Note: Descriptions are shown in the official language in which they were submitted.
CA 02692919 2010-01-08
WO 2009/010231 PCT/EP2008/005638
Process for the production of tertiary alcohols
The invention relates to a process for the production of tertiary alcohol of
formula
OH
Q Rl (I)~
wherein R' is CI-4 alkyl and Q is C1_1o alkyl, C2_lo alkenyl, C3_8 cycloalkyl,
aryl or heteroaryl or
an organic moiety composed of any two or more of the beforementioned, each
C1_1o alkyl, C2_10
1o alkenyl, C3_8 cycloalkyl, aryl and heteroaryl optionally being substituted
with one or more sub-
stituents independently selected from the group consisting of hydroxy,
fluorine, chlorine, amino,
CI-4 alkylamino and di(C1-4 alkyl)amino.
Tertiary alcohols having two lower alkyl groups at the carbinol carbon are
valuable inter-
mediates in the syntheses of several pharmaceutically active compounds. For
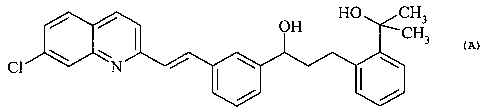
example,
(aS)-a-[3-[(lE)-2-(7-chloro-2-quinolinyl)ethenyl]phenyl]-2-(1-hydroxy-l-
methylethyl)-
benzenepropanol of formula
HO CH3
OH CH3
\ / I \ \
C1 N
is a key intermediate in the synthesis of the pharmaceutically active compound
known as
montelukast (1-[[[(1R)-1-[3-[(lE)-2-(7-chloro-2-quinolinyl)ethenyl]phenyl]-3-
[2-(1-hydroxy-
1-methylethyl)phenyl]propyl]thio]methyl]cyclopropaneacetic acid).
A well known synthesis of tertiary alcohols is the reaction of carboxylic
esters with two
equivalents of a Grignard reagent. However, the yields are often not
satisfactory as undesired
reactions compete with the formation of the alcohol and result in the
formation of byproducts,
in particular when an alkylmagnesium chloride is used as Grignard reagent. It
has recently been
found that "nearly anhydrous" activated cerium trichloride has a beneficial
effect on the above
reaction, which has been postulated to be due to suppression of the
enolization of the ketone
intermediate (D. A. Conlon et al., Adv. Synth. Catal. 2004, 346, 1307-1315).
The water content
and activation method of the cerium trichloride as well as its crystal habit
have been found to be
critical. Moreover, the activation of the cerium chloride is somewhat tedious
and the activated
cerium chloride is sparingly soluble in ethereal solvents such as
tetrahydrofuran which results
in a heterogeneous reaction mixture. In the preparation of the above
montelukast intermediate
the starting material (which is available as a monohydrate) has first to be
carefully dried (e.g. by
CONFIRMATION COPY
CA 02692919 2010-01-08
WO 2009/010231 PCT/EP2008/005638
-2-
azeotropic distillation), but nevertheless, about 5 equivalents of
methylmagnesium chloride are
required, instead of the theoretical amount of 3 equivalents (WO 95/18107 Al).
EP-A-1 759 765 discloses solutions of anhydrous lanthanide salts of formula
MX3=z LiA, such
as LaC13=2 LiCI, and their use in Grignard-type reactions, in particular with
ketones and imines.
In the case of ketones, said lanthanide salts are employed in equimolar
amounts and examples
are given where carboxylic ester moieties are unaffected. The addition of a
trace of water to the
reaction mixture is said to initiate a precipitation of the lanthanide salt.
It is an object of the present invention to provide an improved method for the
preparation of
to tertiary alcohols from carboxylic esters and Grignard reagents which gives
high yields of the
desired product even if the chloride form of the Grignard reagent is used and
even if the starting
material is used in its hydrate form. The method should not involve tedious
activation steps,
heterogeneous reaction mixtures and cumbersome work-up procedures.
