrogen with an alkylating agent.
The compounds of the present invention may be formulated in a wide variety of
oral administration dosage forms and carriers. Oral administration can be in
the form of
tablets, coated tablets, dragees, hard and soft gelatine capsules, solutions,
emulsions,
syrups, or suspensions. Compounds of the present invention are efficacious
when
administered by other routes of administration including continuous
(intravenous drip)
topical parenteral, intramuscular, intravenous, subcutaneous, transdermal
(which may
include a penetration enhancement agent), buccal, nasal, inhalation and
suppository
administration, among other routes of administration. The preferred manner of
administration is generally oral using a convenient daily dosing regimen which
can be
adjusted according to the degree of affliction and the patient's response to
the active
ingredient.
A compound or compounds of the present invention, as well as their
pharmaceutically useable salts, together with one or more conventional
excipients,
carriers, or diluents, may be placed into the form of pharmaceutical
compositions and
unit dosages. The pharmaceutical compositions and unit dosage forms may be
comprised of conventional ingredients in conventional proportions, with or
without
additional active compounds or principles, and the unit dosage forms may
contain any
suitable effective amount of the active ingredient commensurate with the
intended daily
dosage range to be employed. The pharmaceutical compositions may be employed
as
solids, such as tablets or filled capsules, semisolids, powders, sustained
release
formulations, or liquids such as solutions, suspensions, emulsions, elixirs,
or filled
capsules for oral use; or in the form of suppositories for rectal or vaginal
administration;
or in the form of sterile injectable solutions for parenteral use. A typical
preparation will
contain from about 5% to about 95% active compound or compounds (w/w). The
term
"preparation" or "dosage form" is intended to include both solid and liquid
formulations
of the active compound and one skilled in the art will appreciate that an
active ingredient
can exist in different preparations depending on the target organ or tissue
and on the
desired dose and pharmacokinetic parameters.
CA 02687747 2009-11-19
WO 2008/145562 PCT/EP2008/056165
- 25 -
The term "excipient" as used herein refers to a compound that is useful in
preparing a pharmaceutical composition, generally safe, non-toxic and neither
biologically nor otherwise undesirable, and includes excipients that are
acceptable for
veterinary use as well as human pharmaceutical use. The compounds of this
invention
can be administered alone but will generally be administered in admixture with
one or
more suitable pharmaceutical excipients, diluents or carriers selected with
regard to the
intended route of administration and standard pharmaceutical practice.
"Pharmaceutically acceptable" means that which is useful in preparing a
pharmaceutical composition that is generally safe, non-toxic, and neither
biologically nor
otherwise undesirable and includes that which is acceptable for human
pharmaceutical
use.
A "pharmaceutically acceptable salt" form of an active ingredient may also
initially
confer a desirable pharmacokinetic property on the active ingredient which
were absent
in the non-salt form, and may even positively affect the pharmacodynamics of
the active
ingredient with respect to its therapeutic activity in the body. The phrase
"pharmaceutically acceptable salt" of a compound means a salt that is
pharmaceutically
acceptable and that possesses the desired pharmacological activity of the
parent
compound. Such salts include: (1) acid addition salts, formed with inorganic
acids such
as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric
acid, and
the like; or formed with organic acids such as acetic acid, propionic acid,
hexanoic acid,
cyclopentanepropionic acid, glycolic acid, pyruvic acid, lactic acid, malonic
acid,
succinic acid, malic acid, maleic acid, fumaric acid, tartaric acid, citric
acid, benzoic
acid, 3-(4-hydroxybenzoyl)benzoic acid, cinnamic acid, mandelic acid,
methanesulfonic
acid, ethanesulfonic acid, 1,2-ethane-disulfonic acid, 2-hydroxyethanesulfonic
acid,
benzenesulfonic acid, 4-chlorobenzenesulfonic acid, 2-naphthalenesulfonic
acid, 4-
toluenesulfonic acid, camphorsulfonic acid, 4-methylbicyclo[2.2.2]-oct-2-ene-1-
carboxylic acid, glucoheptonic acid, 3-phenylpropionic acid, trimethylacetic
acid,
tertiary butylacetic acid, lauryl sulfuric acid, gluconic acid, glutamic acid,
hydroxynaphthoic acid, salicylic acid, stearic acid, muconic acid, and the
like; or (2) salts
formed when an acidic proton present in the parent compound either is replaced
by a
metal ion, e.g., an alkali metal ion, an alkaline earth ion, or an aluminum
ion; or
coordinates with an organic base such as ethanolamine, diethanolamine,
triethanolamine,
tromethamine, N-methylglucamine, and the like.
CA 02687747 2009-11-19
WO 2008/145562 PCT/EP2008/056165
- 26 -
Solid form preparations include powders, tablets, pills, capsules, cachets,
suppositories, and dispersible granules. A solid carrier may be one or more
substances
which may also act as diluents, flavoring agents, solubilizers, lubricants,
suspending
agents, binders, preservatives, tablet disintegrating agents, or an
encapsulating material.
In powders, the carrier generally is a finely divided solid which is a mixture
with the
finely divided active component. In tablets, the active component generally is
mixed
with the carrier having the necessary binding capacity in suitable proportions
and
compacted in the shape and size desired. Suitable carriers include but are not
limited to
magnesium carbonate, magnesium stearate, talc, sugar, lactose, pectin,
dextrin, starch,
gelatin, tragacanth, methylcellulose, sodium carboxymethylcellulose, a low
melting wax,
cocoa butter, and the like. Solid form preparations may contain, in addition
to the active
component, colorants, flavors, stabilizers, buffers, artificial and natural
sweeteners,
dispersants, thickeners, solubilizing agents, and the like.
Liquid formulations also are suitable for oral administration include liquid
formulation including emulsions, syrups, elixirs, aqueous solutions, aqueous
suspensions.
These include solid form preparations which are intended to be converted to
liquid form
preparations shortly before use. Emulsions may be prepared in solutions, for
example, in
aqueous propylene glycol solutions or may contain emulsifying agents such as
lecithin,
sorbitan monooleate, or acacia. Aqueous solutions can be prepared by
dissolving the
active component in water and adding suitable colorants, flavors, stabilizing,
and
thickening agents. Aqueous suspensions can be prepared by dispersing the
finely divided
active component in water with viscous material, such as natural or synthetic
gums,
resins, methylcellulose, sodium carboxymethylcellulose, and other well known
suspending agents.
The compounds of the present invention may be formulated for parenteral
administration (e.g., by injection, for example bolus injection or continuous
infusion) and
may be presented in unit dose form in ampoules, pre-filled syringes, small
volume
infusion or in multi-dose containers with an added preservative. The
compositions may
take such forms as suspensions, solutions, or emulsions in oily or aqueous
vehicles, for
example solutions in aqueous polyethylene glycol. Examples of oily or
nonaqueous
carriers, diluents, solvents or vehicles include propylene glycol,
polyethylene glycol,
vegetable oils (e.g., olive oil), and injectable organic esters (e.g., ethyl
oleate), and may
contain formulatory agents such as preserving, wetting, emulsifying or
suspending,
stabilizing and/or dispersing agents. Alternatively, the active ingredient may
be in
CA 02687747 2009-11-19
WO 2008/145562 PCT/EP2008/056165
- 27 -
powder form, obtained by aseptic isolation of sterile solid or by
lyophilisation from
solution for constitution before use with a suitable vehicle, e.g., sterile,
pyrogen-free
water.
