Note: Descriptions are shown in the official language in which they were submitted.
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
PIGF-1 COMPANION DIAGNOSTIC METHODS AND PRODUCTS
RELATED APPLICATION INFORMATION
This application claims the benefit of U.S. Application No. 12/485,114, filed
on
June 16, 2009, the benefit of U.S. Application Serial No. 61/110,063 filed
October 31,
2008, U.S. Application No. Number 61/073,624 filed on June 18, 2008 and U.S.
Serial
No. 61/089,172 filed on August 15, 2008, the contents of each of which are
herein
incorporated by reference.
REFERENCE TO JOINT RESEARCH AGREEMENT
Contents of this disclosure are under a joint research agreement entered into
by
and between Genentech and Abbott Laboratories on June 22, 2007, directed to N-
[4-(3-
amino-1 H-indazol-4-yl)phenylJ-N'-(2-fluoro-5-methylphenyl)urea (also known as
ABT-869).
TECHNICAL FIELD
The present disclosure relates, among other things, to methods for determining
whether a subject receiving treatment with a drug (e.g., such as for cancer)
has obtained
an efficacious blood level of the drug by monitoring the levels of biomarkers
of
angiogenesis. Moreover, the present disclosure also relates to methods of
determining
whether a subject predisposed to or suffering from a disease will benefit from
treatment
with a drug, and the response of a subject receiving treatment (e.g., such as
for cancer)
by monitoring biomarkers of angiogenesis. In particular, the disclosure
relates to
P/GF-1 companion diagnostic methods and products.
BACKGROUND
Neoangiogenesis is crucial for tumor growth. Specifically, neoangiogenesis is
a
complex process that involves the imbalance of antiangiogenic and
proangiogenic
molecules such as vascular endothelial growth factors secreted from tumor
cells,
macrophages and stromal cells that interact with vascular endothelial growth
factor
receptor (VEGFR) I and 2 to activate endothelial cells from existing
microvasculature
and circulating endothelial progenitor cells (See, Dvorak, HF, J Clin Oncol.,
20:4368-
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
4380 (2002)). Tumor vasculature is disorganized and more leaky than normal
vasculature, thereby resulting in increased permeability, raised interstitial
pressure and
poorer chemotherapy distribution into the tumor (See, Ferrara, N., VEGF
Oncology,
69: S 11-S 16, (2005)(suppl 3)). Platelet derived growth factor through
binding to its
receptor (PDGFR) activates tumor growth and enhances angiogenesis by
facilitating
pericyte coverage of new microvessels (See, Carmeliet, P., Nat Med., 9:653-660
(2003); Benjamin LE, et al., J Clin Invest., 103:159-165 (1999)), while VEGFR-
3
contributes to the process by facilitating lymphangiogenesis, and may be
potentially
important for tumor metastases (Sundar, SS, et al., JClin Oncol., 25:4298-4307
(2007)). Overactivation of these pathways promotes growth, tumor survival and
metastasis of cancer cells (See, Blume-Jensen P, et al., Nature, 411:355-365
(2001)). In
view thereof, VEGFR-1,-2,-3 and PDGFRs are important targets for inhibiting
angiogenesis and lymphangiogenesis as well as the restoration of normalized
vasculature. It is believed that the combined inhibition of VEGFR-1,-2,-3 and
PDGFRs
might disrupt tumor growth, survival and metastases more effectively than the
specific
inhibition of each receptor alone. Sorafenib and sunitinib are examples of
small
molecule receptor tyrosine kinase (RTK) inhibitors that are used for treating
metastatic
renal cell carcinoma (See, Batchelor, TT, et al., Cancer Cell, 11:83-95
(2007);
Escudier, B, et al., N Engl JMed 356:125-134 (2007)), in.which the loss of the
von
Hippel Lindau gene results in aberrant activation of hypoxic inducible factor
complex
and consequent VEGF and PDGF over-expression (Motzer, RJ, et al., NEngl JMed.,
356:115-124 (2007)).
A number of biomarkers associated with specific types of diseases are known.
In this era of personalized medicine (involving the use of new methods of
analysis and
bioinformatics to better address and manage a patient's disease or
predisposition to
disease) and pharmacogenomics (combining the science of drugs and genomics)
have
promoted the use and interrogation of so-called "companion diagnostics", which
are
diagnostic products intended for use in conjunction with a therapeutic product
to better
inform treatment selection, initiation, dose customization, or avoidance.
Establishing a
correlation between biomarkers and the pharmacological effect and biologic
activity of
a drug or pharmaceutical composition via assessment of plasma levels of these
biomarkers thus can be used to individualize patient prognostication and
treatment
2
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
strategies. In this regard, a number of biomarkers are associated with
angiogenesis.
These include, but are not limited to, VEGF-A, soluble VEGFR-I (sVEGFR-1, also
known as sFlt-1), soluble VEGFR-2 (sVEGFR-2 also known as sKDR), soluble
VEGFR-3 (sVEGFR-3), placenta growth factor (PIGF, a member of the VEGF family
of growth factors and a specific ligand of VEGFR-1), erythropoietin, etc.
Thereupon, there is a need in the art for methods for monitoring or
determining
whether a subject receiving treatment with a drug (e.g., such as for cancer)
has obtained
an efficacious blood level of the drug by monitoring biomarkers of
angiogenesis,
particularly P1GF. Specifically, there is also a need in the art for methods
of identifying
whether a subject, after receiving just a single dose of a drug (e.g., such as
for cancer)
would benefit from continued treatment with said drug. Moreover, there is also
a need
in the an for methods of monitoring the response of a subject receiving
treatment for a
tumor (e.g., such as for cancer) by monitoring biomarkers of angiogenesis,
particularly
PIGF, and especially P1GF-1. Still further, there is also a need in the art
for methods of
using pharmacodynamic based dosing information to bring subjects into the
necessary
drug exposure range (e.g., area under the curve ("AUC")) that is been
associated or
correlated with the efficacy of a specific drug. These and other objects of
the
disclosure will be apparent from the description following herein.
SUMMARY
In one embodiment, the present disclosure provides a method of monitoring
whether a subject being administered a drug has obtained an efficacious blood
level of
the drug in order to optimize dosing or scheduling, the method comprising the
steps of
(a) contacting a test sample obtained from a subject being administered N-[4-
(3-amino-IH-indazol-4-yl)phenyl]-N'-(2-fluoro-5-methylphenyl)urea or an analog
of
N-[4-(3-amino-lH-indazol-4-yl)phenyl]-N'-(2-fluoro-5-methylphenyl)urea with a
first
capture antibody that binds to human P1GF-1 or a human P1GF-1 fragment to form
a
first capture antibody-human P/GF-1 complex,
(b) contacting the first capture antibody-human P1GF-1 complex with a second
antibody that binds to human P1GF-1 or human P1GF-1 fragment and that has been
conjugated to a detectable label ("detection antibody") to form a second
capture
antibody-human PIGF-1 detection complex;
3
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
(c) determining the amount of the second capture antibody-human P1GF-1
detection complex formed in step (b) by detecting the detectable label,
wherein the
amount of the second complex formed is the amount of human P1GF-1 or human
PIGF-
1 fragment contained in the test sample; and ,
(d) comparing the amount of human P1GF-1 or human PIGF-1 fragment in the
test sample determined in step (c) with a predetermined level, wherein if the
concentration of human P1GF-1 or human P/GF-1 fragment determined in step (c)
is
lower than the predetermined level, then the subject is considered not to be
receiving an
efficacious amount of N-[4-(3-amino-1 H-indazol-4-yl)phenyl]-N'-(2-fluoro-5-
methylphenyl)urea or an analog ofN-[4-(3-amino-lH-indazol-4-yl)phenyl]-N'-(2-
fluoro-5-methylphenyl)urea and further wherein, if the concentration of human
PIGF-1
or human P1GF-1 fragment determined in step (c) is the same as or higher than
the
predetermined level, then the subject is considered to be receiving an
efficacious
amount of N-[4-(3-amino-lH-indazol-4-yl)phenyl]-N'-(2-fluoro-5-
methylphenyl)urea
or analog ofN-[4-(3-amino-lH-indazol-4-yl)phenyl]-N'-(2-fluoro-5-
methylphenyl)urea.
In one aspect of such a method, the capture antibody is monoclonal antibody
264 and the detection antibody is polyclonal antibody pB264. Optionally, the
predetermined level is about 30 picograms per milliliter at about 24 hours
after the
subject first receives treatment with N-[4-(3-amino-lH-indazol-4-yl)phenyl]-N'-
(2-
fluoro-5-methylphenyl)urea or an analog ofN-[4-(3-amino-lH-indazol-4-
yl)phenyl]-
N'-(2-fluoro-5-methylphenyl)urea. Alternately, optionally, the predetermined
level is
about 40 picograms per milliliter to about 75 picograms per milliliter at when
the
patient has achieved steady state concentrations following treatment with N-[4-
(3-
amino-lH-indazol-4-yl)phenyl]-N'-(2-fluoro-5-methylphenyl)ureaor an analog of
N-
[4-(3-amino-lH-indazol-4-yl)phenyl]-N'-(2-fluoro-5-methylphenyl)urea.
In another aspect of such a method, the capture antibody is monoclonal
antibody 826 and the detection antibody is monoclonal antibody 255. In one
aspect of
this method, when the capture antibody is a monoclonal antibody and the
detection
antibody is a monoclonal antibody and the concentration of human PIGF-1 or
human
PIGF-1 fragment determined in step (c) is increased by about 60 picograms per
milliliter to about 150 picograms per milliliter when compared to the
predetermined
level at the steady state about either 8 or 15 days after the subject first
receives
4
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
treatment with N-[4-(3-amino-1 H-indazol-4-yl)phenyl]-N'-(2-fluoro-5-
methylphenyl)urea or an analog ofN-[4-(3-amino-lH-indazol-4-yl)phenyl]-N'-(2-
fluoro-5-methylphenyl)urea.
The methods as described herein can be employed wherein the subject is being
treated for cancer selected from the group consisting of lung cancer, breast
cancer,
stomach cancer, bladder cancer, colon cancer, pancreatic cancer, ovarian
cancer,
prostate cancer, renal cancer, hepatocellular cancer, rectal cancer,
hematopoietic
malignancies, glioblastoma and infantile hemangioma, among others. In one
embodiment, the method is adapted for use in an automated system or semi-
automated
system.
In one aspect of the method, when the capture antibody is a monoclonal
antibody and the detection antibody is a polyclonal antibody, the dose or
schedule for
treatment with N-[4-(3-amino-1 H-indazol-4-yl)phenyl]-N'-(2-fluoro-5-
methylphenyl)urea or an analog of N-[4-(3-amino-lH-indazol-4-yl)phenyl]-N'-(2-
fluoro-5-methylphenyl)urea can be adjusted to place the patient.in the range
of about 40
picograms per milliliter to about 75 picograms per milliliter based on the
comparison in
step (d).
In another aspect of the method, when the capture antibody is a monoclonal
antibody and the detection antibody is a monoclonal antibody, the dose or
schedule for
treatment with N-[4-(3-amino-lH-indazol-4-yl)phenyl]-N'-(2-fluoro-5-
methylphenyl)urea or an analog ofN-[4-(3-amino-lH-indazol-4-yl)phenyl]-N'-(2-
fluoro-5-methylphenyl)urea can be adjusted to place the patient in the range
of about 60
picograms per milliliter to about 150 picograms per milliliter based on the
comparison
in step (d).
Accordingly provided by the description herein is a method of monitoring a
response of a subject receiving treatment for cancer with an anti-cancer drug,
the
method comprising the steps of.
(a) contacting a test sample obtained from a subject receiving treatment with
N-
[4-(3-amino-IH-indazol-4-yl)phenyl]-N'-(2-fluoro-5-methylphenyl)urea or an
analog of
N-[4-(3-amino-lH-indazol-4-yl)phenyl]-N'-(2-fluoro-5-methylphenyl)urea with a
first
capture antibody that binds to human PIGF-1 or human P/GF-1 fragment to form a
first
capture antibody-human P/GF-I complex;
5
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
(b) contacting the first capture antibody-human P1GF-1 complex with a second
antibody that binds to human PIGF-1 and that has been conjugated to a
detectable label
("detection antibody") to form a second capture antibody-human P1GF-1
detection
complex;
(c) determining the amount of the second capture antibody-human P/GF-1
detection complex formed in step (b) by detecting the detectable label,
wherein the
amount of the second complex formed is the amount of human P/GF-1 or human
P/GF-
1 contained in the test sample; and
(d) comparing the amount of human P/GF-1 or human PIGF-1 in the test
sample determined in step (c) with a predetermined level, wherein if the
concentration
of human P1GF-1 or human P/GF-1 fragment determined in step (c) is lower than
the
predetermined level, then the subject is considered not to be responding to
treatment
with the N-[4-(3-amino-IH-indazol-4-yl)phenyl]-N'-(2-fluoro-5-
methylphenyl)urea or
an analog of N-[4-(3-amino-lH-indazol-4-yl)phenyl]-N'-(2-fluoro-5-
methylphenyl)urea
and treatment with N-[4-(3-amino-IH-indazol-4-yl)phenyl]-N-(2-fluoro-5-
methylphenyl)urea or an analog ofN-[4-(3-amino-lH-indazol-4-yI)phenyl]-N'-(2-
fluoro-5-methylphenyl)urea is discontinued and the subject is administered an
anti-
cancer drug other than N-[4-(3-amino-lH-indazol-4-yl)phenyl]-N'-(2-fluoro-5-
methylphenyl)urea or an analog ofN-[4-(3-amino-lH-indazol-4-yl)phenyl]-N'-(2-
fluoro-5-methylphenyl)urea and further wherein, if the concentration of human
P/GF-1
or human P/GF-1 fragment determined in step (c) is the same as or higher than
the .
predetermined level, then the subject is considered to be responding to
treatment with
the N-[4-(3-amino-1 H-indazol-4-yl)phenyl]-N'-(2-fluoro-5-methylphenyl)urea or
analog of N-[4-(3-amino-1 H-indazol-4-yl)phenyl]-N'-(2-fluoro-5-
methylphenyl)urea.
In one aspect of this method, optionally, the capture antibody is monoclonal
antibody 264: and the detection antibody is polyclonal antibody pB264. In
another
aspect of this method, optionally, the capture antibody is monoclonal antibody
826 and
the detection antibody is monoclonal antibody 255. The method can be employed
where the cancer is selected from the group consisting of lung cancer, breast
cancer,
stomach cancer, bladder cancer, colon cancer, pancreatic cancer, ovarian
cancer,
prostate cancer, renal cancer, hepatocellular cancer, rectal cancer, rectal
cancer,
hematopoietic malignancies, glioblastoma and infantile hemangioma, among
others. In
6
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
one embodiment, the method is adapted for use in an automated system or semi-
automated system.
Further provided herein is another method that provides for determining
whether a subject who is predisposed to a disease or who is suffering from a
disease
will benefit from receiving treatment with a drug. Optionally the method
comprises the
steps of
(a) contacting a test sample obtained from a subject predisposed to a disease
or
suffering from at least one disease and administered N-[4-(3-amino-lH-indazol-
4-
yl)phenyl]-N'-(2-fluoro-5-methylphenyl)urea or an analog ofN-[4-(3-amino-lH-
indazol-4-yl)phenyl]-N'-(2-fluoro-5-methylphenyl)urea with a first capture
antibody
that binds to human P1GF-1 or human P1GF-1 fragment to form a first capture
antibody-human PIGF-I complex;
(b) contacting the first capture antibody-human PIGF-1 complex with a second
antibody that binds to human PIGF-1 and that has been conjugated to a
detectable label
("detection antibody") to form a second capture antibody-human PIGF-1
detection
complex;
(c) determining the amount of the second capture antibody-human P1GF-1
detection complex formed in step (b) by detecting the detectable label,
wherein the
amount of the second complex formed is the amount of human PIGF-1 or human
P1GF-
1 contained in the test sample; and
(d) comparing the amount of human PIGF-1 or human P1GF- I in the test
sample determined in step (c) with a predetermined level, wherein if the
concentration
of human P1GF-1 or human P1GF-1 fragment determined in step (c) is lower than
the
predetermined level, then a determination is made that the subject will not
benefit from
receiving further or continued treatment with the N-[4-(3-amino-lH-indazol-4-
yl)phenyl]-N'-(2-fluoro-5-methylphenyl)urea or an analog of N-[4-(3-amino- I H-
indazol-4-yl)phenyl]-N'-(2-fluoro-5-methylphenyl)urea and further wherein, if
the
concentration of human PIGF-1 or human PIGF-1 fragment determined in step (c)
is the
same as or higher than the predetermined level, then a determination is made
that the
subject will benefit from receiving further or continued treatment with the N-
[4-(3-
amino-1 H-indazol-4-yl)phenyl]-N'-(2-fluoro-5-methylphenyl)urea or analog of N-
[4-(3-
amino-1 H-indazol-4-yl)phenyl]-N'-(2-fluoro-5-methylphenyl)urea.
7
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
In one aspect of this method, optionally, the capture antibody is monoclonal
antibody 264 and the detection antibody is polyclonal antibody pB264. In
another
aspect of this method, optionally, the capture antibody is monoclonal antibody
826 and
the detection antibody is monoclonal antibody 255. In yet another aspect,
optionally at
least one disease is cancer selected from the group consisting of lung cancer,
breast
cancer, stomach cancer, bladder cancer, colon cancer, pancreatic cancer,
ovarian
cancer, prostate cancer, renal cancer, hepatocellular cancer, rectal cancer,
rectal cancer,
hematopoietic malignancies, glioblastoma and infantile hemangioma. In one
embodiment, the method is adapted for use in an automated system or semi-
automated
system.
Also provided herein is a method of predicting the likelihood of response of a
subject to treatment, wherein the method comprises the steps of
(a) obtaining a first test sample prior to initiation of treatment obtained
from a
subject suffering from a disease and being administered N-[4-(3-amino-lH-
indazol-4-
yl)phenyl]-N'-(2-fluoro-5-methylphenyl)urea or an analog ofN-[4-(3-amino-lH-
i ndazol-4-yl)phenyl]-N'-(2-fluoro-5-methylphenyl)urea;
(b) contacting the first test sample with a first capture antibody that binds
to
human PIGF-1 or a human P1GF-l fragment to form a first capture antibody-human
P1GF-1 complex,
(c) contacting the first capture antibody-human P1GF-1 complex with a second
antibody that binds to human P1GF-1 or human PIGF-1 fragment and that has been
conjugated to a detectable label ("detection antibody") to form a second
capture
antibody-human P1GF-1 detection complex;
(d) determining the amount of the second capture antibody-human P1GF-1
detection complex formed in step (b) by detecting the detectable label,
wherein the
amount of the second complex formed is the concentration of human PIGF-1 or
human
PIGF-I fragment contained in the first test sample;
(e) obtaining a second test sample obtained from the subject suffering from
the
disease and being administered N-[4-(3-amino-lH-indazol-4-yl)phenyl]-N'-(2-
fluoro-5-
methylphenyl)urea or an analog ofN-[4-(3-amino-lH-indazol-4-yl)phenyl]-N'-(2-
fluoro-5-methylphenyl)urea at a period in time after obtaining the first test
sample in
step (a);
8
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
(f) contacting the second test sample with a first capture antibody that binds
to
human P/GF-1 or a human P/GF-1 fragment to form a first capture antibody-human
P1GF-1 complex;
(g) contacting the first capture antibody-human P1GF-I complex with a second
antibody that binds to human P1GF-1 or human P1GF-1 fragment and that has been
conjugated to a detectable label ("detection antibody") to form a second
capture
antibody-human P/GF-1 detection complex;
(h) determining the amount of the second capture antibody-human P/GF-I
detection complex formed in step (g) by detecting the detectable label,
wherein the
amount of the second complex formed is the concentration of human P/GF-1 or
human
P1GF-I fragment contained in the second test sample; and
. (i) comparing the concentration of human P1GF-1 or human P1GF-I fragment
in step (d) with the concentration of human P1GF-1 or human PIGF-1 fragment in
step
(h), wherein if the concentration of human P/GF-I or human P/GF-1 fragment
determined in step (h) has increased when compared to the concentration of
human
P/GF-1 or human P1GF-1 fragment determined in step (d), then the patients with
higher
levels of PIGF-1 or human P1GF-1 fragment are more likely to respond if the
concentration of human P/GF-I or human P/GF-1 fragment determined in step (h)
is
unchanged or higher when compared to the concentration of human P1GF-1 or
human
P1GF-1 fragment in step (d). .
