Note: Descriptions are shown in the official language in which they were submitted.
CA 02698067 2010-02-26
WO 2009/038753 PCT/US2008/010880
1
METHODS AND APPARATUS FOR THE SYNTHESIS
OF USEFUL COMPOUNDS
This application claims the benefit of U.S. provisional patent application no.
60/994,854 filed on September 20, 2007, which is incorporated herein and made
a part hereof
by reference for all purposes as if set forth in its entirety.
BACKGROUND OF THE INVENTION
The present invention relates to methods and apparatus for the activation of a
low
reactivity, non-polar chemical compound. More specifically, the present
invention relates to
process for the synthesis of useful compounds from non-polar compounds such as
carbon
dioxide and the like.
The chemical reduction of carbon dioxide using molecular hydrogen is not
thermodynamically viable. However, the possibility to use activated hydrogen-
containing
compounds for the preparation of useful products from carbon dioxide is
intriguing.
Some catalysts, e.g., transition metal complexes, have been shown to catalyze
the
reduction of carbon dioxide via hydride complexes, in which the origin of the
activated
hydrogen is water. Such reactions result usually in a partial reduction of
carbon dioxide to
carbon monoxide. However, the possibility of the further reduction to
formaldehyde,
methanol and/or methane is potentially very significant. Such reduction
products are
particularly important in chemical manufacture (formaldehyde and methanol), as
well as fuels
(methanol and methane). [see, e.g., "Thermodynamic, Kinetic and Product
Considerations in
Carbon Dioxide Reactivity", F.R. Keene, Chapter 1 in monograph
"Electrochemical and
Electrocatalytic Reactions of Carbon Dioxide" (B.P. Sullivan, K. Krist, and
H.E. Guard, eds.);
Elsevier (Amsterdam), 1993].
In particular, formaldehyde and its derivatives serve a wide variety of end
uses such as
for plastics and coatings. Formaldehyde is considered one of the world's most
important
industrial and research chemicals, owing to the vast number of chemical
reactions it can
participate in.
CA 02698067 2010-02-26
WO 2009/038753 PCT/US2008/010880
2
As formaldehyde polymerizes readily in the presence of minute amounts of
impurities,
the commercial forms usually available comprise:
- the polymer form, which can be reversibly converted to a monomer by the
reaction
of heat or an acid:
H-(OCH2-)-õ-OH
- the cyclic trimeric form, called trioxane; and
- the aqueous solution in which over 99 of formaldehyde is present as hydrate
or a
mixture of oxymethylene glycol oligomers.
It would be advantageous to provide methods and apparatus for activation of a
low
reactivity, non-polar chemical compound. In particular, it would be
advantageous to provide
methods and apparatus for the reduction of carbon dioxide without the need to
use molecular
hydrogen. It would be further advantageous to enable the reduction of carbon
dioxide using
water or steam as the source of hydrogen. It would also be advantageous to
enable the
oxidation or reduction of benzene to derivative compounds, such as
acetophenone, a phenol,
cyclohexane, or other benzene derivatives. Another advantageous possibility is
to provide the
capability of achieving a further reduction of formaldehyde-derived polymers
to higher
molecular mass alcohols and to olefins.
The_methods and apparatus of the present invention provide the foregoing and
other
advantages.
CA 02698067 2010-02-26
WO 2009/038753 PCT/US2008/010880
3
SUMMARY OF THE INVENTION
The present invention relates to methods and apparatus for activation of a low
reactivity, non-polar chemical compound. In one example embodiment, the method
comprises
introducing the low reactivity chemical compound to a catalyst. At least one
of (a) an
oxidizing agent or a reducing agent, and (b) a polar compound is provided to
the catalyst and
the chemical compound. An.alternating current is applied to the catalyst to
produce an
activation reaction in the chemical compound. This activation reaction
produces a useful
product.
The activation reaction may comprise one of a reduction or an oxidation
reaction. The
polar compound may comprise one of water or steam. One of ammonia, nitric
oxide, carbon
monoxide, methane, or the like may be added to the water or steam.
In another example embodiment, the polar compound may comprise one of water,
ammonia, nitric oxide, and carbon monoxide. Those skilled in the art will
appreciate that
other polar compounds may be used with the present invention.
In a further example embodiment, the chemical compound and the at least one of
the
oxidizing agent or the reducing agent and the polar compound may be introduced
into a
chamber containing the catalyst.
In one example embodiment, the low reactivity chemical compound may comprise
CO2. In such an embodiment, the useful product may comprise formaldehyde in at
least one
of a monomeric and a polymeric form. In other example embodiments, the useful
product
may comprise at least one of an aldehyde, trioxane, ethane, ethylene,
formaldehyde, and
paraformaldehyde. The useful products may contain at least one of carbon,
hydrogen, and
oxygen. Still further, the useful products may comprise at least one of an
alcohol compound
and an olefin.
