Note: Descriptions are shown in the official language in which they were submitted.
CA 02719070 2010-09-20
WO 2009/121914 PCT/EP2009/053898
1
Substituted piperidines as therapeutic compounds
Field of the Invention
The present invention relates to the use of substituted piperidines as beta-
secretase-,
cathepsin D-, plasmepsin II- and/or HIV-protease-inhibitors.
Background of the Invention
With regard to beta-secretase-, cathepsin D-, plasmepsin II- and/or HIV-
protease-
inhibition, there is still a need for highly potent active ingredients. In
this context, the
improvement of the pharmacokinetic properties is at the forefront. These
properties
directed towards better bioavailability are, for example, absorption,
metabolic stability,
solubility or lipophilicity.
Alzheimer Disease aspartyl protease: Beta-Secretase
Alzheimer's disease (AD) is a progressive degenerative disease of the brain.
The
symptoms of AD include progressive memory loss, language difficulty and
ultimately
loss of basic neural function and death. The biomarkers in the central nervous
system for AD include amyloid plaques, intracellular neurofibrillary tangles
and
activated microglia. The appearance of these three markers is likely to
contribute to
the neuronal cell death and memory loss observed in AD.
Beta-amyloid is a defining feature of AD and now believed to be a causative
precursor
in the development of the disease. Amyloidogenic plaques and vascular amyloid
angiopathy also characterize the brains of individuals with Trisomy 21 (Down's
Syndrome), Hereditary Cerebral Hemorrhage with Amloidosis of the Dutch-Type
(HCHWA-D) and other neurodegenerative disorders.
Beta-amyloid plaques are predominantly composed of amyloid beta peptide (A-
beta,
also sometimes designated betaA4). The A-beta peptide is derived by
proteolysis of
the beta amyloid precursor protein (APP). Beta-APP is processed by three
distinct
ordered enzymatic activities. The bulk of beta-APP is processed via alpha-
secretase
in a non-amyloidogenic pathway. A small fraction of beta-APP is cleaved by
beta-
secretase activity to generate the membrane-bound C-terminal fragment C99.
CA 02719070 2010-09-20
WO 2009/121914 PCT/EP2009/053898
2
Gamma-secretase cleaves C99 to generate the amyloidogenic A-beta peptide of
39-42 amino acids. The aspartyl protease activity of beta-secretase has been
disclosed using varied nomenclature, including BACE (beta-site APP cleaving
enzyme), Asp and memapsin.
The significance of beta-secretase cleavage of beta-APP as a critical step in
the
generation of AD is underscored by the observation that human mutations at the
beta-secretase cleavage subsites (Swedish mutations) of beta-APP lead to
increased
A-beta production and early onset familial AD. Furthermore, BACE1 - knockout
mice
fail to produce A-beta peptide and present a normal phenotype. When crossed
with
transgenic mice that overexpress APP, the progeny show reduced amounts of A-
beta
in brain extracts as compared with control animals. This evidence supports the
proposal that inhibition of beta-secretase activity and reduction of A-beta
peptide
deposits in the brain provides a therapeutic strategy for the treatment of AD
and
other beta amyloid disorders as described by Verdile et al. (2004) in
Pharmacol.
Res 50, 397-409.
Compounds that are effective inhibitors of beta-secretase may inhibit beta-
secretase-
mediated cleavage of APP and the production of A-beta peptide. The pharma-
cological inhibition of A-beta peptide generation may reduce amyloid beta
deposits,
respectively the formation of plaques. Beta-secretase inhibiting compounds as
discussed by Thompson et al. (2005) in Curr. Pharm. Des. 11, 3383-3404 are
therefore useful to treat or to prevent diseases that are characterized by
amyloid beta
deposits or plaques such as AD.
The present invention also relates to methods of treating subjects who have,
or in
preventing subjects from developing a disease or condition selected from the
group
consisting of AD, for helping prevent or delay the onset of AD, for helping to
slow the
proression of AD, for treating subjects with mild cognitive impairment (MCI)
and
preventing or delaying the onset of AD in those who could progress form MCI to
AD,
for treating Down's syndrome, for treating humans who have HCHWAD, for
treating
cerebral amyloid angiopathy, and for treating degenerative dementias
CA 02719070 2010-09-20
WO 2009/121914 PCT/EP2009/053898
3
Alzheimer's Disease aspartyl protease: Cathepsin D
Human cathepsin D is an intracellular aspartic peptidase found mainly in
lysosomes.
It has a number of housekeeping functions, including the degradation of
cellular and
phagocytosed proteins. The enzymes may be involved in a variety of disease
states,
including cancer and Alzheimer's disease (AD). Clinical studies have shown
that
cathepsin D is overexpressed in breast cancer cells and this seems to be
associated
with an increased risk for metastasis due to enhanced cell growth. Cathepisn D
is
also thought to be involved in formation of the beta-amyloid peptide in AD.
Recently,
several genetic association studies linked cathepsin D with amyloid pathology
and
Alzheimer's disease as described for example by Davidson et al., (2006) in J.
Neurol.
Neurosurg. Psychiatry 77, 515-517. The availability of selective and potent
inhibitors
will help to further define the role of cathepsin D in disease and possibly
lead to
therapeutic agents.
Malaria Aspartyl Protease: Plasmepsin I and II
Malaria is considered as one of the most serious infectious diseases in the
world,
affecting approximately 500 million people. The disease is spread by the
anopheles
mosquito that is mostly found in tropical regions. The species plasmodium
falci-
parum is responsible for more than 95% of malaria-related morbidity and
mortality.
Increasingly, plasmodium falciparum is becoming resistant to existing
therapies such
as chloroquine, mefloquine and sulfadoxime/ pyrimethamine. Thus there is an
urgent
need for new treatments.
In the erythrocytic stage of the parasite's life cycle the parasite invades
the red blood
cells of its host consuming up to 80% of the hemoglobin as a source of
nutrients for
growth and development. Hemoglobin degradation takes place in an acidic
vacuole
of the parasite and many of the current antimalarial drugs appear to disrupt
important
vacuolar functions. The food vacuole contains aspartic, cysteine and metallo-
proteases, which are all considered to play a role in the process of
hemoglobin
degradation. At least 10 genes encoding aspartic proteases have been
identified in
the plasmodium genome. Four of the aspartic proteases have been localized in
the
CA 02719070 2010-09-20
WO 2009/121914 PCT/EP2009/053898
4
acidic food vacuole of the parasite, namely plasmepsin I, II, IV and HAP, a
histo-
aspartic protease. Inhibitors of plasmepsin I and II have shown efficacy in
cell and
animal models of malaria, indicating that these enzymes may represent targets
for
drug discovery as described for example by Coombs et al. (2001) Trends
Parasitol
17, 532-537. Indeed, a non-selective inhibitor of aspartic proteases,
pepstatin,
inhibits the growth of plasmodium falciparum in vitro. Similar results have
been
obtained with analogs of pepstatin or with immunodeficiency virus protease
inhibitors
indicating that inhibition of aspartic proteases interferes with the life
cycle of
plasmodium falciparum as noted for example by Andrews et al. (2006) in
Antimicrob.
Agents Chemother 50, 639-648.
The present invention relates to the identification of low molecular weight,
non-
peptidic inhibitors of the plasmodium falciparum protease plasmepsin II or
other
related aspartic proteases to treat and/or to prevent malaria.
HIV aspartyl protease: HIV-1 peptidase
First reported in 1981 in a small number of patients, Acquired
immunodeficiency
syndrome (AIDS) has now become a major epidemic with more than 38 million
people infected worldwide, including approximately 1 million in the United
States,
580,000 in Western Europe and more than 25 million in Sub-Saharan Africa
(http://www.unaids.org). Since AIDS was first clinically identified,
scientific and
therapeutic progress has been extraordinary. However, AIDS remains out of
control,
especially in developing countries.
