Note: Descriptions are shown in the official language in which they were submitted.
CA 02759196 2011-10-17
WO 2010/131035 PCT/GB2010/050772
-1-
NOVEL CRYSTALLINE POLYMORPH OF SITAGLIPTIN
DIHYDROGEN PHOSPHATE
Field of the invention
The present invention relates to a novel anhydrous crystalline form of
sitagliptin
dihydrogenphosphate (I), to processes for its preparation and to its use in
pharmaceutical
compositions.
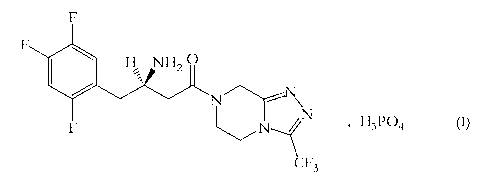
F
F
H NI-12
N
N--// N H3P04 (I)
CF3
Background of the invention
The manufacturing process for many pharmaceuticals is hindered by the fact
that the
organic compound, which is the active pharmaceutical ingredient (API), has
handling
difficulties during the manufacturing process and may impart undesirable
properties to the
final drug or dosage form. In addition it can be difficult to control the
polymorphic form
of the API throughout the manufacturing process.
For pharmaceuticals in which the API can exist in more than one polymorphic
form, it is
particularly important to ensure that the manufacturing process for the API
affords a
single, pure polymorph with a consistent level of polymorphic purity. If the
manufacturing
process leads to a polymorph with varying degrees of polymorphic purity and/or
where the
process does not control polymorphic interconversion, serious problems in
dissolution
and/or bioavailability can result in the finished pharmaceutical composition
comprising the
API.
CA 02759196 2011-10-17
WO 2010/131035 PCT/GB2010/050772
-2-
Sitagliptin dihydrogenphosphate, represented by structural formula (I), is
chemically named
as (2R)-4-oxo-4-[3-(trifluoromethyl)-5,6-dihydro[1,2,4]triazolo[4,3-a]pyrazin-
7(8H)-yl]-1-
(2,4,5-trifluorophenyl)butan-2-amine dihydrogenphosphate.
Sitagliptin is an oral antihyperglycemic of the dipeptidyl peptidase-IV (DPP-
IV) inhibitor
class. Inhibition of dipeptidyl peptidase-IV, an enzyme that inactivates both
glucose-
dependent insulinotropic peptide (GIP) and glucagon-like peptide 1 (GLP-1),
represents a
recent approach to the treatment and prevention of type-2 diabetes, also known
as non-
insulin dependent diabetes mellitus (NIDDM).
Therefore the novel crystalline form of the present invention can be used for
the
preparation of pharmaceutical compositions for the treatment and prevention of
diseases
and conditions for which an inhibitor of dipeptidyl peptidase-IV is indicated,
in particular
type-2 diabetes, hyperglycemia, insulin resistance, obesity, and high blood
pressure. The
novel crystalline form of the present invention can be used in combination
with one or
more other active ingredients if necessary.
Various structural analogues and salts of sitagliptin are disclosed in patent
US 6,699,871,
but no polymorphic data is given.
Solvate forms and three anhydrate polymorphs of sitagliptin
dihydrogenphosphate (forms
I, II and III) are disclosed in patent application US 2006/0287528. However,
desolvated
form II converts spontaneously to form I or III or a mixture thereof.
A process for the preparation of sitagliptin dihydrogenphosphate is disclosed
in patent
application US 2005/0032804 wherein the salt is prepared in isopropyl alcohol
and water to
afford sitagliptin dihydrogenphosphate monohydrate. However, this monohydrate
form
converts to an unstable dehydrated form at temperatures above 40 C.
In addition, crystalline anhydrate form IV is disclosed in patent application
US
2007/0021430, which is prepared from the sitagliptin dihydrogenphosphate
monohydrate
by heating at 120 C for 2 hours. However, form IV is metastable and converts
into the
CA 02759196 2011-10-17
WO 2010/131035 PCT/GB2010/050772
-3-
crystalline monohydrate slowly under ambient conditions or rapidly under high
relative
humidity.
