Note: Descriptions are shown in the official language in which they were submitted.
CA 02738195 2011-03-23
WO 2010/034128 PCT/CA2009/001372
Title
SpylA As A Diagnostic and Prognostic Marker Of Cancer
Field of the Invention
The present invention relates to the use of SpylA as a marker, and in
particular, to the use of SpylA as a diagnostic and prognostic marker for
cancer.
Background of the Invention
Members of the Speedy/RINGO family are unique cyclin-like regulators
of the cell division cycle. In Xenopus oocytes, X-Spyl was shown to
prematurely
activate CDK2 and CDK1, and to allow progression through the G2/M
checkpoint via activation of the MAPK pathway, thus promoting rapid oocyte
maturation (6, 18). The human Spyl homologue SpylAl, herein referred to as
SpylA, is expressed constitutively in most human tissues; it shortens the GUS
transition through activation of CDK2 and is essential for cell proliferation
to
occur (24). Activation of the CDKs by Spyl/RINGO proteins is thought to occur
in an atypical fashion, independent of cyclin binding and in the absence of
CDK
phosphorylation within the T-loop (16). In addition, SpylA can prevent the
inhibitory effects of the CDK inhibitor, p27K'P' (p27) by directly promoting
p27
degradation (19, 25). SpylA-induced proliferation requires endogenous p27, but
whether the direct interaction between SpylA and p27 is required for all SpylA-
mediated proliferative effects is not known. Research also shows that SpylA
plays a role in the DNA damage response, functioning to enhance cell survival
and promote cell proliferation in lieu of apoptosis (2, 8). Importantly,
recent
observations have demonstrated that SpylA is capable of promoting precocious
development and tumorigenesis in the mammary gland and that SpylA protein
levels are implicated in invasive ductal carcinoma of the breast (36). Hence,
determining how SpylA protein levels are regulated may reveal novel
information regarding the dynamics of cell cycle control during normal and
abnormal growth conditions.
1
CA 02738195 2011-03-23
WO 2010/034128 PCT/CA2009/001372
Cyclin proteins are tightly regulated temporally and spatially, controlled
on a fundamental level by the ubiquitin-proteasome system (UPS). The UPS is
the primary mechanism involved in the selective degradation of intracellular
and
membrane-bound proteins, and aberrations in this critically important system
are
correlated to many diseases such as breast cancer (4, 27). The ubiquitination
process involves the conjugation of ubiquitin, a highly conserved protein of
76
amino acids, to a substrate protein; this event can signal the substrate for
degradation by the proteasome (14). Ubiquitination is orchestrated by members
of three classes of enzymes: the ubiquitin-activating enzyme El, the ubiquitin-
conjugating enzyme E2, and the ubiquitin-protein ligase E3. The El first forms
a
thiolester bond between a cysteine residue on itself and a glycine residue on
the
C-terminal of ubiquitin, which is followed by the transfer of ubiquitin to a
cysteine residue on an E2 enzyme. Another transfer of the ubiquitin
polypeptide
then takes place, resulting in a thiolester bond between ubiquitin and a
cysteine
residue of an E3 enzyme. The E3 enzyme can then catalyze the formation of a
bond between the C-terminal glycine of ubiquitin and either a lysine residue
on
the target protein resulting in monoubiquitination, or a lysine residue to a
previously added ubiquitin molecule creating a chain of ubiquitin molecules
known as polyubiquitination (13, 27). Single-site monoubiquitination, multiple
sites of monoubiquitination and K63-linked ubiquitin chains have non-
proteolytic
regulatory roles involved in activities such as DNA repair, transcription,
endocytosis, and protein trafficking. On the other hand, polyubiquitination at
K48
of ubiquitin targets the substrate protein for degradation by the 26S
proteasome
(30). Prior to ubiquitination, the substrate protein is usually modified to
accommodate recognition by the E3, often by phosphorylation (4).
X-Spyl is regulated in Xenopus oocytes by the UPS system and the
activity of two different ubiquitin ligases which function at different stages
of the
cell cycle: SCFOTTCP and Siah-2 (10). Prior to meiosis, G2 arrest of the
oocyte is
maintained by SCFOT`CP-mediated cleavage of the C-terminal of X-Spyl ; this is
initiated by phosphorylation on S233 and S237 by protein kinase A (PKA) and
2
CA 02738195 2011-03-23
WO 2010/034128 PCT/CA2009/001372
glycogen synthase (kinase-3(3). It is believed that the processed fragment of
X-
Spy 1 may inhibit meiotic maturation either by forming inactive heterodimers
with unprocessed copies of X-Spyl, or by competitively interacting with CDKs.
On the other hand, Siah-2 has been shown to be responsible for leading to the
subsequent degradation of phosphorylated X-Spy l between meiosis I and meiosis
II. It was further demonstrated that timely degradation of X-Spyl during the
transition to meiosis II was essential for preventing DNA synthesis during
meiosis. In mammals, SpylA mRNA is known to be up-regulated during G1/S;
however whether this protein is also regulated via protein degradation is
currently
not known (24). Solomon et. al. have suggested that the N-terminal region of
SpylA may be involved in protein stability, however data in this regard has
not
been presented nor has this been further explored (5). Based on the cyclin-
like
function of SpylA, and the known regulation of X-Spyl, it is a valid
hypothesis
that SpylA may also be subject to ubiquitin-mediated proteolysis.
Summary of the Invention
In one aspect, the present invention provides a method diagnosing cancer,
said method comprising: determining the relative amount of Spyl A protein in a
sample cell; determining the relative amount of SpylA protein in a normal
cell;
and comparing the relative amount of SpylA protein in the sample cell to the
relative amount of protein in the normal cell.
In another aspect, the present invention provides a method of diagnosing
cancer, said method comprising: determining the relative amount of Nedd4
protein in a sample cell; determining the relative amount of Nedd4 protein in
a
normal cell; and comparing the relative amount of Nedd4 protein in the sample
cell to the relative amount of Nedd4 protein in the normal cell.
In another aspect, the present invention provides a method of treating or
preventing cancer, said method comprising phosphorylating one or more amino
acid residues in a SpylA protein in a target cell.
3
CA 02738195 2011-03-23
WO 2010/034128 PCT/CA2009/001372
In this publication, we demonstrate that Spy IA degradation occurs via
ubiquitin-mediated proteolysis following entry into mitosis but prior to the
anaphase transition. We determine that the N-terminal 1-57 amino acids within
SpylA are essential to support regulated degradation of the protein. We
further
resolve that the E3 ligase, Nedd4, is capable of binding to SpylA during G2/M
phase of the cell cycle, and that dominant negative forms of Nedd4 reduce
ubiquitination and degradation of SpylA. Additionally, we identify 3 key amino
acids within the N-terminal region of SpylA: T15, S22 and T33, which are
essential for Spy1A degradation. Collectively, our data demonstrates that
SpylA,
like the cyclin family with which it resembles, is tightly regulated at the
protein
level. Importantly, we provide a novel link between SpylA and Nedd4, two
proteins previously implicated in tumorigenesis. Additionally we show that non-
degradable forms of SpylA do not prevent cell cycle progression and instead
promote cell proliferation, further implicating that SpylA may contribute to
uncontrolled growth in tumorigenesis.
Brief Description of the Drawings
Figure. 1. SpylA protein is degraded in a cell cycle dependent fashion.
(A) MCF7 cells were either untreated (Cntl), blocked by serum starvation (SS)
or
blocked and then released into media containing serum and nocodozole (NT) and
analyzed by flow cytometry. Top panel shows flow cytometry profiles and the
bottom panel shows % of cells in each phase of the cell cycle as determined by
CPX analysis. (B) Cell lysates from each population described in A were lysed
and analyzed using 10% SDS PAGE followed by immunoblotting with SpylA
antibody (top panel), Cyclin E as cell cycle marker (middle panel) and Actin
(lower panel). (C) 293T cells were either untreated (Cntl), blocked by double
thymidine block (TB) or blocked and then released into media containing serum
and nocodazole (NT) and analyzed by flow cytometry. Top panel shows flow
cytometry profiles and the bottom panel shows % of cells in each phase of the
cell cycle as determined by CPX analysis. (D) Cell lysates from each
population
described in C were lysed and analyzed using 10% SDS PAGE followed by
4
CA 02738195 2011-03-23
WO 2010/034128 PCT/CA2009/001372
immunoblotting with SpylA antibody (top panel) and Actin as a loading control
(lower panel). (E) 293T cells were transfected with Cyclin B 1 wild-type (CycB
1),
empty vector negative control (GFP) and a non-degradable cyclin B I (CycB-D).
Cell lysates from untransfected (Cntl) or transfected cells were analyzed with
10% SDS PAGE followed by immunoblotting with SpylA antibody.
Figure 2. SpylA degradation is proteosome dependent. (A) 293T cells
were used for an in vivo ubiquitination assay. Cells were transfected with HA
ubiquitin (Ha-Ub), Myc-SpylA-PCS3 (Myc-Spy 1 A) and PCS3 empty vector
(Myc-Cntl). An equal amount of protein was immunoprecipitated for Myc and
analyzed using SDS-PAGE followed by immunoblotting with HA antibody. Ha-
Ub-SpylA is depicted in the upper panel. The membrane was stripped and then
re-probed with monoclonal Myc antibody (lower panel). (B) 293T cells were
treated with a calpain inhibitor (LLNL; lane 3) and a proteasome inhibitor
(MG 132; lane 4) as well as the vehicle control for LLNL (ETOH; lane 2) and
the
vehicle control for MG132 (DMSO; lane 1). Equal amount of protein was
analyzed with 10% SDS-PAGE followed by immunoblotting with SpylA
antibody (upper panel) and Actin as a loading control (lower panel).
