Note: Descriptions are shown in the official language in which they were submitted.
WO 2010/117436 PCT/US2010/001019
HYDROPYROLYSIS OF BIOMASS FOR PRODUCING HIGH
QUALITY LIQUID FUELS
BACKGROUND OF THE INVENTION
Field of the Invention
[00011 This invention relates to an integrated process for thermochemically
transforming biomass into high quality liquid fuels. In one aspect, this
invention relates to
a substantially self-sustaining process for creating high quality liquid fuels
from biomass.
In another aspect, this invention relates to a multi-stage hydropyrolysis
process for creating
high quality liquid fuels from biomass. In another aspect, this invention
relates to a
hydropyrolysis process for transforming biomass into high quality liquid fuels
in which all
of the process fluids are provided by the biomass. In another aspect, this
invention relates
to a hydropyrolysis process for transforming biomass into high quality liquid
fuels in which
the process outputs are substantially only liquid product and CO2.
Description of Related Art
[00021 Conventional pyrolysis of biomass, typically fast pyrolysis, does not
utilize
or require H2 or catalysts and produces a dense, acidic, reactive liquid
product that contains
water, oils, and char formed during the process. Because fast pyrolysis is
most typically
carried out in an inert atmosphere, much of the oxygen present in biomass is
carried over into
the oils produced in pyrolysis, which increases their chemical reactivity. The
unstable liquids
produced by conventional pyrolysis tend to thicken over time and can also
react to a point
where hydrophilic and hydrophobic phases form. Dilution ofpyrolysis liquids
with methanol
or other alcohols has been shown to reduce the activity and viscosity of the
oils, but this
approach is not considered to be practical or economically viable, because
large amounts of
unrecoverable alcohol would be required to produce and transport large amounts
of pyrolysis
liquids.
[00031 In conventional pyrolysis carried out in an inert environment, the
water
miscible liquid product is highly oxygenated and reactive, with total acid
numbers (TAN) in
the range of 100-200, has low chemical stability for polymerization, is
incompatible with
petroleum hydrocarbons due to water miscibility and very high oxygen content,
on the order
of about 40% by weight, and has a low heating value. As a result, transport
and utilization
of this product are problematic and it is difficult to upgrade this product to
a liquid fuel due
1
WO 2010/117436 PCT/US2010/001019
to the retrograde reactions that typically occur in conventional pyrolysis and
in conventional
fast pyrolysis. In addition, the removal of char generated by conventional
pyrolysis from the
liquid pyrolysis product presents a technical challenge due to the large
amounts of oxygen
and free radicals in the pyrolysis vapors which remain highly reactive and
form a pitch-like
material when they come in intimate contact with char particles on the surface
of a filter.
Consequently, filters used to separate the char from the hot pyrolysis vapors
blind quickly
due to the reactions of char and oil that occur on and within the layer of
char on the surface
of the filter.
[0004] The upgrading of pyrolysis oils produced by conventional fast pyrolysis
through hydroconversion consumes too much H2, and extreme process conditions
make it
uneconomical. The reactions are inherently out of balance due to the high
pressures required,
thereby creating too much water and consuming too much H2. In addition,
hydroconversion
reactors often plug due to coke precursors present in the pyrolysis oils or
from coke product
as a result of catalysis.
[0005] In general, hydropyrolysis is a catalytic pyrolysis process carried out
in the
presence of molecular hydrogen. Typically, the objective of conventional
hydropyrolysis
processes has been to maximize liquid yield in one step, and even in one known
case in
which a second stage reaction was added, the objective was to maximize yield
while
obtaining high oxygen removal. However, even this approach compromises
economy,
creates a system which requires an external source of H2, and must be carried
out at
excessive internal pressures. In addition to requiring a continuous input of
hydrogen, such
conventional hydropyrolysis processes produce excessive H2O which must then be
disposed
of.
SUMMARY OF THE INVENTION
[0006] Accordingly, it is one object of this invention to provide a self-
sustaining,
balanced process for conversion of biomass to liquid product using
hydropyrolysis. By self-
sustaining, we mean that, once initiated, the process requires no input of
additional reactants,
heat, or energy from external sources.