An amorphous form of sitagliptin dihydrogenphosphate salt is disclosed in
patent
application US 2007/0281941. However amorphous forms are not ideally suited
for
commercial production and crystalline forms are generally preferred.
As discussed above, the six polymorphic forms of sitagliptin
dihydrogenphosphate
disclosed in the prior art suffer from several disadvantages which do not make
them ideal
forms for pharmaceutical development. In particular, the disadvantages
associated with the
prior art forms can include discolouration, polymorphic impurities and
instability. The
processes to prepare the respective prior art polymorphs suffer from the
disadvantages of
being inconsistent and difficult to reproduce. Consequently, the prior art
processes can
often produce polymorphically impure products. In addition, the prior art
processes are
particularly inconvenient for large scale production.
If crystalline and amorphous forms are made with polymorphic impurities, this
causes
instability and it can accelerate significant interconversion to another
polymorphic form.
Therefore, for commercial production, it is crucial to produce forms,
particularly crystalline
forms, with very high polymorphic purity to avoid or minimize this
interconversion.
In view of the importance acquired by sitagliptin for the treatment of
diabetes, there is a
great need for developing an alternative, relatively simple, economical and
commercially
feasible process for the synthesis of sitagliptin crystalline forms with
commercially
acceptable yield, high polymorphic purity and polymorphic stability.
Object of the invention
Therefore an object of the invention is to provide a new polymorphic form of
sitagliptin
dihydrogenphosphate, which is convenient to manufacture and has improved
properties
suitable for a marketed pharmaceutical composition.
Summary of the invention
CA 02759196 2011-10-17
WO 2010/131035 PCT/GB2010/050772
-4-
The term "sitagliptin" as used herein throughout the description and claims
means
sitagliptin and/or any salt, hydrate, solvate or tautomer thereof unless
specified otherwise.
A first aspect of the present invention provides sitagliptin
dihydrogenphosphate form M,
characterised by an XRPD spectrum comprising the following degrees 20 peaks:
5.0, 14.3,
18.6, 24.0 0.2 degrees 20. Preferably sitagliptin dihydrogenphosphate form M
is
characterised by an XRPD spectrum comprising four or more (preferably five or
more,
preferably six or more, preferably seven or more, preferably eight or more,
preferably nine
or more, preferably ten or more, preferably eleven or more, preferably twelve
or more,
preferably thirteen or more, preferably fourteen or more, preferably fifteen)
of the
following degrees 20 peaks: 5.0, 9.7, 13.7, 14.3, 15.4, 18.6, 19.5, 19.7,
20.3, 22.4, 24.0, 24.5,
25.7, 27.0, 27.3 0.2 degrees 20.
Preferably, the first aspect of the present invention provides sitagliptin
dihydrogenphosphate form M, characterised by an XRPD spectrum substantially
comprising the following degrees 20 peaks ( 0.2 degrees 20):
values d-values Intensity %
5.0 17.68 100.0
9.7 9.13 17.8
13.7 6.41 25.9
14.3 6.16 56.7
15.4 5.72 40.8
18.6 4.75 84.2
19.5 4.53 46.7
19.7 4.49 40.4
20.3 4.35 31.6
22.4 3.95 41.0
24.0 3.70 67.3
24.5 3.61 49.3
25.7 3.45 44.0
27.0 3.29 43.6
27.3 3.25 44.3
CA 02759196 2011-10-17
WO 2010/131035 PCT/GB2010/050772
-5-
Preferably, the first aspect of the present invention provides sitagliptin
dihydrogenphosphate form M, characterised by an XRPD spectrum substantially as
shown
in Figure 1.
Preferably, the first aspect of the present invention provides sitagliptin
dihydrogenphosphate form M, characterised by a DSC thermogram with an
endothermic
peak at about 216.3 2.0 C, preferably characterised by a DSC thermogram
with an
endothermic peak at about 216.3 1.0 C.
Preferably, the first aspect of the present invention provides sitagliptin
dihydrogenphosphate form M, characterised by a DSC thermogram substantially as
shown
in Figure 2.
Preferably, the first aspect of the present invention provides sitagliptin
dihydrogenphosphate form M, characterised by a TGA curve substantially as
shown in
Figure 3.