In a further example embodiment, the chemical compound may comprise an
aromatic
,compound. The aromatic compound may comprise benzene or a benzene derivative.
In such
an embodiment, a reducing agent such as hydrogen may be provided to the
catalyst and the
aromatic compound, and the useful product may comprise cyclohexane or a
benzene
CA 02698067 2010-02-26
WO 2009/038753 PCT/US2008/010880
4
derivative. Alternatively, an oxidizing agent such as oxygen may be provided
to the catalyst
and the aromatic compound, and the useful product may comprise at least one of
acetophenone, a phenol, or a benzene derivative.
The catalyst may comprise one of a precious metal, a semi-conducting oxide, a
semi-
conducting cermet, and a varistor. Examples of catalysts that may be used with
the present
invention include, but are not limited to catalysts comprising platinum,
platinum black,
rhodium, rhodium black, palladium, palladium black, silver, manganese oxide, a
manganese
oxide derivative, molybdenum oxide, a molybdenum oxide derivative, iron oxide,
an iron
oxide derivative, cerium oxide, a cerium oxide derivative, titanium oxide,
doped titanium
oxide and related compounds, cobalt oxide, rhodium oxide, zinc oxide, and the
like.
In one example embodiment, the catalyst may comprise a catalyst layer applied
to a
porous ceramic substrate. The catalyst layer may be supported by a layer of a
solid electrolyte.
The solid electrolyte layer may be one of a continuous layer or a
discontinuous layer. The
solid electrolyte may comprise one of stabilized zirconia (stabilized with,
e.g., gadolinium
oxide, samarium oxide, lanthanum oxide, ytterbium oxide, yttrium oxide or
other adequate
materials known to those skilled in the art), Nafion, other hydrogen ion
conducting materials,
beta aluminas, or the like
The alternating current may be applied across a three-phase boundary at an
interface
between the catalyst and the solid electrolyte layer. In order to apply the
alternating current to
the catalyst layer, three electrodes may be provided. For example, a reference
electrode may
be applied to the solid electrolyte layer, a counter electrode may be applied
between the
catalyst and the solid electrolyte layer, and a working electrode may be
applied to the catalyst
layer.
In a further example embodiment, a polarization impedance of the supported
catalyst
layer may be monitored. The polarization impedance may be controlled by
varying the
alternating current, enabling optimization of the activation reaction.
In addition, a controlled oxygen partial pressure environment may be provided
at a
level of the supported catalyst layer. The partial pressure of the oxygen at a
level of the
catalyst layer may be monitored. The monitoring of the partial pressure of the
oxygen may
CA 02698067 2010-02-26
WO 2009/038753 PCT/US2008/010880
comprise monitoring an interfacial impedance of the supported catalyst layer.
The partial
pressure of oxygen at a level of the catalyst layer may then be determined as
a function of the
interfacial impedance. Altemately, the polarization impedance of the supported
catalyst layer
may be monitored, and the partial pressure of oxygen at the level of the
catalyst layer may be
5 determined as a function of the monitored polarization impedance.
In addition, a momentary value of the alternating current may be determined as
a
function of the monitored polarization impedance.
The amount of the at least one of the oxidizing agent, the reducing agent, and
the polar
compound provided may be controlled in order to optimize the activation
reaction. Further, a
ratio of an amount of the chemical compound to an amount of the at least one
of the oxidizing
agent, the reducing agent, and the polar compound provided may be controlled
in order to
optimize the activation reaction.
In a further example embodiment, heat may be applied to the catalyst in order
to
optimize the activation reaction.
The present invention also generally includes a method for activation of a
chemical
compound. The chemical compound is introduced to a catalyst. An oxidizing
agent or a
reducing agent is provided to the catalyst and the chemical compound. An
alternating current
is applied to the catalyst to produce an activation reaction in the chemical
compound. This
activation reaction produces a useful product. For example, the chemical
compound may
comprise a polar compound and the oxidizing or reducing agent may comprise a
polar
reactant or a nonpolar reactant. Additionally, the chemical compound may
comprise a
nonpolar chemical compound and the oxidizing or, reducing agent may comprise a
polar
reactant or a nonpolar reactant.
The present invention also encompasses apparatus for activation of a low
reactivity,
non-polar chemical compound which can be used to carry out the various
embodiments of the
methods discussed above. The apparatus may comprise a catalyst, a means for
introducing the
low reactivity chemical compound to the catalyst, a means for providing at
least one of (a) an
oxidizing agent or a reducing agent, and (b) a polar compound to the catalyst
and the chemical
compound, and means for applying an alternating current to the catalyst to
produce an
CA 02698067 2010-02-26
WO 2009/038753 PCT/US2008/010880
6
activation reaction in the chemical compound, such that the activation
reaction produces a
useful product.
