Note: Descriptions are shown in the official language in which they were submitted.
CA 02692776 2010-01-07
WO 2009/009002 PCT/US2008/008260
HYDROGEN CHLORIDE SALT OF A SUBSTITUTED 5-OXAZOL-2-YL-QUINOLINE
COMPOUND AND A PROCESS FOR THE PRODUCTION THEREOF
CROSS REFERENCE TO RELATED APPLICATIONS
This application is based on and claims the priority of U.S. Provisional
Patent
Application No. 60/959,479, filed July 10, 2007, the description of which is
incorporated herein by reference in its entirety.
FIELD OF THE INVENTION
The present invention relates to the hydrogen chloride salt of 1-[[5-(1(S)-
aminoethyl)-2-[8-methoxy-2-(trifluoromethyl)-5-quinolyl]-4-oxazolyl]carbonyl]-
4(R)-
[(cydopropyl-carbonyl)amino]-L-proline, ethyl ester, pharmaceutical
compositions
comprising said salt, and methods of treating upper and lower obstructive
diseases of
the airways by inhalation of said salt.
BACKGROUND OF THE INVENTION
Identification of any publication, patent, or patent application in this
section or any section of this application is not an admission that such
publication is
prior art to the present invention.
Phosphodiesterases are known to regulate cyclic AMP, and phosphodiesterase
4 (PDE4) has been shown to be the predominant regulator of cyclic AMP in
respiratory smooth muscle and inflammatory cells. Inhibitors of PDE4 are
useful in
treating a variety of diseases, including allergic and inflammatory diseases,
diabetes,
central nervous system diseases, pain, and viruses that produce TNF.
Amino-substituted quinolyl PDE4 inhibitors are disclosed in US 5,804,588;
sulfonamide-substituted quinolyl PDE4 inhibitors are disclosed in US
5,834,485; and
(benzo-fused)heteroaryl-substituted PDE4 inhibitors are disclosed in US
6,069,151.
Oxazolyl-substituted quinolyl PDE4 inhibitors are disclosed in
PCT/US2005/017134.
A process for the preparation of the compound of Formula Ia, 1-[[5-(1(S)-
aminoethyl)-
2-[8-methoxy-2-(trifluoromethyl)-5-q uinolyl]-4-oxazolyl]carbonylj-4(R )-
[(cycl opropyl-
carbonyl)amino]-L-proline, ethyl ester, is described in published U.S. Patent
CA 02692776 2010-01-07
WO 2009/009002 PCT/US2008/008260
Application 2006/0106062, published May 18, 2006, which is incorporated herein
in its
entirety by reference.
OCH3
NCF3
UH
,\N
O N
H2NN O
CH3 O
O OCH2CH3 Formula Ia
SUMMARY OF THE INVENTION
One aspect of the present invention is the hydrogen chloride salt of 1-[[5-
(1(S)-
aminoethyl)-2-[8-methoxy-2-(trifluoromethyl)-5-quinolyl]-4-oxazolyl]carbonyl]-
4(R)-
[(cyclopropyl-carbonyl)amino]-L-proline, ethyl ester, the compound of Formula
I:
OCH3
U N` CF3
( ~ H
O ~N ,\
N
H2N~ N 0 HCl
CHg 0 0 OCH2CH3 Formula I
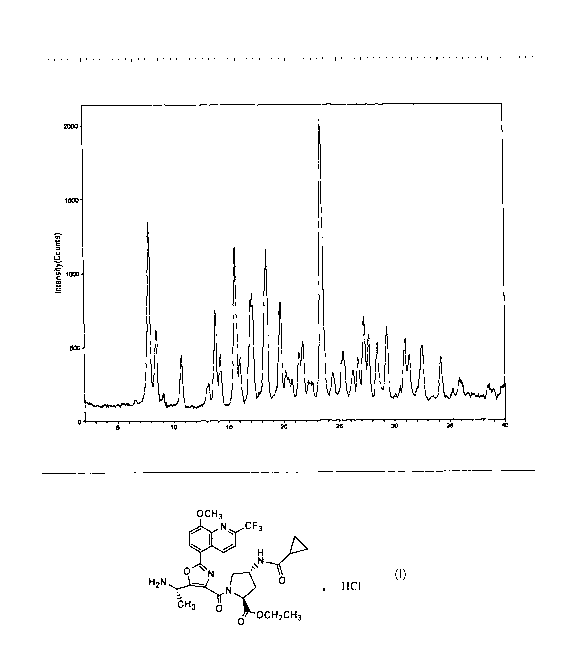
exhibiting a powder x-ray diffraction pattern (PXRD pattern) substantially
similar to
that of Figure I, which has as its four most characteristic peaks appearing at
diffraction
angles of 7.9, 15.6, 18.5, and 23.4 degrees 20.
Another aspect of the present invention is crystalline form of the compound of
Formula I (hydrogen chloride salt of the compound of Formula Ia) having a PXRD
pattem which has it's eight most characteristic peaks appearing at 7.9, 13.8,
15.6,
17.2, 18.5, 19.7, 23.4, and 27.2 degrees 20.
Another aspect of the present invention is a crystalline form of the compound
of
Formula I (hydrogen chloride salt of the compound of Formula Ia) having a PXRD
pattem which has it's twelve most characteristic peaks appearing at 7.9, 8.5,
13.8,
15.6, 17.2, 18.5, 19.7, 23.4, 27.2, 27.6, 29.3 and 30.9 degrees 20.
2
CA 02692776 2010-01-07
WO 2009/009002 PCT/US2008/008260
Another aspect of the present invention is a method of treating upper or lower
obstructive diseases of the airways in a patient in need of such treatment
comprising
administering to said patient by inhalation an effective amount of a
medicament
comprising the compound of Formula I having a PXRD containing peaks at
diffraction
angles of 7.9, 15.6, 18.5, and 23.4 degrees 20, optionally in combination with
at least
one additional agent useful for treating upper or lower obstructive diseases
of the
airway, preferably an additional agent selected from beta-agonists, muscarinic
antagonists or corticosteroids.