In one aspect of this method, the period of time between obtaining the first
test
sample and the second test sample is a time period selected from the group
consisting
of about 1 minute, about 5 minutes, about 10 minutes, about 15 minutes, about
30
minutes, about 45 minutes, about 60 minutes, about 2 hours, about 3 hours,
about 4
hours, about 5 hours, about 6 hours, about 7 hours, about 8 hours, about 9
hours, about
10 hours, about 11 hours, about 12 hours, about 13 hours, about 14 hours,
about 15
hours, about 16 hours, about 17 hours, about 18 hours, about 19 hours, about
20 hours,
about 21 hours, about 22 hours, about 23 hours, about 24 hours, about 2 days,
about 3
days, about 4 days, about 5 days, about 6 days, about 7 days, about 2 weeks,
about 3
weeks, about 4 weeks, about 5 weeks, about 6 weeks, about 7 weeks, about 8
weeks,
about 9 weeks, about 10 weeks, about 11 weeks, about 12 weeks, about 13 weeks,
about 14 weeks, about 15 weeks, about 16 weeks, about 17 weeks, about 18
weeks,
9
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
about 19 weeks, about 20 weeks, about 21 weeks, about 22 weeks, about 23
weeks,
about 24 weeks, about 25 weeks, about 26 weeks, about 27 weeks, about 28
weeks,
about 29 weeks, about 30 weeks, about 31 weeks, about 32 weeks, about 33
weeks,
about 34 weeks, about 35 weeks, about 36 weeks, about 37 weeks, about 38
weeks,
about 39 weeks, about 40 weeks, about 41 weeks, about 42 weeks, about 43
weeks,
about 44 weeks, about 45 weeks, about 46 weeks, about 47 weeks, about 48
weeks,
about 49 weeks, about 50 weeks, about 51 weeks, about 52 weeks, about 1.5
years,
about 2 years, about 2.5 years, about 3.0 years, about 3.5 years, about 4.0
years, about
4.5 years, about 5.0 years, about 5.5. years, about 6.0 years, about 6.5
years, about 7.0
years, about 7.5 years, about 8.0 years, about 8.5 years, about 9.0 years,
about 9.5
years, and about 10.0 years. Optionally, the capture antibody is monoclonal
antibody
264 and the detection antibody is polyclonal antibody pB264. Alternatively,
optionally,
the capture antibody is monoclonal antibody 826 an d the detection antibody is
monoclonal antibody 255. Further, optionally, the at least one disease is
cancer
selected from the group consisting of lung cancer, breast cancer, stomach
cancer,
bladder cancer, colon cancer, pancreatic cancer, ovarian cancer, prostate
cancer, renal
cancer, hepatocellular cancer, rectal cancer, rectal cancer, hematopoietic
malignancies,
glioblastoma and infantile hemangioma. Further optionally, in one aspect, the
method
is adapted for use in an automated system or semi-automated system.
In still yet another embodiment, the present invention relates to a method of
treating a subject suffering from at least one cancer selected from the group
consisting
of lung cancer, breast cancer, stomach cancer, bladder cancer, colon cancer,
pancreatic
cancer, ovarian cancer, prostate cancer, renal cancer, hepatocellular cancer,
rectal
cancer, hematopoietic malignancies, glioblastoma and infantile hemangioma.
Such a
method comprises the steps of
(a) obtaining a test sample from the subject suffering from cancer and who is
receiving treatment with a predetermined amount ofN-[4-(3-amino-lH-indazol-4-
yl)phenyl]-N'-(2-fluoro-5-methylphenyl)urea or an analog of N-[4-(3-amino- I H-
indazol-4-yl)phenyl]-N'-(2-fluoro-5-methylphenyl)urea;
(b) contacting the test sample with a first capture antibody that binds to
human
PIGF-1 or a human PIGF-1 fragment to form a first capture antibody-human PIGF-
1
complex,
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
(c) contacting said first capture antibody-human P1GF-1 complex with a second
antibody that binds to human P1GF-1 or human P1GF-1 fragment and that has been
conjugated to a detectable label ("detection antibody") to form a second
capture
antibody-human PIGF-1 detection complex;
(d) determining the amount of the second capture antibody-human PIGF-1
detection complex formed in step (c) by detecting the detectable label,
wherein the
amount of the second complex formed is the amount of human P1GF-1 or human
P1GF-
1 fragment contained in the test sample;
(e) comparing the amount of human P1GF-1 or human PIGF-1 fragment in the
test sample determined in step (d) with a predetermined level; and
(f) treating a subject having a concentration of human PIGF-I or human PIGF-1
fragment determined in step (d) that is lower than the predetermined level
with: (i) an
adjusted amount of N- [4-(3-amino-IH-indazol-4-yl)phenyl]-N'-(2-fluoro-5-
methylphenyl)urea or an analog ofN-[4-(3-amino-1H-indazol-4-yl)phenyl]-N'-(2-
fluoro-5-methylphenyl)urea that is higher than the predetermined amount of N-
[4-(3-
amino-iH-indazol-4-yl)phenyl]-N'-(2-fluoro-5-methylphenyl)urea or an analog
ofN-
[4-(3-amino-IH-indazol-4-yl)phenyl]-N'-(2-fluoro-5-methylphenyl)urea recited
in step
(a); (ii) a drug other N-[4-(3-amino-IH-indazol-4-yl)phenyl]-N'-(2-fluoro-5-
methylphenyl)urea or an analog ofN-[4-(3-amino-1H-indazol-4-yl)phenyl]-N'-(2-
fluoro-5-methylphenyl)urea; or (iii) combinations of (i) and (ii).
In the above method, the capture antibody is monoclonal antibody 264 and the
detection antibody is polyclonal antibody pB264.
Moreover, in the above method, optionally, the predetermined level when the
capture antibody is a monoclonal antibody and the detection antibody is a
polyclonal
antibody assay is about 30 picograms per milliliter at about 24 hours after
the subject
first receives treatment with N-[4-(3-amino- I H-indazol-4-yl)phenyl]-N'-(2-
fluoro-5-
methylphenyl)urea or an analog ofN-[4-(3-amino-IH-indazol-4-yl)phenyl]-N'-(2-
fluoro-5-methylphenyl)urea. Still further optionally, in the above method, the
predetermined level when the capture antibody is a monoclonal antibody and the
detection antibody is a polyclonal antibody assay is about 40 picograms per
milliliter to
about 75 picograms per milliliter at about 15 days after the subject first
receives
treatment with N-[4-(3-amino-IH-indazol-4-yl)phenyl]-N'-(2-fluoro-5-
11
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
methylphenyl)urea or an analog ofN-[4-(3-amino-lH-indazol-4-yl)phenyl]-N'-(2-
fluoro-5-methylphenyl)urea.
Alternatively, in the above method, the capture antibody is monoclonal
antibody
826 and the detection antibody is monoclonal antibody 255.
Still further, in the above method, optionally, when the capture antibody is a
monoclonal antibody and the detection antibody is a monoclonal antibody assay,
the
predetermined level is In one aspect of this method, when the capture antibody
is a
monoclonal antibody and the detection antibody is a monoclonal antibody and
the
concentration of human P1GF-1 or human PIGF-1 fragment determined in step (e)
is
increased by about 60 picograms per milliliter to about 150 picograms per
milliliter
when compared to the predetermined level at the steady state about either 8 or
15 days
after the subject first receives treatment with- N-[4-(3-amino-lH-indazol-4-
yl)phenyl]-
N'-(2-fluoro-5-methylphenyl)urea or an analog of N-[4-(3-amino-lH-indazol-4-
yl)phenyl] -N'-(2-fluoro-5-methylphenyl)urea.
In still another embodiment, provided herein is a kit for use in determining
whether a subject receiving treatment with a drug has obtained an efficacious
blood
level of the drug, wherein the drug is N-[4-(3-amino-lH-indazol-4-yl)phenyl]-
N'-(2-
fluoro-5-methylphenyl)urea or an analog ofN-[4-(3-amino-lH-indazol-4-
yl)phenyl]-
N'-(2-fluoro-5-methylphenyl)urea, the kit comprising:
(a) at least antibody selected from the group consisting of monoclonal
antibody 264, polyclonal antibody pB264 and combinations thereof, and
(b) instructions for using the kit.
Further provided is a kit for use in determining whether a subject receiving
treatment with a drug has obtained an efficacious blood level of the drug,
wherein the
drug is N-[4-(3-amino-lH-indazol-4-yl)phenyl]-N'-(2-fluoro-5-methylphenyl)urea
or an
analog of N-[4-(3-amino-1 H-indazol-4-yl)phenyl]-N'-(2-fluoro-5-
methylphenyl)urea,
the kit comprising:
(a) at least antibody selected from the group consisting of monoclonal
antibody 826, monoclonal antibody 255 and combinations thereof, and
(b) instructions for using the kit
Further provided is a kit for use in monitoring a response of a subject
receiving
treatment for cancer with an anti-cancer drug, wherein the drug is N-[4-(3-
amino-lH-
12
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
indazol-4-yl)phenyl]-N'-(2-fluoro-5-methylphenyl)urea or an analog ofN-[4-(3-
amino-
1H-indazol-4-yl)phenyl]-N'-(2-fluoro-5-methylphenyl)urea, the kit comprising:
(a) at least antibody selected from the group consisting of monoclonal
antibody 264, polyclonal antibody pB264 and combinations thereof, and
(b) instructions for using the kit.
Still yet further provide is a kit for use in monitoring a response of a
subject
receiving treatment for cancer with an anti-cancer drug, wherein the drug is N-
[4-(3-
amino-I H-indazol-4-yl)phenyl]-N'-(2-fluoro-5-methylphenyl)urea or an analog
of N-
[4-(3-amino- I H-indazol-4-yl)phenyl]-N'-(2-fluoro-5-methylphenyl)urea, the
kit
comprising:
(a) at least antibody selected from the group consisting of monoclonal
antibody 826, monoclonal antibody 255 and combinations thereof, and
(c) instructions for using the kit.
In yet another embodiment, provided herein is a kit for use in determining
whether a subject who is predisposed to a disease or who is suffering from a
disease
will respond to treatment with a drug, wherein the drug is N-[4-(3-amino-lH-
indazol-4-
yl)phenyl]-N'-(2-fluoro-5-methylphenyl)urea or an analog ofN-[4-(3-amino-lH-
indazol-4-yl)phenyl]-N'-(2-fluoro-5-methylphenyl)urea, the kit comprising:
(a) at least antibody selected from the group consisting of monoclonal
antibody 264, polyclonal antibody pB264 and combinations thereof, and
(b) instructions for using the kit.
In still yet another embodiment, provided herein is a kit for use in
determining
whether a subject who is predisposed to a disease or who is suffering from a
disease
will respond to treatment with a drug, wherein the drug is N-[4-(3-amino-lH-
indazol-4-
yl)phenyl]-N'-(2-fluoro-5-methylphenyl)ureaor an analog ofN-[4-(3-amino-lH-
indazol-4-yl)phenyl]-N'-(2-fluoro-5-methylphenyl)urea, the kit comprising:
(a) at least antibody selected from the group consisting of: monoclonal
antibody 826, monoclonal antibody 255 and combinations thereof, and
(b) instructions for using the kit.
Further provided herein is a kit for use in monitoring progression of disease
in a
subject being administered with N-[4-(3-amino-lH-indazol-4-yl)phenyl]-N'-(2-
fluoro-
13
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
5-methylphenyl)urea or an analog ofN-[4-(3-amino-lH-indazol-4-yl)phenyl]-N'-(2-
fluoro-5-methylphenyl)urea, the kit comprising:
(a) at least antibody selected from the group consisting of monoclonal
antibody 264, polyclonal antibody pB264 and combinations thereof; and
(b) instructions for using the kit.
Still further provided herein is a kit for use in monitoring progression of
disease
in a subject being administered with N-[4-(3-amino-lH-indazol-4-yl)phenyl]-N'-
(2-
fluoro-5-methylphenyl)urea or an analog ofN-[4-(3-amino-lH-indazol-4-
yl)phenyl]-
N'-(2-fluoro-5-methylphenyl)urea, the kit comprising:
(a) at least antibody selected from the group consisting of monoclonal
antibody 826, monoclonal antibody 255 and combinations thereof, and
(b) instructions for using the kit.
DESCRIPTION OF THE FIGURES
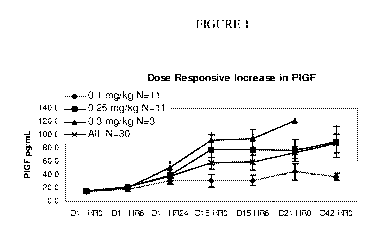
Figure 1 shows the dose responsive increase in human P1GF-1 at various time
points as described in Example 1. In this Figure, -=- is 0.1 mg/kg (N=11), -m -
is 0.25
mg/kg (N=11), -A- is 0.3 mg/kg (N=3), and - *-is all patients (N=30).
Figure 2 shows the optimal human P1GF-1 threshold which stratified patients
for time on therapy (indicative of therapeutic benefit) as described in
Example 1.
Groups are shown for the 24 hour time point after initiation of treatment with
ABT-869
as all patients (shown as the dotted (...) line (N=31) (with the Median TTP
equal to
133)), greater than 30 pg/mL (shown as the dashed and dotted (-=-=-) line
(N=20) (with
the Median TTP equal to 144)) or less than 30 pg/mL (shown as the thick solid
(-)
line (N=1 1) with the Median TTP equal to 82)). Clear differences in the time
to
progression for each group are shown.
Figure 3 shows the optimal human P1GF-1 threshold which stratified patients
for time on therapy (indicative of therapeutic benefit) as described in
Example 1.
Groups are shown at the steady state time point after initiation of treatment
with ABT-
869 as either all patients (shown as the dotted (...) line (N=30) (with the
Median TTP
equal to 133)), greater than 40 pg/mL (shown as the dashed and dotted (-=-=-)
line
(N=17) (with the Median TTP equal to 328)) or less than 40 pg/mL (shown as the
thick
14
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
solid (-) line (N=13) with the Median TTP equal to 96)). Clear differences in
the time
to progression for each group are shown.
Figure 4 shows the human PIGF-1 time course over a variety of time points as
described in Example 1.
DETAILED DESCRIPTION
It is known in the art that human PIGF-1 expression in tumor tissue correlates
with poor prognosis and survival in subjects suffering from colorectal cancer
(See, Wei,
et al., Gut, 54:666-672 (2005)). Thus, one skilled in the art might anticipate
that an
effective therapy capable of reducing disease burden, causing remission or
extending
progression free survival, would result in decreasing plasma concentrations of
human
PIGF-1. Unexpectedly, the inventors of the present disclosure discovered that
increasing human PIGF-1 concentrations in subjects suffering from a disease,
such as
cancer, can serve as a marker associated with effective therapy and improved
time to
progression, particularly in subjects suffering from cancer.
Thus, the present disclosure relates, among other things, to a human PIGF-1 or
human PIGF-1 fragment companion diagnostic assay that can be used in concert
with
subjects receiving treatment with ABT-869 or an analog of ABT-869. More
specifically, the present disclosure relates to methods for determining
whether a subject
receiving treatment with ABT-869 or an analog of ABT-869 has obtained an
efficacious blood level of drug by monitoring the levels of PIGF-1 or human
PIGF-1
fragment in said subject during the course of treatment. Moreover, the present
disclosure also relates to methods of determining whether a subject
predisposed to or
suffering from a disease will benefit from treatment with a drug, and the
response of a
subject receiving treatment (e.g., such as for cancer) by monitoring
biomarkers of
angiogenesis. In particular, the disclosure relates to a PIGF-1 companion
diagnostic
methods and products. Optionally, the methods described herein are adapted for
use on
automated or semi-automated systems.
Finally, the present disclosure relates to methods of treating subjects
suffering
from one or more types of cancer or one or more types of autoimmune diseases.
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
A. Definitions
As used herein, the singular forms "a," "an" and "the" include plural
referents
unless the context clearly dictates otherwise. For the recitation of numeric
ranges
herein, each intervening number there between with the same degree of
precision is
explicitly contemplated. For example, for the range 6-9, the numbers 7 and 8
are
contemplated in addition to 6 and 9, and for the range 6.0-7.0, the numbers
6.0, 6.1, 6.2,.
6.3, 6.4, 6.5, 6.6, 6.7, 6.8, 6.9 and 7.0 are explicitly contemplated.
a) ABT-869
As used herein, the term "ABT-869" refers to N-[4-(3-amino-lH-indazol-4-
yl)phenyl]-N'-(2-fluoro-5-methylphenyl)urea which is an ATP-competitive
receptor
tyrosine kinase inhibitor that has potent inhibitory activity against VEGFR 1
(IC50
30nM), 2 (IC50 8.5nM), 3 (IC50 40nM), PDGFR[3 (IC50 25nM), CSF-1R (IC50 5.3nM)
and Flt-3 kinase (IC50 9.5nM) in kinase enzyme assays. ABT-869, methods for
making
ABT-869 (See, Example 5), types of formulations containing ABT-869 (e.g.,
capsules
or tablets; powders or granules; solutions or suspensions in aqueous or non-
aqueous
liquids; edible foams or whips; or oil-in-water liquid emulsions or water-in-
oil
emulsions), routes of administration (e.g., oral (including buccal or
sublingual), rectal,
nasal, topical (including buccal, sublingual, or transdermal), vaginal, or
parenteral
(including subcutaneous, intramuscular, intravenous, or intradermal) route)
are all
described in U.S. Patent No. 7,297,709, the contents of which are herein
incorporated
by reference.
b) Analogs of ABT-869 or ABT-869 Analogs
As used herein, the phrases "analogs of ABT-869" or "ABT-869 analogs" as
used interchangeably herein refers pharmacologically active analogs,
including, but not
limited, to salts, esters, amides, prodrugs, conjugates, active metabolites,
and other such
derivatives, analogs, and related compounds of ABT-869. Exemplary analogs of
ABT-
869 include, but are not limited to, compounds having the below structural
formulas (I-
XX):
16
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
CH3
0 F 0 1I p0 00
CI 'a Hx NH H~ H~NH CI ` Y 'H `NH I..I~ I H NH
F
FF NHz F FF H l i i
)OH FOH NHZ F NHz NHZ
O O 1/
F F O
K i H or
CH3 CH3
11 III IV
HyC 0 F ` I 0 0
H3C NNxNH
pxNH HyC NH Br NH H
H I
F NHz
NHZ FOH NHz NHZ N
0 d o
a '~ ,moo
H3C H jC
V VI Vii Vii i
F O Fya NxNH Nx H3C I NxNH F F H
HyC NH F F H3C F
o
NHZ NHZ NHZ NH,
N I N N of
cH3
ix x xi xii
F F F
~ ~ ~ 1 , 1 , I
HNAH Z I CH3 HN 3 CH3 HNAr ~ 'CH3 HNxp" ~CH3
IB 0 0 I~ O
F NH2 F NHZ F3CAOH F NHz F3CANHZ F3Cx
OH F OH
N N N I \ `N
xll l Fi xlv xv xvl
C H3
0
F p i
A I A I HNxN 1 FF HNxN CH 3
CH3 H F H 3
H CH3 HN N
H
0 i 0
F
N
NHZ NH2 F3C OH NH2 3CAOH N HZ
`N N N
N
N I N N
N N N N CH3
CH3
xvil xvllI xlx xx
17
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
c) Antibody
As used herein, the term "antibody" refers to an immunoglobulin molecule or
immunologically active portion thereof, namely, an antigen-binding portion.