CA 02698067 2010-02-26
WO 2009/038753 PCT/US2008/010880
7
BRIEF DESCRIPTION OF THE DRAWINGS
The present invention will hereinafter be described in conjunction with the
appended
drawing figures, wherein like reference numerals denote like elements, and:
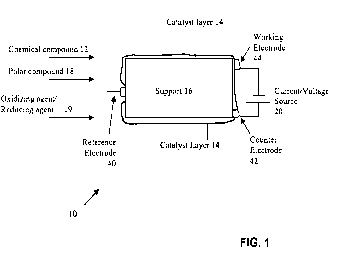
Figure 1 shows an example embodiment of an apparatus in accordance with the
present
invention;
Figure 2 shows a further example embodiment of an apparatus in accordance with
the present
invention;
Figure 3 shows an example embodiment of an electrode arrangement in accordance
with the
present invention;
Figure 4 shows NMR analysis results for the output achieved with one example
embodiment
of the present invention;
Figure 5 shows NMR analysis results for the output achieved with a further
example
embodiment of the present invention;
Figures 6 and 7 show scanning electron microscopy images of the catalyst
assembly at
different resolutions, respectively, in accordance with an example embodiment
of the
invention;
Figure 8 shows an NMR spectrum for the output achieved with a further example
embodiment of the present invention;
Figure 9 shows a polarization Bode spectrum from one example embodiment of the
present
invention; and
Figure 10 shows a single frequency EIS (Electrochemical Impedance
Spectroscopy) spectrum
from one example embodiment of the present invention.
CA 02698067 2010-02-26
WO 2009/038753 PCT/US2008/010880
8
DETAILED DESCRIPTION
The ensuing detailed description provides exemplary embodiments only, and is
not
intended to limit the scope, applicability, or configuration of the invention.
Rather, the
ensuing detailed description of the exemplary embodiments will provide those
skilled in the
art with an enabling description for implementing an embodiment of the
invention. It should
be understood that various changes may be made in the function and arrangement
of elements
without departing from the spirit and scope of the invention as set forth in
the appended
claims.
The present invention is the product of a joint research agreement between
Catelectric
Corp. (Catelectric) and The University of Connecticut and relates to methods
and apparatus
for activation of a low reactivity, non-polar chemical compound in order to
produce useful
products. In particular, the present invention relates to methods and
apparatus for the
preparation of useful products; such as, e.g., paraformaldehyde, via the
activation (e.g.,
reduction or oxidation) of carbon dioxide, using water as the source of
hydrogen and oxygen.
However, as will be explained in detail below, the present invention is not
limited to such
reactions and products. The reaction is activated via the DECANTM process
developed by
Catelectric. The DECANTm process is described in Catelectric's U.S. patent no.
7,325,392
issued on February 5, 2008 and entitled "Control Systems for Catalytic
Processes" and in
Catelectric's pending in U.S. patent application no. 11/588,113 filed on
October 25, 2006
entitled. "Methods and Apparatus for Controlling Catalytic Processes,
Including Catalyst
Regeneration and Soot Elimination" (published as 2007/0095673), both of which
are
incorporated herein and made a part hereof by reference.
The present invention relates to methods and apparatus for activation of a low
reactivity, non-polar chemical compound. Figure 1 shows an example embodiment
of an
apparatus 10 for activation of a low reactivity, non-polar chemical compound.
A low
reactivity chemical compound 12 is introduced to a catalyst (e.g., catalyst
layer 14). The
catalyst layer 14 may be supported on a support 1.6. At least one of (a) an
oxidizing agent or a
reducing agent 19, and (b) a polar compound 18 is provided to the catalyst 14
and the
chemical compound 12. An alternating current (e.g., from current/voltage
source 20) is
CA 02698067 2010-02-26
WO 2009/038753 PCT/US2008/010880
9
applied to the catalyst 14 to produce an activation reaction in the chemical
compound 12. This
activation reaction produces a useful product.
It should be appreciated that the term "non-polar chemical compound" as used
herein
denotes a chemical compound which, as a whole, has a zero permanent dipole
moment. For
example, by this definition, CO2 is considered to be non-polar, even though it
has polar bonds
between the individual molecules. Accordingly, the term "polar compound" as
used herein
denotes a compound that, as a whole, has a non-zero dipole moment. -
The activation reaction may comprise one of a reduction or an oxidation
reaction. The
polar compound 18 may comprise one of water or steam. One of ammonia, nitric
oxide,
carbon monoxide, methane, or the like may be added to the water or steam.
In another example embodiment, the polar compound 18 may comprise one of
water,
ammonia, nitric oxide, and carbon monoxide. Those skilled in the art will
appreciate that
other polar compounds may be used with the present invention. Further, those
skilled in the
art will appreciate that the use of water (or steam) will facilitate both an
oxidation and a
reduction reaction.
