Note: Descriptions are shown in the official language in which they were submitted.
CA 02718830 2010-09-16
- 1 -
SPECIFICATION
METHOD FOR PROLIFERATION OF PLURIPOTENT STEM CELLS
TECHNICAL FIELD
[0001] The present invention relates to a method for proliferation of
pluripotent stem cells,
the use of laminin-5 as a cell-supporting material, and a culture kit for
pluripotent stem cells.
[0002] The present application claims priority based on Japanese Patent
Application Nos.
2008-93350 (filed on March 31, 2008) and 2008-225686 (filed on September 3,
2008).
BACKGROUND ART
[0003] Method for proliferation of pluripotent stem cells
Pluripotent stem cells are stem cells having the ability to differentiate into
cells of
every tissue type (differentiation pluripotency). Cells currently known as
pluripotent stem
cells include embryonic stem cells (ES cells), induced pluripotent stem cells
(iPS cells) which
are prepared from somatic cells by introducing and expressing a combination of
specific
factors (e.g., a combination of Oct3/4, Sox2, K1f4 and c-Myc), embryonic germ
cells (EG
cells) which are prepared from primordial germ cells, and germline stem cells
(GS cells)
which are prepared from germ cells in the testis.
[0004] Embryonic stem cells (ES cells) are pluripotent stem cells established
from the inner
cell mass (ICM) of blastocysts at the early developmental stage (Nature. 292,
154-156, 1981,
Proc. Natl. Acad. Sci. USA. 78, 7634-7638, 1981, Science. 282, 1145-1147,
1998). Mouse
ES cells can retain their pluripotency in the presence of leukemia inhibitory
factor (LIE)
(Nature. 336, 684-687, 1988, Nature. 336, 688-690, 1988). For their
maintenance culture,
in general, LIF-producing cell lines or mouse embryonic fibroblasts (MEFs) are
used as
feeder cells, or alternatively, a LIF-supplemented medium and an appropriate
supporting
material are used instead.
[0005] In the culture of mouse ES cells, feeder cells are responsible for
providing a scaffold
for cell adhesion and supplying growth factors required for ES cells and LIF.
It should be
noted that the culture solution may further be supplemented with LIF, in
addition to LIF
CA 02718830 2015-04-02
- 2 -
supplied from feeder cells. In embodiments where LIF is added, LIF per se or
the
supernatant of a LIF-producing cell line may be added to the culture solution.
On the other
hand, in the case of feeder cell-free systems, mouse ES cells are cultured in
a culture solution
supplemented with LIF by using various extracellular matrixes (e.g., gelatin)
as supporting
materials.
[0006] In contrast, the culture of human ES cells requires the presence of
basic fibroblast
growth factor (FGF2) to maintain their pluripotency (Dev Biol. 227, 271-278,
2000), and also
requires additional factors supplied from MEFs for this purpose. Human ES
cells can be
maintained and cultured in a state retaining their pluripotency, either by
using MEFs as
feeder cells in the presence of FGF2 or by using the supernatant of MEFs in
combination
with an appropriate supporting material (Nature Biotech. 19, 971-974, 2001).
[0007] More specifically, human ES cells may be cultured in the presence of
serum in
addition to FGF2 or may be cultured in a serum-free medium. Currently, it is
more
common to use a serum-free medium for culture of human ES cells, and serum
replacements
used for this purpose include KnockoutTM serum replacement (KSR, Invitrogen)
and so on.
Culture systems commonly used for human ES cells include those in which MEFs
are used as
feeder cells and the culture solution is supplemented with FGF2, or those in
which various
extracellular matrix proteins (e.g., MatrigelTM) are used as supporting
materials and the cells
are cultured in the supernatant of MEFs supplemented with FGF2. It should be
noted that
not only mouse fibroblasts, but also human fibroblasts can be used as feeder
cells.
[0008] Further, recent reports have shown that induced pluripotent stem cells
(iPS cells)
having properties very similar to those of embryonic stem cells were
established from
somatic cells in mice and humans (Cell. 126, 663-672, 2006, Cell. 131, 861-
872, 2007,
Science. 318, 1917-1920, 2007, W02007/069666). iPS cells are somatic cell-
derived
pluripotent stem cells established by introducing 0ct3/4, Sox2, K1f4, c-Myc
and/or other
factors into somatic cells. In general, the presence of feeder cells is also
required for
maintenance culture of iPS cells, as in the case of ES cells.
[0009] iPS cells can be cultured in the same manner as used for ES cells.
Mouse iPS cells
CA 02718830 2010-09-16
- 3 -
can be maintained and cultured on STO cells (cell line derived from mouse SIM
fibroblasts
stably producing LIF) using the supernatant of LIF-producing cells. On the
other hand, in
feeder cell-free systems, mouse iPS cells can be maintained and cultured on
gelatin-coated
plates when LIF is added to their medium. It should be noted that STO cells
are known to
be effective in stably maintaining mouse-derived ES cells, EG cells and EC
cells.
[0010] Human iPS cells can also be cultured in FGF2-supplemented systems by
using
mouse fibroblasts as feeder cells. It should be noted that the group of Shinya
Yamanaka et
al. (Kyoto University) uses SNL cells (cell line derived from mouse
fibroblasts co-expressing
LIF and the G418 resistance gene) for culture of human iPS cells (Cell. 131,
861-872, 2007).
In the case of using feeder cell-free systems for maintenance culture of human
iPS cells,
Matrigel is used as a supporting material and FGF2 is added to the supernatant
of MEFs.
[0011] Somatic cell-derived iPS cells have fewer ethical problems than early
embryo-
derived embryonic stem cells, and are free from the problem of immunological
rejection
because they can be prepared from patients' own cells. Their application to
regenerative
medicine is expected.
[0012] Embryonic germ cells (EG cells) are cells established from primordial
germ cells by
being cultured in the presence of Steel Factor (Kit-Ligand), LIF and FGF2, and
are known to
have substantially the same properties as ES cells, as demonstrated by
experiments in mice.
For maintenance culture of EG cells, it is possible to use the same culture
method as used for
ES cells. Namely, EG cells can be cultured in LIF-supplemented systems in the
presence of
feeder cells such as MEFs or STO cells (Cell 70:841-847, 1992, Development
120, 3197-
3120, 1994).
[0013] Germline stem cells (GS cells) prepared from germ cells in the testis
are cell lines of
spermatogonial stem cells (sperm stem cells) designed to allow in vitro
culture under culture
conditions containing at least GDNF (Glial cell-line derived neurotrophic
growth factor), and
they can form sperms when injected into seminiferous tubules in the testis.
Prolonged
culture of GS cells can be accomplished on MEFs (as feeder cells) by using a
medium
supplemented with GDNF, FGF2, EGF (epidermal growth factor) and LIF. It has
also been
CA 02718830 2010-09-16
- 4 -
reported that GS cells were cultured in feeder cell-free systems; GS cells can
be maintained
by being cultured on laminin-coated plates (Biology of Reproduction 69:612-
616, 2003,
Biology of Reproduction 72:985-991, 2005).
[0014] Among GS cells, mGS cells (multipotent germline stem cells)
particularly have the
same properties as ES cells and also have differentiation pluripotency. mGS
cells are
established by further converting the established GS cells into pluripotent
stem cells in
culture systems for ES cells. The established mGS cells can also be cultured
in the same
manner as used for ES cells, i.e., can be cultured in systems using a LIF-
supplemented
medium in the presence of feeder cells (Cell 119:1001-1012, 2004, Nature
440:1199-1203,
2006).
[0015] Such pluripotent cells as described above are expected to have
applications to
regenerative medicine, etc. However, particularly for their clinical
applications, it is
necessary to avoid the problem of immunological rejection and potential risks
such as
contamination with unknown viruses; and hence there is a need to develop a
maintenance
culture system in which no animal-derived substance is used whatsoever.
Namely, it is
predicted that when animal-derived sialic acids, which cannot be found in
humans, are taken
up into human ES cells or iPS cells, the antigenicity of these sialic acids
will become a
problem and will cause immunological rejection in the regenerated tissue.
Moreover, even
in the case of using human proteins, naturally occurring proteins prepared
from placenta and
other tissues may have potential risks such as contamination with AIDS virus
(HIV), hepatitis
C virus (HCV) and other unknown viruses.
[0016] For these reasons, while maintaining the pluripotency of human ES
cells, iPS cells
and other pluripotent stem cells, efforts have been made to search for the
composition of
culture solution capable of supporting their proliferation and/or appropriate
supporting
materials serving as substitutes for MEFs (Nature Biotech. 24, 185-187, 2006,
Stem Cell. 24,
2649-2660, 2006). However, even when MEFs are used as feeder cells, the cell
adhesion
efficiency obtained is as low as a few percent, and this efficiency further
decreases in feeder
cell-free systems currently under study. In the maintenance culture of
pluripotent stem
CA 02718830 2010-09-16
- 5 -
cells, it is desired to develop a system in which no animal-derived substance
is used
whatsoever, although low cell adhesion efficiency as described above is one of
the great
problems. Thus, there is a demand for human-derived supporting materials,
which exert
effective adhesion activity while maintaining pluripotency, and such
supporting materials
may serve as useful tools to obtain cellular materials that can be used in
clinical applications
such as regenerative medicine.
[0017] Laminin-5
Laminin, which is localized primarily on the basement membranes of various
tissues, is an extracellular matrix protein playing an important role in
maintenance of tissue
structure and in control of cell functions (Matrix Biol., 18:19-28, 1999, Dev.
Dyn., 218:213-
234, 2000).
[0018] The structure of laminin is a heterotrimer molecule composed of a, (3
and y chains
linked to each other via disulfide linkages, which takes a characteristic
cross-structure.
Each chain is composed of multiple domains, and domains I and II form a triple
helix.
Before the filing date of the present application, at least 15 isoforms of
laminin molecules
have been identified according to different combinations of 5 types of a
chains (al to a5), 3
types of (3 chains ((31 to (33) and 3 types of y chains (y1 to y3), and it is
suggested that there
are actually several times that number of isoforms (Miyazaki et al., Jikken
Igaku
(Experimental Medicine) Vol. 16 No. 16 (extra issue), 1998, pages 114-119,
Dev. Dyn.,
218:213-234, 2000, J. Neurosci., 20:6517-6528, 2000, Physiol Rev. 85, 979-
1000, 2005).
These a, 13 and y chains are encoded by different genes, respectively; and the
individual
laminin isoforms have specific sites of localization and specific functions,
and mainly
regulate cell adhesion, proliferation, motility, differentiation and so on
through the cell
membrane receptor integrin (Dev. Dyn. 218, 213-234, 2000, Physiol. Rev. 85,
979-1000,
2005).
[0019] Table 1 shows 15 laminin molecular species and their subunit structure.
[0020]
CA 02718830 2014-01-07
- 6 -
[Table 1]
Laminin molecular species and subunit structure
Name Structure Also called
Laminin-1 al[31y1 EHS laminin
Laminin-2 a2131y1 Merosin
Laminin-3 a1132y1 S-Laminin
Laminin-4 a2132y1 S-Merosin
Laminin-5 a3 133y2 Ladsin/epiligrin/
kalinin/nicein
Laminin-6 a3131y1 K-Laminin
Laminin-7 a3132y1 KS-Laminin
Laminin-8 a4131y1
Laminin-9 a4132y1
Laminin-10 a5131y1
Laminin-11 a5132y1
Laminin-12 a2131y3
Laminin-13 a3(32y3
Laminin-14 a4132y3
Laminin-15 a5P2y3
[0021] Laminin molecules construct the basement membrane by associating with
each other
at the amino (N) terminal portion (short arm) of the triple strand or by
associating with other
matrix molecules. On the other hand, laminin molecules each have 5 homologous
globular
domains (GI-GS domains or LG1-LG5) at the carboxy (C) terminal of the a chain,
and bind
to integrin or other receptors mainly at this site.
[0022] Laminin-5 (also called kalinin, epiligrin, nicein or ladsin) is one of
the laminin
isoforms, which is composed of a3, 133 and y2 chains, and was found by
multiple research
institutes under different circumstances (J. Cell Biol. 114, 567-576, 1991,
Cell 65, 599-610,
CA 02718830 2010-09-16
-7-
1991, J. Invest Dermatol. 101, 738-743, 1993, Proc. Natl. Acad. Sci. USA. 90,
11767-11771,
1993).
[0023] Laminin-5 is reported to have strong cell adhesion activity, cell
dispersion activity,
cell proliferation activity and the like on various cells (Proc. Natl. Acad.
Sci. USA. 90,
11767-11771, 1993, J. Biochem. 116, 862-869, 1994, J. Cell Biol. 125, 205-214,
1994, Mol.
Biol. Cell. 16, 881-890, 2005, Stem Cell. 24, 2346-2354, 2006). W02007/023875
discloses
culture techniques for mesenchymal stem cells using laminin-5.