The compounds of the present invention may be formulated for administration as
suppositories. A low melting wax, such as a mixture of fatty acid glycerides
or cocoa
butter is first melted and the active component is dispersed homogeneously,
for example,
by stirring. The molten homogeneous mixture is then poured into convenient
sized
molds, allowed to cool, and to solidify.
The compounds of the present invention may be formulated for vaginal
administration. Pessaries, tampons, creams, gels, pastes, foams or sprays
containing in
addition to the active ingredient such carriers as are known in the art to be
appropriate.
When desired, formulations can be prepared with enteric coatings adapted for
sustained or controlled release administration of the active ingredient. For
example, the
compounds of the present invention can be formulated in transdermal or
subcutaneous
drug delivery devices. These delivery systems are advantageous when sustained
release
of the compound is necessary and when patient compliance with a treatment
regimen is
crucial. Compounds in transdermal delivery systems are frequently attached to
an skin-
adhesive solid support. The compound of interest can also be combined with a
penetration enhancer, e.g., Azone (1-dodecylaza-cycloheptan-2-one). Sustained
release
delivery systems are inserted subcutaneously into to the subdermal layer by
surgery or
injection. The subdermal implants encapsulate the compound in a lipid soluble
membrane, e.g., silicone rubber, or a biodegradable polymer, e.g., polyactic
acid.
Suitable formulations along with pharmaceutical carriers, diluents and
excipients
are described in Remington: The Science and Practice of Pharmacy 1995, edited
by E.
W. Martin, Mack Publishing Company, 19th edition, Easton, Pennsylvania. A
skilled
formulation scientist may modify the formulations within the teachings of the
specification to provide numerous formulations for a particular route of
administration
without rendering the compositions of the present invention unstable or
compromising
their therapeutic activity.
The modification of the present compounds to render them more soluble in water
or other vehicle, for example, may be easily accomplished by minor
modifications (salt
formulation, esterification, etc.), which are well within the ordinary skill
in the art. It is
CA 02687747 2009-11-19
WO 2008/145562 PCT/EP2008/056165
- 28 -
also well within the ordinary skill of the art to modify the route of
administration and
dosage regimen of a particular compound in order to manage the
pharmacokinetics of the
present compounds for maximum beneficial effect in patients.
The term "therapeutically effective amount" as used herein means an amount
required to reduce symptoms of the disease in an individual. The dose will be
adjusted to
the individual requirements in each particular case. That dosage can vary
within wide
limits depending upon numerous factors such as the severity of the disease to
be treated,
the age and general health condition of the patient, other medicaments with
which the
patient is being treated, the route and form of administration and the
preferences and
experience of the medical practitioner involved. For oral administration, a
daily dosage
of between about 0.01 and about 1000 mg/kg body weight per day should be
appropriate
in monotherapy and/or in combination therapy. A preferred daily dosage is
between
about 0.1 and about 500 mg/kg body weight, more preferred 0.1 and about 100
mg/kg
body weight and most preferred 1.0 and about 10 mg/kg body weight per day.
Thus, for
administration to a 70 kg person, the dosage range would be about 7 mg to 0.7
g per day.
The daily dosage can be administered as a single dosage or in divided dosages,
typically
between 1 and 5 dosages per day. Generally, treatment is initiated with
smaller dosages
which are less than the optimum dose of the compound. Thereafter, the dosage
is
increased by small increments until the optimum effect for the individual
patient is
reached. One of ordinary skill in treating diseases described herein will be
able, without
undue experimentation and in reliance on personal knowledge, experience and
the
disclosures of this application, to ascertain a therapeutically effective
amount of the
compounds of the present invention for a given disease and patient.
In embodiments of the invention, the active compound or a salt can be
administered in combination with another antiviral agent, such as a nucleoside
reverse
transcriptase inhibitor, another non-nucleoside reverse transcriptase
inhibitor or HIV
protease inhibitor. When the active compound or its derivative or salt are
administered
in combination with another antiviral agent the activity may be increased over
the parent
compound. When the treatment is combination therapy, such administration may
be
concurrent or sequential with respect to that of the nucleoside derivatives.
"Concurrent
administration" as used herein thus includes administration of the agents at
the same time
or at different times. Administration of two or more agents at the same time
can be
achieved by a single formulation containing two or more active ingredients or
by
CA 02687747 2009-11-19
WO 2008/145562 PCT/EP2008/056165
- 29 -
substantially simultaneous administration of two or more dosage forms with a
single
active agent.
It will be understood that references herein to treatment extend to
prophylaxis as
well as to the treatment of existing conditions, and that the treatment of
animals includes
the treatment of humans as well as other animals. Furthermore, treatment of a
HIV-1
infection, as used herein, also includes treatment or prophylaxis of a disease
or a
condition associated with or mediated by HIV-1 infection, or the clinical
symptoms
thereof.
The pharmaceutical preparations are preferably in unit dosage forms. In such
form,
the preparation is subdivided into unit doses containing appropriate
quantities of the
active component. The unit dosage form can be a packaged preparation, the
package
containing discrete quantities of preparation, such as packeted tablets,
capsules, and
powders in vials or ampoules. Also, the unit dosage form can be a capsule,
tablet,
cachet, or lozenge itself, or it can be the appropriate number of any of these
in packaged
form.
The following examples illustrate the preparation and biological evaluation of
compounds within the scope of the invention. These examples and preparations
which
follow are provided to enable those skilled in the art to more clearly
understand and to
practice the present invention. They should not be considered as limiting the
scope of the
invention, but merely as being illustrative and representative thereof.
Example 1
3- [6-Bromo-3-(2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-2-fluoro-
phenoxy] -5-chloro-benzonitrile (1-3)
Preparation of 3-chloro-5-hydroxy-benzonitrile
step 1 - A 100 mL round bottom flask was charged under a stream of nitrogen
with
3,5-dichlorobenzonitrile (7.0 g, 40.69 mmol) and anhydrous DMF (75 mL). To the
solution was added sodium methoxide (2.26 g, 44.76 mmol) and resulting
solution was
stirred further at RT for 24 h. When the reaction was complete, aqueous 10%
HC1 added
dropwise to the reaction vessel. The crude mixture was extracted with Et0Ac
and
sequentially washed with aqueous acid, water and brine. The Et0Ac extracts
were dried
(Na2SO4), filtered and the solvent was removed in vacuo to afford a crude
solid which
CA 02687747 2009-11-19
WO 2008/145562 PCT/EP2008/056165
- 30 -
was recrystallized from hexane/acetone to afford 5.9 g (86%) of 5-chloro-3-
methoxy-
benzonitrile.
step 2 - A 250 mL flask was charged with 5-chloro-3-methoxy-benzonitrile (7.0
g,
41.766 mmol) and 2,4,6-collidine (100 mL). The mixture was heated to 170 C and
Lil
(16.76 g, 125.298 mmol) was added and the reaction mixture was heated for 4 h.
When
the methyl ether was consumed the reaction was cooled to RT and quenched with
10%
aqueous HC1. The resulting mixture was extracted with Et0Ac and washed with
water
and brine. The Et0Ac extract was dried over (Na2SO4) and filtered. The solvent
was
removed in vacuo to afford a yellow oil which was purified by silica gel
chromatography
eluting with Et0Ac/hexane (10:90) to afford 6.0 g (94%) of 3-chloro-5-hydroxy-
benzonitrile.