In a further example embodiment as shown in Figure 2, the chemical compound 12
and the at least one of the oxidizing agent or the reducing agent 19 and the
polar compound
18-may be introduced into a chamber 22 containing the catalyst 14. The chamber
22 may
comprise a tubular reactor. The alternating current may be controlled by an
electronic control
device 24. The chemical compound 12 (e.g., C02) may be introduced to the
chamber 22 from
a gas tank 11. The polar compound 18 (e.g., water) may be introduced to the
chamber 22 from
a peristaltic pump 17. The oxidizing agent (e.g., oxygen) or the reducing
agent (e.g.
hydrogen) 19 may be introduced from tank 21. After passing the chemical
compound 12 and
at-least one of the oxidizing agent or the reducing agent 19 and the polar
compound 18
through the chamber 22 containing the catalyst 14 and applying the alternating
current
thereto, the resulting products of the reaction may be passed through an ice-
water trap 26
and/or a dry ice/liquid nitrogen trap 28 before being separated in a molecular
sieve 30 prior to
computer analysis (such as gas chromatography-mass spectrometry (GC-MS), high
CA 02698067 2010-02-26
WO 2009/038753 PCT/US2008/010880
performance liquid chromatography-mass spectrometry (HPLC-MS), nuclear
magnetic
resonance (NMR) and other analysis techniques) at analyzer 32.
In one example embodiment, the low reactivity chemical compound 12 may
comprise
CO2. In such an embodiment, the useful product may comprise formaldehyde in at
least one
of a monomeric and a polymeric form. In other example embodiments, the useful
product
may comprise at least one of an aldehyde, trioxane, ethane, ethylene,
formaldehyde, and
paraformaldehyde. The useful products may contain at least one of carbon,
hydrogen, and
oxygen. Still further, the useful products may comprise at least one of an
alcohol compound
and an olefin. Also, oxygen (02) may be a result of the reaction.
In a further example embodiment, the chemical compound 12 may comprise an
aromatic compound. The aromatic compound may comprise benzene or a benzene
derivative.
In such an embodiment, the reducing agent 19 (such as hydrogen) may be
provided to the
catalyst and the aromatic compound, arid the useful product may comprise
cyclohexane or a
benzene derivative. Alternatively, an oxidizing agent 19 (such as oxygen) may
be provided to
the catalyst and the aromatic compound, and the useful product may comprise at
least one of
acetophenone, a phenol, or a benzene derivative.
The catalyst 14 may comprise one of a precious metal, a semi-conducting oxide,
a
semi-conducting cermet, and a varistor. Examples of catalysts that may be used
with the
present invention include, but are not limited to catalysts comprising
platinum, platinum
black, rhodium, rhodium black, palladium, palladium black, silver, manganese
oxide, a
manganese oxide derivative, molybdenum oxide, a molybdenum oxide derivative,
iron oxide,
an iron oxide derivative, cerium oxide, a cerium oxide derivative, titanium
oxide, doped
titanium oxide and related compounds, cobalt oxide, rhodium oxide, zinc oxide,
and the like.
Further examples for catalyst material may generally include oxides of alkali
metals, alkaline
earths, lanthanides, actinides, transition metals, and nonmetals.
In one example embodiment as shown in Figure 3, the catalyst 14 may comprise a
catalyst layer applied to a support 16 such as porous ceramic substrate. For
example, the
catalyst layer 14 may be supported by a layer 16 of a solid electrolyte. In
certain
embodiments, the catalyst 14 may be applied to the solid electrolyte layer 16,
which in tum
CA 02698067 2010-02-26
WO 2009/038753 PCT/US2008/010880
11
may be applied onto a separate support (not shown). The solid electrolyte
layer 16 may be one
of a continuous layer or a discontinuous layer. The solid electrolyte 16 may
comprise one of
stabilized zirconia (stabilized with, e.g., gadolinium oxide, samarium oxide,
lanthanum oxide,
ytterbium oxide, yttrium oxide or other adequate materials known to those
skilled in the art),
Nafion, other hydrogen ion conducting materials, beta aluminas, or the like.
The temperature
range of the reactor will be determined by the specific properties of these
materials, known to
those skilled in the art.
The alternating current may be applied across a three-phase boundary at an
interface
between the catalyst 14 and the solid electrolyte layer 16 via the electronic
control device 24.
In order to apply the alternating current to the catalyst layer 14, three
electrodes may be
provided. For example, a reference electrode 40 may be applied to the solid
electrolyte layer
16, a counter electrode 42 may be applied to the solid electrolyte layer 16,
and a working
electrode 44 may be applied to the catalyst layer 14.
In a further example embodiment, a polarization impedance of the supported
catalyst
layer 14 may be monitored. In order to monitor the polarization impedance, the
electronic
control device 24 may include means for determining the applied current and
voltage. The
determination of the polarization impedance from the sensed current is
explained in detail in
Catelectric's-U.S. patent no. 7,325,392. The polarization impedance may be
controlled by
varying the alternating current from electronic control device 24, enabling
optimization of the
activation reaction.