The prognosis of AIDS patients who have full access to current therapies has
completely changed since the first cases of AIDS were reported. Today, the
median
survival for HIV-positive patients receiving treatment exceeds 8 years. The
life
expectancy for AIDS patients was less than 1 year before AZT was introduced in
1987. This dramatic change is due to the development of effective therapies,
to early
detection of HIV-positive individuals, and to a sustained effort to analyze
and
understand viral-resistance mechanisms, which can be overcome by rational drug
development and combination therapy.
CA 02719070 2010-09-20
WO 2009/121914 PCT/EP2009/053898
FDA-approved therapies target three steps of the HIV life cycle: reverse
transcription,
proteolytic maturation and fusion. Triple therapy, commonly referred to as
HIGHLY
ACTIVE ANTIRETROVIRAL THERAPY (HAART), is now the standard for treatment.
It consists of a protease inhibitor or a non-nucleoside reverse transcriptase
inhibitor
in combination with two nucleoside reverse transcriptase inhibitors.
Translation of human immunodeficiency virus type-1 (HIV-1) genomic RNA results
in
the production of two polyprotein precursors, Gag and Gag-Pol. The 55-kDa Gag
precursor contains the structural proteins and the 160-kDa Gag-Pol polyprotein
contains the functional viral enzymes protease, reverse transcriptase, and
integrase.
Gag and Gag-Pol polyproteins are transported to the plasma membrane where
assembly of type-C retroviruses and lentiviruses typically occurs. During
particle
assembly, the viral protease cleaves the Gag and Gag-Pol precursors into the
structural and functional proteins required for viral replication. The
protease activity
within the cytoplasma of infected cells allows for the formation of virions
which can be
released from the cell in the last stages of budding.
The mature HIV-1 protease is an obligatory dimer of identical 11-kDa subunits,
each
contributing one of the two catalytic aspartic residues. In contrast, the cell-
derived
members of the aspartic protease family are monomeric enzymes with two Asp-Thr-
Gly-containing domains. The unique dimeric structure of the retroviral
protease is
mainly stabilized by an antiparallel beta-sheet formed by the interdigitation
of the
amino- and carboxyl-terminal beta-strands of each monomer.
The activation of HIV-1 protease i.e. the dimerization and autocatalytic
release from
Gag-Pol, is a critical step in the viral life cycle. Inhibition of protease
activation causes
a severe defect in Gag polyprotein processing and a complete loss of viral
infectivity.
As such, the viral protease has become a target for HIV therapeutics,
resulting in many
HIV protease inhibitors reaching clinical trials as reviewed by Rana et al.
(1999) in
Pharmacotherapy 19, 35-59 and Morse et al., (2006) in Lancet Infect. Dis. 6,
215-225.
Most of these drugs are substrate-based inhibitors, whose design has been
facilitated
by an abundance of crystal structure data for both the native enzyme and
enzyme-
inhibitor complexes. Additionally, there are now extensive biochemical data
detailing
both the catalytic mechanism and the molecular basis for substrate selection.
CA 02719070 2010-09-20
WO 2009/121914 PCT/EP2009/053898
6
Detailed Description of the Invention
Firstly, the present invention relates to compounds of the general formula
I
O
H
N
R~`,, N
O
O
O i~\O
for the inhibition of beta-secretase, cathepsin D, plasmepsin II and/or HIV-
protease
and their pharmaceutically acceptable salts, in which
R1 is straight-chain C1_8-alkanoyloxy, straight-chain C1_8-alkoxy, straight-
chain C1_8-
alkoxy-straight-chain-C1_8-alkoxy, straight-chain C1_8-alkoxycarbonylamino,
straight-
chain C0_8-alkylcarbonylamino, optionally N-mono- or N,N-di-C1_8-alkylated
amino or
hydroxy or straight-chain omega-hydroxy-Cl_8-alkyl.
A straight-chain is also sometimes referred to in the literature as linear or
un-branched.
As used herein, straight-chain C1_8-alkanoyloxy is straight-chain C0_7-
alkylcarbonyloxy
such as formyloxy, acetyloxy, propionyloxy and butyryloxy. Examples of
straight-chain
C1_8-alkyl are methyl, ethyl, n-propyl, n-butyl, n-pentyl and n-hexyl
respectively.
Examples of straight-chain omega-hydroxy-C1_8-alkyl are hydroxymethyl, 2-
hydroxy-
ethyl, 3-hydroxy-n-propyl, 4-hydroxy-n-butyl, 5-hydroxy-n-pentyl and 6-hydroxy-
n-hexyl
respectively. Examples of straight-chain C1_8-alkoxy are radicals such as
methoxy,
ethoxy, n-propoxy and n-butoxy. Examples of straight-chain Co-C8-
alkylcarbonylamino
are for example formylamino (formamido), acetylamino, propionylamino and
CA 02719070 2010-09-20
WO 2009/121914 PCT/EP2009/053898
7
butylcarbonylamino. Optionally N-mono- or N,N-di-Cl_8-alkylated amino is
preferably
optionally N-mono- or N,N-di-straight-chain-C1_8-alkylated amino and may, for
example, be amino, methylamino, dimethylamino, ethylamino, methylethylamino,
n-propylamino, n-butylamino, n-pentylamino or n-hexylamino.
The compounds of the formula (I) have at least three asymmetric carbon atoms
and
may therefore exist in the form of optically pure diastereomers, mixtures of
diastereomers, diastereomeric racemates, mixtures of diastereomeric racemates
or
as meso compounds. The invention encompasses all these forms.
Mixtures of diastereomers, diastereomeric racemates or mixtures of
diastereomeric
racemates can be fractionated by conventional methods, e.g. by column chromato-
graphy, thin-layer chromatography, HPLC and the like.
Salts are primarily the pharmaceutically acceptable or nontoxic salts of
compounds of
formula (I). The term "pharmaceutically acceptable salts" encompasses salts
with
inorganic or organic acids, such as hydrochloric acid, hydrobromic acid,
nitric acid,
sulfuric acid, phosphoric acid, citric acid, formic acid, maleic acid, acetic
acid,
succinic acid, tartaric acid, methanesulfonic acid, p-toluenesulfonic acid and
the like.
Salts of compounds having salt-forming groups are in particular acid addition
salts,
salts with bases, or, in the presence of a plurality of salt-forming groups,
in some
cases also mixed salts or internal salts.