Figure 3. SpylA degradation relies on the N-terminal region. (A) A
schematic diagram for the different SpylA truncation mutants is depicted and
restriction sites used for cleavage of the region are indicated. (B) 293T
cells were
transfected with Myc-SpylA-PCS3 (wt; lane 1) or the different truncation
mutant
TMA-TMZ (A-Z; lanes 3-7) or PCS3 empty vector (-ve; lane 2). Transfected
cells were treated with 70 ng of nocodazole for 16 hrs. The cells were lysed
and
subjected to 10% SDS-PAGE followed by immunoblotting with Myc antibody
(upper panel), or Actin as a loading control (lower panel). (C) 293T cells
were
transfected with Myc-SpylA-PCS3 (wt and -ve; lanes 1 & 2) and TMA-TMZ (A-
Z; lanes 3-7). Cells were either treated with orthophosphoric acid 32P (lanes
1, 3-
7) or left untreated (-ve; lane 2). The cells were lysed and an equal amount
of
protein was immunoprecipitated with Myc antibody. The immunoprecipitates
were analyzed with 10% SDS-PAGE and analyzed by phosphoimager analysis.
5
CA 02738195 2011-03-23
WO 2010/034128 PCT/CA2009/001372
Upper panel shows phosphorylated c-Myc band, and middle panel shows c-Myc
immunoblot. Lower panel shows quantified analysis of the blot. (D) Alignment
of
SpylA and X-Spy 1 N-terminal region (top panel) and C-terminal region (lower
panel) are depicted. Regions known to play a role in the degradation of each
are
indicated in bold. Phosphorylation sites known to direct X-Spy 1 processing at
T8
and S12 are also depicted by +. Essential residues for mediating the X-Spyl-
SCFOT`CP binding, S233 and S237, are also depicted by =. The essential
phosphorylation site for mediating Siah-2 binding, S243, is also depicted by
*.
The potential PPxxxxY binding region for WW domain proteins within SpylA is
also shaded in grey. Potential phosphorylation sites within the N-terminal
region
of SpylA are also indicated by amino acid numbers above the sequence.
Figure 4. Nedd4 is the ubiquitin ligase for SpylA. (A) 293T cells were
transfected with empty vectors (PCS3 or PCEP), Nedd4-PCEP (Nedd4) or Myc-
SpylA-PCS3 (Myc- SpylA). Equal amounts of protein were immunoprecipitated
with Myc antibody and then the precipitates were analyzed using 10% SDS
PAGE followed by immunoblotting with Nedd4 antibody (upper panel). Lysates
from NIH 3T3 cells served as positive control for Nedd4 expression (+; lane
5).
The membrane was stripped and re-probed with Myc antibody (lower panel). (B)
293T cells were transfected with empty vectors (PCS3 or PCEP), Nedd4-PCEP
(Nedd4), Myc-SpylA-PCS3 (Myc-SpylA) or Nedd4-PCEP Dominant Negative
(Nedd4DN). Equal amounts of protein were immunoprecipitated with Myc
antibody and analyzed using 10% SDS PAGE. Immunoblotting with HA
antibody detected HA-Ub-tagged SpylA (Ha-Ub-Spy 1 A; upper panel). The
membrane was stripped and re-probed with Myc antibody (lower panel).
Densitometry was carried out using AlphaEaseFC software. (C) 293T cells were
transfected with PCEP empty vector control (lane 1) and Nedd4-PCEP (Nedd4;
lanes 2 & 3). Cells were either treated with MG132 (lane 2) or DMSO (lanes 1 &
3). Cell lysates were analyzed by 10% SDS PAGE followed by immunoblotting
with SpylA antibody (upper panel). The membrane was stripped and re-probed
with Actin antibody (lower panel). Densitometry was carried out using
6
CA 02738195 2011-03-23
WO 2010/034128 PCT/CA2009/001372
AlphaEaseFC software.
Figure 5. Phosphorylation on T15, T33 and S22 is needed for SpylA
degradation. 293T cells were transfected with SpylA wild-type (WT), SpylA-
T15A (T15A), SpylA-T33A (T33A), SpylA-S22A (S22A). (A) Cells were treated
with 70 ng Nocadazole for 16 hrs. (G2 population; left panels) or untreated
control (Asynchronous population; right panel). Half of the population was
kept
for flow cytometry analysis (see D) and the remainder were lysed and subjected
to 10% SDS-PAGE followed by immunoblotting with Myc antibody (SpylA;
upper panels) and Actin as a loading control (lower panel). (B) Transfected
cells
were co-transfected with Ha-Ub followed by treatment with MG 132 for 16 hrs.
Lysates were immunoprecipitated with Myc and immunoblotted for Ha (Ha- Ub-
SpylA; upper panel), Nedd4 (middle panel) or Myc (lower panel). (C) Triplicate
transfections were counted for alive and dead cells at 36 hrs. post
transfection
using trypan blue exclusion. Counts from the 3 separate transfections were
used
for statistical analysis. Error bars reflect standard deviation and a standard
T-test
was performed assuming equal variance. Statistical data shown reflects
comparisons between the WT transfected cells and mutant transfected cells
which
were * p. 0.05, ** 135_ 0.01. (D) Cells from (A) were analyzed by flow
cytometry. CPX analysis was carried out to determine the % of cells in each
population. These numbers are depicted above the schematic of the cell cycle
profiles.
Figure 6 illustrates example data of Spyl levels detected in tumour tissue
vs control from a tissue microarray detection and quantification of Spyl
protein
levels.
Figure 7 illustrates Spyl levels in tumour tissue vs control from a tissue
protein extraction and Western Blot protocol for SpylA protein levels.
Figure 8 illustrates: (A) expression of Spyl in proliferative mammary
tissues (pregnancy) as well as regenerating mammary tissues (involution); (B)
Western Blot analysis of the same tissues; (C) RT-PCR analysis of tissues.
7
CA 02738195 2011-03-23
WO 2010/034128 PCT/CA2009/001372
Figure 9 illustrates: (A) A schematic diagram for the different SpylA
deletion mutants is depicted and restriction sites used for cleavage of the
region
are indicated. (B) 293 cells were transfected with Myc-SpylA-PCS3 (wt) or the
different deletion mutant DMA-DMZ (A-Z). Transfected cells were treated with
nocodazole (left hand panels; synchronous) or no treatment (right hand panels;
asynchronous) for 16 hrs. post transfection. Lysates were immunoblotted with a-
Myc or a-Actin.
Figure 10 (A) to (D) illustrates the results of blotting experiments to
show that residues T15, S22 and T33 are essential for SpylA degradation.
Figure 11 illustrates a graph showing that aberrant SpylA degradation
enhances cell proliferation.
Figure 12 illustrates experiments relating to anchorage independent
growth in soft agar.
Figure 13. (A) Sequence composition of five shRNA constructs, each
uniquely designed to trigger RNAi-mediated destruction of the SpylA mRNA
transcript to variying degrees (1-5). Also included is the coding sequence for
the
scrambled negative control shRNA construct. (B) Depiction of the mouse SpylA
(mSpylA) protein isoform and its respective amino acid positions that
correspond to each shRNA sequence.
Figure 14. (A) Twenty-four hours post-transfection, alterations in cell
proliferation were quantified through trypan blue exclusion analysis and cell
counting, demonstrating a significant decrease in cell number upon transient
SpylA inhibition. (B) SpylA knockdown was confirmed at the protein level
through immunoblotting techniques (10% SDS-PAGE), and appeared to
downregulate c-Myc in addition to SpylA when normalized to Actin.
Figure 15. Whole mouse dissections displaying fourth inguinal
mammary glands following mammary fat pad transplantation of control
(left) and SpylA knockdown (right) stable tumor cell lines. Murine subjects
8
CA 02738195 2011-03-23
WO 2010/034128 PCT/CA2009/001372
were allowed a recuperation period of 3 days (A), or 6 days (B) prior to fresh
dissection of No. 4 inguinal mammary glands. Sacrifice of test subjects at Day
3
(A) and Day 6 (B) post-surgery revealed a significant decrease in the rate of
mammary tumor formation in those glands exhibiting decreased SpylA activity.
Arrows depict No. 4 mammary glands following differential shRNA treatment.
Detailed Description of the Preferred Embodiments
Cell culture. Human mammary breast cancer cells, MCF7 (ATCC) and
human embryonic kidney cells, HEK 293T (293T; ATCC), were maintained in
DMEM medium (Sigma) containing 2mM L-glutamine (Sigma), penicillin
(Invitrogen), and streptomycin (Invitrogen), and were cultured in a 5% CO2
environment. MCF7 cells were supplemented with 10% (vol/vol) fetal calf serum
(Sigma) and 293T cells were supplemented with 10% fetal bovine serum.
Plasmids and mutagenesis. The Nedd4-PCEP plasmid (Nedd4),
dominant negative Nedd4-PCEP plasmid (Nedd4 N) and empty vector control
(PCEP) were provided by Dr. Dale S. Haines (Temple University School of
Medicine). HA-Ubiquitin (HA-Ub) was provided by Dr. Sylvain Meloche
(Universite de Montreal). A Cyclin B 1 mutant lacking a portion of the D-box
(Cyc BAD) was provided by Dr. Sylvain Meloche with permission from Dr.
Michael Brandeis (The Hebrew University, Israel) and the GFP-Cyclin B1-CMX
vector was provided by Dr. Ed Harlow (Harvard Medical School). Creation of
Myc-SpylA-PCS3 vector was described previously (Porter et al 2002).
