Note: Descriptions are shown in the official language in which they were submitted.
CA 02724630 2010-11-16
WO 2009/140598 PCT/US2009/044148
TARGETED COAGULATION FACTORS AND
METHOD OF USING THE SAME
[001] This application claims benefit of U.S. Provisional Application Serial
No.
61/053,932; filed on May 16, 2008, the contents of which are incorporated
herein by
reference in their entirety.
FIELD OF THE INVENTION
[002] The invention relates to targeted coagulation factors having increased
efficacy.
The invention further provides methods of treating patients suffering from a
coagulation
factor deficiency disorder by selectively targeting coagulation factors to
their biological
sites of action, such as by targeting Factor VIII (FVIII) to red blood cells
and platelets.
Pharmaceutical compositions comprising the targeted coagulation factors
according to the
invention are also provided.
BACKGROUND OF THE INVENTION
[003] The effectiveness of biological drugs is often limited by their duration
of action in
patients, particularly when the disease requires constant modulation by the
drug.
Consequently, enhancement of pharmacokinetic properties is often more critical
to the
success of a therapeutic agent in the clinic than is optimization of the
drug's potency. One
approach to protect drugs from various mechanism of clearance so to prolong
the half-life
is to add targeting domains that promote drug binding to long-lived proteins
in circulation
such as matrix proteins, or to the surface of cells, such as blood cells or
endothelial cells.
For example, localization of therapeutic peptides or proteins to blood cell
surfaces has
been shown to prolong their circulation half-life by preventing normal
clearance
mechanisms (Chen, et al., Blood 105(10):3902-3909, 2005). A wide variety of
molecules
may be used as the targeting domain.
[004] In another instance, when the Kunitz-type protease inhibitor (KPI)
domain of tick
anticoagulant protein was linked with an anionic phospholipid, phosphatidyl-L-
serine (PS)
binding protein, annexin V (ANV), the fusion protein (ANV-KPI) was shown to be
more
active and possess higher in vivo antithrombotic activities than the non-
fusion counterpart
(Chen, et al., 2005). Because ANV has strong affinities for PS and
phosphatidylethanolamine (PE), it is hypothesized that the fusion protein ANV-
KPI can be
CA 02724630 2010-11-16
WO 2009/140598 PCT/US2009/044148
specifically targeted to the PS/PE-rich anionic membrane-associated
coagulation enzyme
complexes present at sites of thrombogenesis. Similarly, Dong, et al.,
reported fusing the
fibrin-selective Desmodus rotundus salivary PA al (dsPA al) to a urokinase
(uPA)/anti-P-
selectin antibody (HuSZ5 1) to produce a fusion protein that is fully
functional with similar
antithrombotic activities as the non-fusion counterpart in in vitro assays.
Furthermore, the
fusion protein HuSZ51-dsPA al was shown to bind to thrombin-activated human
and dog
platelets (Dong, et al., Thromb. Haemost. 92:956-965, 2004).
[005] Other efforts have been made in targeting anticoagulants to prevent
clots and to
reduce mortality associated with thrombotic diseases (see, e.g., WO 94/09034).
A more
recent development is demonstrated by Stoll, et al., (Arterioscler. Thromb.
Vase. Biol.
27:1206-1212, 2007), in which a Factor Xa (FXa) inhibitor, tick anticoagulant
peptide
(TAP), was targeted to ligand-induced binding sites (LIBS) on GPIIb/IIIa, a
glycoprotein
abundantly expressed on the platelet surface, via an anti-LIBS single-chain
antibody
(scFvann_LIBS). The fusion protein scFvaõn_LjBs-TAP was shown to possess an
effective
anticoagulation activity even at low doses at which the non-targeted
counterpart failed.
[006] The aforementioned targeted anticoagulants were fusion proteins designed
to target
specific cells. According to Stoll, et al., the targeted anticoagulant should
be a small
molecule with a highly potent coagulation inhibition activity that is retained
while fused to
an antibody. The release of the anticoagulant from the fusion proteins in its
targeted sites
was not discussed.
[007] The present invention focuses on targeting therapeutic proteins for the
treatment of
hematological diseases such as hemophilia. For example, current treatment of
hemophilia
A patients with FVIII concentrates or recombinant FVIII is limited by the high
cost of
these factors and their relatively short duration of action. Hemophilia A
patients are
currently treated by intravenous administration of FVIII on demand or as a
prophylactic
therapy administered several times a week. For prophylactic treatment, FVIII
is
administered three times a week. Unfortunately, this frequency is cost
prohibitive for
many patients. Because of its short half-life in man, FVIII must be
administered
frequently. Despite its large size of greater than 300 kD for the full-length
protein, FVIII
has a half-life in humans of only about 11-18 (average 14) hours (Gruppo, et
al.,
Haemophila 9:251-260, 2003). For those who can afford the frequent dosaging
recommended, it is nevertheless very inconvenient to frequently intravenously
inject the
2
CA 02724630 2010-11-16
WO 2009/140598 PCT/US2009/044148
protein. It would be more convenient for the patients if a FVIII product could
be
developed that had a longer half-life and therefore required less frequent
administration.
Furthermore, the cost of treatment could be reduced if the half-life were
increased because
fewer dosages may then be required. It is therefore desirable to have more
efficient forms
of FVIII that can lower the effective dose or have a prolonged duration of
action to
significantly improve treatment options for hemophiliacs.
[008] Also, a sustained plasma concentration of targeted FVIII may reduce the
extent of
adverse side effects by reducing the trough to peak levels of FVIII, thus
eliminating the
need to introduce super-physiological levels of protein at early time-points.
Therefore, it
is desirable to have forms of FVIII that have sustained duration and a longer
half-life than
current marketed forms.
[009] An additional disadvantage to the current therapy is that about 25-30%
of patients
develop antibodies that inhibit FVIII activity (Saenko, et al., Haemophilia
8:1-11, 2002).
Antibody development prevents the use of FVIII as a replacement therapy,
forcing this
group of patients to seek an even more expensive treatment with high-dose
recombinant
Factor VIIa (FVIIa) and immune tolerance therapy. A less immunogenic FVIII
replacement product is therefore desirable.
[010] One approach in improving the treatment for hemophiliacs involves gene
therapy.
Ectopically targeting FVIII to platelets by directing FVIII expression in
platelets can have
therapeutic effects in the treatment of hemophilia A (Shi, et al., J. Clin.
Invest.
116(7):1974-1982, 2006).
