Note: Descriptions are shown in the official language in which they were submitted.
CA 02718750 2010-09-16
WO 2010/014176 PCT/US2009/004273
PREPARATION METHOD OF BIODEGRADABLE MICRO-PARTICLES
CONTAINING DRUGS
Background
A variety of dosage forms have been used for drugs that require long-term
administration. To reduce the number of doses that need to be given, and to
provide a
stable level of the drug in the body, these drugs are preferably given in a
sustained-
release formulation. One type of sustained-release drug formulation that has
been used
is biodegradable microspheres containing drug trapped inside the microsphere.
One such
product is LUPRON Depot, which is biodegradable microspheres containing
leutinizing
hormone-releasing hormone (leuprolide or LHRH). Leuprolide is used for the
treatment
of hormone-dependent cancers, particularly prostate cancer, and precocious
puberty.
Microparticles are particles with a diameter of approximately 1 to 1000
microns.
For injection purposes, microparticles smaller than 125 microns are preferred.
Microparticles of this size can be injected with a standard hypodermic needle,
instead of
surgically implanted. One type of microparticle is composed of a network of a
biodegradable polymer that entraps a drug. As the polymer biodegrades in the
body, the
drug is released. The most commonly used biodegrabable polymers are polylactic
acid
and a copolymer of lactic acid and glycolic acid.
The most widely used methods to prepare biodegrabable microparticles are phase
separation, spray drying, and solvent evaporation. Phase separation, also
known as
coacervation, uses a decrease of the polymer solubility by the addition of a
non-solvent.
In a typical procedure, biodegradable polymer is dissolved in an organic
solvent (e.g.,
dichloromethane). Lipophilic drugs are dissolved in the polymer solution.
Hydrophilic
drugs are dissolved in water and then dispersed in the polymer solution (water
in oil
(w/o) emulsion) or dispersed as a solid powder. A non-solvent (typically
silicon oil) is
gradually added. Two phases form: a polymer-rich silicon oil phase and a
polymer-
depleted liquid organic solvent phase. As the organic solvent is extracted or
evaporates,
polymer microparticles with entrapped drug solidify in the silicon oil phase.
The
coacervate (silicon oil) adsorbs to the polymer microparticles.
In spray drying, the biodegradable polymer is dissolved in volatile organic
solvent, such as dichloromethane. The drug is dissolved or dispersed in the
polymer
solution. The solution or dispersion is sprayed in heated air. The solvent
evaporates,
resulting in the formation of solid microparticles.
CA 02718750 2010-09-16
WO 2010/014176
PCT/US2009/004273
Solvent evaporation is the most commonly used method of preparing
microparticles. In this method a drug-containing organic polymer solution is
emulsified
into a dispersion medium that is typically aqueous but may be oil. The methods
can be
further classified into oil in water (o/w), water in oil in water (w/o/w), and
oil in oil (0/0)
emulsion methods.
In an o/w method, drug and polymer are dissolved in an organic solvent, such
as
dichloromethane or a methanol/dichloromethane mixture. The drug-polymer-
organic
solvent solution is dispersed in an aqueous phase. An emulsifier, typically
poly(vinyl
alcohol), is included in the aqueous phase to help form small organic solvent
droplets in
the aqueous phase. The organic solvent evaporates with stirring, and with the
evaporation, the droplets solidify into polymer microparticles with entrapped
drug.
In a w/o/w double emulsion, an aqueous drug solution is prepared and dispersed
into a solution of the polymer in an organic solvent to form a water-in-oil
emulsion
containing the drug and polymer. The w/o polymer-drug emulsion is then
emulsified
into an aqueous phase to form a w/o/w emulsion. With stirring, the organic
solvent
evaporates, allowing the polymer-drug droplets in the emulsion to solidify
into
microparticles.
In an o/o emulsion method, drug and polymer are dissolved in a water-miscible
solvent (e.g., acetonitrile). That solution is emulsified into an oily phase
in the presence
of an emulsifier such as SPAN 80 to form an oil-in-oil emulsion. The organic
solvent is
extracted by the oil and microparticles can be harvested by filtration.