[0007] It is another object of this invention to provide a process for
conversion of
biomass to a liquid product using hydropyrolysis wherein the total output of
the overall
process is substantially only liquid product and CO2. As used herein, the term
"liquid
2
WO 2010/117436 PCT/US2010/001019
product" refers to hydrocarbon products, typically -C5 + liquids, produced by
the process of
this invention.
[0008] These and other objects of this invention are addressed by a multi-
stage, self-
sustaining process for producing liquid products from biomass in which the
biomass is
hydropyrolyzed in a reactor vessel containing molecular hydrogen and a
deoxygenating
catalyst, producing a partially deoxygenated pyrolysis liquid, char, and first-
stage process
heat. The partially deoxygenated pyrolysis liquid is hydrogenated using a
hydroconversion
catalyst, producing a substantially fully deoxygenated pyrolysis liquid, a
gaseous mixture
comprising CO and light hydrocarbon gases (C, - C4), and second-stage process
heat. The
gaseous mixture is then reformed in a steam reformer, producing reformed
molecular
hydrogen. The reformed molecular hydrogen is then introduced into the reactor
vessel for
the hydropyrolysis of additional biomass.
[0009] To provide a self-sustaining, fully balanced process, the
hydropyrolysis and
hydroconversion steps are operated at conditions under which about 40-60% of
oxygen in
the biomass is converted to H2O and about 40-60% of the oxygen is converted to
CO and
CO2. That is, the ratio of oxygen in H2O produced therein to the oxygen in the
CO and CO2
produced therein equals about 1 (i.e. H2O/(CO + C02) Z 1). Preferably, process
pressures for
the hydropyrolysis and hydroconversion steps are in the range of about 300
psig to about 800
psig and are about the same for both steps. Pressures greater than about 800
psig result in
a higher liquid product yield, which is the driving force behind the operating
parameters
employed by conventional processes for maximizing liquid product yield;
however, such
higher pressures also produce higher amounts of water, as a result of which
the overall
process is driven out of balance, requiring, for example, the introduction of
additional
hydrogen into the hydropyrolysis reactor vessel from an external source to
complete the
process. In addition, the excess water produced at the higher pressures must
then be purified
and disposed of. Preferably, temperatures for the hydropyrolysis and
hydroconversion steps
are in the range of about 650 F to about 900 F.
BRIEF DESCRIPTION OF THE DRAWINGS
[0010] These and other objects and features of this invention will be better
understood from the following detailed description taken in conjunction with
the drawings
wherein:
3
WO 2010/117436 PCT/US2010/001019
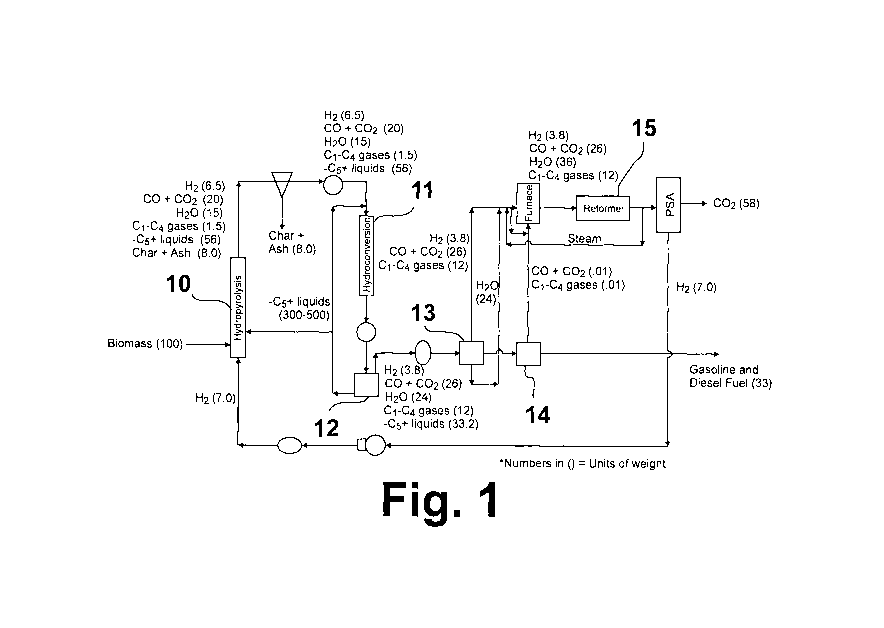
[00111 Fig. 1 is a schematic flow diagram of the self-sustaining process for
producing
liquid fuels from biomass in accordance with one embodiment of this invention.