CITATION LIST
PATENT LITERATURE
[0024] [PTL 1] W02007/069666
[PTL 2] W02007/023875
[PTL 3] JP 2001-172196 A
NON PATENT LITERATURE
[0025] [NPL 1] Nature. 292, 154-156, 1981
[NPL 2] Proc. Natl. Acad. Sci. USA. 78, 7634-7638, 1981
[NPL 3] Science. 282, 1145-1147, 1998
[NPL 4] Nature. 336, 684-687, 1988
[NPL 5] Nature. 336, 688-690, 1988
[NPL 6] Dev. Biol. 227, 271-278, 2000
[NPL 7] Nature Biotech. 19, 971-974, 2001
[NPL 8] Cell. 126, 663-672, 2006
[NPL 9] Science. 318, 1917-1920, 2007
[NPL 101 Nature Biotech. 24, 185-187, 2006
[NPL 11] Stem Cell. 24, 2649-2660, 2006
[NPL 12] Matrix Biol., 18:19-28, 1999
[NPL 13] Dev. Dyn., 218:213-234, 2000
[NPL 14] Miyazaki et al., Jikken Igaku (Experimental Medicine) Vol. 16, No. 16
(extra issue), 114-119, 1998
CA 02718830 2010-09-16
- 8 -
[NPL 15] J. Neurosci., 20:6517-6528, 2000
[NPL 16] Physiol. Rev. 85, 979-1000, 2005
[NPL 17] J. Cell Biol. 114, 567-576, 1991
[NPL 18] Cell. 65, 599-610, 1991
[NPL 19] J. Invest Dermatol. 101, 738-743, 1993
[NPL 20] Proc. Natl. Acad. Sci. USA. 90, 11767-11771, 1993
[NPL 211 J. Biochem. 116, 862-869, 1994
[NPL 22] J. Cell Biol. 125, 205-214, 1994
[NPL 23] Mol. Biol. Cell. 16, 881-890, 2005
[NPL 24] Stem Cell. 24, 2346-2354, 2006
[NPL 25] Dev. Biol. 163: p. 288-292, 1994
[NPL 26] Dev. Biol. 127: p. 224-227, 1988
[NPL 27] Reprod. Fertil. Dev. 6: p. 563-568, 1994
[NPL 28] Reprod. Fertil. Dev. 6: p. 553-562, 1994
[NPL 29] Proc. Natl. Acad. Sci. USA 92: p. 7844-7848, 1995
[NPL 30] Proc. Natl. Acad. Sci. USA 95:13726-13731, 1998
[NPL 31] Nature Biotech., 18, p. 399-404, 2000
[NPL 32] Nature 439: 216-219, 2006
[NPL 33] Cell Stem Cell 2: 113-117, 2008
[NPL 34] Stem Cells 24: 2669-2676, 2006
[NPL 35] Curr. Biol., 11: p. 1553-1558, 2001
[NPL 36] Nature Biotechnol 26:101-106, 2008
[NPL 37] Cell Stem Cell 2:10-12, 2008
[NPL 38] Cell 131:861-872, 2007
[NPL 39] Takahashi and Yamanaka, Saibo Kogaku (Cell Technology), Vol. 27, No.
3, 252-253, 2008
[NPL 40] J. Biol. Chem., 280 (2005), 14370-14377
[NPL 41] J. Biol. Chem. 269: p. 22779-22787, 1994
CA 02718830 2010-09-16
. .
- 9 -
[NPL 42] J. Biol. Chem. 269: p. 11073-11080, 1994
[NPL 43] J. Cell. Biol. 119: p. 679-693, 1992
[NPL 44] Nucleic Acids Res. 25:3389-3402, 1997
[NPL 45] J. Mol. Biol. 215:403-410, 1990
[NPL 46] J. Mol. Biol. 147:195-197, 1981
[NPL 47] Cell 70:841-847, 1992
[NPL 481 Development 120, 3197-3120, 1994
[NPL 49] Biology of Reproduction 69:612-616, 2003
[NPL 50] Biology of Reproduction 72:985-991, 2005
[NPL 51] Cell 119:1001-1012, 2004
[NPL 52] Nature 440:1199-1203, 2006
[NPL 53] Mol. Reprod. Dev. 36: p. 424-433, 1993
[NPL 54] Nature 454:646-650, 2008
[NPL 55] Cell 136:411-419, 2009
[NPL 561 Cell Stem Cell 3:568-574, 2008
[NPL 57] Science 322:945-949, 2008
[NPL 58] Science 322:949-953, 2008
SUMMARY OF THE INVENTION
[0026] The object of the present invention is to provide a technique for
efficient
proliferation of pluripotent stem cells in a system free from any animal-
derived substance
such as feeder cells or serum.
[0027] As a result of extensive and intensive efforts made to achieve the
above object, the
inventors of the present invention have found that the use of laminin-5, an
extracellular
matrix molecule, allows pluripotent stem cells to proliferate in an
undifferentiated state
without the need to use feeder cells or serum. This finding led to the
completion of the
present invention.
[0028] Accordingly, to achieve the above object, the present invention
provides a method
for proliferation of pluripotent stem cells, which comprises culturing the
pluripotent stem
CA 02718830 2015-04-02
- 10 -
cells in a medium free from both feeder cells and serum in a system containing
laminin-5.
[0029] The present invention also provides the use of laminin-5 as a cell-
supporting
material for proliferation of pluripotent stem cells.
[0030] The present invention further provides a culture kit for pluripotent
stem cells, which
comprises a laminin-5-treated culture vessel and a serum replacement.
[0031] The present invention includes the following embodiments as preferred
ones.
[Embodiment 1]
[0032] A method for proliferation of pluripotent stem cells, the method
comprising
culturing the pluripotent stem cells in a medium free from both feeder cells
and serum in the
presence of laminin-5.
[Embodiment 2]
[0033] The method of Embodiment 1, wherein the pluripotent stem cells are
induced
pluripotent stem cells.
[Embodiment 3]
[0034] The method of Embodiment 1 or 2, wherein the induced pluripotent stem
cells are
human induced pluripotent stem cells.
[Embodiment 4]
[0035] The method of Embodiment 1, wherein the pluripotent stem cells are
embryonic
stem cells, embryonic germ cells, or germline stem cells.
[Embodiment 5]
[0036] The method of Embodiment 4, wherein the pluripotent stem cells are
embryonic
stem cells.
[Embodiment 6]
[0037] The method of any one of Embodiments 1 to 5, wherein the culture medium
comprises a serum replacement.
[Embodiment 7]
[0038] The method of Embodiment 6, wherein the serum replacement comprises:
(a) an
amino acid which is: glycine, histidine, isoleucine, methionine,
phenylalanine, proline,
CA 02718830 2015-04-02
- ii -
hydroxyproline, serine, threonine, tryptophan, tyrosine, valine, or any
combination thereof;
(b) a vitamin which is: thiamine, ascorbic acid, or a combination thereof; (c)
a trace metal
element which is: silver, aluminum, barium, cadmium, cobalt, chromium,
germanium,
manganese, silicon, vanadium, molybdenum, nickel, rubidium, tin, zirconium, or
any
combination thereof; (d) a halogen element which is: bromine, iodine,
fluorine, or any
combination thereof; and (e) an ingredient which is: albumin, reduced
glutathione,
transferrin, insulin, sodium selenite, or any combination thereof
[Embodiment 8]
[0039] The method of any one of Embodiments 1 to 7, wherein the pluripotent
stem cells
are cultured in the presence of laminin-5 and an additional extracellular
matrix protein.
[Embodiment 9]
[0040] The method of Embodiment 8, wherein the additional extracellular matrix
protein is
collagen.
[Embodiment 10]
[0041] The method of any one of Embodiments 1 to 9, wherein the pluripotent
stem cells
are cultured in a laminin-5-treated culture vessel.
[Embodiment 11]
[0042] The method of Embodiment 10, wherein the laminin-5 is human laminin-5.
[Embodiment 12]
[0043] The method of any one of Embodiments I to 11, wherein the pluripotent
stem cells
do not differentiate during culture.
[Embodiment 13]
[0044] Use of laminin-5 as a cell-supporting material for the proliferation of
pluripotent
stem cells according to the method of any one of Embodiments Ito 12.
[Embodiment 14]
[0045] A culture kit for use in the proliferation of pluripotent stem cells
according to the
method of any one of Embodiments 1 to 12, the kit comprising a laminin-5-
treated culture
CA 02718830 2015-04-02
- 12 -
vessel and a serum replacement.
ADVANTAGEOUS EFFECTS OF INVENTION
[0046] The present invention enables efficient proliferation of pluripotent
stem cells while
maintaining them in an undifferentiated state, without the need to use feeder
cells or serum.
BRIEF DESCRIPTION OF DRAWINGS
[0047] Figure 1 shows electrophoresis of purified recombinant human laminin-5
on an SDS
polyacrylamide gel. It should be noted that the right lane in Figure 1 shows
the results of
electrophoresis of 1 pg recombinant human laminin-5.
Figure 2 shows a comparison between recombinant human laminin-5 and various
extracellular matrix proteins for their effect on the adhesion effect on ES
cells. Figure 2A
shows the results of adhesion assay after culture for 30 minutes, while Figure
2B shows the
results of adhesion assay after culture for 60 minutes.
Figure 3 shows a comparison of extracellular matrix proteins for their effect
on the
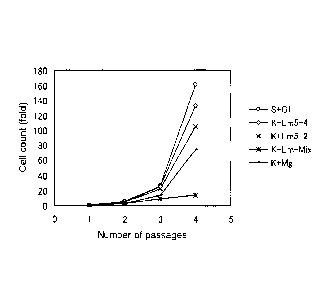
proliferation of ES cells in the absence of feeder cells. Figure 3 shows the
results of
proliferation assay, where open circles represent S+Gl, open diamonds
represent K+Lm5-4,
crosses represent K+Lm5-2, asterisks represent K+Lm-Mix, and small solid
squares
represent K+Mg.
Figure 4 shows a comparison of extracellular matrix proteins for their effect
on the
prolonged subculture of ES cells in the absence of feeder cells. Figure 4
shows the results
of proliferation assay, where open circles represent S+Gl, open diamonds
represent K+Lm5-
4, crosses represent K+Lm5-2, and open triangles represent K+Lm5-4 S+Gl.
Figure 5 shows the morphology of ES cells when subcultured for a long period
of
time on recombinant human laminin-5-coated plates in a serum-free medium and
when
returned to culture on gelatin-coated plates in a serum medium. Figure 5
shows, from the
top, the results of S+Gl, K+Lm5-4, and K+Lm5-4 --> S+Gl, respectively.
Figure 6 shows the results of RT-PCR compared for the expression of various
undifferentiation markers under each culture conditions in ES cells when
cultured in a KSR-
,
CA 02718830 2014-01-07
- 13 -
supplemented medium.
Figure 7 shows embryoid bodies upon culture in a LIF-free maintenance medium,
observed for ES cells cultured in a KSR-supplemented medium. In Figure 7, the
scale bar
represents 250 ptm.
Figure 8 shows the results studied for the differentiation potency of ES cells
in a
culture system of serum medium on gelatin-coated plates (S+G1) (Example 5). In
Figure 8,
the upper panel shows the results of S+G1 (LIF+), while the lower panel shows
the results of
S+G1 (LIF-). In Figure 8, although the scale bar represents 100 m, a single
asterisk (*)
indicates that the scale bar represents 50 m, and a double asterisk (**)
indicates that the
scale bar represents 25 m.
Figure 9 shows the results studied for the differentiation potency of ES cells
when
cultured in a culture system of KSR-supplemented serum-free medium on gelatin-
coated
plates (K+G1) and then returned to a culture system of serum medium (S+GI)
(Example 5).
In Figure 9, the upper panel shows the results of K+G1 = S+G1 (LIF+), while
the lower panel
shows the results of K+G1 = S+G1 (LIF-). In Figure 9, although the scale bar
represents
100 m, a double asterisk (**) indicates that the scale bar represents 25 m.
Figure 10 shows the results studied for the differentiation potency of ES
cells when
cultured in a culture system of KSR-supplemented serum-free medium on 4 g/m1
recombinant human laminin-5-coated plates (K+Lm5-4) and then returned to a
culture
system of serum medium (S+G1). In Figure 10, the upper panel shows the results
of
K+Lm5-4 = S+G1 (LIF+), while the lower panel shows the results of K+Lm5-4 =
S+G1
(LIF-). In Figure 10, although the scale bar represents 100 m, a double
asterisk (**)
indicates that the scale bar represents 25 m.
Figure 11 shows the results studied for the differentiation potency of ES
cells when
cultured in a culture system of KSR-supplemented serum-free medium on 2 g/m1
recombinant human laminin-5-coated plates (K+Lm5-2) and then returned to a
culture
system of serum medium (S+GI). In Figure 11, the upper panel shows the results
of
K+Lm5-2 S+GI (LIF+), while the lower panel shows the results of K+Lm5-2 S+G1
CA 02718830 2010-09-16
- 14 -
(LIF-). In Figure 11, although the scale bar represents 100 Inn, a single
asterisk (*)
indicates that the scale bar represents 50 [tm, and a double asterisk (**)
indicates that the
scale bar represents 25 inn.
Figure 12 shows a comparison of extracellular matrix proteins for their effect
on the
prolonged subculture of ES cells in the absence of feeder cells and using a
medium (medium
Y) supplemented with the serum replacement shown in Example 6. Figure 12 shows
the
results of proliferation assay, where open triangles represent Y+Gl, crosses
represent
Y+Lm5-4, asterisks represent Y+Lm5-2, open circles represent K+Lm-Mix, and
plus signs
(+) represent Y+Mg.
Figure 13 shows the results of RT-PCR compared for the expression of various
undifferentiation markers under each culture conditions in ES cells when
cultured in medium
Y.
Figure 14 shows embryoid bodies observed for ES cells when cultured in medium
Y
and then cultured in a LIF-free maintenance medium. In Figure 14, the scale
bar represents
250 p.m.
