Note: Descriptions are shown in the official language in which they were submitted.
CA 02720344 2010-09-30
WO 2009/129622
PCT/CA2009/000538
1
PRODUCTION OF HYDROGEN FROM OXYGENATED HYDROCARBONS
TECHNICAL FIELD
This invention relates to the production of hydrogen from oxygenated
hydrocarbons, preferably those derived from biomass. More particularly, the
invention relates to the production of hydrogen from oxygenated hydrocarbons
by
aqueous phase reforming procedures.
BACKGROUND ART
Hydrogen fuel cells have emerged as promising devices for clean and
efficient generation of power for global energy needs. Although hydrogen fuel
cells
have a low impact on the environment, current hydrogen production technologies
rely on high-temperature steam reforming of non-renewable hydrocarbon
feedstocks.
Greater environmental benefits of generating power from hydrogen fuel cells
could
be achieved if hydrogen could be produced from renewable resources, such as
biomass. However, current technologies for generating hydrogen from biomass,
such
as enzymatic decomposition of sugars, steam reforming of bio-oils, and
gasification
of biomass, all suffer from low hydrogen production rates and poor economics.
Recently, Dumesic et al. (References 1, 2, 3, 4, 5, 6, 7, 8, 9 and 10 ¨ see
References section at the end of this description) have reported that hydrogen
can be
produced at relatively low temperatures, e.g. around 277 C, over supported
metal
catalysts by a single step aqueous-phase reforming of biomass-derived
oxygenated
hydrocarbons, such as methanol, ethylene glycol, glycerol, sorbitol, xylose
and
glucose. In addition to utilizing renewable feedstocks, aqueous phase
reforming of
oxygenated hydrocarbons eliminates the need to vaporize water and the
oxygenated
hydrocarbon (which reduces the energy requirements and CO2 generation for
producing hydrogen). Generating 1 ton of hydrogen (11,120 m3 at STP) from
glycerol (derived from biomass) displaces 5.5 tons of CO2 from fossil origin
(when
employing steam methane reforming) (see References 11 and 12). In addition,
the
low reaction temperature and the absence of water vaporization eliminate 0.6
ton of
CO2 / ton H2 produced (References 11 and 12). The production of H2 and CO2 by
CA 02720344 2010-09-30
WO 2009/129622
PCT/CA2009/000538
2
aqueous phase reforming also leads to the production of low levels of CO
(<1000
ppm) in a single catalytic process.
Nevertheless, important selectivity challenges govern hydrogen production by
aqueous phase reforming because the mixture of H2 and CO2 formed in this
process
is thermodynamically unstable at low temperatures with respect to formation of
methane (Reference 1). Accordingly, the selective formation of hydrogen
represents
a classic problem in heterogeneous catalysis and reaction engineering: namely
the
identification of a catalyst and the design of equipment and conditions to
maximize
the yields of desired products at the expense of undesired byproducts formed
in series
and/or in parallel reaction pathways. Several types of catalysts, including
supported
metal and Sn-modified Raney Ni catalysts, have been tested for aqueous phase
reforming in order to identify the effect of different catalytically active
metals, metal
alloy components and supports on H2 selectivity (Reference 1). Among them, the
Pt/y-A1203 and Sn-modified Raney Ni catalysts were the most promising
(References
1, 2, 3, 5, 6, 7, 8, 9 and 10). The 3 wt % Pt/y-A1203 catalysts were found to
have the
best hydrogen selectivity, yield and production rate (References 11 and 12).
However, little work has been done on reactor configuration to maximize
hydrogen selectivity, yield and production rate. In the current aqueous phase
reforming development, only fixed-bed tubular reactors have been used for
activity
testing (References 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 and 12). Nevertheless,
aqueous
phase reforming involves multiple phases: reactants in the liquid phase,
catalysts in
the solid phase, and the desired hydrogen product in the gas phase. Therefore,
the
interphase and intraparticle mass transfer is one of the critical issues that
need to be
addressed for measuring and optimizing catalyst activity. To measure the
catalyst
intrinsic activity, very fine particles between 63-125 gm were tested in fixed
bed
tubular reactors by Durnesic et al. (References 3 and 6). For example, for the
kinetic
study of aqueous phase reforming of ethylene glycol over an alumina-supported
platinum catalyst, a maximum particle size of 130 gm was used to ensure that
the
intraparticle and interphase mass transfers were not limiting (Reference 6).
In theory, such fine particles are not recommended in fixed bed tubular
reactors because of the resulting high pressure drop along the catalyst bed in
reaction
conditions. Also it is easy for small particles to be washed out by the liquid
stream,
CA 02720344 2010-09-30
WO 2009/129622
PCT/CA2009/000538
3
which implies potential catalyst loss and undesirable secondary pollution to
the liquid
(Reference 13). Moreover, the use of fixed bed tubular reactors for liquid
solid phase
reaction has some other disadvantages, such as poor wetting of catalyst,
although co-
current upflow has been used to improve the wetting of catalyst (Reference
14).
Various patents and patent applications have been published in this field.
Most notable are the patents and patent applications of Cortright and Dumesic,
assigned to the University of Wisconsin-Madison, e.g. U.S. patent 6,699,457
issued
on March 2, 2004, U.S. patent 6,964,757 issued on November 15, 2005, and U.S.
patent 6,964,758 issued on November 15, 2005, as well as published
applications
2003/0220531 and 2005/0207971. These patents and applications describe the use
of
fixed bed tubular reactors (plug flow reactors) as exemplary systems. As noted
above, such reactors are not ideal for hydrogen generation involving aqueous
phase
reforming.
A different approach has been taken by Patrick Grimes et al. in Canadian
patent 787,831 of June 18, 1968 and Canadian patent application Serial No.
2,613,497 filed June 23, 2006. In these publications, an organic compound is
reacted
with water in a closed reactor in the presence of an electrolyte (preferably
an alkaline
compound such as KOH) and an electronically conductive catalyst. An electrical
potential is applied between electrodes (or via an electrode) and gaseous
hydrogen is
produced (with carbon dioxide reacting to form a carbonate that remains in
solution).