QuikChange Multi-Site-Directed Mutagenesis (SDM; Stratagene) was used to
incorporate new silent sites into the original Spyl -pJT0013 vector (24) in
order to
facilitate the cloning of truncation mutants A (TMA), B (TMB), C (TMC), G
(TMG) and Z (TMZ). A BglII site was inserted by altering nucleotide 256 from
T to C using the primers #A043 5'-
GACGATTTAATTCAAGATCTCTTGTGGATGGACTGCTGC-3' and #A044
5'- GCAGCAGTCCATCCACAAGAGATCTTGAATTAAATCGTC-3' to
construct the pRAO1 vector. Using the pRAO1 plasmid a Mlu site was also added
9
CA 02738195 2011-03-23
WO 2010/034128 PCT/CA2009/001372
by altering nucleotide 175 from C to G using #A004 5'-
CAACAAATCTAAACGCGTCAAAGGACCTTGTCTGG-3' and #A005 5'-
CCAGACAAGGTCCTTTGACGCGTTTAGATTTGTTG-3' to make the vector
pRA02. The pRS2 vector was constructed from Spy]-pJT0013 by creating an
Ndel site just after the stop codon using the primers #A045 5'-
GTCTTGTGTCCATATGTGTTTTGTGGTGACCC-3' and #A046 5'-
GGGTCACCACAAAACACATATGGACACAAGAC-3'. The pRS I vector was
constructed by creating a MluI site in the Spyl-pJT0013 plasmid by altering
nucleotide nucleotide 175 from C to G using primers #A004 5'-
CAACAAATCTAAACGCGTCAAAGGACCTTGTCTGG-3' and #A005 5'-
CCAGACAAGGTCCTITGACGCGTTTAGATTTGTTG-3'. TMA was created
by digesting wild-type Spy IA (in pRS 1) with Ndet and MIuI in order to remove
the first 57 amino acids of the protein. TMB was created by digesting wild-
type
SpylA (in pRA02) with Mlut and BgIII in order to remove 27 amino acids. TMC
was created by digesting wild-type SpylA (in pRAO 1) with BgIII and Ncol in
order to remove 61 amino acids. TMG was created by digesting wild-type SpylA
(in pJT0013) with Ncol and BbsI in order to remove 94 amino acids. Finally,
TMZ was created by digesting wild-type SpylA (in pRS1) with Bbsl and Ndel in
order to remove the last 47 amino acids. Gel electrophoresis of these
digestions
was run on a 1 % agarose gel; the desired band was excised and gel-extracted
(Bio
Basics) for ligation using T4 DNA ligase (Fermentas). For all five truncation
mutants, linkers containing a silent restriction site, Pstl, and complementary
sticky ends were designed, commercially synthesized (Sigma), annealed and
utilized in the ligations. In each case, 20 L ligation reactions were carried
out at
22 C for 2-4 hrs. containing a 1:3 vector to linker ratio. Ligations were
transformed into DH5a cells and selected for ampicillin resistance, mini-
prepped,
and digested with Pstl (Fermentas) to detect the correct ligation. The five
SpylA
truncation mutants (depicted in FIG. 3A), spanning the length of the gene,
were
moved from the pJT0013 into pCS3 using EcoRI and XbaI sites flanking the
gene. Successful cloning was determined by DNA sequencing (Robarts
Sequencing Facility; Univ. of Western Ontario).
CA 02738195 2011-03-23
WO 2010/034128 PCT/CA2009/001372
SDM was also carried out using the PCS3 vector to generate the SpylA-
T15A, SpylA-T33A and SpylA-S22A mutants. SpylA-T15A was designed using
the primers #A151 5' -
GAGACACCACCTACTGTCGCTGTTTATGTAAAATCAG-3' and #A 152 5'-
CTGATTTTACATAAACAGCGACAGTAGGTGGTGTCTC-3'; SpylA-T33A
was designed using the primers #A-153 5'-
CAGCCTAAAAAGCCCATTGCACTGAAGCGTCCTATTTG-3' and #A 154
5'- CAAATAGGACGCTTCAGTGCAATGGGCTTTTTAGGCTG-3'; Spy IA-S
22A was designed using the primers #A139 5'-
GTTTATGTAAAATCAGGGGCCAATAGATCACATCAGC-3' and #A140 5'-
GCTGATGTGATCTATTGGCCCCTGATTTTACATAAAC-3.
Inhibitors and antibodies. The following antibodies were used: SpylA
(NB 100-2521; Novus), Nedd4 (ab14592; Abcam), Myc (9E10 and C19; Santa
Cruz), HA (Y11 and F7; Santa Cruz), Actin (MAB1501R; Chemicon), Cyclin E
(551157; BD Pharmingen). The calpain inhibitor N-Acetyl-L-leucyl-L-leucyl-L-
norleucinal N-Acetyl-Leu-Leu-Norleu-al (LLNL), the proteasome inhibitor
MG132 and nocodazole were all purchased from Sigma.
Transfections. Calcium Phosphate Precipitation transfections were
carried out in 293T cells using 10 g of DNA per 10 cm tissue culture plate.
250
L CaC12 was incubated with the DNA for 30 sec., 250gL 2x BBS at pH 7.01
was added while vortexing and the solution was incubated for 10 min. The
mixture was added slowly to the cells and then incubated in 3% CO2 for 12-16
hrs. Media was then changed and plates were returned to 5% CO2 for 24 hrs.
prior to harvest.
Cell synchronization and flow cytometry. 293T cells were
synchronized using double thymidine block. Briefly, cells were cultured in
media containing 2 mM thymidine for 16 hrs., followed by release into normal
media for 8 hrs. and then a second thymidine block for 14 hrs., and then
release
into media containing 70 ng nocodazole. MCF7 cells were synchronized by
11
CA 02738195 2011-03-23
WO 2010/034128 PCT/CA2009/001372
being cultured in a serum-free media for 48 hrs., followed by release into
media
containing serum and 7Ong nocodazole. 293T and MCF7 cells were trypsinized
at specified times, washed twice in PBS, and then either used immediately or
fixed and stored at -20 C. Fixation was carried out by resuspending cells at 2
x
106 cells in 1 mL of PBS, followed by slow addition of an equal amount of 100%
ethanol. Within 1 week, fixed cells were pelleted, washed, and resuspended in
300 }LL of PBS. Samples were then prepared for flow cytometry by treating with
I L of 10 mg/mL stock of DNase free RNase (Sigma) and 50 L of 500 mg/mL
propidium iodide stock solution. Data was collected using a Beckman Coulter
FC500 (Biology Dept.; U of Windsor) and cell cycle profiles were analyzed
using
CPX Beckman Coulter FC500 software.
Immunoblotting. Cells were lysed in 0.1% NP-40 lysis buffer (5
mL10% NP-40, 10 mL 1M Tris pH 7.5, 5 mL 0.5M EDTA, 10 mL 5M NaCl up
to 500 mL RO water) containing protease inhibitors (PMSF 100 g/mL, aprotinin
5 g/rL, leupeptin 2 g/mL) for 30 min on ice. Bradford Reagent was used to
determine the protein concentration following the manufacturer's instructions
(Sigma). Aliquots of lysates containing 20-30 gg protein were subjected to
electrophoresis on denaturing SDS-10% polyacrylamide gels and transferred to
PVDF-Plus transfer membranes (Osmonics Inc.) for 2 hrs. at 30V using a wet
transfer method. Blots were blocked for 2 hrs. in TBST containing 3% non-fat
dry milk (blocker) at room temperature. Primary antibodies were reconstituted
in
blocker and incubated over night at 4 C at a 1:1000 dilution for all
antibodies,
and secondary antibodies were used at a 1:10,000 dilution in blocker for 1 hr
at
room temperature. Blots were washed three times with TBST following
incubation with both the primary and secondary antibodies. Washes were 6 min
each following the primary antibody and 10 min each following the secondary
antibody. Chemilumiminescent Peroxidase Substrate was used for visualization
following the manufacturer's instructions (Pierce). Chemiluminescence was
quantified on an Alpha Innotech HD2 (Fisher) using AlphaEase FC software.
12
CA 02738195 2011-03-23
WO 2010/034128 PCT/CA2009/001372
Immunoprecipitation reactions were carried out using equal amounts of
protein (200 g/mL) incubated with 2 g of primary antisera, as indicated,
overnight at 4 C. This was followed by the addition of protein A-Sepharose
(Sigma) and incubated at 4 C with gentle rotation for an additional 2 hrs.
Complexes were washed extensively with 0.1% NP-40 lysis buffer and resolved
by 10% SDS-PAGE.
In vivo labeling. 293T cells were treated with IORM MG 132 and 70 ng
nocadozol for 14 hrs. followed by incubation in phosphate-free media for 2
hrs.
and then addition of ImCi of [32P]-orthophosphoric acid (GE healthcare) for 4
hrs. Cells were lysed and immunoprecipitated with Myc antisera.
Immunoprecipitations were washed rigorously with TBS and samples were
analyzed by 10% SDS page gel. Gels transferred to PVDF membranes were
visualized using a Cyclone phosphoimager and quantified using OptiQuant
software (Perkin Elmer; Biology Dept.; U of Windsor).
In Vivo Ubiquitination Assays. 293 cells were plated and transfected
appropriately in a 100-mm dish. 24 hrs. after transfection cells were treated
with
10 M MG132 for 14 hrs. Cells were then collected, pelleted by centrifugation,
lysed in 200 l of preboiled lysis buffer [50 mM Tris-HCI (pH 7.5),0.5 mM
EDTA, 1 % SDS, and 1 mM DTT], and further boiled for an additional 10 min.
Lysates were clarified by centrifugation at 13,000 rpm on a microcentrifuge
for
10 min. Supernatant was diluted 10 times with 0.5% NP40 buffer and
immunoprecipitated with anti-Myc antibody. Immunoprecipitates were washed 3
times and resolved by 10% SDS-PAGE, followed by immunoblotting with anti-
HA antibody.
siRNA Knockdown Process. siRNA against Nedd4-1 was synthesized
by inserting the oligo 5'GATGAAGCCACCATGTATA into the pSUPER-basic
vector, as previously described. As a control, LacZ siRNA (siCntl) was
synthesized and inserted into pSUPER-basic vector as described.293 cells were
transfected using 12 g of either Neddd4 siRNA or siCntl per 100 mm tissue
13
CA 02738195 2011-03-23
WO 2010/034128 PCT/CA2009/001372
culture plate and total protein was isolated from cell cultures and resolved
using
12% SDS polyacrylamide gels as described above.
Tissue Microarray Detection and Quantification of Spyl Protein
levels
De-paraffinization:
Notice: Before deparaffinization in xylene, tissue (microarray) section
slides shall be baked in oven at 60 C for 30 minutes on a vertical rack to
melt the
extra layer of coated paraffin. Trial slides do not have paraffin coating so
it can
be deparaffinized without baking. All of our tissue (microarray) section
slides,
unless otherwise specified, were baked at 60 C for two hours after sectioning
and
are stored at 4 C.