Another aspect of the present invention is the provision of an inhalable
pharmaceutical composition comprising an effective amount of the compound of
Formula I having a PXRD containing peaks at diffraction angles of 7.9, 15.6,
18.5, and
23.4 degrees 20, optionally in combination with at least one additional agent
useful for
treating upper or lower obstructive diseases of the airway, preferably an
additional
agent selected from beta-agonists, muscarinic antagonists or corticosteroids.
Another aspect of the present invention is a process for preparing the
crystalline form of the compound of Formula I having a PXRD containing peaks
at
diffraction angles of 7.9, 15.6, 18.5, and 23.4 degrees 20, the process
comprising:
a) providing an ethanol solution of the compound of Formula Ia:
OCH3
NUCF3
H
~N ,N
O
:' N
HZN O
CHg O
O OCH2CH3 Formula Ia;
b) reacting the solution from Step "a" with an isopropanol solution of HCI
at refluxing temperature;
C) cooling the mixture provided in Step "b" to form a salt precipitate; and
d) optionally isolating the precipitate by filtering the mixture then washing
and drying the resulting solid to yield the compound of Formula I (a
hydrogen chloride salt of the compound of Formula Ia).
3
CA 02692776 2010-01-07
WO 2009/009002 PCT/US2008/008260
Other aspects and advantages of the present invention will become apparent
from following Detailed Description.
BRIEF DESCRIPTION OF THE DRAWINGS
FIG. 1 is a graph of a powder x-ray diffraction (PXRD) pattern of the
crystalline
form of the compound of Formula I, generated using an X-ray diffractometer.
The
graph plots the intensity of the peaks as defined by counts per second versus
the
diffraction angle 2 0 in degrees.
FIG. 2 is a plot of the thermal analysis of the crystalline form of the
compound
of Formula I generated by Differential Scanning Calorimetry (DSC).
DETAILED DESCRIPTION
The free base compound of Formula Ia, having the structure:
OCH3
N\ CF3
) ~ H
O `\\N
H2N N N O
0
0 OCH2CH3 Formula Ia
is described in US 2006/0106062 Al, published May 18, 2006 (the '062
publication),
which application is incorporated herein by reference in its entirety. In
particular, the
'062 publication describes a batch process for the preparation of the compound
of
Formula Ia on pages 83 to 86 (preparative Examples 5 to 7, in preparation of
the
example compound 26-347, which is illustrated on page 193), which process is
incorporated by reference herein in its entirety. A process for preparing the
compound of Formula la is also described in an application filed herewith on
July 10,
2007 under attomey's docket no. CD06670L01 US, which is incorporated herein by
reference.
4
CA 02692776 2010-01-07
WO 2009/009002 PCT/US2008/008260
The compound of Formula I is a crystalline hydrogen chloride salt form of the
compound of Formula Ia. The compound of Formula I can be provided in a
crystalline
form which exhibits a PXRD substantially similar the X-ray powder pattern
shown in
Figure I, and has its four most characteristic peaks at diffraction angles of
7.9, 15.6,
18.5, and 23.4 degrees 20.
It is believed that this crystalline form of the compound of Formula I
demonstrates
superior stability over some other salts, and improved solubility without
unacceptable
hygroscopic properties. Accordingly, it is believed that the compound of
Formula I
may provide medicaments having increased shelf life and improved solubility,
and
thereby improved bioavailability, when compared to some other salts of the
compound
of Formula Ia. Since the intended use of the compound of Formula I is as a
therapeutically active pharmaceutical agent, salt forms of the compound of
Formula Ia
having notable stability and bioavailability are of great interest.
The unique crystalline material comprising the compound of Formula I. can be
prepared as described herein in the Examples, in particular step 5 of Example
2.
As used throughout the specification, the following terms, unless otherwise
indicated, shall be understood to have the following meanings:
"Patient" includes both human and other animals.
"Mammal" includes humans and other mammalian animals.
"Alcohol" means an organic compound containing a hydroxyl group (-OH).
"Excipient" means an essentially inert substance used as a diluent or to give
form or consistency to a formulation.
"Effective" or "therapeutically effective" is meant to describe a polymorph of
a
compound or a composition of the present invention effective as a PDE4
inhibitor and
thus producing the desired therapeutic, ameliorative, inhibitory or
preventative effect.
"Effective amounY' or "therapeutically effective amount" is meant to describe
an
amount of polymorph or a composition of the present invention effective as a
PDE4
inhibitor and thus producing the desired therapeutic, ameliorative, inhibitory
or
preventative effect.
Upper and lower airway obstructive disease treated by the compound of
Formula I include asthma, COPD (chronic obstructive pulmonary disease),
chronic
bronchitis, cystic fibrosis, allergic rhinitis, non-allergic rhinitis,
rhinosinusitis, adult
respiratory disease, acute respiratory distress syndrome, respiratory viruses,
cough,
CA 02692776 2010-01-07
WO 2009/009002 PCT/US2008/008260
interstitial pneumonitis, chronic sinusitis, airflow obstruction, airway
hyperresponsiveness (i.e., airway hyperreactivity), bronchiectasis,
bronchiolitis,
bronchiolitis obliterans (i.e., bronchiolitis obliterans syndrome), dyspnea,
emphysema,
hypercapnea, hyperinflation, hypoxemia, hyperoxia-induced inflammations,
pulmonary
fibrosis, pulmonary hypertension, small airway disease, wheeze and colds.
The compound of Formula I is preferably useful in treating asthma, COPD,
cough, airflow obstruction, airway hyperresponsiveness (i.e., airway
hyperreactivity),
bronchiolitis, chronic bronchitis, emphysema, pulmonary fibrosis, pulmonary
hypertension, small airway disease, wheeze and allergic rhinitis.
More preferably, the compound of Formula I is useful for treating COPD and
asthma.
Other agents for treating an obstructive airway disease (e.g., COPD or asthma)
for use in combination with the compound of Formula I are selected from the
group
consisting of: steroids (e.g. glucocorticoids), 5-lipoxygenase inhibitors, 0-2
adrenoceptor agonists, a-adrenergic receptor agonists, muscarinic Ml
antagonists,
muscarinic M3 antagonists, muscarinic M2 antagonists, LTB4 antagonists,
cysteinyl
leukotriene antagonists, bronchodilators, PDE4 inhibitors, elastase
inhibitors, MMP
inhibitors, phospholipase A2 inhibitors, phospholipase D inhibitors, histamine
H1
antagonists, histamine H3 antagonists, dopamine agonists, adenosine A2
agonists,
NK1, NK2 and NK3 antagonists, GABA-b agonists, nociceptin agonists,
expectorants,
mucolytic agents, decongestants, mast cell stabilizers, antioxidants, anti-IL-
8 anti-
bodies, anti-IL-5 antibodies, anti-IgE antibodies, anti-TNF antibodies, IL-10,
adhesion
molecule inhibitors, growth hormones and other PDE4 inhibitors.