A second aspect of the present invention provides a process for the
preparation of
sitagliptin dihydrogenphosphate form M, comprising contacting sitagliptin base
with
orthophosphoric acid at -10 to 100 C in an organic solvent and crystallisation
of the
resultant product.
Preferably, the organic solvent is an alcohol, more preferably the alcohol is
a C1 to C6
alcohol which can be either straight chain, branched or cyclic. Preferably,
the alcohol is
selected from methanol, ethanol, 1-propanol, 2-propanol, n-butanol, 2-butanol,
tert-
butanol, 2-pentanol, 3-pentanol, 4-penten-2-ol, 1,6-hexanediol, 1-hexanol, 5-
hexen-l-ol,
glycerol, 1-heptanol, 2-heptanol, 1-octanol, 2-octanol, 3-octanol or mixtures
thereof. Most
preferably, the alcohol is 2-propanol.
Preferably, in a process according to the second aspect of the invention, the
organic solvent
is mixed with water. Preferably, the amount of water is less than 30% v/v with
respect to
the organic solvent, more preferably less than 20% v/v with respect to the
organic solvent,
preferably less than 10% v/v with respect to the organic solvent, preferably
less than 5%
CA 02759196 2011-10-17
WO 2010/131035 PCT/GB2010/050772
-6-
v/v with respect to the organic solvent. Most preferably, the amount of water
is about 15-
20% v/v with respect to the organic solvent.
Most preferably, the solvent is a mixture of 2-propanol and water.
Preferably, in a process according to the second aspect of the invention, 1 to
10 molar
equivalents of orthophosphoric acid is used. More preferably, 1.5 to 3.0 molar
equivalents
of orthophosphoric acid is used.
A third aspect of the present invention provides sitagliptin
dihydrogenphosphate form M
as prepared by a process according to the second aspect of the present
invention.
Preferably, the sitagliptin dihydrogenphosphate form M of the first or third
aspect of the
present invention comprises less than:
(i) 10% of sitagliptin dihydrogenphosphate in other polymorphic forms; and/or
(ii) 5% of sitagliptin dihydrogenphosphate in other polymorphic forms; and/or
(iii) 1% of sitagliptin dihydrogenphosphate in other polymorphic forms; and/or
(iv) 0.5% of sitagliptin dihydrogenphosphate in other polymorphic forms;
and/or
(v) 0.2% of sitagliptin dihydrogenphosphate in other polymorphic forms; and/or
(vi) 0.1% of sitagliptin dihydrogenphosphate in other polymorphic forms;
as measured by - RPD.
A fourth aspect of the present invention provides sitagliptin
dihydrogenphosphate form M
comprising less than 10% of sitagliptin dihydrogenphosphate in other
polymorphic forms
(as measured by XRPD). Preferably, the sitagliptin dihydrogenphosphate form M
comprises less than 5% of sitagliptin dihydrogenphosphate in other polymorphic
forms,
more preferably less than 1% of sitagliptin dihydrogenphosphate in other
polymorphic
forms, more preferably less than 0.5% of sitagliptin dihydrogenphosphate in
other
polymorphic forms, more preferably less than 0.2% of sitagliptin
dihydrogenphosphate in
other polymorphic forms, and most preferably less than 0.1% of sitagliptin
dihydrogenphosphate in other polymorphic forms (as measured by 1XRPD).
CA 02759196 2011-10-17
WO 2010/131035 PCT/GB2010/050772
-7-
Preferably, the sitagliptin dihydrogenphosphate form M of the first, third or
fourth aspect
of the present invention has a chemical purity of 99% or more, 99.5% or more,
or 99.85%
or more (as measured by HPLC).
Preferably, the sitagliptin dihydrogenphosphate form M of the first, third or
fourth aspect
of the present invention is suitable for use in medicine, preferably for
treating or preventing
a disease or condition for which an inhibitor of dipeptidyl peptidase-IV is
effective,
preferably for treating or preventing diabetes, hyperglycemia, insulin
resistance, obesity, or
high blood pressure, preferably for treating or preventing diabetes type-2.
