Note: Descriptions are shown in the official language in which they were submitted.
CA 02692748 2010-01-06
WO 2009/009025 PCT/US2008/008326
MIXED MICELLES INCLUDING AMPHIPATHIC CONJUGATES
OF RNA AGENTS, AND USES THEREOF
RELATED APPLICATION
This application claims benefit of priority to U.S. Provisional Application
No. 60/948,433, filed July 6, 2007.
FIELD OF THE INVENTION
The invention relates to the field of pharmaceutical sciences, and
particularly
formulations employing mixed micelles, and specifically the formulation of
mixed micelles
1o including amphipathic conjugates of RNA agents, such as siRNAs, and uses
thereof.
BACKGROUND OF THE INVENTION
Many diseases (e.g., cancers, hematopoietic disorders, endocrine disorders,
and immune
disorders) arise from the abnormal expression or activity of a particular gene
or group of genes.
Similarly, disease can result through expression of a mutant form of protein,
as well as from
expression of viral genes that have been integrated into the genome of their
host. The
therapeutic benefits of being able to selectively silence these abnormal or
foreign genes are
obvious.
Oligonucleotide compounds have important therapeutic applications in medicine.
Oligonucleotides can be used to silence genes that are responsible for a
particular disease. Gene-
silencing prevents formation of a protein by inhibiting translation.
Importantly, gene-silencing
agents are a promising alternative to traditional small, organic compounds
that inhibit the
function of the protein linked to the disease. Antisense RNA, micro-RNA and
small interfering
RNA (siRNA) are different types of oligonucleotides that prevent the formation
of corresponding
proteins by gene-silencing.
Antisense methodology is the complementary hybridization of relatively short
oligonucleotides (e.g., 13-30 nucleotides) to mRNA or DNA such that the
normal, essential
t
CA 02692748 2010-01-06
WO 2009/009025 PCT/US2008/008326
functions, such as protein synthesis, of these intracellular nucleic acids are
disrupted. The
hybridization is by sequence-specific hydrogen bonding via Watson-Crick base
pairs of
oligonucleotides to RNA or single-stranded DNA, and interferes with the
transcription and/or
translation of the target nucleic acid sequence.
Micro-RNAs are a large group of small RNAs produced naturally in organisms, at
least
some of which regulate the expression of target genes. Micro-RNAs are formed
from an
approximately 70 nucleotide single-stranded hairpin precursor transcript by
the ribonuclease
Dicer (Ambros et al. (2003), Current Biology 13(10):807-818), which cleaves
the precursor to
form 21-23 nucleotide double-stranded micro-RNAs. In many instances, the micro-
RNA is
transcribed from a portion of the DNA sequence that previously had no known
function. Micro-
RNAs are not translated into proteins but, rather, they bind to specific
messenger RNAs blocking
translation. It is thought that micro-RNAs base-pair imprecisely with their
targets to inhibit
translation.
RNA interference or "RNAi" is a term initially coined by Fire and co-workers
to describe
the observation that double-stranded RNA (dsRNA) can block gene expression
when it is
introduced into worms (Fire et al. (1998), Nature 391:806-811). Short dsRNA
directs gene-
specific, post-transcriptional silencing in many organisms, including
vertebrates, and has
provided a new tool for studying gene function. RNAi is mediated by an RNA-
induced silencing
complex (RISC), a sequence-specific, multicomponent nuclease that destroys
messenger RNAs
2o homologous to the silencing trigger.
Treatment with dsRNA has become an important method for analyzing gene
functions in
invertebrate organisms. For example, Dzitoveva et al. showed that RNAi can be
induced in adult
fruit flies by injecting dsRNA into the abdomen of anesthetized Drosophila,
and that this method
can also target genes expressed in the central nervous system (Dzitoveva et
al. (2001), Mol.
Psychiatry 6(6):665-670). Both transgenes and endogenous genes were
successfully silenced in
adult Drosophila by intra-abdominal injection of their respective dsRNA.
Moreover, Elbashir et
al. provided evidence that the direction of dsRNA processing determines
whether sense or
antisense target RNA can be cleaved by an siRNA-protein complex (Elbashir et
al. (2001),
Genes Dev. 15 (2):188-200).
Since the first report of the phenomenon of RNA interference (RNAi) in 1998
(Fire et al.
(1998), Nature 391:806-811), there has been a wave of interest in the
development of diagnostic
2
CA 02692748 2010-01-06
WO 2009/009025 PCT/US2008/008326
and therapeutic strategies based on the use of synthetic siRNAs, since those
are clearly seen as a
potential new class of pharmaceutical drugs (Bumcrot et al. (2006), Nat. Chem.
Biol. 2:711-719).
Several RNAi-based in vivo strategies have been reported in the literature for
the treatment of a
wide range of conditions from viral infections (Giladi et al. (2003), Mol.
Ther. 8:769-776; Song
et al. (2003), Nat. Med. 9:347-351) to cancer (Duxbury et al. (2003), Biochem.
Biophys. Res.
Commun. 311:786-792; Duxbury et al. (2004), Oncogene 23:1448-1456) and even
neurological
conditions (Dorn et al. (2004), Nucleic Acids Res. 32:e49; Thakker et al.
(2005), Mol. Psychiatry
10:782-789, 714).
Naked siRNA has been administered intravenously (Zender et al. (2003), Proc.
Natl.
lo Acad. Sci. USA 100:7797-7802), intrathecally (Dorn et al. (2004), Nucleic
Acids Res. 32:e49),
intraperitoneally (Filleur et al. (2003), Cancer Res. 63:3919-3922) and, where
applicable, by
direct injection into the target tissue (Zender et al. (2003), Proc. Natl.
Acad. Sci. USA 100:7797-
7802; Aharinejad et al. (2004), Cancer Res. 64:5378-5384; Lingor et al.
(2005), Brain 128:550-
558; Pille et al. (2005), Mol. Ther. 11:267-274). Appreciable levels of
activity, especially in
liver tissue in the case of hydrodynamic IV injection, have been reported
(Giladi et al. (2003),
Mol. Ther. 8:769-776; Song et al. (2003), Nat. Med. 9:347-351; Zender et al.
(2003), Proc. Natl.
Acad. Sci. USA 100:7797-7802; Bradley et al. (2005), Pancreas 31:373-379;
Hamar et al.
(2004), Proc. Natl. Acad. Sci. USA 101:14883-14888; Hino et al. (2006),
Biochem. Biophys. Res.
Commun. 340:263-267; Tompkins et al. (2004), Proc. Natl. Acad. Sci. USA
101:8682-8686). A
general progress towards in vivo use of siRNAs has been recently reviewed in
(Behlke (2006),
Mol. Ther. 13:644-670), and new RNAi-based therapeutics are actively
developing (Liu et al.
(2007), Histol. Histopathol. 22:211-217). Unfortunately, the applicability of
many of the
alternative routes mentioned is limited to only a few tissues and, in the case
of direct injection,
further limited to easily accessible tissues (Aigner (2006), J. Biomed.
Biotechnol.
2006(4):71659). Further, high doses and repeated administration are often
necessary for activity.
While naked siRNA approaches continue to be pursued for clinical application,
there is
now a growing acceptance that a major hurdle now facing the application of
siRNA based
strategies to practical clinical therapy is the need for efficient delivery of
siRNA to the site of
intended action (Aigner (2006), J. Biomed. Biotechnol. 2006(4):71659).
Although chemical
modification of siRNA molecules (including their modification with cholesteryl
residues) to
achieve better stability and confer tissue-specific targeting has attracted
some attention
3
CA 02692748 2010-01-06
WO 2009/009025 PCT/US2008/008326
(Soutschek et al. (2004), Nature 432:173-178; Manoharan (2004), Curr. Opin.
Chem. Biol.
8:570-579; Morrissey et al. (2005), Nat. Biotechnol. 23:1002-1007), the major
focus has been on
the use of carrier systems to improve the delivery of siRNA. As was noted by
Bumcort et al.
"Effective delivery is the most challenging remaining consideration for
successful translation of
RNAi to the clinic" (Bumcort et al. (2006), Nat. Chem. Biol. 2:711).
Approaches towards improved siRNA delivery have understandably borrowed much
from the already established field of DNA delivery, with both viral and non-
viral based strategies
being explored. The drawbacks of viral approaches already identified from DNA
based therapy,
including immune responses and possible oncogenesis, are potentially more
limiting in the
1o delivery of siRNA because of the short duration of action and the need for
repeated
administration (Bartlett et al. (2006). Nucleic Acids Res. 34:322-333). Non-
viral systems are
comparatively more flexible in design and are easier to formulate. The
potential for repeated
administration is greater due to improved biocompatibility, and non-viral
systems can be used to
deliver both DNA coding for siRNA products or the siRNA duplexes themselves.
Polymer-based siRNA delivery systems, particularly polyethyleneimine(PEI)-
based ones,
have been actively investigated (Aigner (2006), J. Biomed. Biotechnol.
2006(4):71659; Putnam
et al. (2006), Crit. Rev. Ther. Drug Carrier Syst. 23:137-164), and cationic
polymers,
particularly PEI, have been used with some success to improve the efficacy of
siRNA activity in
vivo (Ge et al. (2004), Proc. Natl. Acad. Sci. USA 101:8676-8681; Leng et al.
(2005), Cancer
2o Gene Ther. 12:682-690; Urban-Klein et al. (2005), Gene Ther. 12:461-466;
Yin et al. (2005), J.
Mol. Cell. Cardiol. 39:681-689), although the toxicity of such carriers
remains an issue.
Polymeric nanoparticles have also been considered as potential siRNA carriers
(Toub et al.
(2006), Pharmacother. 60:607-620). Nanoparticle carrier systems based on
polymer or metal
nanoparticles are also being explored for siRNA delivery (Schiffelers et al.
(2004), Nucleic Acids
Res. 32:e149; Derfus et al. (2007), Bioconjug. Chem. 18(5):1391-6).
Lipidic carriers of siRNA have naturally drawn a lot of attention (Spagnou et
al. (2004),
Biochemistry (Mosc). 43:13348-13356). Liposomes appear to be the most widely
explored of
the non-viral systems for siRNA therapy (Aigner (2006), J. Biomed. Biotechnol.
2006(4):71659).
Liposome formulations of siRNA have been used to improve specific gene knock-
down in the
treatment of solid tumors (Landen et al. (2005), Cancer Res. 65:6910-6918),
metastatic cancer
(Yano et al. (2004), Clin. Cancer Res. 10:7721-7726) and a variety of other
conditions in animal
4
CA 02692748 2010-01-06
WO 2009/009025 , - PCT/US2008/008326
models (Nogawa et al. (2005), J. Clin. Invest. 115:978-985). Our own
experience with the
Iiposome-based siRNA delivery systems involving cell-penetrating peptide-
mediated
intracellular delivery (Zhang et al. (2006), J. Control. Release 112:229-239),
although resulting
in some success, was associated with reproducibility problems as well as with
issues relating to
preparation and scalability. First attempts to prepare ligand-targeted
nanocarrier-based siRNA
delivery systems have also. been described (Ikeda et al. (2006), Pharm. Res.
23:1631-1640).
However, none of the strategies tested to date has resulted in clinically
significant systems.
siRNA has been shown to be extremely effective as a potential anti-viral
therapeutic with
numerous published examples appearing recently. siRNA molecules directed
against targets in
lo the viral genome dramatically reduce viral titers by orders of magnitude in
animal models of
influenza (Ge et al. (2004), Proc. Natl. Acad. Sci. USA 101:8676-8681;
Tompkins et al. (2004),
Proc. Natl. Acad. Sci. USA 101:8682-8686; Thomas et al. (2005), Expert Opin.
Biol. Ther.
5:495-505 (2005)), respiratory syncytial virus (RSV) (Bitko et al. (2005),
Nat. Med. 11:50-55),
hepatitis B virus (HBV) (Morrissey et al. (2005), Nat. Biotechnol. 23:1002-
1007), hepatitis C
virus (Kapadia (2003), Proc. Natl. Acad. Sci. USA 100:2014-2018; Wilson et al.
(2003), Proc.
Natl. Acad. Sci. USA 100:2783-2788) and SARS coronavirus (Li et al. (2005),
Nat. Med. 11:944-
951). In addition, research is currently underway to develop interference RNA
therapeutic
agents for the treatment of many diseases including central-nervous-system
diseases,
inflammatory diseases, metabolic disorders, oncology, infectious diseases, and
ocular disease.
Despite advances in antisense RNA, micro-RNA and siRNA, technologies, one of
the
major hurdles is the intracellular delivery of these oligonucleotides into the
cells. In particular,
there is substantial nuclease activity in both the extracellular and
intracellular environments
which rapidly degrades RNA administered in vivo. As a result, only a very
small proportion of
the RNA which is administered to a subject reaches the desired target in
cells. Although a wide
variety of delivery strategies have been investigated in the art (see, e.g.,
Gilmore et al. (2006),
Current Drug Delivery 3:147-145), many important issues for making active and
clinically
acceptable preparations of antisense RNA, micro-RNA and siRNA remain
unresolved, and there
remains a need for a simple, universal, and easily made delivery system for
RNA-based
therapeutic agents.