[011] It is an object of the invention to provide targeted coagulation factors
that have
prolonged duration of action, greater efficacy, fewer side effects, and less
immunogenicity
compared to the untargeted protein.
[012] Another object of the invention is to reduce side effects associated
with therapeutic
protein administration by having the protein targeted to the specific site of
desired action
and thereby reducing the exposure of the protein to other potential
biologically active sites
that may result in undesired side effects.
[013] A further object of the present invention is to obtain further
advantages by
designing targeted therapeutic coagulation factors in which the therapeutic
protein is
released from the targeting domain in the immediate vicinity of its site of
action in vivo. A
3
CA 02724630 2010-11-16
WO 2009/140598 PCT/US2009/044148
high local concentration of the non-fusion, activated proteins may be
achieved. Thus, the
therapeutic efficacy of the proteins is enhanced.
SUMMARY OF THE INVENTION
[014] The targeted coagulation factors according to the present invention
comprise a
coagulation factor linked with at least one domain that specifically binds to
a membrane
protein on a blood cell. A pharmaceutical composition comprising the newly
disclosed
targeted coagulation factors and a method for treating hematological diseases
using the
targeted coagulation factors is also provided. The present invention further
provides a
method for targeting a coagulation factor to the surface of a blood cell by
using the newly
disclosed targeted coagulation factors to increase the efficiency of treating
hematological
disease with coagulation factors.
DESCRIPTION OF THE DRAWINGS
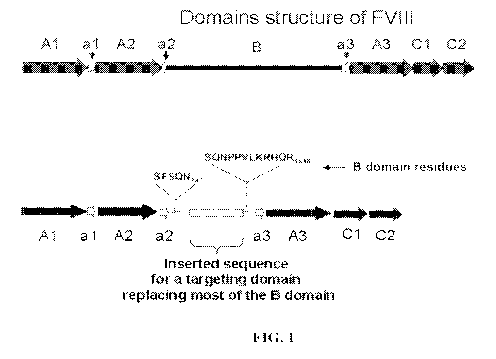
[015] Figure 1: Schematic drawings of full-length FVIII ("Full Length FVIII)
and B-
domain deleted FVIII ("FVIII-BDD-TD") in which a targeting domain ("TD") is
inserted
into the B-domain and most of the B-domain is removed.
[016] Figure 2: Structures of modified cyclic peptide integrilin, "BHRF-1" (A)
and
"BHRF-3" (B), for linking to FVIII through the B-domain cysteine.
[017] Figure 3: Binding affinity of BHRF-1 and BFRH-3 to immobilized
GPIIa/IIIb.
[018] Figure 4: BHRF-I-FVIII binding assay to immobilized GPIIa/IIIb.
[019] Figure 5: In vitro clotting activity of BHRF-1-FVIII as compared with
FVIII.
[020] Figure 6: In vitro binding of BHRF-I-FVIII to human platelets.
[021] Figure 7: In vitro binding of BHRF-I-FVIII to mouse platelets.
DESCRIPTION OF THE INVENTION
[022] The present invention is directed to targeting a coagulation factor to
its site or sites
of action, such as to blood cells. In one embodiment, a targeted coagulation
factor is
provided that is specifically targeted to a blood cell through linking the
factor to at least
one domain that binds to a membrane protein on the blood cell. The domain for
targeting
4
CA 02724630 2010-11-16
WO 2009/140598 PCT/US2009/044148
the coagulation factor to the blood cell may be without limitation an antibody
fragment, an
antibody, a peptide, a receptor ligand, a carbohydrate, or a small molecule
that has a high
affinity to a membrane protein on the surface of the blood cell. The blood
cell for
example is a red blood cell or a platelet.
[023] As used herein, "coagulation factor" refers to a protein that is
involved in the
coagulation cascade and has predominantly procoagulant activity. Coagulation
factors are
well known in the art and include without limitation coagulation factors I,
II, V, VI, VII,
VIII, IX, X, XI, XII, and XIII, and protein S. The coagulation factors may be
concentrated
from plasma or may be recombinantly produced. If recombinantly produced, the
coagulation factors may have an amino acid structure that varies from the
natural structure
as long as sufficient procoagulant activity is maintained such that the
variant is
therapeutically useful. In one embodiment, the coagulation factor is a
functional FVIII
polypeptide, such as without limitation a FVIII concentrate from plasma or
recombinantly
produced FVIII, or Factor IX (FIX).
[024] "Functional FVIII polypeptide" as used herein denotes a functional
polypeptide or
combination of polypeptides that are capable, in vivo or in vitro, of
correcting human
FVIII deficiencies, characterized, for example, by hemophilia A. FVIII has
multiple
degradation or processed forms in the natural state. These are proteolytically
derived from
a precursor, one chain protein. A functional FVIII polypeptide includes such
single chain
protein and also provides for these various degradation products that have the
biological
activity of correcting human FVIII deficiencies. Allelic variations likely
exist. The
functional FVIII polypeptides include all such allelic variations,
glycosylated versions,
modifications and fragments resulting in derivatives of FVIII so long as they
contain the
functional segment of human FVIII and the essential, characteristic human
FVIII
functional activity. Those derivatives of FVIII possessing the requisite
functional activity
can readily be identified by straightforward in vitro tests described herein.
Furthermore,
functional FVIII polypeptide is capable of catalyzing the conversion of Factor
X (FX) to
FXa in the presence of Factor IXa (FIXa), calcium, and phospholipid, as well
as correcting
the coagulation defect in plasma derived from hemophilia A affected
individuals. From
the published sequence of the human FVIII amino acid sequence and the
published
information on its functional regions, the fragments that can be derived via
restriction
enzyme cutting of the DNA or proteolytic or other degradation of human FVIII
protein
CA 02724630 2010-11-16
WO 2009/140598 PCT/US2009/044148
will be apparent to those skilled in the art. Specifically included within
functional FVIII
polypeptides without limitation is full-length human FVIII (e.g., SEQ ID NO: 1
and SEQ
ID NO: 2) and B-domain deleted factor VIII (e.g., SEQ ID NO: 3 and SEQ ID NO:
4) and
having the amino acid sequences as disclosed in WO 2006/053299.
[025] "Procoagulant activity" of FVIII refers to the activity of FVIII in the
coagulation
cascade. FVIII itself does not cause coagulation, but plays an essential role
in the
coagulation cascade. The role of FVIII in coagulation is to be activated to
FVIIIa, which
is a catalytic cofactor for intrinsic FX activation (Thompson, Semin. Thromb.
Hemost.
