Note: Descriptions are shown in the official language in which they were submitted.
CA 02759044 2017-01-24
Hydroconyersion Multi-Metallic Catalyst and Method for Making Thereof
CROSS-REFERENCE TO RELATED APPLICATIONS
[001] This application claims priority to US Patent Application Serial Nos.
12/432,730, 12/432,728, and 12/432,723 all with a filing date of April 29,
2009. This
application claims priority to and benefits from the foregoing.
BACKGROUND
[002] The invention relates generally to a hydroprocessing catalyst precursor,
processes for preparing the catalyst precursor, multi-metallic catalysts
prepared using the
catalyst precursor, and hydroconversion processes employing the multi-metallic
catalysts.
TECHNICAL FIELD
[003] The petroleum industry is increasingly turning to heavy crudes, resids,
coals and tar sands, i.e., lower grade hydrocarbon ("heavy oil"), as sources
for feedstocks.
The upgrading or refining of these feedstocks is accomplished by treating the
feedstocks
with hydrogen in the presence of catalysts to effect conversion of at least a
portion of the
feeds to lower molecular weight hydrocarbons, or to effect the removal of
unwanted
components, or compounds, or their conversion to innocuous or less undesirable
compounds.
[004] The pore structure of catalysts is usually formed in the crystallization
stage
or in subsequent treatment. Depending on their predominant pore size, the
solid
materials are classified according to size into three categories; micropores
(dimension
smaller than 3.5 nm), mesopores (dimension ranging from 3.5 ¨ 500 nm) and
macropores
(dimension larger than 500 nm). The use of macroporous solids as adsorbents
and
catalysts is relatively limited due to their low surface area and large non-
uniform pores.
Microporous and mesoporous solids, however, are widely used in adsorption,
separation
technology and catalysis. Owing to the need for higher accessible surface area
and pore
volume for efficient chemical processes, there is a growing demand for new
highly stable
mesoporous catalysts. However, a catalyst that is highly porous does not
necessarily
mean that the catalyst has a lot of surface area. The catalyst may be too
porous, having
CA 02759044 2017-01-24
very little in terms of surface area and correspondingly, low catalytic
activity in terms of
reactive sites.
[005] It is known in the prior art for the making of zeolites and supported
sulfide
catalysts with mesoporous structure for increased surface area. There is still
a need for a
bulk / unsupported catalyst for use in the hydroconversion of lower grade
hydrocarbon
with improved performance, i.e., providing high yield conversions with optimum
porosity
and surface area. There is also a need for a bulk multi-metallic catalyst
having sufficient
pore volume / size for hydrotreating heavy oil feeds.
SUMMARY
[006] In one aspect, the invention relates to a stable bulk multi-metallic
catalyst
formed from a catalyst precursor having a Type IV isotherms with a H3-type
hysteresis
loop. In one embodiment, the mesopores are characterized as being tunable. In
another
embodiment, the catalyst precursor is characterized as having a poorly
crystalline
structure with disordered stacking layers, i.e., the stacking of the layers is
highly random.
[007] In another aspect, the invention relates to a method for making a stable
bulk multi-metallic catalyst formed from a catalyst precursor having a Type TV
isotherm
with a H3-type hysteresis loop. The manufacturing method comprises: a) forming
a
precipitate comprising at least a promoter metal precursor, at least a Group
VIB metal
precursor, optionally a ligating agent, and optionally at least a diluent,
wherein the
promoter metal precursor is selected from Group VIII, Group JIB, Group IIA,
Group IVA
and combinations thereof; b) removing at least 50% of liquid from the
precipitate
forming a filter cake; c) adding to the filter cake at least one of a shaping
aid agent, a pore
forming agent, a peptizing agent, a diluent, and combinations thereof, forming
a batch
mixture; d) shaping the batch mixture into a shaped catalyst precursor via any
of
pelletizing, extrusion, tableting, molding, tumbling, pressing, spraying and
spray drying;
and b) sulfiding the shaped catalyst precursor forming a bulk multi-metallic
catalyst. In
one embodiment, the amount of ligating agent is controlled to vary or tune the
mesopores
of the catalyst precursor. In another embodiment, the additives to the shaping
step are
varied and controlled to tune the mesopores of the catalyst precursor.
[007a] In another aspect, there is provided a catalyst precursor, upon
sulfidation,
forms a bulk multi-metallic catalyst for hydrotreating a hydrocarbon feed
under
2
=
= CA 2759044 2017-03-09
=
hydroprocessing conditions, the catalyst precursor is formed by reacting to
form a
precipitate: at least two Group VIB metal compounds; at least a promoter metal
compound; at least a ligating agent L; optionally at least a diluent, the
promoter metal
compound is nickel and the at least two Group VIB metal compounds are
molybdenum
and tungsten; wherein the precipitate is formed without addition of any sulfur
compounds
to the reaction; wherein the catalyst precursor is a hydroxide; and wherein
steps before
sulfiding the catalyst precursor to form the bulk multi-metallic catalyst are
carried out at a
temperature of 200 C or less for the catalyst precursor to remain a hydroxide
before
sulfidation to form the bulk multi-metallic catalyst; and wherein the catalyst
precursor is
characterized as having a poorly crystalline structure with disordered
stacking layers with
a type IV adsorption-desorption isotherms of nitrogen, with a hysteresis
starting point
value P/P0 of about 0.35.
[0071)] In an aspect, the optional at least a diluent is selected from the
group of
titania, sodium silicate, potassium silicate, silica gels, silica sols,
hydronium- or
ammonium-stabilized silica sols, sodium aluminate, potassium aluminate,
aluminum
sulfate, aluminum nitrate, magnesium aluminosilicate clay, magnesium metal,
magnesium
hydroxide, magnesium halides, magnesium sulfate, magnesium nitrate, zirconia,
cationic
clay, anionic clays, zinc oxide, zinc sulfide, tetraethyl orthosilicate,
silicic acid, niobia,
titania, and combinations thereof.