Figure 15 shows the results studied for the differentiation potency of ES
cells in a
culture system of serum medium on gelatin-coated plates (S+G1) (Example 7). In
Figure 15,
the upper panel shows the results of S+GI (LIF+), while the lower panel shows
the results of
S+G1 (LIF-). In Figure 15, although the scale bar represents 100 pm, a double
asterisk (**)
indicates that the scale bar represents 25 1.1.m.
Figure 16 shows the results studied for the differentiation potency of ES
cells when
cultured in a culture system of KSR-supplemented serum-free medium on gelatin-
coated
plates (K+G1) and then returned to a culture system of serum medium (S+G1)
(Example 7).
In Figure 16, the upper panel shows the results of K+G1 S+G1 (LIF+), while
the lower
panel shows the results of K+G1 S+G1 (LIF-). In Figure 16, although the
scale bar
represents 100 pm, a double asterisk (**) indicates that the scale bar
represents 25
Figure 17 shows the results studied for the differentiation potency of ES
cells when
cultured in a culture system of medium Y on gelatin-coated plates (Y+G1) and
then returned
CA 02718830 2010-09-16
- 15 -
to a culture system of serum medium (S+G1). In Figure 17, the upper panel
shows the
results of Y+G1 = S+G1 (LIF+), while the lower panel shows the results of Y+G1
S+G1
(LIF-). In Figure 17, although the scale bar represents 100 [tm, a double
asterisk (**)
indicates that the scale bar represents 25
Figure 18 shows the results studied for the differentiation potency of ES
cells when
cultured in a culture system of medium Y on 4 tg/m1 recombinant human laminin-
5-coated
plates (Y+Lm5-4) and then returned to a culture system of serum medium (S+G1).
In
Figure 18, the upper panel shows the results of Y+Lm5-4 S+G1
(LIF+), while the lower
panel shows the results of Y+Lm5-4 S+G1
(LIF-). In Figure 18, although the scale bar
represents 100 ttm, a double asterisk (**) indicates that the scale bar
represents 25
Figure 19 shows the results studied for the differentiation potency of ES
cells when
cultured in a culture system of medium Y on 2 vg/m1 recombinant human laminin-
5-coated
plates (Y+Lm5-2) and then returned to a culture system of serum medium (S+G1).
In
Figure 19, the upper panel shows the results of Y+Lm5-2 S+G1
(LIF+), while the lower
panel shows the results of Y+Lm5-2 = S+G1 (LIF-). In Figure 19, although the
scale bar
represents 100 vm, a double asterisk (**) indicates that the scale bar
represents 25 [tin.
Figure 20 shows the results studied for the differentiation potency of ES
cells when
cultured in a culture system of medium Y in the presence of Lm-Mix (Y+Lm-Mix)
and then
returned to a culture system of serum medium (S+GI). In Figure 20, the upper
panel shows
the results of Y+Lm-Mix S+G1 (LIF+), while the lower panel shows the
results of Y+Lm-
Mix S+G1 (LIF-). In Figure 20, although the scale bar represents 100 [im, a
double
asterisk (**) indicates that the scale bar represents 25
Figure 21 shows the results studied for the differentiation potency of ES
cells when
cultured in a culture system of medium Y in the presence of Mg (Y+Mg) and then
returned to
a culture system of serum medium (S+G1). In Figure 21, the upper panel shows
the results
of Y+Mg S+G1 (LIF+), while the lower panel shows the results of Y+Mg S+GI (LIF-
).
In Figure 21, although the scale bar represents 100 [t.m, a double asterisk
(**) indicates that
the scale bar represents 25 tun.
CA 02718830 2010-09-16
- 16 -
Figure 22 shows the morphology of human iPS cells on feeder cells. In Figure
22,
the left panel shows the morphology of 201B2, while the right panel shows the
morphology
of 201B7. In Figure 22, the scale bar represents 1 mm.
Figure 23 shows a comparison between recombinant human laminin-5 and various
extracellular matrix proteins for their effect on the adhesion effect on human
iPS cells, as
analyzed by adhesion assay.
Figure 24 shows the morphology of adhered cells observed in a comparison
between
recombinant human laminin-5 and various extracellular matrix proteins for
their effect on the
adhesion effect on human iPS cells, as analyzed by adhesion assay. In Figure
24, the scale
bar represents 250 1..tm.
Figure 25 shows a comparison between recombinant human laminin-5 and various
extracellular matrix proteins for their effect on the colony formation of
human iPS cells, as
analyzed by colony assay. In Figure 25, the upper panel shows the results of
Single, while
the lower panel shows the results of Clump.
Figure 26A shows the results studied for maintenance of an undifferentiated
state in
human iPS cell colonies formed from single cells on recombinant human laminin-
5 and
various extracellular matrix proteins. Figure 26A shows the results of
immunostaining
obtained after colony assay in Single state, and the scale bar represents 250
[Am.
Figure 26B shows the results studied for maintenance of an undifferentiated
state in
human iPS cell colonies formed from cell clumps on recombinant human laminin-5
and
various extracellular matrix proteins. Figure 26B shows the results of
immunostaining
obtained after colony assay in Clump state, and the scale bar represents 250
Figure 27 shows the morphology of human iPS cells at 5 weeks of culture during
prolonged subculture of human iPS cells formed on recombinant human laminin-5
and
various extracellular matrix proteins. The scale bar represents 1 mm (left
panel) and
100 [tm (right panel) for each extracellular matrix.
Figure 28 shows the results studied for the expression of various
undifferentiation
markers in human iPS cells when cultured in the presence of recombinant human
laminin-5
CA 02718830 2010-09-16
- 17 -
and various extracellular matrix proteins.
Figure 29 shows the morphology of human iPS cells when induced to
differentiate
after culture in the presence of recombinant human laminin-5 and various
extracellular matrix
proteins. In Figure 29, the scale bar represents 1 mm.
Figure 30 shows the results studied for the expression of various
differentiation
markers in human iPS cells when induced to differentiate after culture in the
presence of
recombinant human laminin-5 and various extracellular matrix proteins.
DESCRIPTION OF EMBODIMENTS
[0048] Details and additional features and advantages of the present invention
will be
described in more detail below on the basis of embodiments.
[0049] 1. Method for proliferation of pluripotent stem cells
The present invention provides a method for proliferation of pluripotent stem
cells.
The method of the present invention comprises culturing the pluripotent stem
cells in a
medium free from both feeder cells and serum in a system containing laminin-5.
[0050] Pluripotent stem cells
As used herein, the term "pluripotent stem cells" is intended to collectively
refer to
stem cells having the ability to differentiate into cells of any tissue type
(differentiation
pluripotency). Although ES cells are used for study in the Example section
described later,
pluripotent stem cells that can be proliferated by the method of the present
invention include
not only embryonic stem cells, but also all pluripotent stem cells derived
from, e.g., cells of
adult mammalian organs or tissues, bone marrow cells, blood cells, and
embryonic or fetal
cells, as long as their characters are similar to those of embryonic stem
cells. In this case,
characters similar to those of embryonic stem cells can be defined by cell
biological
properties specific to embryonic stem cells, including the presence of surface
(antigen)
markers specific to embryonic stem cells, the expression of genes specific to
embryonic stem
cells, or the ability to form teratomas.
[0051] Specific examples of cells that can be proliferated by the method of
the present
invention include, but are not limited to, embryonic stem cells (ES cells),
induced pluripotent
CA 02718830 2010-09-16
,
=
- 18 -
stem cells (iPS cells), embryonic germ cells (EG cells), germline stem cells
(GS cells) and so
on. It should be noted that pluripotent stem cells preferred in the present
invention are ES
cells and iPS cells. iPS cells are particularly preferred, for example,
because they have no
ethical problem.
[0052] As used herein, the term "ES cells" is intended to mean cell lines
designed to allow
in vitro culture, which are prepared from pluripotent stem cells at the early
developmental
stage having the ability to differentiate into all tissue cells constituting
the whole body. ES
cells can be expanded to virtually unlimited numbers while retaining their
ability to
differentiate into all cells constituting the whole body, as in the case of
pluripotent stem cells
in early embryos.
[0053] More specifically, mouse ES cells are the first ES cells reported in
1981 (Proc. Natl.
Acad. Sci. USA 78, 7634-7638, 1981, Nature 292, 154-156, 1981). ES cells have
pluripotency and can generate all tissue and cell types constituting the whole
body.
[0054] Pluripotent embryonic stem cells have been isolated from a wide variety
of species,
including rats (Iannaconns et al., Dev. Biol. 163, 288-292, 1994), hamsters
(Dev. Biol. 127,
224-227, 1988), rabbits (Mol. Reprod. Dev. 36, 424-433, 1993), birds, fish,
pigs (Reprod.
Fertil. Dev. 6, 563-568, 1994), cattle (Reprod. Fertil. Dev. 6, 553-562,
1994), as well as
primates (Proc. Natl. Acad. Sci. USA 92, 7844-7848, 1995).
[0055] In addition, some research teams have succeeded in isolating ES cells
and ES cell-
like stem cells from embryonic human tissue. Their early successes are as
described in
Science 282, 1145-1147, 1998, Proc. Natl. Acad. Sci. USA 95, 13726-13731,
1998, Nature
Biotech., 18, 399-404, 2000. These ES cell lines have been established from
ICM separated
from blastocysts by being cultured on feeder cells. Other recent studies have
indicated that
embryos and embryonic cells can be obtained when nuclei derived from embryos
and mature
mammalian cells are transplanted into enucleated oocytes.
[0056] In 2006, the group of Robert Lanza et al. (Advanced Cell Technology,
Inc.)
succeeded in establishing mouse and human ES cells by using only single
blastomeres from
embryos at the cleavage stage before the blastocyst stage in embryonic discs,
without
CA 02718830 2010-09-16
- 19 -
impairing the developmental potency of embryos (Nature 439: 216-219, 2006,
Cell Stem Cell
2, 113-117, 2008). The development of this technique enabled the establishment
of ES cells
without destructing fertilized eggs. In the same year, the group of Miodrag
Stojkovic et al.
(University of Newcastle) succeeded in establishing ES cells from human
embryos whose
development was arrested (Stem Cells 24, 2669-2676, 2006). This allowed excess
eggs to
be used, which had been discarded in infertility treatment.
[0057] Moreover, in 2004, the group of Yury Verlinsky et al. (Reproductive
Genetics
Institute of Chicago) succeeded in establishing 20 ES cell lines from human
embryos having
hereditary diseases. These are the first ES cells that can be used for
therapeutic studies on
serious hereditary diseases. In addition to them, Yury Verlinsky now possesses
200 or more
ES cell lines having different genes, which can be used for screening of
pharmaceuticals or
other purposes.
[0058] In the method of the present invention, any established ES cell line
can be used.
On the other hand, to avoid immunological rejection which will occur when ES
cells
prepared by the method of the present invention are applied to an individual,
it is effective to
use the subject's somatic cells to create clone embryos, from which ES cell
lines are then
established. This technique allows the establishment of ES cells having the
same genetic
elements as the individual.
[0059] On the other hand, during creation of somatic cell clones, a phenomenon
called
"reprogramming" would occur, in which somatic cell nuclei introduced into ova
would enter
the same state as the nuclei of fertilized eggs. ES cells are reported to also
have activity
similar to such activity as observed in ova (Curr. Biol., 11, 1553-1558,
2001). Namely, it is
expected that fusion between individual's somatic cells and ES cells allows
the somatic cells
to be converted into ES cell-like cells. Since ES cells can be genetically
manipulated in
vitro, it is expected that when ES cells pre-treated to modify factors
responsible for
immunological rejection (e.g., groups of MHC genes) are used for this purpose,
rejection
reaction can be avoided without using techniques such as creation of somatic
cell clone
embryos.
CA 02718830 2010-09-16
- 20 -
[0060] In the present invention, "ES cells" are preferably human ES cells.
Established
human ES cell lines are currently available, for example, from the Institute
for Frontier
Medical Sciences, Kyoto University.
[0061] Alternatively, ES cells can also be prepared as described in the
various documents
cited herein above.
[0062] As used herein, the term "iPS cells" is intended to mean cells having
differentiation
pluripotency similar to that of ES cells, which are obtained from somatic
cells by introducing
genes for transcription factors (e.g., 0ct3/4, Sox2, Klf4, c-Myc). Thus, as in
the case of ES
cells, iPS cells can also be expanded to unlimited numbers while retaining
their
differentiation pluripotency.
[0063] To isolate only somatic cells which have been converted into ES-like
cells, the
group of Shinya Yamanaka et al. (Kyoto University) focused on a gene called
Fbx15, which
is expressed only in ES cells but is not required for maintenance of their
differentiation
pluripotency. At this gene locus, they introduced the neomycin resistance gene
by
homologous recombination techniques and supplemented the medium with G418,
which is
detoxicated by the action of this resistance gene, to construct an
experimental system by
which only ES-like cells expressing Fbx15 would acquire G418 resistance and
hence survive,
whereas normal somatic cells not expressing Fbx15 would be killed. Using this
experimental system, they found that 4 genes, Oct3/4, Sox2, K1f4 and c-Myc,
were sufficient
to establish iPS cells (Cell. 126, 663-672, 2006).
[0064] Further, the group of Shinya Yamanaka et al. also succeeded in
establishing human
iPS cells from fibroblasts by using OCT3/4, SOX2, KLF4 and C-MYC, which are
human
homologs of the mouse genes used for establishment of mouse iPS cells (Cell.
131, 861-872,
2007).