Such salts are formed, for example, from compounds of formula (I) with an
acidic
group, for example a carboxyl or sulfonyl group, and are, for example, the
salts
thereof with suitable bases such as non-toxic metal salts derived from metals
of
group Ia, Ib, Ila and IIb of the Periodic Table of the Elements, for example
alkali
metal, in particular lithium, sodium, or potassium, salts, alkaline earth
metal salts, for
example magnesium or calcium salts, and also zinc salts and ammonium salts,
including those salts which are formed with organic amines, such as optionally
hydroxy-substituted mono-, di- or trialkylamines, in particular mono-, di- or
tri(lower
CA 02719070 2010-09-20
WO 2009/121914 PCT/EP2009/053898
8
alkyl)amines, or with quaternary ammonium bases, e.g. methyl-, ethyl-, diethyl-
or
triethylamine, mono-, bis- or tris(2-hydroxy(lower alkyl))amines, such as
ethanol-,
diethanol- or triethanolamine, tris(hydroxymethyl)methylamine or 2-hydroxy-
tert-
butylamine, N,N-di(lower alkyl)-N-(hydroxy(lower alkyl))amine, such as N,N-di-
N-
dimethyl-N-(2-hydroxyethyl)amine, or N-methyl-D-glucamine, or quaternary
ammonium hydroxides such as tetrabutyl ammonium hydroxide. The compounds of
formula (I) having a basic group, for example an amino group, may form acid
addition
salts, for example with suitable inorganic acids, e.g. hydrohalic acid such as
hydrochloric acid, hydrobromic acid, sulfuric acid with replacement of one or
both
protons, phosphoric acid with replacement of one or more protons, e.g. ortho-
phosphoric acid or metaphosphoric acid, or pyrophosphoric acid with
replacement of
one or more protons, or with organic carboxylic, sulfonic or phosphonic acids
or
N-substituted sulfamic acids, e.g. acetic acid, propionic acid, glycolic acid,
succinic
acid, maleic acid, hydroxymaleic acid, methylmaleic acid, fumaric acid, malic
acid,
tartaric acid, gluconic acid, glucaric acid, glucuronic acid, citric acid,
benzoic acid,
cinnamic acid, mandelic acid, salicylic acid, 4-aminosalicylic acid, 2-
phenoxybenzoic
acid, 2-acetoxybenzoic acid, embonic acid, nicotinic acid, isonicotinic acid,
and also
amino acids, for example the alpha-amino acids mentioned above, and also
methanesulfonic acid, ethanesulfonic acid, 2-hydroxyethanesulfonic acid,
ethane-1,2-
disulfonic acid, benzenesulfonic acid, 4-methylbenzenesulfonic acid,
naphthalene-2-
sulfonic acid, 2- or 3-phosphoglycerate, glucose 6-phosphate, N-
cyclohexylsulfamic
acid (with formation of the cyclamates) or with other acidic organic compounds
such
as ascorbic acid. Compounds of formula (I) having acidic and basic groups may
also
form internal salts.
Salts obtained may be converted to other salts in a manner known per se, acid
addition salts, for example, by treating with a suitable metal salt such as a
sodium,
barium or silver salt, of another acid in a suitable solvent in which an
inorganic salt
which forms is insoluble and thus separates out of the reaction equilibrium,
and base
salts by release of the free acid and salt reformation.
CA 02719070 2010-09-20
WO 2009/121914 PCT/EP2009/053898
9
The compounds of formula (I), including their salts, may also be obtained in
the form
of hydrates or include the solvent used for the crystallization.
Pharmaceutically unsuitable salts may also be used for isolation and
purification.
Prodrug derivatives of the compounds described herein are derivatives thereof
which
on in vivo use liberate the original compound by a chemical or physiological
process.
A prodrug may for example be converted into the original compound when a
physio-
logical pH is reached or by enzymatic conversion. Possible examples of prodrug
derivatives are esters of freely available carboxylic acids, S- and O-acyl
derivatives of
thiols, alcohols or phenols, the acyl group being defined as herein. Preferred
derivatives are pharmaceutically acceptable ester derivatives which are
converted by
solvolysis in physiological medium into the original carboxylic acid, such as,
for
example, lower alkyl esters, cycloalkyl esters, lower alkenyl esters, benzyl
esters,
mono- or disubstituted lower alkyl esters such as lower omega-(amino, mono- or
dialkylamino, carboxy, lower alkoxycarbonyl) - alkyl esters or such as lower
alpha-(alkanoyloxy, alkoxycarbonyl or dialkylaminocarbonyl) - alkyl esters;
con-
ventionally, pivaloyloxymethyl esters and similar esters are used as such.
Because of the close relationship between a free compound, a prodrug
derivative
and a salt compound, a particular compound in this invention also includes its
prodrug derivative and salt form, where this is possible and appropriate. The
definitions mentioned apply within the scope of general chemical principles
such as,
for example, the usual valencies of atoms.
The compounds of the formula (I) also include those compounds in which one or
more
atoms are replaced by their stable, non-radioactive isotopes; for example, a
hydrogen
atom by deuterium.
A preferred group of compounds of the formula (I) and the pharmaceutically
acceptable salts thereof, are compounds in which
CA 02719070 2010-09-20
WO 2009/121914 PCT/EP2009/053898
R1 is hydroxy or straight-chain omega-hydroxy-Cl_8-alkyl, more preferably
hydroxy or
straight-chain omega-hydroxy-C1_4-alkyl.
A further preferred group of compounds of the formula (I) and the
pharmaceutically
acceptable salts thereof, are compounds in which
R1 is straight-chain C1_8-alkoxy or straight-chain C1_8-alkoxy-straight-chain-
C1_8-
alkoxy, more preferably straight-chain C1_4-alkoxy or straight-chain C1_4-
alkoxy-
straight-chain-C1_4-alkoxy.
A further preferred group of compounds of the formula (I) and the
pharmaceutically
acceptable salts thereof, are compounds in which
R1 is straight-chain C1_8-alkanoyloxy, more preferably straight-chain C1_4-
alkanoyloxy.
A further preferred group of compounds of the formula (I) and the
pharmaceutically
acceptable salts thereof, are compounds in which
R1 is straight-chain C0_8-alkylcarbonylamino, more preferably straight-chain
C0_3-
alkylcarbonylamino.
A further preferred group of compounds of the formula (I) and the
pharmaceutically
acceptable salts thereof, are compounds in which
R1 is optionally N-mono- or N,N-di-C1_8-alkylated amino, more preferably
optionally N-
mono- or N,N-di-C1_4-alkylated amino,.
A further preferred group of compounds of the formula (I) and the
pharmaceutically
acceptable salts thereof, are compounds in which
R1 is optionally N-mono- or N,N-di-straight-chain-C1_8-alkylated amino, more
preferably optionally N-mono- or N,N-di-straight-chain-C1_4-alkylated amino.
R1 is very particularly preferably hydroxy, methoxy, 2-methoxy-ethoxy,
acetyloxy
formamido, methylcarbonylamino or ethylcarbonylamino.
CA 02719070 2010-09-20
WO 2009/121914 PCT/EP2009/053898
11
The compound groups mentioned above are not to be regarded as closed, but
rather
parts of these compound groups may be exchanged with one another or with the
definitions given above or omitted in a sensible manner, for example to
replace
general by more specific definitions. The definitions are valid in accordance
with
general chemical principles, such as, for example, the common valences for
atoms.
The compounds of formula (I) can be prepared in an analogous manner to
preparation
processes disclosed in the literature. Similar preparation processes are
described for
example in WO 97/09311. Details of the specific preparation variants can be
found in
the examples.
The compounds of formula (I) may also be prepared in optically pure form. The
separation into antipodes can be effected by procedures known per se, either
preferably at an earlier synthetic stage by salt formation with an optically
active acid,
for example (+)- or (-)-mandelic acid and separation of the diastereomeric
salts by
fractional crystallization, or preferably at a relatively late stage by
derivatizing with a
chiral auxiliary building block, for example (+)- or (-)-camphanoyl chloride,
and
separation of the diastereomeric products by chromatography and/or
crystallization
and subsequent cleavage of the bonds to give the chiral auxiliary. The pure
diastereomeric salts and derivatives may be analysed to determine the absolute
configuration of the piperidine present with common spectroscopic procedures,
and
X-ray spectroscopy on single crystals constitutes a particularly suitable
procedure.
The compounds of formula (I), and their pharmaceutically acceptable salts
reveal
inhibitory activities on the enzymes beta-secretase, cathepsin D, plasmepsin
II and/or
H IV-protease.
The activitiy of inhibitors of beta-secretase, cathepsin D, plasmepsin II
and/or HIV
protease can be assessed experimentally with following in vitro assays.