Non-limitative examples of antihistamines that can be used in combination with
the compound of Formula I include astemizole, azatadine, azelastine,
acrivastine,
brompheniramine, certirizine, chlorpheniramine, clemastine, cyclizine,
carebastine,
cyproheptadine, carbinoxamine, descarboethoxyloratadine, doxylamine,
dimethindene, ebastine, epinastine, efletirizine, fexofenadine, hydroxyzine,
ketotifen,
loratadine, levocabastine, mizolastine, equitazine, mianserin, noberastine,
meGizine,
norastemizole, picumast, pyrilamine, promethazine, terfenadine,
tripelennamine,
temelastine, trimeprazine and triprolidine.
6
CA 02692776 2010-01-07
WO 2009/009002 PCT/US2008/008260
Non-limitative examples of histamine H3 receptor antagonists include:
thioperamide, impromidine, burimamide, clobenpropit, impentamine, mifetidine,
S-
sopromidine, R-sopromidine, SKF-91486, GR-175737, GT-2016, UCL-1199 and
clozapine. Other compounds can readily be evaluated to determine activity at
H3
receptors by known methods, including the guinea pig brain membrane assay and
the guinea pig neuronal ileum contraction assay, both of which are described
in U.S.
Patent 5,352,707. Another useful assay utilizes rat brain membranes and is
described by West et al., "Identification of Two-H3-Histamine Receptor
Subtypes,"
Molecular Pharmacology, Vol. 38, pages 610-613 (1990).
The term "leukotriene inhibitor" includes any agent or compound that inhibits,
restrains, retards or otherwise interacts with the action or activity of
leukotrienes.
Non-limitative examples of leukotriene inhibitors include montelukast and its
sodium
salt; 1-(((R)-(3-(2-(6,7-difluoro-2-quinolinyl)ethenyl)phenyl)-3-(2-(2-hydroxy-
2-propyl)
phenyl)thio) methylcyclopropaneacetic acid, and its sodium salt, described in
U.S.
Patent 5,270,324; 1-(((1(R)-3(3-(2-(2,3-dichlorothieno[3,2-b]pyridin-5-yl)-(E)-
ethenyl)
phenyl)-3-(2-(1-hydroxy-1-methylethyl)phenyl)propyl)thio)methyl)cyclo-
propaneacetic
acid, and its sodium salt, described in U.S. Patent 5,472,964; pranlukast;
zafirlukast;
and [2-[[2(4-tert-butyl-2-thiazolyl)-5-benzofuranyl]oxymethyl]phenyl] acetic
acid,
described in U.S. Patent 5,296,495.
Non-limitative examples of R-adrenergic receptor agonists include: albuterol,
bitolterol, isoetharine, mataproterenol, perbuterol, saimeterol, terbutaline,
isoproterenol, ephedrine and epinephrine. Non-limitative examples of a-
adrenergic
receptor agonists include arylalkylamines, (e.g., phenylpropanolamine and
pseudephedrine), imidazoles (e.g., naphazoline, oxymetazoline,
tetrahydrozoline, and
xylometazoline), and cycloalkylamines (e.g., propylhexedrine).
A non-limitative example of a mast cell stabilizer is nedocromil sodium. A non-
limitative example of an expectorant is guaifenesin. Non-limitative examples
of
decongestants are pseudoephedrine, phenylpropanolamine and phenylephrine.
Non-limitative examples of other PDE4 inhibitors include roflumilast,
theophylline, rolipram, piclamist, cilomilast and CDP-840. Examples of
steroids
include prednisolone, fluticasone, triamcinolone, beclomethasone, mometasone,
budisamide, betamethasone, dexamethasone, prednisone, flunisolide and
cortisone.
7
CA 02692776 2010-01-07
WO 2009/009002 PCT/US2008/008260
Non-limitative examples of NK1, NK2 and NK3 tachykinin receptor antagonists
include CP-99,994 and SR 48968. Non-limitative examples of muscarinic
antagonists include ipratropium bromide and tiatropium bromide.
Non-limiting examples of GABAB agonists include baclofen and 3-
aminopropyl-phosphinic acid. Dopamine agonists include quinpirole, ropinirole,
pramipexole, pergolide and bromocriptine.
"5-lipoxygenase inhibitors" include any agent or compound that inhibits,
restrains, retards or otherwise interacts with the enzymatic action of 5-
lipoxygenase.
Non-limitative examples of 5-lipoxygenase inhibitors include zileuton,
docebenone,
piripost, ICI-D2318, and ABT 761.
The compound of Formula I was prepared by the procedure outlined in
Schemes 1 or 2 and detailed in the following Examples 1 or 2. In Example 1 and
elsewhere in the application, Et means ethyl, Me means methyl, THF is
tetrahydrofuran, DMF is N,N-dimethylformamide, t-BOC and BOC mean t-
butoxycarbonyl, RT is room temperature, HATU is N-[(dimethylamino)-1H-1,2,3-
triazolo[4,5-b]pyridin-l-ylmethylene]-N-methylmethanaminium
hexafluorophosphate
N-oxide, MS means mass spectra or mass spectrum In the application, ETOH means
ethanol, NMR means Nuclear Magnetic Resonance, DMSO means dimethyl sulfoxide,
Et3N means triethylamine, NaHMDS is sodiumbis(trimethylsilyl)amide, HOBT is
hydroxybenztriazole, IPA is isopropanol (isopropyl alcohol), EDCI HCI is 1-
ethyl-3-[3-
dimethylamino)propyl]-carbodimide hydrochloride, NMP is N-methylpyn-olidinone,
ca
is circa (about), KF is Karl Fisher, and EtOAc is ethyl acetate.