5
CA 02692748 2010-01-06
WO 2009/009025 PCT/US2008/008326
SUMMARY OF THE INVENTION
The present invention depends, in part, upon the recognition that certain
mixed micelles
can be produced, which include both amphipathic conjugates of RNA moieties and
micelle-
forming amphipathic molecules having radially extending hydrophilic moieties,
such that the
radially extending hydrophilic moieties form a layer which surrounds or masks
at least a portion
of the RNA moieties, and thereby increases the in vivo stability of siRNA by
sterically hindering
and decreasing the digestion of the RNA moieties by nucleases. In some
embodiments, the
invention provides mixed micelles in which a hydrophilic layer has sufficient
thickness and
density to substantially surround or mask the RNA moieties and thereby
substantially protect
1o them from digestion by nucleases. As a result, the invention provides
improved methods for
delivering RNA to therapeutic targets in cells, with substantially decreased
digestion by
extracellular nucleases, and substantially increased RNA half-life in the
blood, cerebrospinal
fluid or local extracellular microenvironment. In addition, the surface of
such mixed micelles
can be modified with moieties that target the iRNA-containing micelles to
desired target cells
and/or provide enhanced intracellular penetration.
Thus, in one aspect, the invention provides pharmaceutical formulations of an
iRNA
agent including (1) a mixed micelle including (a) a plurality of amphipathic
iRNA conjugates,
each of which includes a hydrophilic moiety and a hydrophobic moiety, and each
of which
hydrophilic moieties includes an iRNA moiety of the iRNA agent; (b) a
plurality of micelle-
forming amphipathic molecules, each of which includes a hydrophilic moiety and
a hydrophobic
moiety, and each of which hydrophilic moieties includes at least one
hydrophilic chain which
extends radially from the center of the mixed micelle; and (2) a
pharmaceutically acceptable
carrier.
In some embodiments of the pharmaceutical formulation, the plurality of
hydrophilic
chains forms a hydrophilic layer extending radially from the center of said
mixed micelle; and
the amphipathic iRNA conjugates are positioned in the mixed micelle such that
at least a portion
of the surface of the iRNA moiety is within the hydrophilic layer.
In some embodiments of the pharmaceutical formulation, no portion of the
surface of the
iRNA moiety extends radially outward beyond the hydrophilic layer.
In some embodiments of the pharmaceutical formulation, the plurality of
hydrophilic
chains have an average backbone length between 200 and 500 A. In other
embodiments, the
6
CA 02692748 2010-01-06
WO 2009/009025 PCT/US2008/008326
plurality of hydrophilic chains have an average backbone length between 100-
1,000 A, and
between 50-2,000 A.
In some embodiments of the pharmaceutical formulation, the plurality of
hydrophilic
chains have an average molecular weight between 1,500 and 5,000 g/mole. In
other
embodiments, the plurality of hydrophilic chains have an average molecular
weight between 500
and 15,000, and between 1,000-10,000 g/mole.
In some embodiments of any of the foregoing, the hydrophobic moieties of the
micelle-
forming amphipathic molecules are selected from radicals of a long-chain fatty
acid, a
phospholipid, a lipid, and a glycolipid.
In some embodiments of any of the foregoing, the hydrophobic moieties of the
amphipathic iRNA conjugates are selected from radicals of a cholesterol, a
long chain fatty acid,
a lipid, a phospholipid, and a glycolipid.
In some of the foregoing embodiments, the fatty acid is selected from butanoic
acid,
hexanoic acid, octanoic acid, decanoic acid, dodecanoic acid, tetradecanoic
acid, hexadecanoic
acid, octadecanoic acid, eicosanoic acid, docosanoic acid, tetracosanoic acid
and unsaturated
congeners thereof.
In some of the foregoing embodiments, the phospholipid is selected from
phosphatidyl
ethanolamine (PE), phosphatidyl choline (PC), phosphatidyl glycerol (PG),
phosphatidyl inositol
(PI), phosphatidyl serine(PS), phosphatidic acid (PA), and sphingomyelin.
In some of the foregoing embodiments, the glycolipid is selected from a
galactolipid, a
sulfolipid, a cerebroside, and a ganglioside.
In some of embodiments of any of the foregoing, the hydrophilic moieties of
the micelle-
forming amphipathic molecules are selected from radicals of a PEG, a PEI, a
polyvinylpyrrolidone, a polyacrylamide, a polyvinyl alcohol, a polyoxazolines,
a
polymorpholines, a chitosan, and a water-soluble peptide.
In some of the foregoing embodiments, the hydrophilic moieties are PEG
radicals having
an average molecular weight between 1,500 and 5,000 g/mole. In other
embodiments, the PEG
radicals have an average molecular weight between 500 and 15,000, and between
1,000-10,000
g/mole.
In another aspect, the invention provides methods of increasing the delivery
of an iRNA
agent to an intracellular target by formulating the iRNA agent in one of the
pharmaceutical
7
CA 02692748 2010-01-06
WO 2009/009025 PCT/US2008/008326
formulations described herein, and administering the pharmaceutical
formulation extracellularly,
such that delivery of the iRNA agent to the intracellular target is increased
relative to delivery of
the iRNA agent in the pharmaceutically acceptable carrier.
In another aspect, the invention provides methods of decreasing extracellular
nuclease
degradation of an iRNA agent by formulating the iRNA agent in one of the
pharmaceutical
formulations described herein, and administering the pharmaceutical
formulation extracellularly,
such that extracellular nuclease degradation of the iRNA agent in the
pharmaceutical formulation
is decreased relative to degradation of the iRNA agent in the pharmaceutically
acceptable carrier.
The details of one or more embodiments of the invention are set forth in the
io accompanying drawings and the description below. Other features, objects,
and advantages of
the invention will be apparent from the description and drawings, and from the
claims.
BRIEF DESCRIPTION OF FIGURES
FIGURE 1 is a graph showing the size distribution of siRNA-Chol/PEG-PE
micelles
made in accordance with the invention.
FIGURE 2 is epifluorescence micrographs of MCF7 (human breast adenocarcinoma)
cells stained with Hoechst33342 nuclear stain. FIGURES 2A and 2B: Cells
exposed to Cy3-
siRNA-Chol/PEG2-PE micelles for 2 hours. FIGURES 2C and 2D: Untreated cells.
FIGURES
2A and 2C: UV Channel. FIGURES 2B and 2D: Red channel.
FIGURE 3 is a graph showing cell-associated Cy3 fluorescence measured at
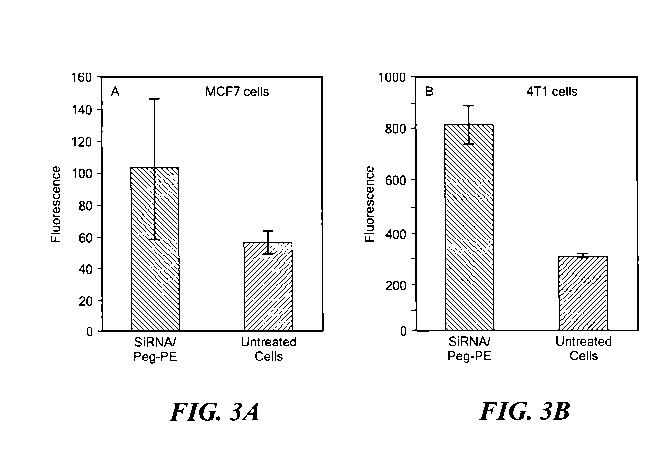
Ex554,
Em570 with MCF7 cells (A) and 4T1 cells (B) after the incubation for 2 h with
Cy3-siRNA-
Chol/PEG-PE micelles; n=3.
FIGURE 4 is a graph showing decrease of GFP production after incubation of C
166-GFP
mouse endothelial cells with Col-SiRNA and Chol-SiRNA-PEG-PE micelles (same
siRNA
quantity).
FIGURE 5 illustrates schemes for siRNA-Cholesterol synthesis. FIG. 5A:
preparation of
mercaptyl-cholesterol. FIG 513: conjugation of inercaptyl cholesterol and
siRNA via SPDP.
FIGURE 6 illustrates schemes for Cy3-siRNA-Cholesterol synthesis.
8
CA 02692748 2010-01-06
WO 2009/009025 PCT/US2008/008326
DETAILED DESCRIPTION
1. Introduction
The present invention depends, in part, upon the recognition that certain
mixed micelles
can be produced, including amphipathic conjugates of RNAs and micelle-forming
amphipathic
molecules having radially extending hydrophilic moieties, such that the
radially extending
hydrophilic moieties form a layer which surrounds or masks at least a portion
of the RNA
moieties and thereby decreases digestion of the RNA moieties by nucleases. In
some
embodiments, the invention provides mixed micelles in which a hydrophilic
layer has sufficient
thickness and density to substantially surround or mask the RNA moieties and
thereby
1o substantially protect them from digestion by nucleases. As a result, the
invention provides
improved methods for delivering RNA to therapeutic targets in cells, with
substantially
decreased digestion by extracellular nucleases, and substantially increased
RNA half-life in the
blood, cerebrospinal fluid, or local extracellular microenvironment.
2. References and Definitions
The patent and scientific literature referred to herein establishes knowledge
that is
available to those of skill in the art. The issued U.S. patents, allowed
applications, published
foreign applications, and references, that are cited herein are hereby
incorporated by reference to
the same extent as if each was specifically and individually indicated to be
incorporated by
2o reference.
As used herein, the term "RNA agent" means an unmodified ribonucleic acid
(RNA),
modified RNA, or nucleoside surrogate.
As used herein with respect to RNA agents, the term "modified" means a
molecule that
differs in chemical structure from a naturally-occurring ribonucleic acid by
one or more of the
following changes: addition, subtraction or substitution of one or more atoms
in the
phosphodiester backbone (e.g., replacement of a phosphodiester linkage with a
phosphorothioate
or peptide nucleic acid linkage); addition, subtraction or substitution of one
or more atoms in the
ribose unit (e.g., replacement of a ribose unit with a deoxyribose unit); and
addition subtraction
or substitution of one or more atoms in the nucleoside base unit (e.g.,
replacement of a base unit
with a base analog, as described herein). While referred to as "modified RNA,"
this term may
include molecules which are not technically RNAs (e.g., because ribose has
been replaced).
9
CA 02692748 2010-01-06
WO 2009/009025 PCT/US2008/008326
As used herein, the term "nucleoside surrogate" means a molecule which differs
from a
naturally-occurring ribonucleic acid in that the ribophosphate backbone is
replaced with a non-
ribophosphate construct that allows the bases to the presented in the correct
spatial relationship
such that hybridization is substantially similar to what is seen with a
ribophosphate backbone
(e.g., non-charged mimics of a ribophosphate backbone).
As used herein, the term "iRNA agent" means an RNA agent which can, or which
can be
cleaved into an RNA agent which can, down regulate the expression of a target
gene, preferably
an endogenous or pathogen target RNA. iRNA agents include antisense RNA, micro-
RNA and
siRNA. While not wishing to be bound by theory, an iRNA agent may act by one
or more of a
1o number of mechanisms, including post-transcriptional cleavage of target
mRNA (sometimes
referred to in the art as RNAi), or pre-transcriptional or pre-translational
mechanisms. An iRNA
agent can include a single strand or can include more than one strand (e.g., a
double-stranded
iRNA agent). If the iRNA agent is a single strand, it is particularly
preferred that it include a 5'
modification which includes one or more phosphate groups or one or more
analogs of a
phosphate group.
As used herein, the term "amphipathic" means comprised of both hydrophilic and
hydrophobic moieties, and is synonymous with "amphiphilic." Amphipathic
molecules are
capable of forming micelles when dispersed in aqueous solutions at an
appropriate temperature
and concentration.
As used herein, the term "critical micelle concentration" and the abbreviation
"CMC"
mean the concentration of an amphipathic compound at which micelles begin to
spontaneously
form in an aqueous solution. A "low" critical micelle concentration is less
than 10 5 M.
As used herein, the term "molecular backbone length" refers to the sum of the
bond
lengths between the atoms constituting the longest continuous chain of atoms
in a molecule,
irrespective of bond angles. Thus, _merely as an example, the molecular
backbone length of
isobutane is the sum of the bond lengths of three C-C bonds and the two C-H
bonds (i.e., 3 x
1.43 A + 2 x 1.09 A). Because this calculation ignores both bond angles and
the fact that
molecules can assume secondary structures (e.g., globular structures), it over-
estimates the
physical length of the molecules in situ. Nonetheless, it is a useful
approximation for comparing
the relative sizes of siRNA agents and hydrophilic chains for purposes of the
invention.
CA 02692748 2010-01-06
WO 2009/009025 PCT/US2008/008326
As used herein, the term "decrease" means to decrease by at least 5% from a
reference
amount, as determined by a method and sample size that achieves statistical
significance (i.e.,
p <0.1).
As used herein, the term "increase" means to increase by at least 5% from a
reference
amount, as determined by a method and sample size that achieves statistical
significance.
As used herein, the recitation of a numerical range for a variable is intended
to convey
that the invention may be practiced with the variable equal to any of the
values within that range.