CA 02719070 2010-09-20
WO 2009/121914 PCT/EP2009/053898
12
The protease inhibitory activity of compounds can be tested with an assay kit
using
the fluorescence resonance energy transfer (FRET) technology and a recombinant
i.e. baculovirus expressed enzyme preparation. The FRET is used to monitor the
cleavage of the peptide substrate. The principle of the assay is as follows
relies on a
measurable energy difference, quantitatively depending on the presence of a
peptide
sequence. The peptide substrate is synthesized with two terminal fluorophores,
a
fluorescent donor and quenching acceptor. The distance between these two
groups
is selected so that upon light excitation, the donor fluorescence energy is
significantly
quenched by the acceptor through resonance energy transfer. Upon cleavage by
the
protease, the fluorophore is separated from the quenching group, restoring the
fluorescence yield of the donor. Thus a weakly fluorescent peptide substrate
becomes highly fluorescent upon enzymatic cleavage; the increase in
fluorescence is
linearly related to the rate of proteolysis.
The FRET assay was performed in white polysorp plates. The assay buffer
consisted of 50 mM sodium acetate pH 5, 392 mM sodium chloride, 12.5% glycerol
and 0.1 % BSA. The incubates per well were composed of 160 pl buffer, 10 pl
inhibitor in DMSO, 10 pl peptide substrate in DMSO and 20 pl enzyme-solution.
The
inhibitors are tested in a concentration range of 1 pM to 1 mM. The
fluorescently
marked donor and acceptor peptide substrates are generated by solid phase
peptide
synthesis (Applied Biosystems). The beta-secretase peptide substrate Rh-Glu-
Val-
Asn-Leu-Asp-Ala-Glu-Phe-Lys-Quencher is obtained from Invitrogen, Carlsbad,
CA,
USA. The cathepsin D peptide substrate of the sequence DABCYL-Pro-Thr-Glu-
Phe-Phe-Arg-Leu-OXL, the plasmepsin peptide substrate of the sequence DABCYL-
Glu-Arg-Nle-Phe-Leu-Ser-Phe-Pro-OXL and the HIV protease peptide substrate of
the sequence DABCYL-His-Lys-Ala-Arg-Val-Leu-Tyr-Glu-Ala-Nle-Ser-EDANS are all
obtained from AnaSpec Inc, San Jose, CA, USA. The recombinantly expressed
enzyme preparations are added in various amounts to the assay systems eg the
beta-sectrase concentration is 1 unit/ml incubation volume, the cathepsin D
concentration is 100 ng/ml, the HIV protease concentration is 500 ng/ml and
the
plasmepsin II concentration is 50 ng/ml. The reaction is started upon addition
of the
enzyme solution. The incubation occurs at 37 C over 30-120 min ie specifically
the
CA 02719070 2010-09-20
WO 2009/121914 PCT/EP2009/053898
13
beta-secretase incubation lasts 60 min, the cathepsin D incubation 120 min,
the
plasmepsin II incubation 40 min and the HIV protease incubation 40 min. The
reactions are stopped by the addition of 20 pl of a 1.0 M Tris Base solution.
The
enzymatic substrate to product conversion is assessed by fluorescence
measurements at 460 nm wave length.
In vitro enzyme inhibitory activities
The compounds of the present invention revealed structure-dependent and enzyme-
specific inhibitory activities. The inhibitory activities were measured as
IC50 values.
Thus the beta-secretase inhibitory activity ranged between 1 pM and 1 mM; the
values for cathepsin D ranged between 1 pM and 1 mM, for plasmepsin II between
1 pM and 1 mM and for HIV-protease between 1 pM and 1 mM.
The compounds of the formula (I) and the pharmaceutically acceptable salts
thereof
may find use as medicines, for example in the form of pharmaceutical
preparations.
The pharmaceutical preparations may be administered enterally, such as orally,
for
example in the form of tablets, coated tablets, sugar-coated tablets, hard and
soft
gelatine capsules, solutions, emulsions or suspensions, nasally, for example
in the
form of nasal sprays, rectally, for example in the form of suppositories, or
trans-
dermally, for example in the form of ointments or patches. The administration
may
also be parenteral, such as intramuscular or intravenous, for example in the
form of
injection solutions.
To prepare tablets, coated tablets, sugar-coated tablets and hard gelatine
capsules,
the compounds of the formula (I) and pharmaceutically usable salts thereof may
be
processed with pharmaceutically inert, inorganic or organic excipients. Such
excipients
used, for example for tablets, coated tablets and hard gelatine capsules, may
be
lactose, corn starch, or derivatives thereof, talc, stearic acid or salts
thereof etc.
Suitable excipients for soft gelatine capsules are, for example, vegetable
oils, waxes,
fats, semisolid and liquid polyols, etc.
Suitable excipients for preparing solutions and syrups are, for example,
water,
CA 02719070 2010-09-20
WO 2009/121914 PCT/EP2009/053898
14
polyols, sucrose, invert sugar, glucose, etc.
Suitable excipients for injection solutions are, for example, water, alcohols,
polyols,
glycerol, vegetable oils, bile acids, lecithin, etc.
Suitable excipients for suppositories are, for example, natural or hardened
oils,
waxes, fats, semisolid or liquid polyols, etc.
The pharmaceutical preparations may additionally also comprise preservatives,
solubilizers, viscosity-increasing substances, stabilizers, wetting agents,
emulsifiers,
sweeteners, colorants, flavourings, salts for altering the osmotic pressure,
buffers,
coatings or antioxidants. They may also comprise other therapeutically
valuable
substances.
Subject of the present invention is also the use of the compounds of formula
(I), and
their pharmaceutically acceptable salts for the prevention, delay of
progression or the
treatment of Alzheimer Disease, malaria or HIV infection.
Subject of the present invention is also the use of the compounds of formula
(I), and
their pharmaceutically acceptable salts for the manufacture of a medication
for the
prevention, delay of progression or the treatment of Alzheimer Disease,
malaria or
HIV infection.
Subject of the present invention is also the method for the prevention, delay
of pro-
gression or the treatment of Alzheimer Disease, malaria or HIV infection,
whereby a
therapeutically effective dose of a compound of the general formula (I) or a
pharma-
ceutically acceptable salt thereof is applied.
Subject of the present invention is also a pharmaceutical preparation that
contains for
the inhibition of beta-secretase, cathepsin D, plasmepsin and/or HIV-protease
a
compound of the general formula (I), or a pharmaceutically acceptable salt
thereof as
well as commonly used ingredients.
CA 02719070 2010-09-20
WO 2009/121914 PCT/EP2009/053898
Subject of the present invention is also a pharmaceutical preparation for the
pre-
vention, delay of progression or treatment of Alzheimer Disease, malaria and
HIV
infection that contains a compound of the general formula (I), or a
pharmaceutically
acceptable salt thereof as well as commonly used ingredients.
The dose may vary within wide limits and has of course to be adapted to the
individual
circumstances in each individual case. In general, for oral administration, a
daily dose
of about 3 mg to about 3 g, preferably about 10 mg to about 1 g, for example
about
300 mg, per adult (70 kg), divided into preferably 1-3 individual doses which
may, for
example, be of equal size, may be appropriate, although the upper limit
specified may
also be exceeded if this should be found to be appropriate; typically,
children receive a
lower dose according to their age and body weight.
Examples
The following examples illustrate the present invention. All temperatures are
stated in
degrees Celsius and pressures in mbar. Unless mentioned otherwise, the
reactions
take place at room temperature. The abbreviation "Rf = xx (A)" means for
example
that the Rf is found in solvent system A to be xx. The ratio of amounts of
solvents to
one another is always stated in parts by volume. Chemical names for final
products
and intermediates have been generated on the basis of the chemical structural
formulae with the aid of the AutoNom 2000 (Automatic Nomenclature) program.