2a
CA 02759044 2011-10-17
WO 2010/127130
PCT/US2010/032989
BRIEF DESCRIPTION OF THE DRAWINGS
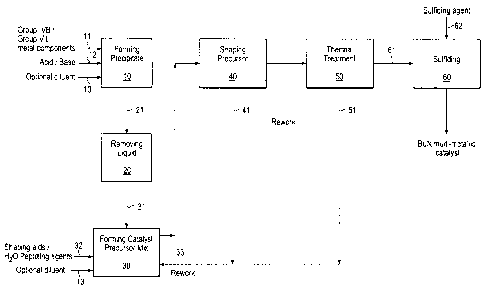
[008] Figure 1 is block diagram showing an embodiment of a process for making
a multi-metallic catalyst from a mesoporous catalyst precursor having a Type
IV isotherm
with a H3-type hysteresis loop.
[009] Figure 2 is a graph illustrating the N2 adsorption (+) and desorption (-
e-)
isotherrns of one embodiment of the catalyst precursor.
[010] Figure 3 is a graph illustrating the N2 adsorption (+) and desorption (-
e-)
isotherms of the catalyst precursor of Figure 2 in the relative pressure range
of 0.35 to
1.00.
[011] Figure 4 is a graph illustrating the N2 adsorption (+) and desorption (-
e-)
isotherms of another embodiment of the catalyst precursor, showing a broad
type H3
desorption hysteresis loop.
[012] Figure 5 is a graph illustrating the N2 adsorption (+) and desorption (-
8-)
isotherms of the catalyst precursor of Figure 4 in the relative pressure range
of 0.35 to
1.00.
[013] Figure 6 is a graph illustrating the N2 adsorption (+) of a catalyst
precursor that does not have a type IV isotherms.
DETAILED DESCRIPTION
[014] The following terms will be used throughout the specification and will
have the following meanings unless otherwise indicated.
[015] SCF / BBL (or scf / bbl, or scfb or SCFB) refers to a unit of standard
cubic
foot of gas (N2, H2, etc.) per barrel of hydrocarbon feed.
[016] LHSV means liquid hourly space velocity.
[017] The Periodic Table referred to herein is the Table approved by IUPAC and
the U.S. National Bureau of Standards, an example is the Periodic Table of the
Elements
by Los Alamos National Laboratory's Chemistry Division of October 2001.
[018] As used here, the term "bulk catalyst" may be used interchangeably with
"unsupported catalyst," meaning that the catalyst composition is NOT of the
conventional
catalyst form which has a preformed, shaped catalyst support which is then
loaded with
metals via impregnation or deposition catalyst. In one embodiment, the bulk
catalyst is
formed through precipitation. In another embodiment, the bulk catalyst has a
binder
3
CA 02759044 2011-10-17
WO 2010/127130
PCT/US2010/032989
incorporated into the catalyst composition. In yet another embodiment, the
bulk catalyst
is formed from metal compounds and without any binder.
[019] As used herein, the phrases "one or more of" or "at least one of" when
used to preface several elements or classes of elements such as X, Y and Z or
X1-X, Y1-
Yn and Z1.-Z., is intended to refer to a single element selected from X or Y
or Z, a
combination of elements selected from the same common class (such as Xi and
X2), as
well as a combination of elements selected from different classes (such as X1,
Y2 and Zn).
[02.0] As used herein, "hydroconversion" or "hydroprocessing" is meant any
process that is carried out in the presence of hydrogen, including, but not
limited to,
to methanation, water gas shift reactions, hydrogenation, hydrotreating,
hydrodesulphurization, hydrodenitrogcnation, hydrodemetallation,
hydrodcaromatization,
hydroisomerization, hydrodewaxing and hydrocracking including selective
hydrocracking. Depending on the type of hydroprocessing and the reaction
conditions,
the products of hydroprocessing can show improved viscosities, viscosity
indices,
saturates content, low temperature properties, volatilities and
depolarization, etc.
[021] As used herein, 700 F+ conversion rate refers to the conversion of an
oil
feedstock having a boiling point of greater than 700 F+ to less than 700 F
(371. C)
boiling point materials in a hydroconvcrsion process, computed as (100% * (wt.
%
boiling above 700 F materials in feed - wt. % boiling above 700 F materials in
products) / wt. % boiling above 700 F materials in feed)).
[022] As used herein, "I,D50" is the amount of a material, given all at once,
causes the death of 50% (one half) of a group of test animals. LD-50 measures
the short-
term poisoning potential (acute toxicity) of a material with the testing being
done with
smaller animals such as rats and mice (in mg/Kg).
[023] As used herein, "shaped catalyst precursor" means catalyst precursor
formed (or shaped) by spray drying, pelleting, pilling, granulating, beading,
tablet
pressing, bricketting, using compression method via extrusion or other means
known in
the art or by the agglomeration of wet mixtures. The shaped catalyst precursor
can be in
any form or shape, including but not limited to pellets, cylinders, straight
or rifled
(twisted) trilobes, multiholed cylinders, tablets, rings, cubes, honeycombs,
stars, tri-lobes,
quadra-lobes, pills, granules, etc.
[024] Pore porosity and pore size distribution in one embodiment can be
measured using mercury intrusion porosimetry, designed as ASTM standard method
D
4
CA 02759044 2011-10-17
WO 2010/127130 PCT/US2010/032989
4284. In another embodiment, pore porosity and size distribution are measured
via the
nitrogen adsorption method.
[025] Layered or textural porosity is the porosity that can be attributed to
voids
between layers or platters of catalyst precursors. One skilled in the art of
transmission
electron spectroscopy (TEM) can determine the existence of layers or platters
of catalyst
precursors from high resolution TEM images. One skilled in the art of
adsorption can
distinguish and evaluate the layered porosity by the specific adsorption
behavior of the
catalyst precursor. One way to detect and assess layered or textural
mesoporosity is
evidenced by the presence of a type IV adsorption-desorption isotherm
exhibiting well-
defined hysteresis loop in the region of relative pressure P/Po > 0.40 (Sing
et al., Pure
App!. Chem., vol. 57, 603-619 (1985)).