29 :11-22, 2003). FVIII is proteolytically activated by thrombin or FXa, which
dissociates
it from von Willebrand factor (vWf) and activates its procoagulant function in
the cascade.
In its active form, FVIIIa functions as a cofactor for the FX activation
enzyme complex in
the intrinsic pathway of blood coagulation, and it is decreased or
nonfunctional in patients
with hemophilia A.
[026] "FIX" means coagulation factor IX, which is also known as human clotting
factor
IX, or plasma thromboplastin component.
[027] As used herein, the term "targeted coagulation factor" refers to a
coagulation factor
that is coupled with at least one domain that specifically binds to a membrane
protein on a
blood cell. The targeted coagulation factor should bind potently to the blood
cells, for
example, with a half maximal binding < 10 nM. Binding should be specific to
the targeted
blood cells, for example, through binding to membrane proteins selectively
expressed on
the targeted cell. "Domain" or "targeting domain" as used herein refers to a
moiety that
has a high affinity for membrane proteins on target cells. Domains suitable
for the present
invention include, but are not limited to, antibodies, antibody fragments,
such as single
chain antibodies (svFv) or FAB fragments, antibody mimetics, and peptides or
small
molecules with high affinity for membrane proteins on the surface of the blood
cells. In
one aspect, a single chain antibody fragment or a peptide is used because its
coding
sequence can be linked with the FVIII coding sequence and a fusion protein can
be
produced using recombinant technology.
[028] The coagulation factor can be coupled with the domain either chemically
or by
recombinant expression of a fusion protein. Chemical linkage can be achieved
by linking
together chemical moieties present on the coagulation factor and the targeting
domain,
including chemical linkages using moieties such as amino, carboxyl, sulfydryl,
hydroxyl
6
CA 02724630 2010-11-16
WO 2009/140598 PCT/US2009/044148
groups, and carbohydrate groups. A variety of homo- and hetero-bifunctional
linkers can
be used that have groups that are activated, or can be activated to link to
attach these
moieties. Some useful reactive groups on linker molecules include maleimides,
N-
hydroxy-succinamic esters and hyrazides. Many different spacers of different
chemical
composition and length can be used for separating these reactive groups
including, for
example, polyethylene glycol (PEG), aliphatic groups, alkylene groups,
cycloalkylene
groups, fused or linked aryl groups, peptides and/or peptidyl mimetics of one
to 20 amino
acids or amino acid analogs in length. For example, the domain may be linked
with the
coagulation factor in such a way that in vivo a functional form of the
coagulation factor
would be released from its targeted domain or the release occurs at or near
the site of
biological activity of the coagulation factor in the body.
[029] Accordingly, in one embodiment of the invention, a targeted coagulation
factor is
provided wherein the linkage attaching the coagulation factor to the domain
for targeting
the coagulation factor to the blood cell can be cleaved or degraded thereby
releasing the
coagulation factor from the conjugate.
[030] The release of the coagulation factors from their conjugate form (i.e.,
from the
targeted coagulation factor) can be achieved by linking the targeting domain
to a site on
the coagulation factor that is removed during its activation process, or by
using a linker
that degrades in a controlled manner by enzymes in the blood. For example,
sugar
polymers or peptides can be used that are susceptible to general blood
proteases or
hydrolases. A variety of such technologies is known in the art and has been
used to make
pro-drugs. The linker could be further engineered to be cleaved specifically
at sites where
the coagulation factors are most needed, such as sites of inflammation or
blood
coagulation triggered through trauma. For example, the linker may be
susceptible to
specific proteases produced only at the desired site of action, such as
proteases released by
the inflammation process or generated by the blood coagulation cascade. This
selective
release of the therapeutic protein may lower the potential for side effects
and increase the
efficiency of the protein at its site of action.
[031] A variety of membrane proteins on blood cells can be targeted according
to the
present invention. To specifically and efficiently target a coagulation factor
to a blood cell,
however, it is preferable that the targeted membrane protein is present
abundantly on the
7
CA 02724630 2010-11-16
WO 2009/140598 PCT/US2009/044148
blood cell surface. For example, the glycoprotein GPIIb/IIIa is found to be
one of the
most abundantly expressed molecules on the platelet surface.
[032] Accordingly, in one embodiment, the coagulation factor is targeted to a
platelet
through a domain that binds specifically to a platelet membrane protein such
as the
glycoprotein GPIIb/IIIa. Examples of such domains to target the coagulation
factor to
GPIIb/IIIa include, but are not limited to, RGD containing peptides and
mimetics (linear
peptides, snake venom peptides, and cyclic peptides) such as integrilin
9containing the
RGD mimetic sequence, homo-arginine, glycine aspartic acid), non-peptide RGD
mimetics, and anti-GPIIb/IIIa antibodies. If an antibody is used as the
targeting domain, a
single chain fragment of the antibody, such as svFv or FAB fragment, can be
used.
Targeting FVIII and FIX
[033] Targeting FVIII and FIX to the surface of blood cells, such as platelets
or red
blood cells, may serve to slow the clearance of these coagulation factors.
Targeting FVIII
to the surface of platelet cells is of particular interest. FVIII is a
critical cofactor in the
FIX-mediated activation of FX, which takes place predominantly on the surface
of
activated platelet cells that accumulate at clot sites. Activation of
platelets triggers
binding of these coagulation factors to its surface to form a complex that
facilitates FXa
generation. Platelets have an average lifespan in circulation of about 9 days.
In contrast,
FVIII in plasma (largely bound to von Willebrand's factor) displays a half-
life of about 14
hours. Thus, binding of FVIII to platelets has the potential to greatly extend
the
circulation time of the molecule. Targeting FVIII to the surface of platelet
cells via a
targeting domain according to the present invention increases the efficiency
of FVIII
action and is anticipated to prolong the half-life of FVIII.
[034] In addition to GPIIb/IIIa, other proteins on platelets could serve as
receptors for
targeted FVIII, such as GPla and Anexin V. The glycoprotein GPIIb/IIIa is
preferred
because it is one of the most abundantly expressed molecules on the platelet
surface. The
concentration of GPIIb/IIIa in blood is estimated to be about 75 nM based on
its surface
density on platelets. This represents a 100-fold excess over the maximum
concentration of
FVIII achieved after therapeutic application of the FVIII (Cmax about 0.7 nM).