(SCHEME A) step 1 - To a solution 3-chloro-5-hydroxy-benzonitrile (153 mg, 1
mmol) and DMA (1 mL) was added NaH (42 mg, 1.05 equiv., 60% mineral oil
dispersion) and the resulting mixture was stirred at 50 C for 30 min. To the
solution was
added A-1 (2.7 g, 10 mmol) and the resulting mixture was heated at 125 C for
2 h. The
solution was cooled and diluted with Et0Ac and the resulting solution washed
with an
equal volume of 10% H2504. The organic extract was dried (Mg504), filtered and
concentrated in vacuo. The crude product was purified by 5i02 chromatography
eluting
with 10% Et0Ac/hexane to afford 331 mg (82%) of A-2a.
step 2 - To a solution of A-2a (2.00g, 4.93mL) in PhMe (40mL) maintained under
an Ar atmosphere and cooled to -78 C was added a solution of i-PrMgC1 (2M in
THF,
3.08 mL, 6.16 mmol). The solution was stirred for 1 h then a solution of
CuCN.2LiC1
(1M in THF, 0.1mL) was added. The resulting solution was stirred at -50 C for
2 h and
then the reaction mixture was cannulated into a flask containing DMF (0.57 mL,
7.4
mmol) and PhMe (10 mL) which was cooled to -78 C. The mixture was warmed to
RT
and quenched by the addition of saturated aqueous NH4C1 solution. The organic
phase
was separated, washed with brine, dried (Mg504) and evaporated to dryness in
vacuo to
afford 1.50 g (86%) of A-2b as an off-white solid.
step 3 - Sodium borohydride was added in portions to a stirred solution of A-
2b in
THF (5 mL) and Me0H (5 mL) at RT. After stirring for 24 h, the reaction
mixture was
quenched by the addition of saturated aqueous NH4C1. The organics were
extracted with
Et0Ac, washed with brine, dried (Mg504) and evaporated to dryness under in
vacuo.
CA 02687747 2009-11-19
WO 2008/145562 PCT/EP2008/056165
- 31 -
The product was purified by Si02 chromatography eluting with an Et0Ac/hexane
gradient (10 to 50% Et0Ac) to afford 0.25 g (31%) of A-2c.
step 4 - To a stirred solution of A-2c (3.00g, 8.41 mmol) in DCM (100 mL) was
added a solution of PBr3 (1M in DCM, 9.3 mL). After stirring at RT under N2
for 24 h
the reaction mixture was quenched by the addition of saturated aqueous NaHCO3.
The
organic phase was separated, washed with brine, dried (MgSO4) and evaporated
in vacuo.
The product was purified by Si02 chromatography eluting with an Et0Ac/hexane
gradient (20 to 50% Et0Ac) to afford 2.0 g (57%) of A-2d as white crystals.
step 5 - A solution of A-2d (0.12g, 0.286 mmol), TMSC1 (2 drops),
hexamethyldisilazane (0.3 mL), DCE (2 mL), iodine (cat. 1 chip), and uracil
(0.16 g, 5
eq) were combined under nitrogen and stirred for 18h at 80 C. The reaction
was cooled,
the organics evaporated, and the residue dissolved in DCM and washed with H20.
The
organic solution was dried (MgSO4), filtered and evaporated. The crude product
was
purified by Si02 chromatography eluting with a Me0H/DCM gradient (1%-7% Me0H)
to afford 0.07g (55%) of I-3.
3-[6-Bromo-2-fluoro-3-(5-methy1-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-
ylmethyl)-phenoxy]-5-chloro-benzonitrile was prepared analogously except in
step 5,
uracil was replaced by thymine. The compound was purified by Si02
chromatography
eluting with a Me0H/DCM gradient (1%-6% Me0H) to afford 1-2.
3-[6-Bromo-3-(5-chloro-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-2-
fluoro-phenoxy]-5-chloro-benzonitrile was prepared analogously except in step
5, uracil
was replaced with 5-chloro-1H-pyrimidine-2,4-dione. The compound was purified
by
Si02 chromatography eluting with a Me0H/DCM gradient (1%-7% Me0H) to afford I-
4.
3-[6-Bromo-3-(5-ethy1-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-2-fluoro-
phenoxy]-5-chloro-benzonitrile was prepared analogously except in step 5,
uracil was
replaced with 5-ethyl-1H-pyrimidine-2,4-dione. The compound was purified by
Si02
chromatography eluting with a Me0H/DCM gradient (1%-7% Me0H) to afford I-5.
3-[6-Bromo-2-fluoro-3-(5-fluoro-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-
ylmethyl)-phenoxy]-5-chloro-benzonitrile was prepared analogously except in
step 5,
uracil was replaced with 5-fluoro-1H-pyrimidine-2,4-dione to afford 1-6.
CA 02687747 2009-11-19
WO 2008/145562 PCT/EP2008/056165
- 32 -
3-[6-Bromo-3-(2,4-dioxo-5-trifluoromethy1-3,4-dihydro-2H-pyrimidin-1-
ylmethyl)-2-fluoro-phenoxy]-5-chloro-benzonitrile was prepared analogously
except in
step 5, uracil was replaced with 5-trifluoromethy1-1H-pyrimidine-2,4-dione
(CASRN 54-
20-6) to afford 1-7.
5-[6-Bromo-2-fluoro-3-(5-methy1-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-
ylmethyl)-phenoxy]-isophthalonitrile, 5-[6-bromo-3-(5-chloro-2,4-dioxo-3,4-
dihydro-
2H-pyrimidin-1-ylmethyl)-2-fluoro-phenoxy]-isophthalonitrile and 5-[6-bromo-3-
(5-
ethy1-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-2-fluoro-phenoxy]-
isophthalonitrile are prepared analogously except in step 1, 3-chloro-5-
hydroxy-
benzonitrile is replaced with 5-hydroxy-isophthalonitrile and thymine, 5-
chloro-uracil
and 5-ethyl-uracil, respectively, were used in place of uracil in step 5.
Example 2
3- [3-(4-Amino-2-oxo-2H-pyrimidin-1-ylmethyl)-6-bromo-2-fluoro-phenoxyl -5-
chloro-benzonitrile (I-12)
A mixture of cytosine (0.04 g, 3 eq), chlorotrimethylsilane (ldrop), and
hexamethyldisilazane (0.15 mL) were heated to 120 C for lh. The reaction was
cooled
to 80 C and then A-2d (0.05 g, 0.119 mmol) and iodine (1 chip) were added.
The
reaction was heating at 80 C for 18 h. The reaction was then cooled, the
organics
evaporated, and the resulting solid dissolved in DCM and washed with H20 and
the
resulting organic phase dried (MgSO4), filtered and evaporated. The crude
product was
purified by Si02 chromatography eluting with a Me0H/DCM gradient (1%-7% Me0H)
to afford 0.036 g (67%) of I-12.
3-[3-(4-Amino-5-methy1-2-oxo-2H-pyrimidin-1-ylmethyl)-6-bromo-2-fluoro-
phenoxy]-5-chloro-benzonitrile (1-13) was prepared analogously except cytosine
was
replaced with 5-methyl cytosine.
Example 3
3- {6-Bromo-2-fluoro-3-[2-(5-methy1-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-y1)-
ethyl] -pheno xy} -5 -chloro-benzonitrile (1-14)
CA 02687747 2009-11-19
WO 2008/145562 PCT/EP2008/056165
- 33 -
H
F F OyNNe0
Ar0 0 R step 3
Ar0 NMe
_2N. 01
Br Br
1-14
step 1 I¨ 10a: R = CO H
2
1=1"
step 2 10b: R = CH2OH
1_,...