It was stated that the conducting electrode may be replaced by suspending
particles
of any electronically conducting material in the liquid reactants, but the
presence of
the electronically conductive material is absolutely necessary because,
without it, the
reaction will not proceed or will proceed so slowly that it could not be of
any
possible commercial interest even at elevated temperatures. Thus, these
procedures
adopt an electro-chemical approach and require the presence of an electrolyte
and a
suspended electronically conductive material.
There is a need for improved processes and equipment for the aqueous phase
reforming reaction used for the production of hydrogen from oxygenated
hydrocarbons, particularly those obtained from biomass.
CA 02720344 2013-03-15
4
DISCLOSURE OF THE INVENTION
According to one exemplary embodiment of the invention, there is provided a
process of producing hydrogen in which water is reacted with an oxygenated
hydrocarbon in the condensed phase in the presence of a metal catalyst
supported on
a carrier material dispersed in said condensed phase, characterized in that
the reaction
is carried out in a stirred tank reactor in the absence of an electrolyte at a
temperature
in the range of 230 to 285 C and at a pressure sufficiently high to maintain
said water
and said oxygenated hydrocarbon in a liquid phase at said temperature.
Liquid reactants (water and oxygenated hydrocarbon) are fed into the reactor
on a continuous or semi-batch basis. The vessel is normally closed so that it
may be
placed under moderately elevated pressure (e.g. 350 to 1350 psig, and more
preferably about 900 psig), and has a heater to raise and maintain the
temperature of
the reactants to within a low temperature range, e.g. a temperature of 230 to
285 C.
An even more preferred temperature range is 255 to 285 C.
The use of electrolytes in the reaction liquids is avoided because such
materials may react with the intended products of the process, e.g. carbon
dioxide to
form carbonates. The reaction also proceeds adequately at initial pH values
that are
substantially neutral (e.g. pH 7 + 25%) when the oxygenated hydrocarbon
starting
material is initially dissolved in water, so strong acids and bases are
avoided. During
the reaction, the liquid phase may tend to acidify due to the presence of acid
byproducts or intermediates.
Stirred tank reactors (particularly continuous stirred tank reactors) have
high
heat capacity, which makes for good temperature stability and good heat
transfer.
The catalyst and reactants have good contact since the system is well mixed
(preferably homogeneously mixed) by the stirrer. Also, the catalyst particles
may be
made very small, with a minimum size limited by filtering, so that the largest
possible solid/liquid interface is obtained, particularly when the catalyst
support is
porous and the metal catalyst is deposited within the pores.
The reaction may be carried out with various oxygenated hydrocarbons used
as starting materials, e.g. as methanol, ethylene glycol, glycerol, sorbitol,
xylose and
CA 02720344 2010-09-30
WO 2009/129622 PCT/CA2009/000538
glucose or other sugars, provided they are capable of undergoing an aqueous
phase
reformation resulting in the production of hydrogen. Glycerol is particularly
preferred. It is also preferred from economic and environmental reasons that
the
oxygenated hydrocarbon material be obtained from biomass, and especially waste
5 biomass, and the production of such oxygenated hydrocarbons from biomass may
if
desired be considered a preliminary step of the process of the invention.
Catalytic aqueous phase reforming has been found attractive because of
significantly lower energy requirements due to low reaction temperatures
compared
with conventional steam methane reforming and the elimination of the need for
reactant vaporization. It has also been found that the use of a stirred tank
reactor
(especially a continuous stirred tank reactor) gives a higher hydrogen
selectivity and
a better rate of conversion of the starting material than conventional fixed
bed tubular
(plug flow) reactors. Rapid separation of the gaseous products (hydrogen and
carbon
dioxide) from the condensed phase helps reduce reverse reaction or side
reactions
caused by the build up of reaction products.
It is most preferred for the reaction to use platinum and nickel catalysts
supported on alumina, silica, activated carbon, zeolite or the like, having
particle
sizes in a range of 150-850 gm, although other supported metals such as
palladium,
ruthenium, rhodium and iridium may be employed. For the conversion of
glycerol, a
temperature of about 265 C and a pressure of about 900 psig was found to be
effective. Under these conditions, an undesirable formation of alkanes was
impeded.
DEFINITIONS
The term "stirred tank reactor" means an agitated reactor vessel (preferably
homogeneously stirred) using a mechanically-operating device such as an
impeller,
propeller, pump, magnetic stirrer, or the like, where the liquid reactants are
converted
to very high conversion levels in presence of catalyst in suspension.
The term "continuous stirred tank reactor" (CSTR) refers to any reactor
vessel having contents that may be stirred and wherein material is
continuously or
continually added to the reactor and material is removed from the reactor at
about the
same rate as material is added thereto. The contents of the reactor are
maintained by
stirring in a generally homogeneous condition both from the composition and
the
CA 02720344 2013-03-15
6
temperature points of view. Gaseous products are continuously removed from the
reactor as they are produced, optionally with the assistance of an unreactive
gas
introduced into the reactor and used to sweep the product gases from the
headspace
of the reactor. The solid catalyst in particulate form is normally introduced
into the
reactor before the reaction is started, but may be introduced in the stream of
liquid
starting materials. Solid catalyst escaping in the liquid stream removed from
the
reactor (if any) may be collected by means of a decanter and/or filter and may
be
recycled back to the reactor. The vessel is normally closed so that it may be
placed
under elevated pressure, and may have a heater of some kind to raise and
maintain
the temperature of the reactants to within the reaction range.
The term "semi-batch stirred tank reactor" refers to any reactor vessel where
only gases are continuously fed and removed from it, but not liquids or solid
catalyst
particles. The semi-batch reactor can test the effect on product selectivity
and yield,
of separating gaseous products (H2, CO2, etc.) from the catalyst particles and
the
liquid phase as it is achieved in a CSTR.