Therefore,
targeting of FVIII to platelets would occupy roughly I% or less of available
GPIIb/IIIa
sites on platelets. This low level of occupancy would not be expected to alter
platelet
function, which requires a much large fraction (i.e., >50-60%) of GPIIb/IIIa
molecules to
8
CA 02724630 2010-11-16
WO 2009/140598 PCT/US2009/044148
be blocked. The high concentration of GPIIb/IIIa would also drive the
equilibrium
binding of targeted FVIII to the platelet surface.
[035] Without restricting the invention in any way, it is believed that
targeting FVIII to
GPIIb/IIIa may also have the benefit that some of the coagulation factors may
be
internalized through endocytosis and recycling of GPIIb/IIIa through the open
intracanicular system of platelets. This FVIII can end up in alpha granules
and be re-
released upon platelet activation, providing a source of FVIII when it is
needed for
coagulation. Bound or internalized FVIII targeted to platelets may be
protected from
inhibitors (e.g., FVIII antibodies) that are present in many patients. Thus,
targeted FVIII
may offer a treatment option for this important group of patients.
[036] For targeted FVIII to promote coagulation, the molecule must be capable
of being
processed to a functional form (FVIIIa), and be released from its GPIIb/IIIa
binding site.
In one embodiment, this is achieved by linking the GPIIb/IIIa targeting domain
to the B-
domain of FVIII. The B-domain is removed in a pro-coagulant environment by
thrombin
or FXa mediated proteolysis, producing the mature FVIIIa molecule. Thus, upon
activation, FVIIIa will be released from GPIIb/IIIa and be available for
formation of the
FX activation complex.
[037] The linkage between FVIII and the targeting domain can be achieved by
covalently
binding the targeting domain to reactive groups on FVIII, including amino,
sulfhydryl,
carboxyl groups and carbonyl groups using cross-linking approaches described
herein.
Targeting domains can also be coupled to carbohydrate present mostly on the B-
domain of
the FVIII molecule. For example, mild oxidation of FVIII with periodate
produces
aldehydes on carbohydrate chains, which can then be reacted with amines or
hyrazides,
followed optionally by reduction to form more stable linkages.
[038] Free cysteine can be selectively generated on the B-domain of
recombinant FVIII
through mild reduction with Tris(2-carboxyethyl)phosphine (TCEP), allowing
specific
linking of the B-domain with a targeting domain that reacts with a free
cysteine, such as a
domain containing a thiol, triflate, tresylate, aziridine, oxirane, S-pyridyl,
or maleimide
moiety. Furthermore, FVIII can be modified to replace an amino acid residue
with
cysteine to provide a specific location for attachment to a targeting domain.
If a B-domain
deleted FVIII is used, a variety of cysteine muteins of B-domain deleted
FVIII, such as
those disclosed in WO 2006/053299, can be used to link FVIII with a targeting
domain
9
CA 02724630 2010-11-16
WO 2009/140598 PCT/US2009/044148
through chemical binding at a surface cysteine residue. Examples of amino acid
residues
that may be modified to replace an amino acid residue with cysteine include,
but not
limited to, 81, 129, 377, 378, 468, 487, 491, 504, 556, 570, 1648, 1795, 1796,
1803, 1804,
1808, 1810, 1864, 1911, 2091, 2118, and 2284 (the amino acid residue is
designated by its
position in the sequence of full-length FVIII).
[039] The coagulation factor may also be coupled to the targeting domain using
recombinant technology. Host cells may be transfected with a vector comprising
a fusion
protein of FVIII and the targeting domain. In one embodiment, the targeting
domain may
be inserted into the B-domain of FVIII and most of the B-domain is deleted
with only
portions of the B-domain left at the carboxy and amino terminals to allow for
the
biological processing of the B-domain to delete it from the full-length
molecule. As
illustrated in Fig. 1, the remaining portions of the B-domain are specified
that allow for
biological processing and removal of the B-domain under physiological
conditions.
[040] The host cell line may be any cell known to those skilled in the art as
useful for
producing a coagulation factor such as without limitation for FVIII CHO cells,
HEK cells,
BHK cells, and HKB 11 cells (a hybrid of a human embryonic kidney cell line,
HEK293
and a human Burkitt B cell lymphoma line, 2B8).
[041] A number of domains can be linked chemically to FVIII, or recombinantly
expressed with FVIII, to target FVIII to GPIIb/IIIa on the surface of
platelets. Examples
of such domains include, but are not limited to, antibodies against
GPIIb/IIIa, RGD
peptides, peptide mimetics, or small molecule mimetics targeting GPIIb/IIIa.
Antibodies,
such as single chain antibodies (svFv) or FAB fragments targeting GPIIb/IIIa,
are
particularly useful as targeting domains.
[042] It has been shown that the B-domain of FVIII can be removed without loss
of
FVIII function. Additionally, it has been also shown that various B-domain
truncated
forms of FVIII and B-domain fusions with other protein domains can yield
functionally
active FVIII. In one aspect, the invention involves targeting domains that can
be
engineered to insert into, replace, or partially replace the B-domain of FVIII
without
blocking the normal processing of the molecule to yield active FVIII. For
example, using
recombinant DNA technology, a FVIII molecule can be produced in which single
chain
antibody fragments are fused to the C-terminus of the B-domain of FVIII.
Alternatively,
svFv fragments can also be used to replace the whole or a part of the B-domain
of FVIII.
CA 02724630 2010-11-16
WO 2009/140598 PCT/US2009/044148
This can be achieved through insertion of the DNA sequence encoding the svFv
fragments,
in frame, after the B-domain coding sequence, or replacing some or all of the
B-domain
coding sequence. This strategy will preserve thrombin cleavage sites required
for normal
proteolyic activation of FVIII. A variety of antibodies against GPIIb/IIIa
which localize
efficiently to platelets are known (see, e.g., Schwarz, et al., Circ. Res.
99(1):25-33, 2006;
Jacobin, et al., Clin. Immunol. 108(3):199-210, 2003; Christopoulos, et al.,
Blood Coagul.
Fibrinolysis 4(5):729-37, 1993; and Chung, et al., FASEB J. 18(2):361-363,
2004).
[043] Likewise, RGD or RGD mimetic containing peptides are also useful ligands
for
targeting FVIII since many of such peptides have been described to have high
binding
affinity to GPIIb/IIIa. These include linear peptides, snake venom peptides,
and cyclic
peptides. Non-peptide RGD mimetics could also be used. Similar to the antibody
fragments, RGD peptides can be chemically coupled to FVIII. Alternatively, RGD
sequences can be inserted into the B-domain coding sequence or used to
replace, in whole
or in part, the B-domain coding sequence of FVIII and expressed using
recombinant DNA
technology.