[0065] Concurrently, the group of James Thomson et al., who were the first
researchers in
the world to establish human ES cells, succeeded in establishing human iPS
cells when 4
genes, OCT3/4, SOX2, NANOG and LIN28, among genes expressed specifically in
human
ES cells were introduced into fetal lung-derived fibroblasts or neonatal
foreskin-derived
CA 02718830 2010-09-16
- 21 -
fibroblasts by using the same strategy as that of Shinya Yamanaka et al.
(Kyoto University)
used for successful establishment of mouse iPS cells (Science 318:1917-1920,
2007).
[0066] Moreover, in December 2007, the group of Shinya Yamanaka et al. (Kyoto
University) demonstrated that only three factors, Oct-4, Sox2 and K1f4, were
sufficient to
establish iPS cells in mice and humans without introducing the c-Myc gene, and
thus
succeeded in suppressing the conversion of iPS cells into cancer cells (Nat.
Biotechnol.
26:101-106, 2008). Almost at the same time, the group of Rudolf Jaenisch et
al.
(Massachusetts Institute of Technology) also succeeded in similar experiments
in mice (Cell
Stem Cell 2:10-12, 2008).
[0067] Moreover, to avoid risks such as carcinogenesis, attempts have been
made to further
reduce the number of factors to be introduced. As a result, Hans R Scholer et
al. (Max
Planck Institute for Biochemistry) have succeeded in establishing iPS cells by
introducing
two genes, i.e., either Oct4 and K1f4 or Oct4 and c-Myc into adult mouse
neural stem cells
(Nature 454:646-650, 2008). Recently, Hans R Scholer et al. have further
reported that the
introduction of Oct4 alone is sufficient to prepare iPS cells from neural stem
cells (Cell
136:411-419, 2009).
[0068] Further, Sheng Ding et al. have reported that some genes can be
compensated by
small-molecule compounds during induction of iPS cells (Cell Stem Cell 3, 568-
574, 2008).
They have demonstrated that the use of small-molecule compounds such as BIX
and BayK
enables the induction of iPS cells by introducing only two genes, Oct4 and
K1f4, into mouse
embryonic fibroblasts.
[0069] Furthermore, attempts have also been made to improve gene transfer
techniques for
induction of iPS cells. Although viruses (e.g., retrovirus) with a high
potential to integrate a
transgene into the chromosome are used widely at present, there are reports of
techniques
using adenovirus (Science 322, 945-949, 2008) or plasmid vectors (Science 322,
949-953,
2008), which appear to be less integrated.
[0070] In the present invention, "iPS cells" are preferably human iPS cells.
Established
human iPS cell lines are currently available, for example, from Kyoto
University or RIKEN
CA 02718830 2010-09-16
- 22 -
BioResource Center.
[0071] Alternatively, iPS cells may also be prepared by reference to the
documents shown
below. For example, induced pluripotent stem cells can be prepared according
to the
procedures described in the documents by the group of Professor Shinya
Yamanaka (Kyoto
University) (Cell 131, 861-872, 2007, Nat. Biotechnol. 26, 101-106, 2008) or
in the
document by the group of Thomson (University of Wisconsin) (Science 318, 1917-
1920,
2007).
[0072] More specifically, any type of somatic cells may be introduced with at
least one or
more genes selected from Oct3/4, Sox2, c-Myc, K1f4, Nanog and LIN28, and then
screened
by detecting the expression of genes or proteins specific to pluripotent stem
cells to prepare
iPS cells.
[0073] As in the case of ES cells, the iPS cells thus prepared can be cultured
together with
basic fibroblast growth factor in the presence of mouse fibroblasts whose
proliferation has
been inactivated or alternatives thereof, and can also be used as pluripotent
stem cells.
[0074] These iPS cells have been found to have the same properties as ES cells
in relation
to features of differentiation into various tissues and features of gene
expression in the cells
(Cell. 126, 663-672, 2006, Cell 131:861-872, 2007, Science 318, 1917-1920,
2007), and
conditions for culturing ES cells and conditions for inducing differentiation
from ES cells
into various tissues can be applied directly to iPS cells (Takahashi and
Yamanaka, Saibo
Kogaku (Cell Technology), Vol. 27, No. 3, 252-253, 2008).
[0075] As used herein, the term "EG cells" is intended to mean any type of
embryonic germ
cells prepared from primordial germ cells, and their origin is not limited in
any way.
[0076] As used herein, the term "GS cells" refers to germline stem cells
prepared from
germ cells in the testis, i.e., cell lines of spermatogonial stem cells (sperm
stem cells)
designed to allow in vitro culture (Cell. 119, 1001-1012, 2004). Among GS
cells, mGS
cells (multipotent germline stem cells) are particularly preferred because
they have the same
properties as ES cells and also have differentiation pluripotency. When used
herein, the
term "GS cells" means mGS cells, depending on the context.
CA 02718830 2010-09-16
- 23 -
[0077] Laminin-5
The method of the present invention is directed to the culture of pluripotent
stem
cells and its most remarkable feature lies in culturing the pluripotent stem
cells in a system
containing laminin-5.
[0078] Laminin-5 is reported to have stronger adhesion activity on many cell
types than
various extracellular matrix proteins including other laminin isoforms (J.
Biochem. 116, 862-
869, 1994, J. Cell Biol. 125, 205-214, 1994, Mol Biol Cell. 16, 881-890,
2005).
[0079] As shown in Table 1, laminin-5 is a laminin molecule composed of a3,
(33 and y2
chains, which plays a dominant role in binding between epidermis and corium,
and binds
preferentially to integrin a3131 in most cells and also binds to integrin
a6131 or a6134 in some
cells. In laminin-5, it has been elucidated that the a3G2A sequence
(RERFNISTPAFRGCMKNLKKTS) in the a3 chain G2 domain and the KRD sequence in
the G3 domain are major binding sites for integrin.
[0080] It is also known that laminin-5, after being secreted as a trimer,
receives limited
hydrolysis by protease to remove G4 and G5 domains located at the C-terminal
of the a3
chain, and is thereby converted from 190 kDa (non-truncated) into 160 kDa
(truncated).
Laminin-5 isolated in a standard manner does not have G4 and G5 domains. Such
a3
chain-truncated laminin-5 is known to have higher stimulatory activities on
cell adhesion,
motility and neuranagenesis, when compared to non-truncated laminin-5 (J.
Biol. Chem., 280
(2005), 14370-14377).
[0081] Laminin-5 in the present invention is not limited in any way, and may
be either in a
non-truncated form containing G4 and G5 domains or in a truncated form free
from all or
part of G4 and G5 domains.
[0082] Moreover, the laminin-5 protein may be either naturally occurring or
modified to
have one or more modified amino acid residues while retaining its biological
activities,
particularly stimulatory activity on cell adhesion. Moreover, the laminin-5
protein in the
present invention may be of any origin and may be prepared in any manner, as
long as it has
the features described herein. Namely, the laminin-5 protein of the present
invention may
CA 02718830 2010-09-16
, .
,
- 24 -
be naturally occurring, expressed from recombinant DNA by genetic engineering
procedures,
or chemically synthesized.
[0083] The laminin-5 protein may be of any origin, preferably of human origin.
In a case
where human pluripotent stem cells are cultured in order to obtain materials
for regenerative
medicine, etc., it is preferred to use laminin-5 of human origin in the sense
of avoiding the
use of materials derived from other animals.
[0084] SEQ ID NOs: 1 to 6 in the Sequence Listing herein show the nucleotide
and amino
acid sequences of human laminin-5 a3, 133 and y2 chains, respectively. The
laminin-5
protein to be used in the present invention is preferably a protein composed
of the following
subunits: an a3 chain having the amino acid sequence of SEQ ID NO: 2 or an
amino acid
sequence comprising deletion, addition or substitution of one or more amino
acids in the
sequence of SEQ ID NO: 2 (amino acid residues 1-1713) (J. Biol. Chem. 269,
22779-22787,
1994), a 133 chain having the amino acid sequence of SEQ ID NO: 4 or an amino
acid
sequence comprising deletion, addition or substitution of one or more amino
acids in the
sequence of SEQ ID NO: 4 (amino acid residues 1-1170) (J. Biol. Chem. 269,
11073-11080,
1994), and a y2 chain having the amino acid sequence of SEQ ID NO: 6 or an
amino acid
sequence comprising deletion, addition or substitution of one or more amino
acids in the
sequence of SEQ ID NO: 6 (amino acid residues 1-1193) (J. Cell. Biol. 119, 679-
693, 1992).
[0085] Globular domains (G1 to G5 domains) in the a3 chain correspond to amino
acid
residues 794-970, 971-1139, 1140-1353, 1354-1529 and 1530-1713, respectively,
in SEQ ID
NO: 1.
[0086] Each chain of laminin-5 may have an amino acid sequence comprising
deletion,
addition or substitution of one or more amino acid residues in the amino acid
sequence
shown in the corresponding SEQ ID NO. Such proteins having amino acid
sequences
homologous to naturally occurring proteins can also be used in the present
invention. The
number of amino acids which may be modified is not limited in any way in the
respective
amino acid sequences of a3, 133 and y2 chains, but it is preferably 1 to 300
amino acid
residues, 1 to 200 amino acid residues, 1 to 150 amino acid residues, 1 to 120
amino acid
CA 02718830 2010-09-16
- 25 -
residues, 1 to 100 amino acid residues, 1 to 80 amino acid residues, 1 to 50
amino acid
residues, 1 to 30 amino acid residues, 1 to 20 amino acid residues, 1 to 15
amino acid
residues, 1 to 10 amino acid residues, or 1 to 5 amino acid residues. More
preferred is a
possible number of amino acid residues which may be modified by known site-
directed
mutagenesis, for example, 1 to 10 amino acid residues, or 1 to 5 amino acid
residues.
[0087] It is well known in the art that conservative substitution of amino
acids can be used
to obtain proteins or polypeptides retaining their original functions. Such
substitution
includes replacement of an amino acid with another residue having similar
physical and
chemical properties, as exemplified by replacement of one fatty acid residue
(Ile, Val, Leu or
Ala) with another, or replacement between basic residues Lys and Arg, between
acidic
residues Glu and Asp, between amide residues Gln and Asn, between hydroxyl
residues Ser
and Tyr, or between aromatic residues Phe and Tyr.
[0088] Laminin-5 to be used in the present invention may also be a protein
sharing at least
80%, 85%, 90%, 95%, 98% or 99% identity with the amino acid sequences shown in
SEQ ID
Nos: 2, 4 and 6 and having the ability to stimulate cell adhesion activity.
When the
structure of constituent subunits is compared between laminin-5 and laminin-1,
the amino
acid sequence homology in each subunit is 50% or less. In particular, the
homology in the
above a chain G domains is as low as about 25%.
[0089] Identity is calculated as follows: the number of identical residues is
divided by the
total number of residues in a corresponding known sequence or a domain
therein, and then
multipled by 100. Computer programs available for use in the determination of
sequence
identity using standard parameters include, for example, Gapped BLAST PSI-
BLAST
(Nucleic Acids Res. 25, 3389-340, 1997), BLAST (J. Mol. Biol. 215:403-410,
1990), and
Smith-Waterman (J. Mol. Biol. 147:195-197, 1981). In these programs, default
settings are
preferably used, but these settings may be modified, if desired.
[0090] The laminin-5 protein in the present invention may be of any origin and
may be
prepared in any manner, as long as it has the features described herein.
Namely, the
laminin-5 protein of the present invention may be a naturally occurring
laminin-5 protein as
,
CA 02718830 2010-09-16
. =
- 26 -
found in or purified from the supernatant of human or animal cells secreting
laminin-5.
However, laminin-5 can be effectively produced as a gene recombinant protein
by expressing
each subunit using recombinant DNA technology known in the art. It is
particularly
preferred to obtain laminin-5 as a human recombinant protein, in the sense of
avoiding
unwanted factors derived from other animals.
[0091] For this purpose, primers may be designed based on a DNA sequence
comprising
nucleic acid residues 1-5139 in SEQ ID NO: 1 (encoding the laminin-5 a3 chain)
and
nucleotide sequences of nucleic acid residues 121-3630 in SEQ ID NO: 3
(encoding the 13
chain) and nucleic acid residues 118-3696 in SEQ ID NO: 5 (encoding the y2
chain), and an
appropriate cDNA library may be used as a template in polymerase chain
reaction (PCR) to
amplify desired sequences. Such PCR procedures are well known in the art and
can be
found, e.g., in "PCR Protocols, A Guide to Methods and Applications," Academic
Press,
Michael, et al., 1990.
[0092] DNA encoding a gene for each chain of laminin-5 may be integrated into
an
appropriate vector and then introduced into either eukaryotic or prokaryotic
cells by using an
expression vector that allows expression in each host, whereby the respective
chains are
expressed to obtain a desired protein. Host cells which can be used to express
laminin-5 are
not limited in any way and include prokaryotic host cells such as E. coli and
Bacillus subtilis,
as well as eukaryotic hosts such as yeast, fungi, insect cells and mammalian
cells. It should
be noted that human fetal kidney cell line HEI(293 used in the Example section
described
later is particularly preferred as a host cell.
[0093] A vector constructed to express laminin-5 can be introduced into the
above host
cells by transformation, transfection, conjugation, protoplast fusion,
electroporation, particle
gun technique, calcium phosphate precipitation, direct microinjection or other
techniques.
The cells containing the vector may be grown in an appropriate medium to
produce a
laminin-5 protein to be used in the present invention, which may then be
purified from the
cells or medium to obtain the laminin-5 protein. Purification may be
accomplished, for
example, by size exclusion chromatography, HPLC, ion exchange chromatography,
CA 02718830 2015-04-02
- 27 -
immunoaffinity chromatography, etc.