Thus, for a variable which is inherently discrete, the variable can be equal
to any integer value
within the numerical range, including the end-points of the range. Similarly,
for a variable which
is inherently continuous, the variable can be equal to any real value within
the numerical range,
including the end-points of the range. As an example, and without limitation,
a variable which is
described as having values between 0 and 2 can take the values 0, 1 or 2 if
the variable is
inherently discrete, and can take the values 0.0, 0.1, 0.01, 0.001, . . . ,
0.9, 0.99, 0.999, or any
other real values > 0 and <_ 2, if the variable is inherently continuous.
As used herein, unless specifically indicated otherwise, the word "or" is used
in the
inclusive sense of "and/or" and not the exclusive sense of "either/or."
3. Micelles Including Amphipathic Coniugates of iRNA Agents
Micelles are aggregates of amphipathic molecules dispersed in an aqueous
medium
forming what is called a colloidal solution. Mixed micelles are micelles
comprising more than
one type of amphipathic molecule. Commonly, micelles are formed from fatty
acids (e.g.,
sodium stearate) and phospholipids (e.g., phosphatidyl ethanolamine). In
aqueous solution, a
micelle forms with the hydrophilic "head" regions at the exterior surface in
contact with the
surrounding solvent, and the hydrophobic "tail" regions buried in the interior
of the micelle,
isolated from the solvent. Micelles are generally spherical in shape but,
because they are fluid,
are deformable when subjected to hydrodynamic forces. Other shapes, such as
ellipsoids and
cylinders are also possible, depending upon the molecular geometry of its
constituent
molecule(s) and conditions such as amphipathic molecule concentration(s),
temperature, pH, and
ionic strength.
The present invention depends, in part, upon the recognition that mixed
micelles can be
produced that include iRNA moieties as part of an amphipathic conjugate, and
in which the other
11
CA 02692748 2010-01-06
WO 2009/009025 PCT/US2008/008326
amphipathic molecules forming the micelle have radially extending hydrophilic
moieties that
form a hydrophilic layer, and such that the iRNA moieties are positioned in
said mixed micelle
with at least a portion of their surfaces within the hydrophilic layer. This
positioning of the
iRNA moieties within the hydrophilic layer physically shields them from other
molecules, and
thereby decreases degradation of the iRNA moieties by nucleases in the local
microenvironment
of the micelle.
In order for the iRNA moieties to be positioned within, or at least partially
within, the
hydrophilic layer, the hydrophilic moieties or head regions of the micelle-
forming amphipathic
molecules must extend radially outward a distance sufficient to form a
hydrophilic layer which
can at least partially surround or mask the iRNA moiety. Thus, simple,
unmodified fatty acids
(e.g., stearic acid, palmitic acid), which have only carboxyl groups as
hydrophilic moieties, are
not useful in the invention. Similarly, most simple, unmodified phospholipids
(e.g., phosphatidyl
ethanolamine (PE), phosphatidyl choline (PC), phosphatidyl serine (PS)), which
have small
hydrophilic moieties, are not useful in the invention. Rather, useful micelle-
forming
amphipathic molecules include at least one hydrophilic chain which extends
radially from the
center of said mixed micelle. These hydrophilic chains can align themselves
radially to form a
hydrophilic layer.
Examples of hydrophilic chains include hydrophilic polymers, including but not
limited
to the following: polyethylene glycol (PEG), polyethyleneimine (PEI),
polyvinylpyrrolidone,
polyacrylamide, polyvinyl alcohol, polyoxazolines, polymorpholines, chitosan,
and water-
soluble peptides. These hydrophilic polymers can be of varying lengths,
determined by the
number of repeating polymer units, and can be described by the number of
repeating units (e.g.,
PEG having 44 ethylene glycol units), the molecular weight in grams/mole
(e.g., PEG having a
molecular weight of 2000 grams/mole), or by the length of the molecular
backbone (e.g., PEG
having a molecular backbone of 200 A).
Using standard chemistries well known in the art, micelle-forming molecules
with small
head regions can be modified by the addition of hydrophilic chains to produce
micelle-forming
amphipathic molecules useful in the invention. For example, the head regions
of fatty acids or
phospholipids can be covalently modified by esterification or other covalent
reactions with
3o hydrophilic chains. Thus, for example, hydrophilic chains such as PEG can
form esters with
12
CA 02692748 2010-01-06
WO 2009/009025 PCT/US2008/008326
fatty acids such as stearic acid to form amphipathic molecules such as stearyl-
PEG, or with
phospholipids such as PE to form amphipathic molecules such as PE-PEG.
As noted above, in order to be useful in the invention, the hydrophilic chains
must extend
radially outward a sufficient distance to at least partially surround or mask
the iRNA moiety.
Therefore, the required or desired length of the hydrophilic chains is a
function of the size of the
iRNA agent. Thus, if an iRNA agent extends radially outward a greater distance
from the
micelle center, it is desirable to form the mixed micelle using a micelle-
forming amphipathic
molecule with hydrophilic chains which also extend outward a greater distance.
For both siRNAs and micro-RNAs, which comprise double-stranded, 21-23 bp iRNA
lo moieties, the molecular backbone length (measured along the ribophosphate
backbone) is
approximately 192-210 A (i.e., 9.15 A per nucleotide times 21-23 nucleotides).
If such an iRNA
moiety is conjugated to a hydrophobic moiety by one of its 5' or 3' termini,
the opposite end of
the iRNA moiety can be expected to project radially outward from the
hydrophobic core of the
mixed micelle by approximately 192-210 A. If the iRNA moiety is attached to
the hydrophobic
moiety of the iRNA conjugate by a linker of any length, the iRNA moiety may
project even
further from the hydrophobic core of the mixed micelle. Therefore, for an iRNA
moiety which is
an siRNA or micro-RNA moiety, the hydrophilic layer must, at a minimum, be
approximately
192-210 A in molecular backbone length in order to surround or mask the iRNA
moiety on all
sides. If a linker causes the iRNA moiety to project even further, a
commensurately longer
2o hydrophilic chain must be used. However, because RNA agents are subject to
digestion from
their ends by exonucleases, it may be desirable for the hydrophilic layer to
extend further, such
that the ends of the hydrophilic chains extend beyond the iRNA moiety, and the
ends of the
iRNA moiety are fully masked or buried within the hydrophilic layer. Thus, for
example,
hydrophilic chains having molecular backbone lengths of 200-2,000 A may be
employed in the
invention. On the other hand, shorter hydrophilic chains may be employed
(e.g., 50-200 A) if
the iRNA moiety adopts a coiled configuration which shortens its effective
length, if partially
surrounding the iRNA moiety is adequate to decrease digestion by nucleases, or
if only partial
protection from nucleases is required.
Antisense RNAs, which typically comprise single-stranded, 13-30 nucleotide
iRNA
moieties, the molecular backbone length (measured along the ribophosphate
backbone) is
approximately 119-275 A (i.e., 9.15 A per nucleotide times 13-30 nucleotides).
Therefore, for an
13
CA 02692748 2010-01-06
WO 2009/009025 PCT/US2008/008326
iRNA moiety which is an antisense RNA moiety, depending upon the length of the
particular
antisense RNA, the hydrophilic layer must, at a minimum, be approximately 119-
275 A in
molecular backbone length in order to surround or mask the iRNA moiety on all
sides. As
before, the presence of linkers between the iRNA moieties and the hydrophobic
moieties of the
iRNA conjugate may require longer chains in the hydrophilic layer, and longer
chains (e.g., 300-
2,000 A) or shorter chains (e.g., 50-200 A) may be desired for the reasons set
forth above.
Thus, in one aspect, the invention provides pharmaceutical formulations of an
iRNA
agent including (1) a mixed micelle including (a) a plurality of amphipathic
iRNA conjugates,
each of which includes a hydrophilic moiety and a hydrophobic moiety, and each
of which
1o hydrophilic moieties includes an iRNA moiety of the iRNA agent; (b) a
plurality of micelle-
forming amphipathic molecules, each of which includes a hydrophilic moiety and
a hydrophobic
moiety, and each of which hydrophilic moieties includes at least one
hydrophilic chain which
extends radially from the center of the mixed micelle; and (2) a
pharmaceutically acceptable
carrier.
In some embodiments of the pharmaceutical formulation, the plurality of
hydrophilic
chains forms a hydrophilic layer extending radially from the center of said
mixed micelle; and
the amphipathic iRNA conjugates are positioned in the mixed micelle such that
at least a portion
of the surface of the iRNA moiety is within the hydrophilic layer.
In some embodiments of the pharmaceutical formulation, no portion of the
surface of the
iRNA moiety extends radially outward beyond the hydrophilic layer.
In another aspect, the invention provides methods of increasing the delivery
of an iRNA
agent to an intracellular target by formulating the iRNA agent in one of the
pharmaceutical
formulations described herein, and administering the pharmaceutical
formulation extracellularly,
such that delivery of the iRNA agent to the intracellular target is increased
relative to delivery of
the iRNA agent in the pharmaceutically acceptable carrier. In some
embodiments, the average
amount of iRNA delivered to the intracellular target is increased by at least
10%, in some
embodiments it is increased 10%-50%, in some embodiments it is increased 50%-
100%, and in
some embodiments it is increased greater than 2x.
In another aspect, the invention provides methods of decreasing extracellular
nuclease
3o degradation of an iRNA agent by formulating the iRNA agent in one of the
pharmaceutical
formulations described herein, and administering the pharmaceutical
formulation extracellularly,
14
CA 02692748 2010-01-06
WO 2009/009025 PCT/US2008/008326
such that extracellular nuclease degradation of the iRNA agent in the
phannaceutical formulation
is decreased relative to degradation of the iRNA agent in the pharmaceutically
acceptable carrier.
In some embodiments, the amount of iRNA degraded by nuclease within the first
hour of
administration is decreased by at least 10%, in some embodiments it is
decreased 10%-50%, in
some embodiments it is decreased 50%-100%, and in some embodiments it is
decreased greater
than 2x.
4. Hydrophobic and Hydrophilic Moieties
A broad variety of monomers may be used to build polymeric hydrophobic cores
as well
lo as polymeric hydrophlic chains for use in the invention. See, for example,
Torchilin (2007),
"Micellar nanocarriers: pharmaceutical perspectives," Pharm Res. 24(1):1-16.
For example, and without limitation, propylene oxide, aspartic acid, (3-
benzoyl-L-
aspartate, y-benzyl-L-glutamate, caprolactone, D,L-lactic acid, spermine, and
many others can be
used to form hydrophobic moieties using standard chemistries. Some of these
monomers form
hydrophobic polymeric blocks that can be used to produce the hydrophobic core
of a micelle,
while other compounds (e.g., lysine, spermine) form hydrophilic polymeric
blocks which can
bind oppositely charged hydrophobic substances to form a hydrophobic
electrostatic complex,
and the complex can form the core of a micelle. Block copolymers of poly(ortho
esters) and
PEG form 40-70 nm micelles with a CMC of around 10-4 g/l and can be
lyophilized. Micelle-
forming ABC-type triblock copolymers composed of monomethoxy-PEG, poly(2-
(dimethylamino)ethyl methacrylate) and poly(2-(diethylamino)ethyl
methacrylate), with the last
component forming a hydrophobic c6re, can also be used and allow for the slow
release of
poorly soluble compounds which can be incorporated into the core. Polylactone-
PEG double
and triple block copolymers can be used as micelle-forming polymers, as well
as poly(2-ethyl-2-
oxazoline-block-poly(epsilon-caprolactone), which forms 20 nm micelles.
Chitosan grafted with
hydrophobic groups, such as palmitoyl, can also be used to prepare
pharmaceutical micelles, and
is highly biocompatible. Other materials which can be to prepare
pharmaceutical micelles
include copolymers of PEG and macromolecules, such as scorpion-like polymers
and some star-
like and core-shell constructs.
Phospholipid residues can also be successfully used as hydrophobic core-
forming
moieties. The use of lipid moieties as hydrophobic blocks capping hydrophilic -
polymer chains
CA 02692748 2010-01-06
WO 2009/009025 PCT/US2008/008326
(e.g., PEG) can provide additional advantages for particle stability when
compared with
conventional amphiphilic polymer micelles due to the existence of two
extremely hydrophobic
fatty acid acyls, which can contribute considerably to an increase in the
hydrophobic interactions
between the polymeric chains in the micelle's core. A variety of conjugates of
lipids with water-
soluble polymers are commercially available, or can be easily synthesized.