10c: R = CH2Br
step 1 - Trimethylsilyldiazomethane (9.75 mL, 1.5 equiv) was added to a
solution
of 10a (5.0 g, 13 mmol) in a mixture of Me0H (30 mL) and DCM (30 mL) at RT.
The
volatile materials were removed to afford the methyl ester which was dissolved
in THF
(116 mL) and Me0H (12 mL), and NaBH4(3.87 g, 8 equiv) was added slowly. The
mixture was heated to 80 C for 3 h, cooled to 0 C, and acidified by adding
4M HC1
dropwise. The mixture was extracted with ether, washed with water, and dried
(MgSO4),
filtered and evaporated. The crude product was purified by Si02 chromatography
eluting
with an Et0Ac/hexane gradient (0% to 30% Et0Ac) to afford 1.8 g (38%) of 10b.
step 2 - Triphenylphosphine (1.54 g, 1.5 equiv) was added in portions to a
solution
of 10b (1.45 g, 3.9 mmol) and CBr4 (1.62 g, 1.25 equiv) in DCM (15 mL) at 0
C. The
mixture was stirred for 1 h, concentrated under vacuum and Et20 was added. The
resultant solids were removed by filtration and the filtrate was concentrated
in vacuo.
The residue was purified by Si02 chromatography eluting with an Et0Ac/hexane
gradient (0% to 30% Et0Ac) to afford 1.62 g, (95%) of 10c.
step 3 - Thymine (0.09 g, 3 eq), HMDS (0.3 mL), and chlorotrimethylsilane (2
drops) were heated to 120 C for 2h. The reaction mixture was cooled, and DCE
(1.0
mL), 10c (100 mg, 0.23 mmol), and iodine (lchip) was added. The reaction was
stirred
for 16 h at 78 C. The reaction was cooled to ambient temperature and then the
organic
solvents evaporated. The resulting crude material was dissolved in DCM and the
organic
layer washed sequentially with water and brine, dried (MgSO4), filtered and
evaporated.
The product was purified by Si02 chromatography eluting with a Me0H/DCM
gradient
(1% to 7% Me0H) to afford 0.30 g (27%) of I-14.
Example 4
3-Chloro-546-chloro-2-fluoro-3-(5-methy1-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-
ylmethyl)-phenoxy]-benzonitrile (1-17, SCHEME B)
CA 02687747 2009-11-19
WO 2008/145562 PCT/EP2008/056165
- 34 -
step 1 - To a solution of 3-chloro-5-fluorobenzonitrile (10 g, 64.28 mmol) and
6-
chloro-2-fluoro-3-methyl-phenol (9.38 g, 58.44 mmol) in DMA (100 mL) was added
Cs2CO3 (1.9 g, 5.84 mmol) followed by K2CO3 (8.9 g, 64.28 mmol). The mixture
was
heated to 120 C (oil bath) under argon for 5.5 h . The reaction was cooled to
RT and
water (150 mL) was added. The mixture was extracted with Et0Ac (150 mL) and
the
aqueous phase back extracted with Et0Ac (2 x 100 mL). The Et0Ac phase was
dried
(MgSO4), filtered and concentrated in vacuo afford 11.1 g (75% purity) of B-3a
as a
white crystalline solid.
step 2 - To a solution of B-3a (11.1 g, 75% pure, 28 mmol) in CC14 (100 mL)
was
added NBS (5.4 g, 30 mmol) followed by AIBN (450 mg, 2.74 mmol). The mixture
was
heated to just below reflux temperature for 5 h. Additional NBS (2.7 g) and
AIBN (200
mg) were added and heating continued for an additional 5 h. The material was
cooled to
ambient and filtered to remove precipitated succinimide. The filtrate was
condensed and
the remainder taken up in Et0Ac (100 mL) and shaken with brine (100 mL). The
Et0Ac
phase was collected and the aqueous phase back extracted with Et0Ac (2 x 80
mL). The
combined organic extract were dried (MgSO4), filtered and concentrated in
vacuo. The
product was purified by Si02 chromatography eluting with an Et0Ac/hexane
gradient
(1.5% - 8% Et0Ac) to afford 6.8 g (65%) of B-3b as a white crystalline solid.
step 3 - To a suspension of thymine (353 mg, mmol) in hexamethyldisilazane
(1.2
mL) was added trimethylsilylchloride (4 drops) and the mixture was heated to
120 C for
1 h. To this solution was added B-3b (300 mg, 0.8 mmol) followed by DCE (5 mL)
and
then iodine (1 chip, catalytic) and the mixture was heated at 80 C overnight.
The
mixture was cooled to RT and then concentrated in vacuo. The residue was
partitioned
between DCM (40 mL) and water (40 mL). The phases were separated and the
aqueous
phase was back extracted with DCM (40 mL). The combined organic extracts were
dried
(MgSO4), filtered and evaporated. The product was purified by Si02
chromatography
eluting with an Et0Ac/hexane gradient (50% to 85% Et0Ac) to afford 0.090 g
(27%) of
1-17 as a white solid.
Example 5
3-[6-Bromo-3-(2,4-dioxo-imidazolidin-1-ylmethyl)-2-fluoro-phenoxy]-5-chloro-
benzonitrile (1-20, SCHEME C)
CA 02687747 2009-11-19
WO 2008/145562 PCT/EP2008/056165
- 35 -
step 1 - Glycine benzyl ester (925 mg, 1.0 equiv) was dissolved in DCE (25 mL)
and A-2h (2 g, 5.6 mmol) and NaBH(OAc)3 (1.66 g, 1.4 equiv) were added
sequentially.
After stirring overnight the reaction mixture was quenched with sat. Na2CO3,
and
extracted with Et20. The organic layers were then washed with brine, dried
over
(MgSO4), concentrated in vacuo. The crude product was purified by Si02
chromatography eluting with an Et0Ac/hexane gradient (20% to 30% Et0Ac) to
afford
1.60 g (57%) of C-la.
step 2 - Trimethylsilylisocyanate (336 ilL, 2.5 equiv) and DMAP (12 mg, 0.10
equiv) were added to a solution of C-la (510 mg, 1.01 mmol) and THF (5 mL).
The
resulting solution was heated at 50 C for 20 h, cooled to RT and concentrated
in vacuo.
The crude product as purified by Si02 chromatography eluting with an
Et0Ac/hexane
gradient (33% to 66% Et0Ac) to afford 0.280 g (51%) of C-lb.
step 3 - NaH (20 mg, 1.1 equiv, 60% in mineral oil) was added to a solution of
C-
lb (250 mg, 0.48 mmol) in DMF (2.5 mL) at 0 C. The reaction mixture was
stirred to
RT for 2 h at which point the reaction mixture was poured into of sat. NH4C1
(20 mL)
and extracted Et0Ac. The organic layers were then washed with brine, dried
(MgSO4)
and concentrated in vacuo to afford 0.190 g (95%) of I-20.
3-[6-Bromo-2-fluoro-34(R)-5-methyl-2,4-dioxo-imidazolidin-1-ylmethyl) -
phenoxy]-5-chloro-benzonitrile (1-21) was prepare analogously except in step
1, glycine
benzyl ester was replaced with (L)-alanine benzyl ester.