8
CA 02692776 2010-01-07
WO 2009/009002 PCT/US2008/008260
Scheme 1
OH H OH Br
HN step 1 HN step 2 BOCN step 3 gOCN
-~ -~ ->
COOH 2 COOEt 3 COOEt 4 COOEt
1
step4
HN NH2 N3
0
step 7 BOCN step BOCN step p 5 BOCN
7 COOEt 6 COOEt 5 COOEt
Me Me
I N CF3 N
HN ~ CF3
O
+ step 8 H
HN O \ --~ N O N \N
COOEt O
BOCNH~OH BOCNH~N
8
9 Me O 10 Me O C OO Et
OMe
OMe jNCF3
~ N\ CF3
/ _ / H step 9
~
O N HCf ~N '~N
N O
H2N N
O
H2N N
O COOEt
Me O COOEt Me
Formula I Formula la
Example 1
Step 1:
To a mechanically stirred suspension of compound 1 (100.6 g, 0.767 mol) in
EtOH (1000 ml) and cooled to 0 C was added SOCI2 (136.9 g, 1.15 mol, 84.0 ml)
dropwise via addition funnel such that the internal temperature was < 15 C.
The
reaction mixture was heated at reflux for 2.5 h, then cooled to 0 C. Ether
(1000 ml)
was added, and a white solid precipitated. The solid was isolated by vacuum
filtration
and washed with ether. The product 2 (HCI salt) was dried in a vacuum oven to
give
9
CA 02692776 2010-01-07
WO 2009/009002 PCT/US2008/008260
146.3 g (97%) of a white solid. MS (M+1): m/e 160. 'H-NMR (DMSO) 81.25 (t,
3H),
2.05 (m, 1 H), 2.20 (m, 1 H), 3.05 (d, 1 H), 3.40 (dd, 1 H), 4.20 (q, 2H),
4.45 (m, 2H),
5.65 (broad s, 1 H).
Step 2:
To a solution of compound 2 (HCI salt, 146.2 g, 0.747 mol) dissolved in CH2CI2
(1600 ml) and EtOH (100 ml) and cooled to 0 C was added Et3N (113.4 g, 1.12
mol,
156.2 ml). t-BOC anhydride (195.6, 0.90 mol) was added portionwise. The
reaction
mixture was stirred at 0 C for 15 min, then at RT for 16 h. The resulting
mixture was
concentrated to - 800 ml volume and washed with water. The organic solution
was
dried (MgSO4), filtered, and concentrated. Purification by silica gel
chromatography
(eluant: 20% EtOAc - CH2CI2) gave the product 3 (193.7 g, 100%) as a yellow
oil. MS
(M+Na): m/e 282. 'H-NMR (CDCI3) 81.30 (t, 3H), 1.45 (s, 9H), 1.75 (m, 1 H),
2.10 (m,
1 H), 2.30 (m, 1 H), 3.45 and 3.55 (d, 1 H for two rotamers), 3.65 (dd, 1 H),
4.25 (m, 2H),
4.40 and 4.45 (t, 1 H for two rotamers), 4.55 (broad s, 1 H).
Step 3:
To a solution of compound 3 (36.5 g, 0.141 mol) and triphenyl phosphine (46.2
g, 0.176 mol) dissolved in dry THF (1000 ml) and cooled to 0 C was added
diethyl
azodicarboxylate (30.7 g, 0.176 mol) dropwise via addition funnel. The
reaction
mixture was stirred at 0 C for 5 min, then LiBr (61.1 g, 0.704 mol) was added
in one
portion. The resulting mixture was stirred at RT for 16 h. The solvent was
evaporated, water (1500 ml) was added, and the aqueous solution was extracted
with
CH2CI2. The combined organic extracts was dried (MgSO4), filtered, and
concentrated. Purification by silica gel chromatography (eluant: 2% EtOAc -
CH2CI2
to 5% EtOAc - CH2CI2) gave the product 4 (31.8 g, 70%) as a yellow oil. MS
(M+1):
m/e 322 and 324. 1H-NMR (CDCI3) S 1.30 (m, 3H), 1.45 and 1.50 (s, 9H for two
rotamers), 2.45 (m, 1 H), 2.85 (m, 1 H), 3.75 (m, 1 H), 4.05 - 4.40 (m, 5H).
Step 4:
To a solution of compound 4(41.2 g, 0.128 mol) dissolved in dry DMSO (300
ml) was added NaN3 (9.15 g, 0.141 mol). The reaction mixture was stirred at RT
for
CA 02692776 2010-01-07
WO 2009/009002 PCT/US2008/008260
16 h. Water (300 ml) was added, and the aqueous solution was extracted with
ether.
The combined organic extracts was dried (MgSO4), filtered, and concentrated to
give
the product 5 (36.4 g, 100%) as an oil. MS (M+Na): m/e 307. 1H-NMR (CDCI3) S
1.30
(t, 3H), 1.45 and 1.50 (s, 9H for two rotamers), 2.20 (m, 1 H), 2.35 (m, 1 H),
3.50 and
3.60 (m, 1 H for two rotamers), 3.75 (m, 1 H), 4.15 - 4.45 (m, 4H).
Step 5:
To a solution of compound 5 (36.4 g, 0.128 mol) dissolved in THF (800 ml) was
added 10% palladium on carbon catalyst (10.0 g). The reaction mixture was
shaken
on a Parr shaker under 40 psi of hydrogen pressure for 16 h. The catalyst was
removed by filtration and washed with isopropanol. The filtrate was
concentrated.
Purification by silica gel chromatography (eluant: CH2CI2 then 10% MeOH with
NH3 -
CH2CI2) gave the product 6 (24.2 g, 73%) as a light gray solid. MS (M+1): m/e
259.
'H-NMR (CDCI3) S 1.30 (t, 3H), 1.45 and 1.50 (3, 9H for two rotamers), 2.00
(m, 1 H),
2.15 (m, 1 H), 3.10 and 3.20 (m, 1 H for two rotamers), 3.70 (m, 2H), 4.20 (m,
2H), 4.35
and 4.40 (m, 1 H for two rotamers).
Step 6:
To a solution of compound 6 (12.0 g, 0.0464 mol) dissolved in dry CH2CI2 (300
ml) was added Et3N (9.4 g, 0.093 mol, 13.0 ml) then cyclopropanecarbonyl
chloride
(5.3 g, 0.051 mol, 4.64 ml). The reaction mixture was stirred at RT for 16 h.