[026] The bulk catalyst of the present invention is made from a randomly
stacking layered or textural mesoporous catalyst precursor, i.e., a poorly
crystalline
structured catalyst precursor exhibiting type IV isotherms. In one embodiment,
the
catalyst precursor for forming the bulk catalyst is characterized as having a
H3-type
hysteresis loop.
[027] Catalyst Product: The catalyst precursor with a type IV adsorption-
desorption isotherm is converted into a catalyst (becoming catalytically
active) upon
sulfidation, for subsequent use in hydrodesulfurization (HDS),
hydrodearomatization
(HDA), and hydrodenitrification (HDN) processes. The catalyst precursor can be
a
hydroxide or oxide material, prepared from at least a Promoter metal precursor
and at
least a Group VIB metal precursor.
[028] In one embodiment, the catalyst precursor in the form of a bulk multi-
metallic oxide comprising of at least one Group VIII non-noble material and at
least two
Group VIB metals. In one embodiment, the ratio of Group VIB metal to Group
VIII
non-noble metal ranges from about 10:1 to about 1:10. In another embodiment,
the oxide
catalyst precursor is of the general formula: (X)b(Mo),(W)d 0,; wherein X is
Ni or Co,
thc molar ratio of b: (c+d) is 0,5/1 to 3/1, thc molar ratio of c: d is >
0.01/1, and z = [2b+
6 (c + d)]/2. In one embodiment, the oxide catalyst precursor further
comprises one or
more ligating agents L.
[029] The term "ligand" may be used interchangeably with "ligating agent,"
"chelating agent" or "complexing agent" (or chelator, or chelant), referring
to an additive
that combines with metal ions, e.g., Group VIB and / or Promoter metals,
forming a larger
5
CA 02759044 2011-10-17
WO 2010/127130 PCT/US2010/032989
complex, e.g., a catalyst precursor, and facilitating thc tuning or adjustment
of the
porosity of the mesopores.
[030] In one, the catalyst precursor is in the form of a hydroxide comprising
of at
least one Group VHI non-noble material and at least two Group VIB metals. In
another
embodiment, the hydroxide compound is of the -general formula A,[(MP) (OH) x
(L)n ylz
(Mv1B0.4). wherein A is one or more monovalent cationic species, M refers to
at least a
metal in their elemental or compound form, and L refers to one or more
ligating agents.
[031] In one embodiment, the catalyst precursor is prepared from a process
with
the inclusion of at least a diluent, for the precursor to have the formula A,
RmilA)s(mviii)(
(Al)õ (OH)v (Si(i_y)Aly02), (Mv1B04), wherein A is one or more monovalent
cationic species, MI" is one or more group IIA metals, Mvin is one or more
Group VIII
metals, Al is aluminum, L is one or more ligating agents, (Si(l_y)Aly02) is a
silica-alumina
moiety, Mvm is one or more Group VIB metals with the atomic ratio of Nem: Mvi5
between 100:1 and 1:100. In one embodiment, A is at least one of an alkali
metal cation,
an ammonium, an organic ammonium and a phosphonium cation. In one embodiment,
A
is selected from monovalent cations such as NH4+, other quaternary ammonium
ions,
organic phosphonium cations, alkali metal cations, and combinations thereof
[032] In one embodiment, L is one or more optional ligating agents. In another
embodiment, L is charge neutral or has a negative charge n <= 0. In another
embodiment, L is a non-toxic organic oxygen containing ligating agent with an
LD50 rate
(as single oral dose to rats) of greater than 500 mg/Kg. The term "charge-
neutral" refers
to the fact that the catalyst precursor carries no net positive or negative
charge.
Examples include but are not limited to polydentate as well as monodentate,
e.g., NH3 as
well as alkyl and aryl amines; carboxylates, carboxylic acids, aldehydes,
ketones, the
enolate forms of aldehydes, the enolate forms of ketones and hemiacetals;
organic acid
addition salts such as formic acid, acetic acid, propionic acid, maleic acid,
malic acid,
cluconic acid, fumaric acid, succinic acid, tartaric acid, citric acid, oxalic
acid, glyoxylic
acid, aspartic acid, alkanc sulfonic acids, aryl sulfonic acids;
arylcarboxylic acids;
carboxylate containing compounds; and combinations thereof
[033] MP is at least a promoter metal. In one embodiment, Mr has an oxidation
state of either +2 or +4 depending on the Promoter metal(s) being employed. MP
is
selected from Group VIII, Group IIB, Group IIA, Group IVA and combinations
thereof.
In one embodiment, MP is at least a Group VIII metal and MP has an oxidation
state P of
6
CA 02759044 2011-10-17
WO 2010/127130
PCT/US2010/032989
+2. In another embodiment, MP is selected from Group IIB, Group IVA and
combinations thereof. In one embodiment, the Promoter metal MP is at least a
Group
VIII metal with MP having an oxidation state of +2 and the catalyst precursor
is of the
formula Av[(MP) (OH)õ (L)" yl, (MVIB04) to have (v -2+2 z¨x* z+n* y* z) = 0.
In
one embodiment, the Promoter metal MP is a mixture of two Group VIII metals
such as
Ni and Co. In yet another embodiment, MP is a combination of three metals such
as Ni,
Co and Fe. In one embodiment where MP is a mixture of two group IIB metals
such as Zn
and Cd, the catalyst precursor is of the formula A,[(ZnaCcl.) (OFI)x (L)A,
(mviso4).