HPLC gradients on Hypersil BDS C-18 (5 um); column: 4 x 125 mm
I 90% water/10% acetonitrile* to 0% water/100% acetonitrile* in 5 minutes +
2.5 minutes (1.5 ml/min)
II 95% water/5% acetonitrile* to 0% water/100% acetonitrile* in 40 minutes
(0.8 ml/min)
contains 0.1 % trifluoroacetic acid
The following abbreviations are used:
Rf ratio of distance migrated by a substance to the distance of the solvent
CA 02719070 2010-09-20
WO 2009/121914 PCT/EP2009/053898
16
front from the starting point in thin-layer chromatography
Rt retention time of a substance in HPLC (in minutes)
M.P. melting point (temperature)
Nr. Strukture Aspect Rf Rt
(System) (Method)
1 o yellow oil 0.14 (B) 3.78 (I)
H
HOV
O
0 ~\O/
2 light yellow oil 0.21 (C) 3.75 (I)
H
H N
O
O
O\O/
3 ! turbid yellow oil 0.20 (D) 3.90 (I)
H
H
H H
O O
O~\O
4 yellow oil 0.28 (A) 4.21 (I)
N
N
0
CA 02719070 2010-09-20
WO 2009/121914 PCT/EP2009/053898
17
Nr. Strukture Aspect Rf Rt
(System) (Method)
! yellow oil -- 4.11 (I)
N
1 0 O \ N
O
O~\O/
6 O yellow oil 0.25 (B) 4.19(l)
r yellow oil 0.25 (B) 4.19 (I)
N
N
I
O
O~\O~
Thin-film chromatography eluent systems:
A dichloromethane-methanol-25% ammonia conc. = 200:20:1
B dichloromethane-methanol-25% ammonia conc. = 200:10:1
C dichloromethane-methanol-25% ammonia conc. = 200:30:1
D dichloromethane-methanol-25% ammonia conc. = 100:10:1
General procedure A: (N-Cbz-deprotection, N-(1-phenyl-ethyl)-deprotection or N-
benzyl-deprotection )
To a stirred solution of 1 mmol of "N-protected derivative" in 15 ml of
tetrahydrofuran
(or methanol) are added 0.1 mmol Pd/C 10% and the reaction mixture is hydro-
genated at room temperature. The reaction mixture is filtered and concentrated
under
reduced pressure. The residue is purified by flash chromatography (Si02 60F)
to
afford the title compound.
General procedure B: (N-Tos-deprotection)
To a stirred solution of 0.09 mmol "tosylamide" in 10 ml of methanol are added
0.44
mmol sodium dihydrogenphosphate and 0.90 mmol of sodium amalgam (10% Na) at
room temperature. The reaction mixture is stirred for 2-18 hours, diluted with
water
and extracted with ethyl acetate (3X). The organic phases are combined, washed
CA 02719070 2010-09-20
WO 2009/121914 PCT/EP2009/053898
18
with brine and dried over sodium sulfate. The solvent is concentrated under
reduced
pressure and the residue is purified by flash chromatography (Si02 60F) to
afford the
title compound.
General procedure C: (BH3-reduction)
To a stirred solution of 1 mmol of "lactam" in 3 ml of tetrahydrofuran is
admixed with
2-4 mmol of borane tetrahydrofuran (1 M in tetrahydrofuran) and heated to 50 C
for
2-8 hours.
The reaction mixture is quenched by addition of 10 ml of methanol and
concentrated
under reduced pressure. The title compound is obtained from the residue by
means
of flash chromatography (Si02 60F).
General procedure D: (0-alkylation I)
1.2 mmol mmol of sodium hydride (60% dispersion in oil) and 0.1 mmol of
tetrabutyl-
ammonium iodide are added to a solution of 1 mmol of "alcohol" and 1.1 mmol of
"benzyl halide" in 2.0 ml of N,N-dimethylformamide while stirring at -10 C.
The
reaction mixture is stirred at -10 C for 1 hour and at room temperature for 18
hours.
The mixture is poured into 1 M aqueous sodium bicarbonate solution and
extracted
with tert-butyl methyl ether (2X). The organic phases are washed successively
with
water and brine, dried with sodium sulfate and evaporated. The title compound
is
obtained from the residue by means of flash chromatography (Si02 60F).
General Method E: (0-alkylation II)
1.1 mmol of sodium hydride (60% dispersion in oil) are added to a solution of
1 mmol
of "alcohol" and 1.0-2.0 mmol of "benzyl halide" in 2.0 ml of N,N-
dimethylformamide
while stirring at -10 C. The reaction mixture is stirred at -10 C for 1 hour
and at room
temperature for 18 hours. The mixture is poured into 1 M aqueous sodium
bicarbonate
solution and extracted with tert-butyl methyl ether (2X). The combined organic
phases
are washed successively with water and brine, dried with sodium sulfate and
eva-
porated. The title compound is obtained from the residue by flash
chromatography
(Si02 60F).
CA 02719070 2010-09-20
WO 2009/121914 PCT/EP2009/053898
19
General Method F: (alcohol desilylation)
A solution of 1 mmol of "silyl ether" in 5 ml of tetrahydrofuran is mixed with
1.5-2.0 mmol of tetrabutylammonium fluoride (1 M solution in tetrahydrofuran)
and the
solution is stirred at room temperature for 1-2 hours. The reaction solution
is then
diluted with water and extracted with tert-butyl methyl ether (2X). The
combined
organic phases are dried with sodium sulfate and evaporated. The title
compound is
obtained from the residue by flash chromatography (Si02 60F).
Example 1
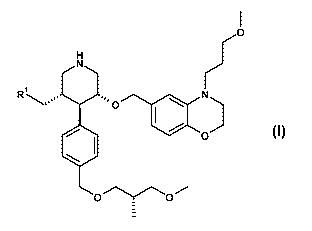
{(3S,4R,5R)-4-f4-((S)-3-Methoxy-2-methyl-propoxymethyl)-phenyll-5-f4-(3-
methoxy-
propyl)-3,4-dihydro-2H-benzof 1,41oxazin-6-ylmethoxyl-piperidin-3-yl}-methanol
According to general procedure A, (3S,4R,5R)-3-hydroxymethyl -4-[4-((S)-3-
methoxy-
2-methyl-propoxymethyl)-phenyl]-5-[4-(3-methoxy-propyl)-3,4-dihydro-2H-
benzo[1,4]oxazin-6-ylmethoxy]-piperidine-1-carboxylic acid benzyl ester is
used to
afford the title compound.
The starting material(s) is(are) prepared as follows:
a) (3S,4R,5R)-3-Hvdroxvmethvl-4-f4-((S)-3-methoxv-2-methyl -propoxymethyl)-
phenyll-5-f4-(3-methoxv-propel)-3,4-dihvdro-2H-benzof1,41oxazin-6-ylmethoxyl-
piperidine-1-carboxylic acid benzyl ester
According to general procedure C, (3S,4R,5R)-3-hydroxymethyl-4-[4-((S)-3-
methoxy-
2-methyl-propoxymethyl)-phenyl]-5-[4-(3-methoxy-propyl)-3-oxo-3,4-dihydro-2H-
benzo[1,4]oxazin-6-ylmethoxy]-piperidine-1-carboxylic acid benzyl ester is
used to
afford the title compound as a turbid oil. Rf = 0.15 (EtOAc-heptane 2:1); Rt =
5.22
(gradient I).
b) (3S,4R,5R)-3-Hydroxymethyl -4-f4-((S)-3-methoxv-2-methyl -propoxymethyl)-
phenyll-5-f4-(3-methoxv-propel)-3-oxo-3,4-dihvdro-2H-benzof1,41oxazin-6-
ylmethoxyl-piperidine-1-carboxylic acid benzyl ester
A solution of 8.526 mmol of (3R,4R,5S)-4-[4-((S)-3-methoxy-2-methyl-propoxy-
methyl)-phenyl]-3-[4-(3-methoxy-propyl)-3-oxo-3,4-dihydro-2H-benzo[1,4]oxazin-
6-
ylmethoxy]-5-trityloxym ethyl-piperidine-1-carboxylic acid benzyl ester in 20
ml of
tetrahydrofuran and 80 ml of methanol is treated with 12.789 mmol of toluene-4-
CA 02719070 2010-09-20
WO 2009/121914 PCT/EP2009/053898
sulfonic acid =monohydrate. After stirring for 15 hours, the reaction mixture
is basified
with saturated aqueous sodium bicarbonate solution and concentrated under
reduced pressure to remove most of the methanol and tetrahydrofuran. The
residue
is extracted with dichloromethane (3X) -- the combined organic layers are
washed
successively with water and brine, dried with sodium sulfate and evaporated.