Example 6
5-[6-Bromo-3-(2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-2-fluoro-
phenoxy]-isophthalonitrile (1-11)
CA 02687747 2009-11-19
WO 2008/145562 PCT/EP2008/056165
- 36 -
0SiMe2tBu OSiMe2tBu OR
step 2
.1110. SO step 3
--Dr.
0 0 step 5
Br R NC CHO NC
Oi
ha: R = Br
step 1 = 13
111." llb: R=CN ste 4 L...1¨
15a: R= TBDMS
p
15b: R= H
C. R R1 0 CH OH 2 Ar AI 0 CH2Br
Br
step 8 Br step 11 Br
CN
CN
.....1¨ 12a: R = Br 16
step 61 14a: R1= CHO
12b: R = CHO step 9
14b: R1= CH=NH(OH)
step 7 eil- 12c: R = CH2OH step 10 õõ,, 14c: R1= CN
Cresol cyanate - Bromine (100 mL; 1.06 eq) was placed under H20 (350 mL) in a
reactor and coolant was circulated through the jacket. An ice-water bath was
used for
cooling. In a separate vessel, a solution of NaCN (100 g, 1.11 equiv) in H20
(350 mL)
5 was prepared and this was solution added to the bromine/water at a rate
that maintained
the temperature at < 30 C. The resulting slurry of cyanogen bromide is added
to a
solution of o-cresol (209 g, 1.00 equiv.) in toluene (900 mL). The biphasic
mixture is
stirred vigorously and cooled below 10 C. TEA (270 mL, 0.98 equiv.) is added
while
maintaining the temperature at < 10 C. The stirring was suspended and the
aqueous
10 phase withdrawn and replaced with heptane (540 mL). The organic phase
was
sequentially washed with dilute NaOH (1.20 equiv.), water, 2M HC1 (0.4 equiv.)
water,
saturated NaHCO3, and water while the temperature was maintained at < 15 C.
The
heptane solution is dried by brief vacuum distillation (temperature < 35 C)
and tested by
Karl Fischer analysis. The organic phase solution was stored until further
use.
step 1 - A degassed reactor was charged with a THF solution of iso-PrMgC1
(1.14
equiv., 2M solution in THF) and ha (495.2 g, 1.352 mol; CASRN 136386-79-3) was
pumped into the reactor while the temperature below 65 C with a water bath.
After the
exothermicity subsided, the reaction was stirred at RT until metallation was
complete
(Aliquots removed, quenched with dil. H2SO4 and assayed by gas
chromatography). The
resulting solution containing the aryl Grignard reagent was added to a heptane
solution of
cresol cyanate (supra; CASRN 1123-89-3) while maintaining the reaction
temperature
below 10 C. The reaction was monitored by removing aliquots, quenching with
dilute
and H2SO4 and assaying the cresol/cyanate ratio. When the cyanate was consumer
the
reaction mixture was added to a dilute H2SO4 solution (86.5 g H2SO4 and 2.15 L
H20).
CA 02687747 2009-11-19
WO 2008/145562 PCT/EP2008/056165
- 37 -
The aqueous layer was separated and the remaining organic phase was diluted
with
heptane and washed sequentially with ice cold aqueous NaOH (320 g of 50% NaOH
and
1 kg of ice), water, saturated NH4C1 and water. The solution was dried by
azeotropic
distillation and the product was purified by vacuum distillation to afford
395.7 g (93.7%)
llb contaminated with 3-6% 3-(tert-butyl-dimethylsilyloxy)-bromobenzene.
steps 2-4 - A reactor was charged with a solution of llb (36 kg) and
toluene/heptane (65 kg). and the solution was cooled to less then -50 C by
direct
injection of liquid N2 under the surface of the solution. A solution of iso-
propylmagnesium chloride (70 kg, 2.0 M in THF) was added at a rate that the
reaction
temperature was maintained below -20 C (liquid N2 was added as required to
maintain
the desired temperature). The addition required ca. 50 min. A -20 C cooling
solution
was circulated through the vessel jacket and the resulting reaction mixture
was stirred at -
C for at least 1 h. The progress of the metallation was monitored by removing
and
quenching aliquots with dilute H2SO4 and assaying by HPLC. DMF (ca. 30 kg)
cooled to
15 <-10 C and transferred at a rate that maintained the temperature below
0 C during the
transfer step. The reaction was slowly warmed to 20 C and aliquots removed,
quenched
and analyzed by hplc. The reaction was recooled to 0 C and a solution of 8.2
kg H2SO4
and 90 L of H20 was added while maintained the reaction mixture below 10 C.
The
reaction vessel was charged with MTBE (50 kg) an agitated for at least 15 min.
The
20 phases were separated and the aqueous phase was withdrawn from the
vessel. The
remaining organic solution was again washed with H20 (110 L) and the aqueous
phase
discarded.
The reaction vessel was fitted with a condenser cooled to 5 C and a Dean-
Stark
trap which could be switched from reflux to full take-off The vessel was
purged with N2
and p-Ts0H (0.5 kg), ethylene glycol (22 kg) and ethylene glycol diacetate (22
kg) were
added sequentially. The THF and MTBE were removed by distillation (jacket
temperature between 80 and 95 C). After the distillation was complete the
Dean-Stark
trap was set to reflux and the jacket temperature was raised to ca. 100 C and
ethylene
glycol and water removed azeotropically. Additional toluene could be added as
required.
Azeotropic removal of water was continued until less than 1% of the aldehyde
was
detected by HPLC. The reaction mixture was cooled to 25 C and a saturated
solution of
NaHCO3 (25 kg) and water (75 L) were added, the solutions agitated, allowed to
separate
and the aqueous solution withdrawn. The residual organic phase was washed with
H20
CA 02687747 2009-11-19
WO 2008/145562 PCT/EP2008/056165
- 38 -
(100 L). The reaction vessel was fitted for distillation and solvents were
removed,
initially at atmospheric pressure, then under vacuum with the jacket warmed to
60 C.
When only toluene and R-3a remained, the reaction was cooled to 25 C and DME
(70 kg) was added. The solution was cooled to between -10 c and -20 C and a
15%
aqueous NaOH cooled to 10 C was added over about 30 min (maintaining the
reaction
temp at < -10 C). Aliquots of the reaction were removed and when desilylation
was
complete the reaction mixture was diluted with H20 (80 L), cooled to <0 C and
the pH
of the reaction mixture was adjusted to 6-7 with cold 6.0 M H2SO4 (13.2 kg con
H2SO4
and 22 L H20). The mixture was partitioned into MTBE (130 kg). The aqueous
layer
was withdrawn and back extracted MTBE. The combined organic extracts were
washed
with H20, the aqueous layer withdrawn and the volatile solvents distilled
until the
reaction volume was about 50-70 L. The residual organic phase was diluted with
heptane (20 kg) and the resulting precipitated phenol filtered and dried in a
Nutsche filter
to afford 15b.
step 5 - A solution of 15b (6.0 g, 31.38 mmol), K2CO3 (4.76 g, 34.52 mmol) and
DMA (48 mL) was stirred for 5 min. To the solution was added 1,4-dibromo-2,3-
difluoro-benzene (85.33 g, 0.3138 mol) and the solution was heated at 125 C
for 55 min.
HPLC analysis indicated the starting material had been consumed. The reaction
mixture
was diluted with H20 (73 mL), stirred well then the bottom organic layer was
withdrawn.