A fifth aspect of the present invention provides a pharmaceutical composition,
comprising
the sitagliptin dihydrogenphosphate form M according to the first, third or
fourth aspect of
the present invention or as prepared by a process according to the second
aspect of the
present invention.
A sixth aspect of the present invention provides a use of the sitagliptin
dihydrogenphosphate form M according to the first, third or fourth aspect of
the present
invention or as prepared by a process according to the second aspect of the
present
invention or a use of the pharmaceutical composition according to the fifth
aspect of the
present invention, in the manufacture of a medicament for the treatment or
prevention of a
disease or condition for which an inhibitor of dipeptidyl peptidase-IV is
effective.
Preferably, the medicament is for the treatment or prevention of diabetes,
hyperglycemia,
insulin resistance, obesity, or high blood pressure. More preferably, the
medicament is for
the treatment or prevention of diabetes type-2.
A seventh aspect of the present invention provides a method of treating or
preventing a
disease or condition for which an inhibitor of dipeptidyl peptidase-IV is
effective, the
method comprising administering to a patient in need thereof a therapeutically
or
prophylactically effective amount of the sitagliptin dihydrogenphosphate form
M according
to the first, third or fourth aspect of the present invention or as prepared
by a process
according to the second aspect of the present invention or a therapeutically
or
prophylactically effective amount of the pharmaceutical composition according
to the fifth
aspect of the present invention. Preferably, the method is for the treatment
or prevention
CA 02759196 2011-10-17
WO 2010/131035 PCT/GB2010/050772
-8-
of diabetes, hyperglycemia, insulin resistance, obesity, or high blood
pressure. More
preferably, the method is for the treatment or prevention of diabetes type-2.
Preferably the
patient is a mammal, preferably a human.
In the use according to the sixth aspect of the present invention or in the
method
according to the seventh aspect of the present invention, optionally the
sitagliptin
dihydrogenphosphate form M is used in combination with one or more other
active
pharmaceutical ingredients, wherein the other active ingredient(s), which may
be
administered separately or in the same pharmaceutical composition, may be
selected from
insulin sensitizers such as glitazones (such as troglitazone, pioglitazone,
englitazone and
roogglitazone); fenofibric acid derivatives (such as gemfibrozil, clofibrate,
fenofibrate and
bezafibrate); biguanides (such as metformin and phenformin); sulfonylureas
(such as
glipizide); or mixtures thereof.
Brief description of the accompanying figures
Figure 1 shows an X-ray powder diffraction X PD) spectrum of sitagliptin
dihydrogenphosphate form M.
Figure 2 shows a differential scanning calorimetry (DSC) thermogram of
sitagliptin
dihydrogenphosphate form M.
Figure 3 shows a thermo-gravimetric analysis (TGA) curve of sitagliptin
dihydrogenphosphate form M.
Detailed description of the invention
As outlined above, the present invention provides a new crystalline form of
sitagliptin
dihydrogenphosphate, form M, which is non-hygroscopic, polymorphically pure
and stable,
and has beneficial properties which avoid the problems associated with prior
art forms.
A major advantage of this invention is the reproducible conditions of the
process to obtain
the novel polymorph and the polymorphic purity and stability of the form M.
The
CA 02759196 2011-10-17
WO 2010/131035 PCT/GB2010/050772
-9-
polymorphic form of the present invention also allows sitagliptin
dihydrogenphosphate to
be easily purified and obtained in very high chemical purity.
The present inventors have surprisingly found that, if required, crystalline
sitagliptin
dihydrogenphosphate form M can be conveniently used to prepare other known
crystalline
forms of sitagliptin or salts of sitagliptin, such as sitagliptin
dihydrogenphosphate, with very
high chemical and/or polymorphic purity.
Sitagliptin dihydrogenphosphate form M can be prepared by reacting sitagliptin
free base
and orthophosphoric acid in an organic solvent optionally in the presence of
water. The
orthophosphoric acid is used preferably at 1.0 to 3.0 molar equivalents and
more preferably
at about 1.5 molar equivalents. The orthophosphoric acid can be used in solid
form or as a
solution, such as an aqueous solution or a solution in an alcohol such as 2-
propanol.