[0094] Laminin-5 is described in detail in JP 2001-172196 A.
[0095] Method for proliferation of pluripotent stem cells
In the present invention, pluripotent stem cells are cultured in a system
containing
laminin-5. As used herein, the phrase "system containing laminin-5" is
intended to mean
that a culture system for pluripotent stem cells contains laminin-5 in some
fashion, and
embodiments thereof are not limited in any way.
[0096] In the present invention, it is a preferred embodiment to use a laminin-
5-treated
culture vessel for culture of pluripotent stem cells in a system containing
laminin-5.
However, the "system containing laminin-5" intended in the present invention
is not limited
to this embodiment, and also includes other embodiments where laminin-5 is
added to the
medium for culture of pluripotent stem cells.
[0097] As used herein, the phrase "laminin-5-treated culture vessel" is
intended to mean a
culture vessel whose surface is treated with laminin-5, e.g., by coating. The
"culture vessel"
intended in the present invention is not limited in any way, and a vessel of
any material and
any shape may be used as long as it is sterilized to avoid bacterial
contamination and is
suitable for cell culture. Examples of such a culture vessel include, but are
not limited to,
culture dishes, culture flasks, culture petri dishes, culture plates (e.g., 96-
well, 48-well, 12-
well, 6-well, 4-well plates), culture bottles and so on, all of which are
commonly used in the
art. Techniques for coating laminin over the surface of a culture vessel are
known in the art,
and those skilled in the art would be able to select any type of culture
vessel suitable for the
object of the present invention, treat the vessel with laminin-5, and use the
laminin-5-treated
vessel to culture pluripotent stem cells by the method of the present
invention.
[0098] The amount of laminin-5 used for culture vessel treatment is not
limited in any way.
When treated with a laminin-5 solution of 0.05 g/m1 or more, preferably 0.5
to 15 g/ml,
more preferably 3.75 to 15 g/ml, good results are obtained.
[0099] As shown in the Example section described herein later, the inventors
of the present
I
CA 02718830 2010-09-16
- 28 -
invention have found that recombinant human laminin-5 shows stronger adhesion
activity on
mouse ES cells than Matrigel, a laminin mixture or other extracellular matrix
proteins, and
thus have arrived at the present invention. Further, in the present invention,
it has been
found that mouse ES cells can be maintained and cultured on plates coated with
recombinant
human laminin-5, even in a LIF-free and serum-free medium and in the absence
of MEFs.
The culture of mouse ES cells in the absence of MEFs has been conventionally
accomplished
by using gelatin-coated plates and in the presence of bovine fetal serum
(FBS). When
cultured by the method of the present invention, mouse ES cells were confirmed
to
proliferate at a level equal to that in conventional methods even under MEF-
free and FBS-
free conditions and also to retain their undifferentiated state. In addition,
when cultured by
the method of the present invention, mouse ES cells were found to retain
pluripotency.
[0100] Studies were also conducted on human iPS cells, indicating that
recombinant human
laminin-5 also showed strong adhesion activity on human iPS cells, as
demonstrated in the
Example section described later. In addition, when cultured on plates coated
with
recombinant human laminin-5, human iPS cells were found to form colonies even
under
serum-free conditions, thus indicating that they were able to be maintained
and cultured
under serum-free conditions. Furthermore, human iPS cell cultured by the
method of the
present invention were found to retain their undifferentiated state.
[0101] Thus, the method of the present invention comprises culturing
pluripotent cells in a
medium free from both feeder cells and serum, more preferably in a medium free
from any
substances derived from non-human animals.
[0102] For human ES cells, various extracellular matrix proteins (e.g.,
Matrigel, fibronectin,
laminin-1, type 1 collagen, type 4 collagen) were tested in the past as
supporting materials
alternative to MEFs, but the adhesion activity was as low as a few percent or
less in each case
(Stem Cell. 24, 2649-2660, 2006). In the present invention, by applying the
strong adhesion
activity of laminin-5 to maintenance culture of pluripotent stem cells (e.g.,
human ES cells,
human iPS cells), which are very poor in cell adhesion efficiency,
improvements can be
achieved in both adhesion efficiency and proliferation efficiency in the
absence of MEFs.
CA 02718830 2010-09-16
- 29 -
Moreover, to ensure the use of human ES cells or human iPS cells for
regenerative medicine,
recombinant human laminin-5 is also useful as a material constructing a
culture system
completely free from animal-derived substances.
[0103] Thus, in the present invention, treatment with laminin-5 allows an
improvement in
the adhesion efficiency of pluripotent stem cells onto a culture vessel and
also enables
efficient proliferation of pluripotent stem cells without the need to use
feeder cells generally
required for their culture.
[0104] As used herein, the term "feeder cells" is intended to mean additional
cells playing a
role as an aid, which are used to adjust culture conditions for target
pluripotent stem cells to
be proliferated or differentiated. In the case of pluripotent cells such as ES
cells or iPS
cells, in commonly used conventional methods, mouse-derived primary cultured
fibroblasts
are used as feeder cells and nutrients such as growth factors are supplied
from the feeder cells
to the pluripotent cells, whereby the pluripotent cells can be cultured.
According to the
present invention, the strong adhesion efficiency of laminin-5 allowed the
culture of
pluripotent stem cells such as ES cells or iPS cells without the need to use
these feeder cells.
[0105] As used herein, the term "supporting material" is intended to mean a
proteinous
factor used to aid cell proliferation, and laminin-5 is used as a supporting
material in the
present invention. As shown in the Example section described later, laminin-5
has higher
adhesion ability than various extracellular matrixes and is excellent as a
supporting material
for pluripotent stem cells.
[0106] When used herein to describe pluripotent stem cells, the term
"differentiation" is
intended to mean a change that causes the pluripotent stem cells to lose their
differentiation
pluripotency (i.e., potential ability to differentiate into all tissues) and
to have characters as
cells constituting a specific tissue. For example, undifferentiation markers
of pluripotent
stem cells, such as Ecat 1, ERas, Nanog, Oct4, Rexl, Sox2 and Utfl, may be
measured to
thereby evaluate whether pluripotent stem cells do not differentiate during
culture. As
shown in Examples 3 and 4 described later, ES cells cultured by the method of
the present
invention were evaluated for passage-induced differentiation, indicating that
they did not
CA 02718830 2010-09-16
. =
- 30 -
differentiate and remained in an undifferentiated state even after 10 or more
passages of
subculture. Further, as shown in Example 5 described later, ES cells cultured
by the method
of the present invention were evaluated for maintenance of pluripotency during
subculture,
indicating that they also retained pluripotency even after passages.
[0107] Furthermore, as shown in Example 11 described later, human iPS cells
cultured by
the method of the present invention were also found not to differentiate and
to remain in an
undifferentiated state even after subculture for 5 weeks. Moreover, as shown
in Example 12
described later, human iPS cells cultured by the present invention were also
able to be
induced to differentiate.
[0108] In one embodiment of the present invention, a culture vessel may be
treated with
laminin-5, for example, by applying laminin-5 onto the inner surface of the
culture vessel and
then drying. Such a laminin-5-treated culture vessel is charged with medium
(e.g., GMEM
or DMEM) commonly used for culture of pluripotent stem cells, and pluripotent
stem cells
are added to the medium. Then, the pluripotent stem cells are cultured under
known
appropriate culture conditions, for example but not limited to, under gas
phase conditions of
37 C and 5% CO,.
[0109] In a preferred embodiment of the present invention, it is more
preferable to add
appropriate additives to the culture medium, in addition to the use of a
system containing
laminin-5. Such additives are preferably those other than serum, and more
preferably
exclude substances derived from non-human animals.
[0110] A non-limiting example of such additives is a serum replacement. A
serum
replacement is an artificial liquid composition designed to have ingredients
similar to those
of serum, and it allows cells to proliferate even in the absence of serum. As
an example, it
is possible to use a serum replacement comprising various amino acids,
inorganic salts,
vitamins, albumin, insulin, transferrin, and antioxidative ingredients.
Various amino acids
include, for example, glycine, L-alanine, L-asparagine, L-cysteine, L-aspartic
acid, L-
glutamic acid, L-phenylalanine, L-histidine, L-isoleucine, L-lysine, L-
leucine, L-glutamine,
L-arginine, L-methionine, L-proline, L-hydroxyproline, L-serine, L-threonine,
L-tryptophan,
CA 02718830 2010-09-16
, =
- 31 -
L-tyrosine, and L-valine. Inorganic salts include, for example, AgNO3, A1C13-
6H70,
Ba(C71-1302)2, CdSO4.8H20, CoC17.6E120, Cr2(SO4)3.1H20, Ge02, Na2Se03, H2Se03,
KBr,
KI, MnC11.4H20, NaF, Na?SiO3.9H2O, NaV03, (NH4)6Mo7024.4H/O, NiSO4.6H20, RbC1,
SnC19, ZrOCI28H20, and sodium selenite. Vitamins include, for example,
thiamine and
ascorbic acid. Antioxidative ingredients include, for example, reduced
glutathione.
[0111] Likewise, KnockoutTM serum replacement (KSR) is a serum replacement for
ES
cells, which is commercially available from Invitrogen, Corp. As shown in
Examples 3 and
4 described later, it is a preferred embodiment of the present invention that
pluripotent stem
cells are cultured in a medium supplemented with about 10% KSR.
[0112] Further, it is also a preferred embodiment of the present invention to
use a serum
replacement of the composition shown in Example 6 described later. In Example
6, the
detailed composition of a serum replacement is disclosed, which comprises
amino acids (e.g.,
glycine, histidine, isoleucine, methionine, phenylalanine, proline,
hydroxyproline, serine,
threonine, tryptophan, tyrosine, valine), vitamins (e.g., thiamine, ascorbic
acid), trace metal
elements (e.g., silver, aluminum, barium, cadmium, cobalt, chromium,
germanium,
manganese, silicon, vanadium, molybdenum, nickel, rubidium, tin, zirconium),
halogen
elements (e.g., bromine, iodine, and fluorine), as well as other ingredients
(e.g., albumin,
reduced glutathione, transferrin, insulin, sodium selenite). However,
ingredients
constituting the serum replacement intended in the present invention are not
limited to those
listed above, and various modifications may be made, for example, by
replacement with other
similar ingredients. Moreover, the content of each ingredient contained in the
serum
replacement is not limited to that shown in Example 6, and may be adjusted as
appropriate
depending on the properties of cells and/or the purpose of experiments.
[0113] Any serum replacement may be used as long as it has ingredients and
functions
similar to those of KSR.
[0114] 2. Use of laminin-5 as a cell-supportin_g material
The present invention is also directed to the use of laminin-5 as a cell-
supporting
material for proliferation of pluripotent stem cells. As has been described
above, laminin-5
CA 02718830 2015-04-02
- 32 -
has a strong effect on cell adhesion and is therefore useful in culturing
pluripotent stem cells
in a medium free from both feeder cells and serum.
[0115] 3. Culture kit for pluripotent stem cells
The present invention is further directed to a culture kit for pluripotent
stem cells,
which comprises a laminin-5-treated culture vessel and a serum replacement.
Such a serum
replacement is preferably KnockoutTM serum replacement (KSR). Alternatively,
it is also
possible to use a serum replacement of the composition shown in Example 6.
[0116] This kit can further comprise culture medium for pluripotent stem
cells, such as
GMEM or DMEM. If necessary, this kit may further comprise other additives
required for
cell culture and/or pluripotent stem cells pre se to be cultured. Examples of
such additives
include nonessential amino acids, sodium pyruvate, mercaptoethanol,
antibiotics and so on.
Such a kit may be provided in the form of a single package or may be provided
in the form of
multiple packages in which only pluripotent stem cells required to be stored
at low
temperature are packaged separately.
EXAMPLES
[0117] The present invention will now be described in more detail below on the
basis of the
following examples, which are not intended to limit the scope of the
invention.
[0118] Example 1: Preparation of recombinant human laminin-5 (rLm5)
In this example, a recombinant human laminin-5 protein was prepared in a known
manner.
[0119] From human fetal kidney cell line HEK293 modified to carry cDNAs for a3
chain
(SEQ ID NO: 1), P3 chain (SEQ ID NO: 3) and y2 chain (SEQ ID NO: 3) (Lm5-
HEK293), the
serum-free supernatant was collected and centrifuged at 4 C at 3000 rpm for 5
minutes. The
human fetal kidney cell line HEK293 was obtained as described in J. Biochem.
132, 607-612
(2002). The supernatant was then applied to Heparin sepharoseTM CL-6B (GE
healthcare) and
eluted. The rLm5-containing fractions were passed through an antibody column,
in which
mouse anti-Lm-a3 (anti-laminin a3) monoclonal antibody (BG5) was covalently
bonded to
ProteinA sepharoseTM CL-6B (GE healthcare), and then eluted. It should be
noted
I
,
CA 02718830 2010-09-16
- 33 -
that monoclonal antibody BG5 is an antibody prepared by the inventors of the
present
invention using an N-terminal fragment of the laminin a3B chain as an antigen
according to
known procedures for monoclonal antibody preparation.
[0120] Purified rLm5 (1 pg) was denaturated under reducing conditions and then
subjected
to SDS polyacrylamide gel electrophoresis on a 5-20% gel to confirm the size
and purity of
a3, (33 and y2 chains. As a result, bands of 160 kDa, 135 kDa and 105 kDa were
detected,
respectively. Figure 1 shows a photograph of SDS polyacrylamide gel
electrophoresis
obtained for purified rLm5.