1. Immerse slide in xylene for 10 minutes. Repeat once in new xylene for 10
minutes.
2. Immerse array slide in 100% ethanol for 5 minutes.
3. Immerse in 95% ethanol for 5 minutes.
4. Immerse in 70% ethanol for 5 minutes.
5. Rinse for 5 minutes in water or PBS buffer.
Immunostaining using fluorescent probes
1. Deparaffinize and dry array slide as referred to in protocol of
deparaffinization.
2. Rinse array slide twice with PBST for 5 min each in a Coplin jar.
3. Antigen retrieval (formalin-fixed, paraffin-embedded tissue sections).
Boiling Bath. Heat the buffer (1mM EDTA, pH 8.0 or 0.O1M sodium citrate
buffer, pH 6.0) to about 95 C, and then put array slides in the buffer for
1015
14
CA 02738195 2011-03-23
WO 2010/034128 PCT/CA2009/001372
min. Do NOT let the medium boil when you have array slide in. Avoid the
slide drying during the procedure.
4. Rinse array slide in PBST for 5 min.
5. Apply the blocking antibody (normal goat serum- 150u1/l Oml), incubate
for 40 min at room temperature, and throw off residual fluid (don't wash.).
6. Apply the primary antibody 60 min.at RT (Spyl Novus 1:50) usually
200ul volume to cover all the cores.
7. Rinse array slide twice for 5 min each in a Coplin jar on the orbital
rotator
8. Incubate array slide with a fluorophore-conjugated secondary antibody at
2037 C (in a humidity chamber) for 20 min. (Alexa488 1:1200)
9. Rinse array slide twice in PBST for 5min each in a Coplin jar on the
orbital rotator.
10. Incubate array slide with TOTO-3 nuclear stain (0.75u1:1000) in PBST at
RT 30 min.
11. Rinse array slide 3 times in PBST for 5min each in a Coplin jar on the
orbital rotator.
12. Dehydration and transparency of array slide.
1. Immerse in 70% ethanol for 5 minutes.
2. Immerse in 95% ethanol for 5 minutes.
3. Immerse array slide in 100% ethanol for 5 minutes.
4. Immerse slide in xylene for 10 minutes. Repeat once in new
xylene for 10 minutes
CA 02738195 2011-03-23
WO 2010/034128 PCT/CA2009/001372
13. Mount array slides with the Vectashield mounting media and cover with a
square or rectangulat cover slip no.1.
14. The TMA slide is being placed in the slide scanner. The edges and
surface of the cover slip have to be clean and dry in order to place the slide
in the slide scanner. The fluorescent signal for the secondary antibody
fluorophore and nuclear stain is detected and quantified by ScanArray Express
software (Perkin Elmer Inc.)
15. The mean the fluorophore signal intensity values are normalized to the
mean of the nuclear stain signal.
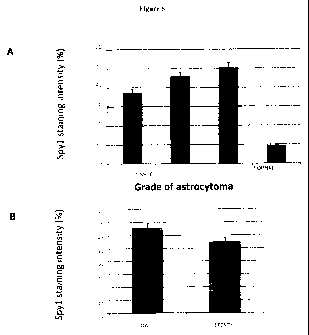
The results are shown in Figure 6.
To correlate the expression levels of Spy I with the type and
grade of brain tumour we used tissue microarrays (TMAs)
consisting of 103 individual patient cores from malignant and benign
human brain tumour samples and normal matching brain tissues. The
TMAs were probed with anti-Spy I primary antibody followed by
staining with Alexa-488 conjugated secondary antibody. ToTo-3 stain
was used as nuclear control. The fluorescent signal was detected and
quantified by ScanArray Express (Perkin Elmer Inc.) The Spyl signal
intensity was normalized to nuclear stain signal. We found that Spy 1
expression levels were highly elevated in tumour tissues comparing to
normal brain tissue (Fig 6A). Moreover the proportion of Spy I positive
cells increased with tumour grade for both oligodendro- and astrocytic
gliomas and correlated with higher expression in malignant tumours
(Fig 6A,B).
Tissue protein extraction and Western Blot protocol for SpylA
protein levels:
1. Tissue samples were stored at -80'C.
16
CA 02738195 2011-03-23
WO 2010/034128 PCT/CA2009/001372
2. Each sample was allowed to thaw on ice for 10min.
3. Tissue lysis buffer:
= 50mM Tris-HCI, pH 7.4;
= I% NP-40;
= 0.25% sodium deoxycholate;
= 150mM NaCl;
= 1 mM EGTA;
= 1mM PMSF;
= 1 ug/ml each aprotinin, leupeptin,
= Antifoam 50u1
Make sure the buffer is cold!
4. Tissues were homogenized in Eppendorf tubes using plastic pestles 20min
and incubated another 45 min on the ice.
5. The samples were centrifuged at 13000 rpm for 15 min and the
supernatant containing the protein extract was stored at -20'C.
6. The protein concentration was estimated using Bradford assay.
7. Samples for the SDS-PAGE analysis were prepared using 4x
sample/loading buffer containing SDS and glycerol.
8. 10% Polyacrylamide gel was prepared according to standard protocol.
9. Prior to loading samples were boiled for 5min.
10. Gel ran at 110V 25mA for 3.5h in a lx SDS running buffer (25mM Tris-
HCI; 200mM Glycine; 0.1% SDS)
11. PVDF membrane was activated in methanol and soaked in transfer buffer
for 3 min.
17
CA 02738195 2011-03-23
WO 2010/034128 PCT/CA2009/001372
12. The gel was taken out from the cast and soaked in the transfer buffer for
10min.
13. "Western Blot sandwich" was assembled and placed in the transferring
chamber.
14. The transfer ran at 30 V for 2hr.
15. The membrane was blocked in 2.5% milk in TBST for lhr with shaking.
16. The membrane was incubated o/n at 4'C on a shaker with the proper
primary antibody (Spy1 Novus 3u1/ml; p27 Calbiochem 1:1000; Actin Santa
Cruz 1:1000) diluted in 2.5% milk.
17. The membrane was washed 3x 15 min in TBST and incubated with the
proper secondary antibody 1:10 000 diluted in 2.5% milk for lhr at RT on a
shaker.
18. The was washed 3x 5 min with TBST.
19. Detection by ECL. The blot was exposed using Alphalnnotech Imager
and software.
The results are shown in Figure 7.
We obtained glioblastoma (Fig 7C), oligodendroglioma (Fig 7B),
oligoastrocytoma (Fig 7A) and pair matched normal tissues samples from the
Ontario Tumour Tissue Bank. We tested samples obtained from biopsies taken
from the tumour center (i), peritumour (ii-iv) samples and normal brain tissue
(v). Total protein was extracted from frozen tissues and subjected to western
blot analysis. Samples were probed using Spy 1, actin, and p27 antibodies.
Spy1
levels were found to be elevated in the tumour center as compared to the
normal
tissue in 9 of 12 patient samples.
Immunohistochemistry detection of SpylA protein levels
18
CA 02738195 2011-03-23
WO 2010/034128 PCT/CA2009/001372
1. Sections placed on slides were deparaffinized in Xylene (2x5min in a
Coplin jar)
2. Rehydrated in 100% ethanol (lx5min)
3. Blocked in 3% H202 in 100% methanol (1x20min)
4. Rehydration continued
= 100% ethanol (lx5min)
= 95% ethanol (lx5min)
= 70% ethanol (lx5min)
= DI water (lx5min)
= 1xPBS (lx5min)
5. Antigen retrieval. Citrate buffer pH 6.0; 600m1 in 2L beaker
Slides were placed in a wheaton glass slide holder and subjected to 4
rounds x 5 min in 1000W microwave on high. Lost volume of watr was replaced
with dd water. The slides were cooled for 20min and washed in water 4x2min
and lx2min in 1xPBS.
6. The sections were blocked using 10% normal goat serum in PBST (0.1%
Tween) minimum 1 hr. The blocker was drained not washed.
7. The sections were incubated with the primary antibody (1:100) for lhr.
8. The antibody was washed away with PBST 3x5min
9. The secondary Ab was applied (1:200) goat anti-mouse biotin conjugated
and incubated for 45' at RT.
10. The sections were washed 3x5min.
11. ABC solution was applied for 45' at RT and washed 2xPBST.
19
CA 02738195 2011-03-23
WO 2010/034128 PCT/CA2009/001372
12. The sections were washed IxPBS
13. The sections were treated with the DAB solution and observed under the
dissection microscope till they reached proper staining level.
14. The DAB was washed with 1xPBS for 5min.
15. The sections were counterstained with hematoxylin for 40" and washed
in water.
16. The sections were dehydrated
= 70% ethanol lx5min
= 95% ethanol lx5min
= 100% ethanol lx5min
= 100% ethanol lx5min
= Xylene 3x 5min
17. The slides were mounted with Permount followed by cover slip.
The results are shown in Figure 8.
Generation of deletion mutant A and the non-degradable point
mutations.
DMA was created by digesting wild-type SpylA with Ndel and Mlul in
order to remove the first 57 amino acids of the protein. Gel electrophoresis
of
these digestions was run on a 1% agarose gel; the desired band was excised and
gel-extracted (Bio Basics) for ligation using T4 DNA ligase (Fermentas).
Linkers
containing a silent restriction site, PstI, and complementary sticky ends were
designed, commercially synthesized (Sigma), annealed and utilized in the
ligations. In each case, 20 L ligation reactions were carried out at 22 C for
2-4
hrs. containing a 1:3 vector to linker ratio. Ligations were transformed into
DH5a
cells and selected for ampicillin resistance, mini-prepped, and digested with
Pstl
(Fermentas) to detect the correct ligation. The five SpylA deletion mutants
CA 02738195 2011-03-23
WO 2010/034128 PCT/CA2009/001372
(depicted in Fig. 9), spanning the length of the gene, were moved from the
pJT0013 into pCS3 using EcoRI and Xbal sites flanking the gene.