Prior art methods of forming biodegradable polymer drug-containing
microparticles have some disadvantages. Emulsifier or oil can adhere to the
microparticles and contaminate them. Some methods are difficult to scale up.
New methods of forming biodegradable drug-containing microparticles are
needed.
Summary
The inventor has found new methods of forming microparticles. In one method,
leuprolide is dissolved in methanol and PLGA is dissolved in dichloromethane.
The
leuprolide and PLGA solutions are mixed to form a drug-polymer-organic solvent
solution. The drug-polymer solution is added to a larger aqueous phase
containing in-
situ-formed hyroxyapatite (Calo(PO4)6(OH)2 gel. The hyroxyapatite gel appears
to coat
the organic droplets to maintain small droplet size and prevent droplets
coalescing. The
2
CA 02718750 2010-09-16
WO 2010/014176 PCT/US2009/004273
organic solvent evaporates, just as with a standard oil-in-water emulsion
method using an
organic emulsifier, leaving behind solidified polymer microparticles with
entrapped
leuprolide. HCI is then added to the suspension, which dissolves the
hydroxyapatite.
The microparticles can be recovered by centrifugation or filtration from the
otherwise
clear solution.
One embodiment of the invention provides a method of preparing drug-
containing microparticles comprising: (a) dissolving a biodegradable polymer
in organic
solvent to form a polymer solution; (b) dissolving or dispersing a drug in the
polymer
solution to form a polymer-drug-solvent phase; (c) mixing the polymer-drug-
solvent
phase with an aqueous suspension comprising an inorganic gel to form a
dispersion
comprising polymer-drug droplets dispersed in the aqueous suspension; (d)
evaporating
the organic solvent from the dispersion to convert the polymer-drug droplets
to drug-
containing microparticles; and (e) recovering the drug-containing
microparticles from the
dispersion.
Preferably the ingorganic gel can be dissolved by acid, and the step of
recovering
the drug-containing microparticles from the dispersion comprises adding acid
to the
dispersion to dissolve the inorganic gel.
Other embodiments of the invention provide drug-containing microparticles
prepared by the methods described herein.
Brief Description of the Drawings
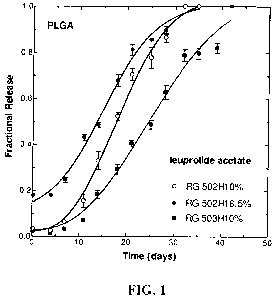
FIG. I is a graph of fractional release of leuprolide acetate from PLGA
microspheres prepared as described in Example 1 versus time.
FIG. 2 is a graph showing a comparison of the kinetic profile of drug release
of
leuprolide acetate from FIG. 1 using 10% PLGA microspheres as prepared in
Example 1
(0), and for comparison the published time release profile of leuprolide
acetate from
conventionally prepared microspheres (*) (D'Souza SS et al., AAPS PHARM Sci.
Tech.
6(4), E553-564, 2005).
Detailed Description
This disclosure provides a method of preparing drug-containing microparticles
comprising: (a) dissolving a biodegradable polymer in organic solvent to form
a polymer
solution; (b) dissolving or dispersing a drug in the polymer solution to form
a polymer-
drug-solvent phase; (c) mixing the polymer-drug-solvent phase with an aqueous
3
CA 02718750 2010-09-16
WO 2010/014176 PCT/US2009/004273
suspension comprising an inorganic gel to form a dispersion comprising polymer-
drug
droplets dispersed in the aqueous suspension; (d) evaporating the organic
solvent from
the dispersion to convert the polymer-drug droplets to drug-containing
microparticles;
and (e) recovering the drug-containing microparticles from the dispersion.
The step of dissolving a biodegradable polymer in organic solvent to form a
polymer solution may occur simultaneously with the step of dissolving or
dispersing a
drug in the polymer solution to form a polymer-drug-solvent phase. For
instance, a
hydrophobic drug could be dissolved into dichloromethand, and then solid
polymer could
be added to the drug-dichloromethane solution. As the polymer dissolves, the
drug of
course disperses in the polymer solution.