The organic solvent may be a protic or aprotic solvent. Preferably the organic
solvent is an
alcohol, a ketone, an ether, an alkane, a cycloalkane, a formamide, an
acetate, a halogenated
solvent, or a mixture thereof, optionally in the presence of water.
Preferably, the organic solvent is an alcohol, preferably a straight chain,
branched or cyclic
C1 to C6 alcohol. More preferably, the alcohol is selected from one or more of
methanol,
ethanol, 1-propanol, 2-propanol, n-butanol, 2-butanol, tert-butanol, 2-
pentanol, 3-pentanol,
4-penten-2-ol, 1,6-hexanediol, 1-hexanol, 5-hexen-l-ol, glycerol, 1-heptanol,
2-heptanol, 1-
octanol, 2-octanol, or 3-octanol, preferably in presence of water. The most
preferred
solvent is 2-propanol in the presence of water.
Preferred embodiments of the process according to the present invention
involve
dissolution of sitagliptin free base in about 5 to 50 volumes of 2-propanol
and about 2 to
10 volumes of water to obtain a clear solution. Preferably, about 8 to 12 or
about 8 to 10
volumes of 2-propanol and about 2 to 4 volumes of water are used to obtain a
clear
solution. To the clear solution, orthophosphoric acid is preferably added at
about -10 to
85 C, preferably at about 15-30 C. The suspension or clear solution is
preferably stirred at
about 70-75 C for up to 10 hours until completion of the reaction. The
precipitated
product is preferably further stirred for 15 minutes to 12 hours at about 25-
30 C until
CA 02759196 2011-10-17
WO 2010/131035 PCT/GB2010/050772
-10-
complete precipitation. The precipitated product is preferably maintained at 0-
5 C for 1
hour and then preferably filtered at 0-5 C. The isolated product is preferably
dried under
reduced pressure at 45-50 C for 1-10 hours, more preferably for 1-3 hours. The
product
can be recrystallised if necessary.
Preferably, the mixture is heated to dissolve the sitagliptin. Preferably, the
mixture is heated
between 40-100 C, more preferably at about 70-75 C.
Preferably, the mixture is cooled before isolation of the sitagliptin
dihydrogenphosphate
form M. Preferably, the mixture is cooled to between -5 to 30 C, more
preferably to about
0-5 C.
The crystalline sitagliptin dihydrogenphosphate form M preferably comprises
less than
0.2% of sitagliptin dihydrogenphosphate in other polymorphic forms.
The crystalline sitagliptin dihydrogenphosphate form M obtained is preferably
dried under
vacuum until a constant weight is obtained. Preferably, the crystalline form M
obtained is
dried until the moisture content falls below 1%, preferably to below about
0.5%.
As discussed above, sitagliptin dihydrogenphosphate monohydrate is disclosed
in patent
application US 2005/0032804. However, this monohydrate form converts to an
unstable
dehydrated form at temperatures above 40 C. The present inventors have
surprisingly
found that by reducing the amount of water used in the organic solvent medium,
a novel,
stable, anhydrous, solvent-free, crystalline form M can be reproducibly
prepared. A solvent
mixture comprising a ratio of 10:2 v/v 2-propanol:water (about 16% water v/v),
afforded
exclusively novel anhydrous form M. Even the wet cake obtained by following
the process
of the present invention exhibited an XRPD pattern identical to that of form
M.
The XRPD d-values of the sitagliptin dihydrogenphosphate form M thus obtained
are
different from those of the reported forms I to IV and the reported
monohydrate form and
this 1ZRPD data is summarised in Table 2.
CA 02759196 2011-10-17
WO 2010/131035 PCT/GB2010/050772
-11-
Table 2: XRPD d-values of prior art forms and form M
Form I Form II Form III Form IV Monohydrate Form M
2.66
2.71
3.04
3.25
3.29
3.45 3.45
3.52
3.61
3.67
3.69
3.70
3.73
3.95
3.96
3.99
4.02
4.19
4.26
4.30
4.35
4.46
4.48
4.49
4.53
4.55
4.65
4.71
4.75 4.75
5.08
5.21
5.27
5.48
5.71
5.72
5.78
5.82
5.85
6.06
6.16 6.16
CA 02759196 2011-10-17
WO 2010/131035 PCT/GB2010/050772
-12-
Form I Form II Form III Form IV Monohydrate Form M
6.26
6.30
6.32
6.41
6.50
7.09
7.42
7.95
9.06
9.13
9.35
9.43
13.69
17.68
17.88
17.94
18.42
18.56
Typical XRPD 20 values and intensity of form M are illustrated in Table 3.