The title
compound is obtained as a yellow oil from the residue by flash chromatography
(Si02
60F). Rt = 4.86 (gradient I).
c) (3R,4R,5S)-4-f4-((S)-3-Methoxv-2-methyl-propoxymethyl)-phenyll-3-f4-(3-
methoxy-propel)-3-oxo-3,4-dihydro-2H-benzof 1,41oxazin-6-ylmethoxyl-5-
trityloxymethyl-piperidine-1-carboxylic acid benzyl ester
According to general procedure D, (3R,4R,5S)-3-hydroxy-4-[4-((S)-3-methoxy-2-
methyl-propoxymethyl)-phenyl]-5-trityloxymethyl -piperidine-1-carboxylic acid
benzyl
ester and 6-chloromethyl -4-(3-methoxy-propyl)-4H-benzo[1,4]oxazin-3-one
[857272-
02-7] are used to afford the title compound as a yellow oil. Rt = 6.58
(gradient I).
d) (3R,4R,5S)-3-Hydroxy-4-f4-((S)-3-methoxy-2-methyl-propoxymethyl)-phenyll-5-
trityloxymethyl-piperidine-1-carboxylic acid benzyl ester
8.478 mmol of benzyl chloroformate are added dropwise to a mixture of 8.478
mmol
of (3R,4R,5S)-4-[4-((S)-3-methoxy-2-methyl-propoxymethyl)-phenyl]-5-trityloxy-
methyl-piperidin-3-ol (L)-(+)-mandelate in 150 ml of ethyl acetate and 150 ml
of
saturated aqueous sodium bicarbonate solution at 0 C. After 1 hour, the
reaction
mixture is partitioned between saturated aqueous sodium carbonate solution and
ethyl acetate -- the organic layer is washed successively with water and
brine. The
combined aqueous layers are back-extracted with ethyl acetate -- the combined
organic layers are dried with sodium sulfate and evaporated. The crude title
compound is obtained as a white foam. Rt = 6.14 (gradient I).
e) (3R,4R,5S)-4-f4-((S)-3-Methoxv-2-methyl-propoxymethyl)-phenyll-5-
trityloxymethyl-piperid in-3-ol (L)-(+)-mandelate
According to general procedure A, (3R,4R,5S)-1-benzyl-4-[4-((S)-3-methoxy-2-
methyl-propoxymethyl)-phenyl]-5-trityloxymethyl-piperidin-3-ol (L)-(+)-
mandelate is
used to afford the title compound as a white foam. Rt = 4.92 (gradient I).
CA 02719070 2010-09-20
WO 2009/121914 PCT/EP2009/053898
21
f) (3R,4R,5S)-1-Benzvl-4-[4-((S)-3-methoxv-2-methyl -propoxymethyl)-phenyll-5-
trityloxymethyl -piperid in-3-ol (L)-(+)-mandelate
2.36 mmol of (L)-(+)-mandelic acid are added to a solution of 5.90 mmol of
(rac-
3R,4R,5S)-1-benzyl-4-[4-((S)-3-methoxy-2-methyl-propoxymethyl)-phenyl]-5-
trityloxy-
methyl-piperidin-3-ol in 36 ml of tetrahydrofuran at 600C (oil bath
temperature). 36 ml
of n-hexane are slowly added dropwise at 60 C. The mixture is slowly cooled to
room
temperature over the course of 3 hours and, after a brief treatment in an
ultrasonic
bath, then cooled at 0 C for 2 hours. The precipitate is filtered off and
washed with 1:4
tetrahydrofuran/n-hexane to afford the title compound as a white solid. Rt =
5.36
(gradient I).
For analytical purposes, a small amount of the salt is dissolved in ethyl
acetate and
washed with saturated aqueous sodium carbonate solution (2X). The organic
phase
is washed with brine, dried with sodium sulfate and evaporated to afford the
title
compound as the free base (white solid). HPLC; Rt = 12.81 (Daicel Chiralpak AD
0.46 x 25 cm; 95% hexane/5% isopropanol; 0.7 ml/minute for 60 minutes).
g) (rac-3R,4R,5S)-1-Benzvl-4-[4-((S)-3-methoxv-2-methyl-propoxymethyl)-phenyll-
5-trityloxymethyl -piperid in-3-ol
86.852 mmol of a solution of borane-tetrahydrofuran (1 M/THF) are added
dropwise
to a solution of 43.426 mmol of 1-benzyl-4-[4-((S)-3-methoxy-2-methyl-propoxy-
methyl)-phenyl]-3-(R,S)-trityloxymethyl -1,2,3,6-tetrahydro-pyridine in 220 ml
of
tetrahydrofuran at room temperature. After overnight stirring, the reaction
mixture is
cooled to 10 C and successively treated dropwise with a solution of 251.871
mmol of
potassium hydroxide in 60 ml of water and then with 86.852 mmol of hydrogen
peroxide (30%/water). The reaction mixture is slowly warmed to 65 C, stirred
for
3 hours and then re-cooled to room temperature. The reaction mixture is
partitioned
between tert-butyl methyl ether and ice-water -- the organic layer is washed
with
brine. The combined aqueous layers are extracted with tert-butyl methyl ether
(2X) --
the combined organic layers are dried with sodium sulfate and evaporated. The
residue is purified by flash chromatography (Si02 60F) to afford the title
compound as
a yellow oil. Rf = 0.15 (EtOAc-heptane 1:1); Rt = 5.36 (gradient I).
CA 02719070 2010-09-20
WO 2009/121914 PCT/EP2009/053898
22
h) 1-Benzvl-4-[4-((S)-3-methoxv-2-methyl-propoxymethyl)-phenyll-3-(R,S)-
trityloxymethyl-1,2,3,6-tetrahydro-pyrid ine
102.636 mmol of thionyl chloride are slowly added to a solution of 85.53 mmol
of
1 -benzyl-4-[4-((S)-3-methoxy-2-methyl-propoxymethyl)-phenyl]-3-(R,S)-
trityloxy-
methyl-piperidin-4-(R,S)-ol in 250 ml of pyridine at 0 C, taking care that the
internal
temperature stays below 10 C. After 5 minutes, the reaction mixture is
quenched by
adding 50 ml of 4N NaOH solution and concentrated under reduced pressure. The
residue is dissolved in ethyl acetate and washed successively with saturated
aqueous sodium bicarbonate solution, water and brine, dried with sodium
sulfate and
evaporated. The residue is purified by flash chromatography (Si02 60F) to
afford the
title compound as a yellow oil. Rf = 0.28 (EtOAc-heptane-25% ammonia conc.