[0121] When analyzed with a CS-Analyzer, purified rLm5 was found to have a
purity of
about 98%. rLm5 thus prepared was used in the following examples.
[0122] Example 2: Adhesion assay using mouse ES cell line EB3
This example shows the results of adhesion assay using mouse ES cell line EB3,
obtained with the use of various cell-supporting materials.
[0123] GMEM (SIGMA) supplemented with 10% fetal bovine serum (FBS), 0.1 mM
nonessential amino acids (Gibco), 1 mM sodium pyruvate (Gibco), 1000 U/ml
ESGRO
(Chemicon) and 104 M 2-mercaptoethanol (WAKO) was used as a maintenance medium
for
mouse ES cell line, EB3 cells. The EB3 cells were provided by the Division of
Stem Cell
Regulation Research, Area of Molecular Therapeutics, Course of Advanced
Medicine G6,
Graduate School of Medicine, Osaka University.
[0124] In the adhesion assay, a serum-free medium was used, whose composition
was the
same as that of the maintenance medium, except for not containing FBS. The
cell-
supporting materials used were bovine gelatin (SIGMA), rLm5, Matrigel (BD),
human
vitronectin (SIGMA), human type IV collagen (BD), human fibronectin (BD),
human
laminin-2 (Chemicon) and human laminin (SIGMA). Further, plates were prepared
by
being treated with these respective cell-supporting materials, and were
subjected to adhesion
assay for each cell-supporting material, as described below.
[0125] 96-well plates (Corning) were treated with various extracellular matrix
proteins
prepared at desired concentrations, and then blocked with a 1.2% BSA (SIGMA)
solution at
CA 02718830 2015-04-02
- 34 -
37 C for 1 hour. The various extracellular matrix proteins were prepared at
the following
concentrations: 1.5 mg/ml for gelatin (G1), 3.75 pg/m1 for laminin-2 (Lm2),
3.75 g/m1 for a
laminin mixture (Lm-Mix), 150 pg/ml for Matrigel (Mg), 15 pg/m1 for collagen
(Co), 15
lAg/m1 for fibronectin (Fn), and 151A.g/m1 for vitronectin (Vn). On the other
hand, rLm5
(Lm5) was prepared by two-fold serial dilution at three concentrations: 3.75
pg/ml, 7.5 g/ml
and 15 p,g/ml.
[0126] After washing with the serum-free medium, EB3 cells were seeded at
30000
cells/well in the plates and cultured at 37 C under gas phase conditions of 5%
CO2 and 95%
air for 30 or 60 minutes. After culture, the plates were gently shaken with a
vortex mixer to
release less adhered cells from the plate surface, followed by treatment with
PercollTM (GE
healthcare) to remove these cells. The adhered cells were fixed with 25%
glutaraldehyde
(Nacalai) and stained with 2.5% crystal violet (Nacalai) for comparison of
relative cell
counts.
[0127] Figure 2 shows the results evaluated for the effect of the various
extracellular matrix
proteins on EB3 cell adhesion after culture for 30 or 60 minutes, as analyzed
by 0D595
measurement. The results shown in Figure 2 indicated that rLm5 showed stronger
adhesion
activity on EB3 cells than the other various extracellular matrix proteins
including Lm-Mix.
[0128] It should be noted that human ES cells, which are poor in adhesion
efficiency, are
known to achieve an adhesion efficiency of less than 1% in the case of using
EHS-derived
laminin, and their adhesion efficiency is as low as 3% even in the case of
Matrigel (50-60%
EHS-derived laminin, 30% type IV collagen, 10% entactin) which is most widely
used at
present. In this example, laminin-5 was found to show strong adhesion activity
on mouse
ES cells, when compared to Matrigel composed primarily of EHS laminin, laminin-
2, and
placenta-derived laminin (Lm-Mix) deemed to be rich in laminin-10/11. This is
regarded as
a remarkable effect of laminin-5 used in the present invention.
[0129] Example 3: Proliferation assay using mouse ES cell line EB3
This example shows the results of proliferation assay using mouse ES cell line
EB3,
obtained with the use of various cell-supporting materials.
CA 02718830 2010-09-16
=
- 35 -
[0130] The maintenance medium used for EB3 cells was the same as that of
Example 2.
In the proliferation assay, a medium supplemented with 10% KnockoutTM serum
replacement
(KSR, Invitrogen) in place of 10% FBS was used (KSR-GMEM). In 12-well plates
(NUNC) which had been treated with various extracellular matrix proteins
prepared at
desired concentrations, EB3 cells were seeded at 40000 cells/well. After
culture at 37 C
under gas phase conditions of 5% CO-, and 95% air for 2 days, the cells were
collected by
enzyme treatment and counted with a hemacytometer.
[0131] The EB3 cells were seeded again at 40000 cells/well in 12-well plates
which had
been treated with the same various extracellular matrix proteins prepared at
desired
concentrations. By repeating this procedure, the various extracellular
matrixes were
compared for their proliferative effect on EB3 cells. The culture medium used
was
maintenance medium (S) or KSR-GMEM (K). The cell-supporting materials were
prepared
at the following concentrations: 1 mg/ml for GI, 4 ig/m1 for rLm5 (Lm5-4), 2
A,g/m1 for
rLm5 (Lm5-2), 4 mg/m1 for Lm-Mix, and 150 ikg/m1 for Mg. In this experiment,
the EB3
cells were acclimated before use by having been subcultured for several
passages in KSR-
GMEM.
[0132] Figure 3 shows the results studied for the effect of the various
extracellular matrixes
on EB3 cell proliferation upon culture in the absence of feeder cells in the
maintenance
medium (S) or KSR-GMEM (K). The results shown in Figure 3 indicated that
during the
first 2 or 3 passages, K+Lm-Mix and K+Mg resulted in slower proliferation than
the other
experimental groups.
[0133] For this reason, all the experimental groups except for K+Lm-Mix and
K+Mg were
further subcultured to study the effect of the extracellular matrixes on EB3
cell proliferation
upon prolonged subculture in the absence of feeder cells. The theoretical fold
increase in
the number of cells finally proliferated was calculated and the results
obtained are shown in
Figure 4. The results shown in Figure 4 indicated that rLm5 showed a
proliferative effect
on EB3 cells at the same level as the control group (S+G1), i.e., a widely
used conventional
maintenance culture system for ES cells.
,
CA 02718830 2010-09-16
,
,
- 36 -
[0134] Figure 5 shows the morphology of EB3 cells upon prolonged subculture in
a system
of maintenance culture (S+G1), a system of rLm5 (K+Lm5) and a system of rLm5
followed
by maintenance culture (K+Lm5-4 ¨> S+G1). As shown in Figure 5, the
experimental group
of K+Lm5 showed a gradual change into epithelial cell-like morphology, but
colony
formation was observed again when returned to a system of normal maintenance
culture
(K+Lm5-4 ¨> S+G1). In general, undifferentiated ES cells are known to form
colonies.
Thus, the above results suggested that EB3 cells retained their
undifferentiated state even
when their morphology was changed to epithelial cell-like morphology upon
prolonged
subculture under experimental conditions of K+Lm5.
[0135] Example 4: Detection of undifferentiation markers
In this example, Ecatl, ERas, Nanog, Oct4, Rex 1, Sox2 and Utfl, which are
known
as undifferentiation markers of mouse ES cells, were measured by RT-PCR on
their genes to
study whether rLm5 had the effect of allowing EB3 cells to remain in an
undifferentiated
state.
[0136] From the EB3 cells subcultured for 10 or more passages in Example 3,
total RNA
was extracted using TRIZOL (Invitrogen). After extraction, a ThermoScript RT-
PCR
System (Invitrogen) was used to synthesize cDNA by reverse transcription
reaction. The
synthesized cDNA was used as a template to perform PCR with the primers shown
in Table
2. Each gene was denatured at 94 C for 30 seconds, annealed for 30 seconds,
and elongated
at 72 C for 20 seconds. Annealing was performed at a temperature of 64 C for
Ecat 1, 61 C
for ERas, Oct4 and Utfl, 59 C for Rex 1, and 54 C for Nanog and Sox2.
[0137] Figure 6 shows the results studied for the expression of various
undifferentiation
markers in each culture. As a result, even under experimental conditions where
the cells
were cultured on rLm5-coated plates in the presence of KSR (K+Lm5-4, K+Lm5-2),
EB3
cells were found to express all the undifferentiation markers analyzed, at the
same levels as
observed under experimental conditions normally maintained (S+G1). Moreover,
when
returned to a system of normal maintenance culture (K+Lm5-4 --> S+G1), the
expression of
these undifferentiation markers was found to return to levels
indistinguishable from those
CA 02718830 2010-09-16
, =
- 37 -
observed when the cells were continued to be cultured in the system of normal
maintenance
culture (S+G1). The above results suggested that EB3 cells retained their
undifferentiated
state even after 10 or more passages of subculture in K+Lm5.
[0138] [Table 2]
RT-PCR primers
Ecatl
5'-TGTGGGGCCCTGAAAGGCGAGCTGAGAT-3' (SEQ ID NO: 7)
5'-ATGGGCCGCCATACGACGACGCTCAACT-3' (SEQ ID NO: 8)
ERas
5'-ACTGCCCCTCATCAGACTGCTACT-3' (SEQ ID NO: 9)
5'-CACTGCC1TGTACTCGGGTAGCTG-3' (SEQ ID NO: 10)
Nanog
5'-AAGCAGAAGATGCGGACTGT-3' (SEQ ID NO: 11)
5'-ACCACTGG 1-11T1CTGCCAC-3' (SEQ ID NO: 12)
Oct4
5'-TC CCACCAGGCCCCCGGCTC-3' (SEQ ID NO: 13)
5'-TGCGGGCGGACATGGGGAGATCC-3' (SEQ ID NO: 14)
Rex1
5'-ACGAGTGGCAG __________________ ITI CTTCTTGGGA-3' (SEQ ID NO: 15)
5'-TATGACTCACTTCCAGGGGGCACT-3' (SEQ ID NO: 16)
Sox2
5'-TAGAGCTAGACTCCGGGCGATGA-3' (SEQ ID NO: 17)
5'-TTGCCTTAAACAAGACCACGAAA-3' (SEQ ID NO: 18)
tiff].
5'-GGATGTCCCGGTGACTACGTCTG-3' (SEQ ID NO: 19)
5'-GGCGGATCTGGTTATCGAAGGGT-3' (SEQ ID NO: 20)
Gapdh
5'-CACCATGGAGAAGGCCGGGG-3' (SEQ ID NO: 21)
5'-GACGGACACATTGGGGGTAG-3' (SEQ ID NO: 22)
CA 02718830 2015-04-02
- 38 -
[0139] Example 5: Study on differentiation potency into three germ layers
This example shows the results studied for maintenance of differentiation
potency in
mouse cell line EB3 when cultured with the use of various cell-supporting
materials.
[0140] After EB3 cells were cultured under KSR-supplemented and serum-free
conditions
in Example 3, the cells were acclimated again under serum conditions by being
subcultured
for an additional 5 passages under maintenance culture conditions (S+Gl)
before use in the
differentiation-inducing test. For culture in a serum-free medium, a system of
gelatin-
coated plates (K+G1) and a system of recombinant human laminin-5-coated plates
(K+Lm5-4
or K+Lm5-2) were used. Experimental groups in which culture in these systems
was
followed by acclimation under serum conditions are expressed as K+G1 = S+Gl,
K+Lm5-4
S+Gl, and K+Lm5-2 S+Gl, respectively.
[0141] Induction of differentiation was accomplished as follow. The cells were
suspended
in a LIF (ESGRO)-free maintenance medium to prepare hanging drops (1000
cells/drop), in
which embryoid bodies were formed for 2 days. The formed embryoid bodies were
transferred to bacterial culture plates and cultured under floating conditions
in the LIF-free
maintenance medium for an additional 5 days. Figure 7 shows the embryoid
bodies
observed at that time.
[0142] The embryoid bodies after floating culture were transferred to 1 mg/ml
GI-coated
chamber slides (NUNC) and cultured for 3 days under adhesion conditions,
followed by
immunostaining to detect differentiation markers, thereby studying the
differentiation
potency of ES cells (i.e., differentiation into cells of the endodermal,
mesodermal and
ectodermal lineages).
[0143] In this study, the markers used were a-fetoprotein (AFP) for cells of
the endodermal
lineage, a-smooth muscle actin (a-SMA) for cells of the mesodermal lineage,
and 3-Ill
tubulin (tubulin) for cells of the ectodermal lineage. For immunostaining, the
cells were
fixed with 4% formaldehyde and then blocked with a 5% FBS solution
supplemented with
0.1% Triton-X100Tm (Nacalai). After blocking, the cells were treated with
primary and
secondary antibodies, stained with DAPI (Nacalai), embedded in VECTASHIELD
CA 02718830 2010-09-16
=
- 39 -
(VECTOR Laboratories) and then observed under a fluorescent microscope to
detect marker
expression.
[0144] The primary antibodies used in immunostaining were anti-AFP polyclonal
antibody
(DAKO) for detection of a-fetoprotein, anti-a-SMA monoclonal antibody (DAKO)
for
detection of a-smooth muscle actin, and anti-13-III tubulin monoclonal
antibody (Chemicon)
for detection of tubulin. The secondary antibodies used were anti-
rabbit IgG
polyclonal antibody (Santa Cruz Biotechnology) and anti-mouse IgG polyclonal
antibody
(Santa Cruz Biotechnology).