This is analogous to an oil-in-water emulsion method for forming
microparticles,
but without the need for an organic emulsifier. In a traditional evaporation
method using
an oil-in-water emulsion, a biodegradable polymer is dissolved in organic
solvent, and
drug is either dissolved in the same solvent in the polymer solution or is
dispersed in the
polymer solution. The drug may be dissolved in an aqueous solution and the
aqueous
solution dispersed in the polymer organic solvent solution, or the drug may be
dispersed
in the form of a dry powder in the polymer solution.
In a conventional oil-in-water emulsion method, the drug-polymer oil phase is
dispersed into an aqueous solution with the assistance of a surfactant or
emulsifier. The
surfactant or emulsifier is necessary to keep droplet size, and thus
microparticle size,
small.
The inventor has found that hydroxyapatite gel can also protect drug-polymer
phase droplets from coalescing, giving consistent and small microparticles.
The drug-
containing microparticles are produced in good yield, with high encapsulation
efficiency
of the drug. The particle size is consistent. Since no poly(vinyl alcohol) or
other
emulsifier needs to be used, the microparticles can be prepared so they are
not
contaminated with adherent poly(vinyl alcohol). The method uses inexpensive
materials
and is easily scaled up. The solidified microparticles can be recovered simply
by
dissolving the hydroxyapatite with acid, and then recovering microparticles by
centrifugation or filtration from the clear aqueous solution.
The method can be used to entrap peptide drugs, protein drugs, and small
molecule drugs. Both cationic and anionic small molecule drugs have been
successfully
entrapped in microparticles using the method.
4
CA 02718750 2010-09-16
WO 2010/014176 PCT/US2009/004273
Hydroxyapatite is a preferred inorganic gel for use in the method. But other
inorganic gels can also be used.
Preferably the inorganic gel can be dissolved by acid. Preferably the step of
recovering the drug-containing microparticles from the dispersion comprises
adding acid
to the dispersion to dissolve the inorganic gel.
Other apatatites than hydroxyapatite can also be used as the inorganic gel. In
particular embodiments, the inorganic gel is fluorapatatite (Ca5(PO4)3F),
chlorapatite
(Ca5(PO4)3CI), iodapatite (Ca5(PO4)30, or carbapatite (Caio(PO4)6CO3).
Mg(OH)2 and Al(OH)3 have also been tested, and performed well as the
to inorganic gel. In other specific embodiments, the inorganic gel is
dihydroxyaluminum
aminoacetate [a basic salt of aluminum and glycine, (NH2CH2C00)A1(OH)2] or
aluminum phosphate (AIP04).
Aluminum phosphate forms by adding phosphoric acid to a solution of aluminum
hydroxide. It forms a gel at a lower pH than does hydroxyapatite, specifically
at about
pH 6 to 7. It also dissolves at a low pH of about pH I or 2. Thus, it is
suited for
encapsulation of anionic drugs at low pHs where carboxyl groups on the drugs
are
partially or fully protonated.
In specific embodiments, the dispersion does not comprise an organic
surfactant
or organic emulsifier that assists dispersion of polymer-drug droplets in the
aqueous
suspension. A surfactant or emulsifier is not necessary because the inorganic
gel serves
the same function. Furthermore, the inorganic gel completely dissolves with
acid, and is
easy to separate from the microparticles. In contrast, organic surfactants and
emulsifiers
are found bound to the microparticles.
Thus, in particular embodiments, the dispersion does not comprise poly(vinyl
alcohol).
In some embodiments, an antistatic agent is added to the dispersion before
adding
acid to dissolve the inorganic gel. Examples of suitable antistatic agents are
poly(vinyl
alcohol) and poly(vinylpyrolidone-co vinyl acetate). The antistatic agent is
added to
prevent aggregation of the microparticles. Without the antistatic agent, the
microparticles may aggregate and they are often most conveniently recovered by
filtration. With the antistatic agent, the microparticles do not aggregate as
much, which
means the microparticles are smaller. In that case, the microparticles are
usually most
conveniently recovered by centrifugation. Aggregation does not change the size
of the
microparticles, and the aggregated microparticles can be disaggregated by
physical shear,
5
CA 02718750 2010-09-16
WO 2010/014176 PCT/US2009/004273
such as passing through a hypodermic needle in aqueous suspension several
times. They
can also be disaggregated by resuspending the microparticles in an aqueous
solution that
contains a surfactant. Typically a Tween 20, Tween 40, or Tween 80 are
included in the
solution in which microparticles are resuspended for injection. Microparticles
are also
disaggregated by lyophilization, and typically the harvested microparticles
are
lyophilized for storage.