SDM was also carried out using the PCS3 vector to generate the SpylA-
T15A, SpylA-T33A, SpylA-S22A and SpylA-S247A mutants. SpylA-T15A
was designed using the pruners #A151 5'-
GAGACACCACCTACTGTCGCTGTTTATGTAAAATCAG-3' and #A 152 5'-
CTGATTTTACATAAACAGCGACAGTAGGTGGTGTCTC-3'; SpylA-T33A
was designed using the primers #A-153 5'-
AGCCTAAAAAGCCCATTGCACTGAAGCGTCCTATTTG-3' and #A 154 5'-
CAAATAGGACGCTTCAGTGCAATGGGCTTTTTAGGCTG-3'; SpylA-
S22A was designed using the primers #A139 5'-
GTTTATGTAAAATCAGGGGCCAA TAGATCACATCAGC-3' and #A 140
5'- CTGATGTGATCTATTGGCCCCTGATTTTACATAAAC-3; SpylA-S247A
was designed using the primers #A 143 5'-
GGATTGTCTTCATCATCAGCGTTATCCAGTCATACTGCAGGGGTG-3'
and #A 144 5'-
CACCCCTGCAGTATGACTGGATAACGCTGATGATGAAGACAATCC-3'.
Successful cloning in all cases was determined by DNA sequencing (Robarts
Sequencing Facility; Univ. of Western Ontario).
The results are shown in Figure 9.
SpylA degradation depends on phosphorylation within the N-
terminal region. Using a panel of SpylA deletion mutants (Fig. 9A), we began
to narrow down the region within the SpylA protein that was necessary for
degradation. We first determined whether deletion of any of the regions of
SpylA
would result in stabilization of the protein. 293 cells were transfected with
wild-
type SpylA or deleted versions of the SpylA protein, DMA-DMZ. Cells were
synchronized at G2/M and levels of SpylA were monitored by immunoblotting
(Fig. 9B; upper panel). All deletion mutants of SpylA were degraded by G2/M
phase with the exception of the mutant lacking the first 57 amino acids (DMA).
21
CA 02738195 2011-03-23
WO 2010/034128 PCT/CA2009/001372
Asynchronous cells demonstrate that all deletion mutants were expressed (Fig.
9B; lower panel). Collectively, these data demonstrate that the N-terminal
region
of SpylA is essential to mediate degradation of the protein and that unlike
the
Xenopus homolog of Spyl the C-terminal region is dispensable for degradation.
Residues T15, S22, and T33 are essential for SpylA degradation. We
have demonstrated that the N-terminal region of SpylA is essential for
mediating
degradation. Hence, we focused on elucidating sites within this region that
may
target the protein for degradation. Utilizing the NetPhos 2.0 Server tool
residues
T15, S22, and T33 were isolated as potential phosphorylation sites. Site-
directed
mutagenesis was performed to alter SpylA residues T15, S22, and T33 to non-
phosphorylatable alanines. Additionally we generated a similar mutation at
S247
in the C-terminal region to serve as a control. 293 cells were transfected
with the
relevant constructs prior to synchronization at G2/M. Surprisingly, mutation
of all
of T15, S22, and T33 to a non-phosphorylatable alanine prevented degradation
and ubiquitination of SpylA (Figs. 1OA & B). Blotting asynchronous cell
populations revealed that protein expression was not affected (Fig. 10A; right
panel). This suggests that phosphorylation, or maintenance of charge of all
three
of T15, S22, and T3 3 is essential in regulating the turnover of Spy IA. To
further
assess the effect of these mutations on SpylA degradation, 293 cells were
transfected and then treated with 50 g/ml cyclohexamide 16hrs. post-
transfection. Immunoblotting for SpylA showed that cells transfected with the
mutants have stabilized SpylA levels (Fig. IOC). Quantifying 3 separate
experiments demonstrate that indeed all 3 mutations significantly enhance the
stability of SpylA protein (Fig. IOC; right hand panel). To assess whether
these
sites are phosphorylated in vivo a triple mutant (SpylA-TST) was created where
all 3 elucidated sites were mutated to a nonphosphorylatable alanine (T15A,
S22A and T33A). Phosphorylation of SpylA-TST at G2/M was compared to that
of wt-SpylA using an orthophosphate labeling experiment. A significant
decrease in phosphorylation was observed with the triple mutant (Fig. I OD),
demonstrating that SpylA is phosphorylated at residues T15, S22 and T33 during
22
CA 02738195 2011-03-23
WO 2010/034128 PCT/CA2009/001372
G2 phase of the cell cycle.
The results are shown in Figure 10.
Aberrant SpylA degradation enhances cell proliferation. To test the effects
of ablating SpylA degradation on cell proliferation live and dead cell
populations
were monitored by trypan blue analysis. SpylA and mutant constructs
significantly enhance cell proliferation as compared to mock with p values of
0.01 for mock: WT, 0.001 for mock: Spy l A-T 15A, 0.0004 for mock: Spy l A-
T33A, and 0.001 for mock:SpylA-S22A. There was no statistical change in the
number of dead cells from one transfection to another (Fig. 11; grey bars).
Interestingly, Spyl degradation mutants statistically enhanced proliferation
over
SpylA alone by 20-60% (Fig. 11; black columns). p-values for these
comparisons were 0.009 for WT:SpylA-T15A, 0.002 for WT:SpylA-T33A and
0.03 for WT:SpylA-S22A.
The results are shown in Figure 11.
Oncogenic effects of TST
Anchorage Independent Growth in Soft Agar. It was examined
whether non-degradable Spyl is able to acquire a transformed phenotype
through a soft agar assay testing for anchorage independence. The assay was
done in triplicates, including 4 transfections (WT, TST, PCS3 and RASV12). The
assay was conducted 3 times as three separate experiments. Overall, the assay
was incubated for 10-14 days before the colonies were photographed.
Consistent with anchorage independent growth and invasive phenotype in
vitro, RASV12 yielded numerous colonies grown in soft agar as it was expected.
(Figure 12A, 12B and 12C) Surprisingly, when TST was plated and allowed to
grow in soft agar, numerous colonies were observed as well. (Figure 12A, 12B
and 12C) However when WT Spyl and PCS3 were grown in soft agar, no
apparent colonies formed and the few colonies that did form for WT Spyl were
23
CA 02738195 2011-03-23
WO 2010/034128 PCT/CA2009/001372
not the same size or magnitude as the RASV 12 or TST formed colonies.(Figure
12B).
The results are shown in Figure 12.
Screening for TST in different cancerous cell lines. To examine if these
mutations are existing in some cancerous cell lines fife cell lines were
utilized;
HCT p21-/-, HCT p53 -/-, MCF7, HTB 231 and HTB 126. Using sequencing
primers A455 and 457 we were able to PCR DMA and purify it using Biobasic
PCR purification kit and sequence at Robart sequencing facility at University
of
Western Ontario. The sequencing results showed that amino acids from 1-61
(DMA) are not mutated in all the cell lines being tested.
MRHNQMCCETPPTVTVYVKSGSNRSHQPKKPITLKRPICKDNWQAFEKNT
..................................................
..................................................
MRHNQMCCETPPTVTVYVKSGSNRSHQPKKPITLKRPICKDNWQAFEKNT
10 20 30 40 50
160 170
HNNNKSKRPKGPC
HNNNKSKRPKGPC
Tool Development for shRNA Knockdown of Spyl Protein Levels.
Available online as free software (http://www.oligoengine.com), the program
OligoEngine Workstation 2 was utilized to design five novel shRNA constructs
25 directed against unique regions of the mouse SpylA isoform, in addition to
a
scrambled shRNA construct chosen for its inability to recognize and target
SpylA for RNAi-mediated degradation. Constructs flanked by HpaI and Xhol
restriction sites were synthesized by Sigma-Genosys Canada (Sigma-Aldrich,
Ontario) and subsequently subcloned into pLB (Addgene, MA, USA), a lentiviral
24
CA 02738195 2011-03-23
WO 2010/034128 PCT/CA2009/001372
vector that utilizes the mouse U6 promoter to dually express shRNA and green
fluorescent protein (GFP) in mammalian cell systems. Thus, GFP fluorescence
served as a positive indicator for induction of RNAi.
The results are shown in Figure 13.
Transient SpylA Knockdown in Primary c-Myc Overexpressing
Tumor Cell Lines. The MMTV-Myc mouse model (MMHCC, NCI), well
documented for its ability to form aggressive mammary tumors, was utilized to
derive a previously uncharacterized tumor cell line engineered to overexpress
the
proto-oncogene c-Myc. Polyethylenimine (PEI) was utilized to transfect
immortalized primary cell cultures with control or SpylA knockdown vectors.
Stable Cell Line Production & Mammary Fat Pad Transplantation:
In vivo Knockdown of SpylA. PEI-mediated transfection of the HEK-293T
producer cell lines was utilized to introduce control or SpylA-shRNA vectors
to
a series of pre-optimized packaging vectors that code for the necessary viral
components required to form lentiviral particles upon transduction of
mammalian
cell lines. Following production of infectious lentivirus, control and SpylA
knockdown particles were each concentrated and titered as previously outlined
in
Welm, B.E., et al. (2008), Cell Stem Cell 2(l): 90-102, proceeded by infection
of
primary tumor cell lines with respective viral concoctions. Subsequently,
stable
cell lines were passaged three times, after which they were utilized to
perform
mammary fat pad transplantations in 28-day old FVB female mice. In summary,
number four glands were cauterized to prevent endogenous stem cell populations
from colonizing the mammary fat pad, which in turn served as an environmental
substrate to support transplanted stable cells of either type. Left cleared
fat pads
were injected with control shRNA stable cells, whereas right cleared fat pads
were injected with SpylA shRNA stable cells (Fig.15).