DETAILED DESCRIPTION OF THE PRESENTLY
PREFERRED EMBODIMENTS
[0012] The process of this invention, shown in Fig. 1, is a compact, balanced,
integrated, multi-stage process for thermochemically transforming biomass into
gasoline plus
diesel liquid product suitable for use as a transportation fuel without the
need for externally
provided H21 CH4, or water. The first reaction stage of this process employs a
pressurized,
catalytically-enhanced, hydropyrolysis reactor vessel 10 to create a low-char,
partially
deoxygenated, hydropyrolysis liquid product from which the char is removed.
The second
reaction stage (subsequent to char removal) employs a hydroconversion reactor
vessel 11 in
which a hydroconversion process is carried out at substantially the same
pressure as the first
reaction stage. The product from the second reaction stage is then cooled and
separated into
liquid and gaseous fractions using high pressure separators 12, 13 and low
pressure separator
14. CO Plus C1 - C4 light gases produced in the two stages are then steam
reformed in a
steam reformer 15 to produce H2 using water which is also produced in the
process. A key
aspect of this invention is that the heat energy required in the process is
supplied by the heat
of reaction of the deoxygenation reaction, which is exothermic, occurring in
both the first and
second stages. Another key aspect of this invention is that the biomass feed
need not be
severely dried and, in fact, the addition of water either in the feed or as a
separate feed is
advantageous to the process because it enhances in-situ H2 formation through a
water-gas-
shift reaction.
[0013] The integrated, balanced process of this invention is carried out under
conditions which balance the levels of decarboxylation, decarbonylation, and
hydrodeoxygenation so that 40-60% of the oxygen present in the biomass is
rejected as CO
and CO2 and the remaining 40-60% of the oxygen in the biomass is rejected as
H2O at the
end of the process where it is easily separated from the hydrophilic liquid
products produced
by the process for use in the reforming process. Overall, after reforming of
the light gases
produced by the first two stages of the process with water produced by the
process, over 95%
of the oxygen in the process is rejected as CO2.
[0014] The unique balancing of reactions is critical to the process of this
invention
4
WO 2010/117436 PCT/US2010/001019
and is achieved through the selection of appropriate catalysts and process
conditions in each
step. Although each step of the process of this invention can yield a variety
of products
depending on the catalyst, pressure, temperature, and time on stream employed,
only when
these processes are integrated in the specific series of steps and process
conditions of this
invention is it possible to provide a balanced process wherein all of the
H21CH4, and water
demands of the overall process are supplied by the biomass, which is critical
for creating a
fungible fuel that can be sold at a reasonable cost.
[00151 In the first step of the process of this invention shown in Fig. 1,
biomass and
molecular hydrogen are introduced into a reactor vessel 10 containing a
deoxygenation
catalyst in which vessel the biomass undergoes hydropyrolysis, producing an
output
comprising a low-char, partially deoxygenated, hydropyrolysis liquid product,
pyrolysis
vapors (C, - C4 gases), H201 CO, C02, and H2 Although any reactor vessel
suitable for
hydropyrolysis may be employed, the preferred reactor vessel is a fluidized
bed reactor. The
hydropyrolysis process employs a rapid heat up of the biomass fuel such that
the residence
time of the pyrolysis vapors in the reactor vessel is less than about 5
minutes. In contrast
thereto, the residence time of the char is relatively long because it is not
removed through the
bottom of the reactor vessel and, thus, must be reduced in particle size until
the particles are
sufficiently small to enable them to be carried out with the vapors exiting
proximate the top
of the reactor vessel.