Poly(vinyl alcohol) and other polymers can serve either as antistatic agents
or as
emulsifiers. But an emulsifier is added to the aqueous suspension before
adding the
polymer-drug-solvent phase in conventional prior art oil-in-water emulsions.
It must be
to present to emulsify the polymer-drug organic solvent droplets before
they solidify into
microparticles. In contrast, an antistatic agent can be added after the
microparticles are
formed, immediately before harvesting the microparticles.
Thus, in specific embodiments, the dispersion does not comprise an organic
surfactant or organic emulsifier before the drug-containing microparticles are
formed
(that is, before the polymer-drug droplets solidify into microparticles with
evaporation of
the organic solvent).
The amount of poly(vinyl alcohol) used as antistatic agent is much less than
as an
emulsifier. In conventional prior art oil-in-water emulsions, poly(vinyl
alcohol) is at a
concentration of 0.25%-0.5% (w/v) in the aqueous suspension for use as
emulsifier. The
inventor has added 0.025 volumes of 0.5% (w/v) poly(vinyl alcohol) solution to
the
dispersion immediately before adding HCI to dissolve the inorganic gel, where
poly(vinyl alcohol) is used as an antistatic agent. Thus, the final
concentration of
poly(vinyl alcohol) in the dispersion in this case is 0.0125%, much less than
when it is
used as an emulsifier.
Thus, in specific embodiments, the dispersion comprises no more than 0.05% by
weight or no more than 0.02% by weight of organic surfactant, organic
emulsifier, or
organic antistatic agent.
Two preferred biodegradable polymers for use in the methods of the invention
are polylactic acid (PLA) and poly(lactic acid-co-glycolic acid) (PLGA). In
other
embodiments, the biodegradable polymer is polyglycolic acid. In specific
embodiments,
the biodegradable polymer is a polyanhydride or a polyorthoester.
The method is effective with both hydrophobic and hydrophilic drugs.
Hydrophobic drugs can be codissolved with the polymer in the organic solvent.
Hydrophilic drugs may be dissolved first in a more polar organic solvent, such
as
6
CA 02718750 2010-09-16
WO 2010/014176 PCT/US2009/004273
methanol, and then mixed with the polymer dissolved in a less polar solvent,
such as
dichloromethane. Alternatively, hydrophilic drugs can be dissolved in an
aqueous
solution, and the aqueous drug solution can be dispersed into an organic
solvent solution
containing the polymer. This forms a water-in-oil dispersion for the drug-
polymer
phase. Hydrophilic drugs may also be directly dispersed as a solid powder in a
polymer
solution in an organic solvent.
Any suitable organic solvent can be used for the polymer-drug phase. These
include dichloromethane, ethyl acetate, acetonitrile, and methanol, and
mixtures thereof.
The organic solvent should at least include an organic solvent that is
immiscible with
to water, such as dichloromethane or ethyl acetate. Water-miscible solvents
such as
methanol or acetonitrile can be mixed with the water-immiscible solvent. A
water-
miscible solvent may be used to assist in dissolving hydrophilic drugs to get
the drug into
the oil phase in the dispersion.
The inventor has found that the best encapsulation efficiency and
microparticle
yield with basic amine-containing drugs, including leuprolide and verapamil
HCI,
nicardipine HCI, was achieved at a pH of about 9.0 to 10Ø Above pH 10, the
PLA and
PLGA polymers begin to hydrolyze and solubilize. This decreases the
microparticle
yield. The basic drugs are in a less ionized state at pH 9 to 10 than they are
at lower pHs,
and this is believed to cause them to stay associated with the polymers
better. But even
at pH 7.0, the encapsulation efficiency for leuprolide was 90%. So a variety
of pHs of
the aqueous suspension may be used.