Examples
of immunologically active portions of immunoglobulin molecules include F(ab)
and
F(ab')2 fragments which can be generated, e.g., by treating an antibody with
an enzyme
such as pepsin. Examples of antibodies that can be used in the present
disclosure
include, but are not limited to, polyclonal antibodies, monoclonal antibodies,
chimeric
antibodies, human antibodies, humanized antibodies, recombinant antibodies,
single-
chain Fvs ("scFv"), affinity maturated antibodies, single chain antibodies,
single
domain antibodies, F(ab) fragments, F(ab') fragments, disulfide-linked Fvs
("sdFv"),
and antiidiotypic ("anti-Id") antibodies, among others, and functionally
active epitope-
binding fragments of any of the above. The antibody may be of classes IgG,
IgM, IgA,
IgD or IgE, or fragments or derivatives thereof. For simplicity sake, an
antibody
against an analyte is frequently referred to as being either an "anti-analyte
antibody", or
merely an "analyte antibody" (e.g., a human P1GF-1 antibody). The antibody may
be
derivatized by the attachment of one or more chemical, peptide, or polypeptide
moieties
known in the art. The antibody may be conjugated with a chemical moiety.
d) Human PIGF-1
The phrases "human P1GF-1" or "human PIGF-1 polypeptide" as used
interchangeably herein refer to any full length (i.e., not a fragment thereof)
human
PIGF-1 sequence, either with or without a signal peptide. For example, the
full length
human P/GF-1 can be a 149 amino acid immature polypeptide with an 18 amino
acid
signal sequence having a centrally located PDGF-like domain with 8 conserved
cysteine residues that form a cysteine knot structure. Alternatively, the
human P1GF-1
can be a 131 amino acid mature polypeptide that does not contain the 18 amino
acid
signal sequence (such as described in the literature). The PIGF may exist in
at least
four alternatively spliced forms: PIGF-1, P1GF-2, P1GF-3 and P/GF-4. PIGF-2
and
PIGF-4 may differ from other forms by the insertion of a heparin-binding
domain in
P1GF-2 and P/GF-4 that may result in increased association with the cell
membrane or
altered affinities for P1GF receptors. The amino acid sequence of PIGF is
described in
EP 0550519, the contents of which are herein incorporated by reference. Human
P/GF
18
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
polypeptide (e.g., polyamino acid) sequences are as found in nature, based on
sequences found in nature, isolated, synthetic, semi-synthetic, recombinant,
or other. In
particular, P1GF as referred to herein is P1GF-1.
The disclosure herein encompasses a multitude of different human PIGF-1
polypeptide sequences as present and/or produced in a prokaryotic and/or
eukaryotic
background (e.g., with consequent optimization for codon recognition). In sum,
the
polypeptide and polynucleotide sequences may or may not possess or encode: (a)
a
signal peptide; and (b) other variations such as would be apparent to one
skilled in the
art.
e) Human P/GF-1 Fragment
As used herein, the term "human P1GF-1 fragment" refers to a polypeptide that
comprises a part that is less than the entirety (i.e., not full length) of a
human P1GF-1
(131 amino acids, referred to by some as the mature protein) or PIGF-1
including a
signal peptide (149 amino acids, referred to by some as the immature protein).
In
particular, a human P1GF fragment comprises at least about 5 contiguous amino
acids
of human PIGF, at least about 10 contiguous amino acids residues of human
P1GF; at
least about 15 contiguous amino acids residues of amino acids of human PIGF;
at least
about 20 contiguous amino acids residues of human P1GF; at least about 25
contiguous
amino acids residues of human P1GF, at least about 30 contiguous amino acid
residues
of amino acids of human P1GF, at least about 35 contiguous amino acid residues
of
human P1GF, at least about 40 contiguous amino acid residues of human PIGF, at
least
about 45 contiguous amino acid residues of human P1GF, at least about 50
contiguous
amino acid residues of human P1GF, at least about 55 contiguous amino acid
residues
of human P/GF, at least about 60 contiguous amino acid residues of human P1GF,
at
least about 65 contiguous amino acid residues of human P1GF, at least about 70
contiguous amino acid residues of human P1GF, at least about 75 contiguous
amino
acid residues of human P1GF, at least about 80 contiguous amino acid residues
of
human PIGF, at least about 85 contiguous amino acid residues of human P1GF, at
least
about 90 contiguous amino acid residues of human P1GF, at least about 95
contiguous
amino acid residues of human P1GF, at least about 100 contiguous amino acid
residues
of human PIGF, at least about 105 contiguous amino acid residues of human
PIGF, at
least about 110 contiguous amino acid residues of human PIGF, at least about
115
19
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
contiguous amino acid residues of human P1GF, at least about 120 contiguous
amino
acid residues of human P/GF, at least about 125 contiguous amino acid residues
of
human PIGF, at least about 130 contiguous amino acid residues of human PIGF,
at least
135 contiguous amino acid residues of human P1GF, at least 140 contiguous
amino acid
residues of human PIGF or 144 contiguous amino acid residues of human PIGF.
An exemplary human P1GF-1 fragment comprises residues 21-149 of human
PIGF-1, which is commercially available from R&D Systems, Inc., Minneapolis,
Minnesota (Catalog Number #P49763). This human P1GF-1 fragment can be used as
a
calibrator or control in an immunoassay for human PIGF-1 fragment or human
P1GF-1
fragment.
f) Identical
"Identical" or "identity" as used herein in the context of two or more
polypeptide or polynucleotide sequences, may mean that the sequences have a
specified
percentage of residues that are the same over a specified region. The
percentage may be
calculated by optimally aligning the two sequences, comparing the two
sequences over
the specified region, determining the number of positions at which the
identical residue
occurs in both sequences to yield the number of matched positions, dividing
the number
of matched positions by the total number of positions in the specified region,
and
multiplying the result by 100 to yield the percentage of sequence identity. In
cases
where the two sequences are of different lengths or the alignment produces one
or more
staggered ends and the specified region of comparison includes only a single
sequence,
the residues of single sequence are included in the denominator but not the
numerator
of the calculation.
g) Monoclonal Antibody 264
As used herein, the phrase "monoclonal antibody 264" or "MAB264" refers to
an unconjugated, mouse anti-human PIGF-1 monoclonal antibody from Clone 37203
which is commercially available from R&D Systems, Inc., Minneapolis, Minnesota
(Catalog Number MAB264).
h) Monoclonal Antibody 255
As used herein, the phrase "monoclonal antibody 255" or "MAB255" refers to
an unconjugated, mouse anti-human PIGF-1 monoclonal antibody produced from
murine hybridoma cell line 1-255-713 having American Type Culture Collection
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
Accession No. PTA-8536,-which is owned by Abbott Laboratories. Murine
hybridoma
cell line 1-255-713 was deposited with the American Type Culture Collection,
10801
University Blvd., Manassas, VA 20110-2209 on July 12, 2007 and assigned
Accession
No. PTA-8536. Monoclonal antibody 255 is described in U.S. Serial Number
61/073,624 filed on June 18, 2008 and U.S. Serial No. 61/089,172 filed on
August 15,
2008, the contents of each of which are herein incorporated by reference.
The phrase "monoclonal antibody 255" or "MAB255" also refers to an
unconjugated, mouse anti-human PIGF-1 monoclonal antibody that is produced by
a
subclone of murine hybridoma cell line 1-255-713, including monoclonal
antibody
produced by the murine hybridoma cell line 1-255-2675. Monoclonal antibody
produced by murine hybridoma cell line 1-255-2675 is identical to the
monoclonal
antibody produced by murine hybridoma cell line 1-255-713. Murine hybridoma
cell
line 1-255-2675 is described in U.S. Serial No. 12/485,114 filed on June 16,
2009, the
contents of which are herein incorporated by reference. When employed as
conjugate
(i.e., detection antibody), MAB255 can be used as a whole molecule or a
fragment
thereof (e.g., Fab'2 fragment).
i) Monoclonal Antibody 826
As used herein, the phrase "monoclonal antibody 826" or "MAB826" refers to
an unconjugated, mouse anti-human P1GF-1 monoclonal antibody produced from
20, murine hybridoma cell line 2-826-335 having American Type Culture
Collection
Accession No. PTA-8539, which is owned by Abbott Laboratories. Murine
hybridoma
cell line was deposited with the American Type Culture Collection, 10801
University
Blvd., Manassas, VA 20110-2209 on July 12, 2007 and assigned Accession No. PTA-
8536. Monoclonal antibody 826 is described in U.S. Serial Number 61/073,624
filed
on June 18, 2008 and U.S. Serial No. 61/089,172 filed on August 15, 2008, the
contents
of each of which are herein incorporated by reference.
j) Pharmaceutical Composition
As used herein, the term "pharmaceutical composition" refers to any agent or
drug, whether a small molecule (e.g., a drug containing an active agent,
typically a non-
peptidic) or biologic (e.g., a peptide or protein based drug, including any
with
modifications, such as, but not limited to pegylation) that can be used to
treat a subject
suffering from a disease or condition that requires treatment. Examples of
21
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
pharmaceutical compositions, include, but are not limited to, ABT-869, analogs
of
ABT-869, hyperlipidemia drugs (including, but not limited to, niacin, fibrates
(e.g.,
clofibrate, fenofibrate, fenofibric acid, simfrate, salts of fenofibric acid
and any
combinations thereof), ezetimibe, HMG-CoA reductase inhibitors (e.g., statins,
such as,
but not limited to rosuvastatin, simvastatin, and combinations thereof
(including
combinations with other hyperlipidemia drugs (e.g., simvastatin and
ezetimibe)), anti-
inflammatories, natriuretic peptide derivatives, etc. as well as any
combinations thereof
k) Polyclonal Antibody pB264
The phrases, "polyclonal antibody pB264", "pB264", "AF-264-PB", "PAB264"
or AB-264-PB as used interchangeably herein, refer to an unconjugated, goat
anti-
human PIGF-1 polyclonal antibody which is available from R&D Systems, Inc.,
Minneapolis, Minnesota (Catalog Number AF-264-PB). Polyclonal antibody pB264
is
produced in goats immunized with purified, E. coli-derived, recombinant human
placenta growth factor. P1GF-1-specific IgG was purified by human P1GF
affinity
chromatography.
1) Predetermined Level
As used herein, the term "predetermined level" refers generally at an assay
cutoff value that is used to assess diagnostic results by comparing the assay
results
against the predetermined level, and where the predetermined level already
that has
been linked or associated with various clinical parameters (e.g., monitoring
whether a
subject being treated with a drug has achieved an efficacious blood level of
the drug,
monitoring the response of a subject receiving treatment for cancer with an
anti-cancer
drug, monitoring the response of a tumor in a subject receiving treatment for
said
tumor, etc.). The predetermined level may be either an absolute value (such as
in
monoclonal antibody/polyclonal assays described in more detail herein) or a
value
normalized by subtracting the value obtained from a patient prior to the
initiation of
therapy (such as in monoclonal antibody/monoclonal antibodies as described in
more
detail herein). An example of a predetermined level that can be used is a
baseline level
obtained from one or more subjects that may optionally be suffering from one
or more
diseases or conditions. The present disclosure provides exemplary
predetermined
levels, and describes the initial linkage or association of such levels with
clinical
parameters for exemplary immunoassays as described herein. However, it is well
22
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
known that cutoff values may vary dependent on the nature of the immunoassay
(e.g.,
antibodies employed, etc.). It further is well within the ordinary skill of
one in the art
to adapt the disclosure herein for other immunoassays to obtain immunoassay-
specific
cutoff values for those other immunoassays based on this description.
m) Substantially Identical
"Substantially identical," as used herein may mean that a first and second
sequence are at least from about 50% to about 99% identical over a region of
from
about 8 to about 100 or more residues (including, in particular, any range
within from
about 8 to about 100 residues).
n) Subject or Patient
As used herein, the terms "subject" and "patient" are used interchangeably. As
used herein, the terms "subject" and "subjects" refer to an animal, in one
aspect, a bird
(for example, a duck or goose), in another aspect, a shark or whale, or in a
further
aspect, a mammal including, a non-primate (for example, a cow, pig, camel,
llama,
horse, goat, rabbit, sheep, hamsters, guinea pig, feline, canine, rat, and
murine) and a
primate (for example, a monkey, such as a cynomologous monkey, chimpanzee, and
a
human).
o) Test Sample
As used herein, the term "test sample" or "sample" generally refers to a
biological material being tested for and/or suspected of containing an analyte
of
interest, such as a human P1GF-1 or human P1GF- I fragment. The test sample
may be
derived from any biological source, such as, a physiological fluid, including,
but not
limited to, whole blood, serum, plasma, interstitial fluid, saliva, ocular
lens fluid,
cerebral spinal fluid, sweat, urine, milk, ascites fluid, mucous, nasal fluid,
sputum,
synovial fluid, peritoneal fluid, vaginal fluid, menses, amniotic fluid, semen
and so
forth. The test sample may be used directly as obtained from the biological
source or
following a pretreatment to modify the character of the sample. For example,
such
pretreatment may include preparing plasma from blood, diluting viscous fluids
and so
forth. Methods of pretreatment may also involve filtration, precipitation,
dilution,
distillation, mixing, concentration, inactivation of interfering components,
the addition
of reagents, lysing, etc. Moreover, it may also be beneficial to modify a
solid test
sample to form a liquid medium or to release the analyte.
23
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
p) Toxicity Level
As used herein, the phrase "toxicity level" refers to a value in which a
subject
being treated with a pharmaceutical composition may begin to experience one or
more
adverse events or adverse side effects as a result of the treatment with said
pharmaceutical composition.
q) Variant
"Variant" as used herein may mean a peptide or polypeptide that differs in
amino acid sequence by the insertion, deletion, or conservative substitution
of amino
acids, but retain at least one biological activity. For purposes of this
disclosure,
"biological activity" includes the ability to be bound by a specific antibody.
A
conservative substitution of an amino acid, i.e., replacing an amino acid with
a different
amino acid of similar properties (e.g., hydrophilicity, degree and
distribution of charged
regions) is recognized in the art as typically involving a minor change. These
minor
changes can be identified, in part, by considering the hydropathic index of
amino acids,
as understood in the art. Kyte et al., J. Mol. Biol., 157:105-132 (1982). The
hydropathic index of an amino acid is based on a consideration of its
hydrophobicity
and charge. It is known in the art that amino acids of similar hydropathic
indexes can
be substituted and still retain protein function. In one aspect, amino acids
having
hydropathic indexes off 2 are substituted. The hydrophilicity of amino acids
can also
be used to reveal substitutions that would result in proteins retaining
biological
function. A consideration of the hydrophilicity of amino acids in the context
of a
peptide permits calculation of the greatest local average hydrophilicity of
that peptide, a
useful measure that has been reported to correlate well with antigenicity and
immunogenicity. U.S. Patent No. 4,554,101, incorporated herein by reference.
Substitution of amino acids having similar hydrophilicity values can result in
peptides
retaining biological activity, for example immunogenicity, as is understood in
the art.
In one aspect, substitutions are performed with amino acids having
hydrophilicity
values within 2 of each other. Both the hydrophobicity index and the
hydrophilicity
value of amino acids are influenced by the particular side chain of that amino
acid.
Consistent with that observation, amino acid substitutions that are compatible
with
biological function are understood to depend on the relative similarity of the
amino
24
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
acids, and particularly the side chains of those amino acids, as revealed by
the
hydrophobicity, hydrophilicity, charge, size, and other properties.
Variant may also refer to a protein that is (i) a portion of a referenced
protein
which may be from about 8 to about 100 or more amino acids (including; in
particular,
any range within from about 8 to about 100 residues); or (ii) a protein that
is
substantially identical to a referenced protein. A variant may also be a
differentially
processed protein, such as by proteolysis, phosphorylation, or other post-
translational
modification.
10, B. PIGF-1 Assays and Methods of Treatment
The present disclosure also relates to assays for determining human PIGF-1 or
human P1GF-1 fragment concentration in a test sample obtained from a subject.
Assays
contemplated include immunoassays (such as sandwich and competitive
immunoassays), clinical chemistry assays and enzymatic assays. Preferably the
human
PIGF-1 or human P1GF-1 fragment measurement is done using an immunoassay, and
more preferably, a sandwich immunoassay, which will be discussed in more
detail
herein.
Assays for determining human PIGF-1 or human P/GF-1 fragment
concentration in a test sample obtained from a subject can comprise the steps
of (a)
providing a test sample obtained from a subject; and (b) determining the
concentration
of human P1GF-1 or human P1GF-1 fragment in the test sample. A specific type
of
assay that can be performed for determining human P1GF-1 or human P1GF-1
fragment
concentration is an immunoassay. Immunoassays can be conducted using any
format
known in the art, such as, but not limited to, a sandwich format, a
competitive
inhibition format (including both forward or reverse competitive inhibition
assays) or in
a fluorescence polarization format. As mentioned above, preferably, the
immunoassay
is in a sandwich format. Specifically, in one aspect of the present
disclosure, at least
two antibodies are employed to separate and quantify the human P1GF-1 or human
PIGF-1 fragment in a test sample. More specifically, the at least two
antibodies bind to
certain epitopes of human P1GF-1 or human PIGF-1 fragment forming an immune
complex which is referred to as a "sandwich". Generally, in the immunoassays
one or
more antibodies can be used to capture the human P1GF-1 or human P1GF-1
fragment
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
in the test sample (these antibodies are frequently referred to as a "capture"
antibody or
"capture" antibodies) and one or more antibodies can be used to bind a
detectable
(namely, quantifiable) label to the. sandwich (these antibodies are frequently
referred to
as the "detection antibody", "detection antibodies", a "conjugate" or
"conjugates"). In a
sandwich assay, it is preferred that both antibodies binding to the human PIGF-
1 or
human PIGF-1 fragment are not diminished by the binding of any other antibody
in the
assay to its respective binding site. In other words, antibodies should be
selected so
that the one or more first antibodies brought into contact with a test sample
or test
sample extract suspected of containing human PIGF-1 or human PIGF-1 fragment
do
not bind to all or part of the binding site recognized by the second or
subsequent
antibodies, thereby interfering with the ability of the one or more second
detection
antibodies to bind to human PIGF-1 or human P1GF-1 fragment.