The term "low temperature" as used herein means a temperature that is
elevated above ambient temperature but low in comparison with temperatures
currently required for the commercial production of hydrogen (typically 850 C
or
higher). Temperatures in the range of 230 to 285 C are often suitable.
The term "electrolyte" as used herein refers to acids, bases and salts that
are
more or less dissociated in aqueous solution and whose ions act, to some
extent, as
free, independent entities or particles. The prseence of such electrolytes is
avoided in
the process of the present invention.
The term "heterogeneous catalytic reaction mechanism" means a process of
achieving or accelerating a chemical reaction by means of a solid catalyst
having
active sites at the surface where reactants are adsorbed and converted to
adsorbed
products which then desorb. Such a process requires neither the application of
electrical potential, nor the transfer of electrical species.
CA 02720344 2013-03-15
7
BRIEF DESCRIPTION OF THE DRAWINGS
Exemplary embodiments of the invention are described in greater detail in the
following in which reference is made to the accompanying drawings. In the
drawings:
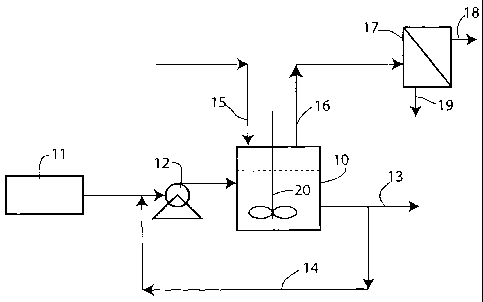
Fig. 1 is a flow diagram of a preferred process and apparatus for carrying out
exemplary embodiments of the present invention; and
Figs. 2a to 2d and Figs. 3a to 3d are graphs showing the results of
experiments carried out in the following Examples.
BEST MODES FOR CARRYING OUT THE INVENTION
Exemplary embodiments of the invention involve the production of hydrogen
from any oxygenated at least partially water-soluble hydrocarbon starting
material
(e.g. glycerol, methanol, ethylene glycol, xylose, sorbitol and sugars) by
aqueous
phase reforming. In particular, C5 and C6 sugars, which can be derived from
cellulose and hemicellulose, and glycerol (glycerin), which is a byproduct of
biodiesel production (the transesterification of triglycerides), are
especially preferred
as starting materials as they are inexpensive and abundant. The production of
hydrogen in this way involves reacting the oxygenated hydrocarbon with water
at
low temperatures and moderately elevated pressures using a suitable supported
particulate catalyst dispersed in the aqueous reaction mixture. In exemplary
embodiments, the reaction is carried out in the condensed phase (i.e. the
water and
oxygenated hydrocarbons are kept in the liquid phase) and the pressure is
adjusted to
ensure that this is the case. Higher pressures are generally not required but
are not
harmful. This means that if the temperature is increased, the pressure is
increased
accordingly to maintain the condensed phase. The actual temperatures employed
may be relatively low (e.g. 230 to 285 C, and more preferably 255 to 285 C)
and the
pressures are adjusted accordingly and are generally in the range of 350 to
1350 psig,
and more preferably about 900 psig (e.g. 900 + 175 psig). Of course, as noted
above,
the temperature and pressure combinations should be selected to ensure that
the
water and oxygenated hydrocarbons are maintained in the liquid phase.
The reaction is carried out in a stirred tank reactor (i.e. a semi-batch, or
more
preferably a continuous, stirred tank reactor) employing a suitable metal
catalyst
CA 02720344 2010-09-30
WO 2009/129622
PCT/CA2009/000538
8
supported on a solid support (e.g. porous alumina) to form solid catalyst
particles.
The solid support used for dispersing the catalytic sites may be any
(generally inert,
high surface-area and normally porous) solid material commonly employed as a
catalyst support, for example alumina, silica-alumina, silica, titania and
various
forms of carbon. The metallic sites may be supported on the solid support
using
conventional techniques to obtain a catalyst having well-dispersed active
metal sites
distributed over the catalyst surface in order to increase conversion and
hydrogen
selectivity. Suitable catalytic metals may be chosen from (for example)
platinum,
nickel, palladium, ruthenium, rhodium, iridium and tin-promoted nickel.
Platinum
and nickel are particularly preferred.
The catalyst particles are preferably made as small as possible without
causing undue problems of catalyst loss and recovery. Smaller particles show
better
selection for hydrogen production and conversion of the starting material. A
particularly preferred particle size range is 150-300 microns (especially 150-
250
microns, or 250-300 microns), although particle sizes up to 850 microns or
more still
show good conversion and selectivity, and other preferred particle size ranges
are
300-590 microns and 590 to 850 microns.
The stirring required in the continuous stirred tank reactor may be carried
out
in any suitable way and should be sufficient to keep the reaction mixture
substantially homogenous and equal in temperature throughout the reactor. The
amount of catalyst employed is generally the minimum amount required for the
desired reaction and an amount that can be blended homogenously within the
contents of the reactor.
The ratio of oxygenated hydrocarbon material to water used for the starting
material is preferably one which permits the oxygenated hydrocarbon to
dissolve in
the water and preferably does not exceed 35 wt.%. In the case of glycerol as
the
oxygenated hydrocarbon, the ratio may be up to 35 wt.%, and is preferably in
the
range of 10 to 30 wt.%. In general, ratios of oxygenated hydrocarbon to water
of 1
to 10 wt.% are normally suitable for any of the starting materials.
The reactants may be kept within the reactor for an average time effective to
drive the reaction to completion. For example, weight hourly space velocities
(the
inverse of residence times) preferably range from 1 to 1011-1.
CA 02720344 2010-09-30
WO 2009/129622
PCT/CA2009/000538
9
The reaction results in the production of hydrogen and carbon dioxide. An
inert gas (preferably nitrogen) may be used to sweep the gaseous reaction
products
from the reactor, although this is by no means essential. The hydrogen can be
separated from the carbon dioxide (and inert gas, if present) by any known
means,
e.g. membrane separation.