[044] A targeted FIX can be prepared using a similar procedure. For example,
targeting
domains can be linked to an activation domain of a FIX molecule (amino acid
residues
191-226 or 145-180, depending on preferences, that is, +/- signal sequence),
which is
proteolytically removed in the activation of FIX to FIXa. The domain can be
linked
chemically using cross-linkers reactive with amino acid side chain groups such
as
sulfhydryls, amines, and carboxyl groups in the activation domain, or linked
through
carbohydrate chains, as was discussed above for FVIII. A fusion molecule can
also be
made using recombinant technology where an amino acid sequence of a targeting
domain
is inserted into the FIX activation peptide, or replacing parts of the
activation peptide
sequence. The inserted targeting domain sequences can code for a single chain
antibody,
or other platelet binding peptide sequence, such as an RGD binding peptide.
Pharmaceutical Compositions and Uses
[045] The invention also concerns pharmaceutical compositions comprising
therapeutically effective amounts of the targeted coagulation factors of the
invention and a
pharmaceutically acceptable excipient or carrier. "Pharmaceutically acceptable
excipient
or carrier" is a substance that may be added to the active ingredient to help
formulate or
stabilize the preparation and causes no significant adverse toxicological
effects to the
II
CA 02724630 2010-11-16
WO 2009/140598 PCT/US2009/044148
patient. Examples of such excipients or carriers are well known to those
skilled in the art
and include water, sugars such as maltose or sucrose, albumin, salts, etc.
Other excipients
or carriers are described, for example, in Remington's Pharmaceutical Sciences
(Mack
Publishing Co., Easton, Pa., 20th edition, 2000). Such compositions will
contain an
effective amount of the targeted coagulation factors together with a suitable
amount of
excipients or carriers to prepare pharmaceutically acceptable compositions
suitable for
effective administration to a patient in need thereof.
[046] For example, the conjugate may be parenterally administered to subjects
suffering
from hemophilia A at a dosage that may vary with the severity of the bleeding
episode.
The average doses administered intraveneously is in the range of 40 units per
kilogram for
pre-operative indications, 15 to 20 units per kilogram for minor hemorrhaging,
and 20 to
40 units per kilogram administered over an 8-hours period for a maintenance
dose.
[047] In one embodiment, the present invention concerns a method for treating
hematological diseases comprising administering an therapeutically effective
amount of
the aforementioned targeted coagulation factor to a patient in need thereof.
[048] As used herein, "therapeutically effective amount" means an amount of a
targeted
coagulation factor that is need to provide a desired level of the targeted
factor (or
corresponding unconjugated factor released from the targeted form) in the
bloodstream or
in the target tissue. The precise amount will depend upon numerous factors,
including, but
not limited to the components and physical characteristics of the therapeutic
composition,
intended patient population, individual patient considerations, and the like,
and can readily
be determined by one skilled in the art.
[049] As used herein, "patient" refers to human or animal individuals
receiving medical
care and/or treatment.
[050] The polypeptides, materials, compositions, and methods described herein
are
intended to be representative examples of the invention, and it will be
understood that the
scope of the invention is not limited by the scope of the examples. Those
skilled in the art
will recognize that the invention may be practiced with variations on the
disclosed
polypeptides, materials, compositions and methods, and such variations are
regarded as
within the ambit of the invention.
12
CA 02724630 2010-11-16
WO 2009/140598 PCT/US2009/044148
[051] The following examples are presented to illustrate the invention
described herein,
but should not be construed as limiting the scope of the invention in any way.
EXAMPLES
[052] In order that this invention may be better understood, the following
examples are
set forth. These examples are for the purpose of illustration only, and are
not to be
construed as limiting the scope of the invention in any manner. All
publications
mentioned herein are incorporated by reference in their entirety.
Example 1: Modified RGD Peptides with High Affinity for GPIIb/IIIa Binding
[053] Cyclic peptides have been described to bind potently and selectively to
GPIIb/IIIa.
One such peptide, integrilin, was used as a targeting domain to link with
FVIII as it has
been shown that integrilin can selectively bind to GPIIb/IIIa. Integrilin was
modified by
adding a short PEG linker ending in a maleimide moiety that can selectively
couple to free
cysteine residues in proteins. The modified integrilin is termed BHRF-1 with
the linker
only (Fig. 2A), and BHRF-3 with the linker and a fluorescein (FITC) (Fig. 2B).
As shown
in Fig. 3, the modified integrilins retain affinity for GPIIb/IIIa as they
potently blocked
fibrinogen (Fbn) binding to immobilized GPIIa/IIIb.
[054] Peptide binding to GPIIb-IIIa was measured using a solid phase binding
assay in
which competition of fibrinogen binding by testing compounds is measured. The
assay
was performed as follows. Purified GPIIb-IIIa (Innovative Research, Novi, MI)
was
coated onto 96-well Immulon-B plates at O.mL/well x 2 gg/mL, diluted in Buffer
A (20
mM Tris pH 7.5, 0.15 M NaCl, and 1 mM each of MgC12, CaC12, and MnC12). After
overnight incubation at 4 C, the plate was blocked for 1 hour at 30 C, with
3.5% BSA in
Buffer B (50 mM Tris pH 7.5, 0.1 M NaCl, and 1 mM each of MgC12, CaC12, and
MnClz).
After washing 3 times with Buffer B, diluted peptide or protein solutions were
combined
with 3.5 nM biotinylated fibrinogen in 0.1% BSA/Buffer B and added to the
wells,
incubating at 30 C for 2 hr. After washing (3 times, Buffer B), 1:4000
streptavidin-
horseradish peroxidase (HRP) was added (Pierce Chemical Co., Rockford, IL) for
1 hour
at 30 C. After a final washing step (3 times, Buffer B), the plate was
developed with Ultra
TMB (3,3',5,5'-tetramentylbenzidine) (Pierce Chemical Co., Rockford, IL) for 5
minutes,
13
CA 02724630 2010-11-16
WO 2009/140598 PCT/US2009/044148
stopping with an equal volume of 2 M sulfuric acid. Plate absorbances were
read at 450
nm, and IC50 values determined using a 4-parameter logistic fit.
[055] The modified integrilin peptide (BHRF1) is then coupled with FVIII via
the
cysteine (Cys) residue located in the B-domain of FVIII.
Example 2: Coupling GPIIb/IIIa Binding Peptides to FVIII
[056] The polypeptide sequence of the full-length FVIII is known in the art
(see, e.g.,
SEQ ID NO: 1, SEQ ID NO: 2, and as disclosed in WO 2006/053299.