100:200:1); Rt = 5.64 (gradient I).
i) 1-Benzvl-4-[4-((S)-3-methoxv-2-methyl-propoxymethyl)-phenyll-3-(R,S)-
trityloxy-
methyl-piperid in-4-(R,S)-ol
2.32 mmol of 1,2-dibromoethane are added to a suspension of 156.518 mmol of
magnesium in 20 ml of tetrahydrofuran under argon at room temperature. The
reaction mixture is warmed until the magnesium starts to react and then 3 ml
of a
solution of 151.509 mmol of 1-bromo-4-((S)-3-methoxy-2-methyl -propoxymethyl)-
benzene in 170 ml of tetrahydrofuran, followed by the rest of the solution, is
added
while maintaining a gentle reflux. After 2 hours, the reaction mixture is
cooled to room
temperature and then a solution of 125.214 mmol of 1-benzyl-3-trityloxymethyl-
piperidin-4-one [234757-27-8] in 170 ml of tetrahydrofuran is slowly added,
taking
care to keep the internal reaction temperature under 40 C. After overnight
stirring at
room temperature, the reaction mixture is quenched with saturated aqueous
ammonium chloride solution. Ethyl acetate is added to the reaction mixture and
the
phases separated -- the organic phase is washed successively with water and
brine.
The combined aqueous phases are back-extracted with ethyl acetate -- the
combined
organic phases are dried with sodium sulfate and evaporated. The residue is
purified
by flash chromatography (Si02 60F) to afford the title compound as a yellow
oil.
Rf = 0.30 (EtOAc-heptane-25% ammonia conc. 100:100:1); Rt = 5.43 (gradient I).
CA 02719070 2010-09-20
WO 2009/121914 PCT/EP2009/053898
23
j) 1-Bromo-4-((S)-3-methoxv-2-methyl-propoxymethyl)-benzene
According to general method E, 1-bromo-4-chloromethyl-benzene [589-17-3] and
(R)-3-methoxy-2-methyl-propan-1-ol are used to afford the title compound as a
yellow oil. Rf = 0.44 (EtOAc-heptane 1:6); Rt = 5.29 (gradient I).
k) (R)-3-Methoxv-2-methyl-propan-1-ol
According to general method F, triisopropyl -(3-methoxy-2(S)-m ethyl
propoxy)siIane is
used to afford the title compound as a yellow oil. Rf = 0.42 (dichloromethane-
diethylether 1:1).
I) Triisopropyl-((S)-3-methoxv-2-m ethyl propoxy)siIane
3.09 g of sodium hydride (60% dispersion in oil) are added to a solution of
9.55 g of
(S)-2-methyl-3-triisopropylsilanyloxypropan-1-ol [256643-28-4] and 7.3 ml of
methyl
iodide in 70 ml of N,N-dimethylformamide at 0 C. After 60 hours at room
temperature, the reaction mixture is diluted with tert-butyl methyl ether and
washed
successively with water and brine, dried with sodium sulfate and evaporated.
The
residue is purified by flash chromatography (Si02 60F) to afford the title as
a yellow
oil. Rf = 0.51 (EtOAc-heptane 1:10).
Example 2
N-{(3S,4R,5R)-4-f4-((S)-3-Methoxv-2-methyl-propoxymethyl)-phenyll-5-f4-(3-
methoxy-propel)-3,4-dihvdro-2H-benzof1,41oxazin-6-ylmethoxyl-piperidin-3-
ylmethyl}-
acetamide
According to general procedure A, (3R,4R,5R)-3-(acetylamino-methyl)-4-[4-((S)-
3-
methoxy-2-methyl-propoxymethyl)-phenyl]-5-[4-(3-methoxy-propyl)-3,4-dihydro-2H-
benzo[1,4]oxazin-6-ylmethoxy]-piperidine-1-carboxylic acid benzyl ester is
used to
afford the title compound.
The starting material(s) is(are) prepared as follows:
a) (3R,4R,5R)-3-(Acetylamino-methyl)-4-f4-((S)-3-methoxv-2-methyl-
propoxymethyl)-phenyll-5-f4-(3-methoxv-propel)-3,4-dihvdro-2H-
benzof1,41oxazin-6-ylmethoxyl-piperidine-1-carboxylic acid benzyl ester
CA 02719070 2010-09-20
WO 2009/121914 PCT/EP2009/053898
24
5.936 mmol of acetyl chloride are added to a solution of 5.396 mmol of
(3R,4R,5R)-3-
aminomethyl-4-[4-((S)-3-methoxy-2-methyl-propoxymethyl)-phenyl]-5-[4-(3-
methoxy-
propyl)-3,4-dihydro-2H-benzo[1,4]oxazin-6-ylmethoxy]-piperidine-1-carboxylic
acid
benzyl ester and 6.475 mmol of triethylamine in 80 ml of dichloromethane under
argon at 0 C. After 30 minutes, the reaction mixture is quenched with
saturated
aqueous sodium bicarbonate solution and extracted with tert-butyl methyl ether
(2X).
The combined organic layers are washed successively with water and brine,
dried
with sodium sulfate and evaporated. The residue is purified by flash
chromatography
(Si02 60F) to afford the title compound as a yellow oil. Rt = 4.98 (gradient
I).
b) (3R,4R,5R)-3-Aminomethyl-4-f4-((S)-3-methoxv-2-methyl -propoxymethyl)-
phenyll-5-f4-(3-methoxv-propel)-3,4-dihvdro-2H-benzof1,41oxazin-6-ylmethoxyl-
piperidine-1-carboxylic acid benzyl ester
7.128 mmol of triphenylphosphine are added to a solution of 5.940 mmol of
(3S,4R,5R)-3-azidomethyl -4-[4-((S)-3-methoxy-2-methyl-propoxymethyl)-phenyl]-
5-
[4-(3-methoxy-propyl)-3,4-dihydro-2H-benzo[1,4]oxazin-6-ylmethoxy]-piperidine-
1-
carboxylic acid benzyl ester in 20 ml of methanol, 20 ml of tetrahydrofuran, 5
ml of
water and 3.56 ml of concentrated aqueous ammonia (25%) at room temperature.
After overnight stirring, the reaction mixture is partitioned between tert-
butyl methyl
ether and 5:1 water/saturated aqueous sodium bicarbonate solution. The aqueous
layer is extracted with tert-butyl methyl ether -- the combined organic layers
are
washed successively with water and brine, dried with sodium sulfate and
evaporated.
The residue is purified by flash chromatography (Si02 60F) to afford the title
compound as a yellow oil. Rf = 0.42 (dichloromethane-methanol-25% ammonia
conc.