Water
(200 ml) was added, and the aqueous solution was extracted with CH2CI2. The
combined organic extracts was dried (MgSO4), filtered, and concentrated.
Purification
by silica gel chromatography (eluant: 5% MeOH with NH3 - CH2CI2) gave the
product
7 (14.3 g, 94%) as an oil. MS (M+Na): m/e 349. 'H-NMR (CDCI3) 6 0.75 (d, 2H),
1.00
(broad s, 2H), 1.30 (t, 3H), 1.35 (m, 1 H), 1.45 and 1.50 (s, 9H for two
rotamers), 2.25
and 2.30 (m, 2H for rotamers), 3.30 and 3.45 (dm, 1 H for rotamers), 3.80 (m,
1 H),
4.15 - 4.45 (m, 3H), 4.55 (m, 1 H), 5.95 and 6.10 (broad singlet, 1 H for
rotamers).
Step 7:
To a solution of compound 7 (40.0 g, 0.123 mol) dissolved in CH2C12 (550 ml)
was added 4 N HCI in dioxane (153 ml, 0.613 mol). The reaction mixture was
stirred
11
CA 02692776 2010-01-07
WO 2009/009002 PCT/US2008/008260
at RT for 4 h then concentrated to give the product 8 (32.2 g, 100%) as a
colorless
foam. MS (M+1): m/e 227. 'H-NMR (CDCI3) S 0.75 (d, 2H), 0.90 (m, 2H), 1.30 (t,
3H),
1.55 (m, 1 H), 2.35 (m, 1 H), 2.55 (m, 1 H), 3.70 (m, 2H), 4.25 (m, 2H), 4.75
(m, 2H),
8.35 (d, 1 H), 9.05 (broad s, 1 H).
Step 8:
To a mixture of compound 8 (5.5 g, 20.8 mmol) and carboxylic acid 9 (10.0 g,
20.8 mmol) in dry DMF (300 ml) was added 3 A sieves (10.0 g), Et3N (6.3 g,
62.3
mmol, 8.7 ml), then HATU (15.8 g, 41.6 mmol). The reaction mixture was stirred
at
RT for 21 h then the solvent was concentrated. Water (400 ml) was added, and
the
aqueous solution was extracted with CH2CI2. The combined organic extracts were
dried (MgSOa), filtered, and concentrated. Purification by silica gel
chromatography
(eluant: 20% EtOAc - CH2CI2 to 60% EtOAc - CH2CI2) gave the product 10 (14.0
g,
98%) as a colorless foam. MS (M+1): m/e 690. See FIG 3 for the NMR spectrum.
Step 9:
To a solution of compound 10 (42.1 g, 0.061 mol) dissolved in CH2CI2 (600 ml)
and cooled to 0 C was added 4 N HCI in dioxane (76 ml, 0.305 mol). The
reaction
mixture was then stirred at RT for 5 h and then concentrated. The crude
product was
dissolved in 1:1 EtOH:H20 (120 ml) and made basic (pH = 9-10) with 25% aqueous
NaOH. CHZCI2 (700 ml) was added, and the reaction mixture was stirred until
all
solids dissolved. The layers were separated, and the aqueous solution was
extracted
with CH2CI2. The combined organic extracts was washed with brine, dried
(MgSO4),
filtered, and concentrated. Additional CH2CI2 was added, and the mixture was
concentrated again. Ether was added, and the mixture was concentrated to give
compound of Formula Ia (34.4 g, 96%) as a light yellow solid. MS (M+1): m/e
590.
12
CA 02692776 2010-01-07
WO 2009/009002 PCT/US2008/008260
Scheme 2.
~N Cf3 \ 3 ~ CH3
I i i step 2 f3
I~ step 1
step 3
----------- D--
H3CS N ~ ~-
\COzEt gpcHN J"Z
12 C02g axNNH
13 9
H3 CH~
N f3 N f' OCH3
~ N f3
i
N O smp' `N HN g~PS
BocHN~N` J O ~' Q ~N H~
O
O ,C(OxEt ~NN~ ~N~! lrN LHcij
COiEt
0 C02g
Formula la Formula I
Example 2
Step 1:
Into a 50 L Hastelloy reactor equipped with a thermocouple, N2 inlet and feed
tank was charged 8.8 kg (46.5 moles, 2 eq) of (S)-2-tert-butoxycarbonylamino-
propionic acid and 90 liters dry tetrahydrofuran (THF, KF < 0.05%) to dissolve
the
acid. Into the reactor was slowly charged 8.5 kg (46.9 moles, 2 eq) of
dicyclohexylamine over about 30 minutes while maintaining the temperature or
the
reaction mixture from about [-5 C] to about [+5 C]. The mixture was agitated
for about
minutes while maintaining the reaction mixture from about [-5 C] to about [+5
C].
At the end of the agitation period, 5.7 kg (47.3 moles, 2 eq) of
trimethylacetylchloride
was charged into the reaction mixture over about 30 minutes while maintaining
the
temperature of the reaction mixture from about [-5 C] to about [+5 C]. The
mixture
was agitated for about 3 hours while maintaining the temperature of the
reaction
mixture from about [-5 C] to about [+5 C]. At the end of the agitation period
the
reactor was charged with 27 liters of heptane, followed by 4.5 kg of celite.
The
reaction mixture was filtered under N2, and the filter cake thus obtained was
washed
with 30 % v/v THF in heptane. The filtrate and washes were combined and
concentrated by distillation under vacuum to a batch volume of about 36
liters. The
13
CA 02692776 2010-01-07
WO 2009/009002 PCT/US2008/008260
concentrated reaction mixture was diluted with 27 liters of THF and the
temperature of
the mixture was adjusted to a temperature between 20 C and 30 C. A sample of
the
reaction mixture was tested for residual water by Karl Fischer titration and
found to be
less than about 0.06 ppm. The mixed anhydride/THF solution thus obtained was
used
in the next step without further purification.