In addition, a controlled oxygen partial pressure environment may be provided
at a
level of the supported catalyst layer. The oxygen may be produced from the
solid electrolyte
layer 16 under the voltage applied between the working electrode 44 and the
reference
electrode 40, and is a function of the DECANTM process. Alternately, the
oxygen may be
provided from tank 21 (Figure 2). The partial pressure of the oxygen at a
level of the catalyst
layer 14 may be monitored. The determining of the partial pressure of oxygen
may also be
achieved via the electronic control device 24 as a function of a voltage
measurement. For
example, a monitoring of the partial pressure of the oxygen may comprise
monitoring an
interfacial impedance of the supported catalyst layer 14. The partial pressure
of oxygen at a
CA 02698067 2010-02-26
WO 2009/038753 PCT/US2008/010880
12
level of the catalyst layer 14 may then be determined as a function of the
interfacial
impedance. Alternately, the polarization impedance of the supported catalyst
layer 14 may be
monitored as discussed above, and the partial pressure of oxygen at the level
of the catalyst
layer 14 may be determined as a function of the monitored polarization
impedance (e.g.,
achieved via the electronic control device 24).
In addition, a momentary value of the alternating current may be determined by
the
electronic control device 24 as a function of the monitored polarization
impedance.
The amount of the oxidizing agent or the reducing agent 19 and/or the polar
compound 18 provided may be controlled in order to optimize the activation
reaction. Further,
a ratio of an amount of the chemical compound 12 to an amount of the oxidizing
agent or the
reducing agent 19 and/or the polar compound 18 provided may be controlled in
order to
optimize the activation reaction.
In a further example embodiment, heat may be applied to the catalyst in order
to
optimize the activation reaction. Heat may be applied via heating element 34,
which is
controlled by temperature control unit 36 (Figure 2). Oxygen 19 may be applied
from an
oxygen source (e.g., tank 21) or may be generated by controlling the voltage
applied to the
solid electrolyte layer, as discussed above.
The present invention also generally includes-a method-for the activation of a
chemical
compound. The chemical compound 12 is introduced to a catalyst 14. An
oxidizing agent or a
reducing agent 19 is provided to the catalyst 14 and the chemical compound 12.
An
alternating current is applied to the catalyst 14 to produce an activation
reaction in the
chemical compound 12. This activation reaction produces a useful product. For
example, the
chemical compound may comprise a polar compound 12 and the oxidizing or
reducing agent
19 may comprise a polar reactant (e.g., water or steam) or a non-polar
reactant (oxygen or
hydrogen). Additionally, the chemical compound may comprise a non-polar
chemical
compound 12 (as discussed above) and the oxidizing or reducing agent 19 may
comprise a
polar reactant (e.g., water or steam) or a non-polar reactant (oxygen or
hydrogen). For
example, one polar compound like methanol could react with another polar
compound like
CA 02698067 2010-02-26
WO 2009/038753 PCT/US2008/010880
13
ethanol to form products of value, or one non-polar compound like benzene
could react with
another nonpolar compound like methane to form products of value.
The examples below illustrate example embodiments of a process for the
reduction of
carbon dioxide using water as the source of hydrogen in accordance with the
present
invention. The examples below were carried out using the apparatus described
above in
connection with Figure 2. However, it should be appreciated by those skilled
in the art that the
inventive process is not limited by the following examples and may be
implemented for the
reduction of other molecules, e.g., higher molecular mass alcohols to olefins
and other
compounds.
Example 1
a. Substrates: Commercial Calcia Fully Stabilized Zirconia (FSZ) porous
ceramics from
Vesuvius Hi-Tech Ceramics was used as the solid electrolyte layer 16.
b. Deposition of the catalyst: Liquid-Phase Chemical Vapor Deposition (LP-CVD)
was used
for coating of the catalyst layer 14 (platinum). Pt(acac)2 (Strem Chemicals
Inc.) was used as
the platinum precursor. The temperature of the precursor was set at 120-150 C,
while the
temperature of the FSZ (calcia) was set at 400-500 C. Argon was used as the
carrier gas. The
carrier gas flow rate of the precursor was 500-1000 sccm/min, and the carrier
gas was heated
to 100-150 C before being introduced into the CVD synthesis tube. Oxygen was
used as an
oxidant. The oxygen flow rate was set at 80-200 sccm/cm. The total pressure of
the CVD
reactor was controlled at 5-20 KPa. The platinum deposition time was 1-4
hours.
c. Assembling of tliree electrodes: Three electrodes were deposited on the FSZ
(calcia)
ceramic catalyst as described above in connection with Figure 3. The three
electrodes each
comprise 0.25 mm platinum wires (Alfa Aesar). The three platinum wires were
assembled on
the FSZ (calcia) using platinum paste (from Engelhard / BASF) and then treated
in air at 900
C. The reference electrode 40 was directly connected to the support 16 without
contact with
the platinum layer 14. The counter electrode 42 was assembled before the
deposition of the
catalyst layer 14 of LP-CVD of platinum, and is in contact with the FSZ
support layer 16. The
CA 02698067 2010-02-26
WO 2009/038753 PCT/US2008/010880
14
working electrode 44 was deposited on the platinum LP-CVD catalyst layer 14.