In
yet another embodiment, MP is a combination of three metals such as Zn, Cd and
Hg, and
the catalyst precursor is of the formula Av[(Zn.Cda.Hga-) (OH),, (L). di 04
VIB 0)
[034] In one embodiment, the Promoter metal MP is selected from the group of
11B and VIA metals such as zinc, cadmium, mercury, germanium, tin or lead, and
combinations thereof, in their elemental, compound, or ionic form. In yet
another
embodiment, the Promoter metal MP further comprises at least one of Ni, Co, Fe
and
combinations thereof, in their elemental, compound, or ionic form. In another
embodiment, the Promoter metal is a Group IIA metal compound, selected from
the
group of magnesium, calcium, strontium and barium compounds which are at least
partly
in the solid state, e.g., a water-insoluble compound such as a carbonate,
hydroxide,
fumarate, phosphate, phosphite, sulphide, molybdate, tungstate, oxide, or
mixtures
thereof.
[035] In one embodiment, Mv1B is at least a Group VIB metal having an
oxidation state of +6. In one embodiment, MP Mv1B has an atomic ratio between
100:1
0 <v<2; 0
< z. In one embodiment, Mv1B is molybdenum. In yet another embodiment, mVIB is
a
mixture of at least two Group VIB metals, e.g., molybdenum and tungsten.
[036] Methods for Making Catalyst: Reference will be made to Figure 1, which
is a block diagram schematically illustrating an embodiment of a general
process for
making the bulk catalyst out of a catalyst precursor exhibiting type IV
adsorption
isotherms.
[037] Forming a Precipitate or Cogel: The first step 10 in the process is a
precipitation or cogellation step, which involves reacting in a mixture of the
metal
precursors 11, e.g., Promoter metal component(s) and the Group VIB metal
component to
obtain a precipitate or cogel. The term "cogel" refers to a co-precipitate (or
precipitate)
7
CA 02759044 2017-01-24
of at least two metal compounds. The metal precursors can be added to the
reaction mixture as a
solid, in solution, suspension, or a combination thereof. If soluble salts are
added as such, they
will dissolve in the reaction mixture and subsequently be precipitated or
cogelled, or forming a
suspension. The solution can be heated optionally under vacuum to effect
precipitation and
evaporation of the liquid.
[038] The precipitation (or cogelation) is carried out at a temperature and pH
under
which the Promoter metal compound and the Group VIB metal compound precipitate
or form a
cogel. In one embodiment, the temperature at which the cogel is formed is
between 25 - 350 C.
In one embodiment, the catalyst precursor is formed at a pressure between 0 to
3000 psig. In a
second embodiment, between 10 to 1000 psig. In a third embodiment, between 30
to 100 psig.
The pH of the mixture can be changed to increase or decrease the rate of
precipitation or
cogelation depending on the desired characteristics of the product. In one
embodiment, the
mixture is left at its natural pH during the reaction step(s). In another
embodiment, the pH is
maintained in the range of 0 - 12. In another embodiment, the pH is maintained
in the range of 7
- 10. Changing the pH can be done by adding base or acid 12 to the reaction
mixture, or adding
compounds, which decompose upon temperature increase into hydroxide ions or Fr
ions that
respectively increase or decrease the pH. In another embodiment, adding
compounds which
participate in the hydrolysis reaction.
[039] In one embodiment, at least a ligating agent L can be optionally added
as one of
the reagents forming the precipitate (prior to the precipitation or cogelation
of the promoter metal
compounds and / or Group VIB metal compounds). In another embodiment, the
litigating agent
L is added after the precipitate is formed. In one embodiment, the ligating
agent L added after the
precipitation step is different from the ligating agent added prior to the
precipitation step.
[040] It should be noted that the mesoporosity of the catalyst precursor can
be
controlled or tuned with the selection of the ligating agent and / or the
amount added. In one
embodiment, it is observed that the incorporation of the ligating agent L
significantly increases
the porosity of the catalyst precursor.
[041] In one embodiment, instead of or in addition to the ligating agent L,
diluent
amounts from 5-95 wt. % of the total composition of the catalyst precursor can
also be added to
this step, depending on the envisaged catalytic application. These materials
can be applied
before or after the precipitation or cogelation of the metal
8
CA 02759044 2011-10-17
WO 2010/127130
PCT/US2010/032989
precursors. Examples include but are not limited to zinc oxide; zinc sulfide;
niobia;
tetraethyl orthosilicate; silicic acid; titania; silicon components such as
sodium silicate,
potassium silicate, silica gels, silica sols, silica gels, hydronium- or
ammonium-stabilized
silica sols, and combinations thereof; aluminum components useful in the
process of the
present invention include, but are not limited to, sodium -aluminate,
potassium aluminate,
aluminum sulfate, aluminum nitrate, and combinations thereof; magnesium
components
such as magnesium aluminosilicate clay, magnesium metal, magnesium hydroxide,
magnesium halides, magnesium sulfate, and magnesium nitrate; zirconia;
cationic clays
or anionic clays such as saponite, bentonite, kaoline, sepiolite or
hydrotalcitc, or mixtures
thereof. In one embodiment, titania is used as a diluent in an amount of
greater than 50
wt. %, on a final catalyst precursor basis (as an oxide or hydroxide).
[042] Liquid Removal: In the next step 20, at least 50 wt. % of liquid
(supernatant / water) is removed from the precipitate (or suspension) via
separation
processes known in the art, e.g., filtering, decanting, centrifuging, etc. In
one
embodiment, liquid in the precipitate is removed via filtration with vacuum
techniques or
equipment known in the art, giving a wet filter cake. A wet filter cake is
generally
defined as filter cake having approximately 10 to 50 wt. % liquid, thus being
generally
free of water or other solvent such as methanol and the like.
[043] In one embodiment, optional drying of the filter cake is performed under
atmospheric conditions or under an inert atmosphere such as nitrogen, argon,
or vacuum,
and at a temperature sufficient to remove water but not removal of organic
compounds.
In one embodiment, the drying is performed at about 50 to 120 C until a
constant weight
of the catalyst precursor is reached. In another embodiment, the drying is
done at a
temperature between 50 C to 200 C for a period ranging from 1A. hour to 6
hours. Drying
can be done via thermal drying techniques known in the art, e.g., flash
drying, belt drying,
oven drying, etc.