Into a 50 gallon glass lined reactor equipped with a thermocouple, N2 inlet
and
feed tank was charged 9.0 kg (23.3 moles, 1 eq) of the compound (12) and 126
liters
dry tetrahydrofuran (THF, KF < 0.05%), with agitation to dissolve the compound
of
Formula (12). The reaction mixture was concentrated by distillation at 1
atmosphere
to a batch volume of about 81 liters. The temperature of the concentrated
reaction
mixture was adjusted and maintained at a temperature of from [-60 C] to [-70
C]. Into
the reaction mixture was charged NaHMDS (2M in THF, 2.70 kg, 5.9 moles, 0.25
eq)
over about 15 minutes while maintaining the temperature of the reaction
mixture. At
the end of the addition period the reaction mixture was agitated for 5 minutes
while
continuing to maintain the temperature. Following agitation the reaction
mixture was
charged over a period of 15 mintues with the mixed anhydride/THF solution
prepared
previously (0.83 kg active anhydride, 3.2 moles, 0.14 eq) while maintaining
the
reaction mixture at a temperature of from [-60 C] to [-70 C], following which
the
mixture was agitated for about 10 minutes while continuing to maintain the
temperature. Two additional charges of (NaHMDS 2M in THF), each followed by a
charge of the mixed anhydride/THF solution were preformed, followed by five
(5)
additional charges of the anhydride/THF solution for a total of eight (8) sets
of charges
or until the conversion is ? 70 %. With each charge an agitation period was
carried
out and the temperature of the reaction mixture was maintained at a
temperature of
from [-60 C] to [-70 C] throughout the charging and agitating period.
Charging
NaHMDS (2M in THF) followed by the mixed anhydride/THF solution in the same
ratio
based on the amount of starting material remaining was until the a conversion
of z 94
% was observed. When the conversion exceeded about 94%, the reaction mixture
was transferred slowly, over about 15 minutes, to an aqueous solution of 13.5
kg
KH2PO4 dissolved in 90 liters H20 whilst maintaining the batch temperature
below
30 C. At the end of the addition period, to the resulting mixture was charged
59 liters
ethyl acetate and the mixture was agitated for about 15 minutes, then the
layers were
allowed to settle. The layers were separated and the aqueous layer extracted
with 45
14
CA 02692776 2010-01-07
WO 2009/009002 PCT/US2008/008260
liters ethyl acetate. The ethylacetate wash was separated and the combined
wotj the
separated organic layer. The combined organics were washed two times with 32
liters
10% aqueous w/v NaCI then concentrated at 1 atmosphere to a volume of about 45
liters. To the concentrate was charged 90 liters methyltertbutylether (MTBE)
and the
mixture was concentrated at 1 atmosphere to a batch volume of about 54 liters.
To
the concentrate was charged 45 liters methyltertbutylether followed by 108
liters of
heptanewhile maintaining the reaction mixture at a temperature of from 55 C
to 65 C.
The temperature of the reaction mixture was adjusted to a temperature of from
45 C
to 55 C and agitate for about 30 minutes while maintaining the temperature. At
the
end of the agitation period the temperature of the reaction mixture was
adjusted to a
temperature of from [-5 C] to [+5 C] over a 1 hour period and agitated for an
additional
30 minutes while maintaining the temperature. At the end of the agitation
period the
reaction mixture was filtered and the filter cake thus obtained was washed
with 33%
v/v methyltertbutylether in heptane. The filter cake was dried in a vacuum
oven for 12
hours at 45 to 55 C affording 8.4 kg (72.2 %) of the compound of Formula (13)
as a
solid with an ee of >99.0 %.
' H NMR (400 MHz, CDCI3); 9.89 (1 H, d); 8.56 (1 H, d); 7.94 (1 H, d); 7.22 (1
H, d); 5.91
(1 H, s,b); 5.58 (1 H, s, b); 4.47 (2H, q); 4.43 (3H, s); 3.75 (2H, t); 1.47
(9H, s); 1.19
(9H, s).
Step 2:
Into a 500 mL three-neck round bottom flask fitted with a mechanical stirrer,
an
additional funnel and a thermocouple was placed 20 g (39.3 mmol, 1 eq) of
compound
(13) followed by 60 ml of THF, 20 mL of EtOH, and 100 mL of water. The flask
was
then charged with 8 mL of 25% sodium hydroxide solution and agitated for about
4
hours while maintaining the the temperature of the reaction mixture at 40 C.
Upon
judging the reaction complete by HPLC assay, 100 ml of water was added to the
reaction mixture and the reaction mixture was heated to 50 C. Once at 50 C, to
the
reaction mixture was added 30 ml 1 N HCI solution over 30 minutes. At the end
of the
addition period the reaction mixture was stirred for an additional 30 minutes
while
maintaining the temperature at 50 C, following which another 24 ml 1 N HCI
solution
was added to the reaction mixture over 30 minutes. At the end of the agitation
period
60 ml of water was added to the reaction mixture over 30 minutes while
continuing to
CA 02692776 2010-01-07
WO 2009/009002 PCT/US2008/008260
maintain the temperature of the reaction mixture at 50 C. At the end of the
addition
period the reaction mixture was cooled to room temperature over 1 hour,
precipitating
a product. The precipitated solids were collected from the reaction mixture by
suction
filtration and the wet cake collected was washed with 40 ml 1:5 v/v mixed
ethanol and
water. The solids were dried under vacuum at 60 C for 12h affording 16.8 g
(90%) of
compound (9) as an off white solid.
' H NMR (400 MHz, d6-DMSO): 9.97 (1 H, d), 8.42 (1 H, d), 8.20 (IH, d), 7.48
(1 H, d),
5.40 (1 H, m), 4.07 (3H, s), 1.45 (3H, d), 1.30 (9H, s)
Step 3:
Part A:
Into a vessel was placed 60g (184 mmol, 1 eq) (2R, 4S)-
4(cyclopropanecarbonyl-amino)-pyrrolidine-1,2-dicarboxylic acid -1-tert-butyl
ester 2-
ethyl ester (BP) dissolved in 1.2 L EtOAc. This solution was sampled for use
as an
HPLC 100% standard. The solution was cooled to a temperature of from 20 C to
35 C and 36 g (980 mM, 5.3 eq) of HCI(g) was added while maintaining the
reaction
mixture temperature at a temperature of from 20 C to 35 C, forming an HCI
salt
precipitate. When the entire amount of HCI was charged, the reaction mixture
was
heated to a temperature of from 20 C to 30 C and agitated for 1 h. After 1
h, the
progress of the reaction was checked for completion by HPLC response of the
reaction mixture in comparison with the standard initially sampled. The
reaction was
continued and sampling repeated until the amount of (BP) relative to standard
was
s0.5% area. The reaction mixture was concentrated under vacuum by distillation
with
the reaction mixture maintained at a temperature of from 35 C to 45 C to a
volume
of 600 mL, forming a thick slurry. NMP (280 mL) was then added to the reaction
mixture and it was further concentrated under the batch under vacuum by
distillation
with the reaction mixture maintained at a temperature of from 35 C to 45 C
to a
volume of about 560mL forming a clear solution. This solution was used
directly in the
Part B coupling step.