[0145] In parallel with induction of differentiation in the experimental
groups of S+Gl,
K+GI S+Gl, K+Lm5-4 S+Gl and K+Lm5-2 = S+Gl as described above, negative
controls were prepared for these experimental groups by being cultured in the
presence of
LIF in a system of normal maintenance culture (S+Gl) without conducting a
series of
differentiation-inducing processes, including embryoid body formation. These
negative
controls were also immunostained in the same manner.
[0146] In these four experimental groups, differentiation markers AFP, a-SMA
and 13411
tubulin were compared for their expression between each experimental group
induced to
differentiate and the corresponding negative control not induced to
differentiate (Figures 8 to
11). Figure 8 shows the results obtained for the experimental group of S+Gl,
Figure 9
shows the results obtained for the experimental group of K+G1
S+Gl, Figure 10 shows the
results obtained for the experimental group of K+Lm5-4 = S+Gl, and Figure 11
shows the
results obtained for the experimental group of K+Lm5-2 = S+Gl.
[0147] As a result, in all of the experimental groups, three signals of AFP, a-
SMA and 13411
tubulin were all observed in the experimental groups induced to differentiate
(lower panel of
Figure 8, lower panel of Figure 9, lower panel of Figure 10, lower panel of
Figure 11). In
contrast, three signals of AFP, a-SMA and 0-In tubulin were not observed in
the negative
controls cultured in the system of normal maintenance culture, thus indicating
that the cells
were not induced to differentiate (upper panel of Figure 8, upper panel of
Figure 9, upper
panel of Figure 10, upper panel of Figure 11).
CA 02718830 2010-09-16
- 40 -
[0148] This result means that EB3 cells were induced to differentiate only
after a series of
differentiation-inducing processes and they became differentiated cells of
three germ layers
expressing AFP, aSMA, p-m tubulin or other markers. Taken together with the
results of
Example 4, the results of this example suggested that ES cells maintained in
the presence of
Lm5 not only remained in an undifferentiated state, but also retained
pluripotency, i.e., the
ability to differentiate into a wide range of cells upon induction.
[0149] Example 6: Preparation of serum replacement of another composition than
KSR
A serum replacement of another composition was prepared, whose composition was
different from that of KSR mentioned above. The composition of the serum
replacement
prepared in this example is shown in Table 3.
CA 02718830 2010-09-16
- 41 -
[0150] [Table 3]
Composition of serum replacement whose composition is known
Glycine (Nacalai) 150 mg/1
Histidine (Nacalai) 940 mg/1
Isoleucine (Nacalai) 3400 mg/1
Methionine (Nacalai) 90 mg/1
Phenylalanine (Nacalai) 1800 mg/1
Proline (Nacalai) 4000 mg/1
Hydroxyproline (Nacalai) 100 mg/1
Serine (Nacalai) 800 mg/1
Threonine (Nacalai) 2200 mg/1
Tryptophan (Nacalai) 440 mg/1
Tyrosine (SIGMA) 77 mg/1
Valine (Nacalai) 2400 mg/1
Thiamine (Nacalai) 33 mg/1
Ascorbic acid (SIGMA) 330 mg/1
Reduced glutathione (SIGMA) 10 mg/1
Human transferrin (Nacalai) 55 mg/1
Bovine insulin (SIGMA) 100 mg/1
Sodium selenite (SIGMA) 0.07 mg/1
BSA (Invitrogen) 83000 mg/1
AgNO3 (Mediatech Inc.) 0.0017 mg/1
AlC13.6H20 (Mediatech Inc.) 0.012 mg/1
Ba(C2H302)2 (Mediatech Inc.) 0.0255 mg/1
CdC12 (Mediatech Inc.) 0.0228 mg/1
CoC17=6H20 (Mediatech Inc.) 0.0238 mg/1
Cr2C13 (Mediatech Inc.) 0.0032 mg/1
Ge02 (Mediatech Inc.) 0.0053 mg/1
KBr (Mediatech Inc.) 0.0012 mg/1
KI (Mediatech Inc.) 0.0017 mg/1
MnSO4.1-120 (Mediatech Inc.) 0.0017 mg/I
NaF (Mediatech Inc.) 0.042 mg/1
Na2SiO3.9H20 (Mediatech Inc.) 1.4 mg/1
NH4V03 (Mediatech Inc.) 0.0065 mg/1
(NH4)6Mo7024=4H20 (Mediatech Inc.) 0.0124 mg/1
NiSO4=6H20 (Mediatech Inc.) 0.0013 mg/1
RbC1(Mediatech Inc.) 0.0121 mg/1
SnC12 (Mediatech Inc.) 0.0012 mg/1
ZrOC12-8F120 (Mediatech Inc.) 0.0322 mg/1
CA 02718830 2010-09-16
- 42 -
[0151] In distilled water, the various amino acids shown in Table 3 (glycine,
histidine,
isoleucine, methionine, phenylalanine, methionine, phenylalanine, proline,
hydroxyproline,
serine, threonine, tryptophan, tyrosine, valine) were dissolved at 3-fold
concentrations to
prepare a 3-fold concentrated amino acid solution. To this 3-fold concentrated
amino acid
solution, thiamine, ascorbic acid and reduced glutathione were each added in
an amount
required to give a 3-fold concentration. This solution is designated as
solution A. Next,
human transferrin and BSA were dissolved at 2-fold concentrations in distilled
water to
prepare a 2-fold concentrated BSA solution. This solution is designated as
solution B.
Further, sodium selenite was dissolved in a required amount in distilled water
to prepare a
7% sodium selenite solution. This solution is designated as solution C. To the
solutions
A, B and C thus prepared, a bovine insulin solution (SIGMA) and trace metal
elements were
added to prepare the serum replacement of this example. It should be noted
that the trace
metal elements added here are commercially available products, Trace Elements
B and C
(Mediatech Inc.) whose composition is known. The serum replacement prepared in
this
example is not commercially available, but its composition is known.
[0152] Example 7: Study using medium supplemented with serum replacement of
Example 6
In this example, the serum replacement prepared in Example 6 was added to GMEM
as an alternative to serum, as in the case of KSR, and the resulting medium
was used as a
maintenance medium for culture of mouse ES cells. The medium thus prepared is
designated as medium Y. Using medium Y, proliferation assay was performed as
described
in Example 3. Further, undifferentiation markers were detected as described in
Example 4.
Furthermore, differentiation potency into three germ layers was studied as
described in
Example 5.
[0153] (1) Proliferation assay
The results of proliferation assay using medium Y are shown in Figure 12. The
experiment was performed in the same manner as shown in Example 3, except that
medium
Y was used as a maintenance medium. As a result of proliferation assay using
mouse ES
CA 02718830 2010-09-16
- 43 -
cell line EB3, the experimental groups where the cells were cultured on rLm5-
coated plates
in medium Y (Y+Lm5-4, Y+Lm5-2) were found to show a proliferative effect on
mouse ES
cells. In addition, the use of Lm5 resulted in better proliferation, when
compared to the
experimental groups using other extracellular matrixes (G1, Lm-Mix, Mg).
[0154] (2) Detection of undifferentiation markers
After proliferation assay as described above, undifferentiation markers were
studied
by RT-PCR for their expression in each experimental group. The experiment was
performed in the same manner as shown in Example 4, except that medium Y was
used as a
maintenance medium. Figure 13 shows the results of 7 undifferentiation markers
detected
in the same manner as shown in Example 4. In the case of using medium Y for
culture of
mouse ES cells, all the undifferentiation markers were also confirmed to be
expressed in the
experimental groups where the cells were cultured on rLm5-coated plates
(Y+Lm5). Thus,
it was suggested that EB3 cells retained their undifferentiated state even
when subcultured
for 10 or more passages in a system of rLm5 using medium Y.
(3) Study on differentiation potency into three germ layers
[0155] A differentiation-inducing test was also performed in the same manner
as shown in
Example 5, except that medium Y was used as a maintenance medium. All of the
experimental groups induced to differentiate were found to form embryoid
bodies
morphologically closely resembling those observed in the control group (S+G1).
The results
obtained are shown in Figure 14.
[0156] After induction of differentiation, markers for differentiation into
three germ layers
(AFP, a-SMA, tubulin) were detected. In the experimental groups of S+G1
(Figure
15), K+G1 = S+G1 (Figure 16), Y+G1 S+G1 (Figure 17), Y+Lm5-4 = S+G1 (Figure
18),
Y+Lm5-2 S+G1 (Figure 19), Y+Lm-Mix = S+G1 (Figure 20) and Y+Mg S+G1 (Figure
21), the differentiation markers were studied for their expression between
each experimental
group induced to differentiate (lower panel, LIF-) and the corresponding
negative control not
induced to differentiate (upper panel, LIF+).
[0157] As shown in lower panels (LW-) of Figures 15 to 21, immunostaining
after
CA 02718830 2010-09-16
- 44 -
induction of differentiation showed the expression of all three markers AFP, a-
SMA and [3-
III tubulin in each experimental group. In contrast, as shown in the upper
panels (LIF+) of
Figures 15 to 21, no expression of AFP, a-SMA and (3-111 tubulin was observed
in the
negative controls cultured in a system of normal maintenance culture without
induction of
differentiation, thus indicating that the cells were not induced to
differentiate.
[0158] The above results suggested that in the case of using medium Y, rLm5
also
supported the proliferation of mouse ES cells, and the proliferated mouse ES
cells also
retained their undifferentiated state. This indicated that rLm5 may serve as a
useful
supporting material for mouse ES cells under feeder cell-free and serum-free
conditions.
[0159] Example 8: Adhesion assay using human iPS cells
This example shows the results of adhesion assay using human iPS cells,
obtained
with the use of various cell-supporting materials.
[0160] The human iPS cells used were those of cell line 201B2 or 201B7
established by the
Department of Stem Cell Biology, Institute for Frontier Medical Sciences,
Kyoto University.
In Figure 22, the left panel shows the morphology of cell line 201B2, while
the right panel
shows the morphology of cell line 201B7.
[0161] Human iPS cells were cultured for 1 hour in the presence of 1011M Y-
27632
(WAKO) before being released from the dishes, and then used in the experiment.
It should
be noted that the same treatment was also performed in the experiments of
Example 8 and the
subsequent examples where human iPS cells were used.
[0162] For maintenance of human iPS cells, the supernatant of mouse embryonic
fibroblasts prepared in DMEM/F12 medium containing 20% KSR, 2 mM glutamine, 1%
nonessential amino acids and 10-4 M 2-mercaptoethanol was supplemented with 4
ng/ml
bFGF (WAKO) and used as a maintenance medium (MEF-CM).
[0163] The cell-supporting materials used were mitomycin C-treated SNL feeder
cells
(Fd) as well as various extracellular matrixes (GI, Lm5, Mg, Vn, Co, Fn, Lm2,
Lm-Mix).
Further, plates were prepared by being treated with these respective cell-
supporting
materials, and were subjected to adhesion assay for each cell-supporting
material using
CA 02718830 2010-09-16
. =
- 45 -
human iPS cell line 201B7, as described below.
[0164] 96-well plates (Corning) were treated with various extracellular matrix
proteins
prepared at desired concentrations, and then blocked with a 1.2% BSA (SIGMA)
solution at
37 C for 1 hour. The various extracellular matrix proteins were prepared at
the following
concentrations: 5 mg/ml and 1.25 mg/ml for Gl, 50 ig/m1 and 12.5 ig/m1 for
Lm2, 50 ig/m1
and 12.5 ttg/m1 for Lm-Mix, 500 vg/m1 and 125 ig/m1 for Mg, 50 [tg/m1 and 12.5
Ag/m1 for
Co, 50 ig/m1 and 12.5 11g/m1 for Fn, and 50 1.1g/m1 and 12.5 lig/Int for Vn.
rLm5 was
prepared by two-fold serial dilution at five concentrations: 50 Wml, 25 ig/ml,
12.5 [ig/ml,
6.25 ttg/m1 and 3.125 pg/ml. Moreover, an additional experimental group
(Lm5+Co) was
also prepared by being treated with both 25 ig/m1 Lm5 and 50 [tg/m1 Co.
[0165] Human iPS cells were treated with trypsin and dispersed into single
cells, and then
seeded at 20000 cells/well in the plates and cultured at 37 C under gas phase
conditions of
5% CO-, and 95% air for 60 minutes. After culture, the plates were tapped
gently to release
less adhered cells from the plate surface, followed by treatment with Percoll
(GE healthcare)
to remove these cells. The adhered cells were fixed with 25% glutaraldehyde
(Nacalai) and
stained with 2.5% crystal violet (Nacalai) for comparison of relative cell
counts.
[0166] Figure 23 shows the results evaluated for the effect of the various
extracellular
matrix proteins on human iPS cell adhesion after culture for 60 minutes, as
analyzed by
OD595 measurement. Further, Figure 24 shows the morphology of cells after the
assay in
each experimental group.
[0167] In Figure 23, the experimental groups of rLm5 were found to show higher
0D595
values than the experimental groups of the other extracellular matrix
proteins, indicating that
rLm5 showed strong adhesion activity on human iPS cells. Also in Figure 24,
many
adhered cells were observed in the experimental groups of rLm5. Moreover, rLm5
was
found to exert stronger adhesion activity when used in combination with Co
(rLm5+Co) than
when used alone.
[0168] Example 9: Colony assay using human iPS cells
This example shows the results of colony assay using human iPS cell line
201B2,
CA 02718830 2010-09-16
- 46 -
obtained with the use of various cell-supporting materials.