Diacyl-lipid-PEG
conjugates have been introduced into the area of controlled drug delivery as
polymeric surface-
modifiers for liposomes. However, the diacyl-lipid-PEG molecule itself
represents a
characteristic amphiphilic polymer with a bulky hydrophilic (PEG) portion and
a very short but
very hydrophobic diacyl-lipid moiety. Similar to other PEG-containing
amphiphilic block-
1o copolymers, diacyl-lipid-PEG conjugates form micelles in an aqueous
environment. A series of
PEG-phosphatidylethanolamine (PEG-PE) conjugates have been synthesized using
PE and N-
hydroxysuccinimide esters of methoxy-PEG succinates (molecular weight of 2
kDa, 5 kDa and
12 kDa). All versions of PEG-PE conjugates form micelles with the size of 7 to
35 nm. No
dissociation into individual polymeric chains was found following the
chromatography of the
serially diluted samples of PEG(5 kDa)-PE up to a polymer concentration of
approximately I
g/ml, which corresponds to a micromolar CMC value, which is at least 100-fold
lower than
those of conventional detergents. With the size of PEG blocks going above 15
kDa, the stability
of PEG-PE micelles begins to decrease. Preparation of lipid-based micelles by
a detergent or
water-miscible solvent removal method results in formation of particles with
very similar
2o diameters. Usually, such micelles have a spherical shape and uniform size
distribution. Another
important issue is that PEG2000-PE and PEG5000-PE micelles retain the size
characteristic for
micelles even after 48 h incubation in the blood plasma, i.e. the integrity of
PEG-PE micelles
should not be immediately affected by components of biological fluids upon
parenteral
administration.
Amphiphilic PVP-lipid conjugates with PVP block size between 1,500 and 8,000
Da
have also been prepared, which easily form micelles in an aqueous environment.
CMC values
and the size of micelles formed depend on the length of the PVP block and vary
between 104
and 10-6 M and 5 and 20 nm, respectively. Micelles prepared from a similar
lipidated polymer,
polyvinyl alcohol substituted with oleic acid, can also be used.
In some embodiments, the amphipathic micelle-forming molecule is a PEG-PE co-
polymer. Polyethylene glycol (PEG) refers to an oligomer or polymer of
ethylene oxide.
16
CA 02692748 2010-01-06
WO 2009/009025 PCT/US2008/008326
Phosphatidyl ethanolamine (PE) is frequently the main lipid component of
microbial membranes.
It is a neutral or Zwitterionic phospholipid (at least in the pH range 2 to
7). In animal tissues,
phosphatidyl ethanolamine tends to exist in diacyl, alkylacyl and alkenylacyl
forms. In general,
animal phosphatidyl ethanolamine tend to contain higher proportions of
arachidonic and
docosahexaenoic acids than the other Zwitterionic phospholipid, phosphatidyl
choline. These
polyunsaturated components are concentrated in position sn-2 with saturated
fatty acids most
abundant in position sn-1.
As noted above, in some embodiments of the pharmaceutical formulation, the
plurality of
hydrophilic chains forms a hydrophilic layer extending radially from the
center of said mixed
micelle; and the amphipathic iRNA conjugates are positioned in the mixed
micelle such that at
least a portion of the surface of the iRNA moiety is within the hydrophilic
layer. In other
embodiments, no portion of the surface of the iRNA moiety extends radially
outward beyond the
hydrophilic layer.
In some embodiments of the pharmaceutical formulation, the plurality of
hydrophilic
chains have an average backbone length between 200 and 500 A. In other
embodiments, the
plurality of hydrophilic chains have an average backbone length between 100-
1,000 A, and
between 50-2,000 A.
In some embodiments of the pharmaceutical formulation, the plurality of
hydrophilic
chains have an average molecular weight between 1,500 and 5,000. In other
embodiments, the
plurality of hydrophilic chains have an average molecular weight between 500
and 15,000, and
between 1,000-10,000.
In some embodiments of any of the foregoing, the hydrophobic moieties of the
micelle-
forming amphipathic molecules are selected from radicals of a long-chain fatty
acid, a
phospholipid, a lipid, and a glycolipid.
In some embodiments of any of the foregoing, the hydrophobic moieties of the
amphipathic iRNA conjugates are selected from radicals of a cholesterol, a
long chain fatty acid,
a lipid, a phospholipid, and a glycolipid.
In some of the foregoing embodiments, the fatty acid is selected from butanoic
acid,
hexanoic acid, octanoic acid, decanoic acid, dodecanoic acid, tetradecanoic
acid, hexadecanoic
3o acid, octadecanoic acid, eicosanoic acid, docosanoic acid, tetracosanoic
acid and unsaturated
congeners thereof.
17
CA 02692748 2010-01-06
WO 2009/009025 PCT/US2008/008326
In some of the foregoing embodiments, the phospholipid is selected from
phosphatidyl
ethanolamine (PE), phosphatidyl choline (PC), phosphatidylglycerol (PG),
phosphatidyl inositol
(PI), phosphatidyl serine(PS), phosphatidic acid (PA), and sphingomyelin.
In some of the foregoing embodiments, the glycolipid is selected from a
galactolipid, a
sulfolipid, a cerebroside, and a ganglioside.
In some of embodiments of any of the foregoing, the hydrophilic moieties of
the micelle-
forming amphipathic molecules are selected from radicals of a PEG, a PEI, a
polyvinylpyrrolidone, a polyacrylamide, a polyvinyl alcohol, a polyoxazolines,
a
polymorpholines, a chitosan, and a water-soluble peptide.
In some of the foregoing embodiments, the hydrophilic moieties are PEG
radicals having
an average molecular weight between 1,500 and 5,000 g/mole. In other
embodiments, the PEG
radicals have an average molecular weight between 500 and 15,000, and between
1,000-10,000.
In some embodiments the siRNA is conjugated to a lipophilic moiety such as
cholesterol,
another lipid or phospholipid.
In some preferred embodiments the delivery system is a mixed micelle
comprising PEG-
PE and siRNA-lipid conjugate.
5. Double-Stranded iRNA Agents and Moieties
Double-stranded RNA (dsRNA), including siRNA and micro-RNA, directs the
sequence-
specific silencing of mRNA through a process known as RNA interference (RNAi).
The process
occurs in a wide variety of organisms, including mammals and other
vertebrates.
It has been demonstrated that 21-23 nt fragments of dsRNA are sequence-
specific
mediators of RNA silencing, e.g., by causing RNA degradation. While not
wishing to be bound
by theory, it may be that a molecular signal, which may be merely the specific
length of the
fragments, present in these 21-23 nt fragments recruits cellular factors that
mediate RNAi.
Complementarity, or degree of homology with the target strand, is most
critical in the
antisense strand. While perfect complementarity is often desired, particularly
in the antisense
strand, some embodiments can include, particularly in the antisense strand,
one or more but
preferably 6, 5, 4, 3, 2, or fewer mismatches (with respect to the target
RNA). The mismatches,
particularly in the antisense strand, are most tolerated in the terminal
regions and if present are
preferably in a terminal region or regions, e.g., within 6, 5, 4, or 3
nucleotides of the 5' and/or 3'
18
CA 02692748 2010-01-06
WO 2009/009025 PCT/US2008/008326
terminus. The sense strand need only be sufficiently complementary with the
antisense strand to
maintain the over all double strand character of the molecule.
As discussed elsewhere herein, an iRNA agent will often be modified. Single
stranded
regions of an iRNA agent will often be modified or include nucleoside
surrogates, e.g., the
unpaired region or regions of a hairpin structure, e.g., a region which links
two complementary
regions, can have modifications or nucleoside surrogates. Modification to
stabilize one or more
3'- or 5'-terminus of an iRNA agent, e.g., against exonucleases, or to favor
the antisense RNA
agent to enter into RISC are also favored. Modifications can include C3 (or
C6, C7, C 12) amino
linkers, thiol linkers, carboxyl linkers, non-nucleotidic spacers (C3, C6, C9,
C12, abasic,
lo triethylene glycol, hexaethylene glycol), special biotin or fluorescein
reagents that come as
phosphoramidites and that have another DMT-protected hydroxyl group, allowing
multiple
couplings during RNA synthesis.
Exemplary modifications include the following:
(a) backbone modifications, e.g., modification of a backbone P, including
replacement of
P with S, or P substituted with alkyl or allyl, e.g., Me, and dithioates (S-
P=S); these
modifications can be used to promote nuclease resistance;
(b) 2'-O alkyl, e.g., 2'-OMe, 3'-O alkyl, e.g., 3'-OMe (at terminal and/or
internal
positions); these modifications can be used to promote nuclease resistance or
to enhance binding
of the sense to the antisense strand, the 3' modifications can be used at the
5' end of the sense
strand to avoid sense strand activation by RISC;
(c) 2'-5' linkages (with 2'-H, 2'-OH and 2'-OMe and with P=0 or P=S) these
modifications can be used to promote nuclease resistance or to inhibit binding
of the sense to the
antisense strand, or can be used at the 5' end of the sense strand to avoid
sense strand activation
by RISC;
(d) L sugars (e.g., L ribose, L-arabinose with 2'-H, 2'-OH and 2'-OMe); these
modifications can be used to promote nuclease resistance or to inhibit binding
of the sense to the
antisense strand, or can be used at the 5' end of the sense strand to avoid
sense strand activation
by RISC;
(e) modified sugars (e.g., locked nucleic acids (LNA's), hexose nucleic acids
(HNA's)
3o and cyclohexene nucleic acids (CeNA's)); these modifications can be used to
promote nuclease
19
CA 02692748 2010-01-06
WO 2009/009025 PCT/US2008/008326
resistance or to inhibit binding of the sense to the antisense strand, or can
be used at the 5' end of
the sense strand to avoid sense strand activation by RISC;
(f) nucleobase modifications (e.g., C-5 modified pyrimidines, N-2 modified
purines, N-7
modified purines, N-6 modified purines), these modifications can be used to
promote nuclease
resistance or to enhance binding of the sense to the antisense strand;
(g) cationic groups and Zwitterionic groups (preferably at a terminus), these
modifications can be used to promote nuclease resistance;
(h) conjugate groups (preferably at terminal positions), e.g., naproxen,
biotin, cholesterol,
ibuprofen, folic acid, peptides, and carbohydrates; these modifications can be
used to promote
1o nuclease resistance or to target the molecule, or can be used at the 5' end
of the sense strand to
avoid sense strand activation by RISC.
Multiple asymmetric modifications can be introduced into either or both of the
sense and
antisense sequence. A sequence can have at least 2, 4, 6, 8, or more
modifications and all or
substantially all of the monomers of a sequence can be modified
6. Ligand-Conjugates, Carriers and Targeting
In order to target the pharmaceutical compositions of the invention to
specific cell types,
to increase the half-life of the pharmaceutical compositions (e.g., in the
general circulation, or in
cerebrospinal or lymphatic spaces), to increase the bioavailability of the
pharmaceutical
compositions, and/or to increase the cell-penetration/uptake of the
pharmaceutical compositions,
the pharmaceutical compositions can further comprise a ligand, carrier or
targeting moiety which
is (a) bound to the iRNA agent, (b) bound to a separate hydrophobic moiety or
amphipathic
moiety and included in the mixed micelle, and/or (c) bound to the hydrophilic
chains of the
hydrophilic layer. In some preferred embodiments, the ligand, carrier or
targeting moiety is
bound to the hydrophilic chains of the hydrophilic layer, and preferably the
ends of the
hydrophilic chains, so that it is exposed to the solvent or extracellular
microenvironment, and is
accessible. For example, the distal tips of the hydrophilic chains may be
activated by various
known chemistries, including without limitation p-nitrophenyl (pNP) carbonyl
groups which
may be used with PEG and other soluble block-forming polymers.
Thus, an iRNA agent, amphipathic moiety and/or hydrophilic chain can
optionally
contain a ligand-conjugated monomer subunit. The carrier (also referred to in
some
CA 02692748 2010-01-06
WO 2009/009025 PCT/US2008/008326
embodiments as a "linker") can be a cyclic or acyclic moiety and includes two
"backbone
attachment points" (e.g., hydroxyl groups) and a ligand. The ligand can be
directly attached
(e.g., conjugated) to the carrier or indirectly attached (e.g., conjugated) to
the carrier by an
intervening tether (e.g., an acyclic chain of one or more atoms; or a
nucleobase, e.g., a naturally
occurring nucleobase optionally having one or more chemical modifications,
e.g., an unusual
base; or a universal base). In one embodiment, the carrier is hydroxyprolinol.
When bound to the iRNA agent, a ligand-conjugated monomer subunit may be the
5' or
3' terminal subunit of the iRNA molecule. Alternatively, the ligand-conjugated
monomer subunit
may occupy an internal position. More than one ligand-conjugated monomer
subunit may be
lo present in an iRNA agent. Preferred positions for inclusion of a tethered
ligand-conjugated
monomer subunits, e.g., one in which a lipophilic moiety, e.g., cholesterol,
is tethered to the
carrier are at the 3' terminus, the 5' terminus, or an internal position of
the sense strand.
In some embodiments, a ligand alters the distribution, targeting or lifetime
of an iRNA
agent into which it is incorporated. In some embodiments a ligand provides an
enhanced affinity
for a selected target, e.g., molecule, cell or cell type, compartment, e.g., a
cellular or organ
compartment, tissue, organ or region of the body, as, e.g., compared to a
species absent such a
ligand.
Ligands can improve transport, hybridization, and specificity properties and
may also
improve nuclease resistance of the resultant natural or modified
oligoribonucleotide, or a
polymeric molecule comprising any combination of monomers described herein
and/or natural or
modified ribonucleotides.