In one aspect of the present invention, excellent (namely, highly robust or
high
quality) immunoassays, particularly, sandwich assays, can be performed using a
monoclonal antibody as a capture antibody and a polyclonal antibody as a
detection
antibody or a polyclonal antibody as a capture antibody and a monoclonal
antibody as a
detection antibody (referred to herein as "mono/poly assays"). An example of
an
exemplary immunoassay is one that employs as a capture antibody, monoclonal
antibody 264, and, as the detection antibody, polyclonal antibody pB264.
Optionally, a
different commercially available antibody (e.g., other than monoclonal
antibody 264)
can be used as the first capture antibody and monoclonal antibody 264 can be
used as a
second or subsequent capture antibody. Alternatively, if monoclonal antibody
264 is
being used as a first capture antibody, a different antibody (other than an
monoclonal
antibody 264, namely, other commercially available antibodies) can be used as
a
second capture antibody. Also optionally, a second detection antibody can be
used in
addition to polyclonal antibody pB264. Any commercially available antibody can
be
used as the second detection antibody. For example, polyclonal antibody pB264
can be
used as a second detection antibody.
In another aspect of the present invention, excellent (namely, highly robust
or
high quality) immunoassays, particularly, sandwich assays, can be performed
using a
monoclonal antibody as both a capture antibody and as a detection antibody
(referred to
herein as "mono/mono assays"). An example of an exemplary immunoassay is one
that
26
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
employs as a capture antibody, monoclonal antibody 826 and, as the detection
antibody, monoclonal antibody 255. Optionally, a different antibody, (e.g.,
other than
monoclonal antibody 826) can be used as the first capture antibody and
monoclonal
antibody 826 can be used as a second or subsequent capture antibody. For
example,
monoclonal antibody 264 can be used as a first capture antibody and monoclonal
antibody 826 can be used as the second or subsequent capture antibodies.
Alternatively, if monoclonal antibody 826 is being used as a first capture
antibody, a
different antibody (other than an monoclonal antibody 826, namely, other
commercially
available antibodies) can be used as a second capture antibody. For example,
monoclonal antibody 826 can be used as the first capture antibody and
monoclonal
antibody 264 can be used as the second capture antibody. Also optionally, a
second
detection antibody can be used in addition to monoclonal antibody 255. Any
commercially available antibody can be used as the second detection antibody,
including a polyclonal antibody, provided that the first detection antibody is
a
monoclonal antibody. For example, polyclonal antibody pB264 can be used as a
second detection antibody, where a monoclonal antibody is used as a first
detection
antibody.
The sample being tested for (for example, suspected of containing) human
PIGF-I or human P/GF-1 fragment can be contacted with at least one capture
antibody
(or antibodies) and at least one detection antibody (which is either a second
detection
antibody or a third detection antibody) either simultaneously or sequentially
and in any
order. For example, the test sample can be first contacted with at least one
capture
antibody and then (sequentially) with at least one detection antibody.
Alternatively, the
test sample can be first contacted with at least one detection antibody and
then
(sequentially) with at least one capture antibody. In yet another'altemative,
the test
sample can be contacted simultaneously with a capture antibody and a detection
antibody.
In the sandwich assay format, a test sample suspected of containing human
P1GF-1 or human P1GF-1 fragment is first brought into contact with an at least
one first
capture antibody under conditions which allow the formation of a first
antibody-human
PIGF-1 complex. If more than one capture antibody is used, a first multiple
capture
antibody-human P/GF-1 (or human PIGF-1 fragment) complex is formed. In a
27
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
sandwich assay, the antibodies, preferably, the at least one capture antibody,
are used in
molar excess amounts of the maximum amount of human P1GF-1 expected in the
test
sample. For example, from about 5 pg/mL to about 1 mg/mL of antibody per mL of
buffer (e.g., microparticle coating buffer) can be used.
Optionally, prior to contacting the test sample with the at least one capture
antibody (for example, the first capture antibody), the at least one capture
antibody can
be bound to a'solid support or solid phase which facilitates the separation
the first
antibody-human P1GF-1 complex from the test sample. Any solid support known in
the
art can be used, including but not limited to, solid supports made out of
polymeric
materials in the forms of wells of a reaction tray, test tubes or beads (for
example,
polystyrene beads, magnetic beads), nitrocellulose strips, membranes,
microparticles
(for example, latex particles, sheep and DURACYTES (Abbott Laboratories,
Abbott
Park, IL; DURACYTES are red blood cells that have been "fixed" by pyruvic
aldehyde and formaldehyde)).
The solid phase also can comprise any suitable porous material with sufficient
porosity to allow access by detection antibodies and a suitable surface
affinity to bind
antigens. Microporous structures generally are preferred, but materials with
gel
structure in the hydrated state may be used as well. Such useful solid
supports include,
but are not limited to, nitrocellulose and nylon. Such porous solid supports
are
preferably in the form of sheets of thickness from about 0.01 to 0.5 mm,
preferably
about 0.1 mm. The pore size may vary within wide limits, and preferably is
from about
0.025 to about 15 microns, especially from about 0.15 to about 15 microns. The
surface
of such supports may be activated by chemical processes which cause covalent
linkage
of the antigen or antibody to the support. The irreversible binding of the
antigen or
antibody is obtained, however, in general, by adsorption on the porous
material by
poorly understood hydrophobic forces.
The antibody (or antibodies) can be bound to the solid support or solid phase
by
adsorption, by covalent bonding using a chemical coupling agent or by other
means
known in the art, provided that such binding does not interfere with the
ability of the
antibody to bind to human P1GF-1 or human P/GF-1 fragment. Alternatively, the
antibody (or antibodies) can be bound with microparticles that have previously
coated
with streptavidin or biotin (for example, using Power-BindTM-SA-MP
streptavidin
28
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
coated microparticles, available from Seradyn, Indianapolis, Indiana, with
antibodies
that have been biotinylated using means known in the art). Alternatively, the
antibody
(or antibodies) can be bound using microparticles that have been previously
coated with
anti-species specific monoclonal antibodies. Moreover, if necessary, the solid
support
can be derivatized to allow reactivity.with various functional groups on the
antibody.
Such derivatization requires the use of certain coupling agents such as, but
not limited
to, maleic anhydride, N-hydroxysuccinimide and 1-ethyl-3-(3-
dimethylaminopropyl)carbodiimide.
After the test sample being tested for and/or suspected of containing human
P/GF-l or human PIGF-1 fragment is brought into contact with the at least one
capture
antibody (for example, the first capture antibody), the mixture is incubated
in order to
allow for the formation of a first antibody (or multiple antibody)-human P1GF-
1
complex. The incubation can be carried out at a pH of from about 4.5 to about
10.0, at
a temperature of from about 2 C to about 45 C, and for a period from at least
about one
(1) minute to about eighteen (18) hours, preferably from about 1 to 20
minutes, most
preferably from about 2-6 minutes. The immunoassay described herein can be
conducted in one step (meaning the test sample, at least one capture antibody
and at
least one detection antibody are all added sequentially or simultaneously to a
reaction
vessel) or in more than one step, such as two steps, three steps, etc.
After formation of the (first or multiple) capture antibody-human P1GF-1
complex, the complex is then contacted with at least one detection antibody
(under
conditions which allow for the formation of a (first or multiple) capture
antibody-
human P1GF-1-(second or multiple) antibody detection complex). The at least
one
detection antibody can be the second, third, fourth, etc. antibodies used in
the
immunoassay. If the capture antibody-human P1GF-1 complex is contacted with
more
than one detection antibody, then a (first or multiple) capture antibody-human
P1GF-1-
(multiple) detection antibody complex is formed. As with the capture antibody
(e.g.,
the first capture antibody), when the at least second (and subsequent)
detection
antibody is brought into contact with the capture antibody-human P1GF-1
complex, a
period of incubation under conditions similar to those described above is
required for
the formation of the (first or multiple) capture antibody-human P1GF-1-(second
or
multiple) detection antibody complex. Preferably, at least one detection
antibody
29
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
contains a detectable label. The detectable label can be bound to the at least
one
detection antibody (e.g., the second detection antibody) prior to,
simultaneously with or
after the formation of the (first or multiple) capture antibody-human PIGF-1-
(second or
multiple) detection antibody complex. Any detectable label known in the art
can be
used. For example, the detectable label can be a radioactive label, such as,
3H, 125I, 355,
14C, 32P, 33P, an enzymatic label, such as horseradish peroxidase, alkaline
phosphatase,
glucose 6-phosphate dehydrogenase, etc., a chemiluminescent label, such as,
acridinium (e.g., acridium esters, acridinium SPSP (N I 0-(3-sulfopropyl)-N-(3-
sulfopropyl, etc.), luminol, isoluminol, thioesters, sulfonamides,
phenanthridinium
esters, etc. a fluorescence label, such as, fluorescein (5-fluorescein, 6-
carboxyfluorescein, 3'6-carboxyfluorescein, 5(6)-carboxyfluorescein, 6-
hexachloro-
fluorescein, 6-tetrachlorofluorescein, fluorescein isothiocyanate, etc.),
rhodamine,
phycobiliproteins, R-phycoerythrin, quantum dots (zinc sulfide-capped cadmium
selenide), a thermometric label or an immuno-polymerase chain reaction label.
An
introduction to labels, labeling procedures and detection of labels is found
in Polak and
Van Noorden, Introduction to Immunocytochemistry, 2nd ed., Springer Verlag,
N.Y.
(1997) and in Hauglarid, Handbook of Fluorescent Probes and Research Chemicals
(1996), which is a combined handbook and catalogue published by Molecular
Probes,
Inc., Eugene, Oregon.
The detectable label can be bound to the antibodies either directly or through
a
coupling agent. An example of a coupling agent that can be used is EDAC (1-
ethyl-3-
(3-dimethylaminopropyl) carbodiimide, hydrochloride) that is commercially
available
from Sigma-Aldrich, St. Louis, MO. Other coupling agents that can be used are
known
in the art. Methods for binding a detectable label to an antibody are known in
the art.
Additionally, many detectable labels can be purchased or synthesized that
already
contain end groups that facilitate the coupling of the detectable label to the
antibody,
such as, N 10-(3-sulfopropyl)-N-(3-carboxypropyl)-acridinium-9-carboxamide,
otherwise known as CPSP-Acridinium Ester or N 10-(3-sulfopropyl)-N-(3-
sulfopropyl)-
acridinium-9-carboxamide, otherwise known as SPSP-Acridinium Ester.
The (first or multiple) capture antibody-human PIGF-1-(second or multiple)
detection antibody complex can be, but does not have to be, separated from the
remainder of the test sample prior to quantification of the label. For
example, if the at
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
least one capture antibody (e.g., the first capture antibody) is bound to a
solid support
or solid phase, such as, but not limited to, a well of a reaction tray, a bead
or a
microparticle, separation can be accomplished by removing the fluid (of the
test
sample) from contact with the solid support. Alternatively, if the at least
first capture
antibody is bound to a solid support it can be simultaneously contacted with
the human
P1GF-1 (or human P1GF-1 fiagment)-containing sample and the at least one
second
detection antibody to form a first (multiple) antibody-human P/GF-1-second
(multiple)
antibody complex, followed by removal of the fluid (test sample) from contact
with the
solid support. If the at least one first capture antibody is not bound to a
solid support,
then the (first or multiple) capture antibody-human P1GF-1-(second or
multiple)
detection antibody complex does not have to be removed from the test sample
for
quantification of the amount of the label.
After formation of the labeled capture antibody-human P1GF-1-detection
antibody complex (e.g., the first capture antibody-human P/GF-1-second
detection
antibody complex), the amount of label in the complex is quantified using
techniques
known in the art. For example, if an enzymatic label is used, the labeled
complex is
reacted with a substrate for the label that gives a quantifiable reaction such
as the
development of color. If the label is a radioactive label, the label is
quantified using a
scintillation counter. If the label is a fluorescent label, the label is
quantified by
stimulating the label with a light of one color (which is known as the
"excitation
wavelength") and detecting another color (which is known as the "emission
wavelength") that is emitted by the label in response to the stimulation. If
the label is a
chemiluminescent label, the label is quantified detecting the light emitted
either
visually or by using luminometers, x-ray film, high speed photographic film, a
CCD
camera, etc. Once the amount of the label in the complex has been quantified,
the
concentration of human P1GF-1 or human P1GF-1 fragment in the test sample is
determined by use of a standard curve that has been generated using serial
dilutions of
human PIGF-1 or human P1GF-1 fragment of known concentration. Other than using
serial dilutions of human P1GF-1 or human P1GF-1 fragment, the standard curve
can be
generated gravimetrically, by mass spectroscopy and by other techniques known
in the
art.
31
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
It goes without saying that the methods and kits as described herein
necessarily
encompass other reagents and methods for carrying out the immunoassay. For
instance, encompassed are various buffers such as are known in the art and/or
which
can be readily prepared or optimized to be employed, e.g., for washing, as a
conjugate
diluent, and/or as a calibrator diluent. An exemplary conjugate diluent is
ARCHITECTS Human P1GF-1 conjugate diluent (Abbott Laboratories, Abbott. Park,
IL) containing 2-(N-morpholino)ethanesulfonic acid (MES), another salt,
protein
blockers, an antimicrobial and detergent. An exemplary calibrator diluent is
ARCHITECTS Human P/GF-1 calibrator diluent (Abbott Laboratories, Abbott Park,
IL), which comprises a buffer containing MES, another salt, a protein blocker
and an
antimicrobial.
Furthermore, as previously mentioned, the methods and kits optionally are
adapted for use on an automated or semi-automated system. Some of the
differences
between an automated or semi-automated system as compared to a non-automated
system (e.g., ELISA) include the substrate to which the capture antibody is
attached
(which can impact sandwich formation and analyte reactivity), and' the length
and
timing of the capture, detection and/or any optional wash steps. Whereas a non-
automated format such as an ELISA may include a relatively longer incubation
time
with sample and capture reagent (e.g., about 2 hours) an automated or semi-
automated
format (e.g., ARCHITECTS) may have a relatively shorter incubation time (e.g.,
approximately 18 minutes for ARCHITECT ). Similarly, whereas a non-automated
format such as an ELISA may incubate a detection antibody such as the
conjugate
reagent (Pb264) for a relatively longer incubation time (e.g., about 2 hours),
an
automated or semi-automated format (e.g., ARCHITECTS) may have a relatively
shorter incubation time (e.g., approximately 4 minutes for the ARCHITECTS).
The assays of the present disclosure can be used to monitor whether a subject
being administered a drug, such as ABT-869 or an analog of ABT-869, has
obtained an
efficacious (or optimal) blood level of said drug. In other words, the assays
of the
present disclosure allow the treating physician to determine whether or not
the subject
has received a sufficient amount of ABT-869 or an analog of ABT-869 to
effectuate
treatment. Such an assay involves contacting a first capture antibody that
binds to
human P1GF-1 or human P/GF-1 fragment with a test sample obtained from a
subject
32
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
receiving treatment with ABT-869 or an analog of ABT-869 to form a first
capture
antibody-human P1GF-1 complex. In mono/poly assays, the capture antibody
optionally is monoclonal antibody 264. In mono/mono assays, the capture
antibody
optionally is monoclonal antibody 826. After the formation of the first
capture
antibody-human P1GF-1 complex, the test sample is then contacted with a second
antibody that binds to human P1GF-1 or human P1GF-1 fragment and that has been
conjugated to a detectable label to form a second capture antibody-human P1GF-
1
detection complex. In mono/poly assays, the detection antibody optionally is
polyclonal antibody pB264. In mono/mono assays, the detection antibody
optionally is
monoclonal antibody 225. The amount of the capture antibody-human P1GF-1
detection complex that has been formed is then determined by detecting the
detectable
label. The amount of capture antibody-human P1GF-1 detection complex present
in the
test sample correlates with the amount of human P1GF-1 or human PIGF-1
fragment in
the test sample. The amount of human P1GF-1 or human P1GF-1 fragment in the
test
sample is then compared with a predetermined level. Specifically, if the
concentration
of human P1GF-1 or human P1GF-1 fragment determined in the test sample is
lower
than the predetermined level, then the subject is considered not to be
receiving an
efficacious (or optimal) amount of ABT-869 or analog of ABT-869. The treating
physician may then make a decision to increase the amount of ABT-869 or analog
of
ABT-869 administered to the subject. However, if the concentration of human
P1GF-1
or human P1GF-1 fragment determined in the test sample is the same as or
higher than
the predetermined level, then the subject is considered to be receiving an
efficacious (or
optimal) amount of ABT-869 or analog of ABT-869. For example, in mono/mono
assays, a subject demonstrating an. increase from the predetermined level (or
baseline
level (such as prior to treatment with ABT-869 or an analog of ABT-869)) of
about 60
picograms per milliliter would be considered to be receiving an efficacious
(or optimal)
amount of ABT-869 or analog of ABT-869. More specifically, a subject
demonstrating
an increase from the predetermined level (such as a base line level (such as
prior to
treatment with ABT-869 or an analog of ABT-869)) in the range of about 60
picograms
per milliliter to about 150 picograms per milliliter would be considered to be
receiving
an efficacious (or optimal) amount of ABT-869 or analog of ABT-869. An
increase
from the predetermined level (such as a baseline level) of 66.5 picograms per
milliliter
33
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
above the predetermined level (such as a baseline level) at about either 8 or
15 days
after the subject first receives treatment with ABT-869 or an analog of ABT-
869 at the
steady state (namely at least about 3 days, at least about 5 days, at least
about 7 days, at
least about 10 days at least about 15 days, at least 20 days, at least 30
days, at least 35
days, at least 40 days, at least 45 days, at least 50 days, at least 60 days,
etc.) after the
subject first receives treatment with ABT-869 or an analog of ABT-869. The
treating
physician may then make the decision not to change or alter the amount of ABT-
869 or
analog of ABT-869 being administered to the subject; although, the treating
physician
could also decide to lower the amount of ABT-869 or analog of ABT-869 being
administered to the subject as well for the reasons discussed further herein.
Additionally, in another aspect, in the mono/mono assays, if the toxicity
level of
the subject being treated with ABT-869 or an analog of ABT-869 reaches a range
of
130 picograms per milliliter to 160 picograms per milliliter, the treating
physician can
reduce the amount of ABT-869 or analog of ABT-869 being administered to the
subject. An exemplary toxicity level at which a treating physician can reduce
the
amount of ABT-869 or analog of ABT-869 being administered to the subject is
150
picograms per milliliter.
An exemplary predetermined level in mono/poly assays can be about 30
picograms per milliliter at about 24 hours after the subject first receives
treatment with
ABT-869 or an analog of ABT-869. Alternatively, in mono/poly assays, the
predetermined level can be about 40 picograms per milliliter to about 75
picograms per
milliliter at the steady state (namely at least about 3 days, at least about 5
days, at least
about 7 days, at least about 10 days at least about 15 days, at least 20 days,
at least 30
days, at least 35 days, at least 40 days, at least 45 days, at least 50 days,
at least 60
days, etc.) after the subject first receives treatment with ABT-869 or an
analog of ABT-
869. An exemplary predetermined level in mono/mono assays can be about 66.5
picograms per milliliter at about 24 hours after the subject first receives
treatment with
ABT-869 or an analog of ABT-869.