Applicants have found that tertiary alcohols of formula
OH
Q 1 Rl (I)~
wherein R' is C1-4 alkyl and Q is C1_10 alkyl, C2_10 alkenyl, C3_8 cycloalkyl,
aryl or heteroaryl or
an organic moiety composed of any two or more of the beforementioned, each
C1_10 alkyl,
C2_10 alkenyl, C3_8 cycloalkyl, aryl and heterocyclyl, optionally being
substituted with one or
more substituents independently selected from the group consisting of hydroxy,
fluorine,
chlorine, amino, C1-4 alkylamino and di(C1-4 alkyl)amino can be prepared by
reacting a carbo-
xylic ester of formula
O
Q O "."R (II),
wherein R is C1_lo alkyl, aryl or arylalkyl,
with a Grignard reagent of formula
R'MgX (III),
wherein R' is as defined above and X is chlorine, bromine or iodine,
in an ethereal solvent in the presence of lanthanum trichloride and lithium
chloride.
Here and hereinbelow, the term "C1_õ alkyl" is to be understood to comprise
any linear or
branched alkyl group having from 1 to n carbon atoms. For example, the term
"C1-4 alkyl" com-
prises methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl and tert-
butyl. In addition to
CA 02692919 2010-01-08
WO 2009/010231 PCT/EP2008/005638
-3-
the beforementioned, the term "Cl_lo alkyl" comprises groups such as pentyl,
isopentyl, neo-
pentyl, hexyl, isohexyl, heptyl, octyl, nonyl, decyl and the like.
The term "C2_10 alkenyl" comprises any linear or branched hydrocarbyl group
having from 2 to
carbon atoms and at least one carbon-carbon double bond.
5 The term "C3_8 cycloalkyl" is to be understood to comprise any mono- or
bicyclic cycloali-
phatic group having from 3 to 8 carbon atoms, such as cyclopropyl, cyclobutyl,
cyclopentyl,
cyclohexyl, cycloheptyl, cyclooctyl, norbornyl, norcaryl and the like.
The term "aryl" is to be understood to comprise any mono-, bi- or
polycarbocyclic group com-
prising at least one aromatic ring, such as phenyl, naphthyl, anthracenyl,
phenanthryl, biphenyl-
10 yl, fluorenyl, tetrahydronaphthalenyl and the like. A preferred meaning of
"aryl" is phenyl.
The term "heterocyclyl" comprises any aromatic and non-aromatic heterocyclic
groups, such as
tetrahydropyranyl, tetrahydrofuranyl, piperidinyl, pyrrolidinyl, morpholinyl,
pyranyl, furanyl,
thiophenyl, pyrrolyl, pyridyl, pyrazinyl, pyrimidinyl, oxazolyl, thiazolyl,
indolyl, quinolinyl,
carbazolyl and the like. Preferred meanings of "heterocyclyl" are pyridyl and
quinolinyl.
The expression "organic moiety composed of any two or more of the
beforementioned" is to be
understood to mean any organic moiety having one free (open) valency that
comprises two or
more of the beforementioned groups, for example arylalkyl or alkylaryl,
(arylalkyl)aryl, (aryl-
alkenyl)aryl, [(alkenylaryl)alkyl]aryl, [[(heterocyclylalkenyl)aryl]alkyl]aryl
and the like.
Each C1_10 alkyl, C2_lo alkenyl, C3_8 cycloalkyl, aryl and heteroaryl
occurring alone or as a com-
ponent of an organic moiety composed of two or more of these groups, as
described above, may
independently be substituted with one or more substituents selected from the
group consisting
of hydroxy, fluorine and chlorine.
The term "ethereal solvent" is to be understood to include any solvent or
solvent mixture com-
prising a substantial amount of an acyclic or cyclic ether that is liquid at
the reaction tempera-
ture, such as diethyl ether, dibutyl ether, methyl tert-butyl ether,
dimethoxyethane, tetrahydro-
furan (THF), 2-methyltetrahydrofuran, 1,4-dioxane and the like. It also
includes cyclic acetals
such as 1,3-dioxolane or 1,3-dioxane.