The organic phase diluted with H20 (900 mL) then the excess dibromo-difluoro-
benzene
was removed by steam distillation. The remaining solution was extracted with
DCM (50
mL) and the organic phase separated and diluted with Me0H (115 mL). The flask
was
fitted for distillation and solvent distilled until thermometer was steady at
65 C for 10
min. The reaction mixture was slowly cooled to 6 C and the resulting solid
filtered and
twice washed with Me0H. The while solid was dried in vacuo to afford 9.7 g of
12a.
step 6 ¨ iso-PrMgC1 (15.6 mL, 1.4 equiv) was added dropwise to a solution of R-
4
(10 g, 22.6 mmol) and toluene (140 mL) cooled to -78 C. The reaction mixture
was
stirred at -78 C for 4 h, warmed briefly to -20 C, and then re-cooled to -78
C. DMF
(3.4 mL) was added to the reaction mixture and the reaction was warmed to RT,
quenched with NH4C1, and extracted with Et0Ac. The crude product was purified
by
Si02 chromatography eluting with 25% Et0Ac/hexanes to afford 5.93 g (68%) of
12b.
step 7 - NaBH4 (1.14 g, 2 equiv) was added to a solution of 12b (5.93 g, 15.1
mmol) in a mixture of THF (25 mL) and Et0H (25 mL). The reaction was stirred
at RT
CA 02687747 2009-11-19
WO 2008/145562 PCT/EP2008/056165
- 39 -
for 2 h, then stored at 0 C overnight. The mixture was quenched with H20,
extracted
with Et0Ac, dried (MgSO4), and concentrated in vacuo. The crude product was
purified
by Si02 chromatography (45% Et0Ac/hexanes) to afford 5.4 g (91%) of 12c as a
clear
oil/foamy solid.
step 8 - An aqueous solution of Ts0H (0.14 gin 6 mL H20, 0.06 equiv) was added
to a solution of 12c (5.4 g, 13.7 mmol) in MeCN (20 mL) and H20 (20 mL). The
mixture was heated to 70 C for 2 h, then stirred at RT overnight. The mixture
was
extracted with Et0Ac, and the combined organic extracts were washed with
NaHCO3,
brine, dried (MgSO4) and concentrated in vacuo to afford 4.1 g (87%) of 14a.
step 9 - Hydroxylamine hydrochloride (2.1 g, 1.05 equiv) was added in three
portions to a solution of NaHCO3 (2.55 g, 1.05 equiv) in H20 (168 mL). A
solution of
14a (10.12 g, 28.9 mmol) in THF (168 mL) was added, and the reaction was
stirred at
RT. When the reaction was complete (ca. 3 h), the mixture was separated, the
aqueous
layer was washed with NH4C1 solution, diluted HC1, and extracted with Et0Ac.
The
combined organic layers were dried (MgSO4), filtered, and concentrated in
vacuo. The
crude product was purified by Si02 chromatography eluting with Et0Ac/hexanes
to
afford 8.62 g (82%) of 14b as an oil that slowly solidified.
step 10 - TFAA (6.5 mL, 2 equiv) was added to a solution of 14b (8.62 g, 24
mmol) in a mixture of pyridine (11.5 mL, 6 equiv) and dioxane (57 mL) cooled
to 0 C.
The reaction mixture was heated to 65 C for several hours, then cooled to RT
and stirred
overnight. The dark yellow mixture was diluted with DCM, and washed with water
and
diluted HC1. The organic layer was dried (MgSO4), filtered and concentrated in
vacuo to
afford a yellow oil that was purified by Si02 chromatography eluting with 40%
Et0Ac/Hexanes to afford a mixture of the alcohol and the corresponding
trifluoroacetate
(5.91 g). This mixture was dissolved in THF, and a H20 solution of LiOH (840
mgs, ca.
1.5 equiv) was added dropwise at 0 C. The mixture was stirred at 0 C for 1
h, quenched
with 1 N HC1, and extracted with Et0Ac. The combined organic layers were dried
(MgSO4), filtered, and concentrated to afford 4.9 g (59%) of 14c as a white
solid slightly
contaminated with the starting ester.
step 11 - A solution of PBr3 (15 mL of a 1.0 M solution in DCM, 1.1 equiv) was
added to a solution of 14c (4.81 g, 13.9 mmol) in DCM (23 mL). The solution
was
stirred at RT for 2 h. The mixture was quenched with NaHCO3, extracted with
DCM,
dried (MgSO4), filtered and concentrated to afford a yellow oil. The product
was
CA 02687747 2009-11-19
WO 2008/145562 PCT/EP2008/056165
- 40 -
purified by Si02 chromatography eluting with 20% Et0Ac/hexanes to afford 1.9 g
of 16
as a white solid.
5-[6-Bromo-3-(2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-2-fluoro-
phenoxy]-isophthalonitrile was prepared from 16 and uracil utilizing the
procedure
described in step 5 of example 1.
Example 7
3- [6-Bromo-2-fluoro-3-(3-methy1-2,5-dioxo-imidazolidin-4-ylmethyl)-phenoxyl -
5-chloro-benzonitrile (1-32)
Boc-N-sarcosine methyl ester (18) was prepared by treating a solution of Boc-
Sar-
OH (1.0 g, 5.3 mmol) in 1:1 DCM/Me0H (25 mL) cooled to 0 C portionwise with
diazomethane (2M solution, excess) until the reaction mixture turned slight
yellow. The
reaction was kept at 0 C for 30 min and then HOAc (2 drops) was added. The
organic
solvents were removed to afford 1.0 g (90%) of 18.
step 1 ¨ A solution of di-isopropylamine (0.14 mL, 1.2 eq) in THF was cooled
to -
78 C and then n-butyllithium (1 equiv) was added. The reaction mixture was
warmed to
0 C then stirred for 15 min. To the resulting solution was added 18 (0.2 g,
1.2 eq) and
the resulting solution stirred for 30 min. The enolate solution was cooled to -
78 C and a
THF solution of D-la (Ar = 3-chloro-5-cyano-phenyl, R4 = Br, 0.3 5g, 0.833
mmol) was
added. The reaction was warmed up to RT and stirred for 4 h. The reaction
mixture was
poured into aqueous NH4C1 and extracted with Et0Ac. The organic layer was
washed
with H20, then dried (Na2SO4) and purified by Si02 chromatography to afford
0.2 g
(44%) of D-lb (Ar = 3-chloro-5-cyano-phenyl, R4 = Br).
step 2 ¨ A solution of D-lb, TFA (1 mL) and DCM was stirred at 0 C for 5h..
The
organic solvents were removed in vacuo and then redissolved in DCM. The
organic
layer was washed with saturated Na2CO3 and then brine. The organic layer was
dried
(Na2SO4) and the solvents evaporated to afford 0.15 g (92%) of D-1c.
step 3 ¨ To a solution of D-lc (0.1g, 0.226mmo1) and DMAP (4mg, 0.15eq) in
THF (1mL) was added TMSNCO (ca. 0.9mL, 2.5 eq) and the reaction mixture was
heated to 50 C for 18h. The volatiles were removed in vacuo and the residue
dissolved
in DCM and Me0H. The organic layer was washed with saturated NaHCO3 and then
brine. The organic layer was dried (Na2SO4) and the organic solvents were
evaporated.
CA 02687747 2009-11-19
WO 2008/145562 PCT/EP2008/056165
- 41 -
The crude product was purified by preparative TLC to afford 0.030 g (30%) of1-
32 (D-2;
Ar = 3-chloro-5-cyano-phenyl, R4 = Br).