Table 3: XRPD 20 values and intensity of form M
No. 20 values d-spacing Intensity %
1 5.0 17.68 100.0
2 9.7 9.13 17.8
3 13.7 6.41 25.9
4 14.3 6.16 56.7
15.4 5.72 40.8
6 18.6 4.75 84.2
7 19.5 4.53 46.7
8 19.7 4.49 40.4
9 20.3 4.35 31.6
22.4 3.95 41.0
11 24.0 3.70 67.3
12 24.5 3.61 49.3
13 25.7 3.45 44.0
14 27.0 3.29 43.6
27.3 3.25 44.3
CA 02759196 2011-10-17
WO 2010/131035 PCT/GB2010/050772
-13-
The pharmaceutical composition according to the fifth aspect of the present
invention can
be a solution or a suspension, but is preferably a solid oral dosage form.
Preferred oral
dosage forms in accordance with the invention include tablets, capsules and
the like which,
optionally, may be coated if desired. Tablets can be prepared by conventional
techniques,
including direct compression, wet granulation and dry granulation. Capsules
are generally
formed from a gelatine material and can include a conventionally prepared
granulate of
excipients in accordance with the invention.
The pharmaceutical composition according to the present invention typically
comprises
one or more conventional pharmaceutically acceptable excipient(s) selected
from the group
comprising a filler, a binder, a disintegrant, a lubricant, and optionally
further comprises at
least one excipient selected from colouring agents, adsorbents, surfactants,
film-formers
and plasticizers.
If the solid pharmaceutical formulation is in the form of coated tablets, the
coating may be
prepared from at least one film-former such as hydroxypropyl methyl cellulose,
hydroxypropyl cellulose or methacrylate polymers which optionally may contain
at least
one plasticizer such as polyethylene glycols, dibutyl sebacate, triethyl
citrate, and other
pharmaceutical auxiliary substances conventional for film coatings, such as
pigments and
fillers.
Preferably the pharmaceutical compositions according to the present invention
are in unit
dosage form comprising sitagliptin in an amount of from 1 mg to 500 mg, such
that the
amount of sitagliptin administered is from 0.1 mg to 100 mg per kg per day.
Preferably, the pharmaceutical compositions according to the fifth aspect of
the present
invention are for use in the treatment and prevention of diseases and
conditions for which
an inhibitor of dipeptidyl peptidase-IV is effective. Preferably, the use is
in the treatment of
diabetes, hyperglycemia, insulin resistance, obesity, or high blood pressure.
More
preferably, the use is in the treatment of diabetes type-2.
CA 02759196 2011-10-17
WO 2010/131035 PCT/GB2010/050772
-14-
The novel crystalline form of the present invention can be used in combination
with other
active ingredients. Examples of other active ingredients that may be
administered in
combination with the crystalline form of the present invention and either
administered
separately or in the same pharmaceutical composition, include but are not
limited to other
dipeptidyl peptidase IV (DP-IV) inhibitors; insulin sensitizers such as
glitazones (such as
troglitazone, pioglitazone, englitazone and rosiglitazone); fenofibric acid
derivatives (such
as gemfibrozil, clofibrate, fenofibrate and bezafibrate); biguanides (such as
metformin and
phenformin); sulfonylureas (such as glipizide); or mixtures thereof.
The details of the invention, its objects and advantages are illustrated below
in greater detail
by a non-limiting example.
Example
Sitagliptin Dihydrogenphosphate Form M
Sitagliptin free base (10.0 gm, 24.5 mmol) was charged in 2-propanol (80.0 ml,
8.0 vol) and
water (20.0 ml, 2.0 vol) at 25-30 C. The clear solution was stirred for 10
minutes.