16
CA 02692776 2010-01-07
WO 2009/009002 PCT/US2008/008260
Part B:
Into a 1 L 3-neck round-bottom flask was placed 320 mL of EtOAc, and
dissolved therein 80 g of compound (9) (166 mmol, 1 eq), 28 g HOBT=H2O(182
mmol,
1.1 eq) and 48 g EDCI-HCI (250 mmol, 1.4 eq) in NMP (320 mL). The reaction
mixture was stirred for 40 minutes while maintaining the reaction mixture at a
temperature of 25 C. The entire amount of the solution of BP prepared in part
A
(above) was added to the reaction mixture with stirring. The reaction mixture
was
stirred for 10 min and 80 mL of N-methyl morpholine (724 mmol, 4.4 eq) was
added to
reaction at a rate which maintained the reaction mixture at a temperature
below 35 C.
The reaction was monitored by HPLC until a complete reaction was indicated,
and
320 mL of EtOAc and 800 mL of water was added to the reaction mixture. The
resultant mixture was stirred for 15 min. additional and the layers were
separated.
The organic layer was washed with 1 M HCI (400 mL), followed by 10% K2C03 (400
mL) and then water (400 mL). The organics were concentrated to a volume of 160
mL
and 800 mL of acetone was added. The mixture was concentrated to -240 mL by
distillation under reduced pressure while maintaining the reaction mixture at
a
temperature of from 40 C to 50 C. The mixture was diluted with another 800
mL of
acetone and again concentrated to a volume of 240 mL by distillation under the
same
conditions. Following concentration, 800 mL of heptanes was added to the
concentrate while maintaining its temperature at 40 C, precipitating a
product. The
product solids were collected by filtration and dried under vacuum at 50 C for
12 h to
afford (103 g, 90%) of (10) as an off white solid.
NMR (400 MHz, d6-DMSO): 9.55, 9.03, 8.18, 7.90, 7.77, 7.66, 7.10, 7.04, 6.70,
6.66,
6.10, 5.76, 5.36, 4.91, 4.80, 4.4-3.5, 2.58, 2.30, 1.82, 1.56, 1.47, 1.31,
1.07, 1.001.84,
0.74. Note: due to the presence of rotomers, the observed peaks are listed as
observed only.
Step 4:
Compound (10) (20 g, 29 mmol, 1 eq) was charged to a flask then dissolved in
60 mL of THF, and the solution was cooled to 0-10 C. Concentrated HCI (20m1)
was
added slowly to maintain the temperature at 0-20 C. At the end of the charge,
the
solution was warmed and maintained at a temperature of from 20 C to 30 C.
The
reaction mixture was agitated for 4 hours, until the reaction was determined
to be
17
CA 02692776 2010-01-07
WO 2009/009002 PCT/US2008/008260
complete by HPLC analysis. The reaction mixture was diluted with 2-Me-THF
(120m1)
and THF (40m1) and the reaction was quenched with 20% K2C03 (110mI) until a pH
of
8-8.5 was observed. After adjusting pH, more water (80ml) was added and the
batch
was heated to about 30 C to achieve a clean phase split. The batch was
settled for
about 15 min, the lower aqueous layer separated, and the organic layer was
washed
with water (80m1). The organic phase was diluted with 2-Me-THF (200m1) and
then
concentrated under reflux at atmospheric pressure to about 100mI. The solid
product
was observed at this volume. The batch was then cooled to 0-10 C and
filtered. The
wet cake was washed 2 times with 2-Me-THF (40m1 each time). The wet cake was
dried for at least 12 h at 60 C under vacuum affording 13.50g (79%) of the
compound
of Formula (Ia) as a white solid.
'H NMR (spectrum indicates rotomers, only chemical shift is reported, not
integration
or peak muitiplicity; 400 MHz, d6-DMSO) 6 9.82, 9.62, 8.51, 8.38, 8.07, 7.45,
5.46,
4.69, 4.57, 4.33, 4.15, 4.08, 3.99, 3.83, 2.39, 2.26, 2.16, 1.56, 1.44, 1.22,
0.82, 0.69;
MSES+ mlz (relative intensity) 590 (M+H).
Step 5:
Crystalline Form: A solution of The compound of Formula (Ia) (22.0 g, 0.0373
mol) dissolved in hot EtOH (440 ml) was filtered and rinsed with EtOH (44 ml).
The
solution was heated to reflux and HCI in IPA (5-6N, 10.2 ml) was added. After
refluxing for about 30 minutes, the mixture was cooled to about 50 C and
vacuum
distilled to about 220 ml. The mixture was then cool to 0 C over about 50
minutes and
agitated for about 30 minutes. The mixture was filtered and washed with EtOH
(132
ml). The solid was dried under vacuum at 45 C to give 20.8 g (89%) of the
compound
of Formula I as a white solid.
18
CA 02692776 2010-01-07
WO 2009/009002 PCT/US2008/008260
Hydrogen Chloride salt formation:
A hydrogen chloride salt having a preferred crystalline form can be prepared
according to the following method:
a) providing a solution of the compound of Formula Ia in ethanol at
reflux;
b) adding HCI in IPA to the solution provided in Step "a";
c) refluxing the mixture for about 30 minutes;
d) allowing the mixture to cool to about 50 C and applying vacuum
distillation to remove excess ethanol;
e) cooling the mixture to 0 C; and
f) filtering the mixture and washing and drying to solid to yield the
compound of Formula I (a hydrogen chloride salt of the compound of
Formula Ia).