[00161 Hydropyrolysis is carried out in the reactor vessel at a temperature in
the range
of about 800 F to about 950 F and a pressure in the range of about 300 psig to
about 800
psig. In conventional hydropyrolysis processes as previously noted, the
objective is to
maximize liquid product yield, which requires operation at substantially
higher pressures, e.g.
2000 psig. This is because decarboxylation is favored at lower pressures
whereas
hydrodeoxygenation is favored at higher operating pressures. By maintaining
pressures in
the process of this invention in the range of 300 to 800 psig, most preferably
at about 500
psig, decarboxylation and dehydrodeoxygenation are balanced, but liquid
product yield is
reduced. At higher pressures, hydrodeoxygenation is favored and the reactions
become
unbalanced.
[00171 As previously indicated, in the hydropyrolysis process of this
invention, the
solid biomass feed is rapidly heated, preferably in a hot fluidized bed,
resulting in liquid
WO 2010/117436 PCT/US2010/001019
product yields comparable to and possibly better than yields obtained with
conventional fast
pyrolysis. However, the pyrolysis vapors now are in the presence of a catalyst
and a high
partial pressure of H2 within the fluidized bed, which provides hydrogenation
activity and
also some deoxygenation activity. Hydrogenation activity is very desirable for
preventing
reactive olefins from polymerizing, thereby reducing the formation of unstable
free radicals.
Similarly, deoxygenation activity is important so that the heat of reaction
from pyrolysis is
supplied by the exothermic deoxygenation reaction, thereby obviating the need
for external
heating. The advantage of hydropyrolysis over existing pyrolysis processes is
that
hydropyrolysis avoids the retrograde reactions of pyrolysis, which is usually
carried out in
an inert atmosphere, most certainly in the absence of H2 and usually in the
absence of a
catalyst, thereby promoting the undesirable formation of polynuclear
aromatics, free radicals
and olefinic compounds that are not present in the original biomass.
[0018] The first stage hydropyrolysis process of this invention operates at a
temperature hotter than is typical of a hydroconversion process, as a result
of which the
biomass is rapidly devolatilized. Thus, the process requires an active
catalyst to stabilize the
hydropyrolysis vapors, but not so active that it rapidly cokes. Although any
deoxygenation
catalyst suitable for use in the temperature range of this process may be
employed in the
hydropyrolysis process, catalysts in accordance with preferred embodiments
ofthis invention
are as follows:
[0019] Glass-ceramic catalysts - Glass-ceramic catalysts are extremely strong
and
attrition resistant and can be prepared as thermally impregnated (i.e.
supported) or as bulk
catalysts. When employed as a sulfided NiMo, Ni/NiO, or Co-based glass-ceramic
catalyst,
the resulting catalyst is an attrition resistant version of a readily
available, but soft,
conventional NiMo, Ni/NiO, or Co-based catalyst. Glass-ceramic sulfided NiMo,
Ni/NiO,
or Co-based catalysts are particularly suitable for use in a hot fluidized bed
because these
materials can provide the catalytic effect of a conventional supported
catalyst, but in a much
more robust, attrition resistant form. In addition, due to the attrition
resistance of the
catalyst, the biomass and char are simultaneously ground into smaller
particles as
hydropyrolysis reactions proceed within the reaction vessel. Thus, the char
that is ultimately
recovered is substantially free of catalyst contaminates from the catalyst due
to the extremely
high strength and attrition resistance of the catalyst. The attrition rate of
the catalyst will
6
WO 2010/117436 PCT/US2010/001019
typically be less than about 2 weight % per hour, preferably less than 1
weight % per hour
as determined in a standard, high velocity jet cup attrition test index test.
[0020] Nickel phosphide catalyst - Ni Phosphide catalysts do not require
sulfur to
work and therefore will be just as active in a sulfur-free environment as in
an environment
containing H2S1 COS and other sulfur-containing compounds. Therefore, this
catalyst will
be just as active for biomass which has little or no sulfur present as with
biomass which does
contain sulfur (e.g. corn stover). This catalyst may be impregnated on carbon
as a separate
catalyst or impregnated directly into the biomass feedstock itself.