Fig. 1 is a flow diagram showing an example of the apparatus and process of
the exemplary embodiments. The reaction is carried out in a continuous stirred
tank
reactor 10 acting as a pressure vessel or autoclave. A blend of oxygenated
hydrocarbon in water 11 is fed continuously into the reactor at a suitable
rate by a
pump 12. Waste water containing organic byproducts is removed via conduit 13
and
part of the waste may be recycled to the inlet feed via conduit 14. Nitrogen
gas is
optionally introduced into the reactor via conduit 15 and the product gas
(containing
mostly hydrogen and carbon dioxide, some light hydrocarbons and traces of CO,
and
optionally nitrogen) is removed via conduit 16 and subjected to gas separation
in
membrane separator 17, resulting in a hydrogen-rich stream withdrawn via
conduit 18 and a CO2-rich stream withdrawn via conduit 19. The reactor 10
contains
a stirrer 20, e.g. an impeller, to keep the contents of the reactor in a
suitable state of
homogeneity. Not shown are heating means provided to raise the temperature of
the
contents of the reactor to the desired reaction temperature, but these may be
conventional.
EXAMPLES
To illustrate the effectiveness of the process, glycerol was used as an
exemplary oxygenated hydrocarbon starting material and tests were carried out
as
follows with a view to maximizing the hydrogen production rate and yield, and
to
compare the process with those using a fixed bed tubular reactor (operated at
in a
temperature range of 255 to 285 C, a pressure range of 770 to 1120 psi, a WHSV
range of 0.1 to 1.1 111, and feedstocks of 10, 20 and 30 wt.% glycerol). It
should be
kept in mind that the following details are merely exemplary and other
starting
materials and conditions may be employed successfully.
CA 02720344 2010-09-30
WO 2009/129622
PCT/CA2009/000538
REACTIONS
According to Davda et al. (1), aqueous phase reforming of glycerol takes
place according to the following stoichiometric reactions:
C3H803 <=> 3C0 + 4H2 [1]
5 Water-gas shift reaction: CO + H20 <=> CO2 + H2 [2]
Overall reaction: C3H803+ 3H20 .(=> 3CO2 + 7H2 [3]
Side methanation reaction: CO + 3H2 <=> CH4+ H20 [4]
Catalyst activity and selectivity are defined as:
Glycerol conversion to gaseous products (%) = [(total moles of C in gas
10 products) / (total moles of C in the feed solution)] x 100 [5]
H2 selectivity (%) = [(moles of H2 produced /moles of C in gas phase) x
(7/3)] x 100 [6]
Allcane selectivity (%) = [(moles of C in gaseous alkanes/total moles of C in
gas products)] x 100 [7]
Yield of H2 (L/100 g glycerol) = [(volume of H2 produced) / (initial weight of
glycerol)] x 100 g glycerol [8]
Hydrogen production rate (L H2/100 g cat/h) = {[(volume of H2
produced)/hour] / weight of catalyst} x 100 g catalyst [9]
To compare with the fixed bed tubular reactor, the integral activity and
selectivity of the stirred tank reactor needs to be calculated because the
fixed bed
tubular reactor is an integrated reactor and it is impossible to measure gas
and liquid
concentrations along the catalyst bed (15). Therefore, only integral reaction
rate and
selectivity obtained in the stirred tank reactor can be used to compare with
fixed bed
tubular reactor (t is time-on-stream).
t
Integral H2 production rate = SH2 production rate(t)dt I t
[10]
0
t
Integral H2 selectivity = SI-12
selectivity(t)dt I t [11]
0
t
Integral Alkane selectivity = fHC selectivity(t)dt I t [12]
0
CA 02720344 2010-09-30
WO 2009/129622 PC
T/CA2009/000538
11
i
Integral H2 yield = j1-12 yield(t)dt I t
[13]
0
According to Cao and Fung (16), weight hourly space velocity (WHSV)
parameters are defined in Equations 14 and 15 for both fixed bed tubular
reactor and
stirred tank reactor. Catalyst activities in both reactors can be compared by
plotting
conversion as a function of WHSV. At the same conversion level, the difference
in
the WHSV values indicates different amounts of glycerol can be converted with
the
same weight of catalyst and the same contact time by different reactors.
WHSV of the fixed bed tubular reactor (based on C3H803) = [feed rate of
glycerol (weight/hour)] / (weight of catalyst) [14]
WHSV of the stirred tank reactor (based on C3H803) = (initial weight of
glycerol in the reactor) / (time on stream) / (weight of catalyst) [15]
Residence time (fixed bed tubular reactor and semi-batch stirred tank reactor)
=1/WHSV [16]
CATALYST PREPARATION
The inventors prepared 3 wt % Pt/y-A1203 and 40 wt % Ni/y-A1203 catalysts
for testing in a semi-batch stirred tank reactor. The advantages of the use of
a stirred
tank reactor compared to a fixed bed reactor were demonstrated by comparing
the
performance of the two reactors in terms of hydrogen production rate, hydrogen
selectivity, alkane selectivity and hydrogen yield.
High surface area 7-alumina (220 m2/g, pore volume 0.62 mL/g, medium pore
diameter 7 nm, Alfa Aesar ) was used as catalyst support. The 7-alumina
support
was crushed and sieved to different mesh sizes in order to investigate the
effect of
catalyst particle size. Preliminary tests indicated that a broad mesh size
significantly
affected the experimental results. Table 1 summarizes different catalyst
particle
sizes.