SpylA protein degradation occurs in a cell cycle dependent fashion. It
is known that spylA is a low copy number gene which is constitutively
expressed
in most human tissues and that the mRNA is specifically up-regulated in the G1
CA 02738195 2011-03-23
WO 2010/034128 PCT/CA2009/001372
phase of the cell cycle (5, 24) unpublished data). Herein we wished to
determine
whether levels of the protein are regulated in a cell cycle dependent fashion,
or
whether levels vary independently of cell cycle stage. In order to address
this
question, MCF7 and 293T cells were blocked at G, phase of the cell cycle using
serum starvation (SS) or a double thymidine block (TB), respectively, followed
by release into nocadozol containing media. A G2 population was collected at
16
hrs following release for both cell lines (nocadozole treatment; NT). Flow
cytometry analysis was used to confirm the cell cycle stage for cells
harvested
during the block (GI) or following release (G2) (FIG. IA and C; upper panels).
Asynchronous populations were also used as a control (Cntl). Percentages of
cells in each phase of the cell cycle, as determined by flow cytometry
analysis
software, are shown in graphic form (FIG. IA and C; lower panels).
Immunoblotting of cell lysates from different stages o the cell cycle showed
that
Spy 1 A protein levels are greatly decreased at the G2/M border (FIG. 1 B and
D;
upper panels). Cyclin E was assessed in order to ensure that cells were in the
proper stage of the cell cycle (FIG. 1B; middle panel), and Actin was used as
the
loading control (FIG. lB and D; lower panels). This demonstrates that SpylA,
like many important cell cycle proteins, is tightly regulated in a cell cycle
dependent fashion.
To further narrow down where in the cell cycle SpylA was being
degraded, we utilized a non-degradable Cyclin B1 mutant (CycBAD). It is well
established that Cyclin B1 must be degraded in order for cells to successfully
complete the metaphase-to-anaphase transition (34). The CycBAD contains a 9
residue motif mutation within the destruction box, thus disrupting the region
responsible for the association between Cyclin B 1 and the ubiquitin ligase,
APC
(35). Hence, the CycBAD mutant of Cyclin B1 arrests cells at the metaphase-to-
anaphase transition. 293T cells were transfected with wild-type Cyclin B1-
PEGFP cDNA (CycB1), CycBAD (CycB-D) or an empty PEGFP vector (GFP).
Cell lysates from the transfections or an untransfected control (Cntl) were
analyzed by immunoblotting. Cells transfected with CycB1 and GFP contained
26
CA 02738195 2011-03-23
WO 2010/034128 PCT/CA2009/001372
similar levels of SpylA as non-transfected cell lysates (FIG. IE; lanes 1, 2,
and
3). However, there was no SpylA protein detected in the lysates from cells
overexpressing the CycBAD mutation (FIG. IE; upper panel; lane 4). Actin was
used as a loading control (FIG. 1 E; lower panel). This data demonstrates that
the
degradation of SpylA takes place prior to the anaphase transition during
mitosis.
SpylA degradation is proteasome dependent. After determining the
timing of SpylA degradation during cell cycle progression, we set out to
investigate the mechanism by which this occurs. Gutierrez et al. have reported
that X-Spyl is being processed and degraded via the ubiquitin proteolytic
pathway, and as such, this was the pathway we began to investigate. 293T cells
were transiently transfected with HA-tagged ubiquitin (Ha-Ub) and Myc-tagged
SpylA (Myc-SpylA), followed by immunoprecipitation with anti-Myc antibody
(FIG. 2A; lower panel). Immunoblotting with anti-HA antibody revealed that
SpylA was labeled with HA-ubiquitin in vivo (FIG. 2A; upper panel). To verify
the involvement of the proteasome machinery in SpylA turnover, we studied
293T cells in the presence or absence of the proteasome inhibitor MG132 or the
calpain inhibitor LLNL. SpylA protein levels were significantly elevated in
the
presence of the proteasome inhibitor MG 132 (FIG. 2B; upper panel; lane 4) but
not in the presence of the calpain inhibitor or the vehicle controls (FIG. 2B;
upper
panel; lanes 1-3). Actin was used as a loading control (FIG. 2B; lower panel).
This data implicates that SpylA protein degradation is proteasome dependent.
SpylA degradation depends on phosphorylation within the N-
terminal region. Using a panel of SpylA truncation mutants (depicted in FIG.
3A), we began to narrow down the region within the SpylA protein that was
necessary for degradation. We first determined whether deletion of any of the
regions of SpylA would result in stabilization of the protein. 293T cells were
transfected with wild-type SpylA (wt), empty PCS3 vector (-ve), or truncated
versions of the SpylA protein, TMA-TMZ (A-Z). Cells were synchronized at
G2/M with nocadozole, and levels of SpylA were monitored by immunoblotting
(FIG. 3B; upper panel); Actin was used as a loading control (FIG. 3B; lower
27
CA 02738195 2011-03-23
WO 2010/034128 PCT/CA2009/001372
panel). This figure demonstrates that all truncation mutants of SpylA were
degraded in the G2/M phase with the exception of TMA (FIG. 3B; upper panel;
lane 3). This suggests that the N-terminal region of SpylA is essential to
mediate
the degradation. Furthermore, the molecular weight of the resolved SpylA was
53 kDa, which is the size of the wild-type tagged SpylA protein. This
demonstrates that either the SpylA protein is not processed prior to
degradation,
as is the case with X-Spy 1, or that the N-terminal region of SpylA is
required for
both processing and degradation of the SpylA protein.
Phosphorylation is often the key event regulating recognition of the
substrate protein by the E3 (4). To determine whether TMA was altering the
phosphorylation status of SpylA during G2, orthophosphate labeling was
performed using the panel of SpylA truncation mutants. A significant decrease
in the incorporation of orthophosphate was observed for TMA (FIG. 3C; upper
panel; lane 3), surprisingly however no substantial decrease in
phosphorylation
was observed for any of the other truncation mutants (FIG. 3C; upper panel;
lanes
4-7). From this information, we conclude that there is at least one
phosphorylation site present within the N-terminal region of SpylA that may
play
a significant role in regulating SpylA stability. We have presented an
alignment
of the SpylA and X-Spyl proteins showing the regions known to be essential for
processing and degradation of the X-Spyl protein (FIG. 3D). Notably the
majority of regions associated with X-Spyl degradation reside within the C-
terminal region, a region which was dispensable for mediating SpylA
degradation
(FIG. 3B).
Furthermore, the sites required for phosphorylation of X-Spyl to initiate
processing, T8 and S12, are not conserved in the mammalian homologue (FIG.
3D; bold, +). Additionally, the phosphorylation sites known to be essential
for
mediating binding of the SCFPT`CP ligase (S233 and S237) are also not
completely
conserved in SpylA. The ubiquitin ligase Siah-2, which regulates the final
degradation process in X-Spy 1, depends on S243 phosphorylation by CDK2 for
binding. This recognition site requires the adjacent proline at residue 244,
which
28
CA 02738195 2011-03-23
WO 2010/034128 PCT/CA2009/001372
is not conserved in the mammalian homologue (FIG. 3D, bold **). This supports
that degradation of SpylA in the somatic cell likely occurs via a different
mechanism than that of the Xenopus homologue during oocyte maturation.
Nedd4 is the E3 ligase responsible for SpylA degradation. There are
many different E3 ubiquitin ligase enzymes that are able to function in the
ubiquitination pathway, therefore we set out to determine which one played a
role
in the degradation of SpylA. A protein blast for the N-terminal region of
SpylA
revealed a weak potential interaction region for WW domain containing
proteins,
PPxxxxY (FIG. 3D; grey shading). It is known that the WW domain-containing
ligase, Nedd4 (product of neuronal precursor cell-expressed developmentally
down-regulated gene 4), while preferring the canonical PPxY sequence, also
binds to a variety of proline rich regions with phosphorylated threonine or
serine
residues to trigger ubiquitination and subsequent degradation (12, 30, 31).
Due to
this we investigated the potential role of Nedd4 in SpylA degradation. Nedd4
is a
family of conserved E3 ubiquitin ligases required for ubiquitination of a
large
number of target proteins (30). Isoforms of Nedd4 exist in the simplest yeast
cells to the most complex mammals, and have been shown to assist with
regulating developmentally-expressed proteins (15, 22). Co-immunoprecipitation
assays in cells overexpressing exogenous Nedd4 as well as Myc-tagged SpylA
(Myc-SpylA) demonstrate that Nedd4 interacts with SpylA in vivo (FIG. 4A;
upper panel; lane 3). To further investigate whether Nedd4 is functioning as
an
ubiquitin ligase for SpylA, we repeated the co-immunoprecipitation experiment
using overexpression of wild-type Nedd4 (Nedd4) or dominant negative Nedd4
(Nedd4 DN) in the presence of HA-tagged Ubiquitin (Ha-Ub). Immunoblotting for
HA-tagged Ubiquitin, followed by quantification, revealed that SpylA
incorporated 41 % HA-Ub in the presence of Nedd4, and that this was
significantly decreased (by 18%) in the presence of Nedd4DN (FIG. 4B; upper
panel; lane 3). Quantification is depicted in FIG. 4B; right graph. To further
assess the effect of Nedd4 on endogenous SpylA, Nedd4 was transfected into
293T cells in the presence and absence of the proteasome inhibitor MG132, and
29
CA 02738195 2011-03-23
WO 2010/034128 PCT/CA2009/001372
endogenous levels of SpylA were measured. SpylA protein levels were
decreased by 20% when Nedd4 was transfected in the absence of MG132 (FIG.
4C; upper panel; lane 3) as compared to when MG 132 was present (FIG. 4C;
upper panel; lane 2). Densitometry was performed and equalized for protein
loading as determined by Actin (FIG. 4C; lower panel; right graph).
Collectively,
this data demonstrates that SpylA binds to the E3 Nedd4 and that Nedd4 is
capable of enhancing ubiquitination of SpylA and promoting the degradation of
SpylA.
SpylA phosphorylation at T15, S22, and T33 regulates SpylA
degradation. Cell cycle regulatory proteins which are targeted to the UPS rely
on signal transduction mechanisms to control the timing of this essential
event.