With acidic carboxyl-containing drugs, including piroxicam, naproxen acid, and
salicylic acid the inventor has found that the best encapsulation efficiency
is obtained at
about pH 5Ø It is believed that this is because at more acidic pHs the acid
drugs are less
ionized, and in the nonionized state they associate with the polymers better
and do not
tend to partition into the aqueous phase as much.
Below about pH 5, hydroxyapatite crystallizes and in the crystal form it loses
the
ability to prevent coalescence of the drug-polymer phase droplets. Thus, with
hydroxyapatite at least, the aqueous suspension is preferably at a pH of about
5 or above.
In the Example below, 2 or 3 ml of drug-polymer solution containing 20% PLGA
and 10% leuprolide acetate was mixed with 400 ml of aqueous suspension
containing
hydroxyapatite prepared with 6 g of CaO. Higher amounts of drug-polymer
solution can
be mixed with the inorganic gel aqueous suspension, but higher concentrations
of
inorganic gel must also be used. The inventor has used as much as 100 ml of
drug-
7
CA 02718750 2010-09-16
WO 2010/014176 PCT/US2009/004273
polymer solution mixed with 400 ml of aqueous suspension containing higher
amounts
of hydroxyapatite.
In mixing the drug-polymer phase with the aqueous suspension, a high mixing
rate is used initially to form small droplets. In the Example below 5,000 rpm
or higher
was used for 5 minutes. After this short period of rapid mixing to form a
dispersion with
small drug-polymer droplets, the stirring rate is decreased to, e.g., 600 rpm.
Stirring is
continued for a longer period of time, typically one hour or longer, to allow
evaporation
of the organic solvent. During this time, solidified microparticles form as
the solvent
evaporates.
The microparticle size can be varied by methods known in the art. A higher
stirring rate during the mixing phase of mixing the drug-polymer phase with
the aqueous
suspension produces smaller droplets and thus smaller microparticles. A lower
polymer
concentration in the drug-polymer phase produces smaller particles because the
lower
polymer concentration produces a less viscous solution that tends to form
smaller
droplets. Longer polymers will produce a more viscous solution than shorter
polymers at
the same concentration, and thus longer polymers will tend to produce larger
microparticles. The organic solvent also affects microparticle size. If the
polymer is
highly soluble in the solvent, the drug-polymer phase will be less viscous and
smaller
microparticles will be formed. If the polymer is less soluble in the organic
solvent,
larger microparticles will be formed.
Any suitable drug can be formulated into these drug-containing microparticles.
In one embodiment, the drug is a protein. In another embodiment, the drug is a
peptide
(e.g., a peptide of 2 to 50 amino acids in length). In another embodiment, the
drug is a
small molecule, e.g., a molecule with a molecular weight of less than 1000 or
less than
500. In some embodiments, the small molecule is non-peptidyl.
In one embodiment, the drug is a peptide analog of leutenizing hormone-
releasing hormone (LHRH). Examples of suitable LHRH peptide analogs are
leuprolide,
triptorelin, and goserelin, and pharmaceutically acceptable salts thereof.
In specific embodiments, the drug is risperidone, octreotide, somatostatin,
human
growth hormone, deslorelin, buserelin, felypressin, gondorelin, oxytocin,
vasopressin,
fertirelin, histrelin, nafarelin, sincalide, thymopentin acetate, naltrexone,
or a
pharmaceutically acceptable salt thereof.
In other embodiments, the drug is verapamil, nicardipine, piroxicam, naproxen
acid, salicylic acid, or a pharmaceutically acceptable salt thereof.