Example 8
3- [6-Bromo-2-fluoro-3-(1-methy1-2,4-dioxo-1,2,3,4-tetrahydro-pyrimidin-5-
ylmethyl)-phenoxy]-5-chloro-benzonitrile (1-31, SCHEME D)
step 4 ¨ To a suspension of NaH (0.14g, 3eq) and DMF (3mL) cooled to 0 C was
added tert-butyl ethyl malonate. The reaction mixture was warmed to RT and
stirred for
30 min. To the resulting solution was cooled to 0 C was added a solution of D-
la (Ar =
3-chloro-5-cyano-phenyl, R4 = Br, 0.5g, 1.19mmol) in DMF (2mL). The reaction
was
stirred at RT for 1.5 h then diluted with ethyl acetate and washed
sequentially with
saturated NH4C1, water and brine. The organic layer was dried (Na2SO4),
filtered and
evaporated to afford 0.62 g (99%) of D-1d.
step 5 ¨ To a solution of D-ld and DCM (2.5 mL) cooled to 0 C. was added TFA
(2mL) and the reaction stirred at RT for 6 h. The volatiles were removed in
vacuo and
the residue diluted with DCM and the organic layer washed sequentially with
aqueous
NaHCO3, water, and brine. The aqueous wash was acidified with HC1 and re-
extracted
with DCM. The combined organic layers were washed with water, dried (Na2SO4),
filtered and evaporated. The crude residue was dissolved in DMF (2mL) and H20
(0.07mL) and heated in the microwave for 20 min at 160 C. The reaction
mixture was
diluted with Et0Ac and washed sequentially with water and brine. The organic
layer
was dried (Na2SO4), filtered and evaporated. The crude product was purified by
Si02
chromatography eluting with a Et0Ac/hexane gradient (0-30% Et0Ac) to afford
0.180 g
(38%) of D-le.
step 6 ¨ A solution of D-le (0.18g, 0.42mmol) and ethyl formate (0.07mL, 2.2
eq)
in Et20 (1 mL) was added dropwise to a solution of potassium tert-butoxide in
Et20
(2.5mL). The reaction mixture was warmed to RT and stirred for 18 h. The
volatiles
were removed in vacuo and the residue dissolved in IPA (5mL). Thiourea was
added,
and the reaction mixture was heated to 80 C for 4 h. The volatiles were
removed in
vacuo and the residue was washed with ether. The residue was dissolved in H20,
and the
solution was acidified with HOAc. The resulting precipitate was filtered and
washed
with water. The residue was dissolved in Et20 and the organic layer washed
with water
and brine. The organic solvents were evaporated to afford 0.1 g (50%) of D-3a.
CA 02687747 2009-11-19
WO 2008/145562 PCT/EP2008/056165
- 42 -
step 7 ¨ To a solution of D-3a (0.1g, 0.21mmol) and HOAc was added a 20%
aqueous solution of chloroacetic acid (1mL). The reaction mixture is heated to
100 C
for 6h. The reaction mixture was cooled to 0 C and H20 (2mL) was added and
the
resulting mixture was stirred. The resulting precipitate was filtered and
washed with
water and Et20 to afford 0.030 g (31%) of D-3b.
step 8- N,0-bis-(trimethylsilypacetamide (0.1mL, 6 eq) was added slowly to D-
3b
(30mg, 0.067mmol) in DCM (0.9mL) at RT and stirred for 2h. Methyl iodide
(0.145mL,
35 eq) was added, and the reaction heated to 28 C for 18h. The volatiles were
removed
in vacuo, and the organic residue was dissolved in Et0Ac. The organic layer
was
washed with water and then brine, dried (Na2SO4), filtered and evaporated. The
crude
product was purified by Si02 chromatography eluting with a Me0H/DCM gradient
(0-
4% Me0H) to afford 0.015 g (48%) of D-3c (Ar = 3-chloro-5-cyano-phenyl, R4 =
Br).
Example 9
3- [6-Bromo-2-fluoro-3-(5-methy1-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-
ylmethyl)-phenoxyl-5-difluoromethyl-benzonitrile (1-27)
C. 0 R OHC 0 CH2 OAc Ar0 0 CH2X
0 I. [Os _4. 001 lo
----VW
Br
step 2 Br step 3 Br
CN CN
20 22a: R = OH
step 1 ii.12c: R = CH2OH
step 4 1,..
18: R = CH20Ac 22b: R = Br
Ar = 3-cyano-5-difluoromethyl-phenyl
step 1 - Acetic anhydride (0.93 g, 1.5 equiv) was added to a solution of the
12c (2.4
g, 6.1 mmol) and TEA (0.93 g, 1.5 equiv) in MeCN (10 mL). The solution was
stirred at
RT for 1 h, diluted with Et0Ac, washed with NaHCO3 solution, dried (Na2SO4),
filtered
and concentrated to afford 2.2 g (82%) of 18 as a clear oil.
step 2 - A solution of p-Ts0H (60 mg, .06 equiv) in H20 (6 mL) was added to a
solution of the 18 (2.2 g, 5.0 mmol) in MeCN (8 mL). The resulting solution
was heated
at 70 C for 5 h. The solution was then cooled to RT, diluted with Et0Ac, and
washed
with saturated NaHCO3 solution and brine. The organic layer was dried (Na2SO4)
and
concentrated. The crude product was purified by Si02 chromatography eluting
with
Et0Ac/hexane to afford 1.3 g (62%) of 20 as a clear oil.
CA 02687747 2009-11-19
WO 2008/145562 PCT/EP2008/056165
- 43 -
step 3 - To a solution of the 20 (0.12 g, 0.3 mmol) in DCM (1 mL) cooled to 0
C
was added a drop of Et0H followed by DAST (0.92 g, 1.2 equiv). The solution
was
warmed to RT and left at this temperature overnight. The mixture was then
carefully
poured onto ice. Saturated NaHCO3 was added, and the mixture was extracted
with
DCM, dried (Na2504), filtered and concentrated. This product was dissolved in
THF (10
mL) and a of 2M LiOH (1.75 mL) in H20 was added and the mixture was stirred
for 3 h.
The reaction was quenched with 1N HC1, extracted with Et0Ac, dried (Na2504),
filtered
and concentrated. Purification of the residue by 5i02 chromatography afforded
0.074 g
(67%) of 22a.
step 4 - The benzyl alcohol 22a can be converted to the corresponding benzyl
bromide 22b as described in step 4 of Example 1.
step 5 - 1-27 was prepared from 22b as described in step 5 of example 1 except
uracil was replaced by thymine
Example 10
HIV-1 Reverse Transcriptase Assay
RNA-dependent DNA polymerase activity was measured using a biotinylated
primer oligonucleotide and tritiated dNTP substrate. Newly synthesized DNA was
quantified by capturing the biotinylated primer molecules on streptavidin
coated
Scintillation Proximity Assay (SPA) beads (Amersham). The sequences of the
polymerase assay substrate were: 18nt DNA primer, 5'-Biotin/GTC CCT GTT CGG
GCG CCA-3'; 47nt RNA template, 5'-GGG UCU CUC UGG UUA GAC CAC UCU
AGC AGU GGC GCC CGA ACA GGG AC-3'. The biotinylated DNA primer was
obtained from the Integrated DNA Technologies Inc. and the RNA template was
synthesized by Dharmacon. The DNA polymerase assay (final volume 50 1)
contained
32 nM biotinylated DNA primer, 64 nM RNA substrate, dGTP, dCTP, dTTP (each at
5
M), 103 nM [3F1]-dATP (specific activity = 29 Ci/mmol), in 45 mM Tris-HC1, pH
8.0,
45 mM NaCl, 2.7 mM Mg(CH3C00)2, 0.045% Triton X-100 w/v, 0.9 mM EDTA. The
reations contained Sul of serial compound dilutions in 100% DMSO for IC50
determination and the final concentrations of DMSO were 10%. Reactions were
initiated by the addition of 30 1 of the HIV-RT enzyme (final concentrations
of 1-3 nM).