CA 02720344 2010-09-30
WO 2009/129622
PCT/CA2009/000538
12
Table 1
Summary of catalysts having different particle sizes
Catalyst Mesh Particle
series size diameter (p,m)
Series I 100-60 150-250
Series II 60-50 250-300
Series III 30-20 590-850
Gamma-alumina supported catalysts were prepared by incipient wetness
impregnation (1.0 mL solution per gram of support) with the desired aqueous
solution. For ?-alumina supported 3 wt % Pt catalysts, tetra-amine platinum
nitrate
(Pt (NH4)4(NO3)2, Alfa Aesare) was used. After impregnation, samples were
dried
at 373 K (100 C) overnight. Samples were subsequently heated to 673 K (400 C)
at
1 K/min in a mixture of 10% 02/N2 and held at 673 K (400 C) for 2 hours.
For y-alumina supported 40 wt % nickel catalysts, a two-step impregnation of
nickel nitrate hexahydrate (Ni(NO3)2.6H20, Fisher Chemical), was performed due
to
the high loading of Ni. The above drying and calcination procedures were
performed
between impregnation steps.
The catalysts were reduced in H2 (GHSV = 733 If') in a tubular reactor prior
to loading in the semi-batch stirred tank reactor. The catalyst bed was heated
to
563 K (290 C) for Pt catalyst or 723 K (450 C) for Ni catalyst at 1 K/min and
held
for 2 hours. After reduction, the catalyst bed was cooled to room temperature
while
purging with H2. Then, the reduced catalyst was transferred to the stirred
tank reactor
using a glycerol solution to protect the active sites from air oxidation.
CATALYST TESTING
The stirred tank reactor used by the inventors for the aqueous phase
reforming of glycerol was a laboratory scale apparatus and consisted of a 300
mL
autoclave (Parr series 4560 Bench Top Mini reactor). After loading the desired
amount of glycerol aqueous solution (approximately 100 mL of a 10 wt %
glycerol
aqueous solution) and a catalyst (about 1 to 4 g) in the reactor, the reactor
was purged
with N2 then pressurized to the desired reaction pressure with N2. Operating
pressures and temperatures were 900 psig and 538 K (265 C) or 548 K (275 C),
CA 02720344 2010-09-30
WO 2009/129622
PCT/CA2009/000538
13
respectively. The stirring rate was set at 500 rpm to disperse the catalyst
into the
liquid phase, maximize mass transfer and ensure uniform temperature and
concentration.
The product gas stream was carried by a nitrogen purge gas through a cold
trap and was analyzed with an Hewlett-Packard 5890 Series II chromatograph
acting
as an on-line gas chromatograph. This gas chromatograph was configured with
three
detectors, i.e. two thermal conductivity detectors (TCD) and one FrD. Hydrogen
was analyzed on one thermal conductivity detector with N2 as carrier gas. CO2,
C2
hydrocarbons, N2, 02, CH4 and CO were analyzed on the other thermal
conductivity
detector with He as a carrier gas. Also, C1 to C6 hydrocarbons were analyzed
on the
FID. Effluent gases were analyzed every 15 minutes. A vertical condenser was
provided to avoid the passage of glycerol aqueous solution out of reactor by
entrainment in the purge gas. The reactor gas outlet inner diameter was
enlarged to
ensure any entrained liquid flowed back to the reactor. The N2 purge flow was
high
enough to ensure the line and sampling loop had the same gas concentration as
the
reactor gas flow.
EXAMPLE 1
Performance of Pt Catalysts
To compare two types of catalytic reactors, the performance of glycerol
aqueous phase reforming on 3 wt % Pt/y-A1203 catalysts in both the semi-batch
stirred tank reactor and a fixed bed tubular reactor is plotted in Figs 2a to
2d.
Fig. 2a is a graph showing conversion versus WHSV using Pt catalysts (10
wt% glycerol aqueous solution as reactant at 538 K (265 C) and 900 psig); the
filled
and empty symbols stand for two repeat runs using 3 wt % Pt/y-A1203 catalysts;
symbols 0 and = show the results for 150-250 gm particles; symbols 0 and =
show
the results for 250-300 gm particles; symbols A and A show the results for 590-
850
gm particles; symbol + shows the results for 3 wt % Pt/y-A1203 catalyst (300-
850
gm) tested in fixed bed tubular reactor (FBTR); and symbol ¨ shows the results
for 1
wt % Pt/y-A1203 catalyst (300-850 gm) tested in the FBTR.
CA 02720344 2010-09-30
WO 2009/129622
PCT/CA2009/000538
14
Fig. 2b is a graph showing integral hydrogen selectivity versus glycerol
conversion using Pt catalysts (the symbols and conditions being the same as in
Fig. 2a).
Fig. 2c is a graph showing alkane selectivity versus glycerol conversion using
Pt catalysts (10 wt % glycerol aqueous solution as reactant, 538K (265 C), 900
psig).
The filled and empty symbols stand for two repeat runs using 3 wt % Pt/y-A1203
catalysts. Symbols 0 and = show the results for 150-250 pm particles; symbols
o
and = show the results for 250-300 pm particles; symbols A and = show the
results
for 590-850 pm particles; symbol + shows the results for 3 wt % Pt/y-A1203
catalyst
(300-850 pm) tested in FBTR; and symbol ¨ shows the results for 1 wt % Pt/7-
A1203
catalyst (300-850 p.m) tested in the fixed bed tubular reactor.
Fig. 2d is a graph showing the yield of hydrogen versus glycerol conversion
using Pt catalysts (the symbols and conditions being the same as in Fig. 2c).
To demonstrate the effect of catalyst particle size on stirred tank reactor
rate,
the performance of catalysts supported on different sizes of y-A1203 particles
is also
plotted in Figs. 2a to 2d.
Figure 2a shows that in the semi-batch stirred tank reactor glycerol
conversion was strongly dependent on catalyst particle size. At the same WHSV
(calculated with Equation 15), higher conversions, thus higher activity, were
obtained
for catalysts having smaller particle size. Moreover, the average conversions
obtained for catalysts having 250-350 pm particle size are very close to those
obtained for catalysts having 150-250 ptm particle size and are higher than
those
obtained for larger catalyst having 590-850 pm particle size.