8
CA 02718750 2010-09-16
WO 2010/014176 PCT/US2009/004273
Example
Materials and Methods:
Leuprolide acetate and poly(lactic acid-co-glycolic acid) (PLGA) RG502H or
PLA R202H were dissolved in a 24/76 (v/v) methanol/ dichloromethane mixture at
a
concentration of 20% polymer and 10% leuprolide (w/v) to prepare the polymer-
drug
solution. An aqueous suspension of hyroxyapatite was prepared by dissolving 6
g of
CaO in 400 ml water. Then phosphoric acid was added to adjust the pH of the
aqueous
suspension to between 9.0 and 10Ø Hydroxyapatite gel was formed in situ in
the
aqueous suspension under these conditions by the reaction 10 CaO + 6 H3PO4 =
Calo(PO4)6(OH)2 + 8 H20. The drug/polymer solution (2 or 3 ml) was dispersed
into the
aqueous suspension medium. The dispersion was initially homogenized by a high-
shear
mixer at 5,000 or more rpm for 5 minutes. The solvents were then evaporated by
stirring
at 600 rpm for 1 hour or longer. Then concentrated HCI was added slowly until
the
hydroxyapatite dissolved and the suspension cleared. Hydroxyapatite is
dissolved by the
reaction Caio(PO4)6(OH)2 + 20 HCI = 10 CaC12 + 6 H3PO4 + 2 1-120. The
solidified
microparticles were recovered by centrifugation and filtration.
Results and Discussion:
PLA and PLGA microparticles were examined and photographed
microscopically (data not shown). Most of the microparticles were found to be
in the
size range of 10-20 microns, which is a size suitable for injection.
Encapsulation efficiency of leuprolide acetate was 95% or higher when the pH
of
the suspending medium was pH 9.0 to pH 10Ø Encapsulation efficiency refers
to the
mass of drug recovered in microparticles divided by the starting mass of drug
in the
polymer-drug phase. To quantify drug in microparticles, the microparticles
were first
dissolved in an ethanol / dichloroethane (28/72 v/v) mixture. The drug content
of the
solution was then measured by ultraviolet absorption.
In other experiments, the pH of the suspending medium was varied. The
encapsulation efficiency of leuprolide acetate decreased to 90% at pH 7.0, and
decreased
further at lower pHs. Small anionic and cationic small molecule drugs were
also tested.
Encapsulation of amine-containing cationic drugs was highest at approximately
pH 9.0
to 1 0Ø It is believed that this is because the amine groups are largely
unprotonated at
elevated pH. At lower pHs where the drug amine groups are protonated, the
drugs are
9
CA 02718750 2015-12-09
=
more water soluble and thus partition more away from the polymer-drug
droplets. With
anionic drugs containing carboxyl groups, the pH relationship is reversed. The
carboxyl
groups are non-ionized at lower pHs, and therefore these drugs are less water-
soluble at
lower pHs. Thus, the anionic drugs have been found to have better
encapsulation
efficiency at lower pHs, e.g., about pH 5Ø Verapamil HC1 and nicardipine HCI
were
the small-molecule amine-containing drugs tested. Piroxicam, lidocaine, and
salicylic
acid were the small molecule anionic drugs tested.
Above pH 10 PLA and PLGA polymers are hydrolyzed to an extent, and this
decreases the yield of microparticles. Yield is defined herein as the
recovered mass of
microparticles divided by the starting mass of (polymer plus drug). At pHs
below about
5, hydroxyapatite crystallizes, instead of forming a gel. Without the
hydroxyapatite gel,
the polymer-drug droplets coalesce and the solid particles formed are too
large.
The release kinetics of leuprolide acetate from the PLGA microparticles
prepared
as described herein was determined and the results are shown in FIG. 1.
Concentration
of drug in the supernatant was measured by ultraviolet absorption. The release
of
leuprolide acetate from PLGA microparticles shows a typical tri-phase. Initial
burst
release occurs due to the fast dissolution of peptide located at the
microparticle surface
followed by a time lag of 4-5 days. After 4-5 days the microparticles have
hydrated
significantly and then erode due to the breakdown of the polymer. During the
middle
portion of the curve, peptide release occurs at a near constant rate.
FIG. 2 compares the release curve of FIG. 1 for 10% RG 502H with a published
release curve for leuprolide acetate from microparticles formed by
conventional oil-in-
water emulsion with poly(vinyl alcohol) as emulsifier (D'Souza et al., 2005,
AAPS
PHARR Sci. Tech. 6(4), E553). The release curves are nearly identical.
10