200:20:1); Rt = 4.64 (gradient I).
c) (3S,4R,5R)-3-Azidomethyl -4-f4-((S)-3-methoxv-2-methyl-propoxymethyl)-
phenyll-
5-f4-(3-methoxv-propel)-3,4-dihvdro-2H-benzof 1,41oxazin-6-ylmethoxyl-
piperidine-1-carboxylic acid benzyl ester
31.265 mmol of sodium azide are added to a solution of 6.253 mmol of
(3R,4R,5S)-4-
[4-((S)-3-methoxy-2-methyl-propoxymethyl)-phenyl]-3-[4-(3-methoxy-propyl)-3,4-
d ihydro-2H-benzo[1, 4]oxazin-6-ylmethoxy]-5-(toluene-4-sulfonyloxymethyl)-
piperidine-1-carboxylic acid benzyl ester in 50 ml of 1,3-dimethyl-tetrahydro-
CA 02719070 2010-09-20
WO 2009/121914 PCT/EP2009/053898
pyrimidin-2-one (DMPU) under argon at 45 C. After 5 hours, the reaction
mixture is
cooled to room temperature, diluted with tert-butyl methyl ether, washed with
water
and brine, dried with sodium sulfate and evaporated. The crude title compound
is
obtained as a yellow oil. Rf = 0.58 (EtOAc-heptane 2:1); Rt = 5.96 (gradient
I).
d) (3R,4R,5S)-4-f4-((S)-3-Methoxv-2-methyl-propoxymethyl)-phenyll-3-f4-(3-
methoxy-propel)-3,4-dihvdro-2H-benzof 1,41oxazin-6-vlmethoxvl-5-(toluene-4-
sulfonyloxymethyl)-piperidine-1-carboxylic acid benzyl ester
7.077 mmol of 4-methyl-benzenesulfonyl chloride are added to a solution of
6.583
mmol of (3S,4R,5R)-3-hydroxymethyl-4-[4-((S)-3-methoxy-2-methyl-propoxymethyl)-
phenyl]-5-[4-(3-methoxy-propyl)-3,4-dihydro-2H-benzo[1,4]oxazin-6-ylmethoxy]-
piperidine-1-carboxylic acid benzyl ester (Example 1 a), 9.875 mmol of
triethylamine
and 0.329 mmol of dimethyl-pyridin-4-yl-amine in 65 ml of dichloromethane
under
argon at 0 C. After overnight stirring at room temperature, the reaction
mixture is
diluted with tert-butyl methyl ether, washed successively with saturated
aqueous
sodium bicarbonate solution, water and brine, dried with sodium sulfate and
evaporated. The crude title compound is obtained as a yellow oil. Rf = 0.44
(EtOAc-
heptane 2:1); Rt = 5.94 (gradient I).
According to the procedures described in example 2, the following compounds
are
prepared in an analogous manner:
3 N-{(3S,4R,5R)-4-f4-((S)-3-Methoxv-2-methyl-propoxymethyl)-phenyll-5-f4-(3-
methoxy-propel)-3,4-dihvdro-2H-benzof1,41oxazin-6-vlmethoxvl-piperidin-3-
ylmethyl}-propionam ide
using propionyl chloride instead of acetyl chloride in step a.
7 N-{(3S,4R,5R)-4-f4-((S)-3-Methoxv-2-methyl-propoxymethyl)-phenyll-5-f4-(3-
methoxy-propel)-3,4-dihvdro-2H-benzof1,41oxazin-6-vlmethoxvl-piperidin-3-
ylmethyl}-formamide
replacing step a with the following procedure:
CA 02719070 2010-09-20
WO 2009/121914 PCT/EP2009/053898
26
1.4 mmol of 4-nitrophenylformiate are added to a solution of 1 mmol of
(3R,4R,5R)-3-
aminomethyl -4-[4-((S)-3-methoxy-2-methyl -propoxymethyl)-phenyl]-5-[4-(3-
methoxy-
propyl)-3,4-dihydro-2H-benzo[1,4]oxazin-6-ylmethoxy]-piperidine-1-carboxylic
acid
benzyl ester (example 2b) in 10 ml of dichloromethane under argon followed by
the
addition of 1 mmol of triethylamine. After 60 minutes, the reaction mixture is
evaporated. The residue is purified by flash chromatography (Si02 60F) to
afford the
title compound, which is identified based on the Rf value.
Example 4
6-{(3R,4R,5S)-5-Methoxymethyl -4-[4-((S)-3-methoxv-2-methyl-propoxymethyl)-
phenyll-piperidin-3-yloxymethyl}-4-(3-methoxv-propel)-3,4-dihvdro-2H-
benzo[1,41oxazine
According to general procedure A, (3S,4R,5R)-3-methoxymethyl-4-[4-((S)-3-
methoxy-
2-methyl-propoxymethyl)-phenyl]-5-[4-(3-methoxy-propyl)-3,4-dihydro-2H-
benzo[1,4]oxazin-6-ylmethoxy]-piperidine-1-carboxylic acid benzyl ester is
used to
afford the title compound.
The starting material(s) is(are) prepared as follows:
a) (3S,4R,5R)-3-Methoxymethyl -4-[4-((S)-3-methoxv-2-methyl -propoxymethyl)-
phenyll-5-[4-(3-methoxv-propel)-3,4-dihvdro-2H-benzo[1,41oxazin-6-ylmethoxyl-
piperidine-1-carboxylic acid benzyl ester
2.568 mmol of methyl iodide are added to a suspension of 0.642 mmol of
(3S,4R,5R)-3-hydroxymethyl -4-[4-((S)-3-methoxy-2-methyl -propoxymethyl)-
phenyl]-
5-[4-(3-methoxy-propyl)-3,4-dihydro-2H-benzo[1,4]oxazin-6-ylmethoxy]-
piperidine-1-
carboxylic acid benzyl ester (Example 1 a) and 0.963 mmol of sodium hydride
(60%
dispersion in oil) under argon at 0 C. After stirring for 1 hour at 0 C and 1
hour at
room temperature, the reaction mixture is partitioned between tert-butyl
methyl ether
and saturated aqueous sodium bicarbonate solution. The aqueous layer is
extracted
with tert-butyl methyl ether (2X) -- the combined organic layers are washed
successively with water and brine, dried with sodium sulfate and evaporated.
The
residue is purified by flash chromatography (Si02 60F) to afford the title
compound as
a yellow oil. Rf = 0.41 (EtOAc-heptane 1:1); Rt = 5.87 (gradient I).
CA 02719070 2010-09-20
WO 2009/121914 PCT/EP2009/053898
27
According to the procedures described in example 4, the following compound is
prepared in an analogous manner:
6 6-{(3R,4R,5S)-5-(2-Methoxy-ethoxymethyl)-4-[4-((S)-3-methoxv-2-methyl-
propoxymethyl)-phenyll-piperid in-3-yloxymethyl}-4-(3-methoxv-propel)-3,4-
dihydro-2H-benzof 1,41oxazine
using 1 -bromo-2-methoxy-ethane (instead of methyl iodide) and 1 equivalent of
tetrabutylammonium iodide in step a.
Example 5
Acetic acid (3S,4R,5R)-4-f4-((S)-3-methoxv-2-methyl-propoxymethyl)-phenyll-5-
f4-(3-
methoxv-propel)-3,4-dihvdro-2H-benzof1,41oxazin-6-ylmethoxyl-piperidin-3-
ylmethyl
ester
According to general procedure A, (3S,4R,5R)-3-acetoxymethyl -4-[4-((S)-3-
methoxy-
2-methyl-propoxymethyl)-phenyl]-5-[4-(3-methoxy-propyl)-3,4-dihydro-2H-
benzo[1,4]oxazin-6-ylmethoxy]-piperidine-1-carboxylic acid benzyl ester is
used to
afford the title compound.
The starting material(s) is(are) prepared as follows:
a) (3S,4R,5R)-3-Acetoxymethyl -4-f4-((S)-3-methoxv-2-methyl -propoxymethyl)-
phenyll-5-f4-(3-methoxv-propel)-3,4-dihvdro-2H-benzof1,41oxazin-6-ylmethoxyl-
piperidine-1-carboxylic acid benzyl ester
Analogously to Example 2a, (3S,4R,5R)-3-hydroxymethyl -4-[4-((S)-3-methoxy-2-
methyl-propoxymethyl)-phenyl]-5-[4-(3-methoxy-propyl)-3,4-dihydro-2H-
benzo[1,4]oxazin-6-ylmethoxy]-piperidine-1-carboxylic acid benzyl ester
(Example
1 a) and acetyl chloride are used to afford the title compound as a colorless
oil.
Rf = 0.20 (EtOAc-heptane 1:1); Rt = 5.69 (gradient I).