Powder X-Ray Diffraction Sample Preparation
The crystalline form of the compound of Formula I was analyzed as a dry
powder for powder x-ray diffraction ("PXRD") analyses.
Powder X-Ray Diffraction (PXRD):
X-ray powder diffraction patterns were collected on Rigaku Miniflex
Dffractometer with CuKl source (X=1.5406A) at 30 kV, 15 mA and a solid state
detector. A continuous scan was recorded with a step size of 0.02 20 and a
step time
of 2 /min.
Using the method and equipment described above, the compound of Formula I
was analyzed by PXRD (Figure I) and the characteristic peaks of the powder
pattern
shown in Figure I are tabulated in Table I, below. The intensity of the peaks
(y-axis is
in counts per second) is plotted versus the 20 angle (x-axis is in degree 2
0). In Table
2, the four most characteristic peaks from Table I are shown as group I, the
eight most
characteristic peaks from Table I are shown as group 2, and the 12 most
characteristic peaks from Table I are shown as group 3.
19
CA 02692776 2010-01-07
WO 2009/009002 PCT/US2008/008260
Table I
PXRD Peak Positions for the Compound of Formula I
Peak Location Intensity
(deg. 20) (Cps)
7.9 2073
8.5 848
9.1 151
10.7 587
13.1 263
13.8 1089
14.2 584
15.6 1794
16.0 517
17.2 1189
18.3 1117
18.5 1675
19.7 1068
20.1 307
20.4 222
20.7 203
21.4 512
21.7 629
22.2 182
22.5 172
23.4 3130
24.4 259
25.4 513
26.3 296
26.7 439
27.2 914
27.6 709
28.4 611
29.3 808
30.5 148
30.9 677
31.3 497
32.0 135
CA 02692776 2010-01-07
WO 2009/009002 PCT/US2008/008260
32.5 628
34.2 491
35.3 120
35.9 243
38.4 140
39.0 102
Table 2
Characteristic PXRD Peak Locations for HCI Salt Form Compound of Formula I
Peak Location Group Peak Location
Number (deg. 20)
7.9
15.6
1
18.5
23.4
7.9
13.8
15.6
17.2
2
18.5
19.7
23.4
27.2
7.9
8.5
13.8
15.6
17.2
18.5
3
19.7
23.4
27.2
27.6
29.3
30.9
21
CA 02692776 2010-01-07
WO 2009/009002 PCT/US2008/008260
It will be appreciated exact PXRD peak locations and intensities for a given
crystalline form of the same compound will vary within a margin of error
depending
upon differences in sample preparation, instrumentation, analytical technique,
and
other factors, however, when these factors are taken into account, PXRD
analysis of a
compound will yield powder pattems having substantially the same
characteristic
peaks.
Differential Scanning Calorimetry (DSC):
Differential scanning calorimetry data (FIG. 2) was generated with a Q100
Differential Scanning Calorimeter from TA instruments. Samples were sealed in
hermetic aluminum pans and two pinholes were punched in the lids of the sample
pans. Analysis was conducted under a nitrogen purge with a heating rate of 10
C per
minute.
The heat flow which was normalized by a sample weight was plotted versus the
measured sample temperature. The data were reported in units of watts/gram
("W/g"). The plot was made with the endothermic peaks pointing down.
The DSC profile for the crystalline form of the compound of Formula I is shown
in FIG 2. A single melting endotherm was observed with onset temperature of
252 C
and peak temperature of 263 C. The heat of fusion cannot be determined due to
the
onset temperature of decomposition in TGA data prior to the completion of
melting.
The premise for treatment by inhalation is to deliver the drug directly to the
site
of action (the lungs) with minimal systemic side effects. Therefore, an
inhaled
compound should exhibit a pharmacokinetic profile with low blood concentration
(AUC) due to low oral bioavailability and / or high clearance when given by
inhalation
or oral dosing routes. It is important that oral AUC be low in order to
minimize the
effect of any swallowed drug during inhalation.
Pharmaceutical Compositions
For preparing pharmaceutical compositions from the compound of Formula I,
preferably the crystalline hydrogen chloride salt described by this invention,
inert,
pharmaceutically acceptable carriers can be either solid or liquid. Examples
of
pharmaceutically acceptable carriers and methods of manufacture for various
22
CA 02692776 2010-01-07
WO 2009/009002 PCT/US2008/008260
compositions may be found in A. Gennaro (ed.), Remington's Pharmaceutical
Sciences, 18th Edition, (1990), Mack Publishing Co., Easton, Pennsylvania.
Liquid form preparations include solutions, suspensions and emulsions for
intranasal administration.
Aerosol preparations suitable for inhalation may include solutions and solids
in
powder form, which may be in combination with a pharmaceutically acceptable
carrier,
such as an inert compressed gas, e.g. nitrogen.
Dosages
The quantity of active compound in a unit dose of preparation may be varied or
adjusted from about 0.01 g to about 100 mg, preferably from about 0.01 g to
about
75 mg, more preferably from about 0.01 g to about 50 mg, and most preferably
from
about 0.01 g to about 25 mg, according to the particular application.
The actual dosage employed may be varied depending upon the requirements
of the patient and the severity of the condition being treated. Determination
of the
proper dosage regimen for a particular situation is within the skill of the
art. For
convenience, the total dosage may be divided and administered in portions
during the
day as required.
The amount and frequency of administration of the compounds of the invention
and/or the pharmaceutically acceptable salts thereof will be regulated
according to the
judgment of the attending clinician considering such factors as age, condition
and size
of the patient as well as severity of the symptoms being treated. A typical
recommended daily dosage regimen for inhalation can range from about 0.04 g
/day
to about 400 mg/day, in one to four divided doses.
Other than as shown in the operating examples or as otherwise indicated, all
numbers used in the specification and claims expressing quantities of
ingredients,
reaction conditions, and so forth, are understood as being modified in all
instances by
23
CA 02692776 2010-01-07
WO 2009/009002 PCT/US2008/008260
the term "about." The above description is not intended to detail all
modifications and
variations of the invention. It will be appreciated by those skilled in the
art that
changes can be made to the embodiments described above without departing from
the inventive concept. It is understood, therefore, that the invention is not
limited to
the particular embodiments described above, but is intended to cover
modifications
that are within the spirit and scope of the invention, as defined by the
language of the
following claims.
24