[044] Forming Catalyst Precursor Mix For Shaping: In this step 30, the fitter
cake is mixed together with water and other optional materials including but
not limited
to shaping aids 32, peptizing agents, pore forming agents, and diluent
materials 13. In
one embodiment, rework material in the form of filter cake material,
extrudable dough
and / or dry particles / pieces of precursor materials from previous runs can
be optionally
included the materials to form a new batch of catalyst precursor mix. In one
embodiment, the amount of water and / or the amount of! type of optional
materials is
9
CA 02759044 2011-10-17
WO 2010/127130
PCT/US2010/032989
varied to control and / or tune the mesoporosity of the catalyst precursor
formed. In one
embodiment, the addition of water helps increase the surface area of the
catalyst
precursor.
[045] The precursor batch mixture is mixed for a sufficient period of time to
- 5 obtain a mixture that is substantially uniform or homogeneous. The
mixing time depends
on the type and efficiency of the mixing technique, e.g., milling, kneading,
slurry mixing,
dry or wet mixing, or combinations thereof and the mixing apparatus used,
e.g., a pug
mill, a blender, a double-arm kneading mixer, a rotor stator mixer, or a mix
mutter. In
one embodiment, the mixing time ranges from 0.1 to 10 hours. In one
embodiment, a
shaping aid agent is added in a ratio of between 100:1 and 10:1 (wt. %
catalyst precursor
to wt. % shaping aid). Examples of shaping aid agents include but are not
limited to
organic binders of the cellulose ether type and / or derivatives, polyakylene
glycols,
saturated or unsaturated fatty acid (such as politic acid, satiric acid or
oleic acid) or a salt
thereof, a polysaccharide derived acid or a salt thereof, graphite, starch,
alkali stearate,
ammonium stearate, stearic acid, mineral oils, and combinations thereof.
[046] In one embodiment, a peptizing agent may be added to the mixture. The
peptizing agent may be an alkali or an acid, e.g., ammonia, formic acid,
citric acid, nitric
acid, maleie acid, carboxylic acid, etc. In another embodiment, a pore forming
agent is
also added to the mixture along with the rework. Examples of pore forming
agents
include but are not limited to mineral oils, steric acid, polyethylene glycol
polymers,
carbohydrate polymers, methacrylates, cellulose polymers, and carboxylatcs
which
decompose upon being heated. In yet another embodiment, diluent materials can
be
added. The diluent materials added in this step can be the same as or
different from any
diluent materials that may have been added to the step of forming the
precipitate from
metal precursors above.
[047] In one embodiment wherein the catalyst precursor is to be shaped via
pelletizing, extrusion, or pressing, a sufficient amount of water is added to
the mixing
batch to adjust the batch viscosity to a convenient level for plasticizing and
shaping, i.e., a
mixture of dough consistency. In one embodiment, a sufficient amount of water
is added
for the mixture to have between 50 to 90 % solids (L01). In another
embodiment,
between 60 to 70 % solids (LOT).
[048] Shaping Process: In this step 40, the catalyst precursor mix is shaped
into
formed particles, such as spheroids, pills, tablets, cylinders, irregular
extrusions, merely
CA 02759044 2011-10-17
WO 2010/127130
PCT/US2010/032989
loosely bound aggregates or clusters, etc., using any of the methods known in
the art
including pelletizing, extrusion, tableting, molding, tumbling, pressing,
spraying and
spray drying.
[049] In one embodiment, a shaped catalyst precursor is formed via extrusion,
using extrusion equipment known in the art, e.g., single screw extruder, ram
extruder,
twin-screw extruder, etc. In another embodiment, the shaping is done via spray
drying
at an outlet temperature ranging from 100 C to 320 C. In one embodiment,
shaped
catalyst precursor is extruded into extrudate having a diameter from about
1/16 to 1/6 of
an inch. After extrusion the extrudate can be cut to suitable lengths, e.g.,
1/16-inch to
5/16-inch, to produce cylindrical pellets.
[050] Thermal Treatment: In one embodiment, the shaped catalyst precursor
undergoes a thermal treatment step 50. In one embodiment, the catalyst
precursor is air
(or nitrogen) dried in a directly or indirectly heated oven, tray drier, or
belt drier at about
50 C. to 325 C. for about 15 minutes to 24 hours, and wherein the temperature
is room
temperature to drying temperature at a rate of 1-50 C. per minute. In one
embodiment,
the temperature is ramped up at a slow rate of 1-2 C. per minute. In a second
embodiment, air drying is performed at a fast ramp up rate of at least 25 C.
per minute.
In one embodiment, the drying is at a temperature at or below 100 C.
[051] Generally, it is known that the higher temperature of the heat
treatment,
the higher the densities of the catalyst precursor and therefore, upon
sulfidation, resulting
in a catalyst that also has a low shrinkage rate. In some embodiments, low
(less than
10%) volumetric shrinkage is obtained with the thermal treatment at a low
temperature,
e.g., less than 325 C, less than 200 C, and even at a temperature at or below
100 C.
[052] In one embodiment, after an optional thermal treatment, the shaped
catalyst can be optionally calcined at a temperature in the range of about 350
C. to 750 C
in a suitable atmosphere, e.g., inerts such as nitrogen or argon, or steam. In
yet another
embodiment, the calcination is carried out at a temperature between 350 C. to
600 C. In
the calcination process, the catalyst precursor gets converted into an oxide.
In one
embodiment, the oxide catalyst precursor is of the general formula:
(X)b(Mo),(W)d Oz;
wherein X is Ni or Co, the molar ratio of b: (c+d) is 0.5/1 to 3/1, the molar
ratio of c: d is
>0.01/1, and z = [2b + 6 (c + d)1/2.