Furthermore, the inventors found that the catalyst was significantly more
active in the stirred tank reactor than in the fixed bed tubular reactor. This
is
illustrated in Figure 2a where the conversion level dropped much faster in the
fixed
bed tubular reactor than in the semi-batch stirred tank reactor. A fixed bed
tubular
reactor operating at the same high WHSV value as the semi-batch stirred tank
reactor
would convert very little glycerol and produce very little H2.
Table 2 below summarizes the integral H2 production rates for the semi-batch
stirred tank reactor and the fixed bed tubular reactor using 3 wt % Pt
catalysts.
Table 2 shows that at the same conversion, the H2 production rates for
catalysts
CA 02720344 2010-09-30
WO 2009/129622 PCT/CA2009/000538
having the largest particle size (590-850 pm, Series III) obtained in the semi-
batch
stirred tank reactor were one order of magnitude higher than those for the
fixed bed
tubular reactor. For example, for 3 wt % Pt/y-A1203catalysts having 590-850 pm
particle size (Series III) and glycerol conversion of 67%, the 112 production
rate was
5 180 L/100 g cat/h in the semi-batch stirred tank reactor while the 112
production rate
was 15 L/100 g cat/h in the fixed bed tubular reactor. At 54% conversion, the
H2
production rate was 208 L/100 g cat/h in the semi-batch stirred tank reactor
while the
H2 production rate was 36 L/100 g cat/h in the fixed bed tubular reactor.
The WHSV parameter indicates that with the same weight of catalyst and the
10 same contact time, stirred tank reactor can convert 10 times more glycerol
than fixed
bed tubular reactor with large catalyst particles (590-850 jtm, Series III)
and 18.5
times more glycerol with small catalyst particles (150-250 Jim, Series I). As
shown
in Table 2, WHSV and 112 production rates for catalysts having 150-250 pm
particle
size (Series I) for the stirred tank reactor were more than one order of
magnitude
15 higher than those for the fixed bed tubular reactor.
Table 2
Integral 142 production rates obtained for fixed bed tubular reactor
and semi-batch stirred tank reactor using 3 wt % Pt catalysts
_________________________________________________________________________
Conversion, % WHSV, h1 Integral H2 production
rate,
L/100 g cat/h
fixed bed semi- semi- fixed bed semi-
semi-
tubular batch batch tubular batch
batch
reactor stirred stirred reactor stirred
stirred
tank tank tank
tank
reactor reactor reactor reactor
(590-850 (150-250 (590-850 (150-250
p,m) pm) inn) 1-
un)
67 0.27 2.8 5 15 180 287
54 0.5 4 7 36 208 296
Figure 2b and 2c show that H2 and alkane selectivities changed with glycerol
conversion and were strongly dependent on catalyst particle size. Hydrogen
selectivity increased with the decrease in catalyst particle size, while
alkane
selectivity decreased. For example, at 60% glycerol conversion, H2 selectivity
for
the semi-batch stirred tank reactor was 80%, 75% and 65% using catalysts
having
CA 02720344 2012-06-15
16
150-250 lam (Series I), 250-300 p.m (Series II) and 590-850 1.tm (Series III)
particle
size respectively, and alkane selectivity was 15%, 15% and 20% respectively.
The H2 and alkane selectivities were less dependent on glycerol conversion
with the semi-batch stirred tank reactor compared with fixed bed tubular
reactor. In
other words, H2 and alkane selectivities obtained using the semi-batch stirred
tank
reactor did not change significantly with conversion compared with the fixed
bed
tubular reactor. Moreover, at high conversions, H2 selectivity was higher for
the
semi-batch stirred tank reactor and alkane selectivity was lower. For example,
at 80%
conversion, H2 selectivity was 55% for the fixed bed tubular reactor while for
the
semi-batch stirred tank reactor, it reached 76% with the smallest particle
size (150-
250 pm). At 80% conversion, the alkane selectivity was significantly lower for
the
semi-batch stirred tank reactor compared with fixed bed tubular reactor: 23%
vs
33%.
Figure 2d shows H2 yield as a function of glycerol conversion. At low
conversions (< 60%), H2 yields were similar for both fixed bed tubular reactor
and
semi-batch stirred tank reactor, while at high conversions, H2 yield for the
semi-batch
stirred tank reactor was higher than that for the fixed bed tubular reactor.
For
example, at 60% conversion, H2 yield was 90 L/100 g glycerol for the semi-
batch
stirred tank reactor, about 10% higher than the fixed bed tubular reactor (82
L/100 g
glycerol). At 80% conversion, H2 yield was 105 L/100 g glycerol for the
stirred tank
reactor, i.e., at least 40% higher than for fixed bed tubular reactor (75
L/100 g
glycerol). The increased H2 yield at the same conversion was due to increased
H2
selectivity using the stirred tank reactor. As discussed later, catalyst
wetting is better
in the stirred tank reactor and less H2, CO and CO2 re-adsorb on the catalyst
for
methanation reaction. Figure 2d also shows that, at the same conversion, H2
yield
was higher on the small size catalyst than that obtained for the large size
catalyst
because the H2 selectivity on the small particle catalyst was higher than that
obtained
on the large size catalyst particle. This is due to better mass transfer in
the semi-
batch stirred tank reactor.
EXAMPLE 2
Performance of Nickel Catalysts
CA 02720344 2010-09-30
WO 2009/129622
PCT/CA2009/000538
17
Catalysts 20 wt % and 40 wt % Ni/?-A1203 were also tested using the semi-
batch stirred tank reactor. Based on results obtained for 3 wt % Pt catalysts
indicating that the performances of catalysts having small particle size were
better,
y-A1203 support between 150-250 gm size were used to prepare 20 wt % and 40 wt
% Ni/?-A1203 catalysts. Experimental results are plotted in Figures 3a to 3d
which
are described as follows.