Protein concentrations were adjusted to provide linear product formation for
at least 30
min of incubation. After incubation at 30 C for 30 min, the reaction was
quenched by
CA 02687747 2009-11-19
WO 2008/145562 PCT/EP2008/056165
- 44 -
addition of 50 1 of 200 mM EDTA (pH 8.0) and 2 mg/mL SA-PVT SPA beads
(Amersham, RPNQ0009, reconstituted in 20 mM Tris-HC1, pH 8.0, 100 mM EDTA and
1% BSA). The beads were left to settle overnight and the SPA signals were
counted in a
96-well top counter-NXT (Packard). ICsovalues were obtained by sigmoidal
regression
analysis using GraphPad and are tabulated in TABLE I.
Example 11
Antiviral assay method:
Anti-HIV antiviral activity was assessed using an adaptation of the method of
Pauwels et al. {Pauwels et al., 1988, J Virol Methods 20:309-321}. The method
is based
on the ability of compounds to protect HIV-infected T lymphoblastoid cells
(MT4 cells)
from cell-death mediated by the infection. The endpoint of the assay was
calculated as
the concentration of compound at which the cell viability of the culture was
preserved by
50% (50% inhibitory concentration', IC50). The cell viability of a culture was
determined by the uptake of soluble, yellow 344,5-dimethylthiazol-2-y1]-2,5-
diphenyltetrazolium bromide (MTT) and its reduction to a purple insoluble
formazan
salt. After solubilization, spectrophotometric methods were employed to
measure the
amount of formazan product.
MT4 cells were prepared to be in logarithmic-phase growth and a total of 2 x
106
cells infected with the HXB2-strain of HIV at a multiplicity of 0.0001
infectious units of
virus per cell in a total volume of between 200-500 microliters. The cells
were incubated
with virus for one hour at 37 C before removal of virus. The cells are then
washed in
0.01 M phosphate buffered saline, pH 7.2 before being resuspensed in culture
medium
for incubation in culture with serial dilutions of test compound. The culture
medium
used was RPMI 1640 without phenol red, supplemented with penicillin,
streptomycin, L-
glutamine and 10% fetal calf serum (GM10).
Test compounds were prepared as 2 mM solutions in dimethyl sulfoxide (DMSO).
Four replicate, serial 2-fold dilutions in GM10 were then prepared and 50
microliters
amounts placed in 96-well plates over a final nanomolar concentration range of
625 ¨
1.22. Fifty microliters GM10 and 3.5 x 104 infected cells were then added to
each well.
Control cultures containing no cells (blank), uninfected cells (100%
viability; 4
replicates) and infected cells without compound (total virus-mediated cell
death; 4
CA 02687747 2009-11-19
WO 2008/145562 PCT/EP2008/056165
- 45 -
replicates) were also prepared. The cultures were then incubated at 37 C in a
humidified
atmosphere of 5% CO2 in air for 5 days.
A fresh solution of 5 mg/mL MTT was prepared in 0.01 M phosphate buffered
saline, pH 7.2 and 20 microliters added to each culture. The cultures were
further
incubated as before for 2 hours. They were then mixed by pipetting up and down
and
170 microliters of Triton X-100 in acidified isopropanol (10% v/v Triton X-100
in 1:250
mixture of concentrated HC1 in isopropanol). When the formazan deposit was
fully
solubilized by further mixing, the absorbance (OD) of the cultures was
measured at
540nm and 690nm wavelength (690 nm readings were used as blanks for artifacts
between wells). The percent protection for each treated culture was then
calculated from
the equation:
(OD drug treated cultures) - (OD untreated virus control cultures)
% Protection ¨ __________________________________________________ x 100%
(OD uninfected cultures) - (OD untreated virus control cultures)
The IC50 can be obtained from graph plots of percent protection versus logi0
drug
concentration.
In both assays, compounds of formulas I range in activity from an IC50 of
about 0.5
to about 10000 nM or 0.5 to about 5000 nM, with preferred compounds having a
range of
activity from about 0.5 to about 750 nM, more preferably about 0.5 to 300 nM,
and most
preferably about 0.5 to 50 nM.
TABLE II
Compound Antiviral Assay
IC50 (1-11\4)
1-2 0.0031
1-20 0.0042
Example 12
Pharmaceutical compositions of the subject Compounds for administration via
several routes were prepared as described in this Example.
Composition for Oral Administration (A)
Ingredient % wt./wt.
Active ingredient 20.0%
Lactose 79.5%
Magnesium stearate 0.5%
CA 02687747 2009-11-19
WO 2008/145562 PCT/EP2008/056165
- 46 -
The ingredients are mixed and dispensed into capsules containing about 100 mg
each; one capsule would approximate a total daily dosage.
Composition for Oral Administration (B)
Ingredient A) wt./wt.
Active ingredient 20.0%
Magnesium stearate 0.5%
Crosscarmellose sodium 2.0%
Lactose 76.5%
PVP (polyvinylpyrrolidine) 1.0%
The ingredients are combined and granulated using a solvent such as methanol.
The formulation is then dried and formed into tablets (containing about 20 mg
of active
compound) with an appropriate tablet machine.
Composition for Oral Administration (C)
Ingredient A) wt./wt.
Active compound 1.0 g
Fumaric acid 0.5 g
Sodium chloride 2.0 g
Methyl paraben 0.15 g
Propyl paraben 0.05 g
Granulated sugar 25.5 g
Sorbitol (70% solution) 12.85 g
Veegum K (Vanderbilt Co.) 1.0 g
Flavoring 0.035 mL
Colorings 0.5 mg
Distilled water q.s. to 100 mL
The ingredients are mixed to form a suspension for oral administration.
The features disclosed in the foregoing description, or the following claims,
expressed in their specific forms or in terms of a means for performing the
disclosed
function, or a method or process for attaining the disclosed result, as
appropriate, may,
separately, or in any combination of such features, be utilized for realizing
the invention
in diverse forms thereof.
CA 02687747 2015-02-18
WO 2008/145562 PCT/EP2008/056165
- 47 -
The foregoing invention has been described in some detail by way of
illustration
and example, for purposes of clarity and understanding. It will be obvious to
one of skill
in the art that changes and modifications may be practiced within the scope of
the
appended claims. Therefore, it is to be understood that the above description
is intended
to be illustrative and not restrictive. The scope of the invention should,
therefore, be
determined not with reference to the above description, but should instead be
determined
with reference to the following appended claims, along with the full scope of
equivalents
to which such claims are entitled.
Any conflict between any reference cited herein and the specific teachings of
this specifications shall be resolved in favor of the latter. Likewise, any
conflict
between an art-understood definition of a word or phrase and a definition of
the word
or phrase as specifically taught in this specification shall be resolved in
favor of the
latter.