[053] In one embodiment, the catalyst precursor is nitrogen stable. As used
herein, the term nitrogen stable means that the properties (after the catalyst
precursor is
11
CA 02759044 2011-10-17
WO 2010/127130
PCT/US2010/032989
sulfided to form a catalyst) are not affected by the drying agent, i.e.,
whether drying in a
nitrogen or oxygen environment.
[054] Sulfiding Step: The shaped catalyst precursor (with optional rework
materials) can be sulfided in a sulfiding step 60 to form an active catalyst,
with the use of
at least a sulfiding agent 62 selected from the group of: elemental sulfur by
itself; a
sulfur-containing compound which under prevailing conditions, is decomposable
into
hydrogen sulphide; H2S by itself or H2S in any inert or reducing environment,
e.g., H2.
Examples of sulfiding agents include ammonium sulfide, ammonium polysulfide
(RNI-14)2Sx), ammonium thiosulfate ((NH4)2S203), sodium thiosulfatc (Na2S203),
thiourea CSN2H4, carbon disulfide, dimethyl disulfide (DMDS), dimethyl sulfide
(DMS),
dibutyl polysulfide (DBPS), mercaptanes, tertiarybutyl poly-sulfide (PSTB),
tertiarynonyl
polysulfide (PSTN), and the like. In one embodiment, hydrocarbon feedstock is
used as
a sulfur source for performing the sulfidation of the catalyst precursor.
[055] In the sulfiding step, shaped catalyst precursor is converted into an
active
catalyst upon contact with the sulfiding agent at a temperature ranging from
25 C. to
500 C, from 10 minutes to 15 days, and under a H2-containing gas pressure. The
total
pressure during the sulfidation step can range between atmospheric to about 10
bar
(1MPa). If the sulfidation temperature is below the boiling point of the
sulfiding agent,
the process is generally carried out at atmospheric pressure. Above the
boiling
temperature of the sulfiding agent / optional components (if any), the
reaction is generally
carried out at an increased pressure.
[056] Use of the Catalyst: As catalyst precursors sometimes can be sulfided in-
situ, e.g., in the same hydrotreating reactors during hydrotreatment, catalyst
performance
can be characterized by the properties of the catalyst precursors before
sulfidation.
[057] In one embodiment, the catalyst precursor for preparing the bulk
catalyst is
characterized as having a poorly crystalline structure with disordered
stacking layers, with
a type IV adsorption-desorption isotherms of nitrogen. The point at which the
relative
pressure P/Põ of N2 adsorption and desorption isotherms begins to diverse
defines the
adsorption capacity of the sulfided catalyst product. The N2 adsorption
desorption
isotherms of the catalyst precursor of the invention forms a close hysteresis
cycle which
enclosed area is proportional to the specific volume of the mesopores. The
lower the 13/130
is, the larger the area enclosed by the hysteresis cycle and consequently the
greater the
12
CA 02759044 2011-10-17
WO 2010/127130
PCT/US2010/032989
adsorption capacity. Põ is the N2 saturation pressure. In one embodiment, the
catalyst
precursor has a P/PO hysteresis starting point value of about 0.35.
[058] In one embodiment, the precursor is characterized as having a type H3
hysteresis loop. In one embodiment, the hysteresis loop is characterized as
having a well
developed plateau above P/Po of about 0.55.
[059] The precursor is also characterized as having a mesoporous structure
with
an average pore size (width) ranging from 2 nm to 200 nm in one embodiment;
from 5 to
150 nm in a second embodiment, from 10 nm to 125 nm in another embodiment, and
from 15 nm to 100 nm in a fourth embodiment. Thc pore volume in one embodiment
is
more than 0.01 cm'/g. In yet another embodiment, the pore volume ranges from
0.01 to
0.50 cm/g. In a third embodiment ranging from 0.02 to 0.20 cm3/g, and in a
fourth
embodiment ranging from 0.05 to 0.15 cm'/g. The surface area measured by the
BET
method, using nitrogen as adsorbate, ranges from 25 to 400 m2/g in one
embodiment;
from 40 to 200 m2/g in a second embodiment; and from 60 to 150 m2/g in a third
embodiment.
[060] As the catalyst precursor and the sulfided bulk metallic catalyst formed
therefrom have sufficient mesopore sites and large pore volume to overcome the
diffusion limitations of heavy petroleum feeds, the bulk metallic catalyst in
one
embodiment is particularly suitable for hydrotreating heavy petroleum feeds
having an
atmospheric residue (AR) boiling point in the range of 343 C. (650 F.) - to
454 C. (850
'F.) and particularly above 371 C. (700 F.). Heavy oil feeds having a boiling
point
greater than 343 C. (650T.) are commonly characterized as having relatively
high
specific gravity, low hydrogen-to-carbon ratios, and high carbon residue. They
contain
large amounts of asphaltenes, sulfur, nitrogen and metals, which increase
hydrotreating
difficulty with their large molecular diameter.
[061] In one embodiment, the bulk catalyst formed from the precursor with
disordered stacking layers and a type IV adsorption-desorption isotherms is
characterized
as being very stable. A catalyst's stability can be evaluated based on the
residual
geometric volume shrinkage of the catalyst precursor, measured as the change
in the
geometric volume of the shaped catalyst precursor before and after it is
sulfided. The
volumetric shrinkage measured after the sulfidation step can be used as an
indication of a
catalyst's mechanical integrity under severe hydroprocessing conditions, as
precursors are
often sulfided in-situ in the same reactor as the hydroprocessing reactor. In
one
13 .
CA 02759044 2011-10-17
WO 2010/127130
PCT/US2010/032989
embodiment, the catalyst precursor with a type IV adsorption-desorption
isotherms is
characterized as having a residual geometric volume shrinkage of less than
about 12%
upon exposure to a temperature of at least 100 C. for at least 30 minutes in a
sulfiding
step. In a second embodiment, the volume shrinkage is less than about 10%. In
a third
embodiment, the volume shrinkage is less than about 8%. In a fourth
einbodiment, less
than 5%.