[0021] Bauxite - Bauxite is an extremely cheap material and, thus, may be used
as
a disposable catalyst. Bauxite may also be impregnated with other materials
such as Ni, Mo,
or be sulfided as well.
[0022] Small size spray-dried silica-alumina catalyst impregnated with low
amounts
of NiMo or CoMo and sulfided to form a low activity hydroconversion catalyst -
Commercially available NiMo or CoMo catalysts are normally provided as large
size 1/8-
1/16 tablets for use in fixed or ebullated beds. In the instant case, NiMo is
impregnated on
spray dried silica alumina catalyst and used in a fluidized bed. This catalyst
exhibits lower
activity with lower NiMo loadings than a conventional NiMo catalyst but would
be of the
right size for use in a fluidized bed.
[0023] In between the hydropyrolysis and hydroconversion processes, char is
removed from the pyrolysis liquid product. Char removal has been a major
barrier in
conventional fast pyrolysis because the char tends to coat filters and react
with oxygenated
pyrolysis vapors to form viscous coatings which can blind hot process filters.
Char may be
removed in accordance with the process of this invention by filtration from
the vapor stream,
or by way of filtering from a wash step - ebullated bed. Backpulsing may be
employed in
removing char from filters, as long as the hydrogen used in the process of
this invention
sufficiently reduces the reactivity of the pyrolysis vapors. Electrostatic
precipitation or a
virtual impactor separator may also be used to remove char and ash particles
from the hot
vapor stream before cooling and condensation of the liquid product.
[0024] In accordance with one embodiment of this invention, hot gas filtration
may
be used to remove the char. In this case, because the hydrogen has stabilized
the free radicals
and saturated the olefins, the dust cake caught on the filters should be more
easily cleaned
7
WO 2010/117436 PCT/US2010/001019
than char removed in the hot filtration of the aerosols produced in
conventional fast
pyrolysis. In accordance with another embodiment of this invention, the char
is removed by
bubbling first stage product gas through a recirculating liquid. The
recirculated liquid used
is the high boiling point portion of the finished oil from this process and is
thus a fully
saturated (hydrogenated), stabilized oil having a boiling point above 650 F.
Char or
catalyst fines from the first reaction stage are captured in this liquid. A
portion of the liquid
may be filtered to remove the fines and a portion may be recirculated back to
the first stage
hydropyrolysis reactor. One advantage of using a recirculating liquid is that
it provides a way
to lower the temperature of the char-laden process vapors from the first
reaction stage to the
temperature desired for the second reaction stage hydroconversion process
while removing
fine particulates of char and catalyst. Another advantage of employing liquid
filtration is that
the use of hot gas filtration with its attendant, well-documented problems of
filter cleaning
is completely avoided.
[00251 In accordance with one embodiment of this invention, large-size NiMo or
CoMo catalysts, deployed in an ebullated bed, are used for char removal to
provide further
deoxygenation simultaneous with the removal of fine particulates. Particles of
this catalyst
should be large, preferably about 1/8-1/16 inch in size, thereby rendering
them easily
separable from the fine char carried over from the first reaction stage, which
is typically less
than 200 mesh (-70 micrometers).
[00261 After removal of the char, the pyrolysis liquid, together with H21 CO,
C021
H2O, and C, - C4 gases from the first reaction stage hydropyrolysis step is
introduced into a
hydroconversion reactor vessel 11 in which it is subjected to a second
reaction stage
hydroconversion step, which preferably is carried out at a lower temperature
(600-800 F)
than the first reaction stage hydropyrolysis step to increase catalyst life
and at substantially
the same pressure (300 - 800 psig) as the first reaction stage hydropyrolysis
step. The liquid
hourly space velocity (LHSV) of this step is in the range of about 0.3 to
about 0.7. The
catalyst used in this step should be protected from Na, K, Ca, P, and other
metals present in
the biomass which can poison the catalyst, which will tend to increase
catalyst life. This
catalyst also should be protected from olefins and free radicals by the
catalytic upgrading
carried out in the first reaction stage process. Catalysts typically selected
for this step are
high activity hydroconversion catalysts, e.g. sulfided NiMo and sulfided CoMo
catalysts. In
8
WO 2010/117436 PCT/US2010/001019
this reaction stage, the catalyst is used to catalyze a water-gas-shift
reaction of CO+H20 to
make CO2 + H21 thereby enabling in-situ production of hydrogen in the second
reaction stage
reactor 11, which, in turn, reduces the hydrogen required for hydroconversion.