The lithium chloride solubilizes the lanthanum trichloride, resulting in a
true solution of the two
salts in the ethereal solvent and thus in a homogeneous reaction mixture. In a
preferred embodi-
ment, lanthanum trichloride and lithium chloride are present in a molar ratio
of 1:2 or less. A
THF solution of LaC13 and LiCl in a molar ratio of 1:2 is commercially
available from Che-
metall GmbH, Frankfurt (Main), Germany.
The alkyl group R' of the Grignard reagent III is preferably methyl.
The halogen component X of the Grignard reagent III is preferably chlorine.
CA 02692919 2010-01-08
WO 2009/010231 PCT/EP2008/005638
-4-
In a preferred embodiment, the organic moiety Q of the tertiary alcohol I and
the carboxylic
ester II comprises at least one aryl group. More preferably, the carboxylate
group of the carbo-
xylic ester II is directly bound to an aryl group.
In a still more preferred embodiment, Q is the group of formula
OH
C1 N
and the carboxylic ester II is
OH O, R
Cl N
wherein R is as defined above,
to yield the tertiary alcohol I of formula
HO CH3
OH CH3
C1 N
Most preferably, the secondary alcohol groups of the above structures have S-
configuration to
make them suitable as intermediates in the synthesis of (R)-montelukast.
In a preferred embodiment, the preferred carboxylic ester depicted above is
used in the mono-
hydrate form, thus rendering a separate drying step superfluous. The water of
crystallization
simply reacts with one equivalent of the Grignard reagent to yield the
corresponding alkane and
magnesium hydroxyhalide. This is surprising in view of EP-A-1 759 765 which
stated that even
traces of water initiate precipitation of the lanthanide salt.
When the monohydrate form of the ester is used as starting material, the
lanthanum trichloride
is advantageously used in a molar ratio of lanthanum trichloride to carboxylic
ester (II) of from
1.5:1 to 1:2.
CA 02692919 2010-01-08
WO 2009/010231 PCT/EP2008/005638
-5-
In another preferred embodiment, the preferred carboxylic ester depicted above
is used in the
anhydrous form which may be obtained by azeotropic dehydration of the
monohydrate using a
suitable entraining agent such as toluene. It has been found that it is
possible to directly use the
solution obtained by azeotropic removal of the water of crystallization and to
add said solution
to a solution comprising the Grignard reagent, the lanthanum trichloride and
the lithium
chloride.
When the anhydrous form of the ester is used as starting material, the amount
of lanthanum
trichloride can be reduced to a preferred molar ratio of lanthanum trichloride
to carboxylic ester
lo (II) of from 1:1 to 1:10, more preferably from 1:2 to 1:10 or from 1:3 to
1:10.
The starting carboxylic ester II is preferably a methyl ester.
The ethereal solvent used in the process of the invention is preferably
tetrahydrofuran alone or a
mixture of tetrahydrofuran and an inert solvent such as an aliphatic or
aromatic hydrocarbon.
Also preferred are 2-methyltetrahydrofuran and 1,3-dioxolane.
The reaction temperature can be in the range that is commonly employed in
Grignard reactions,
it is preferably between -20 C and room temperature, more preferably from -10
C to +10 C.
The work-up of the reaction mixture can be accomplished according to the
methods commonly
used in the art, e.g. by quenching with water or weak aqueous acids and
extracting the product
with a suitable solvent.
The following non-limiting examples will illustrate the process of the
invention.