[062] In one embodiment, the bulk catalyst is particularly suited for
hydrotreating heavy petroleum feeds having an average molecular diameter
ranging from
0.9 nm to 1.7 nm (9 to 17 angstrom), providing an HDN conversion level of>
99.99%
(700 F+ conversion), lowering the sulfur level in fraction above 700 F.
boiling point to
less than 20 ppm in one embodiment, and less than 10 ppm in a second
embodiment. In
one embodiment, the bulk catalyst is particularly suited for hydrotreating a
heavy
petroleum feed having an average molecular diameter ranging from 0.9 nm to 1.7
nm.
In yet another embodiment, the bulk catalyst is particularly suitable for
treating a heavy
oil feed having an average molecular weight Mn ranging from 300 to 400 g/mole.
[063] In one embodiment, the precursor for forming the catalyst also exhibits
other desirable properties, including a compact bulk density (CBD) of at most
1.6 g/cc; a
crush strength of at least about 4 lbs; and an attrition loss of less than 7
wt.%. Attrition
loss is the loss to fine amount measured when tumbled one-half hour in a
rotating drum.
In another embodiment, the attrition loss is less than 5 wt. 'A. In a third
embodiment,
the CBD is at most 1.4 g/cc. In a fourth embodiment, the CBD is at most 1.2
g/cc. In a
fifth embodiment, a CBD in the range of 1.2 glee to 1.4 glee. In one
embodiment, the
crush strength is at least 6 lbs. In one embodiment, the catalyst precursor
has a particle
density of equal or less 2.5 glee. In another embodiment, the particle density
is equal or
less than 2.2 g/cc.
[064] The bulk multi-metallic catalyst can be used in virtually all
hydroprocessing processes to treat a plurality of feeds under wide-ranging
reaction
conditions such as temperatures of from 200 to 450 C., hydrogen pressures of
from 15 to
300 bar, liquid hourly space velocities of from 0.05 to 10 If' and hydrogen
treat gas rates
of from 35.6 to 2670 I113 / m3 (200 to 15000 SCF/B ¨ or "Standard Cubic Feet
per Barrel"
of hydrocarbon compound feed to the reactor). The catalyst is also
characterized by
excellent catalytic activity, as giving an almost full HDN conversion rate
(>99.99%) in
the hydrotreating of heavy oil feedstock such as VG0.
14
CA 02759044 2011-10-17
WO 2010/127130
PCT/US2010/032989
[065] EXAMPLES: The following illustrative examples are intended to be non-
limiting. In the examples, the pore structure was characterized by measuring
the N2
adsorption desorption isotherms using standard continuous sorption procedures.
The
specific surface area and the total pore volume can be calculated from the
isotherms
following IUPAC recommendations. The volume of pores corresponding to the
textural
mesopores can be evaluated from the upper inflection point of the low P/Po
hysteresis
loop.
[066] Example 1 - Ni-Mo-W-maleate catalyst precursor. A catalyst precursor
!AA-
of the formula (NH4) l[Ni2.6(OH)2.0g (C4H2..,ri N )0 06 J 1,,v100.35
W0.6504)2} was prepared as
follows: 954.8g of ammonium heptamolybdatc (NH4)6Mo7024 4H20 was dissolved in
4.8L of deionized water at room temperature. The pH of the resulting solution
was
within the range of 2-3. 1334g of ammonium metatungstate (NH4)6142%2040
.4.7H20
was dissolved in 1.3 L water. The molybdate and tungstate solutions were added
to 34.9 L
deionized water. To this mixed molybdate and tungstate solution 2.03 L of a
7.0 wt-%
NH4OH (ammonia) solution was added, and the temperature was increased to 77 C
with
constant stirring. The solution had a pH in the range of 8-10. A second
solution was
prepared containing 3149g of Ni(NO3)2-6H20 dissolved in 2.7 L deionized water.
To this
nickel solution were added 1.2 L of 28 wt-% NH4OH solution followed by a
solution of
108 g maleic acid in 0.25 L deionized water. The nickel solution was then
added in 10
minutes to the molybdate/ tungstate solution while maintaining the temperature
at 77 C.
The resulting mixture was kept at 77 C and stirring continued for an hour. The
pH of the
suspension was in the range of 6-7. After addition of 0.72 L of a 7.0 wt-%
NH4OH
solution, and cooling to 60 C a blue-green precipitate was collected by
filtration and dried
by pressing it at 150 psi in a filter press. The collected and pressed
precipitate was aged
in a sealed container at 50 C for 15 hours. After ageing the precipitate was
mixed with 4
wt-% Methocel, and dried at 50 C until it exhibited a Loss On Ignition (LOI)
of 45 wt-%
and a carver of 1500 psi, and extruded in a Wolf screw extruder with NAQ dies.
[067] The N2 adsorption desorption isotherm of the precursor is shown in
Figures 2-3, composed of a well defined hysteresis loop corresponding to the
presence of
mesoporosity. Other pore characteristics of the precursor include: Sample
density of 1
gfm3. Surface area characteristics including single point surface area at Pir
= 0.20 of
110.4051 m2/g; BET surface area of 112.5688 m2./g; BJH (Barret-Joyner-Halenda)
adsorption cumulative surface area of pores between 17 and 3000 angstrom width
of
CA 02759044 2011-10-17
WO 2010/127130
PCT/US2010/032989
73.137 m2/g; BJH desorption of 75.886 m2/g. Pore volume characteristics
include single
point desorption total pore volume of pores less than 2278 angstrom at PIP =
0.99 of
0.089960 cm3/g; BJH adsorption cumulative surface area of pores between 17 and
3000
angstrom width of 0.068115 cm3/g; and BJH desorption of 0.074654 em3/g. Pore
size
characteristics including desorption average pore width (4 V/A by BET) of
31.9662
angstrom; BJH adsorption average pore width of 37.254 angstrom; and BJH
desorption
average pore width of 39.350 angstrom.