Fig. 3a is a graph showing conversion versus WHSV using Ni catalysts (10
wt % glycerol aqueous solution as reactant at 548 K (275 C) and 900 psig). The
filled and empty symbols stand for two repeat runs. Symbols A and A show
results
for 40 wt % Ni catalysts (150-250 gm); symbols o and = show the results for 20
wt
% Ni catalyst (150-250 gm); symbol o shows the results for 20 wt % Ni/y-A1203
catalyst (300-850 gm) tested in fixed bed tubular reactor (FBTR); and symbol *
shows the results for 40 wt % Ni/y-A1203 catalyst (300-850 gm) tested in FBTR;
Fig. 3b is a graph showing integral hydrogen selectivity versus conversion of
glycerol using Ni catalysts (the symbols and conditions are the same as for
Fig. 3a).
Fig. 3c is a graph showing integral alkane selectivity versus glycerol
conversion using Ni catalysts (10 wt % glycerol aqueous solution as reactant
at
548 K (275 C) and 900 psig. Filled and empty symbols stand for two repeat
runs.
The symbols A and = show results for 40 wt % Ni catalysts (150-250 gm);
symbols
o and = show the results for 20 wt % Ni catalyst (150-250 gm); symbol o shows
the
results for 20 wt % Ni/y-A1203 catalyst (300-850 gm) tested in FBTR; and
symbol *
shows the results for 40 wt % Ni/y-A1203 catalyst (300-850 gm) tested in FBTR.
Fig. 3d is a graph showing integral hydrogen yield versus the conversion of
glycerol to gas using Ni catalysts (the symbols and conditions are the same as
in Fig.
3c).
Like 3 wt % Pt catalysts, the inventors found that Ni catalysts were
significantly more active for the semi-batch stirred tank reactor than for the
fixed bed
tubular reactor. This is illustrated in Figure 3a where the conversion level
dropped
much faster for fixed bed tubular reactor than for semi-batch stirred tank
reactor and
a fixed bed tubular reactor operating at the same high WHSV value as the semi-
batch
stirred tank reactor would convert very little glycerol and produce very
little H2.
CA 02720344 2010-09-30
WO 2009/129622
PCT/CA2009/000538
18
Table 3 below summarizes the integral H2 production rates obtained for the
semi-batch stirred tank reactor and the fixed bed tubular reactor using 40wt %
Ni
catalysts. Table 3 shows that at the same conversion, H2 production rates for
small
particle size catalysts (125-2501m, Series I) obtained for the semi-batch
stirred tank
reactor were more than one order of magnitude higher than those for the fixed
bed
tubular reactor. For example, using 40 wt % Ni/y-A1203catalyst having 150-250
um
particle size (Series I) and at a glycerol conversion of 96%, H2 production
rate was
60 L/100 g cat/h for the semi-batch stirred tank reactor while H2 production
rate was
2.6 L/100 g cat/h for the fixed bed tubular reactor. At 43.4% conversion, H2
production rate was 165 L/100 g cat/h for the semi-batch stirred tank reactor
while
H2 production rate was 6.5 L/100 g cat/h for the fixed bed tubular reactor.
The
WHSV parameter indicates that for the same weight of catalyst and the same
contact
time, semi-batch stirred tank reactor can convert at least 20 times more
glycerol than
fixed bed tubular reactor using small catalyst particles (150-250 gm, Series
I).
Table 3
Integral H2 production rates fixed bed tubular reactor
and semi-batch stirred tank reactor using 40 wt % Ni catalysts
Conversion, % WHSV, Integral H2 production rate,
L/100 g cat/h
Fixed bed semi-batch fixed bed semi-batch
tubular reactor stirred tank tubular reactor stirred
tank
reactor reactor
(150-250 gm) (150-250
gm)
96 0.10 2.0 2.6 60
43.4 0.19 13 6.5 165
Figure 3b and 3c show selectivities of H2 and of alkanes as a function of
glycerol conversion. Figure 3b shows that H2 selectivity was higher than that
obtained for the fixed bed tubular reactor at high conversions, and that
alkane
selectivity was lower. For example, at 80% conversion, H2 selectivity was 35%
for
the semi-batch stirred tank reactor and only 18% for fixed bed tubular reactor
while
the alkane selectivity was at 40% for the semi-batch stirred tank reactor
while it
reached 50% for fixed bed tubular reactor (40 wt % Ni catalysts).
CA 02720344 2010-09-30
WO 2009/129622
PCT/CA2009/000538
19
Figure 3d shows H2 yield as a function of glycerol conversion. Figure 3d
shows that, like 3 wt % Pt catalysts, H2 yields were similar for both fixed
bed tubular
reactor and semi-batch stirred tank reactor at low conversion. At 40%
conversion, H2
yield was 25 L/100 g glycerol for the semi-batch stirred tank reactor while
for the
fixed bed tubular reactor the H2 yield was 18 L/100 g glycerol. At high
conversion,
H2 yield for the semi-batch stirred tank reactor was higher than that for the
fixed bed
tubular reactor. At 60% conversion, H2 yield was 40 L/100 g glycerol for the
semi-
batch stirred tank reactor while for the fixed bed tubular reactor H2 yield
was 20
L/100 g glycerol. At 80% conversion, the difference was more significant: 50
L/100
g glycerol vs 23 L/100 g glycerol. The increased H2 yield at high conversions
was
due to an increase in H2 selectivity for the semi-batch stirred tank reactor:
better
mass transfer and lower probability of side reaction.
Figure 2a and Fig. 3a show that, for both Ni and Pt catalysts (150-250
)..trn),
H2 production rates for the semi-batch stirred tank reactor were more than one
order
of magnitude higher than those for the fixed bed tubular reactor. Figure 2a
also
shows that, even for the Pt catalyst having large particle size 590-850 tim,
H2
production rates from the semi-batch stirred tank reactor were one order of
magnitude higher than those for the fixed bed tubular reactor.