NiMo and
CoMo catalysts both catalyze the water-gas-shift reaction. The objective in
this second
reaction stage is once again to balance the deoxygenation reactions. This
balancing is done
by using relatively low pressures (300-800 psig) along with the right choice
of catalyst. In
conventional hydrodeoxygenation processes, pressures in the range of about
2000 psig to
about 3000 psig are typically employed. This is because the processes are
intended to convert
pyrolysis oils, which are extremely unstable and difficult to process at lower
pressures of H2.
[0027] Following the hydroconversion step, the oil product will be
substantially
totally deoxygenated so that it can be directly utilized as a transportation
fuel, after it is
separated by means of high pressure separators 12, 13 and low pressure
separator 14, by
distillation into gasoline and diesel portions. A key aspect of this process
is to adjust
temperature and pressure and space velocity to balance the level of
decarbonylation,
decarboxylation and hydrodeoxygenation so that all the H2 required for the
process can be
made by reforming the light gases that are produced within the process. If too
much
hydrodeoxygenation occurs, then too much H2 will be required for the process
and the system
will be driven out of balance. Likewise, if too much decarboxylation or
decarbonylation
occurs, too much carbon will be lost to CO2 and CO instead of being converted
into liquid
product, as a result of which liquid yields will be reduced.
[0028] After the hydroconversion step, the effluent therefrom is cooled
substantially
so that gasoline and diesel boiling materials condense and only the light
gases remain in the
vapor phase. These gases (containing CO, CO2, CH4, ethane, propane, butanes,
heptanes,
etc.) are sent to the steam reformer 15 together with water from the process
for conversion
into H2 and CO2. A portion of these gases are burned in a furnace or other
combustor to heat
up the remaining portion of gases to the operating temperature of the steam
reformer, about
1700 F. Steam reformers have a 3/1 steam-to-hydrocarbon ratio in their feed to
push the
reaction equilibrium, but this is far more than the amount required for
reaction. The steam
is recovered and recycled around inside the steam reformer. The CO2 is removed
from the
process by pressure swing absorption (PSA) and the H2 is recirculated back to
the first
reaction stage (hydropyrolysis) of the process. The product liquid is
separated into diesel and
9
WO 2010/117436 PCT/US2010/001019
gasoline fraction which are suitable for use as transportation fuels.
[0029] In addition, this process is also balanced with respect to water so
that enough
water is made in the process to provide all the water needed in the steam
reforming step. In
accordance with one embodiment of this invention, the amount of water employed
is such
that the overall process output contains substantially only CO2 and liquid
products, thereby
avoiding an additional process step for excess water disposal. It will be
appreciated by those
skilled in the art that the use of steam reforming in combination with
hydropyrolysis and
hydroconversion steps as set forth herein only makes sense where the objective
is to provide
a self-sustaining process in which the ratio of 02 in H2O to 02 in CO and CO2
produced by
the process is about 1Ø In the absence of such an objective, steam reforming
is not
necessary because H2 required for the hydropyrolysis process would still be
provided by
external sources. If one were to employ steam reforming in the absence of the
objectives
stated herein, one would not end up with the self-sustaining process of this
invention in
which the process output consists essentially of liquid product and CO2.