The above described comparisons (also referred to as informational analysis)
involving the amount of human P/GF-1 or human P1GF-1 fragment in the test
sample
and the predetermined level (such as a baseline level, can be made by an
automated
system, such as a software program or intelligence system that is part of, or
compatible
34
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
with, the automated or semi-automated equipment (e.g., computer platform) on
which
the above described assays are carried out. Alternatively, the above described
comparisons can be done by a physician.
Subjects receiving treatment with ABT-869 or an analog of ABT-869 may be
receiving treatment for cancer. The types of cancer include, but are not
limited to, lung
cancer, breast cancer, stomach cancer, bladder cancer, colon cancer,
pancreatic cancer,
ovarian cancer, prostate cancer, renal cancer, hepatocellular cancer, rectal
cancer,
hematopoietic malignancies, glioblastoma or infantile hemangioma.
Alternatively, the
subject may be suffering from autoimmune diseases, such as rheumatoid
arthritis,
thyroiditis, type 1 diabetes, multiple sclerosis, sarcoidosis, inflammatory
bowel disease,
Crohn's disease, myasthenia gravis and systemic lupus erythematosus;
psoriasis, organ
transplant rejection (e.g., kidney rejection, graft versus host disease),
benign and
neoplastic proliferative diseases or ocular diseases, such as, but not limited
to, macular
degeneration.
In another aspect, the assays of the present disclosure can be used to monitor
the
response of a subject receiving treatment for cancer with an anti-cancer drug.
Such an
assay involves contacting a first capture antibody that binds to human PIGF-1
or human
PIGF-1 fragment with a test sample obtained from a subject suffering from
cancer and
receiving treatment with ABT-869 or an analog of ABT-869 to form a first
capture
antibody-human P1GF-1 complex. For mono/poly assays, the capture antibody is
optionally monoclonal antibody 264. For mono/mono assays, the capture antibody
optionally is monoclonal antibody 826. After the formation of the first
capture
antibody-human P1GF-1 complex, the test sample is then contacted with a second
antibody that binds to human PIGF-1 or human P1GF-1 fragment and that has been
conjugated to a detectable label to form a second capture antibody-human P1GF-
1
detection complex. For mono/poly assays, the detection antibody optionally is
polyclonal antibody pB264. For mono/mono assays, the detection antibody
optionally
is monoclonal antibody 255. The amount of the capture antibody-PIGF-1
detection
complex that has been formed is then determined by detecting the detectable
label. The
amount of capture antibody-human P1GF-1 detection complex present in the test
sample correlates with the amount of human P/GF-1 or human P/GF-1 fragment in
the
test sample. The amount of human PIGF-1 or human P/GF-1 test fragment in the
test
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
sample is then compared with a predetermined level. Specifically, if the
concentration
of human P1GF-1 or human P1GF-1 test fragment determined in the test sample is
lower
than the predetermined level, then the subject is considered not to be
responding to
treatment with ABT-869 or an analog of ABT-869 and the treatment with ABT-869
or
an analog of ABT-869 is discontinued.
The treating physician may then make a decision to increase the amount
of ABT-869 or analog of ABT-869 administered to the subject or to switch the
subject
to an entirely different anti-cancer drug to try and treat the cancer or may
add another
anti-cancer drug to the treatment regimen (e.g., administer to the subject ABT-
869 or
an analog of ABT-869 in combination with another anti-cancer drug).
Additionally, the
subject may then be administered a new (e.g., different) anti-cancer drug
which is an
anti-cancer drug other than ABT-869 or an analog of ABT-869. However, if the
concentration of human P/GF-1 or human PIGF-1 'fragment determined in the test
sample is the same as or higher than the predetermined level, then the subject
is
considered to be responding to treatment with ABT-869 or analog of ABT-869.
For
example, in mono/mono assays, a subject demonstrating an increase from the
predetermined level of about 60 picograms per milliliter would be considered
to be
responding to treatment with ABT-869 or analog of ABT-869. More specifically,
a
subject demonstrating an increase from the predetermined level of about 60
picograms
per milliliter to about 150 picograms per milliliter would be considered to be
responding to treatment with ABT-869 or analog of ABT-869. An increase of 66.5
picograms per milliliter above the predetermined level is particularly
preferred. The
treating physician may then make the decision not to change or alter the
amount of
ABT-869 or analog of ABT-869 being administered to the subject; although, the
treating physician could also decide to lower the amount of ABT-869 or analog
of
ABT-869 being administered to the subject as well. For example, in mono/mono
assays, a subject demonstrating an increase from the predetermined level (or
baseline
level) of about 60 picograms per milliliter would be considered to be
responding to'
treatment with ABT-869 or analog of ABT-869. More specifically, a subject
demonstrating an increase from the predetermined level (such as a base line
level) in
the range of about 60 picograms per milliliter to about 150 picograms per
milliliter
would be considered to be responding to treatment with ABT-869 or analog of
ABT-
36
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
869. An increase from the predetermined level (such as a baseline level) of
66.5
picograms per milliliter above the predetermined level (such as a baseline
level) at
about either 8 or 15 days after the subject first receives treatment with ABT-
869 or an
analog of ABT-869 at the steady state (namely at least about 3 days, at least
about 5
days, at least about 7 days, at least about 10 days at least about 15 days, at
least 20
days, at least 30 days, at least 35 days, at least 40 days, at least 45 days,
at least 50
days, at least 60 days, etc.) after the subject first receives treatment with
ABT-869 or an
analog of ABT-869.
Additionally, also in the mono/mono assays, if the toxicity level of the
subject
being treated with ABT-869 or an analog of ABT-869 reaches a range of 130
picograms per milliliter to 160 picograms per milliliter, the treating
physician can
reduce the amount of ABT-869 or analog of ABT-869 being administered to the
subject. An exemplary toxicity level at which a treating physician can reduce
the
amount of ABT-869 or analog of ABT-869 being administered to the subject is
150
picograms per milliliter.
An exemplary predetermined level in mono/poly assays can be about 30
picograms per milliliter at about 24 hours after the subject first receives
treatment with
ABT-869 or an analog of ABT-869. Alternatively, the predetermined level in
mono/poly can be about 40 picograms per milliliter to about 75 picograms per
milliliter
at the steady state (namely at least about 3 days, at least about 5 days at
least about 7
days, at least about 10 days, at least about 15 days, at least 20 days, at
least 30 days, at
least 35 days, at least 40 days, at least 45 days, at least 50 days, at least
60 days, etc.)
after the subject first receives treatment with ABT-869 or an analog of ABT-
869. An
exemplary predetermined level in mono/mono assays can be about 66.5 picograms
per
milliliter at about 24 hours after the subject first receives treatment with
ABT-869 or an
analog of ABT-869. The above described comparisons (also referred to as
informational analysis) involving the amount of human P1GF-1 or human PIGF-1
fragment in the test sample and the predetermined level (such as a baseline
level, can be
made by an automated system, such as a software program or intelligence system
that is
part of, or compatible with, the automated or semi-automated equipment (e.g.,
computer platform) on which the above described assays are carried out.
Alternatively,
the above described comparisons can be done by a physician. Subjects receiving
37
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
treatment with ABT-869 or an analog of ABT-869 may be suffering from cancers
such
as lung cancer, breast cancer, stomach cancer, bladder cancer colon cancer,
pancreatic
cancer, ovarian cancer, prostate cancer, renal cancer, hepatocellular cancer,
rectal
cancer, hematopoietic malignancies, glioblastoma or infantile hemangioma.
In another aspect, the assays of the present disclosure can be used to
determining whether a subject who is predisposed to a disease (e.g., a type of
cancer) or
who is suffering from a disease will respond to treatment with a drug. Such an
assay
involves contacting a first capture antibody that binds to human P1GF-1 or
human
PIGF-1 fragment with a test sample obtained from a subject who is predisposed
or has a
predisposition to a disease or that is suffering from at least one disease and
receiving
treatment with ABT-869 or an analog of ABT-869 to form a first capture
antibody-
human P1GF-1 complex. In mono/poly assays, the capture antibody optionally is
monoclonal antibody 264. In mono/mono assays, the capture antibody optionally
is
monoclonal antibody 826. After the formation of the first capture antibody-
human
P1GF-1 complex, the test sample is then contacted with a second antibody that
binds to
human PIGF-1 or human P1GF-1 fragment and that has been conjugated to a
detectable
label to form a second capture antibody-human P1GF-1 detection complex. In
mono/poly assays, the capture antibody optionally is polyclonal antibody
pB264. In
mono/mono assays, the detection antibody optionally is monoclonal antibody
255. The
amount of the capture antibody-P1GF-1 detection complex that has been formed
is then
determined by detecting the detectable label. The amount of capture antibody-
human
PIGF-1 detection complex present in the test sample correlates with the amount
of
human PIGF-I or human P1GF-1 fragment in the test sample. The amount of human
PIGF-I or human P1GF-1 test fragment in the test sample is then compared with
a
predetermined level. Specifically, if the concentration of human P1GF-1 or
human
PIGF-1 test fragment determined in the test sample is lower than the
predetermined
level, then a determination is made that the subject will not benefit from
further or
continued treatment with ABT-869 or an analog of ABT-869. The treating
physician
may then make a decision to see if the subject is eligible for further or
continued
treatment with a different drug. However, if the concentration of human PIGF-1
or
human PIGF-1 fragment determined in the test sample is the same as or higher
than the
predetermined level, then a determination is made that the subject would or
will benefit
38
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
from further or continued treatment with ABT-869 or analog of ABT-869.
Subjects
that can be tested to determine whether or not they are eligible for treatment
with a drug
are subjects suffering from cancers such as lung cancer, breast cancer,
stomach cancer,
bladder cancer, colon cancer, pancreatic cancer, ovarian cancer, prostate
cancer, renal
cancer, hepatocellular cancer, rectal cancer, hematopoietic malignancies,
glioblastoma
or infantile hemangioma. Alternatively, the subject may be suffering from
autoimmune
diseases, such as rheumatoid arthritis, thyroiditis, type 1 diabetes, multiple
sclerosis,
sarcoidosis, inflammatory bowel disease, Crohn's disease, myasthenia gravis
and
systemic lupus erythematosus; psoriasis, organ transplant rejection (e.g.,
kidney
rejection, graft versus host disease), benign and neoplastic proliferative
diseases or
ocular diseases, such as, but not limited to, macular degeneration.
The above assay can be used to monitor the progression of disease in subjects
suffering from acute conditions. Acute conditions, also known as critical care
conditions, refer to acute, life threatening diseases or other critical
medical conditions
involving the cardiovascular system (including, but not limited to, sepsis),
central
nervous stem and/or respiratory system. Typically, critical care conditions
refer to
those conditions requiring acute medical intervention in a hospital based
setting
(including, but not limited to, the emergency room, intensive care unit,
trauma center or
other emergent care setting) or administration by a paramedic or other field-
based
medical personnel. For critical care conditions, repeat monitoring is
generally done
within a shorter time frame, namely, minutes, hours or days (e.g., about 1
minute, about
5 minutes, about 10 minutes, about 15 minutes, about 30 minutes, about 45
minutes,
about 60 minutes, about 2 hours, about 3 hours, about 4 hours, about 5 hours,
about 6
hours, about 7 hours, about 8 hours, about 9 hours, about 10 hours, about 11
hours,
about 12 hours, about 13 hours, about 14 hours, about 15 hours, about 16
hours, about
17 hours, about 18 hours, about 19 hours, about 20 hours, about 21 hours,
about 22
hours, about 23 hours, about 24 hours, about 2 days, about 3 days, about 4
days, about
5 days, about 6 days or about 7 days), and the initial assay likewise is
generally done
within a shorter timeframe, e.g., about minutes, hours or days of the onset of
the disease
or condition.
The above assay can also be used to monitor the progression of disease in
subjects suffering from chronic, or non-acute conditions. Non-critical care
or, non-
39
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
acute conditions, refers to conditions other than acute, life threatening
disease or other
critical medical conditions involving the cardiovascular system, central
nervous system
and/or respiratory system. Typically, non-acute conditions include those of
longer-term
or chronic duration, and include, e.g., ophthalmic conditions and cancer. For
non-acute
conditions, repeat monitoring generally is done with a longer timeframe, e.g.,
hours,
days, weeks, months or years (e.g., about 1 hour, about 2 hours, about 3
hours, about 4
hours, about 5 hours, about 6 hours, about 7 hours, about 8 hours, about 9
hours, about
hours, about 11 hours, about 12 hours, about 13 hours, about 14 hours, about
15
hours, about 16 hours, about 17 hours, about 18 hours, about 19 hours, about
20 hours,
10 about 21 hours, about 22 hours, about 23 hours, about 24 hours, about 2
days, about 3
days, about 4 days, about 5 days, about 6 days, about 7 days, about 2 weeks,
about 3
weeks, about 4 weeks, about 5 weeks, about 6 weeks, about 7 weeks, about 8
weeks,
about 9 weeks, about 10 weeks, about 11 weeks, about 12 weeks, about 13 weeks,
about 14 weeks, about 15 weeks, about 16 weeks, about 17 weeks, about 18
weeks,
about 19 weeks, about 20 weeks, about 21 weeks, about 22 weeks, about 23
weeks,
about 24 weeks, about 25 weeks, about 26 weeks, about 27 weeks, about 28
weeks,
about 29 weeks, about 30 weeks, about 31 weeks, about 32 weeks, about 33
weeks,
about 34 weeks, about 35 weeks, about 36 weeks, about 37. weeks, about 38
weeks,
about 39 weeks, about 40 weeks, about 41 weeks, about 42 weeks, about 43
weeks,
about 44 weeks, about 45 weeks, about 46 weeks, about 47 weeks, about 48
weeks,
about 49 weeks, about 50 weeks, about 51 weeks, about 52 weeks, about 1.5
years,
about 2 years, about 2.5 years, about 3.0 years, about 3.5 years, about 4.0
years, about
4.5 years, about 5.0 years, about 5.5. years, about 6.0 years, about 6.5
years, about 7.0
years, about 7.5 years, about 8.0 years, about 8.5 years, about 9.0 years,
about 9.5 years
or about 10.0 years), and the initial assay likewise generally is done within
a longer
time frame, e.g., about hours, days, months or years of the onset of the
disease or
condition.
In another aspect, the assays of the present invention can be used to monitor
the
response of a subject receiving treatment for cancer (e.g., the tumor) with a
drug. Such
an assay involves contacting a first capture antibody that binds to PIGF-1
with a test
sample obtained from a subject that has one or more tumors and is receiving
treatment
with ABT-869 to form a first capture antibody-PIGF-1 complex. In mono/poly
assays,
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
the capture antibody optionally is monoclonal antibody 264. In mono/mono
assays, the
capture antibody is optionally monoclonal antibody 826. After the formation of
the
first capture antibody-PIGF complex, the test sample is then contacted with a
second
antibody that binds to PIGF-1 and that has been conjugated to a detectable
label to form
a second capture antibody-P1GF-1 detection complex. In mono/poly assays, the
detection antibody is optionally polyclonal antibody pB264. In mono/mono
antibodies,
the detection antibody is optionally, monoclonal antibody 255. The amount of
the
capture antibody-PIGF-1 detection complex that has been formed is then
determined by
detecting the detectable label. The amount of capture antibody-PIGF-1
detection
complex present in the test sample correlates with the amount of PIGF-1 in the
test
sample. The amount of PIGF-1 in the test sample is then compared with a
predetermined level. Specifically, if the concentration of P1GF-1 determined
in the test
sample is lower than the predetermined level, then the patient is considered
not to be
responding to treatment with ABT-869. The treating physician may then make a
decision to increase the amount of ABT-869 administered to the subject or to
switch the
subject to an entirely different drug to try and treat the cancer (e.g.,
tumor) or may add
another drug to the treatment regiment (e.g., administer to the subject ABT-
869 in
combination with another drug). However, if the concentration of PIGF-1
determined
in the test sample is the same as or higher than the predetermined level, then
the patient
is considered to be responding to treatment with ABT-869. The treating
physician may
then make the decision not to change or alter the amount of ABT-869 being
administered to the subject; although, the treating physician could also
decide to lower
the amount of ABT-869 being administered to the subject as well. Subjects
receiving
treatment with ABT-869 may be suffering cancers (e.g., tumors) in connection
with
cancers such as lung cancer, breast cancer, stomach cancer, bladder cancer,
colon
cancer, pancreatic cancer, ovarian cancer, prostate cancer, renal cancer,
hepatocellular
cancer, rectal cancer, hematopoietic malignancies, glioblastoma or infantile
hemangioma. Alternatively, the subject may be suffering from autoimmune
diseases,
such as rheumatoid arthritis, thyroiditis, type 1 diabetes, multiple
sclerosis, sarcoidosis,
inflammatory bowel disease, Crohn's disease, myasthenia gravis and systemic
lupus
erythematosus; psoriasis, organ transplant rejection (e.g., kidney rejection,
graft versus
41
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
host disease), benign and neoplastic proliferative diseases or ocular
diseases, such as,
but not limited to, macular degeneration.
In another aspect, the assays of the present disclosure can be used to confirm
the
biological activity of ABT-869 or an analog of ABT-869. Such an assay involves
contacting a first capture antibody that binds to human PIGF-1 or human PIGF-1
fragment with a test sample obtained from a subject administered ABT-869 or an
analog of ABT-869 to form a first capture antibody-human P1GF-1 complex. In
mono/poly assays, the capture antibody is optionally monoclonal antibody 264.
In
mono/mono assays, the capture antibody optionally is monoclonal antibody 826.
After
the formation of the first capture antibody-human P1GF-1 complex, the test
sample is
then contacted with a second antibody that binds to human P1GF-1 or human P1GF-
1
fragment and that has been conjugated to a detectable label to form a second
capture
antibody-human P1GF-1 detection complex. In mono/poly assays, the detection
antibody is optionally polyclonal antibody pB264. In mono/mono assays, the
detection
antibody optionally is monoclonal antibody 255. The amount of the capture
antibody-
P1GF-1 detection complex that has been formed is then determined by detecting
the
detectable label. The amount of capture antibody-human P1GF-1 detection
complex
present in the test sample correlates with the amount of human P1GF-1 or human
P1GF-
1 fragment in the test sample. The amount of human P1GF-1 or human P1GF-1 test
fragment in the test sample is then compared with a predetermined level (such
as a
baseline level prior to the subject being treated with ABT-869 or an analog of
ABT-
869). Specifically, if the concentration of human P1GF-1 or human P1GF-1 test
fragment determined in the test sample is lower than the predetermined level,
then a
determination is made ABT-869 or analog of ABT-869 does not exhibit biological
activity in the subject. However, if the concentration of human P1GF-1 or
human
P1GF-1 fragment determined in the test sample is the same as or higher than
the
predetermined level, then a determination is made that ABT-869 or analog of
ABT-869
exhibits biological activity in the subject.
In still yet another aspect, the present disclosure relates to methods of
treating
subjects suffering from one or more types of cancer or one or more types of
autoimmune diseases. For example, the cancer that can be treated according to
the
methods of the present invention can be one or more cancers selected from the
group
42
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
consisting of lung cancer, breast cancer, stomach cancer, bladder cancer,
colon cancer,
pancreatic cancer, ovarian cancer, prostate cancer, renal cancer,
hepatocellular cancer,
rectal cancer, hematopoietic malignancies, glioblastoma and infantile
hemangioma.