Concentration of FVIII and uncapping of free sulfhydryl groups
[057] The Cys residue located in the B-domain of recombinant FVIII can be
capped by
cysteine present in the media during protein expression, but it can be readily
removed by
treatment with reducing agents, such as TCEP, as follows. FVIII (20 mL) was
thawed and
concentrated in two Amicon -15 cartridges (Millipore, Billerica, MA), spun at
2000 x g
(about 3153 rpm) for 25 minutes in the cold. The concentration of the 2.8 mL
retentate is
about 0.8-0.9 mg/mL by A280 using a NanoDrop spectrophotometer (ThermoFisher
Scientific, Waltham, MA). The buffer was then exchanged using a 10 mL Zeba
desalting
cartridge, pre-equilibrated with 50 mM Tris, 150 mM NaCl, 2.5 mM CaC12 and 100
ppm
Tween -80 (polyoxyethylenesorbitan monooleate). A protein solution of 2.8 mL
with a
concentration of 0.88 mg/mL was obtained. TCEP was then added to a final
concentration
of 0.68 mM and the mixture was gently turned end-over-end at 4 C for about 3
hours.
TCEP was removed by two successive Zeba cartridge spins, and the FVIII was
allowed to
re-oxidize for at least 30 minutes before addition of the peptide. After the
removal of
TCEP, the FVIII concentration was measured at 0.768 mg/mL ("KG-R").
Coupling of the RGD targeting peptide
[058] To couple the modified integrilin peptide BHRF-l to FVIII, 0.294 mg of
the
peptide (M.W. 1225) was added to 48 gL dry dimethyl sulfoxide (DMSO) to make a
mM stock solution. This stock solution (34.4 L) was then added to 2.8 mL KG-
R. The
reaction was quenched by addition of an equi-molar amount of cysteine after 80
minutes.
The reaction mixture was then extensively dialyzed against starting Tris
buffer (2 liters).
The final concentration of BHRF-1-FVIII was 0.74 mg/mL and the yield was 2 mg.
A
similar procedure was also used to prepare BHRF-3-FVIII.
14
CA 02724630 2010-11-16
WO 2009/140598 PCT/US2009/044148
[059] As shown in Fig. 3, the modified integrilin peptides, BHRF-1 and BHRF-3,
retain
affinity for GPIIb/IIIa as they potently blocked fibrinogen (Fbn) binding to
immobilized
GPIIa/IIIb. FVIII coupled to BHRF-1 (FVIII-BHRF-1) showed high potency for
inhibition of fibrinogen binding to immobilized GPIIb/IIIa (IC50 = 0.043 +/-
0.05 nM (N =
3)). This was even more potent than the parent BHRF-1 peptide. Results are
shown in
Table 1.
TABLE 1
Conjugate Moiety nM (N)
Integrelin 1.3+/-1.0 4
BHRF-1 (+ linker) 1.2+/-0.6 2
BHRF-3 1.5+/-1.3 3
(+ linker + FITC)
Coupling of the RGD targeting peptide to B-domain deleted FVIII
[060] If a B-domain deleted FVIII ("BDD") is used for coupling, a variety of
Cys
muteins of B-domain deleted FVIII as disclosed in WO 2006/053299 can be used
to
couple BDD to a targeting domain such as the modified RGD peptides as
disclosed herein.
Example 3: BHRF-1-FVIII Binds to Immobilized GPIIb/IIIa
[061] To test the binding activity of BHRF-I-FVIII to GPIIb/IIIa, biotinylated
GPIIb/IIIa
was immobilized on streptavidin plates and treated with either BHRF- 1-F VIII
or
unmodified FVIII, both in binding buffer (50 mM Tris, pH 7.5, 100 mM NaC12, 1
mM
CaC12, 1 mM MgC12, 1 mM MnClz and 1 mg/mL BSA). The unbound protein was
removed by washing three times with binding buffer. Assay buffer (25 L) was
added to
the plate, and FVIII activity was determined using a chromogenic assay kit
(Coatest SP4,
Chromogenix, Lexington, MA). As shown in Fig. 4, there was binding of BHRF-I-
FVIII,
while only little binding of unmodified FVIII was detected. The increased
binding of
BHRF- 1-F VIII was completely eliminated by addition of a cyclic RGD peptide
(GpenGRGDSPCA; SEQ ID NO: 5) that competes for BHRF-1 binding to GPIIb/IIIa.
Furthermore, only low background levels of either protein bound when no
GPIIb/IIIa was
immobilized on the plate. These data show that BHRF-I-FVIII can be targeted to
GPIIIIIIIa through the peptide targeting domain.
CA 02724630 2010-11-16
WO 2009/140598 PCT/US2009/044148
[062] Because unconjugated FVIII was not removed from the preparations of
BHRF1-
FVIII, experiments were performed to determine the amount of unconjugated
FVIII
present. BHRF 1-F VIII activity was depleted using beads containing excess
levels of
immobilized GPIIb/IIIa. Roughly 80% of the activity of BHFR1-FVIII can be
depleted,
indicating about 20% of the FVIII activity in the preparation came from
unconjugated
FVIII.
Example 4: In vitro Whole Blood Clotting Activity Assay with BHRF-1-FVIII and
F VIII
[063] To assess the effect of platelet binding of BHRF- 1-F VIII on hemostatic
activity, its
activity was compared to that of unconjugated FVIII using a Rotational
Thromboelastometry (ROTEM , Pentapharm GmbH) system as described in
Landskroner,
et al., (Haemophilia 11:346-352, 2005). Unlike measures of clotting activity
such as the
Coatest chromogenic assay or the activated partial thromboplastin time (aPTT)
assay,
the ROTEM assay depends on the function of the platelets and therefore, can
show
effects of BHRF-I-FVIII binding to platelets. To perform the assay, citrated
hemophilia A
mouse whole blood was mixed with an equal dose of BHRF-I-FVIII (1 mIU) or
unconjugated FVIII (based on the Coatest chromogenic assay) at room
temperature.
Samples were recalcified by dispensing 300 gL treated blood with an automated
pipette
into ROTEM cups with 20 gL CaC12 (200 mmol) without exogenous activator
(NATEM). Measurement was started immediately after the last pipetting and
blood clot
formation was continuously monitored for 2 hours (7200 seconds) at 37 C.