However, Figures 2a and 3a show that, at high conversion, H2 selectivities
obtained for both Ni and Pt catalyst from the semi-batch stirred tank reactor
were
higher than those obtained for the fixed bed tubular reactor without modifying
the
catalyst active site. Hence the results show that the reactor configuration
can affect
H2 selectivity.
Table 4 below compares the maximum yields over 3 wt % Pt/y-A1203 for both
reactors. A maximum H2 yield of 108 L/100 g glycerol was obtained in the semi-
batch stirred tank reactor at 91.0% glycerol conversion. This is 32.0% higher
than
the maximum H2 yield obtained in a fixed bed tubular reactor using the same
catalyst
(81.6 L/100 g glycerol). The higher maximum H2 yield achieved in the semi-
batch
stirred tank reactor was due to higher glycerol conversion, higher H2
selectivity and
lower alkane selectivity. At 91.5% glycerol conversion, H2 selectivity in the
semi-
batch stirred tank reactor hardly decreased. It was still very close to 69.7%
obtained
in the fixed bed tubular reactor at 68.8% conversion. At the same operating
CA 02720344 2010-09-30
WO 2009/129622
PCT/CA2009/000538
conditions for maximum yield in the semi-batch stirred tank reactor, the H2
production rate was 162 L/100 g cat/h. This rate is 9.6 times higher than 15.3
L/100
g cat/h in the fixed bed tubular reactor.
5 Table 4
Comparison of 3 wt % Pt/y-A1203 performances
in the semi-batch stirred tank reactor and the fixed bed tubular reactor
fixed bed tubular semi-batch stirred
reactor tank reactor
WHSV, (based on C314803) 0.2 1.8 =
H2 yield, L/100 g C3H803 81.6 107.7
H2 production rate, L/100 g cat/h 15.3 162.0
Glycerol conversion, % (to 68.8 91.5
gaseous products)
H2 selectivity, % 69.7 69.0
Alkane selectivity, % 18.4 24.7
CA 02720344 2010-09-30
WO 2009/129622
PCT/CA2009/000538
21
REFERENCES
1. Davda, R.R., Shabaker, J.W., Huber, G.W., Cortright, R.D. and Dumesic, J.A.
"A
review of catalytic issues and process conditions for renewable hydrogen and
alkanes by aqueous-phase reforming of oxygenated hydrocarbons over supported
metal catalysts" Applied Catalysis B: Environmental 56:1-2:171:186, 2005.
2. Huber, G.W., and Dumesic, J.A. "An overview of aqueous-phase catalytic
processes for production of hydrogen and alkanes in a biorefinery" Catalysis
Today 111:1-2:119:132, 2006.
3. Cortright, R.D., Davda, R.R.and Dumesic, J.A. "Hydrogen from catalytic
reforming of biomass-derived hydrocarbons in liquid water" Nature 418:6901:
964:967, 2002.
4. Davda, R.R., and Dumesic, J.A. "Catalytic reforming of oxygenated
hydrocarbons for hydrogen with low levels of carbon monoxide" Angewandte
Chemie - International Edition 42:34:4068:4071, 2003.
5. Huber, G.W., Shabaker, J.W., Evans, S.T. and Dumesic, J.A. "Aqueous-phase
reforming of ethylene glycol over supported Pt and Pd bimetallic catalysts"
Applied Catalysis B: Environmental 62:3-4:226:235, 2006.
6. Shabaker, J.W., Davda, R.R., Huber, G.W., Cortright, R.D. and Dumesic J.A.
"Aqueous-phase reforming of methanol and ethylene glycol over alumina-
supported platinum catalysts" Journal of Catalysis 215:2:344:352, 2003.
7. Shabaker, J.W., and Dumesic, J.A. "Kinetics of aqueous-phase reforming of
oxygenated hydrocarbons: Pt/A1203 and Sn-modified Ni catalysts" Industrial and
Engineering Chemistry Research 43:12:3105-3112, 2004.
8. Shabaker, J.W., Huber, G.W., Davda, R.R., Cortright, R.D. and Dumesic, J.A.
"Aqueous-phase reforming of ethylene glycol over supported platinum catalysts"
Catalysis Letters 88:1-2:1:8, 2003.
9. Shabaker, J.W., Huber, G.W. and Dumesic, J.A. "Aqueous-phase reforming of
oxygenated hydrocarbons over Sn-modified Ni catalysts" Journal of Catalysis
222:1:180:191, 2004.
CA 02720344 2010-09-30
WO 2009/129622
PCT/CA2009/000538
22
10. Shabaker, J.W., Simonetti, D.A., Cortright, R.D. and Dumesic, J.A. "Sn-
modified
Ni catalysts for aqueous-phase reforming: characterization and deactivation
studies" Journal of Catalysis 231:1:67:76, 2005.
11. Monnier, J., Tourigny, G., Sulimma, H. and Pelletier, L. "Low temperature
production of hydrogen from glycerol and sugars" 18th Canadian Symposium on
Catalysis, Montreal, Quebec, Canada, May 16-19, 2004,
12. Monnier, J., Tourigny, G., Sulimma, H. and Pelletier, L. "Evaluation of
catalyst
performance for low temperature production of hydrogen from glycerol and
sugars" 19th North American Catalysis Society Meeting, Philadelphia,
Pennsylvania USA, May 22-27, 2005.
13. Guo, J. and Al-Dajjan, M. "Activity and stability of iron-containing
pillared clay
catalysts for wet air oxidation of phenol" Applied Catalysis A: General
299:175:184, 2006.
14. Perry R. and Green, D. "Perry's chemical engineer's Handbook" McGraw-Hill
Education, 1998.
15. Pitault, I., Fongarland, P., Mitrovic, M., Ronze, D. and Forissier, M.
"Choice of
laboratory scale reactors for HDT kinetic studies or catalyst tests" Catalysis
Today 98:31:42, 2004.
16. Cao, C. and Fung, S. "Comparison of heterogeneous catalyst activities in
various
reactor types and reaction conditions" Chemical Engineering Technology
29:3:307:312, 2006.