After
assembling the three electrodes, the catalyst assembly with three electrodes
was placed in a
quartz tube and reduced in 8% hydrogen/helium mixed gas at 600-800 C for 4-6
hours.
d. Catalytic reaction - reactor and reaction parameters: The supported Pt-FSZ
catalyst
assembly, with the three electrodes, was placed in a quartz tube reactor
(e.g., tubular reactor
22 of Figure 2). The reactor was purged of air and was thereafter operated at
slightly positive
pressure of about 5-14 psig. The tube reactor temperature was set at 600 to
950 C.
It should be noted that the present invention is not limited to the foregoing
description.
For example, the temperature may be as low as room temperature or higher than
950 C; the
solid electrolyte can be Nafion, and the catalyst can be platinum black. Other
materials for use
as the solid electrolyte or catalyst will be apparent to those skilled in the
art.
Further, the solid electrolyte layer 16 can be deposited on a support
comprising an
inert ceramic substrate (e.g., cordierite catalyst supports provided by
Corning Inc. or St.
Gobain Co) via any of the appropriate methods known to those skilled in the
art. Similarly,
the catalyst 14 can be deposited on the solid electrolyte layer 16 via any of
the appropriate
methods known to those skilled in the art.
In addition, the implementation of the process does not require a continuity
of the
solid electrolyte layer 16 or of the catalyst layer 14. What is necessary is a
preponderance of
grain boundaries where the catalyst 14 is in contact with the solid
electrolyte 16 and sufficient
open porosity to allow for the access of the reacting phases to the
catalytically active
interfaces.
Carbon dioxide (C02) used was zero grade gas from Airgas. Water used was de-
ionized water. Water was injected by a peristaltic pump 17, and evaporated by
a heated
ceramic tube. CO2 was used as the carrier gas provided from tank 11. The molar
ratio of CO2
to water was set at 10 to 1 or 5 to 1. The flow of CO2 was monitored by a mass
flow meter
and was varied between 200 scc/minute and 1600 scc/minute. It should be noted
that the
water / CO2 ratio can take any values within the interval 1/1000 to 1000/1,
and even outside
this range.
CA 02698067 2010-02-26
WO 2009/038753 PCT/US2008/010880
The system was polarized (via the electronic control device 24 and three
electrodes 40,
42, and 44) with a pulsed current at about 1 kHz at average voltages ranging
from 0.03 to 0.1
V rms. The current passed averaged between 0.03 and 0.13 mA. This process is
described in
detail in U.S. patent application no. 11/588,113 mentioned above.
5 Eight runs of polarization were applied, each lasting about 15 minutes.
An unexpected result of this process was that a substantial amount of a white
powder
was formed, which was collected at the cold areas of the reactor 22, as well
as in the water
trap 26 and liquid nitrogen trap 28. The gas phase was analyzed by gas
chromatography (e.g.,
analyzer 32) with thermal and flame ionization detectors.
10 The powder was dispersed in the water samples collected by the traps, which
were
then analyzed by Nuclear magnetic resonance spectroscopy (NMR) and High-
Pressure Liquid
Chromatography (HPLC). With the reactor temperature set at 900 C data
collected was
consistent with the presence in these samples of paraformaldehyde and small
amounts of
trioxane. The result of the NMR analysis is shown in Figure 4.
Example 2
15 The catalyst 14 used in this example was the same as that for example 1.
The temperature of
the quartz tube reactor was set at 600 C. The main product identified by NMR
was
paraformaldehyde, as shown in Figure 5.
Example 3
a. Substrates: Commercial Calcia Fully Stabilized Zirconia (FSZ) porous
ceramics from
Vesuvius Hi-Tech Ceramics was used as the solid electrolyte layer 16. *
b. Deposition of the catalyst: A catalyst layer 14 of octahedral manganese
oxide OMS-2 was
prepared as follows: 5.6g K2SO4, 8.81g K2S208 and 3.77g MnSOa and 70m1 DI
water were
added into a 125ml autoclave and put into a 4748 Parr acid digestion bomb for
96 hours; the
temperature was maintained at 250 C. The solid was washed repeatedly with de-
ionized
water. The suspension was filtered and stirred overnight at 85 C into a beaker
with 300 ml de-
CA 02698067 2010-02-26
WO 2009/038753 PCT/US2008/010880
16
ionized water. The suspension was coated on the Vesuvius porous ceramic body
and was
dried at 120 C for 12 hours.
c. Assembly of electrodes: Three platinum electrodes were positioned as
described in
Example 1. Platinum paste (Engelhard BASF) was applied to assemble the
electrodes. Then
the catalytic assembly was reduced in 6% Hydrogen/helium mixed gas for 2 hours
at 150-
300 C.
The as-prepared catalytic assembly was placed in a tube quartz reactor
(tubular reactor
22) and connected with the electronic control device 24. The tube quartz
reactor 22 was
sealed and isolated with an air environment. CO2 (zero grade from Air gas) was
introduced
from tank 11 and. controlled with a flow meter. Water was injected with a pre-
calibrated
peristaltic pump 17. Water was heated by a ceramic tube at above 130 C. Then
the reactor 22
was purged with CO2.