Examples of autoimmune diseases that can be treated according to the methods
of the
present invention include, but are not limited to, rheumatoid arthritis,
thyroiditis, type 1
diabetes, multiple sclerosis, sarcoidosis, inflammatory bowel disease, Crohn's
disease,
myasthenia gravis and systemic lupus erythematosus; psoriasis, organ
transplant
rejection (e.g., kidney rejection, graft versus host disease), benign and
neoplastic
proliferative diseases or ocular diseases, such as, but not limited to,
macular
degeneration.
The methods of treatment of the present disclosure involve obtaining a test
sample from the subject suffering from at least one cancer or at least one
autoimmune
disease and who is receiving treatment with a predetermined amount ofN-[4-(3-
amino-
1 H-indazol-4-yl)phenyl]-N'-(2-fluoro-5-methylphenyl)urea or an analog of N-[4-
(3-
amino-lH-indazol-4-yl)phenyl]-N'-(2-fluoro-5-methylphenyl)urea. The
predetermined
amount of N-[4-(3-amino-1H-indazol-4-yl)phenyl]-N'-(2-fluoro-5-
methylphenyl)urea
or an analog ofN-[4-(3-amino-lH-indazol-4-yl)phenyl]-N'-(2-fluoro-5-
methylphenyl)urea being administered to the subject for whom the test sample
is being
obtained is the amount determined by the treating physician to be appropriate
for the
cancer or autoimmune disease being treated. For example, the predetermined
amount
of N-[4-(3-amino-IH-indazol-4-yl)phenyl]-N'-(2-fluoro-5-methylphenyl)urea or
an
analog ofN-[4-(3-amino-1H-indazol-4-yl)phenyl]-N'-(2-fluoro-5-
methylphenyl)urea
being administered or dosed to the subject can be 0.01 mg/kg, 0.05 mg/kg, 0.10
mg/kg,
0.25 mg/kg, 0.50 mg/kg, 0.75 mg/kg, 1.00 mg/kg, 1.25 mg/kg, 1.50 mg/kg, 1.75
mg/kg,
2.0 mg/kg, 2.25 mg/kg, 2.50 mg/kg, 2.75 mg/kg, 3.0 mg/kg, 3.25 mg/kg, 3.50
mg/kg,
3.75 mg/kg, 4.0 mg/kg, 4.25 mg/kg, 4.50 mg/kg, 4.75 mg/kg, 5.0 mg/kg, 5.25
mg/kg,
5.50 mg/kg, 5.75 mg/kg, 6.0 mg/kg, 6.25 mg/kg, 6.50 mg/kg, 6.75 mg/kg, 7.0
mg/kg,
7.25 mg/kg, 7.50 mg/kg, 7.75 mg/kg, 8.0 mg/kg, 8.25 mg/kg 8.50 mg/kg, 8.75
mg/kg,
9.0 mg/kg, 9.25 mg/kg, 9.50 mg/kg, 9.75 mg/kg, 10.00 mg/kg, etc.
Once the test sample has been obtained from the subject, the test sample is
contacted with a first capture antibody that binds to PIGF-1 to form a first
capture
antibody-PIGF-1 complex. In mono/poly assays, the capture antibody optionally
is
43
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
monoclonal antibody 264. In mono/mono assays, the capture antibody is
optionally
monoclonal antibody 826. After the formation of the first capture antibody-
PIGF
complex, the test sample is then contacted with a second antibody that binds
to PIGF-1
and that has been conjugated to a detectable label to form a second capture
antibody-
PIGF-1-detection complex. In mono/poly assays, the detection antibody is
optionally
polyclonal antibody pB264. In mono/mono antibodies, the detection antibody is
optionally, monoclonal antibody 255. The amount of the capture antibody-P1GF-1
detection complex that has been formed is then determined by detecting the
detectable
label. The amount of capture antibody-PIGF-1 detection complex present in the
test
sample correlates with the amount of P1GF-1 in the test sample. The amount of
PIGF-1
in the test sample is then compared with a predetermined level (such as a
baseline level
before the subject is treated with ABT-869 or an analog of ABT-869).
Specifically, if
the concentration of P1GF-1 determined in the test sample is lower than the
predetermined level, then the patient is considered not to be responding to
treatment
with ABT-869. The treating physician may then make a decision to treat the
subject by
(a) increasing or adjusting the amount of ABT-869 or analog of ABT-869 being
administered to the subject such that the amount of ABT-869 or analog of ABT-
869 is
higher then the predetermined amount of ABT-869 or analog of ABT-869 that has
been
previously been administered to the subject (For example, if the predetermined
amount
of ABT-869 or analog of ABT-869 being administered to the subject at the time
the test
sample is obtained is 0.05 mg/kg, the treating physician made decide to treat
the subject
by increasing the dosage of ABT-869 or analog of ABT-869 from 0.05 mg/kg to
0.25
mg/kg), (b) switching the subject to an entirely different drug to treat the
cancer (e.g.,
tumor) or autoimmune disease; or (c) may add another drug to the treatment
regimen
(e.g., administer to the subject ABT-869 in combination with another drug).
However,
if the concentration of PIGF-1 determined in the test sample is the same as or
higher
than the predetermined level, then the patient is considered to be responding
to
treatment with ABT-869. The treating physician may then make the decision not
to
further treat the patient by changing or altering the amount of ABT-869 being
administered to the subject; although, the treating physician can also decide
to treat the
patient by lowering the amount of ABT-869 being administered to the subject as
well.
44
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
The above described comparisons (also referred to as informational analysis)
involving the amount of human P1GF-1 or human P/GF-1 fragment in the test
sample
and the predetermined level (such as a baseline level, can be made by an
automated
system, such as a software program or intelligence system that is part of, or
compatible
with, the automated or semi-automated equipment (e.g., computer platform) on
which
the above described assays are carried out. Alternatively, the above described
comparisons can be done by a physician.
Additionally, also in the mono/mono assays, if the toxicity level of the
subject
being treated with ABT-869 or an analog of ABT-869 reaches a range of 130
picograms per milliliter to 160 picograms per milliliter, the treating
physician can
reduce the amount of ABT-869 or analog of ABT-869 being administered to the
subject. An exemplary toxicity level at which a treating physician can reduce
the
amount of ABT-869 or analog of ABT-869 being administered to the subject is
150
picograms per milliliter.
C. Kits
The present disclosure also contemplates kits for detecting the presence of
human P1GF-1 or human P1GF-1 fragment in a test sample and to be employed as a
companion diagnostic for ABT-869 or an analog of ABT-869. Such kits can
comprise
one or more antibodies, including one or more of the antibodies described
herein. More
specifically, if the kit is a kit for performing an immunoassay, the kit
optionally can
contain (1) at least one capture antibody that specifically binds to human
PIGF-1 or
human P/GF-1 fragment; (2) at least one conjugate; and (3) one or more
instructions for
performing the immunoassay in connection with ABT-869 or an analog of ABT-869
(e.g., including for patient selection and/or dosage optimization). The
antibodies
described herein can be included in such a test kit as a capture antibody, as
a detection
antibody or both as a capture antibody and a detection antibody. For example,
for use
in mono/poly assays, monoclonal antibody 264 can be included in the kit as a
capture
antibody and polyclonal antibody pB264 can be included in the kit as a
detection
antibody. Alternatively, polyclonal antibody pB264 can be included in the kit
as a
capture antibody and monoclonal antibody 264 can be included in the kit as a
detection
antibody. In still yet another alternative, monoclonal antibody 264 or
polyclonal
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
antibody pB264 can be included in the kit as a capture antibody and a
different
antibody included in the kit as a detection antibody. In still yet another
alternative,
monoclonal antibody 264 or polyclonal antibody pB264 can be included in the
kit as a
detection antibody and a different antibody included in the kit as a capture
antibody.
Moreover, for example, for use in mono/mono assays, monoclonal antibody 826
can be
included in the kit as capture antibody and monoclonal antibody 255 can be
included in
the kit as a detection antibody. Alternatively, monoclonal antibody 255 can be
included in the kit as a capture antibody and monoclonal antibody 826 can be
included
in the kit as a detection antibody. In still yet another alternative,
monoclonal antibody
826 or monoclonal antibody 255 can be included in the kit as a capture
antibody and a
different antibody included in the kit as a detection antibody. In still yet
another
alternative, monoclonal antibody 826 or monoclonal antibody 255 can be
included in
the kit as a detection antibody and a different antibody included in the kit
as a capture
antibody.
Optionally, the kit can also contain at least one calibrator or control. Any
calibrator or control can be included in the kit. Preferably, however, the
calibrator or
control is a human P1GF-1 fragment. More preferably, the calibrator or control
comprises an isoform of human P1GF-1 that comprises residues 21-149 of human
PIGF-1 and is available from R&D Systems, Inc., Minneapolis, Minnesota
(Catalog
Number #P49763). Optionally, the kit can also contain at least one sample
collection
tube.
Thus, the present disclosure further provides for diagnostic and quality
control
kits comprising one or more antibodies described herein. Optionally the
assays, kits
and kit components of the invention are optimized for use on commercial
platforms
(e.g., immunoassays on the Prising, AxSYM , ARCHITECT and EIA (Bead)
platforms of Abbott Laboratories, Abbott Park, IL, as well as other commercial
and/or
in vitro diagnostic assays). Additionally, the assays, kits and kit components
can be
employed in other formats, for example, on electrochemical or other hand-held
or
point-of-care assay systems. The present disclosure is, for example,
applicable to the
commercial Abbott Point of Care (i-STAT , Abbott Laboratories, Abbott Park,
IL)
electrochemical immunoassay system that performs sandwich immunoassays for
several cardiac markers, including TnI, CKMB and BNP. Immunosensors and
methods
46
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
of operating them in single-use test devices are described, for example, in
U.S. Patent
Applications 20030170881, 20040018577, 20050054078 and 20060160164 which are
incorporated herein by reference. Additional background on the manufacture of
electrochemical and other types of immunosensors is found in U.S. Patent
5,063,081
which is also incorporated by reference for its teachings regarding same.
Optionally the kits include quality control reagents (for example, sensitivity
panels, calibrators, and positive controls). Preparation of quality control
reagents is well
known in the art, and is described, e.g., on a variety of immunodiagnostic
product insert
sheets.
In another embodiment, the present disclosure provides for a quality control
kit
comprising one or more antibodies described herein for use as a sensitivity
panel to
evaluate assay performance characteristics and/or to quantitate and monitor
the
integrity of the antigen(s) used in the assay.
The kits can optionally include other reagents required to conduct a
diagnostic
assay or facilitate quality control evaluations, such as buffers, salts,
enzymes, enzyme
co-factors, substrates, detection reagents, and the like. Other components,
such as
buffers and solutions for the isolation and/or treatment of a test sample
(e.g.,
pretreatment reagents), may also be included in the kit. The kit may
additionally
include one or more other controls. One or more of the components of the kit
may be
lyophilized and the kit may further comprise reagents suitable for the
reconstitution of
the lyophilized components.
The various components of the kit optionally are provided in suitable
containers. As indicated above, one or more of the containers may be a
microtiter plate.
The kit further can include containers for holding or storing a sample (e.g.,
a container
or cartridge for a blood or urine sample). Where appropriate, the kit may also
optionally contain reaction vessels, mixing vessels and other components that
facilitate
the preparation of reagents or the test sample. The kit may also include one
or more
instruments for assisting with obtaining a test sample, such as a syringe,
pipette,
forceps, measured spoon, or the like.
The kit further can optionally include instructions for use, which may be
provided in paper form or in computer-readable form, such as a disc, CD, DVD
or the
like.
47
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
Exemplary kits according to the present invention can be used to determine
whether a subject receiving treatment with a drug has obtained an efficacious
blood
level of that drug, to monitor response of a subject receiving treatment for
cancer with
an anti-cancer drug, to determine whether a subject who is predisposed to
disease or
who is suffering from a disease will respond to treatment with a drug, to
monitor
progression of disease in a subject being treated with a drug, to confirm
biological
activity of a drug like ABT-869 or analog of ABT-869 in a subject or as part
of a
treatment regimen to treat a subject suffering from cancer or an autoimmune
disease.
D. Adaptations of the Methods of the Present Disclosure
The invention as described herein also can be adapted for use in a variety of
automated and semi-automated systems (including those wherein the solid phase
comprises a microparticle), as described, e.g., in U.S. Patent Nos. 5,089,424
and
5,006,309, and as, e.g., commercially marketed by Abbott Laboratories (Abbott
Park,
IL) including but not limited to Abbott's ARCHITECT , AxSYM , IMX, PRISM,
and Quantum II instruments, as well as other platforms. Moreover, the
invention
optionally is adaptable for the Abbott Laboratories commercial Point of Care
(i-
STATTM) electrochemical immunoassay system for performing sandwich
immunoassays. Immunosensors, and their methods of manufacture and operation in
single-use test devices are described, for example in, U.S. Patent No.
5,063,081, U.S.
Patent Application 2003/0170881, U.S. Patent Application 2004/0018577, U.S.
Patent
Application 2005/0054078, and U.S. Patent Application 2006/0160164, which are
incorporated in their entireties by reference for their teachings regarding
same.
By way of example and not of limitation, examples of the present disclosure
shall now be given.
Example 1: Study Involving Patients with Refractory Solid Malignancies
Receiving Treatment with ABT-869 including Human PIGF-1 Analysis
A. Patients and Methods
Eligible patients were 18 years or older with histologically confirmed
advanced
non-haematologic malignancy refractory to or with no standard therapy. Other
criteria
48
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
included having ECOG (Eastern Cooperative Oncology Group) Performance Score of
0-2, measurable disease by CT or MRI, and laboratory values fulfilling the
following
criteria: haemoglobin >_9.0 g/dL, platelet >:100,000/4L, absolute neutrophil
count
(ANC) >_1,000/ L, creatinine <_1.5 x upper normal limit (ULN), bilirubin, AST
and
ALT 51.5x ULN of institution's normal range.
The main exclusion criteria were: anticancer therapy within the previous 28
days; life expectancy of less than 12 weeks; history of central nervous system
metastases; significant proteinuria; uncontrolled hypertension; left
ventricular ejection
fraction <50%; active signs of bleeding; or anticoagulation therapy for
therapeutic
intent. The study received approval by the institutional ethics review board
and all
patients provided written informed consent.
B. Study Design and drug treatment
This study was designed as a single-arm, open-label Phase I trial and was
conducted in three segments (A, B and Q. Segment A was a sequential dose-
escalation
study with primary intent to.define the maximum tolerable dose (MTD), segment
B
involved expansion of the next lower dose to a total of 12 patients to further
evaluate
tolerability, and segment C was to study the tolerability and pharmacodynamics
of a
lower dose cohort to better define dose-effect relationships. In all three
segments,
patients received ABT-869 until tumor progression or occurrence of dose-
limiting
toxicity (DLT).
The starting dose of 10 mg was obtained by applying a safety factor of 5 to
the
no-observed-adverse-event-level dosage used in the one-month rat study, which
was
the more sensitive species. A dose of 10 mg was selected as the projected Cmax
and
AUC (0.05 g/ml and 0.75 g=hr/ml, respectively) had a safety margin for
observed
toxicity of at least 4.2 for a 70 kg person based on a body surface area
scaling. ABT-
869 was self-administered as a continuous daily oral dosage at night (except
on days 1
and 15 when drug was administered in the morning for assessment of PK) in
treatment
periods of 21 days. No drug was administered on day 14. As ABT-869 lacks high
aqueous solubility, the study drug was diluted in 60 mLs of Ensure Plus .
Preliminary
PK at doses if 10 mg showed a modest correlation between oral clearance and
body-
weight; thus subsequent dose escalations in segment A were based on body-
weight.
49
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
Dose escalation was planned in cohorts of three patients each, and cohort
expansion to six patients was planned if DLT occurred in I of 3 patients
during the first
treatment period. DLT was defined as follows: grade 3 fatigue; grade 3
proteinuria;
persistent grade 3 hypertension despite intervention; grade 3 neutropenia with
fever;
grade 4 neutropenia > 7 days; grade 4 thrombocytopenia; any other related
grade 3 or 4
adverse events; and any unexpected grade 2 toxicity of possible or probable
relationship to treatment, which required dose modification or delay of more
than one
week. Dose escalation was stopped if 2 or more patients out of 6 in a dose
cohort
experienced DLT within the first treatment period; that dose would be
considered the
MTD.
C. Platelet-free Plasma for Human P/GF-1 Analysis
Approximately 4 mL of blood was collected by venipuncture into a 4 mL
EDTA (purple cap) on TP 1 D 1 pre-dose and at 6 and 24 hour post-dose and TP 1
D 15
pre-dose and at 6 hour post-dose. Additional specimens were collected on TP2D
1 (Day
22) and TP3D 1 (Day 43) and at the end of TP 4, 6 and 8. A sample was
collected at the
time of the Final Visit. A sample was also collected at the time of a Grade 3
toxicity of
asthenia. The collection tube was inverted (2-3 times) to reduce the
likelihood of clot
formation. The tube was centrifuged at 1500 x g for 15min. at 2-8 C within
30 minutes of collection. The plasma was split into two 1.5 mL aliquots and
centrifuged at 10,000 x g for 10 minutes at 2-8 C to complete platelet
removal. The
plasma was immediately transferred (supernatant) to two appropriately labeled
2 mL
cryovials and frozen at -70 C. The samples were stored at -70 C until
notification
was received to ship the batches. The complete process of centrifugation,
transfer to
cryovial and freezing was accomplished in less than 1 hour from blood draw.
D. Tumor Response Evaluation and Safety
Baseline CT imaging (CT) was performed within 4 weeks before the ABT-869
treatment, and repeated every two treatment periods (6 weeks). Lesions were
evaluated
using RECIST (Response Evaluation Criteria in Solid Tumors) . Adverse events
were
recorded and graded according to CTC (Common Terminology Criteria) version
3Ø
Physical examination, complete blood counts, serum chemistries including
troponin T,
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
urinalysis and serial electrocardiograms were assessed at least weekly for the
first 4
treatment periods. ACTH tests and Multiple Gated Acquisition Scan scans were
done
at baseline and after every four treatment periods to assess adrenal and
cardiac safety
with repeated dosing.
E. Pharmacokinetic Assessment
PK sampling was performed on days 1, 15 at the following time points: pre-
dose, 0.25, 0.5, 1, 2, 3, 4, 6, 8 and 24 hours post-dosing. ABT-869 and its
metabolite
concentrations in plasma were determined using a validated method based on
triple
quadrupole tandem mass spectrometry with a lower limit of quantification of
1.Ong/mL.
Pharmacokinetic parameters and metabolites were analyzed with non-
compartmental
methods based on WINNONLIN (Pharsight Corp., Cary, NC). The area under the
concentration-time curve (AUC) was estimated using the log-linear trapezoidal
option
and by extrapolation of the terminal curve to infinity including at least the
last 3
concentration points; oral clearance, half-life (tl/2) and volume of
distribution at steady
state (VDss) were calculated.