Ligands in general can include therapeutic modifiers, e.g., for enhancing
uptake;
diagnostic compounds or reporter groups e.g., for monitoring distribution;
cross-linking agents;
nuclease-resistance conferring moieties; and natural or unusual nucleobases.
General examples
include lipophiles, lipids, steroids (e.g., uvaol, hecigenin, diosgenin),
terpenes (e.g., triterpenes,
e.g., sarsasapogenin, Friedelin, epifriedelanol derivatized lithocholic acid),
vitamins (e.g., folic
acid, vitamin A, biotin, pyridoxal), carbohydrates, proteins, protein binding
agents, integrin
targeting molecules, polycationics, peptides, polyamines, and peptide mimics.
Ligands can include a naturally occurring substance, (e.g., human serum
albumin (HSA),
low-density lipoprotein (LDL), or globulin); carbohydrate (e.g., a dextran,
pullulan, chitin,
chitosan, inulin, cyclodextrin or hyaluronic acid); amino acid, or a lipid.
The ligand may also be
21
CA 02692748 2010-01-06
WO 2009/009025 PCT/US2008/008326
a recombinant or synthetic molecule, such as a synthetic polymer, e.g., a
synthetic polyamino
acid. Examples of such polymers include polyamino acid, such as polylysine
(PLL),
poly L-aspartic acid, poly L-glutamic acid, and styrene-maleic acid anhydride
copolymer,
poly(L-lactide-co-glycolide) copolymer, divinyl ether-maleic anhydride
copolymer, N-(2-
hydroxypropyl)methacrylamide copolymer (HMPA), polyethylene glycol (PEG),
polyvinyl
alcohol (PVA), polyurethane, poly(2-ethylacryllic acid), N-isopropylacrylamide
polymers, or
polyphosphazine. Example of polyamines include: polyethylenimine, polylysine
(PLL),
spermine, spennidine, polyamine, pseudopeptide-polyamine, peptidomimetic
polyamine,
dendrimer polyamine, arginine, amidine, protamine, cationic moieties, e.g.,
cationic lipid,
lo cationic porphyrin, quatemary salt of a polyamine, or an alpha helical
peptide.
Ligands can also include targeting groups, e.g., a cell or tissue targeting
agent, e.g., a
lectin, glycoprotein, lipid or protein, e.g., an antibody, that binds to a
specified cell type such as a
kidney cell. A targeting group can be a thyrotropin, melanotropin, lectin,
glycoprotein, surfactant
protein A, Mucin carbohydrate, multivalent lactose, multivalent galactose, N-
acetyl-
galactosamine, N-acetyl-glucosamine multivalent mannose, multivalent fucose,
glycosylated
polyamino acids, multivalent galactose, transferrin, bisphosphonate,
polyglutamate,
polyaspartate, a lipid, cholesterol, a steroid, bile acid, folate, vitamin
B12, biotin, or an RGD
peptide or RGD peptide mimetic.
Other examples of ligands include dyes, intercalating agents (e.g.,
acridines), cross-
linkers (e.g., psoralene, mitomycin C), porphyrins (TPPC4, texaphyrin,
sapphyrin), polycyclic
aromatic hydrocarbons (e.g., phenazine, dihydrophenazine), artificial
endonucleases (e.g.,
EDTA), lipophilic molecules, e.g., cholesterol, cholic acid, adamantane acetic
acid, 1-pyrene
butyric acid, dihydrotestosterone, glycerol (e.g., esters and ethers thereof,
e.g., Clo, C>>, C12, C13,
C14, C15, C16, C17, C18, C19, or C20 alkyl; e.g., 1,3-bis-
O(hexadecyl)glycerol, 1,3-bis-
O(octadecyl)glycerol), geranyloxyhexyl group, hexadecylglycerol, borneol,
menthol, 1,3-
propanediol, heptadecyl group, palmitic acid, myristic acid, O3-
(oleoyl)lithocholic acid, 03-
(oleoyl)cholenic acid, dimethoxytrityl, or phenoxazine)and peptide conjugates
(e.g.,
antennapedia peptide, Tat peptide), alkylating agents, phosphate, amino,
mercapto, PEG (e.g.,
PEG-40K), MPEG, [MPEG]2, polyamino, alkyl, substituted alkyl, radiolabeled
markers,
3o enzymes, haptens (e.g., biotin), transporC/absorption facilitators (e.g.,
aspirin, vitamin E, folic
acid), synthetic ribonucleases (e.g., imidazole, bisimidazole, histamine,
imidazole clusters,
22
CA 02692748 2010-01-06
WO 2009/009025 PCT/US2008/008326
acridine-imidazole conjugates, Eu3+ complexes of tetra-azamacrocycles),
dinitrophenyl, HRP, or
AP.
For example, dyes can be conjugated to the pharmaceutical formulations of the
invention
to act as imaging agents. Such imaging agents are useful for detecting the
uptake of the iRNA
agents of the invention by targeted cells, and for studying the
pharmacokinetics of the
pharmaceutical formulations of the invention. Such imaging agents can be
produced by, for
example, conjugating a chelating group to the hydrophilic chains of the
hydrophilic layer, and
chelating a heavy metal (e.g., 111 In, 99Tc, Gd, Mn, Fe) to the chelating
group for gamma or NMR
imaging. As noted herein, fluorescent dyes (e.g., Cy3) can also be conjugated
to the
lo pharmaceutical formulations. Thus, in another aspect, the invention
provides methods and
products for detecting or imaging the uptake, localization, and/or
pharmacokinetics of the mixed
micelles of the invention.
Ligands can be proteins, e.g., glycoproteins, or peptides, e.g., molecules
having a specific
affinity for a co-ligand, or antibodies, e.g., an antibody, that binds to a
specified cell type such as
a cancer cell, endothelial cell, or bone cell. Ligands may also include
hormones and hormone
receptors. They can also include non-peptidic species, such as lipids,
lectins, carbohydrates,
vitamins, cofactors, multivalent lactose, multivalent galactose, N-acetyl-
galactosamine, N-acetyl-
glucosamine multivalent mannose, or multivalent fucose. The ligand can be, for
example, a
lipopolysaccharide, an activator of p38 MAP kinase, or an activator of NF-xB.
The ligand can be a substance, e.g., a drug, which can increase the uptake of
the iRNA
agent into the cell, for example, by disrupting the cell's cytoskeleton, e.g.,
by disrupting the cell's
microtubules, microfilaments, and/or intermediate filaments. The drug can be,
for example,
taxon, vincristine, vinblastine, cytochalasin, nocodazole, japlakinolide,
latrunculin A, phalloidin,
swinholide A, indanocine, or myoservin.
The ligand can increase the uptake of the iRNA agent into the cell by
activating an
inflammatory response, for example. Exemplary ligands that would have such an
effect include
tumor necrosis factor alpha (TNFalpha), interleukin-I beta, or gamma
interferon.
In one aspect, the ligand is a lipid or lipid-based molecule. Such a lipid or
lipid-based
molecule preferably binds a serum protein, e.g., human serum albumin (HSA). An
HSA binding
ligand allows for distribution of the conjugate to a target tissue, e.g., a
non-kidney target tissue of
the body. For example, the target tissue can be the liver, including
parenchymal cells of the liver.
23
CA 02692748 2010-01-06
WO 2009/009025 PCT/US2008/008326
Other molecules that can bind HSA can also be used as ligands. For example,
neproxin or
aspirin can be used. A lipid or lipid-based ligand can (a) increase resistance
to degradation of the
conjugate, (b) increase targeting or transport into a target cell or cell
membrane, and/or (c) can be
used to adjust binding to a serum protein, e.g., HSA.
In another aspect, the ligand is a moiety, e.g., a vitamin, which is taken up
by a target cell,
e.g., a proliferating cell. These are particularly useful for treating
disorders characterized by
unwanted cell proliferation, e.g., of the malignant or non-malignant type,
e.g., cancer cells.
Exemplary vitamins include vitamin A, E, and K. Other exemplary vitamins
include B vitamin,
e.g., folic acid, B12, riboflavin, biotin, pyridoxal or other vitamins or
nutrients taken up by
lo cancer cells. Also included are HSA and low density lipoprotein (LDL).
In another aspect, the ligand is a cell-permeation or cell-penetration agent.
For example,
Torchilin (2008), Adv Drug Deliv Rev. 60(4-5):548-58, discloses Tat peptide-
mediated
intracellular delivery of pharmaceutical nanocarriers. In some embodiments,
the agent is
amphipathic. Exemplary agents include a peptides such as TAT, antennopedia,
penetratin, or
arginine oligomers (e.g., R8 or R9). If the agent is a peptide, it can be
modified, such as a
peptidylmimetic or an invertomer, or include non-peptide or pseudo-peptide
linkages, or the use
of D-amino acids. A helical agent can be an alpha-helical agent, which in some
embodiments
can have a lipophilic and a lipophobic phase.
Peptides that target markers enriched in proliferating cells can be used. For
example,
2o RGD containing peptides and peptidomimetics can target cancer cells, in
particular cells that
exhibit an I0,93 integrin. Thus, one could use RGD peptides, cyclic peptides
containing RGD,
RGD peptides that include D-amino acids, as well as synthetic RGD mimics. In
addition to
RGD, one can use other moieties that target the I, 93 integrin ligand.
Generally, such ligands
can be used to control proliferating cells and angiogenesis. Conjugates of
this type include a
pharmaceutical composition that targets PECAM-1, VEGF, or a cancer gene, e.g.,
a cancer gene
described herein.
A targeting agent that incorporates a sugar, e.g., galactose and/or analogues
thereof, can
also be useful. These agents target, in particular, the parenchymal cells of
the liver. For
example, a targeting moiety can include more than one or preferably two or
three galactose
moieties, spaced about 15 A from each other. The targeting moiety can
alternatively be lactose
(e.g., three lactose moieties), which is glucose coupled to a galactose. The
targeting moiety can
24
CA 02692748 2010-01-06
WO 2009/009025 PCT/US2008/008326
also be N-Acetyl-Galactosamine, N-Ac-Glucosamine. A mannose or mannose-6-
phosphate
targeting moiety can be used for macrophage targeting.
The ligand can be a peptide or peptidomimetic. A peptidomimetic (also referred
to as an
oligopeptidomimetic) is a molecule capable of folding into a defined three-
dimensional structure
similar to a natural peptide. The attachment of peptide and peptidomimetics to
the
pharmaceutical compositions of the invention can affect pharmacokinetic
distribution of the
composition, such as by enhancing cellular recognition and absorption. The
peptide or
peptidomimetic moiety can be about 5-50 amino acids long, e.g., about 5, 10,
15, 20, 25, 30, 35,
40, 45, or 50 amino acids long. A peptide or peptidomimetic can be, for
example, a cell
1o permeation peptide, cationic peptide, amphipathic peptide, or hydrophobic
peptide (e.g.,
consisting primarily of Tyr, Trp or Phe). The peptide moiety can be a
dendrimer peptide,
constrained peptide or crosslinked peptide.
An RGD peptide moiety can be used to target a tumor cell, such as an
endothelial tumor
cell or a breast cancer tumor cell (Zitzmann et al. (2002), Cancer Res.
62:5139-43). An RGD
peptide can facilitate targeting to tumors of a variety of other tissues,
including the lung, kidney,
spleen, or liver (Aoki et al. (2001), Cancer Gene Therapy 8:783-787). In some
embodiments,
the RGD peptide will facilitate targeting of the pharmaceutical composition to
the kidney. The
RGD peptide can be linear or cyclic, and can be modified, e.g., glycosylated
or methylated to
facilitate targeting to specific tissues. For example, a glycosylated RGD
peptide can deliver a
pharmaceutical composition to a tumor cell expressing avf33 (Haubner et al.
(2001), J. Nucl.
Med. 42:326-336).
A "cell permeation peptide" is capable of permeating a cell, such as a
microbial cell (e.g.,
a bacterial or fungal cell) or a mammalian cell (e.g., a human cell). A
microbial cell-permeating
peptide can be, for example, an a-helical linear peptide (e.g., LL-37 or
Ceropin PI), a disulfide
bond-containing peptide (e.g., a -defensin, P-defensin or bactenecin), or a
peptide containing
only one or two dominating amino acids (e.g., PR-39 or indolicidin). A cell
permeation peptide
can also include a nuclear localization signal (NLS). For example, a cell
permeation peptide can
be a bipartite amphipathic peptide, such as MPG, which is derived from the
fusion peptide
domain of HIV-1 gp41 and the NLS of SV40 large T antigen (Simeoni et al.
(2003), Nucleic
3o Acids Res. 31:2717-2724).
CA 02692748 2010-01-06
WO 2009/009025 PCT/US2008/008326
In some embodiments, the ligand can be a substituted amine, e.g.,
dimethylamino. In
certain embodiments the substituted amine can be rendered cationic, e.g., by
quaternization, e.g.,
protonation or alkylation. In certain embodiments, the substituted amine can
be at the terminal
position of a relatively hydrophobic chain, e.g., an alkylene chain.