The system was set at slightly higher atmosphere pressure (for example 5 kpa).
The
electronic control device 24 supplied polarized current or voltage to the
catalytic assembly via
electrodes 40, 42, and 44. The tube reactor was set at 250-450 C.
The products were analyzed by NMR and GC techniques.
The Pt-OMS-2 catalyst 14 was tested in the C02-H20 system starting from 250 C
and up to 450 C. When the reaction started at 250 C, it was slow. After 4
hours, the sample
was analyzed from the first ice water trap 26 by NMR. The resultant NMR
patterns did not
show any product. The concentration of products may have been out of the limit
or the
product yield may have been very low. The second test was done at 300 C. The
resultant
NMR proton patterns showed a low concentration,of paraformaldehyde (about 0.5-
1.0% in
molar). In particular, the NMR results showed a weak peak of paraformaldehyde
at this
temperature. The third test was done at 400 C. The resultant NMR patterns
from the ice water
trap 26 and the NMR patterns of the dry ice trap 28 showed stronger peaks of
paraformaldehyde at this temperature. The concentration of paraformaldehyde
was about 1.0-
1.5% in molar. The fourth test was done at 450 C. The resultant NMR proton
patterns
showed higher concentrations of paraformaldehyde at this temperature. The
concentration of
paraformaldehyde was about 3.0-5.0% in molar.
CA 02698067 2010-02-26
WO 2009/038753 PCT/US2008/010880
17
For the above four tests, the CO2 flow rate used was 200.sccm, and the water
injection rate
was 9.16 ml/min. The flow rate of C02/H20 was 2.37.
Based on the above results, the CO2 conversion rate at different temperatures
is shown in
Table 1 below.
Table 1. Conversion rate of carbon dioxide in the reactions at different
temperatures
Tem erature C Conversion Rate %
250 Low
300 0.5-1.0%
400 1.0-1.5%
450 3.0-5.0%
Example 4
Synthesis of ZnO Catalyst: A low-pressure chemical vapor deposition (LPCVD)
technique
was used to deposit a catalyst layer 14 of ZnO on a calcium fully stabilized
zirconia (FSZ)
support 16. The Zn precursor was Zn(CHCOO)2(98+%, Aldrich). The temperature of
the FSZ
template was set at 300 T. The temperature of precursor was set at 160 C. The
deposition
pressure was controlled at 3 kPa. The sample was coated two times. In the
second run, the
position of the sample was reversed (front to back and top bottom of reactor)
to get better
uniformity of coating. Each coating time was 4 hours. The total CVD coating
time was 8
hours. After LPCVD, the sample was heated with a ramp rate at 5 C/min and
calcined at
600 C for 12 hours in air.
Reactor and Electrodes: Three electrodes were assembled on the ZnO-coated FSZ
support as
described above in connection with Figure 3. After the ZnO coated FSZ catalyst
assembly
was calcined, an area of 25 mm2 at the end was pretreated with 5M HCL to
remove ZnO. A
Platinum reference electrode 40 was assembled at this area. At another end of
the cylinder
sample, the same method as above was used to remove the ZnO layer, and a
platinum wire
was connected with the FSZ support layer 16 directly as the counter electrode
42. The
working electrode 44 was attached to the ZnO catalyst layer 14. Platinum paste
(6082 from
CA 02698067 2010-02-26
WO 2009/038753 PCT/US2008/010880
18
BASF) was applied to enable the platinum electrodes to have good contact with
the catalyst
assembly.
After the electrodes were assembled, the resistance between the electrodes was
measured with a Digital Multimeter (HDM350). The results are shown in Table 2
below.
Table 2: Resistance between electrodes at different tem eratures
Resistance between Resistance between Resistance between
working electrode working electrode and counter electrode and
and reference counter electrode reference electrode
Temp. electrode
200 C 20M 135K >20M
500 C 10.5M 19.6K 10.1M
600 C 1.06M 5.85K 0.55M
The COZ flow from tank 11 was measured with a flowmeter (OMEGA FL-3504G).
Water injection was measured by a calibrated peristaltic pump 17 (Watson
Marlow Sci400).
Water was.dropped on heated ceramic frit (>130 C) and evaporated in a T tube.