[068] Example 2: Another embodiment of a Ni-Mo-W-maleate catalyst
precursor. A catalyst precursor of the formula (NH4) {[NI2.6 (OH)2.08
(C4I12042)o.061
(Moo.35W0.6504)2} was prepared as follows: 477.2g of ammonium heptamolybdate
(NH4)6Mo7024 -4H20 was dissolved in 2.9L of deionized water at room
temperature.
666.6g of ammonium metatungstate (NH4)6H2W120.40 -4.7H20 was dissolved in 0.67
L
water. The molybdate and tungstate solutions were added to 15.4 L deionized
water. To
this mixed molybdate and tungstate solution 1.9 L of a 7.0 wt-% NH4OH
(ammonia)
solution was added so as to reach a pH in the range of 9-10. After this, the
temperature
was increased to 76 C with constant stirring. A second solution was prepared
containing
1575g of Ni(NO3)2-6H20 dissolved in 1.5 L deionized water. The nickel solution
was then
added in 25 minutes to the molybdate/ tungstate solution while maintaining the
temperature at 76 C. The resulting mixture was kept at 76 C and stirred for
half an hour.
Subsequently, 95 g of maleic acid was added to the suspension and stirring was
continued
for another half an hour. The pH of the suspension was in the range of 5-6.
After cooling
to 60 C a blue-green precipitate was collected by filtration and dried by
pressing it for 30
minutes at 150 psi in a filter press. At a Loss On Ignition (LO!) of 50 wt-%
and a carver
of 800 psi, the precipitate was mixed with 4 wt-% MethoeelTM and extruded in a
Wolf
screw extruder with NAQ dies.
[069] The N2 adsorption desorption isotherm of the precursor of Example 2 is
shown in Figures 4-5, also composed of a well defined hysteresis loop
corresponding to
the presence of mcsoporosity. Other pore characteristics of this precursor
include:
Sample density of 1 g/cm3. Surface area characteristics including single point
surface
area at P/P = 0.20 of 56.1297 m2/g; BET surface area of 58.1421 m2/g; BJH
(Barret-
Joyner-Halenda) adsorption cumulative surface area of pores between 17 and
3000
angstrom width of 56.2515 m2/g; BJH desorption of 59.6379 m2/g. Pore volume
characteristics include single point desorption total pore volume of pores
less than 2008
16
CA 02759044 2011-10-17
WO 2010/127130
PCT/US2010/032989
angstrom at 13/13 = 0.99 of 0.149469 cm3/g; BJH adsorption cumulative surface
area of
pores between 17 and 3000 angstrom width of 0.145741 cm3/g; and BJH desorption
of
0.148929 cm/g. Pore size characteristics including desorption average pore
width (4
V/A by BET) of 102.8301 angstrom; BJH adsorption average pore width of 103.635
angstrom; and BJH desorption average pore width of 99.889 angstrom.
[070] Example 3 ¨ A third embodiment of a Ni-Mo-W-maleate catalyst
precursor. A catalyst precursor of the formula (NH4) {[Ni2.6 (OH)2 (C4H2042
)0.061
AMoo.35%..6504)2} was prepared as follows: 954.4g of ammonium heptamolybdate
(NH4)6Mo7024 '4H20 was dissolved in 5.8L of dcionized water at room
temperature.
1333g of ammonium metatungstate (Nf14)6H2W12040 .4.7H20 was dissolved in 1.3 L
water. The molybdatc and tungstatc solutions were added to 15.0 L deionized
water. To
this mixed molybdate and tungstate solution 5.0 L of a 7.0 wt-% N H4OH
(ammonia)
solution was added until the pH reached 9.8. A second solution was prepared
containing
2835g of Ni(NO3)2.6H20 dissolved in 6.38 L deionized water. A third solution
was
prepared by dissolving 284.9 g Ni(SO4)=6H20 in 2.0 L water, and by
subsequently
adjusting the pH to 1.0 with concentrated sulfuric acid. After combination of
the two
nickel solutions, 110.0 g maleic acid dissolved in 0.60 L water was added to
the nickel
solution. The mixed molybdatc/tungstate solution was combined with the nickel
solution
through an in-line, high-shear mixer which discharged the combined solution
into 9.78 L
deionized water. The resulting suspension was continuously stirred and
maintained at 77
C. The pH of this suspension was raised to 6.5 through addition of an 7.0 wt-%
NH4OH
solution, and aged for 90 minutes with continuous stirring at 77 C. A blue-
green
precipitate was collected by filtration and dried at 115 C until Carver of
5000 psi.
Subsequently the paste was wetted to a Carver of 1500 psi, 4 wt-% Methoceffm
was
added, and the paste was extruded in a Wolf screw extruder with NAQ dies.
[071] Figure 6 is a graph showing the isotherms of catalyst precursor prepared
in
Example 3, which do not fall into the pattern of a type IV isotherms.
[072] For the purposes of this specification and appended claims, unless
otherwise indicated, all numbers expressing quantities, percentages or
proportions, and
other numerical values used in the specification and claims, are to be
understood as being
modified in all instances by the term "about." Accordingly, unless indicated
to the
contrary, the numerical parameters set forth in the following specification
and attached
claims are approximations that can vary depending upon the desired properties
sought to
17
CA 02759044 2017-01-24
be obtained by the present invention. It is noted that, as used in this
specification and the
appended claims, the singular forms "a," "an," and "the," include plural
references unless
expressly and unequivocally limited to one referent. As used herein, the term
"include"
and its grammatical variants are intended to be non-limiting, such that
recitation of items
in a list is not to the exclusion of other like items that can be substituted
or added to the
listed items.
[073] This written description uses examples to disclose the invention,
including
the best mode, and also to enable any person skilled in the art to make and
use the
invention. The patentable scope is defined by the claims, and can include
other examples
that occur to those skilled in the art. Such other examples are intended to be
within the
scope of the claims if they have structural elements that do not differ from
the literal
language of the claims, or if they include equivalent structural elements with
insubstantial
differences from the literal languages of the claims.
18