[0169] The cell-supporting materials used were GI, Lm5, Mg, Vn, Co, Fn, Lm2
and Lm-
Mix. Further, 60 mm dishes (IWAKI) were prepared by being treated with these
respective
cell-supporting materials, and were subjected to colony assay for each cell-
supporting
material, as described below.
[0170] The various extracellular matrix proteins were prepared at the
following
concentrations: 1 mg/ml for GI, 30 [tg/ml, 15 lg/ml, 8 ig/ml, 4 [ig/m1 and 2
1.1g/m1 for Lm5,
30 1.tg/ml, 15 1.1,g/m1, 8 Ag/ml, 4 tg/m1 and 2 11g/m1 for Lm2, 30 p.g/ml, 15
Ag/ml, 8 g/ml,
4 1.1g/m1 and 2 ig/m1 for Lm-Mix, 300 lig/m1 for Mg, 30 [tg/m1 for Co, 30
lAg/m1 for Fn, and
[tg/m1 for Vn.
[0171] In this example, human iPS cells were prepared in a state of single
cells dispersed by
trypsin treatment (hereinafter referred to as "Single") and in a state of cell
clumps prepared
by gentle pipetting while preventing dispersion (hereinafter referred to as
"Clump"), both of
which were subjected to the assay. Namely, human iPS cells were seeded in
Single or
Clump state at a density corresponding to 1000 cells per 60 mm dish. Culture
was
performed at 37 C under gas phase conditions of 5% CO2 and 95% air for 13 days
(Single) or
6 days (Clump). After culture, the number of formed colonies was counted for
each case.
[0172] The results obtained are shown in Figure 25. In Figure 25, the upper
panel shows
the results obtained for Single, while the lower panel shows the results
obtained for Clump.
[0173] In Single, colony formation was observed in the experimental groups of
rLm5, Lm-
Mix, Vn and Mg, and the number of colonies formed in the experimental group of
8 1,tg,/m1
Lm5 or 30 g/m1 Lm-Mix was close to that in the positive control, i.e., the
experimental
group using SNL feeder cells (Fd). On the other hand, in Clump, colony
formation was
observed in the experimental groups of rLm5, Lm2, Lm-Mix, Vn, Mg and Co, and
the
experimental groups of rLm5 showed colony formation comparable to or better
than that
observed in Fd.
[0174] Further, the formed colonies were immunostained to detect a marker of
human
pluripotent stem cells, thereby evaluating whether the cultured human iPS
cells were in an
CA 02718830 2010-09-16
- 47 -
undifferentiated state. The marker of human pluripotent stem cells used for
this purpose
was a cell surface antigen marker, SSEA3.
[0175] For immunostaining, the cells were fixed with 4% formaldehyde and then
blocked
with 1% BSA-containing PBS. After blocking, the cells were treated with
primary and
secondary antibodies, stained with Hoechist 33342 (Invitrogen) and then
observed under a
fluorescent microscope to detect marker expression. The primary and secondary
antibodies
used in immunostaining were anti-SSEA3 monoclonal antibody and anti-rat IgM
antibody
(Jackson ImmunoResearch), respectively.
[0176] The results of immunostaining are shown in Figure 26. Figure 26A shows
the
results of Single, while Figure 26B shows the results of Clump. The ES cell
marker SSEA3
was detected in colonies formed from both Single and Clump. This suggested
that colonies
formed from human iPS cells retained their undifferentiated state.
[0177] The above results suggested that rLm5 supported the colony formation of
human iPS
cells comparably to or better than the other extracellular matrixes, and the
formed colonies
also retained their undifferentiated state.
[0178] Example 10: Maintenance culture test using human iPS cells
This example shows the results of maintenance culture test using human iPS
cell
line 201B7, obtained with the use of various cell-supporting materials.
[0179] The cell-supporting materials used were GI, Lm5, Lm2, Vn, Co and Fn.
Further,
6-well plates (Falcon) were prepared by being treated with these respective
cell-supporting
materials, and were subjected to colony assay for each cell-supporting
material, as described
below. The maintenance medium used was MEF-CM as mentioned in Example 8.
[0180] The various extracellular matrix proteins were prepared at the
following
concentrations: 1 mg/ml for Gi, 2 1..tg/m1 for Lm5, 30 ig/m1 for Lm2, 30 ig/m1
for Co,
30 [ig/ml for Fn, and 10 [tg/m1 for Vn.
[0181] In this example, human iPS cells were assayed only in Clump state. Once
a week,
1/9 of all the cells were re-seeded in newly prepared 6-well plates treated
with the same
various extracellular matrixes. The cells were cultured at 37 C under gas
phase conditions
CA 02718830 2010-09-16
=
- 48 -
of 5% CO, and 95% air for 5 weeks. Figure 27 shows the morphology of cells
cultured for
weeks.
[0182] As a result, in the experimental groups of the various extracellular
matrix proteins
tested, cell proliferation was observed upon culture for 1 week after
subculture at a level
comparable to that in the positive control, i.e., the experimental group of
SNL feeder cells
(Fd). As shown in Figure 27, this tendency was also observed after culture for
5 weeks.
This result indicated that rLm5 and Fd were able to almost equally support the
proliferation
of human iPS cells. In the case of Lm2, colony formation disappeared during
the course of
culture.
[0183] Example 11: Detection of undifferentiation markers in human iPS cells
In this example, NANOG, OCT4 and SOX2, which are know as undifferentiation
markers of human pluripotent stem cells, were detected by RT-PCR on their
genes to study
whether rLm5 had the effect of allowing human iPS cells to remain in an
undifferentiated
state.
[0184] From the human iPS cells subcultured for 5 weeks in Example 10, total
RNA was
extracted using TRIZOL (Invitrogen). After extraction, a ThermoScript RT-PCR
System
(Invitrogen) was used to synthesize cDNA by reverse transcription reaction.
The
synthesized cDNA was used as a template to perform PCR with the primers shown
in Table
4. Each gene was denatured at 94 C for 10 seconds, annealed for 15 seconds,
and elongated
at 72 C for 30 seconds. Annealing was performed at a temperature of 60 C for
OCT4 and
GAPDH, and 55 C for NANOG and SOX2.
[0185]
CA 02718830 2010-09-16
- 49 -
[Table 4]
RT-PCR primers
OCT4
5'-GACAGGGGGAGGGGAGGAGCTAGG-3' (SEQ ID NO: 23)
5'-CTTCCCTCCAACCAGTTGCCCCAAAC-3' (SEQ ID NO: 24)
NANOG
5'-CAGCCCTGATTCTTCCACCAGTCCC-3' (SEQ ID NO: 25)
5'-TGGAAGGTTCCCAGTCGGGTTCACC-3' (SEQ ID NO: 26)
SOX2
5'-GGGAAATGGGAGGGGTGCAAAAGAGG-3' (SEQ ID NO: 27)
5'-TTGCGTGAGTGTGGATGGGATTGGTG-3' (SEQ ID NO: 28)
GAPDH
5'-GTGGACCTGACCTGCCGTCT-3' (SEQ ID NO: 29)
5'-GGAGGAGTGGGTGTCGCTGT-3' (SEQ ID NO: 30)
[0186] The results of RT-PCR are shown in Figure 28. All of the experimental
groups
showed the expression of the three tested factors. This suggested that when
cultured using
rLm5 as a cell-supporting material, human iPS cells retained their
undifferentiated state.
[0187] Example 12: Differentiation-inducing test in human iPS cells
This example shows the results studied for maintenance of differentiation
potency in
human iPS cells when cultured with the use of various cell-supporting
materials. In human
iPS cells induced to differentiate, differentiation markers were detected by
RT-PCR to study
whether human iPS cells cultured with the use of various cell-supporting
materials retained
differentiation potency.
[0188] After culture for 3 weeks, i.e., during the third passage in Example
10, a part of the
cells were collected and used in the differentiation-inducing test. Induction
of
differentiation was accomplished as follow.
[0189] Clumps, which had been prepared from human iPS cells under culture by
being
CA 02718830 2010-09-16
=
- 50 -
released from the plates, were cultured using low adsorption plates (NUNC)
under floating
conditions for 8 days in DMEM/F12 medium supplemented with 20% KSR, 2 mM
glutamine, 1% nonessential amino acids and 104 M 2-mercaptoethanol. The
embryoid
bodies thus formed were re-seeded in 1 mg/ml Gl-coated 6-well plates (Falcon)
and cultured
under adhesion conditions for an additional 8 days.
[0190] Figure 29 shows the morphology of cells after adhesion culture. The
cells after
being induced to differentiate in the bFGF-free medium exhibit various
morphologies, which
suggests that the cells have differentiated. The cell morphologies observed in
this study
clearly differ from those observed during normal maintenance culture in the
presence of
bFGF, as shown in Figure 27.
[0191] From the human iPS cells induced to differentiate, RNA was extracted
and cDNA
was synthesized in the same manner as shown in Example 11, and the synthesized
cDNA was
used as a template to perform PCR with the primers shown in Table 5. The
markers used
were SOX17 and AFP as endodermal markers, BRACHYURY and MSX1 as mesodermal
markers, PAX6 as an ectodermal marker, and CDX2 as a trophectodermal marker.
Each
gene was denatured at 94 C for 10 seconds, annealed for 10 seconds, and
elongated at 72 C
for 30 seconds. Annealing was performed at a temperature of 63 C for SOX17,
BRACHYURY, MSX1 and PAX6, 65 C for AFP, and 55 C for CDX2. Only in the case of
CDX2, elongation was performed at 72 C for 15 seconds.
[0192] The results of RT-PCR are shown in Figure 30. The experimental group of
rLm5
showed the expression of all the six tested factors, as in the case of the
positive control, i.e.,
the experimental group of Fd. Thus, it is suggested that rLm5 had the effect
of maintaining
pluripotency on human iPS cells. In contrast, the experimental groups of some
other
extracellular matrixes (e.g., Vn, Co) were found to show very low expression
of the
mesodermal marker MSX1. This indicated that these extracellular matrixes may
cause
human iPS cells to partially lose their differentiation potency or may induce
resistance to
differentiation.
[0193]
=
CA 02718830 2010-09-16
- 51 -
[Table 5]
RT-PCR primers
SOX17
5'-CGC Fri CATGGTGTGGGCTAAGGACG-3' (SEQ ID NO: 31)
5'-TAGTTGGGGTGGTCCTGCATGTGCTG-3' (SEQ ID NO: 32)
AFP
5'-GAATGCTGCAAACTGACCACGCTGGAAC-3' (SEQ ID NO: 33)
5'-TGGCATTCAAGAGGG __________________________________________________
ITITCAGTCTGGA-3' (SEQ ID NO: 34)
BRACHYURY
5'-GCCCTCTCCCTCCCCTCCACGCACAG-3' (SEQ ID NO: 35)
5'-CGGCGCCGTTGCTCACAGACCACAGG-3' (SEQ ID NO: 36)
MSX1
5'-CGAGAGGACCCCGTGGATGCAGAG-3' (SEQ ID NO: 37)
5'-GGCGGCCATCTTCAGCTTCTCCAG-3' (SEQ ID NO: 38)
PAX6
5'-ACCCATTATCCAGATGTG _______________________________________________
ITTGCCCGAG-3' (SEQ ID NO: 39)
5'-ATGGTGAAGCTGGGCATAGGCGGCAG-3' (SEQ ID NO: 40)
CDX2
5'-GCAGAGCAAAGGAGAGGAAA-3' (SEQ ID NO: 41)
5'-CAGGGACAGAGCCAGACACT-3' (SEQ ID NO: 42)
[0194] The present invention allowed pluripotent stem cells to proliferate in
an
undifferentiated state, without the need to use feeder cells or serum, when
culturing them in a
system containing laminin-5, an extracellular matrix molecule. According to
the method of
the present invention, pluripotent stem cells can be cultured without using
any animal-
derived material such as feeder cells or serum, which eliminates risks of
immunological
rejection, virus infection and so on. Because of their totipotency, human-
derived
pluripotent stem cells have a great potential to be used as cellular materials
in regenerative
CA 02718830 2010-09-16
- 52 -
medicine. In particular, human induced pluripotent stem cells (iPS cells) have
no ethical
problem and are also free from the problem of immunological rejection because
they can be
prepared from patients' own cells.
SEQUENCE LISTING FREE TEXT
[0195] <SEQ ID NO: 1>
SEQ ID NO: 1 shows the nucleotide sequence of human laminin a3 chain.
<SEQ ID NO: 2>
SEQ ID NO: 2 shows the amino acid sequence of human laminin a3 chain.
<SEQ ID NO: 3>
SEQ ID NO: 3 shows the nucleotide sequence of human laminin (33 chain.
<SEQ ID NO: 4>
SEQ ID NO: 4 shows the amino acid sequence of human laminin 133 chain.
<SEQ ID NO: 5>
SEQ ID NO: 5 shows the nucleotide sequence of human laminin y2 chain.
<SEQ ID NO: 6>
SEQ ID NO: 6 shows the amino acid sequence of human laminin y2 chain.
<SEQ ID NOs: 7 to 22>
SEQ ID NOs: 7 to 22 show the nucleotide sequences of RT-PCR primers used for
undifferentiation marker detection in ES cells.
<SEQ ID NOs: 23 to 30>
SEQ ID NOs: 23 to 30 show the nucleotide sequences of RT-PCR primers used for
undifferentiation marker detection in human iPS cells.
<SEQ ID NOs: 31 to 42>
SEQ ID NOs: 31 to 42 show the nucleotide sequences of RT-PCR primers used for
differentiation marker detection in human iPS cells.