In some embodiments, the ligand can be one of the following triterpenes:
O
H O H H H H H H
Fi fi
HO O HO
H
Sarsasapogenin Friedelin Epifriedelanol
H2N '~~rO-R
COOH O, O N-_T_---O"R
H O`R
R = C18H37
,.
HO~,= HO` H
Lithocholic acid
HQ
~ O N~_O-R
--------- - ~NI1ODMT H O'R
----------- - H
O N yO` H
O
In some embodiments the ligand is cholesterol or another lipid or
phospholipid.
iRNA agents include: molecules that are long enough to trigger the interferon
response
(which can be cleaved by the ribonuclease Dicer (Bernstein et al. (2001),
Nature 409:363-366)
1o and enter a RISC (RNAi-induced silencing complex)); and, molecules which
are sufficiently
short that they do not trigger the interferon response (which molecules can
also be cleaved by
Dicer and/or enter a RISC), e.g., molecules which are of a size which allows
entry into a RISC,
e.g., molecules which resemble Dicer-cleavage products. Molecules that are
short enough that
26
CA 02692748 2010-01-06
WO 2009/009025 PCT/US2008/008326
they do not trigger an interferon response are termed sRNA agents or shorter
iRNA agents
herein. "sRNA agent or shorter iRNA agent" as used herein, refers to an iRNA
agent, e.g., a
double stranded RNA agent or single strand agent, that is sufficiently short
that it does not
induce a deleterious interferon response in a human cell, e.g., it has a
duplexed region of less
than 60 but preferably less than 50, 40, or 30 nucleotide pairs. The sRNA
agent, or a cleavage
product thereof, can down regulate a target gene, e.g., by inducing RNAi with
respect to a target
RNA, preferably an endogenous or pathogen target RNA.
Each strand of a sRNA agent can be equal to or less than 30, 25, 24, 23, 22,
21, or 20
nucleotides in length. The strand is preferably at least 19 nucleotides in
length. For example,
lo each strand can be between 21 and 25 nucleotides in length. Preferred sRNA
agents have a
duplex region of 17, 18, 19, 29, 21, 22, 23, 24, or 25 nucleotide pairs, and
one or more
overhangs, preferably one or two 3' overhangs, of 2-3 nucleotides.
In addition to homology to target RNA and the ability to down regulate a
target gene, an
iRNA agent can have one or more of the following properties:
(1) if single stranded it can have a 5' modification which includes one or
more
phosphate groups or one or more analogs of a phosphate group;
(2) it can, despite modifications, even to a very large number, or all of the
nucleosides, have an antisense strand that can present bases (or modified
bases) in the proper
three dimensional framework so as to be able to form correct base pairing and
form a duplex
structure with a homologous target RNA which is sufficient to allow down
regulation of the
target, e.g., by cleavage of the target RNA;
(3) it can, despite modifications, even to a very large number, or all of the
nucleosides, still have "RNA-like" properties, i.e., it can possess the
overall structural, chemical
and physical properties of an RNA molecule, even though not exclusively, or
even partly, of
ribonucleotide-based content. For example, an iRNA agent can contain, e.g., a
sense and/or an
antisense strand in which all of the nucleotide sugars contain e.g., 2' fluoro
in place of 2'
hydroxyl. This deoxyribonucleotide-containing agent can still be expected to
exhibit RNA-like
properties. While not wishing to be bound by theory, the electronegative
fluorine prefers an
27
CA 02692748 2010-01-06
WO 2009/009025 PCT/US2008/008326
axial orientation when attached to the C2' position of ribose. This spatial
preference of fluorine
can, in turn, force the sugars to adopt a C3'-endo pucker. This is the same
puckering mode as
observed in RNA molecules and gives rise to the RNA-characteristic A-family-
type helix.
Further, since fluorine is a good hydrogen bond acceptor, it can participate
in the same hydrogen
bonding interactions with water molecules that are known to stabilize RNA
structures.
(Generally, it is preferred that a modified moiety at the 2' sugar position
will be able to enter into
H-bonding which is more characteristic of the OH moiety of a ribonucleotide
than the H moiety
of a deoxyribonucleotide. For example, in some embodiments an iRNA agent can:
exhibit a C3-
endo pucker in all, or at least 50, 75, 80, 85, 90, or 95 % of its sugars;
exhibit a CY-endo pucker
lo in a sufficient amount of its sugars that it can give rise to a the RNA-
characteristic A-family-type
helix; may have no more than 20, 10, 5, 4, 3, 2, or I sugar which is not a CY-
endo pucker
structure. These limitations may be preferable in the antisense strand;
(4) regardless of the nature of the modification, and even though the RNA
agent
can contain deoxynucleotides or modified deoxynucleotides, particularly in
overhang or other
single strand regions, it may be preferred that DNA molecules, or any molecule
in which more
than 50, 60, or 70 % of the nucleotides in the molecule, or more than 50, 60,
or 70 % of the
nucleotides in a duplexed region are deoxyribonucleotides, or modified
deoxyribonucleotides
which are deoxy at the 2' position, are excluded from the definition of RNA
agent.
A "single strand iRNA agent" as used herein, is an iRNA agent which is made up
of a
single molecule. It may include a duplexed region, formed by intra-strand
pairing, e.g., it may
be, or include, a hairpin or pan-handle structure. Single strand iRNA agents
are preferably
antisense with regard to the target molecule. In preferred embodiments single
strand iRNA
agents are 5' phosphorylated or include a phosphoryl analog at the 5' prime
terminus. 5'-
phosphate modifications include those which are compatible with RISC mediated
gene silencing.
Suitable modifications include: 5'-monophosphate ((HO)2(O)P-O-5'); 5'-
diphosphate
((HO)2(O)P-O-P(HO)(O)-0-5'); 5'-triphosphate ((HO)2(O)P-O-(HO)(O)P-O-P(HO)(O)-
0-5');
5'-guanosine cap (7-methylated or non-methylated) (7m-G-O-5'-(HO)(O)P-O-
(HO)(O)P-O-
P(HO)(O)-0-5'); 5'-adenosine cap (Appp), and any modified or unmodified
nucleotide cap
structure (N-O-5'-(HO)(O)P-O-(HO)(O)P-O-P(HO)(O)-0-5'); 5'-monothiophosphate
(phosphorothioate; (HO)2(S)P-O-5'); 5'-monodithiophosphate
(phosphorodithioate;
28
CA 02692748 2010-01-06
WO 2009/009025 PCT/US2008/008326
(HO)(HS)(S)P-O-5'), 5'-phosphorothiolate ((HO)2(O)P-S-5'); any additional
combination of
oxygen/sulfur replaced monophosphate, diphosphate and triphosphates (e.g. 5'-
alpha-
thiotriphosphate, 5'-gamma-thiotriphosphate, etc.), 5'-phosphoram i dates
((HO)2(O)P-NH-5',
(HO)(NH2)(O)P-O-5'), 5'-alkylphosphonates (R=alkyl=methyl, ethyl, isopropyl,
propyl, etc., e.g.
RP(OH)(O)-0-5'-, (OH)2(O)P-5'-CH2-), 5'-alkyletherphosphonates
(R=alkylether=methoxymethyl (MeOCH2-), ethoxymethyl, etc., e.g. RP(OH)(O)-0-5'-
). (These
modifications can also be used with the antisense strand of a double stranded
iRNA.)
A single strand iRNA agent should be sufficiently long that it can enter the
RISC and
participate in RISC mediated cleavage of a target mRNA. A single strand iRNA
agent is at least
1o 14, and more preferably at least 15, 20, 25, 29, 35, 40, or 50 nucleotides
in length. It is
preferably less than 200, 100, or 60 nucleotides in length.
Hairpin iRNA agents will have a duplex region equal to or at least 17, 18, 19,
29, 21, 22,
23, 24, or 25 nucleotide pairs. The duplex region will preferably be equal to
or less than 200,
100, or 50, in length. Preferred ranges for the duplex region are 15-30, 17 to
23, 19 to 23, and 19
to 21 nucleotides pairs in length. The hairpin will preferably have a single
strand overhang or
terminal unpaired region, preferably the 3', and preferably of the antisense
side of the hairpin.
Preferred overhangs are 2-3 nucleotides in length.
A "double stranded (ds) iRNA agent" as used herein, is an iRNA agent which
includes
more than one, and preferably two, strands in which interchain hybridization
can form a region
of duplex structure.
The antisense strand of a double stranded iRNA agent should be equal to or at
least, 14,
15, 16 17, 18, 19, 25, 29, 40, or 60 nucleotides in length. It should be equal
to or less than 200,
100, or 50, nucleotides in length. Preferred ranges are 17 to 25, 19 to 23,
and 19 to 21
nucleotides in length.
The sense strand of a double stranded iRNA agent should be equal to or at
least 14, 15,
16 17, 18, 19, 25, 29, 40, or 60 nucleotides in length. It should be equal to
or less than 200, 100,
or 50, nucleotides in length. Preferred ranges are 17 to 25, 19 to 23, and 19
to 21 nucleotides in
length.
29
CA 02692748 2010-01-06
WO 2009/009025 PCT/US2008/008326
The double strand portion of a double stranded iRNA agent should be equal to
or at least,
14, 15, 16 17, 18, 19, 20, 21, 22, 23, 24, 25, 29, 40, or 60 nucleotide pairs
in length. It should be
equal to or less than 200, 100, or 50, nucleotides pairs in length. Preferred
ranges are 15-30, 17
to 23, 19 to 23, and 19 to 21 nucleotides pairs in length.
In many embodiments, the ds iRNA agent is sufficiently large that it can be
cleaved by an
endogenous molecule, e.g., by Dicer, to produce smaller ds iRNA agents, e.g.,
sRNAs agents
It is preferred that the sense and antisense strands be chosen such that the
ds iRNA agent
includes a single strand or unpaired region at one or both ends of the
molecule. Thus, a ds iRNA
agent contains sense and antisense strands, preferable paired to contain an
overhang, e.g., one or
1o two 5' or 3' overhangs but preferably a 3' overhang of 2-3 nucleotides.
Most embodiments
will have a 3' overhang. Preferred sRNA agents will have single-stranded
overhangs, preferably
3' overhangs, of 1 or preferably 2 or 3 nucleotides in length at each end. The
overhangs can be
the result of one strand being longer than the other, or the result of two
strands of the same length
being staggered. 5' ends are preferably phosphorylated.
Preferred lengths for the duplexed region is between 15 and 30, most
preferably 18, 19,
20, 21, 22, and 23 nucleotides in length, e.g., in the sRNA agent range
discussed above. sRNA
agents can resemble in length and structure the natural Dicer processed
products from long
dsRNAs. Embodiments in which the two strands of the sRNA agent are linked,
e.g., covalently
linked are also included. Hairpin, or other single strand structures which
provide the required
2o double stranded region, and preferably a 3' overhang are also within the
invention.
The isolated iRNA agents described herein, including ds iRNA agents and sRNA
agents
can mediate silencing of a target RNA, e.g., mRNA, e.g., a transcript of a
gene that encodes a
protein. For convenience, such mRNA is also referred to herein as mRNA to be
silenced. Such
a gene is also referred to as a target gene. In general, the RNA to be
silenced is an endogenous
gene or a pathogen gene. In addition, RNAs other than mRNA, e.g., tRNAs, and
viral RNAs,
can also be targeted.
As used herein, the phrase "mediates RNAi" refers to the ability to silence,
in a sequence
specific manner, a target RNA. While not wishing to be bound by theory, it is
believed that
CA 02692748 2010-01-06
WO 2009/009025 PCT/US2008/008326
silencing uses the RNAi machinery or process and a guide RNA, e.g., an sRNA
agent of 21 to 23
nucleotides.
As used herein, "specifically hybridizable" and "complementary" are terms
which are
used to indicate a sufficient degree of complementarity such that stable and
specific binding
occurs between a compound of the invention and a target RNA molecule. Specific
binding
requires a sufficient degree of complementarity to avoid non-specific binding
of the oligomeric
compound to non-target sequences under conditions in which specific binding is
desired, i.e.,
under physiological conditions in the case of in vivo assays or therapeutic
treatment, or in the
case of in vitro assays, under conditions in which the assays are performed.
The non-target
1o sequences typically differ by at least 5 nucleotides.
In one embodiment, an iRNA agent is "sufficiently complementary" to a target
RNA,
e.g., a target mRNA, such that the iRNA agent silences production of protein
encoded by the
target mRNA. In another embodiment, the iRNA agent is "exactly complementary"
(excluding
the RRMS containing subunit(s))to a target RNA, e.g., the target RNA and the
iRNA agent
anneal, preferably to form a hybrid made exclusively of Watson-Crick base
pairs in the region of
exact complementarity. A "sufficiently complementary" target RNA can include
an internal
region (e.g., of at least 10 nucleotides) that is exactly complementary to a
target RNA.
Moreover, in some embodiments, the iRNA agent specifically discriminates a
single-nucleotide
difference. In this case, the iRNA agent only mediates RNAi if exact
complementary is found in
the region (e.g., within 7 nucleotides of) the single-nucleotide difference.
As used herein, the term "oligonucleotide" refers to a nucleic acid molecule
(RNA or
DNA) preferably of length less than 100, 200, 300, or 400 nucleotides.