Then water
was introduced into the reactor with the CO2 carrier gas. ZnO-FSZ catalyst
assembly was
placed into a 2-inch quartz tube reactor (e.g., tubular reactor 22). The
reactor 22 was heated to
600-700 C with a tube furnace (Thermolyne 21100) or via heating element 34.
The ZnO-FSZ
catalyst assembly was connected with the three electrodes to the electronic
control device 24
and polarized by a voltage or a current controlled by the electronic control
device 24. The
outflow products were cooled by an ice-water trap 26 and a dry ice trap 28.
The gas from the
reactor was dried by a molecular sieve column 30, then the gas composition was
analyzed
with an analyzer 32 (e.g., a gas chromatograph (SRI 8610C)).
A voltage of -2.5 V to 2.5V was applied for the polarization tests for with a
potentiostatic EIS mode or single frequency mode. The temperature of CO2 and
H20 was set
at 600 and 700 C. The flow rate of C02 was between 200-500 sccm. The ratio of
CO2/H2O
was set at 1:1 and 1:3 respectively. With different polarization, each EIS
spectrum was taken
by a Gamry Reference 600.
The ZnO coated FSZ assembly was investigated by scanning electron microscopy
(SEM). The morphology of the ZnO catalyst layer 14 is shown in Figure 6
(x50000) and
CA 02698067 2010-02-26
WO 2009/038753 PCT/US2008/010880
19
Figure 7(x100000). Based on SEM images, the morphologies suggest that the ZnO
catalyst
layer 14 is continuous and the ZnO particle size is about 20-50 nm.
The products of CO2 and H20 activation were separated into two phases: liquid
phase
and gas phase. Liquid phase products were characterized by NMR and gas phases
were
analyzed with an SRI 8610C gas chromatograph. Other techniques such as HPLC-MS
and
GC-MS may also be employed. Figure 8 is a proton NMR spectrum of the
synthesized
products. The CO2 flow rate was set at 320-450 sccm; water was injected with a
flow rate of
mL/hour ( or 207sccm/min). The CO2/HZO molar ratio was 1.6-2.2. Based on the
results
shown in Figure 8, one major product was synthesized. The NMR chemical shift
is between
10 4.75 to 5.20 ppm. Small amounts of formaldehyde were present at a chemical
shift of 8.25
ppm.
The polarization voltage was set at -1.2 V to -1.5 V. The typical polarization
Bode
spectrum is shown in Figure 9. The "A" line is without polarization and the
"B" line is with
-1.2 V polarization. In the polarization condition, the Zmod decreased. For
example, Zmod
decreased from 3.825 kS2 to 3.573 kSZ at a frequency of 500 kHz. These data
suggest that the
reaction is fast when the catalytic cell was polarized.
Figure 10 is a single frequency EIS spectrum. With the fixed frequency of
500KHz,
the Zmod was shown to change with-time. This change reflected the dynamic
reactions at the
surface of the catalytic assembly. The comparison tests showed that if
alternating negative
and positive polarizations were used, the Zmod would decrease after negative
polarization,
which increases the reaction rate.
Gas chromatography (GC) online analysis of the products of the reaction found
new
broad peaks at 14.5-20.5min. These peaks were assigned to ethylene and ethane.
The foregoing examples are meant to illustrate the function and applicability
of the
present invention without limiting its scope. Those. skilled in the art will
appreciate that the
present invention has numerous applications and that the parameters,
materials, chemical
compounds, products, and other variables mentioned in the examples above can
be modified
or changed depending on the application and desired result.
CA 02698067 2010-02-26
WO 2009/038753 PCT/US2008/010880
From the foregoing examples those skilled in the art will appreciate that the
present
invention encompasses methods, processes, and apparatus for the activation of
the reaction
between low-reactivity, non-polar molecules (such as C02) with polar molecules
/ species
(such as water or steam), leading to products useful in the production of
polymers, in organic
5 synthesis reactions. For example, in accordance with the present invention a
process is
provided which leads to the activation of the reaction of carbon dioxide (and
of other similar
low-reactivity, non-polar molecules) with polar compounds (such as water,
steam, or others)
in a heterogeneous catalytic reaction. For example, the present invention may
be used to
activate the following reactions (among others):
10 COZ + H20
CO2 + H2O + CH4
CO2 + NO
CO2 + NO + CH4
CO2 + NH3
15 C6H6 + H2O
C6H6 + C6H6 + CHa
C6H6 + H20 + CH4
C6H6+ CH3OH and similar compounds
C6H6 + NO
20 C6H6 + NH3
Those skilled in the art will appreciate that the foregoing list of reactions
is not
intended to be limiting, and that the present invention may be used to
facilitate other
reactions, as discussed in detail above.
It should now be appreciated that the present invention provides advantageous
methods and apparatus for the activation of carbon dioxide and other low-
reactivity
molecules.
Although the invention has been described in connection with various
illustrated
embodiments, numerous modifications and adaptations may be made thereto
without
departing from the spirit and scope of the invention as set forth in the
claims.