[064] ROTEM analysis parameters for hemostasis include Clotting Time (CT),
the
time required to obtain clot firmness of 2 mm following the initiation of
measurement,
Clot Formation Time (CFT), the time from clot firmness of 2 mm till clot
strength of
20 mm, and a-angle, the velocity of clot formation.
[065] As shown in Fig. 5, BHRF-1-FVIII required less time to form a clot in
the
ROTEM assay than an equal dose (based on a chromogenic assay) of unconjugated
FVIII, indicating a higher efficiency of clotting. The difference in CT was
about 400
seconds, which corresponds to roughly 2-3 fold more FVIII activity, based on
FVIII
standard curves.
[066] Hemostatic activity and pharmacokinetic parameter of targeted
coagulation factors
can be assessed in vivo using the hemophilia A mouse model. Targeted
coagulation
16
CA 02724630 2010-11-16
WO 2009/140598 PCT/US2009/044148
factors can be administered by tail vein intravenous injection. At multiple
time points
after the treatment, blood will be collected in % sodium citrate and
hemostatic activity will
be measured using ROTEM over 48 hours post infusion period which is equivalent
to > 6
half-life of FVIII (t1/2) in mice.
Example 5: In vitro Binding Assay to Human and Mouse Platelets
Binding of FVIII-BHRF-1 to human platelets
[067] Human platelets were obtained from Allcells (Emeryville, CA) at 5 x 109
platelets/tube in 14 mL plasma. The platelets and all washes, buffers,
reagents, and
centrifuges were warmed to room temperature and maintained at room temperature
during
the course of the experiment. The wash buffer (WB) for the platelets is
Tyrode's buffer
supplemented with 20 mM HEPES, 0.5% BSA, and 50 ng/mL PGE1 and 2.5 U/mL
apyrase, pH 7.4.
[068] The cells were centrifuged at 700 x g for 15 minutes at 25 C, and then
the
supernatant was carefully removed and 14 mL WB was added. The cells were
gently re-
suspended in the WB and centrifuged as described.
[069] Following the second centrifugation, the supernatant was removed and the
platelets
were re-suspended in 15 mL WB. At this point, the cells were split into three
equal
aliquots of 5 mL each. The three aliquots were centrifuged as described
earlier, and then
the three platelet pellets were re-suspended in either:
A. 5 mL binding buffer + 5 mg/mL BSA (BBB, 50 mM Tris, 100 mM NaCl, 1
mM each CaC12, MgC12, and MnC12)
B. 5 mL HemA plasma which lacks FVIII, but vWF is present
C. 5 mL immuno-depleted plasma lacking both FVIII and vWF.
[070] For buffer (A) or plasma (B or C), the following conditions were used:
1. buffer/plasma alone + 2.5 nM BHRF-I-FVIII (containing about 20%
uncongugated FVIII (see Example 3))
2. buffer/plasma + platelet + 2.5 nM BHRF-I-FVIII (containing about 20%
uncongugated FVIII)
3. buffer/plasma alone + 2.5 nM recombinant FVIII
4. buffer/plasma + platelet + 2.5 nM recombinant FVIII
17
CA 02724630 2010-11-16
WO 2009/140598 PCT/US2009/044148
[071] For each condition 1-4, 100 gL A, B, or C was pipetted into a microfuge
tube at
room temperature, then the BHRF-I-FVIII or unconjugated FVIII was added to the
tube.
The tubes were incubated at 37 C for 1.5 hours (without shaking). Following
the
incubation period, the tubes were centrifuged at maximum speed (16,000 rpm)
for
minutes to pellet the platelets. The supernatant was collected to assay for
FVIII activity.
The amount of activity in the supernatant reflects the amount of unbound FVIII
or BHRF-
1-FVIII. The data demonstrate binding of the BHRFI-FVIII to human platelets in
all
conditions (shown in Fig. 6). Since the BHRF-I-FVIII contains roughly 20%
unconjugated FVIII for conditions A and C, the data indicate that a high
percentage of
conjugate was bound. There was no binding of FVIII observed for conditions A
and B,
while 35% of the FVIII activity was bound in condition C. The figure also
shows the level
of FVIII activity remaining for condition C corrected for the 35% non-specific
binding of
FVIII were observed for this condition (i.e., the starting FVIII activity was
reduced by
35% to calculate the percentage bound).
Binding of FVIII-BHRF-1 to mouse platelets
[072] BHRF-I-FVIII also bound to mouse platelets as shown in Fig. 7. A similar
binding assay was performed as described for human platelets except that
citrated mouse
blood was centrifuged 200 x g for 15 minutes to harvest platelet rich plasma
(PRP). The
PRP was diluted with citrate wash buffer (11 mM glucose, 128 mM NaCl, 4.3 mM
NaH2PO4, 7.5 mM Na2HPO4, 4.8 mM Na-citrate, 2.4 mM citric acid, 0.35% BSA, pH
6.5)
+ 50 ng/mL PGE1, and washed twice in citrate wash buffer + 50 ng/mL PGE1 (by
centrifuging at 1200 x g for 10 minutes). The platelets were finally re-
suspended in
binding buffer (50 mM Tris, 100 mM NaCl, 1mM each CaC12, MgC12, and MnC12) +
5 mg/mL BSA. Un-conjugated FVIII and BHRF- 1-F VIII were added to the
platelets and
after 2 hours at 37 C, the platelets were removed by centrifugation, and the
unbound FVIII
activity in the supernatant determined.
[073] As shown in the Fig. 7, 59% of the activity of unconjugated FVIII bound
to the
platelets. To calculate the percentage of the added BHRF-1-FVIII activity
binding to
platelets through the BHRF-1 peptide, the amount of starting FVIII activity
was corrected
by 59% to reflect the level of non-specific binding of FVIII (not occurring
through the
peptide). The corrected value for BHRF-I-FVIII was 31% unbound (69% bound).
When
100 uM integrilin was added to complete for peptide binding, unbound activity
rose to
18
CA 02724630 2010-11-16
WO 2009/140598 PCT/US2009/044148
82% unbound (18% bound) (also corrected for nonspecific FVIII binding). These
data
demonstrate that BHRF-I-FVIII can bind to mouse platelets through the BHRF-1
targeting
domain.
Example 6: Pharmacokinetic Study
[074] The level of FVIII in blood at various times after injection into
hemophilia A mice
is determined using a whole blood coagulation assay such as ROTEM described
above,
which reflects FVIII activity in both plasma and bound to cells (e.g.,
platelets).