F. Pharmacodynamic Assessments
Samples for biomarkers of angiogenesis were collected on days 1 (baseline and
6, 24 hours post dosing), 15, 21 and 42 and at the end of the 4fl' and 6th
treatment
periods. Plasma VEGF was measured using commercial ELISA kits from R& D
Systems (Minneapolis, MN). A prototype, automated immunoassay designed to
measure PIGF-1 on the Abbott ARCHITECT instrument system (Abbott
Laboratories, Abbott Park, IL) was used in this study. The immunoassay is
configured
in a two-step sandwich assay format using monoclonal antibody 264 as the
capture
antibody and polyclonal antibody pB264 as the detection antibody, and using
recombinant human PIGF-1 (R&D Systems, Inc., Minneapolis, Minnesota, Catalog
Number 264-PG/CF) as calibrator. The circulating (or free) P1GF-1 present in
blood
specimens was captured using anti-P1GF-1 monoclonal antibody 264 coated
paramagnetic microparticles (which were made using routine techniques known in
the
art) with detection via affinity purified anti-PIGF-1 polyclonal antibodies
pB264 that
were labeled with acridinium. The dynamic range of the assay was 0 - 1500
pg/mL.
51
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
Generally, paramagnetic latex microparticles (4.7 M), derivatized with
carboxyl functional groups, were coated with anti-P1GF-1 monoclonal antibody
at an
antibody concentration demonstrated to be sufficient to maximize the level of
antibody
absorbed to the surface area of the microparticles (2% solids by weight) in 50
mM 2-
(N-morpholino)ethanesulfonic acid (MES), pH 5.5. After 10 minutes, the non-
absorbed
antibody was removed by washing the particles multiple times with MES buffer.
Following washing of the particles, EDAC (1-ethyl-3-(3-dimethylaminopropyl)
carbodiimide, hydrochloride, Sigma-Aldrich, St. Louis, MO) was added and
allowed to
react, forming a covalent coupling of the antibody molecules to the particles.
The
particles were then washed with phosphate buffered saline to stop the reaction
and
remove unreacted EDAC. The particles were then diluted to 0.1% in buffer for
use in
an automated immunochemical analyzer.
Acridinium-labeled anti-P1GF-1 antibody was prepared by incubating a
polyclonal antibody (alternatively a monoclonal antibody could be used) with
an
.
acridinium designated SPSP (N10-(3-sulfopropyl)-N-(3-sulfopropyl) acridinium-9-
carboxamide at a molar ratio of acridinium to antibody ranging from 1 to 100.
Unconjugated acridinium was then separated from the acridinium-labeled
antibody
conjugate by size chromatography. The purified conjugate was then diluted in
buffer to
a concentration yielding the maximum signal to noise ratio in the assay.
Circulating populations of activated, apoptotic and progenitor endothelial
cells
were measured using a modified flow cytometry-based method previously
described in
Mancuso P, et al:, Blood 97:3658-3661 (2001) and Yee, K.W., et al., Clin
Cancer Res.,
11: 6615-6624 (2005).
G. Statistical Analyses
For PIGF-1 analysis, the statistical analysis was performed using the JMP
7Ø1
statistical analysis program produced by the SAS Institute. Univariate
Survival
Analysis with log rank analysis was performed to identify an optimal P/GF-1
threshold
which stratified patients for time on therapy (indicative of therapeutic
benefit). Groups
are shown for the Day 15 time point as either all patients, > 40 pg/mL or <40
pg/mL,
with clear differences in the time to progression for each group.
52
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
Patients were classified as having poor effect if tumor progression occurred
at
or before treatment period 4 and good effect if progression had not occurred
at that
time.
H Results
Patient Characteristics
Thirty-three patients were recruited into the study (See, Table 1, below).
53
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
Table 1
Patient Characteristic
Sex (Males/Females) 16/17
Age Median (range) years 56(29-76)
Eastern Cooperative Group Performance Status
0 18
1 13
2 2
Tumour Sites
Non-small cell lung 8
Colorectal 7
Hepatocellular 4
Ovary 3
Breast 2
Neuroendocrine 2
Endometrial Sarcoma 2
Others* 5
Prior lines of treatment
0-2 13
>2 20
*Other tumor types included one patient each with urachal carcinoma, soft
tissue
sarcoma, renal cell carcinoma, nasopharyngeal carcinoma and primitive
neuroectodermal tumor.
Treatment was discontinued due to disease progression and adverse events in 18
patients , of which 13 subjects had radiographic disease progression; 5
patients
discontinued due to adverse events related to ABT-869; 1 patient discontinued
due to
an adverse event unrelated to ABT-869; 1 patient discontinued due to an
adverse event
with an unknown relationship to ABT-869; 4 patients continued to receive
treatment
with clinical benefit at the time of writing. Median overall treatment
duration
(excluding days without taking ABT-869) for all dose cohorts was 84 days
(range 4-
694 days). There were no treatment-related deaths.
54
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
1. Toxicity and tolerability of repeated dosing
The most common drug-related adverse events were fatigue, proteinuria,
hypertension, myalgia, skin toxicity (hand and foot blisters) and oral
hypersensitivity,
and these toxicities increased in frequency and intensity with increasing
doses (See,
Tables 2A and 2B and 3, below).
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
o 00 - 'fl C'4 0 0 0 0 0 0 0 0
M M
N
4-4
O
~jGG O .~ ~n 0 0 0 0 0 0 0
za~
8 00 t- to N M M O [~ v1 p~
00 I- \.D Vl M M M O, N --V
M M M
O M N w 00 "D "t N N~~ O C\ 'n
cl "o .0 0
56
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
II O `.-. O ry 0 0 O
N
y 7I7I O N N N O
8 N C
O
00
00
E
II -- 0 0 0 0 0 0 0 0
O -j
N
N O -- O O
II O O O O
F"
U -~
F-~ M M N M N M M N M
a
R
~ o.
y ~ tom~y.
tz
0
iii 111
57
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
a;
00 00
O O o O 00 ~O o h O U
O O O N N O O O O--~ O
O et O rn r O a M O~ N m O 0
M N M N ri N N M N Wl 11
1
O O O N O O
U Caj
o
L
00 O O N 0 N M O ~ O O 00
O O O N O 1 O O N C
41 ii +1 41 1-1 -H +1 4i -H +1 +1 +1
v) n h O. 00 O -. N m O,
N N N R 00 M C.1 M 4 M 4Ly:
C O O O O O O
o
o ~ S -o
C> Q
1 -H t1 -i +j 'o +1 -H +i t1 +1 Oo u
--0 M M N N
O 0
O O O .O
Q 4
0 N OO O D~O O O O O $-
+1 +1 +1 +1 +1 DN O L
O O O C O 00 )
Q Cd
-
4a 12
0c
=L rn
2 x go 3 Sn 0
=L =L
cdd
E v)
ea ~ U
6Li 4 =~ Q
q =~ O
U ct
U
fs. U
58
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
The most common drug-related adverse events were fatigue, proteinuria,
hypertension, myalgia, skin toxicity (hand and foot blisters) and oral
hypersensitivity,
and these toxicities increased in frequency and intensity with increasing
doses (Tables
2B). DLTs in the first treatment period (3 weeks) in segment A were grade 3
fatigue in
1 of 6 patients at 10mg/day, none at 0.25mg/kg/day, grade 3 hypertension in I
patient
and grade 3 proteinuria in another patient at the MTD of 0.3mg/kg/day. In
segment B,
the cohort of 0.25mg/kg/day was expanded to a total of 12 patients, with a
first
treatment period DLT of grade 3 proteinuria observed in a patient with a
diagnosis of
quiescent systemic lupus erythematosis, and one patient with grade 3
hypertension.
Repeated dosing at 0.25mg/kg/day after the first treatment period resulted in
dose
reductions for drug-related toxicity in 7 patients, including one patient with
both grade
3 proteinuria and grade 2 hypertension (Cycle 3), one patient with grade 3
foot blisters
(Cycle 2), one patient with symptomatic grade 2 hypertension (Cycle 2), one
patient
with grade 3 gastroenteritis (Cycle 5) and one with grade 3 abdominal pain
(Cycle 3).
In addition two patients (one each in Cycle 1 and Cycle 3) experienced grade 2
or grade
3 pneumothorax attributed to therapy induced cavitation of lung nodules.
Since the starting dose of 10mg achieved or exceeded the minimum PK targeted
for efficacy and antitumor effects were observed, a lower dose of 0.1mg/kg/day
dose
was studied in an additional 12 patients in segment C. Compared to
0.25mg/kg/day, the
0.1mg/kg/day dose had fewer episodes of grade 2 or higher toxicities, with DLT
observed in I patient who experienced grade 3 proteinuria in the first
treatment period
and a patient each with grade 3 hypertension and proteinuria after treatment
period 1.
The incidence of hypertension was dose-dependent, with an incidence of
hypertension
of at least grade 2 of 25% at 0.1mg/kg/day compared to 50% at 0.25mg/kg/day.
In
addition, the change in mean blood pressure correlated with dose (data not
shown).
Hypertension responded to standard antihypertensive therapy with angiotensin
converting enzyme inhibitors, (3 blockers and calcium channel blockers and no
patient
developed hypertensive crisis. Skin blisters and proteinuria resolved after
reduction or
discontinuation of ABT-869. No significant hematological or cardiac toxicities
were
observed in any of the patients treated. Overall, 14 patients (42%) required
dose
59
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
reductions due to adverse events. Four patients have been on the study for >_
12 months
without major cumulative toxicities after dose stabilization.
J. Pharmacokinetic Evaluation
Pharmacokinetic data was available for 32 and 31 patients on days I and 15,
respectively. Doses above 0.1 mg/kg achieved plasma exposures (>2.7 p.g =h/mL)
that
were relevant for antitumor activity based on a preclinical murine HT 1080
fibrosarcoma model. (See, Albert, et al., Molec. Cancer Ther. 5(4), 996-1006
(2006).
Over the studied dose range , absorption and elimination of ABT-869 were
linear (See,
Table 3) and pharmacokinetics of ABT-869 were dose-proportional and time-
invariant.
The mean time to Cm., t1n and oral clearance was 2.9h (range 2-8h), 18.6
5.7h and
2.7 1.2L/h, respectively. Day 15 accumulation ratio was 1.1 0.4. The main
systemic metabolite was the carboxylate metabolite. From the urinary recovery
analysis of 4 patients, <10% of ABT-869 dose was recovered in the urine as
unchanged
drug and carboxylate metabolite. At final analysis, CL/F did not correlate
with body
weight. No correlation with BSA, creatinine clearance, or baseline
AST/ALT/Albumin
was observed.
K. Efficacy
Three (10%) out of 29 patients with one post baseline CT scan achieved partial
response (PR); two had non-small cell lung cancer (NSCLC) treated at 0.3
mg/kg/day
and 10 mg/day respectively, and one had colorectal cancer (CRC) treated at 0.1
mg/kg/day (Figures 1 and 2a). An additional sixteen patients had stable
disease lasting
longer than 12 weeks, among which were patients with CRC (5), NSCLC (2),
ovarian
cancer (2), hepatocellular carcinoma (HCC) (2) and neuroendocrine tumour (2).
Prolonged stable disease lasting more than 12 months with minimal toxicity was
observed in four patients: alveolar soft part sarcoma (27 months), CRC (19
months),
HCC (17 months), and renal cell carcinoma (18 months). The patient with CRC
received 0.25 mg/kg/day of ABT-869 and developed cavitation of lung nodules,
despite
previous bevacizumab treatment, but required 2 dose reductions for grade 3
abdominal
discomfort to 0.1 mg/kg. With a planned protocol deviation to allow increase
in dose
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
to 0.15 mg/kg/day, further tumor reduction and cavitation was observed after
11
months, suggestive of a dose response for antiangiogenic activity from ABT-
869.
L. Pharmacodynamic Analyses
Human PIGF-1 (n = 31) increased from 15.6 4.8 pg/mL at baseline to 20.7
9.4pg/mL at 6 hours (p=0.02) and remained significantly elevated at 82.0
70.3pg/mL
on day 42 (p=0.0001) (See, Figures 1-4). Plasma VEGF (n=12) at baseline was
68.2
82.4pg/mL and was significantly increased by day 15 at 124.8 80.7pg/mL
(p=0.004).
Comparison of these biomarkers between 0.1 mg/kg/day and 0.25 mg/kg/day doses
showed significantly higher human P1GF-1 (paired t-test p=0.017), percentage
apoptotic CECs (p=0.028) and lower percentage activated CECs (p=0.027) on day
15
of treatment. In addition, percentage increase in P1GF-1 correlated positively
with
ABT869-AUCinf (r=0.57 p=0.0001) and ABT869-Cmax (r=0.42 p=0.0001).
Example 2:
Three phase 2 single agent multicenter clinical trials were run in
hepatocellular
carcinoma (HCC), renal cell carcinoma (RCC) and non-small cell lung cancer
(NSCLC), each of which will be summarized separately.
HCC
An open-label, randomized, multicenter phase 2 trial (M06-879) of oral ABT-869
at 0.25 mg/kg daily in Child-Pugh A (hereinafter "C-PA") or every other day
("QOD") in
Child-Pugh B (hereinafter referred to as "C-PB") patients (hereinafter
referred to as "pts")
until progressive disease (hereinafter referred to as "PD") or intolerable
toxicity, is
ongoing. Key eligibility criteria included unresectable or metastatic HCC; up
to one prior
line of systemic treatment; and at least one measurable lesion by computed
tomography
(hereinafter "CT") scan. The primary endpoint was the progression free
(hereinafter "PF")
rate at 16 weeks. Secondary endpoints included objective response rate
(hereinafter
"ORR"), time to progression (hereinafter "TTP"), progression free survival
(hereinafter
"PFS") and overall survival (hereinafter "OS"). CT scans were assessed
centrally and by
the investigators; presented results are from central assessment. Treatment
will continue
61
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
until disease progression or intolerable toxicity, or for up to 18 months
after the.last patient
enrollment. Safety (graded by NCI CTCAE, Ver. 3.0) was evaluated at study
visits
Inclusion criteria: Adults diagnosed with unresectable or metastatic HCC,
measurable lesion by Response Evaluation Criteria In Solid Tumors ("RECIST"),
received no more than 1 prior line of systemic treatment for HCC, had an ECOG
performance status 0-2, no other malignancies within previous 5 years, and
adequate organ
function. Exclusion criteria: received anti-cancer therapy within 21 days (5
half-lives)
before dosing or VEGF/PDGF/TKI therapy (prior Avastin allowed), untreated
brain or
meningeal metastases, Child-Pugh C hepatic impairment, proteinuria,
uncontrolled
hypertension, left ventricular Ejection Fraction <50%, >Grade 2 encephalopathy
(CTC
criteria), clinically significant uncontrolled conditions, and/or receiving
therapeutic
anticoagulation therapy or anti-retroviral therapy. The baseline
characteristics and medical
history of the patients are shown below in Table 4.
Table 4
All Child-Pugh A Child-Pugh
Baseline characteristics Patients n=38 n=6
n=44
Median age, yrs (range) 62.5 (20 61.5 (20 - 81) 64.5 (36-69)
-81)
Men, n (%) 36(82) 31(82) 5 (83)
Asian, n (%) 39 (89) 33 (87) 6 (100)
ECOG status, n (%)
0 22 (50) 21 (55) 1 (17
1 19 (43) 15 (40) 4 (67
2 3 2(5) 1 17
Medical history at screening
Alcohol use, ever, n (%) 22(50) 22(58) 1 (17)
Hepatitis, n (%)
B 27 (61) 23 (61) 4 (67
C 4 (9) 4 (11) 0
Other, % 13(30) 11(29) 2(33)
Extrahepatic spread, n (%) 26 (59) 23 (61) 3 (50)
Prior local regional therapies, n (%) 24 (55) 20 (53) 4 (67)
Prior systemic therapies, n (%)
0 37 (84) 31 (82) 6 (100
1 6 (14) 6 (16) 0
2 1(2) 1(3) 0
62
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
Results: 44 pts were enrolled from September 2007 to August 2008 at 6 centers
internationally. There were 38 C-PA patients (median age, 63.5 y [range, 20-
81]) and 6 C-
PB patients (median age, 64.5 y [range, 36-69]) and 73.5% received no prior
systemic
therapy. 33/44 (75%) patients were followed for at least 16 weeks for disease
progression.
For the 38 evaluable C-PA patients included in the per-protocol interim
analysis, 13
(34.1%) were progression free at 16 weeks [95% Cl 19.6, 51.4]. The estimated
ORR was
7.9% [95% Cl, 1.7, 21.4] for the 38 C-PA patients and 0% for the 6 C-PB
patients who had
at least one post-baseline CT scan reviewed by central imaging. For all
44patients, median
TTP was 5.4 months [95% CI, 3.7, -not reached] and median OS was 9.3 months
[95% Cl,
6.0, 11.0]. The most common adverse events (AEs) for all pts were hypertension
(41%),
fatigue (47%), diarrhea (38%), rash (35%), proteinuria (24%), vomiting (24%),
cough
(24%) and oedema peripheral (24%). The most common grade 3/4 AEs for all pts
were
hypertension (20.6%) and fatigue (11.8%). Most AEs were mild/moderate and
reversible
with interruption/dose reductions/or discontinuation of ABT-869.
Conclusions: ABT-869 appears to benefit HCC patients, with an estimated TTP of
112 days and an acceptable safety profile.
NSCLC
This ongoing, open-label, randomized, multicenter phase 2 trial of ABT-869 at
0.10
mg/kg daily (Arm A) and 0.25 mg/kg daily (Arm B) until progressive disease
(PD) or
intolerable toxicity, was initiated to assess antitumor activity and toxicity
of ABT-869 in
patients with NSCLC. Eligibility criteria included locally advanced or
metastatic NSCLC;
> I prior systemic treatment, and >1 measurable lesion by RECIST criteria. The
primary
endpoint was the progression free (hereinafter "PF") rate at 16 wks. Secondary
endpoints
were objective response rate (hereinafter "ORR"), time to progression
(hereinafter "TTP"),
progression free survival (hereinafter "PFS") and overall survival
(hereinafter "OS"). CT
scans were assessed by the investigator and centrally; central assessment
results are
provided. Patients were randomized 1:1 to ABT-869, stratified by dose and
Asian status.
ABT-869 0.10 or 0.25 mg/kg was self-administered as a daily oral dose under
fasting
conditions. Patients receiving the 0.10 mg/kg dose, whose disease progressed,
were
allowed to cross-over to the 0.25 mg/kg dose within 30 days from the last 0.10
mg/kg dose.
Safety (graded by NCI CTCAE, Ver. 3.0) was evaluated at study visits.
63
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
Inclusion criteria: Adults diagnosed with locally advanced or metastatic
NSCLC,
had at least 1 measurable non-radiated lesion by RECIST on CT scan, received
at least 1 to
2 prior lines of systemic treatment for NSCLC, and no neo-adjuvant or adjuvant
chemotherapy for NSCLC, ECOG performance status 0-2, no other active
malignancy
within the previous 5 years, adequate organ function. Exclusion criteria:
received anti-
cancer therapy within 21 days or 5 half-lives before dosing, radiation or
major surgery
within previous 21 days before dosing, or targeted VEGF/PDGF/TKI therapy
(prior
Avastin allowed), had untreated brain or meningeal metastases, history of >10%
weight
loss during 6 weeks before study entry, significant central thoracic lesions
invading/abutting heart or major blood vessels, clinically relevant
hemoptysis, proteinuria,
symptomatic or persistent uncontrolled hypertension, left ventricular Ejection
Fraction
<50%, >Grade 2 encephalopathy (CTC criteria), had clinically significant
uncontrolled
conditions, was receiving therapeutic anticoagulation therapy or anti-
retroviral therapy.
The baseline characteristics and medical history of the patients are shown
below in Table 5.