Example 1
(aS)-a-[3-[(lE)-2-(7-Chloro-2-quinolinyl)ethenyl] phenyl]-2-(1-hydroxy-l-
methylethyl)-
benzenepropanol
In a 50 mL three necked flask equipped with magnetic stirrer, a solution of
lanthanum trichlo-
ride and lithium chloride (molar ratio 1:2) in THF (3.09 g of a 16 wt.%
solution, 2.00 mmol)
was diluted with THF (6.0 mL). Methyl 2-[(35)-3-[3-[(1E)-2-(7-chloro-2-
quinolinyl)ethenyl]-
phenyl]-3-hydroxypropyl]benzoate monohydrate (0.916 g, 2.00 mmol; prepared
according to
EP 0 480 717 Al, Example 146, Step 2) was added, and after 1 h stirring at
room temperature
under nitrogen the mixture was cooled to -5 C. Methylmagnesium chloride (3 Nt
solution in
THF, 3.4 mL, 10 mmol) was added dropwise while the temperature was not allowed
to exceed
-5 C. The mixture was then stirred at 0 C for 12 h and after warming to room
temperature
CA 02692919 2010-01-08
WO 2009/010231 PCT/EP2008/005638
-6-
saturated aqueous ammonium chloride solution (10 mL) was added at such a rate
as to maintain
the temperature below 25 C. Water (10 mL) and Toluene (20 mL) were added and
the result-
ing suspension was filtered through a sintered glass filter. The phases were
separated and the
aqueous phase was extracted with toluene (20 mL) and discarded. The combined
organic phases
were washed with water (5 mL) and evaporated in vacuo (50 mbar, 40 C) to
leave a residue of
3.1 g. The residue was triturated at 50 C with heptane (3 mL) and then cooled
to 20 C during
3 h. The precipitated product was isolated by filtration at 20 C, washed
first with heptane/tolu-
ene (1:1 v/v, 4 mL), then with heptane (4 mL), and finally dried at 40 C to
yield 0.63 g of the
desired product.
'H NMR (DMSO-d6, 500 MHz): S= 1.51 (s, 3H); 1.52 (s, 3H); 2.00 (m, 2H), 2.96
(m, 1H);
3.10 (m, 1 H); 4.72 (m, 1 H); 4.94 (s, 1 H); 5.3 6 (d, J= 4.4 Hz, 1 H); 7.09
(t, J= 7.6 Hz, 1 H);
7.14 (t, J= 7.8 Hz, 1 H); 7.18 (d, J= 6.4 Hz, 1 H); 7.41 (m, 2H); 7.44 (d, J=
7.9 Hz, 1 H);
7.49 (d, J= 16.6 Hz, 1 H); 7.5 6 (dd, J= 8.3, 2.2 Hz, 1 H); 7.62 (d, J= 6.8
Hz, 1 H); 7.77 (bs, 1 H);
7.91 (d, J= 16.6 Hz, 1H); 7.92 (d, J= 8.7 Hz, 1 H); 7.99 (d, J= 8.8 Hz, 1H);
8.03 (d, J= 2.0 Hz,
1 H); 8.3 8 (d, J= 8.4 Hz, 1 H).
13C NMR (DMSO-d6, 126 MHz): 8 = 29.82, 31.55, 31.57, 42.34, 71.60, 72.31,
120.24, 124.73,
124.88, 125.24, 125.51, 125.71, 126.22, 126.54, 127.16, 128.04, 128.49,
129.65, 130.82, 134.23,
135.20, 135.67, 136.43, 140.25, 146.66, 146.93, 147.99, 156.78.
Examples 2-4
The procedure of Example 1 was repeated using different amounts of lanthanum
trichloride (0.5,
1.0 and 1.5 molar equivalents) and 10 molar equivalents of methylmagnesium
chloride (instead
of 5 molar equivalents). The yield of the desired product was determined by
HPLC.
The observed yields were as follows:
0.5 equivalents LaC13: 88.3%
1.0 equivalents LaC13: 94.9%
1.5 equivalents LaC13: 98.5%
Example 5
(aS)-a-[3-[(1E)-2-(7-Chloro-2-quinolinyl)ethenyl] phenyl]-2-(1-hydroxy-l-
methylethyl)-
benzenepropanol
In a first 500 mL reaction vessel a 14.7 wt.% solution of LaC13/LiCI in THF
(14.03 g, 8.4 mmol,
0.2 equiv) was diluted with THF (30 mL). A 3.0 tvt solution of MeMgCI (71.43
g, 210 mmol,
5 equiv) was added to the solution at room temperature to obtain a first
reaction mixture. The
solution was cooled to -9 C. In a second 500 mL reaction vessel a mixture of
methyl
2-[(3S)-3-[3-[(lE)-2-(7-chloro-2-quinolinyl)ethenyl]phenyl]-3-
hydroxypropyl]benzoate mono-
CA 02692919 2010-01-08
WO 2009/010231 PCT/EP2008/005638
-7-
hydrate (20.0 g, 42.02 mmol, 1 equiv) was added to toluene (200 mL). Water in
the reaction
mixture was removed by azeotropic distillation (50 C, 100 mbar) until the
volume was reduced
to 60 mL. THF (40 mL) was added to the distillation residue in order to obtain
a clear solution.