[0030] In accordance with one embodiment of this invention, the heat generated
in
the second reaction stage may be used to supply all or part of the heat needed
to drive the
hydropyrolysis process in the first reaction stage. In accordance with one
embodiment of this
invention, the process also employs recirculation of the heavy finished
products as a wash
liquid in the second step as stated herein above to capture process fines
exiting the first stage
pyrolysis reactor and control the heat of reaction. In accordance with one
embodiment of this
invention, this liquid is also recirculated to the hydroconversion and
possibly to the first stage
hydropyrolysis step to regulate the generation of heat in each step. The rate
of recirculation
is preferably in the range of about 3-5 times the biomass feed rate. This is
necessary because
hydrodeoxygenation is a strongly exothermic reaction.
[0031] In accordance with one embodiment of this invention, the biomass feed
is a
high lipid containing biomass such as algae, enabling production of the same
deoxygenated
diesel oil which would be made from lipids extracted from the algae plus
additional gasoline
and diesel which can be made from the remainder of the algae biomass. This is
particularly
attractive because lipid extraction is expensive. By contrast, conventional
fast pyrolysis of
algae biomass would be very unattractive because the uncontrolled thermal
reactions
characteristic of fast pyrolysis would degrade these lipids. Thus, the
integrated process of this
WO 2010/117436 PCT/US2010/001019
invention is ideal for algae conversion because it can be carried out on algae
which are
usually only partially dewatered and still produce high quality diesel and
gasoline product.
[0032] The process of this invention provides several distinct advantages over
conventional fast pyrolysis-based processes in that it produces a negligible
to low-char,
partially deoxygenated, stabilized product from which residual char can be
easily separated
by hot gas filtration or contacting with a recirculated liquid; clean, hot
hydropyrolysis oil
vapors can be directly upgraded to a final product in a close-coupled second
catalytically-
enhanced process unit operated at almost the same pressure as was employed
upstream; and
upgrading is carried out quickly before degradation can occur in the vapor
produced from the
hydropyrolysis step.
[0033] The liquid product produced by this process should contain less than 5%
oxygen and preferably less than 2% oxygen with a low total acid number (TAN)
and exhibit
good chemical stability to polymerization or a reduced tendency to reactivity.
In the preferred
embodiment of this invention wherein the total oxygen content of the product
is reduced
below 2%, the water and hydrocarbon phases will easily separate out in any
normal
separation vessel because the hydrocarbon phase has become hydrophobic. This
is a
significant advantage when compared to conventional pyrolysis in which the
water is
miscible with and mixed in with the highly oxygenated pyrolysis oil. Table 1
presents an
estimated material balance for a balanced hydropyrolysis + hydroconversion
process in
accordance with this invention utilizing a mixed hardwood feed. Because the
fungible fuels
produced in the proposed process have low oxygen content, any excess water
produced from
this process is relatively free of dissolved hydrocarbons and will likely
contain less than
2000ppm dissolved total organic carbon (TOC), rendering it suitable for
irrigation in and
areas. Additionally, the finished hydrocarbon product is now easily
transportable, has low
total acid number (TAN), and excellent chemical stability. In conventional
fast pyrolysis, the
pyrolysis oils typically contain 50-60% oxygen in the form of oxygenated
hydrocarbons and
25% dissolved water. Therefore, final products transportation costs for the
integrated
hydropyrolysis + hydroconversion process of this invention are less than half
of the costs for
conventional fast pyrolysis. Furthermore, water produced in the proposed
process becomes
a valuable byproduct especially for and regions.
11
WO 2010/117436 PCT/US2010/001019
Table 1. Estimated Material Balance for a Balanced Hydropyrolysis +
Hydroconversion Process Utilizing a Mixed Hardwood Feed*
Hydropyrolysis + Overall system process
hydroconversion balance, Wt% balance, Wt%
Biomass feed 100 100
H2 feed 3.7 -
Gasoline + diesel product 29 29
Char product 8 8
Water 22.5 .7
CO2 27.5 59.4
Hydrocarbon gas 16.7 2.9
* All H2 is made by reforming light gases and no external natural gas is
required
[00341 While in the foregoing specification this invention has been described
in
relation to certain preferred embodiments thereof, and many details have been
set forth for
purpose of illustration, it will be apparent to those skilled in the art that
the invention is
susceptible to additional embodiments and that certain of the details
described herein can be
varied considerably without departing from the basic principles of the
invention.
12