Table 5
All Patients .10mg/kg ABT-86 .25mg/kg ABT-86
aseline characteristics =139 =65 1=74
Median age, yrs (range) 62 61 2
Men, n (%) 82 (59.0) 1 (63.1) 1 (55.4)
sian ethnicity, n (%) 49 (35.3) 3 (35.4) 26(35.1)
COG status, n (%)
0 45 (33)23 (35)22 (30
1 88 (63)38 (58)50 (67
2 6 4 4 7 2(3)
Medical history at screening
Smoker, n (%) 95 (68)41 (63)54 (73
on-smoker, n (%) 44(32) 24(37) 20(27)
Squamous, n . (%) 17 (12 8 (12 9 (12
on-s uamous, n (%) 122(88) 57(88) 65(88)
rior systemic therapies, n (%)
1 56 (40 5 (39)31 (42
2 72 (52 4 (52)38 (51
>2 11 8 6 9 15(7)
All Patients .10 mg/k .25 mg/kg ABT-869
n=139 ABT-869 In=74
Endpoints n=65
64
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
Primary FR at 16 Weeks 8 (34.5) 1 (32.3)27 (36.5)
(%) [95% CI] [26.7, 43.1] [21.2, 45.1] [25.6, 48.5]
Secondary PFS, median 3.6 [3.0, 3.9] .5 [2.0, 4.3] 3.7 [3.1, 4.9]
no [95% Cl]
OS, median .0 [6.9, 12.8] 10.7 [6.9, --] 8.6 [5.6, 12.8]
f o [95% CI]
stimated O (1.4) 1. 7, 5.1] 0 (2.7) [0.3, 9.4]
/ t (%) [95% CI]
FR based on radiographic assessment by the central imaging center and on
clinical assessment
by the investigator
--not reached
Table 6 below shows the most Common Treatment-related Adverse Events
Table 6
0.10 mg/kg 0.25 mg//kg
All ABT-869 ABT-869
ABT-869-related n=139 n=65 n=74 P-value
Any grade, >20% of all patients
Diarrhea 36 (25.9) 10 (15.4) 26 (35.1) 0.011*
Nausea 32 (23.0) 11 (16.9) 21 (28.4) ---
Fatigue 55 (39.6) 19 (29.2) 36 (48.6) 0.024*
Anorexia 42 (30.2) 13 (20.0) 29 (39.2) 0.016*
Proteinuria 28 (20.1) 5(7.7) 23 (31.1) <0.001*
Hypertension 47 (33.8) 12 (18.5) 35 (47.3) <0.001*
Grade 3 or 4, > 10% of
all patients
Hypertension 18 (12.9) 1 (1.5) 17 (23.0) <0.001 *
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
P-value for comparison between the high vs. low dose groups using Fisher's
Exact test
*Statistically significant difference
Results: 138 patients (pts) were enrolled from 08/07-10/08 from 27 centers
120/139
(86%) patients were followed for at least 16 weeks for disease progression. A
total of 10 pts
had squamous cell histology (all 10 were included in the interim analysis).
The remainder
had non-squamous histology. Median age was 64 years and 62 years in Arm A and
B
respectively. For the interim analysis population (Arm A, n=24; Arm B, n=24),
16 (33.3%)
pts were PF at 16 wks: 7 (29.2%) in Arm A and 9 (37.5%) in Arm B. The ORR in
Arm A
(n=30) was 0% and 7.3% in Arm B (n=41).- The median TTP and median PFS were
110
and 109 days, and 112 days and 108 days in Arm A and B, respectively. The most
common
adverse events (AEs) in Arm A were fatigue (35%), nausea (21%), and anorexia
(21%),
and in Arm B were hypertension (51%), fatigue (51%), diarrhea (43%), anorexia
(41%),
nausea (31%), proteinuria (31%) and vomiting (26%). The most common grade 3/4
toxicities in the Arm A were fatigue (7%), ascites (5%), dehydration (5%),
pleural effusion
(5%), and in the Arm B were hypertension (23%), fatigue (8%), PPE syndrome
(8%),
dyspnoea (6%) and stomatitis (6%). Most AE's were mild/moderate and reversible
with
interruptions/dose reduction/or discontinuation of ABT-869.
Conclusions: ABT-869 demonstrates an acceptable safety profile and appears to
be
active in NSCLC patients.
RCC
Phase II, single-arm, open-label, multicenter trial in adults with advanced
RCC,
previously treated with sunitinib. Oral dose of ABT-869 0.25 mg/kg every day
("QD")
was self-administered by patients, under fasting conditions. Treatment
continued until
disease progression or intolerable toxicity. Efficacy endpoints: primary:
objective
response rate (hereinafter "ORR"); secondary: progression-free survival
(hereinafter
"PFS"), overall survival (hereinafter "OS"), and progression-free rate
(hereinafter
"PFR") at week 16.
Inclusion Criteria: adults diagnosed with locally recurrent or metastatic RCC,
at
least 1 unidimensionally measurable lesion by RECIST, previous nephrectomy,
received >2 cycles (12 wks) of treatment with sunitinib for RCC and stopped
therapy
due to disease progression within 100 days before screening, ECOG performance
status
0-1, no history of other active cancer within the previous 5 years, life
expectancy of at
66
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
least 4 months, adequate organ function. Exclusion Criteria: received anti-
cancer
therapy within 21 days (5 half-lives) before dosing, major surgery within 21
days
before dosing, TKI therapy targeting VEGFR and/or PDGFR other than sunitinib
or
sorafenib (prior bevacizumab allowed), or anti-retroviral therapy for HIV,
proteinuria,
symptomatic or persistent uncontrolled hypertension, left ventricular ejection
fraction
<50%, known autoimmune disease with renal involvement, clinically significant
uncontrolled conditions. Tumor response was assessed at 8-week intervals using
RECIST. Safety was evaluated through physical examinations, laboratory tests,
assessment of ECOG performance status, and AEs. AEs were graded using the
National
Center Institute Common Terminology Criteria for AEs, version 3Ø
53 patients enrolled in the study from August 2007 to October 2008, across 12
centers in the US and Canada47/53 (89%) patients either developed disease
progression
or were followed for at least 24 weeks for disease progression. Baseline
characteristics
for the patients is shown in Table 7.
Table 7
aei aa~ei= c e
Median age, yrs (range) 1.0 (40-80)
4en, n (%) 42(79)
COG status 0, n (%) 19 (36)
COG status 1, n(%) 34(64)
Histology, n (%)
Clear cell 43(81)
Non-clear cell 10(19)
Prior systemic therapies, n (%)
1 6 (49)
2 17(32)
>2 10(19)
rior therapies, n (%)
Sunitinib 53(100)
Cytokine 12 (23)
Sorafenib 10 (19)
Temsirolimus 2 (4)
Bevacizumab 9(17)
Pest response (PR) to prior sunitinib, % 13.2
Reasons for treatment discontinuation
-30 patients due to Progressive Disease (PD) (clinical, radiographic,
or AE related to PD)
-7 patients due to AEs not related to PD
67
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
-1 patient for other reasons
-At the time of this analysis, 15 patients remained on
The response of the patients is shown in Table 8 and AEs are shown below in
Table 9.
68
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
Table 8
All Patients, n=53
Endpoints
Central Imaging Site Assessment
rimary ORR, % [95% CI] .4 [3.1, 20.7] 17.0 [8.1, 29.81
Secondary FS Median .4 [3.6, 6.3] 5.8 [3.9, 7.3]
no [95% CI]
FR at 16 Weeks 19.1 [35.1, 63.2] 4.7 [40.4, 68.4]
% [95% CI]
OS Median 11.6 [10.1,--l
o 195% CI]
- not reached
Table 9
ABT-869-related All patients (n=53), n (%)
Any grade, >20% of all patients
Diarrhea 37 (69.8)
Fatigue 37 (69.8)
Hypertension 30 (56.6)
Nausea 22 (41.5)
Hand-foot skin 18 (34.0)
reaction
Weight loss 18 (34.0)
Anorexia 17 (32.1)
Proteinuria 17 (32.1)
Vomiting 16 (30.2)
Mucosal inflammation 11 (20.8)
Rash 11 (20.8)
Grade 3 or 4, >10% of all patients
Hypertension 15 (28.3)
69
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
Fatigue 10 (18.9)
Diarrhea 9(17.0)
Hand-foot skin reaction* 9 (17.0)
* Coded as palmar-plantar erythrodysesthesia
Analysis of P1GF-1 concentrations in the above described three Phase 2
monotherapy studies was performed using the optimized P1GF-1 assay described
in
Example 3, with greater sensitivity which necessitated the identification of
new
efficacy and toxicity thresholds. A greater variation in baseline
concentrations of
P1GF-1 was noted in this larger patient population therefore analysis of the
P1GF-1 was
performed using values normalized by baseline subtraction (in which the
pretreatment
number was subtracted from the post treatment number and used to obtain the
resulting
value). The baseline values of P1GF-1 were determined to be predictive of
response
with those having P1GF-1 concentrations lower than 24 pg/mL ultimately
performing
better (Time to Progression (TTP) 168 versus 112 days, p=0.03). Changes in
P1GF-1
following administration of ABT-869 were analyzed using either an early sample
(Day
8 and/or 15), taken prior to any dose reduction/interruption or using a mean
P1GF-1
AUC (the AUC was derived using all P1GF-1 values for a given patient during
the
dosing period including values derived during dose interruptions) to reflect
the dose
intensity over the first 3 weeks of treatment and resulted in the
determination of
optimized P1GF-1 efficacy thresholds. For Child-Pugh A patients, the early
P1GF-1
threshold of 81.5 pg/mL was associated with improved overall survival (316
versus 266
days, p=0.03). This same P1GF-1 threshold was also predictive patients for
improved
median TTP (211 versus 58 days, p=0.05) and OS (352 versus 284 days, p=.3) in
a
Phase 2 study of ABT-869 in renal cell carcinoma (RCC). The AUC threshold for
the
first 22 days similarly segregates patients for improved response (less than
66.5 pg/mL,
OS =268 days and greater than 66.5 pg/mL the median was not reached, p=0.04).
Many of the patients on these studies experienced toxicities that necessitated
either a dose interruption and/or a dose reduction. Using the early predose
reduction
samples, a clear PIGF-1 toxicity cutoff of 148 pg/mL above which 87% of the
Hepatocellular Cancer (HCC) patients had "toxicity" (grade 3/4 events that
result in a
dose reduction/interruption, or grade 2 toxicities that result in dose
interruptions) was
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
identified. ` his threshold was predictive of toxicity in all 3 studies.
Utilization of this
toxicity threshold could play a role in proactive toxicity management.
Interestingly,
the patients with the best overall survival had high P1GF-1 values within the
first two
weeks of treatment and a subsequent dose reduction. Analysis of the mean AUC
P1GF-
1 concentration through day 56 in HCC and day 112 in RCC takes into account
both the
initial dose and the impact of dose reductions at timepoints relevant to a
majority of
patients for the respective studies. Application of the P1GF-1 efficacy
threshold
(generated from the mean AUC through day 22) of 66.5 pg/mL and the 148 pg/mL
toxicity threshold to this data set demonstrated that an optimal PIGF-I
concentration
could be identified, and resulted in increased OS in HCC (316 versus 193 days,
p=0.015) and in RCC (median not determined versus 405 days, p=0.035).
Example 3:
Analysis of PIGF-1 concentrations in the above described three Phase 2
monotherapy studies in Example 2 were performed using a P1GF-1 assay as
described
in greater detail in Example 15 (referencing Example 11) of U.S. Application
Serial
No. 12/485,114 filed June 16, 2009 (incorporated by reference for its
teachings
regarding same). Briefly, this assay employs an ARCHITECTS immunoassay format
for detection of human P1GF-1, and utilizes anti-P/GF-1 monoclonal antibody
from the
2-826-335 hybridoma cell line (MAB826) as the capture reagent immobilized on
paramagnetic microparticles, and uses anti-P1GF-1 monoclonal antibody from the
1-
255-713 or 1-255-2675 hybridoma cell lines (MAB255) as the conjugate reagent
labeled with acridinium. The conjugate reagent was either an intact IgG
monoclonal
antibody, an F(ab')2, or Fab fragment. The ARCHITECT assay was run as
described
in Example 11 of U.S. Application Serial No. 12/485,114 using a range of
sample
volumes from 50 to 100 microliters for optimization of the assay. The optimum
sample
volume of the assay is 50 microliters. PIGF-1 purchased from R&D Systems was
used
as calibrator material for the assay. P1GF-1 was first diluted in a buffer
matrix called
`calibrator diluent' (a buffer containing 2-(N-morpholino)ethanesulfonic acid
(MES),
other salt, a protein blocker and an antimicrobial) to obtain concentrated
intermediate
stock solutions. Calibrators were prepared gravimetrically on an analytical
balance by
diluting the intermediate stock with the calibrator diluent. Calibrators were
prepared at
71
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
PIGF-1 concentrations of 0, 10, 30, 60, 500, and 1,500 pg/mL (Cal A, Cal B,
Cal C, Cal
D, Cal E, and Cal F, respectively). Specimens were collected in either EDTA
plasma
or serum collection tubes.
The immunoassay was carried out by automated ARCHITECT i2000 analyzer
(Abbott Laboratories, Abbott Park, IL). Briefly, the assay involved the
following steps:
1. Mixing 50 gL of human sample with 50 gL of microparticles coated with
anti-human PIGF-l antibody (namely, MAB826). Similarly, calibrator solutions
can be
used in this step in place of the human sample to prepare a standard curve and
calibrate
the assay.
2. Incubating the reaction mixture for approximately 18 minutes at a
maintained
ambient temperature of 33 - 38 C. The human PIGF-1 antigen in the sample
bound to
the anti-human PIGF-1 antibody on the microparticles.
3. Unbound human PIGF-1 was separated from magnet-detained microparticle
and drained into waste. The microparticles were washed with a phosphate
buffer.
4. Adding 50 pL of the detection antibody labeled with an acridinium ester
(monoclonal antibody MAb 255) to the reaction mixture.
5. Incubating the reaction mixture for approximately 4 minutes at 33 - 38 C.
The anti-human P1GF-1 antibody-acridinium molecule forms a sandwich with human
P1GF-1 captured by antibody immobilized on the microparticle.
6. Washing the microparticles with a phosphate buffer.
7. Adding Pre-trigger (acid solution with hydrogen peroxide) and Trigger
(basic
solution) to cause the captured human P1GF-1 to emit light, which was measured
by the
instrument as Relative Light Units (RLUs). RLUs are the designation for the
optical
unit of measurement utilized on the ARCHITECT systems. The ARCHITECT
optics system is essentially a photomultiplier tube (PMT) that performs photon
counting on the light emitted by the chemiluminescent reaction. The amount of
light
generated by the chemiluminescent reaction is proportional to the amount of
acridinium
tracer present in the reaction mixture, and thereby allows quantitation of the
sample
analyte that is also proportional to the amount of acridinium remaining in the
reaction
mixture at the time the chemiluminescent reaction occurs. The term "Relative
Light
Units" comes from the relation of the photon counting to a certain amount of
acridinium. Each optics module is calibrated with a set of acridinium
standards. When
72
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
the chemiluminescent reaction occurs, light is emitted and the photons are
measured
over a 3 second time period. The PMT converts the photons counted to digital
signal,
which is then sent to a circuit board for processing. The optics circuit board
converts
the digital signal from the PMT to an analog signal that is proportional to
the photons
counted, which is in turn proportional to the amount of acridinium present.
This analog
signal is then further processed to produce an RLU value. This relationship
was
established to produce a standard for calibration of the optics module, where
the
different acridinium standards have RLU values assigned to them. So, while the
RLU
unit itself is arbitrary, it is proportional (i.e., relative) to a certain
amount of acridinium.
A range of sample volumes from 50 to 100 microliters was used during
optimization of the assay. The preferred sample volume is 50 to 75 microliters
and the
optimal sample volume is 50 microliters. PIGF-1 purchased from R&D Systems was
used as calibrator material for the assay. Calibrators are prepared at PIGF-1
concentrations of 0, 10, 30, 60, 500, and 1,500 pg/mL.
The preferred immunoassay format was used to measure PIGF-1 in 400
apparently normal individuals. The specimens were purchased from ProMedDx, LLC
(Norton, MA) and comprised of 200 males and 200 females. The specimens were
collected in either EDTA plasma or serum collection tubes. The results are
shown in
Table 10, where the median P1GF concentration is 16.0 pg/mL and the upper 97.5
percentile is 25.8 pg/mL. The lowest sample is 7.9 pg/mL and the highest value
is 29.8
pg/mL in this sample set.
Table 10
Sample size 400
Median 16.0
Lowest value 7.9
Highest value 29.8
Geometric mean 16.1
Kolmogorov-Smirnov test accept Normality (P=0.751)
for Normal distribution
Percentiles
0.5 9.7
2.5 10.7
5 11.1
13.9
75 18.6
95 23.6
97.5 25.8
99.5 28.0
Values back-transformed after logarithmic transformation.
73
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
The preferred immunoassay format was used to measure P1GF in pregnant
individuals with gestational age ranging from 4.5 to 39 weeks. The specimens
were
collected in EDTA plasma. The results are shown in Figure 5 (N = 1,490
specimens),
where a steady increase in PIGF value is observed with increasing gestational
age up to
approximately 30 weeks. After approximately 32 weeks, the PIGF values are
widely
scattered. The P1GF concentration in these specimens ranges from approximately
7.0
pg/mL to approximately 4,500 pg/mL. Specimens with initial values greater than
1,500
pg/mL were retested after a 4-fold dilution to provide a result within the
calibration
range.
These results show that the preferred immunoassay format is able to detect
human PIGF-1 in pregnant individuals. Studies have also been done with the
preferred
immunoassay format in individuals with preeclampsia, patients with cardiac
conditions,
and patients with carcinoma such as renal cell carcinoma, hepatocellular
carcinoma,
and non small cell lung carcinoma.
One skilled in the art would readily appreciate that the present disclosure is
well
adapted to carry out the objects and obtain the ends and advantages mentioned,
as well
as those inherent therein. The molecular complexes and the methods,
procedures,
treatments, molecules, specific compounds described herein are presently
representative of preferred embodiments, are exemplary, and are not intended
as
limitations on the scope of the invention. It will be readily apparent to one
skilled in the
art that varying substitutions and modifications may be made to the invention
disclosed
herein without departing from the scope and spirit of the invention.
All patents and publications mentioned in the specification are indicative of
the
levels of those skilled in the art to which the invention pertains. All
patents and
publications are herein incorporated by reference to the same extent as if
each
individual publication was specifically and individually indicated to be
incorporated by
reference.
The invention illustratively described herein suitably may be practiced in the
absence of any element or elements, limitation or limitations which is not
specifically
disclosed herein. Thus, for example, in each instance herein any of the terms
"comprising," "consisting essentially of and "consisting of' may be replaced
with
either of the other two terms. The terms and expressions which have been
employed are
74
CA 02728449 2010-12-17
WO 2009/155381 PCT/US2009/047714
used as terms of description and not of limitation, and there is no intention
that in the
use of such terms and expressions of excluding any equivalents of the features
shown
and described or portions thereof, but it is recognized that various
modifications are
possible within the scope of the invention claimed. Thus, it should be
understood that
although the present disclosure has been specifically disclosed by preferred
embodiments and optional features, modification and variation of the concepts
herein
disclosed may be resorted to by those skilled in the art, and that such
modifications and
variations are considered to be within the scope of this invention as defined
by the
appended claims.