Subsequently, the solution was cooled to 20 C and transferred into the first
reaction mixture
while keeping the internal temperature of the first reaction vessel in the
range of -9 C to -5 C.
The reaction mixture was allowed to stand for an additional 1.5 h while the
reaction progression
was monitored by HPLC. After completion of the reaction the solution was
cooled to -15 C
and was quenched by addition of 4 Nt aqueous acetic acid (128 mL) while
keeping the internal
temperature below 10 C. The resulting biphasic system was kept to 10 C.
Toluene (50 mL)
was added and the system was stirred for 15 min at 10 C, settled for 5 min at
10 C to give a
clear two phase separation. The organic layer was then separated and washed
with a 10 wt.%
solution of Na2CO3 (104 mL) at 10 C and with a 10 wt.% solution of NaCI (104
mL) at 10 C.
The organic phase was concentrated (to 60 mL) in vacuo (40 C, 150 mbar). The
distillation
residue was heated to 60 C and heptane (15 g) was added over 10 min followed
by seeding
with crystals of the desired product in order to initiate crystallisation at
60 C. The suspension
was stirred for an additional 2 h at 60 C. Heptane (60 g) was added over 10 h
at 60 C. The
suspension was cooled over 1 h to 0 C and the precipitated product was
isolated on a glass
filter funnel, washed with heptane (50 mL) at 20 C and vacuum dried at 45 C.
The drying was
monitored by Karl Fischer titration.
Yield: 18.3 g(94.1 %) of dry product (assay: 98.7%).
Example 6
2-Phenyl-2-propanol
A 13.9 wt.% solution of LaC13/LiCl in THF (14.84 g, 8.4 mmol, 0.2 equiv) was
diluted with
THF (50 mL). A 3.0 ivt solution of methylmagnesium chloride (42.86 g, 126
mmol, 3 equiv)
was added to the solution at room temperature. The solution was then cooled to
-9 C and a
mixture of ethyl benzoate (6.30 g, 42.9 mmol, 1 equiv) in toluene (7 mL) was
added to the first
solution over 60 min within a temperature range of -9 C to -5 C. After an
additiona130 min
an in-process control with GLC showed no starting material present. The
reaction mixture was
cooled to -20 C and was quenched by addition of 4 M aqueous acetic acid (128
mL) while the
temperature was kept below 10 C. The resulting biphasic system was allowed to
warm to
20 C. Toluene (50 mL) was added and the system was agitated for 15 min at 20
C and settled
for 5 min at 20 C to give a clear phase separation. The organic layer was
washed at 20 C with
a 10 wt.% aqueous Na2CO3 solution (104 mL), followed by a 10 wt.% aqueous NaCI
solution.
Then the organic phase was dried over Na2S04 and concentrated in vacuo to give
the product as
a yellow oil. The structure of the product was confirmed by 'H NMR.
Yield: 89.7 %, purity (GLC): 98%.
CA 02692919 2010-01-08
WO 2009/010231 PCT/EP2008/005638
-8-
Example 7
2-Phenyl-2-propanol
The procedure of Example 6 was repeated using 5 equivalents of MeMgCI.
The isolated yield was 93.9% with 99.6% purity.
Example 8
2-Methylundecan-2-ol
The procedure of Example 6 was repeated using methyl decanoate instead of
ethyl benzoate.
The isolated yield was 76.6 % with 97.6% purity.