Example 7: Chromogenic Assay for the Assessment of FVIII Activity
[075] FVIII activity of purified proteins and conjugates was assessed using
the Coatest
SP assay kit (Chromogenix, Lexington, MA). The assay was performed following
the
manufacturer's instructions in a 96-well plate format. Briefly, diluted
samples containing
FVIII or conjugate were combined in order with a mixture of activated
FIX/FX/phospholipid, followed by 25 mM CaClz and chromogenic substrate S-
2765/I-
2581. Between each reagent addition, the samples were incubated at 37 C for 5
minutes.
After the final addition of chromogenic substrate, the reaction was stopped
after 5 minutes
with 20% acetic acid and the plate absorbances were read at 405 nm, normalized
against a
490 nm background. Sample absorbances were calibrated against a WHO/NIBSC
plasma-
derived FVIII standard curve with an operating range of 0.3-40 mIU/mL.
Example 8: In vivo Efficacy Assay in Hemophilic Mice
[076] To show the efficacy of targeted FVIII molecules in promoting blood
clotting and
to assess the duration of these effects, the tail clip injury or tail vein
transection models
which use hemophilic (HemA) mice, can be used as described below.
Tail Clip Injury Model
[077] Test samples are administrated to the mice via a tail vein injection.
Following
administration, the mice are anesthetized intraperitoneal (IP) with
ketamine/xylazine
(100 mg/kg, 10 mg/kg). When the animals are fully anesthetized, the tails are
placed
individually in 13 mL 37 C pre-warmed saline for approximately 10 minutes. A
tail cut is
made with a sharp scalpel and the tail is placed immediately in a new tube
with 9 mL 37 C
warm saline. Blood is collected continuously for 30 minutes. Blood loss volume
is
19
CA 02724630 2010-11-16
WO 2009/140598 PCT/US2009/044148
determined either by weight gain of the blood collection tube or determined by
the optical
density of the blood/saline mixture in the blood collection tube.
Tail Vein Transection
[078] HemA male mice are randomized into different treatment groups by their
body
weight. Mice are dosed by tail vein injection 24 hours prior to the tail vein
transaction.
Before the tail vein transection, mice are anesthetized (IP) with a cocktail
containing
50 gg/kg of ketamine and 1 mg/kg of medetomidine. The tail is marked at a
diameter of
2.7 mm using a french catheter. The anesthetic effect of medetomidine is
reversed with
1 mg/kg of atipamezole by IP injection. The tail vein is transected with a
scalpel blade.
The tail is then submerged into 37 C saline tube, and the tube is rotated to
rinse away the
blood from the cut. When the saline becomes too opaque to visualize, it is
replaced with a
new tube until the tail stops bleeding. The time it takes to stop bleeding is
recorded as the
acute clotting time. The mouse is then returned to its individual clean cage
with white
paper bedding placed on top of a 4 x 8 inch heating pad. The time to re-bleed
and
moribund is monitored hourly for the next 9-11 hours for excessive blood loss.
Example 9: Recombinant Expression of Targeted FVIII
[079] In one embodiment, HKB 11 cells are grown in suspension culture on an
orbital
shaker (100-125 rpm) in a 5% CO2 incubator at 37 C in a protein-free media and
maintained at a density between 0.25 and 1.5 x 106 cells/mL. HKB 11 cells for
transfection are collected by centrifugation then resuspended in an expression
medium
such as FreeStyleTM 293 Expression Medium (Invitrogen, Carlsbad, CA) at 1.1 x
106
cells/mL. The cells are seeded in 6-well plates (4.6 mL/well) and incubated on
an orbital
rotator (125 rpm) in a 37 C CO2 incubator. For each well, 5 g plasmid DNA is
mixed
with 0.2 mL Opti-MEMO I medium (Invitrogen, Carlsbad, CA). For each well, 7 L
293fectinTM reagent (Invitrogen, Carlsbad, CA) is mixed gently with 0.2 mL
Opti-MEMO
I medium and incubated at room temperature for 5 minutes. The diluted
293fectinTM is
added to the diluted DNA solution, mixed gently, incubated at room temperature
for 20-30
minutes, and then added to each well that has been seeded with 5 x 106 (4.6
mL) HKB11
cells. The cells are then incubated on an orbital rotator (125 rpm) in a C02
incubator at
37 C for 3 days after which the cells are pelleted by centrifugation at 1000
rpm for
minutes and the supernatant is collected.
CA 02724630 2010-11-16
WO 2009/140598 PCT/US2009/044148
[080] Stable transfection of HKB11 cells is obtained using the following
procedure.
HKB11 cells are transfected with plasmid DNA using 293fectinTM reagent as
described in
transient transfection. The transfected cells are split into 100-mm culture
dishes at various
dilutions (1:100, 1:1000, 1; 10,000) and maintained in DMEM-F 12 medium
supplemented
with 5% FBS and 200 ug/mL hygromicin (Invitrogen, Carlsbad, CA) for about 2
weeks.
Individual single colonies are picked and transferred into 6-well plates using
sterile
cloning disks (Scienceware , Sigma-Aldrich, St. Louis, MO). The clones are
established
and banked. These clones are screened for high expression of the fusion
protein by FVIII
activity assays (e.g., Coatest and aPTT assays) as well as by FVIII ELISA.
[081] Factor VIII activity levels in culture supernatants and purification
fractions may be
determined using a commercial chromogenic assay kit (Coatest SP4 FVIII,
Chromogenix, Lexington, MA) in a 96-well format as described above. Factor
VIII
coagulation activity may also be determined using an aPTT assay in FVIII-
deficient
human plasma by an Electra 18000 automatic coagulation analyzer (Beckman
Coulter,
Fullerton, CA). Briefly, three dilutions of supernatant samples in coagulation
diluent are
created by the instrument and 100 L is then mixed with 100 L FVIII-deficient
plasma
and 100 L automated aPTT reagent (rabbit brain phospho lipid and micronized
silica,
Biomerieux, Durham, NC). After the addition of 100 L 25mM CaC12 solution, the
time
to clot formation is recorded. A standard curve is generated for each run
using serial
dilutions of the same purified FVIII used as the standard in the ELISA assay.
[082] While the present invention has been described with reference to the
specific
embodiments and examples, it should be understood that various modifications
and
changes may be made and equivalents may be substituted without departing from
the true
spirit and scope of the invention. The specification and examples are,
accordingly, to be
regarded in an illustrative rather then a restrictive sense. Furthermore, all
articles, patent
applications and patents referred to herein are incorporated herein by
reference in their
entireties.
21