RNA agents discussed herein include otherwise unmodified RNA as well as RNA
which
have been modified, e.g., to improve efficacy, and polymers of nucleoside
surrogates.
Unmodified RNA refers to a molecule in which the components of the nucleic
acid, namely
sugars, bases, and phosphate moieties, are the same or essentially the same as
that which occur in
nature, preferably as occur naturally in the human body. The art has referred
to rare or unusual,
but naturally occurring, RNAs as modified RNAs, see, e.g., Limbach et al.
(1994), Nucleic Acids
Res. 22: 2183-2196. Such rare or unusual RNAs, often termed modified RNAs
(apparently
31
CA 02692748 2010-01-06
WO 2009/009025 PCT/US2008/008326
because they are typically the result of a post transcriptionally
modification) are within the term
unmodified RNA, as used herein.
References
General References
The oligoribonucleotides and oligoribonucleosides used in accordance with this
invention
may be with solid phase synthesis, see for example "Oligonucleotide synthesis,
a practical
approach", Ed. M. J. Gait, IRL Press, 1984; "Oligonucleotides and Analogues, A
Practical
Approach", Ed. F. Eckstein, IRL Press, 1991 (especially Chapter 1, Modern
machine-aided
methods of oligodeoxyribonucleotide synthesis, Chapter 2, Oligoribonucleotide
synthesis,
1o Chapter 3, 2'-O--Methyloligoribonucleotide- s: synthesis and applications,
Chapter 4,
Phosphorothioate oligonucleotides, Chapter 5, Synthesis of oligonucleotide
phosphorodithioates,
Chapter 6, Synthesis of oligo-2'-deoxyribonucleoside methylphosphonates, and
Chapter 7,
Oligodeoxynucleotides containing modified bases. Other particularly useful
synthetic
procedures, reagents, blocking groups and reaction conditions are described in
Martin, P., Helv.
Chim. Acta, 1995, 78, 486-504; Beaucage, S. L. and Iyer, R. P., Tetrahedron,
1992, 48, 2223-
2311 and Beaucage, S. L. and Iyer, R. P., Tetrahedron, 1993, 49, 6123-6194, or
references
referred to therein.
Modification described in WO 00/44895, WO01/75164, or W002/44321 can be used
herein.
Phosphate Group References
The preparation of phosphonate oligoribonucleotides is described in U.S. Pat.
No.
5,508,270. The preparation of alkyl phosphonate oligoribonucleotides is
described in U.S. Pat.
No. 4,469,863. The preparation of phosphoramidite oligoribonucleotides is
described in U.S.
Pat. No. 5,256,775 or U.S. Pat. No. 5,366,878. The preparation of
phosphotriester
oligoribonucleotides is described in U.S. Pat. No. 5,023,243. The preparation
of borano
phosphate oligoribonucleotide is described in U.S. Pat. Nos. 5,130,302 and
5,177,198. The
preparation of 3'-Deoxy-3'-amino phosphoramidate oligoribonucleotides is
described in U.S. Pat.
No. 5,476,925. 3'-Deoxy-3'-methylenephosphonate oligoribonucleotides is
described in An, H,
et al. J. Org. Chem. 2001, 66, 2789-2801. Preparation of sulfur bridged
nucleotides is described
in Sproat et al. Nucleosides Nucleotides 1988, 7,651 and Crosstick et al.
Tetrahedron Lett. 1989,
30, 4693.
32
CA 02692748 2010-01-06
WO 2009/009025 PCT/US2008/008326
Sugar Groug References
Modifications to the 2' modifications can be found in Verma et al. (1998),
Annu. Rev.
Biochem. 67:99-134 and all references therein. Specific modifications to the
ribose can be found
in the following references: 2'-fluoro (Kawasaki et al., J. Med. Chem., 1993,
36, 831-841), 2'-
MOE (Martin, P. Helv. Chim. Acta 1996, 79, 1930-1938), "LNA" (Wengel, J. Acc.
Chem. Res.
1999, 32, 301-310).
Replacement of the Phosj2hate Group References
Methylenemethylimino linked oligoribonucleosides, also identified herein as
MMI linked
oligoribonucleosides, methylenedimethylhydrazo linked oligoribonucleosides,
also identified
lo herein as MDH linked oligoribonucleosides, and methylenecarbonylamino
linked
oligonucleosides, also identified herein as amide-3 linked
oligoribonucleosides, and
methyleneaminocarbonyl linked oligonucleosides, also identified herein as
amide-4 linked
oligoribonucleosides as well as mixed backbone compounds having, as for
instance, alternating
MMI and PO or PS linkages can be prepared as is described in U.S. Pat. Nos.
5,378,825,
5,386,023, 5,489,677 and in published PCT applications PCT/US92/04294 and
PCT/US92/04305 (published as WO 92/20822 WO and 92/20823, respectively).
Formacetal and
thioformacetal linked oligoribonucleosides can be prepared as is described in
U.S. Pat. Nos.
5,264,562 and 5,264,564. Ethylene oxide linked oligoribonucleosides can be
prepared as is
described in U.S. Pat. No. 5,223,618. Siloxane replacements are described in
Cormier, J.F. et al.
Nucleic Acids Res. 1988, 16, 4583. Carbonate replacements are described in
Tittensor, J.R. J.
Chem. Soc. C 1971, 1933. Carboxymethyl replacements are described in Edge,
M.D. et al. J.
Chem. Soc. Perkin Trans. 1 1972, 1991. Carbamate replacements are described in
Stirchak, E.P.
Nucleic Acids Res. 1989, 17, 6129.
Replacement of the Phosphate-Ribose Backbone References
Cyclobutyl sugar surrogate compounds can be prepared as is described in U.S.
Pat. No.
5,359,044. Pyrrolidine sugar surrogate can be prepared as is described in U.S.
Pat. No.
5,519,134. Morpholino sugar surrogates can be prepared as is described in U.S.
Pat. Nos.
5,142,047 and 5,235,033, and other related patent disclosures. Peptide Nucleic
Acids (PNAs) are
known per se and can be prepared in accordance with any of the various
procedures referred to in
Peptide Nucleic Acids (PNA): Synthesis, Properties and Potential Applications,
Bioorganic &
33
CA 02692748 2010-01-06
WO 2009/009025 PCT/US2008/008326
Medicinal Chemistry, 1996, 4, 5-23. They may also be prepared in accordance
with U.S. Pat. No.
5,539,083.
Terminal Modification References
Terminal modifications are described in Manoharan, M. et al. Antisense and
Nucleic Acid
Drug Development 12, 103-128 (2002) and references therein.
Bases References
N-2 substituted purine nucleoside amidites can be prepared as is described in
U.S. Pat.
No. 5,459,255. 3-Deaza purine nucleoside amidites can be prepared as is
described in U.S. Pat.
No. 5,457,191. 5,6-Substituted pyrimidine nucleoside amidites can be prepared
as is described in
lo U.S. Pat. No. 5,614,617. 5-Propynyl pyrimidine nucleoside amidites can be
prepared as is
described in U.S. Pat. No. 5,484,908. Additional references can be disclosed
in the above
section on base modifications.
7. Delivery Modules
The pharmaceutical compositions of the invention can be complexed to a
delivery agent
that features a modular complex. The complex can include a carrier 'agent
linked to one or more
of (preferably two or more, more preferably all three of): (a) a condensing
agent (e.g., an agent
capable of attracting, e.g., binding, a nucleic acid, e.g., through ionic or
electrostatic
interactions); (b) a fusogenic agent (e.g., an agent capable of fusing and/or
being transported
through a cell membrane, e.g., an endosome membrane); and (c) a targeting
group, e.g., a cell or
tissue targeting agent, e.g., a lectin, glycoprotein, lipid or protein, e.g.,
an antibody, that binds to
a specified cell type such as a kidney cell.
Thus, the pharmaceutical compositions described herein, can be linked, e.g.,
coupled or
bound, to the modular complex. The pharmaceutical compositions can interact
with the
condensing agent of the complex, and the complex can be used to deliver an
iRNA agent to a
cell, e.g., in vitro or in vivo. For example, the complex can be used to
deliver an iRNA agent to a
subject in need thereof, e.g., to deliver an iRNA agent to a subject having a
disorder, e.g., a
disorder described herein, such as a disease or disorder of the kidney.
The fusogenic agent and the condensing agent can be different agents or the
one and the
same agent. For example, a polyainino chain, e.g., polyethyleneimine (PEI),
can be the
fusogenic and/or the condensing agent.
34
CA 02692748 2010-01-06
WO 2009/009025 PCT/US2008/008326
The delivery agent can be a modular complex. For example, the complex can
include a
carrier agent linked to one or more of (preferably two or more, more
preferably all three of):
(a) a condensing agent (e.g., an agent capable of attracting, e.g., binding, a
nucleic acid,
e.g., through ionic interaction),
(b) a fusogenic agent (e.g., an agent capable of fusing and/or being
transported through a
cell membrane, e.g., an endosome membrane), and
(c) a targeting group, e.g., a cell or tissue targeting agent, e.g., a lectin,
glycoprotein, lipid
or protein, e.g., an antibody, that binds to a specified cell type such as a
kidney cell. A targeting
group can be a thyrotropin, melanotropin, lectin, glycoprotein, surfactant
protein A, mucin
lo carbohydrate, multivalent lactose, multivalent galactose, N-acetyl-
galactosamine, N-acetyl-
glucosamine multivalent mannose, multivalent fucose, glycosylated polyamino
acids,
multivalent galactose, transferrin, bisphosphonate, polyglutamate,
polyaspartate, a lipid,
cholesterol, a steroid, bile acid, folate, vitamin B12, biotin, Neproxin, or
an RGD peptide or
RGD peptide mimetic.
7.1 Carrier agents
The carrier agent of a modular complex described herein can be a substrate for
attachment of one or more of: a condensing agent, a fusogenic agent, and a
targeting group. The
carrier agent would preferably lack an endogenous enzymatic activity. The
agent would
preferably be a biological molecule, preferably a macromolecule. Polymeric
biological carriers
2o are preferred. It would also be preferred that the carrier molecule be
biodegradable..
The carrier agent can be a naturally occurring substance, such as a protein
(e.g., human
serum albumin (HSA), low-density lipoprotein (LDL), or globulin); carbohydrate
(e.g., a
dextran, pullulan, chitin, chitosan, inulin, cyclodextrin or hyaluronic acid);
or lipid. The carrier
molecule can also be a recombinant or synthetic molecule, such as a synthetic
polymer, e.g., a
synthetic polyamino acid. Examples of synthetic polymers include polyamino
acids, such as
polylysine (PLL), poly L-aspartic acid, poly L-glutamic acid, and styrene-
maleic acid anhydride
copolymer, poly(L-lactide-co-glycolide) copolymer, divinyl ether-maleic
anhydride copolymer,
N-(2-hydroxypropyl)methacry lamide copolymer (HMPA), polyethylene glycol
(PEG), polyvinyl
alcohol (PVA), polyurethane, poly(2-ethylacryllic acid), N-isopropylacrylamide
polymers, or
polyphosphazine. Other useful carrier molecules can be identified by routine
methods.
CA 02692748 2010-01-06
WO 2009/009025 PCT/US2008/008326
A carrier agent can be characterized by one or more of: (a) is at least 1 Da
in size; (b) has
at least 5 charged groups, preferably between 5 and 5000 charged groups; (c)
is present in the
complex at a ratio of at least 1:1 carrier agent to fusogenic agent; (d) is
present in the complex at
a ratio of at least 1:1 carrier agent to condensing agent; (e) is present in
the complex at a ratio of
at least 1:1 carrier agent to targeting agent.
7.2 Fusogenic agents
A fusogenic agent of a modular complex described herein can be an agent that
is
responsive to, e.g., changes charge depending on, the pH environment. Upon
encountering the
pH of an endosome, it can cause a physical change, e.g., a change in osmotic
properties which
lo disrupts or increases the permeability of the endosome membrane. In some
embodiments, the
fusogenic agent changes charge, e.g., becomes protonated, at pH lower than
physiological range.
For example, the fusogenic agent can become protonated at pH 4.5-6.5. The
fusogenic agent
can serve to release the iRNA agent into the cytoplasm of a cell after the
complex is taken up,
e.g., via endocytosis, by the cell, thereby increasing the cellular
concentration of the iRNA agent
in the cell.
In some embodiments, the fusogenic agent can have a moiety, e.g., an amino
group,
which, when exposed to a specified pH range, will undergo a change, e.g., in
charge, e.g.,
protonation. The change in charge of the fusogenic agent can trigger a change,
e.g., an osmotic
change, in a vesicle, e.g., an endocytic vesicle, e.g., an endosome. For
example, the fusogenic
2o agent, upon being exposed to the pH environment of an endosome, will cause
a solubility or
osmotic change substantial enough to increase the porosity of (preferably, to
rupture) the
endosomal membrane.
The fusogenic agent can be a polymer, such as a polyamino chain, e.g.,
polyethyleneimine (PEI). The PEI can be linear, branched, synthetic or
natural. The PEI can be,
e.g., alkyl substituted PEI, or lipid substituted PEI.