Example 9
(aS)-a-[3-[(1E)-2-(7-Chloro-2-quinolinyl)ethenyl] phenyl]-2-(1-hydroxy-l-
methylethyl)-
benzenepropanol
Methyl 2-[(3S)-3-[3-[(1 E)-2-(7-chloro-2-quinolinyl)ethenyl]phenyl]-3-
hydroxypropyl]benzoate
monohydrate (20.00 g, 42.01 mmol) and 2-methyltetrahydrofuran (100 mL) were
charged to a
250 mL reactor. To remove water, 60 mL of solvent was distilled from the
solution at 79 C and
normal pressure. Karl Fischer analysis showed that the water content was 0.04%
(1 mL of
solution was drawn). The solution was stored at 20-30 C. Methylmagnesium
chloride (59.04 g,
175.24 mmol) was charged under nitrogen to a second 250 mL reactor and cooled
to -10 C.
Then, 14.10 g of a 16.06 wt.% solution of LaC13=2 LiCI in tetrahydrofuran was
added within
0.5 h. The resulting suspension was cooled to -15 C. The solution in the
first reactor was
syringed into the second reactor at such a rate as to maintain the temperature
below -10 C.
The reaction was monitored by HPLC. After the reaction was completed, 4 m
acetic acid
(90 mL) was added slowly, while maintaining the temperature below 0 C (pH of
water phase:
5-6). The mixture was heated to 20 C. The organic phase was separated and
washed twice
with 10 wt.% aqueous Na2CO3 (90 mL) and twice with 10 wt.% aqueous NaCl (60 mL
each).To remove water and tetrahydrofuran, 15.0 g of solvent was distilled
from above solution.
Then 2-methyltetrahydrofuran (32.0 g) was added, followed by distillation of
another 24.0 g of
solvent. The residue was cooled to 30 C and then n-heptane (22.4 g) was added
to form a
saturated solution. To the solution, 0.4 g of the diol product was added as
seed crystals and the
resulting suspension was stirred overnight. n-Heptane (75.0 g) was added
within 1.5 h, and the
suspension was cooled to -2 C within 1 h and kept at this temperature for 3
h. The product
was isolated by filtration. The filter cake was washed with n-heptane (30 mL)
and dried at
C/<100 mbar.
Yield: 17.5 g (87%), purity 98.4% (ketone content 0.6%).
CA 02692919 2010-01-08
WO 2009/010231 PCT/EP2008/005638
-9-
Example 10
(aS)-a-[3-[(1E)-2-(7-Chloro-2-quinolinyl)ethenyl] phenyl]-2-(1-hydroxy-l-
methylethyl)-
benzenepropanol
Methyl 2-[(3S)-3-[3-[(1 E)-2-(7-chloro-2-quinolinyl)ethenyl]phenyl]-3-
hydroxypropyl]benzoate
monohydrate (10.0 g, 21.62 mmol) followed by 1,3-dioxolane (50 mL) were
charged to a
100 mL reactor.To remove water, 30 mL of solvent was distilled from the
solution at 79 C and
normal pressure. Karl Fischer analysis showed that water content was 0.07% (1
mL of solution
was drawn). The solution was stored at 20-30 C. Methylmagnesium chloride
(30.10 g,
89.34 mmol) was charged under nitrogen to a 250 mL reactor and cooled to -10
C. Then
LaCl3/LiCI solution in THF (16.06 wt.%, 7.75 g) was added within 0.5 h. The
suspension was
cooled to -15 C. The solution in the first reactor was syringed into the
second reactor at such a
rate as to maintain the temperature below -10 C. The reaction was monitored
by HPLC. After
the reaction was completed, 4 m acetic acid (40 mL) was added slowly, while
maintaining the
reaction mixture below 0 C (pH value of water phase: 5-6). The organic phase
was separated
and washed with aqueous Na2CO3 (5 wt,%, 30 mL) and NaCl (10 wt.%, 30 mL). The
solvent
was removed in vacuo and then the residue was dissolved in toluene (10 mL).
The solution was
heated to 45 C and n-heptane (3 mL) was added. Seed crystals of the desired
product (0.4 g)
were added to induce crystallization. The suspension formed was heated to 50
C and stirred for
3 h. n-Heptane (30 mL) was added within 3 h, followed by cooling to 0 C. The
product was
isolated by filtration. The filter cake was washed with n-heptane (15 mL) and
dried at
40 C/<100 mbar.
Yield: 8.9 g (83.3%), purity 98.4% (ketone content 0.6% ).