We have demonstrated that the N-terminal region of SpylA (depicted in FIG. 3D)
is essential for mediating degradation. Hence, we focused on elucidating
potential phosphorylation sites within the N-terminal region of SpylA that may
target the protein for degradation. Utilizing the NetPhos 2.0 Server tool,
which
predicts potential serine, threonine, and tyrosine phosphorylation sites,
residues
T15, S22, and T33 were shown to have a high probability of being potential
phosphorylation sites (3). Site-directed mutagenesis was subsequently
performed
to alter SpylA residues T15, S22, and T33 to non-phosphorylatable alanines
(T15A, S22A, and T33A respectively). 293T cells were then transfected with
wild-type SpylA (wt), SpylA-T15A (T15A), SpylA-S22A (S22A), and SpylA-
T33A (T33A) prior to synchronization at G2/M. Surprisingly, mutation of any of
the three sites T15, S22, and T33 to a non-phosphorylatable alanine prevented
degradation of SpylA (FIG. 5A; left, upper panel; lanes 1-3). Blotting
asynchronous cell populations revealed that protein expression was not
affected
(FIG. 5A; right, upper panel), Actin blotting determined even loading (FIG.
5A;
left & right lower panels). This suggests that phosphorylation at all three of
T15,
S22, and T33 is an essential event in regulating the turnover of SpylA. Cells
transfected with wild-type SpylA (wt) and each of the mutants (S22A, T33A,
T15A) were also subjected to a number of other experiments. In the presence of
CA 02738195 2011-03-23
WO 2010/034128 PCT/CA2009/001372
MG 132, to prevent degradation, cell lysates were immunoprecipitated with c-
Myc antibody and immunoblotted for HA (Ha-Ub-SpylA; FIG. 513; upper panel)
and Myc (FIG. 5B; lower panel). These results demonstrate that wild-type SpylA
is ubiquitinated, however all of the three mutations had no detectable
incorporation of Ha-Ub (FIG. 5B; upper panel; lane 4 vs. lanes 1-3).
Similarly,
this membrane was immunoblotted for Nedd4 (FIG. 5B; middle panel). This
panel demonstrates that Nedd4 immunoprecipitates with wild-type SpylA and
SpylA-T33A (FIG. 5B; middle panel; lanes 4 and 2), however the SpylA-S22A
and SpylA-T15A mutants no longer demonstrate binding to Nedd4 (FIG. 5B;
middle panel; lanes 1 and 3). These data demonstrate that S22 and T15 on SpylA
are essential phosphorylation sites to mediate Nedd4 binding. This further
implies
that loss of ubiquitination seen with the SpylA-T33A mutant (FIG. 5B; upper
panel; lane 2) might represent the phosphorylation site which mediates
ubiquitination. To test the effects of ablating SpylA degradation on cell
growth
characteristics, wild-type SpylA (WT) and the mutants were transfected into
293T cells, and cell proliferation and flow cytometry were carried out. FIG.
5C
depicts the results of 3 separate transfections of each eDNA counted in
triplicate.
Wild-type SpylA and mutant constructs were shown to significantly enhance cell
proliferation over mock with p values of 0.01 for mock:WT, 0.001 for
mock: Spy l A-T 15A, 0.0004 for mock: Spy l A-T3 3 A, and 0.001 for mock: Spy
l A-
S 22A (these stats are not reflected in FIG. 5B). Preventing the degradation
of
SpylA through the alteration of T15, T33, S33 to non-phosphorylatable alanines
statistically enhanced proliferation over SpylA alone by 20-60% (FIG. 5C;
black
columns). p-values for these comparisons were 0.009 for WT:SpylA-T15A, 0.002
for WT:SpylA-T33A and 0.03 for WT:SpylA-S22A; these p-values are reflected
in FIG. 5C. There was no statistical change in the number of dead cells from
one
transfection to another (FIG. 5C; grey bars). Flow cytometry analysis was
performed using cells from FIG. 5A. Profiles were collected for WT or mutant
transfected cells in either an asynchronous population, to determine whether
preventing the degradation of SpylA altered the normal cell cycle profile, or
in
nocodazole treated population, to determine if SpylA overexpression could
31
CA 02738195 2011-03-23
WO 2010/034128 PCT/CA2009/001372
override a G2/M arrest (FIG. 5D). WT and mutant SpylA constructs
demonstrated very similar cell cycle profiles, demonstrating that preventing
the
degradation of SpylA in this cell type did not trigger any normal cellular
responses to prevent cell cycle progression. Collectively, this data
demonstrates
that residues T15, S22, and T33 within the N-terminal region of SpylA are
important phosphorylation sites for mediating the degradation of the protein;
T15
and S22 appear to be essential sites mediating the binding of Nedd4, and T33
may be a potential phosphorylation site to trigger ubiquitination events.
Furthermore we demonstrate that preventing the degradation of SpylA in the
293T cell line does not trigger cell cycle arrest or prevent nocodazol induced
G2/M arrest,however it does result in enhanced cell proliferation.
Importance of SpylA Degradation in Cell Cycle Regulation. Tight
regulation over the protein levels of cyclins is known to be one essential
mechanism by which the cell ensures the proper timing of cell cycle events.
This
regulation is also accomplished through the regulated activity of the CDKs
(23).
More recently it has come to light that CDKs can also be activated by members
of the Speedy/RINGO family. These proteins lack any sequence homology with
cyclins however our data demonstrates that, like the cyclins, SpylA is tightly
regulated at the protein level through the cell cycle. It has been
demonstrated that
X-Spyl can induce cleavage arrest in embryos when overexpressed post meiosis
(10); this suggests that, like the cyclins, regulation of X-Spyl levels is
necessary
to maintain normal procession through the cell cycle. Our data in 293T cells
demonstrates that preventing the degradation of SpyI A did not trigger a cell
cycle arrest of the somatic cell cycle. This may reflect a bone fide species
or cell
type difference in regulation of this protein, whereby SpylA is not
functioning
like a core cyclin in mediating the somatic cell cycle. However, this requires
further experimentation in non-immortalized cells. The significance of
Speedy/RINGO proteins in the cell cycle is irrefutable, and their tight
control is
absolutely essential for normal progression; indeed, deregulated levels of
Speedy/RINGO proteins and their binding partners are found in many cancer
32
CA 02738195 2011-03-23
WO 2010/034128 PCT/CA2009/001372
types (19, 36). Therefore, our data demonstrates that preventing the
degradation
of SpylA enhances cell proliferation, further supporting a potential role for
SpylA in tumorigenesis.
The SpylA-Nedd4 Interaction. Herein, we demonstrate a novel
interaction between SpylA and the E3 ligase Nedd4 which mediates the
degradation of SpylA. The domain structure of Nedd4 family members are very
similar and contain a series of typically two to four WW domains which
function
as recognition sites for specific substrates or adaptor proteins (1, 15). The
WW
domains of Nedd4 preferentially recognize PPxY motifs in their substrates
(Murillas, 2001). The N-terminal region of SpylA lacks this consensus site;
however SpylA does contain a potential PpxxxxY site and it is known that
Nedd4 can also interact with phosphorylated threonine or serine residues to
trigger ubiquitination and subsequent degradation (30) (31). Following
mutagenesis of 3 potential phosphorylation sites within the N-terminal region
of
SpylA, we have determined that phosphorylation in the region of amino acids
15-33 is generally important for SpylA degradation. Our data has demonstrated
that phosphorylation at T15 is particularly important for mediating
degradation:
T15 is completely conserved among the mammalian SpylA homologues, and is
preceded by a highly conserved proline rich region (PPTV). Potential
phosphorylation sites at position 15, 22 and 33, or amino acids which mimic
phosphorylation are also found among other members of the Speedy/RINGO
family in mammals: Spyl/RINGO-B, which is only found in the testes, has a
conserved S22 site and contains a glutamic acid at position 15 which may mimic
phosphorylation, however it lacks flanking prolines; Spyl/RINGO-C, which is
found in the liver, kidney, bone marrow, placenta and testes has a conserved
serine at position 15 with two prolines at positions 11 and 13, and a
conserved
T33; whether these sites are involved in proteolysis of other Spyl/RINGO
family
members remains to be determined.
Furthermore, the Nedd4 family consists of nine members, all containing
WW domains. We know from overexpression assays using Nedd4-1 eDNA that
33
CA 02738195 2011-03-23
WO 2010/034128 PCT/CA2009/001372
this member of the Nedd4 family is capable of interacting with SpylA and
promoting the ubiquitination and degradation; however, whether other members
of the Nedd4 family are also capable of regulating the degradation of Spyl A
is
currently not known. Assays using a dominant negative form of Nedd4-1 did not
completely abolish ubiquitination of SpylA in vivo, however this may reflect
an
issue of transiently transfecting the dominant negative vector. Future studies
into
the sufficiency of Nedd4-1 to regulate SpylA degradation is required.
SpylA-Nedd4 Interaction in Cancer. From the current catalogue of
known Nedd4 substrates it appears that Nedd4 can act as both a proto-oncogene,
as well as a tumor suppressor under different circumstances. For example,
Nedd4 has been shown to mediate the degradation of the vascular endothelial
growth factor receptor 2 (VEGF-R2) (21). VEGF-R2 is a positive regulator of
cell proliferation, migration, and angiogenesis (7), and it is known to be up-
regulated in colon (9), brain (26), and breast cancer (29). In addition, Nedd4
has
been shown to lead to the down-regulation of the insulin-like growth factor 1
receptor (IGF-1 R) (32), which has been implicated in both the initiation and
development of many human cancers types (20). Our data provides further
evidence that Nedd4 can function like a tumor suppressor to regulate the
levels of
proteins stimulating cell growth mechanisms. Conversely, Nedd4, or Nedd4
family members, have been shown to regulate the degradation and function of
important tumor suppressor genes such as the phosphatase and tensin homolog 1
(PTEN), p53 and the p53 family member, p73 (17, 33)(28).
Taken together, these conflicting findings underscore the critical
importance of resolving the factors that mediate Nedd4 substrate specificity.
In
addition to understanding the regulatory factors mediating Nedd4 interactions
and
regulating Nedd4 expression itself, it is also important to resolve the
subcellular
localization of Nedd4 and its specific substrates. Although primarily
cytoplasmic,
Nedd4 is known to enter the nucleus, within which it continues its role as a
ligase
(11). SpylA, on the other hand, is primarily nuclear however unpublished data
has demonstrated that SpylA also shuttles under different circumstances.