Meanwhile, a clear solution of orthophosphoric acid (85%) was prepared at 25-
30 C [2.40
gm (1.0 eq, 24.5 mmol) in 20.0 ml (2.0 vol) of 2-propanol]. Slow addition of
the solution of
orthophosphoric acid to the solution of sitagliptin free base was carried out
at 25-30 C.
The addition was completed within 15-20 minutes. The thick solution was
further diluted
with 2-propanol (20.0 ml, 2.0 vol) and the white suspension was further heated
at 70-75 C
and maintained for 1 hour before the reaction mixture was allowed to cool
gradually at 25-
C within 1.5 hour. The white suspension was stirred at 25-30 C for 1 hour
until a white
25 thick solution was obtained. After 1 hour of stirring at 25-30 C, the
mixture was cooled to
0-5 C and maintained at 0-5 C for 1 hour. The product was filtered at 0-5 C
and washed
with 2-propanol (50.0 ml, 5.0 vol). The product was suction filter dried for
30 minutes and
then dried at 40-45 C at reduced pressure for 1-2 hours to obtain crude
sitagliptin
dihydrogenphosphate. XRPD, DSC and TGA analysis data confirmed that the crude
30 sitagliptin dihydrogenphosphate obtained is form M.
Molar yield: 81 % (10.0 gm)
Chemical purity: >99.50% (measured by HPLC)
Polymorphic purity: no levels of other polymorphic forms detected (measured by
XRPD)
CA 02759196 2011-10-17
WO 2010/131035 PCT/GB2010/050772
-15-
The crude sitagliptin dihydrogenphosphate (10.0 gm) was further crystallised
from a
mixture of 2-propanol (100.0 ml, 10.0 vol) and water (20.0 ml, 2.0 vol) to
obtain very pure
sitagliptin dihydrogenphosphate. XXRPD, DSC and TGA analysis data confirmed
that the
pure sitagliptin dihydrogenphosphate obtained is form M.
Molar yield: 85% (8.5 gm)
Chemical purity: >99.85% (measured by HPLC)
Polymorphic purity: no levels of other polymorphic forms detected (measured by
XRPD)
XRPD and DSC analysis data for the crude and pure products obtained from the
example
(see Figures 1 and 2) confirmed that the products obtained were a novel
polymorph of
sitagliptin dihydrogenphosphate. The novel polymorph obtained, crystalline
form M, was
substantially pure polymorphically with no levels of other forms detected.
TGA analysis data (see Figure 3) showed that the novel crystalline anhydrous
form of
sitagliptin dihydrogenphosphate form M displays no weight loss in the
temperature range
of 50 C to 225 C.
The XRPD was recorded on a Bruker D8 Advance Instrument, using copper
radiation as
the X-ray source and LynxEye as the detector, with a 20 range of from 3 to 50
, a step-
size of 0.05 and a time/step of 1 sec.
The DSC was recorded on a Perkin Elmer Pyris 6, with a temperature range of
from 25 C
to 250 C and a rate of heating of 10 C/min.
The TGA was recorded on a Perkin Elmer Pyris 1, with a temperature range of
from 25 C
to 250 C and a rate of heating of 10 C/min.
The crude and pure products were also subjected to two months accelerated
stability
studies, monitoring chemical and polymorphic purities, and it was found that
the sitagliptin
dihydrogenphosphate form M is stable. The accelerated stability studies were
performed as
follows. Sitagliptin dihydrogenphosphate form M was kept in a single
polyethylene bag in a
triple laminated aluminium pouch. This pouch was kept in a HDPE container.
This
CA 02759196 2011-10-17
WO 2010/131035 PCT/GB2010/050772
-16-
container was kept in a stability chamber at a temperature of 40 C 2 C and a
relative
humidity of 75% 5% for two months. The sitagliptin dihydrogenphosphate form
M was
found to be very stable chemically and very stable polymorphically with no
conversion over
time to other polymorphs.
It will be understood that the present invention has been described above by
way of
example only. The example is not intended to limit the scope of the invention.
Various
modifications and embodiments can be made without departing from the scope and
spirit
of the invention, which is defined by the following claims only.
>0