In other embodiments, the fusogenic agent can be polyhistidine, polyimidazole,
polypyridine, polypropyleneimine, mellitin, or a polyacetal substance, e.g., a
cationic polyacetal.
In some embodiments, the fusogenic agent can have an alpha helical structure.
The fusogenic
agent can be a membrane disruptive agent, e.g., mellittin.
A fusogenic agent can have one or more of the following characteristics: (a)
is at least
1 Da in size; (b) has at least 10 charged groups, preferably between 10 and
5000 charged groups,
36
CA 02692748 2010-01-06
WO 2009/009025 PCT/US2008/008326
more preferably between 50 and 1000 charged groups; (c) is present in the
complex at a ratio of
at least 1:lfusogenic agent to carrier agent; (d) is present in the complex at
a ratio of at least 1:1
fusogenic agent to condensing agent; (e) is present in the complex at a ratio
of at least 1:1
fusogenic agent to targeting agent.
Other suitable fusogenic agents can be tested and identified by a skilled
artisan. The
ability of a compound to respond to, e.g., change charge depending on, the pH
environment can
be tested by routine methods, e.g., in a cellular assay. For example, a test
compound is
combined or contacted with a cell, and the cell is allowed to take up the test
compound, e.g., by
endocytosis. An endosome preparation can then be made from the contacted cells
and the
lo endosome preparation compared to an endosome preparation from control
cells. A change, e.g.,
a decrease, in the endosome fraction from the contacted cell vs. the control
cell indicates that the
test compound can function as a fusogenic agent. Alternatively, the contacted
cell and control
cell can be evaluated, e.g., by microscopy, e.g., by light or electron
microscopy, to determine a
difference in endosome population in the cells. The test compound can be
labeled. In another
type of assay, a modular complex described herein is constructed using one or
more test or
putative fusogenic agents. The modular complex can be constructed using a
labeled nucleic acid
instead of the iRNA. The ability of the fusogenic agent to respond to, e.g.,
change charge
depending on, the pH environment, once the modular complex is taken up by the
cell, can be
evaluated, e.g., by preparation of an endosome preparation, or by microscopy
techniques, as
2o described above. A two-step assay can also be performed, wherein a first
assay evaluates the
ability of a test compound alone to respond to, e.g., change charge depending
on, the pH
environment; and a second assay evaluates the ability of a modular complex
that includes the test
compound to respond to, e.g., change charge depending on, the pH environment.
7.3 Condensing agent
The condensing agent of a modular complex described herein can interact with
(e.g.,
attracts, holds, or binds to) a pharmaceutical composition of the invention to
(a) condense, e.g.,
reduce the size or charge of the pharmaceutical composition and/or (b) protect
the
pharmaceutical composition, e.g., protect the iRNA agent against degradation.
The condensing
3o agent can include a moiety, e.g., a charged moiety, that can interact with
a hydrophilic moiety,
e.g., a hydrophilic chain or an iRNA agent, e.g., by ionic interactions. The
condensing agent
37
CA 02692748 2010-01-06
WO 2009/009025 PCT/US2008/008326
would preferably be a charged polymer, e.g., a polycationic chain. The
condensing agent can be
a polylysine (PLL), spermine, spermidine, polyamine, pseudopeptide-polyamine,
peptidomimetic
polyamine, dendrimer polyamine, arginine, amidine, protamine, cationic lipid,
cationic
porphyrin, quaternary salt of a polyamine, or an alpha helical peptide.
A condensing agent can have the following characteristics: (a) at least IDa in
size; (b)
has at least 2 charged groups, preferably between 2 and 100 charged groups;
(c) is present in the
complex at a ratio of at least 1:1 condensing agent to carrier agent; (d) is
present in the complex
at a ratio of at least 1:1 condensing agent to fusogenic agent; (e) is present
in the complex at a
ratio of at least 1:1 condensing agent to targeting agent.
Other suitable condensing agents can be tested and identified by a skilled
artisan, e.g., by
evaluating the ability of a test agent to interact with a hydrophilic moiety
or an iRNA agent. The
ability of a test agent to interact with a hydrophilic moiety or an iRNA
agent, e.g., to condense or
protect the iRNA agent, can be evaluated by routine techniques. In one assay,
a test agent is
contacted with a nucleic acid, and the size and/or charge of the contacted
nucleic acid is
evaluated by a technique suitable to detect changes in molecular mass and/or
charge. Such
techniques include non-denaturing gel electrophoresis, immunological methods,
e.g.,
immunoprecipitation, gel filtration, ionic interaction chromatography, and the
like. A test agent
is identified as a condensing agent if it changes the mass and/or charge
(preferably both) of the
contacted hydrophilic moiety or nucleic acid, compared to a control. A two-
step assay can also
be performed, wherein a first assay evaluates the ability of a test compound
alone to interact
with, e.g., bind to, e.g., condense the charge and/or mass of, a hydrophilic
moiety or nucleic cid;
and a second assay evaluates the ability of a modular complex that includes
the test compound to
interact with, e.g., bind to, e.g., condense the charge and/or mass of, a
hydrophilic moiety or
nucleic acid.
Methods of preparing delivery systems:
A delivery system described herein, e.g., a mixed micelle, can be prepared by
co-
suspending an amphipathic micelle-forming molecule and an iRNA-conjugate
together, for
example, in buffer. The amphipathic micelle-forming molecule and an iRNA-
conjugate can be
suspended together in various ratios, for example, in a molar ratio of 1:1. In
some preferred
3o embodiments, the micelle is prepared by suspending a 1:1 molar ratio of PEG-
PE with a
conjugated siRNA such as siRNA-Chol.
38
CA 02692748 2010-01-06
WO 2009/009025 PCT/US2008/008326
A preferred buffer includes a buffer having a pH of about 7, such as HBS
buffer having a
pH of about 7.4.
7.4 Second-therapeutic conjuizates
A pharmaceutical composition of the invention can be coupled, e.g., covalently
coupled,
to a second therapeutic agent. For example, an iRNA agent used to treat a
particular disorder can
be coupled to a second therapeutic agent, e.g., an agent other than the iRNA
agent.
Alternatively, the second therapeutic agent can be coupled to a hydrophilic
moiety in the
hydrophilic layer of the mixed micelle, or to a hydrophobic moiety which is
included in the
1o hydrophobic core of the mixed micelle or, if it is hydrophobic, can be
dissolved in the
hydrophobic core of the micelle. The second therapeutic agent can be one which
is directed to
the treatment of the same disorder. For example, in the case of an iRNA used
to treat a disorder
characterized by unwanted cell proliferation, e.g., cancer, the iRNA agent or
hydrophilic chains
can be coupled to a second agent which has an anti-cancer effect. For example,
the second
therapeutic agent which stimulates the immune system, e.g., a CpG motif, or
more generally an
agent that activates a toll-like receptor and/or increases the production of
gamma interferon.
The invention is further illustrated by the following examples, which should
not be
construed as further limiting.
EXAMPLES
PEG-PE Micelles for siRNA Delivery
Effective intracellular delivery of siRNA is essential for its therapeutic
activity.
However, native siRNA doesn't always demonstrate the required high
intracellular penetration.
Various siRNA derivatives have been suggested to enhance its activity, such as
siRNA-Chol.
Polymeric micelles were prepared with high load of siRNA to enhance its uptake
by the cells.
To make such micelles, a mixed micelle system consisting of siRNA-Chol and PEG-
PE
conjugate was used, which demonstrated a very low CMC value and the ability to
form stable
mixed micelles with various amphiphiles. Cholesteryl-modified siRNA forms
stable mixed
micelles with PEG-PE, having a CMC value of approximately 6xl0-6M (see Fig.
1), an average
size of about 10 nm, and a narrow size distribution (see Fig. 2). The CMC
values for the mixed
siRNA-Chol/PEG-PE micelles remains the same when the siRNA moiety in siRNA-
Chol is
additionally modified with Cy3 dye. When incubated with MCF7 or 4T1 cancer
cells, mixed
39
CA 02692748 2010-01-06
WO 2009/009025 PCT/US2008/008326
siRNA-Chol/PEG-PE micelles with high siRNA load (approx. 50mol%) and
fluorescently
labeled with Cy3 dye demonstrated effective cellular uptake within just 2
hours of incubation
(see Figs. 2, 3) and may serve as a convenient means to deliver siRNA into
cells (Figs. 2, 3).
Preliminary experiments on the silencing property of micellar siRNA-Chol
demonstrated
that, in addition to intracellular uptake, siRNA-Chol/PEG-PE micelles were
able to provoke a
significant silencing of the target gene and decrease the GFP production in
pGFP-transfected
cells (Fig. 4). The first generation siRNA delivery system used in this
experiment can be
significantly optimized to make it highly effective.
Preparation of mixed micelles from PEG-PE and Chol-siRNA.
A commercial preparation of siRNA (silencing the GFP gene) can be used
(Silencer
GFP (eGFP) siRNA cat#AM4626 from Ambion). An siRNA-Chol conjugate can be
prepared by
any standard chemistries, as described below. Mixed polymeric micelles can be
prepared by co-
suspending PEG-PE and Chol-siRNA (a small fraction of Chol-siRNA can be
fluorescently
labeled with Cy3 dye) in different molar ratios in HBS buffer, pH 7.4. The
labeling of siRNA
with Cy3 can be used for tracking purposes.
Synthesis of siRNA-Chol. Double stranded siRNA can be derivatized on both the
5'-end
and 3'-end of the sense strand. Various functional groups bearing fluorescence
(Chiu et al.
(2002), Mol. Cell 10:549-561; Harborth et al. (2003), Antisense Nucleic Acid
Drug Dev. 13:83-
105) or radioactivity (Braasch et al. (2004), Bioorg. Med. Chem. Lett. 14:1139-
1143) as well as
groups facilitating cellular uptake and transport through membranes (e.g.,
cholesterol, litocholic
acid, lauryl acid or long alkyl chains) can be conjugated at this position
(Soutschek et al. (2004),
Nature 432:173-178; Lorenz et al. (2004), Bioorg. Med. Chem. Lett. 14:4975-
4977). Delivery of
cholesterol-modified siRNAs has been proven to be effective both in vitro
(Lorenz et al. (2004),
Bioorg. Med. Chem. Lett. 14:4975-4977) and in vivo (Soutschek et al. (2004),
Nature 432:173-
178), without addition of polycationic transfection reagents. Pendant groups
such as puromycin
(Chiu et al. (2002), Mol. Cell 10:549-561), biotin (Chiu et al. (2002), Mol.
Cell 10:549-561),
inverted nucleosides (Morrissey et al. (2005), Nat. Biotechnol. 23:1002-1007;
Morrissey et al.
(2005), Hepatology 41:1349-1356), allyl residue (Amarzguioui et al. (2003),
Nucleic Acids Res
31:589-595) or aminoalkanes (Czauderna et al. (2003), Nucleic Acids Res
31:2705-2716) have
3o been attached at the 3'-ends of siRNA molecules without any significant
loss of silencing
activity of resulting constructs. The 3'-amino-activated oligonucleotide can
be conjugated to
CA 02692748 2010-01-06
WO 2009/009025 PCT/US2008/008326
mercaptyl cholesterol via a disulfide linkage according to a published
procedure (Bandgar et al.
(2000), Chem. Lett. 29:1304-1305). For example, mercaptyl cholesterol can be
prepared by
direct synthesis of thiols from alcohols using trifluoroacetic anhydride and
polymer supported
hydrosulfide, under mild conditions. See, Fig. 5A. 3'amino-siRNA can be
conjugated to
mercaptyl cholesterol using the heterofunctional spacer N-Succinimidyl-3-(2-
pyridyldithio)
propionate (SPDP) (Kim et al. (2006), J. Control. Release 116:123-129). See,
Fig. 5B.
The characterization of siRNA-S-S-Chol can be performed by: (1) UV analysis
for
conjugation yield evaluation (X=260 nm) (Lee et al. (2007), Biochem. Biophys.
Res. Commun.
357:511-516); (2) Checking stability against the nuclease attack (Kim et al.
(2006), J. Control.
lo Release 116:123-129): using RNase protection assay, since it cleaves
phosphodiester bond
between any two ribonucleotides; (3) Evaluation of cleavability of the
disulfide linkage in
simulated intracellular conditions by incubation with a glutathione solution
and subsequent
analysis by electrophoresis; (4) Evaluation of serum stability by
electrophoresis.
Labeling of siRNA-Chol with Cy3. When required, Cholesterol and Cy3 can be
attached
to the same siRNA moiety. Cholesterol-siRNA can be conjugated on the available
phosphate
group at a 5' end using the water soluble carbodiimide EDC (1-ethyl-3-(3-
dimethylamino-
propyl) carbodiimide hydrochloride) producing a highly reactive phosphodiester
intermediate;
further reaction with ethylene diamine can provide a terminal amino reactive
group. The
activated carboxylic derivative Cy3B NHS Ester can be conjugated with the 5'-
NH2-3'-
Cholesterol-siRNA to obtain Cy3 fluorescently labeled micelles. See Fig. 6.
41