34
CA 02738195 2011-03-23
WO 2010/034128 PCT/CA2009/001372
Furthermore, resolving the expression levels of Nedd4 and Nedd4 family
members in specific cell types throughout development may resolve apparent
conflicting data with regard to the role of Nedd4 mediated actions on cell
proliferation. Therefore, in addition to understanding the regulation of
temporal
control of Nedd4, it is important to also study the spatial subcellular
localization of these interactions through real time. Future studies with
regard to
the regulation of Nedd4 may aid in understanding how SpylA participates in
regulating both normal and abnormal development.
CA 02738195 2011-03-23
WO 2010/034128 PCT/CA2009/001372
REFERENCES
1. Anan, T., Y. Nagata, H. Koga, Y. Honda, N. Yabuki, C. Miyamoto, A.
Kuwano, I. Matsuda, F. Endo, H. Saya, and M. Nakao. 1998. Human
ubiquitin-protein ligase Nedd4: expression, subcellular localization and
selective
interaction with ubiquitin-conjugating enzymes. Genes Cells 3:751-63.
2. Barnes, E. A., L. A. Porter, J. L. Lenormand, R. W. Dellinger, and D.
J. Donoghue. 2003. Human Spy 1 promotes survival of mammalian cells
following DNA damage. Cancer Res 63:3701-7.
3. Blom, N., S. Gammeltoft, and S. Brunak. 1999. Sequence and structure-
based prediction of eukaryotic protein phosphorylation sites. J Mol Biol
294:1351-62.
4. Chen, C., A. K. Seth, and A. E. Aplin. 2006. Genetic and expression
aberrations of E3 ubiquitin ligases in human breast cancer. Mol Cancer Res
4:695-707.
5. Cheng, A., W. Xiong, J. E. Ferrell, Jr., and M. J. Solomon. 2005.
Identification and Comparative Analysis of Multiple Mammalian Speedy/Ringo
Proteins. Cell Cycle 4.
6. Ferby, I., M. Blazquez, A. Palmer, R. Eritja, and A. R. Nebreda.
1999. A novel p34(cdc2)-binding and activating protein that is necessary and
sufficient to trigger G(2)/M progression in Xenopus oocytes. Genes Dev
13:2177-89.
7. Ferrara, N. 2005. VEGF as a therapeutic target in cancer. Oncology 69
Suppl 3:11-6.
8. Gastwirt, R. F., D. A. Slavin, C. W. McAndrew, and D. J. Donoghue.
2006. Spyl expression prevents normal cellular responses to DNA damage:
Inhibition of apoptosis and checkpoint activation. J Biol Chem.
36
CA 02738195 2011-03-23
WO 2010/034128 PCT/CA2009/001372
9. Giatromanolaki, A., M. I. Koukourakis, E. Sivridis, G. Chlouverakis,
E. Vourvouhaki, H. Turley, A. L. Harris, and K. C. Gatter. 2007. Activated
VEGFR2/KDR pathway in tumour cells and tumour associated vessels of
colorectal cancer. Eur J Clin Invest.
10. Gutierrez, G. J., A. Vogtlin, A. Castro, I. Ferby, G. Salvagiotto, Z.
Ronai, T. Lorca, and A. R. Nebreda. 2006. Meiotic regulation of the CDK
activator RINGO/Speedy by ubiquitin-proteasome-mediated processing and
degradation. Nat Cell Biol 8:1084-94.
11. Hamilton, M. H., I. Tcherepanova, J. M. Huibregtse, and D. P.
McDonnell. 2001. Nuclear import/export of hRPFI/Nedd4 regulates the
ubiquitin-dependent degradation of its nuclear substrates. J Biol Chem
276:26324-31.
12. Harvey, K. F., and S. Kumar. 1999. Nedd4-like proteins: an emerging
family of ubiquitin-protein ligases implicated in diverse cellular functions.
Trends Cell Biol 9:166-9.
13. Hershko, A., and A. Ciechanover. 1998. The ubiquitin system. Annu
Rev Biochem 67:425-79.
14. Hershko, A., and A. Ciechanover. 1992. The ubiquitin system for
protein degradation. Annu Rev Biochem 61:761-807.
15. Ingham, R. J., G. Gish, and T. Pawson. 2004. The Nedd4 family of E3
ubiquitin ligases: functional diversity within a common modular architecture.
Oncogene 23:1972-84.
16. Karaiskou, A., L. H. Perez, I. Ferby, R. Ozon, C. Jessus, and A. R.
Nebreda. 2001. Differential regulation of Cdc2 and Cdk2 by RINGO and
cyclins. J Biol Chem 276:36028-34.
37
CA 02738195 2011-03-23
WO 2010/034128 PCT/CA2009/001372
17. Laine, A., and Z. Ronai. 2007. Regulation of p53 localization and
transcription by the HECT domain E3 ligase WWP1. Oncogene 26:1477-83.
18. Lenormand, J. L., R. W. Dellinger, K. E. Knudsen, S. Subramani,
and D. J. Donoghue. 1999. Speedy: a novel cell cycle regulator of the G2/M
transition. Embo J 18:1869-77.
19. McAndrew, C. W., R. F. Gastwirt, A. N. Meyer, L. A. Porter, and D.
J. Donoghue. 2007. Spyl enhances phosphorylation and degradation of the cell
cycle inhibitor p27. Cell Cycle 6:1937-45.
20. Miller, B. S., and D. Yee. 2005. Type I insulin-like growth factor
receptor as a therapeutic target in cancer. Cancer Res 65:10123-7.
21. Murdaca, J., C. Treins, M. N. Monthouel-Kartmann, R. Pontier-Bres,
S. Kumar, E. Van Obberghen, and S. Giorgetti-Peraldi. 2004. Grb10 prevents
Nedd4-mediated vascular endothelial growth factor receptor-2 degradation. J
Biol
Chem 279:26754-61.
22. Murillas, R., K. S. Simms, S. Hatakeyama, A. M. Weissman, and M.
R. Kuehn. 2002. Identification of developmentally expressed proteins that
functionally interact with Nedd4 ubiquitin ligase. J Biol Chem 277:2897-907.
23. Nurse, P. M. 2002. Nobel Lecture. Cyclin dependent kinases and cell
cycle control. Biosci Rep 22:487-99.
24. Porter, L. A., R. W. Dellinger, J. A. Tynan, E. A. Barnes, M. Kong, J.
L. Lenormand, and D. J. Donoghue. 2002. Human Speedy: a novel cell cycle
regulator that enhances proliferation through activation of Cdk2. J Cell Biol
157:357-66.
25. Porter, L. A., M. Kong, and D. J. Donoghue. 2003. Speedy interacts
with p27Kipl to allow Gl/S progression. Mol Biol Cell 14:3664-3674.
38
CA 02738195 2011-03-23
WO 2010/034128 PCT/CA2009/001372
26. Puputti, M., 0. Tynninen, H. Sihto, T. Blom, H. Maenpaa, J. Isola, A.
Paetau, H. Joensuu, and N. N. Nupponen. 2006. Amplification of KIT,
PDGFRA, VEGFR2, and EGFR in gliomas. Mol Cancer Res 4:927-34.
27. Robinson, P. A., and H. C. Ardley. 2004. Ubiquitin-protein ligases. J
Cell Sci 117:5191-4.
28. Rossi, M., V. De Laurenzi, E. Munarriz, D. R. Green, Y. C. Liu, K. H.
Vousden, G. Cesareni, and G. Melino. 2005. The ubiquitin-protein ligase Itch
regulates p73 stability. Embo J 24:836-48.
29. Ryden, L., B. Linderholm, N. H. Nielsen, S. Emdin, P. E. Jonsson, and
G. Landberg. 2003. Tumor specific VEGF-A and VEGFR2/KDR protein are co-
expressed in breast cancer. Breast Cancer Res Treat 82:147-54.
30. Shearwin-Whyatt, L., H. E. Dalton, N. Foot, and S. Kumar. 2006.
Regulation of functional diversity within the Nedd4 family by accessory and
adaptor proteins. Bioessays 28:617-28.
31. Sutterluty, H., E. Chatelain, A. Marti, C. Wirbelauer, M. Senften, U.
Muller, and W. Krek. 1999. p45SKP2 promotes p27Kip 1 degradation and
induces S phase in quiescent cells. Nat Cell Biol 1:207-14.
32. Vecchione, A., A. Marchese, P. Henry, D. Rotin, and A. Morrione.
2003. The Grb10/Nedd4 complex regulates ligand-induced ubiquitination and
stability of the insulin-like growth factor I receptor. Mol Cell Biol 23:3363-
72.
33. Wang, X., L. C. Trotman, T. Koppie, A. Alimonti, Z. Chen, Z. Gao, J.
Wang, H. Erdjument-Bromage, P. Tempst, C. Cordon-Cardo, P. P. Pandolfi,
and X. Jiang. 2007. NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN.
Cell 128:129-39.
34. Yamano, H., J. Gannon, and T. Hunt. 1996. The role of proteolysis in
cell cycle progression in Schizosaccharomyces pombe. Embo J 15:5268-79.
39
CA 02738195 2011-03-23
WO 2010/034128 PCT/CA2009/001372
35. Yamano, H., J. Gannon, H. Mahbubani, and T. Hunt. 2004. Cell
cycle-regulated recognition of the destruction box of cyclin B by the APC/C in
Xenopus egg extracts. Mol Cell 13:137-47.
36. Zucchi, I., E. Mento, V. A. Kuznetsov, M. Scotti, V. Valsecchi, B.
Simionati, E. Vicinanza, G. Valle, S. Pilotti, R. Reinbold, P. Vezzoni, A.
Albertini, and R. Dulbecco. 2004. Gene expression profiles of epithelial cells
microscopicallyisolated from a breast-invasive ductal carcinoma and a nodal
metastasis. Proc Natl Acad Sci U S A 101:18147-52.